

Abstract
Ineffective drug delivery and poor prognosis are two major challenges in the treatment of pancreatic ductal adenocarcinoma (PDAC). While there is significant downregulation of tumor suppressor microRNA-34a (miR-34a), which targets many oncogenes related to proliferation, apoptosis, and invasion, high expression level of Polo-like kinase 1 (PLK1) is closely associated with short survival rates of pancreatic cancer patients. Therefore, the objective is to codeliver miR-34a mimic and small molecule PLK1 inhibitor volasertib (BI6727) using poly(ethylene glycol)–poly[aspartamidoethyl(p-boronobenzyl)diethylammonium bromide] (PEG-B-PAEBEA). This polymer could self-assemble into micelles of ~100 nm with 10% drug loading of volasertib and form a complex with miR-34a at the N/P ratio of 18 and higher. Combination treatment of volasertib and miR-34a displayed the synergistic effect and superior antiproliferative activity along with an enhanced G2/M phase arrest and suppression of colony formation, leading to cell death due to potential c-myc targeting therapeutics. Orthotopic pancreatic tumor bearing NSG mice were scanned for fluorescence by IVIS after systemic administration of micelles encapsulating volasertib and miR-34a at doses of 5 and 1 mg/kg, respectively. Cy5.5 concentration in plasma and major organs was determined by measuring fluorescence intensity. There was significant reduction in tumor volume, and histological examination of major organs suggested negligible systemic toxicity. In conclusion, PEG-B-PAEBEA micelles carrying volasertib and miR-34a mimic have the potential to treat pancreatic cancer.
Keywords: ROS-responsive micelles, Polypeptide, miR-34a, volasertib, pancreatic cancer
Graphical Abstract

1. INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) is one of the most aggressive cancers, where 85% of patients have metastatic disease making surgical interventions ineffective. The high mortality of pancreatic cancer patients is due to late-stage prognosis and development of chemoresistance upon prolonged treatment with chemotherapeutic drug(s).1 Gemcitabine is the first line Food and Drug Administration (FDA) approved drug used alone or in combination with others to improve the quality and life span of PDAC patients.2 However, pancreatic tumors present a complex and highly obstructive microenvironment that is characterized by dysregulation of miRNAs and development of chemoresistance.3 Alteration of miRNA expression and replenishment of anticancer miRNAs may effectively restore chemosensitivity by decreasing the expression of essential genes involved in cell growth or survival.4 Meanwhile, polo-like kinase 1 (PLK1) emerges as a promising target due to the mediation of gemcitabine resistance and regulation of mitosis.5,6
MicroRNAs (miRNAs) are 17–25 nucleotides long and highly conserved, and noncoding RNA products play critical roles in the regulation of drug resistance, cancer stem cell (CSC) proliferation, and cancer metabolism.7,8 The advantage of miRNAs lies in their ability to modulate multiple cellular pathways simultaneously, which is difficult to be accomplished using small molecules and therapeutic proteins.9 Cellular stimuli, glucose level, and hypoxia critically orchestrate miRNA activity in various pathologies, including pancreatic cancer.10 Among many miRNAs, miR-34a is downregulated in PDAC patients and has a key role in chemosensitization.11 Increase in miR-34a expression level contributes to the suppression of Bcl-2, a well-known antiapoptotic gene, leading to decrease in migration, invasion and proliferation, and enhancement in apoptosis of tumor cells,12,13 thereby overcoming drug resistance and treating PDAC more efficiently.
Polo-like kinase 1 (PLK1) plays a key role in mitosis and meiosis, and its overexpression has been found in various cancer types, including pancreatic cancer. Moreover, higher PLK1 expression correlates with tumor aggressiveness and a poor prognosis. Therefore, PLK1 is an attractive target for treating pancreatic cancer. Recently, several PLK1 inhibitors, including volasertib and GSK461364, have been investigated in various cancers. Volasertib is an ATP-competitive kinase inhibitor with higher potency and selectivity than those of its predecessor, BI 2536. Treatment with volasertib14 results in the inhibition of G2/M cell cycle arrest in cancer cells.15,16 Most importantly, combination treatment with Bcl-2 and PLK1 inhibitors specifically induces synergistic cell death due to the depletion of c-myc, a common target miR-34a and PLK1.17 Here, we hypothesized that combination of miR-34a and volasertib could synergistically and effectively treat PDAC.
Considering the challenges that miRNAs and hydrophobic drugs face, including off-target effects, low transfection efficiency, poor aqueous solubility, and burst release, recent efforts have been focused on developing nanocarriers for delivery of both of miRNAs and hydrophobic drugs.18 Compared with conventional drug delivery systems which have poor bioavailability, adverse side effects, and low therapeutic indices,19 polymeric nanoparticle delivery systems have generated considerable commercial and translational attention due to their potential for extended drug release kinetics and enhanced stability.20 Cationic polymers and lipids are widely used for miRNA delivery.21,22 Unfortunately, the highly positive charge systems with high density of amines will generate severe and intense toxicity, impeding their clinical translation.
We previously reported methoxypoly(ethylene glycol)-block-poly(2-methyl-2-carboxylpropylene carbonate-graft-dodecanol-graf t-tetraethylenepentamine) (mPEG-b-PCC-g-DC-g-TEPA) conjugated polycarbonate-based polymer for codelivery of miRNA and small molecules with high drug loading and efficient complex formation with miRNA for treating type I diabetes and pancreatic cancer.23–25 However, the polymer synthesis requires multiple steps, and polycarbonate is subjected to degradation when being modified under alkali conditions. Nonspecific hydrophobic interactions between the drug and polymer often lead to drug aggregation and decrease in drug loading.26 Moreover, the intracellular dissociation and release of miRNAs and drugs from delivery systems is well-known to be the prerequisite for effective transfection and therapy, whereas miRNA/polymer complexes are thermodynamically unstable and the drug–polymer conjugates linked with chemical bonds are even more resistant to release cargos under physiological environment. Therefore, it is highly desirable to develop a nanocarrier that can not only coload both genes and drugs but also effectively dissociate and rapidly release the cargos in cancer cells. To date, low pH in acidic endocytic vesicles and increased level of glutathione (GSH) as well as oxidative species in cancer cells have encouraged the development of polymers which are sensitive to specific stimuli for bioimaging and cancer treatment.27 Inspired by these recent advances, we hypothesize the overproduced reactive oxygen species (ROS) can be exploited as a cancer related stimulus to mediate successfully cargo release from nanocarriers in the cytoplasm.28 Taken together, we present a synthetic polymeric delivery system of miR-34a and volasertib for treating pancreatic cancer. We synthesized poly(ethylene glycol)–poly[aspartamidoethyl(p-boronobenzyl)diethylammonium bromide] (PEG-B-PAEBEA), which could form stable micelles in aqueous solution with both of miR-34a and volasertib through electrostatic interaction of nucleic acid and quaternary ammonium and intense nitrogen–boron (N–B) coordination of tertiary amines and boronic acid, respectively. Upon oxidation by ROS, boronic acid would be first detached, followed by a self-immolative process to produce tertiary amines and p-quinone methide, leading to rapid release of miR-34a and volasertib, thereby improving the potential of their antitumor effects and minimal systemic toxicity (Scheme 1). In this study, the biodistribution, therapeutic efficacy and systemic toxicity of miR-34a and volasertib loaded micelles were determined after systemic administration into orthotopic pancreatic tumor-bearing NSG mice.
Scheme 1.

Illustration of Combination Therapy of Small Molecule PLK1 Inhibitor and MicroRNA-34a in Pancreatic Cancer
2. RESULTS
2.1. Copolymer Synthesis and Characterization.
A reactive oxygen species (ROS)-labile charge-reversal polymer PEG-B-PAEBEA was synthesized by a four-step process and characterized by 1H NMR. Briefly, BLA-NCA was produced from the reaction of L-aspartic acid β-benzyl ether, followed by copolymerization with mPEG-NH2 (Mw = 5000) to produce PEG–PBLA. Afterward, N, N-diethylethylenediamine (DEEDA) replaced the benzyl groups through aminolysis, affording PEG–PAsp(DIE). Finally, the reaction between 4-(bromomethyl)phenylboronic acid and PEG–PAsp(DIE) gave the final product PEG-B-PAEBEA (Figure 1A,B). The molecular weight of PEG–PBLA was 12 594 Da with a PDI of 1.04 according to GPC determination (Figure 1C). PEG-B-PAEBEA had strong interactions with the GPC column due to the positive charge, so their molecular weight could not be measured by the GPC and were calculated from their precursor PEG–PBLA. According to the 1H NMR spectrum of PEG-B-PAEBEA in Figure 1B, 15 4-methylphenylboronic acids were attached to tertiary amines of PEG–PAsp(AIE), as based on the integrated areas of the peak at 5.28 ppm (assigned to methylene hydrogens of -PEG). Thus, the molecular weight of PEG-B-PAEBA is 14 611 Da.
