

Summary
Homologous recombination (HR) is essential for high-fidelity DNA repair during mitotic proliferation and meiosis. Yet, context-specific modifications must tailor the recombination machinery to avoid (mitosis) or enforce (meiosis) the formation of reciprocal exchanges—crossovers—between recombining chromosomes. To obtain molecular insight into how crossover control is achieved, we affinity purified 7 DNA-processing enzymes that channel HR intermediates into crossovers or noncrossovers from vegetative cells or cells undergoing meiosis. Using mass spectrometry, we provide a global characterization of their composition and reveal mitosis- and meiosis-specific modules in the interaction networks. Functional analyses of meiosis-specific interactors of MutLγ-Exo1 identified Rtk1, Caf120, and Chd1 as regulators of crossing-over. Chd1, which transiently associates with Exo1 at the prophase-to-metaphase I transition, enables the formation of MutLγ-dependent crossovers through its conserved ability to bind and displace nucleosomes. Thus, rewiring of the HR network, coupled to chromatin remodeling, promotes context-specific control of the recombination outcome.
Keywords: DNA repair, crossing-over, Mlh1-Mlh3-Exo1, Sgs1(BLM)-Top3-Rmi1, Mus81-Mms4(EME1), Yen1(GEN1), Srs2(RTEL1), Slx1-Slx4(BTDB12), Mph1(FANCM), Holliday junction
Graphical Abstract
Highlights
-
•
Affinity proteomics reveals the composition and interaction landscape of HR enzymes
-
•
Interaction network rewiring of HR enzymes during mitotic proliferation and meiosis
-
•
Chd1, Rtk1, and Caf120 regulate meiotic crossing-over
-
•
Chd1 remodels chromatin to enable formation of MutLγ-Exo1-dependent crossovers
Wild et al. used affinity proteomics to characterize the composition and interaction landscape of 7 DNA repair enzymes during mitotic proliferation and meiosis. They report a concerted and context-specific rewiring of the interactomes and reveal meiosis-specific network components with roles in crossing-over.
Introduction
The repair of DNA lesions by homologous recombination (HR) can lead to the formation of recombinant chromosomes in which large regions are reciprocally exchanged through crossing-over. During meiosis, crossovers break haplotypes and enable the disjunction of maternal and paternal centromeres at anaphase I (Petronczki et al., 2003). HR also fulfills essential functions outside the germline. In mitotically dividing cells, however, the formation of crossovers can be highly detrimental. Although relatively rare, inter-homolog crossovers can lead to the loss of heterozygosity of tumor suppressor genes, which is frequently linked to the development and progression of cancer (Moynahan and Jasin, 2010). Hence, to limit the potentially harmful effect of crossovers during mitotic proliferation and to avoid uncontrolled crossing-over during meiosis, HR also produces gene conversion events without reciprocal exchange, called noncrossovers (Heyer et al., 2010, Symington et al., 2014). How mitotic and meiotic cells modify the recombination machinery to avoid or enforce crossing-over remains poorly understood.
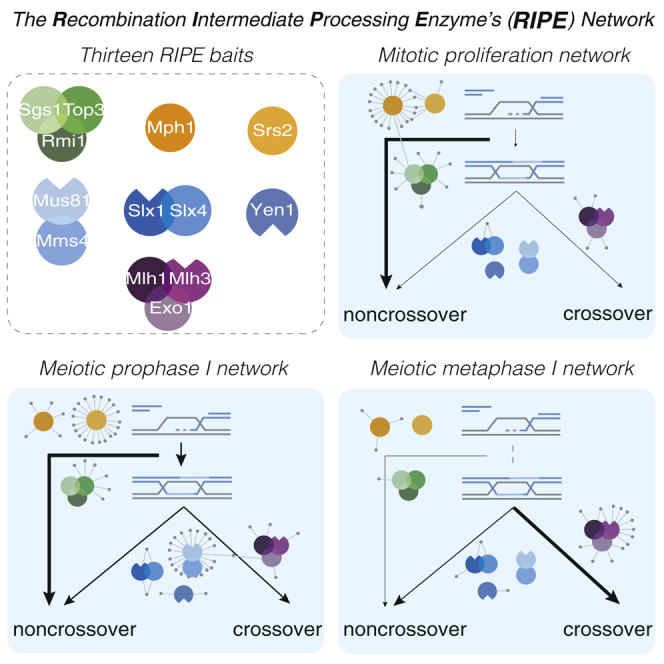
One universal feature in the repair of broken chromosomes by HR is that pairing and strand exchange reactions result in the formation of DNA joint molecule intermediates (Figure 1A). To achieve efficient disengagement of joint molecules while allowing a flexible HR outcome, eukaryotic cells contain up to 7 recombination intermediate processing enzymes (RIPEs) (Figure 1A). In mitotically dividing budding yeast cells, the DNA helicases Sgs1(BLM [Bloom’s helicase] in humans), Mph1(FANCM [Fanconi anemia complementation group M]), and Srs2 target HR intermediates to generate noncrossovers (Figure 1A) (Bzymek et al., 2010, Ira et al., 2003, Prakash et al., 2009). Sgs1 also promotes the formation of noncrossovers during meiosis (Kaur et al., 2015, Tang et al., 2015), while the contributions of Mph1 and Srs2 are less clear. Genetic experiments suggest that all of the functions of Sgs1 require the strand-passage proteins Top3 and Rmi1, which can associate with Sgs1 to form the STR complex. Conversely, Top3 and Rmi1 have roles in joint molecule processing, which are independent of Sgs1 (Fasching et al., 2015, Kaur et al., 2015, Tang et al., 2015).
Figure 1.

The Expression Levels of RIPE Components Do Not Change Significantly during Mitotic Proliferation Versus Meiosis
(A) Simplified model depicting the enzymes responsible for DNA joint molecule processing during mitotic and meiotic recombination. In several organisms, SLX4 coordinates MUS81 and SLX1 nucleases to resolve Holliday junctions. Srs2 can also regulate HR through the disassembly of Rad51 filaments on single-stranded DNA (ssDNA) (not depicted).
(B) Western blot analysis of the expression levels of all RIPE components depicted in (A), during exponential growth and throughout a meiotic time course. Samples were collected during asynchronous proliferation (As) or at 2-h intervals after transfer into SPM. Cdc5 accumulation marks the exit from pachytene. Variations in the kinetics of Cdc5 accumulation reflect experimental variation in the synchronous release of cells from G1 to undergo meiosis. Crm1 is a protein normalization control.
As persistent inter-sister or inter-homolog DNA connections can interfere with chromosome segregation, mitotic cells use structure-selective endonucleases to safeguard joint molecule resolution (Dehé and Gaillard, 2017, Matos and West, 2014). Mature joint molecules containing Holliday junctions that escape STR are resolved by Mus81-Mms4(EME1 [essential meiotic structure-specific endonuclease 1]), Slx1-Slx4(FANCP), and Yen1(GEN1) nucleases (Boddy et al., 2001, Dehé and Gaillard, 2017, Fricke and Brill, 2003, Ip et al., 2008). However, the nucleolytic resolution of Holliday junctions by structure-selective endonuclease yields noncrossovers as well as crossovers (Argueso et al., 2004, Dayani et al., 2011, Ho et al., 2010, Wechsler et al., 2011).
While mitotic crossovers occur sporadically, most meiotic crossovers (type I) are highly regulated, and only a small proportion derives from structure-selective endonucleases (type II) (de los Santos et al., 2003, De Muyt et al., 2012, Zakharyevich et al., 2012). Spatial patterning of type I crossovers involves the positive selection of nascent joint molecules in a process called “crossover designation.” Crossover designation is supported by meiosis-specific ZMM proteins, which promote the maturation of nascent joint molecules into double Holliday junctions (Allers and Lichten, 2001, Börner et al., 2004, Lynn et al., 2007, Schwacha and Kleckner, 1994, Snowden et al., 2004). Crossover formation is also temporally controlled. The final nucleolytic resolution of Holliday junctions is linked to the expression of Ndt80, a transcription factor that promotes the exit from pachytene and progression into the metaphase of meiosis I (Allers and Lichten, 2001, Chu and Herskowitz, 1998). Ndt80 controls the accumulation of M phase cyclins and Polo kinase Cdc5, which in turn elicits Holliday junction resolution throughout the genome (Clyne et al., 2003, Sourirajan and Lichten, 2008). Cdc5-mediated phosphorylation hyper-activates Mus81-Mms4 to initiate the formation of type II crossovers (Matos et al., 2011), but how Cdc5 triggers the formation of type I crossovers is unknown.
Elegant genetic experiments have established that the formation of type I crossovers requires the mismatch repair factors Mlh1-Mlh3 (MutLγ) and Exo1 (Figure 1A) (Zakharyevich et al., 2012). Moreover, in vitro reconstitution approaches have succeeded in demonstrating that MutLγ has endonuclease activity, which relies on the ability of Mlh1-Mlh3 heterodimers to polymerize on DNA (Manhart et al., 2017, Ranjha et al., 2014, Rogacheva et al., 2014). It is, however, unclear whether MutLγ polymerization is relevant in vivo or subject to regulation. In addition, the cleavage of model Holliday junctions by MutLγ has not been observed in vitro, suggesting that essential components of the pathway remain unknown.
As highlighted above, significant progress has been made in the delineation of the pathways that channel recombination intermediates toward the formation of crossovers or noncrossovers. However, the mechanistic basis for the differential, context-specific hierarchies in pathway usage remains poorly understood. For example, we do not know whether RIPEs engage in mitosis- or meiosis-specific interactions, which could influence enzyme function and control HR outcome. This is in part due to the significant technical challenges posed by meiotic cultures, which have limited the use of biochemical approaches to investigate protein function. In this study, we have established conditions to affinity purify each of the individual subunits of the 7 RIPEs from meiotically and mitotically dividing cells. Using mass spectrometry (MS), we have characterized their composition across different cellular environments and reveal context-specific rewiring of the interaction networks. Functional dissection of meiosis-specific binding partners of MutLγ-Exo1 implicated 3 factors—Chd1, Rtk1, and Caf120—to play important roles in crossing-over. Finally, in-depth analysis of Chd1 function suggests that the remodeling of meiotic chromatin, specifically at the prophase-to-metaphase I transition, constitutes a fundamental regulatory step in the formation of type I crossovers by MutLγ-Exo1.
Results
The Protein Abundance of the Individual RIPE Components Does Not Change Significantly during Mitotic Proliferation Versus Meiosis
We hypothesized that mitosis- and/or meiosis-specific changes in the expression or composition of RIPEs may play important roles in the control of HR outcome. To address these possibilities, we set out to generate a library of yeast strains expressing endogenously tagged versions of each RIPE (a total of 13 proteins; Figure 1A). Given that many RIPEs are found in low abundance in cells, we chose the 6xFLAG tag. Spore viability, colony growth, and crossover frequency assays ensured that the C-terminal fusions interfered minimally with protein function (Figures S1A–S1E; Table S1). Western blot analyses showed that with the exception of Exo1, whose protein levels appeared to be lower during G1 (0 h in sporulation medium [SPM]), all 13 RIPE components were expressed at comparable levels in vegetative cells and throughout meiosis (Figure 1B). Therefore, changes in the abundance of RIPEs are unlikely to play a major role in the specialized regulation of mitotic and meiotic HR. However, we detected stage-specific changes in the electrophoretic mobility for Sgs1, Srs2, and Exo1, suggestive of post-translational modifications (Figure 1B; data not shown). In agreement with previous work, we also observed transient changes in the mobility of Mms4, Slx4, and Yen1, as a consequence of cell-cycle stage-specific phosphorylation (Matos et al., 2011).
