

Abstract
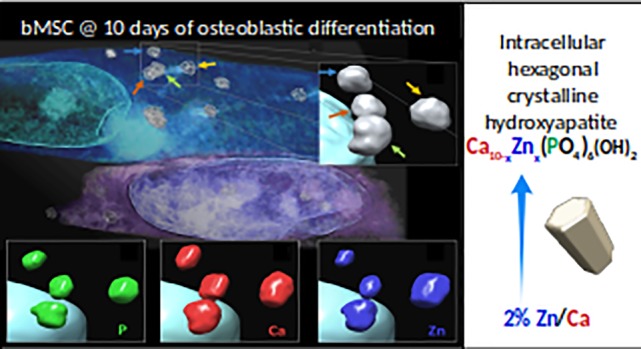
The core knowledge about biomineralization is provided by studies on the advanced phases of the process mainly occurring in the extracellular matrix. Here, we investigate the early stages of biomineralization by evaluating the chemical fingerprint of the initial mineral nuclei deposition in the intracellular milieu and their evolution toward hexagonal hydroxyapatite. The study is conducted on human bone mesenchymal stem cells exposed to an osteogenic cocktail for 4 and 10 days, exploiting laboratory X-ray diffraction techniques and cutting-edge developments of synchrotron-based 2D and 3D cryo-X-ray microscopy. We demonstrate that biomineralization starts with Zn–hydroxyapatite nucleation within the cell, rapidly evolving toward hexagonal hydroxyapatite crystals, very similar in composition and structure to the one present in human bone. These results provide experimental evidence of the germinal role of Zn in hydroxyapatite nucleation and foster further studies on the intracellular molecular mechanisms governing the initial phases of bone tissue formation.
Short abstract
Biomineralization starts with Zn−hydroxyapatite nucleation within the cell, rapidly evolving toward hexagonal hydroxyapatite crystals, similar in composition and structure to the one of human bone.
Introduction
Biomineralization, the process by which living systems generate organized mineral crystals, is an extremely widespread phenomenon common to all six taxonomic kingdoms. Hydroxyapatite (HA) is a naturally occurring mineral form of calcium apatite with the general formula (Ca10–xYx)(PO4)6(OH)2–p(CO3)p, where Y indicates the typical substituting metals (Zn, Mg, Sr) found in many tissues including bones1 and cartilage.2 Biomineralization is a lifelong process, fundamental in bone formation, repair, and remodeling. Chemical–physical reactions orchestrated by the cells convert ions in solution into biominerals, which nucleate and grow within an organic matrix framework, thus generating complex composite materials with a wide range of properties. Bone shows a complex structure organized across multiple length scales, from the molecular to the macroscopic level. On the nanometric scale, collagen microfibrils direct the formation of nanosized HA platelets oriented parallel to the collagen fibril axis,3−8 for interfibrillar mineralization.9,10 Together with collagen, various highly acidic noncollagenous proteins contribute to the mineralization process by binding minerals. Alongside these organic components, calcium phosphate, an amorphous mineral precursor, plays a fundamental role in the growth of HA nanocrystals.11
Biomineralization depends upon the complex crosstalk between osteoclasts, osteoblasts, and osteocytes. Osteoclasts digest bone matrix while osteoblasts synthesize it and orchestrate its mineralization.12 Osteoblasts are derived from bone mesenchymal stem cells (bMSCs), and once they are trapped within the bone they have formed, they terminally differentiate into osteocytes. Osteocytes are interconnected stellar cells, which integrate mechanical and biochemical cues and govern bone deposition and resorption. As the precursor of osteoblasts and osteocytes, bMSCs are heavily investigated to unveil the molecular basis of osteogenesis, to translate basic knowledge to application in tissue engineering and regenerative medicine.13
Calcium is a crucial mineral for the bone and is present in the extracellular mineralized matrix as an integral component of hydroxyapatite crystals.3−5 Very little is known about the intracellular Ca concentration, distribution, and homeostasis in bMSCs, and even less about the progression of the extracellular Ca phosphate and polyphosphate deposition during osteoblast differentiation. In fact, this core knowledge is provided by studies on the advanced phases of biomineralization, which mainly occur in the extracellular matrix, while studies on the early stages of this process are scarce. Here, we report findings on the early stages of biomineralization, i.e., the genesis and evolution of the mineral nuclei and their elemental composition during the osteogenic differentiation of human bMSCs. To tackle the challenge to characterize the complex phenomenon of the early stages of the biomineralization, we exploited the remarkable versatility of X-rays generated by high-brilliance synchrotron sources. Over the past decade, cutting-edge X-ray synchrotron-based microscopy techniques have been improved to overcome the limitations in spatial resolution14,15 and in sensitivity, becoming more and more accessible and common in life science studies. These breakthroughs in synchrotron-based microscopy allow us to study the biomineralization in single cells at the intracellular level and in the extracellular matrix where part of this phenomenon takes place. In particular, in this work we exploited the high sensitivity and elemental selectivity of X-ray fluorescence microscopy to determine the intracellular elemental distribution, with a spatial resolution in the range of 70 nm, and the chemical composition of mineral nuclei, down to 15 nm. Moreover, since biomineralization culminates with crystalline HA depositions, we employed synchrotron-based X-ray spectromicroscopy and laboratory diffraction techniques to resolve the atomic scale structural properties of bone mineral nuclei and to study their genesis toward mature hydroxyapatite. The aforementioned techniques limit their investigation to two dimensions, averaging the results over the sample thickness. We carried out combined X-ray fluorescence and phase-contrast nanotomography, to precisely localize the mineral nuclei depositions and to determine their elemental composition in the cellular environment. From the combined results, we propose a possible mechanism of the genesis and the evolution of hexagonal hydroxyapatite.
