

3D biomimetic pancreatic cancer on a chip reveals endothelial ablation via activin-ALK7, a mechanism for tumor hypovascularity.
Abstract
Pancreatic ductal adenocarcinoma (PDAC) is an aggressive, lethal malignancy that invades adjacent vasculatures and spreads to distant sites before clinical detection. Although invasion into the peripancreatic vasculature is one of the hallmarks of PDAC, paradoxically, PDAC tumors also exhibit hypovascularity. How PDAC tumors become hypovascular is poorly understood. We describe an organotypic PDAC-on-a-chip culture model that emulates vascular invasion and tumor–blood vessel interactions to better understand PDAC-vascular interactions. The model features a 3D matrix containing juxtaposed PDAC and perfusable endothelial lumens. PDAC cells invaded through intervening matrix, into vessel lumen, and ablated the endothelial cells, leaving behind tumor-filled luminal structures. Endothelial ablation was also observed in in vivo PDAC models. We also identified the activin-ALK7 pathway as a mediator of endothelial ablation by PDAC. This tumor-on-a-chip model provides an important in vitro platform for investigating the process of PDAC-driven endothelial ablation and may provide a mechanism for tumor hypovascularity.
INTRODUCTION
Although the diagnosis and treatment of cancer in its earliest stages has substantially improved outcomes in many tumors, survival rates in patients with tumors that have spread to distant sites remain dismal (1). Hence, the vast majority of cancer mortalities stem from metastasis and its complications (2). Metastasis is a final product of a chain of multiple complex steps, including local spread of cancer cells at primary sites of origin, invasive entry into nearby vasculature (intravasation), exit from vasculature (extravasation), and growth at distant organ sites (3). To complete the metastasis cascade, tumor cells need to extensively interact with the vasculature. However, the interactions between cancer cells and blood vessels in particular are poorly understood.
One of the many examples of poorly understood tumorendothelium interactions is in pancreatic ductal adenocarcinoma (PDAC). PDAC is a highly metastatic cancer whose cancer cells have been shown to escape the tumor and enter the circulation at the very earliest stages of tumor progression (4, 5). The vast majority of patients once diagnosed with PDAC are already at the later stages of the disease (6). At the same time, tumor masses from these patients generally exhibit hypovascularity or a paucity of capillary vessels within these tumors (7). Hence, there is an unusual yet poorly understood transition in interactions between tumor and the blood vessels in PDAC: At early stages, nearby blood vessels are essential in providing a means for tumor cells to gain access to the circulation, but at later stages, they are absent and can limit chemotherapeutic drug delivery to the tumors. Thus, understanding these tumor-endothelium interactions within PDAC will provide important insights into PDAC tumor biology.
In part, a lack of detailed understanding of tumor-vessel interactions in PDAC is due to difficulties in observing and studying these interactions in traditional models of tumor invasion. Although a few histologic studies of patient samples have observed the extensive invasion of tumor cells into the vasculature (8–10), follow-up in vivo studies in mice have not advanced our understanding of this phenomenon owing to the complexity of animal models (11) and challenges in spatiotemporal imaging of tumor-vessel interactions in internal organs, such as pancreas (12). Alternative to in vivo approaches, recent advances in microfluidic microphysiological systems have allowed the generation of blood vessel–tumor interfaces in vitro (13–18). Early studies using these in vitro cultures to model the interactions between tumor cells with two-dimensional (2D) planar or 3D vessel-like structures have demonstrated the value of juxtaposing tumor cells with the vascular compartment, especially in the context of investigating extravasation and intravasation properties (14, 15, 19–21). However, so far, these models have not been used to study interactions between malignant PDAC and blood vessels.
To achieve a deeper understanding of tumor-endothelium interactions in PDAC, we describe here a model system in which a biomimetic ductal channel containing PDAC cells is juxtaposed to a rudimentary blood vessel consisting of an endothelialized, perfused lumen. Using this model, we observed that PDAC tumor cells can invade and remove the vascular endothelium to leave behind tumor-lined and tumor-filled luminal structures, a process we refer to as endothelial ablation. We further validated our findings in in vivo PDAC models and identified a crucial role for activin-ALK7 signaling in mediating endothelial ablation in PDAC. On the basis of our studies, we propose that endothelial cell ablation by tumor cells via the activin-ALK7 signaling pathway may be a potential mechanism to explain hypovascularity in PDAC.
RESULTS
An organotypic model of PDAC exhibits 3D invasion and ablation of endothelial cells
To examine the process of PDAC invasion, we engineered an organotypic model of PDAC building on a previously developed vessel-on-a-chip (18). Briefly, our PDAC organotypic model is composed of two hollow cylindrical channels, which are completely embedded into 3D collagen matrix (Fig. 1A). In one of the channels, we seeded endothelial cells to form a biomimetic blood vessel, as previously described (18). In a parallel channel, we seeded primary mouse pancreatic cancer cells PD7591 and allowed them to adhere to form a monolayer of epithelial cells to mimic a ductal compartment of a pancreatic duct. To study the interactions of PDAC cells with the blood vessels, we performed a screening experiment wherein different chemotactic agents were introduced into the biomimetic blood vessel and found that a gradient of fetal bovine serum (FBS) most efficiently stimulated invasion of pancreatic cancer cells into the collagen matrix (fig. S1, A to D). Characterization of the gradient using 21-kDa fluorescein isothiocyanate dextran (FITC-dextran) showed a gradual loss in the steepness of the gradient (fig. S1E), prompting us to administer FBS daily in the endothelial channel to maintain the FBS gradient during the course of the experiments.
Fig. 1. Organotypic model for PDAC-on-a-chip to capture pancreatic tumor vascular invasion.
