

Abstract
In many lichen-forming fungi, molecular phylogenetic analyses lead to the discovery of cryptic species within traditional morphospecies. However, in some cases, molecular sequence data also questions the separation of phenotypically characterised species. Here we apply an integrative taxonomy approach – including morphological, chemical, molecular, and distributional characters – to re-assess species boundaries in a traditionally speciose group of hair lichens, Bryoria sect. Implexae. We sampled multilocus sequence and microsatellite data from 142 specimens from a broad intercontinental distribution. Molecular data included DNA sequences of the standard fungal markers ITS, IGS, GAPDH, two newly tested loci (FRBi15 and FRBi16), and SSR frequencies from 18 microsatellite markers. Datasets were analysed with Bayesian and maximum likelihood phylogenetic reconstruction, phenogram reconstruction, STRUCTURE Bayesian clustering, principal coordinate analysis, haplotype network, and several different species delimitation analyses (ABGD, PTP, GMYC, and DISSECT). Additionally, past population demography and divergence times are estimated. The different approaches to species recognition do not support the monophyly of the 11 currently accepted morphospecies, and rather suggest the reduction of these to four phylogenetic species. Moreover, three of these are relatively recent in origin and cryptic, including phenotypically and chemically variable specimens. Issues regarding the integration of an evolutionary perspective into taxonomic conclusions in species complexes, which have undergone recent diversification, are discussed. The four accepted species, all epitypified by sequenced material, are Bryoria fuscescens, B. glabra, B. kockiana, and B. pseudofuscescens. Ten species rank names are reduced to synonymy. In the absence of molecular data, they can be recorded as the B. fuscescens complex. Intraspecific phenotype plasticity and factors affecting the speciation of different morphospecies in this group of Bryoria are outlined.
Keywords: chemotypes, cryptic species, haplotypes, incomplete lineage sorting, integrative taxonomy, microsatellites, speciation, species concepts
INTRODUCTION
Accurate identification and characterization of species is the basis of communication, conservation, resources management, and material used in biological research. However, in groups of relatively recent origin, species delimitation is often difficult (Jakob & Blattner 2006, Leavitt et al. 2011, Lumley & Sperling 2011). Organisms are always evolving, changing in response to either selective pressures or genetic drift, so that delimiting units to accord species names is not always clear (Naciri & Linder 2015). Several phenomena can hinder species delimitation: phylogenetic/phenotypic mismatches (Articus et al. 2002, Mark et al. 2016, Pino-Bodas et al. 2016), ‘intermediate’ specimens between generally accepted taxa (Seymour et al. 2007), hybridization (Konrad et al. 2002, Steinová et al. 2013), an absence of delimited clades (Jakob & Blattner 2006, Lumley & Sperling 2011), or incomplete lineage sorting (Saag et al. 2014, Leavitt et al. 2016). Long-term reproductive isolation may produce structured, non-overlapping lineages, whereas an intraspecific phylogeny, as well as a recent or contemporary speciation event, may produce reticulated lineages (Abbott et al. 2016).
The family Parmeliaceae is one of the most studied amongst lichenised fungi. It contains many genera with species delimitation problems, such as Cetraria aculeata (Lutsak et al. 2017); Letharia (Altermann et al. 2014), the Parmotrema reticulatum complex (Del-Prado et al. 2016), and Pseudephebe (Boluda et al. 2016). In some cases, a lack of correlation between genotypes and phenotypes has led to the recognition of cryptic species within morphologically indistinguishable or scarcely indistinguishable morphospecies (Molina et al. 2011a, b, Leavitt et al. 2012a, b, Singh et al. 2015, Boluda et al. 2016, Del-Prado et al. 2016), and so far, more than 80 cryptic lineages have been detected in Parmeliaceae (Crespo & Lumbsch 2010, Divakar et al. 2010). However, in other cases there is a mismatch between lineages revealed by standard DNA-barcoding markers and long-accepted morphospecies (Articus et al. 2002, Seymour et al. 2007, Velmala et al. 2014, Mark et al. 2016, Kirika et al. 2016a, b, McMullin et al. 2016).
In the morphologically similar ‘beard’ and ‘hair’ lichens of the Alectoria sarmentosa, Bryoria sect. Implexae, and Usnea barbata species complexes (Velmala et al. 2014, Mark et al. 2016, McMullin et al. 2016), DNA sequences from standard barcoding markers show that what were considered well delimited morphospecies are found admixed in a single lineage that may be interpreted as a single phylogenetic species. In such situations, many processes may be operative, including environmental plasticity (Boluda et al. 2016), hybridisation, ancestral polymorphisms, incomplete lineage sorting (Joly et al. 2009), limited value of neutral markers (Bekessy et al. 2003), or morphological variability mediated by low selective pressure, genetic drift, or huge population sizes (Hartl & Clark 2007). In these cases, the use of additional markers, especially highly variable ones (e.g., microsatellites), may contribute to an explanation of the underlying phenomena.
Chemical characters, mainly the production of polyketides, were accorded major importance in species delimitation in lichen-forming fungi in the 1960s and 1970s (Hawksworth 1976, Lumbsch 1988). These compounds are formed by the fungal partner, and that expression can differ according to the position in a thallus or in pure culture. For almost 50 years, chemical products, generally linked to minor morphological differences, have been used to circumscribe species in Bryoria (Hawksworth 1972, Brodo & Hawksworth 1977, Myllys et al. 2011, Velmala et al. 2014). The advent of molecular phylogenetics has enabled such species concepts to be tested, and they have proved particularly wanting in one group of species, those placed in Bryoria sect. Implexae (Myllys et al. 2011, Velmala et al. 2014, Boluda et al. 2015). Velmala et al. (2014) provided DNA sequence data for 11 species in the section, and with the exception of B. glabra, all the other species were intermixed in clades with diverse, and not concordant, chemical and morphological features. Genetically indistinguishable taxa (with the markers used), maintain distinctive phenotypes even when growing in physical contact with one another (Velmala et al. 2014, Boluda et al. 2015), so the variation cannot be attributed solely to ecological factors.
A study on the morphospecies B. fuscescens in central Spain (Boluda et al. 2015) revealed specimens with the same nuclear internal transcribed spacer sequence (nuITS) but different extrolites (compounds formed on the surface of or excreted from hyphae). Subsequent fieldwork across Europe has revealed further combinations of extrolites, and also specimens sharing characters of additional morphospecies. In order to understand the evolutionary processes involved in B. fuscescens and related species we have adopted an integrative approach including morphological, distributional, and chemical data together with DNA sequences from three standard loci (Schoch et al. 2012), two newly tested loci, and eighteen microsatellite (SSRs) markers (Nadyeina et al. 2014). We then analysed these datasets in a rigorous statistical framework to effectively integrate an evolutionary perspective into a revised and defensible taxonomic treatment. These studies are reported here, and we anticipate that the experience gained in this group of lichens will inform how other species complexes with similarly discordant datasets can be addressed.
MATERIALS AND METHODS
Sampling
We examined 142 specimens from 14 countries in Europe, the Mediterranean Basin, and North and South America, representing 11 named morphospecies in Bryoria sect. Implexae (Table 1). Our dataset included 91 of the 97 specimens used by Velmala et al. (2014) in their revision of B. sect. Implexae. Newly obtained sequences are shown in bold in Table 1. Bryoria furcellata was used as outgroup to root the tree (Velmala et al. 2014). Names used in the analyses follow the species concepts adopted in Velmala et al. (2014).
Specimen information and GenBank accession numbers of the Bryoria sect. Implexae samples used in this study. Newly obtained sequences are in bold.
| Taxon | Locality | Source/Voucher | Lab. code | Fig. 2 and Appendix 4 code | Chemistry1 | GenBank accession numbers |
||||
|---|---|---|---|---|---|---|---|---|---|---|
| ITS | IGS | GAPDH | FRBi15 | FRBi16 | ||||||
| Bryoria capillaris | Canary Islands, Tenerife | Velmala et al. (2014) | S192 | 87 | Ale., Bar. | GQ996289 | KJ396490 | GQ996261 | KY026810 | KY002720 |
| Canary Islands, Tenerife | MAF-Lich. 20683 | L15.15 | 95 | Ale., Bar. | KY026899 | KY026945 | KY026992 | KY026807 | KY002718 | |
| Finland, Etelä-Häme | Velmala et al. (2014) | L141 | 84 | Ale., Bar. | FJ668493 | FJ668455 | FJ668399 | KY026806 | KY002697 | |
| Finland, Etelä-Savo | Velmala et al. (2014) | L211 | 85 | Ale., Bar. | GQ996287 | KJ396487 | GQ996259 | KY026809 | KY002711 | |
| Finland, Uusimaa | Velmala et al. (2014) | S2 | 88 | Ale., Bar | KJ396433 | KJ396489 | KJ954306 | KY026811 | KY002729 | |
| Greece, Peloponese | MAF-Lich. 19670 | L06.10 | 91 | Ale., Bar. | KY026894 | KY026940 | KY026987 | KY026801 | KY002715 | |
| Norway, Nord-Trøndelag | Velmala et al. (2014) | L270 | 86 | Bar. | GQ996288 | KJ396488 | GQ996260 | – | – | |
| Spain, Lérida | MAF-Lich. 19672 | L07.15 | 90 | Ale., Bar. | KY026895 | KY026941 | KY026988 | KY026802 | KY002716 | |
| Spain, Madrid | MAF-Lich. 19664 | L01.17 | 89 | Ale., Bar. | KY026893 | KY026939 | KY026986 | KY026800 | KY002694 | |
| Spain, Navarra | MAF-Lich. 19674 | L08.12 | 92 | Ale., Bar. | KY026896 | KY026942 | KY026989 | KY026803 | KY002695 | |
| Spain, Teruel | MAF-Lich. 20682 | L14.02 | 94 | Ale., Bar., Fum., Pso. | KY026898 | KY026944 | KY026991 | KY026805 | KY002717 | |
| Sweden, Västerbotten | MAF-Lich. 19685 | L13.03 | 93 | Ale., Bar. | KY026897 | KY026943 | KY026990 | KY026804 | KY002696 | |
| Switzerland, Bivio | MAF-Lich. 20687 | L16.21 | 96 | Ale., Bar., Nor., Pso. | KY026900 | KY026946 | KY026993 | KY026808 | KY002719 | |
| B. friabilis | Canada | Velmala et al. (2014) | L407 | 15 | Gyr. | KJ396435 | KJ396492 | KJ954308 | KY026812 | KY002751 |
| Canada, British Columbia | Velmala et al. (2014) | L355 | 14 | Gyr. | KJ396434 | KJ396491 | KJ954307 | – | – | |
| Canada, British Columbia | MAF-Lich. 20602 | fri_02 | 17 | Gyr. | – | KY083555 | – | – | – | |
| USA, Alaska | Velmala et al. (2014) | S395a | 16 | Gyr. | KJ576728 | KJ396493 | KJ599481 | – | – | |
| B. furcellata | Finland, Etelä-Savo | Velmala et al. (2014) | L147 | – | Fum. | HQ402722 | KJ396494 | HQ402627 | – | – |
| B. fuscescens | Canada | Velmala et al. (2014) | S259 | 71 | Fum. | KJ396441 | KJ396506 | KJ954313 | – | – |
| Canada, Alberta | Velmala et al. (2014) | S256 | 70 | Fum. | GQ996307 | KJ396505 | GQ996280 | KY026825 | KY002702 | |
| Canada, Alberta | Velmala et al. (2014) | S260a | 72 | Fum. | KJ396442 | KJ396507 | KJ954314 | – | – | |
| Canada, Alberta | Velmala et al. (2014) | S261 | 73 | Fum. | KJ396443 | KJ396509 | KJ954315 | – | – | |
| Canada, Alberta | Velmala et al. (2014) | S267 | 74 | Fum. | KJ576716 | KJ396510 | KJ599469 | – | – | |
| Canada, Alberta | Velmala et al. (2014) | S272 | 75 | Fum. | KJ576717 | KJ396511 | KJ599470 | – | – | |
| Canada, Alberta | Velmala et al. (2014) | S369 | 77 | Fum. | KJ396444 | KJ396514 | KJ954316 | – | – | |
| Canada, Alberta | Velmala et al. (2014) | S379 | 78 | Fum. | KJ396445 | KJ396515 | KJ954317 | – | – | |
| Canada, Alberta | Velmala et al. (2014) | S380 | 79 | Fum. | KJ396446 | KJ396516 | KJ954318 | – | – | |
| Canada. Alberta | Velmala et al. (2014) | S274 | 76 | Abs. | GQ996303 | KJ396512 | GQ996276 | – | – | |
| Canary Islands, Tenerife | MAF-Lich. 20684 | L15.21 | 83 | Fum. | KY026901 | KY026949 | KY026996 | KY026817 | – | |
| Finland, Ahvenanmaa | Velmala et al. (2014) | L149 | 61 | Fum. | GQ996290 | KJ396496 | GQ996262 | KY026816 | KY002698 | |
| Finland, Etelä-Savo (epitype) | Velmala et al. (2014) | L139 | 60 | Fum. | KJ396436 | KJ396495 | KJ954309 | KY026815 | – | |
| Finland, Koillismaa | Velmala et al. (2014) | S24 | 69 | Fum. | KJ576715 | KJ396501 | KJ599468 | KY026824 | – | |
| Finland, Koillismaa | Velmala et al. (2014) | S56 | 80 | Fum. | GQ996291 | KJ396502 | GQ996263 | – | KY002732 | |
| Finland, Oulun Pohjanmaa | Velmala et al. (2014) | L189 | 63 | Fum. | GQ996305 | KJ396498 | GQ996278 | KY026819 | KY002699 | |
| Finland, Pohjois-Karjala | Velmala et al. (2014) | S109 | 67 | Fum. | KJ396440 | KJ396503 | KJ954312 | KY026822 | KY002701 | |
| Greenland | Velmala et al. (2014) | L232 | 65 | Abs. | GQ996304 | KJ396500 | GQ996277 | – | KY002700 | |
| Norway, Sogn og Fjordane | Velmala et al. (2014) | L224 | 64 | Fum. | KJ396437 | KJ396499 | KJ954310 | KY026820 | KY002731 | |
| Norway, Telemark | Velmala et al. (2014) | L305 | 66 | Fum. | KJ396438 | – | – | KY026821 | – | |
| Norway, Troms | MAF-Lich. 19681 | L12.03 | 81 | Fum. | KY026902 | KY026947 | KY026994 | KY026813 | – | |
| Norway, Troms | MAF-Lich. 19682 | L12.05 | 82 | Fum. | KY026903 | KY026948 | KY026995 | KY026814 | KY002730 | |
| Russia, Perm Territory | Velmala et al. (2014) | S157 | 68 | Fum. | GQ996306 | KJ396504 | GQ996279 | KY026823 | KY002742 | |
| Sweden, Södermanland | Velmala et al. (2014) | L160 | 62 | Fum. | GQ996300 | KJ396497 | GQ996272 | KY026818 | – | |
| B. glabra | Chile, IX Region | MAF-Lich. 20595 | Bg1 | 5 | Fum. | KY026904 | KY026950 | KY026997 | – | – |
| Chile, IX Region | MAF-Lich. 20596 | Bg2 | 6 | Fum. | KY026905 | KY026951 | KY026998 | – | KY002693 | |
| Chile, IX Region | MAF-Lich. 20597 | Bg3 | 7 | Fum. | KY026906 | KY026952 | KY026999 | – | KY002691 | |
| Chile, IX Region | MAF-Lich. 20598 | Bg4 | 8 | Fum. | KY026907 | KY026953 | KY027000 | – | – | |
| Chile, IX Region | MAF-Lich. 20599 | Bg5 | 9 | Fum. | KY026908 | KY026954 | KY027001 | – | KY002690 | |
| Finland, Koillismaa | Velmala et al. (2014) | L186 | 1 | Abs. | FJ668494 | FJ668456 | FJ668400 | – | KY002688 | |
| USA, Alaska (epitype) | Dillman 11May11:1 (UBC) | L406 | 2 | Abs. | KY026909 | KY083556 | KY026955 | – | KY002692 | |
| B. glabra (cont.) | USA, Alaska | Dillman 26July11:4 (UBC) | L414 | 3 | Abs. | KY026910 | KY083557 | KY026956 | – | – |
| USA, Washington | Björk 1546 (UBC) | S388 | 4 | Fum. | KY026911 | – | – | – | KY002689 | |
| B. implexa | Cyprus, Troodos | MAF-Lich. 19683 | L11.15 | 105 | Pso. | KY026915 | KY026960 | KY027005 | KY026829 | KY002704 |
| Finland, Koillismaa | Velmala et al. (2014) | S22 | 98 | Pso. | GQ996294 | KJ396517 | GQ996266 | KY026832 | KY002714 | |
| Finland, Koillismaa | Velmala et al. (2014) | S36 | 99 | Pso. | KJ576719 | KJ396518 | KJ599472 | KY026833 | – | |
| Finland, Koillismaa | Velmala et al. (2014) | S39 | 100 | Pso. | GQ996293 | KJ396519 | GQ996265 | KY026834 | KY002733 | |
| Finland, Koillismaa | Velmala et al. (2014) | S67 | 101 | Pso. | KJ396447 | KJ396520 | KJ954319 | KY026835 | – | |
| Greece, Peloponese | MAF-Lich. 19669 | L06.05 | 103 | Fum., Pso. | KY026913 | KY026958 | KY027003 | KY026827 | – | |
| Morocco, Rif | MAF-Lich. 19679 | L10.03 | 104 | Pso. | KY026914 | KY026959 | KY027004 | KY026828 | KY002721 | |
| Russia, Murmansk | Velmala et al. (2014) | S168 | 97 | Pso. | KJ396448 | KJ396521 | KJ954320 | KY026831 | KY002705 | |
| Spain, Madrid | MAF-Lich. 19663 | L01.01 | 102 | Pso. | KY026912 | KY026957 | KY027002 | KY026826 | KY002703 | |
| Switzerland, Bivio | MAF-Lich. 20685 | L16.15 | 106 | Pso. | KY026916 | KY026961 | KY027006 | KY026830 | – | |
| B. inactiva | Canada, British Columbia | Velmala et al. (2014) | L206 | 18 | Abs. | GQ996283 | KJ396522 | GQ996255 | – | KY002760 |
| Canada, British Columbia | Velmala et al. (2014) | L323b | 19 | Abs. | KJ396449 | KJ396523 | KJ954321 | KY026836 | – | |
| Canada, British Columbia | Velmala et al. (2014) | L358 | 21 | Abs. | KJ396451 | KJ396525 | KJ954323 | KY026838 | – | |
| Canada, British Columbia | Velmala et al. (2014) | S239a | 22 | Abs. | GQ996284 | KJ396526 | GQ996256 | KY026839 | – | |
| Canada, British Columbia | Velmala et al. (2014) | S392a | 24 | Abs. | KJ396452 | KJ396528 | KJ954324 | – | – | |
| Canada, British Columbia (holotype) | Velmala et al. (2014) | L347 | 20 | Abs. | KJ396450 | KJ396524 | KJ954322 | KY026837 | KY002761 | |
| USA, Alaska | Velmala et al. (2014) | S384 | 23 | Abs. | KJ576724 | KJ396527 | KJ599479 | – | – | |
| B. kockiana | USA, Alaska (holotype) | Velmala et al. (2014) | L394 | 10 | Pso. | KJ396453 | KJ396529 | KJ954325 | KY026840 | KY002764 |
| USA, Alaska | Velmala et al. (2014) | L396 | 11 | Pso. | KJ396454 | KJ396530 | KJ954326 | KY026841 | KY002765 | |
| B. kuemmerleana | Iran, East Azarbaijan | Velmala et al. (2014) | L244a | 107 | Nor. | GQ996295 | KJ396531 | GQ996267 | KY026846 | – |
| Morocco, Middle Atlas | MAF-Lich. 19677 | L09.04 | 113 | Nor. | KY026918 | KY026963 | KY027008 | KY026843 | KY002743 | |