Affinity Purification of RIPEs from Large Meiotic and Mitotic Cultures for Proteomic Analyses
Next, we reasoned that context-specific protein-protein interactions could modulate the function of RIPEs. Therefore, we set out to affinity purify all of the individual RIPE components for subsequent characterization using MS (Figures 2A–2C). Since most meiotic noncrossovers and crossovers arise sequentially, during prophase I and at the prophase-to-metaphase I transition (Allers and Lichten, 2001, Clyne et al., 2003, Matos et al., 2011), we analyzed RIPEs from cultures synchronized in prophase I, in metaphase I, and from mitotically dividing cells (Figures 2A, 2B, and S2A). While metaphase I cells will have resolved most HR intermediates, they maintain the Ndt80-driven M phase environment that triggers crossing-over, including high levels of Cdc5 and the M phase cyclins Clb1 and Clb4 (Clyne et al., 2003, Lee and Amon, 2003). To obtain synchronous cultures, first we generated a library of strains carrying FLAG-tagged RIPE genes in combination with mutations in the transcription factor NDT80 (ndt80Δ) or the anaphase-promoting complex (APC/C) activator CDC20 (PCLB2-CDC20). When released to undergo meiosis, ndt80Δ mutants arrest meiotic progression in pachytene, while cells depleted of Cdc20 accumulate in metaphase I (Figure 2A) (Lee and Amon, 2003, Xu et al., 1995). Second, we constructed yeast fermenters that allow the preparation of large mitotic and meiotic cultures (Figures 2A and 2B). We then purified the 13 bait proteins from the 3 cellular contexts in 2 biological replicates and performed 15 control purifications from the parental untagged strains (Figures 2C and S2A). Before the final MS analyses, we verified by western blotting that each bait protein was similarly enriched in the 6 independent purifications (Figures 2D, S2B, and S2C; data not shown). Proteins in the anti-FLAG immune complexes were then analyzed on a high-resolution high-accuracy mass spectrometer, with a false discovery rate of <1%. To obtain a high-confidence set of interactors, the total number of spectral counts was processed by SAINT (Significance Analysis of INTeractome) probability scoring (Mellacheruvu et al., 2013). Applying a stringent score of ≥0.9 resulted in a total of 165 high-confidence RIPE-associated proteins across the 3 cellular contexts (Figure 2E; Tables S2, S3, and S4). Hierarchical clustering of the high-confidence binders was sufficient to group all of the purifications according to the predicted formation of protein complexes (Figure 2E; Table S5). Notably, the filtered datasets contained previously known interactors of the baits (Figure S3A), as well as numerous unanticipated interactions for most RIPEs (Figures 3A–3G). As an additional data curation step, we analyzed the dataset by 3 independent criteria: (1) annotated subcellular localization of the preys to the cytoplasm; (2) removal of proteins in the top 20th percentile of cellular abundance; and (3) prevalence in the Contaminant Repository for Affinity Purification Mass for anti-GFP and anti-hemagglutinin (HA) purifications. While applying these filters did not change the overall landscape of interactions, it did reduce the size of the Mus81 and Srs2 networks (Figure S4). This was particularly evident when proteins that localize predominantly to the cytoplasm were subtracted (Figure S4; Table S6).
Figure 2.

Affinity Proteomics of RIPEs during Mitotic Proliferation and Meiosis
(A) Workflow with the key steps in the generation of large and synchronous meiotic and mitotic cultures for subsequent affinity proteomics.
(B) Fluorescence-activated cell sorting (FACS) analysis of DNA content after the transfer of ndt80Δ and PCLB2-CDC20 cells into SPM. To follow chromosome synapsis and accumulation in pachytene, Zip1 is stained on chromosome spreads. The typical fraction (%) of cells with fully synapsed chromosomes, 8 h after transfer to SPM, is shown. The accumulation of cells in metaphase I is evaluated by the immunofluorescence analysis of spindle morphology. The typical percentage of cells with a bipolar spindle after 8 h in SPM is indicated; the observed value for each individual culture analyzed by MS is detailed in Figure S2A.
(C) Flowchart of the affinity-purification (AP)-MS approach used for this study.
(D) Protein extracts and immuno-affinity purified material (immunoprecipitates [IPs]) from large mitotic (asynchronously cycling) or meiotic (prophase I or metaphase I) cultures expressing Slx1-FLAG, Sgs1-FLAG, or untagged controls were analyzed by western blotting for the indicated proteins. Two independent biological replicates were analyzed per condition.
(E) Heatmap generated from the hierarchical clustering of 165 high-confidence interacting proteins identified by MS for the 13 RIPE components. The interactome of each bait protein was filtered using 5 independent negative controls per cellular context (5 mitotic, 5 prophase I, and 5 metaphase I). Preys with a SAINT score ≥0.9 are shown. The gradient scale at top left corresponds to the log2 enrichment of the preys based on spectral counts.
Figure 3.

Context-Specific Interaction Network of the RIPEs during Mitotic Proliferation and Meiosis
(A) High-confidence interactors with a SAINT score ≥0.9 are shown for Mph1. Left: composite with all of the interactions detected during mitotic proliferation, meiotic prophase I, and meiotic metaphase I. The highlighted lines depict the interactions previously reported in BioGRID: green, genetic interaction; red, physical interaction; blue, both genetic and physical. Center and right: context-specific network components. IPAs, number of interaction partners.
(B) As in (A) for Srs2.
(C) As in (A) for Sgs1, Top3, and Rmi1.
(D) As in (A) for Mus81 and Mms4.
(E) As in (A) for Yen1.
(F) As in (A) for Slx1-Slx4.
(G) As in (A) for Mlh1, Mlh3, and Exo1.
See also Figures S3 and S4 and Tables S2, S3, S4, S5, and S6.
Identification of Context-Specific Interactions of RIPEs
Mph1 and Srs2
MS analyses of FLAG-affinity purifications identified 61 high-confidence interactors of Mph1-FLAG (Figure 3A), including the histone fold proteins Mhf1 and Mhf2 (Xue et al., 2015), the Forkhead transcription factor Fkh1 (Dummer et al., 2016), and the telomere maintenance factor Mte1 (Silva et al., 2016, Xue et al., 2016, Yimit et al., 2016). Among the interactors we found Fkh2, which was not previously shown to associate with Mph1. We also detected the binding of several DNA repair factors known to be genetically linked to Mph1, including Rad52, Pif1, and the RIPE helicase Sgs1 (Figure S3A). Besides the identification of a high number of previously unknown interactions, our approach revealed a profound rewiring of the Mph1 interactome between mitotic proliferation and meiosis: 61 interactors during mitotic proliferation, 5 in prophase I, and only 3 in metaphase I (Figure 3A). These data suggest that Mhf1-Mhf2 and Mte1, which bind Mph1 constitutively, may have roles in regulating Mph1 function during meiosis. In contrast, delimited association of all of the other factors may be important for the control of context-specific functions of Mph1.
By contrast, we found a total of only 28 interactors in purifications of Srs2. With the exception of Cog1 (León Ortiz et al., 2011), no previous links have been established with the other Srs2 binders (Figures 3B and S3A). In contrast to Mph1, the vast majority of Srs2 interactors were detected during meiotic prophase I (22 of 28). In mitotically dividing cells we detected only 6 interactions, and none could be found during meiotic metaphase I (Figures 3B and S3A). Among the prophase I interactors, we noticed the presence of several proteins involved in DNA or RNA metabolism, including Rfc1, Ioc2, Ioc3, Tfg1, Tfg2, and Spo14. Since recent reports indicate that Srs2 functions during prophase I to ensure normal DNA joint molecule metabolism (Hunt et al., 2019, Sasanuma et al., 2019), it will be interesting to investigate whether these functions require its ability to interact with the above factors.
Sgs1-Top3-Rmi1
For the STR complex, we detected 20 interaction partners across the 3 cellular contexts analyzed (Figure 3C). The number of interactions did not change between mitotic proliferation and prophase I (9 interactors), but the identity of the proteins was markedly different. Analogous to the trends observed for Mph1 and Srs2, we observed a substantial reduction in interactions in metaphase I (3 interactors).
Genetic experiments have established that Sgs1, Top3, and Rmi1 share both anti- and pro-crossover functions during meiosis (Kaur et al., 2015, Tang et al., 2015). However, formation of the STR complex during meiosis has not been formally shown. Analyses of Sgs1, Top3, and Rmi1 purifications from different contexts strongly indicate that STR forms both during prophase I, when noncrossovers are made, and during metaphase I, when most crossovers are generated (Figure 3C). However, Sgs1 was not significantly enriched in Rmi1 and Top3 purifications from prophase I, whereas Rmi1 and Top3 could be readily detected in Sgs1 purifications (Figure 3C). This raised the possibility that only a small fraction of Top3-Rmi1 stably associates with Sgs1 (Figure S3B). In agreement with this idea, SYPRO ruby staining of the respective FLAG-affinity purifications showed that Rmi1-FLAG, which is significantly more abundant than Sgs1-FLAG, binds more Top3 than Sgs1-FLAG (Figures S3C and S3D). Besides demonstrating that our approach is capable of detecting changes in complex stoichiometry, these data are consistent with Top3-Rmi1 having Sgs1-independent functions during prophase I (Fasching et al., 2015, Kaur et al., 2015, Tang et al., 2015).
Structure-Selective Endonucleases: Mus81-Mms4, Slx1-Slx4, and Yen1
For Mus81-Mms4 we were unable to identify high-confidence interactors during mitotic proliferation and meiotic metaphase I. However, we detected 34 proteins enriched in Mus81 purifications from meiotic prophase I (Figure 3D). Several of the prophase I interactors have functions in regulating chromatin dynamics (e.g., Isw2, Hda1, Abs1, Pol3) and cell-cycle control (e.g., Mad1, Rim15, Spc110). However, none of them was shared with Mms4. This unexpected observation raises the possibility that Mus81-FLAG may not be fully functional (e.g., mislocalize to the cytoplasm) or that Mus81 may have Mms4-independent roles during prophase I.
The only interactor of Yen1 validated to date is the B-type cyclin Clb5, which promotes Yen1 phosphorylation during S phase (Blanco et al., 2014, Eissler et al., 2014). We identified Clb5 as a high-confidence interactor of Yen1 during both prophase I and metaphase I (Figure 3E). We also found Ypl088w to be significantly enriched in prophase I purifications of Yen1 (Figure 3E). Even though the function of this protein remains unknown, a previous systematic study reported that ypl088wΔ mutants display reduced spore viability and defective meiotic chromosome segregation (Marston et al., 2004). Thus, it will be interesting to determine whether Ypl088w and Yen1 share a functional relation.
We were unable to identify new interactors of Slx1-Slx4, but we did detect binding to both Rtt107 and Dpb11, which had been previously shown to physically associate with Slx4 during mitotic proliferation (Gritenaite et al., 2014, Ohouo et al., 2010) (Figure 3F). Both Dpb11 and Rtt107 could be found in purifications from prophase I and metaphase I, suggesting that they may also contribute to the function of Slx1-Slx4 nuclease during meiosis.