Figure 1.

Synthesis and characterization of PEG-B-PAEBEA. (A) Synthesis scheme for PEG-B-PAEBEA. (B) 1H NMR spectrum of PEG-B-PAEBEA in TFA-d6. (C) Molecular weight of PEG–PBLA confirmed by GPC.
2.2. Preparation and Characterization of Micelles.
Volasertib loaded micelles were prepared by nanoprecipitation using PEG-B-PAEBEA and coating drug particles through N–B coordination, thereby allowing the micelles to achieve a positively charged surface for complex formation with miR-34a via ionic interaction. The mean particle size of micelles was 90.1 ± 0.3 nm with volasertib loading of 10.8 ± 0.1%, volasertib encapsulation efficiency of 96.7 ± 0.8%, and ζ potential of 35.5 ± 2.7 mV (Figure 2A). The polymer effectively condensed miR-34a at N/P ratios above 18, as probed by gel electrophoresis, and the ζ potential of micelles decreased to 10.4 ± 1.6 mV with the mean particle size increased to 101.4 ± 0.7 nm. TEM of micelles presented spherical particles. Upon oxidation of the boronic acid group by ROS such as H2O2 solution at 0.25 mM for 1 h, miR-34a could be released from the micelles, and their morphologies were collapsed as shown in TEM image, demonstrating the ROS-responsive release of miR-34a (Figure 2B,C). We investigated the ROS-triggered degradation of micelles by 1H NMR as well. Micelles in D2O showed high PEG peak (Figure 2D); however, peaks of 4-hydroxymethylphenol appeared, the intensity increased with incubation time, and the ζ potential altered to 3.67 ± 0.28 mV, indicating the disassembly of micelles (Figure 2D). Additionally, elevated ROS production induced by volasertib can in turn promote the degradation of micelles and further increase the release of small molecules and genes (Figure 2E). Moreover, the polymeric micelles improved intracellular delivery of miRNA, compared to the commercial transfection reagent, lipofectamine 2000 (Figure 2F). The critical micelle concentration (CMC) of micelles was determined to be 2.0 μg/mL using pyrene as a fluorescent probe.
Figure 2.

Characterization of micelles. (A) Particle size distribution and morphology of nanoparticles. (B) ROS responsiveness confirmed by a transmission electron microscope (TEM). (C) Gel electrophoresis to confirm miRNA condensation and ROS-responsive miRNA release. (D) 1H NMR spectra of PEG-B-PAEBEA micelles after H2O2 treatment in D2O. (E) ROS production. (F) Cellular uptake of lipofectamine/FITC-miRNA and polymer/FITC-miRNA. (G) FRET effect is demonstrated via the reduction fluorescence intensity of donor at 520 nm with increased fluorescence of acceptor at 600 nm.
To further ascertain miR-34a loading in micelles, FRET measurement was performed, where FITC was utilized as a donor conjugated to miR-34a (FITC-miR-34a) and rhodamine was employed as an acceptor loaded in micelles. The maximal fluorescence intensity at 520 nm of FITC (donor) from dual-labeled micelles carrying miR-34a and volasertib was significantly reduced, whereas the fluorescent intensity at 600 nm was increased compared to FITC-labeled miR-34a, which demonstrates an effective energy transfer from the donor to acceptor in the micelles (Figure 2G).
2.3. Synergistic Effect of Volasertib and miR-34a and Apoptosis.
PEG-B-PAEBEA showed low cytotoxicity and was suitable for codelivery of volasertib and miR-34a (Figure S4). The cytotoxicity of volasertib and its combination with miR-34a on pancreatic cancer cell lines was performed as a function of volasertib or miR-34a concentrations. As shown in Figure 3A–D, cell viability after transfection with volasertib was dose-dependent, and the capability of killing cancer cells by volasertib remained in the nanomolar level. Most importantly, the IC50 value of volasertib in combination with miR-34a was decreased by ~2–4.5-fold compared to volasertib alone and the combination of volasertib and scrambled miRNA (Figure 3E–H). Meanwhile, CI values obtained after combination treatment of volasertib and miR-34a were 0.1–0.7 (less than 1), indicating the synergistic effect of the small molecule and miRNA. Quantitative apoptosis analysis by flow cytometry using Annexin V-Cy5/PI apoptosis detection kits also indicated that combination of volasertib and miR-34a with total apoptotic cells of 55% exhibited higher apoptotic activity in contrast to other groups (Figure 3I and Figure S5). Accordingly, micelles carrying miR-34a and volasertib were capable of inducing apoptosis with high efficiency. However, once the ROS was eliminated by a scavenger dithiothreitol (DTT), micelles carrying miR-34a and volasertib displayed no efficiency compared to the group without DTT pretreatment, which also suggested the ROS-responsive release of volasertib and miR-34a contributed to the therapeutic effect in pancreatic cancer cells (Figure 3J,K).
Figure 3.

Effect of volasertib and miR-34a on pancreatic cancer. (A–D) Cytotoxicity induced by volasertib alone. (E–H) Cytotoxicity caused by the combination of volasertib and miR-34a. (I) Effect of miR-34a and volasertib on apoptosis. MIA PaCa-2R cells were treated with micelles loaded with miR-34a, volasertib or their combination. Results are presented as the mean ± SD (n = 3). (J, K) Effect of ROS inhibition triggered by DTT on cytotoxicity in pancreatic cancer cells.
2.4. Colony Assay and Cell Cycle.
To determine whether the combination of volasertib and miR-34a has a greater effect on long-term anticancer therapy, we studied volasertib, miR-34a, and their combination using scrambled miRNA as a negative control in MIA PaCa-2R cell line with final concentration of 100 nM for volasertib and 100 nM for miR-34a, respectively. A significant decrease in the density of colony was observed with the combination treatment compared to small molecule volasertib alone (Figure 4A). The lower colonies indicated an improved therapeutic efficacy of the combination of volasertib and miR-34a due to their synergistic effect.
Figure 4.

Colony formation assay, cell cycle, effect on tumor spheroids, and migration assays. (A) Colony assay after treatment of MIA PaCa-2R cells with micelles carrying miR-34a, volasertib, or their combination. The cells treated with micelles carrying scrambled miRNA and volasertib were also used control. (B) Cell cycle analysis in MIA PaCa-2R cells. (C) Effect of different formulations on 3D tumor spheroids. Migration inhibition investigated by scratch wound healing assay (D) and transwell chamber assay (E).
PLK1 regulates the cell division cycle during late G2 and M phase, and volasertib is a highly potent PLK1 inhibitor, resulting in G2/M cell cycle arrest followed by programmed cell death in cancer cell lines. Combination of volasertib and miR-34a demonstrated the strongest ability in arresting MIA PaCa-2R cells in the G2/M phase among all tested samples. Volasertib and combination group progressively induced G2/M arrest in MIA PaCa-2R cells, with significantly parallel decrease of G1 population (Figure 4B and Figure S6).
2.5. Therapeutic Efficacy in 3D Tumor Spheroids and Migration Inhibition.
The 3D tumor spheroid sizes in control and scrambled miRNA groups were increased after treatment. In contrast, miR-34a could slightly decrease the size of tumor spheroid. Notably, free volasertib and micelles encapsulating volasertib exhibited similar results in inhibiting the growth of tumor spheroids, while micelles formulating miR-34a and volasertib could significantly reduce the spheroid size and consequently lead to the collapse of tumor spheroids (Figure 4C). These results indicated that miR-34a and volasertib loaded micelles displayed significant inhibition on tumor spheroid growth.
