

Abstract
First described clinically in 1906, Alzheimer's disease (AD) is the most common neurodegenerative disease and form of dementia worldwide. Despite its prevalence, only five therapies are currently approved for AD, all dealing with the symptoms rather than the underlying causes of the disease. A multitude of experimental evidence has suggested that the once thought inconsequential process of neuroinflammation does, in fact, contribute to the AD pathogenesis. One such CNS cell type critical to this process are microglia. Plastic in nature with varied roles, microglia are emerging as key contributors to AD pathology. This review will focus on the role of microglia in the neuroinflammatory response in AD, highlighting recent studies implicating aberrant changes in microglial function in disease progression. Of critical note is that with these advances, a reconceptualization of the framework in which we view microglia is required.
Linked Articles
This article is part of a themed section on Therapeutics for Dementia and Alzheimer's Disease: New Directions for Precision Medicine. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v176.18/issuetoc
Abbreviations
- 5XFAD
5X familial Alzheimer's disease mutation
- AAV
adeno‐associated virus
- ABI3
ABI gene family member 3
- ACH
amyloid cascade hypothesis
- AD
Alzheimer's disease
- APOE
apolipoprotein E
- APP
amyloid precursor protein
- APPSWE/PS1ΔE9
APP containing Swedish mutation and PS1 containing δ E9 mutation
- CD
cluster of differentiation
- CK‐p25
cyclin‐dependant kinase 5 protein 25
- CLU
clusterin
- CR1
complement receptor 1
- DAM
damage‐associated microglia
- DAMP
damage‐associated molecular pattern
- GWAS
genome‐wide association study
- IBA1
ionized calcium‐binding adapter molecule 1
- IL1RAP
IL‐1 receptor accessory protein
- iPSC
induced pluripotent stem cell
- IRF
IFN regulatory factor
- LOAD
late onset Alzheimer's disease
- MCI
mild cognitive impairment
- MV
microvesicle
- PS1
presenilin 1
- RIPK1
receptor‐interacting serine/threonine‐protein kinase 1
- SP
senile plaque
- TLR
Toll‐like receptor
- TREM2
triggering receptor expressed on myeloid cells 2
Alzheimer's disease
Pathologically, Alzheimer's disease (AD) is characterized by the death of pyramidal neurons and synapses in the cerebral cortex and certain subcortical regions, with classical hallmarks of senile plaque (SP) deposits and hyperphosphorylation of the microtubule‐associated protein tau. In particular, this loss occurs in the hippocampus resulting in gross atrophy of the brain and increased ventricular spaces. This presents clinically as a progressive pattern of worsening cognition and behavioural function. This pattern is divided into three stages: preclinical, mild cognitive impairment (MCI) and dementia (Jack et al., 2011). Despite identification of genetic mutations that confer significant risk factors for disease, the underlying cause of AD remains unproven and as such raises difficulties in definitive diagnoses. Indeed, the MCI stage of AD, considered a prodromal stage of disease, is often attributed to normal ageing (Dubois and Albert, 2004).
The amyloid cascade hypothesis (ACH) was developed following the identification that SPs are composed of amyloid beta (Aβ) and that these deposits are unique to AD. The ACH posits that Aβ accumulation is a critical event in AD pathology. First articulated in 1992 by Hardy and Higgins (1992), the ACH originally postulated that Aβ accumulation is causative of AD pathology, although this has now evolved to contend that Aβ accumulation is a central event in AD pathology (Pimplikar, 2009). Regardless of the validity of the ACH, it has driven AD research and aided in our understanding of underlying biological mechanisms.
However, this has not translated into clinical trial results. All Aβ‐directed therapies have failed in clinical trials, many in multiple attempts (Cummings et al., 2014). Similarly, the vast majority of non‐Aβ‐directed therapies have also failed. This results in AD having one of the highest failure rates of any disease at 99.5–99.6% (Cummings et al., 2014; Calcoen et al., 2015). Pfizer has even gone as so far to abandon the field entirely, citing money can be better spent elsewhere (Hawkes, 2018). This has not been unnoticed by those outside of the scientific sphere with individuals becoming disconcerted with the scientific process (Cummings et al., 2014; Mannix, 2018).
To address this, it is clear that a re‐evaluation of the precise role of the various pathologies seen in AD is required; one that accounts for the complex milieu observed. In concordance, our therapeutic approaches should also follow a re‐evaluation. Once such revaluation involves investigating the role of neuroinflammation within AD.
Neuroinflammation
The term ‘neuroinflammation’ refers to immune‐related responses, in particular, the innate immune response that occurs within the CNS. Neuroinflammation is normally seen throughout the CNS with roles in both cellular and tissue homeostasis as well as proper neuronal functioning (Xanthos and Sandkuhler, 2014). Under normal physiological conditions, this response is self‐limiting. This acute form of neuroinflammation contrasts with the chronic form observed in AD that sees a failure of the immune clearance mechanism (Maderna and Godson, 2003; Shastri et al., 2013).
Neuroinflammation is a complex process that involves a number of cell–cell interactions within the CNS (Skaper et al., 2018). The inflammatory cascade is principally initiated through two glial cell types: microglia and astrocytes (Perry et al., 2010). Upon recognition of an initial stimulus, these cells secrete a number of hallmark pro‐inflammatory cytokines including TNFα, IL‐1β and IL‐6, as well as a number of chemokines that recruit further glial cells. These pro‐inflammatory mediators can then facilitate the neurodegeneration of otherwise healthy neighbouring neuronal populations, with the cellular contents and damage‐associated molecular patterns (DAMPs) produced and secreted by these dying neurons further contributing to the inflammatory milieu.
Neuroinflammation in AD is considered to be a chronic and detrimental process (Eikelenboom et al., 2006). Human post mortem examination of AD individuals consistently reports enhanced levels of microgliosis and astrogliosis surrounding Aβ plaques (Heneka et al., 2014). This is in concordance with increased levels of inflammatory cytokines and chemokines (Heneka et al., 2014). Furthermore, levels of neuroinflammation have been shown to correlate with disease progression and impairments in cognition (Morris et al., 2014; Heneka et al., 2014). Recent studies suggest that microglia are the critical cell type that contributes to this neuroinflammatory‐related pathology (Sarlus and Heneka, 2017).
Microglia
Microglia are a class of innate immune cells within the CNS. Unlike other CNS glial cell types, they share a myeloid lineage with their peripheral counterparts macrophages (Ginhoux et al., 2013). Critically, microglia are formed from primitive macrophage progenitors that infiltrate the brain during development (Ginhoux et al., 2010). These microglia have a defined core gene set that distinguishes them from related peripheral cells (Butovsky and Weiner, 2018). The median lifespan for a single microglial cell is 15 months, over half the lifetime of a mouse (Fuger et al., 2017). However, when genetically depleted, microglia can repopulate to normal cell levels within 5 days (Bruttger et al., 2015). Under homeostatic conditions, microglial repopulation is shown to be exclusively from CNS residing progenitor cells (Butovsky and Weiner, 2018). However, it is now appreciated that under certain conditions, infiltration of peripheral myeloid cells can indeed differentiate into microglia (Cronk et al., 2018; Lund et al., 2018). These cells do express a subset of the unique microglial gene signature but have differential functional roles (Butovsky and Weiner, 2018). Microglial proliferation is increased in a number of neurodegenerative conditions in mice, including AD (Gomez‐Nicola et al., 2013).
Microglia exhibit a number of cell surface markers allowing for their identification and distinction from other immune cell types. Both microglia and peripheral macrophages are macrophage‐1 antigen and cluster of differentiation 11b (CD11b) positive, whereas they can be distinguished based on being CD45low and CD45high respectively (D'Mello et al., 2009). Ionized calcium‐binding adapter molecule 1 (IBA1) is also used as a microglial marker and has historically been used as a marker for microglial ‘activation’ (Ito et al., 1998). IBA1 has been shown to be involved in actin remodelling and subsequently phagocytosis, a key functional role that microglia are involved in (Ohsawa et al., 2004). CD68 expression has also been used with IBA1 to identify ‘phagocytic’ microglia (Boche et al., 2013).
