

SUMMARY
The NRL-PRL murine model, defined by mammary-selective transgenic rat prolactin ligand rPrl expression, establishes spontaneous ER+ mammary tumors in nulliparous females, mimicking the association between elevated prolactin (PRL) and risk for development of ER+ breast cancer in postmenopausal women. Whole-genome and exome sequencing in a discovery cohort (n = 5) of end-stage tumors revealed canonical activating mutations and copy number amplifications of Kras. The frequent mutations in this pathway were validated in an extension cohort, identifying activating Ras alterations in 79% of tumors (23 of 29). Transcriptome analyses over the course of oncogenesis revealed marked alterations associated with Ras activity in established tumors compared with preneoplastic tissues; in cell-intrinsic processes associated with mitosis, cell adhesion, and invasion; as well as in the surrounding tumor environment. These genomic analyses suggest that PRL induces a selective bottleneck for spontaneous Ras-driven tumors that may model a subset of aggressive clinical ER+ breast cancers.
In Brief
Campbell et al. identify highly recurrent Kras-activating mutations in spontaneous prolactin-induced ER+ mammary tumors in female mice. This mimics the association between elevated prolactin and risk of ER+ breast cancer in postmenopausal women, providing a useful model for an important subset of aggressive clinical ER+ breast cancers.
Graphical Abstract
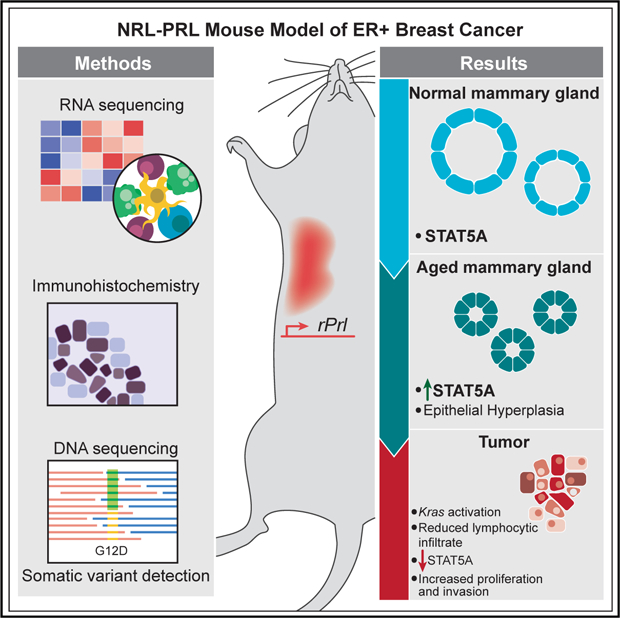
INTRODUCTION
Epidemiologic evidence has linked higher levels of circulating prolactin (PRL) with increased risk for estrogen receptor alpha (ER+) breast cancers, particularly in postmenopausal women (Tikk et al., 2014; Tworoger and Hankinson, 2008; Tworoger et al., 2013). However, the role of PRL in tumor progression is more controversial (Hachim et al., 2016; Shemanko, 2016; Tworoger and Hankinson, 2008). Moreover, activation of STAT5A, the downstream mediator of canonical PRL signals, is associated with a better prognosis (Peck et al., 2012; Tworoger and Hankinson, 2008). To distinguish the contributions of PRL to breast cancer from its actions in pregnancy, we generated the NRL-PRL murine model, where transgenic rat PRL (rPRL) is secreted by mammary epithelia to mimic the local production of PRL reported in women (Marano and Ben-Jonathan, 2014; McHale et al., 2008; O’Leary et al., 2015; Rose-Hellekant et al., 2003). In young adult nulliparous NRL-PRL females, prior to evidence of detectable mammary lesions, ductal epithelia proliferate more rapidly and exhibit increased progenitor activity and Wnt signaling, resulting in reduced maturation of luminal progenitors compared with wild-type (WT) littermates (O’Leary et al., 2017; Rose-Hellekant et al., 2003). With age, NRL-PRL females develop hyperplastic lesions that strongly express ER and progesterone receptor (PR) and eventually develop histologically diverse and metastatic carcinomas (O’Leary et al., 2015; Rose-Hellekant et al., 2003). In contrast to the preneoplastic lesions, established tumors express variable ER and low PR, resembling the luminal B subtype of clinical breast cancer (Arendt et al., 2011; O’Leary et al., 2015).
To understand the mechanisms that underlie the ability of PRL to drive the development of cancers in this model, we used comprehensive genomic analyses to identify genomic alterations and patterns of gene expression associated with cancer progression. In a discovery set of five independent, histologically diverse carcinomas and matched adjacent tumor-free mammary glands from aged NRL-PRL females, we found that all tumors contained somatic alterations in Kras, including canonical activating mutations (i.e., G12, G13, and Q61) or amplifications that resulted in elevated Ras pathway activation. Kras was not altered in any preneoplastic mammary glands. Our findings were validated in an extension set of 22 tumors and 4 cell lines (derived from two additional tumors), demonstrating activating Ras alterations in 79% of tumors. Ras activation was associated with increased phosphorylation of ERK1/2 and transcripts for Ras target genes and some pathway mediators, but variable rPrl transgene expression and reduced canonical downstream mediators of PRL signaling. These findings coincide with recent reports that elevated Ras signaling drives many clinical luminal B cancers and is associated with a poor prognosis (Olsen et al., 2017; Wright et al., 2015). In contrast to Kras-activated tumors, preneoplastic mammary glands maintained constitutive expression of rPrl and displayed elevated transcripts for inflammatory cytokines. Together, these data indicate that constitutive PRL signaling in mammary glands induces a pro-tumor environment, facilitating Ras pathway activation and spontaneous tumorigenesis.
RESULTS
To understand the molecular events underlying development of the prolactin-induced carcinomas in NRL-PRL females, we interrogated a discovery set from 10 NRL-PRL nulliparous female mice, consisting of five young adults and five aged tumor-bearing mice. Mammary cell preparations (MCPs) were harvested from the caudal glands of NRL-PRL females (n = 5) at 12 weeks (young adults), prior to morphologic evidence of pathology. Following the formation of spontaneous tumors at 15–20 months of age, three tissues from these mice were collected: (1) end-stage mammary tumors, (2) adjacent non-tumor mammary tissue (aged mammary glands [AMGs]), and (3) matched tail tissue (Figure 1A; Table S1). Mammary glands from 12-week-old mice contained primarily simple ductal structures and expressed ER and PR at levels similar to WT age-matched females (Figure 2A). In contrast, mammary glands of aged NRL-PRL females contained hyperplastic epithelial structures and expressed ER/PR similar to ductal structures of young glands of either genotype (Figure 2B). The ER+ tumors that develop in this model are histotypically diverse, including more differentiated adenocarcinomas (such as T2 and T3) and less differentiated spindle cell carcinomas (such as T1, T4, and T5). These end-stage tumors expressed lower levels of PR than structures in nondysplastic or hyperplastic AMGs (Figure 2B).
Figure 1. Sample Cohort Summary.

(A) For our discovery set, mammary cell preparations (MCPs) were isolated from caudal glands of 12-week-old NRL-PRL females (MCPs A–E, n = 5). Nulliparous mice with matched end-stage tumors (T) and adjacent aged mammary glands (AMGs) were collected following development of spontaneous ER+ tumors (15–20 months; T1–T5, AMG1–AMG5, n = 5). All samples were examined by RNA-seq. Tumors and matched tail DNA samples underwent WGS and WES.
(B) In our extension set, we evaluated 22 additional tumors from NRL-PRL females (T6–T27) and 4 cell lines (derived from 2 independent tumors).
(C) Summary of samples interrogated in this study. The number of mice associated with each group or the sample identifiers are indicated in parentheses. *Four cell lines were derived from 2 additional independent tumors (2 each).
See Table S1 for further details.
Figure 2. Histology of Sequenced Mammary Glands and Histologically Diverse Carcinomas.

(A) Mammary glands of 12-week-old NRL-PRL females did not exhibit marked morphological differences and did not express different levels of ER and PR from wild-type (WT) age-matched females.
(B) Histology of tumors sequenced and characterized in the discovery set and representative adjacent glands.
ER, estrogen receptor alpha; PR, progesterone receptor. Scale bars, 50 μm.