On the other hand, the migration of cancer cells was predominant element leading to cancer recurrence. Compared to PBS control group, scrambled miRNA did not suppress cell migration and partially closed the wound after 48 h. In contrast, miR-34a and combination of miR-34a and volasertib displayed little migration after 24 h and caused reduction of migration after 48 h (Figure 4D,E), thereby indicating the efficient intracellular delivery of miR-34a to cancer cells.
2.6. Molecular Mechanisms of Volasertib and miR-34a.
miR-34a expression was significantly lower in pancreatic cancer cell lines compared to normal pancreatic cancer HPDE cell line (Figure 5A). After transfection with miR-34a mimics, miR-34a expression level was enhanced by 6–10-fold in MIA PaCa-2 cells (Figure 5B–E). On the other hand, the PLK1 expression level was profoundly suppressed in MIA PaCa-2 cells after incubation with volasertib in 5 and 80 nM, respectively, while higher concentration (80 nM) was needed to inhibit PLK1 expression level in MIA PaCa-2R cells (Figure 5F,G).
Figure 5.

Molecular mechanism for the synergistic effect of volasertib and miR-34a. (A–E) miR-34a expression level determined by qPCR after treatment of HPDE, MIA PaCa-2, MIA PaCa-2R, Capan-1, and Capan-1R cell lines. (F) Western blotanalysis of Bcl-2, PLK1, and c-myc in MIA PaCa-2 and MIA PaCa-2R cell lines. (G, H) PLK1 and c-myc mRNA expression levels determined by qPCR. PBS was used as control (n = 3, *p < 0.05, **p < 0.01, and ***p < 0.001).
Bcl-2 has shown to have a dominant role in the survival of cancer cells. miR-34a downregulated Bcl-2 expression, further leading to inhibition of proliferation and enhancement of apoptosis in cancer cells (Figure 5F). The treatment of volasertib, miR-34a, and their combination contributed to significant suppression of c-myc expression as well in pancreatic cancer cell lines (Figure 5F,H), indicating that c-myc was the common target of volasertib and miR-34a, leading to the synergistic effect of volasertib and miR-34a in pancreatic cancer.
2.7. Biodistribution Studies.
Following systemic administration to NSG mice harboring orthotopic pancreatic tumors, we compared the delivery and retention of free Cy5.5 and Cy5.5 labeled micelles to evaluate whether micelles could accumulated in solid tumors. Because of the short half-life of Cy5.5,29 the NIR signal intensity was dimer and disappeared rapidly in mice injected with free Cy5.5. In contrast, strong fluorescence was obtained at the tumor sites within 4 h of Cy5.5-labeled micelles injection, following which the signal intensity lasted for 24 h (Figure 6A). In addition, the liver, kidney, and spleen were also major sites of micelles distribution (Figure 6B–D).
Figure 6.
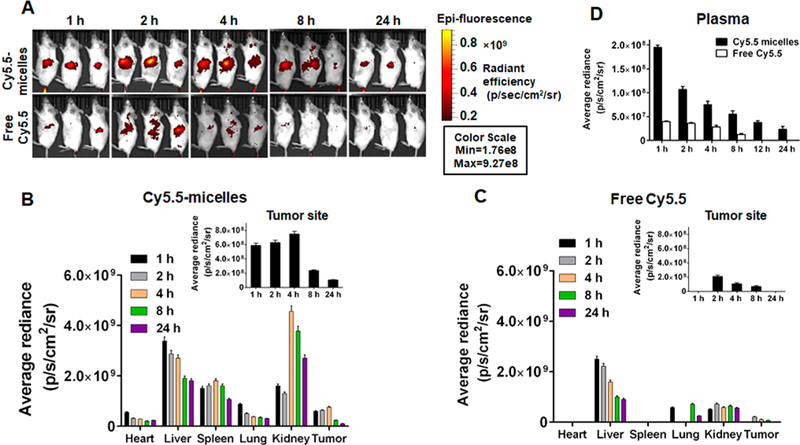
Biodistribution of micelles in vivo. (A) Real-time biodistribution of free Cy5.5 and Cy5.5-labeled micelles at 1, 2, 4, 8, and 24 h (n = 3). Quantified accumulation of (B) Cy5.5-labeled micelles and (C) free Cy5.5 in major organs, including heart, liver, spleen, lung, kidney, and tumor (n = 3). (D) Quantified accumulation of Cy5.5-labeled micelles and free Cy5.5 in plasma (n = 3). In plasma, Cy5.5 signal intensity in Cy5.5-micelle injected mice may be from both Cy5.5 micelles and cleaved Cy5.5 dye.
2.8. Antitumor Activity.
Systemic administration was repeated every 3 days for up to seven repetitions. Orthotopic tumor growth was monitored by IVIS bioluminescence imaging, demonstrating significantly slower tumor growth by treatment with combination micelles compared to that with saline, free volasertib, miR-34a, or volasertib loaded micelles (Figure 7A,B). This suggested that combination of miR-34a and volasertib elicited a potent antitumor efficacy. No reduction in mouse body weight or morbidity was observed (Figure 7C), indicating that these micellar formulation of volasertib and miR-34a were well tolerated. The pathological changes in major organs were compared and H&E staining suggested negligible systemic toxicity of the formulations as no damage or inflammation was observed (Figure S7).
Figure 7.
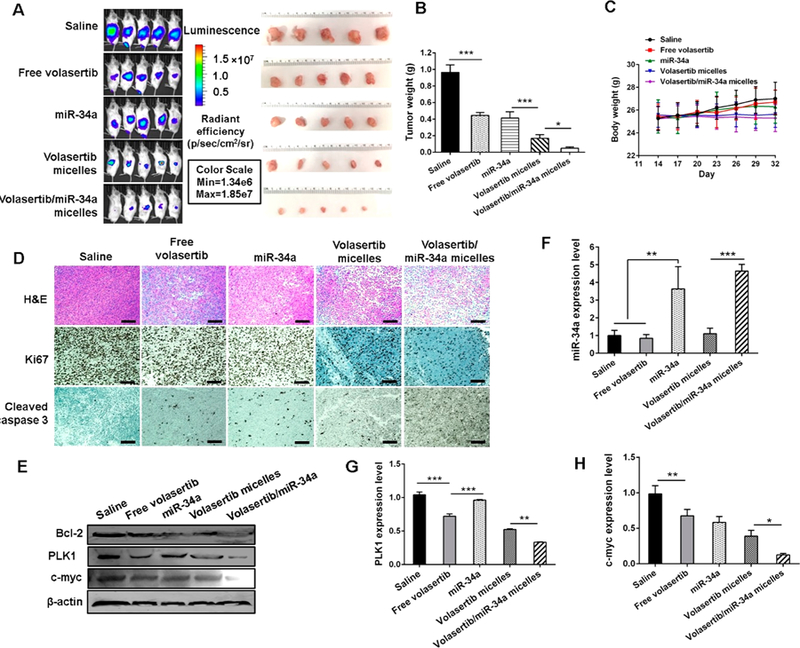
Antitumor efficiency in vivo. (A) Bioluminescence images of tumors and isolated tumor images at the end of the experiment. (B) Tumor weight after treatment with the various formulations (n = 5, *P < 0.05, **P < 0.01, and ***P < 0.001). (C) Body weight. (D) H&E staining, Ki67, and cleaved caspase 3 analysis of the tumor specimens. (E) Bcl-2, PLK1, and c-myc expression determined by Western blot assay. (F) miR-34a, (G) PLK1, and (H) c-myc expression level in tumor was determined by PCR assay (n = 3, **P < 0.01 and ***P < 0.001).
