

Abstract
Background
Skeletal muscle atrophy is defined as a reduction of muscle mass caused by excessive protein degradation. However, the development of therapeutic interventions is still in an early stage. Although glucagon‐like peptide‐1 receptor (GLP‐1R) agonists, such as exendin‐4 (Ex‐4) and dulaglutide, are widely used for the treatment of diabetes, their effects on muscle pathology are unknown. In this study, we investigated the therapeutic potential of GLP‐1R agonist for muscle wasting and the mechanisms involved.
Methods
Mouse C2C12 myotubes were used to evaluate the in vitro effects of Ex‐4 in the presence or absence of dexamethasone (Dex) on the regulation of the expression of muscle atrophic factors and the underlying mechanisms using various pharmacological inhibitors. In addition, we investigated the in vivo therapeutic effect of Ex‐4 in a Dex‐induced mouse muscle atrophy model (20 mg/kg/day i.p.) followed by injection of Ex‐4 (100 ng/day i.p.) for 12 days and chronic kidney disease (CKD)‐induced muscle atrophy model. Furthermore, we evaluated the effect of a long‐acting GLP‐1R agonist by treatment of dulaglutide (1 mg/kg/week s.c.) for 3 weeks, in DBA/2J‐mdx mice, a Duchenne muscular dystrophy model.
Results
Ex‐4 suppressed the expression of myostatin (MSTN) and muscle atrophic factors such as F‐box only protein 32 (atrogin‐1) and muscle RING‐finger protein‐1 (MuRF‐1) in Dex‐treated C2C12 myotubes. The suppression effect was via protein kinase A and protein kinase B signalling pathways through GLP‐1R. In addition, Ex‐4 treatment inhibited glucocorticoid receptor (GR) translocation by up‐regulating the proteins of GR inhibitory complexes. In a Dex‐induced muscle atrophy model, Ex‐4 ameliorated muscle atrophy by suppressing muscle atrophic factors and enhancing myogenic factors (MyoG and MyoD), leading to increased muscle mass and function. In the CKD muscle atrophy model, Ex‐4 also increased muscle mass, myofiber size, and muscle function. In addition, treatment with a long‐acting GLP‐1R agonist, dulaglutide, recovered muscle mass and function in DBA/2J‐mdx mice.
Conclusions
GLP‐1R agonists ameliorate muscle wasting by suppressing MSTN and muscle atrophic factors and enhancing myogenic factors through GLP‐1R‐mediated signalling pathways. These novel findings suggest that activating GLP‐1R signalling may be useful for the treatment of atrophy‐related muscular diseases.
Keywords: Skeletal muscle atrophy, GLP‐1R agonists, Dexamethasone, Glucocorticoid receptor, Chronic kidney disease, Duchenne muscular dystrophy
Introduction
Skeletal muscle atrophy is a pathological condition caused by excessive muscle protein degradation by diverse factors: genetic factors, various diseases, disuse, and aging. Patients either with chronic kidney disease (CKD) or cancer or who are getting older are highly susceptible to the development of skeletal muscle atrophy.1, 2, 3, 4, 5 It disrupts the quality of life and increases mortality and morbidity.6 Thus, the demand for the development of specialized medicine for muscle atrophy has been increasing and becoming an important research topic worldwide. The understanding of molecular mechanisms implicated in muscle atrophy has substantially progressed for the last few decades; however, there are still no effective pharmacological and therapeutic approaches up to now, except for exercise regimens.1 In addition, the use of exercise regimen is severely limited in the elderly or bed‐ridden population or those with acute illnesses.1 Currently, myostatin (MSTN) and activin A antagonists are considered the most promising candidates for the treatment of muscle atrophy, but they still have obstacles (such as off‐targeting to other transforming growth factor family members) to overcome for use on human patients.1, 7, 8
Glucocorticoid (GC) is implicated in protein and glucose metabolism in skeletal muscle.9, 10, 11 Circulating GC levels increase during fasting/starvation (catabolic conditions) or in various diseases (e.g. CKD, Cushing's syndrome, and cancer) and in the elderly. It is a known risk factor for the development of muscle atrophy.1, 11 Chronic or excessive exposure to GC induces muscle atrophy by up‐regulating the expression of MSTN and muscle‐specific E3 ubiquitin ligases such as F‐box only protein 32 (atrogin‐1) and muscle RING‐finger protein‐1 (MuRF‐1).11
Glucagon‐like peptide‐1 (GLP‐1) is a 30‐amino‐acid peptide hormone synthesized and secreted from the intestinal endocrine L‐cells. It has numerous physiological actions through its receptor, GLP‐1R, including promoting glucose‐induced insulin secretion, elevating β‐cell survival, inhibiting glucagon production, and delaying gastric emptying.12, 13, 14 Therefore, GLP‐1‐based drugs have been developed as an anti‐diabetic therapy. Although some studies have implicated the possible effects of GLP‐1‐based drugs on muscle mass, metabolism, and function, little is known about the precise mechanisms underlying its effects.15, 16, 17 Therefore, we investigated whether GLP‐1‐based drugs have a therapeutic effect in muscle wasting.
In the current study, we examined whether a GLP‐1R agonist, exendin‐4 (Ex‐4), regulates muscle atrophic and myogenic factors and, if so, what mechanisms are involved. In addition, we also investigated whether GLP‐1R agonists attenuate muscle atrophy in different types of in vivo models of muscle atrophy.
Materials and methods
Animals
In order to generate a dexamethasone (Dex)‐induced muscle atrophy mouse model, we administered Dex (D4902, Sigma‐Aldrich, MO, USA) intraperitoneally at 20 mg/kg/day to 10‐week‐old C57BL/6 male mice (Orient Bio Inc, Gyeonggi, Korea) for 8 days followed by injection of Ex‐4 (1933, Tocris Bioscience, Bristol, UK) at 100 ng/day for 12 days. To generate a CKD‐induced muscle atrophy model, 10‐week‐old C57BL/6 mice with similar body weights underwent subtotal nephrectomy in two stages, as described previously.18 Briefly, in the first stage, ~70% of the right kidney was removed. Seven days later, the entire left kidney was removed. CKD developed in 2 weeks, as shown by the increased urea nitrogen and creatinine in blood, indicating kidney dysfunction. Sham mice underwent surgery without damaging the kidneys. Ex‐4 (100 ng) or an equal amount of phosphate‐buffered saline (PBS) was injected intraperitoneally every day for 8 weeks. DBA/2J‐mdx mice (Duchenne muscular dystrophy model) were purchased from the Jackson Laboratory (MA, USA). Dulaglutide (Trulicity®, Eli Lilly and Company, IN, USA) was injected subcutaneously at 1 mg/kg into 7‐week‐old male DBA/2J‐mdx mice once a week for 3 weeks. Body weight and food intake were assessed. All mice were housed at ~23 ± 1°C with 12 h light/dark cycles with free access to water and a normal diet. All animal experiments were performed according to the animal protocols approved by the Institutional Animal Care and Use Committee at Gachon University.
To verify the expression of GLP‐1R in muscle tissue, we purchased floxed Glp1r mice (#09030) on a C57BL/6 background from the European Mouse Mutant Archive (MRC, UK), which allowed Cre‐mediated recombination of GLP‐1R. We crossed the floxed Glp1r mice with the Cre‐expressing mouse line, β‐actin Cre, to obtain wild‐type (Glp1r +/+ Cre −) and heterozygous (Glp1r flox/+ Cre +) mice. These heterozygous (Glp1r flox/+ Cre +) mice were mated to obtain homozygous (Glp1r flox/flox Cre +) mice.
Cell culture
C2C12 cells (CRL‐1772, ATCC®, VA, USA) were grown in Dulbecco's modified Eagle's medium (LM001‐05, Welgene, Daegu, Korea) supplemented with 10% foetal bovine serum (S001‐07, Welgene, Daegu, Korea), 0.2 mM glutamine, 100 IU/mL penicillin, and 0.1 mg/mL streptomycin. To differentiate myoblasts from myotubes, C2C12 cells were seeded at 1 × 106 cells per 6‐well plate, and then the medium was replaced with a differential medium containing 2% horse serum (16050‐122, Gibco, UK), 100 IU/mL penicillin, and 0.1 mg/mL streptomycin for 5 days. In order to determine whether Ex‐4 acts through GLP‐1R, the C2C12 myotubes were pretreated with 20 nM exendin‐9 (Ex‐9), GLP‐1R antagonist (E7269, Sigma‐Aldrich, MO, USA) for 10 min and then treated with 20 nM Ex‐4 for 30 min. To further investigate the molecular mechanism of Ex‐4 action, C2C12 myotubes were first pretreated with 1 μM of Dex 6 h before treatment with 20 nM Ex‐4, 20 nM MG132 (C2211, Sigma‐Aldrich, MO, USA), or 20 nM RU486 (M8046, Sigma‐Aldrich, MO, USA) for further 6 h.
Transfection
C2C12 cells were seeded in 6‐well plates with 1 × 106 cells per well and then differentiated into myotubes for 5 days using a differentiation medium. At 4 days during differentiation, the cells were transiently transfected with siRNA control (Bioneer, Daejeon, Korea) or siRNA GLP‐1R (Bioneer, Daejeon, Korea) using Lipofectamine® RNAiMAX reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. After 6 h of transfection, the medium was replaced with differentiation medium, and then the cells were kept until 5 days. The cells were treated 20 nM Ex‐4 for 30 min at 5 days.
Reverse transcriptase‐quantitative polymerase chain reaction
Total RNA was isolated from muscle tissue and C2C12 myotubes with RNAiso (9109, Takara, Japan) reagent according to the manufacturer's protocol. The cDNA was synthesized from 2 μg total RNA using the PrimeScript 1st strand cDNA synthesis kit (6110A, Takara, Japan). Quantitative real‐time PCR (qRT‐PCR) was performed using CFX384 Touch™ Real‐Time PCR Detection System (Bio‐Rad, USA). The relative gene expression levels were normalized by cyclophilin RNA expression. The primer sequences used are shown in Table 1.
Table 1.