| Morocco, Middle Atlas | MAF-Lich. 19678 | L09.07 | 114 | Nor. | KY026919 | KY026964 | KY027009 | KY026844 | KY002744 | |
| Norway, Nord-Trøndelag | Velmala et al. (2014) | L274 | 108 | Nor. | GQ996296 | KJ396532 | GQ996268 | KY026847 | – | |
| Norway, Nord-Trøndelag | Velmala et al. (2014) | L275 | 109 | Nor. | KJ396455 | KJ396533 | KJ954327 | – | – | |
| Russia, Perm Territory | Velmala et al. (2014) | S160 | 111 | Nor. | KJ396456 | KJ396535 | KJ954328 | KY026849 | – | |
| Spain, Zamora | MAF-Lich. 19667 | L04.03 | 112 | Nor. | KY026917 | KY026962 | KY027007 | KY026842 | – | |
| Sweden, Härjedalen | Velmala et al. (2014) | S128 | 110 | Nor. | KJ576720 | KJ396534 | KJ599473 | KY026848 | KY002706 | |
| Switzerland, Bivio | MAF-Lich. 20686 | L16.17 | 115 | Nor., Pso. | KY026920 | – | KY027010 | KY026845 | – | |
| B. pikei | Canada, Alberta | Velmala et al. (2014) | S382 | 34 | Ale., Bar. | KJ396466 | KJ396547 | KJ954338 | KY026864 | KY002752 |
| Canada, Alberta | Velmala et al. (2014) | S383a | 35 | Ale., Bar. | KJ396467 | KJ396548 | KJ954339 | – | KY002759 | |
| Canada, British Columbia | Velmala et al. (2014) | L197 | 25 | Ale., Bar. | KJ396457 | KJ396536 | KJ954329 | – | – | |
| Canada, British Columbia | Velmala et al. (2014) | L210 | 26 | Ale., Bar. | KJ576714 | KJ396539 | KJ599467 | – | KY002762 | |
| Canada, British Columbia | Velmala et al. (2014) | L421 | 27 | Ale. | KJ396462 | KJ396543 | KJ954334 | KY026850 | KY002753 | |
| Canada, British Columbia | Velmala et al. (2014) | L374 | 28 | Ale., Gyr. | KJ396459 | KJ396540 | KJ954331 | KY026851 | – | |
| Canada, British Columbia | Velmala et al. (2014) | L376 | 29 | Ale., Gyr. | KJ396460 | KJ396541 | KJ954332 | KY026852 | KY002754 | |
| Canada, British Columbia | Velmala et al. (2014) | L377 | 30 | Ale., Gyr. | KJ396461 | KJ396542 | KJ954333 | KY026853 | KY002755 | |
| Canada, British Columbia | Velmala et al. (2014) | S221 | 31 | Ale., Bar. | KJ396463 | KJ396544 | KJ954335 | – | – | |
| Canada, British Columbia | Velmala et al. (2014) | S362 | 32 | Ale., Bar. | KJ396464 | KJ396545 | KJ954336 | – | – | |
| Canada, British Columbia | Velmala et al. (2014) | S368 | 33 | Ale., Bar. | KJ396465 | KJ396546 | KJ954337 | KY026863 | KY002756 | |
| Canada, British Columbia | MAF-Lich. 20601 | pik_c | 51 | Ale. | – | – | – | – | – | |
| Canada, Nova Scotia | MAF-Lich. 20600 | pik_a | 49 | Ale., Bar. | – | – | – | – | – | |
| Canada, Prince Edward Island | MAF-Lich. 20603 | pik_02 | 38 | Ale., Bar. | KY026925 | KY026971 | KY027014 | KY026857 | KY002749 | |
| Canada, Prince Edward Island | MAF-Lich. 20606 | pik_04 | 39 | Ale., Bar. | KY026926 | KY026972 | KY027015 | KY026858 | KY002745 | |
| Canada, Prince Edward Island | MAF-Lich. 20607 | pik_05 | 40 | Ale., Bar. | KY026927 | KY026973 | KY027016 | KY026859 | KY002727 | |
| Canada, Prince Edward Island | MAF-Lich. 20609 | pik_09 | 42 | Ale., Bar. | – | KY026975 | – | KY026861 | KY002724 | |
| Canada, Prince Edward Island | MAF-Lich. 20612 | pik_10 | 43 | Ale., Bar. | KY026921 | KY026965 | – | – | – | |
| B. pikei (cont.) | Canada, Prince Edward Island | MAF-Lich. 20610 | pik_11 | 44 | Ale., Bar. | KY026922 | KY026966 | KY027011 | KY026854 | KY002750 |
| Canada, Prince Edward Island | MAF-Lich. 20622 | pik_12 | 45 | Ale., Bar. | – | KY026967 | – | – | – | |
| Canada, Prince Edward Island | MAF-Lich. 20611 | pik_13 | 46 | Ale., Bar. | KY026923 | KY026968 | KY027012 | KY026855 | KY002726 | |
| Canada, Prince Edward Island | MAF-Lich. 20613 | pik_14 | 47 | Ale., Bar. | KY026924 | KY026969 | KY027013 | KY026856 | KY002728 | |
| Canada, Prince Edward Island | MAF-Lich. 20614 | pik_d | 52 | Ale., Bar. | – | – | – | – | KY002725 | |
| Canada, Quebec | MAF-Lich. 20608 | pik_07 | 41 | Ale., Bar. | – | KY026974 | KY027017 | KY026860 | KY002723 | |
| Canada, Quebec | MAF-Lich. 20605 | pik_15 | 48 | Ale., Bar. | – | KY026970 | – | – | KY002741 | |
| Canada, Quebec | MAF-Lich. 20604 | pik_b | 50 | Ale., Bar. | KY026928 | – | KY027018 | KY026862 | KY002748 | |
| USA, Alaska | Velmala et al. (2014) | S390 | 36 | Ale., Bar. | KJ396468 | KJ396549 | KJ954340 | – | – | |
| USA, Oregon | Velmala et al. (2014) | S394 | 37 | Ale., Bar. | KJ576727 | KJ396550 | KJ599480 | – | – | |
| B. pseudofuscescens | Canada, British Columbia (epitype) | Velmala et al. (2014) | S222 | 53 | Nor. | KJ396469 | KJ396551 | KJ954341 | KY026865 | KY002757 |
| Canada, British Columbia | Velmala et al. (2014) | S232 | 54 | Nor. | KJ396470 | KJ396552 | KJ954342 | KY026866 | – | |
| Canada, British Columbia | Velmala et al. (2014) | S370 | 55 | Nor. | KJ396471 | KJ396553 | KJ954343 | – | – | |
| Canada, British Columbia | Velmala et al. (2014) | S371 | 56 | Nor. | KJ396472 | KJ396554 | KJ954344 | – | – | |
| USA, Alaska | Velmala et al. (2014) | S377 | 57 | Nor. | KJ396473 | KJ396555 | KJ954345 | – | – | |
| USA, Alaska | Velmala et al. (2014) | S386 | 58 | Nor. | KJ576725 | KJ396556 | KJ599478 | – | KY002707 | |
| USA, Alaska | Velmala et al. (2014) | S387 | 59 | Nor. | KJ576726 | KJ396557 | KJ599477 | KY026867 | KY002758 | |
| B. sp. | USA, Alaska | Velmala et al. (2014) | L395 | 12 | Abs. | KJ396486 | KJ396581 | KJ954358 | KY026869 | KY002766 |
| USA, Alaska | Velmala et al. (2014) | L392 | 13 | Abs. | KJ396485 | KJ396580 | KJ954357 | KY026868 | KY002763 | |
| B. vrangiana | Canada, Alberta | Velmala et al. (2014) | S385 | 124 | Fum. | KJ396484 | KJ396578 | KJ954356 | KY026886 | KY002739 |
| Finland, Kainuu | Velmala et al. (2014) | S341b | 123 | Abs. | KJ396483 | KJ396577 | KJ954355 | KY026885 | – | |
| Finland, Kainuu | Velmala et al. (2014) | S72 | 131 | Abs. | KJ396481 | KJ396573 | KJ954353 | KY026892 | KY002740 | |
| Finland, Koillismaa | Velmala et al. (2014) | S10 | 119 | Gyr. | GQ996297 | KJ396564 | GQ996269 | KY026882 | – | |
| Finland, Koillismaa | Velmala et al. (2014) | S42 | 125 | Gyr. | KJ396478 | KJ396566 | KJ954350 | KY026887 | KY002710 | |
| Finland, Koillismaa | Velmala et al. (2014) | S45 | 126 | Abs. | GQ996302 | KJ396568 | GQ996275 | KY026888 | – | |
| Finland, Koillismaa | Velmala et al. (2014) | S57 | 127 | Fum. | KJ576722 | KJ396570 | KJ599475 | – | – | |
| Finland, Koillismaa | Velmala et al. (2014) | S59 | 128 | Fum. | KJ396480 | KJ396571 | KJ954352 | KY026889 | – | |
| Finland, Oulun Pohjanmaa | Velmala et al. (2014) | S196a | 122 | Abs. | KJ396482 | KJ396576 | KJ954354 | KY026884 | – | |
| Finland, Uusimaa | Velmala et al. (2014) | S6 | 130 | Fum. | KJ396477 | KJ396563 | KJ954349 | KY026890 | – | |
| Finland, Varsinais-Suomi | Velmala et al. (2014) | S62 | 129 | Gyr. | KJ576721 | KJ396572 | KJ599474 | KY026891 | – | |
| Italy, Sicily | MAF-Lich. 19668 | L05.17 | 135 | Fum. | KY026931 | KY026978 | KY027021 | KY026872 | KY002709 | |
| Morocco, Rif | MAF-Lich. 19680 | L10.13 | 140 | Abs. | KY026936 | KY026983 | KY027026 | KY026877 | KY002722 | |
| Norway, Nord-Trøndelag | Velmala et al. (2014) | L272 | 116 | Gyr. | GQ996299 | KJ396558 | GQ996271 | KY026880 | KY002747 | |
| Norway, Nord-Trøndelag | Velmala et al. (2014) | L273 | 117 | Abs. | KJ396474 | KJ396559 | KJ954346 | – | – | |
| Norway, Nord-Trøndelag | Velmala et al. (2014) | L300 | 118 | Abs. | GQ996301 | KJ396562 | GQ996274 | KY026881 | KY002746 | |
| Norway, Oppland | Velmala et al. (2014) | L307 | 132 | Gyr. | KJ396439 | – | KJ954311 | – | – | |
| Norway, Troms | MAF-Lich. 19684 | L12.11 | 141 | Fum., Gyr. | KY026937 | KY026984 | KY027027 | KY026878 | KY002738 | |
| Russia, Perm Territory | Velmala et al. (2014) | S164 | 120 | Abs. | GQ996285 | KJ396574 | GQ996257 | KY026883 | KY002712 | |
| Russia, Perm Territory | Velmala et al. (2014) | S166 | 121 | Fum. | GQ996308 | KJ396575 | GQ996273 | – | KY002713 | |
| Spain, Asturias | MAF-Lich. 19666 | L03.07 | 134 | Fum. | KY026930 | KY026977 | KY027020 | KY026871 | KY002708 | |
| Spain, Cáceres | MAF-Lich. 19665 | L02.20 | 133 | Fum. | KY026929 | KY026976 | KY027019 | KY026870 | KY002734 | |
| Spain, Lérida | MAF-Lich. 19671 | L07.03 | 136 | Fum. | KY026932 | KY026979 | KY027022 | KY026873 | KY002735 | |
| Spain, Lérida | MAF-Lich. 19673 | L07.19 | 137 | Abs. | KY026933 | KY026980 | KY027023 | KY026874 | KY002736 | |
| Spain, Navarra | MAF-Lich. 19675 | L08.19 | 138 | Abs. | KY026934 | KY026981 | KY027024 | KY026875 | KY002737 | |
| Spain, Navarra | MAF-Lich. 19676 | L08.20 | 139 | Fum. | KY026935 | KY026982 | KY027025 | KY026876 | – | |
| Sweden, Västerbotten | MAF-Lich. 19686 | L13.12 | 142 | Gyr. | KY026938 | KY026985 | KY027028 | KY026879 | – | |
1 Abs. = No substances detected; Ale. = Alectorialic acid; Bar. = Barbatolic acid; Fum. = Fumarprotocetraric acid; Gyr. = Gyrophoric acid; Nor. = Norstictic acid; Pso. = Psoromic acid.
Morphology and chemistry
The newly studied specimens (Table 1) were examined morphologically under a Nikon SMZ-1000 dissecting microscope, and hand-cut sections studied with a Nikon Eclipse-80i compound microscope equipped with bright field and differential interference contrast (DIC). Habit photographs were taken with a Nikon 105 mm f/2.8D AF Micro-Nikkor Lens coupled to a Nikon D90 camera with daylight. Spot tests (K, C, and PD) and TLC were carried out following Orange et al. (2010). Solvent system C (200 ml toluene / 30 mL acetic acid) was used for TLC, with concentrated acetone extracts at 50 °C spotted onto silica gel 60 F254 aluminium sheets (Merck, Darmstadt, Germany). Spotted sheets were dried for 10 min in an acetic acid atmosphere to maximize resolution. Segments from the same lichen branch were used for both TLC and DNA extraction to avoid the possible risk of taking samples from mixed collections. Morphological and thin layer chromatographic (TLC) analyses of the samples used in Velmala et al. (2014; Table 1) were taken from that study.
DNA dataset
The molecular dataset comprised DNA sequences and SSRs frequencies. DNA extraction was performed with the DNeasy Plant Mini Kit (Qiagen, Barcelona, Spain), following the manufacturer’s instructions.
Eighteen fungal-specific microsatellites markers (Bi01, Bi02, Bi03, Bi04, Bi05, Bi06, Bi07, Bi08, Bi09, Bi10, Bi11, Bi12, Bi13, Bi14, Bi15, Bi16, Bi18 and Bi19) were amplified following Nadyeina et al. (2014) using fluorescently labelled primers. Fragment lengths were determined on an ABI PRISM® 3130 Genetic Analyser (Life Technologies, Carlsbad, CA, USA). Genotyping was performed using GeneScan-500 LIZ as the internal size standard and GeneMapper v. 3.7 (Applied Biosystems, Foster City, CA, USA).
For DNA sequencing, five loci were selected (Table 2), three commonly used as standard markers in fungi (ITS, IGS, and GAPDH), which were also used in Velmala et al. (2014), and two microsatellite flanking regions tested here for the first time (FRBi15 and FRBi16). Microsatellite flanking regions are variable non-coding DNA fragments that can contain phylogenetical signal through a neutral molecular evolution (Zardoya et al. 1996, Chatrou et al. 2009). To explore this possibility, the flanking regions of the 18 microsatellite markers were checked upstream and downstream in the 454 pyrosequencing contigs used for microsatellite searching in Nadyeina et al. (2014). The variability of each region was assessed with the number of variable sites in contigs supported by 2–16 copies. From the 36 regions (two for each of the 18 microsatellites), the most variable flanking regions were in Bi15 and Bi16, and specific primers were designed for those loci (Table 2).
Table 2.
Primer information used in Bryoria sect. Implexae.
| Marker | Description | Primer forward (5’–3’) | Source | Primer reverse (5’–3’) | Source |
|---|---|---|---|---|---|
| ITS | Internal transcribed spacers of the nuclear rDNA including the 5.8S region | ITS1-F: | Gardes & Bruns (1993) | ITS4: | White et al. (1990) |
| CTTGGTCATTTAGAGGAAGTAA | TCCTCCGCTTATTGATATGC | ||||
| IGS | Intergenic spacer of the nuclear rDNA | IGS12b: | Printzen & Ekman (2002) | SSU72R: | Gargas & Taylor (1992) |
| AGTCTGTGGATTAGTGGCCG | TTGCTTAAACTTAGACATG | ||||
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase gene partial sequence | Gpd1-LM: | Myllys et al. (2002) | Gpd2-LM: | Myllys et al. (2002) |
| ATTGGCCGCATCGTCTTCCGCAA | CCACTCGTTGTCGTACCA | ||||
| FRBi15 | Flanking region of Bryoria sect.Implexae microsatellite marker 15 | FRBi15f: | This paper | FRBi15r: | This paper |
| GTCATAAGGGTATCAATCC | TGAAAAGGTTTGGTGACTC | ||||
| FRBI16 | Flanking region of Bryoria sect. Implexae microsatellite marker 16 | FRBi16f: | This paper | FRBi16r: | This paper |
| CGAGGTTTCAGGAAAGGGAA | AGGAAGTGATGTCGAGGT |
New DNA sequences (Table 1) were obtained using polymerase chain reactions (PCRs) as follows: a reaction mixture of 25 μL, containing 12 μL sterile water, 9 μL JumpStartTM REDTaq ReadyMix PCR Reaction Mix (Sigma-Aldrich, St Louis, MI, USA), 1.25 μL of each primer (forward and reverse) at 10 μM, and a 1.5 μL DNA template. Cycling conditions for ITS, GAPDH, FRBi15, and FRBi16 were 2 min at 94 °C; 35 cycles of 30 s at 94 °C; 30 s at 56 °C; 2 min at 72 °C; and a final extension of 5 min at 72 °C. For IGS, the cycling process was: 2 min at 94 °C; 15 cycles of 30 s at 94 °C, 30 s at 55 °C (decreasing 1 °C each cycle down to 40 °C), 2 min at 72 °C, then 35 cycles of 30 s at 94 °C, 30 s at 55 °C; 90 s at 72 °C, and a final extension of 5 min at 72 °C. PCR products were checked and quantified on 1 % agarose gel stained with ethidium bromide and cleaned using Exonuclease I and FastAP Thermosensitive Alkaline Phosphatase (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions. Sequencing was performed with labelling using BigDye Terminator v. 3.1 Kit (Applied Biosystems) as follows: 25 cycles of 20 s at 96 °C, 5 s at 50 °C, and 2 min at 60 °C. PCR products were cleaned-up with the BigDye XTerminator Purification Kit (Applied Biosystems) according to the manufacturer’s instructions. Sequences were obtained in an ABI PRISM 3130 Genetic Analyser (Life Technologies) and manually adjusted using DNA Workbench v. 6 (CLC bio, Aarhus, Denmark) and MEGA5 (Tamura et al. 2011). Newly generated sequences were deposited in GenBank (Table 1 in bold).
Clustering methodologies
Phenetic analyses
Two presence/absence (1/0) matrices were constructed, one for the extrolites detected by TLC, and another with morphology and geography data (Appendix 1). Morphological characters scored comprised those traditionally used to separate morphospecies in the group:
pale/dark thallus colour;
branching angles (acute/obtuse/mixed);
soralia (absent/fissural/tuberculate/both); and
pseudocyphellae (conspicuous/inconspicuous).
For distributions, Old World vs New World was used. The R package cluster (Maechler et al. 2013) was used to obtain the dissimilarity matrix, and then the pvclust package (Suzuki & Shimodaira 2006) was run to obtain a phenogram (Zamora et al. 2013). Multiscale bootstrap resampling with 10 000 bootstrap (bp) replicates was used to obtain approximately unbiased (au) p-values for branch supports. Groups were considered as supported when bp values exceeded 70 or au values exceeded 95.
Phylogenetic tree
Alignments for each locus were performed using MAFFT v. 7 (http://mafft.cbrc.jp/alignment/server/; Katoh & Standley 2013) with the G-INS-i alignment algorithm, a ‘1PAM/K = 2’ scoring matrix, with an offset value of 0.1, and the remaining parameters set as default. Alignments were deposited in TreeBASE under accession nos TB2:S20007 (ITS, IGS, and GAPDH), TB2:S20005 (FRBi15), and TB2:S20004 (FRBi16). RDP v. 4 (Martin et al. 2010) was used to detect potential recombination events, through the methods RDP (Martin & Rybicki 2000), GENECONV (Padidam et al. 1999), Chimaera (Posada & Crandall 2001), Maxchi (Maynard-Smith 1992), Bootscan (Gibbs et al. 2000, Martin et al. 2005), SiScan (Weiller 1998, Gibbs et al. 2000), PhylPro (Weiller 1998), and 3Seq (Boni et al. 2007). Partitionfinder (Lanfear et al. 2012) was used to detect possible intra-locus substitution model variability, resulting in the splitting of the ITS region into ITS1, 5.8S, and ITS2, and coding each codon position separately in GAPDH. Models of DNA sequence evolution for each locus partition were selected with jModeltest v. 2.0 (Darriba et al. 2012), using the Akaike information criterion (AIC, Akaike 1974). The best-fit model of evolution obtained was: ITS1 = TIM2, 5.8S = K80, ITS2 = TIM2ef + G, IGS = TrN + I, GAPDH 1st position = TrN + I, GAPDH 2nd position = F81 + I, GAPDH 3th position = TPM3uf, FRBi15 = TPM3uf + I, FRBi16 = TPM3uf + G. To detect possible topological conflicts among loci, the CADM test (Legendre & Lapointe 2004, Campbell et al. 2011) was performed using the function ‘CADM.global’ implemented in the library ‘ape’ of R (Paradis et al. 2004). As loci FRBi15 and FRBi16 were not congruent among them and neither with the remaining loci, three alignments were used, resulting in three trees, one for each FRBi region and another for the concatenated dataset including loci ITS, IGS and GAPDH. For the concatenated matrix, specimens with more than one missing locus were excluded. Datasets were analysed using maximum likelihood (ML) and Bayesian (B/MCMCMC) approaches with gaps treated as missing data.