Microglia fulfil a number of varied roles within the CNS including the immune response, maintenance of homeostasis, extracellular signalling, phagocytosis, antigen presentation and synaptic pruning (Kettenmann et al., 2011; Walker et al., 2014). The varied roles of microglia may be attributed to their limited self‐renewal under normal physiological conditions (Lawson et al., 1992).
It follows that microglial populations are heterogeneous in nature; populations isolated from different regions within the brain under different conditions will ultimately differ in phenotype, associated markers and function (Wes et al., 2016). In an effort to characterize microglia, the M1/M2 dichotomy was adopted from their peripheral macrophage counterparts, representing ‘classical’ and ‘alternative’ activation states. Whilst popular for some time, this is now considered an oversimplification of the vast array of phenotypes microglia can adopt. Ransohoff (2016) provides the most compelling argument for this. Briefly, these microglial subtypes are based on specific cytokines that induce each phenotype as described in vitro, which fails to account for the complex milieu seen in the CNS. Furthermore, the suggested spectrum view of microglia with ‘M1 like’ and ‘M2 like’ phenotypes as bookends also fails, with differing disease states failing to identify an axis on which polarization occurs. This is further compounded with the original notion of M1/M2 macrophages also under debate (Martinez and Gordon, 2014). Extending this, describing microglia as either ‘activated’ or ‘resting/quiescent’ further hinders our understanding of these cells and conclusions we subsequently draw. A more appropriate view of microglia (Figure 1) shifts away from such pigeonholing and rather views them as an extensive array of distinct, though overlapping phenotypes with corresponding functions. This is not to entirely dismiss previous research. Rather, a more holistic view is required when drawing inferences about microglial phenotype and subsequent function.
Figure 1.
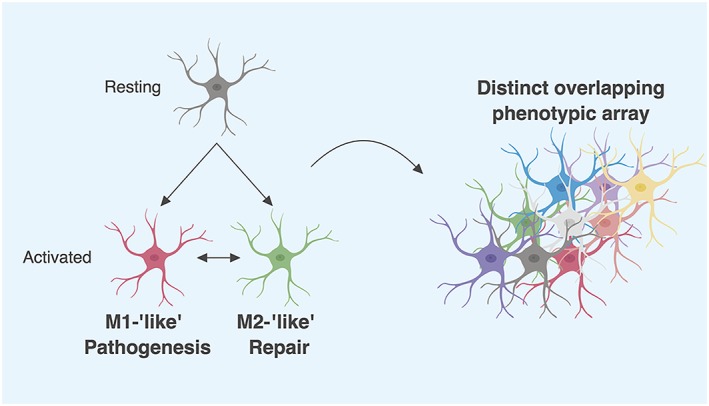
A proposed new framework for microglia characterization. The M1/M2 paradigm for microglial characterization is now recognized as a vast oversimplification of microglial phenotypes and functions. Rather than pigeonholing these cells as either ‘good’ or ‘bad’, a context‐dependant label, a more appropriate view is to recognize them as a distinct array of overlapping phenotypes. Microglia can in fact have altered expression levels of both classical M1 and M2 markers. This framework also includes shifting away from notions of resting/quiescent and active microglia. Whilst this may seem an issue of semantics, not only does it not dismiss these phenotypes entirely as previously done in the past, it also allows for a more holistic view when interpreting data. For simplicity, morphological differences have not been illustrated. Created with BioRender.
Microglia transcriptomic changes in AD
A number of efforts have been made to accurately characterize microglia primarily through transcriptomic approaches. Although the first microglial‐specific characterization is less than 5 years old, this approach has now expanded and aided in understanding the complexity that is microglial phenotype (Orre et al., 2014). By utilizing direct RNA sequencing (RNAseq), the microglial ‘sensome’ has been identified as a distinct group of transcripts encoding for ligand sensing proteins that changes in ageing (Hickman et al., 2013). In the cyclin‐dependant kinase 5 protein 25 (CK‐p25) inducible mouse model of neurodegeneration, which shows similar transcript profiles to the 5X familial Alzheimer's disease mutation (5XFAD) AD model, Mathys et al. (2017) utilized a single‐cell RNAseq to characterize microglial heterogeneity (Hargis and Blalock, 2017). Clustering of transcription profiles revealed a number of disease‐specific microglial states that were not observed in wild‐type controls. In a separate study in the 5XFAD AD model, a single‐cell RNAseq was used to profile all immune cell types within the brain, after which a cluster of microglial cells termed the damage‐associated microglia (DAM) was identified. These cells were associated with plaque, involved the down‐regulation of inhibitory checkpoints and were conserved in humans (Keren‐Shaul et al., 2017). Evidence has shown that both triggering receptor expressed on myeloid cells 2 (TREM2) and apolipoprotein E (APOE), known AD risk factors, act as regulators for the phenotypic switching of microglia (Krasemann et al., 2017). Utilizing the double transgenic AD mouse model containing amyloid precursor protein (APP) with the Swedish mutation and presenilin (PS) 1 with the delta E9 mutation (APPSWE/PS1ΔE9), in parallel to models of multiple sclerosis and amyotrophic lateral sclerosis, mRNA counts using Nanostring technology identified a molecular signature of DAM dependant on both TREM2 and APOE signalling.
Ageing alone is known to alter microglial phenotype deleteriously, yet its exact contribution to disease remains unknown (Olah et al., 2018; Silvin and Ginhoux, 2018). This includes observations of microglial priming, in where aged microglial populations exhibit an increased pro‐inflammatory response upon insult (Norden and Godbout, 2013; Rawji et al., 2016). In aged mice, peripheral injections of LPS resulted in increased levels of pro‐inflammatory cytokine levels when compared to young mice (Godbout et al., 2005; Henry et al., 2009). In addition, ex vivo microglia isolated from adult mice show greater secretion of both IL‐6 and TNFα (Njie et al., 2012). Gene ontology analysis on transcriptomic data from young and aged mice reveals that these changes are largely IFN‐dependant (Deczkowska et al., 2017). These changes are accompanied by morphological alterations, most notably with observations of hypertrophy (Conde and Streit, 2006). In addition to altered immune‐related functions, phagocytotic functions are also altered, with the ability to phagocytose Aβ1–42 decreasing with age (Floden and Combs, 2011). However, whether this is indeed bona fide senescence has not been determined (Baker and Petersen, 2018). Furthermore, issues arise when attempts are made to distinguish the individual contributions that either AD or normal ageing makes on observed dysfunction.
The extent that these findings translate to humans is unclear. Whilst whole brain transcriptional profiles for humans and rodents in ageing alone are concordant, when AD humans and AD mouse models are examined concordance is not observed (Hargis and Blalock, 2017). Different rodent models of AD reproduce different features and states of disease. Critically, all are based on familial disease states. Additionally, whilst similar profiles are seen within matched AD individuals, separate rodent models result in separate transcriptomic profiles (Hargis and Blalock, 2017). Furthermore, examination of whole tissue or entire cell populations has the potential to mask distinct populations. In an AD model containing APPSWE and the N141I mutation in PS2, genes that were altered in whole brain tissue were found to be highly enriched in microglia and astrocytes; however, only a fraction of these genes was in fact up‐regulated when examined in these cell types as a specific population. Of these, microglia accounted for 87% (Kim et al., 2016). Single‐cell RNAseq continually reports multiple glial cell populations, even in wild‐type mice (Zeisel et al., 2015). However, the extent that individual microglial populations translate across both models and species is yet to be fully explored. Epigenomic signals do however appear to be conserved between mice and humans. In the CK‐p25 AD‐like model, conserved transcriptional changes were shown to be associated with immune‐related genes, as well as conserved chromatin dynamics (Gjoneska et al., 2015).
One such technique to overcome this is the use of induced pluripotent stem cells (IPSCs) from humans. Although a recent technique, these cells are indeed microglial‐like and have been validated as a viable research tool (Abud et al., 2017; Pocock and Piers, 2018). The generation of microglial‐like iPSCS from APOE4 carriers shows transcriptomic changes alongside impaired phagocytosis of Aβ (Lin et al., 2018). Microglial IPSCs have recently been used to investigate specific missense mutations in TREM2, a newly identified AD risk factor (Garcia‐Reitboeck et al., 2018).