The list of primers sequences
| No. | Gene symbol | Sequences | |
|---|---|---|---|
| 1 | Mouse cyclophilin | Sense | 5′ TGGAGAGCACCAAGACAGACA 3′ |
| Anti‐sense | 3′ TGCCGGAGTCGACAATGAT 5′ | ||
| 2 | Mouse MSTN | Sense | 5′ GGCCATGATCTTGCTGTAAC3′ |
| Anti‐sense | 3′ TTGGGTGCGATAATCCAGTC 5′ | ||
| 3 | Mouse atrogin‐1 | Sense | 5′ GCAAACACTGCCACATTCTCTC 3′ |
| Anti‐sense | 3′ CTTGAGGGGAAAGTGAGACG 5′ | ||
| 4 | Mouse MuRF‐1 | Sense | 5′ TGACCACAGAGGGTAAAG 3′ |
| Anti‐sense | 3′ TGTCTCACTCATCTCCTTCTTC 5′ | ||
| 5 | Mouse MyoD | Sense | 5′ CTTCTATCGCCGCCACTC 3′ |
| Anti‐sense | 3′ AAGTCGTCTGCTGTCTCAA 5′ | ||
| 6 | Mouse MyoG | Sense | 5′ CCAACCCAGGAGATCATTTG 3′ |
| Anti‐sense | 3′ ACGATGGACGTAAGGGAGTG 5′ | ||
| 7 | Mouse KLF15 | Sense | 5′ CACAAATGCACTTTCCCAGG 3′ |
| Anti‐sense | 3′ TTGACAACTCATCTGAGCGG 5′ | ||
| 8 | Mouse Sesn1 | Sense | 5′ TATGGCCATGCACAAAGATG 3′ |
| Anti‐sense | 3′ TTCCAAACATGCAGTGGATA 5′ | ||
| 9 | Mouse REDD1 | Sense | 5′ CCAGAGAAGAGGGCCTTGA 3′ |
| Anti‐sense | 3′ CCATCCAGGTATGAGGAGTCTT 5′ | ||
| 10 | Mouse p85α | Sense | 5′ CAAAGCGGAGAACCTATTGC 3′ |
| Anti‐sense | 3′ ATAGCAGCCCTGCTTACTGC 5′ | ||
| 11 | Mouse FoxO3a | Sense | 5′ GGAAATGGGCAAAGCAGA 3′ |
| Anti‐sense | 3′ AAACGGATCACTGTCCACTTG 5′ | ||
Western blotting
Total protein was isolated using mammalian protein extract buffer (28‐9712‐79, GE Life Sciences, NY, USA) containing a protease inhibitor cocktail (P8340, Sigma‐Aldrich, MO, USA). The cytosolic extract (CE) and nuclear extract (NE) were isolated from treated C2C12 myotubes as previously described.19 Western blotting was conducted with equal amounts of protein and reacted with the following antibodies: anti‐MSTN (ab203076, Abcam, MA, USA), anti‐atrogin‐1 (ab74023, Abcam, MA, USA), anti‐MuRF‐1 (ab172479, Abcam, MA, USA), anti‐actin (#8457, Cell Signaling Technology, MA, USA), anti‐lamin A/C (#4777, Cell Signaling Technology, MA, USA), anti‐GC receptor (GR) (ab55400, Abcam, MA, USA), anti‐heat shock protein (HSP)70 (ab2787, Abcam, MA, USA), anti‐HSP90 (ab13792, Abcam, MA, USA), anti‐FK506‐binding protein 4 (FKBP52) (ab59460, Abcam, MA, USA), anti‐prostaglandin E synthase 3 (p23) (sc‐376725, Santa Cruz Biotechnology, CA, USA), anti‐26S proteasome (ab182576, Abcam, MA, USA), anti‐GLP‐1R (sc‐390774, Santa Cruz Biotechnology, CA, USA), anti‐p‐protein kinase B (AKT) (#9271, Cell Signaling Technology, MA, USA), anti‐AKT (#9272, Cell Signaling Technology, MA, USA), anti‐p‐nuclear factor (NF)‐κB (#3033, Cell Signaling Technology, MA, USA), anti‐NF‐κB (sc‐372, Santa Cruz Biotechnology, CA, USA), anti‐p‐protein kinase A (PKA) (#4781, Cell Signaling Technology, MA, USA), anti‐PKA (sc‐903, Santa Cruz Biotechnology, CA, USA), anti‐heat shock factor (HSF)‐1 (sc‐17757, Santa Cruz Biotechnology, CA, USA), anti‐myosin heavy chain (MHC) (sc‐376157, Santa Cruz Biotechnology, CA, USA), anti‐tropomyosin (sc‐58868, Santa Cruz Biotechnology, CA, USA), and anti‐desmin (sc‐23879, Santa Cruz Biotechnology, CA, USA). The target binding was detected using the Chemidoc™ XRS+ system with Image Lab™ Software (#1708265, Bio‐Rad, CA, USA), and band density was quantified by the Image J program.
Measurement of protein synthesis
Protein synthesis was measured using the surface sensing of translation technique, as previously described.20 In brief, C2C12 cells were differentiated for 5 days. At 5 days, the cells were treated with or without 1 μM Dex for 6 h. After 6 h, 20 nM Ex‐4 was added and incubated for further 6 h, followed by 1 μM puromycin (P8833‐10MG; Sigma‐Aldrich, St Louis, MO, USA) incubation. Cycloheximide, protein synthesis inhibitor (CHX, C7698‐1G, Sigma‐Aldrich), was used as a negative control treatment. Puromycin‐labelled proteins were assessed by immunoblotting with an anti‐puromycin antibody (MABE343; Millipore, Burlington, MA, USA) using an equal amount of total protein per sample.
Immunofluorescent staining
Immunofluorescent (IF) staining was performed as previously described.21 Briefly, the cells were fixed in 100% methanol or 10% neutral buffered formalin (NBF) for 20 min at RT and washed with PBS. The fixed cells were permeabilized at 25°C with PBS containing 0.27% Triton X‐100 for all staining except GLP‐1R staining. Cells were incubated with protein blocking solution (Dako, CA, USA) at 25°C for 1 h. The cells were exposed to anti‐GR antibodies overnight at 4°C and then incubated with fluorescein isothiocyanate‐conjugated secondary antibody (sc‐2012, Santa Cruz Biotechnology, CA, USA) followed by 4′,6‐diamidino‐2‐phenylindole staining. Fluorescent images of the cells were taken using laser scanning confocal microscopy (A1 plus, Nikon, Tokyo, Japan).
Measurement of cyclic adenosine monophosphate production
C2C12 myotubes were pretreated with 20 nM Ex‐9 for 10 min and then treated with 20 nM Ex‐4 for 30 min. The cells were washed and incubated with 0.1 M HCl for 30 min at 25°C, and then the supernatant was harvested after centrifugation. The cyclic adenosine monophosphate (cAMP) levels were determined using a Direct cAMP ELISA kit (ADI‐901‐006, Enzo Life Sciences, Lausen, Switzerland) according to the manufacturer's protocol.
Immunoprecipitation assay
In order to perform the immunoprecipitation (IP) assay, cell lysates were first pre‐cleaned, and then 400 μg of protein was incubated with 2 μg of primary antibodies (anti‐GR antibody, anti‐HSP70 antibody, anti‐HSP90 antibody, anti‐FKBP52 antibody, or anti‐p23 antibody) overnight at 4°C. Subsequently, the cell lysates were further incubated with protein A/G‐plus agarose beads (sc‐2003, Santa Cruz Biotechnology, CA, USA) for 4 h at 4°C and then washed five times with mammalian protein extract buffer. The samples were stored at −80°C until use.
Grip strength/four‐limb hanging test
Limb grip strength was measured in mice using a grip strength meter (BIO‐G53, BIOSEB, FL, USA). Mice were lifted and held by their tail so that their limbs could grasp a wire grid. The mice were then gently pulled backward by the tail with their posture parallel to the surface of the table until they released the grid. The peak force applied by the limbs of the mouse was recorded in grams (g). Each mouse was tested three times, and the average value was used for statistical analysis. Four‐limb hanging tests were performed as previously described.22 Briefly, each mouse was placed on a grid so that it grasped the grid with its four paws. The grid was inverted, and then the time the mouse spent hanging was measured for up to 10 min. The longer trial was used for analysis.
Tissue collection
At the end of each experiment, mice were weighed and humanely sacrificed. Blood samples were collected immediately before sacrifice after 4 h of fasting. Subsequently, the serum was separated and stored at −80°C until use. Skeletal muscles and other tissues were weighed and stored at −80°C for further analysis. For histological analysis, some parts of muscle tissues were embedded in optimal cutting temperature compound (4583, Tissue‐Tek, CA, USA) or were fixed in 10% NBF.
Serum analysis
The levels of blood urea nitrogen (BUN) and creatinine in the serum were measured using the AU680 chemistry system (AU680, Beckman, CA, USA) as described in the creatinine (OSR6178, Beckman, CA, USA) and BUN (OSR6234, Beckman, CA, USA) kit instructions.
Immunohistochemistry
Frozen serial transverse cryosections (7 μm) from the tibialis anterior (TA) muscle of Dex and CKD mice were mounted on glass slides. Slides were fixed with pre‐cooled 10% NBF for 20 min at 4°C and then washed three times with PBS. The slides were stained with haematoxylin and eosin (H&E) and observed under the light microscopy. Paraffin serial transverse sections (7 μm) of TA muscle of DBA/2J‐mdx mice were also stained with H&E. The cross‐sectional area (CSA) was assessed using the Image J program.
Frozen sections (7 μm) of muscle tissue were immunohistochemically stained for the expression of MSTN. The slides were fixed with pre‐cooled 10% NBF for 20 min at 4°C and washed three times with PBS. To block endogenous peroxidase activity, the slides were dipped in 0.3% H2O2 solution in PBS and then incubated with a protein blocking solution (X0909, Dako, CA, USA) for 1 h at 25°C. Subsequently, the slides were exposed to anti‐MSTN (ab996, Abcam, MA, USA) antibody overnight at 4°C followed by incubation for 30 min with biotinylated antibodies. The bound antibodies were detected with the DAB substrate chromogen system (K346811, Dako, CA, USA) and observed under the light microscope.
Statistical analysis
All values are expressed as the mean ± standard error. Statistical analysis was performed using one‐way or two‐way analysis of variance test of the IBM SPSS Statistics 19 program (IBM, NY, USA). Statistical significance was set at P < 0.05. All in vitro experiments were conducted at least three times independently, unless otherwise stated.