For ML tree reconstruction, we used RAxML v. 8.2.10 (Stamatakis 2006) implemented in CIPRES Science Gateway (https://www.phylo.org/; Miller et al. 2010) with the GTRGAMMA model (Stamatakis 2006, 2014, Stamatakis et al. 2008). Support values were assessed using the ‘rapid bootstrapping’ option with 1 000 replicates. For the Bayesian reconstruction, MrBayes v. 3.2.1 (Ronquist & Huelsenbeck 2003) was used. Two simultaneous runs with 10 M generations each, starting with a random tree and employing 12 simultaneous chains, were executed. Every 500th tree was saved to a file. Preliminary analysis resulted in an overestimation of branch lengths and to correct this we used the uniform compound Dirichlet prior brlenspr = unconstrained : gammadir (1, 1, 1, 1; Zamora et al. 2015). We plotted the log-likelihood scores of sample points against generations using Tracer v. 1.5 (Rambaut et al. 2014) and determined that stationarity had been achieved when the log-likelihood values of the sample points reached an equilibrium and ESS values exceeded 200 (Huelsenbeck & Ronquist 2001). Posterior probabilities (PPs) were obtained from the 50 % majority rule consensus of sampled trees after excluding the initial 25 % as burn-in. The phylogenetic tree was drawn with FigTree v. 1.4 (Rambaut 2009).
STRUCTURE
STRUCTURE v. 2.3.4 (Pritchard et al. 2000, Falush et al. 2003) was run with the SSRs data matrix. Analysis was computed with 100 000 burn-in generations and 100 000 iterations using a K value from 1 to 12 (i.e., the putative number of species we may have) and 20 replicates for each K. To combine the 20 runs of each K in a single result, CLUMMP v. 1.1.2 (Jakobsson & Rosenberg 2007) was used and visualised replacing the CLUMMP output values in a STRUCTURE output of the same K, and then plotted using the STRUCTURE software. To show the probability of each K value, STRUCTURE HARVESTER (Earl & Von Holdt 2012), with the ΔK method (Evanno et al. 2005) was used, considering the most probable K the first one that appears close to 0 in the output graphic.
Principal coordinate analysis
Principal coordinate analysis (PCoA) was carried out with the SSRs length data in GenAlEx 6.5. The results of the three first axes were plotted in a three-axis graph using The Excel 3D Scatter Plot v. 2.1, in which the graphic can be moved in 3D to obtain a better understanding of how the plots are distributed in the space. Since the projection of this 3D graph on a paper is necessarily confusing, PCoA results were plotted on two different 2D graphs showing axes 1 and 2, and 1 and 3, respectively.
Haplotype network
Haplotype network reconstruction was performed using TCS v. 1.2.1 (Clement et al. 2000) with the concatenated sequences matrix, excluding the outgroup, using gaps as missing data, and a 95 % connection limit. Specimens differing only by missing or ambiguous characters were not counted as haplotypes.
Species delimitation analyses
In order to examine species delimitation, four computational approaches not requiring prior hypothesis of a putative number of species were used:
Automatic Barcode Gap Discovery ABGD (Puillandre et al. 2011) based on barcode gaps using genetic distances;
Poisson Tree Processes PTP (Zhang et al. 2013), based on gene trees;
The Generalized Mixed Yule coalescent approach GMYC, which combines a coalescent model of intraspecific branching with a Yule model for interspecific branching (Pons et al. 2006, Monaghan et al. 2009); and
DISSECT (Jones et al. 2014) based on the multispecies coalescent model for species delimitation.
ABGD and PTP were carried out using the online servers http://wwwabi.snv.jussieu.fr/public/abgd/ and http://species.h-its.org, respectively. GMYC was analysed with the gmyc function in the SPLITS package in R (v. 2.10, www.cran.r-project.org), employing the single (GMYCs) and multiple (GMYCm) threshold methods. Because GMYC needs a strictly ultrametric and bifurcating tree with no zero branch lengths, identical sequences were deleted and an ultrametric tree was generated using BEAST v. 1.8.2 software (Drumond et al. 2012), with the evolutionary models explained in the Bayesian phylogenetic reconstruction. A run of 100 M iterations logging every 1 000th iteration was conducted. Consensus tree was generated with TreeAnotator v. 1.8.2 after discarding the initial 10 % trees as burn-in. ESS values above 200 were ensured using Tracer v. 1.6 (Rambaut et al. 2014).
DISSECT analysis was implemented in STARBEAST (*BEAST, Drumond et al. 2012) using the concatenated DNA matrix after removing identical sequences and following the instructions of Jones et al. (2014). First, we used BEAUti (Drumond et al. 2012) to produce the xml file, with every individual encoded as if it was a separate species. Sites, clocks and trees were released as unlinked. Nucleotide substitution models and other parameters (as in the Bayesian analysis, see above), were encoded using BEAUti if possible, or manually entered. For the ITS locus, a substitution rate of 0.0033 substitutions per site per million years was introduced (Leavitt et al. 2012a, b), setting other loci as estimated with a lognormal relaxed clock. A birth-death-collapse prior that controlled the minimal split heights for the putative resulting species was manually added to the xml file. This prior contained the parameters CollapseHeight (ɛ) with a value of 0.0001 and CollapseWeight (ω), set as estimated using a Beta distribution with values 10 and 1.5. Selected parameters provide a highest probability density around 4–5 clusters, the most probable number of taxa meriting separation according to other analyses performed for this paper. However, this prior is diffuse and allows to obtain a different number of putative taxa if they adjust better to the data. The xml file was executed in BEAST with 250 M MCMC iterations, sampling every 10 000th iteration. Tracer v. 1.6 (Rambaut et al. 2014) was used to assess ESS values above 140. The resulting *BEAST species tree output was then treated with SpeciesDelimitationAnalyzer (Jones et al. 2014), with a burn-in of 5 000 trees (20 % of the total generated), a collapse height of 0.001 (one fraction lower than in the *BEAST analysis) and a simcutoff value of 1 to ignore this parameter, as according to sequence variability, we expected very similar putative species to emerge. The resulting similarity matrix was plotted with R v. 2.15.1 (R Core Team 2014) following the method of Jones et al. (2014).
Divergence time estimation
Two divergence time estimations were performed, one only with the ITS region and a defined substitution rate, and the other with the concatenated data matrix of ITS, IGS, and GAPDH loci. A rate of 3.30 × 10–9 s·s–1·yr–1 for the ITS region as a whole was used, with a GTR + G + I substitution model (Leavitt et al. 2012a, b). In the concatenated matrix analysis, as no previous literature on substitution rates for IGS and GAPDH in lichen-forming fungi is available, these were set as estimated in the Bayesian phylogenetic analysis. A *BEAST analysis was executed, using a relaxed clock model (uncorrelated lognormal), a birth-death model prior for the node heights and unlinked substitution models, clocks and trees for each partition. Clades G, Ko, NA, and WD were selected as potential species, forcing them to remain monophyletic (Fig. 6). No calibration points could be used, as no fossils or previous dating of this species complex are available. To avoid stochastic events, two independent analyses were run, each with 200 million generations, sampling each 5 000 trees, and discarding the first 10 000 trees (25 %) as burn-in. Tracer v. 1.6 (Rambaut et al. 2014) was used to ensure ESS parameter values above 115 in the concatenated matrix, and 185 for the ITS analysis. Different priors were tested but no higher ESS values could be obtained, which we suspect was due to the very similar sequences, and the uncertain topology of the backbone connecting the groups Ko, NA, and WD. The two runs performed for each input were merged with logcombiner v. 1.8.2, and the resulting trees merged in a consensus tree using TreeAnnotator v. 1.8.2 (Drumond et al. 2012). FigTree v. 1.4 (Rambaut 2009) was used to display the ITS and the concatenated dated species trees.
Fig. 6.
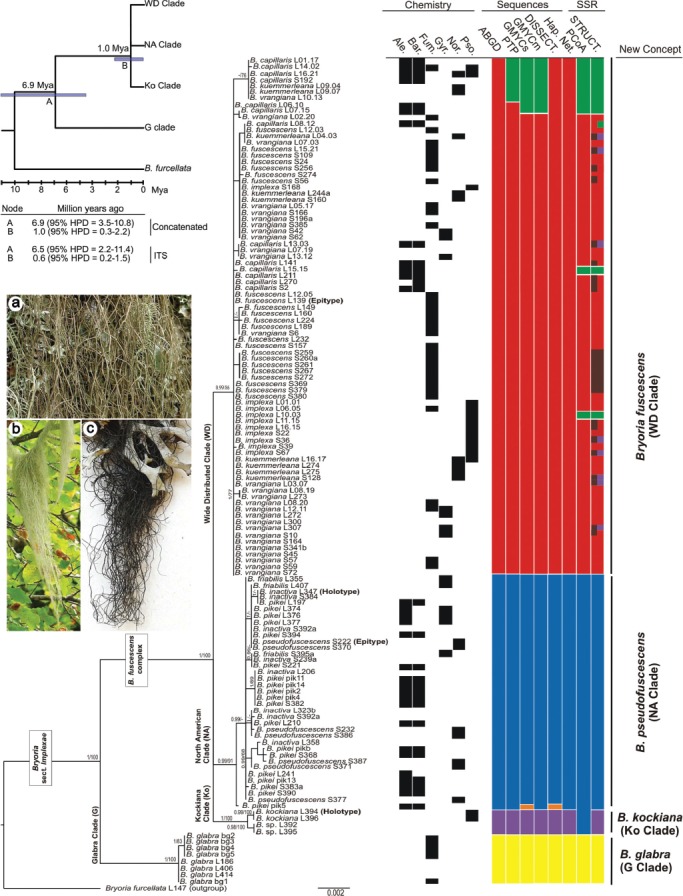
Integrated assessment of results in Bryoria sect. Implexae. Tree topology depicts the result of the Bayesian Markov chain Monte Carlo (B/MCMC) analysis. Posterior probabilities and bootstrap analysis for the supported nodes (≥ 0.95 and ≥ 70 %) are indicated at the main nodes. For each specimen, the extrolites detected, and the putative number of species predicted by the different methodologies is shown. The left top corner tree shows the results of the molecular dating analysis. — a. Bryoria implexa morphotype (Spain, Asturias, 2013, Boluda, MAF-Lich. 20749); b. B. capillaris morphotype (Spain, Navarra, 2013, Boluda & Villagra, MAF-Lich. 20748); c. B. fuscescens morphotype (Morocco, Ifrane, 2013, Boluda, MAF-Lich. 20751). — Ale. = Alectorialic acid; Bar. = Barbatolic acid; Fum. = Fumarprotocetraric acid; G = Glabra clade; Gyr. = Gyrophoric acid; Hap. Net. = Haplotype Network; HPD = Highest Posterior Density; Ko = Kockiana clade; Mya = Million years ago; NA = North American clade; Nor. = Norstictic acid; PCoA = Principal Coordinates Analysis; Pso. = Psoromic acid; STRUCT. = Structure; WD = Wide Distributed clade.
Demography
Changes in population sizes through time were estimated using the Bayesian skyline analysis (Drumond et al. 2005) with BEAST. Only clades Ko, NA, and WD, isolated and merged, were studied, as they show a clock-like tree topology and adequate sampling sizes.
Following the methods used for divergence time estimation analysis, the demography analyses were run using the ITS region without partitioning, with the GTR + G + I model of nucleotide substitution and a substitution rate of 3.30 × 10–9 s·s–1·yr–1, and with a strict molecular clock model (Leavitt et al. 2012a, b). Additionally, the same analysis was repeated with the concatenated data matrix using the ITS substitution rate, estimating the other loci rates with a relaxed clock model and using the nucleotide substitution models for IGS and GAPDH explained in the Bayesian phylogenetic reconstruction. Four independent runs for each input were processed with 50 M MCMC generations, sampling parameter values every 5 000th generation, using the Bayesian Skyline tree prior model, six discreet changes in population size and the linear growth option. ESS values were checked with Tracer v. 1.5 (Rambaut et al. 2014), and the two best of the four runs were combined, obtaining values usually above 200, with some exceptions with a lower limit of 100. Skyline plots were drawn with Tracer v. 1.5.
To support the Bayesian skyline test, a neutrality test was performed to infer if populations are in mutation-drift equilibrium. Tajima’s D (Tajima 1989) and Fu’s Fs (Fu 1997) were calculated with DnaSP v. 5.10 (Librado & Rozas 2009). A significantly positive D is interpreted as a diversifying selection or a recent bottleneck, whereas a negative significant D shows purifying selection or a recent expansion. If D is not significantly different from 0, a mutation-drift equilibrium may be occurring. Fu’s Fs can be interpreted in the same way.
RESULTS
Morphological and chemical clustering
The wide geographical range of collections revealed a combination of characters not previously reported in the Bryoria fuscescens complex, especially those from the previously less-studied Mediterranean Basin. Specimens with intermediate morphologies amongst traditionally accepted species were recognized, and the application of species names according to the current taxonomy was ambiguous. Individuals connecting the phenotypes and chemotypes of the taxa currently recognized as Bryoria fuscescens, B. implexa, B. kuemmerleana, and B. vrangiana were not rare in the Mediterranean Basin. For example, chemotypes thought to be diagnostic for a particular taxon were detected in specimens morphologically belonging to other taxa, as well as specimens producing extrolites characteristic of different taxa in a single thallus. Thin-layer chromatography (TLC) revealed seven different extrolites: alectorialic, barbatolic, fumarprotocetraric, gyrophoric, norstictic, and psoromic acids, atranorin, and sometimes also related substances such as chloroatranorin, protocetraric, or connorstictic acids. Atranorin, a typical accessory substance in the genus Bryoria, was not used in the posterior analyses because it appears in trace amounts in many samples and is often difficult to unequivocally discern if it is present or absent by TLC alone.
The chemical presence/absence matrix resulted in the phenogram shown in Appendix 2a. The matrix included specimens with as many as four extrolites, something not previously reported in the complex. Chemical characters were separated into two main groups:
specimens that contain benzyldepsides (i.e., alectorialic and barbatolic acids), substances traditionally used to separate B. capillaris and B. pikei from other species in the complex; and
specimens without benzyldepsides.
The latter were clustered in two well-supported groups, one with fumarprotocetraric acid as the main substance, and the other without it (including specimens with no detectable substances). If the structural relationships of the compounds were encoded in the presence/absence matrix (benzyldepsides vs depsidones), the same clustering was obtained.
The analysis of combined morphological, geographical, and chemical characters resulted in the phenogram in Appendix 2b. Only terminal branches were supported, including few monophyletic morphospecies, although not well isolated from others. Neither accepted morphospecies nor an unequivocal number of phenotypic groups could be recognized. This ambiguity was largely attributable to phenotypically intermediate specimens, mainly from the Mediterranean Basin, and also by the presence of some shared characters amongst the morphospecies, such as the presence/absence of soralia, pseudocyphellae, extrolite composition, and thallus colour.
Phylogenetic tree
Due to the topological conflict between loci, three DNA matrices were used to generate three phylogenetic trees:
a concatenated matrix including ITS, IGS, and GAPDH with 134 individuals consisting of 1 774 unambiguously aligned nucleotide position characters, with 83 parsimony informative (Pi) sites;
FRBi15 with 93 individuals contained 569 unambiguously aligned nucleotide position characters, with 44 Pi sites; and
FRBi16 with 80 individuals had 632 unambiguously aligned nucleotide position characters, with 160 Pi sites.
No evidence of recombination events was detected in the concatenated matrix. The resulting tree (Fig. 6) had four well-supported main clades, G (Glabra, yellow), Ko (Kockiana, magenta), NA (North American, blue), and WD (Widely Distributed, red + green + brown). Clade G included only material of B. glabra, appearing as an isolated taxon sister to the other three clades, which showed an uncertain topology between them. Clade Ko included material named B. kockiana and two unidentified specimens, all collected in Alaska (USA). Clade NA comprised the previously recognized North American morphospecies group (the ‘North American endemic species’, Velmala et al. 2014) named as B. friabilis, B. inactiva, B. pikei, and B. pseudofuscescens. While these species were mixed in the tree, the group as a whole was resolved as monophyletic. The WD clade included specimens widely distributed but mainly from Europe (the ‘European and globally distributed species’ group, Velmala et al. 2014) under the names B. capillaris, B. fuscescens (syn. B. chalybeiformis and B. lanestris), B. implexa, B. kuemmerleana, and B. vrangiana. None of these previously recognised species formed a monophyletic group.
The phylograms produced using the FRBi15 and FRBi16 markers had a different tree topology, not congruent among them or with that from the preceding concatenated dataset. In the FRBi15 reconstruction (Fig. 1), B. glabra was not represented due to the lack of primer annealing in the PCR process, and the tree could not be rooted. Several well-supported groups were produced, but did not follow any evident geographic, morphological, chemical, or SSR frequency pattern. Bryoria pikei L376 had a sequence with a putative recombination fragment with the B. vrangiana S10 clade in c. 50 % of the total length. This insertion placed the specimen out of the main parental group and it appears as its sister. Although marker FRBi16 (Fig. 1) also produced a well-resolved tree with supported nodes, the clades do not show any phenotypic and/or geographic structure. In this tree reconstruction, B. glabra was an isolated taxon and served to root the tree. In FRBi16 sequences, many putative recombination events were detected, suggesting a reticulate evolution. In both trees in Fig. 1, clade Ko (magenta) was recovered as monophyletic, but embedded between other named morphospecies.
Fig. 1.
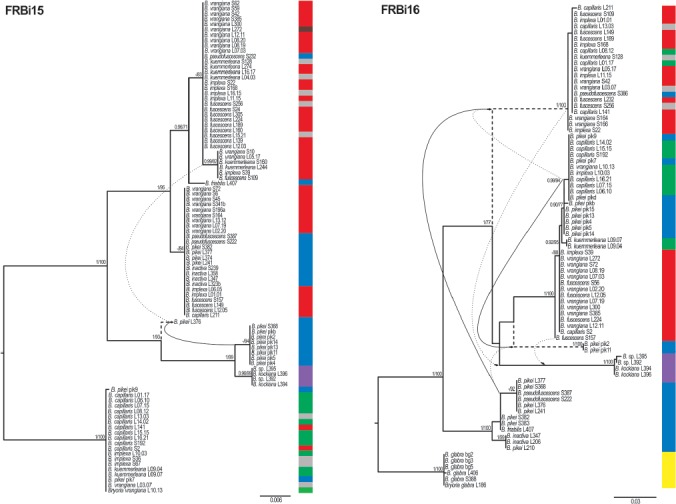
Phylogenetic relationships in Bryoria sect. Implexae based on FRBi15 and FRBi16 loci. Tree topology depicts the result of the Bayesian Markov chain Monte Carlo (B/MCMC) analysis. Posterior probabilities and bootstrap analysis for the supported nodes (≥ 0.95 and ≥ 70 %) are indicated at the main nodes. Lines connecting clades indicate putative recombination events, with main parents (continuous lines) and minor parents (discontinuous lines). Because the clade insertion in the trees is influenced by the recombination, clades with recombination are depicted with a discontinuous branch line. Note that clades with recombination appear as sister or close to the main parent but tending to be deviated towards the minor parent. — The coloured bar corresponds to the SSR genepool from Fig. 2, with specimens intermediate between two or more genepools in grey. The clades obtained, although well-supported, do not follow any evident geographic, morphologic, chemical, or microsatellite pattern.
STRUCTURE clustering
Of the 18 microsatellite markers, the nine that showed more than 95 % successful amplification across the samples were used (number of haplotypes shown in brackets, Appendix 3): Bi01 (17), Bi03 (6), Bi04 (8), Bi05 (5), Bi10 (8), Bi11 (9), Bi12 (12), Bi14 (6), and Bi19 (5). We allowed a maximum of three missing loci per specimen, a value reached only in seven samples. STRUCTURE was allowed to run to K = 12, but from K = 6 the clustering process started to be uninformative (Fig. 2). The likelihood results of the ΔK analysis (Evanno et al. 2005) indicated three as the most probable number of clusters (likelihood = –1232, ΔK = 2.2), the clades G, NA, and WD (Fig. 2, K = 3). Clade Ko, which appeared isolated in the concatenated phylogenetic tree (Fig. 6), could not be accepted as distinct under a K = 3 hypothesis. However, B. glabra, a morphologically delimited taxon, was not isolated at first in STRUCTURE. This could be attributable to the clustering algorithms being influenced by unbalanced sampling sizes, masking clade Ko, which appeared isolated at K = 6. From K = 4 to K = 6, the new groups appeared mainly inside the WD clade, showing that the samples from Europe were much more diverse than those from North America. Indeed, the NA clade was not split into subgroups even at K = 10. Apart from B. glabra, no other named morphospecies formed an exclusive cluster even at high K values.
Fig. 2.
