

Abstract
Mycofactocin (MFT) is a putative ribosomally synthesized and post-translationally modified (RiPP) redox cofactor. The biosynthesis of MFT is encoded by the gene cluster mftABCDEF. While processing of the precursor peptide by MftB, MftC, and MftE has been shown to result in the formation of the small molecule 3-amino-5-[(p-hydroxyphenyl)methyl]-4,4-dimethyl-2-pyrrolidinone (AHDP), no activity has been shown for the putative dehydrogenase MftD and the putative glycosyltransferase MftF. In addition, evidence demonstrating that MFT is a redox cofactor has only been limited to the requirement of mft genes for ethanol assimilation in M. smegmatis mc2155. Here, we demonstrate that MftD catalyzes the oxidative deamination of AHDP, forming an α-keto moiety on the resulting molecule which we call premycofactocin (PMFT). We characterize PMFT by 1D and 2D nuclear magnetic resonance spectroscopy techniques and by high-resolution mass spectrometry data to solve its structure. We further characterized PMFT by cyclic voltammetry and found its midpoint potential to be ~255 mV. Lastly, we demonstrate that PMFT is a biologically active redox cofactor that oxidizes NADH bound by M. smegmatis carveol dehydrogenase (MsCDH) and can be used by MsCDH in the oxidation of carveol. These data demonstrate for the first time that PMFT functions as a biologically active redox mediator and provides the most direct evidence to date that MFT is a RiPP-derived redox cofactor.
Keywords: mycofactocin, ribosomally synthesized and post-translationally modified peptide, redox cofactor, oxidative deamination, carveol dehydrogenase, MftD
Graphical abstract
INTRODUCTION
Ribosomally synthesized and post-translationally modified peptides (RiPPs)have emerged as a structurally and biologically diverse class of secondary metabolites.1 Interest in RiPPs is particularly focused on the immense bioavailability and engineerability of diverse antibiotics.2–5 While the vast majority of characterized RiPPs have been shown to possess antimicrobial activity, others have been shown to have more diverse biological functions such as quorum sensing and redox activity.6–13 Until recently, this latter activity was defined by the sole quintessential member, pyrroloquinoline qui-none (PQQ), which is used as an electron acceptor in bacterial sugar and alcohol dehydrogenases.11,14,15 However, it was proposed that the uncharacterized mycofactocin biosynthetic pathway could encode for the second member of the RiPP-derived redox cofactor family.16 While, the mycofactocin biosynthetic pathway bears a striking resemblance to that of PQQ, the bona fide structure and function of the molecule remains unknown.
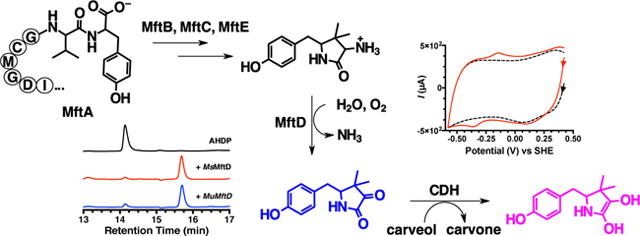
Mycofactocin (MFT) was first identified by a bioinformatic analysis that demonstrated the cooccurrence of the genes mftABCDEF (Figure 1A)16 in over 600 bacterial species, including a score of human pathogens (eg. M. tuberculosis, M. avium, and M. ulcerans).16,17 In addition, the same bioinformatic study indicated that genes encoding for three different dehydrogenase families (TIGR03971, TIGR03989, and TIGR04266) were found in the genome only when mft genes were present. This codependence upon the mft biosynthetic pathway implicates an important role for MFT in the activities of the aforementioned TIGR families. Notably, members of the MFT associated dehydrogenase families were shown to contain non-exchangeable nicotinamide cofactors that were active in vitro with only non-physiologically relevant redox mediators.18,19 This further suggested that MFT could be required for catalytic turnover for these enzymes. In support of this notion, recent gene knockout studies have indicated that mftA-D and mftF, along with the iron-dependent alcohol dehydrogenase msmeg_6242 (TIGR04266), are essential for ethanol assimilation in the model organism M. smegmatis mc2155.20 In addition, the same study demonstrated that disruption of the mft genes led to imbalance of cellular concentrations of NAD+/NADH, indicating for the first time in vivo, that MFT may play a role in redox metabolism. However, direct evidence for the physiological role of MFT has not been provided, in part because its structure remains unknown.
Figure 1 –

Mycofactocin biosynthetic pathway. A) An arrow representation of the mycofactocin biosynthetic pathway showing the association of a short chain dehydrogenase (SDR) and the MuMftA amino acid sequence. B) A condensed reaction scheme of known steps in mycofactocin biosynthesis. Enzymatic modifications are shown in red.
Recent work by our lab, and others, has focused on elucidating the in vitro biosynthesis, and thereby the structure and function, of MFT. It was proposed that MFT is synthesized from the conserved C-terminal region on precursor peptide MftA.16 Validation of this proposal was provided when two independent studies reported that MftC catalyzes the S-adenosylmethionine dependent oxidative decarboxylation of the C-terminus on MftA, in the presence of MftB, resulting in an α/β unsaturated tyramine (Figure 1B, MftA**).21,22 However, upon further investigation it was shown that MftC catalysis also resulted in the a functionally relevant conversion of MftA** to MftA* which contains a bicyclic modification consisting of a 5-membered lactam ring derived from the penultimate valine residue (Figure 1B).23 These results led to the examination of MftE catalysis where it was shown that the enzyme specifically hydrolyzes MftA*, resulting in the formation of 3-amino-5-[(p-hydroxyphenyl)methyl]-4,4-dimethyl-2-pyrrolidinone (AHDP, Figure 1B).24 The remaining gene products, MftD and MftF, encode for putative lactate dehydrogenase and glycosyl transferase respectively, and their function in mycofactocin biosynthesis have been hitherto unknown.
MftD belongs to the aldolase-TIM barrel fold family that is comprised of α-hydroxy acid dehydrogenases which are known to be flavin mononucleotide (FMN) dependent.25–27 This putative FMN dependency led us to speculate that MftD could catalyze the molecular oxygen dependent oxidation of the phenyl ring of AHDP to generate a the corresponding catechol, reminiscent to PQQ and other redox cofactors.28–31 The rationale for this proposal was that if MFT was redox active, it required a physiologically relevant redox center which was not apparent on the AHDP parent molecule. However, without any evidence, this proposal was only speculative. Herein, we present a combination of activity assays, nuclear magnetic resonance studies, and high resolution-mass spectrometry studies to demonstrate conclusively that MftD catalyzes the oxidation of the L-amino moiety on AHDP to form the redox center of MFT. Moreover, we provide compelling evidence that the resulting molecule is redox active and is shown to serve, in vitro, as a catalytically competent cofactor for the putative MFT-dependent carveol dehydrogenase, MsCDH. Together, our experimental data provides strong evidence that supports the notion that MFT is a novel, catalytically-competent, RiPP-derived, redox cofactor.
RESULTS
MftD is an FMN Binding Protein.
The mftD gene (mul_0774) was cloned from M. ulcerans Agy99 and the MuMftD protein was heterologously expressed in and anaerobically purified from Escherichia coli (Figure 2A). Characterization of as-isolated MuMftD protein by UV-visible absorbance spectroscopy indicated the presences of a reduced flavin which is characterized by the broad shoulder between 300 nm – 410 nm (Figure 2B, red). Following exposure to air, the absorbance spectra of MuMftD shifted to that of an oxidized flavin, characterized by the dual absorbance maxima at 360 nm and 445 nm (Figure 2B, blue). To determine which species of flavin was bound, MuMftD was heat precipitated and the soluble fraction was analyzed by HPLC. The retention time of the flavin in the soluble fraction was compared to authentic flavin standards (flavin adenine dinucleotide, FMN, and riboflavin, Figure 2C). From this analysis, it was evident that the absorbance features of the cofactor bound to MuMftD originated from FMN, consistent with other members of the protein-fold family.32 Notably, when MuMftD was purified aerobically, the protein appeared to lose FMN during the isolation process. Consistent with this observation, analysis of MuMftD by analytic HPLC-SEC demonstrated that upon oxidation, the protein releases FMN (Figure S1), in the absence of substrate. In our hands, we observed that the loss of FMN is irreversible.