Microglia morphology changes in AD
A key constituent of the plastic nature of microglia is their capacity for rapid morphological transformations. It has been widely observed that under normal physiological conditions, ‘quiescent’ microglia adopt a ramified morphology, and ‘activated’ microglia appear ameboid in shape (Boche et al., 2013). Furthermore, this transition is stepwise in nature. Multiple morphological measurements available can be used to compare separate microglia. Under normal conditions, microglia exhibit an altered morphology that is region‐dependant (Lawson et al., 1990). Microglia from LPS‐treated mice were shown to differ in measurements of cell perimeter, roundness and soma size (Kongsui et al., 2015). In the dual Indiana and Swedish APP mutation AD model, plaque‐associated microglia show alterations in branch length, area and ramification when compared to wild‐type controls (Plescher et al., 2018). Examination of human AD autopsy samples also reveals distinct morphological microglial populations. In a human cohort, five distinct morphological types were identified in AD and cognitively normal controls. Differences were seen in various regions of the hippocampus (Bachstetter et al., 2015). However, further work is required to elucidate the link between morphology and phenotype. Microglia that exhibit similar morphologies can indeed differ in underlying phenotype (Wes et al., 2016). Although a recent technique, these studies provide evidence for novel inferential analysis of microglial function through morphology.
Microglial function in AD
Functional alterations in microglia are indeed observed contiguous to phenotypic changes, although the translational effect of these changes varies (Figure 2). The initial sensing of the neuroinflammatory trigger by the Aβ peptide and its aggregates via microglia is varied. This sensing occurs through a number of receptors, including Toll‐like receptors (TLRs), NOD‐like receptors, receptors for advanced glycation endproducts, formyl peptide receptors, scavenger receptors, pentraxins and the complement cascade. All of which are involved in the initiation of a neuroinflammatory response through a number of signalling pathways (Salminen et al., 2009). This sensing can also occur through the NOD, leucine‐rich‐containing family, pyrin domain‐containing‐3 inflammasome, multiprotein complexes involved in the maturation and secretion of pro‐inflammatory cytokines IL‐1β and IL‐18 (Halle et al., 2008). The most studied of the receptors involved in this sensing are the TLRs. TLRs belong to a superfamily of pattern recognition receptors recognizing both DAMPs and pathogen‐associated molecular patterns. Of these, the TLR2 and TLR4 subtypes are considered critical in Aβ recognition. Microglial TLR2 recognition of Aβ is considered the principal method that triggers the neuroinflammatory response (Liu et al., 2012). Downstream TLR signalling through NFκB, activator protein 1 and IFN regulatory factor (IRF) pathways lead to pro‐inflammatory gene transcription (Miyake, 2007).
Figure 2.
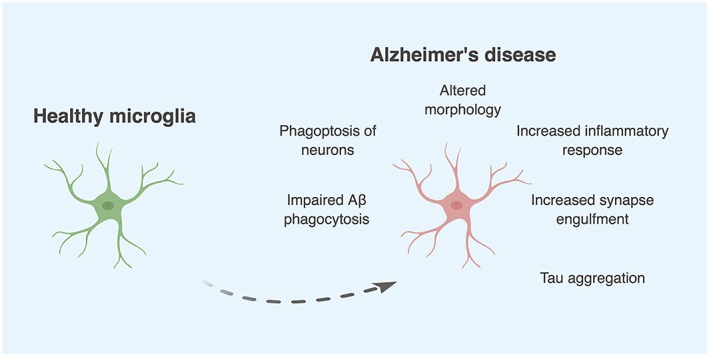
Alterations in microglial phenotype and function as seen in AD. A multitude of experimental evidence has now demonstrated that these changes are indeed deleterious and may in fact contribute to disease progression. Created with BioRender.
Microglia are also involved in the clearance of Aβ through endocytosis. This process is dependent on whether Aβ is in a fibrillar or soluble oligomeric state, resulting in phagocytic or macropinocytic processes respectively. Fibrillar Aβ is recognized through an assembly of receptors to form a cell surface receptor complex, consisting of class A scavenger receptor, class B scavenger receptor, α6β1 integrin, CD14, CD36, CD47, TLR2, TLR4, TLR6 and TLR9 (Bamberger et al., 2003; Lee and Landreth, 2010). Of these receptors, the TLRs appear to be the most crucial, with TLR activation increasing fibrillar Aβ clearance through phagocytosis (Tahara et al., 2006). Furthermore, TLR2 and TLR4 ablation results in a failure of the cell surface receptor complex to recognize Aβ and subsequently initiate phagocytosis (Reed‐Geaghan et al., 2009). Soluble Aβ species are cleared by microglia through a process known as fluid phase macropinocytosis. Soluble Aβ is brought into the cell through an invagination of the cellular membrane, which then closes to form a vesicle. This uptake also occurs in astrocytes, although not as effectively (Mandrekar et al., 2009). Excessive Aβ deposits and neurofibrillary tangles have been shown to impair the Aβ‐related clearance abilities of the recruited microglia and astrocytes, compounding the issue (Yamanaka et al., 2012).
Microglia also perform various functions through the release of extracellular microvesicles (MVs) through exocytosis. MVs have been shown to be potent modulators of inflammation and immunity, and when released, MVs can stimulate synaptic activity both in vitro and in vivo (Sadallah et al., 2011; Antonucci et al., 2012). Critically, MVs, likely of microglial origin, are elevated within AD individuals. Examination of these MVs showed that they present toxicity towards neurons and promote the formation of Aβ (Joshi et al., 2014).
Microglia activity within AD can also affect tau and drive tau‐related pathology (Maphis et al., 2015). Analysis of human brain samples has demonstrated a senescent morphological microglial phenotype that is associated with tau (Streit et al., 2009). In addition, microglial‐mediated inflammation has been linked to the aggregation and phosphorylation of tau. In wild‐type mice, LPS‐induced inflammation has been shown to induce tau aggregation with this further enhanced in transgenic mice lacking the microglial fractalkine receptor CX3CR1. (Bhaskar et al., 2010).
It is known that microglia are involved in synaptic pruning, a process critical not only during development but also in the mature brain (Paolicelli et al., 2011; Hong et al., 2016). Initially thought to engulf whole synapses, recent evidence questions this with the process of ‘trogocytosis’ observed, where partial phagocytosis has been linked to selective pruning (Weinhard et al., 2018). Aberrant engulfment of synapses by microglia can lead to dysfunction. In lupus, a microglial‐mediated synapse loss has been observed, which was rescued upon promoting an anti‐inflammatory phenotype (Bialas et al., 2017). This is now being investigated within AD, where it is suggested that microglia may indeed be involved in such a process (Hong et al., 2016). The genome‐wide association study (GWAS) has identified a risk gene, clusterin (CLU), which encodes for the receptor complement component 3b, and has been shown to be critical in the maturation of developing neural circuits by microglia (Schafer et al., 2012).
Similarly, it has recently been observed that microglia can execute neuronal death through phagocytosis on stressed but otherwise viable neurons, a process termed phagoptosis (Brown and Neher, 2014). This is due to an increase in various ‘eat me’ signals (phosphatidylserine, complement component 1q, calreticulin, de‐sialylated glycoprotein) and/or loss of ‘don't eat me’ signals (glycoprotein, CD47, neuraminidase) presented on neuronal surfaces. In an inflammatory setting, both microglia and astrocytes release milk fat globule EGF8 that is able to bind to exposed phosphatidylserine, opsonising the neuron. In excessive neuroinflammation, this process is skewed where microglia are able to phagocytose healthy neurons (Brown and Neher, 2012). In neuronal‐glial co‐cultures stimulated with either LPS or Aβ, a lack of discrimination occurs between stressed and healthy neurons targeted for phagoptosis by microglia (Neniskyte et al., 2011).