Tumor tissue sections from different treatment groups were examined by H&E staining and immunohistochemistry (IHC) staining. In comparison with the morphological characteristics of H&E stained tissue sections of saline-treated tumors that contained dense carcinoma cells, tumor cell density in combination micelles-treated tumors was significantly decreased with necrotic tumor cell areas (Figure 7D). Utilizing an IHC staining for the detection of cleaved caspase 3, we observed notable apoptotic cell death in combination micelle-treated mice compared to those treated with free volasertib, miR-34a, and volasertib loaded micelles. Representative IHC images are shown in Figure 7D. Ki-67 is a marker of proliferation, and its staining showed marked differences between different treatment groups. There was a remarkable decrease in the number of Ki-67 positive proliferating cells in the tumor of the mice treated with combination micelles than that in other groups (Figure 7D). Therefore, effective induction of the apoptotic cell death and profound inhibition of tumor cell proliferation contributed to the antitumor effect in mice. As depicted in Figure 7F, miR-34a was considerably upregulated in miR-34a complexed micelles and the group treated with micelles loaded with volasertib and miR-34a and that was ascertained the downregulation of Bcl-2 and c-myc in tumor tissues by Western blot and PCR analysis (Figure 7E,G,H). We further confirmed this by quantifying the Western blot bands of Figure 7E by using ImageJ 1.5a (Java 1.8.0_112 (64 bit)), and the results are shown in Figure S8. Moreover, the downregulation of PLK1 induced by volasertib contributed notable to c-myc suppression in tumor sites as well. According to those results, the combination of miR-34a and volasertib can restore the miR-34a expression level, sensitize the cells to chemotherapy, and further exhibit a synergistic effect in pancreatic cancer via c-myc, leading to tumor growth suppression and apoptosis.
3. DISCUSSION
Several nanocarriers have been reported to enhance drug delivery to the tumor after systemic administration. However, most of the current nanoparticle platforms are rarely used for codelivery of small molecules and nucleic acids. Our previously reported nanocarriers are capable of codeliverying small molecules and nucleic acids. Compared to polycarbonate which we used in our previous studies, polyaspartate has the following advantages: (1) it is stable during synthesis and postmodification; (2) it is compatible with ring-opening polymerization (ROP) protocols with functional alcohols; (3) one-step synthesis of monomer for copolymerization; (4) it avoids a two-step reaction that is deprotection with H2 and subsequent coupling in the presence of EDC/HOBt for replacement of pendent benzyl groups by simply mixing polyaspartate and tertiary amines; and (5) micelles can be formed by simply dissolving lyophilized polymer in aqueous solution, making it a highly attractive platform for drug delivery.
Smart polymers capable of responding to the stimuli intrinsic in tumor environment have emerged as efficient nanocarriers for the treatment of cancer.30 Elevated rates of ROS have been detected in most cancers, and the levels of intracellular ROS in cancer cells are unique biological stimuli that can be utilized for efficient and targeted drug delivery to cancer cells.28,31 Boronic acid has been extensively used for detection of H2O2 in vitro and in vivo, and the integration of these functional moieties in the polymeric backbone has been reported.32,33 This mechanism depends on the oxidation-triggered hydrolysis of the boronic ester to a phenol and subsequent degradation of micelles,33 leading to the release of therapeutic agents. Meanwhile, phenylboronic acid (PBA) is also known as Lewis acid, which renders donor–acceptor interaction, also called coordination bonding with various amines. The donor–acceptor bond energy is much stronger than nonspecific hydrophobic interactions, and undesired drug aggregation can be evitable during this process.26
Volasertib with two tertiary amines in the piperazine group could be encapsulated by PEG-B-PAEBEA via specific nitrogen–boronic coordination instead of hydrophobic interaction.26 More importantly, upon the oxidation of boronic acid group of PEG-B-PAEBEA, the positive quaternary amines could alter to tertiary amines with no loading capacity (Figure 2D),26,32 facilitating the release of miR-34a and volasertib simultaneously (Scheme 1). With the elimination of ROS by DTT, a well-known ROS scavenger,34 volasertib micelles exhibited no effect to cancer cells (Figure 3J,K),35,36 further indicating the release of volasertib is in a ROS-dependent manner. Additionally, volasertib could boost ROS levels, which in turn accelerated the degradation of PEG-B-PAEBEA (Scheme 1 and Figure 2E); thus, a positive feedback loop was established to enhance the antitumor efficacy while minimizing systemic toxicity.
Challenges of nanoparticulate systems include rapid clearance by the reticuloendothelial systems (RES), intrinsic cytotoxicity, and release of inflammatory cytokines caused by the activation of toll-like receptors (TLRs).37,38 However, the rational design of the polymer/carrier could be used to address these limitations. For example, micelles with particle size below 100 nm modified by PEG on the surface can prolong the circulation by decreasing serum protein adsorption and preventing recognition by RES.39,40 The micelles designed in this study were decorated by PEG as the hydrophilic surface, with particle size around 100 nm, providing them a great chance for prolonged circulation, thus resulting in higher tumor accumulation.
By whole-body fluorescence imaging, mice injected with free Cy5.5 showed maximal tumor fluorescence at 2 h followed by rapid clearance with time. The positive quandary amine of Cy5.5 might increase its binding to plasma proteins; therefore, biodistribution of free Cy5.5 showed little liver and kidney accumulation, with low tumor retention at 8 h.41,42 In contrast, mice treated with Cy5.5-conjugated micelles showed excellent tumor accumulation of micelles even at 24 h post-systemic administration, demonstrating that the Cy5.5 was not removed from micelles, and the high intensity in the tumor was resulted from high accumulation of micelles, not free Cy5.5.
High expression of PLK1 and low expression of miR-34a is associated with short survival rate in pancreatic cancer patients.14,43,44 PLK1 is essential for stabilizing Myc family oncoproteins, and miR-34a can affect cell proliferation through repression of Myc.45 PLK1-Myc feedforward activation loop participates in PDAC progression, and Myc amplification in pancreatic cancers predicts poor prognosis and resistance to therapy.17,46,47 Altogether, c-myc downregulation can contribute to the inhibition of pancreatic cancer development.14,47 PLK1 inhibitor volasertib could not only downregulate PLK1 to arrest cell cycle of tumor cells but also reduce downstream c-myc phosphoraylation and then inhibit c-myc transcription.46 Meanwhile, miR-34a can target c-myc and Bcl-2 directly.45,48 To date, volasertib and miR-34a were tested alone in clinical trial, but their combination has not been evaluated yet.6,49 Herein, we investigated efficacy of combination of those two anticancer agents encapsulated by PEG-B-PAEBEA in orthotopic pancreatic tumor-bearing mice. Because of the synergistic effect of volasertib and miR-34a, downregulating PLK1 while upregulating miR-34a resulted in significant suppression of their common target c-myc and pronounced tumor inhibition, which suggests the potential of combination of volasertib and miR-34a could be translated as a strategy in future pancreatic cancer therapy.17 Moreover, H&E staining validated the formulations were well tolerated and did not show any signs of tissue or cellular damage such as ballooning and degeneration of hepatocytes, increased alveolar wall thickness or cellular infiltration in lung tissue, myocardial fibrillar loss or vacuolation in heart tissues, edema, tubular vacuolization or tubular dilation with hemorrhagic areas in the kidney, or increased numbers of granulocytes in the spleen (Figure S7).50,51 Those results support that our ROS-triggered polymeric micellar system has potential to overcome the limitations of current nanocarriers.
In conclusion, we have demonstrated a ROS-labile polymer PEG-B-PAEBEA which was strongly positively charged can be self-assembled to micelles and effectively packaged volasertib and miR-34a into micelles but became neutral once triggered by intracellular ROS. The micelles successfully delivered volasertib and miR-34a in a controlled manner so that they can be released into the tumor sites accurately, resulting in high tumor regression compared to single drug loaded micelles. Without complicated polymer design or drug modification, PEG-B-PAEBEA provided a facile and robust strategy with high biocompatibility for the encapsulation of hydrophobic drugs and genes.
4. EXPERIMENTAL SECTION
4.1. Materials.
Dulbecco’s Modified Eagle Medium (DMEM), high glucose medium, Dulbecco’s phosphate buffered saline (DPBS), and 0.25% trypsin were purchased from Hyclone (Logan, UT). Keratinocyte-SFM medium, bovine pituitary extract, and human recombinant EGF were purchased from Gibco (Chevy Chase, MD). Heat inactivated fetal bovine serum (FBS), antibiotic–antimycotic for cell culture, RIPA buffer for cell lysis, halt protease and phosphatase inhibitor cocktail (100×), Pierce BCA protein assay kit, H2DCFDA (D399), and FxCycle PI/RNase staining solution (F10797) were purchased from Thermo Fisher Scientific (Waltham, MA). HEPES buffer was purchased from Millipore Sigma (St. Louis, MO). Human PLK1 primary antibody was purchased from Cell Signaling Technology (Danvers, MA). Human c-myc primary antibody was purchased from Proteintech (Manchester, UK). β-Actin primary antibody was purchased from Santa Cruz Biotechnology (Dallas, TX). Dithiothreitol (DTT), 4× Laemmli buffer, 10× Tris/glycine/SDS protein electrophoresis running buffer (pH 8.3), and 10× TBS buffer were purchased from Bio-Rad (Hercules, CA). Volasertib was purchased from Adooq Bioscience (Irvine, CA). miR-34a was purchased from GE Healthcare Dharmacon, Inc. (Lafayette, CO).