Figure 2 –

A) MftD was purified to homogeneity as determined by SDS-PAGE analysis. Lane 1 contains the Goldbio BLUEstain Protein Ladder standard, Lane 2 contains His6-purified MuMftD, and lane 3 contains His6-purified MsMftD. B) A UV-Vis spectral analysis of anaerobically prepared MftD indicates the presence of a reduced flavin (red). Upon exposure to air of the protein sample, the UV-Vis spectrum shifts considerably, indicating that the flavin is oxidized. C) High pressure liquid chromatography analysis of the flavin containing fraction of protein precipitate (red) indicates that MftD likely binds FMN when compared to retention times of flavin standards.
Reconstitution of MftD Activity.
Our expectation was that AHDP, the product from the MftE reaction, might be the substrate for MftD. To validate this hypothesis, reactions containing AHDP and MftD were carried out aerobically and the reaction mixture was analyzed by HPLC. The aerobic addition of AHDP to MftD and subsequent analysis by HPLC does in fact give rise to a new species at a retention time of ~15.8 min (Figure 3A, blue) with concomitant disappearance of AHDP (~14.1 min); the associated kobs for the single turnover reaction was measured to be 0.010 ± 0.002 min−1 (Figure S2A). Interestingly, the UV-visible absorbance spectrum for the new species is substantially different than that of AHDP, indicating that the resulting modification also impacted the electronics of the molecule (Figure S2B and S2C). To ensure that this activity was not unique to MuMftD, the gene msmeg_1424, encoding for MsMftD, was cloned from M. smegmatis mc2155, the protein was purified from E. coli (Figure 2A), and reactions with AHDP were prepared and analyzed by HPLC. As expected, the HPLC chromatogram for the MsMftD reaction showed similar results to that of MuMftD (Figure 3A, red), indicating that AHDP is most likely the substrate for both Ms and MuMftD. Herein, we worked solely with MuMftD and will refer to the protein as simply MftD.
Figure 3 –

A) HPLC chromatograms of reactions containing AHDP (black), AHDP and MsMftD (red), and AHDP and MuMftD (blue) indicate that AHDP is an active substrate for MftD. B) HRMS analysis of the AHDP (black) and the MftD product (blue) shows an ion with a m/z that is consistent with the loss of -NH3 and the addition of O. C) From the HPLC and HRMS analysis, MftD is proposed to catalyze the oxidative deamination of AHDP (black) to form pre-mycofactocin (blue, PMFT). The theoretical m/z for the molecules are indicated above their structure.
To determine the nature of the MftD catalyzed modification, we analyzed the product of the reaction by high resolution mass spectrometry (HRMS). Consistent with our previous work,24 HRMS analysis of the AHDP [M+H]+ ion, reanalyzed here, was found to have a mass-to-charge ratio (m/z) = 235.1445 (Figure 3B, black), within 4 ppm of the predicted mass (C13H18N2O2, theoretical m/z = 235.1441 Da). HRMS analysis of the ~15.8 min MftD product provided a m/z = 234.1107 (Figure 3B, blue). This mass is consistent with the loss of NH3 and the addition of a single oxygen atom to ADHP (C13H15NO3, theoretical [M+H]+ m/z = 234.1125). These results are inconsistent with our previous proposal suggesting that MftD could catalyze the formation of a catechol on the phenyl ring of AHDP. Instead, our HRMS analysis suggests that MftD might catalyze the oxidative deamination of AHDP, installing an α-keto moiety, resulting in the formation of 5-[(p-hydroxyphenyl)methyl]-4,4-dimethyl-2,3-pyrrolidinedione (Figure 3C, blue) or herein referred to as pre-mycofactocin (PMFT). Precedence for flavin dependent oxidative deamination comes from D-amino acid oxidase (DAAO). DAAO’s function to catalyze the flavin adenine dinucleotide (FAD) dependent and stereospecific oxidation of D-amino acid to form α-keto acids.33,34 However, it should be noted that DAAO and MftD belong to different protein fold families.35,36
Structural Elucidation of the MftD Product.
To validate that MftD catalyzes the oxidative deamination of AHDP and the subsequent formation of PMFT, 1D and 2D NMR studies were carried out on the isolated product. Reactions containing AHDP and MftD were performed on a large scale (~2 mg AHDP) and the HPLC purified PMFT was analyzed by NMR. For 13C NMR, we used synthesized MftA labeled with U-13C valine and U-13C tyrosine at positions Val29 and Tyr30 in reactions with MftB, MftC, MftE, and MftD to generate the U-13C labeled PMFT. This was required to increase the 13C NMR signal due to the limited quantities of isolated PMFT. Although we had already reported on the 1H, 13C, and COSY NMR characterization of AHDP, we further analyzed it by HSQC 2D NMR in this study. This allowed us to directly compare AHDP spectra to that of PMFT.
Overlaid COSY spectra of the lactam region for both AHDP (red) and PMFT (blue) are shown in Figure 4A. Notably, the chemical shifts of the protons within the lactam are modestly shifted down-field by 0.1–0.4 ppm. This downfield shift is accompanied by the disappearance of the Hα in the 1H NMR spectrum for PMFT, suggesting that Cα was the site of modification. Consistent with this observation, 13C NMR indicated a drastic downfield shift of the Cα in AHDP from ~61 ppm to ~210 ppm in PMFT (Figure 4B and see Supplementary Information for full spectra). The chemical shift of Cα is corroborated by HMBC spectra (Figure 4B) which shows long range interaction between the protons on the geminal methyl and the Cα. Lastly, overlaid HSQC spectra of AHDP and PMFT show the loss the proton-carbon coupling associated with H-Cα. Together these NMR data provide strong evidence that MftD catalyzes the oxidative deamination of AHDP to form PMFT.
Figure 4 –

NMR analysis of PMFT. A) Overlaid COSY spectra of AHDP (red) and PMFT (blue) reveals the loss of the Hα signal. B) HMBC spectra for PMFT provides evidence that the Cα has shifted downfield to ~210 ppm. C) Overlaid HSQC spectra of ADHP (red) and PMFT (blue) shows the loss of the H-Cα heteronuclear interaction. D) A reference structure of PMFT with the important carbon annotated. E) A summary of relevant NMR correlations on PMFT.
The Role of Oxygen in the MftD Reaction
Having solved the PMFT structure, we next sought to determine the source of the incorporated oxygen atom. Since MftD catalyzed a similar reaction to DAAO, we turned to its well-characterized mechanism for guidance. The DAAO reaction is described by the two-step oxidative deamination of the D-amino group on the amino acid (Scheme 1A). To begin with, DAAO oxidizes the amino acid to an a-imimo acid through a hydride transfer from the Cα to FAD.37 This is followed by a nucleophilic substitution by an activated water and the subsequent oxidation to form the ketone.33 We rationalized that MftD could carry out a similar reaction mechanism which we describe in Scheme 1B. To determine if water was the source of the inserted oxygen, we carried out MftD reactions in 99% enriched 18OH2 and analyzed the resulting PMFT by HRMS. From this experiment, we expected to observe an enrichment of a PMFT ion that was consistent with a single 18O being incorporated (+2 m/z). As expected, the mass spectrum of the reaction mixture carried out in 18OH2 shows two predominant ions (Figure 5A), one at the expected m/z for unlabeled PMFT ([M+H]+ m/z = 234.1125) and the other at a m/z = 236.1169, within 2 ppm of the expected mass for the 18O-incorporated PMFT ([M+H]+ m/z = 236.1167). The percent incorporation was ~65%, near the expected theoretical maximum of 87%. Conversely, when the reaction was carried out in 18O2, we did not observe significant incorporation of 18O in the product (Figure S3A). The rate of back exchange was determined to not significantly affect the overall incorporation of 18O during the course of this experiment (Figure S3B). Therefore, these results are consistent with water being the source of the inserted oxygen atom in the MftD catalyzed oxidative deamination of AHDP.
Scheme 1 –

A) A condensed reaction scheme for D-amino acid oxidase (DAAO) and B) for MftD.