Results and Discussion
bMSC Osteogenic Differentiation
Our experimental model consists of bone-marrow-derived human bMSCs stimulated to differentiate into osteoblasts using an osteogenic cocktail containing β-glycero-phosphate, ascorbic acid, and vitamin D for 4 and 10 days. In agreement with previous results,16−18 after 4 days of stimuli, no calcium deposits in the extracellular matrix were detectable by Alizarin Red staining (Figure S1A). At the same time point, by reverse transcription polymerase chain reaction (RT-PCR), we observed the overexpression of some osteogenic genes such as RUNX2, the master regulator of osteogenesis, and SP7, coding for Osterix, another transcription factor required for osteoblast differentiation. We also detected increased transcripts for COL1A1, coding for collagen type 1 which is the most abundant extracellular protein in bone, and for SPP1 and BGLAP (Figure S1B). The BGLAP gene encodes osteocalcin, one of the most abundant noncollagenous proteins in bone tissue that is localized in the mineralized matrix of bone. The SSP1 gene encodes osteopontin, which is secreted and binds hydroxyapatite with high affinity. COL1A1 is an early marker of osteoprogenitor cells, while SPP1 and BGLAP are associated with a more mature. In our experimental model, BGLAP and SPP1 were already significantly upregulated on day 4 and maximally expressed at day 10, when we observed a marked extracellular matrix mineralization (Figure S1A). It is noteworthy that osteopontin is a potent protein nucleator of hydroxyapatite.19
We also evaluated the synthesis of RUNX2 and collagen type 1 by enzyme-linked immunosorbent assay (ELISA) and found both of them upregulated. RUNX2 was elevated after 4 days of osteogenic induction and further increased thereafter, while collagen type 1 peaked at day 4 and then decreased.
Chemical Composition of Mineral Nuclei Assessed by X-ray Fluorescence Microscopy
We exploited cryogenic synchrotron X-ray fluorescence microscopy to determine the chemical composition of mineral nuclei at the nanometric scale in the early stages of the biomineralization. After 4 days of osteoblastic induction, some mineral nuclei are present (Figure 1, left panel), which is not the case for the noninduced bMSCs (Figure S2). These data indicate that biomineralization starts within the cell, early after the osteogenic stimuli of bMSCs. The spatial resolution and sensitivity of synchrotron X-ray fluorescence microscopy enabled precise chemical characterization of the mineral nuclei despite their small dimensions, with volumes ranging between 0.01 and 2.5 μm3. The elemental analysis revealed different degrees of Ca, P, and Zn colocalization, as displayed in the composite map shown in Figure 1D. The occurrence of Zn in the early mineral nuclei represents a novel finding, providing experimental evidence of the germinal role of Zn in HA nucleation hypothesized a long time ago.20
Figure 1.

Two-dimensional X-ray fluorescence maps (pixel size 70 nm, expressed in areal mass (g/cm2)) showing the evolution between day 4 and day 10 of bMSC osteogenic differentiation. (A, E) Red: elemental maps of Ca. (B, F) Green: elemental maps of P. (C, G) Blue: elemental maps of Zn. (D, H) Composite elemental distribution of Ca, P, and Zn to better understand the correspondence of elements accumulation.
Results reported in Figure 1A–C suggest a different timing of elemental accumulation in the five detected mineral nuclei (colored arrows). The presence of all three elements (Ca, P, Zn) is clear in two granules (red and yellow arrow, Figure 1A–C), while in the others (white and green arrow, Figure 1 A–C), Ca and Zn, or only Ca was detected (violet arrow, Figure 1A). This suggests that the process of Ca deposition can start as a compound, which is not bound to phosphate. A plausible alternative anion candidate is carbonate, since its content in the bone mineral is known to be about 4–8 wt %.21 According to these results, it is possible that initial mineral nuclei formation starts as Ca-carbonate compounds, which subsequently incorporate Zn, thus starting HA nucleation by integrating phosphates to build the lattice structure of HA.
After 10 days of osteogenic differentiation, the mineral nuclei are massive in terms of both dimension and number (Figure 1E–G). The elements colocalize quite heterogeneously, as shown by the composite map (Figure 1H).
Molecular and Crystalline Structure of Mineral Nuclei Analyzed by X-ray Spectromicroscopy and Diffraction
It is known that biomineralization culminates with crystalline HA depositions; however, crystalline material inside cells has never been described. Therefore, we employed synchrotron-based X-ray absorption near-edge structure (XANES) microscopy and soft X-ray cryo-nanotomography on frozen–hydrated differentiating bMSCs (4 and 10 days) to gain information on the molecular and crystalline structure of the mineral nuclei and their three-dimensional localization. The XANES signal across the Ca L2,3-edges was extracted selectively from the Ca depositions. Panel A of Figure 2 reports the zero angle projections taken at 346 and 349 eV on 10 days differentiating bMSCs showing a mineral deposition visible only at Ca L3 peak maxima (349 eV). With the transmission of the other elements almost the same at these two energies, the difference between these images defines the two-dimensional Ca distribution. In Figure 2B, the spectrum extracted from the corresponding area (shown in panel A) was compared with spectra extracted from (i) the mineral deposition of bMSCs at 4 days; (ii) crystalline HA; and (iii) calcium phosphate. All spectra present two main peaks (L2 and L3 in Figure 2B) located at the same energy positions and a multipeak pattern (named a–d in Figure 2B) located between 346 and 349 eV, which instead varies significantly for the different samples. This multipeak pattern is associated with the crystal field and enables the identification of different calcium phosphate minerals.22−27 The most distinctive and interesting parameter is the energy position of peak d, whose shift to lower energy is very sensitive to crystallinity in calcium phosphate compounds.24,25,28
Figure 2.

(A) Two projections recorded at 346 and 349 eV on 10 days differentiating bMSCs using synchrotron-based energy-resolved soft X-ray transmission microscopy. The image acquired at 349 eV (Ca L3 peak maximum) shows a mineral deposition (black spot) near the bMSC nucleus. (B) XANES spectra obtained from mineral nuclei localized in bMSCs at 4 days (pink spectrum) and 10 days (violet spectrum), HA reference sample (green spectrum), and calcium phosphate (Ca(H2PO4)2, blue spectrum). Spectra are shifted in the vertical direction for the sake of clarity. Best fits (black lines) are superimposed on the corresponding experimental points (colored circles). (C) Highlighted fit curves in the region of the smaller crystal field peaks a–d. (D) Central slice of the 3D reconstructed volume from the soft X-ray cryo-nanotomography measurement of the cell area of panel A showing the same mineral deposition (highlighted in red) and the corresponding color-coded 3D rendering of the nucleus (indigo) and Ca mineral deposition (red) localized in the bMSC cytoplasm.