4.2. Synthesis of Poly(ethylene glycol)–Poly[aspartamidoethyl(p-boronic acid)benzyldiethylammonium Bromide] (PEG-B-PAEBEA).
Step 1.
Triphosgene (2.744 g, 9.24 mmol) dissolved in 10 mL of tetrahydrofuran (THF) was added to a suspension of benzyl aspartate (4.7 g, 21 mmol) in 200 mL of THF with stirring, and the mixture was then heated to 50 °C. Once the suspension became clear (~30 min), the solvent was removed under reduced pressure. The resultant solid was recrystallized from excess hexane. Finally, 4.459 g of β-benzyl-L-aspartate N-carboxyanhydride (BLA-NCA) was obtained with a yield of 85%. 1H NMR (500 MHz, CDCl3, δ): 7.43–7.15 (m, 5H), 5.13 (s, 2H), 4.69 (t, J = 4.5 Hz, 1H), 2.98 (ddd, J = 88.8, 17.8, 4.5 Hz, 2H).
Step 2.
A solution of BLA-NCA (2.739 g, 11 mmol) in N,N-dimethylformamide (DMF) was added to a stirred solution of mPEG-NH2(1.0 g, 0.2 mmol) in DMF under a N2 atmosphere. The solution was heated to 40 °C and kept stirring for 72 h. The resulting mixture was poured to a large amount of diethyl ether. The resulting solid was collected by filtration and dried to afford poly(ethylene glycol)–poly(β-benzyl-L-aspartate) (PEG–PBLA) copolymer (2.99 g, 80%).
Step 3.
PEG–PBLA (2 g) was first dissolved in DMF, followed by the addition of N,N-diethylethylenediamine (4 equiv to BLA unit) and triethylamine (TEA) (4 equiv to BLA unit). The mixture was stirred at 35 °C overnight. Afterward, the mixture was dialyzed against distilled water and lyophilized to produce poly(ethylene glycol)–poly(β-diisoethylaminoethylaspartamide) (PEG–PAsp(DIE)), with a yield of 77%.
Step 4.
In a 50 mL round-bottom flask, (4-(bromomethyl)phenylboronic acid (2.5 equiv of DIE unit) and PEG-Pasp(DIE) (700 mg) were dissolved in DMF and stirred at room temperature for 24 h. Then, the resulting mixture was dialyzed against deionized water, and the final product poly(ethylene glycol)–poly[aspartamidoethyl(p-boronic acid)benzyldiethylammonium bromide] (PEG-B-PAEBEA) was obtained as white solid after lyophilization, with a yield of 69%. The chemical structure of PEG-B-PAEBEA was determined by 1H NMR, and its molecular weight and polydispersity (PDI) were determined by gel permeation chromatography (GPC) under following conditions: Styragel HR 4E column (5 μm, 7.8 × 300 mm2) at 40 °C and flow rate of 1 mL/min. DMF (containing 1% lithium bromide) was used as the mobile phase, and the injection volume was 20 μL.
4.3. Cell Culture.
MIA PaCa-2 and Capan-1 cell lines were purchased from American Type Culture Collection (ATCC, Manassas, VA). MIA PaCa-2 resistant (MIA PaCa-2R) and Capan-1 resistant (Capan-1R) cells were generated from MIA PaCa-2 and Capan-1 by incubating with gemcitabine (GEM), respectively. All the above cell lines were maintained in high glucose DMEM medium supplemented except human pancreatic duct epithelial (HPDE) cells with 10% FBS and 1% penicillin/streptomycin at 37 °C, 5% CO2, and 100% humidity and were split when confluent. HPDE cells were cultured in Keratinocyte-SFM supplemented bovine pituitary extract (BPE) and human recombinant EGF in an identical atmosphere.
4.4. Preparation and Characterization of Micelles.
Volasertib (1 mg/mL) dissolved in acetone (100 μL) was added drop by drop to aqueous solution of the copolymer (1 mg) under stirring at room temperature. To remove the residual acetone, the samples were evaporated under reduced pressure. These micelles were then used for complex formation with miR-34a or scrambled miRNA by mixing at an equal volume and vortexing gently for 30 s followed by incubation at room temperature for 30 min. Rhodamine (Rho)-labeled micelles were prepared by dissolving Rho and volasertib in acetone, which was then mixed with the polymer solution under stirring. FITC-labeled micelles were fabricated by forming complex between FITC-labeled miRNA and nanoparticles. The particle size distribution and ζ potential were determined by dynamic light scattering (DLS) using a Malvern Zetasizer (Worcestershire, UK) at 25 °C. Morphology of these micelles was determined by transmission electron microscopy (TEM, Burlington, NC). The encapsulation efficiency (EE) and drug loading (DL) of volasertib were calculated using the following formulas:
| (1) |
| (2) |
4.5. miRNA Binding Ability.
The miRNA complexation efficiency was assessed by agarose gel retardation assay. Micelles were prepared at various N/P ratios, and miRNA was used as a control. Twenty microliters of micelles was mixed with 6 μL of 6 × loading buffer, and then the mixture was loaded onto 0.8% (w/v) agarose gel containing 0.5 μg/mL of ethidium bromide (EB). Electrophoresis was performed at a constant voltage of 110 V for 20 min in 1× TBE running buffer. The migration of miRNA bands was visualized and photographed with a using GelDoc Ez system (Bio-Rad, Hercules, CA).
4.6. ROS-Triggered Degradation of PEG-B-PAEBEA and Release of miRNA.
PEG-B-PAEBEA (5 mg) was dissolved in D2O (1 mL) with 15 mM H2O2 solution at 37 °C. At given time points (1 and 24 h), the solution was detected by 1H NMR. To confirm the ROS responsiveness release of miRNA, micelles at N/P ratio of 32 were incubated with final H2O2 concentrations at 0.25, 0.5, 0.75, and 1.5 mM. Those solutions were then subjected to the electrophoresis as descried above after incubation for 1 h at 37 °C. Naked miRNA were used as a control.
4.7. Critical Micelle Concentration (CMC).
The critical micelle concentration (CMC) was determined with fluorescent spectroscopy using pyrene as a hydrophobic fluorescent probe.27 Briefly, 1 mL of micelle samples was prepared in DI water with concentration ranges from 1 × 10−7 to 0.5 mg/mL, and pyrene stock solution (1.2 × 10−6 M) was added to micelle samples. The emission fluorescent intensity of 384 and 373 nm was recorded with an excitation wavelength of 334 nm. The intensity ratio (I384/I373) was plotted against the logarithm of micelle concentrations.
4.8. Fluorescence Resonance Energy Transfer.
For fluorescence resonance energy transfer (FRET) determination, FITC and rhodamine were employed as the donor and acceptor, respectively. The dual-labeled micelles carrying FITC labeled miR-34a and volasertib were fabricated by using Rho-labeled polymer. The emission spectra of FITC-miRNA and dual-labeled micelles from 500 to 700 nm were scanned at an excitation wavelength of 490 nm (donor) using a fluorescence spectrometer.
4.9. Measurement of ROS Production.
ROS produced by cells were detected using 2′,7′-dichlorofluorescin diacetate (DCFDA), which can passively diffuse across cell membranes. DCFDA is deacetylated by esterases to dichlorofluorescein (DCFH) and subsequently reacts with ROS to generate the green fluorophore, DCF.52 MIA PaCa-2R cells were seeded in 12-well plates for 24 h and incubated with free volasertib and micelles carrying ether miR-34a, volasertib, or their combination. The concentrations of volasertib and miR-34a were fixed at 50 and 100 nM. After 24 h, cells were removed from growth media, resuspended in prewarmed PBS containing 5 μM DCFDA and incubated 30 min at 37 °C. Then the cells were washed with PBS for three times and resuspended in serum-free media. Fluorescence was measured by flow cytometry (Becton, Dickinson, NJ) with excitation at 490 nm and emission at 520 nm.