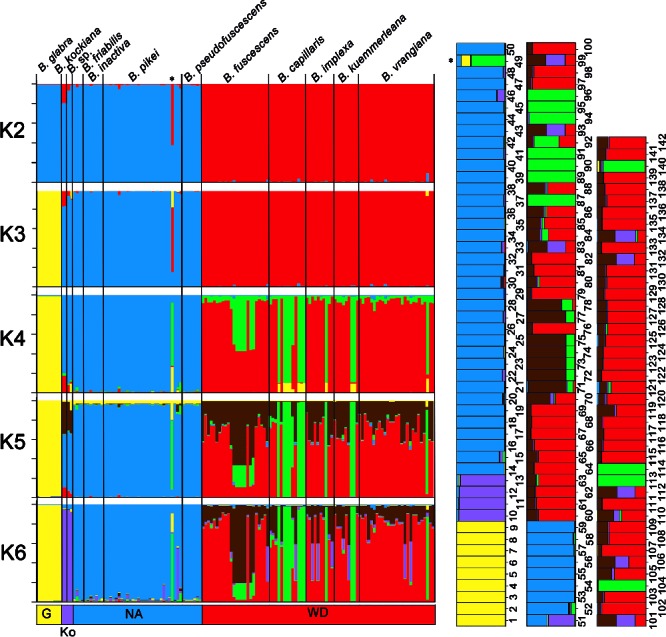
Bayesian inference of population structuring using STRUCTURE v. 2.3.4 (Pritchard et al. 2000, Falush et al. 2003) and nine microsatellite loci in Bryoria sect. Implexae. – Left. Results from the hypothesis of 2–6 clusters. Vertical bars represent specimen assignment probability into a genetic cluster depicted by the colours. Morphospecies names given to the specimens appear at the top. – Right. Detailed columns of the K = 6 hypothesis, the numbers representing the specimens shown in Table 1 to provide a better understanding of the components of each individual. — G = Glabra clade; Ko = Kockiana clade; NA = North American clade; WD = Widely Distributed clade; * = Bryoria pikei specimen 49.
Principal coordinates analysis (PCoA)
The PCoA analysis has a three-dimensional output represented here in two graphs, one comparing axis 1 against 2, and the other 1 against 3 (Fig. 3). The information percentage of each axis was 44.47 %, 15.06 %, and 14.44 %, respectively. Clade G (Fig. 3, yellow) appeared isolated, whereas clade Ko (Fig. 3, magenta) was admixed with NA clade (Fig. 3, blue), forming a single cluster. Clade WD was isolated from the others, but divided into two clusters, one corresponding to the red and brown groups in the K = 6 STRUCTURE output (Fig. 2), and one for the green group. Apart from B. glabra, none of the currently accepted morphospecies formed a defined group. Four reasonably isolated clusters could be distinguished, corresponding to the groups G, WDr (Widely Distributed, Fig. 3, red), WDg (Widely Distributed, Fig. 3, green), and Ko together with NA forming a single cluster.
Fig. 3.
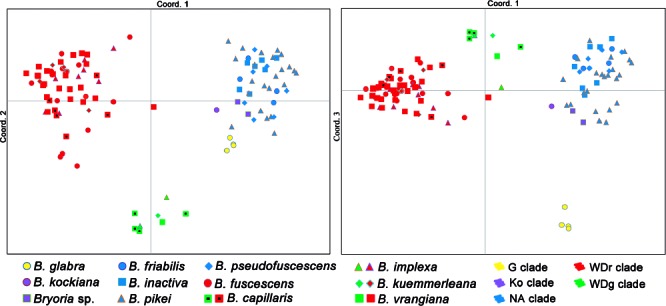
Principal Coordinate Analysis (PCoA) of microsatellite data in Bryoria sect. Implexae. Species names according to Velmala et al. 2014 (shape and colours) and STRUCTURE clusters (colours) for each specimen are represented in the three main coordinates. Note that the Ko clade does not appear isolated from the NA clade in any coordinate axis. — G = Glabra clade; Ko = Kockiana clade; NA = North American clade; WDr = Widely Distributed red clade; WDg = Widely Distributed green clade.
Haplotype network
The haplotype network of the concatenated data matrix, coding gaps as missing data, produced 39 haplotypes. Bryoria glabra specimens (Appendix 4, yellow) formed two haplotypes not connected with other members of the network, indicating genetic isolation of this species. One of the haplotypes was composed exclusively of South American specimens, whereas the other contained European, North American, and South American samples. Clade Ko (Appendix 4, magenta) fell into two haplotypes, one including specimens with psoromic acid and identified as B. kockiana, and the other clustering unidentified samples with no substances detected. This group was connected to the NA clade (Appendix 4, blue) by a long branch with 13 mutation steps. The NA clade was separated by nine mutations from the WD clade (Appendix 4, green, red, and brown). The WD green, red, and brown groups split by STRUCTURE (Fig. 2) formed a unique cluster. Four isolated clusters could be distinguished, corresponding to the groups G, Ko, NA, and WD.
Species delimitation programs
The ABGD, PTP, GMYC, and DISSECT programs (Table 3) use different algorithms, and consequently different numbers of putative species may be predicted. The genetic distance method (ABGD) gave the smallest number of putative species, whereas the coalescence methods (especially GMYC) the largest. Analyses also revealed the contribution of each locus to the postulated species delimitation, GAPDH being the most informative and constant marker. DISSECT analysis (Fig. 4) predicted five species corresponding to G, Ko, NA, and WD clades, and specimen B. pikei 5. Although the GMYC analysis also showed the B. pikei 5 specimen as a separate species, it was grouped in the NA clade in the other analyses. DISSECT showed two internal greyish square groups in WD, but they did not correspond exactly to the WDr and WDg groups in Fig. 2, 3 (STRUCTURE and PCoA analyses).
Table 3.
Species delimitation analysis results for loci ITS, IGS, GAPDH and the concatenated data matrix in Bryoria sect. Implexae. Brackets indicate groups predicted as conspecific. — G = Glabra clade; Ko = Kockiana clade; NA = North American clade; WD = Wide Distributed clade; WDr = Wide Distributed red clade; WDg = Wide Distributed green clade; pik5 = Specimen Bryoria pikei 5.
| Method | ITS | IGS | GAPDH | Concatenated |
|---|---|---|---|---|
| ABGD | 2 = G + (Ko, NA, WD) | 2 = G + (Ko, NA, WD) | 4 = G + Ko + NA + WD | 4 = G + Ko + NA + WD |
| PTP | 2 = G + (Ko, NA, WD) | 2 = G + (Ko, NA, WD) | 4 = G + Ko + NA + WD | 5 = G + Ko + NA + WDr + WDg |
| GMYCs | 4 = G + (Ko, NA, WDg) + WDr + WDr | 3 = G + (Ko, WD) + NA | 4 = G + Ko + NA + WD | 6 = G + Ko + NA + pik5 + WDr + WDg |
| GMYCm | 4 = G + (Ko, NA, WDg) + WDr + WDr | 4 = G + (Ko, WD) + NA1 + NA2 | 4 = G + Ko + NA + WD | 5 = G + Ko + NA + WDr + WDg |
| DISSECT | – | – | – | 5 = G + Ko + NA + pik5 + WD |
Fig. 4.
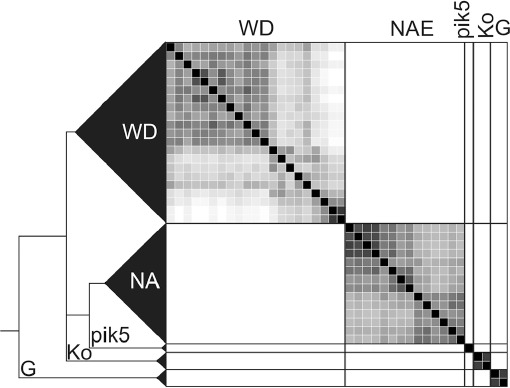
Similarity matrix from DISSECT analysis performed after clone correction in Bryoria sect. Implexae. Squares represent posterior probability (white = 0, black = 1) of pairs of specimens to belong to the same species. Resulting major groups are delimited by lines, which indicate the clade on the collapsed phylogenetic tree.
Node dates and demographic history
The calibrated maximum clade credibility chronogram for the concatenated data matrix is shown in Fig. 6. As only the ITS mutation rate is estimated in previous studies (Leavitt et al. 2012a, b), a second chronogram was prepared using this locus alone. Results from this analysis have to be treated with caution, as the species tree is not strictly clock-like (B. glabra has a shorter branch), and the ITS mutation rate has been taken from Melanohalea, a lichen-forming genus in the same family. Both analyses produced similar values, and the divergence of the B. glabra lineage was estimated at 6.9 Mya (95 % HPD = 3.5–10.8) in the concatenated matrix analysis, and 6.5 Mya (95 % HPD = 2.2–11.4) in the ITS data alone. The Ko, NA, and WD split was estimated at 1.0 Mya (95 % HPD = 0.3–2.2) from the concatenated matrix and 0.6 Mya (95 % HPD = 0.2–1.5) from the ITS data alone.
Bayesian Skyline Plots (Fig. 5, left) indicate a recent population increase in the NA and WD clades. However, the sequences contained few informative mutations and the deepest coalescence was reached in around 700 000 yr, with no population changes detectable further back from this period. Tests of neutrality (Fig. 5, right) are commonly used to support inferences from Bayesian Skyline Plots. As indicated by non-significant Tajima’s D and Fs results, all sampled groups seem in mutation-drift equilibrium, with the exception of the GAPDH locus of the NA clade which had a significant negative D value (Fig. 5 bold). This could indicate a recent expansion or ‘purifying’ selection, as seen in the concatenated Bayesian Skyline analysis, but other loci did not support this hypothesis.
Fig. 5.
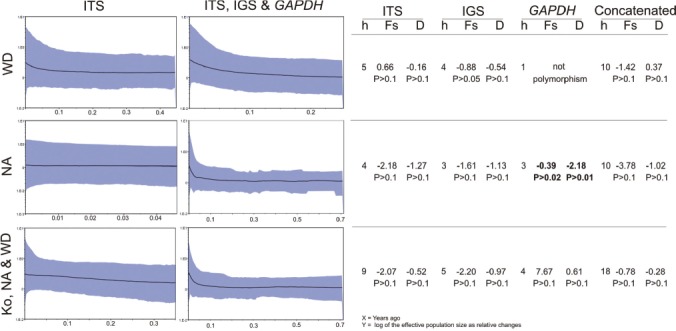
Bryoria sect. Implexae. – Left. Bayesian Skyline Plots for each clade predicted by the ITS marker and the concatenated loci matrix. The X-axis of each graph represent time (in Myr), and the Y-axis represents the value for the log of the effective population size as relative changes, because generation times in Bryoria species are unknown. Grey shadows indicate the upper and lower 95 % credible intervals. – Right. Results from neutrality tests for each marker and clade, indicating (in bold) any statistically significant deviation from neutrality. — h = number of haplotypes; Fs = Fu’s Fs; D = Tajima’s D.
Markers ITS, IGS, and GAPDH indicate population stability over the recent past for clades NA and WD, with putative even more recent small population expansions. Due to the low variability of the loci, and the putative loss of demographic signals, this hypothesis is not confirmed by this analysis.
Integrated assessment of datasets
Depending on the analysis, different numbers of putative species were suggested, ranging from four to six (Table 4). All analyses, however, confirmed that the combination of morphological and chemical characters generally used for species circumscription in the complex was inadequate. GAPDH, despite its low variability, was the only marker tested that supported species-rank assignations for the clades G, Ko, NA, and WD (Table 3). ITS, one of the most used loci for DNA barcoding in lichen-forming fungi, did not unambiguously distinguish those clades. The new markers FRBi15 and FRBi16, despite their higher variability, showed inconclusive results and putative recombination events. The microsatellite data (Fig. 2) supported the DNA sequences results and reflected internal variability not revealed in our sequence data, showing that the WD cluster was much more diverse than NA, which had a particularly low diversity.
Table 4.
Summary of the number of putative species suggested by the different methods used for each dataset in Bryoria sect. Implexae.
| Method | Data | Figure / reference | Number of putative species |
|---|---|---|---|
| Traditional concept | DNA sequences and phenotypic | Velmala et al. (2014) | 12 |
| Chemical | Phenotypic | Appendix 2 Left | c. 4 |
| Morpho-chemical | Phenotypic | Appendix 2 Right | Not conclusive |
| Phylogeny | DNA sequences of ITS, IGS, and GAPDH | Fig. 6 | 4 = G + Ko + NA + WD |
| Phylogeny | DNA sequences of FRBi15 | Fig. 1 | Not conclusive |
| Phylogeny | DNA sequences of FRBi16 | Fig. 1 | Not conclusive |
| STRUCTURE | Microsatellites | Fig. 2 | 5 = G + Ko + NA + WDr + WDg |
| PCoA | Microsatellites | Fig. 3 | 4 = G + (Ko, NA) + WDr + WDg |
| Haplotype Network | DNA sequences of ITS, IGS, and GAPDH | Appendix 4 | 4 = G + Ko + NA + WD |
| ABGD | DNA sequences of ITS, IGS, and GAPDH | Table 3 | 4 = G + Ko + NA + WD |
| PTP | DNA sequences of ITS, IGS, and GAPDH | Table 3 | 5 = G + Ko + NA + WDr + WDg |
| GMYCs | DNA sequences of ITS, IGS, and GAPDH | Table 3 | 6 = G + Ko + NA + pik5 + WDr + WDg |
| GMYCm | DNA sequences of ITS, IGS, and GAPDH | Table 3 | 5 = G + Ko + NA + WDr + WDg |
| DISSECT | DNA sequences of ITS, IGS, and GAPDH | Fig. 4 | 5 = G + Ko + NA + pik5 + WD |
TAXONOMY
Bryoria sect. Implexae (Gyeln.) Brodo & D. Hawksw., Opera Bot. 42: 114. 1977
Basionym. Bryopogon sect. Implexae Gyeln., Feddes Repert. Spec. Nov. Regni Veg. 38: 223, 238. 1935.
Type species. Bryoria implexa (Hoffm.) Brodo & D. Hawksw. 1977. ≡ Usnea [unranked] implexa Hoffm. 1796. = Bryoria fuscescens (Gyeln.) Brodo & D. Hawksw. 1977; but see below.
Species with a fruticose, hair-like, subpendent to mainly pendent thallus, lateral spinules or spinulose branches absent, whitish grey to brown or black, often paler in the basal parts. Angles between branches variable, acute to obtuse or even rounded. Pseudocyphellae absent or present, then frequently inconspicuous, ± fusiform, concolorous or whitish. Soralia absent or present, tuberculate or fissural, white to dark. Isidia or isidioid spinules absent. Apothecia mainly absent, if present, usually afunctional. Chemistry varied, with no detectable or with one or a combination of major substances, including alectorialic, barbatolic, connorstictic, fumarprotocetraric, gyrophoric, norstictic, protocetraric, psoromic and possibly salazinic acids, atranorin, and chloroatranorin. Photobiont Trebouxia ‘hypogymniae’ (Lindgren et al. 2014).
Notes — Most species included in Brodo & Hawksworth (1977) under Bryoria sect. Implexae were transferred to other sections in Myllys et al. (2011). In the light of our results (but see the Discussion later), Bryoria sect. Implexae includes the four species treated below. Comments on particular morphological or chemical traits that may be helpful for distinguishing these taxa are given under each species. Nevertheless, nearly all cited characters are shared by different taxa, so they can be interpreted as ‘cryptic’. The species names adopted here are epitypified by sequenced material here in order to fix their identities at the molecular level. This epitypification is essential to fix the application of these names as no DNA sequences are available and cannot be obtained from old type material of most species names. The old types cannot therefore be critically identified for purposes of the precise application of the names and so epitypes may be designated (Turland et al. 2018: Art. 9.9 and Ex 9).
As molecular data are necessary for unambiguous species level identification in the taxonomy proposed here, we recommend using the collective ‘Bryoria fuscescens complex’ when referring to material lacking molecular data.
Bryoria fuscescens (Gyeln.) Brodo & D. Hawksw., Opera Bot. 42: 83. 1977
Basionym. Alectoria fuscescens Gyeln., Nytt Mag. Naturvidensk. 70: 55. 1932, nom. cons. (cf. Hawksworth & Jørgensen 2013).
Synonyms. Lichen chalybeiformis L., Sp. Pl. 2: 1155. 1753, (nom. cons.) nom. rej. against Bryoria fuscescens (cf. Hawksworth & Jørgensen 2013).
Bryoria chalybeiformis (L.) Brodo & D. Hawksw., Opera Bot. 42: 81. 1977.
Usnea [unranked] implexa Hoffm., Deutschl. Fl., Zweiter Teil: 134. 1796.
Bryoria implexa (Hoffm.) Brodo & D. Hawksw., Opera Bot. 42: 121. 1977.
Parmelia jubata β. [var.] capillaris Ach., Methodus, Sectio post.: 273. 1803.
Bryoria capillaris (Ach.) Brodo & D. Hawksw., Opera Bot. 42: 115. 1977.
Alectoria jubata χ. [var.] lanestris Ach., Lichenogr. Universalis: 593. 1810.
Bryoria lanestris (Ach.) Brodo & D. Hawksw., Opera Bot. 70: 88 1977.
Alectoria kuemmerleana Gyeln., Nytt Mag. Naturvidensk. 70: 49. 1932.
Bryoria kuemmerleana (Gyeln.) Brodo & D. Hawksw., Opera Bot. 42: 155. 1977.
Alectoria prolixa var. subcana Nyl. ex Stizenb., Ann. Naturhist. Mus. Wien 7: 129. 1892, nom. rej. against Bryoria fuscescens (cf. Hawksworth & Jørgensen 2013).
Bryoria subcana (Nyl. ex Stizenb.) Brodo & D. Hawksw., Opera Bot. 42: 91. 1977.
Alectoria vrangiana Gyeln., Magyar Bot. Lapok 31: 46. 1932.
Bryoria vrangiana (Gyeln.) Brodo & D. Hawksw., Opera Bot. 42: 97. 1977.
Type specimens. FINLAND, Tavastia austr., Hollola, ad truncos Pini locis apricioribus in silva, Sept. 1882, J.P. Norrlin (Norrlin, Herb. Lich. Fenn. No. 46) (BP 33947 – lectotype designated by Hawksworth 1972: 217). — FINLAND, Etelä-Savo, Savitaipale, 600 m NW of Mustapää, 61, N1721° E27,6900°, 2005, L. Myllys 464 (HA.H9209715 (L139)) – epitype designated here, MycoBank MBT381730.
Nomenclature — A large number of species rank names belong to this group, and are synonymised, but these have not been epitypified with sequenced material. Further information on synonyms and type materials can be seen in Hawksworth (1972), Brodo & Hawksworth (1977) and Velmala et al. (2014). Although no samples of Bryoria austromontana have been studied, the published description and images (Jørgensen & Galloway 1983) suggest this taxon also belong here.
The earliest species rank epithets amongst these are chalybeiformis dating from 1753 (Lichen chalybeiformis), and implexa dating from 1799 (Usnea implexa). The former has been rejected against Bryoria fuscescens, but not against other species names apart from B. subcana (Hawksworth & Jørgensen 2013). A proposal to add the four earlier names Alectoria capillaris, Usnea implexa, A. kuemmerleana, and A. lanestris to the two against which Alectoria fuscescens is already conserved is being prepared separately. Protection against A. vrangiana is not required as it appeared in the same work as A. fuscescens. While the proposal is under discussion, the name B. fuscescens should be adopted in accordance with Rec. 14A.1 (Turland et al. 2018).
We refrained from epitypifying and taking up any of the earlier and potentially competing names by epitypification primarily as the name B. fuscescens is the most commonly used species name in the complex, is well established, the most widely used* and is already conserved over two earlier species names in the complex. In addition, all the other names have been associated with particular morphotypes or chemotypes since the 1970s, and so their use might be mistaken as applying to a taxon with those particular traits.
If the proposal for rejection of the previously mentioned competing synonyms is not accepted, the principle of priority would rule the use of the earliest and not already rejected, validly published name at the species rank, i.e., Usnea implexa (and then the combination Bryoria implexa), which would require epitypification by sequenced material in order to fix the precise application of that name. The species was first described from Germany but with no named locality, and neotypified by an unlocalised and undated specimen in Hoffmann’s herbarium in Moscow (Hoffmann 8569, MW) which may be part of the original material from Germany or have been collected later and perhaps in Russia (Hawksworth 1969a). As the neotype contained psoromic acid, and the epithet has therefore been applied to that chemotype, a potential sequenced epitype should represent that chemotype and ideally also have been collected in Germany. No such specimen was available to us during this study.
Distribution — Widely distributed, known from cool temperate to boreal and arctic areas of Europe, Asia, North America, and Africa. There are also reports from Antarctica, Oceania, and South America, but material from those regions has not yet been studied molecularly and so we cannot confirm that they belong to this complex.