Figure 5 –

A) High resolution mass spectra of the MftD reaction carried out in 18OH2 shows the enrichment of the 18O incorporated PMFT (red). The structure for natural abundance and enriched PMFT are shown above with their theoretical mass for the [M+H]+ ion. B) HPLC analysis of MftD reactions under anaerobic (blue) and aerobic (red) conditions suggest that molecular oxygen is required for catalytic turnover. The reference chromatogram for AHDP is shown in black.
In the DAAO mechanism and the proposed mechanism for MftD, O2 is the final electron acceptor.33 To verify that molecular oxygen is required for MftD turnover, reactions were carried out anaerobically. To achieve anoxic conditions, reactions were conducted in an anaerobic chamber and supplemented with glucose and glucose oxidase prior to the addition of MftD. Glucose oxidase catalyzes the oxidation of glucose to gluconate and hydrogen peroxide and is often added to reactions to achieve anaerobicity.38,39 The subsequent HPLC chromatogram of the anaerobic reaction indicated only the presence of the starting material AHDP (Figure 5B). This suggests that molecular oxygen is required for the MftD activity. Since anaerobically purified MftD contains FMNH2, and since we did not observe any turnover of MftD, it is likely that O2 is required for the oxidation of FMNH2. Notably, MftD was inactive in reactions carried out under the same conditions but with the addition of NAD+ (Figure S4) suggesting that the nicotinamide cannot fulfill the role of O2. While efforts to measure the existence and stoichiometry of the putative peroxide were not undertaken, the results from this analysis is consistent with O2 being the final electron acceptor in MftD catalysis.
PMFT is Redox and Functionally Active In Vitro.
As noted previously, the structure of AHDP did not have an apparent physiologically relevant redox center. However, following oxidative deamination by MftD, we rationalized that the resulting α-keto-amide moiety could be the active site of MFT. Indeed α-keto acids are a common 2e−/2H+ redox moiety and can be found in biologically important molecules such as pyruvate, α-ketoglurate, and oxaloacetate. To provide insight about the possible redox behavior of PMFT, cyclic voltammetry was used to directly measure the midpoint potential of PMFT. Three-electrode CV experiments were carried out using a glassy carbon working electrode, an Ag/AgCl reference electrode, and a platinum counter electrode. PMFT was non-covalently adsorbed onto the working electrode using single walled carbon nanotubes (SWCNT) as a co-absorbent. CV measurements yielded a single signal that was observed between −100 mV and −400 mV as compared to the buffer/SWCNT control (Figure 6A). From this measurement, the reductive and oxidative potentials were found to be −370 mV and −140 mV respectively, and the overall midpoint potential to be −255 mV. We did not observe significantly differentiated peaks for two single electron transfer events, thus we define the midpoint potential for the 2e−/2H+ transfer as −255 mV.
Figure 6 –

A) Overlaid cyclic voltammogram of PMFT/SWCNT (red) and buffer/SWCNT (black). Voltammetry of PMFT was measured at pH 7.0 and at 22 °C with a scan rate of 50 mV/s. B) Overlaid UV-visible spectra showing the oxidation of MsCDH bound NADH by PMFT. C) HPLC analysis demonstrates that MsCDH is active towards carveol in the presence of PMFT (blue). The chromatogram for PMFT is shown in red. D) HRMS analysis of the ~14.9 min peak shows an ion with a m/z that is consistent with the mass of PMFTH2 (theoretical [M+H]+ m/z = 236.1281). E) The proposed structure of PMFT following a 2e−/2H+ reduction.
Having shown that PMFT is redox active, we next postulated that it could be used to oxidize NADH in MFT-dependent dehydrogenases. M. smegmatis mc2155 carveol dehydrogenase (MsCDH, Msmeg_1410) was chosen as the model MFT-dependent dehydrogenase to test this hypothesis since the R. erythropolis DCL14 homolog has been shown to catalyze the oxidation of carveol to carvone using 2,6-dichlorophenolindophenol (DCPIP) as an electron acceptor19 and since it was known to copurify with NADH.18 This latter property is significant because it could allow us to monitor the PMFT dependent oxidation of NADH to NAD+ by monitoring UV-visible absorbance at 340 nm (ΔεNADH 340nm = 6.22 mM−1cm−1). To demonstrate that PMFT could be used as an oxidant, scanning UV-visible spectroscopic measurements were carried out with purified MsCDH (see Figure S5 for an SDS-PAGE). As shown in Figure 6B, the incremental addition of PMFT resulted in a stoichiometric decrease in absorbance at 340 nm suggesting that PMFT is mediating the oxidation of NADH to NAD+ on MsCDH. It should be noted that we did not observe the same oxidative activity towards free NADH (Figure S6A). Consequently, this implies that PMFT is biologically active with MsCDH and potentially with other MFT dependent dehydrogenases.
After finding that PMFT was capable of oxidizing NADH bound to an MFT-dependent dehydrogenase, we next sought to couple PMFT to the MsCDH catalyzed oxidation of carveol. To do so, MsCDH was incubated with excess amounts of carveol and PMFT in a stoichiometric ratio. The subsequent reaction mixture was analyzed by HPLC (Figure 6C). We observed a change in the retention time of PMFT in the chromatogram, from ~15.8 to ~14.9 min, with an accompanying change in the UV-visible spectrum (Figure S7A and S7B). In addition, we observed a concomitant appearance of new species at a retention time of ~19.5 min corresponding to carvone (for a full HPLC analysis see Figure S6B). We expected that the species at ~14.9 min is the reduced form of PMFT (PMFTH2). To provide evidence for this hypothesis, we analyzed the HPLC purified species by HRMS. As expected, HRMS analysis (Figure 6D) of the ~14.9 min species provided a m/z = 236.1267 which is consistent with the m/z of PMFTH2 (C13H17NO3, theoretical [M+H]+ m/z = 236.1281). Taken together, the observation that PMFT oxidizes the NADH on MsCDH, PMFT facilitates MsCDH carveol oxidation activity, and the observation of a mass consistent with PMFTH2, provides compelling evidence that PMFT is the biologically relevant redox moiety of MFT.
To determine the efficacy of PMFT as a redox mediator, the apparent rate constant (kobs) governing the oxidation of MsCDH bound NADH by PMFT was determined. To do so, time-dependent stopped-flow UV-vis spectrometry experiments were carried out at two PMFT concentrations, in triplicate each, monitoring the oxidation of MsCDH bound NADH at 340 nm (Figure 7). Data were fitted to a single exponential decay which yielded a kobs = 0.8 ± 0.1 s−1. To directly compare this rate to the rate of oxidation of MsCDH by DCPIP, we carried out similar stopped-flow reactions monitoring the absorbance of DCPIP at 600 nm (ΔεDCPIP 600nm = 20.7 mM−1cm−1, Figure S8A) due to spectral overlap with NADH. These reactions were fitted to a linear equation which provided a kobs < 0.01 s−1, substantially slower than that observed for PMFT. Control stopped-flow experiments were carried out to ensure that DCPIP was not oxidizing MsCDH on the millisecond timescale (Figure S8B). While there is a slim possibility that DCPIP could oxidize MsCDH faster than the deadtime of the instrument (3 ms), the more likely scenario is that DCPIP is non-specifically interacting with MsCDH which leads to the slow rate of oxidation. Overall, the >100-fold rate enhancement of PMFT over DCPIP further indicates that (P)MFT is a physiological reductant of MsCDH.
Figure 7 –

Stopped-flow kinetic analysis of a single turnover oxidation reaction with 160 μM MsCDH and 25 μM (red dashes) or 50 μM (magenta dashes) PMFT. The oxidation of NADH at 340 nm was monitored during the reaction, sampling every 2 ms. Each kinetic trace is an average of 3 individual experiments. The averages were fitted to a single exponential decay (black) to determine the rate constant.