Results
Ex‐4 down‐regulates the expression of myostatin and muscle atrophic factors in C2C12 myotubes
MSTN is a negative regulator of muscle mass by up‐regulating muscle atrophy‐related factors such as MuRF‐1 and atrogin‐1.23 Therefore, we first determined whether Ex‐4 regulates MSTN expression in C2C12 myotubes after treatment with various concentrations of Ex‐4 for 6 h. MSTN mRNA levels decreased in a dose‐dependent manner up to 20 nM Ex‐4 (Figure S1A). In a time course study using 20 nM Ex‐4, the reduction of MSTN mRNA expression reached a maximum level at 6 h and gradually increased thereafter (Figure S1B). We then investigated whether Dex‐induced MSTN expression is inhibited by Ex‐4 in C2C12 myotubes. Dex treatment significantly up‐regulated MSTN mRNA and protein expression, which was reduced by Ex‐4 treatment (Figure 1A, B). Similarly, the expression pattern of atrogin‐1 and MuRF‐1 was correlated with MSTN expression at both the mRNA and protein levels (Figure 1C, D). In contrast, myogenic factors including MyoD and myogenin (MyoG) were significantly reduced in Dex‐treated condition, but recovered by Ex‐4 treatment. Ex‐4 treatment alone also increased the expression of myogenic factors per se as shown in Figure 1E. We also measured protein synthesis in C2C12 myotubes by quantifying puromycin‐labelled proteins. Ex‐4 treatment significantly increased protein synthesis compared with that in the control without Ex‐4 treatment. Dex treatment significantly reduced protein synthesis compared with that in the control, but this was reversed by Ex‐4 treatment (Figure 1F).
Figure 1.
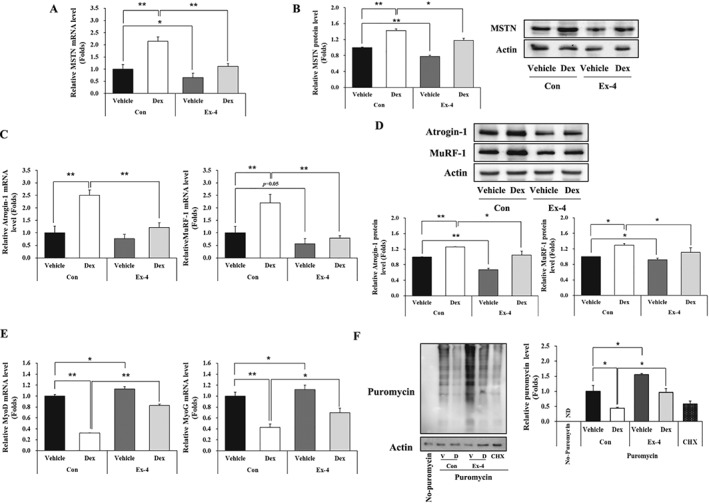
Ex‐4 suppresses MSTN‐mediated muscle atrophic factors in C2C12 myotubes. C2C12 myotubes differentiated for 5 days were treated with 1 μM Dex at 6 h earlier prior to treatment of with or without 20 nM Ex‐4 for 6 h. (A) The mRNA level of MSTN. (B) The protein level of MSTN with a representative blot. (C) The mRNA level of muscle atrophic factors (atrogin‐1 and MuRF‐1). (D) The protein level of muscle atrophic factors (atrogin‐1 and MuRF‐1) with a representative blot. (E) The mRNA level of myogenic factors (MyoD and MyoG). (F) The puromycin‐labelled protein levels with a representative blot. All values are expressed as the mean ± standard error of the fold‐change relative to controls. Significant differences are indicated as **P < 0.01, *P < 0.05 compared with Con + vehicle or Con + Dex. n = 3. Con, control; Dex, dexamethasone; Ex‐4, exendin‐4; CHX, cycloheximide; ND, not detected.
Ex‐4 regulates the expression of myostatin through glucagon‐like peptide‐1 receptor‐mediated protein kinase A and AKT signalling pathways
GLP‐1R expression in muscle tissues and muscle cells is controversial.24, 25 Therefore, we first investigated whether GLP‐1R is expressed in muscle tissue and C2C12 myotubes. GLP‐1R was abundantly expressed in the pancreas and at lower levels in the TA muscle of WT mice. The GLP‐1R expression level decreased by around 50% in heterozygous (Glp1r flox/+ Cre +) mice compared with WT (Glp1r +/+ Cre −) and was barely detectable in homozygous (Glp1r flox/flox Cre +) mice (Figure 2A). GLP‐1R expression was also detected in C2C12 myotubes, as confirmed by the results for both IF staining and western blotting, although the expression level was lower than that of INS‐1 cells (Figure S2A, S2B). Next, we determined whether GLP‐1R‐mediated cAMP production is actually activated by Ex‐4 in C2C12 myotubes. Ex‐4 increased cAMP production by 50% compared with control vehicle but reduced in the presence of the GLP‐1R antagonist Ex‐9, suggesting that Ex‐4 acts through GLP‐1R in C2C12 myotubes (Figure 2B). Increased cAMP production subsequently activated two downstream signalling levels of PKA and AKT signalling as shown by increased phosphorylated PKA (Thr197) and AKT (Ser473) (Figure 2C, D, Figure S3A, S3B). As expected, the activated PKA signalling increased its target protein expression of HSF‐1, whereas the elevated AKT signalling suppressed the activation of NF‐κB (Ser536) (Figure 2C, D, Figure S3A, S3B). In contrast, this activity was attenuated by pretreatment with Ex‐9 (Figure 2C, D; Figure S3A, S3B). It is well known that NF‐κB binds to the promoter region of MSTN, which leads to the induction of its expression.26 Consistent with the reduction of NF‐κB activity by Ex‐4 treatment, MSTN expression was decreased by Ex‐4 treatment, and this decrease was inhibited by Ex‐9 treatment (Figure 2E, Figure S3C). Similarly, GLP‐1R knockdown (Figure 2F, Figure S3D) inhibited the up‐regulation of upstream anabolic signalling pathways (p‐PKA/PKA, p‐AKT/AKT) and its target gene (HSF‐1/actin) and blunted the decrease in p‐NF‐κB signalling pathway activation induced by Ex‐4 (Figure 2G, H, Figure S3E, S3F). In addition, GLP‐1R knockdown abolished the down‐regulation of MSTN expression induced by Ex‐4 (Figure 2I, Figure S3G). Collectively, these results suggest that the effects of Ex‐4 are mediated by the GLP‐1R signalling pathway.
Figure 2.
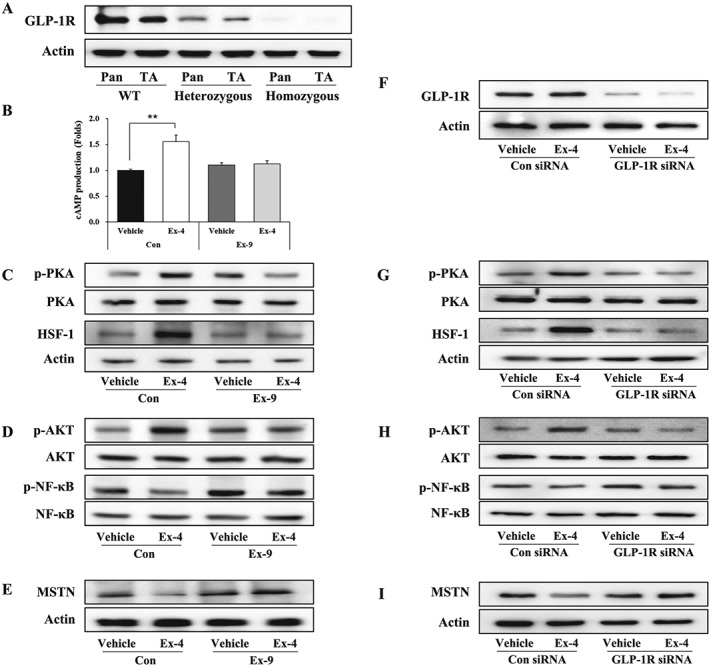
Ex‐4 regulates the expression of MSTN through GLP‐1R‐mediated PKA and AKT signalling pathways. (A) The protein expression level of GLP‐1R in the pancreas and TA muscle from WT (Glp1r +/+ Cre −), heterozygous (Glp1r flox/+ Cre +), and homozygous (Glp1r flox/flox Cre +) mice with a representative blot. (B) C2C12 myotubes were pretreated with 20 nM of Ex‐9 (GLP‐1R antagonist) for 10 min and then were further treated with 20 nM of Ex‐4 for 30 min. cAMP production was measured using cAMP ELISA kit. (C–E) The treated C2C12 myotubes were examined for the activation of GLP‐1R downstream mediators including PKA and HSF‐1 (C) and AKT and NF‐κB (D) signals using western blot. (E) The protein level of MSTN with a representative blot. (F–I) GLP‐1R siRNA‐transfected C2C12 myotubes were treated with 20 nM Ex‐4 for 30 min. (F) Representative blot of GLP‐1R protein levels. (G–I) C2C12 myotubes were examined for the activation of downstream mediators of GLP‐1R, including PKA and HSF‐1 (G) and AKT and NF‐κB (H) by western blotting. (I) Representative blot of MSTN protein levels. All values are expressed as the mean ± standard error. Significant differences are indicated as **P < 0.01 compared with Con + vehicle. n = 3. Con, control; Ex‐4, exendin‐4; Ex‐9, exendin‐9.
Ex‐4 inhibits the translocation of glucocorticoid receptor from the cytosol into the nucleus
GC degrades skeletal muscle proteins through its receptor, GR, by regulating its target genes involved in the ubiquitin proteasome system (UPS).27 To examine whether Ex‐4 affects GR translocation, we performed IF staining in C2C12 myotubes. The GR expression in the nucleus was increased by Dex but decreased with Ex‐4 treatment (Figure 3A). GR protein expression decreased in the CE of Dex‐treated C2C12 myotubes but increased in NE. In contrast, Ex‐4 treatment resulted in a totally opposite trend as shown in Figure 3B and Figure S4A. We further investigated the expression level of GR target genes such as Krüppel‐like factor 15 (KLF15), sestrin 1 (Sesn1), regulated in development and DNA damage responses 1 (REDD1), phosphatidylinositol 3‐kinase regulatory subunit α (p85α), and Forkhead box O3a (FoxO3a)28, 29 in C2C12 myotubes treated with Ex‐4. The KLF15, Sesn1, REDD1, p85α, and FoxO3a mRNA expression levels were up‐regulated in Dex‐treated C2C12 myotubes, whereas they were decreased by Ex‐4 or RU486, a GR inhibitor (Figure 3C), indicating that Ex‐4 inhibits GR translocation from the cytosol into the nucleus, thereby down‐regulating the expression of GR target genes.
Figure 3.