Genomic Characterization
Whole-genome sequencing (WGS) and whole-exome sequencing (WES) were performed on end-stage tumors and matched tails for mice within the discovery set (n = 5). Somatic variants were detected by comparing tumors with matched tail sequencing data. A range of 3–20 nonsilent mutations (median, 12) were identified in each tumor (Figures 3A and S1; Table S2). Kras was the only recurrently mutated gene in this cohort, with missense mutations detected in 4 of 5 tumors. Activating mutations occurred at two known hotspots (three G12D and one Q61L) and were present among the highest fractions (13%–67% DNA variant allele fraction [VAF]) within tumors, suggesting their presence in the founding clone of the tumors. Kras-activating mutations were confirmed in the RNA of end-stage tumors (at 46%–75% VAF); however, they were not present in RNA derived from adjacent AMG tissues. The tumor without an activating mutation (T5) was assessed for copy number and structural variants of Kras, and a focal amplification and structural variant were identified containing the Kras gene locus. There were no large-scale copy number alterations, structural variants, or gene-gene fusions recurrently altered across the discovery set outside of Kras (Figure S1).
Figure 3. Summary of Kras Modifications in NRL-PRL Tumors.

(A) The mutation frequency ranged from 3–20 nonsynonymous coding mutations across the five tumors in the discovery set (median, 12; top bar chart). The mutation burden (mutations per megabase) is shown. The only recurrently mutated gene in our cohort was Kras (top row of the bottom plot). The waterfall plot indicates the types of mutations detected in the remainder of the cohort (as detailed in the legend below C).
(B) Schematic of all mutations, comparing the prevalence of Kras missense mutations in tumors in NRL-PRL mice (n = 29; 20 contained Kras missense mutations) with KRAS missense mutations in human breast cancer: The Cancer Genome Atlas [TCGA], n = 825; METABRIC, n = 2,056; Lefebvre et al. (2016), n = 216; Stephens et al. (2012), n = 100; Banerji et al. (2012), n = 103; Griffith et al. (2018), n = 1,038.
(C) Left: Normalized depth of sequencing (coverage normalized by respective sample total sequencing depth) over the exons spanning the Kras gene locus (in 320- to 920-bp windows) of all tumor and normal samples. Tumor samples are not normalized with respect to matched normal data because matched normal tails were not available for all mice. Right: the average depth of sequencing (all windows) within the Kras locus is indicated on a log2 scale. Amplifications detected in T5 and T23 are labeled.
(D) RNA-seq data were available for T1–T5 (discovery set). The copy number (right panel, C) of Kras is plotted along the x axis, and the gene expression of Kras (in fragments per kilobase transcript per million mapped reads [FPKM]; quantified by Cufflinks, see STAR Methods) is indicated on the y axis. Note, two points nearly overlap. Refer to Figures S1 and S2 and Table S3 for further details.
Confirmation of Kras Activation in rPrl-Induced Tumors
We hypothesized that prolonged PRL exposure was creating a selective bottleneck for activating Kras mutations in the NRLPRL mouse model. To determine whether mutations at this locus were consistent in a larger set of these tumors, Sanger sequencing was performed on Kras exons 2 and 3 (containing the G12, G13, and Q61 hotspot loci) in an extension set of 22 independent PRL-induced tumors (15 archival tumors [T6–T20] and 7 fresh tumors with matched tails [T21–T27]) and 4 cell lines derived from 2 additional PRL-induced tumors (Figure 1B; Table S1). Hotspot mutations were identified in 14 tumors and all cell lines in this set (Figure 3B; Table 1). Overall, the most common alteration was Kras G12D (n = 9).
Table 1.
Summary of Ras Alterations in the Discovery and Extension Sets
| Sample Source | |||
|---|---|---|---|
| Mutation Type | Cell Line | Tumor | Total |
| WT | 6 | 6 | |
| Kras amplification | 2 | 2 | |
| Kras G12 | 2 | 12 | 13 |
| G12C | 3 | 3 | |
| G12D | 9 | 9 | |
| G12V | 2 | 1 | |
| Kras G13 | 1 | 1 | |
| G13A | 1 | 1 | |
| Kras Q61 | 2 | 5 | 6 |
| Q61H | 2 | 1 | 2 |
| Q61L | 1 | 1 | |
| Q61R | 3 | 3 | |
| Nras A59 | 1 | 1 | |
| A59D | 1 | 1 | |
| Number of mutated samples (tumors) | 4 (2)a | 21 | 25 (23) |
| Number of total samples (tumors) | 4 (2)a | 27 | 31 (29) |
Four cell lines were derived from 2 additional independent tumors (2 each). In total, 29 independent tumors were evaluated; 23 (79%) had detectable Ras pathway mutations.
WES was performed on the eight tumors within the extension set that did not contain Kras-activating mutations in exons 2 or 3. The mutational burden in these tumors ranged from 0–12 (median 6). Other mutations were not identified in Kras; however, there was a missense mutation in Nras in one tumor (T21, A59D, 21.3% VAF; Table S2). The copy number landscape was evaluated for Kras copy number alterations, as identified in T5 from the discovery set. Copy number was evaluated by quantifying the depth of exome sequencing and normalized per sample. A putative copy number increase in Kras was identified in one other tumor (T23), with an average copy number of 7.76 (Figure 3C). For reference, the average copy number within the discovery set was 1.34, whereas T5 exhibited an average copy number of 29. Given that an increased copy number is significantly associated with higher Kras expression in T5, we hypothesized that copy number alterations in sample T23 could affect transcript expression (Figure 3D). Together, these data demonstrated that at least 23 of 29 (79%) independent spontaneous NRL-PRL tumors developed alterations in Ras genes. We were unable to identify similar somatic alterations using WES in 6 tumors from the extension set (T6, T7, T12, T13, T17, and T25). Five of these tumors did not have matched normal tails available, reducing confidence in somatic mutation calling and filtering. Additional WGS of these tumors may provide further resolution of the somatic landscape. We focused our subsequent expression analysis on the discovery set, which, like the majority of the tumors, had Kras-activating alterations.
Analysis of Human Genomic Datasets for Ras Mutation Recurrence
We queried previously reported clinical datasets to better understand how the NRL-PRL mouse model can be used to comprehend ER+ human breast cancers. Consistent with the spontaneous tumors that arise in this model, amino acid G12 is the most recurrently mutated position in KRAS (Figure 3B; Banerji et al., 2012; Cancer Genome Atlas Network, 2012; Curtis et al., 2012; Griffith et al., 2018; Lefebvre et al., 2016; Stephens et al., 2012). This analysis was extended to include other RAS proteins and RasGAP tumor suppressors (KRAS, NRAS, HRAS, RASAL2, and DAB2IP) because of recent evidence revealing that a high proportion of luminal B ER+ breast cancers display elevated Ras pathway activity as a result of attenuated expression or somatic loss of RasGAP proteins (Olsen et al., 2017; Wright et al., 2015). Thus, although the frequency of KRAS alterations is low in human breast cancers (0%–6.94%), the Ras pathway is altered in 3.8%–22.9% of patients with ER+ breast tumors (Figure S2; Table S3).
Ras Pathway Activation Differentiates Expression in Tumors and Dysplastic Glands
Gene expression was evaluated across the samples from the discovery set, including five nondysplastic MCPs from young females and dysplastic AMGs and end-stage tumors from five aged tumor-bearing mice. Unsupervised approaches indicated clustering based upon sample source, where end-stage tumors behaved more similar to other tumors than their matched adjacent AMGs (Figure 4A). To understand the varied expression patterns across sample types, we first quantified rPrl transgene expression in each sample. rPrl expression was maintained in both nondysplastic MCPs and dysplastic AMGs but varied in tumors (Figure 4B). Lower-grade, more differentiated tumors (T2 and T3) contained relatively high levels of transgene transcripts, whereas high-grade spindle cell carcinomas (T1, T4, and T5) expressed very low levels of rPrl mRNA. This association suggests that rPrl transgene expression may influence tumor phenotype but is not required for tumor maintenance.
Figure 4. Constitutive Prolactin Signaling Induces a Selective Bottleneck for Ras Pathway Activation.

(A) Unsupervised principal-component analysis was performed across nondysplastic MCPs (n = 5), AMGs (n = 5), and end-stage spontaneous ER+ tumors (n = 5) from the discovery set. The color indicates the sample type.
(B) Kallisto was used to quantify transgene expression in transcripts per million (TPM). Pseudoalignment of RNA reads was performed against the annotated mouse transcriptome and rPrl (ENSRNOT00000023412.4).
(C) Heatmap displaying expression of canonical downstream mediators of prolactin signaling (Jak2 and Stat5a) and Ras pathway mediators (Mapk1/3, Akt1/2/3, Src, Map2k1, and Kras). Fill indicates the expression level in FPKM for AMG and tumor samples from the discovery set.