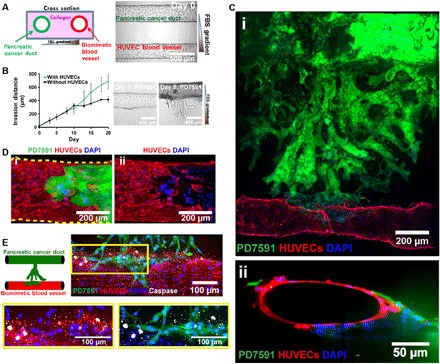
(A) Schematic of PDAC-on-a-chip with a biomimetic blood vessel and a pancreatic cancer duct. The microfluidic device is composed of two hollow cylindrical channels embedded within 3D collagen matrix. One channel was seeded with endothelial cells to form a perfusable biomimetic blood vessel, while the other channel was seeded with pancreatic cancer cells to form a pancreatic cancer duct. Phase-contrast image shows seeded cells in the device before PDAC migration. (B) Average invasion distance of a PDAC cell line, YFP PD7591, toward a gradient of FBS with and without human umbilical vein endothelial cells (HUVECs). The presence of HUVECs increased the migration speed of PD7591 (n = 3 individual experiments). Representative phase-contrast images of PD7591 migration at days 0 and 8 demonstrated the collective migration of PDAC invasion. (C) YFP PD7591 (in green) invaded toward the biomimetic blood vessel (in red), migrated along the vessel (i), and wrapped around the blood vessel, as shown in the cross-sectional image of the biomimetic blood vessel (ii). (D) A confocal image of a section of the blood vessel (in red) invaded by YFP PD7591 (in green) showed that part of the blood vessel was ablated by cancer cells in our organotypic model (i and ii). (E) Apoptosis, marked by cleaved caspase-3 staining (in white), was observed in endothelial cells (in red) during invasion of YFP PD7591 (in green) in the blood vessels in our 3D PDAC organotypic model. Endothelial cells in all images were stained with anti-CD31 antibody. YFP PD7591 was restained with FITC-conjugated anti-GFP (green fluorescent protein) antibody. Cell nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) (in blue). Error bars are SEM.
Upon stimulation with FBS, the PDAC cells in the biomimetic ductal channel began to proliferate to form a multilayer of cells (fig. S2). By day 4, PD7591 cells began to invade into the matrix toward the endothelial lumen. The invasion was collective, with epithelial cells remaining in contact with each other, to form branched structures reminiscent of epithelial morphogenesis (Fig. 1B). The presence of the endothelium increased the migration speed of PD7591 in response to the FBS gradient (Fig. 1B), until reaching the engineered blood vessel. Upon contact with the biomimetic blood vessel, the PDAC cells wrapped around the blood vessel and spread along the length of the blood vessel before invading into the vessel itself (Fig. 1C, i and ii, and movie S1).
During PD7591 invasion into the blood vessel, we observed that part of the blood vessel was occupied by the tumor PD7591 cells (Fig. 1D, i and ii, and movie S2), a finding that was replicated with three additional primary mouse PDAC cell lines and a human pancreatic cancer cell line (fig. S3). As the PDAC cells invaded and occupied the lumen of the biomimetic blood vessels, we also observed apoptotic endothelial cells in proximity to the PDAC cells (Fig. 1E), whereas endothelial cells in the biomimetic blood vessels without tumor invasion exhibited no apoptotic activity (fig. S4A). In our 3D organotypic model, our endothelium deposited a collagen IV layer, while the PDAC cells in the biomimetic pancreatic cancer duct did not deposit collagen IV (fig. S4B). We also observed that, as the tumor cells ablated the endothelial cells in the 3D biomimetic blood vessel, the collagen IV layer deposited by the endothelium gradually disappeared (fig. S4C). We collectively refer to this process as endothelial ablation, where the PDAC tumor cells invade the blood vessels and ablate the endothelial cells, leaving behind tumor-lined and tumor-filled luminal structures.
Endothelial ablation is observed in in vivo tumor models of PDAC
To verify that the used tumor cells PD7591 also ablated endothelial cells in vivo and further confirm that the biomimetic model reproduces an in vivo process that occurs in pancreatic cancers, we subcutaneously inoculated the same tumor cells into mice. After the tumor volume reached approximately 400 mm3, we resected the tumor, including the adjacent area around the tumor, for analysis. Immunohistochemical staining for cleaved caspase-3 and CD31 showed that endothelial cells were apoptotic in the PDAC tumor in vivo (Fig. 2A). In contrast, control mice injected with Matrigel alone had a minimal amount of apoptotic endothelial cells (fig. S5A). Because tumor cells PD7591 did not deposit collagen IV in our in vitro and in vivo experiments (fig. S4, B and D), while blood vessels in vivo were surrounded by collagen IV (fig. S4, E and F), we stained for endomucin or CD31 as endothelial cell markers and collagen IV (a basement membrane protein in vessels) to identify blood vessels in PDAC tumors. A subset of vessel lumens in the tumor-containing regions was mosaically lined by both endothelial and PDAC cells (Fig. 2B, red arrows), and several collagen IV–lined lumens, reminiscent of blood vessels, were lined luminally with only PDAC cells (Fig. 2B, yellow arrows). These data are aligned with our in vitro findings (Fig. 1, D and E), therefore suggesting that our biomimetic model of PDAC captures a relevant in vivo process of PDAC progression.
Fig. 2. Endothelial ablation is observed in in vivo mouse tumor models (subcutaneous tumor implantation model and genetically engineered mouse model).
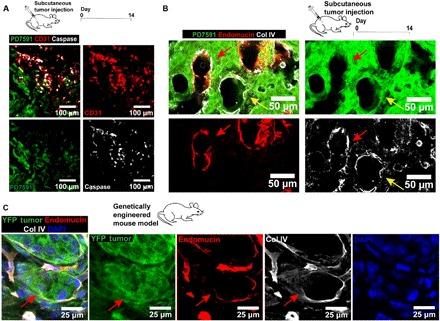
(A) Examination of endothelial cells in the ectopic mouse tumors in vivo. YFP PD7591 (in green) was subcutaneously injected into mice for 14 days. Apoptotic endothelial cells were also observed in the in vivo tumor microenvironment, similarly to the observation in the 3D organotypic model. Apoptotic endothelial cells (in red) were marked by cleaved caspase-3 signal (in white). (B) Endothelial ablation in subcutaneous tumor implantation model. Resected tumors at day 14 exhibited partial ablation of endothelial cells by pancreatic cancer cells (red arrows) in hybrid blood vessels. Some small vessels, decorated with collagen IV (in white), a basement membrane protein, demonstrated complete endothelial ablation by YFP PD7591 in the luminal side of the vessels (yellow arrows). Blood vessels were stained with anti-mouse endomucin antibody (in red). (C) Endothelial ablation was observed in GEMM of PDAC. Partial ablation of endothelium in the blood vessel was indicated by red arrows, where the blood vessel displayed the occupation of yellow fluorescent protein (YFP) tumor cells in place of the endothelium. Endothelial cells in all images were stained with either anti-mouse CD31 antibody or anti-mouse endomucin. YFP PD7591 was restained with FITC-conjugated anti-GFP antibody. Cell nuclei were stained with DAPI (in blue).