Therefore, the position of the peaks a, c, and d in the mineral nuclei spectra obtained at 10 days (purple line of Figure 2C) compared to calcium phosphate spectra (blue line in Figure 2C) and crystalline HA (green line in Figure 2C) suggests that at 10 days the formation of crystalline HA is almost complete. Moreover, the comparison of spectra extracted from mineral nuclei at 10 days versus 4 days of differentiation stimuli, where the multipeak pattern is almost absent, clearly shows a dynamic process of chemical transformation starting from calcium compounds at lower grade of crystallinity and moving toward the formation of HA crystals. This scenario provides a biological support to the chemical mechanism of the calcium phosphate crystal formation proposed by a recent study according to which the existence of amorphous calcium phosphate prenucleation clusters decreases the energy barrier to nucleation, enabling biominerals to mineralize following a nonclassical crystal growth process defined as crystallization by particle attachment.29
Current models of biomineralization hypothesize that the formation of crystal HA occurs in the extracellular matrix where amorphous calcium phosphate formed within the cell is transported out via intracellular vesicles.30 Therefore, to investigate the cellular localization of the mineral nuclei containing crystal HA analyzed by XANES, we performed soft X-ray cryo-nanotomography at 349 eV on the same sample. Results reported in Figure 2D show that the mineral nucleus analyzed was located within the cell cytoplasm at about 2 μm to the cell nucleus. This finding sheds new light on the early stage of biomineralization suggesting that the HA formation starts within the cell.
To determine more precisely the crystalline structure of the mineral nuclei, X-ray diffraction techniques were used. Scanning small-/wide-angle X-ray scattering (SAXS/WAXS) results are reported in Figure 3. The scanning SAXS microscopy signal collected across a 0.9 × 0.9 mm2 area (Figure 3a,b) shows the lateral electron-density inhomogeneity of the samples at 4 and 10 days of differentiation. The red/blue regions indicate regions with the highest/lowest scattering. The spatial resolution is limited in this case by the beam size of 0.2 mm (full width at half-maximum). WAXS data, measured simultaneously to SAXS microscopy and integrated across the explored area, were quantitatively analyzed to determine the crystalline nature of the compounds, and in particular the crystallographic unit cell. Fitting of the WAXS spectra (Figure 3c,d) allowed the identification of the hexagonal HA (red profile best fit) as the unique crystalline structure present in differentiating bMSCs. The WAXS profile collected from the bMSC sample kept for 10 days in osteogenic stimuli, compared to 4 days, reveals the following: (i) a reduction in the amorphous contribution (background under the peaks, Figure 3,f); and (ii) a lower volume (1–3%) of the HA crystallographic unit cell (Table S2, volume contraction factor column). The higher amorphous signal, found for 4 days, which reduces with incubation time, gives evidence of a role in the amorphous mineral precursor in bone mineralization, found in the literature.30 In addition, the volume contraction can be ascribed to the substitution of Ca with Zn atoms in the HA crystal lattice. Indeed, the Zn substitution causes a volume contraction of the unit cell due to the smaller ionic radii of Zn (1.22 Å) with respect to Ca (1.76 Å),31,32 which leads to a smaller distance between atoms.
Figure 3.

(a, b) Scanning SAXS microscopy (scale bar is 0.1 mm). (c, d) Corresponding WAXS spectra. (e) Geometry adopted for SAXS and WAXS data collection. (f) Raw WAXS data superimposed for samples at 4 days and at 10 days. WAXS data identify the hexagonal hydroxyapatite (red markers) as the unique crystalline structure present in the differentiated bMSCs (residual NaCl in the sample is also marked by the blue dashed vertical bars). The best fits (red profiles), obtained for samples of 4 days (c) and 10 days (d), are superimposed on the experimental data (black profiles).
The 1–3% volume decrease of the hexagonal HA unit cell, and its explanation in terms of substitution of Ca with Zn atoms in the HA crystal lattice, is confirmed by X-ray fluorescence microscopy at high spatial resolution (15 nm pixel size) in single mineral nuclei (Figure 4, left panel). Indeed, the Zn/Ca ratio increases in bMSCs exposed to the differentiation cocktail for 10 versus 4 days (Figure 4, right panel). This increase of Zn quantity in mineral nuclei, together with the decrease of the volume of the HA unit cell, supports the interpretation that the process of Zn–Ca substitution is taking place between 4 and 10 days from the beginning of osteogenic differentiation. As mentioned above, Zn is a structural part of the HA crystal of human bone, and it has been reported that its effect in HA nucleation could cause a gain in the critical size of the HA crystals in the initial phase of mineralization.20 Interestingly, at 10 days the Zn/Ca ratio measured by X-ray fluorescence microscopy in mineral nuclei is about 2% (Figure 4, left panel), which is the value that approximates the Zn/Ca ratio in mature human bone.33
Figure 4.

Left: X-ray fluorescence microscopy measurements acquired at 15 nm pixel size (expressed in areal mass (g/cm2)) in a single mineral nucleus formation reporting from top to bottom, the Ca, Zn composition, and Zn/Ca ratio. Right: results of the Zn/Ca ratio in bMSCs exposed to the differentiation cocktail at 10 vs 4 days. To calculate the ratio, the moles of Ca and Zn of the analyzed mineral nuclei were calculated multiplying the respective XRF maps (expressed in areal mass (g/cm2)) for the pixel area and normalizing for respective atomic weight.