4.10. Cytotoxicity, Synergistic Effect, and Apoptosis.
Cytotoxicity of volasertib, miR-34a, and their combination was determined by MTT assay. MIA PaCa-2, MIA PaCa-2R, Capan-1, and Capan-1R cells were plated at a density of 5000 cells/well in a 96-well plate for 24 h incubation. Then, free volasertib and formulations containing volasertib or the combination of volasertib and miR-34a were added to 96-well plates with volasertib concentration ranging from 10 to 1000 nM and miR-34a concentration from 10 to 200 nM, respectively. After 48 h of incubation, 5 mg/mL MTT solution was added, and cells were cultured for another 3 h. The supernatant was discarded, and 200 μL of DMSO was added to each well. Cell viability was determined by a spectrophotometer. The combination index (CI) and dose reduced index (DRI) were calculated using CompuSyn 1.0 software (ComboSyn, Inc., USA). In addition, CI < 1, CI = 1, and CI > 1 indicated synergistic, additive, and antagonistic effects, respectively. To demonstrate ROS-responsive cytotoxicity, cells were treated with dithiothreitol (DTT, 5 mM), a potent ROS scavenger, for 6 h prior to incubation with formulations.
For apoptosis assay, MIA PaCa-2R cells were seeded in 12-well plates for 24 h and incubated with micelles carrying ether miR-34a, volasertib, or their combination. We also used scrambled miRNA complexed micelles as well as free volasertib as controls. Volasertib and miR-34a were fixed at 50 and 100 nM concentrations, respectively. These cells were incubated for 24 h, washed with PBS for three times, and stained with Annexin V-Cy5/PI apoptosis kit according to the manufacturer’s protocol. The early and late apoptosis rates were calculated by flow cytometry (Becton, Dickinson, NJ).
4.11. Colony Formation Assay.
MIA PaCa-2R cells were transfected with volasertib, miR-34a, and their combination for 24 h using scrambled miRNA as control. Transfected cells were reseeded in the 12-well plate at 1000 cells per well. Cells were cultured for 14 days; then plates were washed with PBS, fixed with formalin, stained with 5% crystal violet for 30 min, washed with tapping water, and photographed.
4.12. Cell Cycle.
2 × 105 MIA PaCa-2R cells were seeded in a 6-well plate for 24 h and incubated with volasertib, miR-34a, and their combination using scrambled miRNA as a negative control. After 48 h, cells were harvested through trypsinization, fixed by 70% ethanol, washed three times by PBS, and centrifuged, and then the supernatant was decanted, leaving a pellet of cells in each sample tube. Then, 0.5 mL of FxCycle PI/RNase staining solution was added to each sample tube and incubated for 15 min at room temperature while protecting from light. Finally, flow cytometry was utilized to analyze the samples using excitation at 532 nm and emission at 585 nm.
4.13. 3D Spheroid Tumor Model.
MIA PaCa-2R cells (4 × 103 cells in 40 μL per well) in DMEM medium with 10% FBS were seeded onto Perfecta3D 96-well hanging drop plates (3D Biomatrix, Inc., Ann Arbor, MI) in triplicate and incubated at 37 °C in 5% CO2. On the fourth day, the spheroids were then treated with micelles carrying either miR-34a, volasertib, or their combination. Cells treated with PBS, scrambled miRNA, and free volasertib were used as controls. Morphologies of tumor spheroids were visualized under a microscope on day 2, 4, and 6 to check the cytotoxic efficacy of different formulations.
4.14. Cell Migration in Vitro.
Inhibition of cell migration was evaluated by scratch wound healing assay and Transwell assay, respectively. For scratch wound healing assay, MIA PaCa-2R cells (2 × 105 per well) were plated onto a 12-well plate. Then, cells were treated with micelles carrying either miR-34a, volasertib, or their combination. Cells treated with PBS, scrambled miRNA, and free volasertib were used as controls. At 24 h post-treatment, a wound was created in each well using 200 μL tips, dislodged cells were washed with PBS, and treatment was readded for 24 and 48 h. Cell migration assay was performed using Transwell chambers in MIA PaCa-2R cell lines. MIA PaCa-2R cells were cultured at a density of 4 × 105 cells/well in 6-well plates for 24 h prior to use. The cells were treated with micelles carrying miR-34a, volasertib, or their combination. Cells treated with PBS, scrambled miRNA, and free volasertib were used as controls. These formulations were incubated in 2 mL of culture media without serum for 6 h, followed by two washes with PBS and incubation in DMEM medium with 10% FBS for an additional 24 h. The cells were harvested, and 200 μL of cell suspension in DMEM medium was added into the upper chamber of Transwell. Subsequently, 1 mL of DMEM with 20% FBS as the chemoattractant was added into the lower Transwell chamber, and the cells were incubated for 24 h. Then, cells that did not migrate through the pores were carefully removed using a cotton-tipped swab. The filters were fixed in 4% paraformaldehyde, dried in room temperature, and stained with crystal violet.
4.15. Real-Time RT-PCR.
HPDE, MIA PaCa-2, MIA PaCa-2R, Capan-1, and Capan-1R cells were cultured in 6-well plates at a density of 2 × 105 cells/well for 24 h and transfected with volasertib or miR-34a for 24 h. Cells were then subjected to extraction of total RNA using an RNAeasy isolation kit (Qiagen, Valencia, CA). A reverse transcription reaction of total RNA to cDNA was performed using a miScript II Reverse Transcription kit and Taqman Reverse Transcription reagents (Thermo Fisher, Waltham, MA). miR-34a and PLK1 mRNA levels and c-myc were quantitatively assayed in a real-time PCR system (Roche, Basel, Switzerland) using miScript SYBR Green dye universal master mix, miR-34a primer, and primers of PLK1 and c-myc on a Light Cycler 480 (Roche, Indianapolis, IN). U6 was used as a housekeeping gene, and relative amounts of miR-34a, PLK1, and c-myc were calculated using the crossing point (Cp) value. Primers were designed as PLK1 FWD: 5′-CAGCAAGTGGGTGGA-CTATT-3′, PLK1 REV: 5′-ATCAGTGGGCACAAGATGAG-3′; c-myc FWD: 5′-CACGAAACTTTGCCCATAGC-3′, c-myc REV: 5′-GCAAGGAGAGCCTTTCAGAG-3′.
4.16. Western Blot Analysis.
MIA PaCa-2 and MIA PaCa-2R cells in 6-well plates with a density of 2 × 105 cells/well were incubated with formulations for 24 h, followed by protein isolation with radioimmunoprecipitation assay (RIPA) buffer and determination with a BCA protein assay kit. The cell lysates were then mixed with loading buffer, boiled buffer at 100 °C for 5 min, loaded on the wells of SDS-PAGE gel, transferred by electroporation to poly-(vinylidene difluoride) (PVDF) membrane, incubated with blocking buffer for 1 h at room temperature, and cultured with PLK1, Bcl-2, and c-myc rabbit primary antibodies as well as β-actin primary antibody overnight at 4 °C; IR fluorescent dye labeled secondary antibodies were added, followed by analysis using the Licor Odyssey system (LI-COR Biotechnology, Lincoln, NE). β-actin was used as the loading control.
4.17. Biodistribution and Therapeutic Studies.
Our animal protocol was approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Nebraska Medical Center and performed as per the National Institutes of Health (NIH) use guidelines. The orthotopic human pancreatic cancer mouse model was developed using 6–8 week male NSG mice (24–30 g). Luciferase stably expressing MIA PaCa-2R cells (1 × 106) were mixed with Matrigel (2:1) using ice-cold instruments and syringes. Then mice were anesthetized using isoflurane in an induction chamber, and 20 μL of Matrigel-cell suspension was orthotopically injected into the pancreatic tail. Tumor growth was monitored using an IVIS Spectrum in vivo imaging system (PerkinElmer, Hopkinton, MA) after intraperitoneal administration of 20 mg/mL luciferin to mice (150 μL per mouse). Three weeks after tumor injection, free Cy5.5 and Cy5.5-labeled micelles were injected into the mice via the tail vein at a single of Cy5.5 at the dose of 0.1 mg/kg. To localize Cy5.5-labeled micelles, the mice were observed in the IVIS imaging system at an excitation wavelength of 640 nm and emission wavelength of 710 nm at 1, 2, 4, 8, and 24 h. After in vivo imaging, mice were sacrificed and major organs such as tumor, heart, liver, spleen, lung, and kidney were collected for ex vivo imaging.