(D) Immunohistochemical staining for STAT5A, pERK1/2, and pAKT in two representative tumors of different histotypes with adjacent glands (left and right). Tumor regions are indicated by T in the image, and arrows point to adjacent AMGs. Scale bars, 50 μm.
To determine the downstream effects specific to Kras activation, supervised differential expression analysis was performed, comparing matched tumors with adjacent AMGs. There were 41 genes that were differentially expressed between these two tissue types (q < 0.001) that were also annotated as part of the Kyoto Encyclopedia of Genes and Genomes (KEGG) Ras pathway (mmu04014). This included upregulation of Mapk1, Map2k1, and Src mRNAs in end-stage tumors (Figure 4C), indicating higher levels of transcripts for mediators of the Ras pathway in Kras-driven tumors than in AMGs. Consistently, tumors, but not adjacent AMGs, displayed strong phosphorylation of ERK1/2 and AKT by immunohistochemistry (Figure 4D).
In normal mammary function, most PRL signals are mediated by the JAK2/STAT5A pathway (Oakes et al., 2008). Activation of this signaling cascade results in phosphorylation and dimerization of STAT5A, which then translocates to the nucleus to regulate transcription (Hammarén et al., 2018). Interestingly, Stat5a mRNA was significantly reduced in end-stage tumors compared with AMGs (Wald test, q = 1.54e-5; Figure 4C), particularly in poorly differentiated tumors T1, T4, and T5. Non-tumor mammary structures adjacent to the tumors also displayed strong nuclear STAT5A staining (Figure 4D), suggesting that the constitutive rPrl expression seen consistently across AMGs was activating this signaling cascade.
Tumorigenic Processes and the Microenvironment Differ across Stages of Disease Progression
Subsequent gene set enrichment and pathway analyses using the RNA sequencing (RNA-seq) data from the discovery set revealed multiple changes with respect to disease progression. Upregulation of tumorigenic processes, including cell cycle regulation and cell adhesion, were associated with Ras pathway activation in end-stage tumors (Tables S4 and S5; Figures 5A and 5B). Chromosome organization and chromatin structure, mitotic nuclear division, as well as cadherin and proteins involved in cell adhesion and adherens junctions were significantly upregulated in tumors compared with AMGs or MCPs (Figure 5B). This is consistent with increased mitotic and invasive properties of cancer cells. AMGs displayed increased expression of genes related to fatty acid metabolism and adipo-genesis relative to MCPs and tumors (p < 0.01; Figures 5A and 5B), reflecting the higher proportion of adipocytes in these preparations.
Figure 5. Tumorigenic Processes Differ across Stages of Disease Progression.

(A) Supervised differential expression analysis was performed on all comparisons of nondysplastic MCPs (n = 5), AMGs, and end-stage tumors (n = 5) from the discovery set. Genes that were significantly upregulated (p < 0.0001) in either tumors (compared with AMGs and MCPs) or glands (compared with tumors and MCPs) that were also annotated within significantly upregulated GO annotations (p < 0.001) are shown in the heatmap (n = 229). Fill indicates the gene-median centered expression value, calculated with respect to the entire discovery set. GO annotations were grouped into cellular processes, and genes corresponding to each annotation are noted in the side color bar (STAR Methods).
(B) A representative set of differentially expressed GO processes, categorized as “Cell Cycle” and “Immune Processes” in (A), are displayed based on their q value. Following each GO process, the number of genes included in the annotation is indicated. Comparisons are summarized as those that were upregulated in either MCPs or tumors compared with the other sample types.
(C) seq-ImmuCC was used to predict the relative proportions of immune cell populations (y axis) in MCPs, AMGs, and end-stage tumors from the discovery set. Samples are ordered by identifier (MCP A–E, AMG 1–5, and Tumors 1–5).
Compared with MCPs, both AMGs and tumors showed significantly lower expression of processes related to immune activation, including leukocyte cell-cell adhesion and aggregation, and T cell activation (q < 0.01; Tables S4 and S5; Figures 5A and 5B). These observations led us to hypothesize that the immune microenvironment differed across stages of disease. Markers of hematopoietic and immune cell lineages were significantly downregulated in tumors compared with preneoplastic cells, including Ptprc, Cd8a, Cd4, Tra, Trb, Cd40, and Cd19 mRNAs (p < 0.001), suggesting that tumors have reduced immune infiltrate, specifically those responsible for adaptive immune recognition and rejection (Table S4). We therefore interrogated the RNA-seq data to predict the relative proportions of infiltrating immune cell subpopulations using deconvolution approaches to further resolve the immune landscape across the discovery set (Chen et al., 2018). All samples displayed a mixture of immune cell subpopulations, and each tumor showed considerable diversity in the predicted proportions of these immune cell types (Figure 5C). Macrophages represented the predominant immune cell population across all samples (Figure 5C); however, these algorithmic approaches only determined the relative proportion of immune cell subpopulations without quantifying the absolute levels of immune infiltrate in each sample.
To confirm findings and evaluate trends in the RNA-seq data, we examined select transcripts by RT-PCR in the discovery set. In addition, MCPs were generated from WT 12-week-old females (WT MCPs) and from aged NRL-PRL females (aged mammary cell preparations [AMCPs]), enabling us to assess processes associated with preneoplastic changes in similar mammary cell preparations. Lipid metabolic enzymes (Acadm and Fasn mRNAs) were significantly upregulated in AMGs compared with AMCPs, confirming that the AMG preparations of tissue adjacent to tumors were enriched with adipocytes (Figure 6A).
Figure 6. Validation of RNA-Seq Findings.

(A–F) RT-PCR was used to evaluate transcripts associated with cellular processes altered with oncogenesis (identified in Figure 5) across samples in the discovery set (MCP, AMG, and tumor; indicated by †) as well as aged mammary cell preparations from NRL-PRL females (AMCP). These include fatty acid metabolism (A), hormone receptors (B), signaling pathways (C), cell cycle (D), cell adhesion (E), and immune processes (F). MCPs were also isolated from wild-type young adults (WT MCP) (n = 4–5). Data are represented as mean ± SEM. Significant differences were determined by one-way ANOVA, followed by the Tukey post-test. *p < 0.05, **p < 0.01, and ***p < 0.001.
(G) Immunohistochemistry demonstrating higher levels of CCND1 expression in tumors compared with preneoplastic tissue (i), low levels of CD8+ intratumoral lymphocytes compared with mammary lymphoid tissue (inset, ii), and F4/80+ myeloid cells within tumors and stroma and surrounding preneoplastic structures (inset, iii). Scale bars, 50 μm.
To further elucidate the stages of PRL-induced pathogenesis, hormone receptors and signaling pathways were interrogated. Although transcripts for ERα (Esr1) were not altered across samples, Pgr transcripts fell markedly with disease progression (Figure 6B), confirming detected protein expression (Figure 2).
Acquisition of somatic alterations in Kras in tumors was associated with loss of Stat5a mRNA (Figure 6C), consistent with the reduction in nuclear STAT5A protein (Figures 4C and 4D). Increased Notch activity, indicated by Hes1 transcripts, and Ccne1 mRNA in both AMCPs and tumors were consistent with increased proliferation of preneoplastic epithelial populations and tumors in NRL-PRL females (Zender et al., 2013). However, Ccnd1, Padi4, and Hmga2 transcripts were upregulated only in tumors, indicating high proliferation and chromatin remodeling, well-recognized cell-autonomous effects of elevated Ras activity; the dramatic rise in CCND1 protein expression in tumors was confirmed using immunohistochemistry (IHC) (Figures 6D and 6G, i). Unsurprisingly, tumors also showed striking elevations in transcripts associated with invasion and interaction with the extracellular matrix (Itgb4 and Mmp9) (Figure 6E).
Interrogation of cytokines and immune mediators revealed significant differences between tumors and preneoplastic mammary tissue (Figure 6F). Hyperplastic AMCPs, prior to Kras alterations, displayed elevated levels of Nfkb1, Csf1, Csf2, Ifng, and Tgfb1 mRNAs, encoding inflammatory cytokines. However, most of these transcripts, along with Icam1 mRNA, were precipitously lower in established tumors, depicting an immunosup-pressed environment. This was reflected in the low numbers of CD8+ intratumoral lymphocytes (Figure 6G, ii), confirming the RNA-seq data (Tables S4 and S5; Figure 5C). In contrast, intratumoral F4/80+ myeloid cells were plentiful (Figure 6G, iii; as predicted in Figure 5C). Tgfb1 remained elevated, and Arg1 and Nos2 transcripts were increased in tumors, consistent with shifts in activation of resident myeloid populations, including macrophages and myeloid-derived suppressor cells (Sica and Bronte, 2007). Together, these data support marked changes in the immune microenvironment in PRL-induced mammary pathogenesis.