GWAS microglia
Genetic mutations in either APP or PS1/2 remain the only identified causative agents of early onset AD, with genetic determinants for late onset AD (LOAD) less characterized. Recent GWASs have revealed a number of genetic mutations that confer an increased risk of LOAD. Interestingly, a number of these SNPs reside in genes that directly relate to innate inflammatory function. As such, collections of these variants have the potential to drive pathological neuroinflammation and contribute to the progression and exacerbation of LOAD. These genes include APOE, major histocampaitibility complex class II, complement receptor 1 (CR1), CLU, membrane spanning 4 domain subfamily A and ATP‐binding cassette sub‐family A member 7 (ABCA7) (Villegas‐Llerena et al., 2016). Of the set of inflammatory‐related genes identified by GWAS, a subset has also been shown to play additional roles in microglial function. The genes CD33 (Siglec‐3), TREM2, CR1, PLCγ2 (PLCG2), ABI gene family member 3 (ABI3) and IL‐1 receptor accessory protein (IL1RAP) have been shown to be involved in the regulation of phenotypic switching and Aβ phagocytosis (Ramanan et al., 2015). Of these, the contributions of CD33 and TREM2 to AD pathology have been explored in a rodent model.
CD33 is a type‐I transmembrane receptor and member of the sialic acid‐binding immunoglobulin‐type lectins (Siglec) family and is expressed exclusively on immune cells. CD33 has been shown to mediate cell–cell interactions that inhibit or restrict immune responses, although no precise role for CD33 has been demonstrated in the CNS (Crocker et al., 2012). Carriers of CD33 SNPs have been shown to have higher Aβ deposits, as measured through Aβ deposits in both cortical and hippocampal regions, in addition to decreased soluble Aβ1–42 levels. Furthermore, primary microglia isolated from these knockout mice showed an increased ability to phagocytose Aβ1–42, whilst CD33 overexpression in the BV‐2 murine microglial cell line inhibited phagocytosis (Griciuc et al., 2013). Functioning as a type‐I transmembrane glycoprotein, TREM2 is predominantly expressed on CNS‐residing microglia. Post mortem examination of AD‐affected individuals reported increased levels of TREM2 within microglia surrounding Aβ plaques (Lue et al., 2015). The rs75932628 SNP identified by GWAS leads to a loss of function mutation, with APPSWE/PS1ΔE9 mice overexpressing this mutation demonstrating increased hippocampal Aβ deposits (Ulrich et al., 2014). These mice also exhibited impaired recruitment of microglia to Aβ plaques. Knockout of TREM2 in both APPSWE/PS1ΔE9 and 5xFAD mice leads to increased Aβ deposits and attenuated microglial activity. It was concluded that this effect was due to the infiltration of peripheral macrophages lacking TREM2 enhancing the pathology (Wang et al., 2015). However, a second study investigating TREM2 knockout in 5xFAD mice found that reduced TREM2 expression conferred an increased Aβ load despite a similarly observed attenuated microglial function (Jay et al., 2015). Within the same 5xFAD mice, a recent study has demonstrated that gene dosage increases of TREM2 alters microglia morphology and function. Critically, increases were seen in phagocytosis‐related genes and alterations in plaque types (Lee et al., 2018). The contrasting nature of these two studies is thought to be attributable to the differences in Aβ accumulation rates between mouse models (Leavy, 2015). Despite this, both studies show a crucial role for TREM2 in the production of the pro‐inflammatory cytokines TNFα and IL‐1β.
APOE4 is the most common gene identified by the GWASs undertaken, with this allele conferring the major genetic risk for LOAD. Forty two families with a family history of LOAD were analysed for the E4 allele variant and individuals homozygous for this allele succumbed to LOAD in over 90% of the cases examined (Corder et al., 1993). This gene dosage effect of APOE4 has been replicated in a number of subsequent studies. Indeed, a key comparison was made in a GWAS and the SNPs of the APO allelic variant individuals possess were identified. Utilizing CRISPR/Cas9 editing, Lin et al. (2018) recently generated isogenic IPSC lines containing either APOE3 or APOE4, which were then differentiated to microglial‐like cells. APOE4‐containing cells were shown to have enhanced inflammatory transcriptomes and an impaired uptake of Aβ1–42.
A recently completed GWAS coupled with longitudinal PET scans identified IL1RAP as a potential risk factor. IL1RAP is involved in the phenotypic switching of microglia. Individuals with the rs12053868‐G SNP had a higher chance of progressing from MCI to AD as well as higher accumulation rates of Aβ, with further PET scans revealing lower rates of microglial‐mediated inflammation (Ramanan et al., 2015). These data highlight the intricate link between neuroinflammation and the immune system in AD. An extensive study, in which 85 133 independent samples from both control and LOAD individuals were examined, identified two novel rare coding variants in PLCG2 and ABI3, in addition to a new variant in TREM2 (Sims et al., 2017). PLCG2 encodes the protein PLCγ2, a phospholipase that hydrolyzes inositol 1,4,5‐biphosphate to generate inositol 1,4,5‐trisphosphate and DAG. Interestingly, this variant is protective in AD, with an odds ratio of 0.68. PLCG2 is primarily expressed in microglia, with PLCG2 variants leading to PCLG2‐associated antibody deficiency and immune dysregulation disorders (Milner, 2015). ABI3 is suggested to have a role in the innate immune response, in particular through IFN‐mediated pathways (Sims et al., 2017). CR1 encodes the complement receptor 1, a protein involved in the regulation of the complement system in immunity. Critically, it is involved in the enhancement of phagocytosis of particles that have been opszonized (Fonseca et al., 2016). There are currently four SNPs identified in CR1 that confer increased AD risk (Zhu et al., 2015). Critically, the aforementioned genes as well as a number of other genes identified need to be further explored to confirm an altered functional role in microglia.
Targeting microglia
A considered approach is needed when targeting microglia therapeutically. As the CNS is composed of heterogeneous microglial populations, of which a number are involved in homeostatic roles, broad eliminations of microglial function are inappropriate. Rather, a more appropriate therapeutic strategy is one that results in a net shift of populations to a more protective phenotype. Disease state is also critical in the therapeutic management of microglia. Heightened microglial activity may indeed be beneficial during early stages of disease, yet detrimental during later stages (Butovsky and Weiner, 2018). This is of critical note as clinical trials in AD are moving further towards prodromal stages of disease, aiming to prevent rather than cure disease. Furthermore, issues arise when selecting what specifically to target. Certainly, targeting the associated proteins of immune‐related risk genes is a novel personalized medicine approach for those that possess these mutations. However, for the vast majority of individuals, this is not feasible. The precise biological mechanisms by which these genes alter AD risk remains unknown.
Currently, there are no approved selective therapeutics directed towards microglia. However, a number of clinical trials are underway that work through modulating microglia. Receptor‐interacting serine/threonine‐protein kinase 1 (RIPK1), an enzyme downstream of TNFα signalling, has been shown to mediate microglial responses in AD (Ofengeim et al., 2017). Denali Therapeutics purportedly has an RIPK1 inhibitor that has now entered phase 1 trials (Mullard, 2018). GliaCure has a small molecule that claims to promote microglial phagocytosis through binding of the microglial purinergic P2Y6 receptor. It is currently in phase 1 trials, its safety being examined in both healthy individuals and those with AD (GliaCure, 2014, 2015). GM‐CSF has been shown to reduce amyloid burden and increase microglial numbers in AD mice (Boyd et al., 2010). Sargramostim, a recombinant form of GM‐CSF, is currently in phase 2 trials (University of Colorado & Foundation, 2011). CSP‐1103, a small molecule that binds to the APP intracellular binding domain, has been shown to modulate microglial phenotype both in vitro and in vivo (Porrini et al., 2015). It is currently in phase 3 trials. A number of major pharmaceutical companies are indeed working with microglia and have made significant efforts to increase internal microglial research (Mullard, 2018).
