

Abstract
Lipids, either endogenously synthesized or exogenous, have been linked to human cancer. Here we found that PML is frequently co-deleted with PTEN in metastatic human prostate cancer (CaP). We demonstrated that conditional inactivation of Pml in the mouse prostate morphs indolent Pten-null tumors into lethal metastatic disease. We identified MAPK reactivation, subsequent hyperactivation of an aberrant SREBP prometastatic lipogenic program, and a distinctive lipidomic profile as key characteristic features of metastatic Pml and Pten double-null CaP. Furthermore, targeting SREBP in vivo by fatostatin blocked both tumor growth and distant metastasis. Importantly, a high-fat diet (HFD) induced lipid accumulation in prostate tumors and was sufficient to drive metastasis in a nonmetastatic Pten-null mouse model of CaP, and an SREBP signature was highly enriched in metastatic human CaP. Thus, our findings uncover a prometastatic lipogenic program and lend direct genetic and experimental support to the notion that a Western HFD can promote metastasis.
Life Sciences Reporting Summary.
Further information on experimental design is available in the Life Sciences Reporting Summary.
Although localized CaP is highly curable, metastatic CaP remains invariably fatal1. Although it has been widely postulated that a Western diet can promote CaP progression2,3, direct evidence supporting a strong association between dietary lipids and CaP is still lacking4. Indeed, the rates of cancer mortality associated with metastatic disease are much higher in Western countries for many cancer types, including CaP, and they correlate with lifestyle factors such as diet2,3,5. Moreover, the progression to metastasis is a pivotal event influencing patient outcomes and the available therapeutic options. Thus, understanding the molecular events that underlie progression to metastasis, at both the genetic and environmental levels, has the potential to substantially improve therapeutic options and facilitate preventive interventions. However, to date, CaP metastasis has proven to be particularly challenging to model in vivo, and the progression to metastasis from either primary indolent or advanced-stage disease is rarely observed in most genetically engineered mouse models (GEMMs) of CaP6.
PTEN is among the most frequently lost or mutated tumor-suppressor genes (TSGs) in human cancer7,8. Partial loss of PTEN occurs early in tumorigenesis and is present in up to 70% of localized CaP (LPC)9–11, whereas complete loss of PTEN is linked to metastatic castration-resistant CaP (mCRPC)12–15. PTEN inactivation facilitates aberrant activation of the phosphoinositide-3-kinase (PI3K)-AKT pathway16. Intriguingly, studies from Pten-knockout mouse models have shown that complete inactivation of Pten alone in the mouse prostate leads to indolent tumors with minimally invasive features after a long latency17–19. We and others have previously shown that TSGs induced after Pten loss serve as failsafe mechanisms that restrict cancer progression and that inactivation of such barriers promotes indolent Pten-null tumorigenesis20,21. Therefore, identifying an additional event that cooperates with PTEN loss in driving metastatic progression is crucial to be able to model and study the lethal stage of CaP.
The Ras–Raf–MEK–ERK mitogen-activated protein kinase (MAPK) signaling cascade is another pathway that is often aberrantly activated in advanced metastatic CaP15,22,23. However, little is known about the underlying molecular mechanisms leading to MAPK activation, because genetic alterations in components of MAPK signaling are rare in human CaP14,24–29. Despite the low frequency of Ras and Raf mutations in CaP, recent studies have shown that oncogenic K-Ras p.Gly12Asp or B-Raf p.Val600Glu alterations, which activate MAPK signaling, cooperate with Pten loss in driving metastatic progression of CaP in GEMMs30,31. These findings underscore the critical role of MAPK signaling in cancer progression but do not address how this cascade is activated and what the key cellular events are in most cases of metastatic CaP
Here we identified an sterol regulatory element-binding protein (SREBP) prometastatic lipogenic program as a key downstream effector of MAPK activation. This program is opposed by the PML tumor suppressor (a potent failsafe mechanism that is genetically evaded in human cancer) and can be overcome by a HFD in vivo. Our results thus have potential implications in the prevention and treatment of metastatic CaP with stringent dietary regimens in combination with targeting of lipogenic enzymes.
Results
Concomitant loss of PTEN and PML expression in metastatic human CaP.
To identify cooperating genomic alterations that may be associated with PTEN loss in metastatic human CaP, we took advantage of a recent array-based comparative genomic hybridization dataset14 of 59 LPC and 35 mCRPC samples and evaluated the frequencies of co-deletion of PTEN with 58 high-confidence TSGs32. In this dataset, PTEN was lost in 14% of LPC and 66% of mCRPC samples (Fig. 1a). The frequencies of co-deletion of the other TSGs with PTEN in metastatic disease ranged from 0% to 50% (Supplementary Fig. 1a). Among the top 25 TSGs co-deleted with PTEN, PML was lost in 31% of mCRPC samples but remained intact in LPC samples (Fig. 1b) and was among several TSGs that were co-deleted with PTEN solely in metastatic disease (Supplementary Fig. 1a). Concomitant deletion of PTEN and PML occurred in 20% of mCRPC and was significantly associated with metastatic disease (Fig. 1c), thus suggesting that PML might suppress metastatic CaP in coordination with PTEN. To validate these findings, we analyzed a recent whole-exome sequencing dataset25 of 150 mCRPC samples. PTEN and PML were lost in 38% and 30% of the mCRPC samples, respectively and co-deletion of PTEN and PML occurred in 11% of the mCRPC samples (Supplementary Fig. 1b,c). In contrast to frequent homozygous PTEN deletion in mCRPC (Fig. 1a and Supplementary Fig. 1b), homozygous PML deletion was less common than hemizygous PML deletion, which was frequently observed and was focal in many cases (Fig. 1b and Supplementary Fig. 1b,d).
Fig. 1 |. Co-loss of PTEN and PML expression in advanced and metastatic human CaP.
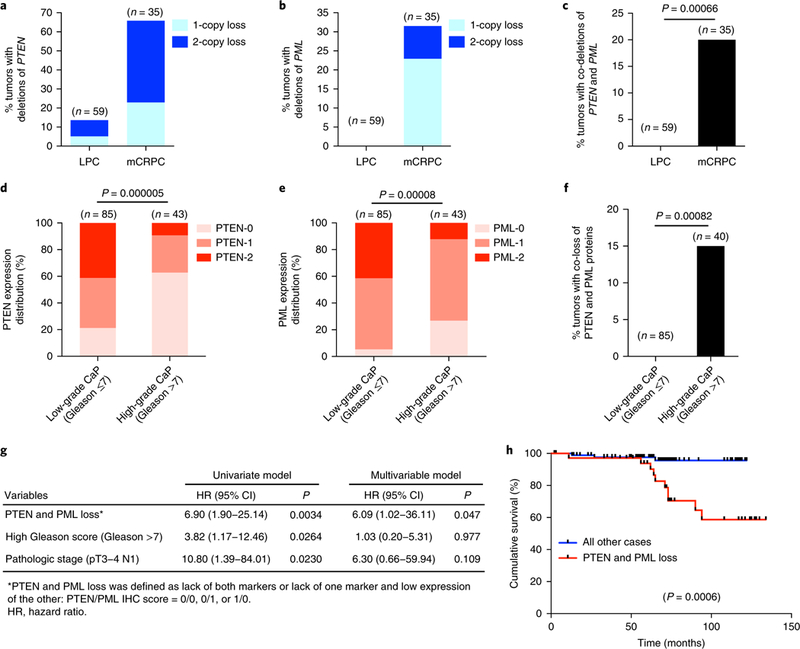
a–c, Bar graph showing the percentage of deletion of PTEN (a), PML (b) or PTEN and PML (c) in LPC and mCRPC samples from the Grasso et al. dataset14. Co-loss events include homozygous, heterozygous or mixed co-loss events. d,e, Bar graph showing the correlation of disease progression with low levels of PTEN (d) or PML (e) protein in primary CaP. The color bar represents the intensity of staining, which was ranked as one of three groups: normal = 2, low = 1 and negative = 0. f, Bar graph showing the percentage of co-loss of PTEN and PML proteins in low-grade and high-grade primary CaP. g, Univariate and multivariable Cox proportional regression analysis of PTEN and PML loss, Gleason score and pathologic stage. h, Kaplan–Meier plot of overall survival data for patients with CaP after radical prostatectomy on the basis of co-loss of PTEN and PML protein. In a–f, n is the number of independent samples. In c and f, Fisher’s exact test (two tailed) was used to determine significance. In d and e, Pearson’s chi-square test was used to determine significance. In h, log-rank test was used to determine significance.
We next evaluated PTEN and PML protein expression in human CaP and sought to identify whether PTEN and PML loss might represent cooperative predictors of overall survival after prostatectomy. Tissue microarray (TMA) analysis was performed in prostatectomy specimens from 144 men with primary CaP (Supplementary Table 1). Loss of PTEN and/or PML was significantly correlated with disease progression (Fig. 1d,e and Supplementary Fig. 1e,f). Complete loss of PTEN and PML at the protein level occurred in 15% of the high-grade CaP samples, but not in the low-grade CaP samples (Fig. 1f). We then applied Cox proportional hazards models to identify risk factors predicting overall survival after prostatectomy. Univariate analysis showed that PTEN and PML loss, Gleason grade (>7 vs. ≤7) and pathologic stage (pT3–4 or N1 vs. pT1–2 N0) were associated with adverse prognosis (Fig. 1g). Importantly, through multivariable analysis, loss of PTEN and PML was confirmed as a statistically significant independent prognostic factor for overall survival (Fig. 1g) and had the most statistically significant power to stratify patient survival time (Fig. 1h and Supplementary Fig. 1g-j). These results further supported a model in which loss of PML and PTEN might cooperatively promote advanced CaP
Pml loss promotes metastatic progression in Pten-null CaP.
The tumor-suppressive function of the PML gene alone or in cooperation with PTEN has, to date, been studied in the context of primary CaP initiation and progression33. However, we postulated that generating conditional Pten and Pml compound inactivation would allow us to conduct a long-term follow-up and model a continuum of lesions beyond minimally invasive Pten-null cancer and to determine whether co-deletion of Pten and Pml would favor metastatic progression. We generated conditional Pmlflox/flox mice (Supplementary Fig. 2a–c) and crossed them with mice carrying Pb-Cre4 and floxed Pten alleles to obtain prostate-epithelium-specific Pten and Pml double-null mice (Ptenpc−/−; Pmlpc−/−).
Immunohistochemistry (IHC) assays showed that Pml protein levels were markedly higher in Ptenpc−/− prostate intraepithelial neoplasia (PIN) than in wild type (WT) prostate epithelium (Fig. 2a and Supplementary Fig. 3a), thus suggesting that Pml might serve as a potential failsafe mechanism restricting cancer progression in Pten-null indolent tumors. Moreover, Pml protein was almost undetectable in Ptenpc−/−; Pmlpc−/− PIN, thus confirming prostate-epithelium-specific inactivation of Pml (Fig. 2a and Supplementary Fig. 3a).
Fig. 2 |. Pml loss renders localized Pten-null tumors lethal and metastatic to lymph nodes.