Three-Dimensional Analysis and Rendering of the Intracellular Mineral Nuclei: “Fantastic Voyage”
To complete the physical characterization of the mineral nuclei, we examined their localization and found that after 10 days of differentiation some of them are clearly outside the cells, while after 4 days they are mainly near the inner part of the plasma membrane (Figure 1A–C). However, these data limit the investigation to two dimensions since the results are averaged over the sample thickness (projections). The inability to derive the elemental distribution over the sample thickness could lead to a misinterpretation of 2D X-ray fluorescence maps.34 Therefore, we acquired X-ray phase-contrast nanotomography on bMSCs to obtain the 3D localization of the mineral nuclei across the cell thickness at both 4 and 10 days of differentiation. The output of the nanotomography reconstruction is a set of 2000 virtual slices through the cell, showing the three-dimensional electron-density distribution.
Browsing through the cells (Movies S1 and S2), it is worthwhile noting that several mineral nuclei are localized in the cytoplasm, at both 4 and 10 days of differentiation (Figure 5A,B), confirming that the first steps of the biomineralization take place intracellularly. This is in agreement with a study on a mouse cell model of bone formation which detected intracellular Ca accumulation employing nanoanalytical electron microscopy techniques.30 However, our results in a human cell model provide the first experimental evidence that intracellular Ca accumulation contains crystalline HA.
Figure 5.

(A, B) Two z-stacks representing three sections along the thickness of cells at 4 and 10 days of differentiation, respectively. Black arrows indicate the intracellular mineral nuclei depositions, red arrows the cell membrane, and N the nucleus. (C) Two slices of the z-stack reconstructed from the phase-contrast tomography. (D) Volume rendering of the phase-contrast nanotomography data; the colored arrows indicate four intracellular depositions. (E) Zoomed 3D solid nanorendering of the four granules highlighted in panel D. (F) Two slices of the z-stack reconstructed from the X-ray fluorescence tomography. (G–I) Zoomed 3D nanorendering of P, Ca, and Zn intracellular depositions, respectively.
The phase-contrast nanotomography allows us to reconstruct both the three-dimensional rendering of the intracellular sub-organelles and mineral nuclei (Figure 5D) as well as to derive the volumetric information on the mineral nuclei. Figure 5E shows a zoomed 3D rendering of four granules located in the cytoplasm of the cell. A virtual trip within the rendered cell is available in the Supporting Information, respectively, at 4 and 10 days of differentiation (Movies S3 and S4).
The same cells at 10 days of differentiation were scanned by cryo-X-ray fluorescence tomography35 to visualize in 3D the elemental distribution (Movies S5 and S6). In Figure 5G–I, renderings of, respectively, the P, Ca, and Zn 3D composition of the same four mineral nuclei are shown. Focusing on this mineral deposition, it is possible to observe that Ca gives the major contribution to these granules, followed by Zn and P. This result confirms, at the three-dimensional level, a different timing of elemental accumulation in the mineral nuclei.
Conclusions
Urist’s pioneering vision of the early stages of osteogenesis proposed back in the 1960s36 finds here a confirmation at the atomic scale. He hypothesized that the process of differentiation of the osteoprogenitor cell is elicited by yet uncharacterized local morphological, though substantial, alterations within the cell at the sub-ultramicroscopic level. Lately, Ripamonti37 refers to Urist’s insight as an “Aristotelian patterning scenario” in which “the language of shape is the language of geometry and the language of geometry is the language of a sequence of inductive and differentiating cascades of molecular and cellular events”. The results reported in this study give the experimental evidence of the visionary hypothesis formulated by Urist. In particular, a chemical fingerprint of Zn–hydroxyapatite in the early stages of differentiating human bMSCs highlights a unique molecular pattern characterized by the presence of a specific molecule.
Our study used the most advanced X-ray microscopy techniques that have only now reached the required spatial resolution and sensitivity to study biomineralization within single cells in three dimensions. All microscopy and spectromicroscopy investigations were conducted on frozen–hydrated samples, keeping the bMSCs as close as possible to their natural hydrated state and preserving the structural and chemical information. The combined X-ray phase contrast and fluorescence microscopy acquisitions have been pushed to their technical and physical limits in terms of spatial resolution, with pixel sizes down to 15 nm in two and three dimensions. We underline that both the phase-contrast nanotomography and X-ray fluorescence tomography have been acquired on the same frozen–hydrated cells, enabling a correlative interpretation of the results.
The combination of different synchrotron microscopy and laboratory X-ray diffraction techniques enabled following the evolution of hydroxyapatite formation in differentiating bMSCs during the early stage of biomineralization (summarized in Figure 6A). We provide a quantitative assessment of the chemical composition of the mineral depositions together with their molecular and crystalline structure characterization. The analysis presented here reveals that biomineralization starts with hydroxyapatite nucleation within the cell, and it rapidly evolves toward a hexagonal hydroxyapatite crystal very similar to the one present in mature human bone, as detected after just 10 days of osteogenic induction. Zn inclusion in the mineral depositions favors the nucleation of the hydroxyapatite crystal lattice, and at 10 days the process of Zn incorporation is almost complete. In Figure 6B we propose a model based on these results, with a possible mechanism of the genesis and the evolution of hexagonal hydroxyapatite. It is worth underlining that the model proposed in Figure 6 refers to 2D culture cells; therefore, a further confirmation will need to be performed in three-dimensional cell culture systems.
Figure 6.

(a) Outline describing the rationale, experimental plan, and results on the study of the early stages of biomineralization. The gray boxes indicate the information provided by the techniques. The ovals show the techniques applied. The yellow boxes describe the main results obtained using different techniques. (b) Graph sketch depicting a model based on the results obtained in this study, hypothesizing a mechanism of the genesis and the evolution of the hexagonal hydroxyapatite. The mineral depositions showing a different elemental composition, as detected by XRFM, are represented by different polygonal shapes. The roman numerals portray the chronological sequence.
The quantitative phenomenological follow-up of the early stages of biomineralization presented here will serve as a reference for further studies on the intracellular molecular mechanisms governing the initial stage of bone tissue formation. Last but not least, the evidence of the chemical fingerprint of Zn–hydroxyapatite in the early stages of bone formation could have translational implications by impacting medical bioengineering and regenerative medicine.
