

Abstract
Objective
To determine the frontal lobe proton magnetic resonance spectroscopy (1H MRS) abnormalities in asymptomatic and symptomatic carriers of microtubule-associated protein tau (MAPT) mutations.
Methods
We recruited patients with MAPT mutations from 5 individual families, who underwent single voxel 1H MRS from the medial frontal lobe at 3T (n = 19) from the Longitudinal Evaluation of Familial Frontotemporal Dementia Subjects (LEFFTDS) Study at the Mayo Clinic site. Asymptomatic MAPT mutation carriers (n = 9) had Frontotemporal Lobar Degeneration Clinical Dementia Rating Sum of Boxes (FTLD-CDR SOB) score of zero, and symptomatic MAPT mutation carriers (n = 10) had a median FTLD-CDR SOB score of 5. Noncarriers from healthy first-degree relatives of the patients were recruited as controls (n = 25). The demographic aspects and 1H MRS metabolite ratios were compared by use of the Fisher exact test for sex and linear mixed models to account for within-family correlations. We used Tukey contrasts for pair-wise comparisons.
Results
Asymptomatic MAPT mutation carriers had lower neuronal marker N-acetylaspartate (NAA)/creatine (Cr) (p = 0.001) and lower NAA/myo-inositol (mI) (p = 0.026) than noncarriers after adjustment for age. Symptomatic MAPT mutation carriers had lower NAA/Cr (p = 0.01) and NAA/mI (p = 0.01) and higher mI/Cr (p = 0.02) compared to noncarriers after adjustment for age. Furthermore, NAA/Cr (p = 0.006) and NAA/mI (p < 0.001) ratios decreased, accompanied by an increase in mI/Cr ratio (p = 0.001), as the ages of carriers approached and passed the age at symptom onset.
Conclusion
Frontal lobe neurochemical alterations measured with 1H MRS precede the symptom onset in MAPT mutation carriers. Frontal lobe 1H MRS is a potential biomarker for early neurodegenerative processes in MAPT mutation carriers.
Frontotemporal lobar degeneration (FTLD) is a progressive neurodegenerative disease associated with behavioral and language disorders, executive dysfunction, and impaired social cognition.1 It is familial in 30% to 50% of patients.2 Mutations in the microtubule-associated protein tau (MAPT) gene were the first to be associated with inherited FTD3–5 and can result in filamentous accumulation of hyperphosphorylated tau in neurons and glia, leading to neurodegeneration and atrophy years before the onset of clinical symptoms.6–8 With the potential disease-modifying treatments that are under development, noninvasive biomarkers that help determine the early brain changes in asymptomatic patients will be critical for tracking disease progression and enrolling the right participants in the clinical trials at the right time in the disease course.
A potential imaging marker for early detection of tau pathology is proton magnetic resonance spectroscopy (1H MRS), which allows a noninvasive assessment of brain biochemistry. 1H MRS metabolite measurements have been sensitive biomarkers of early neurodegenerative pathology in Alzheimer disease (AD), Lewy body dementia, and FTLD.9–14 Single-voxel 1H MRS measurements from the posterior cingulate gyrus have identified neurochemical abnormalities in both asymptomatic and symptomatic carriers of MAPT mutation.15 A decrease in the neuronal integrity marker N-acetylaspartate (NAA) or NAA/creatine (Cr) ratio and an elevation in myo-inositol (mI) or mI/Cr ratio have been found in symptomatic patients with FTLD,12 while only elevation in mI/Cr in posterior cingulate gyrus has been found in asymptomatic MAPT mutation carriers.16 Typically, MAPT mutation carriers have early involvement and greatest lobar rates of atrophy in the temporal and frontal lobes,17 but the neurochemical changes from these regions have not been studied in MAPT mutation carriers.
Our objective was to determine the NAA/Cr, mI/Cr, and NAA/mI abnormalities from the medial frontal lobe single-voxel 1H MRS in both asymptomatic and symptomatic carriers of MAPT mutations. We hypothesized that frontal 1H MRS abnormalities in MAPT mutation carriers precede the clinical symptoms and that the severity of 1H MRS metabolite alterations is associated with the estimated or actual age at disease onset in MAPT mutation carriers.
Methods
Participants
We studied Longitudinal Evaluation of Familial Frontotemporal Dementia Subjects (LEFFTDS) study participants at the Mayo Clinic site. LEFFTDS is a multisite study investigating the biomarkers of disease progression in familial FTLD mutation carriers. The current study included participants who had screened positive for a mutation in MAPT between May 2015 and September 2017 and participated in the 1H MRS study at the Mayo Clinic. Noncarriers from healthy first-degree relatives of the patients were recruited as a control group after DNA screening (n = 25). All participants underwent a clinical examination at the time of magnetic resonance (MR) examination. The behavioral neurologist (B.F.B) who examined all of the participants was blinded to the mutation status and to the 1H MRS findings. None of the participants had structural lesions that could cause cognitive impairment or dementia such as cortical infarction, subdural hematoma, or tumor or had concurrent illness that would interfere with cognitive function other than FTLD at the time of the MR examination.
Nineteen participants from 5 individual families were identified, and their demographics are shown in the table. Of the 19 MAPT mutation carriers, 9 had no clinical symptoms and had a Frontotemporal Lobar Degeneration Clinical Dementia Rating Sum of Boxes (FTLD-CDR SOB)18 score of 0, whom we refer to as asymptomatic MAPT mutation carriers. Ten MAPT mutation carriers were symptomatic and had a mean FTLD-CDR SOB score of 5, including 5 patients with the primary diagnosis of behavioral variant frontotemporal dementia, 4 with mild cognitive impairment (MCI), and 1 with progressive supranuclear palsy (Richardson syndrome). The mutations identified in these symptomatic MAPT mutation carriers were as follows: N279K (n = 5), V337M (n = 3), IVS9-10G>T (n = 1), and R406W (n = 1). Age at disease onset in each family/mutation type was estimated from the median age at symptom onset in symptomatic MAPT mutation carriers of the family obtained from the clinical history and previous publications on these families.16,19–21
Table.