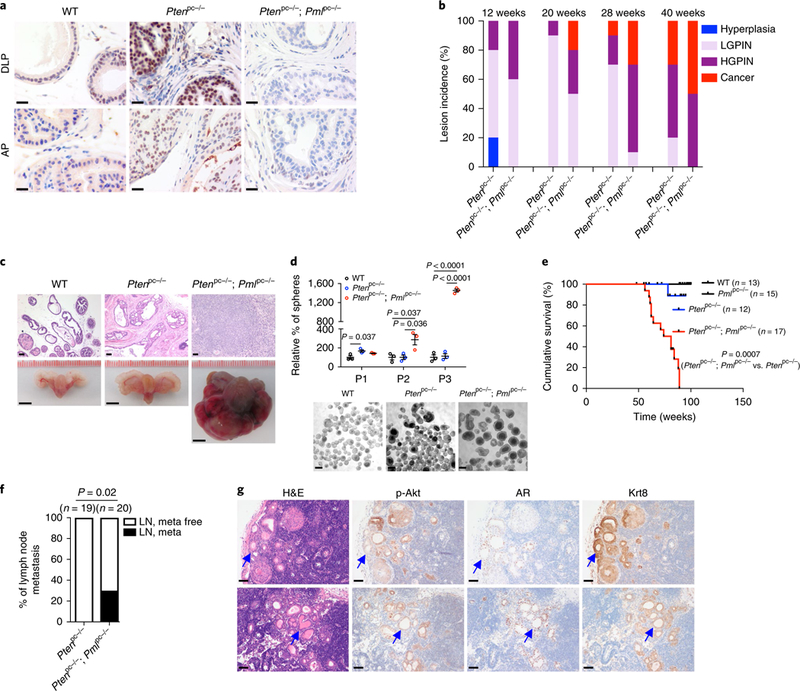
a, IHC staining for Pml in the DLP and anterior prostate (AP) tissues from WT, Ptenpc−/− and Ptenpc−/−; Pmlpc−/− mice at 12 weeks of age. Scale bar, 20 μm. b, Bar graph showing prostate tumor progression in Ptenpc−/− and Ptenpc−/−; Pmlpc−/− mice in different age cohorts (n = 10). The AP was excluded from the analysis because of cystic lesions after the 12-week time point. HGPIN, high-grade PIN; LGPIN, low-grade PIN. c, Hematoxylin-eosin (H&E)-stained mouse prostate tissues and gross anatomy of representative WT, Ptenpc−/− and Ptenpc−/−; Pmlpc−/− urogenital tracts from mice at 13 months of age (n = 10 mice per genotype). Scale bars, 50 ^m (top) and 5 mm (bottom). d, Plot showing the sphere-forming ability of WT, Ptenpc−/− and Ptenpc−/−; Pmlpc−/− prostate epithelia after serial passage. For comparative purposes, the results are shown as percentages relative to the sphere number of the WT (set at 100%) at the passages examined. Representative images of spheres at P1 are shown at bottom. Scale bars, 20 μm. e, Cumulative survival analysis of WT, Pmlpc−/−, Ptenpc−/− and Ptenpc−/−; Pmlpc−/− mice. f, Incidence of lymph node metastasis (meta) in cohorts of Ptenpc−/− and Ptenpc−/−; Pmlpc−/− mice. g, H&E and IHC staining of lumbar lymph node metastases from two representative Ptenpc−/−; Pmlpc−/− mice. Arrows indicate metastases. Scale bars, 50 μm. p, phospho. In d, the results of one representative experiment are shown (n = 5 experiments). Data are from three independent cultures and are mean±s.e.m. Student’s t test (two tailed) was used to determine significance. In e and f, n is the number of mice. In e, log-rank test was used to determine signifcance. In f, Fisher’s exact test (two tailed) was used to determine significance.
We next evaluated the consequences of Pml loss for Pten-null prostate tumorigenesis in mice. Histopathological analyses of prostate tumors were performed in cohorts of Ptenpc−/− mice and Ptenpc−/−; Pmlpc−/− mice at 12 to 40 weeks of age. At 12 weeks of age, Ptenpc−/− mice and Ptenpc−/−; Pmlpc−/− mice displayed low-to high-grade PIN, and higher penetrance of high-grade PIN was observed in Ptenpc−/−; Pmlpc−/− mice (Fig. 2b). In agreement with previously reported findings20, Ptenpc−/− mice developed invasive prostate adenocarcinoma after a 28-to 40-week latency and, in sharp contrast, Ptenpc−/−; Pmlpc−/− mice developed invasive prostate adenocarcinoma as early as 20 weeks and showed increasing penetrance over 28 and 40 weeks (Fig. 2b and Supplementary Fig. 3b). Furthermore, Ptenpc−/−; Pmlpc−/− tumors progressed to aggressive, poorly differentiated adenocarcinoma with little or no glandular structure and with focal features of sarcomatoid carcinoma with high-grade pleomorphic spindle cells at 52–60 weeks, whereas age-matched Ptenpc−/− tumors remained indolent (Fig. 2c and Supplementary Fig. 3c). Culture and serial passage of prostate spheres derived from WT, Ptenpc−/− and Ptenpc−/−; Pmlpc−/− mice showed that Ptenpc−/−; Pmlpc−/− prostate epithelial cells, compared with WT and Ptenpc−/− cells, had significantly enhanced stem/progenitor self-renewal capacity and growth (Fig. 2d). These differences resulted in drastically impaired survival of the Ptenpc−/−; Pmlpc−/− mice compared with the Ptenpc−/− mice. All Ptenpc−/− mice survived beyond 18 months; in contrast, Ptenpc−/−; Pmlpc−/− mice succumbed to localized disease, probably because of bladder obstruction and renal failure, or were euthanized because of extensive tumor burden from 13 months of age. Although 21% (4 out of 19) of the Pmlpc−/− mice exhibited low-grade PIN in ventral (VP) and dorsolateral (DLP) prostates after 12 months of age, none died of the disease (Fig. 2e and Supplementary Fig. 3d,e). These results supported the notion that inactivation of Pml can favor both CaP initiation and progression.
Critically, Pml loss can promote metastasis in Pten-null CaP Although Ptenpc−/− mice have not been found to develop distant metastasis in an 18-month follow-up20, we observed that 30% of Ptenpc−/−; Pmlpc−/− mice developed lumbar lymph node metastasis at 13–15 months of age (Fig. 2f). Histological and molecular pathological analysis indicated that these metastases resembled primary prostate tumors and showed high levels of phospho-Akt as well as nuclear androgen receptor (AR) and CK8 staining (Fig. 2g and Supplementary Fig. 3f).
PML loss reactivates MAPK signaling in PTEN-null cells.
To determine how Pml inactivation might affect Pten-null tumorigenesis and favor a prometastatic switch, we analyzed the expression of CaP-relevant metastatic pathways21,30,31,34. In agreement with previous findings33, compared with Ptenpc−/− prostates, Ptenpc−/−; Pmlpc−/− prostates displayed up to a 1.5-fold increase in the level of Akt phosphorylation, and in stark contrast, a 19-to 35-fold increase in the level of Erk phosphorylation (Fig. 3a). In line with the notion that PTEN loss or AKT–mechanistic target of rapamycin (mTOR) activation leads to a feedback inhibition on MAPK pathway35,36, Ptenpc−/− prostates displayed even lower level of Erk phosphorylation than did WT prostates (Fig. 3a), thus suggesting that Pml loss triggers relief of feedback inhibition and consequent reactivation of MAPK signaling in Pten-null prostates. IHC assays confirmed that Ptenpc−/−; Pmlpc−/−, compared with Ptenpc−/− prostates, showed similar widespread membrane staining for phospho-Akt, along with rare and scattered nuclear phospho-Akt staining, whereas phospho-Erk staining was markedly greater (Fig. 3b and Supplementary Fig. 3g). In contrast, we observed negligible changes in other pathways involved in metastasis21,34 (Supplementary Fig. 3h). In agreement with the notion that PML is haploinsufficient for some of its tumor-suppressive functions37, Ptenpc−/−; Pmlpc+/− prostates also displayed high levels of Erk phosphorylation (Supplementary Fig. 3i). Collectively, the magnitudes of change in MAPK signaling strongly implicated Pml-loss-induced MAPK reactivation in metastatic progression of Ptenpc−/−; Pmlpc−/− tumors.
Fig. 3 |. PML loss triggers MAPK reactivation in PTEN-null cells.
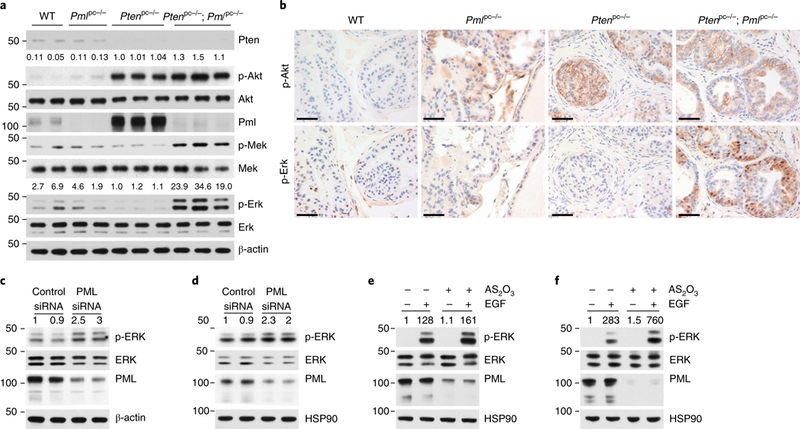
a,b, Immunoblot (a) and IHC (b) analyses of the DLP tissues from WT, Pmlpc−/−, Ptenpc−/− and Ptenpc−/−; Pmlpc−/− mice at 12 weeks of age. Scale bars, 50 μm. c-f, Immunoblot of lysates from LNCaP (c) or PC3 (d) cells transfected with control or PML siRNA for 72 h, and of lysates from LNCaP (e) or PC3 (f) cells pretreated with vehicle or 1 arsenic trioxide (AS2O3) for 12 h, serum starved for 4 h and stimulated with 10 ng/ml EGF for 5 min. In a, numbers above blots indicate the relative ratios to values for Ptenpc−/− mice for phosphoprotein/total protein; in c-f, numbers above blots indicate the relative ratios to values for controls for phosphoprotein/total protein; numbers to left of blots indicate molecular weight (kDa). Uncropped images are shown in Supplementary Fig. 7.
We then investigated the effects of knockdown of PML on MAPK activation via small interfering RNA (siRNA) in the PTEN-null CaP cell lines LNCaP and PC3. Knockdown of PML consistently resulted in MAPK activation (Fig. 3c, d). Moreover, the degradation of PML protein triggered by arsenic trioxide also led to upregulation of epidermal growth factor (EGF)-induced ERK phosphorylation (Fig. 3e, f), thus lending further support to MAPK suppression by PML.
An SREBP-dependent lipogenic program is hyperactivated in Pten and Pml double-null CaP.