DISCUSSION
Nature has used ribosomally synthesized and post-translationally modified peptides (RiPPs) to produce a diverse array of bioactive molecules. In particular, the use of a RiPP biosynthetic pathway to produce a small molecule, such as pyrroloquinoline quinone (PQQ), seems gratuitous considering the high energetic costs associated with the synthesis of the precursor peptide. In the case of PQQ, of the ~30 amino acids on the precursor peptide PqqA, only tyrosine and glutamate are conserved in the final compound.40–42 Likewise, in the case of MFT biosynthesis only two residues from a ~30 amino acid peptide are being used to make MFT. This implies that the function of the molecules contributes more to the physiology of the cell than the energetic cost to make them. As it happens, this is the case for PQQ, which serves to transfer electrons from sugar and alcohol dehydrogenases to the electron transport chain,43 and thus participates in the generation of cellular currency, ATP. By analogy, the same could be predicted for MFT.
Nearly a decade ago it was proposed that MFT is the second member of the RiPP-derived redox cofactor family.16 The assertion of this proposal was solely based on a bioinformatic analysis that concluded that associated dehydrogenases might require MFT for catalytic turnover and that the MFT biosynthetic pathway appeared reminiscent to that of PQQ. Over a short period of time, we have seen the maturation of MFT take place. The discovery that MftC catalyzed a two-step cyclization to from the bicyclic MftA* provided intrigue to the possibility that MFT could be a redox cofactor.21–23 The demonstration of the subsequent hydrolysis of MftA* to form the small molecule AHDP further fueled this hypothesis.24,44 Lastly and significantly, recent work has shown that mft genes are required for the in vivo ethanol assimilation in M. smegmatis mc2155.20 Although, these findings have coalesced to suggest that MFT is a redox cofactor, no direct evidence has been provided to date.
In this work, we have provided evidence that AHDP is an active substrate for MftD. The NMR structural workup of the MftD product, paired with HRMS analysis, provides conclusive evidence that MftD catalyzes the oxidative deamination of AHDP to form the α-keto moiety on PMFT. The existence of the α-keto moiety on PMFT led us to postulate that the molecule could be redox active. To probe for redox activity, we used cyclic voltammetry to measure the midpoint potential of PMFT. The observance of redox activity suggested to us that PMFT is likely the work horse of the MFT molecule, much like F0 is for F420 and riboflavin is for FMN and FAD. Consequently, we considered that PMFT might be recruited and harnessed by MFT-dependent dehydrogenases and therefore we probed for coupled enzymatic reactivity with the putative carveol dehydrogenase MsCDH. Accordingly, PMFT was shown to cycle MsCDH and was shown to be reduced to PMFTH2, clearly indicating that PMFT undergoes a 2e−/2H+ reduction by MsCDH. More importantly, this provides compelling evidence that PMFT, and thereby MFT, is a biologically active redox cofactor.
The discovery of novel FMN-dependent AHDP deaminase activity for MftD is extraordinary. To begin with, it continues to validate our previous findings that MftC is responsible for the two-step modification of MftA and that MftE hydrolyzes MftA* to form AHDP. Although this could be regarded as superficial, the fact that the elucidation of MFT biosynthesis has been successfully executed without any prior structural knowledge of MFT is remarkable. Validation of MFT intermediates (eg. AHDP) has only come from the subsequent enzyme (eg. MftD) recognizing the intermediate as a substrate. In addition to validating previous work, the reconstitution of MftD also pulls in parallels with that of PQQ biosynthesis. In PQQ and MFT biosynthesis, the first biosynthetic step requires a radical-S-adenosylmethionine protein dependent intramolecular C-C bond formation.23,41 In both pathways this step is followed by the subsequent excision of the crosslinked moiety by a protease24 and the oxidation of the resulting small molecule.42,45 The similarities between PQQ and MFT biosynthesis was first identified by Haft through bioinformatics and that analysis appears to have withstood experimental scrutiny.16 It would be worthwhile to use the combined PQQ and MFT biosynthetic pathway architectures as the basis for the discovery of potentially new RiPP derived redox cofactors or interesting small molecules.
Although only RiPP derived redox cofactors have been discussed thus far, it should be noted that other non-RiPP peptide derived redox cofactors are known. The four known examples are trihydroxyphenylalanine quinone (TPQ), tryptophan tryptophylquinone (TTQ), lysyl tyrosine quinone (LTQ) and cysteine tryptophylquinone (CTQ) (Figure 8).14 Although PQQ does belong to the peptide-derived redox cofactor family, there are substantial differences to the remaining quinones. Whereas PQQ is synthesized through dedicated biosynthetic pathways, the aforementioned quinones are synthesized in situ of the functional enzyme.29–31 Here, we propose that MFT should be permanently added to the list of peptide derived redox cofactors. As previously mentioned, PMFT is capable of functioning as a biologically active redox cofactor. However, when compared to the remaining peptide-derived redox cofactors a startling and significant structural difference is observed: PMFT is not a quinone like its brethren. Instead the structure of PMFT consists of an α-keto amide moiety which can be reduced by a 2e−/2H+ process, similar to that of a quinone. Indeed, not only is the functionality of the α-keto amide moiety similar to the quinones but its electrochemical properties are too. The PMFT midpoint potential (−255 mV) is comparable to that of the remaining quinones which have measured potentials in the range of −150 mV to −240 mV.46–48
Figure 8.

The structures of the known peptide derived redox cofactors where TPQ, LTQ, TTQ, and CTQ are formed in situ of the active enzyme and PQQ and PMFT are formed from a dedicated RiPP biosynthetic pathway.
In terms of functionality, we show here that PMFT, and by extension MFT, is likely the physiologically relevant cofactor for MsCDH. It is interesting to note that mft containing genomes also encode a wide variety and quantity of the MFT-associated dehydrogenases.16 For instance, the M. smegmatis mc2 155 genome encodes for eight members of the TIGR03971 family and four members of the TIGR03989 family. The work here provides the only in vitro and direct evidence that MFT is biologically active with MsCDH, a member of the TIGR03971 family. Of the remainder MFT-dependent dehydrogenases, Msmeg_6242, a putative primary alcohol dehydrogenase, is the only other dehydrogenase in M. smegmatis that has a potential activity associated with it.20 Beyond these two examples and their respective homologues, it could be that MFT participates in many biological processes.
Our studies herein now raise the question: What is the final mycofactocin structure? The remaining enzyme in the pathway is the putative glycosyltransferase, MftF. However, the role it is playing in mycofactocin biosynthesis is, more now than ever, convoluted. Considering that PMFT is recognized by at least one MFT-dehydrogenase, it could be that MftF is being used to decorate the p-hydroxy of the phenyl ring. Alternatively, MftF could be used in another unknown function unrelated to MFT biosynthesis and that PMFT could represent the final MFT structure. In summary, we demonstrate for the first time that MftD catalyzes the oxidative deamination of AHDP, forming the redox active center in PMFT. Moreover, we measured the midpoint potential for PMFT and demonstrated that it is biologically active with the carveol dehydrogenase MsCDH. Taken together, we provide the most direct evidence that MFT is the second member of the RiPP derived redox cofactor family.
METHODS
Cloning, Expression, and Purification of MftD from Mycobacterium ulcerans Agy99.