Participants' characteristics

All participants have undergone genetic testing for research. The behavioral neurologists evaluating the participants were blinded to the findings of the genetic testing for research. All symptomatic mutation carriers described in this report have undergone clinical genetic testing; therefore, they and/or their proxies are aware of their mutation status. Most of the asymptomatic mutation carriers included in this report have not undergone clinical genetic testing and thus are not aware of their mutation status. Details for each person have purposefully been excluded to maintain confidentiality.
Standard protocol approvals, registrations, and patient consents
Informed consent was obtained from all participants for participation in the studies, which were approved by the Mayo Institutional Review Board.
1H MRS and MRI
Single-voxel 1H MRS and MRI studies were performed at 3T with an 8-channel phased-array head coil (GE Healthcare, Milwaukee, WI). A 3D high-resolution T1-weighted magnetization-prepared rapid gradient echo (MPRAGE) acquisition with a repetition time/echo time/inversion time of 7/3/900 milliseconds, flip angle of 8°, in-plane resolution of 1.0 mm, and slice thickness of 1.2 mm was performed in the sagittal plane for voxel positioning. 1H MRS studies were performed with the automated MRS package (PROBE/SV; GE Healthcare).1 Point-resolved spectroscopy sequence with a repetition time of 2,000 milliseconds and an echo time of 30 milliseconds with 2,048 data points and 128 excitations was used for the examination. We used outer volume suppression for the suppression of fat signal. An edge mask was set to place outer volume suppression bands around the 6 planes of the voxel.
An 8-cm3 (2 × 2 × 2 cm) voxel was placed by trained MRI technologists on a midsagittal MPRAGE image covering the medial portion of the right and left superior frontal gyrus (figure 1). The frontal lobe voxel had to be sufficiently distanced from the tissue-air-CSF interface at the skull base and the frontal sinuses that are a source of magnetic field inhomogeneity, which reduces 1H MRS quality. The posterior edge of the frontal lobe voxel was in alignment with the posterior border of the genu of the corpus callosum. The posterior lower corner was placed at the border of the corpus callosum. Individual voxel placements were visually evaluated by a trained image analyst for quality control. 1H MR spectra from voxels that were not properly placed according to predetermined anatomic landmarks and those with poor water suppression, lipid contamination, or baseline distortions failed the quality control and were excluded (n = 5; 3 symptomatic and 1 asymptomatic MAPT mutation carriers and 1 noncarrier). After exclusion of poor-quality 1H MR spectra, 44 1H MR spectra were analyzed (19 MAPT mutation carriers and 25 noncarriers).
Figure 1. Voxel location and representative 1H MRS from all participants.

Medial frontal lobe voxel is placed on a midsagittal 3D T1-weighted image (left). Voxel landmarks include the following: (1) the posterior edge of the frontal lobe voxel is in alignment with the posterior border of the genu of the corpus callosum; and (2) the posterior lower corner is at the superior border of the corpus callosum. Examples of proton magnetic resonance spectra (1H MRS) in (A) a noncarrier, (B) an asymptomatic microtubule-associated protein tau (MAPT) mutation carrier, and (C) a patient with behavioral variant frontotemporal dementia (bvFTD) with MAPT mutation. Spectra are scaled to the creatine (Cr) peak as indicated with the dotted red line. The N-acetylaspartate (NAA) peak is decreased in (B) the asymptomatic MAPT mutation carrier and (C) the patient with bvFTD. The myo-inositol (mI) peak is elevated only in (C) the patient with bvFTD.
The prescan algorithm of PROBE makes automatic adjustments to the transmitter and receiver gains and center frequency. The local magnetic field homogeneity is optimized with the 3-plane auto-shim procedure. The flip angle of the third water suppression pulse is adjusted for chemical shift water suppression before point-resolved spectroscopy acquisition. Metabolite intensity ratios are automatically calculated using a previously validated algorithm at the end of each PROBE/SV.22,23 1H MRS metabolite ratios that were analyzed for this study included NAA/Cr, mI/Cr, and NAA/mI.
Genetic analysis
Analysis of MAPT exons 1, 7, and 9 through 13 was performed with primers and conditions that were previously published.3 PCR amplicons were purified with the Multiscreen system (Millipore, Billerica, MA) and then sequenced in both directions using Big Dye chemistry following the manufacturer's protocol (Applied Biosystems, Foster City, CA). Sequence products were purified using the Montage system (Millipore) before being run on an Applied Biosystem 3730 DNA Analyzer. Sequence data were analyzed with either SeqScape (Applied Biosystem) or Sequencher software (Gene Codes, Ann Arbor, MI).
Statistical analysis
Between-group comparisons of the demographic aspects and 1H MRS metabolite ratios were performed with Fisher exact test for sex (we do not expect any family clustering) and linear mixed models to account for within-family correlations. We used Tukey contrasts for pair-wise comparisons. Because age influences metabolite ratios, we used the data from the noncarriers to estimate the age effects on metabolite ratios and adjusted the metabolite ratios in the MAPT mutation carriers for age. The associations between 1H MRS metabolite ratios and time to expected age at onset and time past age at onset were tested with linear mixed models after adjustment for age. We included the interaction between time from symptom onset and group in all metabolites and then reduced to parsimonious models by removing the nonsignificant interactions and then group variables. Because we were interested in specific individual metabolite hypotheses and not a universal null hypothesis and did not want to inflate the probability of a type II error, we did not adjust for multiple comparisons in these analyses.