To gain insight into which effectors located downstream of this aberrant signaling drive metastatic progression in Ptenpc−/−; Pmlpc−/− tumors, we performed microarray transcriptomic analysis on 12-week-old WT, Ptenpc−/− and Ptenpc−/−; Pmlpc−/− prostates (n = 3) (Supplementary Fig. 4a). We identified 101 genes whose expression was significantly upregulated and 158 genes whose expression was significantly downregulated at least 1.5-fold (P < 0.01) in Ptenpc−/−; Pmlpc−/− prostates compared with Ptenpc−/− prostates (Supplementary Table 2). Gene ontology analysis showed that the three most significantly enriched gene categories in the Ptenpc−/−; Pmlpc−/− signature were ‘cell migration’ (P = 0.0017), ‘cell motility’ (P = 0.0014) and ‘lipid-metabolic process’ (P = 0.007) (Fig. 4a). Although enrichment in genes affecting cell migration and cell motility was consistent with the high metastatic potential of Ptenpc−/−; Pmlpc−/− tumors, the lipid-metabolic-process signature was unexpected, because activation of lipogenesis is an early event in the development of CaP, and the potential role of aberrant lipogenesis in metastasis in vivo remains largely elusive38,39.
Fig. 4 |. An SREBP-dependent lipogenic program is hyperactivated in prostate tumors from Ptenpc−/−; Pmlpc−/− mice.
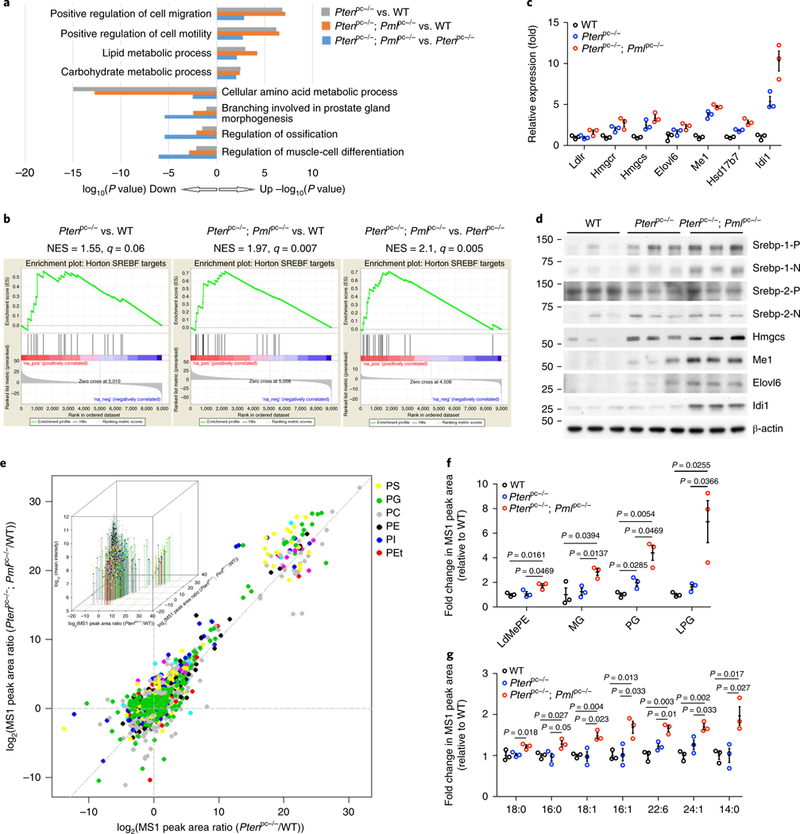
a, Top enriched biological-process categories from Gene Ontology enrichment analysis among WT, Ptenpc−/− and Ptenpc−/−; Pmlpc−/− prostates. b, GSEA enrichment plot for one of the top-scoring gene sets, SREBP targets, among WT, Ptenpc−/− and Ptenpc−/−; Pmlpc−/− prostates. c,d, Validation of the expression changes in SREBP targets through qPCR (c) and immunoblot (d) analyses of the DLP tissues from WT, Ptenpc−/− and Ptenpc−/−; Pmlpc−/− mice at 12 weeks of age. In d, P and N denote the precursor and cleaved nuclear forms, respectively, of SREBP-1 or SREBP-2. Uncropped images are shown in Supplementary Fig. 7. Numbers to left of blot indicate molecular weight (kDa). e, Scatter plot showing the distribution of all 1,743 identifiable lipid ions in prostate tissues from the three mouse genotypes. Each dot represents one lipid ion, and each color represents a class of lipids. Representative lipid classes (6 out of 35) are shown. Inset, same chart with 3D visual effect with z axis plotting the log10(mean intensity) of each lipid ion across the three genotypes. PS, phosphatidylserine; PG, phosphatidylglycerol; PC, phosphatidylcholine; PE, phosphatidylethanolamine; PI, phosphatidylinositol; PEt, phosphatidylethanol. f,g, A statistically significant increase in abundance of various lipid classes (f) or fatty acyl chains (g) in Ptenpc−/−; Pmlpc−/− tumors compared with Ptenpc−/− tumors. The plot represents fold change in relative intensity. LdMePE, lysodimethylphosphatidylethanolamine; MG, monoglyceride; LPG, lysophosphatidylglycerol. In c, the results of one representative experiment are shown (n = 3 experiments). Data are from three independent mice per genotype and are shown as mean± s.e.m. In f and g, n = 3 mice per genotype; data are shown as mean±s.e.m. Student’s t test (two tailed) was used to determine significance.
Additional unbiased gene set enrichment analysis (GSEA) showed that Ptenpc−/−; Pmlpc−/− prostates displayed a significantly upregulated gene set activated by the SREBP family of transcription factors, the master regulator of transcription of genes involved in fatty acid (FA) and cholesterol biosynthesis40 (Fig. 4b, right; Supplementary Fig. 4b and Supplementary Table 3), thus confirming the results of the gene ontology analysis (Fig. 4a). In Ptenpc−/− prostates compared with WT prostates, the lipid-metabolic-process signature and the SREBP signature were also mildly upregulated (Fig. 4a, gray, P < 0.0017 and Fig. 4b, left)—in agreement with the finding that activation of AKT–mTOR–ribosomal protein S6 kinase (S6K) signaling promotes lipogenesis by activating SREBP41,42—but were more extensively enhanced in Ptenpc−/−; Pmlpc−/− prostates (Fig. 4a, orange, P < 0.00005 and Fig. 4b, middle). qPCR analysis confirmed that SREBP-target genes were further upregulated in Ptenpc−/−; Pmlpc−/− prostates than in Ptenpc−/− prostates (Fig. 4c), and western blot assays demonstrated elevated protein expression of Mel, Elovl6, Hmgcs and Idil, four key lipogenesis/cholesterol-biosynthesis enzymes, in Ptenpc−/−; Pmlpc−/− prostate lysates (Fig. 4d). The SREBP family comprises two members: SREBP-1 and SREBP-2. Ptenpc−/−; Pmlpc−/− prostates, compared with WT prostates, displayed higher levels of precursor and nuclear SREBP-1 and, to a lesser extent, nuclear SREBP-2 (Fig. 4d). In contrast, no significant differences in the gene signatures of other lipogenic transcription factors were observed (Supplementary Fig. 4c–f). Therefore, the SREBP pathway is hyperactivated and appears to be the main transcriptional factor responsible for the changes in lipid metabolism in Pten and Pml double-null CaP.
Lipidomic analysis shows that de novo lipogenesis positively correlates with the aggressiveness and metastatic potential of CaP.
To confirm the aberrant alterations in de novo lipogenesis in Ptenpc−/−; Pmlpc−/− tumors, we performed a global lipidomic analysis in 12-week-old WT, Ptenpc−/− and Ptenpc−/−; Pmlpc−/− prostates (n = 3) by using untargeted high-resolution liquid chromatography-tandem mass spectrometry (LC–MS/MS)43–45. These analyses identified 1,743 lipid ions in the mouse prostate (Supplementary Tables 4 and 5), which belonged to 35 classes of lipids (Supplementary Table 6). Among all the identified lipid ions, 127 distinct fatty acyl chains, varying in chain length from 4 to 38 carbons and with 0 to 7 double bonds, were found (Supplementary Table 7). The most abundant lipid classes and fatty acyl chains in the mouse prostate were phosphatidylcholine, sphingomyelin, phosphatidylethanolamine, triglyceride, lysophosphatidylcholine and oleate (C18:1), palmitate (C16:0), linoleate (18:2), arachidonate (20:4) and stearate (C18:0) (Supplementary Fig. 4g,h).
We next performed unsupervised hierarchical clustering to identify the lipid ions with significant changes in abundance among WT, Ptenpc−/− and Ptenpc−/−; Pmlpc−/− prostates. Among the top 70 most significantly differential lipid ions, in agreement with the results of the transcriptome analysis in lipogenic signatures, the abundance of most of the lipid ions was mildly higher in Ptenpc−/− than WT prostates and was more extensively enhanced in Ptenpc−/−; Pmlpc−/− than WT prostates (Supplementary Fig. 4i). To further confirm the alterations in lipid abundance among the three mouse genotypes, we used a scatter plot of the log2 of the MS1 peak-area ratio between Ptenpc−/−; Pmlpc−/− and WT vs. that between Ptenpc−/− and WT, to illustrate the estimated number of down-or upregulated lipid ions while comparing two of the three genotypes of mice at a time44,45. This procedure allowed us to investigate the overall changes in the relative lipid abundance of all 1,743 identified lipid ions (Fig. 4e). Overall, 1,131 out of 1,743 lipid ions had log2(MS1 peak-area ratio between Ptenpc−/− and WT) values greater than zero, whereas 1,261 out of 1,743 lipid ions had log2(MS1 peak-area ratio between Ptenpc−/−; Pmlpc−/− and WT) values greater than zero, thus indicating a prominent upregulation of lipid abundance in both Ptenpc−/− and Ptenpc−/−; Pmlpc−/− prostates compared with WT prostates. Importantly, 1,154 lipid ions were above the diagonal x=y line, thus revealing a further augmentation of lipid abundance in Ptenpc−/−; Pmlpc−/− prostates compared with Ptenpc−/− prostates (Fig. 4e). These data are consistent with the transcriptome analysis showing that aberrantly increased lipogenesis correlates with the aggressiveness and metastatic potential of CaP.
Distinctive alterations in lipid species and lipid saturation in Pten and Pml double-null CaP.
We subsequently examined the qualitative changes in lipid profiles and found that 25 out of 35 lipid classes identified had greater abundance in Ptenpc−/−; Pmlpc−/− prostates than Ptenpc−/− prostates (Supplementary Fig. 4g and Supplementary Table 8). Among them, the increases in the abundance of lysodi-methylphosphatidylethanolamine (LdMePE), monoglyceride (MG), phosphatidylglycerol (PG) and lysophosphatidylglycerol (LPG) were statistically significant (Fig. 4f and Supplementary Fig. 4g and Supplementary Table 8). Among the membrane phospholipids LdMePE, PG and LPG, PG is present at a level of 1–2% in most animal tissues and, together with its hydrolytic product LPG, can serve as the precursor of cardiolipin found in mitochondrial membranes. PG lipids have recently been found to be the most significantly upregulated lipid species in MYC-driven cancer46–48, thus suggesting a possible link between PG lipids and tumorigenesis. Additionally, MG lipids, a class of glycerolipids, can be hydrolyzed by monoacylglycerol lipase and serve as the precursors for synthesis of prometastatic lipid messengers, including lysophosphatidic acid and prostaglandin49.