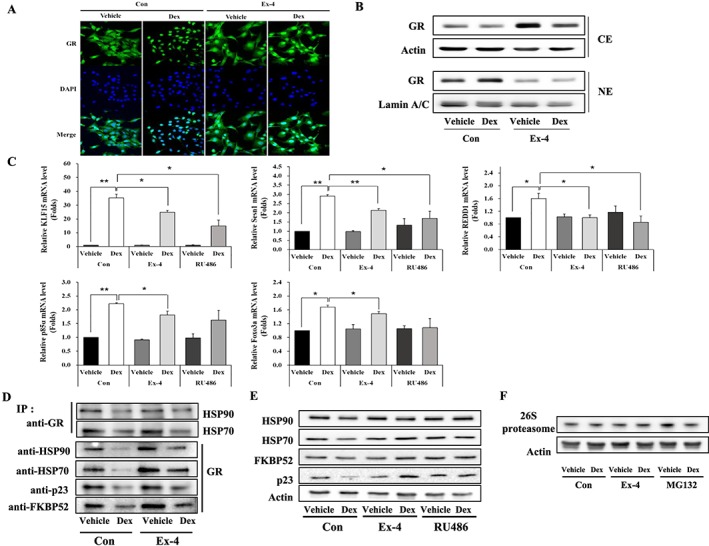
Ex‐4 inhibits the translocation of GR from cytosol into nucleus by up‐regulating the proteins of GR inhibitory complexes. (A) C2C12 myotubes treated with 1 μM Dex at 6 h prior to treatment of with or without 20 nM Ex‐4 for 6 h. GR expression was determined by immunostaining with anti‐GR (green). The nucleus was detected by 4′,6‐diamidino‐2‐phenylindole (DAPI) staining (blue) (magnification ×400). (B) The treated C2C12 myotubes were separated into cytosolic extract (CE) and nuclear extract (NE). The protein level of GR was measured in CE and NE using western blotting. (C) The differentiated C2C12 myotubes treated with 1 μM of Dex at 6 h earlier prior to treatment with 20 nM of either Ex‐4 or RU486, a GR inhibitor, for 6 h. The mRNA level of GR‐targeted genes (KLF15, Sesn1, REDD1, p85α, and FoxO3a) were assessed by RT‐QPCR. (D) The whole cell lysate was isolated and then immunoprecipitated with appropriate antibodies. IP assay of the interaction between GR and GR inhibitory complex proteins (HSP70, HSP90, FKBP52, and p23). (E) The protein level of GR inhibitory complexes in CE. C2C12 myotubes were pretreated with 1 μM Dex and then 6 h later incubated with 20 nM Ex‐4 or RU486 until 12 h. The CE were isolated and then subjected to western blotting and probing with appropriate antibodies. (F) 26S proteasome level was assessed by western blotting. All value expressed as the mean ± standard error. Significant differences are indicated as **P < 0.01, *P < 0.05 compared with Con + vehicle or Con + Dex. n = 3. Con, control; Dex, dexamethasone; Ex‐4, exendin‐4.
Ex‐4 up‐regulates the proteins of glucocorticoid receptor inhibitory complexes in C2C12 myotubes
GR is a ligand‐dependent transcription factor whose inactive form is associated with chaperone complexes, including HSP90, HSP70, FKBP52, and p23.30, 31, 32 Once GC binds to GR within GR inhibitory complexes, the inhibitory complexes are separated from GR and then degraded by 26S proteasome in the cytoplasm, allowing GR translocation to nucleus.33, 34 To investigate whether Ex‐4 affects the interaction of GR and GR inhibitory complex proteins, the affinity of GR to GR inhibitory complex proteins was assessed using the IP assay. The amount of proteins (HSP90, HSP70, FKBP52, or p23) bound to GR was decreased in Dex‐treated C2C12 myotubes but was increased by Ex‐4 (Figure 3D). Examination of the expression of GR inhibitory complex proteins in cytoplasm showed that Dex treatment significantly reduced the expression of HSP90, HSP70, FKBP52, and p23 in cytoplasm. However, this reduction was inhibited by Ex‐4 treatment. Treatment with RU486, a GR inhibitor, yielded similar results (Figure 3E, Figure S4B), suggesting that Ex‐4 up‐regulates the expression of GR inhibitory complex proteins and increases the interaction between GR and GR inhibitory complex proteins, thereby inhibiting GR translocation into the nucleus. 26S proteasome expression was not changed by Ex‐4 treatment (Figure 3F).
Ex‐4 ameliorates muscle wasting in Dex‐induced muscle atrophy mice
To determine whether Ex‐4 affects muscle atrophy in vivo, we treated Dex‐induced muscle atrophy mice with Ex‐4. The body weight decreased after Dex administration, but the Ex‐4 treatment gradually recovered the loss of body weight in Dex‐administered mice (Figure 4A). The Ex‐4 alone injected group had reduced body weight as compared with that of the control vehicle group (Figure 4A). Food intake decreased in all three groups except for the control vehicle group (Figure S5A). Total muscle weight decreased in the Dex‐administered mice but significantly increased with Ex‐4 treatment (Figure 4B). Consistent with this, weights of the gastrocnemius, TA, quadriceps, and extensor digitorum longus were reduced in Dex‐administered mice, but Ex‐4 treatment recovered to the levels of control group. However, the weight of soleus (SOL) was identical among all groups (Figure 4C). In addition, the weight of white adipose tissue (WAT), including epididymal and inguinal adipose tissues, was lower in other three groups (Con + Ex‐4, Dex + Vehicle, and Dex + Ex‐4) than in the control group but comparable within three groups (Figure S5B). The CSA of TA muscle was reduced by ~40% by Dex treatment compared with the control vehicle group. However, this decrease was significantly recovered by as much as the control level by Ex‐4 treatment. Treatment with Ex‐4 alone also significantly increased CSA, compared with the control group (Figure 5A, B). Moreover, the reduced grip strength in Dex‐treated mice was recovered by Ex‐4 treatment, which was positively correlated to the CSA of the TA muscle (Figure 5C). Taken together, these effects of Ex‐4 result in increasing muscle strength and function. In line with aforementioned data, the level of BUN, a catabolism marker in the body, was significantly reduced in the Ex‐4 treatment groups compared with Dex‐administered group (Figure S5C). Mechanistically, Dex up‐regulated the expression of MSTN, atrogin‐1, and MuRF‐1 (Figure 5D, E, F) but suppressed that of myogenic factors including MyoD and MyoG (Figure 5G). In contrast, Ex‐4 administration prevented the atrophic effect of Dex in TA muscle by directly reducing the expression of muscle atrophy factors and increasing the expression of myogenic factors (Figure 5D, E, F, G). In agreement with the results for myogenic factors expression, Ex‐4 restored the expression levels of myofibrillar proteins, such as tropomyosin, MHC, and desmin, which were decreased by Dex treatment in the TA muscle of mice (Figure 5H).
Figure 4.
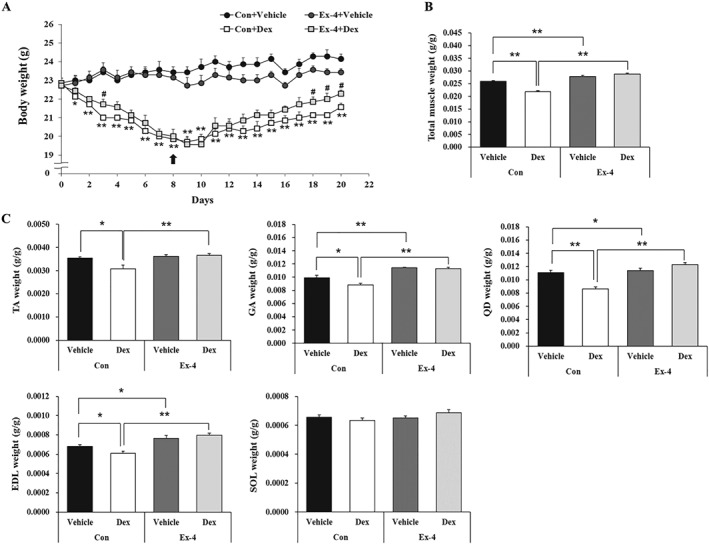
Ex‐4 increases muscle mass in Dex‐induced muscle atrophy mice. (A) Body weight was determined during treatment of period. Ten‐week‐old C57BL/6 male mice were administered the Dex (20 mg/kg i.p.) daily for 8 days and then were administered Ex‐4 (100 ng/mouse i.p.) daily for 12 days. Black arrow indicates the start of Ex‐4 injection. Significant differences are indicated as **P < 0.01, *P < 0.05 compared with Con + vehicle; ##P < 0.01, #P < 0.05 compared with Con + Dex. n = 5–8/group. (B) The weight of total muscle tissue. The total muscle weight was normalized to the final body weight (g). (C) The weight of each muscle, including gastrocnemius (GA), tibialis anterior (TA), quadriceps (QD), extensor digitorum longus (EDL), and soleus (SOL), were measured right after sacrifice and normalized to the final body weight (g). All values are expressed as the mean ± standard error. Significant differences are indicated as **P < 0.01, *P < 0.05 compared with Con + vehicle or Con + Dex. n = 5–8/group. Con, control; Dex, dexamethasone; Ex‐4, exendin‐4.
Figure 5.
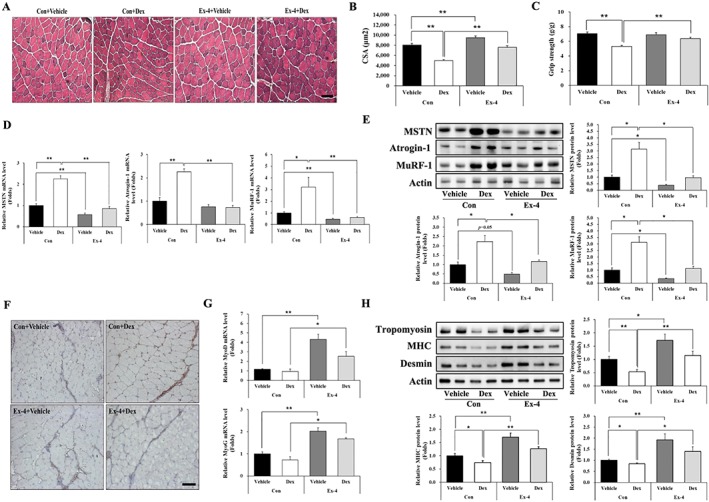
Ex‐4 recovers muscle strength in Dex‐induced muscle atrophy mice. (A) Frozen serial transverse cryosections (7 μm) from TA muscle tissue were stained with H&E and examined under a microscope (magnification ×200). (B) The cross‐sectional area (CSA) of muscle fibre was measured using Image J program. (C) Muscle function was assessed using grip strength measurement. The grip strength was normalized to the final body weight (g). (D–E) The mRNA level of muscle atrophic factors (MSTN, atrogin‐1, and MuRF‐1) (D) and protein (E) levels were assessed using RT‐QPCR and western blotting in TA muscle tissue. (F) The TA muscle tissue from mice administered with Dex was immunostained with anti‐MSTN antibody. The image was taken under a confocal microscope (magnification ×200). Brown colour indicates MSTN expression. (G) The mRNA levels of myogenic factors (MyoD and MyoG) were assessed using RT‐QPCR in TA muscle tissue. (H) The protein levels of myofibrillar proteins (tropomyosin, MHC, and desmin) were measured using western blot. All values are expressed as the mean ± standard error. Significant differences are indicated as **P < 0.01, *P < 0.05 compared with Con + vehicle or Con + Dex, n = 5/group. Con, control; Dex, dexamethasone; Ex‐4, exendin‐4.