MutLγ-Exo1
Despite a well-established collaborative role in promoting crossing-over (Zakharyevich et al., 2012), it was unclear whether Mlh1, Mlh3, and Exo1 physically associate during meiosis. Therefore, we started by examining the interactions between the 3 proteins. Mlh1 and Mlh3 reciprocally associated in all of the cellular contexts investigated. Exo1 could also be detected as a high-confidence interactor of Mlh1 in mitotic and meiotic metaphase I cells, but not during prophase I. Finally, Exo1 and Mlh3 were not part of each other’s direct network of interactors in any of the cellular contexts (Figure 3G). Overall, these data suggest that a complex of MutLγ and Exo1 is likely to form in mitotic cells and during meiotic metaphase I, but may assemble to a lesser extent—or in a different configuration—during prophase I. Since Exo1 and Mlh3 were not detected in the reciprocal purifications, it remains possible that Mlh1 associates with Mlh3 or Exo1 in a mutually exclusive manner (Figure 3G).
Whereas Mlh1 bound to its known interactors Mlh2 and Pms1 in both mitotic and meiotic cells, Exo1 and Mlh3 associated with several factors exclusively during meiosis (Figure 3G). One particularly interesting interactor of Mlh3 was Rtk1, a putative kinase of unknown function, which was equally enriched in metaphase I purifications of Mlh1 (Figure 3G). The network of Exo1 also expanded significantly, with 6 new context-specific interactions identified, including the conserved chromatin remodeler Chd1 (Figure 3G).
Global Rewiring of the RIPE Network during Mitotic Proliferation and Meiosis
To visualize the entire landscape of interactions, we combined the individual interactomes into a global network (Figure 4A), which highlighted 2 key features of the dataset: (1) with the exception of Yen1, all RIPEs showed indirect interconnectivity with at least 1 other RIPE (Figure 4B), so we speculate that network “hubs” may exist to enable the coordinated regulation of multiple pathways and (2) analyses of the global network from different cellular contexts exposed a concerted remodeling of interactomes, which was especially striking for meiotic metaphase I (Figure 4C). While most RIPEs showed a marked loss of interactions, MutLγ-Exo1 displayed a substantial gain (12 metaphase I-specific components). This increase was particularly significant, considering that the entire metaphase I network contained only 36 proteins, substantially fewer than the 86 and 89 components found in mitosis and prophase I, respectively (Figure 4C). A semiquantitative analysis of the 165 RIPE-associated proteins further confirmed the context-specific enrichment of the preys in the respective purifications (Figures S5A–S5C). Thus, we conclude that the RIPE network is extensively rewired according to the cellular context. Overall, this suggests that mitosis- and meiosis-specific interactions are a prime candidate mechanism to regulate pathway use and HR outcome.
Figure 4.

Overview of the RIPE Network during Meiosis and Mitosis
(A) Global RIPE network during mitosis and meiosis. A total of 165 high-confidence interactions (SAINT score ≥0.9) detected after affinity proteomics are shown for all of the bait proteins. IPAs, number of interaction partners.
(B) Subset of the RIPE network from (A) that interacts with >1 bait protein.
(C) Global RIPE network during asynchronous mitotic proliferation, meiotic prophase I, and meiotic metaphase I.
A Functional Screen Identifies Network Components Required for Meiotic Crossing-Over
To investigate whether context-specific network components regulate HR, we focused on the interactors of MutLγ-Exo1 (Figure 5A). Using spore autonomous fluorescence (Figure 5B) (Thacker et al., 2011), we monitored genetic distance, a measure of crossover frequency, at the CEN8-THR1 interval in deletion mutants for several of the genes encoding for metaphase I-enriched preys (Figure 5C). Notably, 3 of the 5 mutants analyzed displayed a reproducible reduction in genetic distance: chd1Δ, rtk1Δ, and caf120Δ (Figures 5C and S6A; Table S1). Moreover, chd1Δ mutants showed a particularly strong phenotype, which was comparable to the deletion of MLH1, MLH3, or EXO1 (Figure 5C) (Arter et al., 2018).
Figure 5.

Chd1 Promotes the Formation of MutLγ-Exo1-Dependent Crossovers Genome-wide
(A) MutLγ-Exo1 interactors selected for functional analysis.
(B) Schematic representation of a fluorescence reporter assay to measure the crossover (CO) recombination at CEN8-THR1. Homologous chromosomes are shown in light and dark gray, with GFP, tdTomato, and CFP reporters represented in green, red, and cyan, respectively.
(C) Meiosis was induced in SPM plates for 48 h at 30°C. Genetic distances at the CEN8-THR1 interval were determined using the fluorescent markers described in (B). More than 600 tetrads were analyzed in 3 independent experiments. The plotted values indicate means ± SDs (2-tailed, unpaired t test; ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, and ∗∗∗∗p < 0.0001). A correction for multiple comparisons using statistical hypothesis testing (Dunnett test) was used.
(D) Western blot analysis of the indicated proteins in soluble extracts and anti-myc IPs from PCLB2-CDC20 strains expressing Chd1-myc9 and Exo1-FLAG or Chd1 and Exo1-FLAG. Samples were collected 8 h after the induction of meiosis.
(E) Genetic distances at the CEN8-THR1 interval were determined for strains with the indicated genotypes. The plotted values indicate means ± SDs (2-tailed, unpaired t test; n.s., not statistically significant p > 0.05; ∗∗∗∗p < 0.0001).
(F) Rationale of genome-wide mapping of recombination events by the analysis of SNPs after sequencing germinated spores resulting from hybrid meiosis.
(G) Scheme of meiotic recombination showing detected crossover (CO) and noncrossover (NCO) outcomes and minority events. In the subsequent analysis, E1 and E4 events classify as NCOs, whereas E2, E3, E5, E5A, E6, and E7 events classify as COs. E8 events may represent 2 overlapping NCOs or may arise from pre-meiotic recombination.
(H) Average number of COs and NCOs in wild type, chd1Δ, and mlh3Δ per tetrad are shown. The mlh3Δ data shown are from Arter et al. (2018). A total of 6 wild-type, 5 chd1Δ, and 4 mlh3Δ tetrads have been analyzed.
(I) The number of events depicted in (G) per tetrad, in strains with the indicated genotypes. The plotted values indicate means ± SDs (2-tailed, unpaired t test; ∗p < 0.05 and ∗∗p < 0.01).
(J) Ratio of COs to NCOs in strains of the indicated genotypes.
(K) The mean number of crossover events detected per chromosome in strains with the indicated genotypes. Chromosomes are distributed along the x axis according to size (kb).
(L) Histogram analysis of the distances between adjacent COs in wild-type cells. Inter-crossover distances are well fit by a γ distribution. The value of the shape parameter γ of the best-fit distribution indicates the strength of interference, with γ > 1 indicating positive interference and γ = 1 indicating random distribution. A total of 6 tetrads were analyzed.
(M) 5 chd1Δ tetrads were analyzed as in (L).
(N) 4 mlh3Δ tetrads were analyzed as in (L).
Since Chd1 physically associated with Exo1 (Figure 5A) and the reciprocal interaction could be confirmed (Figure 5D), we suspected that Chd1 may contribute to the generation of MutLγ-Exo1-dependent crossovers. Chd1 is a highly conserved chromatin remodeler, with important roles in nucleosome assembly and array spacing (Smolle, 2018), yet without any reported functions in meiosis. Consonant with the physical interaction detected, mlh1Δ, chd1Δ, and chd1Δ mlh1Δ double mutants displayed a similar reduction in genetic distance, suggesting that Chd1 is a component of the MutLγ-Exo1 pathway (Figure 5E; Table S1). By contrast, the deletion of CHD1 in an mus81Δ background led to a reduction in genetic distance, placing Chd1 and Mus81 in 2 separate pathways (Figure 5E).
To further investigate whether Chd1 is generally required for crossing-over, we monitored recombination after homothallic switching (HO) endonuclease-mediated double-strand break (DSB) formation in mitotically dividing cells (Ira et al., 2003). chd1Δ cells repaired DSBs efficiently and generated similar levels of ectopic crossover products (XO) as detected for control cells (Figure S6B). These data suggest that Chd1 is dispensable for mitotic crossing-over, which is almost entirely dependent on Mus81-Mms4 and Yen1 (Ho et al., 2010).
Chd1 Is Required for Efficient Crossing-Over, but Dispensable for the Formation of NonCrossovers, Genome-wide
To explore the involvement of Chd1 in regulating meiotic HR genome-wide (Chen et al., 2008, Mancera et al., 2008), we turned to next-generation sequencing (Figures 5F and 5G). In support of the CEN8-THR1 data, chd1Δ mutants exhibited a global decrease in crossover frequency (Figure 5H). This defect, caused by a specific drop in simple crossover events (Figure 5I, E2 events), occurred in the absence of a detectable change in the number of noncrossovers (Figures 5H and 5I, E1 events). Consequently, chd1Δ mutants displayed a reduction in the crossover:noncrossover ratio (Figure 5J). As predicted from the results obtained at CEN8-THR1 (Figure 5C), chd1Δ and mlh3Δ mutants displayed similar phenotypes, mainly characterized by a reduction in E2-type crossovers in all chromosomes (Figure 5K). To our surprise, however, analysis of the inter-crossover distances revealed an interesting difference between chd1Δ and mlh3Δ mutants. mlh3Δ cells showed partially defective spacing of crossovers, as revealed by the γ distribution of inter-joint molecule distances (γ = 1.90 in wild type and γ = 1.41 in mlh3Δ [Arter et al., 2018]). In contrast, chd1Δ cells generated widely spaced crossovers (γ = 2.02), with inter-crossover distances being slightly higher than in wild-type cells (Figures 5L–5N). Since γ is sensitive to changes in crossover (CO) density, we also analyzed interference using the coefficient of coincidence (CoC) method, in which the frequency of COs in 2 intervals is compared with the expected frequency of double COs under an assumption of no interference. Interference, expressed as 1 − CoC, also showed a significant decrease in mlh3Δ, but not in chd1Δ mutants (for the 25-kb bin size: wild type = 0.61, mlh3Δ = 0.50, and chd1Δ = 0.25; Figure S6C). Thus, we infer from these data that Chd1 is not necessary for all of the functions of Mlh3, and presumably also Mlh1 and Exo1. Overall, the data above confirm that Chd1 is required for meiotic recombination by promoting crossing-over genome-wide. However, Chd1 appears to be dispensable for the process of crossover patterning, which is thought to occur long before Holliday junction resolution (Bishop and Zickler, 2004, Hunter, 2015), and is partly dependent on Mlh3.