Notes — Bryoria fuscescens is highly variable in morphology and chemistry, and many of the analysed specimens develop soralia. Further, atranorin, which is not normally detectable in the other three species accepted here, is frequently found in concentrated extracts from both sorediate and esorediate morphs.
Bryoria glabra (Motyka) Brodo & D. Hawksw., Opera Bot. 42: 86. 1977
Basionym. Alectoria glabra Motyka, Fragm. Florist. Geobot. 6: 448. 1960.
Type specimens. USA, Washington, Olympic Peninsula, Clallam Co., Hurricane Ridge, 5800 ft, on trunk of Abies lasiocarpa, 24 July 1950, B.I. Brown & W.C. Muenscher 129 (US – holotype). — USA, Alaska, Mainland, Valley between the Bucher and Gilkey Glaciers, southern end of subalpine valley, on east side of creek running through valley, subalpine forest, N58°47’20.12" W134°30’0.10", 773 m elevation, on Tsuga mertensiana twigs, 4 Aug. 2011, K.L. Dillman 4Aug11:1 (UBC (L406)) – epitype designated here, MycoBank MBT381731.
Distribution — Known from northern Europe (Scandinavia), and North and South America. In North America, it is most abundant in the Pacific North-West.
Notes — Distinguishing features in well-developed specimens are the brownish thallus with a regular branching pattern, generally with obtuse and rounded angles between the branches, and broad oval and usually white soralia. It is, however, difficult to separate poorly developed or small specimens conclusively, so molecular sequences are recommended for unambiguous identifications. Only fumarprotocetraric and protocetraric acids have been detected in this species, and these are characteristically produced in the soralia.
The Alaskan specimen is selected as the epitype here as sequences are available from all loci, whereas the material we have sequenced from Washington state (type locality) only has data on the ITS locus.
Bryoria kockiana Velmala, Myllys & Goward in Velmala et al., Ann. Bot. Fenn. 51: 361. 2014
Type specimen. USA, Alaska, Fairbanks, North Star Borough, 26 July 2011, D. Nossov 20019-1 (UBC (L394) – holotype).
Distribution — Known only from Alaska (USA) and British Columbia (Canada), on conifer branches.
Notes — Few specimens of this species have so far been studied, and these are characterised by the absence of any whitish grey tone in the thallus, the lack of soralia, and greyish to brown branches with conspicuous, white to concolourous, broad, elongate-fusiform, sometimes slightly raised pseudocyphellae. It lacks TLC-detectable substances or produces psoromic acid. The not validly published designation Alectoria krogii D. Hawksw. 1972 may be synonymised here.
Bryoria pseudofuscescens (Gyeln.) Brodo & D. Hawksw., Opera Bot. 42: 127. 1977
Basionym. Alectoria pseudofuscescens Gyeln., Ann. Hist.-Nat. Mus. Natl. Hung. 28: 283. 1934, and Rev. Bryol. Lichénol. 7: 51. 1934.
Synonyms. Bryoria friabilis Brodo & D. Hawksw., Opera Bot 42: 118. 1977.
Bryoria pikei Brodo & D. Hawksw., Opera Bot 42: 125. 1977.
Bryoria inactiva Goward et al., Ann. Bot. Fenn. 51: 360. 2014.
Type specimens. USA, Oregon, Benton County, Corvallis, on old apple trees, Dec. 1931, F.P. Sipe 669 (BP 33958 – holotype of Alectoria pseudofuscescens). — CANADA, British Columbia, 25 Sept. 2006, T. Goward 07-02-2011 (UBC (S222) – epitype selected here, MycoBank MBT381732; British Columbia, Clearwater Valley, 0.5 km S of Philip Creek, ‘Edgewood West’, 715 m, 9 Nov. 2011, T. Goward 11-61 (UBC (L347) – holotype of Bryoria inactiva).
Nomenclature — A number of species rank names are synonymised to this taxon, but these have not been epitypified with sequenced material. All these names, however, are later in date than pseudofuscescens, and so could not have priority over that name. Further information on type materials can be seen in Brodo & Hawksworth (1977) and Velmala et al. (2014). Although no samples of Bryoria salazinica have been studied at molecular level, the published description and images (Brodo & Hawksworth 1977) suggests this taxon also belong here.
Distribution — Only known from North America, growing on bark, branches, rock or soil.
Notes — Characterised by the absence of soralia and detectable atranorin.
DISCUSSION
Phylogenetic relationships
Species concepts in Bryoria sect. Implexae have previously been based primarily on well-characterised northern European and North American specimens (Hawksworth 1972, Brodo & Hawksworth 1977, Velmala et al. 2014). Velmala et al. (2014) recognised 11 species on the basis of morphological and chemical characters, but many of these were not supported by molecular data, and different species names were accepted for taxa that could not be distinguished molecularly. We discovered that these demarcations broke down when specimens from more southern European populations were incorporated. This is shown in a phenetic analysis using only phenotypically diagnostic characters (Appendix 2), where the resulting groups are not resolved as clear-cut morphospecies. Indeed, any character previously used in the group could be used to define the three lineages of the Bryoria fuscescens complex (Fig. 6).
Sexual structures are of major importance in species identification in fungi, but here the rarity of apothecial production has hampered their study in most Bryoria species. Any such features found would in any case be of limited practical value in identification as nearly all samples lack apothecia. Extrolite composition has been accorded a major role in species delimitation in the complex, but many of the substances that were considered to be of diagnostic value are biosynthetically closely related, being produced by the same gene cluster (pks genes; Keller & Hohn 1997), and may be environmentally influenced (Myllys et al. 2016, Lutsak et al. 2017).
Integrative taxonomy, rather than phylogenies based only on neutral markers, are increasingly being used to resolve complex taxonomical groups (e.g., Dayrat 2005, Will et al. 2005, Lumley & Sperling 2011, Zamora et al. 2015, Caparrós et al. 2016). Microsatellites are also now widely used in intraspecific population studies because of their high variability (Widmer et al. 2012, Dal Grande et al. 2014), and in species complexes with diffuse genetic barriers, microsatellite data can improve DNA sequence resolution (Lumley & Sperling 2011, Vanhaecke et al. 2012). It is generally assumed that DNA sequence data reflect the evolution of the species, but these data only reflect the history of the studied loci, which may sometimes be different from the species history overall (Nichols 2001). In this case, we demonstrated that traditionally used loci (ITS, IGS, and GAPDH) and microsatellite data reveal similar clades, whereas other intergenic loci (FRBi15 and FRBi16) produced discrepant but statistically supported lineages. These incongruences may be due to recombination, hybridisation, or incomplete lineage sorting, as documented in many other species groups (e.g., Jakob & Blattner 2006, McGuire et al. 2007, Edwards et al. 2008, Stewart et al. 2014). In lichen-forming fungi, outcrossing and recombination have been demonstrated, for example, in Lobaria pulmonaria (Zoller et al. 1999, Singh et al. 2012, Keller & Scheidegger 2016), Letharia (Kroken & Taylor 2001a, b, Altermann et al. 2014), and Cladonia (Steinová et al. 2013).
Apothecia are usually absent in Bryoria sect. Implexae, and even when present may not contain mature spores. If cryptic sexuality is not occurring, hybridization is unlikely to provide an explanation of our data. In the absence of sexual reproduction, any recombination is improbable, although some fungi lacking sexual structures show recombination events attributable to parasexual cycles (Schoustra et al. 2007). We did detect signals suggesting putative recombination in the FRBi loci, but not in the standard three loci used in the taxonomy adopted here. Recombination signals may reflect some mitotic recombination, actual or ancient sexual reproduction (Douhan et al. 2007) or be merely false positives produced by chance production of similar sequences. In any case, recombination alone is insufficient to explain all the discordances found. For example, only one putative recombination event was detected in FRBi15, and disentangling the FRBi16 recombination points is insufficient to obtain the topology of the three-locus phylogeny. Incongruences may also be caused by the analysis of different paralogs of FRBi15 and FRBi16 amplified with the new primers, but this seems improbable, as no paralogs have been detected in the SSRs of these loci (Boluda et al. unpubl.). However, our results indicate recent diversification and large effective population sizes in this lichenised complex. Thus, incongruences amongst loci seem rather attributable to incomplete lineage sorting.
The different putative species concepts generated by the species delimitation programs (Table 3) may not be only due to the different algorithms applied, but also because some of the programs were designed for use in single-locus phylogenies (e.g., ABGD, PTP, or GMYC). Nevertheless, all the clustering analyses showed a tendency to distinguish four groups, G, Ko, NA, and WD (Table 4; Fig. 6). STRUCTURE was unable to define these groups until reaching the K = 6 hypothesis, which can be attributed to the highly unbalanced sampling sizes; the analyses shows that the WD cluster is much more variable than NA, which was not divided into subgroups until K = 10.
Specimen 49 (identified as B. pikei, Fig. 2 asterisk), probably reflects the impossibility of unequivocally distinguishing that species from B. capillaris; however, as sequence amplification of this specimen failed, we cannot determine if this mismatch was due to misidentification or DNA contamination. Haplotype network analyses have been extensively used in infraspecific population and less frequently closely related species groups (Houbraken et al. 2012, Pino-Bodas et al. 2016). Even this type of nested clade phylogeographic analysis has some critics (e.g., Knowles 2008, Templeton 2009). The resulting network in the present case is concordant with that obtained from other analyses. If two DNA barcoding standard marker networks are obtained from a single analysis with a 95 % parsimony connection limit, members of each network might be considered as different species (Hart & Sunday 2007), showing the clear isolation of B. glabra from the other taxa in the complex. In the case of connections with several mutation steps, as between clades Ko, NA, and WD, taxon delimitation is below the species level, but in any case indicates some degree of genetic isolation.
The relationships amongst the Ko, NA, and WD clades remains unresolved, indicating that the evolutionary history may be too complex to be adequately captured by dichotomous phylogenies based on a few neutral markers. Moreover, the putative presence of shared ancestral polymorphisms amongst the clades may be producing incompatible topologies, which result in clades with low support.
We also performed analyses to estimate changes in past population sizes, which may have affected current clade diversity. Genealogies of most plant and animal species coalesce between 2.58–0.01 Mya (Grant 2015), and our estimated intervals are within this range. However, in our case the dates are relatively recent, with the oldest coalescences at 0.7 Mya. A flat graphic is generally interpreted as population stability but can also be due to a lack of detection power produced by small sample sizes. Moreover, a small rise in the curve near the present, seen in Fig. 5, can be a consequence of the random sampling of the MCMC haplotype trees (Grant 2015); this result must therefore be treated with caution. Our sequences do not bear imprints of ancient population history, but rather more recent population growth, for example by extensions northwards in post-glacial periods. The loss of information may also arise from bottlenecks (i.e., a marked reduction in population size), local extinctions, and subsequent recolonization. Additionally, the use of genes with low levels of polymorphisms, may impede a robust reconstruction of population sizes through time.
Species concept
Some species delimitation analyses, such as STRUCTURE, GMYC, PTP, can overestimate the number of taxa meriting formal recognition, particularly when sampling is uneven or in species with a strong intraspecific genetic structure (Altermann et al. 2014, Modica et al. 2014, Alors et al. 2016, Del-Prado et al. 2016). ABGD, in contrast, has been considered as rather conservative, less prone to species overestimation and less sensitive to unbalanced sampling. While that method only detects discontinuities in DNA sequence variation (Puillandre et al. 2011), it can also be expected to fail in species with strong population genetics structures, for example ones containing exclusive haplotypes. All species delimitation programs will provide a number of reasonably discrete groups (‘evolving lineages’) that should be evaluated for consideration as meriting species rank, but the decision has to be by taxonomists with experience in the group concerned. Some of our analyses suggest that groups such as WDr, WDg, or pik5 might merit species rank, but our experience, together with the results from other analyses shown here, leads us to reject this hypothesis. We conclude that the most pragmatic solution, supported by the general trend of the results from the different analyses we performed is to consider clades G, Ko, NA, and WD as the species Bryoria glabra, B. kockiana, B. pseudofuscescens, and B. fuscescens, respectively.
Clade age can contribute to decisions as to species limits. Divergence time estimates can be robust if the analyses are performed with well-resolved phylogenies and can incorporate fossil calibration points, as in some vertebrates (Perelman et al. 2011). Contrarily, in lichenised fungi, fossils are rare and in many cases enigmatic or with ambiguous relationships (Thomas et al. 2014, Hawksworth 2015, Kaasalainen et al. 2015). In addition, generation times can be expected to be different among species, as is the case with nuITS locus mutation rates between herbaceous and woody plants, or even a difference of almost an order of magnitude between different plant genera (Kay et al. 2006). Here we used a nuITS mutation rate estimated from Melanohalea, a genus in the same family (Leavitt et al. 2012a), species of which frequently grow with Bryoria and reproduce asexually as well as sexually. The split of B. glabra from the other taxa in the B. fuscescens complex is estimated at c. 6.9 Mya, and clearly separated from the much later divergence of the other three species estimated at c. 1 Mya (0.6 Mya if only the nuITS locus is used). This contrasts with other lichenised species considered of recent origin, estimated around 2.5–5 Mya (Pliocene), with any Pleistocene speciation event rare and always older than 1 Mya (Amo de Paz et al. 2012, Leavitt et al. 2012a, b, Molina et al. 2016). As the three B. fuscescens complex clades seem to have diverged more recently, the extent of their reproductive isolation is uncertain, and the discovery of intermediate lineages amongst other named species from unsampled geographical regions, such as continental Asia remains possible.
In the absence of supporting phenotypic, geographic, or ecological differences, the recent divergence, and the possibility of incomplete lineage sorting, clades Ko, NA, and WD may be considered as conspecific evolving lineages. It is, however, important to recognise the lineages formally in order to facilitate their conservation by enabling their threat status to be assessed by IUCN criteria. We decided not to adopt the rank of subspecies as that is now hardly used in mycology, and then not in any consistent way; traditionally this rank was used in plants for morphologically distinguishable populations with geographical differences and where intermediates occurred where they were sympatric (Stuessy 2009).
The formal recognition of cryptic lineages at species level, as suggested by our analyses, emerges as the most appropriate solution. Cryptic speciation is now recognised as a common phenomenon in Parmeliaceae, and our results are in accord with other studies in which molecular markers in combination with statistical tools revealed genetically distinct lineages previously hidden under a single taxon name in this family (e.g., Singh et al. 2015, Alors et al. 2016, Del-Prado et al. 2016, Divakar et al. 2016, Leavitt et al. 2016). Further, this solution is in line with the increasing need to formally recognize cryptic species-level lineages in all fungi (Hibbett 2016); indeed, cryptic speciation may mean that there are on average ten or more fungi masked in formally named species (Hawksworth & Lücking 2017).
Of the lineages recognised here, only the WD clade emerged as cosmopolitan, occurring in Europe, Asia, North America, and Africa (Appendix 5). NA and Ko have been collected so far only in North America, despite our extensive sampling in Europe (Appendix 5). Further sampling, especially in South America, Asia, and Africa, is needed before any finer-scale biogeographic patterns might be detected.
The practical issue of naming older museum specimens and material in ecological surveys could be resolved by recognising the three groups as species within a broad concept, such as an aggregate, complex, or adding ‘s.lat.’. We considered commending the adoption of the suffix ‘agg.’ for material when precise molecular species identifications cannot be made. While this has been done in a few other groups of fungi (e.g., Parnmen et al. 2013, Pažoutová et al. 2015), ‘complex’ has come to be used more widely and was strongly favoured at the Cryptic Speciation in Fungi symposium in Utrecht in September 2017 (report awaited). We therefore suggest the use of ‘complex’ here but recognise some may prefer to use ‘agg.’ or ‘s.lat.’. Where DNA samples can be obtained and analysed, we recommend use of the GAPDH locus, as all the other tested markers are not able to distinguish with confidence the three species we recognize in the complex.
Infraspecific phenotypic variation
While our results support rejection of the morphospecies concept in this group of lichens, two main phenotypes can nevertheless often be distinguished by the naked eye in the field:
the pale grey ‘capillaris’ morphotype (including B. capillaris and B. pikei, Fig. 6b); and
the fuscous brown to dark brown ‘fuscescens’ morphotype, in which most other species names are placed (Fig. 6a, c).
The chemical characters are not always checked by field workers, and while the ‘capillaris’ morphotype typically has benzyldepsides, the ‘fuscescens’ morphotype lacks those compounds and has fumarprotocetraric acid or various depsidones. However, there are dark morphs with benzyldepsides (once called f. fuscidula), and pale grey ones with fumarprotocetraric acid (e.g., B. subcana) or other depsidones (e.g., B. kuemmerleana). It is conceivable that the two morphotypes originated before the separation of B. pseudofuscescens and B. fuscescens, as both colour variants and chemistries appear in both taxa. This phenomenon cannot be explained by a simple ongoing speciation event in which one lineage has originated new adaptations, but is still not isolated from the parental lineages, as neither are monophyletic in a paraphyletic clade.
The difference in phenotype cannot be attributed to different algal partners as all material in the complex shares the same species and even in many cases the same nuITS haplotypes of Trebouxia (Lindgren et al. 2014, Boluda et al. unpubl.). Further, as we used neutral markers to detect gene-flow gaps between lineages, the phenotypes are also not the result of genetic isolation, and other possibilities must be considered.
It has recently been reported that yeast morphs of the lichenicolous and gall-forming basidiomycete genus Cyphobasidium can be abundant in or on the outermost cortical tissues of Bryoria species (Spribille et al. 2016). Spribille et al. (2016) reported a possible relation between Cyphobasidium yeast abundance and vulpinic acid production in two other species of Bryoria, B. fremontii and B. tortuosa, and also visualised these yeasts in material identified as B. capillaris phenotypes. Contrary to the claims of Spribille et al. (2016), these fungi do not appear to be an integral part of the mutualism (Oberwinkler 2017). It is, however, feasible that the yeasts cells are able to develop to a greater extent in ‘capillaris’ morphotypes as the cortices can have lumpier polysaccharide deposits than do those of ‘fuscescens’ (Hawksworth 1969b, Boluda et al. 2014, Esseen et al. 2017). How the occurrence of yeast morphs of these lichenicolous fungi in the surface of the cortex could possibly determine colour morphotypes is obscure.
Material referred to the ‘capillaris’ and ‘fuscescens’ phenotypes has been reported to show slight differences in water holding capacity (Esseen et al. 2015), and also the pigments may provide protection against excesses of light (Färber et al. 2014). Further, in southern Europe particularly, the ‘capillaris’ phenotype tends to be restricted to humid, shaded, and protected or undisturbed environments than the ‘fuscescens’ one, something already recognised by Motyka (1964). Additionally, in northern Europe, dark specimens containing barbatolic acid are much more common than in southern Europe, where they are extremely rare (Myllys et al. 2016, Esseen et al. 2017). As both phenotypes can grow side by side and even intermixed on the same trees, where environmental conditions must be identical, ecological plasticity has to be discounted. However, some unknown epigenetic modification could perhaps have a role in that process, as once a metabolic pathway is activated or silenced, it may be hardly modifiable under more or less neutral environmental conditions, transferring the phenotypes to the clonal offspring. Specimens with dark thalli, containing barbatolic acid, or with pale thalli with traces of barbatolic and also containing other extrolites, could represent transitional specimens.
Molecular and morphological rates of divergence may sometimes be uncoupled. Incomplete lineage sorting arises when an ancestral polymorphism persists through a speciation event and each polymorphism can lead to different alleles being carried among descendants (Maddison 1997, Hartl & Clark 2007). Consequently, different tree topologies may be obtained depending which specimens or loci are used. Rosenberg (2003) has shown that 5.3Ne generations are needed for a species to acquire monophyly at 99 % of its loci given that all loci in the sister species are also monophyletic. That indicates that for a species of 1 M individuals with a generation time of 10 yr, the full monophyly will only be reached 50 M years after speciation, whereas only around 1 000 yr may be needed for species with small populations (Naciri & Linder 2015). Incomplete lineage sorting may be frequent in closely related taxa or during a speciation process (Hobolth et al. 2011, Blanco-Pastor et al. 2012, Saag et al. 2014, Naciri & Linder 2015), as may be considered in our case. The topological incongruence observed among the standard loci, FRBi15 and FRBi16, supports the incomplete lineage sorting hypothesis as one of the main reasons explaining why morphospecies are not monophyletic. While neutral markers are useful for understanding gene-flow patterns, adaptive markers provide the evolutionary pressure that contributes to speciation (Emelianov et al. 2004, Hey 2006, Holderegger et al. 2006). As adaptive markers are under natural selection, certain alleles can be present in some morphospecies and absent in others, even if there is gene flow amongst them (Lumley & Sperling 2011). The use of phylogenomic datasets may provide a more accurate and supported phylogenetic reconstruction, especially if the appropriate scale of loci variability is selected from all the genome (Leavitt et al. 2016). However, if there are high levels of incomplete lineage sorting, it might not be expected that morphospecies would appear forming supported clades. Nevertheless, genomic data may reveal few mutations linked to certain morphospecies, which would be producing adaptive traits. Darwin’s finches are an iconic example of a rapid speciation process, in which there is a mismatch between the phylogenetic species concept and phenotype-based taxonomy; in that case, genomic studies have detected specific loci subjected to selection pressure, which are directly related with the development of taxon-specific phenotypes (Lamichhaney et al. 2015). In Bryoria, supposed adaptive traits may be influenced by the genes involved in the production of certain extrolites or in the epicortical substances (Boluda et al. 2014), which may produce differential selection pressure for each morphotype, at least in some environments. The process might be similar to that of natural selection of the pale and melanic morphs of the Peppered Moth (Biston betularia) in Europe (Majerus 2009), impeding the fixation of a single morphotype in all populations. In our case also, high levels of incomplete lineage sorting mixed with a few phenotypically important genes under variable degrees of selection in different environments, may explain the mismatch observed between phenotypes and genotypes.