To further strengthen our finding of endothelial ablation in PDAC in vivo, we sought to demonstrate the presence of blood vessels occupied by tumor cells in a genetically engineered mouse model (GEMM) of PDAC, in which many aspects of human tumor biology and progression are captured (22). In this GEMM, tumor cells were genetically tagged with a yellow fluorescent protein, and PDAC tumor tissue sections were immunohistochemically stained for endomucin and collagen IV to highlight the endothelium and their basement membrane protein. Similar to the ectopic model, tumor cells in the GEMM were also found at the luminal side of blood vessels (Fig. 2C). Together, our findings in both in vivo tumor mouse models and our 3D biomimetic model suggest that PDAC invades blood vessels, gains access to the luminal space of the blood vessels, and ultimately ablates the endothelium to form tumor cell–lined luminal structures, a process that may contribute to the hypovascularity observed in larger PDAC tumor masses.
Endothelial ablation in PDAC is mediated through a transforming growth factor–β receptor signaling pathway
We next sought to identify which signaling pathways were involved in endothelial ablation. The transforming growth factor–β (TGF-β) signaling pathway has been implicated in tumor progression in many different types of tumors, including PDAC (23). We therefore asked whether the TGF-β receptor signaling pathway was involved in endothelial ablation in PDAC. Using our 3D organotypic model, we allowed pancreatic cancer cells from the pancreatic cancer duct to invade the engineered blood vessel. Once the pancreatic cancer cells reached the blood vessels, we initiated treatment with SB431542 at 5 μM for a duration of 7 days. We observed that inhibition of TGF-β receptor signaling significantly reduced the ablation of endothelial cells by pancreatic cancer cells in our biomimetic blood vessel (Fig. 3A, i and ii, and fig. S8, A and B). Quantification of the endothelial cell vessel area that was ablated by PDAC cells further confirmed our observation (Fig. 3A). Western blot of phosphorylated Smad2 confirmed the effect of SB431542 inhibition on TGF-β receptor signaling in both endothelial cells and PD7591 (Fig. 3A).
Fig. 3. Inhibition of the TGF-β receptor signaling pathway significantly reduced endothelial ablation.
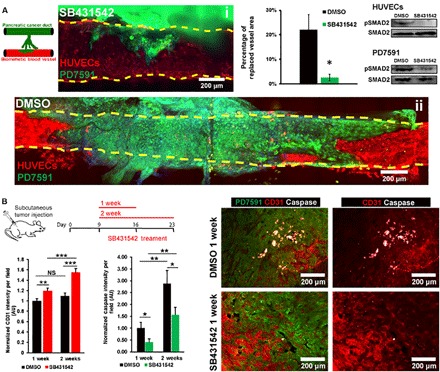
(A) Inhibition of TGF-β receptor signaling pathway with SB431542 in 3D organotypic model. PDAC tumor cells from the pancreatic cancer duct were allowed to invade into the biomimetic vessels. Once the PDAC tumor cells reached the blood vessels, 5 μM SB431542 or dimethyl sulfoxide (DMSO) was administered for 7 days. Representative confocal images of vessels treated with SB431542 (i) and DMSO (ii) in 3D organotypic model. The percentage of endothelium-ablated area showed that SB431542 significantly reduced the endothelial ablation in 3D organotypic model (n = 4 individual experiments). Western blot for phosphorylated Smad2 in endothelial cells and PD7591 in 2D monoculture confirmed the effectiveness of SB431542 to inhibit the TGF-β signaling pathway in endothelial cells and in pancreatic cancer cells PD7591. (B) Inhibition of TGF-β receptor signaling pathway with SB431542 in a mouse tumor model for either 1 or 2 weeks. SB431542 was peritoneally administered into the mice daily for 1 and 2 weeks. Mice were sacrificed at days 16 and 23. Quantification of endothelial cell density revealed a significantly higher endothelial cell density within the tumors treated with SB431542 as compared to vehicle control (n = 5 mice per experimental group) for both 1 and 2 weeks. Cleaved caspase-3 activity in endothelial cells was also less in SB431542-treated tumors versus control tumors in both 1 and 2 weeks. Cleaved caspase-3 activity was significantly increased in tumors treated with SB431542 for 2 weeks versus 1 week. Right: Representative images of tumor samples in vehicle control DMSO and SB431542 conditions for 1-week condition. Endothelial cells were stained with anti-mouse CD31 antibody. YFP PD7591 was restained with FITC-conjugated anti-GFP antibody. Cell nuclei were stained with DAPI (in blue). *P < 0.05, **P < 0.01, and ***P < 0.001 indicate statistical significance. NS, not significant. Two-tailed Student’s t test. Error bars are SEM.
We further investigated whether inhibition of TGF-β receptor signaling also led to reduced endothelial ablation in vivo by subcutaneously injecting the same pancreatic cancer cell line PD7591 into mice. By day 9, when the tumor reached 100 mm3, we administered SB431542 versus control vehicle into two groups of mice through daily peritoneal injections for 1 and 2 weeks. In tumors harvested from SB431542-treated mice, we measured a higher density of vessels by quantifying the CD31 signal intensity within the tumor microenvironment (Fig. 3B and fig. S5B). We also noticed a minimal proliferation of endothelial cells in both control and SB431542-treated tumors (fig. S5C), indirectly suggesting that the increase in CD31 signal intensity in SB431542-treated tumors was likely due to less endothelial cell ablation. Similarly, quantification of cleaved caspase-3 signal intensity showed a less apoptotic signal in endothelial cells in SB431542-treated mice at 1 and 2 weeks versus control mice. However, endothelial cell ablation and apoptosis were not fully inhibited, as the apoptotic caspase signal was still higher at 2 weeks versus 1 week. Of note, the tumor growth rate was not affected by administration of SB431542 in vivo (fig. S5D). Together, our in vitro and in vivo data suggested that inhibiting TGF-β receptor signaling reduces endothelial ablation in PDAC.