Glucagon‐like peptide‐1 receptor agonists increase muscle mass and improves muscle functions in chronic kidney disease and in DBA/2J‐mdx mice
Because the muscle atrophic phenotype is often observed in patients and animal models with CKD,1, 2, 18 we further investigated whether Ex‐4 ameliorates muscle atrophy using mice with partially removed kidney through subtotal nephrectomy. We first verified the CKD mouse model having increased BUN and creatinine levels in the serum because these are known indicators of kidney function and catabolism. We found that treatment with Ex‐4 reduced these levels (Figure 6A). The body weight decreased in CKD mice compared with sham mice; however, Ex‐4 administration to CKD mice, compared with the vehicle‐treated mice, significantly increased body weight (Figure 6B). Body weight was continuously increased in sham + vehicle group and sham + Ex‐4 groups, but the level of increase was lower in sham + Ex‐4 group probably owing to the lower food intake. Total muscle mass was increased by Ex‐4 treatment in CKD mice compared with vehicle‐treated mice (Figure 6C). In addition, Ex‐4 treatment also increased muscle mass in sham‐operated, vehicle‐treated mice. The CSA of TA muscle was significantly decreased in CKD mice but was significantly increased by Ex‐4 treatment (Figure 6D, E). The Ex‐4 treatment also enhanced grip strength in CKD mice compared with vehicle treatment group (Figure 6F), suggesting that Ex‐4 improves muscle function in a mouse model of CKD.
Figure 6.
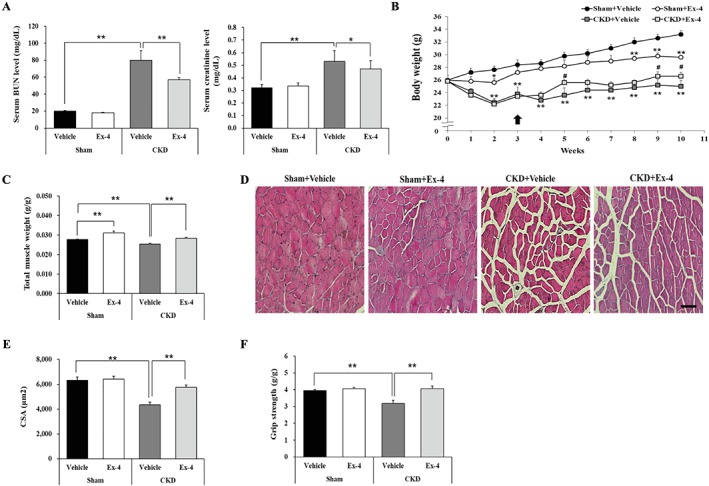
Ex‐4 increases total muscle mass and improves muscle functions in CKD mice. Ten‐week‐old C57BL/6 male mice underwent two‐step subtotal nephrectomy to induce muscle atrophy. (A) The serum level of blood urea nitrogen (BUN) and creatinine. (B) The body weight change during treatment. Black arrow indicates the start of Ex‐4 injection. Significant differences are indicated as **P < 0.01, *P < 0.05 compared with sham + vehicle; ##P < 0.01, #P < 0.05 compared with CKD + vehicle. (C) The weight of total muscle tissue. Total muscle weight was normalized to final body weight (g). (D) Frozen serial transverse cryosections (7 μm) of TA muscle were stained with H&E and examined under a microscope (magnification ×200). (E) The CSA of muscle fibre was measured using Image J program. (F) The grip strength was normalized to the final body weight (g). All values are expressed as the mean ± standard error. Significant differences are indicated as **P < 0.01, *P < 0.05 compared with sham + vehicle or CKD + vehicle. n = 5–7/group.
We then examined the therapeutic effect of GLP‐1R agonist in a neuromuscular disease caused by dystrophin deficiency. Administration of a long‐acting GLP‐1R agonist, dulaglutide (1 mg/kg/week) for 3 weeks, to DBA/2J‐mdx mice did not show the differences in body weight between vehicle and dulaglutide treatment group (not shown data). However, dulaglutide treatment increased the CSA of TA muscle (Figure 7A, B) and grip strength than did vehicle treatment (Figure 7C, D). Dulaglutide also substantially increased the four‐limb hanging time in DBA/2J‐mdx mice (Figure 7E).
Figure 7.
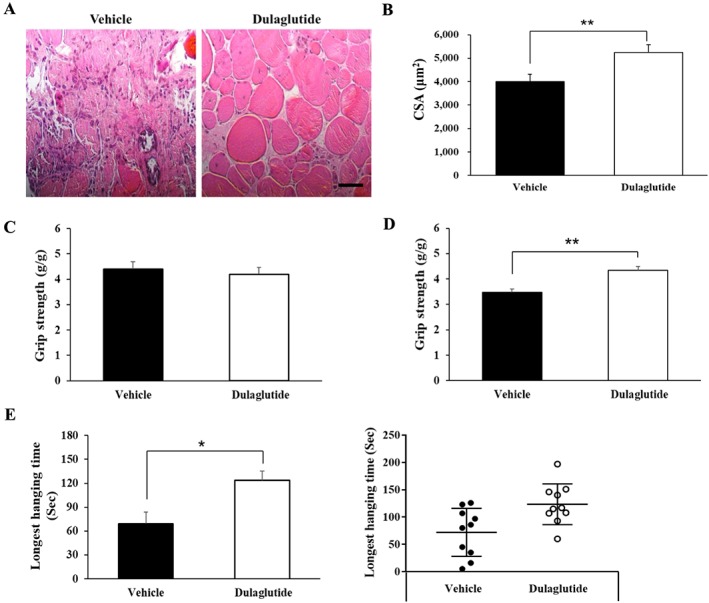
Dulaglutide improves muscle strength in DBA/2J‐mdx mice. Seven‐week‐old DBA/2J‐mdx male mice were subcutaneously administered with 1 mg/kg of dulaglutide once a week for 3 weeks. (A) Sections of TA muscle were stained with H&E and examined under a microscope (magnification ×200). (B) The CSA of muscle fibre was measured using Image J program. (C) The grip strength before administration of dulaglutide. (D) The grip strength after administration of dulaglutide. The grip strength was normalized to the final body weight (g). (E) Four‐limb hanging test after administration of dulaglutide. The left bar graph shows the average of total hanging time of each group; the right dot graph shows the distribution of individual mice for hanging time. All values are expressed as the mean ± standard error. Significant differences are indicated as **P < 0.01, *P < 0.05 compared with vehicle. n = 9–10/group.
Discussion
GLP‐1 and GLP‐1R agonists have been commonly used as anti‐diabetic drugs to lower blood glucose levels.35, 36 In skeletal muscles, Ex‐4 regulates glucose uptake36 and oxygen consumption,37 as well as increases insulin sensitivity.38 However, beyond this glycaemic control activity, the other effects of GLP‐1 or GLP‐1R agonists on muscle are not well understood. In the current study, we have shown for the first time that the GLP‐1R agonist, Ex‐4, attenuates muscle atrophy in a Dex‐induced muscle atrophy mouse model as well as in a CKD‐derived muscle atrophy model. In addition, a long‐acting GLP‐1R agonist, dulaglutide, showed a therapeutic effect in DBA/2J‐mdx mice, a Duchenne muscular disease model.
MSTN is known as a negative regulator of muscle mass, determining both muscle fibre number and size in several muscle atrophy models, through the regulation of MuRF‐1 and atrogin‐1.39, 40, 41 Dex is well known as an anti‐inflammatory drug, but high doses or long‐term use in rodent models induces muscle atrophy by activating the catabolic pathway.40, 42, 43 Dex‐induced and CKD‐induced muscle atrophy models also show the negative correlation between the increased level of MSTN and decreased muscle mass.18, 44 We found that a GLP‐1R agonist, Ex‐4, down‐regulated MSTN mRNA and protein expression in C2C12 myotubes in the presence or absence of Dex. The up‐regulation of muscle atrophic factors and down‐regulation of myogenic factors (MyoD and MyoG) by Dex in C2C12 myotubes were reversed by Ex‐4 treatment, indicating that Ex‐4 may regulate muscle atrophic factors, at least partly, by regulating MSTN expression.
The expression of GLP‐1R in muscle is controversial, because both expression and non‐expression have been reported.25 In order to carefully address this debatable issue, we first tested several commercially available antibodies from different companies and also used muscle tissues from Glp1r −/− mice to check the specificity of the tested antibodies. Our data showed that GLP‐1R is highly expressed in the pancreas and lower expression in muscle tissues. However, it was barely detectible in homozygous Glp1r −/− mice. These data strongly indicate that GLP‐1R is expressed in muscle tissue and C2C12 myotubes, as observed in human muscle satellite cells and cardiomyocytes.25, 45
Silveira et al. reported that activated cAMP/PKA signalling in skeletal muscle suppresses the UPS46 and activated AKT signalling induces protein synthesis in skeletal muscle.47, 48, 49 Our data showed that Ex‐4 treatment activates both PKA and AKT signalling pathways and inhibits phosphorylated NF‐κB protein expression. Activated AKT signalling can directly suppress the phosphorylated protein level of NF‐κB in C2C12 myotubes, which may in turn reduce the binding of NF‐κB to promoter regions of MSTN.50 As expected, Ex‐4 treatment decreased MSTN protein expression and Ex‐9 or GLP‐1R knockdown inhibited this effect, suggesting that the GLP‐1R‐mediated signalling pathway might be involved in regulation of MSTN expression. PKA is known to activate HSF‐1,51 which in turn regulates HSP70 expression through the regulatory heat‐shock elements on its promoter.52 Consistent with these data, our data also showed that Ex‐4 up‐regulated HSF‐1 expression through GLP‐1R signalling, suggesting that Ex‐4 can increase the expression of HSP70, a component of the GR inhibitory complex.
In the cytosol, the increased GCs form complex with GR and then move to the nucleus for binding to the GC response element on the promoter of GR target genes (MSTN, KLF15, and FoxO3a).29, 53, 54, 55 They are well known to activate UPS through E3 ligase that degrades muscle proteins.26, 55, 56 Our data showed that Ex‐4 inhibited GR translocation into the nucleus and down‐regulated GR‐targeted genes (KLF15, FoxO3a, REDD1, p85α, Sesn1), which was similar to the effects of the GR inhibitor, RU486.