Acknowledgments
This work was undertaken with the support of the Spanish Ministerio de Economía y Competitividad projects CGL2011-25003 and CGL2014-55542-P, and the BES-2012-054488 grant to CGB. Microsatellite analyses carried out at WSL were financially supported by the Federal Office for the Environment (FOEN) and SwissBOL (grants to CS), and we acknowledge the Genetic Diversity Centre at ETHZ. We are grateful also to A. Crespo (Madrid), B. Abbott (Arkadias), C. Ruibal (Madrid), H. Holien (Steinkjer), J. Villagra (Madrid), M. Wedin (Stockholm), N. Calpena-Grau (Madrid), and T. Goward (Vancouver) for providing specimens. We are especially grateful to C. Cornejo (Switzerland) for her help with the laboratory work, and for a helpful review of the manuscript.
Appendix 1.
Chemical, geographical and morphological characters used of Bryoria sect. Implexae samples in the phenogram reconstruction (Fig. 1).
| Samples | Alectorialic acid | Barbatolic acid | Fumarprotocetraric acid | Gyrophoric acid | Norstictic acid | Psoromic acid | Old World (0) / New World (1) | Thallus brownish (0) / whitish (1) | Branching angles acute | Branching angles obtuse | Soralia fissural | Soralia tuberculate | Pseudocyphellae inconspicuous (0) / conspicuous (1) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| capillaris_L01-17 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| capillaris_L06-10 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 |
| capillaris_L07-15 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 0 | 0 |
| capillaris_L08-12 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 0 |
| capillaris_L13-03 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 1 | 0 |
| capillaris_L14-02 | 1 | 1 | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 1 | 0 | 0 | 1 |
| capillaris_L141 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 0 |
| capillaris_L15-15 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| capillaris_L16-21 | 1 | 1 | 0 | 0 | 1 | 1 | 0 | 0 | 1 | 1 | 0 | 0 | 0 |
| capillaris_L211 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 0 |
| capillaris_L270 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 0 |
| capillaris_S192 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 0 |
| capillaris_S2 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 0 |
| friabilis_02 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 1 | 1 | 0 | 0 | 0 | 1 |
| friabilis_L355 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 1 | 1 | 0 | 0 | 0 | 1 |
| friabilis_L407 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 1 | 1 | 0 | 0 | 0 | 1 |
| friabilis_S395 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 1 | 1 | 0 | 0 | 0 | 1 |
| fuscescens_L12-03 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 1 | 1 | 0 |
| fuscescens_L12-05 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 1 | 1 | 0 |
| fuscescens_L139 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 1 | 1 | 0 |
| fuscescens_L149 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 1 | 1 | 0 |
| fuscescens_L15-21 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 0 |
| fuscescens_L160 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 1 | 1 | 0 |
| fuscescens_L189 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 1 | 1 | 0 |
| fuscescens_L224 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 1 | 1 | 0 |
| fuscescens_L232 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 1 | 1 | 0 |
| fuscescens_L305 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 1 | 1 | 0 |
| fuscescens_S109 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 1 | 1 | 0 |
| fuscescens_S157 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 1 | 1 | 0 |
| fuscescens_S24 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 1 | 1 | 0 |
| fuscescens_S256 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 1 | 1 | 0 |
| fuscescens_S259 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 1 | 1 | 0 |
| fuscescens_S260a | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 1 | 1 | 0 |
| fuscescens_S261 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 1 | 1 | 0 |
| fuscescens_S267 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 1 | 1 | 0 |
| fuscescens_S272 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 1 | 1 | 0 |
| fuscescens_S274 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 1 | 1 | 0 |
| fuscescens_S369 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 1 | 1 | 0 |
| fuscescens_S379 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 1 | 1 | 0 |
| fuscescens_S380 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 1 | 1 | 0 |
| fuscescens_S56 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 1 | 1 | 0 |
| implexa_L01-01 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 1 | 1 | 1 | 1 | 1 |
| implexa_L06-05 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 1 | 1 | 0 | 0 | 1 | 0 |
| implexa_L10-03 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 1 | 0 | 0 | 1 |
| implexa_L11-15 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 1 | 0 | 1 | 0 |
| implexa_L16-15 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 1 | 0 | 0 | 0 | 1 |
| implexa_S168 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 1 | 1 | 1 | 1 | 1 |
| implexa_S22 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 1 | 1 | 1 | 1 | 1 |
| implexa_S36 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 1 | 1 | 1 | 1 | 1 |
| implexa_S39 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 1 | 1 | 1 | 1 | 1 |
| implexa_S67 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 1 | 1 | 1 | 1 | 1 |
| inactiva_L206 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 1 |
| inactiva_L323b | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 1 |
| inactiva_L347 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 1 |
| inactiva_L358 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 1 |
| inactiva_S239a | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 1 |
| inactiva_S384 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 1 |
| inactiva_S392 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 0 | 1 |
| kockiana_L394 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 1 | 1 | 0 | 0 | 0 |
| kockiana_L396 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 1 | 1 | 0 | 0 | 0 |
| kuemmerleana_L04-03 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 |
| kuemmerleana_L09-04 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 1 | 1 | 0 | 0 | 1 |
| kuemmerleana_L09-07 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 |
| kuemmerleana_L16-17 | 0 | 0 | 0 | 0 | 1 | 1 | 0 | 1 | 1 | 1 | 1 | 1 | 1 |
| kuemmerleana_L244 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 1 |
| kuemmerleana_L274 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 1 |
| kuemmerleana_L275 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 1 |
| kuemmerleana_S128 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 1 |
| kuemmerleana_S160 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 1 |
| pikei_02 | 1 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 0 | 0 | 1 |
| pikei_04 | 1 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 0 | 0 | 1 |
| pikei_05 | 1 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 0 | 0 | 1 |
| pikei_07 | 1 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 0 | 0 | 1 |
| pikei_09 | 1 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 0 | 0 | 1 |
| pikei_10 | 1 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 0 | 0 | 1 |
| pikei_11 | 1 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 0 | 0 | 1 |
| pikei_12 | 1 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 0 | 0 | 1 |
| pikei_13 | 1 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 0 | 0 | 1 |
| pikei_14 | 1 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 0 | 0 | 1 |
| pikei_15 | 1 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 0 | 0 | 1 |
| pikei_a | 1 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 0 | 0 | 1 |
| pikei_b | 1 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 0 | 0 | 1 |
| pikei_c | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 0 | 0 | 1 |
| pikei_d | 1 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 0 | 0 | 1 |
| pikei_L197 | 1 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 0 | 0 | 1 |
| pikei_L210 | 1 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 0 | 0 | 1 |
| pikei_L241 | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 0 | 0 | 1 |
| pikei_L374 | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 1 | 1 | 0 | 0 | 0 | 1 |
| pikei_L376 | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 1 | 1 | 0 | 0 | 0 | 1 |
| pikei_L377 | 1 | 0 | 0 | 1 | 0 | 0 | 1 | 1 | 1 | 0 | 0 | 0 | 1 |
| pikei_S221 | 1 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 0 | 0 | 1 |
| pikei_S362 | 1 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 0 | 0 | 1 |
| pikei_S368 | 1 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 0 | 0 | 1 |
| pikei_S382 | 1 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 0 | 0 | 1 |
| pikei_S383a | 1 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 0 | 0 | 1 |
| pikei_S390 | 1 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 0 | 0 | 1 |
| pikei_S394 | 1 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 0 | 0 | 1 |
| pseudofuscescens_S222 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 1 | 1 | 0 | 0 | 1 |
| pseudofuscescens_S232 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 1 | 1 | 0 | 0 | 1 |
| pseudofuscescens_S370 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 1 | 1 | 0 | 0 | 1 |
| pseudofuscescens_S371 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 1 | 1 | 0 | 0 | 1 |
| pseudofuscescens_S377 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 1 | 1 | 0 | 0 | 1 |
| pseudofuscescens_S386 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 1 | 1 | 0 | 0 | 1 |
| pseudofuscescens_S387 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 1 | 1 | 0 | 0 | 1 |
| sp_L395 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 1 | 0 | 0 | 0 |
| sp_S392 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 1 | 0 | 0 | 0 |
| vrangiana_L02-20 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 1 | 1 | 0 |
| vrangiana_L03-07 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 1 | 0 |
| vrangiana_L05-17 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 1 | 0 |
| vrangiana_L07-03 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 1 | 1 |
| vrangiana_L07-19 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 0 |
| vrangiana_L08-19 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 1 | 1 | 0 |
| vrangiana_L08-20 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 1 | 0 |
| vrangiana_L10-13 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 0 |
| vrangiana_L12-11 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 1 | 0 | 1 | 1 | 1 | 0 |
| vrangiana_L13-12 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 1 | 1 | 0 | 0 | 1 |
| vrangiana_L272 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 1 | 1 | 1 | 0 |
| vrangiana_L273 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 1 | 1 | 0 |
| vrangiana_L300 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 1 | 1 | 0 |
| vrangiana_L307 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 1 | 1 | 1 | 0 |
| vrangiana_S10 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 1 | 1 | 1 | 0 |
| vrangiana_S164 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 1 | 1 | 0 |
| vrangiana_S166 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 1 | 1 | 0 |
| vrangiana_S196a | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 1 | 1 | 0 |
| vrangiana_S341 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 1 | 1 | 0 |
| vrangiana_S385 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 1 | 1 | 0 |
| vrangiana_S42 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 1 | 1 | 1 | 0 |
| vrangiana_S45 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 1 | 1 | 0 |
| vrangiana_S57 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 1 | 1 | 0 |
| vrangiana_S59 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 1 | 1 | 0 |
| vrangiana_S6 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 1 | 1 | 0 |
| vrangiana_S62 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 1 | 1 | 1 | 0 |
| vrangiana_S72 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 1 | 1 | 0 |
Appendix 2.

Phenograms based on a presence/absence distance matrix in Bryoria sect. Implexae from: a. Extrolite composition alone; b. extrolite composition, with geographical, and morphological data. — Bold branches represent supported clades (bootstrap ≥ 70 %, approximately unbiased ≥ 95 %). — Ale. = Alectorialic acid; Bar. = Barbatolic acid; Fum. = Fumarprotocetraric acid; Gyr. = Gyrophoric acid; No subs. = No substances detected; Nor. = Norstictic acid; Pso. = Psoromic acid; * = Except specimen named Bryoria capillaris L14.02.
Appendix 3.
Microsatellite fragment lengths of Bryoria sect. Implexae analysed specimens.
| Sample | Bi01 | Bi03 | Bi04 | Bi05 | Bi10 | Bi11 | Bi12 | Bi14 | Bi19 |
|---|---|---|---|---|---|---|---|---|---|
| capillaris_L01-17 | 103 | 279 | 323 | 128 | 437 | 316 | 100 | 365 | 346 |
| capillaris_L06-10 | 103 | 279 | 323 | 128 | 437 | 316 | 100 | 365 | 346 |
| capillaris_L07-15 | 103 | 279 | 323 | 128 | 437 | 316 | 100 | 365 | 346 |
| capillaris_L08-12 | 103 | 279 | 323 | 128 | 434 | 316 | 103 | 361 | 346 |
| capillaris_L13-03 | 94 | 279 | 325 | 138 | 434 | 318 | 115 | 361 | 346 |
| capillaris_L14-02 | 112 | 279 | 327 | 128 | 437 | 320 | 100 | 365 | 346 |
| capillaris_L141 | 123 | 281 | 323 | 136 | 434 | 316 | 100 | 361 | 346 |
| capillaris_L15-15 | 112 | 279 | 323 | – | 437 | 316 | 100 | 365 | 346 |
| capillaris_L16-21 | 103 | 279 | 323 | 128 | 437 | 316 | 100 | 365 | 346 |
| capillaris_L211 | 112 | 279 | 323 | 136 | 434 | 316 | 103 | 361 | 346 |
| capillaris_L270 | 112 | 279 | 323 | 138 | 434 | 316 | 124 | 361 | 346 |
| capillaris_S192 | 112 | 279 | 323 | 128 | 437 | 316 | 100 | 365 | 346 |
| capillaris_S2 | 94 | 279 | 323 | 138 | 434 | 316 | 124 | 361 | 346 |
| friabilis_02 | 114 | 281 | 317 | 137 | 434 | 310 | 100 | – | 350 |
| friabilis_L355 | 120 | 277 | 316 | 137 | 436 | 310 | 100 | 365 | 350 |
| friabilis_L407 | 132 | 281 | 316 | 137 | 434 | 310 | 131 | 365 | 346 |
| friabilis_S395 | 109 | 277 | 316 | 132 | 438 | 310 | 100 | 365 | 350 |
| fuscescens_L12-03 | 112 | 279 | 323 | 138 | 434 | 316 | 103 | 361 | 352 |
| fuscescens_L12-05 | 94 | 281 | 323 | 138 | 434 | 316 | 103 | 361 | 350 |
| fuscescens_L139 | 94 | 279 | 323 | 138 | 434 | 316 | 103 | 361 | 352 |
| fuscescens_L149 | 123 | 279 | 323 | 138 | 434 | 316 | 103 | 361 | 350 |
| fuscescens_L15-21 | 123 | 277 | 325 | 138 | 434 | 318 | 115 | 361 | 352 |
| fuscescens_L160 | 94 | 281 | 323 | 136 | 434 | 316 | 103 | 361 | 352 |
| fuscescens_L189 | 112 | 279 | 323 | 138 | 434 | 316 | 100 | 361 | 352 |
| fuscescens_L224 | 112 | 279 | 323 | 138 | 434 | 316 | 103 | 361 | 352 |
| fuscescens_L232 | 94 | 279 | 323 | 138 | 434 | 316 | 103 | 361 | 352 |
| fuscescens_L305 | 117 | 279 | 323 | 138 | 434 | 316 | 137 | 361 | 352 |
| fuscescens_S109 | 112 | 281 | 323 | 138 | 434 | 316 | 118 | 361 | 350 |
| fuscescens_S157 | 117 | 279 | 323 | 136 | 434 | 316 | 103 | 361 | 350 |
| fuscescens_S24 | 112 | 281 | 323 | 138 | 434 | 316 | 106 | 361 | 352 |
| fuscescens_S256 | 94 | 281 | 323 | – | 434 | 316 | 124 | 363 | 354 |
| fuscescens_S259 | 94 | 277 | 323 | 138 | 437 | 316 | 103 | 363 | 352 |
| fuscescens_S260a | 82 | 279 | 323 | 138 | 437 | 316 | 103 | 363 | 352 |
| fuscescens_S261 | 82 | 279 | 323 | 138 | 437 | 316 | 103 | 363 | 352 |
| fuscescens_S267 | 82 | 279 | 323 | 138 | 437 | 316 | 103 | 363 | 352 |
| fuscescens_S272 | 82 | 279 | 323 | 138 | 437 | 316 | 103 | 363 | 352 |
| fuscescens_S274 | 94 | 279 | 323 | 138 | 434 | 316 | – | 361 | 350 |
| fuscescens_S369 | – | 279 | 323 | 138 | 437 | 316 | 103 | 363 | 352 |
| fuscescens_S379 | 94 | 279 | 323 | 138 | 437 | 316 | 100 | 363 | 352 |
| fuscescens_S380 | 112 | 279 | 323 | 138 | 434 | 316 | 118 | 361 | 352 |
| fuscescens_S56 | 123 | 279 | 323 | 136 | 434 | 316 | 100 | 361 | 350 |
| glabra_01 | 114 | 283 | 306 | 132 | 426 | 299 | 115 | 369 | 344 |
| glabra_02 | 120 | 283 | 306 | 132 | 426 | 299 | – | 369 | 344 |
| glabra_03 | 120 | 283 | 306 | 132 | 426 | 299 | – | 369 | 344 |
| glabra_04 | 120 | 283 | 306 | 132 | 426 | 299 | – | 369 | 344 |
| glabra_05 | 120 | 283 | 306 | 132 | 426 | 299 | – | 369 | 344 |
| glabra_L186 | 114 | 283 | 304 | 132 | 426 | 297 | 115 | 371 | 344 |
| glabra_L406 | 114 | 283 | 304 | 132 | 426 | 297 | 115 | 371 | 344 |
| glabra_L414 | 114 | 283 | 306 | 132 | 426 | 299 | 115 | 371 | 344 |
| glabra_S388 | 114 | 283 | 306 | 132 | 426 | 299 | 115 | 371 | 344 |
| implexa_L01-01 | 135 | 281 | 323 | 138 | 434 | 316 | 106 | 361 | 350 |
| implexa_L06-05 | 112 | 263 | 323 | 136 | 434 | 316 | 103 | 361 | 350 |
| implexa_L10-03 | 106 | 279 | 323 | 128 | 437 | 316 | 100 | 365 | 346 |
| implexa_L11-15 | 94 | 279 | 323 | 136 | 435 | 316 | 103 | 361 | 352 |
| implexa_L16-15 | 112 | 281 | 325 | 138 | 434 | 318 | 115 | 361 | 352 |
| implexa_S168 | 112 | 279 | 323 | 138 | 434 | 316 | 115 | 361 | 350 |
| implexa_S22 | 94 | 279 | 323 | 136 | 436 | 316 | 118 | 361 | 352 |
| implexa_S36 | 94 | 279 | 325 | 136 | 434 | 318 | 103 | 361 | 346 |
| implexa_S39 | – | – | 323 | 138 | 434 | 316 | – | 361 | 350 |
| implexa_S67 | 94 | 279 | 325 | 136 | 434 | 318 | 103 | 361 | 346 |
| inactiva_L206 | 114 | 277 | 317 | 137 | 434 | 311 | 100 | 365 | 350 |
| inactiva_L323b | 114 | 281 | 316 | 132 | 434 | 310 | 131 | 365 | 350 |
| inactiva_L347 | 114 | 277 | 317 | 132 | 434 | 310 | 124 | 365 | 350 |
| inactiva_L358 | 109 | 281 | 317 | 132 | 436 | 310 | 106 | 365 | 350 |
| inactiva_S239a | 109 | 277 | 314 | 132 | 434 | 308 | 100 | 365 | 350 |
| inactiva_S384 | 114 | 277 | 316 | 137 | 434 | 310 | 100 | 365 | 350 |
| inactiva_S392 | 120 | 281 | 316 | 137 | 434 | 310 | 100 | 365 | 350 |
| kockiana_L394 | 94 | 279 | 317 | 136 | 472 | 310 | 109 | 365 | 344 |
| kockiana_L396 | 94 | 279 | 317 | 136 | 472 | 310 | 109 | 365 | 344 |
| kuemmerleana_L04-03 | 129 | 279 | 325 | 138 | 434 | 318 | 103 | 361 | 352 |
| kuemmerleana_L09-04 | 112 | 281 | 323 | 128 | 437 | 316 | 100 | 365 | 346 |
| kuemmerleana_L09-07 | 112 | 281 | 323 | 128 | 437 | 316 | 100 | 365 | 346 |