Chd1 Functions at the Prophase-to-Metaphase I Transition to Promote Crossing-Over
To examine whether Chd1 functions during meiosis to promote crossing-over, we engineered strains expressing Chd1 from the mitosis-specific promoter PCLB2 (Lee and Amon, 2003). PCLB2-CHD1 cells accumulated Chd1 during mitotic proliferation, but showed an abrupt reduction in protein levels as cells initiated pre-meiotic S phase (Figures 6A and S7A, left panels). Analysis of the genetic distance in PCLB2-CHD1 mutants revealed a significant decrease in crossover frequency, indicating that the meiotic expression of Chd1 is required for crossing-over (Figure 6B; Table S1). Since Chd1 was enriched in Exo1 purifications from metaphase I (Figure 3G), we then asked whether the expression of Chd1 at the prophase-to-metaphase I transition would suffice to support meiotic HR. To this end, we replaced the promoter of CHD1 by the promoter of the M phase cyclin CLB1 (Chu and Herskowitz, 1998). When expressed from PCLB1, Chd1 accumulated with similar kinetics to Polo kinase Cdc5, which drives the exit from pachytene (Figures 6A and S7A, right panels) (Clyne et al., 2003, Sourirajan and Lichten, 2008). PCLB1-CHD1 cells presented wild-type values of genetic distance, indicating that the late expression of Chd1 is sufficient to support crossing-over (Figure 6B). To confirm these data, we also generated strains carrying Chd1 under the control of an inducible promoter (PCUP1-CHD1) (Figure S7B). In agreement with the experiments using PCLB1-CHD1, the induction of Chd1 after joint molecule accumulation (7 h in SPM) was sufficient to restore the genetic distance at CEN8-THR1 (Figures S7B–S7E). Finally, we monitored the kinetics of DSB formation and joint molecule accumulation at the HIS4-LEU2 interval in chromosome III. Consonant with a model in which Chd1 promotes the resolution of joint molecules into crossovers, both DSBs and joint molecules accumulated efficiently in chd1Δ cells (Figures 6C, 6E, 6F, and S7F), while crossover formation, measured genetically, was reduced (Figure 6D). Further strengthening these observations genome-wide, chd1Δ mutants accumulated chromatin-bound Zip3, which is thought to mark future crossover sites (Fung et al., 2004, Zhang et al., 2014) (Figures S7G and S7H).
Figure 6.

Chd1 Remodels Meiotic Chromatin at the Prophase-to-Metaphase I Transition to Enable MutLγ-Exo1-Dependent Processing of Recomination Intermediates
(A) Western blot analysis of the indicated proteins in trichloroacetic acid (TCA) extracts from a meiotic time course of cells expressing PCLB2-ha3-CHD1 (left) or PCLB1-ha3-CHD1 (right). Samples were collected at 2-h intervals after the induction of meiosis by transfer into SPM. As, asynchronously proliferating cells.
(B) Meiosis was induced in strains with the indicated genotypes for 48 h at 30°C. Genetic distances at the CEN8-THR1 interval were determined using the fluorescent markers described in Figure 5B. A total of 600 tetrads were analyzed per strain, in 3 biological replicates. The plotted values indicate means ± SDs.
(C) Illustration of the HIS4-LEU2 hotspot, displaying the flanking markers in chromosome III.
(D) Genetic distances for the interval shown in (C) for strains with the indicated genotypes. The plotted values indicate means ± SEMs.
(E) Southern blot analysis of recombination at the HIS4-LEU2 locus in ndt80Δ CHD1 or ndt80Δ chd1Δ mutants. DNA double-strand break (DSB) formation was quantified as a fraction of the total lane signal from Figure S7F and from a biological replicate. The plotted values show the mean of 2 independent experiments; error bars represent range.
(F) The accumulation of DNA joint molecules was quantified from the cells analyzed in (E).
(G) Genetic distances at CEN8-THR1 for strains expressing Yen1WT, or constitutively active Yen1ON, as in (B).
(H) Chromosome spreads were prepared from cells expressing Chd1-myc9, 7 h after the induction of meiosis, and stained for DNA, Zip1, and Chd1-myc9. Pachytene cells were identified by full Zip1 loading. After image deconvolution, Chd1 appears to be enriched in discrete chromosomal regions, but a basal signal can be detected throughout the whole chromatin.
(I) Analysis of Chd1 foci in chromosome spreads from (H). The horizontal line depicts the median number of Chd1 foci per cell. A total of 20 cells were analyzed. The experiment shown is representative of 2 independent experiments.
(J) The domain architecture of Chd1. The key residues at the boundaries of domains are indicated. The key residues required for the function of the respective domains are highlighted.
(K) Western blot analysis of Chd1-myc9 protein levels in chromodomain (E220L), ATPase motor (D513N), and DNA binding (R1016A, K1020A, and R1255A) mutants expressed from the endogenous locus.
(L) Genetic distances at CEN8-THR1 for strains with the indicated genotypes, as in (K).
The plotted values indicate means ± SDs (2-tailed, unpaired t test; n.s., not statistically significant p > 0.05; ∗∗∗∗p < 0.0001), as in (B).
The Chromatin Remodeling Properties of Chd1 Promote the Formation of MutLγ-Exo1-Dependent Crossovers
The data above indicate that Chd1 plays a specific role in enabling the crossover resolution of joint molecules by MutLγ-Exo1. However, chd1Δ cells still recruited Mlh1, Mlh3, and Exo1 to pachytene chromosomes (Figures S7I and S7J; data not shown). Thus, rather than regulating the expression or bulk chromatin recruitment of MutLγ-Exo1, Chd1 is likely to support the enzymatic activation of MutLγ. If so, then ectopic expression of a constitutively active resolvase should bypass the requirement for Chd1 in promoting crossing-over. To test this model, we generated chd1Δ strains expressing constitutively active Yen1 resolvase (Yen1ON) (Figure S7K) (Arter et al., 2018). chd1Δ YEN1ON double mutants displayed a significantly improved genetic distance, confirming that Chd1 is dispensable for the formation of HR intermediates that act as crossover precursors (Figure 6G; Table S1).
The finding that Yen1 activity can restore crossing-over in chd1Δ mutants means that Chd1 must be specifically required for joint molecule resolution by MutLγ. Since Chd1 is an ATP-dependent chromatin remodeler that binds and shifts nucleosomes (Lusser et al., 2005, Qiu et al., 2017, Smolle, 2018), its function in promoting crossing-over could be to displace nucleosomes to facilitate MutLγ function. One key prediction of this model, however, is that the chromatin-binding and -remodeling properties of Chd1 should be required for crossing-over. To test this prediction, we first asked whether Chd1 associated with meiotic chromosomes on surface spreads. Chd1-myc9 could be readily detected in cells with synapsed chromosomes (Figures 6H, 6I, and S7L). Next, we generated point mutants of CHD1 that interfere with the function of the chromodomain (E220L [Pray-Grant et al., 2005]), the ATPase motor (D513N [Hauk et al., 2010]), or the DNA-binding region (R1016A, K1020A, R1255A [Ryan et al., 2011]) (Figure 6J). While all of the versions of Chd1 were expressed at comparable levels (Figure 6K), only wild-type CHD1 was capable of restoring the genetic distance in chd1Δ mutants (Figure 6L; Table S1). Thus, these data suggest that Chd1 remodels meiotic chromatin—at the prophase-to-metaphase I transition—to enable MutLγ-Exo1-dependent processing of joint molecules into crossovers.
Discussion
In this study, we set out to investigate how cells regulate the processing of recombination intermediates according to the specialized needs of mitotic proliferation and meiosis. To this end, we used a biochemical approach to survey the composition of individual RIPEs in both their mitotic and meiotic environments. This effort, which is of unprecedented scale in the meiosis field, led to the generation of a unique map of interactions, the RIPE network, which we explored as a resource to identify and functionally characterize factors required for HR.
The RIPE Network: A Rich Resource for Regulators of DNA Repair
The first important insight from the RIPE network is that it provides direct evidence for the assembly of the STR and MutLγ-Exo1 complexes during meiosis, as hinted by genetic experiments (Kaur et al., 2015, Tang et al., 2015, Zakharyevich et al., 2012). Our data, however, also suggest that the composition of both complexes is subject to regulation. We find that Mlh1-Mlh3 forms constitutively, whereas the Mlh1-Exo1 association appears to be reduced during meiotic prophase I. Thus, it is tempting to speculate that the specific activation of MutLγ-Exo1 at the onset of meiosis I may be linked to the transient dissociation of Exo1 during prophase I. This could contribute to the temporal control of Holliday junction resolution and Ndt80 and Cdc5-dependent generation of type I crossovers (Allers and Lichten, 2001, Zakharyevich et al., 2012). Our data also establish that prophase I cells contain Top3-Rmi1 complexes that are apparently devoid of Sgs1. This is particularly interesting in light of recent studies showing that Top3 and Rmi1 have Sgs1-independent functions (Fasching et al., 2015, Kaur et al., 2015, Mullen et al., 2005, Tang et al., 2015). Future work should now focus on determining whether Sgs1 may interfere with Top3-Rmi1-specific functions, thus entailing the controlled formation of STR in cells. Of note, our dataset also provides biochemical evidence that Slx1-Slx4, Mph1-Mhf1, Mph1-Mhf2, Mph1-Mte1, Mph1-Fkh1, and Mph1-Fkh2 complexes assemble during meiosis.
Having recovered many previously identified interactions, the RIPE network also includes numerous unanticipated binding partners (146 in total) for most RIPEs. Moreover, the network reveals the existence of interconnectivity between RIPEs (Figure 4B). Although only 1 potentially direct interaction between Mph1 and Sgs1 was detected, many RIPEs shared interactors, suggesting the existence of mechanisms for pathway co-regulation. For instance, the DNA helicases Srs2 and Mph1 have 5 mitosis-specific interactors in common (Figure 4C). Since the known roles of Srs2 and Mph1 in promoting synthesis-dependent strand annealing are limited to mitotic cells (Ira et al., 2003, Prakash et al., 2009), it will be interesting to determine whether this is functionally connected to the absence of such interactions during meiosis.
The RIPE network provides a panoramic view into the interaction landscape of RIPEs in yeast. We have only functionally tested 5 RIPE network genes, which leaves >100 others for future studies. In addition, it is important to consider that the assays used here can be modified to investigate other potential roles of RIPEs. For example, the RIPE network should be of use to identify genes required for mitotic recombination, or, for example, to identify factors generally required for DNA repair or resilience to replication stress. Some of the interactions may also relate to unforeseen functions of RIPE components. These may provide unexpected new insight into the links between the mutation of RIPE genes and human disease, including Fanconi anemia (SLX4/FANCP and FANCM) and BLM.
RIPE Network Rewiring during Mitotic Proliferation and Meiosis
A remarkable feature of the RIPE network is its dynamic rewiring according to cellular context (Figures 4C and S5A–S5C). While the abundance of RIPE components does not change significantly between mitotic proliferation and meiosis, interactomes change extensively and appear to be modified in a concerted manner. For example, during meiotic metaphase I, the detectable RIPE network becomes reduced to 34 components, from the global 165 (Figure 7A). Most of the interactions center around MutLγ-Exo1, the enzyme responsible for generating type I crossovers (Figure 7A) (Zakharyevich et al., 2012). In addition, we verified that 3 of the meiosis-specific interactors of MutLγ-Exo1 were required for efficient crossing-over (Figure 5C). Thus, we propose that remodeling the RIPE interactomes is of fundamental importance in establishing pathway usage and, as such, in regulating HR outcome (Figure 7A).
Figure 7.

Remodeling the RIPE Network for Meiotic Crossover Formation
(A) Simplified model depicting the pathways of DNA joint molecule processing and expected recombination outcomes. Left, global network with interactions detected for RIPEs across different cellular contexts. Right, metaphase I RIPE network, with widespread rewiring of the interactomes. Highlighted interactions of MutLγ-Exo1 are required for crossing-over.
(B) Chd1 remodels chromatin to promote MutLγ-Exo1-dependent crossing-over. In the model depicted, Chd1 displaces nucleosomes to enable the polymerization of MutLγ at crossover-designated sites. MutLγ polymerization leads to Mlh3 nuclease activation, DNA cleavage, and crossing-over. In the absence of Chd1 or in the absence of its ability to bind and displace nucleosomes, MutLγ is unable to channel HR intermediates into crossovers. Ndt80 triggers the exit from pachytene and promotes Exo1-Chd1 association by an unknown mechanism.