The mul_0774 gene sequence encoding for MftD from Mycobacterium ulcerans Agy99 (Uniprot: 0PM50) was cloned into pET28a-TEV using BamHI and XhoI restriction sites. The sequence verified MumftD/pET28a-TEV plasmid was transformed into E. coli ArcticExpress (DE3) (Stratagene) for protein production. An overnight culture of the E. coli ArcticExpress (DE3)-MumftD/pET28a-TEV grown in 50 mL of terrific broth (TB) medium, was used to inoculate 4 L of the same medium containing 50 μg/mL of kanamycin and 10 μg/mL of tetracycline and supplemented with 100 μM riboflavin. The culture was incubated at 37 °C with shaking at 200 rpm until an OD600 ~ 0.6 was attained, at which point MftD production was induced by addition of IPTG to a final concentration of 1 mM. Following a 24 h induction at 13 °C with shaking at 200 rpm, the cells were harvested by centrifugation at 7000 rpm for 10 min and stored at −80 °C until lysis. All purification steps were carried out in Coy Lab anaerobic chamber maintained under an atmosphere of 97% N2 and 3% H2 gases. The harvested cells were thawed and resuspended in five times pellet mass of lysis buffer containing 2X phosphate buffered saline (PBS) buffer (pH 8.0), 30 mM imidazole and 2 mM dithionite (DTH). To the suspension was added 0.1 mg/g of lysozyme, 0.1 mg/g of DNase and 1 % w/v CHAPS and stirred on ice for an additional 30 min. Cells were disrupted by sonication for 5 min to complete the lysis. The resulting lysate was clarified by centrifugation at 13,000 rpm for 10 min and the supernatant was loaded onto a 5 ml HisTrap FF Ni-NTA column (GE Healthcare) pre-equilibrated with lysis buffer using an AKTA Start FPLC (GE Healthcare). The column was washed with 2X PBS buffer (pH 8.0) containing 50 mM imidazole and the bound protein was eluted using 2X PBS buffer (pH 8.0) containing 300 mM imidazole and 2 mM DTH. Fractions containing MftD proteins were immediately buffer exchanged into 2X PBS storage buffer (pH 8.0) containing 10% glycerol and 2 mM DTH using PD-10 columns (GE Healthcare). The resulting volume containing MftD protein was concentrated using 30 kDa spin concentrators (Millipore). Tobacco-etch virus (TEV) protease was added to the protein and incubated at room temperature for 3 h to hydrolyze the N-terminal His-tag. The protein/protease mixture was loaded onto a 5 mL HisTrap FF Ni-NTA column pre-equilibrated with storage buffer. The flow through was collected and the bound protein was eluted with 2X PBS (pH 8.0) containing 50 mM imidazole and 2 mM DTH. Fractions containing MftD proteins were pooled together, buffer exchanged into a fresh 2X PBS storage buffer (pH 8.0) containing 10% glycerol using PD-10 columns (GE Healthcare). The resulting protein fraction was concentrated using 30 kDa spin concentrators and subjected to a final size exclusion chromatography purification step as described below.
Cloning, Expression and Purification of MftD from Mycobacterium smegmatis.
The msmeg_1424 gene encoding for MftD from Mycobacterium smegmatis mc2155 (Uniprot: A0QSB) was cloned into pET28a vector using NdeI and XhoI restriction sites. The sequence verified msmeg_1424/pET28a plasmid was transformed into E. coli Arctic Express (DE3) for protein expression. MsMftD proteins were expressed and purified as described above for MuMftD with thrombin being substituted for TEV.
Flavin Determination in MftD.
To determine the type of flavin contained in MftD, 100 μL of 200 μM of the purified protein was first denatured by heating at 80 °C for 10 min. The denatured protein was centrifuged at 14000 rpm for 10 min to remove all precipitated proteins. The supernatant was pipetted into a 1.5 mL HPLC autosampler vial and analyzed by reverse-phase chromatography on a Shimadzu Prominence-i LC-2030C HPLC using a Jupiter C 18, 5 μm, 4.6 × 250-mm column (Phenomenex), and 10 mM sodium phosphate monobasic, pH 5.5 (Buffer A) and 90% methanol (Buffer B) as the mobile phase. A linear gradient of solvent B from 0% to 35% was applied from 2 to 7 min, which was followed by another linear gradient to 95% solvent B from 7 to 15 min. A third gradient of solvent B from 95% to 100% was applied from 15 to 17 min after which solvent B was held constant at 100% from 17 to 19 min. This was followed by linear decrease to 100% solvent A from 19 to 22 min. Solvent A was held constant for an additional 2 min to re-equilibrate the column before subsequent sample injections. Chromatograms were reported at 450 nm while monitoring wavelengths between 200 and 600 nm. Authentic samples of riboflavin, flavin mononucleotide (FMN) and flavin adenine dinucleotide (FAD) were run as controls.
AHDP Modification Reactions.
AHDP modification reactions were conducted in volumes of 200 μL under aerobic and anaerobic conditions and contained 50 mM sodium phosphate (pH 8.0) as the reaction buffer. Consecutively, 100 μM MftD, 200 μM FMN and 100 μM of AHDP (see Supplementary Information for isolation procedures) were added and the reactions were incubated under aerobic and anerobic conditions for 12 h. Control reactions were set up in a similar fashion except buffer was substituted for individual reagents, protein or substrate. Reactions were then filtered through 0.2 μm spin columns to remove all precipitated proteins. The supernatant was pipetted into 300 μL autosampler HPLC vials and injected directly onto a reverse-phase Jupiter C5, 5 μm, 4.6 × 250-mm column (Phenomenex) using Shimadzu Prominence-i LC-2030C HPLC and 5 mM sodium phosphate, pH 7.5 (Buffer A) and 5 mM sodium phosphate in 70% acetonitrile pH 7.5 (Buffer B). A linear gradient of solvent B from 0% to 35% was applied from 2 to 8 min, which was followed by a linear gradient to 100% solvent B from 8 to 17 min. Solvent B was then held constant at 100% from 17 to 19 min followed by linear decrease to 100% solvent A from 19 to 22 min. Solvent A was held constant for an additional 2 min to re-equilibrate the column before subsequent sample injections. Chromatograms were reported at 280 nm while monitoring wavelengths between 200 and 400 nm. New peaks were isolated, lyophilized and analyzed by NMR and HRMS.
Oxygen Requirement for MftD Reaction with AHDP.
To investigate the dependence of MftD catalysis on oxygen, a total of 200 μL of MftD reaction was set up under anaerobic conditions as follows. To the reaction buffer containing 2X PBS (pH 8.0) were added consecutively10 mM glucose and 10 μM of glucose oxidase (GOX) followed by incubation at room temperature for 10 min to deplete oxygen from the buffer. Sequentially, 100 μM of MftD, 200 μM of FMN, and 200 μM of AHDP were added to the reaction followed by an additional 12 h of incubation period at room temperature. Reactions were filtered through 0.2 μm spin columns to remove all precipitated proteins. The supernatant was pipetted into 300 μL autosampler HPLC vials and injected directly onto a reverse-phase HPLC system. HPLC analysis was carried out as described above.
18O Isotope Labelling of PMFT.
To determine the source of oxygen atom incorporated into the product of MftD catalysis, reactions were carried out in either 18OH2 or 18O2. For reactions carried out in 18OH2, a buffer solution of 25 mM ammonium acetate was first prepared by lyophilizing 500 μl of the buffer solution to powder and re-suspending into an equal volume of 18OH2 (PET grade, Sigma Aldrich). Reactions consisted of 90 μl of 18OH2 buffer, 100 μM of MftD (5 μL), 200 μM of FMN (1 μL) and 100 μM of AHDP (4 μL). Following a 1 h aerobic incubation at room temperature, the reaction was analyzed by HRMS by direct infusion onto a Thermo Scientific Q Exactive. Reactions carried out in the presence of 18O2 proceeded similarly but were perform in 16OH2 buffer that was bubbled with 18O2 for 5-min prior to the addition of MftD. A control reaction was performed to monitor the back-exchange of labeled oxygen. To do so, 16O-PMFT was incubated in 18OH2-buffer for 60 min prior to analysis by HRMS.
Nuclear Magnetic Resonance Spectroscopy.
All NMR spectra were recorded at the at the University of Denver on a Bruker UltraShield 500/54 Plus spectrometer. All Spectra were processed and analyzed using TopSpin v. 2.1 program (Bruker). All peptide NMR samples were prepared in 99.96% deuterated water. Water Suppression signal was applied at a frequency of 2353.37 Hz for all 1H NMR spectra. Signals were integrated, and the coupling constants were calculated in MestReNova v. 10.0.1 program (Mestrelab Research).
High Resolution Mass Spectrometry.
All samples were analyzed at Anschutz Medical Campus by the Biological Mass Spectrometry Facility at the University of Colorado. 18O-labelled samples were infused directly while all other samples were desalted using a C18 ZipTip (EMD Millipore) and subjected to LC-ESI-MS using a Thermo Scientific Q Exactive and a nanoflow liquid chromatography system. The data was analyzed using XCalibur Qual Browser v. 3.0.63 (Thermo Scientific).
Electrochemical Characterization of PMFT.