Endothelial ablation of PDAC is mediated through an activin-ALK7 pathway
Because SB431542 has previously been reported as a broad inhibitor for the TGF-β superfamily receptors ALK4, ALK5, and ALK7 (24), we sought to determine which of these receptors in each of the cell types might contribute to endothelial ablation. To address this question, we designed a simple 2D patterned coculture invasion assay to quickly enable us to identify the molecular mechanism of endothelial cell ablation. Briefly, PDAC cells were plated inside a silicone annulus, while endothelial cells were plated outside the annulus. Peeling the annulus off the substrate left a circular population of PDAC cells surrounded by a monolayer of endothelial cells (Fig. 4A). As time progressed, the PDAC cells invaded into the endothelial cell area and ablated the endothelial cells (movie S3), resulting in an increase in the area of PDAC cells (Fig. 4B and fig. S9A). When SB431542 was added into the coculture of PD7591 and endothelial cells, the PDAC invasion area was significantly decreased (Fig. 4B, fig. S9B, and movie S4), resembling the effects of blocking the TGF-β receptor signaling pathway to prevent endothelial ablation in our 3D organotypic model. We further showed the effects of inhibiting the TGF-β receptor signaling pathway to reduce endothelial ablation in two other human pancreatic cancer cell lines (Panc-1 and BxPC-3; fig. S6, A and B).
Fig. 4. Endothelial ablation required activin-ALK7 signaling in the invasive PDAC.
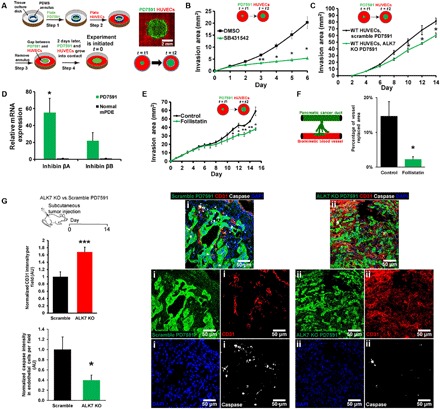
(A) A schematic demonstrates a setup for 2D patterned coculture of PDAC and endothelial cells. Tumor cells were plated inside an annulus, and endothelial cells were plated outside the annulus. Once the annulus was removed, tumor invaded into the endothelial cells, resulting in an expansion of the tumor area. (B) SB431542 reduced endothelial ablation in 2D patterned coculture (n = 3 individual experiments). (C) ALK7 was knocked out in YFP PD7591 by CRISPR-Cas9 and plated in 2D patterned cocultures with wild-type (WT) HUVECs. Invasion area of ALK7 knockout (KO) YFP PD7591 was significantly less than scramble PD7591, suggesting the role of ALK7 in PD7591 to mediate endothelial ablation (n = 3 individual experiments). (D) Inhibin βA and inhibin βB, major subunits of activins, were up-regulated in pancreatic cancer cells as compared to normal mouse pancreatic ductal epithelial cells (mPDE) (n = 3 individual experiments). (E) Inhibition of activin with the endogenous inhibitor follistatin significantly diminished endothelial ablation of PDAC in 2D patterned cocultures (n = 3 individual experiments). (F) Inhibition of activin by follistatin (200 ng/ml) in 3D organotypic model revealed a significant reduction in tumor replaced blood vessel (n = 3 individual experiments). (G) In vivo mouse tumor model to confirm the role of ALK7 in endothelial ablation. ALK7 knockout PD7591 versus scramble cells were subcutaneously implanted into mice for 2 weeks. Cleaved caspase-3 signal indicated a substantial reduction in apoptotic endothelial cells in ALK7 knockout condition versus scrambled PD7591 condition (n = 5 mice per experimental group). A significant increase in endothelial cell density was also observed with ALK7 knockout PD7591 tumors versus scramble PD7591 tumors (n = 5 mice per experimental group). (i) Representative images of tumors implanted with scramble PD7591 depicted less endothelial cell density and high apoptotic signal. (ii) Representative images of tumor implanted with ALK7 knockout PD7591 depicted high endothelial cell density and less apoptotic signal. *P < 0.05, **P < 0.01, and ***P < 0.001 indicate statistical significance. Two-tailed Student’s t test. Error bars are SEM.
Using this 2D patterned coculture invasion assay, we first stained for cleaved caspase-3 activity to examine whether apoptosis coincided with PDAC invasion of the endothelial cell area. In agreement with our findings in the 3D organotypic model, the cleaved caspase-3 signal was only detected in endothelial cells, and the cleaved caspase-3 signal intensity in endothelial cells was the highest at the interface between endothelial and tumor cells and dropped as the endothelial cells were further away from the interface (fig. S6C). Administration of SB431542 significantly reduced the number of apoptotic endothelial cells, particularly at the interface between endothelial and cancer cells (fig. S6C), suggesting that direct contact between endothelial and PDAC cells may be required for endothelial ablation. To test this hypothesis, we monitored the endothelial-PDAC interface using time-lapse microscopy and found that endothelial cells begin to round up and detach from the substrate, two characteristics that are reminiscent of apoptosis, 8 hours after contact with the invading tumor cells (fig. S6D). Treatment of an endothelial monolayer with tumor-conditioned medium did not increase apoptosis, suggesting that the apoptotic signal is not soluble (fig. S7A). In contrast, endothelial cell ablation was reduced when proliferation of tumor cells was inhibited with aphidicolin (fig. S7B). Together, these data suggest that proliferation of the tumor cells and direct contact between endothelial and tumor cells are required for endothelial cell ablation to occur.
Because SB431542 exerted its effects in both endothelial and PDAC cells, we first used this system to investigate whether ALK4, ALK5, or ALK7 in endothelial cells played a role in endothelial ablation. We genetically deleted receptors ALK4, ALK5, and ALK7 in the endothelial cells using CRISPR-Cas9 technology and plated wild-type PD7591 and ALK4, ALK5, or ALK7 knockout human umbilical vein endothelial cells (HUVECs) in the 2D patterned coculture invasion assay. Unexpectedly, the quantified invasion area of PD7591 showed no significant difference between ALK4, ALK5, or ALK7 knockout and scramble HUVEC conditions (fig. S7, C to E). Knockout of ALK4, ALK5, and ALK7 also did not affect the population doubling time of endothelial cells (fig. S7F). These data strongly suggested the involvement of the TGF-β superfamily receptors in pancreatic cancer cells rather than in endothelial cells.