Other microglial‐targeting therapeutics have however failed. Receptor for advanced glycation endproducts is involved in both microglial Aβ recognition and inflammation (Burstein et al., 2018). A small molecule antagonist against this receptor recently failed in phase 3 trials after it failed to meet primary outcomes. Minocycline, an anti‐inflammatory antibiotic which has classically been described as a putative microglial inhibitor, was shown to be ineffective at modifying the disease (Huntington Medical Research, 2012). Indeed, this historical classification of minocycline has been shown to be incorrect, with evidence demonstrating that it is indeed not a bona fide microglial inhibitor (Moller et al., 2016). As AD is indeed a heterogeneous disease, reducing it to a single therapeutic target may indeed be an incorrect approach. Future treatment strategies will most likely be combinatorial, addressing multiple aetiologies. In addition, novel microglial targets have been identified. Homing peptides directed towards microglia that contained siRNA for IRF5 resulted in a decrease in neuropathic pain (Terashima et al., 2018). Injection of microglial precursor cells transduced with TREM2 ex vivo in an EAE model of multiple sclerosis facilitated the clearance of cellular debris and ameliorated the disease (Takahashi et al., 2007).
A shifting of the microglial phenotypes can also be achieved by modulating the overarching process of neuroinflammation through targeting the so called ‘master regulators’. One such regulator is the type‐I IFN, which has been recently but consistently reported to be involved in microglial function. Type‐I IFNs have been shown to have the ability to act as master regulators of the peripheral immune response, with evidence also supporting this role within the CNS (Akira et al., 2006). C57BL/6 mice lacking type‐I IFN signalling exhibit an immunocompetent phenotype but importantly fail to produce a robust neuroinflammatory response in models of traumatic brain injury, Parkinson's disease and AD (Taylor et al., 2014; Karve et al., 2016; Main et al., 2016). Microglial type‐I IFN signalling has shown to be critical to microglial pathology (McDonough et al., 2017). In a number of recent studies aiming to characterize microglial phenotypes, type‐I IFN signalling has been shown to be crucial for specific populations (Hickman et al., 2013; Keren‐Shaul et al., 2017; Mathys et al., 2017). Blocking type‐I IFN signalling in lupus prevents microglial‐mediated synapse loss (Bialas et al., 2017). The most attractive evidence comes from a meta‐analysis of microglial transcripts from 69 individual disease states including AD. Critically, across all datasets, a co‐regulated IFN gene set was observed (Friedman et al., 2018).
Other ‘master regulators’ have also been identified. Removal or inhibition of inducible (i)NOS in APPSWE/PS1ΔE9 mice shifts the microglial phenotype, which results in decreased levels of Aβ. Functionally, this was shown to rescue cognition as measured through the radial arm maze behavioural test (Kummer et al., 2011). Deletion of TNF type 1 death receptor TNFR1, the endogenous receptor for TNFα, in APP23 mice leads to decreased levels of Aβ as well as decreased numbers of CD11b + microglia in the entorhinal cortex (He et al., 2007). Hippocampal injections of an adeno‐associated virus (AAV) expressing IL‐1β into APPSWE/PS1ΔE9 mice leads to increased numbers of arginase‐1‐positive microglia, which are associated with the clearance of Aβ plaques (Cherry et al., 2015). Reductions in microgliosis and improvements in cognition have been reported following AAV delivery of IL‐4 or IL‐10 to hippocampal regions of APPSWE/PS1ΔE9 mice (Kiyota et al., 2010; Kiyota et al., 2012). In APP transgenic mice expressing astrocytic TGFβ, decreased Aβ levels in parallel with increases in overall microgliosis were reported (Wyss‐Coray et al., 2001). This emerging body of work demonstrates that, at least as a proof of principle, targeting microglia does in fact ameliorate AD pathology.
Discussion
Current therapies for AD are symptomatic and, for the most part, ineffective. This is exacerbated by the notion that we are indeed unaware of the underlying and causative disease mechanisms in AD. There is a clear need for therapeutics that are able to target downstream effects of hallmark pathologies and as such manage disease exacerbation and progression. One such downstream effect is neuroinflammation in which a chronic form is seen in AD. The neuroinflammatory process is complex and multifaceted, with a number of considerations required regarding its management and modulation. Critical, however, are the class of resident CNS immune cells, microglia. It is only recently that research has begun to understand and appreciate the multitude of roles they play within AD and how this contributes to the pathology. The emerging literature suggests that targeting these cells presents as a novel therapeutic area for the management of AD.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY Harding et al. (2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a,b,c,d).
Author contributions
Z.M., J.M.T. and P.C. all contributed to the writing of this manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgement
This study was supported by grants from the National Health and Medical Research Council (NHMRC) of Australia.
Moore Z., Taylor J. M., and Crack P. J. (2019) The involvement of microglia in Alzheimer's disease: a new dog in the fight, British Journal of Pharmacology, 176, 3533–3543, 10.1111/bph.14546.
Contributor Information
Juliet M Taylor, Email: juliett@unimelb.edu.au.
Peter J Crack, Email: pcrack@unimelb.edu.au.
References
- Abud EM, Ramirez RN, Martinez ES, Healy LM, Nguyen CHH, Newman SA et al (2017). iPSC‐derived human microglia‐like cells to study neurological diseases. Neuron 94: 278–293.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akira S, Uematsu S, Takeuchi O (2006). Pathogen recognition and innate immunity. Cell 124: 783–801. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA et al (2017a). The Concise Guide To PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 174: S17–S129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017b). THE Concise Guide to PHARMACOLOGY 2017/18: Catalytic receptors. Br J Pharmacol 174: S225–S271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017c). The Concise Guide To PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion NV, Peters JA, Faccenda E, Harding SD et al (2017d). The Concise Guide To PHARMACOLOGY 2017/18: Overview. Br J Pharmacol 174: S1–S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonucci F, Turola E, Riganti L, Caleo M, Gabrielli M, Perrotta C et al (2012). Microvesicles released from microglia stimulate synaptic activity via enhanced sphingolipid metabolism. EMBO J 31: 1231–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachstetter AD, Van Eldik LJ, Schmitt FA, Neltner JH, Ighodaro ET, Webster SJ et al (2015). Disease‐related microglia heterogeneity in the hippocampus of Alzheimer's disease, dementia with Lewy bodies, and hippocampal sclerosis of aging. Acta Neuropathol Commun 3: 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DJ, Petersen RC (2018). Cellular senescence in brain aging and neurodegenerative diseases: evidence and perspectives. J Clin Investig 128: 1208–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamberger ME, Harris ME, McDonald DR, Husemann J, Landreth GE (2003). A cell surface receptor complex for fibrillar β‐amyloid mediates microglial activation. J Neurosci 23: 2665–2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhaskar K, Konerth M, Kokiko‐Cochran ON, Cardona A, Ransohoff RM, Lamb BT (2010). Regulation of tau pathology by the microglial fractalkine receptor. Neuron 68: 19–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bialas AR, Presumey J, Das A, van der Poel CE, Lapchak PH, Mesin L et al (2017). Microglia‐dependent synapse loss in type I interferon‐mediated lupus. Nature 546: 539–543. [DOI] [PubMed] [Google Scholar]
- Boche D, Perry VH, Nicoll JA (2013). Review: activation patterns of microglia and their identification in the human brain. Neuropathol Appl Neurobiol 39: 3–18. [DOI] [PubMed] [Google Scholar]