DISCUSSION
Increased local exposure to PRL in the NRL-PRL model results in spontaneous diverse ER+ mammary cancers after a long latency, mimicking clinical luminal B, ER+ cancers in postmenopausal women. Here we demonstrated that Ras pathway activation in this model, particularly through recurrent somatic alterations in Kras, follows constitutive PRL signaling. Elevated Ras activity was associated with reduced activity in the canonical PRL-JAK2/STAT5A signaling cascade despite continued transgene expression in some tumors. Transcriptome analyses over the course of oncogenesis revealed marked alterations associated with Ras activity in established tumors in cell-intrinsic processes associated with mitosis, cell adhesion, and invasion as well as in the tumor microenvironment, including immune activity.
Mirroring the connection to human disease (Tikk et al., 2014; Tworoger and Hankinson, 2008; Tworoger et al., 2013), PRL has been observed to promote mammary carcinogenesis in multiple genetically engineered mouse models (Arendt and Schuler, 2008). Elevated PRL accelerates tumor development in combination with other oncogenes (Arendt et al., 2006, 2009; O’Leary et al., 2014), and, conversely, its absence increases tumor latency in combination with viral oncogenes (Oakes et al., 2008; Vomachka et al., 2000). Interestingly, BALB/c mice implanted with pituitary isografts, which elevate circulating PRL, develop mutagen-induced mammary tumors that also exhibit mutations in Kras (Guzman et al., 1992). Furthermore, expression of transgenic Kras G12V in beta lactoglobulin-expressing mammary cells during lactation (driven in part by high PRL) results in ER+ tumors in C57BL/6 mice (Andò et al., 2017). The link between PRL and development of Ras-driven mammary tumors in these distinct mouse models in different genetic backgrounds points to strong complementarity of these signals in mammary oncogenesis.
Germline ablation of Stat1 in 129/SvEv mice results in mammary tumors that acquire a truncating mutation in Prlr, resulting in its constitutive activation (Griffith et al., 2016). The well-differentiated tumors in Stat1−/− mice are characterized by strong STAT5 phosphorylation (Chan et al., 2014), similar to tumors that develop in response to transgenic Stat5a (Iavnilovitch et al., 2004). However, in NRL-PRL females, the robust STAT5 activation in preneoplastic mammary structures was markedly absent in established tumors, associated with reduced Stat5a mRNA and acquisition of Ras mutations, demonstrating a transition from the JAK2/STAT5 axis to the Ras signaling pathway. Consistently, tumors that arise in NRL-PRL females lose dependence on JAK2 signaling with disease progression (Sakamoto et al., 2010), whereas mammary tumors in Stat1-deficient mice remain dependent on JAK2 (Chan et al., 2014). Together, these data are congruent with a model whereby JAK2/STAT5A signals promote early tumorigenesis, but their ongoing activity supports well-differentiated, less aggressive luminal cancers that are more likely to respond to anti-estrogen therapies (Peck et al., 2012; Rädler et al., 2017).
Although all tumors in the NRL-PRL discovery set developed alterations in Ras family members, any ongoing role of the JAK2/STAT5 pathway and PRL-initiated signals appeared to differ across individual tumors. In the discovery set, the more differentiated adenocarcinomas (T2 and T3) maintained transgenic rPrl expression and showed slightly higher Stat5a mRNA than the less differentiated spindle cell carcinomas (T1, T4, and T5). Continued STAT5A signals may promote cell adhesion and other differentiated characteristics in these tumors (Nouhi et al., 2006; Sultan et al., 2005), despite the overall reduction of Stat5a transcripts compared with preneoplastic cells. PRL is able to initiate a diverse spectrum of signals apart from JAK2/STAT5, including activation of Src and downstream pathways and crosstalk with the Ras cascade itself (Barcus et al., 2013; Erwin et al., 1995; Martín-Pérez et al., 2015). These observations likely account for the controversy surrounding the role of PRL signals in clinical disease (Hachim et al., 2016; Shemanko, 2016).
Analysis of differentially expressed genes and associated processes with disease progression revealed complex alterations in cell signaling, the cytokine milieu, and the immune microenvironment early in PRL-driven pathogenesis. Together with PRL-augmented mammary epithelial progenitor cell sub-populations (O’Leary et al., 2017), our findings predict elevated inflammatory cytokines prior to acquisition of Ras pathway mutations that may contribute to tumorigenesis. End-stage tumors with genetic alterations in Kras exhibited marked modulation of the immune response, including reduced expression of inflammatory cytokines and increased markers of activated myeloid cells. These findings are consistent with the literature describing immune evasion by Kras activation by multiple mechanisms, including reduced expression of tumor antigens and promotion of recruitment and differentiation of immune populations that inhibit anti-tumor immune activity (Cullis et al., 2018; Dias Carvalho et al., 2018). Future studies will elucidate interactions between preneoplastic or malignant epithelial cells and the immune microenvironment through stages of PRL-induced pathogenesis.
The low rate of somatic mutations, reduced levels of transcripts encoding markers of many immune cell lineages, and low numbers of CD8+ infiltrating lymphocytes in NRL-PRL tumors mimic the relative immune silence of clinical luminal breast cancers (Dieci et al., 2016; Luen et al., 2016; Vonderheide et al., 2017). Recent studies have revealed that a high proportion of luminal B ER+ breast cancers display elevated Ras pathway activity as a result of reduced expression or somatic loss of Ras-GAP tumor suppressors (Griffith et al., 2018; Olsen et al., 2017; Wright et al., 2015). Thus, although the frequency of KRAS alterations is low, collectively, RAS proteins and RasGAP tumor suppressors (KRAS, NRAS, HRAS, RASAL2, and DAB2IP) are altered in a significant subset of ER+ breast tumors in publicly available datasets (Figure S2; Banerji et al., 2012; Cancer Genome Atlas Network, 2012; Curtis et al., 2012; Griffith et al., 2018; Lefebvre et al., 2016; Stephens et al., 2012). RAS itself has been an elusive therapeutic target (McCormick, 2015; Tran et al., 2016). However, targeting RAS-modulated immune regulators (Candido et al., 2018; Mitchem et al., 2013) may intercept T cell-suppressive signaling by pro-tumor myeloid populations. This approach may facilitate responses to T cell-directed therapies, such as the recent exciting anecdotal report of T cell transfer in metastatic breast cancer (Zacharakis et al., 2018).
For many years, it was questioned whether mouse models of mammary cancer could be estrogen responsive and model clinical luminal breast cancers. However, the ER+ cancers that develop in the NRL-PRL model, like the Stat1−/− and Kras (G12V) models (Andò et al., 2017; Chan et al., 2012), demonstrate that the tools available for murine systems can be valuable to study ER+ breast cancer. Even in PRL-induced tumors that have lost dependence on estrogen for growth, ER-mediated signals continue to modulate cell phenotype and cancer stem cell activity (Shea et al., 2018). These preclinical immunocompetent models of estrogen-responsive mammary tumors can provide insight into the biological behavior of these cancers, including metastasis, tumor cell plasticity and cancer stem cell activity, therapeutic responsiveness, and patterns of resistance. We would like to emphasize the critical need for comprehensive genomic studies in relevant experimental models to understand the similarities and differences from human disease and optimize the translational efficacy of these models. Without such genomic profiling, many models are being used and interpreted without understanding the full set of molecular alterations that drive and define the model. Our studies of tumorigenesis in the NRLPRL model describe the ability of constitutive rPrl expression to create a pro-tumor mammary environment converging into a selective bottleneck conducive for Ras pathway activation, which initiates aggressive tumor growth, invasion, and immuno-suppression of the tumor microenvironment, features shared with a subset of clinical ER+ breast cancers.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Obi Griffith (obigriffith@wustl.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
NRL-PRL mice [lines 1647–13, TgN(Nrl-Prl)23EPS; 1655–8, TgN(Nrl-Prl)24EPS] were generated and maintained in the FVB/N strain background (O’Leary et al., 2015; Rose-Hellekant et al., 2003). All tumors in the Discovery Set were TgN(Nrl-Prl)23EPS; the Extension Set included archived tumors of both lines. Mice were housed and handled in accordance with the Guide for Care and Use of Laboratory Animals in AAALAC-accredited facilities, and all procedures were approved by the University of Wisconsin-Madison Animal Care and Use Committee.