For therapeutic study, mice were randomly divided into five groups (n = 5 per group) when the bioluminescence reached 1 × 106. Formulations were administered via tail vein with volasertib 5 mg/kg and miR-34a 1 mg/kg every 3 days for a total of seven injections, according to the animal’s weight. Body weights were measured every time before injection. At the third day after the seventh administration, the animals were euthanized to harvest major organs such as tumor, heart, liver, spleen, lung, and kidney for hematoxylin and eosin (H&E) staining as well as immunohistochemical and Western blot (WB) analysis.
4.18. Statistical Analysis.
Results are presented as the mean ± standard deviation (SD). One-way analysis of variance (ANOVA) was performed to assess the statistical significance of the differences between groups. P < 0.05 indicated significant differences.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge the National Institutes of Health (1R01GM113166) and the Faculty Start-up fund University of Nebraska Medical Center for financial support. Xiaofei Xin is supported by the Chinese Scholarship Council (CSC).
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsami.9b02756.
Figures S1–S7, including 1H NMR spectra, biocompatibility of polymer study, flow cytometry analysis, and histological images of tissues (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Jia Y; Gu D; Wan J; Yu B; Zhang X; Chiorean EG; Wang Y; Xie J The role of GLI-SOX2 signaling axis for gemcitabine resistance in pancreatic cancer. Oncogene 2019, 38, 1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Adamska A; Domenichini A; Falasca M Pancreatic ductal adenocarcinoma: current and evolving therapies. Int. J. Mol. Sci 2017, 18 (7), 1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Baradaran B; Shahbazi R; Khordadmehr M Dysregulation of key microRNAs in pancreatic cancer development. Biomed. Pharmacother 2019, 109, 1008–1015. [DOI] [PubMed] [Google Scholar]
- (4).Singh S; Chitkara D; Kumar V; Behrman SW; Mahato RI miRNA profiling in pancreatic cancer and restoration of chemosensitivity. Cancer Lett 2013, 334 (2), 211–220. [DOI] [PubMed] [Google Scholar]
- (5).Song B; Liu XS; Rice SJ; Kuang S; Elzey BD; Konieczny SF; Ratliff TL; Hazbun T; Chiorean EG; Liu X Plk1 phosphorylation of orc2 and hbo1 contributes to gemcitabine resistance in pancreatic cancer. Mol. Cancer Ther 2013, 12 (1), 58–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Gutteridge REA; Ndiaye MA; Liu X; Ahmad N Plk1 inhibitors in cancer therapy: from laboratory to clinics. Mol. Cancer Ther 2016, 15 (7), 1427–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Garzon R; Marcucci G; Croce CM Targeting microRNAs in cancer: rationale, strategies and challenges. Nat. Rev. Drug Discovery 2010, 9 (10), 775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Rottiers V; Näär AM MicroRNAs in metabolism and metabolic disorders. Nat. Rev. Mol. Cell Biol 2012, 13, 239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Kumar V; Kumar V; Chaudhary AK; Coulter DW; McGuire T; Mahato RI Impact of miRNA-mRNA Profiling and Their Correlation on Medulloblastoma Tumorigenesis. Mol. Ther.–Nucleic Acids 2018, 12, 490–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Lu W; Logsdon C; Abbruzzese J Cancer Metabolism and its Therapeutic Implications. J. Cell Sci. Ther 2013, 4 (2), 143–152. [Google Scholar]
- (11).Li XJ; Ren ZJ; Tang JH MicroRNA-34a: a potential therapeutic target in human cancer. Cell Death Dis 2014, 5, No. e1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Ling H; Fabbri M; Calin GA MicroRNAs and other noncoding RNAs as targets for anticancer drug development. Nat. Rev. Drug Discovery 2013, 12, 847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Bommer GT; Gerin I; Feng Y; Kaczorowski AJ; Kuick R; Love RE; Zhai Y; Giordano TJ; Qin ZS; Moore BB; MacDougald OA; Cho KR; Fearon ER p53-mediated activation of miRNA34 candidate tumor-suppressor genes. Curr. Biol 2007, 17 (15), 1298–1307. [DOI] [PubMed] [Google Scholar]
- (14).Gibori H; Eliyahu S; Krivitsky A; Ben-Shushan D; Epshtein Y; Tiram G; Blau R; Ofek P; Lee JS; Ruppin E; et al. Amphiphilic nanocarrier-induced modulation of PLK1 and miR-34a leads to improved therapeutic response in pancreatic cancer. Nat. Commun 2018, 9 (1), 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Rudolph D; Steegmaier M; Hoffmann M; Grauert M; Baum A; Quant J; Haslinger C; Garin-Chesa P; Adolf GR BI 6727, a Polo-like kinase inhibitor with improved pharmacokinetic profile and broad antitumor activity. Clin. Cancer Res 2009, 15 (9), 3094–3102. [DOI] [PubMed] [Google Scholar]
- (16).Schöffski P; Awada A; Dumez H; Gil T; Bartholomeus S; Wolter P; Taton M; Fritsch H; Glomb P; Munzert G A phase I, dose-escalation study of the novel Polo-like kinase inhibitor volasertib (BI 6727) in patients with advanced solid tumours. Eur. J. Cancer 2012, 48 (2), 179–186. [DOI] [PubMed] [Google Scholar]
- (17).Xiao D; Yue M; Su H; Ren P; Jiang J; Li F; Hu Y; Du H; Liu H; Qing G Polo-like kinase-1 regulates Myc stabilization and activates a feedforward circuit promoting tumor cell survival. Mol. Cell 2016, 64 (3), 493–506. [DOI] [PubMed] [Google Scholar]
- (18).Xin X; Pei X; Yang X; Lv Y; Zhang L; He W; Yin L Rod Shaped Active Drug Particles Enable Efficient and Safe Gene Delivery. Advanced Science 2017, 4 (11), 1700324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Senapati S; Mahanta AK; Kumar S; Maiti P Controlled drug delivery vehicles for cancer treatment and their performance. Signal Transduction and Targeted Therapy 2018, 3 (1), 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Cheng CJ; Tietjen GT; Saucier-Sawyer JK; Saltzman WM A holistic approach to targeting disease with polymeric nanoparticles. Nat. Rev. Drug Discovery 2015, 14, 239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Semple SC; Akinc A; Chen J; Sandhu AP; Mui BL; Cho CK; Sah DW; Stebbing D; Crosley EJ; Yaworski E; Hafez IM; Dorkin JR; Qin J; Lam K; Rajeev KG; Wong KF; Jeffs LB; Nechev L; Eisenhardt ML; Jayaraman M; Kazem M; Maier MA; Srinivasulu M; Weinstein MJ; Chen Q; Alvarez R; Barros SA; De S; Klimuk SK; Borland T; Kosovrasti V; Cantley WL; Tam YK; Manoharan M; Ciufolini MA; Tracy MA; de Fougerolles A; MacLachlan I; Cullis PR; Madden TD; Hope MJ Rational design of cationic lipids for siRNA delivery. Nat. Biotechnol 2010, 28 (2), 172–6. [DOI] [PubMed] [Google Scholar]
- (22).Schulze J; Kuhn S; Hendrikx S; Schulz Siegmund M; Polte T; Aigner A Spray Dried Nanoparticle in Microparticle Delivery Systems (NiMDS) for Gene Delivery, Comprising Polyethylenimine (PEI) Based Nanoparticles in a Poly (Vinyl Alcohol) Matrix. Small 2018, 14 (12), 1701810. [DOI] [PubMed] [Google Scholar]
- (23).Peng Y; Wen D; Lin F; Mahato RI Co-delivery of siAlox15 and sunitinib for reversing the new-onset of type 1 diabetes in non-obese diabetic mice. J. Controlled Release 2018, 292, 1. [DOI] [PubMed] [Google Scholar]