- Boyd TD, Bennett SP, Mori T, Governatori N, Runfeldt M, Norden M et al (2010). GM‐CSF upregulated in rheumatoid arthritis reverses cognitive impairment and amyloidosis in Alzheimer mice. J Alzheimer's Dis: JAD 21: 507–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown GC, Neher JJ (2012). Eaten alive! Cell death by primary phagocytosis: ‘phagoptosis’. Trends Biochem Sci 37: 325–332. [DOI] [PubMed] [Google Scholar]
- Brown GC, Neher JJ (2014). Microglial phagocytosis of live neurons. Nat Rev Neurosci 15: 209–216. [DOI] [PubMed] [Google Scholar]
- Bruttger J, Karram K, Wortge S, Regen T, Marini F, Hoppmann N et al (2015). Genetic cell ablation reveals clusters of local self‐renewing microglia in the mammalian central nervous system. Immunity 43: 92–106. [DOI] [PubMed] [Google Scholar]
- Burstein A, Sabbagh M, Andrews R, Valcarce C, Dunn I, Altstiel L (2018). Development of Azeliragon, an oral small molecule antagonist of the receptor for advanced glycation endproducts, for the potential slowing of loss of cognition in mild Alzheimer's disease. J Prev Alzheimers Dis 5: 149–154. [DOI] [PubMed] [Google Scholar]
- Butovsky O, Weiner HL (2018). Microglial signatures and their role in health and disease. Nat Rev Neurosci 19: 622–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calcoen D, Elias L, Yu X (2015). What does it take to produce a breakthrough drug? Nat Rev Drug Discov 14: 161–162. [DOI] [PubMed] [Google Scholar]
- Cherry JD, Olschowka JA, O'Banion MK (2015). Arginase 1+ microglia reduce Abeta plaque deposition during IL‐1beta‐dependent neuroinflammation. J Neuroinflammation 12: 203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conde JR, Streit WJ (2006). Microglia in the aging brain. J Neuropathol Exp Neurol 65: 199–203. [DOI] [PubMed] [Google Scholar]
- Corder E, Saunders A, Strittmatter W, Schmechel D, Gaskell P, Small G et al (1993). Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science 261: 921–923. [DOI] [PubMed] [Google Scholar]
- Crocker PR, McMillan SJ, Richards HE (2012). CD33‐related siglecs as potential modulators of inflammatory responses. Ann N Y Acad Sci 1253: 102–111. [DOI] [PubMed] [Google Scholar]
- Cronk JC, Filiano AJ, Louveau A, Marin I, Marsh R, Ji E et al (2018). Peripherally derived macrophages can engraft the brain independent of irradiation and maintain an identity distinct from microglia. J Exp Med 215: 1627–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings JL, Morstorf T, Zhong K (2014). Alzheimer's disease drug‐development pipeline: few candidates, frequent failures. Alzheimer's Res Ther 6: 37–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Mello C, Le T, Swain MG (2009). Cerebral microglia recruit monocytes into the brain in response to tumor necrosis factoralpha signaling during peripheral organ inflammation. J Neurosci 29: 2089–2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deczkowska A, Matcovitch‐Natan O, Tsitsou‐Kampeli A, Ben‐Hamo S, Dvir‐Szternfeld R, Spinrad A et al (2017). Mef2C restrains microglial inflammatory response and is lost in brain ageing in an IFN‐I‐dependent manner. Nat Commun 8: 717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois B, Albert ML (2004). Amnestic MCI or prodromal Alzheimer's disease? Lancet Neurol 3: 246–248. [DOI] [PubMed] [Google Scholar]
- Eikelenboom P, Veerhuis R, Scheper W, Rozemuller AJM, Van Gool WA, Hoozemans JJM (2006). The significance of neuroinflammation in understanding Alzheimer's disease. J Neural Transm 113: 1685–1695. [DOI] [PubMed] [Google Scholar]
- Floden AM, Combs CK (2011). Microglia demonstrate age‐dependent interaction with amyloid‐beta fibrils. J Alzheimers Dis 25: 279–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca MI, Chu S, Pierce AL, Brubaker WD, Hauhart RE, Mastroeni D et al (2016). Analysis of the putative role of CR1 in Alzheimer's disease: genetic association, expression and function. PLoS One 11: e0149792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman BA, Srinivasan K, Ayalon G, Meilandt WJ, Lin H, Huntley MA et al (2018). Diverse brain myeloid expression profiles reveal distinct microglial activation states and aspects of Alzheimer's disease not evident in mouse models. Cell Rep 22: 832–847. [DOI] [PubMed] [Google Scholar]
- Fuger P, Hefendehl JK, Veeraraghavalu K, Wendeln AC, Schlosser C, Obermuller U et al (2017). Microglia turnover with aging and in an Alzheimer's model via long‐term in vivo single‐cell imaging. Nat Neurosci 20: 1371–1376. [DOI] [PubMed] [Google Scholar]
- Garcia‐Reitboeck P, Phillips A, Piers TM, Villegas‐Llerena C, Butler M, Mallach A et al (2018). Human induced pluripotent stem cell‐derived microglia‐like cells harboring TREM2 missense mutations show specific deficits in phagocytosis. Cell Rep 24: 2300–2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S et al (2010). Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330: 841–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginhoux F, Lim S, Hoeffel G, Low D, Huber T (2013). Origin and differentiation of microglia. Front Cell Neurosci 7: 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gjoneska E, Pfenning AR, Mathys H, Quon G, Kundaje A, Tsai LH et al (2015). Conserved epigenomic signals in mice and humans reveal immune basis of Alzheimer's disease. Nature 518: 365–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GliaCure I (2014). Safety, tolerability, and pharmacokinetic study of single ascending doses of GC021109 in healthy subjects.
- GliaCure I (2015). Study evaluating safety, tolerability, and PK of multiple ascending doses of GC021109 in subjects with mild to moderate Alzheimer's disease.
- Godbout J, Chen J, Abraham J, Richwine A, Berg B, Kelley K et al (2005). Exaggerated neuroinflammation and sickness behavior in aged mice following activation of the peripheral innate immune system. FASEB J 19: 1329–1331. [DOI] [PubMed] [Google Scholar]
- Gomez‐Nicola D, Fransen NL, Suzzi S, Perry VH (2013). Regulation of microglial proliferation during chronic neurodegeneration. J Neurosci 33: 2481–2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griciuc A, Serrano‐Pozo A, Parrado AR, Lesinski AN, Asselin CN, Mullin K et al (2013). Alzheimer's disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron 78: 631–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T et al (2008). The NALP3 inflammasome is involved in the innate immune response to amyloid‐[beta]. Nat Immunol 9: 857–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy JA, Higgins GA (1992). Alzheimer's disease: the amyloid cascade hypothesis. Science 256: 184–185. [DOI] [PubMed] [Google Scholar]
- Hargis KE, Blalock EM (2017). Transcriptional signatures of brain aging and Alzheimer's disease: what are our rodent models telling us? Behav Brain Res 322: 311–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkes N (2018). Pfizer abandons research into Alzheimer's and Parkinson's diseases. BMJ 360: k122. [DOI] [PubMed] [Google Scholar]
- He P, Zhong Z, Lindholm K, Berning L, Lee W, Lemere C et al (2007). Deletion of tumor necrosis factor death receptor inhibits amyloid beta generation and prevents learning and memory deficits in Alzheimer's mice. J Cell Biol 178: 829–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, Kummer MP, Latz E (2014). Innate immune activation in neurodegenerative disease. Nat Rev Immunol 14: 463–477. [DOI] [PubMed] [Google Scholar]
- Henry CJ, Huang Y, Wynne AM, Godbout JP (2009). Peripheral lipopolysaccharide (LPS) challenge promotes microglial hyperactivity in aged mice that is associated with exaggerated induction of both pro‐inflammatory IL‐1beta and anti‐inflammatory IL‐10 cytokines. Brain Behav Immun 23: 309–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman SE, Kingery ND, Ohsumi TK, Borowsky ML, Wang LC, Means TK et al (2013). The microglial sensome revealed by direct RNA sequencing. Nat Neurosci 16: 1896–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S, Dissing‐Olesen L, Stevens B (2016). New insights on the role of microglia in synaptic pruning in health and disease. Curr Opin Neurobiol 36: 128–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntington Medical Research I (2012). Minocycline in patients with Alzheimer's disease.