Materials and Methods
Isolation and Culture of Human Mesenchymal Stem Cells
Mesenchymal stem cells from the human bone marrow of healthy male volunteers were donated by Prof. Berti (Policlinico, Milan). The cells were isolated after obtaining informed consent from all the subjects at the Policlinico in Milano, in compliance with the Helsinki declaration and with applicable laws and regulations, according to institutional guidelines and regulations of the Ethical Committee of “IRCCS Policlinico” Milano. No unexpected or unusually high safety hazards were encountered. bMSCs were tested for purity by flow cytometry and cultured in Dulbecco’s modified Eagle’s medium with 1000 mg/L glucose, 10% fetal bovine serum (FBS), and 2 mM l-glutamine (culture medium) at 37 °C. When confluent, the cells were detached by treatment with trypsin-EDTA 1× (Sigma–Aldrich), characterized, subcultured, and used at passage numbers 3–5.16
Osteogenic Differentiation of bMSCs
To induce osteogenic differentiation, bMSCs were seeded in 6- or 96-well plates. Once the cells were confluent, an osteogenic cocktail was added to the osteogenic medium. The osteogenic cocktail contains 2 × 10–8 M 1α,25-dihydroxyvitamin D3, 10 mM β-glycerolphosphate, and 0.05 mM ascorbic acid (Sigma–Aldrich).17 To investigate calcium deposition by bMSCs, the cells were rinsed with PBS, fixed (70% ethanol, 1 h), and stained for 10 min with 2% Alizarin Red S (pH 4.2, Sigma–Aldrich).17 The experiment was repeated three times in triplicate. Photographs were taken at 10× magnification (Figure S1A). Alizarin Red S staining was released from the cell matrix by incubation in 10% cetylpyridinium chloride (Sigma–Aldrich) in 10 mM sodium phosphate (pH 7.0), for 15 min, and the absorbance was measured at 562 nm.
Gene Expression Analysis and ELISA
Total RNA was extracted by the PureLink RNA Mini kit (Thermo Fisher Scientific). Single-stranded cDNA was synthesized from 0.2 μg of RNA in a 20 μL final volume using the high-capacity cDNA reverse transcription kit, with RNase inhibitor (Thermo Fisher Scientific) according to the manufacturer’s instructions. Real-time PCR was performed three times in triplicate on the 7500 FAST real time PCR system instrument using TaqMan gene expression assays (Life Technologies, Thermo Fisher Scientific): Hs00231692_m1 (RUNX2), Hs00164004_m1 (COL1A1), Hs01587814_g1 (BGLAP), and Hs00959010_m1 (SPP1).
The housekeeping gene GAPDH (Hs99999905_m1) was used as an internal reference gene. Relative changes in gene expression were analyzed by the 2-ΔΔCt method.17
For the quantitative determination of RUNX2 and collagen type 1, we utilized ELISA (Cusabio) according to the manufacturer’s instructions. ELISAs were performed three times, and each sample was measured in triplicate. Data are shown as the mean ± standard deviation.
ID16A-NI “Nano-Imaging” Beamline
The X-ray fluorescence and X-ray phase-contrast measurements were performed at the ID16A-NI “Nano-Imaging” beamline of the European Synchrotron Radiation Facility (Figure S3).54
The beam was focused using Kirkpatrick–Baez mirrors to a spot of 23 nm × 37 nm, and the selected energy was 17 keV. For the X-ray fluorescence measurements, the sample was placed in the focal plane, and elemental maps were obtained through raster scanning. The selected pixel sizes were 100 nm for noninduced bMSCs, 70 nm for the samples measured at 4 days and 10 days of osteogenic differentiation, and 15 nm for more precise maps of individual mineral nuclei at the 10th day of differentiation. The photon flux was 2.3 × 1011 photons/s; the exposure time was 0.05 s per pixel, and the recorded maps covered areas of 10–20 μm × 10–20 μm, depending on the cell sizes. For each pixel, the fluorescence spectrum was recorded with an energy-resolving detector consisting of six silicon drift diodes, set orthogonal to the incoming beam. The acquired spectra were fitted using the PyMCA38 open source software to obtain quantitative data.
For a region containing stem cells at 10 days of osteogenic differentiation, a tomographic fluorescence scan was acquired with a pixel size of 125 nm, to access the elemental distribution in three dimensions. Two-dimensional maps of cells positioned on the rotation axis were recorded at 28 angles. After fitting with PyMCA and alignment using in-house Octave code, the maps were used for tomographic reconstruction through TomoJ,39 a plugin of ImageJ.40 An iterative method using total variation regularization was selected for tomographic reconstruction. This resulted in 3D maps of Ca, P, and Zn concentrations. The output of the reconstruction, the tomogram, is a stack of virtual slices (Movie S5), which for better visualization has been converted to 3D renderings (Movie S6), obtained after manual segmentation using Chimera41 open source software.
For X-ray phase-contrast nanotomography, the samples with bMSCs after 4 and 10 days of osteogenic differentiation were placed in the cone beam at a distance z1 downstream of the focus. The 2D detector (using a FReLoN charge-coupled device with binned 2048 × 2048 pixels array) was placed at a distance z2 downstream of the sample, achieving a magnification M = (z1 + z2)/z1 and an effective propagation distance D = z2/M. The magnification was set to achieve a pixel size of 50 nm (sample at the 4th day of differentiation) and 15 nm (sample at the 10th day of differentiation). Tomographic scans were recorded at four different distances from the focal spot. For each scan, 2000 projections over 180° with an exposure time of 0.25 s were recorded. For image reconstruction, each set of four holograms corresponding to one rotation angle was brought to the same magnification and aligned before being introduced into a phase-retrieval algorithm.55 These steps of data processing are implemented in Octave. The obtained phase maps were then used for tomographic reconstruction via filtered back-projection using PyHST.56
The output of the holographic nanotomography is a stack of virtual slices (Movies S1 and S2). Three-dimensional renderings (Movies S3 and S4) were realized after a manual segmentation in Chimera software, to better visualize the 3D information given by the reconstructed electron density.