Data availability
Anonymized data can be shared after a formal review of a request from any qualified investigator.
Results
Characteristics of participants in each group are listed in the table. The MAPT mutation carriers and noncarriers did not differ in sex and level of education. The asymptomatic MAPT mutation carriers (age 36.4 ± 10.3 years) were significantly younger than symptomatic MAPT mutation carriers (age 51.8 ± 9.4 years, p = 0.02) and noncarriers (age 48.6 ± 13.7 years, p = 0.02).
Representative spectra from the 3 clinical groups are shown in figure 1. Asymptomatic MAPT mutation carriers had lower neuronal marker NAA/Cr ratio (p = 0.001) and lower NAA/mI (p = 0.026) than noncarriers, but the mI/Cr (p = 0.76) ratios were not different from noncarriers after adjustment for age. Symptomatic MAPT mutation carriers had significantly lower NAA/Cr (p = 0.01) and NAA/mI (p = 0.01) ratios and a higher mI/Cr (p = 0.02) ratio compared to noncarriers after adjustment for age. No 1H MRS metabolite differences were identified between the symptomatic and asymptomatic MAPT mutation carriers (figure 2).
Figure 2. Box plots of Frontal 1H MRS metabolite ratios in MAPT mutation carriers and noncarriers.
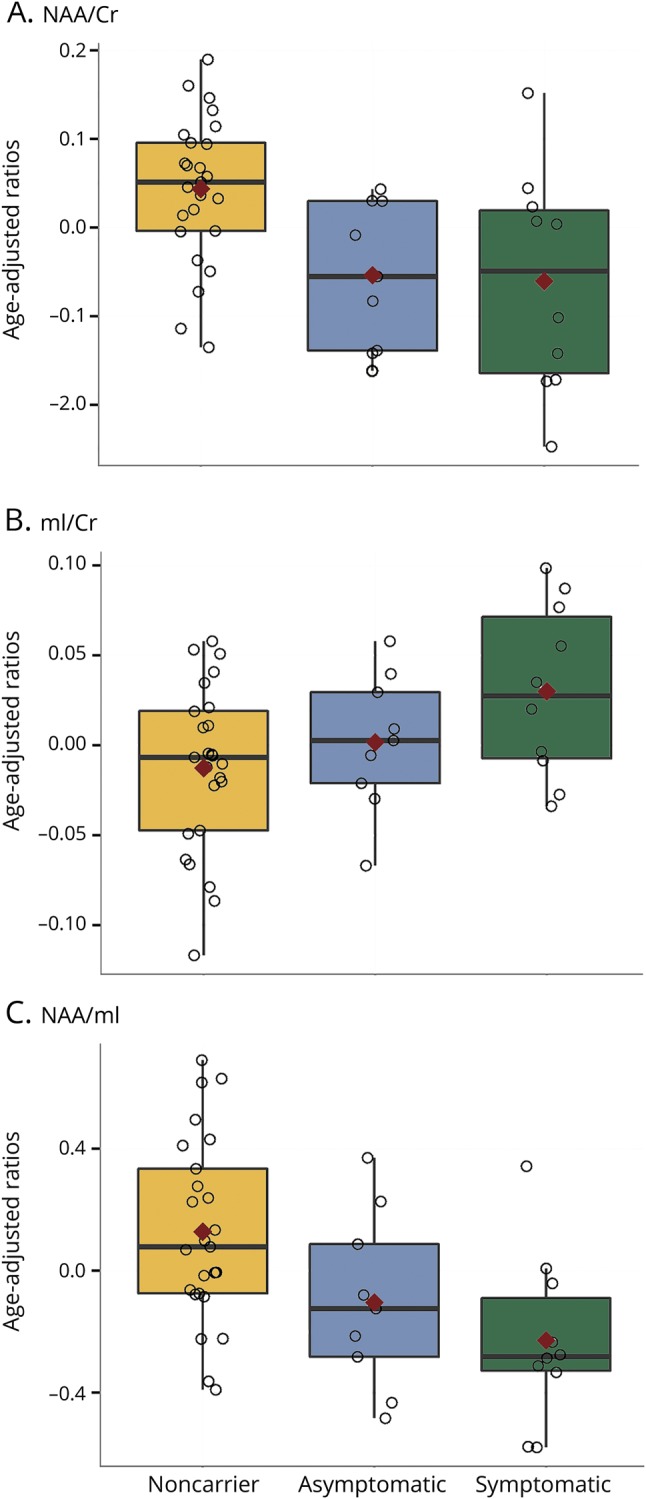
Box plots show the median proton magnetic resonance spectroscopy (1H MRS) metabolite ratios (interquartile range adjusted for age) from the medial frontal lobe voxel in asymptomatic and symptomatic microtubule-associated protein tau (MAPT) mutation carriers and noncarriers. Red marks indicate the mean value adjusted for age for (A) N-acetylaspartate (NAA)/creatine (Cr), (B) myo-inositol (mI)/Cr, and (C) NAA/mI.