- Ito D, Imai Y, Ohsawa K, Nakajima K, Fukuuchi Y, Kohsaka S (1998). Microglia‐specific localisation of a novel calcium binding protein, Iba1. Mol Brain Res 57: 1–9. [DOI] [PubMed] [Google Scholar]
- Jack CR Jr, Albert MS, Knopman DS, McKhann GM, Sperling RA, Carrillo MC et al (2011). Introduction to the recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 7: 257–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jay TR, Miller CM, Cheng PJ, Graham LC, Bemiller S, Broihier ML et al (2015). TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer's disease mouse models. J Exp Med 212: 287–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi P, Turola E, Ruiz A, Bergami A, Libera DD, Benussi L et al (2014). Microglia convert aggregated amyloid‐beta into neurotoxic forms through the shedding of microvesicles. Cell Death Differ 21: 582–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karve IP, Zhang M, Habgood M, Frugier T, Brody KM, Sashindranath M et al (2016). Ablation of type‐1 IFN signaling in hematopoietic cells confers protection following traumatic brain injury. eNeuro 3 ENEURO.0128‐0115.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren‐Shaul H, Spinrad A, Weiner A, Matcovitch‐Natan O, Dvir‐Szternfeld R, Ulland TK et al (2017). A unique microglia type associated with restricting development of Alzheimer's disease. Cell 169: 1276–1290 e1217. [DOI] [PubMed] [Google Scholar]
- Kettenmann H, Hanisch UK, Noda M, Verkhratsky A (2011). Physiology of microglia. Physiol Rev 91: 461–553. [DOI] [PubMed] [Google Scholar]
- Kim J, Kwon J, Kim M, Do J, Lee D, Han H (2016). Low‐dielectric‐constant polyimide aerogel composite films with low water uptake. Polymer J 48: 829–834. [Google Scholar]
- Kiyota T, Ingraham KL, Swan RJ, Jacobsen MT, Andrews SJ, Ikezu T (2012). AAV serotype 2/1‐mediated gene delivery of anti‐inflammatory interleukin‐10 enhances neurogenesis and cognitive function in APP+PS1 mice. Gene Ther 19: 724–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyota T, Okuyama S, Swan RJ, Jacobsen MT, Gendelman HE, Ikezu T (2010). CNS expression of anti‐inflammatory cytokine interleukin‐4 attenuates Alzheimer's disease‐like pathogenesis in APP+PS1 bigenic mice. FASEB J 24: 3093–3102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kongsui R, Johnson SJ, Graham BA, Nilsson M, Walker FR (2015). A combined cumulative threshold spectra and digital reconstruction analysis reveal structural alterations of microglia within the prefrontal cortex following low‐dose LPS administration. Neuroscience 310: 629–640. [DOI] [PubMed] [Google Scholar]
- Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R et al (2017). The TREM2‐APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 47: 566–581.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kummer MP, Hermes M, Delekarte A, Hammerschmidt T, Kumar S, Terwel D et al (2011). Nitration of tyrosine 10 critically enhances amyloid beta aggregation and plaque formation. Neuron 71: 833–844. [DOI] [PubMed] [Google Scholar]
- Lawson LJ, Perry VH, Dri P, Gordon S (1990). Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 39: 151–170. [DOI] [PubMed] [Google Scholar]
- Lawson LJ, Perry VH, Gordon S (1992). Turnover of resident microglia in the normal adult mouse brain. Neuroscience 48: 405–415. [DOI] [PubMed] [Google Scholar]
- Leavy O (2015). Neuroimmunology: TREM2 in Alzheimer disease. Nat Rev Immunol 15: 201–201. [Google Scholar]
- Lee CY, Landreth GE (2010). The role of microglia in amyloid clearance from the AD brain. J Neural Transm (Vienna) 117: 949–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CYD, Daggett A, Gu X, Jiang LL, Langfelder P, Li X et al (2018). Elevated TREM2 gene dosage reprograms microglia responsivity and ameliorates pathological phenotypes in Alzheimer's disease models. Neuron 97: 1032–1048 e1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YT, Seo J, Gao F, Feldman HM, Wen HL, Penney J et al (2018). APOE4 causes widespread molecular and cellular alterations associated with Alzheimer's disease phenotypes in human iPSC‐derived brain cell types. Neuron 98: 1141–1154.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Liu Y, Hao W, Wolf L, Kiliaan AJ, Penke B et al (2012). TLR2 is a primary receptor for Alzheimer's amyloid β peptide to trigger neuroinflammatory activation. J Immunol 188: 1098–1107. [DOI] [PubMed] [Google Scholar]
- Lue LF, Schmitz C, Walker DG (2015). What happens to microglial TREM2 in Alzheimer's disease: immunoregulatory turned into immunopathogenic? Neuroscience 302: 138–150. [DOI] [PubMed] [Google Scholar]
- Lund H, Pieber M, Parsa R, Grommisch D, Ewing E, Kular L et al (2018). Fatal demyelinating disease is induced by monocyte‐derived macrophages in the absence of TGF‐beta signaling. Nat Immunol 19: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maderna P, Godson C (2003). Phagocytosis of apoptotic cells and the resolution of inflammation. Biochimica et Biophysica Acta (BBA) – Mol Basis Dis 1639: 141–151. [DOI] [PubMed] [Google Scholar]
- Main BS, Zhang M, Brody KM, Ayton S, Frugier T, Steer D et al (2016). Type‐1 interferons contribute to the neuroinflammatory response and disease progression of the MPTP mouse model of Parkinson's disease. Glia 64: 1590–1604. [DOI] [PubMed] [Google Scholar]
- Mandrekar S, Jiang Q, Lee CYD, Koenigsknecht‐Talboo J, Holtzman DM, Landreth GE (2009). Microglia mediate the clearance of soluble Aβ through fluid phase macropinocytosis. J Neurosci 29: 4252–4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannix L (2018). What if we have got it wrong on Alzheimer's? In The Age.
- Maphis N, Xu G, Kokiko‐Cochran ON, Jiang S, Cardona A, Ransohoff RM et al (2015). Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain 138: 1738–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez FO, Gordon S (2014). The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000prime Rep 6: 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathys H, Adaikkan C, Gao F, Young JZ, Manet E, Hemberg M et al (2017). Temporal tracking of microglia activation in neurodegeneration at single‐cell resolution. Cell Rep 21: 366–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonough A, Lee RV, Weinstein JR (2017). Microglial interferon signaling and white matter. Neurochem Res 42: 2625–2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner JD (2015). PLAID: a syndrome of complex patterns of disease and unique phenotypes. J Clin Immunol 35: 527–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyake K (2007). Innate immune sensing of pathogens and danger signals by cell surface Toll‐like receptors. Semin Immunol 19: 3–10. [DOI] [PubMed] [Google Scholar]
- Moller T, Bard F, Bhattacharya A, Biber K, Campbell B, Dale E et al (2016). Critical data‐based re‐evaluation of minocycline as a putative specific microglia inhibitor. Glia 64: 1788–1794. [DOI] [PubMed] [Google Scholar]
- Morris GP, Clark IA, Vissel B (2014). Inconsistencies and controversies surrounding the amyloid hypothesis of Alzheimer's disease. Acta Neuropathol Commun 2: 135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullard A (2018). Microglia‐targeted candidates push the Alzheimer drug envelope. Nat Rev Drug Discov 17: 303–305. [DOI] [PubMed] [Google Scholar]
- Neniskyte U, Neher JJ, Brown GC (2011). Neuronal death induced by nanomolar amyloid beta is mediated by primary phagocytosis of neurons by microglia. J Biol Chem 286: 39904–39913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Njie EG, Boelen E, Stassen FR, Steinbusch HW, Borchelt DR, Streit WJ (2012). Ex vivo cultures of microglia from young and aged rodent brain reveal age‐related changes in microglial function. Neurobiol Aging 33: e191–e112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norden DM, Godbout JP (2013). Review: microglia of the aged brain: primed to be activated and resistant to regulation. Neuropathol Appl Neurobiol 39: 19–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ofengeim D, Mazzitelli S, Ito Y, DeWitt JP, Mifflin L, Zou C et al (2017). RIPK1 mediates a disease‐associated microglial response in Alzheimer's disease. Proc Natl Acad Sci U S A 114: E8788–E8797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohsawa K, Imai Y, Sasaki Y, Kohsaka S (2004). Microglia/macrophage‐specific protein Iba1 binds to fimbrin and enhances its actin‐bundling activity. J Neurochem 88: 844–856. [DOI] [PubMed] [Google Scholar]