When ALK7 was knocked out in PD7591 cells, cellular invasion and endothelial ablation of PD7591 in 2D patterned cocultures (Fig. 4C and fig. S7H) were significantly reduced, while CRISPR-mediated knockout of ALK5 in PD7591 cells did not reduce endothelial ablation (fig. S7I). Knockout of ALK4 was not possible in these cancer cells despite multiple attempts with different guide RNA sequences, suggesting a requirement for ALK4 in these cells. Knockout of ALK7, but not ALK5, significantly prolonged the doubling time of PD7591 (fig. S7G), further supporting the notion that proliferation of tumor cells is likely involved in endothelial cell ablation.
Next, we sought to identify the ligand for the ALK7 signaling, as ALK7 can be activated via different ligands, such as nodal and activin. The major subunits of activin (inhibin βA and βB), in particular, were found to be highly up-regulated in the PDAC cells as compared to the normal mouse pancreatic ductal epithelial cells (Fig. 4D). We then exposed PDAC cells to follistatin, a naturally occurring activin inhibitor (25), in our 2D patterned coculture and in our 3D organotypic model. Inhibition of activin resulted in a significant reduction in endothelial ablation in 2D (Fig. 4E) and in a reduction of vessel replaced area in the 3D organotypic model (Fig. 4F and fig. S8, C and D), suggesting the involvement of activin in ALK7 activation during the process of endothelial ablation.
To further confirm the role of ALK7 in endothelial ablation, we knocked out ALK7 expression in PD7591 and subcutaneously implanted these tumor cells into mice. Tumors were allowed to grow for 2 weeks before they were resected. These tumors exhibited slower growth compared to control tumors (fig. S5E). Two weeks after tumor implantation, the tumors were resected and examined for vessel density and apoptotic endothelial cells. The ALK7-deficient tumors displayed a significantly higher vessel density and less apoptotic endothelial cells within the tumor mass as compared to scramble-treated control mice (Fig. 4G). Together, our in vitro and in vivo findings affirmed a role for ALK7 in mediating endothelial ablation during tumor invasion in PDAC.
DISCUSSION
In contrast to many tumors that are highly angiogenic, PDAC is poorly vascularized (7, 26), a paradoxical finding in light of the fact that pancreatic cancer cells are also known to express and secrete a plethora of proangiogenic factors (27). Using our 3D organotypic model to recapitulate invasion of PDAC tumor cells to blood vessels, we observed that PDAC tumor cells rapidly penetrated the lumen of blood vessels and ablated the endothelial cells, ultimately leaving lumens lined by tumor cells. We refer to this process as endothelial ablation. Furthermore, through histologic costaining of vascular basement membranes, endothelial cells, and PDAC cells within mouse tumor models, we observed that endothelial ablation by PDAC tumors occurs in the in vivo setting. Our study is consistent with histologic studies of PDAC patient tumors, which have reported luminal walls of arteriolar vessels that were lined by cuboidal epithelial cells in place of normal endothelium (8–10). Our study suggests that one factor contributing to the apparent microvascular hypovascularity of PDAC is not a result of reduced angiogenesis per se, but instead a result of endothelial ablation triggered by the invading tumor cells. These invading tumor cells exhibited a faster growth rate than the endothelial cells and eventually led to apoptosis in tumor endothelial cells and subsequently diminished the number of functional blood vessels in the PDAC tumor microenvironment. Perhaps, our observation of tumor endothelial cell ablation in PDAC fits a classical concept of cell-cell competition, which describes clearance of “loser cells” by “winner cells” based on their fitness differences within the local environment such as their cellular division potential (28).
The ability of PDAC tumors to rapidly invade into the vessel, ablate the endothelium, and occupy the vessel lumen could explain the high rate of circulating tumor cells and metastatic load of PDAC. A similar mechanism, where tumor cells ablate the stromal cells, has been reported in a study of ovarian cancer (29). In that study, after ovarian cancer spheroids were plated on a monolayer of endometrial cells, the ovarian cancer cells began to spread over the endometrial monolayer and displaced the endometrial cells. A histologic study of ovarian cancer in human patients also reported the absence of the endometrium due to the penetration of ovarian cancer cells into the endometrium (30). In the context of these studies, our work suggests that carcinoma cells have the capacity to clear and ablate luminal compartments within the tumor microenvironment. It would be interesting to examine this phenomenon of endothelial ablation or stromal ablation in other types of cancer.
Apart from proposing a possible mechanism for the apparent hypovascularity in PDAC, our study unveils a crucial role for activin-ALK7 signaling to contribute to endothelial ablation. Although the functional roles of TGF-β and TGF-β receptor signaling have been investigated in cancer progression, little is known about the contribution of activin and ALK7 to pancreatic cancer. In ovarian cancer, hepatoma, and breast cancer, ALK7 serves as a tumor-suppressive receptor to induce apoptosis in cancer cells or prevent cancer progression (31–33). In contrast, overexpression of ALK7 has been reported to be a marker for poor prognosis in gallbladder cancer (34). ALK7 is a receptor for both nodal and activin (35). Nodal has been reported to mediate vascular mimicry in melanomas, wherein cancer cells formed perfused tubular structures and coexpressed endothelial cell markers (36). Additionally, nodal has also been found to initiate cancer stem cells in pancreatic cancer (37). In contrast, the role of activin in tumor progression has remained largely unappreciated, although activin levels in plasma have been associated with poor prognosis (38) and are overexpressed in multiple human pancreatic cancer cells (39). These reports and our findings together suggest that further investigations into the role of the activin-ALK7 axis in PDAC progression are warranted.