Median age at symptom onset in the 5 families ranged from 43 to 50 years. One asymptomatic MAPT mutation carrier was past the median age at symptom onset in that family by 4 years. Symptomatic MAPT mutation carriers were experiencing symptoms for up to 33 years. One patient was diagnosed as having MCI at the time of 1H MRS examination. Linear mixed-effect models found no significant interactions after accounting for family membership of each of the MAPT mutation carriers. One of the symptomatic MAPT mutation carriers had disease duration of 33 years. Because this symptomatic participant was an outlier with long disease duration, we repeated the full analysis by removing this outlier, and the results did not qualitatively change. We fitted parsimonious models with only time from symptom onset as a predictor and found that this was significant for NAA/Cr, mI/Cr, and NAA/mI. The NAA/Cr (R2 = 0.21, p = 0.006) and NAA/mI (R2 = 0.35, p < 0.001) ratios decreased, accompanied by an increase in mI/Cr (R2 = 0.40, p = 0.001), as the ages of MAPT mutation carriers approached and passed the estimated or actual age at symptom onset (figure 3). There were no differences between the slopes of asymptomatic and symptomatic MAPT mutation carriers (p = 0.75 for NAA/Cr, p = 0.93 for mI/Cr, p = 0.99 for NAA/mI) after adjustment for time from symptom onset.
Figure 3. Plots of frontal lobe 1H MRS metabolite ratios and estimated or actual proximity to symptom onset.
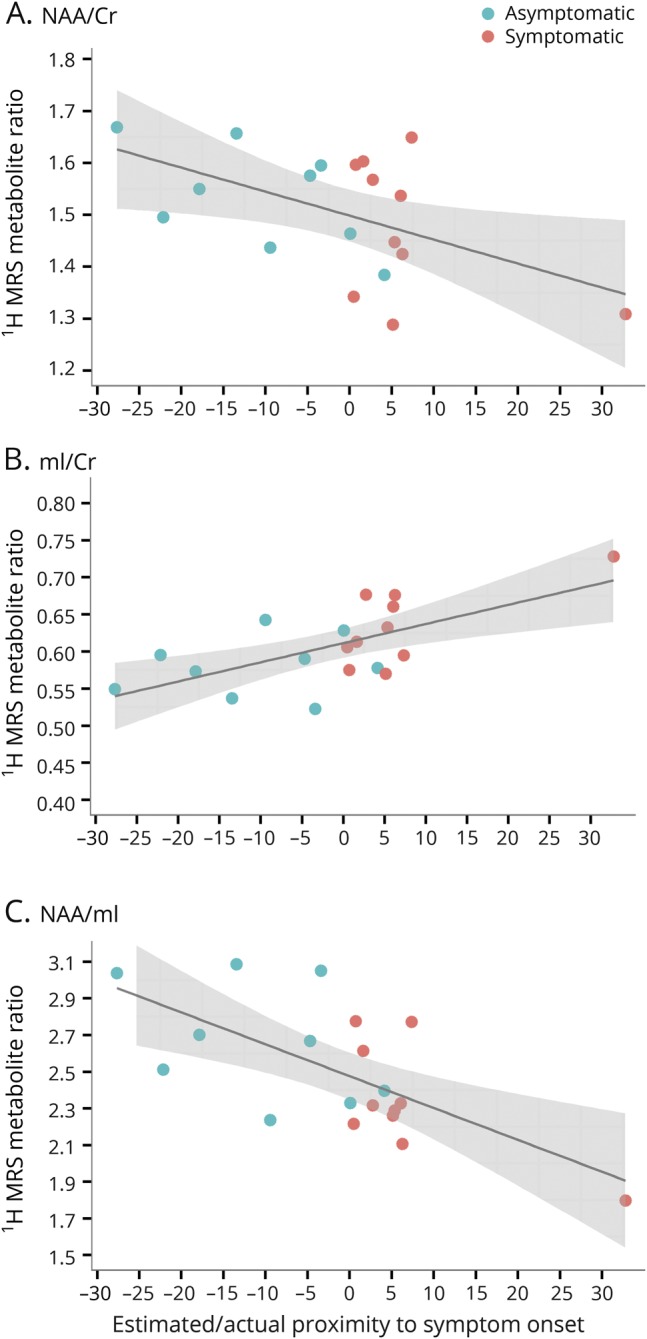
In the horizontal axis, 0 indicates the estimated age at symptom onset for asymptomatic microtubule-associated protein tau (MAPT) mutation carriers based on the median age at symptom onset in symptomatic MAPT mutation carriers from the same family; 0 indicates the age at actual symptom onset in symptomatic MAPT mutation carriers. (A) N-acetylaspartate (NAA)/creatine (Cr), (B) myo-inositol (mI/Cr), and (C) NAA/mI.
Discussion
In this study, we report the medial frontal lobe 1H MRS abnormalities in the asymptomatic and symptomatic carriers of MAPT mutations. We found decreased NAA/Cr and NAA/mI ratios in the asymptomatic MAPT mutation carriers, while symptomatic MAPT mutation carriers were characterized by decreased NAA/Cr and NAA/mI and increased mI/Cr ratios. Furthermore, the severity of 1H MRS metabolite alterations in MAPT mutation carriers was associated with proximity to the estimated or actual age at symptom onset. Medial frontal lobe metabolite alterations in asymptomatic carriers of the causative gene MAPT suggest that early neurodegenerative processes associated with tau pathology precede symptom onset.