- Olah M, Patrick E, Villani AC, Xu J, White CC, Ryan KJ et al (2018). A transcriptomic atlas of aged human microglia. Nat Commun 9: 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orre M, Kamphuis W, Osborn LM, Jansen AHP, Kooijman L, Bossers K et al (2014). Isolation of glia from Alzheimer's mice reveals inflammation and dysfunction. Neurobiol Aging 35: 2746–2760. [DOI] [PubMed] [Google Scholar]
- Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P et al (2011). Synaptic pruning by microglia is necessary for normal brain development. Science 333: 1456–1458. [DOI] [PubMed] [Google Scholar]
- Perry VH, Nicoll JA, Holmes C (2010). Microglia in neurodegenerative disease. Nat Rev Neurol 6: 193–201. [DOI] [PubMed] [Google Scholar]
- Pimplikar SW (2009). Reassessing the amyloid cascade hypothesis of Alzheimer's disease. Int J Biochem Cell Biol 41: 1261–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plescher M, Seifert G, Hansen JN, Bedner P, Steinhauser C, Halle A (2018). Plaque‐dependent morphological and electrophysiological heterogeneity of microglia in an Alzheimer's disease mouse model. Glia 66: 1464–1480. [DOI] [PubMed] [Google Scholar]
- Pocock JM, Piers TM (2018). Modelling microglial function with induced pluripotent stem cells: an update. Nat Rev Neurosci 19: 445–452. [DOI] [PubMed] [Google Scholar]
- Porrini V, Lanzillotta A, Branca C, Benarese M, Parrella E, Lorenzini L et al (2015). CHF5074 (CSP‐1103) induces microglia alternative activation in plaque‐free Tg2576 mice and primary glial cultures exposed to beta‐amyloid. Neuroscience 302: 112–120. [DOI] [PubMed] [Google Scholar]
- Ramanan VK, Risacher SL, Nho K, Kim S, Shen L, McDonald BC et al (2015). GWAS of longitudinal amyloid accumulation on 18F‐florbetapir PET in Alzheimer's disease implicates microglial activation gene IL1RAP. Brain 138: 3076–3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransohoff RM (2016). A polarizing question: do M1 and M2 microglia exist? Nat Neurosci 19: 987–991. [DOI] [PubMed] [Google Scholar]
- Rawji KS, Mishra MK, Michaels NJ, Rivest S, Stys PK, Yong VW (2016). Immunosenescence of microglia and macrophages: impact on the ageing central nervous system. Brain 139: 653–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed‐Geaghan EG, Savage JC, Hise AG, Landreth GE (2009). CD14 and toll‐like receptors 2 and 4 are required for fibrillar A {beta}‐stimulated microglial activation. J Neurosci 29: 11982–11992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadallah S, Eken C, Schifferli JA (2011). Ectosomes as modulators of inflammation and immunity. Clin Exp Immunol 163: 26–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salminen A, Ojala J, Kauppinen A, Kaarniranta K, Suuronen T (2009). Inflammation in Alzheimer's disease: amyloid‐β oligomers trigger innate immunity defence via pattern recognition receptors. Prog Neurobiol 87: 181–194. [DOI] [PubMed] [Google Scholar]
- Sarlus H, Heneka MT (2017). Microglia in Alzheimer's disease. J Clin Invest 127: 3240–3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R et al (2012). Microglia sculpt postnatal neural circuits in an activity and complement‐dependent manner. Neuron 74: 691–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shastri A, Bonifati DM, Kishore U (2013). Innate immunity and neuroinflammation. Mediators Inflamm 2013: 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvin A, Ginhoux F (2018). Microglia heterogeneity along a spatio‐temporal axis: more questions than answers. Glia . [DOI] [PubMed] [Google Scholar]
- Sims R, van der Lee SJ, Naj AC, Bellenguez C, Badarinarayan N, Jakobsdottir J et al (2017). Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial‐mediated innate immunity in Alzheimer's disease. Nat Genet 49: 1373–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaper SD, Facci L, Zusso M, Giusti P (2018). An inflammation‐centric view of neurological disease: beyond the neuron. Front Cell Neurosci 12: 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streit WJ, Braak H, Xue QS, Bechmann I (2009). Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer's disease. Acta Neuropathol 118: 475–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahara K, Kim HD, Jin JJ, Maxwell JA, Li L, Fukuchi K (2006). Role of toll‐like receptor signalling in Abeta uptake and clearance. Brain 129: 3006–3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Prinz M, Stagi M, Chechneva O, Neumann H (2007). TREM2‐transduced myeloid precursors mediate nervous tissue debris clearance and facilitate recovery in an animal model of multiple sclerosis. PLoS Med 4: e124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JM, Minter MR, Newman AG, Zhang M, Adlard PA, Crack PJ (2014). Type‐1 interferon signaling mediates neuro‐inflammatory events in models of Alzheimer's disease. Neurobiol Aging 35: 1012–1023. [DOI] [PubMed] [Google Scholar]
- Terashima T, Ogawa N, Nakae Y, Sato T, Katagi M, Okano J et al (2018). Gene therapy for neuropathic pain through siRNA‐IRF5 gene delivery with homing peptides to microglia. Mol Ther Nucleic Acids 11: 203–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich JD, Finn MB, Wang Y, Shen A, Mahan TE, Jiang H et al (2014). Altered microglial response to Abeta plaques in APPPS1‐21 mice heterozygous for TREM2. Mol Neurodegener 9: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- University of Colorado D, & Foundation TD (2011). Study of the safety & efficacy of Leukine® in the treatment of Alzheimer's disease Available at https://ClinicalTrials.gov/show/NCT01409915.
- Villegas‐Llerena C, Phillips A, Garcia‐Reitboeck P, Hardy J, Pocock JM (2016). Microglial genes regulating neuroinflammation in the progression of Alzheimer's disease. Curr Opin Neurobiol 36: 74–81. [DOI] [PubMed] [Google Scholar]
- Walker FR, Beynon SB, Jones KA, Zhao Z, Kongsui R, Cairns M et al (2014). Dynamic structural remodelling of microglia in health and disease: a review of the models, the signals and the mechanisms. Brain Behav Immun 37: 1–14. [DOI] [PubMed] [Google Scholar]
- Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML et al (2015). TREM2 lipid sensing sustains the microglial response in an Alzheimer's disease model. Cell 160: 1061–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinhard L, di Bartolomei G, Bolasco G, Machado P, Schieber NL, Neniskyte U et al (2018). Microglia remodel synapses by presynaptic trogocytosis and spine head filopodia induction. Nat Commun 9: 1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wes PD, Sayed FA, Bard F, Gan L (2016). Targeting microglia for the treatment of Alzheimer's disease. Glia 64: 1710–1732. [DOI] [PubMed] [Google Scholar]
- Wyss‐Coray T, Lin C, Yan F, Yu G‐Q, Rohde M, McConlogue L et al (2001). TGF‐β1 promotes microglial amyloid‐β clearance and reduces plaque burden in transgenic mice. Nat Med 7: 612–618. [DOI] [PubMed] [Google Scholar]
- Xanthos DN, Sandkuhler J (2014). Neurogenic neuroinflammation: inflammatory CNS reactions in response to neuronal activity. Nat Rev Neurosci 15: 43–53. [DOI] [PubMed] [Google Scholar]
- Yamanaka M, Ishikawa T, Griep A, Axt D, Kummer MP, Heneka MT (2012). PPARγ/RXRα‐induced and CD36‐mediated microglial amyloid‐β phagocytosis results in cognitive improvement in amyloid precursor protein/presenilin 1 mice. J Neurosci 32: 17321–17331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisel A, Munoz‐Manchado AB, Codeluppi S, Lonnerberg P, La Manno G, Jureus A et al (2015). Cell types in the mouse cortex and hippocampus revealed by single‐cell RNA‐seq. Science 347: 1138–1142. [DOI] [PubMed] [Google Scholar]
- Zhu XC, Yu JT, Jiang T, Wang P, Cao L, Tan L (2015). CR1 in Alzheimer's disease. Mol Neurobiol 51: 753–765. [DOI] [PubMed] [Google Scholar]