| kuemmerleana_L16-17 | 112 | 281 | 323 | 138 | 434 | 316 | 106 | 361 | 352 |
| kuemmerleana_L244 | 117 | 279 | 323 | 138 | 434 | 316 | 115 | 361 | 350 |
| kuemmerleana_L274 | 94 | 279 | 323 | 136 | 434 | 316 | 103 | 361 | 352 |
| kuemmerleana_L275 | 94 | 279 | 323 | 136 | 434 | 316 | 103 | 361 | 352 |
| kuemmerleana_S128 | 100 | 279 | 323 | 136 | 434 | 316 | 100 | 361 | 354 |
| kuemmerleana_S160 | 117 | 279 | 323 | 138 | 434 | 316 | 103 | 361 | 350 |
| pikei_02 | 117 | 277 | 317 | 137 | 434 | 310 | 100 | 365 | 344 |
| pikei_04 | 117 | 277 | 317 | 137 | 434 | 310 | 100 | 365 | 344 |
| pikei_05 | 117 | 277 | 317 | 137 | 434 | 310 | 100 | 365 | 344 |
| pikei_07 | 117 | 281 | 317 | 137 | 434 | 311 | 109 | – | 344 |
| pikei_09 | 114 | 281 | 317 | 132 | – | 310 | 100 | – | 344 |
| pikei_10 | 117 | 277 | 317 | 137 | 434 | 311 | 100 | 365 | 344 |
| pikei_11 | 114 | 277 | 317 | 137 | 434 | 310 | 100 | 365 | 344 |
| pikei_12 | 114 | 277 | – | 137 | 434 | – | 100 | – | 344 |
| pikei_13 | – | 277 | 317 | 137 | 434 | 310 | 118 | 365 | 344 |
| pikei_14 | 117 | 277 | 317 | 137 | 434 | 310 | 127 | 365 | 344 |
| pikei_15 | 114 | 281 | 317 | 137 | 436 | 310 | 109 | – | 344 |
| pikei_a | 114 | – | 323 | 132 | 437 | 316 | 100 | – | 346 |
| pikei_b | 117 | 277 | 317 | 137 | 434 | 311 | 100 | 365 | 344 |
| pikei_c | 108 | – | – | 137 | – | 314 | 100 | 326 | 344 |
| pikei_d | 117 | 277 | – | 137 | 437 | 310 | 100 | – | 344 |
| pikei_L197 | 114 | 277 | 316 | 132 | 434 | 310 | 109 | 365 | 344 |
| pikei_L210 | 109 | 277 | 316 | 132 | 434 | 310 | 100 | 365 | 344 |
| pikei_L241 | 126 | 277 | 316 | 137 | 434 | 310 | 109 | – | 352 |
| pikei_L374 | 114 | 277 | 316 | 137 | 434 | 310 | 106 | 365 | 344 |
| pikei_L376 | 132 | 277 | 316 | 137 | 434 | 310 | 127 | 365 | 352 |
| pikei_L377 | 109 | 277 | 316 | 137 | 434 | 310 | 103 | 365 | 344 |
| pikei_S221 | 109 | 277 | 316 | 132 | 436 | 310 | 106 | 365 | 352 |
| pikei_S362 | 114 | 281 | 317 | 137 | 434 | 311 | 100 | 365 | 344 |
| pikei_S368 | 112 | 281 | 316 | 132 | 436 | 310 | 109 | 365 | 344 |
| pikei_S382 | 114 | 281 | 317 | 137 | 434 | 311 | 100 | 365 | 344 |
| pikei_S383a | 114 | 281 | 317 | 132 | 436 | 311 | 100 | 365 | 344 |
| pikei_S390 | 114 | 277 | 316 | 137 | 434 | 310 | 100 | 365 | 344 |
| pikei_S394 | 126 | 277 | 316 | 137 | 434 | 310 | 100 | 365 | 352 |
| pseudofuscescens_S222 | – | 277 | 316 | 132 | – | 310 | 100 | 365 | – |
| pseudofuscescens_S232 | 114 | 277 | 317 | 137 | 436 | 310 | 100 | 365 | 350 |
| pseudofuscescens_S370 | 117 | 277 | 316 | 132 | 434 | 310 | 100 | 365 | 350 |
| pseudofuscescens_S371 | 117 | 281 | 316 | 132 | 434 | 310 | 100 | 365 | 350 |
| pseudofuscescens_S377 | – | 277 | – | 132 | 434 | 311 | 100 | 365 | – |
| pseudofuscescens_S386 | 112 | 277 | 317 | 132 | 438 | 311 | 100 | 365 | 350 |
| pseudofuscescens_S387 | 117 | 277 | 316 | 132 | 434 | 310 | 127 | 365 | 350 |
| sp_L395 | 94 | 273 | 317 | 136 | 460 | 310 | 109 | 365 | 344 |
| sp_S392 | 97 | 283 | 317 | 136 | 472 | 310 | 118 | 365 | 344 |
| vrangiana_L02-20 | 112 | 279 | 323 | 138 | 434 | 316 | 106 | 361 | 352 |
| vrangiana_L03-07 | 94 | 279 | 325 | 136 | 434 | 318 | 115 | 365 | 346 |
| vrangiana_L05-17 | 117 | 281 | 323 | 138 | 434 | 316 | 121 | 361 | 350 |
| vrangiana_L07-03 | 94 | 281 | 323 | 138 | 434 | 316 | 118 | 361 | 352 |
| vrangiana_L07-19 | 123 | 279 | 323 | 138 | 434 | 316 | 103 | 361 | 350 |
| vrangiana_L08-19 | 94 | 281 | 323 | 136 | 436 | 316 | 103 | 361 | 352 |
| vrangiana_L08-20 | 94 | 279 | 323 | 138 | 434 | 316 | 121 | 361 | 352 |
| vrangiana_L10-13 | 123 | 283 | 323 | 128 | 437 | 316 | 100 | 365 | 346 |
| vrangiana_L12-11 | 94 | 279 | 323 | 138 | 434 | 316 | 144 | 361 | 352 |
| vrangiana_L13-12 | 94 | 279 | 323 | 138 | 434 | 316 | 103 | 361 | 352 |
| vrangiana_L272 | 112 | 279 | 323 | 136 | 434 | 316 | – | 361 | 350 |
| vrangiana_L273 | 94 | 279 | 323 | 138 | 436 | 316 | 103 | 361 | 350 |
| vrangiana_L300 | 94 | 281 | 323 | 136 | 434 | 316 | 115 | 361 | 350 |
| vrangiana_L307 | 123 | 279 | 325 | 138 | 434 | 318 | 103 | 361 | 352 |
| vrangiana_S10 | 123 | 279 | 323 | 136 | 434 | 316 | 124 | 361 | 352 |
| vrangiana_S164 | 94 | 279 | 323 | 136 | 434 | 316 | 127 | 361 | 350 |
| vrangiana_S166 | 117 | 279 | 323 | 138 | 434 | 316 | 144 | 365 | 352 |
| vrangiana_S196a | 94 | 281 | 323 | 138 | 434 | 316 | 118 | 361 | 350 |
| vrangiana_S341 | – | 279 | 323 | – | 434 | 316 | – | 361 | 350 |
| vrangiana_S385 | 94 | 279 | 323 | 138 | 434 | 316 | 103 | 361 | 350 |
| vrangiana_S42 | 117 | 279 | 323 | 138 | 434 | 316 | 131 | 361 | 352 |
| vrangiana_S45 | 112 | 279 | 323 | 136 | 434 | 316 | 100 | 361 | 350 |
| vrangiana_S57 | 94 | 279 | 323 | 138 | 434 | 316 | 115 | 361 | 352 |
| vrangiana_S59 | 94 | 279 | 323 | 138 | 434 | 316 | 115 | 361 | 352 |
| vrangiana_S6 | 123 | 279 | 323 | 138 | 434 | 316 | 103 | 361 | 350 |
| vrangiana_S62 | 112 | 279 | 323 | 138 | 434 | 316 | 115 | 361 | 352 |
| vrangiana_S72 | 94 | 279 | 323 | 136 | 436 | 316 | 103 | 361 | 352 |
Appendix 4.
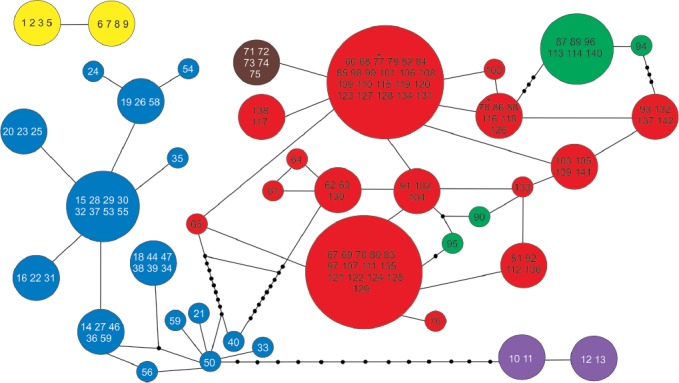
Haplotype network in Bryoria sect. Implexae of a concatenated matrix containing ITS, IGS and GAPDH sequences. The analysis coded gaps as missing data and used a 95 % connection limit. Numbers represent the specimens shown in Table 1 and colours depict the STRUCTURE microsatellites genepool (Fig. 2). Connecting line length do not depict the genetic distance. Each line represents a single mutation connected by black small circles. Circle size is related with the number of analysed specimens. — * = WDb (Wide Distributed brown cluster) specimens.
Appendix 5.
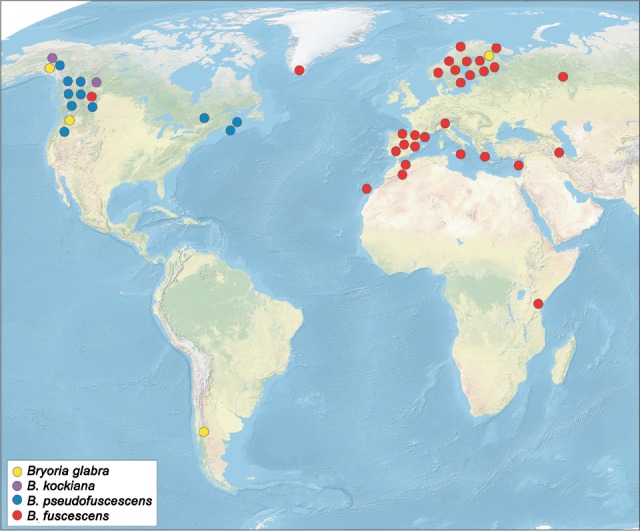
Distribution of Bryoria sect. Implexae specimens examined. Two samples of geographical interest, not analysed in this study, have been added: Bryoria kockiana (Canada, British Columbia, 1982, Goward 82-1040, UBC – paratype; cf. Velmala et al. 2014) and Bryoria fuscescens (Tanzania, Kilimanjaro, 2016, Boluda & Kitara, MAF-Lich. 20750). — Basemap source: U.S. National Park Service (NPS) Natural Earth physical map.
Footnotes
Hits obtained for these names in Google and Google Scholar respectively on 12 April 2018 were: B. fuscescens (12 300 and 1 620), B. capillaris (10 300 and 999), B. implexa (6 020 and 447), B. kuemmerleana (226 and 20), B. lanestris (6 740 and 185), and B. vrangiana (532 and 61).
REFERENCES
- Abbott RJ, Barton NH, Good JM. 2016. Genomics of hybridization and its evolutionary consequences. Molecular Ecology 25: 2325–2332. [DOI] [PubMed] [Google Scholar]
- Akaike H. 1974. A new look at the statistical model identification. IEEE Transactions on Automatic Control 19: 716–723. [Google Scholar]
- Alors D, Lumbsch TH, Divakar PK, et al. 2016. An integrative approach for understanding diversity in the Punctelia rudecta species complex (Parmeliaceae, Ascomycota). PLoS ONE 11: e0146537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altermann S, Leavitt SD, Goward T, et al. 2014. How do you solve a problem like Letharia? A new look at cryptic species in lichen-forming fungi using Bayesian clustering and SNPs from multilocus sequence data. PLoS ONE 5: e97556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amo de Paz G, Cubas P, Crespo A, et al. 2012. Transoceanic dispersal and subsequent diversification on separate continents shaped diversity of the Xanthoparmelia pulla group (Ascomycota). PLoS ONE 7: e39683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Articus K, Mattsson J-E, Tibell L, et al. 2002. Ribosomal DNA and β-tubulin data do not support the separation of the lichens Usnea florida and U. subfloridana as distinct species. Mycological Research 106: 412–418. [Google Scholar]
- Bekessy SA, Ennos RA, Burgman MA, et al. 2003. Neutral DNA markers fail to detect genetic divergence in an ecologically important trait. Biological Conservation 110: 267–275. [Google Scholar]
- Blanco-Pastor JL, Vargas P, Pfeil BE. 2012. Coalescent simulations reveal hybridization and incomplete lineage sorting in Mediterranean Linaria. PLoS ONE 7: e39089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boluda CG, Hawksworth DL, Divakar PK, et al. 2016. Microchemical and molecular investigations reveal Pseudephebe species as cryptic with an environmentally modified morphology. The Lichenologist 48: 527–543. [Google Scholar]
- Boluda CG, Rico VJ, Crespo A, et al. 2015. Molecular sequence data from populations of Bryoria fuscescens s.lat in the mountains of central Spain indicates a mismatch between haplotypes and chemotypes. The Lichenologist 47: 279–286. [Google Scholar]
- Boluda CG, Rico VJ, Hawksworth DL. 2014. Fluorescence microscopy as a tool for the visualization of lichen substances within Bryoria thalli. The Lichenologist 46: 723–726. [Google Scholar]
- Boni MF, Posada D, Feldman MW. 2007. An exact nonparametric method for inferring mosaic structure in sequence triplets. Genetics 176: 1035–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodo IM, Hawksworth DL. 1977. Alectoria and allied genera in North America. Opera Botanica 42: 1–164. [Google Scholar]
- Campbell V, Legendre P, Lapointe FJ. 2011. The performance of the congruence among distance matrices (CADM) test in phylogenetic analysis. BMC Evolutionary Biology 11: 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caparrós R, Lara F, Draper I, et al. 2016. Integrative taxonomy sheds light on an old problem: the Ulota crispa complex (Orthotrichaceae, Musci). Botanical Journal of the Linnean Society 180: 427–451. [Google Scholar]
- Chatrou LW, Escribano MP, Viruel MA, et al. 2009. Flanking regions of monomorphic microsatellite loci provide a new source of data for plant species-level phylogenetics. Molecular Phylogenetics and Evolution 53: 726–733. [DOI] [PubMed] [Google Scholar]
- Clement M, Posada D, Crandall KA. 2000. TCS: a computer program to estimate gene genealogies. Molecular Ecology 9: 1657–1659. [DOI] [PubMed] [Google Scholar]
- Crespo A, Lumbsch HT. 2010. Cryptic species in lichen-forming fungi. IMA Fungus 1: 167–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dal Grande F, Alors D, Pradeep KD, et al. 2014. Insights into intrathalline genetic diversity of the cosmopolitan lichen symbiotic green alga Trebouxia decolorans Ahmadjian using microsatellite markers. Molecular Phylogenetics and Evolution 72: 54–60. [DOI] [PubMed] [Google Scholar]
- Darriba D, Taboada GL, Doallo R, et al. 2012. jModelTest 2: more models, new heuristics and parallel computing. Nature Methods 9: 772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayrat B. 2005. Towards integrative taxonomy. Biological Journal of the Linnean Society 85: 407–415. [Google Scholar]
- Del-Prado R, Divakar PK, Lumbsch HT, et al. 2016. Hidden genetic diversity in an asexually reproducing lichen forming fungal group. PLoS ONE 11: e0161031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divakar PK, Cubas P, Blanco O, et al. 2010. An overview on hidden diversity in lichens: Parmeliaceae. http://taxateca.com/files/Divakar_et_al_2010.pdf. [Google Scholar]
- Divakar PK, Leavitt SD, Molina MC, et al. 2016. A DNA barcoding approach for identification of hidden diversity in Parmeliaceae (Ascomycota): Parmelia sensu stricto as a case study. Botanical Journal of the Linnean Society 180: 21–29. [Google Scholar]
- Douhan GW, Martin DP, Rizzo DM. 2007. Using the putative asexual fungus Cenococcum geophilum as a model to test how species concepts influence recombination analyses using sequence data from multiple loci. Current Genetics 52: 191–201. [DOI] [PubMed] [Google Scholar]
- Drumond AJ, Rambaut A, Shapiro B, et al. 2005. Bayesian coalescent inference of past population dynamics from molecular sequences. Molecular Biology and Evolution 22: 1185–1192. [DOI] [PubMed] [Google Scholar]
- Drumond AJ, Suchard MA, Xie D, et al. 2012. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Molecular Biology and Evolution 29: 1969–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earl DA, Von Holdt B. 2012. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conservation of Genetics Resources 4: 359–361. [Google Scholar]
- Edwards CE, Soltis DE, Soltis PS. 2008. Using patterns of genetic structure based on microsatellite loci to test hypotheses of current hybridization, ancient hybridization and incomplete lineage sorting in Conradina (Lamicaceae). Molecular Ecology 17: 5157–5174. [DOI] [PubMed] [Google Scholar]
- Emelianov I, Marec F, Mallet J. 2004. Genomic evidence for divergence with gene flow in host races of the larch budmoth. Proceedings of the Royal Society of London, Biological Sciences 271: 97–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esseen PA, Olsson T, Coxson D, et al. 2015. Morphology influences water storage in hair lichens from boreal forest canopies. Fungal Ecology 18: 26–35. [Google Scholar]
- Esseen PA, Rönnqvist M, Gauslaa Y, et al. 2017. Externally held water – a key factor for hair lichens in boreal forest canopies. Fungal Ecology 30: 29–38. [Google Scholar]
- Evanno G, Regnaut S, Goudet J. 2005. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Molecular Ecology 14: 2611–2620. [DOI] [PubMed] [Google Scholar]
- Falush D, Stephens M, Pritchard J. 2003. Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics 155: 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Färber L, Solhaug KA, Esseen PA, et al. 2014. Sunscreening fungal pigments influence the vertical gradient of pendulous lichens in boreal forest canopies. Ecology 95: 1464–1471. [DOI] [PubMed] [Google Scholar]
- Fu YX. 1997. Statistical test of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147: 915–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardes M, Bruns TD. 1993. ITS primers with enhanced specificity for basidiomycetes-application to the identification of mycorrhizae and rusts. Molecular Ecology 2: 113–118. [DOI] [PubMed] [Google Scholar]
- Gargas A, Taylor JW. 1992. Polymerase chain reaction (PCR) primers for amplifying andsequencing nuclear 18s rDNA from lichenized fungi. Mycologia 84: 589–592. [Google Scholar]
- Gibbs MJ, Armstrong JS, Gibbs AJ. 2000. Sister-scanning: a Monte Carlo procedure for assessing signals in recombinant sequences. Bioinformatics 16: 573–582. [DOI] [PubMed] [Google Scholar]
- Grant WS. 2015. Problems and cautions with sequence mismatch analysis and Bayesian skyline plots to infer historical demography. Journal of Heredity 16: 1–14. [DOI] [PubMed] [Google Scholar]
- Hart WM, Sunday J. 2007. Things fall apart: biological species form unconnected parsimony networks. Biology Letters 3: 509–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartl DL, Clark AG. 2007. Principles of population genetics. 4th Ed. Sinauer Associates Inc, Sunderland, MA. [Google Scholar]
- Hawksworth DL. 1969a. Chemical and nomenclatural notes on Alectoria (Lichenes) I. Taxon 18: 393–399. [Google Scholar]
- Hawksworth DL. 1969b. The scanning electron microscope, an aid to the study of cortical hyphal orientation in the lichen genera Alectoria and Cornicularia. Journal de Microscopie 8: 753–760. [Google Scholar]
- Hawksworth DL. 1972. Regional studies in Alectoria (Lichenes) II. The British species. The Lichenologist 5: 181–261. [Google Scholar]
- Hawksworth DL. 1976. Lichen chemotaxonomy. In: Brown DH, Hawksworth DL, Bailey LH. (eds), Lichenology: progress and problems: 139–184. Academic Press, London. [Google Scholar]
- Hawksworth DL. 2015. Lichenization: The origins of fungal life-style. In: Upreti DK, Divakar PK, Shukla V, et al (eds), Recent advances in lichenology. Modern methods and approaches in lichen systematics and culture techniques, vol. 2: 1–10. Springer, India. [Google Scholar]
- Hawksworth DL, Jørgensen PM. 2013. (2196) Proposal to conserve the name Alectoria fuscescens (Bryoria fuscescens) against Lichen chalybeiformis and Alectoria subcana (Ascomycota: Lecanorales: Parmeliaceae). Taxon 62: 1057. [Google Scholar]
- Hawksworth DL, Lücking R. 2017. Fungal diversity revisited: 2.2 to 3.8 million species. Microbiology Spectrum 5: FUNK-0052-2016. [DOI] [PubMed] [Google Scholar]
- Hey J. 2006. Recent advances in assessing gene flow between diverging populations and species. Current Opinion in Genetics & Development 16: 592–596. [DOI] [PubMed] [Google Scholar]
- Hibbett DS. 2016. The invisible dimension of fungal diversity. Science 351: 1150–1151. [DOI] [PubMed] [Google Scholar]
- Hobolth A, Dutheil JY, Hawks J, et al. 2011. Incomplete lineage sorting patterns among human, chimpanzee, and orangutan suggest recent orangutan speciation and widespread selection. Genome Research 21: 349–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holderegger R, Kamm U, Gugerli F. 2006. Adaptative vs neutral genetic diversity: implications for landscape genetics. Landscape Ecology 21: 797–807. [Google Scholar]
- Houbraken J, Frisvad JC, Seifert KA, et al. 2012. New penicillin-producing Penicillium species and an overview of section Chrysogena. Persoonia 29: 78–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huelsenbeck JP, Ronquist F. 2001. MrBayes: Bayesian inference of phylogenetic trees. Bioinformatics 17: 754–755. [DOI] [PubMed] [Google Scholar]
- Jakob SS, Blattner FR. 2006. A chloroplast genealogy of Hordeum (Poaceae): Long-term persisting haplotypes, incomplete lineage sorting, regional extinction, and the consequences for phylogenetic inference. Molecular Biology and Evolution 23: 1602–1612. [DOI] [PubMed] [Google Scholar]
- Jakobsson M, Rosenberg NA. 2007. CLUMMP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23: 1801–1806. [DOI] [PubMed] [Google Scholar]
- Joly S, McLenachan PA, Lockhart PJ. 2009. A statistical approach for distinguishing hybridization and incomplete lineage sorting. The American Naturalist 174: E54–E70. [DOI] [PubMed] [Google Scholar]
- Jones G, Aydin Z, Oxelman B. 2014. DISSECT: an assignment-free Bayesian discovery method for species delimitation under the multispecies coalescent. Bioinformatics 31: 991–998. [DOI] [PubMed] [Google Scholar]
- Jørgensen PM, Galloway DJ. 1983. Bryoria (lichenized Ascomycota) in New Zealand. New Zealand Journal of Botany 21: 335–340. [Google Scholar]