Besides Chd1, whose role in meiotic crossing-over we have characterized in greater detail, Rtk1 and Caf120 are interesting candidates for further functional analyses. The former is a particularly exciting factor, as it lacks any reported functions. Rtk1, which associated with both Mlh1 and Mlh3 during metaphase I, has a putative kinase domain. Thus, it will be important to investigate whether Rtk1 is a bona fide kinase and subsequently determine whether it acts to phosphorylate MutLγ-Exo1. Caf120 has been shown to associate with a transcriptional regulatory complex (Chen et al., 2001) and thus may have a more indirect role in regulating HR.
One exciting prediction from our work is the existence of cellular mechanisms that rewire the RIPE network according to cellular context. One of the prime candidate mechanisms is the simple control of protein abundance, which could change for the bait proteins. This is, however, clearly not the only mechanism. For example, Chd1 interacts with Exo1 preferentially during meiotic metaphase I, but its expression levels do not change (Figure S7L). As pointed out above, we noticed that the electrophoretic mobility of Exo1 changes during meiosis, indicating that Exo1 is post-translationally modified just before entry into meiosis I (Figure 1B). It is therefore possible that Exo1 modification controls its ability to interact with Chd1 (as well as with MutLγ). The nature of this modification and its functional relevance will be interesting topics of future research. Related regulatory mechanisms may also apply to Sgs1, which is post-translationally modified at the onset of meiosis I (Figures 1B and 2D).
Despite the multiple stringent controls used to generate the RIPE network, it is important to consider that some of the interactions detected may be indirect, mediated by other components of the network, or occur post-cell lysis. Thus, careful interpretation and further validation of the data are recommended. However, the extensive and context-specific characterization of the contaminant proteome, the FLAG-affinity “CRAPome” (Tables S2, S3, and S4), should help others in filtering specific interactors from purifications performed under similar conditions.
Chd1 Remodels Chromatin to Enable MutLγ-Exo1-Dependent Crossing-Over
Our genetic data strongly suggest that Chd1 promotes the formation of MutLγ-Exo1-dependent crossovers throughout the genome, consistent with the binding to Exo1 during metaphase I. Our results also establish that Chd1 functions specifically at the prophase-to-metaphase I transition (Figures 6A and 6B). Thus, we propose that Chd1 regulates the very last step of HR, enabling the nucleolytic processing of joint molecules by MutLγ. In agreement with this model, premature activation of Yen1 resolvase is sufficient to restore normal crossover levels in the absence of Chd1. Besides confirming that Chd1 is dispensable for the formation of HR intermediates that act as crossover precursors, this result also means that Chd1 is specifically required for joint molecule resolution by MutLγ-Exo1.
How does Chd1 promote MutLγ-Exo1 function? As has been recently proposed by others, the MutLγ complex needs to assemble into a polymer to become competent for DNA nicking in vitro (Figure 7B) (Manhart et al., 2017). For oligomerization to occur in the context of chromatin, one would envisage that protein barriers adjacent to the initial MutLγ-binding sites would have to be displaced to enable polymer growth and nuclease activation. Since Chd1 is a conserved chromatin remodeler (Lusser et al., 2005, Qiu et al., 2017, Smolle, 2018), we suggest that its ATPase motor and chromodomain may act to displace nucleosomes and facilitate MutLγ oligomerization on DNA (Figure 7B). Since the binding of Chd1 to Exo1 and the binding of Exo1 to MutLγ are temporally regulated, this would result in the licensing of “dormant” MutLγ complexes to polymerize specifically at the prophase-to-metaphase I transition, which coincides with the formation of most type I crossovers (Allers and Lichten, 2001) (Figure 7B). This model would be consistent with the observed difference between chd1Δ and mlh3Δ mutants in terms of spatial crossover distribution. Chd1-independent recruitment of MutLγ to maturing joint molecules would contribute to crossover designation and thus promote spatial crossover patterning. In contrast, Chd1 would only support a second—late—function of MutLγ: nuclease activation and nucleolytic processing of crossover-designated HR intermediates.
Finally, one important implication of our model is that nucleosomes may serve as natural barriers to the uncontrolled polymerization of MutLγ on chromatin (Figure 7B). As such, the nucleosome-mediated inhibition of enzyme oligomerization on DNA may constitute a general mechanism to temporally uncouple enzyme-DNA binding from enzyme activation and DNA processing.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rat monoclonal anti-tubulin | Serotec | Cat#MCA78G |
| Rabbit polyclonal anti-Zip1 | Santa Cruz | Cat#sc-33733 |
| Rabbit polyclonal anti-Myc | Santa Cruz | Cat#sc-789 |
| Donkey anti-Rat IgG Alexa Fluor 488 | Invitrogen | Cat#A-21208 |
| Goat anti-Rabbit IgG Alexa Fluor 546 | Invitrogen | Cat#A-11010 |
| Donkey anti-Rabbit IgG Alexa Fluor 488 | Invitrogen | Cat#A-21206 |
| Donkey anti-Mouse IgG Alexa Fluor 555 | Invitrogen | Cat#A-31570 |
| Donkey anti-Mouse IgG Alexa Fluor 647 | Invitrogen | Cat#A-31571 |
| Goat anti-Rat IgG Alexa Fluor 568 | Invitrogen | Cat#A-11077 |
| Rabbit polyclonal anti-Myc (HRP) | Abcam | Cat#ab1326 |
| Mouse monoclonal anti-FLAG (HRP) | Sigma Aldrich | Cat#a8592 |
| Goat polyclonal anti-Cdc5 | Santa Cruz | Cat#sc-6732 |
| Mouse monoclonal anti-Cdc5 (4F10) | MEDIMABS | #MM-0192-1-100 |
| Rabbit polyclonal anti-Crm1 | K. Weis (ETHZ) | N/A |
| Mouse monoclonal anti-Myc 9E10 | Cancer Research UK | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| β-Estradiol | Sigma Aldrich | Cat#E8875 |
| Copper(II) sulfate | Sigma Aldrich | Cat#61230 |
| Trioxsalen | Sigma Aldrich | Cat#T6137 |
| ProLong™ Diamond Antifade Mountant with DAPI | Thermo Fisher | Cat#P36962 |
| NuPAGE sample buffer | Thermo Fisher | Cat#NP0008 |
| 100 kU Nuclease | Pierce Universal Nuclease | Cat#88702 |
| cOmplete protease inhibitor cocktail | Roche | Cat#05056489001 |
| Anti-FLAG M2 Magnetic Beads | Sigma Aldrich | M8823 |
| Critical Commercial Assays | ||
| AminoLink Plus Immobilization Kit | Thermo Fisher | Cat#44894 |
| Qubit dsDNA broad range kit | Thermo Fisher | Cat#Q32850 |
| Deposited Data | ||
| Sequencing datasets | This study | NIH Sequence Read Archive under accession number PRJNA505664 |
| Mass spectrometry datasets | This study | ProteomeXchange Consortium via PRIDE with the dataset identifier PXD012486 |
| Original imaging data: western blots, Southern blots and Immunofluorescence | This study | https://doi.org/10.17632/fshnb5swnv.1 |
| Experimental Models: Organisms/Strains | ||
| All strains used in this study are listed in Table S7 | ||
| Software and Algorithms | ||
| ReCombine | (Anderson et al., 2011) | |
| Stahl lab online tools | (Stahl and Lande, 1995) | http://elizabethhousworth.com/StahlLabOnlineTools |
| Crapome | Mellacheruvu et al., 2013 | http://www.crapome.org |
Lead Contact and Materials Availability
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Joao Matos (joao.matos@bc.biol.ethz.ch).
Experimental Model and Subject Details
All strains were SK1, tGI354, YJM789 or S96 derivatives, as detailed in Table S7. The following alleles have been described previously: ndt80Δ, PGAL1-NDT80, PGPD1-GAL4-ER, mus81Δ, mms4Δ, mlh1Δ, mlh3Δ, yen1Δ, slx1Δ, slx4Δ, sgs1Δ, srs2Δ, PCLB2-CDC20, spore-autonomous fluorescent markers for the live-cell recombination assays, HIS4-LEU2 alleles for physical analysis of recombination, YEN1ON (Arter et al., 2018, Kim et al., 2010, Matos et al., 2011, Petronczki et al., 2006, Thacker et al., 2011). The CLB2 promoter was used for meiosis-specific depletion of Chd1 (Lee and Amon, 2003). The CLB1 promoter was used for Ndt80-dependent expression of Chd1 during meiosis. To this end, we replaced the promoter of CLB2 in pFA6a-KanMX6-pCLB2-3HA, by the promoter of CLB1 (−1000 to +6). Strains carrying copper-inducible CHD1 (PCUP1-CHD1-Myc9) were generated by one-step promoter replacement in a strain carrying CHD1-Myc9. Plasmids carrying chd1E220L, chd1D513N and chd1R1016A, K1020A, R1255A were generated by site-directed mutagenesis of pRS306 carrying CHD1WT under the control of its natural promoter (500 bp upstream of ATG) and terminator (500 bp downstream of STOP) sequences. Reconstitution of chd1Δ strains with CHD1WT (or with chd1 mutants) was performed by integration of the respective pRS306 vector variants into the promoter region of CHD1. For C-terminal PCR-based tagging of chromosomal genes with the His6-6xFLAG (referred to in the text as FLAG) and 9xMyc (referred to as myc9) cassettes were amplified from plasmids as described (Grigaitis et al., 2018, Knop et al., 1999). Gene deletions were introduced into SK1 by PCR-based amplification of cassettes from the yeast knock-out collection.
Method Details
Meiotic time courses and cycling cultures
Meiotic time courses were performed with diploid SK1 strains produced by mating of the MATa and MATα haploids, as previously described (Oelschlaegel et al., 2005, Petronczki et al., 2006). In brief, cells selected on YP2%glycerol plates for two days at 30°C were spread on YPD plates and grown for ∼24 hr to form a lawn. Cells were further expanded on YPD plates and used to inoculate pre-sporulation medium YP2%KAc to OD600 ∼0.3. Cells were grown for either 15 h (25°C) or 11 h (30°C), washed with pre-warmed sporulation medium (SPM, 2% KAc) and inoculated into SPM to OD600 3.5-4.0. This time defines t = 0 hr in all meiotic time course experiments. Large meiotic cultures were prepared after scaling up of the protocol above and using a 10L fermenter system as previously detailed (Grigaitis et al., 2018).
Asynchronous mitotic cultures were generated by inoculating the relevant amount of YPD (50 mL for TCA extracts and 6 L for AP-MS experiments) with an exponentially growing culture (OD600 ∼1.2), to an OD600 of ∼0.2. Cells were grown for ∼2.5 generations and harvested at OD600 ∼1.2. Exponential growth was monitored by FACS analysis of DNA content to ascertain cell cycle stage distribution. Induction of Ndt80 expression in ndt80Δ PGAL1-NDT80 cells was initiated by addition of 1 μM β-estradiol. Induction of Chd1 expression from the copper-inducible promoter was initiated by addition of 1 μM CuSO4.
FACS Analysis of DNA content
Cellular DNA content was determined using a FACSCalibur cytometer (Becton Dickinson) running CellQuest software. Briefly, 1 mL of meiotic culture was collected and cells were fixed in 70% Ethanol. Cells were washed once in 50 mM Tris-HCl pH 7.5 and resuspended in 50 mM Tris-HCl pH 7.5. RNA was digested for at least 4 h at 37°C (2 μl RNase (100 mg/ml)). Cells were washed once in FACS buffer (200 mM Tris-HCl pH 7.5, 211 mM NaCl, 78 mM MgCl2) and sonicated in FACS buffer containing 50 μg/ml propidium iodide. An aliquot (40-60 μl) was diluted in 1 mL 50 mM Tris-HCl pH 7.5 and DNA content measured.