To determine the redox potential of PMFT, a 5 mg/ml suspension of functionalized single walled carbon nanotubes (SWCNTs) was made by dispersing 50 mg of the nanotubes in 10 ml of dimethylformamide and sonicating in ultrasonic bath for 1 h to obtain a uniform suspension. Glassy carbon electrodes were polished with 0.05 μm alumina on nylon and microbroth polishing pads. The electrode was sonicated in 10 ml of water in ultrasonic bath for 5 min followed by 10 ml of ethanol for additional 5 min and allowed to thoroughly dry at room temperature. The dried electrode was coated with 20 μL of the SWCNTs suspension and allowed to dry at room temperature. This was repeated two more times and allowing for thorough drying in between casts. The dried SWCNTs-modified glassy carbon electrode was incubated in phosphate buffer (pH 7.0) as control or in phosphate buffer (pH 7.0) containing 500 μM of PMFT for 3 h prior to running cyclic voltammetry (CV) experiments. A conventional thre-electrode system was used in a water-jacketed glass cell. The counter electrode was platinum wire and Ag/AgCl was the reference electrode. CV measurements were carried out in Coy Lab anaerobic chamber using CHI 600E Series Electrochemical analyzer. Measurements were recorded at 0.05 V/s and a potential sweep window of +0.2 to −0.8 V at 22 °C.
Cloning, Expression, and Purification of Carveol Dehydrogenase (CDH).
The msmeg_1410 gene sequence encoding CDH from Mycobacterium smegmatis mc2155 (Uniport: A0QSA5) was cloned into pET28a using NdeI and HindIII restriction sites. The sequence verified msmeg_1410/pET28a plasmid was transformed into the E. coli BL21 star (DE3) and grown overnight at 37 °C in 50 mL LB containing 50 μg/ml of kanamycin. The overnight culture was used to inoculate 4 L of LB growth medium containing 50 μg/ml of kanamycin. The culture was incubated at 37 °C with shaking at 200 rpm until an OD600 of ~0.6, at which point MsCDH production was induced by addition of IPTG to a final concentration of 1 mM and allowed to grow overnight at 20 °C with shaking at 200 rpm. The cells were harvested by centrifugation at 7000 rpm for 10 min and the pellet was resuspended in five times pellet mass of lysis 50 mM MOPS buffer (pH 7.0) containing, 250 mM NaCl and 30 mM imidazole. To the suspension was added 0.1 mg/g of lysozyme, 0.1 mg/g of DNase and 1 % w/v CHAPS and stirred for 30 min. Cells were disrupted by sonication for 5 min on ice to complete the lysis. The resulting lysate was clarified by centrifugation at 13,000 rpm for 10 min the supernatant was loaded onto a 5 mL HisTrap HP Ni-NTA column pre-equilibrated with lysis buffer using an AKTA Pure FPLC. The column was washed with 50 mM MOPS buffer (pH 7.0) containing 250 mM NaCl and 30 mM imidazole and the bound protein was eluted using 50 mM MOPS buffer (pH 7.0) containing 250 mM NaCl and 300 mM imidazole. Fractions containing MsCDH proteins were pooled together and immediately buffer exchanged into 50 mM MOPS storage buffer (pH 7.0) containing 250 mM NaCl and 10% glycerol using a HiPrep 26/10 desalting column. The resulting volume of protein was concentrated using 10 kDa spin concentrators. Thrombin protease was added to the concentrated protein and incubated at room temperature for 1 h to cleave off the N-terminal His-tag. The protein/protease mixture was loaded onto a Superdex 200 10/300 GL analytical size exclusion chromatographic column using an AKTA Pure FPLC. The mobile phase was storage buffer containing 50 mM MOPS (pH 7.0), 250 mM NaCl, and 10% glycerol at a flow rate of 1.0 mL/min. Protein peaks were collected and concentrated using 10 kDa spin concentrators. The concentrated protein was aliquoted into 1.5 mL vials, flash-frozen in liquid nitrogen and stored at −80 °C until use.
Monitoring MsCDH UV Vis Spectral Changes in the Presence of PMFT.
To determine if PMFT could be used as a cofactor by MsCDH for carveol modification, a UV spectroscopic assay was performed using Shimadzu TCC-240A UV-visible spectroscopy as follows. An initial UV absorbance spectrum of 200 μL reaction buffer containing 50 mM MOPS (pH 7.0), 250 mM NaCl, 10% glycerol and 150 μM MsCDH was taken from 190 nm to 500 nm. Following initial scan, 30 μM PMFT was added to MsCDH, incubated for 5 min, and the UV-visible spectra was measured. This was repeated until a final concentration of 150 μM PMFT was achieved. A control experiment was carried out using the same procedures but with NADH free in solution.
MsCDH-Carveol Modification Reactions.
Carveol modification reaction was conducted in a total volume of 200 μL containing 50 mM MOPS buffer (pH 7.0), 250 μM NaCl, 10% glycerol, 100 μM MsCDH, 500 μM carveol, and 500 μM PMFT. The reaction was incubated at room temperature for an hour. Control reactions were set up in a similar fashion except buffer was substituted for individual reagents, protein or substrate. Reactions were filtered through 3 kDa spin columns to remove all precipitated proteins. The supernatant was pipetted into 300 μL autosampler HPLC vials and injected directly onto a reverse-phase HPLC system. The HPLC analysis was run on a Shimadzu Prominence-i LC-2030C HPLC using a Jupiter C5, 5 μm, 4.6 × 250-mm column (Phenomenex), and 5 mM sodium phosphate, pH 7.5 (Buffer A) and 5 mM sodium phosphate in 70% acetonitrile pH 7.5 (Buffer B). A linear gradient of solvent B from 0% to 35% was applied from 2 to 8 min, which was followed by a linear gradient to 100% solvent B from 8 to 17 min. Solvent B was then held constant at 100% from 17 to 19 min followed by linear decrease to 100% solvent A from 19 to 22 min. Solvent A was held constant for an additional 2 min to re-equilibrate the column before subsequent sample injections. UV absorbance readings were monitored between 200 nm and 400 nm.
Stopped flow spectrophotometry.
A DX.17MV sequential stopped-flow spectrometer (Applied Photophysics, Leatherhead, U.K) with a 3 ms deadtime was used to measure the single turnover oxidation experiments of MsCDH by PMFT and DCPIP. All reactions were carried out in 50 mM Hepes, pH 7.0, 250 mM NaCl, and 10% glycerol. For reactions with PMFT, the absorbance of the MsCDH bound NADH was monitored at 340 nm (ΔεNADH 340nm = 6.22 mM−1cm−1). For reactions with DCPIP, the absorbance of the oxidant was monitored at 600 nm (ΔεDCPIP 600nm = 20.7 mM−1cm−1). Reactions were set up so that the final concentrations were 160 μM MsCDH and 25 or 50 μM PMFT, or 22 or 60 μM DCPIP. Reactions were run in triplicate at each concentration, averaged, and fitted to a single exponential decay or linear equations using the accompanying software.
Supplementary Material
ACKNOWLEDGMENT
We thank Dr. Monika Dzieciatkowska (University of Colorado Anchutz Medical Campus) for performing the HRMS measurements.
Funding Sources
This work was supported by National Institutes of Health Grant GM 124002 to J.A.L.
ABBREVIATIONS
- AHDP
3-amino-5-[(p-hydroxyphenyl) methyl]-4,4-dimethyl-2-pyrrolidinone
- CHAPS
(3-[(3-cholamidopropyl)dime-thylammonio]-1-propanesulfonate
- COSY
correlation spectroscopy
- CV
cyclic voltammetry
- DCPIP
2,3-dichlorophenolindophenol
- DTT
dithiothreitol
- FAD
flavin adenine dinucleotide
- FMN
flavin mononucleotide
- FPLC
fast protein liquid chromatography
- HEPES
2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid
- HMBC
heteronuclear multiple bond correlation
- HPLC
high performance liquid chromatography
- HRMS
high resolution mass spectrometry
- HSQC
heteronuclear single quantum coherence
- IPTG
isopropyl-β-D-1-thiogalctopyanoside
- MFT
mycofactocin
- MOPS
(3-(N-morpholino)propanesulfonic acid)
- NAD+/NADH
nicotinamide dinucleotide
- NMR
nuclear magnetic resonance
- PBS
phosphate buffer saline
- PMFT
premycofactocin
- PQQ
pyrroloquinoline quinone
- SAM
S-adenosylmethionine
- SEC
size exclusion chromatography
- SHE
standard hydrogen electrode
- SWCNT
single walled carbon nanotubes
- TCA
trichloroacetic acid
- TCEP
Tris(2-carboxyethyl)phosphine
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website. Select methods, Figures S1-S8, and NMR spectra (PDF).