In this study, we use a 3D organotypic model to demonstrate that tumor–endothelial cell interactions not only are restricted to intravasation, extravasation, and vascular mimicry (36, 40) but also include the process of endothelial ablation within the blood vessels, a process that requires further study. Our simple model of PDAC and blood vessels provided sufficient complexity to reveal this process, yet allowed us to introduce genetic and spatiotemporal control to isolate signaling pathways involved for each cell type. Going forward, inclusion of vascular mural cells, and immune cells, or varying the composition of extracellular matrices could be introduced in this engineered 3D organotypic model to more faithfully study the roles of the tumor microenvironments during tumor progression in vitro. Although existing in vivo mouse models provide great opportunities to capture the progression of cancer, dissecting the molecular mechanisms and cell-cell interactions is often difficult because of the complexity of in vivo models. Thus, as demonstrated here, 3D organotypic models provide an important complement to understand these complex cellular interactions with more mechanistic insight.
MATERIALS AND METHODS
Our device was microfabricated as previously described (18). Briefly, the polydimethylsiloxane (PDMS) device was assembled by bonding a bilayer PDMS gasket on top of a glass coverslip. Rat tail collagen 1 (Corning) was prepared at 2.5 mg/ml following the manufacturer’s instructions and was injected into the PDMS device to surround two 300-μm acupuncture needles (Hwato). After collagen polymerizes, acupuncture needles were withdrawn. HUVECs (Lonza) were seeded into one of the channels, while pancreatic cancer cells were seeded into the other channel. FBS (Atlanta Biologicals) was added at 10% (v/v) concentration into the HUVEC channel to generate a gradient of chemoattractants to trigger migration of pancreatic cancer.
KrasLSL-G12D; p53L/+; Pdx1-cre; Rosa26YFP/YFP (KPCY) tissues were a gift from B.Z.S.’s laboratory (University of Pennsylvania). The model has been described previously (5). Briefly, mice were palpated and examined for evidence of morbidity twice per week. Tumor-bearing animals were sacrificed when morbid, and primary pancreatic tumors were collected from mice aged 14 to 30 weeks. Tissues were flash-frozen in OCT (optimal cutting temperature) and subsequently processed for immunofluorescent staining.
Detailed explanations of the materials and methods used in this study can be found in the Supplementary Materials.
Supplementary Material
Acknowledgments
We wish to thank A. Rhim, N. Aiello, S. Yuan, and B. Bakir for providing cell lines and advice. We thank J. Yang for technical assistance and T. Mirabella for helpful discussions. Funding: This work was supported, in part, by grants from the NIH (EB00262, UH3EB017103, and UC4DK104196). E.L. acknowledges financial support from a LE&RN postdoctoral grant from Lymphatic Education and Research Network and a BU-CTSI grant (TL1TR001410) from the National Center for Advancing Translational Sciences at the NIH. J.J.-K.L. acknowledges financial support from the Harvard Ludwig Center. Author contributions: D.-H.T.N. and E.L. designed and performed experiments. S.A., R.J.N., A.W., J.J.-K.L., and J.E. performed the experiments and data analysis. D.-H.T.N., E.L., J.E., B.Z.S., and C.S.C. wrote the manuscript. B.Z.S. and C.S.C. supervised the project. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/8/eaav6789/DC1
Fig. S1. Screening for chemoattractants for pancreatic cancer cell invasions and characterization of gradient in the 3D organotypic model.
Fig. S2. PD7591 in the biomimetic duct channel proliferated in response to the FBS gradient introduced from the biomimetic blood vessel.
Fig. S3. Vascular invasion and endothelial ablation were observed in multiple primary murine PDAC cell lines and a human pancreatic cancer cell line in our 3D biomimetic PDAC-on-a-chip model.
Fig. S4. Staining for cleaved caspase-3 and collagen IV (Col IV) in vivo and in 3D organotypic model.
Fig. S5. Tumor growth rate and caspase signal in endothelial cells in subcutaneous tumor implantation model.
Fig. S6. Inhibition of the TGF-β receptor signaling pathway reduced the endothelial ablation in human pancreatic cancer cell lines in 2D patterned coculture invasion assay and caspase staining in 2D patterned coculture.
Fig. S7. Examination of ALK4, ALK5, and ALK7 in endothelial cells and PD7591 in endothelial cell ablation.
Fig. S8. Representative images of vessel area replaced by tumor cells in 3D organotypic model.
Fig. S9. Representative images of tumor cell pattern in 2D coculture pattern assays.
Fig. S10. Schematics describing how CD31 signal intensity and cleaved caspase-3 signal intensity were measured for quantification.
Movie S1. 3D rendering of confocal image z-stack showed that YFP PD7591 (in green) invaded toward the biomimetic blood vessel (stained with CD31 in red) and wrapped around the blood vessel.
Movie S2. YFP PD7591 (in green) invaded toward the biomimetic blood vessel (stained with CD31 in red).
Movie S3. YFP PD7591 (in green) and HUVECs (in phase) were plated in 2D patterned coculture invasion assay in the presence of vehicle control (dimethyl sulfoxide).
Movie S4. YFP PD7591 (in green) and HUVECs (in phase) were plated in 2D patterned coculture invasion assay in the presence of 5 μM SB431542.
Table S1. Oligo sequences for CRISPR.
Table S2. Primer sequences for qPCR.