Anatomically, frontal and temporal lobes are the most severely affected brain regions in MAPT mutation carriers even in the asymptomatic stage.8,24 Frontal lobes have faster rates of atrophy than the temporal lobes in FTLD patients with MAPT mutations,17 which neuroanatomically correspond to a behavioral clinical syndrome in the majority of the cases with familial FTLD. We found that NAA/Cr levels from the frontal lobe were decreased in both asymptomatic and symptomatic MAPT mutation carriers and continued to decline as the participants approached the estimated age at onset and after the actual age at onset. This finding is consistent with previous reports in presymptomatic carriers of the amyloid precursor protein or presenilin 1,25 in healthy children with APOE ε4 and high air pollution exposures,26 and in the patients with FTLD12 and similar neurodegenerative diseases such as AD, progressive supranuclear palsy, and dementia with Lewy bodies.27,28
NAA/Cr is considered a biomarker of neuronal integrity29 and neuronal viability.30,31 In an antemortem 1H MRS and neuropathology correlation study, a decrease in NAA/Cr was associated with loss of synaptic integrity measured with SV2A and early phospho-tau pathology.32 Furthermore, NAA/Cr ratio alteration was observed very early in an animal model of FTLD-taupathy.33 Taken together with our findings, early decrease in NAA/Cr ratio in the frontal lobe and its relationship with the proximity to age at onset suggest that frontal lobe NAA/Cr can be a biomarker for early neurodegeneration due to the underlying tau pathology in MAPT mutation carriers.
The mI/Cr ratio was elevated in the symptomatic MAPT mutation carriers but not in asymptomatic MAPT mutation carriers in the medial frontal lobe. In a previous study, we found elevated mI/Cr in both symptomatic and asymptomatic MAPT mutation carriers in the posterior cingulate gyrus voxel.16 Our findings in the current study imply differences in pathophysiologic processes among frontal lobe and posterior cingulate gyrus regions before symptom onset in patients with MAPT mutations. Although the significance of mI elevation in neurodegenerative diseases is unclear, mI is present in glial cells.34 Elevated mI is thought to be associated with glial proliferation and astrocytic and microglial activation.35,36 A growing body of evidence demonstrates that the mI/Cr ratio is elevated in MCI and mild AD even with normal NAA/Cr.37–40 It is associated with higher β-amyloid burden in both cognitively healthy participants41 and cognitively normal individuals with preclinical AD.42 Patients with FTLD tend to show more severe elevations in mI/Cr and decreases in NAA/Cr in the frontal cortex compared to patients with AD,12,43 suggesting specific regional involvement in neurodegenerative diseases. However, regional differences that are prominent during early stages of a pathologic process may be lost as neurodegenerative pathology involves most of the cerebral cortex in later stages.
We found an association between the frontal lobe metabolite abnormalities and the estimated and actual proximity to age at onset, characterized by decreasing NAA/Cr and NAA/mI ratios and increasing mI/Cr ratio with the approaching estimated age at onset that continues after the age at onset. These findings suggest that the 1H MRS metabolite abnormalities related to neurodegeneration occur years before symptom onset in MAPT mutation carriers. This association of 1H MRS metabolite abnormalities with estimated and actual proximity to age at onset may be used in tracking disease progression in asymptomatic MAPT mutation carriers, potentially in clinical trials. However, longitudinal studies are needed to determine the rate of change of metabolite abnormalities in MAPT mutation carriers during both the asymptomatic and symptomatic stages.
The median age at symptom onset in symptomatic MAPT mutation carriers of the family was used to estimate the time to symptom onset in asymptomatic MAPT mutation carriers. The clinical symptoms may vary between different mutation subtypes or even within a same mutation family. In our cohort, only 1 asymptomatic MAPT mutation carrier has passed 4 years of estimated age at symptom onset without clinical symptoms. Moreover, 1 symptomatic MAPT mutation carrier diagnosed as having behavioral variant FTLD from the same family of V337M mutation has had behavioral symptoms for >30 years, while the rest of the symptomatic MAPT mutation carriers had a symptomatic phase of <10 years. Typically, the disease duration from onset to death in patients with FTLD is <10 years, with an average duration of 8 years.44,45 The unusually delayed symptom onset and long duration were also reported in another family with FTLD with V337M mutation.46 Studies are needed to fully characterize the heterogeneous clinical presentation across different mutations.
Limitations of our study include a small sample size in a single center and the unequal representation of MAPT families in the carrier and noncarrier groups. Although the anterior temporal lobe also is involved with significant neurodegeneration in symptomatic MAPT mutation carriers, we preferred to study the medial frontal lobe. The reason is that the anterior temporal lobes are proximal to the magnetic susceptibility artifacts at the skull base that affect the quality of the 1H MR spectra. The basal frontal lobe is also close to the magnetic susceptibility artifacts, but we were able to acquire good-quality 1H MR spectra from the medial frontal lobe voxel by placing the voxel away from the skull base. It is possible that other regions in the frontal lobe such as the base of the medial frontal lobe and the anterior temporal lobe would show more severe abnormalities. However, this may be offset by a reduction in the quality of the 1H MRS data. In the current study, we used the vendor-provided sequence, which may limit the number of reliably quantified metabolites and absolute quantification of metabolite concentration. With the use of more advanced acquisition methods, it would be possible to quantify metabolites such as glutamine, glutathione, and scyllo-inosital.47 For example, an advanced 1H-MRS protocol comprising semilocalization by adiabatic selective refocusing localization and FAST(EST)MAP shimming detected lower glutamate concentration in patients with amnestic MCI than in clinically normal controls, indicating early AD pathophysiology.48 By taking a ratio of the metabolites, we eliminated the partial volume averaging of the CSF to our findings. In addition, whole-brain and multivoxel MRS could provide metabolite change in more brain regions than single-voxel MRS.27 The current study demonstrates the potential clinical utility of frontal lobe 1H MRS as a biomarker of early neurodegenerative process in MAPT mutation carriers. In the future, 1H MRS alterations in the frontal lobes of MAPT mutation carriers need to be confirmed in larger studies, and longitudinal studies are needed to elucidate the sequential order of alterations in these 1H MRS metabolites during the evolution of FTLD in MAPT mutation carriers.