REFERENCES
- (1).Arnison PG; Bibb MJ; Bierbaum G; Bowers AA; Bugni TS; Bulaj G; Camarero JA; Campopiano DJ; Challis GL; Clardy J; Cotter PD; Craik DJ; Dawson M; Dittmann E; Donadio S; Dorrestein PC.; Entian K-D; Fischbach MA; Garavelli JS; Göransson U; Gruber CW; Haft DH; Hemscheidt TK; Hertweck C; Hill C; Horswill AR; Jaspars M; Kelly WL; Klinman JP; Kuipers OP; Link AJ; Liu W; Marahiel MA; Mitchell DA; Moll GN; Moore BS; Müller R; Nair SK; Nes IF; Norris GE; Olivera BM; Onaka H; Patchett ML; Piel J; Reaney MJT; Rebuffat S; Ross RP; Sahl H-G; Schmidt EW; Selsted ME; Severinov K; Shen B; Sivonen K; Smith L; Stein T; Süssmuth RD; Tagg JR; Tang G-L; Truman AW; Vederas JC; Walsh CT; Walton JD; Wenzel SC; Willey JM; van der Donk WA. Ribosomally Synthesized and Post-Translationally Modified Peptide Natural Products: Overview and Recommendations for a Universal Nomenclature. Nat. Prod. Rep 2013, 30 (1), 108–160. 10.1039/C2NP20085F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Ortega MA; van der Donk WA New Insights into the Biosynthetic Logic of Ribosomally Synthesized and Post-Translationally Modified Peptide Natural Products. Cell Chem. Biol 2016, 23 (1), 31–44. 10.1016/j.chembiol.2015.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).De Veer SJ; Weidmann J; Craik DJ Cyclotides as Tools in Chemical Biology. Acc. Chem. Res 2017, 50 (7), 1557–1565. 10.1021/acs.accounts.7b00157. [DOI] [PubMed] [Google Scholar]
- (4).Craik DJ; Lee MH; Rehm FBH; Tombling B; Doffek B; Peacock H Ribosomally-Synthesised Cyclic Peptides from Plants as Drug Leads and Pharmaceutical Scaffolds. Bioorganic Med. Chem 2018, 26 (10), 2727–2737. 10.1016/j.bmc.2017.08.005. [DOI] [PubMed] [Google Scholar]
- (5).Hudson GA; Mitchell DA RiPP Antibiotics: Biosynthesis and Engineering Potential. Curr. Opin. Microbiol 2018, 45, 61–69. 10.1016/j.mib.2018.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Rea MC; Sit CS; Clayton E; O’Connor PM; Whittal RM; Zheng J; Vederas JC; Ross RP; Hill C Thuricin CD, a Posttranslationally Modified Bacteriocin with a Narrow Spectrum of Activity against Clostridium Difficile. Proc. Natl. Acad. Sci. U. S. A 2010, 107 (20), 9352–9357. 10.1073/pnas.0913554107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Rea MC; Dobson A; O’Sullivan O; Crispie F; Fouhy F; Cotter PD; Shanahan F; Kiely B; Hill C; Ross RP Effect of Broad- and Narrow-Spectrum Antimicrobials on Clostridium Difficile and Microbial Diversity in a Model of the Distal Colon. Proc. Natl. Acad. Sci 2011, 108 (Supplement_1), 4639–4644. 10.1073/pnas.1001224107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Bentley R; Meganathan R Biosynthesis of Vitamin K (Menaquinone) in Bacteria. Microbiol. Rev 1982, 46 (3), 241–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Shelburne CE; An FY; Dholpe V; Ramamoorthy A; Lopatin DE; Lantz MS The Spectrum of Antimicrobial Activity of the Bacteriocin Subtilosin A. J. Antimicrob. Chemother 2007, 59 (2), 297–300. 10.1093/jac/dkl495. [DOI] [PubMed] [Google Scholar]
- (10).Duquesne S; Destoumieux-Garzón D Microcins, Gene-Encoded Antibacterial Peptides from Enterobacteria. Nat. Prod. Rep 2007, 24, 708–734. 10.1039/b516237h. [DOI] [PubMed] [Google Scholar]
- (11).Goodwin PM; Anthony C The Biochemistry, Physiology and Genetics of PQQ and PQQ-Containing Enzymes. Adv. Microb. Physiol 1998, 40, 1–80. [DOI] [PubMed] [Google Scholar]
- (12).Ibrahim M; Guillot A; Wessner F; Algaron F; Besset C; Courtin P; Gardan R; Monnet V Control of the Transcription of a Short Gene Encoding a Cyclic Peptide in Streptococcus Thermophilus: A New Quorum-Sensing System? J. Bacteriol 2007, 189 (24), 8844–8854. 10.1128/JB.01057-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Shen Y-Q; Bonnot F; Imsand EM; RoseFigura JM; Sjölander K; Klinman JP Distribution and Properties of the Genes Encoding the Biosynthesis of the Bacterial Cofactor, Pyrroloquinoline Quinone. Biochemistry 2012, 51 (11), 2265–2275. 10.1021/bi201763d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Klinman JP; Bonnot F The Intrigues and Intricacies of the Biosynthetic Pathways for the Enzymatic Quinocofactors: PQQ, TTQ, CTQ, TPQ and LTQ. Chem. Rev 2014, 114 (8), 4343–4365. 10.1021/cr400475g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Duine JA; Frank J; van Zeeland JK Glucose Dehydrogenase from Acinetobacter Calcoaceticus: A Quinoprotein. FEBS Lett. 1979, 108 (2), 443–446. [DOI] [PubMed] [Google Scholar]
- (16).Haft DH Bioinformatic Evidence for a Widely Distributed, Ribosomally Produced Electron Carrier Precursor, Its Maturation Proteins, and Its Nicotinoprotein Redox Partners. BMC Genomics 2011, 12 (1), 21 10.1186/1471-2164-12-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Ayikpoe R; Ngendahimana T; Langton M; Bonitatibus S; Walker LM; Eaton SS; Eaton GR; Pandelia ME; Elliott SJ; Latham JA Spectroscopic and Electrochemical Characterization of the Mycofactocin Biosynthetic Protein, MftC, Provides Insight into Its Redox Flipping Mechanism. Biohcemistry 2019, 58, 940–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Haft DH; Pierce PG; Mayclin SJ; Sullivan A; Gardberg AS; Abendroth J; Begley DW; Phan IQ; Staker BL; Myler PJ; Marathias VM; Lorimer DD; Edwards TE Mycofactocin-Associated Mycobacterial Dehydrogenases with Non-Exchangeable NAD Cofactors. Sci. Rep 2017, 7 (October 2016), 41074 10.1038/srep41074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Van Der Werf MJ; Van Der Ven C; Barbirato F; Eppink MHM; De Bont JAM; Van Berkel WJH Stereoselective Carveol Dehydrogenase from Rhodococcus Erythropolis DCL14. J. Biol. Chem 1999, 274 (37), 26296–26304. [DOI] [PubMed] [Google Scholar]
- (20).Krishnamoorthy G; Kaiser P; Lozza L; Hahnke K; Mollenkopf H; Kaufmann SH E. Mycofactocin Is Associated with Ethanol Metabolism in Mycobacteria. MBio 2019, 10 (3), 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Khaliullin B; Aggarwal P; Bubas M; Eaton GR; Eaton SS; Latham JA Mycofactocin Biosynthesis: Modification of the Peptide MftA by the Radical S-Adenosylmethionine Protein MftC. FEBS Lett 2016, 590 (16), 2538–2548. 10.1002/1873-3468.12249. [DOI] [PubMed] [Google Scholar]