Elevated levels of saturated fatty acyl chains have been reported to protect cancer cells against oxidative damage by decreasing lipid peroxidation50. We therefore compared the levels of the 30 most abundant fatty acyl chains to assess the alterations in lipid saturation among the three genotypes of mice (Supplementary Fig. 4h and Supplementary Table 9). A total of seven fatty acyl chains had significantly greater abundance in Ptenpc−/−; Pmlpc−/− prostates than in Ptenpc−/− prostates (Fig. 4g and Supplementary Fig. 4h and Supplementary Table 9). Only one was a polyunsaturated FA (22:6), and the other six were saturated and monounsaturated FAs (14:0, 16:0, 18:0, 16:1, 18:1 and 24:1), thus indicating that hyperactivation of lipid metabolism in Ptenpc−/−; Pmlpc−/− prostates leads to higher levels of saturated and monounsaturated fatty acyl chains. These findings are consistent with earlier studies showing that elevated levels of saturated and monounsaturated FAs are associated with aggressive breast cancer51.
SREBP is the downstream target of PML-loss-induced MAPK activation.
Because the levels and transcriptional activity of SREBP are known to be regulated by growth-factor-induced phosphorylation52,53, we tested whether SREBP is a target of the MAPK activation induced by PML loss. PTEN-null CaP cells were cultured in medium supplemented with 10% lipoprotein-deficient serum to limit the availability of exogenous lipids and to increase endogenous synthesis. qPCR analysis showed that treatment of LNCaP cells with the MEK inhibitor U0126 resulted in decreased basal expression of all SREBP targets examined (Fig. 5a). Knockdown of PML led to upregulation of SREBP-target genes. Moreover, U0126 reversed the PML-depletion-induced upregulation of SREBP-target genes (Fig. 5a). Similar results were also obtained in PC3 cells (Fig. 5b). Cell fractionation experiments showed that U0126 strongly down-regulated protein levels of both nuclear SREBP-1 and SREBP-2 and markedly decreased SREBP-target-gene expression (Fig. 5c,d). In contrast, MAPK activation through either knockdown of PML or the presence of constitutively active MEK led to higher levels of nuclear SREBP proteins and of SREBP-target-gene expression. These increases were largely suppressed by U0126 (Fig. 5c,d). We, and others, have previously shown that MAPK signaling inhibits the TSC complex’s tumor-suppressor function to activate Mtor–S6K54,55. In agreement with these results, we found that the levels of both phospho-ERK and phospho-S6K appeared to mirror the changes in SREBP and their target genes. As such, we cannot exclude that SREBP might be an indirect target of MAPK signaling through mTOR–S6K, but we can conclude that PML-loss induced MAPK activation is sufficient to drive an SREBP-dependent lipogenic program.
Fig. 5 |. SREBP is the downstream target of PML-loss-induced MAPK activation.
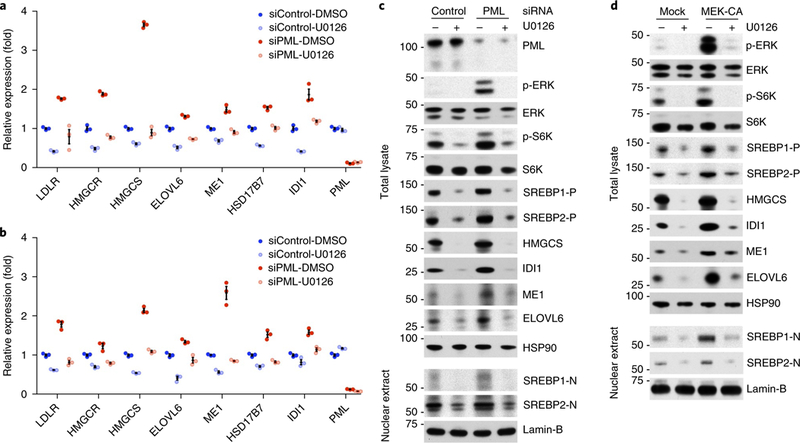
a,b, qPCR of SREBP targets in LNCaP (a) or PC3 (b) cells. CaP cells were transfected with control or PML siRNA for 48 h, then treated for 12 h with DMSO or 20 μM U0126 in medium with 10% lipoprotein-deficient serum. c,d, Immunoblot analysis of total lysates or nuclear extracts from LNCaP cells. LNCaP cells were transfected with control or PML siRNA (c) or empty vector or HA-MEK1p.Ser218Asp/p.Ser222Asp (d) for 24h, then treated for 24h with DMSO or 20μM U0126 in medium with 10% lipoprotein-deficient serum. In a and b, the results of one representative experiment are shown (n = 3 experiments). Data are from three independent cultures and are shown as mean±s.e.m. In c and d, the results of one representative experiment are shown (n = 3 experiments). Uncropped images for c and d are shown in Supplementary Fig. 7. Numbers to left of blots indicate molecular weight (kDa).
SREBP-dependent lipogenesis is critical for PML-loss-induced CaP growth and metastasis.
We then assessed whether the PML-loss-induced prometastatic phenotype might be dependent on an SREBP-mediated lipogenic program. In an in vitro cell migration and invasion assay, knockdown of either SREBP-1 or SREBP-2 by siRNA significantly inhibited LNCaP and PC3 cell migration and invasion (Fig. 6a and Supplementary Fig. 5a,b). Inhibition of FA synthesis by acetyl CoA carboxylase inhibitor (TOFA) or cholesterol synthesis by simvastatin (HMG-CoA reductase inhibitor) mimicked the effect of SREBP knockdown on cell migration and invasion (Fig. 6b). In contrast, CaP cells transfected with PML siRNA showed significantly increased migration and invasion. This effect was nearly abolished when cells were cotransfected with SREBP-1 siRNA (Fig. 6a and Supplementary Fig. 5a).
Fig. 6 |. SREBP-dependent lipogenesis is critical for PML-loss-induced CaP growth and metastasis.
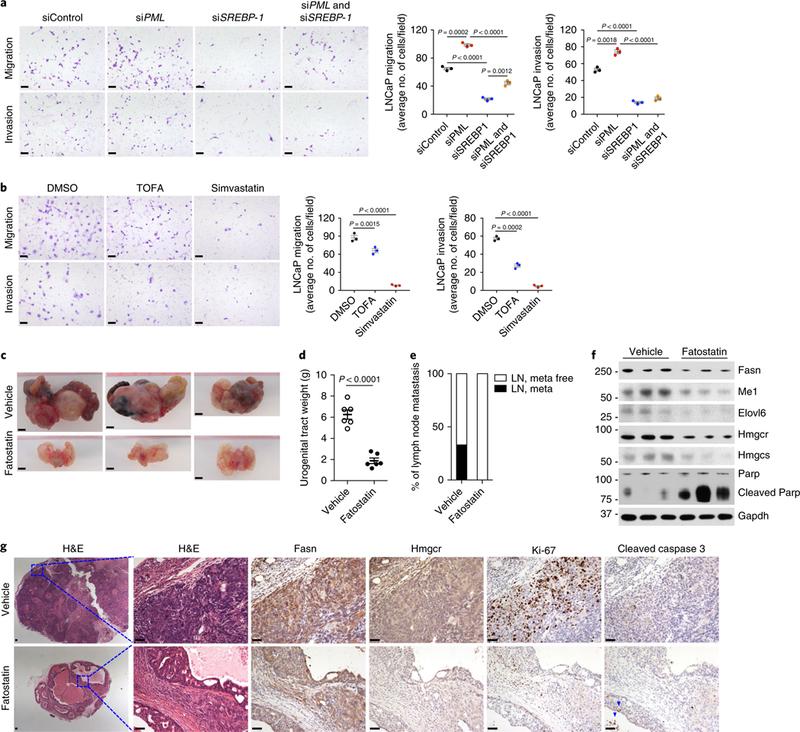
a,b, Representative images and quantification of migrated or invaded LNCaP cells in the migration and invasion assay. LNCaP cells were transfected with the indicated control or target siRNAs against PML or/and SREBP-1 (a) or were pretreated with DMSO, 10 μg/ml TOFA or 10 μM simvastatin (b) for 48 h, then subjected to migration (24 h) or invasion (48 h) assays. Scale bars, 50 μm. c-g, Gross anatomy of representative Ptenpc−/−; Pmlpc−/− tumors (c), quantification of tumor weight (d) and the incidence of metastasis (e) and immunoblot (f) and IHC (g) analyses of tumors from Ptenpc−/−; Pmlpc−/− mice after treatment with vehicle or 15mg/kg fatostatin for two months. Scale bars, 5 mm (c) and 50 μm (g). Arrowheads in g indicate apoptotic cells. In a and b, the results of one representative experiment are shown (n = 5 experiments). Data are from three independent cultures (4 fields per insert). In d, n= 6 mice per treatment. Data shown in a, b and d are mean± s.e.m. Student’s t test (two tailed) was used to determine significance. Uncropped images for f are shown in Supplementary Fig. 7. Numbers to left of blot indicate molecular weight (kDa).
To further corroborate the crucial role of SREBP-dependent lipogenesis in PML-loss-driven CaP growth and metastasis, we carried out in vivo preclinical studies of fatostatin targeting SREBP in Ptenpc−/−; Pmlpc−/− mice. Fatostatin is a recently discovered SREBP chemical inhibitor that directly binds SREBP cleavage-activating protein and blocks the endoplasmic reticulum–Golgi transport and the subsequent activation of SREBP56–58. Treatment with fatostatin for two months in Ptenpc−/−; Pmlpc−/− mice inhibited both prostate tumor growth (Fig. 6c, d) and distant lymph node metastasis (Fig. 6e and Supplementary Fig. 5c). This potent antitumor and antimetastatic activity of fatostatin is presumably due to the suppression of SREBP pathway, because fatostatin-treated Ptenpc−/−; Pmlpc−/− tumors displayed markedly lower expression of SREBP-regulated enzymes for synthesis of FA and cholesterol (Fig. 6f, g). In agreement with the crucial role of lipid metabolism in various aspects of cancer development39, fatostatin-treated Ptenpc−/−; Pmlpc−/− tumors, compared with vehicle-treated tumors, displayed a drastic decrease in the frequency of mitotic cells positive for Ki-67 staining, along with a concomitant induction of apoptosis, as indicated by higher expression of cleaved Parp and cleaved caspase 3 (Fig. 6f, g). These functional data suggested that SREBP-mediated lipogenesis is a key downstream effector of PML-loss-driven CaP growth and metastasis.
A HFD drives metastatic progression and increases lipid abundance in prostate tumors.