Protein purification from mitotic and meiotic cultures
FLAG-affinity purifications were prepared from ∼3-6 L of yeast culture. Cell pellets were resuspended in 80 mL of lysis buffer (25 mM HEPES pH 8.0, 150 mM KCl, 2 mM MgCl2, 1 mM NaF, 0.1 mM EDTA pH 8.0, 0.5 mM EGTA pH 8.0, 15% glycerol, 0.1% NP-40, 20 mM β-glycerphosphate) containing freshly added 1 mM DTT and protease inhibitors (2 mM PMSF, 1 tablet/50 mL inhibitor cocktail (Roche)) and disrupted using a Freezer Mill (SPEX SamplePrep 6870) with the following settings: pre-cool (2 min), run time (3 min), cool time (2 min), cycles (6), rate (15 CPS). The obtained yeast powder was resuspended in 80 mL lysis buffer and, after addition of 100 kU Nuclease (Pierce Universal Nuclease), incubated for 1 h at 4°C while rotating. Cell lysates were cleared in two consecutive centrifugation steps, first at 3220 g for 10 min and then at 38800 g for 30 min at 4°C. After protein normalization (10 mg/ml in 65 ml) FLAG-tagged bait proteins were captured on magnetic anti-FLAG beads (Sigma Aldrich) while rotating for 90 min at 4°C. The immuno-affinity purified material was washed three times with 30 mL lysis buffer and three times with 30 mL wash buffer (25 mM HEPES pH 8.0, 150 mM KCl) prior to analysis by mass spectrometry or western blotting.
On beads digestion and peptide clean-up for MS analysis
The immune-affinity purified material was resuspended in 30 μl 0.1 M ammonium bicarbonate pH 8.0 and incubated with 500 ng of LysC (0.5 μl) for 1 h at 32°C. For disulfide reduction 1 mM TCEP (Tris(2-carboxyethyl) phosphine) was added to the supernatant (eluted peptides) and incubated for 30 min at 37°C. Free sulfhydryl groups were alkylated with 10 mM iodoacetamide for 30 min at room temperature in the dark. Initially, the proteins were digested with 300 ng LysC for 5 h followed by overnight incubation with 250 ng trypsin at 25°C. After acidification with 2% formic acid (pH 2.0), peptide clean-up was achieved using C-18 ZipTips (Millipore). Prior to peptide loading ZipTips were equilibrated with 80% acetonitrile and 0.1% formic acid. Bound peptides were washed with 0.1% formic acid and eluted with 80% acetonitrile/0.1% formic acid.
MS data acquisition and analysis
Peptide samples were analyzed on an Orbitrap Q Exactive Plus mass spectrometer (Thermo Fisher Scientific) equipped with a nanoelectrospray ion source and a nano-flow LC system (Easy-nLC 1000, Thermo Fisher Scientific). Peptides were separated on a 40 cm x 0.75 mmi.d. column (New Objective, PF360-75-10-N-5) packed in house with 1.9 um C18 beads (Dr. Maisch Reprosil-Pur 120). The following gradient of an acetonitrile/water mix was used for separation: linear from 5 to 8% buffer B over 2 minutes, linear from 8 to 25% buffer B over 68 minutes, linear from 25 to 40% buffer B over 10 minutes, linear from 40 to 90% buffer B over 5 minutes and isocratic for 5 minutes. Buffer A was 0.1% formic acid and buffer B was 0.1% formic acid in 100% acetonitrile. The flow rate was 300 nL/min and the column was heated to 50°C. The mass spectrometer was operated in data-dependent acquisition mode.
MS1 spectra were acquired from 350 to 1500 m/z at a resolution of 70000. The 20 most intense precursors were selected for Collision-induced dissociation fragmentation and the corresponding MS2 spectra were acquired at a resolution of 17500 using maximally 100000 ions, collected for maximally 55 ms. All multiply charged ions were used to trigger MS-MS scans followed by a dynamic exclusion for 30 s. Singly charged precursor ions and ions of undefinable charged states were excluded from fragmentation.
The collected DDA spectra were searched against the S. cerevisiae S288C reference proteome Uniprot FASTA database (Version: November 2015) and a list of common protein contaminants (exported from the MaxQuant software proteomics package) using the Sorcerer-SEQUEST database search engine (Thermo Electron). Trypsin was set as the digesting protease with the tolerance of two missed cleavages and not allowing for cleavages of KP and RP peptide bonds. The monoisotopic peptide and fragment mass tolerances were set to 10 p.p.m. and 0.02 Da, respectively. Carbamidomethylation of cysteins (+57.021 Da) was defined as a fixed modification and the oxidation of methionines (+15.995) as a variable modification. Protein identifications were statistically analyzed with Percolator and filtered to a cutoff of a false discovery rate of < 1% calculated based on a target-decoy approach.
The number of peptides observed in each pull-down, calculated as spectral counts, were integrated to quantify the amount of protein present. The protein quantification experiment relative to each bait was normalized to the number of spectral counts detected for the same protein in an experiment performed in the same cellular context using a strain where the protein was not FLAG tagged.
Filtering of the MS datasets was performed using SAINT probability scoring (Mellacheruvu et al., 2013). Additionally, interactions with the ribosomal proteins Rpl35A and Rps17 were manually removed from the dataset. Cytoscape (v 3.7.1) loaded with the DyNet Analyzer App, was used to visualize the interaction networks.
Fluorescence microscopy
Chromosome spreads were processed for immunostaining as described (Matos et al., 2011) using the following antibodies: mouse monoclonal anti-Myc 9E10 (1:100, CRUK), rat anti-tubulin (1:600, MCA78G, AbD Serotec), rabbit anti-Zip1 (1:200, this study), rabbit anti-GFP (1:500). Secondary antibodies conjugated to Alexa555, Alexa488 and Alexa647 were used for detection (1:300, Invitrogen). DNA was stained with 4’,6-diamidino-2-phenylindole (DAPI). Images were acquired using a DeltaVision personalDV multiplexed with a 60x 1.4NA DIC Oil PlanApoN objective and a Roper CoolSnap HQ2 camera under the control of Softworx Version 4.1.0 (Applied Precision) software. Image deconvolution was performed using the Deconvolve tool from Softworx Version 4.1.0 (Applied Precision) software. Images were processed using Fiji or Adobe Photoshop.
Protein analyses by western blotting
TCA extracts were performed as described previously (Matos et al., 2008). Briefly, meiotic cultures (OD600 ∼3.5, 10 ml) were disrupted using glass beads in 10% TCA. Precipitates were collected by centrifugation, resuspended in 2x NuPAGE sample buffer, and neutralized with 1 M Tris. Samples were boiled at 95°C for 10 min, cleared by centrifugation, and separated in NuPAGE 4%–12% Bis-Tris or NuPAGE 3%–8% Tris-Acetate gels (Invitrogen). After gel electrophoresis, proteins were transferred onto PVDF membranes (GE Healthcare). Antibodies targeting the following tags or proteins were used: FLAG HRP-conjugated (1:5000, A8592-1MG, Sigma), rabbit anti-Myc (1:500, ab1326, Abcam), mouse anti-HA (16B12, BioLegend), Crm1 (1:5000, a gift from K. Weis, ETH Zurich), Cdc5 (1:5000, clones 4F10 and 11H12, Medimabs).
Analysis of recombination using spore-autonomous fluorescence
The spore-autonomous fluorescence analysis of recombination was performed as described (Thacker et al., 2011), with minor modifications. Diploid yeast colonies grown on YP2%glycerol plates were expanded in YPD plates and grown for 24 h. Cells were transferred to SPM plates and incubated at 30°C for 48-60 h. Spores were resuspended in water, gently sonicated and transferred onto a microscope slide for imaging. For each strain, 3 colonies were independently expanded, sporulated and imaged. Biological duplicates or triplicates were independently generated and analyzed for selected mutants. > 600 tetrads were analyzed per strain in each experiment. Imaging was performed with a DeltaVision Multiplexed Widefield microscope. To maximize the number of quantifiable tetrads per image, 9-10 Z stacks were collected per field of view at 3 μm intervals.
The pattern of spore fluorescence in tetrads was scored by manual inspection using Fiji. Only tetrads in which each fluorescence marker was detected in two spores were included in the final analysis. Recombination frequency, expressed as map distance in centimorgans with standard error, was calculated using Stahl lab online tools (https://elizabethhousworth.com/StahlLabOnlineTools/)(Stahl and Lande, 1995).
Analysis of recombination at the HIS4-LEU2 locus
Southern-blot analyses of recombination were carried out as described previously (Arter et al., 2018, Kim et al., 2010). In brief, cells from 50-100 mL cultures were treated with psoralen. DNA crosslinking was initiated using a SpectroLinker XL-1500 crosslinker (Spectroline). Crosslinking was carried out for 10 min, with cells being mixed at regular intervals while kept on ice. After genomic DNA preparation, DNA concentrations were determined using the Qubit dsDNA broad range kit. After digestion with the appropriate restriction enzymes, DNA (∼2 μg) was separated by electrophoresis on 0.6% agarose gels. A Typhoon scanner and ImageQuant software were used to image and quantify different recombination intermediates. Manual background subtraction was performed by deducting the signal at time point 0 from all measurements.
Genetic analysis of recombination at HIS4-LEU2 was performed after tetrad microdissection using standard approaches. Map distances and NPD ratios were calculated using Stahl Lab online tools.
Genome-wide analysis of recombination
DNA was prepared for Illumina sequencing using a NextFlex kit (BIOO) with Illumina-compatible indices or as described (Anderson et al., 2011) with 4-base or 8-base inline barcodes. Read alignment, genotyping and recombination mapping were performed using the ReCombine package (Anderson et al., 2011). While running CrossOver.py, the input values for ‘closeCOs’, ‘closeNcoSame’ and ‘closeNCODiff’ were all set to 0. Insertions and deletions were removed from the set of genotyped markers. Recombination events within 5kb of each other were then merged into single events and categorized into seven types as described (Oke et al., 2014).
Analysis of spore viability
Spore viability was determined by microdissection of > 144 spores per strain.
Quantification and Statistical Analysis
Statistical analyses were performed using Microsoft Excel, Prism or RStudio. For multiple comparisons, analysis of variance (one-way ANOVA) was performed with Prism, followed by a correction for multiple comparisons using statistical hypothesis testing (Dunnett test). For pairwise comparisons two-tailed unpaired t tests were used. To test for changes in the distribution of genetic events at HIS4-LEU2, used in the calculation of genetic distance, a Poisson regression was used. The p value for the difference between the logarithmized wild-type counts and the logarithmized counts for a given mutant serves as an indication for a significant difference in counts.
Data and Code Availability
Raw sequence data from the genome-wide analysis of recombination have been deposited in the NIH Sequence Read Archive under Accession: PRJNA505664, ID: 505664. The mlh3Δ data is from (Arter et al., 2018). The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Perez-Riverol et al., 2019) partner repository with the dataset identifier PXD012486. Raw imaging data of western blots, Southern blots and microscopy can be accessed on Mendeley data: https://doi.org/10.17632/fshnb5swnv.1
Acknowledgments
We thank Toni Lehmann for help in building the yeast fermenters, and Scott Keeney, Wolfgang Zachariae, and Fabienne Lampert for strains and plasmids. ScopeM at ETH Zürich provided the imaging facility. P.W. and I.P. were supported by EMBO long-term fellowships (ALTF 475-2015 and ALTF 846-2014, respectively). The Haber lab is supported by NIH grant R35 GM127029. The Fung lab is supported by NIH R01 GM116895. The Picotti lab is supported by ETH Zürich, the European Research Council (FP7-ERC-StG-337965), and the Swiss National Science Foundation (PP00P3_133670/1). The Matos lab is supported by ETH Zürich and the Swiss National Science Foundation (153058, 155823, and 176108).