- (24).Kumar V; Mondal G; Slavik P; Rachagani S; Batra SK; Mahato RI Codelivery of small molecule hedgehog inhibitor and miRNA for treating pancreatic cancer. Mol. Pharmaceutics 2015, 12 (4), 1289–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Mittal A; Chitkara D; Behrman SW; Mahato RI Efficacy of gemcitabine conjugated and miRNA-205 complexed micelles for treatment of advanced pancreatic cancer. Biomaterials 2014, 35 (25), 7077–7087. [DOI] [PubMed] [Google Scholar]
- (26).Lv S; Wu Y; Cai K; He H; Li Y; Lan M; Chen X; Cheng J; Yin L High Drug Loading and Sub-Quantitative Loading Efficiency of Polymeric Micelles Driven by Donor–Receptor Coordination Interactions. J. Am. Chem. Soc 2018, 140 (4), 1235–1238. [DOI] [PubMed] [Google Scholar]
- (27).Lin F; Wen D; Wang X; Mahato RI Dual responsive micelles capable of modulating miRNA-34a to combat taxane resistance in prostate cancer. Biomaterials 2019, 192, 95. [DOI] [PubMed] [Google Scholar]
- (28).Liou G-Y; Storz P Reactive oxygen species in cancer. Free Radical Res 2010, 44 (5), 479–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Zhang Y; Wei J; Liu S; Wang J; Han X; Qin H; Lang J; Cheng K; Li Y; Qi Y; et al. Inhibition of platelet function using liposomal nanoparticles blocks tumor metastasis. Theranostics 2017, 7 (5), 1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Mura S; Nicolas J; Couvreur P Stimuli-responsive nanocarriers for drug delivery. Nat. Mater 2013, 12 (11), 991. [DOI] [PubMed] [Google Scholar]
- (31).Shim MS; Xia Y A reactive oxygen species (ROS) responsive polymer for safe, efficient, and targeted gene delivery in cancer cells. Angew. Chem., Int. Ed 2013, 52 (27), 6926–6929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Qiao C; Yang J; Shen Q; Liu R; Li Y; Shi Y; Chen J; Shen Y; Xiao Z; Weng J; Zhang X Traceable Nanoparticles with Dual Targeting and ROS Response for RNAi Based Immunochemotherapy of Intracranial Glioblastoma Treatment. Adv. Mater 2018, 30 (18), 1705054. [DOI] [PubMed] [Google Scholar]
- (33).Tapeinos C; Pandit A Physical, Chemical, and Biological Structures based on ROS Sensitive Moieties that are Able to Respond to Oxidative Microenvironments. Adv. Mater 2016, 28 (27), 5553–5585. [DOI] [PubMed] [Google Scholar]
- (34).Kováčik J; Klejdus B; Bačkor M Nitric oxide signals ROS scavenger-mediated enhancement of PAL activity in nitrogen-deficient Matricaria chamomilla roots: side effects of scavengers. Free Radical Biol. Med 2009, 46 (12), 1686–1693. [DOI] [PubMed] [Google Scholar]
- (35).Jin R; Liu Z; Bai Y; Zhou Y; Chen X Multiple-Responsive Mesoporous Silica Nanoparticles for Highly Accurate Drugs Delivery to Tumor Cells. ACS Omega 2018, 3 (4), 4306–4315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Zhang F; Lau SS; Monks TJ The cytoprotective effect of N-acetyl-L-cysteine against ROS-induced cytotoxicity is independent of its ability to enhance glutathione synthesis. Toxicol. Sci 2011, 120 (1), 87–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Owens DE III; Peppas NA Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int. J. Pharm 2006, 307 (1), 93–102. [DOI] [PubMed] [Google Scholar]
- (38).Wilhelm S; Tavares AJ; Dai Q; Ohta S; Audet J; Dvorak HF; Chan WC Analysis of nanoparticle delivery to tumours. Nat. Rev. Mater 2016, 1 (5), 16014. [Google Scholar]
- (39).Kommareddy S; Amiji M Antiangiogenic gene therapy with systemically administered sFlt-1 plasmid DNA in engineered gelatin-based nanovectors. Cancer Gene Ther 2007, 14 (5), 488. [DOI] [PubMed] [Google Scholar]
- (40).Qi H; Zhou H; Tang Q; Lee JY; Fan Z; Kim S; Staub MC; Zhou T; Mei S; Han L; et al. Block copolymer crystalsomes with an ultrathin shell to extend blood circulation time. Nat. Commun 2018, 9 (1), 3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Ji X; Kong N; Wang J; Li W; Xiao Y; Gan ST; Zhang Y; Li Y; Song X; Xiong Q; et al. A Novel Top Down Synthesis of Ultrathin 2D Boron Nanosheets for Multimodal Imaging Guided Cancer Therapy. Adv. Mater 2018, 30 (36), 1803031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Song M; Liu T; Shi C; Zhang X; Chen X Bioconjugated manganese dioxide nanoparticles enhance chemotherapy response by priming tumor-associated macrophages toward M1-like phenotype and attenuating tumor hypoxia. ACS Nano 2016, 10 (1), 633–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Li L; Yuan L; Luo J; Gao J; Guo J; Xie X MiR-34a inhibits proliferation and migration of breast cancer through down-regulation of Bcl-2 and SIRT1. Clin. Exp. Med 2013, 13 (2), 109–117. [DOI] [PubMed] [Google Scholar]
- (44).Weichert W; Schmidt M; Jacob J; Gekeler V; Langrehr J; Neuhaus P; Bahra M; Denkert C; Dietel M; Kristiansen G Overexpression of Polo-like kinase 1 is a common and early event in pancreatic cancer. Pancreatology 2005, 5 (2–3), 259–265. [DOI] [PubMed] [Google Scholar]
- (45).Christoffersen N; Shalgi R; Frankel L; Leucci E; Lees M; Klausen M; Pilpel Y; Nielsen F; Oren M; Lund AH p53-independent upregulation of miR-34a during oncogene-induced senescence represses MYC. Cell Death Differ 2010, 17 (2), 236. [DOI] [PubMed] [Google Scholar]
- (46).Murga-Zamalloa C; Polk A; Hanel W; Chowdhury P; Brown N; Hristov AC; Bailey NG; Wang T; Phillips T; Devata S; et al. Polo-like-kinase 1 (PLK-1) and c-myc inhibition with the dual kinase-bromodomain inhibitor volasertib in aggressive lymphomas. Oncotarget 2017, 8 (70), 114474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Lobo VJS-A; Fernández LC; Carrillo-de-Santa-Pau E; Richart L; Cobo I; Cendrowski J; Moreno U; del Pozo N; Megías D; Bréant B; et al. c-Myc downregulation is required for preacinar to acinar maturation and pancreatic homeostasis. Gut 2017, 67 (4), 707–718. [DOI] [PubMed] [Google Scholar]
- (48).Ji Q; Hao X; Zhang M; Tang W; Yang M; Li L; Xiang D; DeSano JT; Bommer GT; Fan D; et al. MicroRNA miR-34 inhibits human pancreatic cancer tumor-initiating cells. PLoS One 2009, 4 (8), No. e6816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Beg MS; Brenner AJ; Sachdev J; Borad M; Kang Y-K; Stoudemire J; Smith S; Bader AG; Kim S; Hong DS Phase I study of MRX34, a liposomal miR-34a mimic, administered twice weekly in patients with advanced solid tumors. Invest. New Drugs 2017, 35 (2), 180–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Zhang M; Xiao B; Wang H; Han MK; Zhang Z; Viennois E; Xu C; Merlin D Edible ginger-derived nano-lipids loaded with doxorubicin as a novel drug-delivery approach for colon cancer therapy. Mol. Ther 2016, 24 (10), 1783–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Xiao B; Zhang M; Viennois E; Zhang Y; Wei N; Baker MT; Jung Y; Merlin D Inhibition of MDR1 gene expression and enhancing cellular uptake for effective colon cancer treatment using dual-surface-functionalized nanoparticles. Biomaterials 2015, 48, 147–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Lin C-W; Lu K-Y; Wang S-Y; Sung H-W; Mi F-L CD44-specific nanoparticles for redox-triggered reactive oxygen species production and doxorubicin release. Acta Biomater 2016, 35, 280–292. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.