Given that HFD feeding stimulates the expression of SREBP and subsequent expression of genes encoding lipogenic enzymes59, and induces lipid accumulation in nonadipose tissues40,60, 12-month-old Ptenpc−/− and Ptenpc−/−; Pmlpc−/− mice were fed a HFD for 3 months to test whether a causal relationship exists among lipogenesis, lipid accumulation and metastasis. To mimic hyperactivated de novo lipogenesis (new lipids enriched in saturated and monounsaturated fatty acyl chains), we chose a lard-based HFD enriched in saturated and monounsaturated FAs and capable of inducing classic HFD effects in rodents61. As expected, HFD-fed mice gained considerably more weight than chow-fed mice (Fig. 7a). Ptenpc−/−; Pmlpc−/− mice displayed limited metastasis to lymph nodes when fed a chow diet but, after HFD feeding, developed lymph node metastases in 6 out of 8 cases, and lung metastasis in 4 out of 8 cases (Fig. 7b,c and Supplementary Fig. 6a). Ptenpc−/− mice are nonmetastatic when fed a chow diet20, but they developed lymph node metastases in 3 out of 8 cases, and lung metastases in 3 out of 8 cases after HFD feeding (Fig. 7d,e and Supplementary Fig. 6b). These metastases resembled primary prostate tumors and displayed high levels of phospho-Akt as well as nuclear AR staining. Strong CK8 staining was also observed in lymph node metastases, but this staining was less intense in lung metastases (Fig. 7b,d and Supplementary Fig. 6a,b). No other distant metastases or substantial effects on survival were observed in HFD-fed Ptenpc−/− or Ptenpc−/−; Pmlpc−/− mice (Supplementary Fig. 6c).
Fig. 7 |. A HFD drives metastatic progression in mouse models of CaP and increases lipid abundance in prostate tumors.
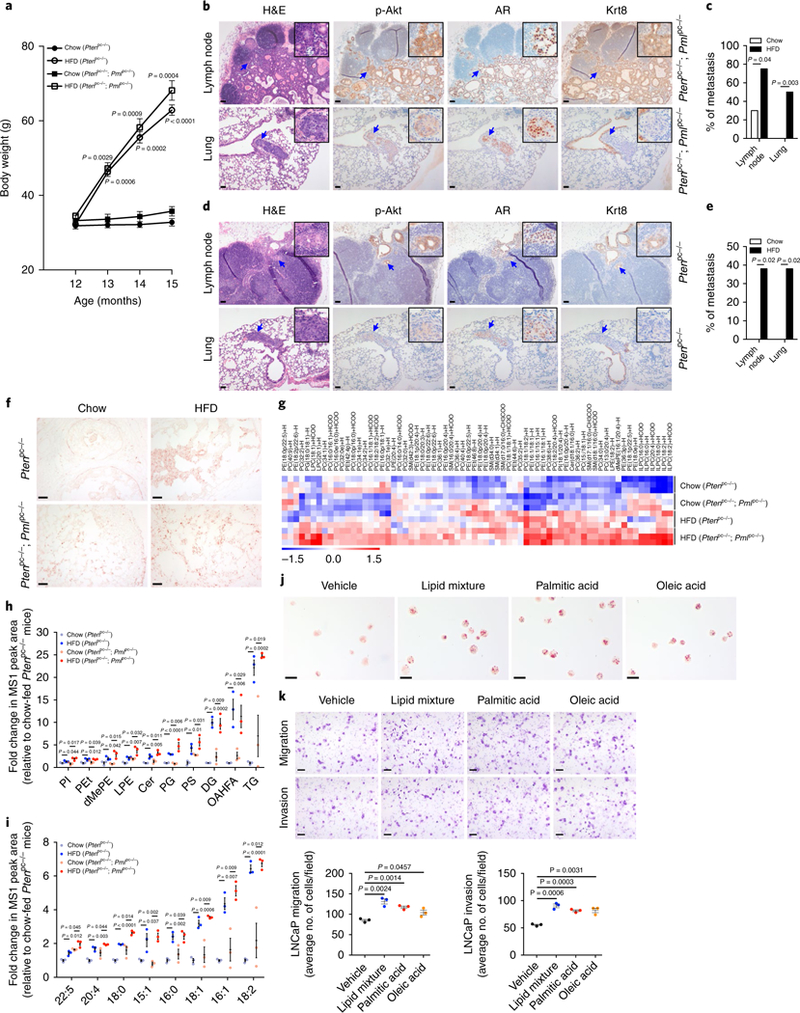
a, HFD-fed mice gain body weight. Mice (n = 8) at 12 months of age were fed chow or HFD for 3 months. Body weights of mice were measured each month. b-e, H&E and IHC staining of metastases in the lumbar lymph node and lung of a representative HFD-fed Ptenpc−/−; Pmlpc−/− mouse (b) or Ptenpc−/− mouse (d) and comparison of the incidence of metastasis between chow-and HFD-fed Ptenpc−/−; Pmlpc−/− mice (c) or Ptenpc−/− mice (e). f, ORO staining of tumors from chow-or HFD-fed Ptenpc−/− and Ptenpc−/−; Pmlpc−/− mice. g-i, Heat map of the top 70 most regulated lipid ions (g), plot of significantly increased lipid classes (h) and plot of fatty acyl chains (i) in tumors from HFD-fed Ptenpc−/− and Ptenpc−/−; Pmlpc−/− mice compared with chow-fed counterparts. dMePE, dimethylphosphatidylethanolamine; LPE, lysophosphatidylethanolamine; Cer, ceramides; DG, diglyceride, OAHFA, (O-acyl)-omega-hydroxy fatty acids; TG, triglyceride. j, ORO staining of vehicle-or dietary-lipid-treated LNCaP cells. k, Representative images and quantification of migrated or invaded LNCaP cells in the migration and invasion assay. LNCaP cells were pretreated with BSA vehicle control, 2% lipid mixture, or 30 μm BSA-conjugated palmitic acid or oleic acid for 7 d, then subjected to migration (24h) or invasion (48 h) assays. Arrows in b and d indicate metastases. In c, chow-fed mice (n = 20) and HFD-fed mice (n = 8). In e, chow-fed mice (n = 19) and HFD-fed mice (n = 8). In h and i, n = 3 mice per group. In k, the results of one representative experiment are shown (n = 5 experiments). Data are from three independent cultures (4 fields per insert). In a, h, i and k, data shown are mean ±s.e.m. Student’s t test (two tailed) was used to determine significance. In c and e, Fisher’s exact test (two tailed) was used to determine significance. Scale bars in all panels, 50 μm.
To further establish the causal relationship between dietary lipids and metastasis, we conducted Oil Red O (ORO) staining to determine whether lipids accumulated in prostate tumors after HFD feeding. Compared with tumors from chow-fed mice, tumors from HFD-fed Ptenpc−/− or Ptenpc−/−; Pmlpc−/− mice showed much stronger ORO staining (Fig. 7f). To validate the results of the ORO staining, we performed an additional global lipidomic analysis in prostate tumors from chow-or HFD-fed Ptenpc−/− and Ptenpc−/−; Pmlpc−/− mice (Supplementary Tables 10–12). Analyses of the top 70 most significantly differential lipid ions (Fig. 7g), significantly increased lipid classes (Fig. 7h, Supplementary Fig. 6d and Supplementary Table 13) and significantly increased fatty acyl chains from the 30 most abundant chain types (Fig. 7i, Supplementary Fig. 6e and Supplementary Table 14) showed that the abundance of most lipids was markedly higher in tumors from Ptenpc−/− or Ptenpc−/−; Pmlpc−/− mice fed a HFD rather than a chow diet. Among the four groups of tumors, the lowest lipid abundance was observed in tumors from chow-fed Ptenpc−/− mice, whereas in response to HFD feeding, Ptenpc−/− tumors displayed higher lipid abundance than that in tumors from chow-fed Ptenpc−/−; Pmlpc−/− mice but a largely similar lipid profile to that in HFD-fed Ptenpc−/−; Pmlpc−/− tumors (Fig. 7g–i). These findings indicated that HFD feeding causes a much greater increase in lipid abundance in prostate tumors than does the de novo lipogenesis driven by Pml inactivation; moreover, they suggested that a HFD affects metastatic progression, at least in part through increased lipid accumulation in prostate tumors.
We then sought to determine whether dietary constituents of the HFD might affect CaP cell invasiveness by recapitulating aspects of the HFD-induced metastatic phenotype. CaP cells were cultured in medium supplemented with a lipid mixture, palmitic acid or oleic acid, representing the main lipid components of the lard-based HFD61. In agreement with the findings in HFD-fed prostate tumors, CaP cells treated with dietary lipids showed markedly greater lipid droplet accumulation than that in vehicle-treated cells, as shown by ORO staining (Fig. 7j and Supplementary Fig. 6f). Moreover, these cells displayed significantly greater cell migration and invasion (Fig. 7k and Supplementary Fig. 6g), thus suggesting that dietary lipids directly affect CaP cells and are sufficient to recapitulate aspects of the in vivo HFD-induced metastatic phenotypes.
A highly enriched SREBP signature in metastatic human CaP.
To assess the relevance of these findings to human CaP, we performed a GSEA of the metastasis-gene signatures in mCRPC vs. LPC from a dataset from Grasso et al.14. Our analysis identified significant enrichment of SREBP-1-target genes in mCRPC compared with LPC (Fig. 8a). Additionally, we analyzed a dataset from Taylor et al.15, which contains 181 primary and 37 metastatic CaP samples. In agreement with the findings for the Grasso et al. dataset, the SREBP-1 signature was highly enriched in metastatic vs. primary CaP (Fig. 8b). Thus, the highly enriched SREBP/lipogenesis signature is frequently observed in metastatic human CaP, a finding consistent with our transcriptomic data showing enrichment of the SREBP signature in Pten and Pml double-null metastatic CaP (Fig. 4b) and our in vivo preclinical studies showing that lipid accumulation promotes metastatic progression (Fig. 7b–i).
Fig. 8 |. An SREBP signature is highly enriched in metastatic human CaP.
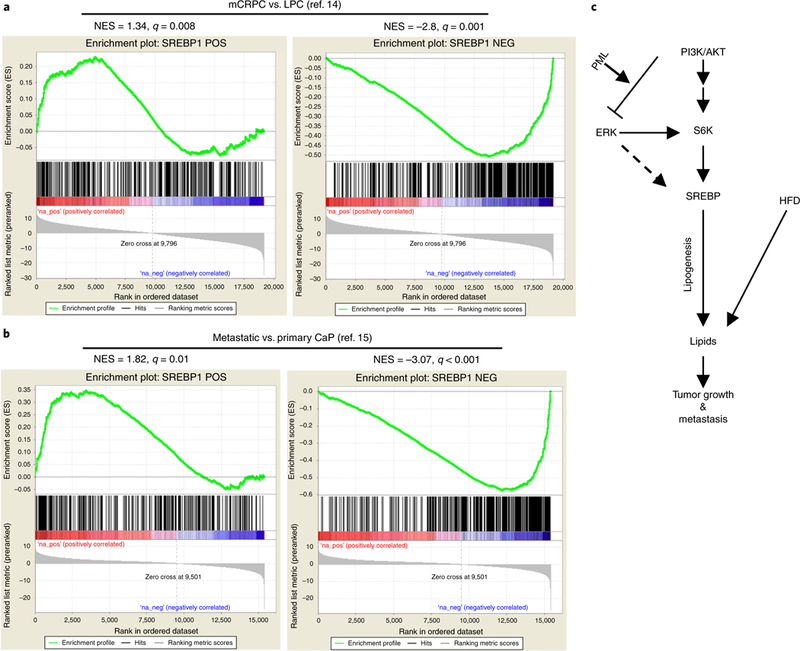
a,b, GSEA enrichment plot for the gene set derived from the SREBP-1 signature. The up-to downregulated genes from the ranked gene list for metastasis vs. primary samples in the Grasso et al.14 (a) or Taylor et al.15 (b) dataset were analyzed with the GSEA algorithm for enrichment of the SREBP-1 signature. The SREBP-1 signature was inferred through an information-theory-based CLR algorithm by using the RNA-seq data from The Cancer Genome Atlas normal prostate samples. The SREBP-1 signature was split into two subsets: positively correlated (POS) and negatively correlated (NEG) between SREBP-1 and its targets. (c) Mechanisms of aberrant lipid metabolism, regulated by PML, and the increased lipid influx by HFD as a potential risk factor for metastatic CaP.