Author Contributions
P.W. and A.S. performed the FLAG-affinity purifications. I.P. and C.D. performed all of the MS analyses. K.C.C., P.W., and K.V. performed the analyses of protein expression during meiosis. A.S. performed the phenotypic analyses using spore fluorescence, with assistance from A.T.H. and K.V. A.S. prepared and analyzed the chromosome spreads. A.O., T.G., and J.C.F. performed genome-wide analysis of recombination. M.Y. and J.E.H. performed and analyzed HO-induced DSB repair. M.A. performed the physical analysis of recombination, with the help of P.W. P.P. supervised all of the MS analyses. J.M. conceived the study and wrote the manuscript, with assistance from P.W. and A.S. All of the authors proofread and contributed to the final manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: July 24, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.molcel.2019.06.022.
Supplemental Information
References
- Allers T., Lichten M. Differential timing and control of noncrossover and crossover recombination during meiosis. Cell. 2001;106:47–57. doi: 10.1016/s0092-8674(01)00416-0. [DOI] [PubMed] [Google Scholar]
- Anderson C.M., Chen S.Y., Dimon M.T., Oke A., DeRisi J.L., Fung J.C. ReCombine: a suite of programs for detection and analysis of meiotic recombination in whole-genome datasets. PLoS One. 2011;6:e25509. doi: 10.1371/journal.pone.0025509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argueso J.L., Wanat J., Gemici Z., Alani E. Competing crossover pathways act during meiosis in Saccharomyces cerevisiae. Genetics. 2004;168:1805–1816. doi: 10.1534/genetics.104.032912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arter M., Hurtado-Nieves V., Oke A., Zhuge T., Wettstein R., Fung J.C., Blanco M.G., Matos J. Regulated Crossing-Over Requires Inactivation of Yen1/GEN1 Resolvase during Meiotic Prophase I. Dev. Cell. 2018;45:785–800.e6. doi: 10.1016/j.devcel.2018.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop D.K., Zickler D. Early decision; meiotic crossover interference prior to stable strand exchange and synapsis. Cell. 2004;117:9–15. doi: 10.1016/s0092-8674(04)00297-1. [DOI] [PubMed] [Google Scholar]
- Blanco M.G., Matos J., West S.C. Dual control of Yen1 nuclease activity and cellular localization by Cdk and Cdc14 prevents genome instability. Mol. Cell. 2014;54:94–106. doi: 10.1016/j.molcel.2014.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boddy M.N., Gaillard P.H., McDonald W.H., Shanahan P., Yates J.R., 3rd, Russell P. Mus81-Eme1 are essential components of a Holliday junction resolvase. Cell. 2001;107:537–548. doi: 10.1016/s0092-8674(01)00536-0. [DOI] [PubMed] [Google Scholar]
- Börner G.V., Kleckner N., Hunter N. Crossover/noncrossover differentiation, synaptonemal complex formation, and regulatory surveillance at the leptotene/zygotene transition of meiosis. Cell. 2004;117:29–45. doi: 10.1016/s0092-8674(04)00292-2. [DOI] [PubMed] [Google Scholar]
- Bzymek M., Thayer N.H., Oh S.D., Kleckner N., Hunter N. Double Holliday junctions are intermediates of DNA break repair. Nature. 2010;464:937–941. doi: 10.1038/nature08868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J., Rappsilber J., Chiang Y.C., Russell P., Mann M., Denis C.L. Purification and characterization of the 1.0 MDa CCR4-NOT complex identifies two novel components of the complex. J. Mol. Biol. 2001;314:683–694. doi: 10.1006/jmbi.2001.5162. [DOI] [PubMed] [Google Scholar]
- Chen S.Y., Tsubouchi T., Rockmill B., Sandler J.S., Richards D.R., Vader G., Hochwagen A., Roeder G.S., Fung J.C. Global analysis of the meiotic crossover landscape. Dev. Cell. 2008;15:401–415. doi: 10.1016/j.devcel.2008.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu S., Herskowitz I. Gametogenesis in yeast is regulated by a transcriptional cascade dependent on Ndt80. Mol. Cell. 1998;1:685–696. doi: 10.1016/s1097-2765(00)80068-4. [DOI] [PubMed] [Google Scholar]
- Clyne R.K., Katis V.L., Jessop L., Benjamin K.R., Herskowitz I., Lichten M., Nasmyth K. Polo-like kinase Cdc5 promotes chiasmata formation and cosegregation of sister centromeres at meiosis I. Nat. Cell Biol. 2003;5:480–485. doi: 10.1038/ncb977. [DOI] [PubMed] [Google Scholar]
- Dayani Y., Simchen G., Lichten M. Meiotic recombination intermediates are resolved with minimal crossover formation during return-to-growth, an analogue of the mitotic cell cycle. PLoS Genet. 2011;7:e1002083. doi: 10.1371/journal.pgen.1002083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de los Santos T., Hunter N., Lee C., Larkin B., Loidl J., Hollingsworth N.M. The Mus81/Mms4 endonuclease acts independently of double-Holliday junction resolution to promote a distinct subset of crossovers during meiosis in budding yeast. Genetics. 2003;164:81–94. doi: 10.1093/genetics/164.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Muyt A., Jessop L., Kolar E., Sourirajan A., Chen J., Dayani Y., Lichten M. BLM helicase ortholog Sgs1 is a central regulator of meiotic recombination intermediate metabolism. Mol. Cell. 2012;46:43–53. doi: 10.1016/j.molcel.2012.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehé P.M., Gaillard P.H. Control of structure-specific endonucleases to maintain genome stability. Nat. Rev. Mol. Cell Biol. 2017;18:315–330. doi: 10.1038/nrm.2016.177. [DOI] [PubMed] [Google Scholar]
- Dummer A.M., Su Z., Cherney R., Choi K., Denu J., Zhao X., Fox C.A. Binding of the Fkh1 Forkhead Associated Domain to a Phosphopeptide within the Mph1 DNA Helicase Regulates Mating-Type Switching in Budding Yeast. PLoS Genet. 2016;12:e1006094. doi: 10.1371/journal.pgen.1006094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eissler C.L., Mazón G., Powers B.L., Savinov S.N., Symington L.S., Hall M.C. The Cdk/cDc14 module controls activation of the Yen1 holliday junction resolvase to promote genome stability. Mol. Cell. 2014;54:80–93. doi: 10.1016/j.molcel.2014.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasching C.L., Cejka P., Kowalczykowski S.C., Heyer W.D. Top3-Rmi1 dissolve Rad51-mediated D loops by a topoisomerase-based mechanism. Mol. Cell. 2015;57:595–606. doi: 10.1016/j.molcel.2015.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fricke W.M., Brill S.J. Slx1-Slx4 is a second structure-specific endonuclease functionally redundant with Sgs1-Top3. Genes Dev. 2003;17:1768–1778. doi: 10.1101/gad.1105203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung J.C., Rockmill B., Odell M., Roeder G.S. Imposition of crossover interference through the nonrandom distribution of synapsis initiation complexes. Cell. 2004;116:795–802. doi: 10.1016/s0092-8674(04)00249-1. [DOI] [PubMed] [Google Scholar]
- Grigaitis R., Susperregui A., Wild P., Matos J. Characterization of DNA helicases and nucleases from meiotic extracts of S. cerevisiae. Methods Cell Biol. 2018;144:371–388. doi: 10.1016/bs.mcb.2018.03.029. [DOI] [PubMed] [Google Scholar]
- Gritenaite D., Princz L.N., Szakal B., Bantele S.C., Wendeler L., Schilbach S., Habermann B.H., Matos J., Lisby M., Branzei D., Pfander B. A cell cycle-regulated Slx4-Dpb11 complex promotes the resolution of DNA repair intermediates linked to stalled replication. Genes Dev. 2014;28:1604–1619. doi: 10.1101/gad.240515.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauk G., McKnight J.N., Nodelman I.M., Bowman G.D. The chromodomains of the Chd1 chromatin remodeler regulate DNA access to the ATPase motor. Mol. Cell. 2010;39:711–723. doi: 10.1016/j.molcel.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyer W.D., Ehmsen K.T., Liu J. Regulation of homologous recombination in eukaryotes. Annu. Rev. Genet. 2010;44:113–139. doi: 10.1146/annurev-genet-051710-150955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho C.K., Mazón G., Lam A.F., Symington L.S. Mus81 and Yen1 promote reciprocal exchange during mitotic recombination to maintain genome integrity in budding yeast. Mol. Cell. 2010;40:988–1000. doi: 10.1016/j.molcel.2010.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt L.J., Ahmed E.A., Kaur H., Ahuja J.S., Hulme L., Chou T.-C., Lichten M., Goldman A.S.H. S. cerevisiae Srs2 helicase ensures normal recombination intermediate metabolism during meiosis and prevents accumulation of Rad51 aggregates. Chromosoma. 2019 doi: 10.1007/s00412-019-00705-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter N. Meiotic Recombination: The Essence of Heredity. Cold Spring Harb. Perspect. Biol. 2015;7:a016618. doi: 10.1101/cshperspect.a016618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ip S.C., Rass U., Blanco M.G., Flynn H.R., Skehel J.M., West S.C. Identification of Holliday junction resolvases from humans and yeast. Nature. 2008;456:357–361. doi: 10.1038/nature07470. [DOI] [PubMed] [Google Scholar]
- Ira G., Malkova A., Liberi G., Foiani M., Haber J.E. Srs2 and Sgs1-Top3 suppress crossovers during double-strand break repair in yeast. Cell. 2003;115:401–411. doi: 10.1016/s0092-8674(03)00886-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur H., De Muyt A., Lichten M. Top3-Rmi1 DNA single-strand decatenase is integral to the formation and resolution of meiotic recombination intermediates. Mol. Cell. 2015;57:583–594. doi: 10.1016/j.molcel.2015.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K.P., Weiner B.M., Zhang L., Jordan A., Dekker J., Kleckner N. Sister cohesion and structural axis components mediate homolog bias of meiotic recombination. Cell. 2010;143:924–937. doi: 10.1016/j.cell.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knop M., Siegers K., Pereira G., Zachariae W., Winsor B., Nasmyth K., Schiebel E. Epitope tagging of yeast genes using a PCR-based strategy: more tags and improved practical routines. Yeast. 1999;15(10B):963–972. doi: 10.1002/(SICI)1097-0061(199907)15:10B<963::AID-YEA399>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Lee B.H., Amon A. Role of Polo-like kinase CDC5 in programming meiosis I chromosome segregation. Science. 2003;300:482–486. doi: 10.1126/science.1081846. [DOI] [PubMed] [Google Scholar]
- León Ortiz A.M., Reid R.J.D., Dittmar J.C., Rothstein R., Nicolas A. Srs2 overexpression reveals a helicase-independent role at replication forks that requires diverse cell functions. DNA Repair (Amst.) 2011;10:506–517. doi: 10.1016/j.dnarep.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lusser A., Urwin D.L., Kadonaga J.T. Distinct activities of CHD1 and ACF in ATP-dependent chromatin assembly. Nat. Struct. Mol. Biol. 2005;12:160–166. doi: 10.1038/nsmb884. [DOI] [PubMed] [Google Scholar]