GR interacts with chaperone proteins (HSP70, HSP90, FKBP52, and p23) in the cytoplasm, which functionally inhibits GR translocation to the nucleus.56 Once GCs bind to GR; these interaction proteins are separated from the GR and then are degraded by the 26S proteasome.57 HSP70, a member of the heat‐shock protein family, is elevated during cellular stress conditions in skeletal muscle. Previous studies report that the overexpression of HSP70 in mouse skeletal muscle protects against muscle damage and age‐related muscle dysfunction58 and reduces MuRF‐1 and atrogin‐1 promoter activity in a rodent model of disused muscle atrophy.59 Ex‐4 treatment increased the affinity of GR and GR inhibitory complexes by increasing the levels of HSP70, HSP90, FKBP52, and p23 in Dex‐treated C2C12 myotubes. However, Ex‐4 per se did not increase the expression of 26S proteasomes, indicating that the accumulation of GR inhibitory complex is not due to a reduction in the level of the 26S proteasome. Collectively, these data suggest that inhibition of the GCs–GR complex translocation to the nucleus is at least partially due to the up‐regulation of inhibitory complexes of proteins.
Inflammation is a risk factor for inducing muscle atrophy by regulating the NF‐κB signalling pathway60, 61 and the suppression of inflammation reverses muscle atrophy.61, 62 In fact, GLP‐1R agonists have shown anti‐inflammatory effects in many different diseases.63 Our result showed that Ex‐4 treatment inhibited the activation of NF‐κB, a major transcription factor involved in the inflammatory response. Therefore, it is quite possible that the anti‐inflammatory effects of Ex‐4 may also contribute to the amelioration of muscle atrophy. Furthermore, GLP‐1R agonists increase the expression of MyoD and MyoG, which are known as major transcription factors involved in the muscle tissue repair of satellite cells.64, 65 The PKA‐mediated cAMP response element binding protein is activated by muscle injury and promotes muscle regeneration,66, 67, 68 suggesting that cAMP/PKA signalling mediated by GLP‐1R might gives beneficial effects on recovery from muscle atrophy. Further studies are required to clearly identify these mechanisms.
We then investigated whether GLP‐1R agonists have therapeutic effect on pharmacologically, surgically, or genetically induced muscle atrophy in vivo. Dex administration in mice reduced the weight of all muscle types assessed in the current study, but these were statistically recovered to the level of control group by Ex‐4 treatment. The exception was the SOL muscle, and this may be due to the lower expression level of MSTN in SOL than other types of muscle, suggesting that MSTN is a potential mediator of Ex‐4 effects.69, 70 The beneficial effects of Ex‐4 were established by the down‐regulation of muscle‐specific E3 ligase proteins such as atrogin‐1 and MuRF‐1, while up‐regulating myogenic factors including MyoD and MyoG. As a consequence, muscle mass and muscle function improved in Dex‐induced muscle atrophy mice. In support of our results, recent studies reported that the inhibitors of DPP4, a GLP‐1 degradation enzyme, are associated with improvement of reduced muscle mass in diabetic and elderly diabetic patients15, 71 and with improvement of mitochondrial biogenesis in a heart failure mouse model.16
CKD is a model of cachexia that is mainly induced by inflammatory cytokines in the human pathological state.18 Similar to the observations in Dex‐treated mice, Ex‐4 significantly increased total muscle mass, CSA of the TA muscle, and muscle function in CKD mice. Ex‐4 and sitaglitin (DPP4 inhibitor) are known to improve kidney function by attenuating inflammation in chronic or diabetic kidney disease models.17, 72 Therefore, it is unclear whether the beneficial effect of Ex‐4 is due to its role in improving kidney function, its direct effect on muscle, or both in the current study. Further studies are needed to clarify this. Duchenne muscular disease is a neuromuscular disease caused by dystrophin deficiency and is the most common and severe form of muscular dystrophy.73 Along with highlighting high clinical potential, our data also showed that a long‐acting GLP‐1R agonist, dulaglutide, substantially improved muscle atrophy in DBA/2J‐mdx mice. Collectively, our findings strongly suggest that GLP‐1R agonists may have high translational value for treating diverse types of muscle atrophy.
The reduction in fat depots and reduction in food intake, which are known effects of GLP‐1R agonists, are not desirable effects in chronic wasting conditions. However, our data showed that Ex‐4 treatment did not affect food intake in Dex‐induced (Figure S5) or CKD‐induced muscle atrophy models (data not shown). In addition, body weight increased after Ex‐4 treatment, although the changes were very modest in both models. Liver weights were comparable between the sham and CKD groups, regardless of Ex‐4 treatment (data not shown). These data suggest that Ex‐4 plays different roles in pathological conditions and that the beneficial effect of Ex‐4 appears to override the negative effect of reduced food intake. However, appropriate precautions are needed for clinical application of Ex‐4.
In conclusion, we found that the GLP‐1R agonist has therapeutic effects on muscle atrophy by suppressing MSTN and muscle atrophic factors and enhancing myogenic factors via GLP‐1R signalling‐mediated regulation and suppression of GR translocation. Our data provide proof‐of‐concept evidence for the therapeutic effects of GLP‐1R agonists on muscle atrophy. These findings highlight the potential application of GLP‐1R agonists for the treatment of muscle wasting diseases.
Conflict of interest
Y.H.H., J.H.L., K.W.J., C.S.C., and H.‐S.J. declare that they have no conflict of interest.
Supporting information
Figure S1. Ex‐4 downregulates MSTN mRNA expression. A‐B. C2C12 myoblasts were differentiated into C2C12 myotubes for 5 days. C2C12 myotubes were treated with A. the indicated concentrations of Ex‐4 for 6 h and B. for the indicated times with 20 nM of Ex‐4. mRNA levels of MSTN were measured using RT‐QPCR. All values are expressed as the mean ± SE. Significant differences are indicated as **p<0.01, *p<0.05 compared with Ex‐4 (0 nM) or Ex‐4 (0 h). n=3.
Figure S2. GLP‐1R is expressed in muscle tissue and C2C12 cells. A. INS‐1 and C2C12 myotubes were immunostained with anti‐GLP‐1R antibody (green) and DAPI (blue for the nucleus). Cells were observed under a confocal microscope (magnification 200×). B. The protein expression levels of GLP‐1R were determined among INS‐1 cells, C2C12 myoblast and myotubes using western blotting.
Figure S3. Ex‐4 regulates the expression of MSTN through GLP‐1R mediated PKA and AKT signaling pathways. A‐C. The quantitative data for the expression of GLP‐1R downstream mediators such as PKA and HSF‐1 (A), AKT and NF‐κB (B) by Ex‐9. C. Quantitative graph of MSTN protein levels. D‐G. GLP‐1R siRNA‐transfected C2C12 myotubes were treated with 20 nM Ex‐4 for 30 min. D. The quantitative data for the protein levels of GLP‐1R and GLP‐1R downstream mediators including PKA and HSF‐1 (E), AKT and NF‐κB (F) by GLP‐1R siRNA. G. Quantitative graph of MSTN protein levels. All values are expressed as the mean ± SE. Significant differences are indicated as **p<0.01, *p<0.05 compared with Con+Vehicle or Con+Ex‐4, compared with Con siRNA+Vehicle or Con siRNA+Ex‐4. n=3. Con, Control; Ex‐4, Exendin‐4.
Figure S4. Ex‐4 inhibits the translocation of GR from cytosol into nucleus by upregulating the proteins of GR inhibitory complexes. A. The quantitative graph for GR protein level in CE and NE using western blotting. B. The proteins level of GR inhibitory complexes. C2C12 myotubes were pretreated with 1 μM Dex, and then 6 h later, incubated with 20 nM Ex‐4 or RU486 until 12 h. The CE were isolated and then subjected to western blotting and probing with appropriate antibodies (HSP70, HSP90, FKBP52 and p23). All values are expressed as the mean ± SE. Significant differences are indicated as **p<0.01, *p<0.05 compared with Con+Vehicle or Con+Dex. n=3. Con, Control; Dex, Dexamethasone; Ex‐4, Exendin‐4.
Figure S5. Ex‐4 reduced food intake and catabolic effects in Dex‐administered mice. A. Food intake. B. Total weight of white adipose tissue (WAT). C. The serum levels of BUN. All values are expressed as the mean ± SE. Significant differences are indicated as **p<0.01, *p<0.05 compared with Con+Vehicle or Con+Dex. n=5‐8/group. Con, Control; Dex, Dexamethasone; Ex‐4, Exendin‐4.
Supplementary Figure legends
Acknowledgements
This study was supported by grants from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health and Welfare, Republic of Korea (grant number HI14C1135), and the National Research Foundation of Korea (NRF 2017R1D1A1B03036210). The authors certify that they comply with the ethical guidelines for authorship and publishing of the Journal of Cachexia, Sarcopenia and Muscle.74
Hong Y., Lee J. H., Jeong K. W., Choi C. S., and Jun H.‐S. (2019) Amelioration of muscle wasting by glucagon‐like peptide‐1 receptor agonist in muscle atrophy, Journal of Cachexia, Sarcopenia and Muscle, 10, 903–918, doi: 10.1002/jcsm.12434.