Sample Acquisition
We examined changes with age and pathogenesis in heterozygotic nulliparous females. Mammary epithelial cells were isolated from 12 week old animals, prior to evidence of morphological abnormalities. Other animals were aged until the primary mammary tumors were 1.5 cm in diameter (end stage), when the tumors, adjacent mammary glands, and tails were collected and flash frozen for subsequent analyses. For the Extension Set, an additional 22 tumors and 2 pairs of cell lines derived from 2 independent tumors were examined. These additional tumors included 7 freshly isolated tumors and matched tails, and 15 archived tumors were equally distributed among the glandular, papillary, and spindle cell carcinoma histotypes. All tumors analyzed developed in the cranial glands, reflecting the predominance of this site in this and many other transgenic mammary cancer models.
METHOD DETAILS
Morphological and Immunohistochemical Analyses
Samples were collected and fixed overnight in 10% neutral buffered formalin and processed into 5 micron sections. Mammary structures were assessed in hematoxylin and eosin (H&E) stained tissue sections, and protein expression was visualized by immunohistochemistry as previously described (Arendt et al., 2011) using antibodies against the following antigens: Cyclin D1, CP236B, Biocare Medical, (1:200); ER, #SC-542, Santa Cruz Biotechnology (1:500); PR, A0098, Dako (1:500); pAKTS473, #3787, Cell Signaling Technologies (1:50); pERK1/2, #9101, Cell Signaling Technologies (1:400); STAT5A, #SC-1081, Santa Cruz Biotechnology (1:5000). For CD8 and F4/80, frozen sections were stained. Tissues were embedded in OCT and frozen in a dry ice/methanol bath. Seven micron sections were cut, fixed with acetone at −20°C for 20 min, blocked with 5% normal goat serum and incubated overnight with either anti-CD8, Clone 53–6.7, Invitrogen (1:1000) or anti-F4/80, Clone BM8, Biolegend, (1:250). Slides were then processed in the same manner as for the paraffin sections.
Library Construction and Sequencing Strategy
Genomic DNA was isolated using the QIAGEN Blood & Cell Culture DNA Mini Kit (#13323; Hilden, Germany). Whole genome sequencing (WGS) libraries for matched tumor and normal tail samples (n = 5) were prepared using the Illumina TruSeq PCR free kit with dual indexed adaptors. WGS libraries were pooled and sequenced across 1 flow cell (8 lanes) on the Illumina HiSeq X platform with paired 2 × 150 bp reads with median 33.35X coverage (27.18–37.52X). Whole exome sequencing (WES) libraries for matched tumor and normal tail samples were pooled and sequenced on 1 lane of an Illumina HiSeq 4000 with paired 2 × 150 bp reads with median 37.99X coverage (10.39–85.45X). Total RNA was isolated using the QIAGEN RNeasy Midi Kit (#75142; Hilden, Germany). RNA sequencing (RNAseq) libraries were prepared using the Illumina TruSeq stranded total RNA kit. Libraries were pooled and sequenced across 4 lanes of a HiSeq 4000 with 2 × 150 bp reads with median 60,072,344 total reads (43,482,392–100,068,565 total reads).
Sanger Validation Sequencing
For the Extension Set and positive controls from the Discovery Set, the regions surrounding Kras hotspots G12/G13 and Q61 were amplified from 5ng of genomic DNA, using Phusion Hot Start Flex DNA polymerase (#M0535; New England Biolabs), and primers which tagged products with M13F(−21) (forward strand) and M13R (reverse strand) sequences (see Table S6 for Primer Sequences). PCR products were purified using the QIAquick PCR Purification Kit (#28104; QIAGEN), and sent to GeneWiz (South Plainfield, NJ) for Sanger Sequencing. Traces were manually assessed for variants within G12, G13, and Q61 using the CodonCode Aligner software (version 7.0.1; CodonCode Corporation).
DNA sequencing and analysis
The Genome Modeling System (GMS) was used for all analysis, including the somatic variant detection and RNA-seq analysis (Griffith et al., 2015). WGS and WES data were processed through SpeedSeq v0.1.0 (Chiang et al., 2015; Faust and Hall, 2014), which aligns reads by BWA-MEM v0.7.10 (Li, 2013) to the mouse reference genome (NCBI build 38, GRCm38) and marks duplicates using SAMBLASTER v0.1.22 (Faust and Hall, 2014). Matched (normal) tail samples were analyzed for all 5 tumors in the Discovery Set and 3 tumors in the Extension Set which did not exhibit Kras G12, G13, or Q61 mutations. Somatic events were identified by individually comparing tumor DNA to matched tail (normal) DNA. Single nucleotide variants (SNVs) were detected by the union of SomaticSniper v1.0.4 (Larson et al., 2012), VarScan2 v2.3.6 (Koboldt et al., 2012), Strelka v1.0.11 (Saunders et al., 2012), and Mutect v1.1.4 (Cibulskis et al., 2013). Small insertions and deletions (indels) were detected by taking the union of GATK Somatic Indel Detector (v5336) (McKenna et al., 2010), Varscan2, and Strelka. 5 tumors from the Extension Set which did not exhibit Kras G12, G13, or Q61 mutations did not have DNA from matched normal tail samples (T6, 7, 12, 13, and 17). For these tumors, SNV/indels were detected by comparing the aligned tumor WES data to the mouse reference genome GRCm38 using Varscan 2 (v2.3.6) (Koboldt et al., 2012).
SNV/indels were filtered using Samtools r982 (Li et al., 2009) ([mpileup-BuDS] filtered by var-filter-snv v1 then false-positive-vcf v1) and annotated by the GMS transcript variant annotator against Ensembl v84. Mutations were filtered to those annotated as nonsynonymous Tier 1 mutations within the protein-coding regions, meeting the following thresholds: minimum 5 reads supporting the variant, minimum 5% variant allele frequency, minimum 20X coverage of the corresponding genomic position in the tumor sample, and minimum 20X coverage of the corresponding genomic position in the normal tail sample (if available). The 8 tail samples were used as a ‘panel of normal’ samples in order to remove common nucleotide polymorphisms and artifacts with the following requirements: SNVs with a minimum 2.5% VAF with 20X coverage detected in at least 2 (25%) of the normal samples or Indels with minimum 2% VAF with 2X coverage detected in at least 2 (25%) of the normal samples. Following this filtering strategy, false positives were manually removed, as previously described (Barnell et al., 2018).
All variants were further filtered by removing those identified in normal mice by the Mouse Genomes Project (v142) (Keane et al., 2011). There were no remaining variants detected in T23, likely indicating that this tumor had low tumor cellularity, making it difficult to differentiate sequencing artifacts from variants detected. There were five tumors (T6, T7, T12, T13, T17) without matched normal tail samples. Recurrent mutations that were identified only in these unmatched tumors were removed as likely germline variants. Furthermore, mutations in olfactory receptor and uncharacterized cDNA regions (Olfr and Rik genes) were removed.
Copy number variants, including Kras amplifications, were identified using CopyCat v0.1 [https://github.com/chrisamiller/copyCat], comparing matched tumor and normal tail samples, if available. In tumors with no matched normal samples, copy number alterations were detected by normalizing the depth of sequencing within each sample over the exons spanning the Kras gene locus (in 320–920 bp windows). While there were no variants detected in T23, there was increased normalized coverage over the Kras gene locus indicating a possible copy number amplification. Structural variants were predicted using Manta on WGS and WES data (Chen et al., 2016), and intergenic fusions were detected using Integrate (Zhang et al., 2016).
Publicly Available Datasets
Clinical and mutational data from five public datasets (Banerji et al., 2012; Cancer Genome Atlas Network, 2012; Curtis et al., 2012; Lefebvre et al., 2016; Stephens et al., 2012) were obtained through cBioPortal (Gao et al., 2013). Samples were filtered to those annotated as ‘Breast Cancer’ (removing ‘Breast Sarcoma’). Patients were annotated as ‘ER+ or HR+/HER2-’ in Figure S2 based upon the ‘ER Status’ and ‘HR+/HER2-’ columns in the clinical annotations. These datasets were specifically queried for mutations and copy number alterations in KRAS, NRAS, HRAS, DAB2IP, and RASAL2. The Griffith dataset (Griffith et al., 2018) contained only HR+/HER2- breast cancers, and was only assessed for KRAS mutations (the other four genes were not evaluated in this targeted gene panel study).
rPrl Expression Analysis
A kallisto index was built to incorporate the annotated Ensembl mouse transcriptome (v92) and the full rPrl transcript (ENSRNOT00000023412.4) (Zerbino et al., 2018). RNaseq reads were pseudoaligned to this kallisto index to quantify read abundance and expression (TPM) of rPrl.