- Lynn A., Soucek R., Borner G.V. ZMM proteins during meiosis: crossover artists at work. Chromosome Res. 2007;15:591–605. doi: 10.1007/s10577-007-1150-1. [DOI] [PubMed] [Google Scholar]
- Mancera E., Bourgon R., Brozzi A., Huber W., Steinmetz L.M. High-resolution mapping of meiotic crossovers and non-crossovers in yeast. Nature. 2008;454:479–485. doi: 10.1038/nature07135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manhart C.M., Ni X., White M.A., Ortega J., Surtees J.A., Alani E. The mismatch repair and meiotic recombination endonuclease Mlh1-Mlh3 is activated by polymer formation and can cleave DNA substrates in trans. PLoS Biol. 2017;15:e2001164. doi: 10.1371/journal.pbio.2001164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marston A.L., Tham W.-H., Shah H., Amon A. A genome-wide screen identifies genes required for centromeric cohesion. Science. 2004;303:1367–1370. doi: 10.1126/science.1094220. [DOI] [PubMed] [Google Scholar]
- Matos J., West S.C. Holliday junction resolution: regulation in space and time. DNA Repair (Amst.) 2014;19:176–181. doi: 10.1016/j.dnarep.2014.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matos J., Lipp J.J., Bogdanova A., Guillot S., Okaz E., Junqueira M., Shevchenko A., Zachariae W. Dbf4-dependent CDC7 kinase links DNA replication to the segregation of homologous chromosomes in meiosis I. Cell. 2008;135:662–678. doi: 10.1016/j.cell.2008.10.026. [DOI] [PubMed] [Google Scholar]
- Matos J., Blanco M.G., Maslen S., Skehel J.M., West S.C. Regulatory control of the resolution of DNA recombination intermediates during meiosis and mitosis. Cell. 2011;147:158–172. doi: 10.1016/j.cell.2011.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellacheruvu D., Wright Z., Couzens A.L., Lambert J.P., St-Denis N.A., Li T., Miteva Y.V., Hauri S., Sardiu M.E., Low T.Y. The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nat. Methods. 2013;10:730–736. doi: 10.1038/nmeth.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moynahan M.E., Jasin M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat. Rev. Mol. Cell Biol. 2010;11:196–207. doi: 10.1038/nrm2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen J.R., Nallaseth F.S., Lan Y.Q., Slagle C.E., Brill S.J. Yeast Rmi1/Nce4 controls genome stability as a subunit of the Sgs1-Top3 complex. Mol. Cell. Biol. 2005;25:4476–4487. doi: 10.1128/MCB.25.11.4476-4487.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oelschlaegel T., Schwickart M., Matos J., Bogdanova A., Camasses A., Havlis J., Shevchenko A., Zachariae W. The yeast APC/C subunit Mnd2 prevents premature sister chromatid separation triggered by the meiosis-specific APC/C-Ama1. Cell. 2005;120:773–788. doi: 10.1016/j.cell.2005.01.032. [DOI] [PubMed] [Google Scholar]
- Ohouo P.Y., Bastos de Oliveira F.M., Almeida B.S., Smolka M.B. DNA damage signaling recruits the Rtt107-Slx4 scaffolds via Dpb11 to mediate replication stress response. Mol. Cell. 2010;39:300–306. doi: 10.1016/j.molcel.2010.06.019. [DOI] [PubMed] [Google Scholar]
- Oke A., Anderson C.M., Yam P., Fung J.C. Controlling meiotic recombinational repair - specifying the roles of ZMMs, Sgs1 and Mus81/Mms4 in crossover formation. PLoS Genet. 2014;10:e1004690. doi: 10.1371/journal.pgen.1004690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Riverol Y., Csordas A., Bai J., Bernal-Llinares M., Hewapathirana S., Kundu D.J., Inuganti A., Griss J., Mayer G., Eisenacher M. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 2019;47(D1):D442–D450. doi: 10.1093/nar/gky1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petronczki M., Siomos M.F., Nasmyth K. Un ménage à quatre: the molecular biology of chromosome segregation in meiosis. Cell. 2003;112:423–440. doi: 10.1016/s0092-8674(03)00083-7. [DOI] [PubMed] [Google Scholar]
- Petronczki M., Matos J., Mori S., Gregan J., Bogdanova A., Schwickart M., Mechtler K., Shirahige K., Zachariae W., Nasmyth K. Monopolar attachment of sister kinetochores at meiosis I requires casein kinase 1. Cell. 2006;126:1049–1064. doi: 10.1016/j.cell.2006.07.029. [DOI] [PubMed] [Google Scholar]
- Prakash R., Satory D., Dray E., Papusha A., Scheller J., Kramer W., Krejci L., Klein H., Haber J.E., Sung P., Ira G. Yeast Mph1 helicase dissociates Rad51-made D-loops: implications for crossover control in mitotic recombination. Genes Dev. 2009;23:67–79. doi: 10.1101/gad.1737809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pray-Grant M.G., Daniel J.A., Schieltz D., Yates J.R., 3rd, Grant P.A. Chd1 chromodomain links histone H3 methylation with SAGA- and SLIK-dependent acetylation. Nature. 2005;433:434–438. doi: 10.1038/nature03242. [DOI] [PubMed] [Google Scholar]
- Qiu Y., Levendosky R.F., Chakravarthy S., Patel A., Bowman G.D., Myong S. The Chd1 Chromatin Remodeler Shifts Nucleosomal DNA Bidirectionally as a Monomer. Mol. Cell. 2017;68:76–88.e6. doi: 10.1016/j.molcel.2017.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranjha L., Anand R., Cejka P. The Saccharomyces cerevisiae Mlh1-Mlh3 heterodimer is an endonuclease that preferentially binds to Holliday junctions. J. Biol. Chem. 2014;289:5674–5686. doi: 10.1074/jbc.M113.533810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogacheva M.V., Manhart C.M., Chen C., Guarne A., Surtees J., Alani E. Mlh1-Mlh3, A Meiotic Crossover and DNA Mismatch Repair Factor, is a Msh2-Msh3-Stimulated Endonuclease. J. Biol. Chem. 2014;289:5664–5673. doi: 10.1074/jbc.M113.534644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan D.P., Sundaramoorthy R., Martin D., Singh V., Owen-Hughes T. The DNA-binding domain of the Chd1 chromatin-remodelling enzyme contains SANT and SLIDE domains. EMBO J. 2011;30:2596–2609. doi: 10.1038/emboj.2011.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasanuma H., Sakurai H.S.M., Furihata Y., Challa K., Palmer L., Gasser S.M., Shinohara M., Shinohara A. Srs2 helicase prevents the formation of toxic DNA damage during late prophase I of yeast meiosis. Chromosoma. 2019 doi: 10.1007/s00412-019-00709-5. [DOI] [PubMed] [Google Scholar]
- Schwacha A., Kleckner N. Identification of joint molecules that form frequently between homologs but rarely between sister chromatids during yeast meiosis. Cell. 1994;76:51–63. doi: 10.1016/0092-8674(94)90172-4. [DOI] [PubMed] [Google Scholar]
- Silva S., Altmannova V., Luke-Glaser S., Henriksen P., Gallina I., Yang X., Choudhary C., Luke B., Krejci L., Lisby M. Mte1 interacts with Mph1 and promotes crossover recombination and telomere maintenance. Genes Dev. 2016;30:700–717. doi: 10.1101/gad.276204.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smolle M.M. Chd1 bends over backward to remodel. Nat. Struct. Mol. Biol. 2018;25:2–3. doi: 10.1038/s41594-017-0014-4. [DOI] [PubMed] [Google Scholar]
- Snowden T., Acharya S., Butz C., Berardini M., Fishel R. hMSH4-hMSH5 recognizes Holliday Junctions and forms a meiosis-specific sliding clamp that embraces homologous chromosomes. Mol. Cell. 2004;15:437–451. doi: 10.1016/j.molcel.2004.06.040. [DOI] [PubMed] [Google Scholar]
- Sourirajan A., Lichten M. Polo-like kinase Cdc5 drives exit from pachytene during budding yeast meiosis. Genes Dev. 2008;22:2627–2632. doi: 10.1101/gad.1711408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl F.W., Lande R. Estimating interference and linkage map distance from two-factor tetrad data. Genetics. 1995;139:1449–1454. doi: 10.1093/genetics/139.3.1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symington L.S., Rothstein R., Lisby M. Mechanisms and regulation of mitotic recombination in Saccharomyces cerevisiae. Genetics. 2014;198:795–835. doi: 10.1534/genetics.114.166140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang S., Wu M.K.Y., Zhang R., Hunter N. Pervasive and essential roles of the Top3-Rmi1 decatenase orchestrate recombination and facilitate chromosome segregation in meiosis. Mol. Cell. 2015;57:607–621. doi: 10.1016/j.molcel.2015.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thacker D., Lam I., Knop M., Keeney S. Exploiting spore-autonomous fluorescent protein expression to quantify meiotic chromosome behaviors in Saccharomyces cerevisiae. Genetics. 2011;189:423–439. doi: 10.1534/genetics.111.131326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wechsler T., Newman S., West S.C. Aberrant chromosome morphology in human cells defective for Holliday junction resolution. Nature. 2011;471:642–646. doi: 10.1038/nature09790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L., Ajimura M., Padmore R., Klein C., Kleckner N. NDT80, a meiosis-specific gene required for exit from pachytene in Saccharomyces cerevisiae. Mol. Cell. Biol. 1995;15:6572–6581. doi: 10.1128/mcb.15.12.6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue X., Choi K., Bonner J.N., Szakal B., Chen Y.-H., Papusha A., Saro D., Niu H., Ira G., Branzei D. Selective modulation of the functions of a conserved DNA motor by a histone fold complex. Genes Dev. 2015;29:1000–1005. doi: 10.1101/gad.259143.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue X., Papusha A., Choi K., Bonner J.N., Kumar S., Niu H., Kaur H., Zheng X.-F., Donnianni R.A., Lu L. Differential regulation of the anti-crossover and replication fork regression activities of Mph1 by Mte1. Genes Dev. 2016;30:687–699. doi: 10.1101/gad.276139.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yimit A., Kim T., Anand R.P., Meister S., Ou J., Haber J.E., Zhang Z., Brown G.W. MTE1 Functions with MPH1 in Double-Strand Break Repair. Genetics. 2016;203:147–157. doi: 10.1534/genetics.115.185454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakharyevich K., Tang S., Ma Y., Hunter N. Delineation of joint molecule resolution pathways in meiosis identifies a crossover-specific resolvase. Cell. 2012;149:334–347. doi: 10.1016/j.cell.2012.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L., Wang S., Yin S., Hong S., Kim K.P., Kleckner N. Topoisomerase II mediates meiotic crossover interference. Nature. 2014;511:551–556. doi: 10.1038/nature13442. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw sequence data from the genome-wide analysis of recombination have been deposited in the NIH Sequence Read Archive under Accession: PRJNA505664, ID: 505664. The mlh3Δ data is from (Arter et al., 2018). The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Perez-Riverol et al., 2019) partner repository with the dataset identifier PXD012486. Raw imaging data of western blots, Southern blots and microscopy can be accessed on Mendeley data: https://doi.org/10.17632/fshnb5swnv.1