Acknowledgment
The authors thank their patients and families for their contributions.
Glossary
- AD
Alzheimer disease
- Cr
creatine
- FTLD
frontotemporal lobar degeneration
- FTLD-CDR SOB
Frontotemporal Lobar Degeneration Clinical Dementia Rating Sum of Boxes
- 1H MRS
proton magnetic resonance spectroscopy
- LEFFTDS
Longitudinal Evaluation of Familial Frontotemporal Dementia Subjects
- MAPT
microtubule-associated protein tau
- MCI
mild cognitive impairment
- mI
myo-inositol
- MPRAGE
magnetization-prepared rapid gradient echo
- MR
magnetic resonance
- NAA
N-acetylaspartate
Author contributions
Q. Chen: design or conceptualization of the study, data collection, analysis and interpretation of the data, drafting the manuscript. B.F. Boeve: data collection, analysis or interpretation of the data, revising the manuscript, and study funding. N. Tosakulwong and T. Lesnick: analysis or interpretation of the data, statistical analysis, revising the manuscript. D. Brushaber and C. Dheel: analysis or interpretation of the data, revising the manuscript. J. Fields, L. Forsberg, R. Gavrilova, D. Gearhart, and D. Haley: data collection, analysis or interpretation of the data, revising the manuscript. J.L. Gunter, J. Graff-Radford, D. Jones, D. Knopman, N. Graff-Radford, and R. Kraft: analysis or interpretation of the data, revising the manuscript. M. Lapid and R. Rademakers: data collection, analysis or interpretation of the data, revising the manuscript. J. Syrjanen: analysis or interpretation of the data, revising the manuscript. Z.K. Wszolek: analysis or interpretation of the data, revising the manuscript, communicating with the research participants, and study funding. H. Rosen and A.L. Boxer: analysis or interpretation of the data, revising the manuscript. K. Kantarci: design or conceptualization of the study, data collection, analysis and interpretation of the data, drafting the manuscript, study funding.
Study funding
This study is funded by U01 AG 45390, R01 AG 40042, and P50 AG 16574. The corresponding author had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Disclosure
Q. Chen reports no disclosures relevant to the manuscript. B. Boeve has served as an investigator for clinical trials sponsored by GE Healthcare and Axovant. He receives royalties from the publication of Behavioral Neurology of Dementia (Cambridge Medicine, 2009, 2017). He serves on the Scientific Advisory Board of the Tau Consortium. He receives research support from NIH, the Mayo Clinic Dorothy and Harry T. Mangurian Jr. Lewy Body Dementia Program, and the Little Family Foundation. N. Tosakulwong, T. Lesnick, D. Brushaber, and C. Dheel report no disclosures relevant to the manuscript. J. Fields, L. Forsberg, and R. Gavrilova receive research support from the NIH. D. Gearhart, D. Haley, and J. Gunter report no disclosures relevant to the manuscript. J. Graff-Radford receives research support from the NIH. D. Jones receives research support from the NIH and the Minnesota Partnership for Biotechnology and Medical Genomics. D. Knopman serves on the Data Safety Monitoring Board of the Dominantly Inherited Alzheimer Network Trials Unit (DIAN-TU) study and is a site principal investigator for clinical trials. N. Graff-Radford receives royalties from UpToDate and has participated in multicenter therapy studies by sponsored by Biogen, TauRx, AbbVie, Novartis, and Lilly. He receives research support from the NIH. R. Kraft and M. Lapid report no disclosures relevant to the manuscript. R. Rademakers receives research funding from the NIH and the Bluefield Project to Cure Frontotemporal Dementia. J. Syrjanen reports no disclosures relevant to the manuscript. Z.K. Wszolek is partially supported by the NIH/National Institute of Neurological Disorders and Stroke (NINDS) P50 NS072187, NIH/National Institute on Aging (NIA) (primary), and NIH/NINDS (secondary) 1U01AG045390-01A1, Mayo Clinic Center for Regenerative Medicine, the gifts from Carl Edward Bolch, Jr., and Susan Bass Bolch, The Sol Goldman Charitable Trust, and Donald G. and Jodi P. Heeringa. H. Rosen has received research support from Biogen Pharmaceuticals, has consulting agreements with Wave Neuroscience and Ionis Pharmaceuticals, and receives research support from the NIH. A. Boxer receives research support from the NIH, the Tau Research Consortium, the Association for Frontotemporal Degeneration, the Bluefield Project to Cure Frontotemporal Dementia, Corticobasal Degeneration Solutions, the Alzheimer's Drug Discovery Foundation, and the Alzheimer's Association. He has served as a consultant for Aeton, Abbvie, Alector, Amgen, Arkuda, Ionis, Iperian, Janssen, Merck, Novartis, Samumed, Toyama, and UCB. He has received research support from Avid, Biogen, BMS, C2N, Cortice, Eli Lilly, Forum, Genentech, Janssen, Novartis, Pfizer, Roche, and TauRx. K. Kantarci serves on the Data Safety Monitoring Board for Takeda Global Research & Development Center, Inc. She receives research funding from the NIH, Alzheimer's Drug and Discovery Foundation, Avid Radiopharmaceuticals, and Eli Lilly. Go to Neurology.org/N for full disclosures.