- (22).Bruender NA; Bandarian V The Radical S -Adenosyl- L-Methionine Enzyme MftC Catalyzes an Oxidative Decarboxylation of the C-Terminus of the MftA Peptide. Biochemistry 2016, 55 (20), 2813–2816. 10.1021/acs.biochem.6b00355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Khaliullin B; Ayikpoe R; Tuttle M; Latham JA Mechanistic Elucidation of the Mycofactocin-Biosynthetic Radical S-Adenosylmethionine Protein, MftC. J. Biol. Chem 2017, 292 (31), 13022–13033. 10.1074/jbc.M117.795682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Ayikpoe R; Salazar J; Majestic B; Latham JA Mycofactocin Biosynthesis Proceeds through 3-Amino-5-[(p-Hydroxyphenyl)Methyl]-4,4-Dimethyl-2-Pyrrolidinone (AHDP); Direct Observation of MftE Specificity toward MftA*. Biochemistry 2018, 57, 5379–5383. 10.1021/acs.biochem.8b00816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Williams E; Cregeen D; Rumsby G Identification and Expression of a CDNA for Human Glycolate Oxidase. Biochim. Biophys. Acta 2000, 1493 (1–2), 246–248. 10.1016/S0167-4781(00)00161-5. [DOI] [PubMed] [Google Scholar]
- (26).Macheroux P; Mulrooney SB; Williams CH; Massey V Direct Expression of Active Spinach Glycolate Oxidase in Escherichia Coli. BBA-Gene Struct. Expr 1992, 1132 (1), 11–16. 10.1016/0167-4781(92)90046-3. [DOI] [PubMed] [Google Scholar]
- (27).Gao C; Wang Y; Zhang Y; Lv M; Dou P; Xu P; Ma C NAD-Independent L-Lactate Dehydrogenase Required for L-Lactate Utilization in Pseudomonas Stutzeri A1501. J. Bacteriol 2015, 197 (13), 2239–2247. 10.1128/JB.00017-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Ayikpoe R; Govindarajan V; Latham JA Occurrence , Function , and Biosynthesis of Mycofactocin. Appl. Microbiol. Biotechnol 2019, 103, 2903–2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Cai D; Klinman JP Evidence of a Self-Catalytic Mechanism of 2,4,5-Trihydroxyphenylalanine Quinone Biogenesis in Yeast Copper Amine Oxidase. J. Biol. Chem 1994, 269 (51), 32039–32042. [PubMed] [Google Scholar]
- (30).Tang C; Klinman JP The Catalytic Function of Bovine Lysyl Oxidase in the Absence of Copper. J. Biol. Chem 2001, 276 (33), 30575–30578. 10.1074/jbc.C100138200. [DOI] [PubMed] [Google Scholar]
- (31).Datta S; Mori Y; Takagi K; Kawaguchi K; Chen ZW; Okajima T; Kuroda S; Ikeda T; Kano K; Tanizawa K; Mathews FS Structure of a Quinohemoprotein Amine Dehydrogenase with an Uncommon Redox Cofactor and Highly Unusual Crosslinking. Proc. Natl. Acad. Sci. U. S. A 2001, 98 (25), 14268–14273. 10.1073/pnas.241429098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Sukumar N; Dewanti A; Merli A; Rossi GL; Mitra B; Mathews FS Structures of the G81A Mutant Form of the Active Chimera of ( S )-Mandelate Dehydrogenase and Its Complex with Two of Its Substrates. Acta Crystallogr. Sect. D 2009, 65, 543–552. 10.1107/S0907444909010270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Molla G; Sacchi S; Bernasconi M; Pilone MS; Fukui K; Pollegioni L Characterization of Human D -Amino Acid Oxidase. FEBS Lett 2006, 580, 2358–2364. 10.1016/j.febslet.2006.03.045. [DOI] [PubMed] [Google Scholar]
- (34).Walsh CT; Schonbrunn A; Abeles RH Studies on the Mechanism of Action of D-Amino Acid Oxidase. J. Biol. Chem 1971, 246 (22), 6855–6866. [PubMed] [Google Scholar]
- (35).Umhau S; Pollegioni L; Molla G; Diederichs K; Welte W; Pilone MS; Ghisla S The X-Ray Structure of D -Amino Acid Oxidase at Very High Resolution Identifies the Chemical Mechanism of Flavin-Dependent Substrate Dehydrogenation. Proc. Natl. Acad. Sci 2000, 97 (23), 12463–12468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Lindqvist Y Refined Structure of Spinach Glycolate Oxidase at 2 A Resolution. J. Mol. Biol 1989, 209, 151–166. [DOI] [PubMed] [Google Scholar]
- (37).Porter DJ; Voet JG; Bright HJ Direct for Carbanions and Covalen N5-Flavin-Carbanion Adducts as Catalytic Intermediates in the Oxidation of Nitroethane by D-Amino Acid Oxidase. J. Biol. Chem 1973, 248 (12), 4400–4416. [PubMed] [Google Scholar]
- (38).Hartman C; Brzovic P; Klinman JP Spectroscopic Detection of Chemical Intermediates in Thereaction of Para-Substituted Benzlamines with Bovine Serium Amine Oxidase. Biochemistry 1993, 32, 2234–2241. 10.1021/bi00060a015. [DOI] [PubMed] [Google Scholar]
- (39).Jonsson T; Glickman MH; Sun S; Klinman JP Experimental Evidence for Extensive Tunneling of Hydrogen in the Lipoxygenase Reaction: Implications for Enzyme Catalysis. J. Am. Chem. Soc 1996, 7863 (10), 10319–10320. 10.1021/ja961827p. [DOI] [Google Scholar]
- (40).Goosen N; Huinen RGM; Van de Putte PA 24-Amino-Acid Polypeptide Is Essential for the Biosynthesis of the Coenzyme Pyrrolo-Quinoline-Quinone. J. Bacteriol 1992, 174 (4), 1426–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Barr I; Latham JA; Iavarone AT; Chantarojsiri T; Hwang JD; Klinman JP The Pyrroloquinoline Quinone (PQQ) Biosynthetic Pathway: Demonstration of de Novo Carbon-Carbon Cross-Linking within the Peptide Substrate (PqqA) in the Presence of the Radical SAM Enzyme (PqqE) and Its Peptide Chaperone (PqqD). J. Biol. Chem 2016, 291 (17), 8877–8884. 10.1074/jbc.C115.699918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Koehn EM; Latham JA; Armand T; Evans RL; Tu X; Wilmot CM; Iavarone AT; Klinman JP Discovery of Hydroxylase Activity for PqqB Provides a Missing Link in the Pyrroloquinoline Quinone Biosynthetic Pathway. J. Am. Chem. Soc 2019, 141, 4398–4405. 10.1021/jacs.8b13453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Ameyama M; Kazunobu M; Ohno Y; Shinagawa E; Adachi O Existence of a Novel Prosthetic Group, PQQ, in Mebrane-bound, Electron Transport Chain-linked, Primary Dehydrogenases of Oxidative Bacteria. FEBS Lett 1981, 130 (2), 179–183. [DOI] [PubMed] [Google Scholar]
- (44).Bruender NA; Bandarian V The Creatininase Homolog MftE from Mycobacterium Smegmatis Catalyzes a Peptide Cleavage Reaction in the Biosynthesis of a Novel Ribosomally Synthesized Post-Translationally Modified Peptide (RiPP). J. Biol. Chem 2017, 292 (10), 4371–4381. 10.1074/jbc.M116.762062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Bonnot F; Iavarone AT; Klinman JP Multistep, Eight-Electron Oxidation Catalyzed by the Cofactorless Oxidase, PqqC: Identification of Chemical Intermediates and Their Dependence on Molecular Oxygen. Biochemistry 2013, 52, 4667–4675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Klinman JP How Many Ways to Craft a Cofactor? Proc. Natl. Acad. Sci 2002, 98 (26), 14766–14768. 10.1073/pnas.011602498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Itoh S; Ohshiro Y; Agawa T Reaction of Reduced PQQ (PQQH2) and Molecular Oxygen. Bulletin of the Chemical Society of Japan. 1986, pp 1911–1914. 10.1246/bcsj.59.1911. [DOI] [Google Scholar]
- (48).Murakami Y; Tachi Y; Itoh S A Model Compound of the Novel Organic Cofactor CTQ (Cysteine Tryptophylquinone) of Quinohemoprotein Amine Dehydrogenase. European J. Org. Chem 2004, No. 14, 3074–3079. 10.1002/ejoc.200400191. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.