Supplementary Methods
REFERENCES AND NOTES
- 1.Steeg P. S., Targeting metastasis. Nat. Rev. Cancer 16, 201–218 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gupta G. P., Massagué J., Cancer metastasis: Building a framework. Cell 127, 679–695 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Tuveson D. A., Neoptolemos J. P., Understanding metastasis in pancreatic cancer: A call for new clinical approaches. Cell 148, 21–23 (2012). [DOI] [PubMed] [Google Scholar]
- 4.Rhim A. D., Thege F. I., Santana S. M., Lannin T. B., Saha T. N., Tsai S., Maggs L. R., Kochman M. L., Ginsberg G. G., Lieb J. G., Chandrasekhara V., Drebin J. A., Ahmad N., Yang Y.-X., Kirby B. J., Stanger B. Z., Detection of circulating pancreas epithelial cells in patients with pancreatic cystic lesions. Gastroenterology 146, 647–651 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rhim A. D., Mirek E. T., Aiello N. M., Maitra A., Bailey J. M., McAllister F., Reichert M., Beatty G. L., Rustgi A. K., Vonderheide R. H., Leach S. D., Stanger B. Z., EMT and dissemination precede pancreatic tumor formation. Cell 148, 349–361 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rehders A., Stoecklein N. H., Güray A., Riediger R., Alexander A., Knoefel W. T., Vascular invasion in pancreatic cancer: Tumor biology or tumor topography? Surgery 152, S143–S151 (2012). [DOI] [PubMed] [Google Scholar]
- 7.Craven K. E., Gore J., Korc M., Overview of pre-clinical and clinical studies targeting angiogenesis in pancreatic ductal adenocarcinoma. Cancer Lett. 381, 201–210 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hong S.-M., Goggins M., Wolfgang C. L., Schulick R. D., Edil B. H., Cameron J. L., Handra-Luca A., Herman J. M., Hruban R. H., Vascular invasion in infiltrating ductal adenocarcinoma of the pancreas can mimic pancreatic intraepithelial neoplasia: A histopathologic study of 209 cases. Am. J. Surg. Pathol. 36, 235–241 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bandyopadhyay S., Basturk O., Coban I., Thirabanjasak D., Liang H., Altinel D., Adsay N. V., Isolated solitary ducts (naked ducts) in adipose tissue: A specific but underappreciated finding of pancreatic adenocarcinoma and one of the potential reasons of understaging and high recurrence rate. Am. J. Surg. Pathol. 33, 425–429 (2009). [DOI] [PubMed] [Google Scholar]
- 10.Liang H., Basturk O., Bandyopadhyay S., Altinel D., Adsay N. V., Pancreatic adenocarcinoma and its mimickers: Traps in diagnosis. Diagn. Histopathol. 14, 275–283 (2008). [Google Scholar]
- 11.Lee E., Song H.-H. G., Chen C. S., Biomimetic on-a-chip platforms for studying cancer metastasis. Curr. Opin. Chem. Eng. 11, 20–27 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tummala P., Junaidi O., Agarwal B., Imaging of pancreatic cancer: An overview. J. Gastrointest. Oncol. 2, 168–174 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zheng Y., Chen J., Craven M., Choi N. W., Totorica S., Diaz-Santana A., Kermani P., Hempstead B., Fischbach-Teschl C., López J. A., Stroock A. D., In vitro microvessels for the study of angiogenesis and thrombosis. Proc. Natl. Acad. Sci. U.S.A. 109, 9342–9347 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jeon J. S., Bersini S., Gilardi M., Dubini G., Charest J. L., Moretti M., Kamm R. D., Human 3D vascularized organotypic microfluidic assays to study breast cancer cell extravasation. Proc. Natl. Acad. Sci. U.S.A. 112, 214–219 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zervantonakis I. K., Hughes-Alford S. K., Charest J. L., Condeelis J. S., Gertler F. B., Kamm R. D., Three-dimensional microfluidic model for tumor cell intravasation and endothelial barrier function. Proc. Natl. Acad. Sci. U.S.A. 109, 13515–13520 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee H., Park W., Ryu H., Jeon N. L., A microfluidic platform for quantitative analysis of cancer angiogenesis and intravasation. Biomicrofluidics 8, 054102 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moya M. L., Hsu Y.-H., Lee A. P., Hughes C. C. W., George S. C., In vitro perfused human capillary networks. Tissue Eng. Part C Methods 19, 730–737 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nguyen D.-H. T., Stapleton S. C., Yang M. T., Cha S. S., Choi C. K., Galie P. A., Chen C. S., Biomimetic model to reconstitute angiogenic sprouting morphogenesis in vitro. Proc. Natl. Acad. Sci. U.S.A. 110, 6712–6717 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ehsan S. M., Welch-Reardon K. M., Waterman M. L., Hughes C. C. W., George S. C., A three-dimensional in vitro model of tumor cell intravasation. Integr. Biol. 6, 603–610 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sobrino A., Phan D. T. T., Datta R., Wang X., Hachey S. J., Romero-Lopez M., Gratton E., Lee A. P., George S. C., Hughes C. C. W., 3D microtumors in vitro supported by perfused vascular networks. Sci. Rep. 6, 31589 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chung M., Ahn J., Son K., Kim S., Jeon N. L., Biomimetic model of tumor microenvironment on microfluidic platform. Adv. Healthc. Mater. 6, 1700196 (2017). [DOI] [PubMed] [Google Scholar]
- 22.Westphalen C. B., Olive K. P., Genetically engineered mouse models of pancreatic cancer. Cancer J. 18, 502–510 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Padua D., Massagué J., Roles of TGFβ in metastasis. Cell Res. 19, 89–102 (2009). [DOI] [PubMed] [Google Scholar]
- 24.Inman G. J., Nicolás F. J., Callahan J. F., Harling J. D., Gaster L. M., Reith A. D., Laping N. J., Hill C. S., SB-431542 is a potent and specific inhibitor of transforming growth factor-β superfamily type I activin receptor-like kinase (ALK) receptors ALK4, ALK5, and ALK7. Mol. Pharmacol. 62, 65–74 (2002). [DOI] [PubMed] [Google Scholar]