Discussion
Our data provide a strong genetic foundation elucidating the mechanisms underlying metastatic progression, and they demonstrate how environmental dietary factors can boost progression from primary to metastatic cancer, intertwining with the genetic makeup of cancer (Fig. 8c). We demonstrated that SREBP-dependent lipogenesis, which can be hyperactivated by concomitant activation of the PI3K–AKT and MAPK pathways, or a HFD regimen, functions as an underlying rheostat toward metastatic cancer progression. Furthermore, we identified PML as a critical mediator of feedback inhibition of MAPK signaling and lipogenesis, and its inactivation propels metastatic progression in cancers driven by PTEN loss and PI3K–AKT activation. Interestingly, Cph2, the yeast homolog of SREBP, has been shown to be involved in invasive/pseudohyphal growth in yeast, thereby suggesting a conserved evolutionary function of SREBP in invasiveness62.
In agreement with findings from earlier studies showing that lipid metabolism is required for metastasis in other cancer systems63,64, our analysis identified a highly enriched SREBP-1-dependent lipogenic signature in metastatic human CaP. Concordantly, a HFD enriched in saturated and monounsaturated FAs triggered metastasis in a nonmetastatic Ptenpc−/− CaP model and further enhanced metastasis in a metastatic Ptenpc−/−; Pmlpc−/− CaP model. In addition to our genetic and experimental evidence in GEMMs of CaP, a HFD has been reported to favor metastatic progression in xenograft models from cell lines of various histological origin63,65,66. Numerous mechanisms have been proposed to explain a possible association between dietary lipids and CaP67, including paracrine mechanisms through secreted cytokines from adipose tissues, endocrine mechanisms through alteration of androgen levels (Supplementary Fig. 6h) and an induction of basal-to-luminal cell differentiation caused by immune-cell infiltration68. However, we showed here that specific genetic perturbations or a HFD are probably able to exert a direct effect on metastasis through increased lipid accumulation. We also characterized the intracellular lipid changes in GEMMs of CaP and detected qualitative changes in four different lipid classes as well as in the saturation of fatty acyl chains. Together, these results established a strong mechanistic and causal link among aberrant lipogenesis, excess lipid accumulation and metastasis, thus providing a compelling rationale for integrating lifestyle data (for example, diet) and tumor genetics into clinical practice to identify patients at high risk of metastasis. Additionally, lipid metabolism itself is an attractive therapeutic target through inhibition of lipogenic enzymes39,69. Notably, such inhibition decreases CaP cell viability only in the absence of an exogenous lipid source such as lipoprotein70, thereby highlighting the importance of integrating pharmacologic approaches with stringent dietary regimens to prevent metastasis. Future studies are warranted to evaluate whether specific lipid subsets/signatures may serve as prognostic biomarkers to distinguish CaP with metastatic potential from indolent disease.
Finally, given that PML is lost in human cancer of multiple histological origins71, our study suggests that PML loss may underlie MAPK activation in cancers lacking genetic alterations in MAPK-signaling components26–28,72. In an accompanying study, we have demonstrated that PP1α is a B-Raf-activating phosphatase genetically amplified in CaP, and that PML suppresses PP1α-dependent activation of MAPK signaling73. Moreover, Ptenpc−/−; Pmlpc−/− tumors displayed a significantly upregulated gene set induced by hypoxia, in agreement with findings from earlier studies showing that PML is a key player orchestrating the cellular response to hypoxia through repression of mTOR74 (Supplementary Table 3 and Supplementary Fig. 4j). This finding has equally important implications for tumorigenesis, because PML loss in the hypoxic core or tumoral lesions not only would activate mTOR, thus resulting in sustained HIF-1 activation, but also would relieve the feedback inhibition of MAPK signaling triggered by mTOR activation, thereby leading to simultaneous activation of both mTOR and MAPK signaling. Together, our results provide a potential roadmap for targeted therapies tailored to individual patients for the prevention and treatment of metastatic cancer.
Methods
Mouse models.
All animal experiments were approved by the Beth Israel Deaconess Medical Center IACUC Committee on Animal Research. The floxed Pml allele was created following the scheme shown in Supplementary Fig. 2a. The Pb-Cre4 transgenic mice and Ptenflox/flox mice have been previously described20. Ptenflox/flox mice were first crossed with Pb-Cre4 mice. The resulting compound mice or Pb-Cre4 transgenic mice were then crossed with Pmlflox/flox mice to generate the conditional knockout of Pten and/or Pml in the prostate epithelium. The three mouse genotypes were maintained on a mixed C57BL/6 (80%) × 129S1/SvImJ (20%) background. Ten mice per genotype were randomly chosen and used to examine the tumor grade at the indicated ages. The histological grade was determined by a pathologist in a blinded manner. For HFD feeding, male mice were given a dietary chow consisting of 60% kcal fat (Teklad Diet TD.06414) starting at the age of 12 months and continuing for 3 months. All other mice were fed standard chow consisting of 17% kcal fat (Lab Diet 5008). Male mice were housed one per cage during preclinical studies of both HFD and fatostatin.
Plasmids, reagents and antibodies.
HA-MEK1 p.Ser218Asp/p.Ser222Asp was purchased from Upstate. Two independent siRNA duplexes targeted to PML, SREBP-1 or SREBP-2 and control nontarget siRNA were purchased from Dharmacon or Sigma. The sequences for the siRNAs are listed in Supplementary Table 15. U0126 was from Selleck Chemicals. TOFA and simvastatin were from Cayman Chemical. Fatostatin was from EMD Millipore. EGF, Lipofectamine 2000, Lipofectamine RNAiMAX, RPMI, DMEM, Opti-MEM reduced-serum medium and FBS were from Invitrogen. TransIT-X2 was from Mirus Bio. FA free BSA, sodium palmitate, oleic acid, lipid mixture, polybrene and puromycin were from Sigma. We used the following primary antibodies: anti-p-ERK (9101), anti-p-ERK (4376, 20G11), anti-ERK (9102), anti-p-MEK (9154), anti-MEK (9126), anti-p-AKT (9271), anti-p-AKT (4060, D9E), anti-AKT (9272), anti-PTEN (9559), anti-EZH2 (5246), anti-PARP (9532), anti-cleaved caspase-3 (9661) and anti-GAPDH (2118) from Cell Signaling Technology; anti-Rb (IF8) and anti-HSP90 (H-114) from Santa Cruz; anti-IDI1 (NBP1–57587) from Novus Biologicals; anti-SREBP1 (2A4) from Active Motif; anti-β-actin (AC-74) from Sigma; anti-SREBP2 (A303–125A), anti-PML (A301–167A), anti-FASN (A301–324A) and anti-HMGCS1 (A304–590A) from Bethyl Laboratories; anti-PML (MAB3738, for detecting mouse Pml protein) from Millipore; anti-AR (EPR1535) and anti-SMAD4 (EP618Y) from Abcam; anti-Ki-67 (SP6) from Thermo Fisher Scientific; anti-CK8 (MMS-162P) from Covance; anti-α-SM-actin (1A4) from Dako; and anti-ELOVL6 (PA5–20520), anti-HMGCR (PA5–37367) and anti-ME1 (PA5–40600) from Invitrogen.
Cell culture and transfection.
All cell lines were obtained from ATCC and were checked for mycoplasma with a MycoAlert Mycoplasma Detection Kit (Lonza). Cells were maintained in DMEM or RPMI supplemented with 10% FBS, 2 mM glutamine, 100 U/ml penicillin and streptomycin (Invitrogen). To study the effects of PML and MAPK signaling on SREBP and SREBP targets, LNCaP or PC3 cells were seeded and cultured in medium with 10% lipoprotein-deficient serum (Kalen Biomedical) throughout the course of the experiments. Transfections were performed with Lipofectamine 2000, Lipofectamine RNAiMAX reagent or TransIT-X2 according to the manufacturer’s instructions. In brief, 50 nM mixtures of two independent siRNA pairs targeting each gene or 1 μg of DNA plasmids were transfected into 1 × 105 cells in a six-well dish. Cells were allowed to recover in complete medium after 12-h transfections and then harvested at the indicated times. For the treatment of dietary lipids, palmitic and oleic acid were conjugated to FA-free BSA as previously described75. CaP cells were cultured with BSA vehicle control, 2% lipid mixture, 30 μm BSA-conjugated palmitic acid or oleic acid for 7 d, then subjected to ORO staining, cell migration and invasion assays.
Western blotting.
For western blotting, prostate tissues or cells were lysed in RIPA buffer (Sigma) supplemented with protease (Roche) and phosphatase (Sigma) inhibitors. Proteins were separated on 4–12% Bis-Tris gradient gels (Invitrogen) and transferred to polyvinylidine difluoride membranes (Immobilon P, Millipore), and the blots were probed with the indicated antibodies. Densitometry quantification was performed in ImageJ. Nuclear/cytoplasmic fractionation was performed as previously described76.
Histology, IHC and ORO staining.
Individual prostate lobes were dissected and fixed in 4% paraformaldehyde for histology and IHC analysis, or were cryoembedded in OCT compound (Sakura) for ORO staining. For staining, the tissues were embedded in paraffin through standard procedures. 5-μm sections were cut and processed for histology or immunostaining. Primary antibodies to the following proteins were used for IHC: PML (MAB3738, 1:300), p-ERK (20G11, 1:100), p-AKT (D9E, 1:100), CK8 (MMS-162P, 1:200), AR (EPR1535, 1:100), Ki-67 (SP6, 1:200), cleaved caspase-3 (9661, 1:300), FASN (A301–324A, 1:100), HMGCR (PA5–37367, 1:100) and smooth muscle α-actin (1A4, 1:1000). For ORO staining, cells were prepared by drying drop on poly-L-lysine slides after 7-d treatment with dietary lipids. Frozen tissues or cells were stained by ORO working solution as previously described77. The stained slides were visualized with a bright-field microscope.
Prostate sphere assay.