RNA Expression Analysis
RNaseq reads were aligned to the mouse reference genome (NCBI build 38, GRCm38) using TopHat (Trapnell et al., 2009). Cufflinks (Trapnell et al., 2010) and HTSeq-count (Anders and Huber, 2010) were used to quantify gene and transcript expression, and differential expression analysis was performed using the DESeq2 R package (Love et al., 2014). Gene Set Enrichment Analysis was performed using the fgsea R package (Sergushichev, 2016). The GAGE R package was used for pathway analysis, and data visualization was performed using the ggplot2 R package (Wickham, 2009) and GenVisR (Skidmore et al., 2016). Immune infiltrate was assessed by seq-ImmuCC using the Illumina RNaseq platform and the SVR method (Chen et al., 2018). Immune populations were summarized as relative frequencies (the default output of the program).
RT-PCR
Total RNA was isolated from samples in the Discovery Set (MCPs, AMGs, tumors), WT age-matched (12–14 week) MCPs, and non-tumor mammary cells from aged (15–16 month) mice isolated using the MCP procedure, using the RNeasy Midi Kit (#75142; QIAGEN). cDNA was synthesized from 1 μg of RNA using M-MLV reverse transcriptase (#M1705; Promega) and random hexamers. Real time PCR was performed using SYBR Green (#4309155; ThermoFisher) on the Applied Biosystems 7300 platform as described (Arendt et al., 2011) using the primer sequences in Table S6. Transcripts of interest were normalized to 18S RNA, and fold changes calculated using the ΔΔCt method.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical Analysis
Gene expression differences and statistics were summarized by the results of supervised comparisons in DESeq2. Specifically, p values were calculated by Negative Binomial generalized linear model fitting and Wald statistics, and adjusted p values (q values) were calculated by the Benjamini Hochberg method. Immune populations were compared using Mann-Whitney tests between groups.
DATA AND CODE AVAILABILITY
The reference-aligned whole genome, exome, and RNA sequencing data and sample details for 36 tumor and non-tumor mouse tissues have been submitted to NCBI SRA study SRP189110, BioProject PRJNA489661.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Cyclin D1 | Biocare Medical | Cat# CP236B |
| Estrogen receptor | Santa Cruz Biotechnology | Cat# SC-542; RRID:AB_631470 |
| pAKT S473 | Cell Signaling Technologies | Cat# 3787; RRID:AB_331170 |
| pERK1/2 | Cell Signaling Technologies | Cat# 9101; RRID:AB_331646 |
| STAT5A | Santa Cruz Biotechnology | Cat# SC-1081; RRID:AB_632448 |
| CD8 | ThermoFisher | Cat# 14008185; RRID:AB_467088 |
| F4/80 | Biolegend | Cat#123102; AB_893506 |
| Progesterone Receptor | Dako | Cat#A0098; RRID:AB_2315192 |
| Biological Samples | ||
| Mammary cell preparations from NRL-PRL mammary glands | This Paper | N/A |
| Critical Commercial Assays | ||
| Blood & Cell Culture DNA Mini Kit | QIAGEN | 13323 |
| TruSeq PCR free WGS library preparation kit | Illumina | 20015962 |
| RNEasy Midi Kit | QIAGEN | 75142 |
| TruSeq Stranded Total RNA library preparation kit | Illumina | 20020596 |
| Phusion Hot Start Flex DNA polymerase | New England Biolabs | M0535 |
| QIAquick PCR Purification Kit | QIAGEN | 28104 |
| M-MLV reverse transcriptase | Promega | M1705 |
| SYBR Green | ThermoFisher | 4309155 |
| Deposited Data | ||
| Mouse reference genome (NCBI build 38, GRCm38) | Genome Reference Consortium | https://www.ncbi.nlm.nih.gov/assembly/GCF_000001635.26 |
| Ensembl reference transcriptome v84 | Ensembl | ftp://ftp.ensembl.org/pub/release-84/gtf/mus_musculus/ |
| Mouse Genomes Project v142 | Keane et al., 2011 | ftp://ftp-mouse.sanger.ac.uk/REL-1505-SNPs_lndels/ |
| Human breast cancer sequencing dataset | Banerji et al., 2012 | http://www.cbioportal.org/ |
| Human breast cancer sequencing dataset | Cancer Genome Atlas Network, 2012 | http://www.cbioportal.org/ |
| Human breast cancer sequencing dataset | Curtis et al., 2012 | http://www.cbioportal.org/ |
| Human breast cancer sequencing dataset | Lefebvre et al., 2016 | http://www.cbioportal.org/ |
| Human breast cancer sequencing dataset | Stephens et al., 2012 | http://www.cbioportal.org/ |
| Human breast cancer sequencing dataset | Griffith et al., 2018 | phs001234.v1.p1 |
| Raw and analyzed data | This Paper | SRP189110 |
| Experimental Models: Organisms/Strains | ||
| Mouse: NRL-PRL [lines 1647–13, TgN(Nrl-Prl)23EPS; 1655–8, TgN(Nrl-Prl)24EPS] | Rose-Hellekant et al., 2003 | N/A |
| Oligonucleotides | ||
| Primers for Real Time PCR, see Table S6 | This Paper | N/A |
| Primers for Sanger Sequencing, see Table S6 | This Paper | N/A |
| Software and Algorithms | ||
| Codon Code Aligner [v 7.0.1] | CodonCode Corporation | https://www.codoncode.com/aligner/ |
| Genome Modeling System | Griffith et al., 2015 | https://github.com/genome/gms |
| BWA-MEMvO.7.10 | Li, 2013 | https://github.com/lh3/bwa |
| SAMBLASTERvO.1.22 | Faust and Hall, 2014 | https://github.com/GregoryFaust/samblaster |
| SomaticSniper v1.0.4 | Larson et al., 2012 | http://gmt.genome.wustl.edu/packages/somatic-sniper/ |
| VarScan2 V2.3.6 | Koboldt et al.,2012 | http://varscan.sourceforge.net/ |
| Strelkav1.0.11 | Saunders et al., 2012 | https://github.com/lllumina/strelka |
| Mutect v1.1.4 | Cibulskis et al., 2013 | https://software.broadinstitute.org/cancer/cga/mutect |
| GATK Somatic Indel Detector v5336 | McKenna et al., 2010 | https://software.broadinstitute.org/gatk/ |
| Samtools r982 | Li et al., 2009 | http://samtools.sourceforge.net/ |
| CopyCat v0.1 | N/A | https://github.com/chrisamiller/copyCat |
| Manta | Chen et al., 2016 | https://github.com/lllumina/manta |
| Integrate | Zhang et al., 2016 | https://sourceforge.net/projects/integrate-fusion/ |
| cBioPortal | Gaoet et al., 2013 | http://www.cbioportal.org |
| Kallisto | Zerbino et al., 2018 | https://pachterlab.github.io/kallisto/ |
| TopHat | Trapnell et al., 2009 | https://github.com/infphilo/tophat |
| Cufflinks | Trapnell et al., 2010 | https://github.com/cole-trapnell-lab/cufflinks |
| HTSeq-count | Anders and Huber, 2010 | https://htseq.readthedocs.io/en/release_0.11.1/ |
| DESeq2 | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Gene set enrichment analysis (fgsea) | Sergushichev, 2016 | https://bioconductor.org/packages/release/bioc/html/fgsea.html |
| Ggplot2 | Wickham, 2009 | https://ggplot2.tidyverse.org/ |
| GenVisR | Skidmore et al., 2016 | https://bioconductor.org/packages/release/bioc/html/GenVisR.html |
| Seq-lmmuCC | Chen et al., 2018 | http://218.4.234.74:3200/immune/ |
| Other | ||
| Manual review standard operating procedures | Barnell et al., 2018 | N/A |
Highlights.