References
- 1. Cohen S, Nathan JA, Goldberg AL. Muscle wasting in disease: molecular mechanisms and promising therapies. Nat Rev Drug Discov 2015;14:58–74. [DOI] [PubMed] [Google Scholar]
- 2. Workeneh BT, Mitch WE. Review of muscle wasting associated with chronic kidney disease. Am J Clin Nutr 2010;91:1128s–1132s. [DOI] [PubMed] [Google Scholar]
- 3. Evans WJ, Morley JE, Argilés J, Bales C, Baracos V, Guttridge D, et al. Cachexia: a new definition. Clin Nutr 2008;27:793–799. [DOI] [PubMed] [Google Scholar]
- 4. Williams A, Sun X, Fischer JE, Hasselgren PO. The expression of genes in the ubiquitin‐proteasome proteolytic pathway is increased in skeletal muscle from patients with cancer. Surgery 1999;126:744–749, discussion 749‐50, 750. [PubMed] [Google Scholar]
- 5. Muscaritoli M, Anker SD, Argiles J, Aversa Z, Bauer JM, Biolo G, et al. Consensus definition of sarcopenia, cachexia and pre‐cachexia: joint document elaborated by Special Interest Groups (SIG) “cachexia‐anorexia in chronic wasting diseases” and “nutrition in geriatrics”. Clin Nutr 2010;29:154–159. [DOI] [PubMed] [Google Scholar]
- 6. Powers SK, Lynch GS, Murphy KT, Reid MB, Zijdewind I. Disease‐induced skeletal muscle atrophy and fatigue. Med Sci Sports Exerc 2016;48:2307–2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Benny Klimek ME, Aydogdu T, Link MJ, Pons M, Koniaris LG, Zimmers TA. Acute inhibition of myostatin‐family proteins preserves skeletal muscle in mouse models of cancer cachexia. Biochem Biophys Res Commun 2010;391:1548–1554. [DOI] [PubMed] [Google Scholar]
- 8. Busquets S, Toledo M, Orpí M, Massa D, Porta M, Capdevila E, et al. Myostatin blockage using actRIIB antagonism in mice bearing the Lewis lung carcinoma results in the improvement of muscle wasting and physical performance. J Cachexia Sarcopenia Muscle 2012;3:37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yi CX, Foppen E, Abplanalp W, Gao Y, Alkemade A, La Fleur SE, et al. Glucocorticoid signaling in the arcuate nucleus modulates hepatic insulin sensitivity. Diabetes 2012;61:339–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Oakley RH, Cidlowski JA. The biology of the glucocorticoid receptor: new signaling mechanisms in health and disease. J Allergy Clin Immunol 2013;132:1033–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kuo T, Harris CA, Wang JC. Metabolic functions of glucocorticoid receptor in skeletal muscle. Mol Cell Endocrinol 2013;380:79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Edwards CM, Todd JF, Mahmoudi M, Wang Z, Wang RM, Ghatei MA, et al. Glucagon‐like peptide 1 has a physiological role in the control of postprandial glucose in humans: studies with the antagonist exendin 9‐39. Diabetes 1999;48:86–93. [DOI] [PubMed] [Google Scholar]
- 13. Abu‐Hamdah R, Rabiee A, Meneilly GS, Shannon RP, Andersen DK, Elahi D. Clinical review: The extrapancreatic effects of glucagon‐like peptide‐1 and related peptides. J Clin Endocrinol Metab 2009;94:1843–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Drucker DJ. The biology of incretin hormones. Cell Metab 2006;3:153–165. [DOI] [PubMed] [Google Scholar]
- 15. Rizzo MR, Barbieri M, Fava I, Desiderio M, Coppola C, Marfella R, et al. Sarcopenia in elderly diabetic patients: role of dipeptidyl peptidase 4 inhibitors. J Am Med Dir Assoc 2016;17:896–901. [DOI] [PubMed] [Google Scholar]
- 16. Takada S, Masaki Y, Kinugawa S, Matsumoto J, Furihata T, Mizushima W, et al. Dipeptidyl peptidase‐4 inhibitor improved exercise capacity and mitochondrial biogenesis in mice with heart failure via activation of glucagon‐like peptide‐1 receptor signalling. Cardiovasc Res 2016;111:338–347. [DOI] [PubMed] [Google Scholar]
- 17. Kalaitzoglou E, Fowlkes JL, Popescu I, Thrailkill KM. Diabetes pharmacotherapy and effects on the musculoskeletal system. Diabetes Metab Res Rev 2019;35:e3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang L, Rajan V, Lin E, Hu Z, Han HQ, Zhou X, et al. Pharmacological inhibition of myostatin suppresses systemic inflammation and muscle atrophy in mice with chronic kidney disease. Faseb J 2011;25:1653–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Baldwin AS Jr. The NF‐kappa B and I kappa B proteins: new discoveries and insights. Annu Rev Immunol 1996;14:649–683. [DOI] [PubMed] [Google Scholar]
- 20. Goodman CA, Mabrey DM, Frey JW, Miu MH, Schmidt EK, Pierre P, et al. Novel insights into the regulation of skeletal muscle protein synthesis as revealed by a new nonradioactive in vivo technique. Faseb j 2011;25:1028–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rodriguez J, Pierre N, Naslain D, Bontemps F, Ferreira D, Priem F, et al. Urolithin B, a newly identified regulator of skeletal muscle mass. J Cachexia Sarcopenia Muscle 2017;8:583–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Aartsma‐Rus A, van Putten M. Assessing functional performance in the mdx mouse model. J Vis Exp 2014; 85:e51303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Thomas M, Langley B, Berry C, Sharma M, Kirk S, Bass J, et al. Myostatin, a negative regulator of muscle growth, functions by inhibiting myoblast proliferation. J Biol Chem 2000;275:40235–40243. [DOI] [PubMed] [Google Scholar]
- 24. Henriksen T, Green C, Pedersen BK. Myokines in myogenesis and health. Recent Pat Biotechnol 2012;6:167–171. [DOI] [PubMed] [Google Scholar]
- 25. Green CJ, Henriksen TI, Pedersen BK, Solomon TP. Glucagon like peptide‐1‐induced glucose metabolism in differentiated human muscle satellite cells is attenuated by hyperglycemia. PLoS One 2012;7:e44284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sriram S, Subramanian S, Sathiakumar D, Venkatesh R, Salerno MS, McFarlane CD, et al. Modulation of reactive oxygen species in skeletal muscle by myostatin is mediated through NF‐kappaB. Aging Cell 2011;10:931–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen S, Wang J, Yu GQ, Liu W, Pearce D. Androgen and glucocorticoid receptor heterodimer formation. A possible mechanism for mutual inhibition of transcriptional activity. J Biol Chem 1997;272:14087–14092. [DOI] [PubMed] [Google Scholar]
- 28. Delany AM, Durant D, Canalis E. Glucocorticoid suppression of IGF I transcription in osteoblasts. Mol Endocrinol 2001;15:1781–1789. [DOI] [PubMed] [Google Scholar]
- 29. Shimizu N, Yoshikawa N, Ito N, Maruyama T, Suzuki Y, Takeda SI, et al. Crosstalk between glucocorticoid receptor and nutritional sensor mTOR in skeletal muscle. Cell Metab 2011;13:170–182. [DOI] [PubMed] [Google Scholar]
- 30. Dittmar KD, Demady DR, Stancato LF, Krishna P, Pratt WB. Folding of the glucocorticoid receptor by the heat shock protein (hsp) 90‐based chaperone machinery. The role of p23 is to stabilize receptor.hsp90 heterocomplexes formed by hsp90.p60.hsp70. J Biol Chem 1997;272:21213–21220. [DOI] [PubMed] [Google Scholar]
- 31. Hutchison KA, Scherrer LC, Czar MJ, Stancato LF, Chow YH, Jove R, et al. Regulation of glucocorticoid receptor function through assembly of a receptor‐heat shock protein complex. Ann N Y Acad Sci 1993;684:35–48. [DOI] [PubMed] [Google Scholar]
- 32. Fuchsl AM, Reber SO. Chronic psychosocial stress and negative feedback inhibition: enhanced hippocampal glucocorticoid signaling despite lower cytoplasmic GR expression. PLoS One 2016;11:e0153164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Riccardi C, Bruscoli S, Migliorati G. Molecular mechanisms of immunomodulatory activity of glucocorticoids. Pharmacol Res 2002;45:361–368. [DOI] [PubMed] [Google Scholar]
- 34. Rekers NV, De Fijter JW, Claas FH, Eikmans M. Mechanisms and risk assessment of steroid resistance in acute kidney transplant rejection. Transpl Immunol 2016;38:3–14. [DOI] [PubMed] [Google Scholar]
- 35. Gedulin BR, Nikoulina SE, Smith PA, Gedulin G, Nielsen LL, Baron AD, et al. Exenatide (exendin‐4) improves insulin sensitivity and {beta}‐cell mass in insulin‐resistant obese fa/fa Zucker rats independent of glycemia and body weight. Endocrinology 2005;146:2069–2076. [DOI] [PubMed] [Google Scholar]
- 36. Arnes L, Moreno P, Nuche‐Berenguer B, Valverde I, Villanueva‐Peñacarrillo ML. Effect of exendin‐4 treatment upon glucose uptake parameters in rat liver and muscle, in normal and type 2 diabetic state. Regul Pept 2009;153:88–92. [DOI] [PubMed] [Google Scholar]
- 37. Choung JS, Lee YS, Jun HS. Exendin‐4 increases oxygen consumption and thermogenic gene expression in muscle cells. J Mol Endocrinol 2017;58:79–90. [DOI] [PubMed] [Google Scholar]
- 38. Idris I, Patiag D, Gray S, Donnelly R. Exendin‐4 increases insulin sensitivity via a PI‐3‐kinase‐dependent mechanism: contrasting effects of GLP‐1. Biochem Pharmacol 2002;63:993–996. [DOI] [PubMed] [Google Scholar]
- 39. Qin J, Du R, Yang YQ, Zhang HQ, Li Q, Liu L, et al. Dexamethasone‐induced skeletal muscle atrophy was associated with upregulation of myostatin promoter activity. Res Vet Sci 2013;94:84–89. [DOI] [PubMed] [Google Scholar]
- 40. Nakao R, Yamamoto S, Yasumoto Y, Oishi K. Dosing schedule‐dependent attenuation of dexamethasone‐induced muscle atrophy in mice. Chronobiol Int 2014;31:506–514. [DOI] [PubMed] [Google Scholar]
- 41. Tobin JF, Celeste AJ. Myostatin, a negative regulator of muscle mass: implications for muscle degenerative diseases. Curr Opin Pharmacol 2005;5:328–332. [DOI] [PubMed] [Google Scholar]
- 42. Lee MK, Kim YM, Kim IH, Choi YH, Nam TJ. Pyropia yezoensis peptide PYP15 protects against dexamethasone‐induced muscle atrophy through the downregulation of atrogin1/MAFbx and MuRF1 in mouse C2C12 myotubes. Mol Med Rep 2017;15:3507–3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cea LA, Balboa E, Puebla C, Vargas AA, Cisterna BA, Escamilla R, et al. Dexamethasone‐induced muscular atrophy is mediated by functional expression of connexin‐based hemichannels. Biochim Biophys Acta 2016;1862:1891–1899. [DOI] [PubMed] [Google Scholar]
- 44. Ma K, Mallidis C, Bhasin S, Mahabadi V, Artaza J, Gonzalez‐Cadavid N, et al. Glucocorticoid‐induced skeletal muscle atrophy is associated with upregulation of myostatin gene expression. Am J Physiol Endocrinol Metab 2003;285:E363–E371. [DOI] [PubMed] [Google Scholar]