- Kaasalainen U, Heinrichs J, Krings M, et al. 2015. Alectorioid morphologies in Paleogene lichens: new evidence and re-evaluation of the fossil Alectoria succini Mägdefrau. PLoS ONE 10: e0129526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Molecular Biology and Evolution 30: 772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay KM, Whittall JB, Scott AH. 2006. A survey of nuclear ribosomal internal transcribed spacer substitution rates across angiosperms: an approximate molecular clock with life history effects. BMC Evolution Biology 6: 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller C, Scheidegger C. 2016. Multiple mating events and spermatia-mediated gene flow in the lichen-forming fungus Lobaria pulmonaria. Herzogia 29: 435–450. [Google Scholar]
- Keller NP, Hohn TM. 1997. Metabolic pathway gene clusters in filamentous fungi. Fungal Genetics and Biology 21: 17–21. [PubMed] [Google Scholar]
- Kirika P, Divakar PK, Crespo A, et al. 2016a. Polyphyly of the genus Canoparmelia – uncovering incongruences between phenotype-based classification and molecular phylogeny within lichenized Ascomycota (Parmeliaceae). Phytotaxa 289: 36–48. [Google Scholar]
- Kirika P, Divakar PK, Crespo A, et al. 2016b. Phylogenetic studies uncover a predominantly African lineage in a widely distributed lichen-forming fungal species. MycoKeys 14: 1–16. [Google Scholar]
- Knowles LL. 2008. Why does a method that fails continue to be used. Evolution 62: 2713–2717. [DOI] [PubMed] [Google Scholar]
- Konrad H, Kiristis T, Riegler M, et al. 2002. Genetic evidence for natural hybridization between the Dutch elm disease pathogens Ophiostoma novo-ulmi ssp. novo-ulmi and O. novo-ulmi ssp. americana. Plant Pathology 51: 78–84. [Google Scholar]
- Kroken S, Taylor JW. 2001a. Outcrossing and recombination in the lichenized fungus Letharia. Fungal Genetics and Biology 34: 83–92. [DOI] [PubMed] [Google Scholar]
- Kroken S, Taylor JW. 2001b. A gene genealogical approach to recognize phylogenetic species boundaries in the lichenized fungus Letharia. Mycologia 93: 38–53. [Google Scholar]
- Lamichhaney S, Berglund J, Markus SA, et al. 2015. Evolution of Darwin’s finches and their beaks revealed by genome sequencing. Nature 518: 371–375. [DOI] [PubMed] [Google Scholar]
- Lanfear R, Calcott B, Ho SY, et al. 2012. Partitionfinder: combined selection of partitioning schemes and substitution models for phylogenetic analyses. Molecular Biology and Evolution 29: 1695–1701. [DOI] [PubMed] [Google Scholar]
- Leavitt SD, Divakar PK, Crespo A, et al. 2016. A matter of time – understanding the limits of the power of molecular data for delimiting species boundaries. Herzogia 29: 479–492. [Google Scholar]
- Leavitt SD, Esslinger TL, Divakar PK, et al. 2012a. Miocene and Pliocene dominated diversification of the lichen-forming fungal genus Melanohalea (Parmeliaceae, Ascomycota) and Pleistocene population expansions. BMC Evolutionary Biology 12: 176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leavitt SD, Esslinger TL, Lumbsch HT. 2012b. Neogene-dominated diversification in neotropical montane lichens: Dating divergence events in the lichen-forming fungal genus Oropogon (Parmeliaceae). American Journal of Botany 99: 1764–1777. [DOI] [PubMed] [Google Scholar]
- Leavitt SD, Grewe F, Widhelm T, et al. 2016. Resolving evolutionary relationships in lichen-forming fungi using diverse phylogenomic datasets and analytical approaches. Scientific Reports 6: 22262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leavitt SD, Johnson L, St. Clair LL. 2011. Species delimitation and evolution in morphologically and chemically diverse communities of the lichen-forming genus Xanthoparmelia (Parmeliaceae, Ascomycota) in Western North America. American Journal of Botany 98: 175–188. [DOI] [PubMed] [Google Scholar]
- Legendre P, Lapointe FJ. 2004. Assessing congruence among distance matrices: Single-malt Scotch whiskies revisited. Australian & New Zealand Journal of Statistics 46: 615–629. [Google Scholar]
- Librado P, Rozas J. 2009. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25: 1451–1452. [DOI] [PubMed] [Google Scholar]
- Lindgren H, Velmala S, Högnabba F, et al. 2014. High fungal selectivity for algal symbionts in the genus Bryoria. The Lichenologist 46: 681–695. [Google Scholar]
- Lumbsch HT. 1988. The use of metabolic data in lichenology at the species and subspecific levels. The Lichenologist 30: 357–367. [Google Scholar]
- Lumley LM, Sperling FAH. 2011. Utility of microsatellites and mitochondrial DNA for species delimitation in the spruce budworm (Choristoneura fumiferana) species complex (Lepidoptera: Tortricidae). Molecular Phylogenetics and Evolution 58: 232–243. [DOI] [PubMed] [Google Scholar]
- Lutsak T, Fernández-Mendoza F, Nadyeina O, et al. 2017. Testing the correlation between norstictic acid content and species evolution in the Cetraria aculeata group in Europe. The Lichenologist 49: 39–56. [Google Scholar]
- Maddison WP. 1997. Gene trees in species trees. Systematic Biology 46: 523–536. [Google Scholar]
- Maechler M, Rousseeuw P, Struyf A, et al. 2013. cluster: Cluster Analysis Basics and Extensions. R package version 1.14.4. [Google Scholar]
- Majerus MEN. 2009. Industrial melanism in the Peppered Moth, Biston betularia: An excellent teaching example of Darwinian evolution in action. Evolution: Education and Outreach 2: 63–74. [Google Scholar]
- Mark K, Saag L, Leavitt SD, et al. 2016. Evaluation of traditionally circumscribed species in the lichen-forming genus Usnea, section Usnea (Parmeliaceae, Ascomycota) using six-locus dataset. Organisms Diversity and Evolution 16: 497–524. [Google Scholar]
- Martin DP, Lemey P, Lott M, et al. 2010. RDP3: a flexible and fast computer program for analyzing recombination. Bioinformatics 26: 2462–2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin DP, Posada D, Crandall KA, et al. 2005. A modified bootscan algorithm for automated identification of recombinant sequences and recombination breakpoints. AIDS Research and Human Retroviruses 21: 98–102. [DOI] [PubMed] [Google Scholar]
- Martin DP, Rybicki E. 2000. RDP: detection of recombination amongst aligned sequences. Bioinformatics 16: 562–563. [DOI] [PubMed] [Google Scholar]
- Maynard-Smith J. 1992. Analyzing the mosaic structure of genes. Journal of Molecular Evolution 34: 126–129. [DOI] [PubMed] [Google Scholar]
- McGuire JA, Linkem CW, Koo MS, et al. 2007. Mitochondrial introgression and incomplete lineage sorting through space and time: Phylogenetics of crotaphytid lizards. Evolution 61: 2879–2897. [DOI] [PubMed] [Google Scholar]
- McMullin RT, Lendemer JC, Braid HE, et al. 2016. Molecular insights into the lichen genus Alectoria (Parmeliaceae) in North America. Botany 94: 1–11. [Google Scholar]
- Miller MA, Pfeiffer W, Schwartz T. 2010. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In: Proceedings of the Gateway Computing Environments Workshop (GCE), November 14: 1–8 USA, New Orleans. [Google Scholar]
- Modica MV, Puillandre N, Castelin M, et al. 2014. A good compromise: rapid and robust species proxies for inventorying biodiversity hotspots using the Terebridae (Gastropoda: Conoidea). PLoS ONE: e102160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina MC, Del-Prado R, Divakar PK, et al. 2011a. Another example of cryptic diversity in lichen-forming fungi: the new species Parmelia mayi (Ascomycota: Parmeliaceae). The Lichenologist 11: 331–342. [Google Scholar]
- Molina MC, Divakar PK, Goward T, et al. 2016. Neogene diversification in the temperate lichen-forming fungal genus Parmelia (Parmeliaceae, Ascomycota). Systematics and Biodiversity 15: 166–181. [Google Scholar]
- Molina MC, Divakar PK, Millanes AM, et al. 2011b. Parmelia sulcata (Ascomycota: Parmeliaceae), a sympatric monophyletic species complex. The Lichenologist 43: 585–601. [Google Scholar]
- Monaghan MT, Wild R, Elliot M, et al. 2009. Accelerated species inventory on Madagascar using coalescent-based models of species delineation. Systematic Biology 58: 298–311. [DOI] [PubMed] [Google Scholar]
- Motyka J. 1964. The North American species of Alectoria. The Bryologist 67: 1–44. [Google Scholar]
- Myllys L, Lindgren H, Aikio S, et al. 2016. Chemical diversity and ecology of the genus Bryoria section Implexae (Parmeliaceae) in Finland. The Bryologist 119: 29–38. [Google Scholar]
- Myllys L, Stenroos S, Thell A. 2002. New genes for phylogenetic studies of lichenized fungi: glyceraldehyde-3-phosphate dehydrogenase and beta-tubulin genes. The Lichenologist 34: 237–246. [Google Scholar]
- Myllys L, Velmala S, Holien H, et al. 2011. Phylogeny of the genus Bryoria. The Lichenologist 6: 617–638. [Google Scholar]
- Naciri Y, Linder HP. 2015. Species delimitation and relationships: The dance of seven veils. Taxon 64: 3–16. [Google Scholar]
- Nadyeina O, Cornejo C, Boluda CG, et al. 2014. Characterization of microsatellite loci in lichen-forming fungi of Bryoria section Implexae (Parmeliaceae). Applied Plant Sciences 2: 1400037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols R. 2001. Gene trees and species trees are not the same. Trends in Ecology & Evolution 16: 358–364. [DOI] [PubMed] [Google Scholar]
- Oberwinkler F. 2017. Yeasts in Pucciniomycotina. Mycological Progress 16: 831–856. [Google Scholar]
- Orange A, James PW, White FJ. 2010. Microchemical methods for the identification of lichens. 2 edn British Lichen Society, London. [Google Scholar]
- Padidam M, Sawyer S, Fauquet CM. 1999. Possible emergence of new geminiviruses by frequent recombination. Virology 265: 218–225. [DOI] [PubMed] [Google Scholar]
- Paradis E, Claude J, Strimmer K. 2004. APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20: 289–290. [DOI] [PubMed] [Google Scholar]
- Parnmen S, Leavitt SD, Rangsiruji A, et al. 2013. Identification of species in the Cladia aggregata group using DNA barcoding (Ascomycota: Lecanorales). Phytotaxa 115: 1–14. [Google Scholar]
- Pažoutová S, Pešicová K, Chudíčková M, et al. 2015. Delimitation of cryptic species inside Claviceps purpurea. Fungal Biology 119: 7–26. [DOI] [PubMed] [Google Scholar]
- Perelman P, Johnson WE, Roos C, et al. 2011. A molecular phylogeny of living primates. PLoS Genetics 7: e1001342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pino-Bodas R, Pérez–Vargas I, Stenroos S, et al. 2016. Sharpening the species boundaries in the Cladonia mediterranea complex (Cladoniaceae, Ascomycota). Persoonia 37: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pons J, Barraclough TG, Gómez-Zurita J, et al. 2006. Sequence based species delimitation for the DNA taxonomy of undescribed insects. Systematic Biology 55: 595–609. [DOI] [PubMed] [Google Scholar]
- Posada D, Crandall KA. 2001. Evaluation of methods for detecting recombination from DNA sequences: computer simulations. Proceedings of the National Academy of Sciences of the United States of America 98: 13757–13762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Printzen C, Ekman S. 2002. Genetic variability and its geographical distribution in the widely disjunct Cavernularia hultenii. The Lichenologist 34: 101–111. [Google Scholar]
- Pritchard JK, Stephens M, Donnelly P. 2000. Inference of population structure using multilocus genotype data. Genetics 155: 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puillandre N, Lambert A, Brouillet S, et al. 2011. ABGD, Automatic Barcode Gap Discovery for primary species delimitation. Molecular Ecology 21: 1864–1877. [DOI] [PubMed] [Google Scholar]
- R Core Team. 2014. R: A language and environment for statistical computing. Vienna, R Foundation for Statistical Computing. [Google Scholar]
- Rambaut A. 2009. FigTree v.1.4. http://tree.bio.ed.ac.uk/software/figtree/. [Google Scholar]
- Rambaut A, Suchard MA, Xie D, et al. 2014. Tracer. Version 1.6. http://beast.bio.ed.ac.uk/Tracer. [Google Scholar]
- Ronquist F, Huelsenbeck JP. 2003. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19: 1572–1574. [DOI] [PubMed] [Google Scholar]
- Rosenberg NA. 2003. The shapes of neutral gene genealogies in two species: Probabilities of monophyly, paraphyly, and polyphyly in a coalescence model. Evolution 57: 1465–1477. [DOI] [PubMed] [Google Scholar]
- Saag L, Mark K, Saag A, et al. 2014. Species delimitation in the lichenized fungal genus Vulpicida (Parmeliaceae, Ascomycota) using gene concatenation and coalescent-based species tree approaches. American Journal of Botany 101: 2169–2182. [DOI] [PubMed] [Google Scholar]
- Schoch CL, Seifert KA, Huhndorf S, et al. 2012. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode for fungi. Proceedings of the National Academy of Sciences of the United States of America 109: 6241–6246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoustra SE, Debets AJM, Slakhorst M, et al. 2007. Mitotic recombination accelerates adaptation in the fungus Aspergillus nidulans. PLoS Genetics 3: e68 APR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seymour FA, Crittenden PD, Wirtz N, et al. 2007. Phylogenetic and morphological analysis of Antarctic lichen-forming Usnea species in the group Neuropogon. Antarctic Science 19: 71–82. [Google Scholar]
- Singh G, Dal Grande F, Cornejo C, et al. 2012. Genetic basis of self-incompatibility in the lichen-forming fungus Lobaria pulmonaria and skewed frequency distribution of mating-type idiomorphs: Implications for conservation. PloS ONE 7: e51402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh G, Dal Grande F, Divakar PK, et al. 2015. Coalescent-based species delimitation approach uncovers high cryptic diversity in the cosmopolitan lichen-forming fungal genus Protoparmelia (Lecanorales, Ascomycota). PLoS ONE 10: e0124625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spribille T, Touvinen V, Resl P, et al. 2016. Basidiomycete yeasts in the cortex of ascomycete macrolichens. Science. doi: 10.1126/science.aaf8287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. 2006. RaxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22: 2688–2690. [DOI] [PubMed] [Google Scholar]
- Stamatakis A. 2014. RaxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30: 1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A, Hoover P, Rougemont J. 2008. A rapid bootstrap algorithm for the RAxML webservers. Systematic Biology 57: 758–771. [DOI] [PubMed] [Google Scholar]
- Steinová J, Stenroos S, Grube M, et al. 2013. Genetic diversity and species delimitation of the zeorin-containing red-fruited Cladonia species (lichenized Ascomycota) assessed with ITS rDNA and β-tubulin data. The Lichenologist 45: 665–684. [Google Scholar]
- Stewart JE, Timmer LW, Lawrence CB, et al. 2014. Discord between morphological and phylogenetic species boundaries: incomplete lineage sorting and recombination results in fuzzy species boundaries in an asexual fungal pathogen. BMC Evolutionary Biology 14: 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuessy TF. 2009. Plant taxonomy. The systematic evaluation of comparative data. Columbia University Press, New York. [Google Scholar]
- Suzuki R, Shimodaira H. 2006. pvclust: an R package for assessing the uncertainty in hierarchical clustering. Bioinformatics 22: 1540–1542. [DOI] [PubMed] [Google Scholar]
- Tajima F. 1989. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123: 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Peterson D, Peterson N, et al. 2011. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Molecular Biology and Evolution 28: 2731–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Templeton AR. 2009. Why does a method that fails continue to be used: the answer. Evolution 63: 807–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas NT, Krings M, Taylor EL. 2014. Fossil fungi. Academic Press, London. [Google Scholar]
- Turland NJ, Wiersema JH, Barrie FR, et al. (eds). 2018. International Code of Nomenclature for algae, fungi, and plants (Shenzhen Code) adopted by the Nineteenth International Botanical Congress Shenzhen, China, July 2017. [Regnum Vegetabile no. 159.] Königstein, Koeltz Botanical Books. [Google Scholar]
- Vanhaecke D, García de Leaniz C, Gajardo G, et al. 2012. DNA barcoding and microsatellites help species delimitation and hybrid identification in endangered galaxiid fishes. PLoS ONE 7: e32939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velmala S, Myllys L, Goward T, et al. 2014. Taxonomy of Bryoria section Implexae (Parmeliaceae, Lecanoromycetes) in North America and Europe, based on chemical, morphological and molecular data. Annales Botanici Fennici 51: 345–371. [Google Scholar]
- Weiller GF. 1998. Phylogenetic profiles: a graphical method for detecting genetic recombination in homologous sequences. Molecular Biology and Evolution 15: 326–335. [DOI] [PubMed] [Google Scholar]
- White TJ, Bruns T, Lee S, et al. 1990. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis MA, Gelfand DH, Sninsky JJ, et al. (eds), PCR protocols: a guide to methods and applications: 315–322. New York: Academic Press. [Google Scholar]
- Widmer I, Dal Grande F, Excoffier L, et al. 2012. European phylogeography of the epiphytic lichen fungus Lobaria pulmonaria and its green algal symbiont. Molecular Ecology 21: 5827–5844. [DOI] [PubMed] [Google Scholar]
- Will KW, Mishler BD, Wheeler QD. 2005. The perils of DNA barcoding and the need for integrative taxonomy. Systematic Biology 54: 844–851. [DOI] [PubMed] [Google Scholar]
- Zamora JC, Calonge FD, Martín MP. 2013. New sources of taxonomic information for earthstars (Geastrum, Geastraceae, Basidiomycota): phenoloxidases and rhizomorph crystals. Phytotaxa 132: 1–20. [Google Scholar]
- Zamora JC, Calonge FD, Martín MP. 2015. Integrative taxonomy reveals an unexpected diversity in Geastrum section Geastrum (Geastrales, Basidiomycota). Persoonia 34: 130–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zardoya R, Vollmer DM, Craddock C, et al. 1996. Evolutionary conservation of microsatellite flanking regions and their use in resolving the phylogeny of cichlid fishes (Pisces: Perciformes). Proceedings of the Royal Society, Biological Sciences 263: 1589–1598. [DOI] [PubMed] [Google Scholar]
- Zhang J, Kapli P, Pavlidis P, et al. 2013. A general species delimitation method with applications to phylogenetic placements. Bioinformatics 29: 2869–2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoller S, Lutzoni F, Scheidegger C. 1999. Genetic variation within and among populations of the threatened lichen Lobaria pulmonaria in Switzerland and implications for its conservation. Molecular Ecology 8: 2049–2059. [DOI] [PubMed] [Google Scholar]