- 25.Harrington A. E., Morris-Triggs S. A., Ruotolo B. T., Robinson C. V., Ohnuma S.-i., Hyvönen M., Structural basis for the inhibition of activin signalling by follistatin. EMBO J. 25, 1035–1045 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Olive K. P., Jacobetz M. A., Davidson C. J., Gopinathan A., McIntyre D., Honess D., Madhu B., Goldgraben M. A., Caldwell M. E., Allard D., Frese K. K., Denicola G., Feig C., Combs C., Winter S. P., Ireland-Zecchini H., Reichelt S., Howat W. J., Chang A., Dhara M., Wang L., Ruckert F., Grutzmann R., Pilarsky C., Izeradjene K., Hingorani S. R., Huang P., Davies S. E., Plunkett W., Egorin M., Hruban R. H., Whitebread N., McGovern K., Adams J., Iacobuzio-Donahue C., Griffiths J., Tuveson D. A., Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 324, 1457–1461 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Korc M., Pathways for aberrant angiogenesis in pancreatic cancer. Mol. Cancer 2, 8 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gogna R., Shee K., Moreno E., Cell competition during growth and regeneration. Annu. Rev. Genet. 49, 697–718 (2015). [DOI] [PubMed] [Google Scholar]
- 29.Davidowitz R. A., Selfors L. M., Iwanicki M. P., Elias K. M., Karst A., Piao H., Ince T. A., Drage M. G., Dering J., Konecny G. E., Matulonis U., Mills G. B., Slamon D. J., Drapkin R., Brugge J. S., Mesenchymal gene program-expressing ovarian cancer spheroids exhibit enhanced mesothelial clearance. J. Clin. Invest. 124, 2611–2625 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Witz C. A., Monotoya-Rodriguez I. A., Schenken R. S., Whole explants of peritoneum and endometrium: A novel model of the early endometriosis lesion. Fertil. Steril. 71, 56–60 (1999). [DOI] [PubMed] [Google Scholar]
- 31.Xu G., Zhou H., Wang Q., Auersperg N., Peng C., Activin receptor-like kinase 7 induces apoptosis through up-regulation of Bax and down-regulation of Xiap in normal and malignant ovarian epithelial cell lines. Mol. Cancer Res. 4, 235–246 (2006). [DOI] [PubMed] [Google Scholar]
- 32.Kim B.-C., van Gelder H., Kim T. A., Lee H.-J., Baik K. G., Chun H. H., Lee D. A., Choi K. S., Kim S.-J., Activin receptor-like kinase-7 induces apoptosis through activation of MAPKs in a Smad3-dependent mechanism in hepatoma cells. J. Biol. Chem. 279, 28458–28465 (2004). [DOI] [PubMed] [Google Scholar]
- 33.Zeng F., Xu G., Zhou T., Yang C., Wang X., Peng C., Zhou H., Reduced expression of activin receptor-like kinase 7 in breast cancer is associated with tumor progression. Med. Oncol. 29, 2519–2526 (2012). [DOI] [PubMed] [Google Scholar]
- 34.Li J., Yang Z., Zou Q., Yuan Y., Li J., Liang L., Zeng G., Chen S., PKM2 and ACVR 1C are prognostic markers for poor prognosis of gallbladder cancer. Clin. Transl. Oncol. 16, 200–207 (2014). [DOI] [PubMed] [Google Scholar]
- 35.Wakefield L. M., Hill C. S., Beyond TGFβ: Roles of other TGFβ superfamily members in cancer. Nat. Rev. Cancer 13, 328–341 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Postovit L.-M., Margaryan N. V., Seftor E. A., Hendrix M. J. C., Role of nodal signaling and the microenvironment underlying melanoma plasticity. Pigment Cell Melanoma Res. 21, 348–357 (2008). [DOI] [PubMed] [Google Scholar]
- 37.Lonardo E., Hermann P. C., Mueller M. T., Huber S., Balic A., Miranda-Lorenzo I., Zagorac S., Alcala S., Rodriguez-Arabaolaza I., Ramirez J. C., Torres-Ruiz R., Garcia E., Hidalgo M., Cebrián D. A., Heuchel R., Löhr M., Berger F., Bartenstein P., Aicher A., Heeschen C., Nodal/Activin signaling drives self-renewal and tumorigenicity of pancreatic cancer stem cells and provides a target for combined drug therapy. Cell Stem Cell 9, 433–446 (2011). [DOI] [PubMed] [Google Scholar]
- 38.Togashi Y., Kogita A., Sakamoto H., Hayashi H., Terashima M., de Velasco M. A., Sakai K., Fujita Y., Tomida S., Kitano M., Okuno K., Kudo M., Nishio K., Activin signal promotes cancer progression and is involved in cachexia in a subset of pancreatic cancer. Cancer Lett. 356, 819–827 (2015). [DOI] [PubMed] [Google Scholar]
- 39.Kleeff J., Ishiwata T., Friess H., Buchler M. W., Korc M., Concomitant over-expression of activin/inhibin β subunits and their receptors in human pancreatic cancer. Int. J. Cancer 77, 860–868 (1998). [DOI] [PubMed] [Google Scholar]
- 40.Reymond N., d'Água B. B., Ridley A. J., Crossing the endothelial barrier during metastasis. Nat. Rev. Cancer 13, 858–870 (2013). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/8/eaav6789/DC1
Fig. S1. Screening for chemoattractants for pancreatic cancer cell invasions and characterization of gradient in the 3D organotypic model.
Fig. S2. PD7591 in the biomimetic duct channel proliferated in response to the FBS gradient introduced from the biomimetic blood vessel.
Fig. S3. Vascular invasion and endothelial ablation were observed in multiple primary murine PDAC cell lines and a human pancreatic cancer cell line in our 3D biomimetic PDAC-on-a-chip model.
Fig. S4. Staining for cleaved caspase-3 and collagen IV (Col IV) in vivo and in 3D organotypic model.
Fig. S5. Tumor growth rate and caspase signal in endothelial cells in subcutaneous tumor implantation model.
Fig. S6. Inhibition of the TGF-β receptor signaling pathway reduced the endothelial ablation in human pancreatic cancer cell lines in 2D patterned coculture invasion assay and caspase staining in 2D patterned coculture.
Fig. S7. Examination of ALK4, ALK5, and ALK7 in endothelial cells and PD7591 in endothelial cell ablation.
Fig. S8. Representative images of vessel area replaced by tumor cells in 3D organotypic model.
Fig. S9. Representative images of tumor cell pattern in 2D coculture pattern assays.
Fig. S10. Schematics describing how CD31 signal intensity and cleaved caspase-3 signal intensity were measured for quantification.
Movie S1. 3D rendering of confocal image z-stack showed that YFP PD7591 (in green) invaded toward the biomimetic blood vessel (stained with CD31 in red) and wrapped around the blood vessel.
Movie S2. YFP PD7591 (in green) invaded toward the biomimetic blood vessel (stained with CD31 in red).
Movie S3. YFP PD7591 (in green) and HUVECs (in phase) were plated in 2D patterned coculture invasion assay in the presence of vehicle control (dimethyl sulfoxide).
Movie S4. YFP PD7591 (in green) and HUVECs (in phase) were plated in 2D patterned coculture invasion assay in the presence of 5 μM SB431542.
Table S1. Oligo sequences for CRISPR.
Table S2. Primer sequences for qPCR.
Supplementary Methods