BL09—Mistral “Soft X-ray Microscopy” Beamline
Energy-resolved soft X-ray transmission microscopy (ER-TXM) was performed at the MISTRAL beamline of the ALBA synchrotron.44,45 Varying the energy of the incident photons on the sample across the Ca L-edge, ER-TXM directly detects Ca and its chemical state, as it provides a full Ca L-edge absorption spectrum at each image pixel, with a spatial resolution of a few tens of nm. Variations in energy positions and in relative intensities of the peaks in the XANES spectra depend on the Ca chemical state and probe the local environment of Ca atoms, thus providing local structural information.22
On selected areas, 2D XANES images (12 s exposure time, effective pixel size 10 nm, field of view (FOV) 13 μm × 13 μm) were collected varying the energy across the Ca L-edge with a variable spectral sampling (0.5–0.1 eV). The chosen value for the fixed focus constant of the variable line spacing plane grating monochromator45 was 1.5 to optimize the compromise between spectral resolution (≈0.2 eV) and the rejection of the grating second harmonic. The objective zone plate lens and the CCD detector positions were automatically adjusted to maintain the sample in focus and constant magnification. The necessary total acquisition time was about 1.5 h per energy scan, including the flat field acquisition at each energy step.
ER-TXM measurements were performed on two bMSCs at 10 days and on two bMSCs at 4 days. For each cell, five fields of view were selected in such a way to almost cover the full area of the cell. As a consequence, it results that five energy scans were acquired for each cell for a total of 20 measurements. The “spectra analysis by peak deconvolution” was performed only on the selected spectra (one from a bMSC at 4 days and one from a bMSC at 10 days). It is worth noting that the bMSCs at 4 days samples showed a bigger spectral variety with respect to the bMSCs at 10 days.
Each image of the energy stack (I(x, y), where I is the transmitted intensity by the sample, and x and y are the 2D coordinates of a generic pixel) is first normalized to unity dividing by the corresponding flat field image (I0(x, y), with I0 being the incident intensity on the sample), taking into account the value of the electron current in the storage ring, and then aligned taking the first image as a reference. The alignment is performed by homemade software selecting a suitable region of interest (ROI). The ROI of a single image is compared with the ROI of the first image, and thanks to the Python library of cv2 (Open Computer Vision: OpenCV), the normalized cross-correlation of both ROIs is used to detect the best matching between them (the function used is “cv2.matchTemplate()”). Once this best matching is detected, the image is shifted by the number of pixels calculated in the precedent operation. This process is repeated for each image of the energy stack. Normally this operation reduces the effective field of view by about 10–15%, i.e., from 13 μm × 13 μm to ≈11 μm × 11 μm. At this point, a full Ca L-edge X-ray absorption spectrum (XAS) at each image pixel can be extracted. In particular, the absorbance for each pixel (A) as a function of the energy can be calculated from the measured intensities using the Beer–Lambert law:
where t is the thickness, and μ is the linear absorption coefficient of the material.
Spectra were extracted only from pixels in the field of view containing Ca. Pixels containing Ca are those pixels with a signal to noise ratio bigger than two, i.e., S/N > 2, with S defined as the absorbance difference between the average absorbance value around the L2 peak maxima (352.3–352.7 eV) and the absorbance average value in the pre-edge energy region (344–346.5 eV), and N is defined as the sigma in the pre-edge energy region. The pixel selection operation was performed using ImageJ and TXMWizard software.46,47
Peak deconvolution was performed to compare the measured spectra. In a first step a linear background correction is performed interpolating the data points in the 344–346.5 eV (pre-edge energy) region with a straight line and subtracting it from the total spectra, to eliminate the contribution of lower energy absorption. This operation is done using a homemade routine implemented in Matlab. In particular, the fit is performed using the Matlab function “polyfit”; i.e., the calculated straight line is the best linear fit of the data points in the pre-edge energy region in a least-squares sense. Then, the edge steps at the Ca L2-, L3-edges resulting from transitions to the continuum were also subtracted using a double arctan function, as described in refs (46 and 47):
with w1 = w2 = 0.2 eV, E1 = L2peakmax and E2 = L2peakmax + 3 eV. The others parameters (h1, h2, and C) were fitted with the data points in the pre-edge (344–346.5 eV) and in the postedge (355.8–360 eV) energy regions.
These two operations (linear background subtraction and double arctan subtraction), applied to the Ca L2,3-edge spectrum of the HA reference sample, are illustrated in Figure S4A. Following,25 the data after this double subtraction (“subtracted data”) are fitted with 7 or 5 Gaussians (the chosen number of Gaussians corresponds to the best fit) using the tool “Peak Analyzer” of Origin software. Fitting results are reported in Figure S4B. Details on the peak fit parameters are reported in Table S1.
Soft X-ray Cryotomography
Soft X-ray cryotransmission tomography measurements of whole frozen hydrated cells were also performed. The cryogenic conditions were maintained during all of the experiment. Soft X-ray cryotransmission tomography was carried out at 349 eV to optimize the contrast between the calcium-rich subject and the surrounding water-rich cytoplasmic solutions without staining, sectioning, or using enhancing agents. For each cell, a tilt series was acquired using an angular step of 1° on a 130° angular range. Data sets were acquired using a zone plate objective with an outermost zone width of 40 nm. The effective pixel size in the images was 11.8 nm. No radiation damage was detected at our spatial resolution. Each transmission projection image of the tilt series was normalized using flat-field images of 1 s acquisition time. The tilt series was manually aligned using eTomo in the IMOD tomography software suite.48 To decrease as much as possible the deviations from an ideal rotation that creates artifacts in the reconstructed tomograms, the rotation of gold nanobeads was followed. The transmission tilt series were finally reconstructed with TomoJ,39 a plugin of ImageJ40 using the SIRT iterative-algorithms.