The culture and passage of prostate spheres were carried out as previously described78. Dissociated prostate epithelial cells were prepared from mice at 12 weeks of age. To initiate sphere formation, unsorted mouse prostate cells were prepared in PrEGM medium (Lonza) at a density of 2.5 × 105 cells per ml. 40 μl of cell suspension was mixed with 60 μl cold Matrigel (BD Bioscience), and pipetted around the rim of a well of a 12-well plate and allowed to solidify at 37 °C for 30 min. 1 ml warm PrEGM was then added to each well. The spheres were cultured and monitored for 14 d, and 50% of the medium was changed every 3 d. To passage spheres, Matrigel was digested with 1 mg/ml dispase solution (StemCell Technologies) for 30 min at 37 °C. Digested cultures were collected, pelleted, resuspended and subjected to sequential digestion by 2 mg/ml type I collagenase (Sigma) for 1 h and 0.05% trypsin/EDTA (Invitrogen) for 5 min at 37 °C, and then passed through a 27-gauge syringe 5–10 times, and filtered through a 40-μm cell strainer. Cells were counted with a hemocytometer and replated at a density of 1 × 104 cells per 12 wells.
Microarray analysis.
RNA was extracted from WT and knockout mice with QIAzol (Qiagen). 200 ng of total RNA was hybridized to Affymetrix Mouse Gene 2.1 ST arrays at the Beth Israel Deaconess Medical Center Genomics and Proteomics Core. The obtained raw intensity.cel files were normalized through robust multichip analysis (Bioconductor release 3.1), and differential expression was determined with the limma Bioconductor package by fitting a linear model. GSEA was conducted with the gene sets from the Molecular Signatures Database (MolSigDB v3.1). Gene Ontology analysis was conducted with the Panther Annotation System (version 9.0).
qPCR.
Total RNA was prepared with TRIzol (Invitrogen). cDNA was obtained with an iScript cDNA Synthesis kit (Bio-Rad). Triplicate samples for qPCR were run in a Lightcycler 480 (Roche) by using SYBR Green I Master Mix (Roche). Each value was adjusted according to the levels of Sdha (for mouse genes) or RPLP0 (for human genes) as a reference (primer sequences in Supplementary Table 16).
Lipidomics by untargeted high-resolution liquid chromatography-tandem mass spectrometry (LC–MS/MS).
The lipidomic analysis was performed as previously described44. Briefly, nonpolar lipids were extracted from 5 mg prostate tissues with MTBE. The upper phase containing the nonpolar lipids was dried in a SpeedVac rotary evaporator with no heat. Lipid samples were resuspended in 35 μl of 50% isopropanol (IPA)/50% MeOH. 10 μl of samples was injected for reversed-phase (C18) LC–MS/MS with a hybrid QExactive Plus Orbitrap mass spectrometer (Thermo Fisher Scientific) coupled to an Agilent 1100 HPLC in DDA mode with positive/negative-ion polarity switching (top 8 in both modes). The lipidomics data were analyzed with LipidSearch 4.1.9 software. The software identifies intact lipid molecules on the basis of their molecular weight and fragmentation pattern by using an internal library of predicted fragment ions per lipid class, the spectra are then aligned on the basis of retention time, and MS1 peak areas are quantified across sample conditions. Excel 2010 was used to calculate intensity, and the R program (version 3.2.5) was used for data manipulation and statistical analyses, including unsupervised hierarchical clustering and heat-map visualization.
Cell migration and invasion assays.
PC3 or LNCaP cells were transfected with the indicated siRNAs or pretreated with 10 μg/ml TOFA or 10 μM simvastatin for 48 h, then detached into single-cell suspensions. LNCaP (1 × 105) or PC3 (5 × 103 and 5 × 104 for migration and invasion assays, respectively) cells were resuspended in 100 μl of RPMI medium containing 0.1% FBS and placed into the top chamber of 8-μm Transwell inserts for migration assays or Matrigel-coated Transwell inserts for invasion assays (BD Biosciences). The bottom wells contained 600 μl RPMI supplemented with 10% FBS. After 24 h or 48 h (for LNCaP invasion assay only), cells on the upper surfaces of the inserts were removed with a cotton swab. Migrated cells were fixed in 10% formalin, then stained with 0.2% crystal violet for 10 min. Cells were counted in four microscopic fields under 20× magnification. Results are representative of three to five independent experiments.
In vivo treatment.
Ptenpc−/−; Pmlpc−/− mice at 12–13 months of age were treated with fatostatin (15 mg/kg) or corn oil (vehicle control) by intraperitoneal (i.p.) injections, every other day for 2 months. Mice were then euthanized, and mouse tissues, including prostate tumors, were dissected, weighed and processed for histopathology and molecular analyses.
Blood sampling and testosterone ELISA.
After 90 d of chow or HFD feeding, blood samples were collected from the mice through cardiac puncture into BD Vacutainer SST Serum Separation Tubes (BD 367986) and immediately mixed by proper inversion. The SST blood specimens were allowed to clot for 30 min and then centrifuged at 1,000 g for 10 min in a swing-bucket centrifuge to recover the serum. Serum levels of testosterone were measured with a competitive ELISA kit according to the manufacturer’s instructions (Abcam 108666).
Array CGH analysis.
We downloaded the data from the GEO database (Grasso et al., GSE35988; Taylor et al., GSE21032) or cBioportal (for the Robinson et al. dataset25), with a focus on the array-based comparative genomic hybridization datasets. We wrote R scripts to process the data and to generate the heat map based on the log2-transformed ratio. The cutoff thresholds used were −0.35 to −0.8 as heterozygous deletions and lower than −0.8 as homozygous deletions.
TMA analysis.
All the prostate specimens were obtained with informed consent and with approval from the Memorial Sloan-Kettering Cancer Center (MSKCC) ethics committee. Clinical histopathological and follow-up information is included in Supplementary Table 1. The study cohort comprised radical prostatectomy specimens from 144 patients with primary CaP. Tumor samples were collected at the time of surgical resection after written informed consent was obtained. The patients were treated and followed at Memorial Sloan-Kettering Cancer Center. PML (Santa Cruz) and PTEN (Cell Signaling Technology) staining were performed as previously described71. Cases that had more than 50% of the core composed of tumor cells were analyzed.
Statistical analysis.
No statistical methods were applied to determine sample size. The mouse studies were randomized. The investigators were not blinded to allocation during experiments and outcome assessment. For analysis of average data, datasets were compared using unpaired two-tailed Student’s t tests. For analysis of categorical data (for example, copy number alteration), 2 × 2 contingency tables were constructed, and datasets were compared with Fisher’s exact test, For the correlation of TMA staining with clinical parameters, datasets were compared using Pearson’s chi-square test. Survival outcomes were evaluated with Kaplan–Meier survival estimates, log-rank (Mantel–Cox) tests and univariate and multivariate Cox proportional hazards models. All tests were two sided, and an α-error of 5% was considered significant. Univariate exploratory analyses showed that grouping PTEN and PML loss (defined as a lack of both markers or a lack of one marker and low expression of the other), Gleason score (≤7 vs. >7) and pathologic stage (pT1–2 N0 vs. pT3–4 or N1) maximized the likelihood-ratio chi-square value for overall survival; these groupings were used in the multivariable model. P values <0.05 were considered to be statistically significant. Statistical tests were executed in the statistical software R (version 3.1.2) or GraphPad Prism software.
Supplementary Material
Acknowledgements
We thank all the members of the laboratory of P.P.P. for critical comments, and L. Southwood and E. Stack for editing the manuscript. We are grateful to G. Augusto dos Santos for insightful discussions. We thank the BIDMC Histology Core facility and M. Yuan in the BIDMC Mass Spectrometry Core for help with the lipidomics experiments. M.C. was supported in part by a DOD Prostate Cancer Research Program (PCRP) Postdoctoral Training Award. This work was supported by NIH grants R01CA142780, R01CA142874 and R35CA197529 to P.P.P. This work was also partially supported by NIH grants P01CA120964 and R35CA197459 to J.M.A.
Footnotes
URLs. Gene Ontology Analysis (GO), http://pantherdb.org/; Gene Set Enrichment Analysis (GSEA), http://software.broadinstitute.org/gsea/msigdb/; NCBI Gene Expression Omnibus (GEO), http://www.ncbi.nlm.nih.gov/geo/.
Methods
Methods, including statements of data availability and any associated accession codes and references, are available at https://doi.org/10.1038/s41588-017-0027-2 .
Competing interests
The authors declare no competing financial interests.
Supplementary information is available for this paper at https://doi.org/10.1038/s41588-017-0027-2.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Data availability. Microarray data from this paper have been deposited in the Gene Expression Omnibus database and are accessible through GEO series accession number GSE98493. All other data supporting the findings of this study are available within the article and its Supplementary Information files and are available from the authors upon reasonable request.