Genomic analyses are performed on prolactin-induced ER+ murine mammary tumors
Kras is recurrently mutated in tumors, suggesting a selective bottleneck
Tumors exhibit reduced STAT5A and low lymphocytic infiltrate
Genomic profiling is required to assess translational effect of murine models
ACKNOWLEDGMENTS
K.M.C. was supported by the Cancer Biology Pathway through the Siteman Cancer Center at Washington University School of Medicine. M.G. was supported by the National Human Genome Research Institute (R00 HG007940). L.A.S. was supported by the NIH (R01 CA157675 and R01 CA179556) and UWCCC core grant NIH P30 CA014520. O.L.G. was supported by the National Cancer Institute (K22 CA188163 and U01 CA209936).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.06.098.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Anders S, and Huber W (2010). Differential expression analysis for sequence count data. Genome Biol. 11, R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andò S, Malivindi R, Catalano S, Rizza P, Barone I, Panza S, Rovito D, Emprou C, Bornert J-M, Laverny G, and Metzger D (2017). Conditional expression of Ki-RasG12V in the mammary epithelium of transgenic mice induces estrogen receptor alpha (ERα)-positive adenocarcinoma. Oncogene 36, 6420–6431. [DOI] [PubMed] [Google Scholar]
- Arendt LM, and Schuler LA (2008). Transgenic models to study actions of prolactin in mammary neoplasia. J. Mammary Gland Biol. Neoplasia 13, 29–40. [DOI] [PubMed] [Google Scholar]
- Arendt LM, Rose-Hellekant TA, Sandgren EP, and Schuler LA (2006). Prolactin potentiates transforming growth factor alpha induction of mammary neoplasia in transgenic mice. Am. J. Pathol 168, 1365–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendt LM, Evans LC, Rugowski DE, Garcia-Barchino MJ, Rui H, and Schuler LA (2009). Ovarian hormones are not required for PRL-induced mammary tumorigenesis, but estrogen enhances neoplastic processes.J. Endocrinol 203, 99–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendt LM, Rugowski DE, Grafwallner-Huseth TA, Garcia-Barchino MJ, Rui H, and Schuler LA (2011). Prolactin-induced mouse mammary carcinomas model estrogen resistant luminal breast cancer. Breast Cancer Res. 13, R11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerji S, Cibulskis K, Rangel-Escareno C, Brown KK, Carter SL, Frederick AM, Lawrence MS, Sivachenko AY, Sougnez C, Zou L, et al. (2012). Sequence analysis of mutations and translocations across breast cancer subtypes. Nature 486, 405–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barcus CE, Keely PJ, Eliceiri KW, and Schuler LA (2013). Stiff collagen matrices increase tumorigenic prolactin signaling in breast cancer cells.J. Biol. Chem 288, 12722–12732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnell EK, Ronning P, Campbell KM, Krysiak K, Ainscough BJ, Ramirez C, Spies N, Kunisaki J, Hundal J, Skidmore ZL, et al. (2018). Standard operating procedure for somatic variant refinement of tumor sequencing data. Genet. Med 21, 972–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Network (2012). Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candido JB, Morton JP, Bailey P, Campbell AD, Karim SA, Jamieson T, Lapienyte L, Gopinathan A, Clark W, McGhee EJ, et al. (2018). CSF1R+ Macrophages Sustain Pancreatic Tumor Growth through T Cell Suppression and Maintenance of Key Gene Programs that Define the Squamous Subtype. Cell Rep. 23, 1448–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan SR, Vermi W, Luo J, Lucini L, Rickert C, Fowler AM, Lonardi S, Arthur C, Young LJ, Levy DE, et al. (2012). STAT1-deficient mice spontaneously develop estrogen receptor a-positive luminal mammary carcinomas. Breast Cancer Res. 14, R16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan SR, Rickert CG, Vermi W, Sheehan KCF, Arthur C, Allen JA, White JM, Archambault J, Lonardi S, McDevitt TM, et al. (2014). Dysregulated STAT1-SOCS1 control of JAK2 promotes mammary luminal progenitor cell survival and drives ERa(+) tumorigenesis. Cell Death Differ. 21, 234–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Schulz-Trieglaff O, Shaw R, Barnes B, Schlesinger F, Källberg M, Cox AJ, Kruglyak S, and Saunders CT (2016). Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 32, 1220–1222. [DOI] [PubMed] [Google Scholar]
- Chen Z, Quan L, Huang A, Zhao Q, Yuan Y, Yuan X, Shen Q, Shang J, Ben Y, Qin FX-F, and Wu A (2018). seq-ImmuCC: Cell-Centric View of Tissue Transcriptome Measuring Cellular Compositions of Immune Microenvironment From Mouse RNA-Seq Data. Front. Immunol 9, 1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang C, Layer RM, Faust GG, Lindberg MR, Rose DB, Garrison EP, Marth GT, Quinlan AR, and Hall IM (2015). SpeedSeq: ultra-fast personal genome analysis and interpretation. Nat. Methods 12, 966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, and Getz G (2013). Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol 31, 213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullis J, Das S, and Bar-Sagi D (2018). Kras and Tumor Immunity: Friend or Foe? Cold Spring Harb. Perspect. Med 8, a031849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis C, Shah SP, Chin S-F, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y, et al. ; METABRIC Group (2012). The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 486, 346–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias Carvalho P, Guimarães CF, Cardoso AP, Mendonça S, Costa ÂM, Oliveira MJ, and Velho S (2018). KRAS Oncogenic Signaling Extends beyond Cancer Cells to Orchestrate the Microenvironment. Cancer Res. 78, 7–14. [DOI] [PubMed] [Google Scholar]
- Dieci MV, Griguolo G, Miglietta F, and Guarneri V (2016). The immune system and hormone-receptor positive breast cancer: Is it really a dead end? Cancer Treat. Rev 46, 9–19. [DOI] [PubMed] [Google Scholar]
- Erwin RA, Kirken RA, Malabarba MG, Farrar WL, and Rui H (1995). Prolactin activates Ras via signaling proteins SHC, growth factor receptor bound 2, and son of sevenless. Endocrinology 136, 3512–3518. [DOI] [PubMed] [Google Scholar]
- Faust GG, and Hall IM (2014). SAMBLASTER: fast duplicate marking and structural variant read extraction. Bioinformatics 30, 2503–2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. (2013). Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 6, pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith M, Griffith OL, Smith SM, Ramu A, Callaway MB, Brummett AM, Kiwala MJ, Coffman AC, Regier AA, Oberkfell BJ, et al. (2015). Genome Modeling System: A Knowledge Management Platform for Genomics. PLoS Comput. Biol 11, e1004274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith OL, Chan SR, Griffith M, Krysiak K, Skidmore ZL, Hundal J, Allen JA, Arthur CD, Runci D, Bugatti M, et al. (2016). Truncating Prolactin Receptor Mutations Promote Tumor Growth in Murine Estrogen Receptor-Alpha Mammary Carcinomas. Cell Rep. 17, 249–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith OL, Spies NC, Anurag M, Griffith M, Luo J, Tu D, Yeo B, Kunisaki J, Miller CA, Krysiak K, et al. (2018). The prognostic effects of somatic mutations in ER-positive breast cancer. Nat. Commun 9, 3476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman RC, Osborn RC, Swanson SM, Sakthivel R, Hwang SI, Miyamoto S, and Nandi S (1992). Incidence of c-Ki-ras activation in N-methyl-N-nitrosourea-induced mammary carcinomas in pituitary-isografted mice. Cancer Res. 52, 5732–5737. [PubMed] [Google Scholar]
- Hachim IY, Shams A, Lebrun J-J, and Ali S (2016). A favorable role of prolactin in human breast cancer reveals novel pathway-based gene signatures indicative of tumor differentiation and favorable patient outcome. Hum. Pathol 53, 142–152. [DOI] [PubMed] [Google Scholar]
- Hammarén HM, Virtanen AT, Raivola J, and Silvennoinen O (2018). The regulation of JAKs in cytokine signaling and its breakdown in disease. Cytokine 118, 48–63. [DOI] [PubMed] [Google Scholar]
- Iavnilovitch E, Cardiff RD, Groner B, and Barash I (2004). Deregulation of Stat5 expression and activation causes mammary tumors in transgenic mice. Int. J. Cancer 112, 607–619. [DOI] [PubMed] [Google Scholar]
- Keane TM, Goodstadt L, Danecek P, White MA, Wong K, Yalcin B, Heger A, Agam A, Slater G, Goodson M, et al. (2011). Mouse genomic variation and its effect on phenotypes and gene regulation. Nature 477, 289–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, and Wilson RK (2012). VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 22, 568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson DE, Harris CC, Chen K, Koboldt DC, Abbott TE, Dooling DJ, Ley TJ, Mardis ER, Wilson RK, and Ding L (2012). SomaticSniper: identification of somatic point mutations in whole genome sequencing data. Bioinformatics 28, 311–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre C, Bachelot T, Filleron T, Pedrero M, Campone M, Soria J-C, Massard C, Lévy C, Arnedos M, Lacroix-Triki M, et al. (2016). Mutational Profile of Metastatic Breast Cancers: A Retrospective Analysis. PLoS Med. 13, e1002201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv, arXiv:1303.3997, https://arxiv.org/abs/1303.3997. [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luen S, Virassamy B, Savas P, Salgado R, and Loi S (2016). The genomic landscape of breast cancer and its interaction with host immunity. Breast 29, 241–250. [DOI] [PubMed] [Google Scholar]