X-ray Micro-Imaging Laboratory
X-ray scanning transmission microscopy, in simultaneous small-angle X-ray scattering (SAXS) and wide-angle X-ray scattering (WAXS) mode, was used to study supra- and submolecular structural ordering of the investigated samples. In SAXS, electron-density inhomogeneity is monitored at nanometric scale and, in WAXS, at atomic scale. Indeed, each material produces a unique X-ray diffraction pattern (intensity versus scattering angle) that is a fingerprint of its crystalline structure. Among the most important data extracted from this quantitative analysis is the crystallographic unit cell, which repeats identically within the lattice to form the crystal.
The SAXS/WAXS scanning microscope of the X-ray Micro-Imaging Laboratory (XMI-L@b) is equipped with a Rigaku Fr-E+ SuperBright rotating anode table-top microsource (Cu Kα, λ = 0.154 05 nm, 2475 W), multilayer focusing optics (Confocal Max-Flux; CMF 15-105), and a three-pinhole camera (SMAX-3000). An image plate (IP) detector with 100 μm pixel size and a 6 mm hole in the center was placed at about 3 cm from the sample to acquire WAXS data, while collecting the SAXS data. The SAXS detector is a Triton 20 gas-filled photon counter with ∼200 μm pixel size, which was placed at about 2.2 m from the sample. Once acquired, 2D data were calibrated by means of the standard reference materials (Si NIST SRM 640b for WAXS and silver behenate for SAXS data) and folded into 1D profiles. A detailed description of the XMI-L@b performances can be found in Altamura et al. and Sibillano et al.49,50 The raw SAXS data, collected in scanning mode, were composed into microscopy, as explained in ref (51).
Scanning SAXS 2D data were recorded, spatially resolved, across a 0.9 × 0.9 mm2 area (Figure S5, left) with the WAXS detector simultaneously collecting the 2D data (Figure S5, right), averaged in the same area. A schematic layout of the setup is shown in panel e of Figure 3. SAXS and WAXS raw 2D data were centered, calibrated, and folded51 into 1D profiles.
For each sample position across the 0.9 × 0.9 mm2 area, the SAXS intensity in the relevant 1D profile was then integrated over the measured scattering range, normalized to the transmitted intensity, and plotted for each sample position as a pixel in the image of Figure 3 for samples at the 4th day (panel a) and 10th day (panel b) of differentiation.
The WAXS 1D profiles, shown in Figure 3 for samples differentiated for 4 days (panel c) and 10 days (panel d), were first qualitatively analyzed with the QUALX program51 to identify the structure that can explain the measured diffraction peaks. All peaks were indexed (see red markers) with the hexagonal hydroxyapatite (a = b = 9.415 Å, c = 6.879 Å, α = β = 90°, γ = 120°, space group P63/m, ICSD code 187840) as the unique crystalline structure, apart from the peak at about 2θ = 45° which was attributed to NaCl contamination (the marked vertical bar corresponds to NaCl contamination and is in the same angular position for both samples relative to the HA peak positions, which can be adopted as internal reference). This qualitative analysis was followed by a quantitative whole profile fitting analysis of the diffraction profiles, performed using the FULLPROF program,52 based on the Rietveld approach. Figure 3 shows the best fits (red profiles) obtained for samples differentiated for 4 days (panel c) and 10 days (panel d), overimposed to the experimental data (black profiles). We analyzed with the same procedure 2 samples incubated for 10 days (bMSC_10days_A and bMSC_10days) and 1 sample for 4 days (bMSC_4days_4C). Fits allowed the determination of the crystal lattice constants (a–c) of an alloy and the corresponding cell volume (V), reported in Table S2. We can observe a decrease of the refined cell volume with the increase of the incubation days. The relative volume contraction factor X, given by the ratio V(10days)/V(4days), is reported in the same Table S2. We computed also a mean value for the contraction:
The 1–3% volume contraction was explained as the result of a substitution of Ca atoms with Zn atoms in the lattice. Indeed, the ionic radius of Zn (0.88 Å), which is smaller than that of Ca (1.14 Å), induces a reduction of the interatomic distances, which justifies the volume contraction. To quantitatively estimate the percentage of substitutional Zn/Ca atoms in the lattice, we rely on the empirical Vegards’s rule which linearly relates the crystal lattice constants of an alloy to the concentrations of the constituent elements.53 Therefore, the Xmean directly relates to the ratio of Zn atoms substitutional to Ca atoms.
Sample Preparation for Synchrotron-Based Techniques
For Synchrotron-based techniques at beamline ID16A-NI, bMSCs were grown on silicon nitride membranes Si3N4 considered as an ideal support for XRF analysis (from Silson Ltd., Northampton (UK), 5 × 5 mm2 size, 4 mm2 membrane area, 200 nm membrane thickness, and 200 μm frame thickness). The cells were plated at a concentration of 1 × 104 cell/cm2 on membrane windows previously sterilized in ethanol, under previously described culture conditions (considered as ideal). After 4 and 10 days from the osteogenic induction, the attachment and spreading of the cells were carefully verified using optical microscopy. Cell culture medium was removed, and the membranes were briefly washed in 100 mM freshly prepared ammonium acetate solution two times, to remove salts and trace metals from the medium. The cells were frozen–hydrated by a rapid plunge freezing (Leica EM GP) in a liquid ethane bath cooled with liquid nitrogen. As vitreous ice upon the samples causes X-ray absorption, excess water is carefully removed before plunge freezing via blotting.
After plunge freezing, a cryogenic workflow is maintained until and during the XRF measurements. Wafers are transferred into the liquid nitrogen bath of a Leica EM VCM (vacuum cryomanipulation system), where they are clamped in a precooled gold-coated VCT sample holder. The sample holder can then be loaded into the Leica EM VCT500 (vacuum cryotransfer system). Finally, a sample shuttle is attached to the ID16A-NI vacuum chamber, and the gold-coated copper cube holding the silicon nitride membrane is transferred onto the sample stage kept at −150 °C. A visible light microscope system with ultralong working distance is used to bring bMSCs in the focus of the X-ray nanobeam.