- 45. Ye Y, Birnbaum Y. Cyclic AMP‐mediated pleiotropic effects of glucagon‐like peptide‐1 receptor activation. Focus on “Exendin‐4 attenuates high glucose‐induced cardiomyocyte apoptosis via inhibition of endoplasmic reticulum stress and activation of SERCA2a”. Am J Physiol Cell Physiol 2013;304:C505–C507. [DOI] [PubMed] [Google Scholar]
- 46. Silveira WA, Gonçalves DA, Graça FA, Andrade‐Lopes AL, Bergantin LB, Zanon NM, et al. Activating cAMP/PKA signaling in skeletal muscle suppresses the ubiquitin‐proteasome‐dependent proteolysis: implications for sympathetic regulation. J Appl Physiol (1985) 2014;117:11–19. [DOI] [PubMed] [Google Scholar]
- 47. Hunter RB, Hunter RB, Stevenson EJ, Koncarevic A, Mitchell‐Felton HE, Essig DA, et al. Activation of an alternative NF‐kappaB pathway in skeletal muscle during disuse atrophy. FASEB J 2002;16:529–538. [DOI] [PubMed] [Google Scholar]
- 48. Rommel C, Bodine SC, Clarke BA, Rossman R, Nunez L, Stitt TN, et al. Mediation of IGF‐1‐induced skeletal myotube hypertrophy by PI(3)K/Akt/mTOR and PI(3)K/Akt/GSK3 pathways. Nat Cell Biol 2001;3:1009–1013. [DOI] [PubMed] [Google Scholar]
- 49. Romanick M, Thompson LV, Brown‐Borg HM. Murine models of atrophy, cachexia, and sarcopenia in skeletal muscle. Biochim Biophys Acta 2013;1832:1410–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Samant SA, Kanwal A, Pillai VB, Bao R, Gupta MP. The histone deacetylase SIRT6 blocks myostatin expression and development of muscle atrophy. Sci Rep 2017;7:11877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Murshid A, Chou SD, Prince T, Zhang Y, Bharti A, Calderwood SK. Protein kinase A binds and activates heat shock factor 1. PLoS One 2010;5:e13830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Krause M, Bock PM, Takahashi HK, De Bittencourt PI, Newsholme P. The regulatory roles of NADPH oxidase, intra‐ and extra‐cellular HSP70 in pancreatic islet function, dysfunction and diabetes. Clin Sci (Lond) 2015;128:789–803. [DOI] [PubMed] [Google Scholar]
- 53. Kuo T, Liu PH, Chen TC, Lee RA, New J, Zhang D, et al. Transcriptional regulation of FoxO3 gene by glucocorticoids in murine myotubes. Am J Physiol Endocrinol Metab 2016;310:E572–E585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Luo J, Liang A, Liang M, Xia R, Rizvi Y, Wang Y, et al. Serum glucocorticoid‐regulated kinase 1 blocks CKD‐induced muscle wasting via inactivation of FoxO3a and Smad2/3. J Am Soc Nephrol 2016;27:2797–2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lutzner N, Kalbacher H, Krones‐Herzig A, Rösl F. FOXO3 is a glucocorticoid receptor target and regulates LKB1 and its own expression based on cellular AMP levels via a positive autoregulatory loop. PLoS One 2012;7:e42166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Echeverria PC, Mazaira G, Erlejman A, Gomez‐Sanchez C, Pilipuk GP, Galigniana MD. Nuclear import of the glucocorticoid receptor–hsp90 complex through the nuclear pore complex is mediated by its interaction with Nup62 and importin beta. Mol Cell Biol 2009;29:4788–4797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Stavreva DA, Wiench M, John S, Conway‐Campbell BL, McKenna MA, Pooley JR, et al. Ultradian hormone stimulation induces glucocorticoid receptor‐mediated pulses of gene transcription. Nat Cell Biol 2009;11:1093–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. McArdle A, Dillmann WH, Mestril R, Faulkner JA, Jackson MJ. Overexpression of HSP70 in mouse skeletal muscle protects against muscle damage and age‐related muscle dysfunction. Faseb J 2004;18:355–357. [DOI] [PubMed] [Google Scholar]
- 59. Senf SM, Dodd SL, McClung JM, Judge AR. Hsp70 overexpression inhibits NF‐kappaB and Foxo3a transcriptional activities and prevents skeletal muscle atrophy. Faseb J 2008;22:3836–3845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Jackman RW, Cornwell EW, Wu CL, Kandarian SC. Nuclear factor‐kappaB signalling and transcriptional regulation in skeletal muscle atrophy. Exp Physiol 2013;98:19–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Yu R, Yu R, Chen JA, Xu J, Cao J, Wang Y, et al. Suppression of muscle wasting by the plant‐derived compound ursolic acid in a model of chronic kidney disease. J Cachexia Sarcopenia Muscle 2017;8:327–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ham DJ, Murphy KT, Chee A, Lynch GS, Koopman R. Glycine administration attenuates skeletal muscle wasting in a mouse model of cancer cachexia. Clin Nutr 2014;33:448–458. [DOI] [PubMed] [Google Scholar]
- 63. Lee YS, Jun HS. Anti‐inflammatory effects of GLP‐1‐based therapies beyond glucose control. Mediators Inflamm 2016;2016:3094642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Aguayo‐Mazzucato C, Bonner‐Weir S. Stem cell therapy for type 1 diabetes mellitus. Nat Rev Endocrinol 2010;6:139–148. [DOI] [PubMed] [Google Scholar]
- 65. Le Grand F, Rudnicki MA. Skeletal muscle satellite cells and adult myogenesis. Curr Opin Cell Biol 2007;19:628–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Rahnert JA, Zheng B, Hudson MB, Woodworth‐Hobbs ME, Price SR. Glucocorticoids alter CRTC‐CREB signaling in muscle cells: impact on PGC‐1alpha expression and atrophy markers. PLoS One 2016;11:e0159181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Stewart R, Flechner L, Montminy M, Berdeaux R. CREB is activated by muscle injury and promotes muscle regeneration. PLoS One 2011;6:e24714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zhang Y, Aguilar OA, Storey KB. Transcriptional activation of muscle atrophy promotes cardiac muscle remodeling during mammalian hibernation. PeerJ 2016;4:e2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wang M, Yu H, Kim YS, Bidwell CA, Kuang S. Myostatin facilitates slow and inhibits fast myosin heavy chain expression during myogenic differentiation. Biochem Biophys Res Commun 2012;426:83–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Allen DL, Cleary AS, Speaker KJ, Lindsay SF, Uyenishi J, Reed JM, et al. Myostatin, activin receptor IIb, and follistatin‐like‐3 gene expression are altered in adipose tissue and skeletal muscle of obese mice. Am J Physiol Endocrinol Metab 2008;294:E918–E927. [DOI] [PubMed] [Google Scholar]
- 71. Bouchi R, Fukuda T, Takeuchi T, Nakano Y, Murakami M, Minami I, et al. Dipeptidyl peptidase 4 inhibitors attenuates the decline of skeletal muscle mass in patients with type 2 diabetes. Diabetes Metab Res Rev 2018;34:e2957. [DOI] [PubMed] [Google Scholar]
- 72. Yin QH, Zhang R, Li L, Wang YT, Liu JP, Zhang J, et al. Exendin‐4 ameliorates lipotoxicity‐induced glomerular endothelial cell injury by improving ABC transporter A1‐mediated cholesterol efflux in diabetic apoE knockout mice. J Biol Chem 2016;291:26487–26501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Petrof BJ, Shrager JB, Stedman HH, Kelly AM, Sweeney HL. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc Natl Acad Sci U S A 1993;90:3710–3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. von Haehling S, Morley JE, Coats AJ, Anker SD. Ethical guidelines for publishing in the journal of cachexia, sarcopenia and muscle: update 2017. J Cachexia Sarcopenia Muscle 2017;8:1081–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Ex‐4 downregulates MSTN mRNA expression. A‐B. C2C12 myoblasts were differentiated into C2C12 myotubes for 5 days. C2C12 myotubes were treated with A. the indicated concentrations of Ex‐4 for 6 h and B. for the indicated times with 20 nM of Ex‐4. mRNA levels of MSTN were measured using RT‐QPCR. All values are expressed as the mean ± SE. Significant differences are indicated as **p<0.01, *p<0.05 compared with Ex‐4 (0 nM) or Ex‐4 (0 h). n=3.
Figure S2. GLP‐1R is expressed in muscle tissue and C2C12 cells. A. INS‐1 and C2C12 myotubes were immunostained with anti‐GLP‐1R antibody (green) and DAPI (blue for the nucleus). Cells were observed under a confocal microscope (magnification 200×). B. The protein expression levels of GLP‐1R were determined among INS‐1 cells, C2C12 myoblast and myotubes using western blotting.
Figure S3. Ex‐4 regulates the expression of MSTN through GLP‐1R mediated PKA and AKT signaling pathways. A‐C. The quantitative data for the expression of GLP‐1R downstream mediators such as PKA and HSF‐1 (A), AKT and NF‐κB (B) by Ex‐9. C. Quantitative graph of MSTN protein levels. D‐G. GLP‐1R siRNA‐transfected C2C12 myotubes were treated with 20 nM Ex‐4 for 30 min. D. The quantitative data for the protein levels of GLP‐1R and GLP‐1R downstream mediators including PKA and HSF‐1 (E), AKT and NF‐κB (F) by GLP‐1R siRNA. G. Quantitative graph of MSTN protein levels. All values are expressed as the mean ± SE. Significant differences are indicated as **p<0.01, *p<0.05 compared with Con+Vehicle or Con+Ex‐4, compared with Con siRNA+Vehicle or Con siRNA+Ex‐4. n=3. Con, Control; Ex‐4, Exendin‐4.
Figure S4. Ex‐4 inhibits the translocation of GR from cytosol into nucleus by upregulating the proteins of GR inhibitory complexes. A. The quantitative graph for GR protein level in CE and NE using western blotting. B. The proteins level of GR inhibitory complexes. C2C12 myotubes were pretreated with 1 μM Dex, and then 6 h later, incubated with 20 nM Ex‐4 or RU486 until 12 h. The CE were isolated and then subjected to western blotting and probing with appropriate antibodies (HSP70, HSP90, FKBP52 and p23). All values are expressed as the mean ± SE. Significant differences are indicated as **p<0.01, *p<0.05 compared with Con+Vehicle or Con+Dex. n=3. Con, Control; Dex, Dexamethasone; Ex‐4, Exendin‐4.
Figure S5. Ex‐4 reduced food intake and catabolic effects in Dex‐administered mice. A. Food intake. B. Total weight of white adipose tissue (WAT). C. The serum levels of BUN. All values are expressed as the mean ± SE. Significant differences are indicated as **p<0.01, *p<0.05 compared with Con+Vehicle or Con+Dex. n=5‐8/group. Con, Control; Dex, Dexamethasone; Ex‐4, Exendin‐4.
Supplementary Figure legends