References
- 1.Bang J, Spina S, Miller BL. Frontotemporal dementia. Lancet 2015;386:1672–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rohrer JD, Warren JD. Phenotypic signatures of genetic frontotemporal dementia. Curr Opin Neurol 2011;24:542–549. [DOI] [PubMed] [Google Scholar]
- 3.Hutton M, Lendon CL, Rizzu P, et al. Association of missense and 5'-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 1998;393:702–705. [DOI] [PubMed] [Google Scholar]
- 4.Poorkaj P, Bird TD, Wijsman E, et al. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol 1998;43:815–825. [DOI] [PubMed] [Google Scholar]
- 5.Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci USA 1998;95:7737–7741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ingram EM, Spillantini MG. Tau gene mutations: dissecting the pathogenesis of FTDP-17. Trends Mol Med 2002;8:555–562. [DOI] [PubMed] [Google Scholar]
- 7.Bunker JM, Kamath K, Wilson L, Jordan MA, Feinstein SC. FTDP-17 mutations compromise the ability of tau to regulate microtubule dynamics in cells. J Biol Chem 2006;281:11856–11863. [DOI] [PubMed] [Google Scholar]
- 8.Cash DM, Bocchetta M, Thomas DL, et al. Patterns of gray matter atrophy in genetic frontotemporal dementia: results from the GENFI study. Neurobiol Aging 2018;62:191–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kantarci K, Knopman DS, Dickson DW, et al. Alzheimer disease: postmortem neuropathologic correlates of antemortem 1H MR spectroscopy metabolite measurements. Radiology 2008;248:210–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kantarci K. Magnetic resonance spectroscopy in common dementias. Neuroimaging Clin N Am 2013;23:393–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Graff-Radford J, Boeve BF, Murray ME, et al. Regional proton magnetic resonance spectroscopy patterns in dementia with Lewy bodies. Neurobiol Aging 2014;35:1483–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ernst T, Chang L, Melchor R, Mehringer CM. Frontotemporal dementia and early Alzheimer disease: differentiation with frontal lobe H-1 MR spectroscopy. Radiology 1997;203:829–836. [DOI] [PubMed] [Google Scholar]
- 13.Waragai M, Moriya M, Nojo T. Decreased N-acetyl aspartate/myo-inositol ratio in the posterior cingulate cortex shown by magnetic resonance spectroscopy may be one of the risk markers of preclinical Alzheimer's disease: a 7-year follow-up study. J Alzheimers Dis 2017;60:1411–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Joe E, Medina LD, Ringman JM, O'Neill J. (1)H MRS spectroscopy in preclinical autosomal dominant Alzheimer disease. Brain Imaging Behav Epub 2018 Jun 16. [DOI] [PMC free article] [PubMed]
- 15.Kizu O, Yamada K, Ito H, Nishimura T. Posterior cingulate metabolic changes in frontotemporal lobar degeneration detected by magnetic resonance spectroscopy. Neuroradiology 2004;46:277–281. [DOI] [PubMed] [Google Scholar]
- 16.Kantarci K, Boeve BF, Wszolek ZK, et al. MRS in presymptomatic MAPT mutation carriers: a potential biomarker for tau-mediated pathology. Neurology 2010;75:771–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Whitwell JL, Boeve BF, Weigand SD, et al. Brain atrophy over time in genetic and sporadic frontotemporal dementia: a study of 198 serial magnetic resonance images. Eur J Neurol 2015;22:745–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Knopman DS, Kramer JH, Boeve BF, et al. Development of methodology for conducting clinical trials in frontotemporal lobar degeneration. Brain 2008;131:2957–2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wszolek ZK, Pfeiffer RF, Bhatt MH, et al. Rapidly progressive autosomal dominant parkinsonism and dementia with pallido-ponto-nigral degeneration. Ann Neurol 1992;32:312–320. [DOI] [PubMed] [Google Scholar]
- 20.Malkani R, D'Souza I, Gwinn-Hardy K, Schellenberg GD, Hardy J, Momeni P. A MAPT mutation in a regulatory element upstream of exon 10 causes frontotemporal dementia. Neurobiol Dis 2006;22:401–403. [DOI] [PubMed] [Google Scholar]
- 21.Arvanitakis Z, Witte RJ, Dickson DW, et al. Clinical-pathologic study of biomarkers in FTDP-17 (PPND family with N279K tau mutation). Parkinsonism Relat Disord 2007;13:230–239. [DOI] [PubMed] [Google Scholar]
- 22.Webb PG, Sailasuta N, Kohler SJ, Raidy T, Moats RA, Hurd RE. Automated single-voxel proton MRS: technical development and multisite verification. Magn Reson Med 1994;31:365–373. [DOI] [PubMed] [Google Scholar]
- 23.Soher BJ, Hurd RE, Sailasuta N, Barker PB. Quantitation of automated single-voxel proton MRS using cerebral water as an internal reference. Magn Reson Med 1996;36:335–339. [DOI] [PubMed] [Google Scholar]
- 24.Jones DT, Knopman DS, Graff-Radford J, et al. In vivo (18)F-AV-1451 tau PET signal in MAPT mutation carriers varies by expected tau isoforms. Neurology 2018;90:e947–e954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Godbolt AK, Waldman AD, MacManus DG, et al. MRS shows abnormalities before symptoms in familial Alzheimer disease. Neurology 2006;66:718–722. [DOI] [PubMed] [Google Scholar]