- Marano RJ, and Ben-Jonathan N (2014). Minireview: Extrapituitary prolactin: an update on the distribution, regulation, and functions. Mol. Endocrinol 28, 622–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martín-Pérez J, García-Martínez JM, Sánchez-Bailón MP, Mayoral-Varo V, and Calcabrini A (2015). Role of SRC family kinases in prolactin signaling. Adv. Exp. Med. Biol 846, 163–188. [DOI] [PubMed] [Google Scholar]
- McCormick F (2015). KRAS as a Therapeutic Target. Clin. Cancer Res. 21, 1797–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHale K, Tomaszewski JE, Puthiyaveettil R, Livolsi VA, and Clevenger CV (2008). Altered expression of prolactin receptor-associated signaling proteins in human breast carcinoma. Mod. Pathol 21, 565–571. [DOI] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, and DePristo MA (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchem JB, Brennan DJ, Knolhoff BL, Belt BA, Zhu Y, Sanford DE, Belaygorod L, Carpenter D, Collins L, Piwnica-Worms D, et al. (2013). Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 73, 1128–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nouhi Z, Chughtai N, Hartley S, Cocolakis E, Lebrun J-J, and Ali S (2006). Defining the role of prolactin as an invasion suppressor hormone in breast cancer cells. Cancer Res. 66, 1824–1832. [DOI] [PubMed] [Google Scholar]
- O’Leary KA, Rugowski DE, Sullivan R, and Schuler LA (2014). Prolactin cooperates with loss of p53 to promote claudin-low mammary carcinomas. Oncogene 33, 3075–3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Leary KA, Shea MP, and Schuler LA (2015). Modeling prolactin actions in breast cancer in vivo: insights from the NRL-PRL mouse. Adv. Exp. Med. Biol 846, 201–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Leary KA, Shea MP, Salituro S, Blohm CE, and Schuler LA (2017). Prolactin Alters the Mammary Epithelial Hierarchy, Increasing Progenitors and Facilitating Ovarian Steroid Action. Stem Cell Reports 9, 1167–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakes SR, Rogers RL, Naylor MJ, and Ormandy CJ (2008). Prolactin regulation of mammary gland development. J. Mammary Gland Biol. Neoplasia 13, 13–28. [DOI] [PubMed] [Google Scholar]
- Olsen SN, Wronski A, Castaño Z, Dake B, Malone C, De Raedt T, Enos M, DeRose YS, Zhou W, Guerra S, et al. (2017). Loss of RasGAP Tumor Suppressors Underlies the Aggressive Nature of Luminal B Breast Cancers. Cancer Discov. 7, 202–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peck AR, Witkiewicz AK, Liu C, Klimowicz AC, Stringer GA, Pequignot E, Freydin B, Yang N, Ertel A, Tran TH, et al. (2012). Low levels of Stat5a protein in breast cancer are associated with tumor progression and unfavorable clinical outcomes. Breast Cancer Res. 14, R130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rädler PD, Wehde BL, and Wagner K-U (2017). Crosstalk between STAT5 activation and PI3K/AKT functions in normal and transformed mammary epithelial cells. Mol. Cell. Endocrinol 451, 31–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose-Hellekant TA, Arendt LM, Schroeder MD, Gilchrist K, Sandgren EP, and Schuler LA (2003). Prolactin induces ERalpha-positive and ERalpha-negative mammary cancer in transgenic mice. Oncogene 22, 4664–4674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto K, Triplett AA, Schuler LA, and Wagner K-U (2010). Janus kinase 2 is required for the initiation but not maintenance of prolactin-induced mammary cancer. Oncogene 29, 5359–5369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders CT, Wong WSW, Swamy S, Becq J, Murray LJ, and Cheetham RK (2012). Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 28, 1811–1817. [DOI] [PubMed] [Google Scholar]
- Sergushichev A (2016). An algorithm for fast preranked gene set enrichment analysis using cumulative statistic calculation. bioRxiv. 10.1101/060012. [DOI] [Google Scholar]
- Shea MP, O’Leary KA, Fakhraldeen SA, Goffin V, Friedl A, Wisinski KB, Alexander CM, and Schuler LA (2018). Antiestrogen Therapy Increases Plasticity and Cancer Stemness of Prolactin-Induced ERα+ Mammary Carcinomas. Cancer Res. 78, 1672–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shemanko CS (2016). Prolactin receptor in breast cancer: marker for meta-static risk. J. Mol. Endocrinol 57, R153–R165. [DOI] [PubMed] [Google Scholar]
- Sica A, and Bronte V (2007). Altered macrophage differentiation and immune dysfunction in tumor development. J. Clin. Invest 117, 1155–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skidmore ZL, Wagner AH, Lesurf R, Campbell KM, Kunisaki J, Griffith OL, and Griffith M (2016). GenVisR: Genomic Visualizations in R. Bioinformatics 32, 3012–3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens PJ, Tarpey PS, Davies H, Van Loo P, Greenman C, Wedge DC, Nik-Zainal S, Martin S, Varela I, Bignell GR, et al. ; Oslo Breast Cancer Consortium (OSBREAC) (2012). The landscape of cancer genes and mutational processes in breast cancer. Nature 486, 400–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultan AS, Xie J, LeBaron MJ, Ealley EL, Nevalainen MT, and Rui H (2005). Stat5 promotes homotypic adhesion and inhibits invasive characteristics of human breast cancer cells. Oncogene 24, 746–760. [DOI] [PubMed] [Google Scholar]
- Tikk K, Sookthai D, Johnson T, Rinaldi S, Romieu I, Tjønneland A, Olsen A, Overvad K, Clavel-Chapelon F, Baglietto L, et al. (2014). Circulating prolactin and breast cancer risk among pre- and postmenopausal women in the EPIC cohort. Ann. Oncol 25, 1422–1428. [DOI] [PubMed] [Google Scholar]
- Tran E, Robbins PF, Lu Y-C, Prickett TD, Gartner JJ, Jia L, Pasetto A, Zheng Z, Ray S, Groh EM, et al. (2016). T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N. Engl. J. Med 375, 2255–2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, and Salzberg SL (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, and Pachter L (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol 28, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tworoger SS, and Hankinson SE (2008). Prolactin and breast cancer etiology: an epidemiologic perspective. J. Mammary Gland Biol. Neoplasia 13, 41–53. [DOI] [PubMed] [Google Scholar]
- Tworoger SS, Eliassen AH, Zhang X, Qian J, Sluss PM, Rosner BA, and Hankinson SE (2013). A 20-year prospective study of plasma prolactin as a risk marker of breast cancer development. Cancer Res. 73, 4810–4819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vomachka AJ, Pratt SL, Lockefeer JA, and Horseman ND (2000). Prolactin gene-disruption arrests mammary gland development and retards T-antigen-induced tumor growth. Oncogene 19, 1077–1084. [DOI] [PubMed] [Google Scholar]
- Vonderheide RH, Domchek SM, and Clark AS (2017). Immunotherapy for Breast Cancer: What Are We Missing? Clin. Cancer Res. 23, 2640–2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H (2009). ggplot2: Elegant Graphics for Data Analysis (Springer Verlag; New York: ). [Google Scholar]
- Wright KL, Adams JR, Liu JC, Loch AJ, Wong RG, Jo CEB, Beck LA, Santhanam DR, Weiss L, Mei X, et al. (2015). Ras Signaling Is a Key Determinant for Metastatic Dissemination and Poor Survival of Luminal Breast Cancer Patients. Cancer Res. 75, 4960–4972. [DOI] [PubMed] [Google Scholar]
- Zacharakis N, Chinnasamy H, Black M, Xu H, Lu Y-C, Zheng Z, Pasetto A, Langhan M, Shelton T, Prickett T, et al. (2018). Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat. Med 24, 724–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zender S, Nickeleit I, Wuestefeld T, Sörensen I, Dauch D, Bozko P, El-Khatib M, Geffers R, Bektas H, Manns MP, et al. (2013). A critical role for notch signaling in the formation of cholangiocellular carcinomas. Cancer Cell 23, 784–795. [DOI] [PubMed] [Google Scholar]
- Zerbino DR, Achuthan P, Akanni W, Amode MR, Barrell D, Bhai J, Billis K, Cummins C, Gall A, Girón CG, et al. (2018). Ensembl 2018. Nucleic Acids Res. 46 (D1), D754–D761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, White NM, Schmidt HK, Fulton RS, Tomlinson C, Warren WC, Wilson RK, and Maher CA (2016). INTEGRATE: gene fusion discovery using whole genome and transcriptome data. Genome Res. 26, 108–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The reference-aligned whole genome, exome, and RNA sequencing data and sample details for 36 tumor and non-tumor mouse tissues have been submitted to NCBI SRA study SRP189110, BioProject PRJNA489661.