References
- 1.Wu JN, Fish KM, Evans CP, Devere White RW & Dall’Era MA No improvement noted in overall or cause-specific survival for men presenting with metastatic prostate cancer over a 20-year period. Cancer 120, 818–823 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Yang M et al. Dietary patterns after prostate cancer diagnosis in relation to disease-specific and total mortality. Cancer Prev. Res. (Phila.) 8, 545–551 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grönberg H Prostate cancer epidemiology. Lancet 361, 859–864 (2003). [DOI] [PubMed] [Google Scholar]
- 4.Markozannes G et al. Diet, body size, physical activity and risk of prostate cancer: an umbrella review of the evidence. Eur. J. Cancer 69, 61–69 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Zhu Y et al. Dietary patterns and colorectal cancer recurrence and survival: a cohort study. BMJ Open 3, e002270 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ittmann M et al. Animal models of human prostate cancer: the consensus report of the New York meeting of the Mouse Models of Human Cancers Consortium Prostate Pathology Committee. Cancer Res. 73, 2718–2736 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Steck PA et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat. Genet. 15, 356–362 (1997). [DOI] [PubMed] [Google Scholar]
- 8.Li J et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 275, 1943–1947 (1997). [DOI] [PubMed] [Google Scholar]
- 9.Yoshimoto M et al. Interphase FISH analysis of PTEN in histologic sections shows genomic deletions in 68% of primary prostate cancer and 23% of high-grade prostatic intra-epithelial neoplasias. Cancer Genet. Cytogenet. 169, 128–137 (2006). [DOI] [PubMed] [Google Scholar]
- 10.Sircar K et al. PTEN genomic deletion is associated with p-Akt and AR signalling in poorer outcome, hormone refractory prostate cancer. J. Pathol. 218, 505–513 (2009). [DOI] [PubMed] [Google Scholar]
- 11.Han B et al. Fluorescence in situ hybridization study shows association of PTEN deletion with ERG rearrangement during prostate cancer progression. Mod. Pathol. 22, 1083–1093 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lunardi A et al. A co-clinical approach identifies mechanisms and potential therapies for androgen deprivation resistance in prostate cancer. Nat. Genet. 45, 747–755 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beltran H et al. Targeted next-generation sequencing of advanced prostate cancer identifies potential therapeutic targets and disease heterogeneity. Eur. Urol. 63, 920–926 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grasso CS et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 487, 239–243 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Taylor BS et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 18, 11–22 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Song MS, Salmena L & Pandolfi PP The functions and regulation of the PTEN tumour suppressor. Nat. Rev. Mol. Cell. Biol. 13, 283–296 (2012). [DOI] [PubMed] [Google Scholar]
- 17.Ma X et al. Targeted biallelic inactivation of Pten in the mouse prostate leads to prostate cancer accompanied by increased epithelial cell proliferation but not by reduced apoptosis. Cancer Res. 65, 5730–5739 (2005). [DOI] [PubMed] [Google Scholar]
- 18.Trotman LC et al. Pten dose dictates cancer progression in the prostate. PLoS Biol. 1, E59 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Svensson RU et al. Slow disease progression in a C57BL/6 pten-deficient mouse model of prostate cancer. Am. J. Pathol. 179, 502–512 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen Z et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 436, 725–730 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ding Z et al. SMAD4-dependent barrier constrains prostate cancer growth and metastatic progression. Nature 470, 269–273 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gao H et al. Combinatorial activities of Akt and B-Raf/Erk signaling in a mouse model of androgen-independent prostate cancer. Proc. Natl. Acad. Sci. USA 103, 14477–14482 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kinkade CW et al. Targeting AKT/mTOR and ERK MAPK signaling inhibits hormone-refractory prostate cancer in a preclinical mouse model. J. Clin. Invest. 118, 3051–3064 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang XS et al. Characterization of KRAS rearrangements in metastatic prostate cancer. Cancer Discov. 1, 35–43 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Robinson D et al. Integrative clinical genomics of advanced prostate cancer. Cell 161, 1215–1228 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Palanisamy N et al. Rearrangements of the RAF kinase pathway in prostate cancer, gastric cancer and melanoma. Nat. Med. 16, 793–798 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moul JW, Friedrichs PA, Lance RS, Theune SM & Chang EH Infrequent RAS oncogene mutations in human prostate cancer. Prostate. 20, 327–338 (1992). [DOI] [PubMed] [Google Scholar]
- 28.Gumerlock PH, Poonamallee UR, Meyers FJ & deVere White RW Activated ras alleles in human carcinoma of the prostate are rare. Cancer Res. 51, 1632–1637 (1991). [PubMed] [Google Scholar]
- 29.Barbieri CE et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat. Genet. 44, 685–689 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang J et al. B-Raf activation cooperates with PTEN loss to drive c-Myc expression in advanced prostate cancer. Cancer Res. 72, 4765–4776 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mulholland DJ et al. Pten loss and RAS/MAPK activation cooperate to promote EMT and metastasis initiated from prostate cancer stem/progenitor cells. Cancer Res. 72, 1878–1889 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Walker EJ et al. Monoallelic expression determines oncogenic progression and outcome in benign and malignant brain tumors. Cancer Res. 72, 636–644 (2012). [DOI] [PubMed] [Google Scholar]
- 33.Trotman LC et al. Identification of a tumour suppressor network opposing nuclear Akt function. Nature 441, 523–527 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Varambally S et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 419, 624–629 (2002). [DOI] [PubMed] [Google Scholar]
- 35.Serra V et al. PI3K inhibition results in enhanced HER signaling and acquired ERK dependency in HER2-overexpressing breast cancer. Oncogene 30, 2547–2557 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carracedo A et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J. Clin. Invest. 118, 3065–3074 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang ZG et al. Role of PML in cell growth and the retinoic acid pathway. Science 279, 1547–1551 (1998). [DOI] [PubMed] [Google Scholar]
- 38.Swinnen JV et al. Overexpression of fatty acid synthase is an early and common event in the development of prostate cancer. Int. J. Cancer 98, 19–22 (2002). [DOI] [PubMed] [Google Scholar]
- 39.Baenke F, Peck B, Miess H & Schulze A Hooked on fat: the role of lipid synthesis in cancer metabolism and tumour development. Dis. Model. Mech. 6, 1353–1363 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goldstein JL & Brown MS A century of cholesterol and coronaries: from plaques to genes to statins. Cell 161, 161–172 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Porstmann T et al. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 8, 224–236 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Düvel K et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell 39, 171–183 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Trevino MB et al. Perilipin 5 regulates islet lipid metabolism and insulin secretion in a cAMP-dependent manner: implication of its role in the postprandial insulin secretion. Diabetes 64, 1299–1310 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Breitkopf SB, Yuan M, Helenius KP, Lyssiotis CA & Asara JM Triomics analysis of imatinib-treated myeloma cells connects kinase inhibition to RNA processing and decreased lipid biosynthesis. Anal. Chem. 87, 10995–11006 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Breitkopf SB et al. A relative quantitative positive/negative ion switching method for untargeted lipidomics via high resolution LC-MS/MS from any biological source. Metabolomics 13, 30 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shroff EH et al. MYC oncogene overexpression drives renal cell carcinoma in a mouse model through glutamine metabolism. Proc. Natl. Acad. Sci. USA 112, 6539–6544 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eberlin LS et al. Alteration of the lipid profile in lymphomas induced by MYC overexpression. Proc. Natl. Acad. Sci. USA 111, 10450–10455 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Perry RH et al. Characterization of MYC-induced tumorigenesis by in situ lipid profiling. Anal. Chem. 85, 4259–4262 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nomura DK et al. Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell 140, 49–61 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rysman E et al. De novo lipogenesis protects cancer cells from free radicals and chemotherapeutics by promoting membrane lipid saturation. Cancer Res. 70, 8117–8126 (2010). [DOI] [PubMed] [Google Scholar]
- 51.Hilvo M et al. Novel theranostic opportunities offered by characterization of altered membrane lipid metabolism in breast cancer progression. Cancer Res. 71, 3236–3245 (2011). [DOI] [PubMed] [Google Scholar]
- 52.Kotzka J et al. Insulin-activated Erk-mitogen-activated protein kinases phosphorylate sterol regulatory element-binding protein-2 at serine residues 432 and 455 in vivo. J. Biol. Chem. 279, 22404–22411 (2004). [DOI] [PubMed] [Google Scholar]
- 53.Abidi P, Zhang F, Li C & Liu J Blockage of the ERK signaling pathway abrogates the SCAP ligand-induced transcriptional activation of the LDL receptor gene in HepG2 cells. Int. J. Mol. Med. 16, 779–785 (2005). [PubMed] [Google Scholar]
- 54.Ma L, Chen Z, Erdjument-Bromage H, Tempst P & Pandolfi PP Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell 121, 179–193 (2005). [DOI] [PubMed] [Google Scholar]
- 55.Roux PP, Ballif BA, Anjum R, Gygi SP & Blenis J Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase. Proc. Natl. Acad. Sci. USA 101, 13489–13494 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Williams KJ et al. An essential requirement for the SCAP/SREBP signaling axis to protect cancer cells from lipotoxicity. Cancer Res. 73, 2850–2862 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li X, Chen YT, Hu P & Huang WC Fatostatin displays high antitumor activity in prostate cancer by blocking SREBP-regulated metabolic pathways and androgen receptor signaling. Mol. Cancer Ther. 13, 855–866 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kamisuki S et al. A small molecule that blocks fat synthesis by inhibiting the activation of SREBP. Chem. Biol. 16, 882–892 (2009). [DOI] [PubMed] [Google Scholar]
- 59.Lin J et al. Hyperlipidemic effects of dietary saturated fats mediated through PGC-1beta coactivation of SREBP. Cell 120, 261–273 (2005). [DOI] [PubMed] [Google Scholar]
- 60.Unger RH Lipotoxic diseases. Annu. Rev. Med. 53, 319–336 (2002). [DOI] [PubMed] [Google Scholar]
- 61.Buettner R et al. Defining high-fat-diet rat models: metabolic and molecular effects of different fat types. J. Mol. Endocrinol. 36, 485–501 (2006). [DOI] [PubMed] [Google Scholar]
- 62.Lane S, Zhou S, Pan T, Dai Q & Liu H The basic helix-loop-helix transcription factor Cph2 regulates hyphal development in Candida albicans partly via TEC1. Mol. Cell. Biol. 21, 6418–6428 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pascual G et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 541, 41–45 (2017). [DOI] [PubMed] [Google Scholar]
- 64.Nieman KM et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 17, 1498–1503 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Park H et al. A high-fat diet increases angiogenesis, solid tumor growth, and lung metastasis of CT26 colon cancer cells in obesity-resistant BALB/c mice. Mol. Carcinog. 51, 869–880 (2012). [DOI] [PubMed] [Google Scholar]
- 66.Kim EJ et al. Dietary fat increases solid tumor growth and metastasis of 4T1 murine mammary carcinoma cells and mortality in obesity-resistant BALB/c mice. Breast. Cancer Res. 13, R78 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sonn GA, Aronson W & Litwin MS Impact of diet on prostate cancer: a review. Prostate Cancer Prostatic Dis. 8, 304–310 (2005). [DOI] [PubMed] [Google Scholar]
- 68.Kwon OJ, Zhang B, Zhang L & Xin L High fat diet promotes prostatic basal-to-luminal differentiation and accelerates initiation of prostate epithelial hyperplasia originated from basal cells. Stem Cell Res. 16, 682–691 (2016). [DOI] [PubMed] [Google Scholar]
- 69.Menendez JA & Lupu R Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 7, 763–777 (2007). [DOI] [PubMed] [Google Scholar]
- 70.Ros S et al. Functional metabolic screen identifies 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 4 as an important regulator of prostate cancer cell survival. Cancer Discov. 2, 328–343 (2012). [DOI] [PubMed] [Google Scholar]
- 71.Gurrieri C et al. Loss of the tumor suppressor PML in human cancers of multiple histologic origins. J. Natl. Cancer Inst. 96, 269–279 (2004). [DOI] [PubMed] [Google Scholar]
- 72.Tilch E et al. Mutations in EGFR, BRAF and RAS are rare in triple-negative and basal-like breast cancers from Caucasian women. Breast Cancer Res. Treat. 143, 385–392 (2014). [DOI] [PubMed] [Google Scholar]
- 73.Chen M et al. Deregulated PPlα phosphatase activity towards MAPK activation is antagonized by a tumor suppressive failsafe mechanism. Nat. Commun. doi: 10.1038/s41467-017-02272-y (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bernardi R et al. PML inhibits HIF-1α translation and neoangiogenesis through repression of mTOR. Nature 442, 779–785 (2006). [DOI] [PubMed] [Google Scholar]
- 75.Das SK, Mondal AK & Elbein SC Distinct gene expression profiles characterize cellular responses to palmitate and oleate. J. Lipid Res. 51, 2121–2131 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Trotman LC et al. Ubiquitination regulates PTEN nuclear import and tumor suppression. Cell 128, 141–156 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mehlem A, Hagberg CE, Muhl L, Eriksson U & Falkevall A Imaging of neutral lipids by oil red O for analyzing the metabolic status in health and disease. Nat. Protoc. 8, 1149–1154 (2013). [DOI] [PubMed] [Google Scholar]
- 78.Lukacs RU, Goldstein AS, Lawson DA, Cheng D & Witte ON Isolation, cultivation and characterization of adult murine prostate stem cells. Nat. Protoc. 5, 702–713 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.