- 26.Calderon-Garciduenas L, Mora-Tiscareno A, Melo-Sanchez G, et al. A critical proton MR spectroscopy marker of Alzheimer's disease early neurodegenerative change: low hippocampal NAA/Cr ratio impacts APOE varepsilon4 Mexico City children and their parents. J Alzheimers Dis 2015;48:1065–1075. [DOI] [PubMed] [Google Scholar]
- 27.Su L, Blamire AM, Watson R, He J, Hayes L, O'Brien JT. Whole-brain patterns of (1)H-magnetic resonance spectroscopy imaging in Alzheimer's disease and dementia with Lewy bodies. Transl Psychiatry 2016;6:e877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guevara CA, Blain CR, Stahl D, Lythgoe DJ, Leigh PN, Barker GJ. Quantitative magnetic resonance spectroscopic imaging in Parkinson's disease, progressive supranuclear palsy and multiple system atrophy. Eur J Neurol 2010;17:1193–1202. [DOI] [PubMed] [Google Scholar]
- 29.Valenzuela MJ, Sachdev P. Magnetic resonance spectroscopy in AD. Neurology 2001;56:592–598. [DOI] [PubMed] [Google Scholar]
- 30.Bates TE, Strangward M, Keelan J, Davey GP, Munro PM, Clark JB. Inhibition of N-acetylaspartate production: implications for 1H MRS studies in vivo. Neuroreport 1996;7:1397–1400. [PubMed] [Google Scholar]
- 31.Petroff OA, Errante LD, Kim JH, Spencer DD. N-acetyl-aspartate, total creatine, and myo-inositol in the epileptogenic human hippocampus. Neurology 2003;60:1646–1651. [DOI] [PubMed] [Google Scholar]
- 32.Murray ME, Przybelski SA, Lesnick TG, et al. Early Alzheimer's disease neuropathology detected by proton MR spectroscopy. J Neurosci 2014;34:16247–16255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim J, Choi IY, Duff KE, Lee P. Progressive pathological changes in neurochemical profile of the hippocampus and early changes in the olfactory bulbs of tau transgenic mice (rTg4510). Neurochem Res 2017;42:1649–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Glanville NT, Byers DM, Cook HW, Spence MW, Palmer FB. Differences in the metabolism of inositol and phosphoinositides by cultured cells of neuronal and glial origin. Biochim Biophys Acta 1989;1004:169–179. [DOI] [PubMed] [Google Scholar]
- 35.Brand A, Richter-Landsberg C, Leibfritz D. Multinuclear NMR studies on the energy metabolism of glial and neuronal cells. Dev Neurosci 1993;15:289–298. [DOI] [PubMed] [Google Scholar]
- 36.Yamada T, McGeer E, Schelper R, et al. Histological and biochemical pathology in a family with autosomal dominant Parkinsonism and dementia. Neurol Psychiatry Brain Res 1993;2:26–35. [Google Scholar]
- 37.Kantarci K, Jack CR, Jr., Xu YC, et al. Regional metabolic patterns in mild cognitive impairment and Alzheimer's disease: a 1H MRS study. Neurology 2000;55:210–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Catani M, Cherubini A, Howard R, et al. (1)H-MR spectroscopy differentiates mild cognitive impairment from normal brain aging. Neuroreport 2001;12:2315–2317. [DOI] [PubMed] [Google Scholar]
- 39.Sheikh-Bahaei N, Sajjadi SA, Manavaki R, McLean M, O'Brien JT, Gillard JH. Positron emission tomography-guided magnetic resonance spectroscopy in Alzheimer disease. Ann Neurol 2018;83:771–778. [DOI] [PubMed] [Google Scholar]
- 40.Huang W, Alexander GE, Chang L, et al. Brain metabolite concentration and dementia severity in Alzheimer's disease: a (1)H MRS study. Neurology 2001;57:626–632. [DOI] [PubMed] [Google Scholar]
- 41.Nedelska Z, Przybelski SA, Lesnick TG, et al. (1)H-MRS metabolites and rate of beta-amyloid accumulation on serial PET in clinically normal adults. Neurology 2017;89:1391–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Voevodskaya O, Sundgren PC, Strandberg O, et al. Myo-inositol changes precede amyloid pathology and relate to APOE genotype in Alzheimer disease. Neurology 2016;86:1754–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mihara M, Hattori N, Abe K, Sakoda S, Sawada T. Magnetic resonance spectroscopic study of Alzheimer's disease and frontotemporal dementia/pick complex. Neuroreport 2006;17:413–416. [DOI] [PubMed] [Google Scholar]
- 44.Cosseddu M, Benussi A, Gazzina S, et al. Mendelian forms of disease and age at onset affect survival in frontotemporal dementia. Amyotroph Lateral Scler Frontotemporal Degener 2018;19:87–92. [DOI] [PubMed] [Google Scholar]
- 45.Hodges JR, Davies R, Xuereb J, Kril J, Halliday G. Survival in frontotemporal dementia. Neurology 2003;61:349–354. [DOI] [PubMed] [Google Scholar]
- 46.Domoto-Reilly K, Davis MY, Keene CD, Bird TD. Unusually long duration and delayed penetrance in a family with FTD and mutation in MAPT (V337M). Am J Med Genet B Neuropsychiatr Genet 2017;174:70–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barbagallo G, Morelli M, Quattrone A, et al. In vivo evidence for decreased scyllo-inositol levels in the supplementary motor area of patients with progressive supranuclear palsy: a proton MR spectroscopy study. Parkinsonism Relat Disord 2019;62:185–191. [DOI] [PubMed] [Google Scholar]
- 48.Zeydan B, Deelchand DK, Tosakulwong N, et al. Decreased glutamate levels in patients with amnestic mild cognitive impairment: an sLASER proton MR spectroscopy and PiB-PET study. J Neuroimaging 2017;27:630–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Anonymized data can be shared after a formal review of a request from any qualified investigator.