For the acquisition at the MISTRAL beamline of the ALBA synchrotron, cells were seeded onto gold quantifoil R 2/2 holey carbon-film microscopy grids and after 4 or 10 days vitrified by plunge freezing (as described previously). The frozen grids were then transferred into the Mistral transmission X-ray microscope under cryogenic conditions. An artificial hydroxyapatite (HA) reference sample was prepared by finely crushing HA powder (Sigma–Aldrich) in a mortar, and laying down the obtained micrometer and submicrometer dust on a Quantifoil Au TEM grid.
Sample Preparation for Diffraction Techniques
The cells were plated at a concentration of 1 × 104 cell/cm2 on silicon membrane windows (from Silson Ltd., Northampton (UK), 5 × 5 mm2 size, 4 mm2 membrane area, 200 nm membrane thickness, and 200 μm frame thickness), previously sterilized in ethanol. After 4 and 10 days from the osteogenic induction, cell culture medium was removed, and the membranes were washed briefly in 100 mM ammonium acetate two times and then fixed in methanol/acetone 1:1 at the temperature of −20 °C for 5 min. Then, the solution of methanol–acetone was removed, and the cells on silicon membranes were dried at room temperature.
Acknowledgments
The ESRF is acknowledged for providing beamtime and support for the experiments at the nano-imaging beamline ID16A in the frame of proposals LS-2490 and LS-2634.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.9b00509.
Alizarin Red staining, and real-time PCR, 2D X-ray fluorescence maps, linear background subtraction, double arctan subtraction, scanning SAXS, WAXS, fitting results of XANES spectra analyses, crystallographic unit cell parameters, cell volume (V), and relative volume contraction factor X (PDF)
Movie S1: Virtual stack of X-ray phase-contrast tomography of bMSCs after 4 days of the osteogenic induction (pixel size 50 nm) (AVI)
Movie S2: Virtual stack of X-ray phase-contrast tomography of bMSCs after 10 days of the osteogenic induction (pixel size 15 nm) (AVI)
Movie S3: 3D nanorendering of X-ray phase-contrast tomography of bMSCs after 4 days of differentiation (AVI)
Movie S4: 3D nanorendering of phase-contrast tomography of bMSCs after 10 days of differentiation (AVI)
Movie S5: Virtual stack of X-ray fluorescence projections of Ca, P, and Zn of bMSCs after 10 days of the osteogenic induction (pixel size 125 nm) (AVI)
Movie S6: Zoomed 3D nanorendering of Ca (red spots), P (green spots), and Zn (blue spots) of bMSCs after 10 days of the osteogenic induction (AVI)
Author Contributions
A. Procopio and E. Malucelli equally contributed to this work. J. A. M. Maier and S. Iotti share senior authorship.
The authors declare no competing financial interest.
Supplementary Material
References
- Roy D. M.; Linnehan S. K. Hydroxyapatite Formed from Coral Skeletal Carbonate by Hydrothermal Exchange. Nature 1974, 247 (5438), 220–222. 10.1038/247220a0. [DOI] [PubMed] [Google Scholar]
- Ohira T.; Ishikawa K. Hydroxyapatite Deposition in Articular Cartilage by Intra-Articular Injections of Methylprednisolone. A Histological, Ultrastructural, and x-Ray-Microprobe Analysis in Rabbits. Journal of Bone & Joint Surgery 1986, 68 (4), 509–520. 10.2106/00004623-198668040-00005. [DOI] [PubMed] [Google Scholar]
- Weiner S.; Wagner H. D. THE MATERIAL BONE: Structure-Mechanical Function Relations. Annu. Rev. Mater. Sci. 1998, 28 (1), 271–298. 10.1146/annurev.matsci.28.1.271. [DOI] [Google Scholar]
- Prockop D. J.; Fertala A. The Collagen Fibril: The Almost Crystalline Structure. J. Struct. Biol. 1998, 122 (1), 111–118. 10.1006/jsbi.1998.3976. [DOI] [PubMed] [Google Scholar]
- Cedola A.; Mastrogiacomo M.; Burghammer M.; Komlev V.; Giannoni P.; Favia A.; Cancedda R.; Rustichelli F.; Lagomarsino S. Engineered Bone from Bone Marrow Stromal Cells: A Structural Study by an Advanced x-Ray Microdiffraction Technique. Phys. Med. Biol. 2006, 51 (6), N109–N116. 10.1088/0031-9155/51/6/N02. [DOI] [PubMed] [Google Scholar]
- Betts F.; Blumenthal N. C.; Posner A. S.; Becker G. L.; Lehninger A. L. Atomic Structure of Intracellular Amorphous Calcium Phosphate Deposits. Proc. Natl. Acad. Sci. U. S. A. 1975, 72 (6), 2088–2090. 10.1073/pnas.72.6.2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nudelman F.; Pieterse K.; George A.; Bomans P. H. H.; Friedrich H.; Brylka L. J.; Hilbers P. A. J.; de With G.; Sommerdijk N. A. J. M. The Role of Collagen in Bone Apatite Formation in the Presence of Hydroxyapatite Nucleation Inhibitors. Nat. Mater. 2010, 9 (12), 1004–1009. 10.1038/nmat2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaglione S.; Giannoni P.; Bianchini P.; Sandri M.; Marotta R.; Firpo G.; Valbusa U.; Tampieri A.; Diaspro A.; Bianco P.; et al. Order versus Disorder: In Vivo Bone Formation within Osteoconductive Scaffolds. Sci. Rep. 2012, 2, 274. 10.1038/srep00274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reisinger A. G.; Pahr D. H.; Zysset P. K. Sensitivity Analysis and Parametric Study of Elastic Properties of an Unidirectional Mineralized Bone Fibril-Array Using Mean Field Methods. Biomech. Model. Mechanobiol. 2010, 9 (5), 499–510. 10.1007/s10237-010-0190-1. [DOI] [PubMed] [Google Scholar]
- Giannini C.; Siliqi D.; Bunk O.; Beraudi A.; Ladisa M.; Altamura D.; Stea S.; Baruffaldi F. Correlative Light and Scanning X-Ray Scattering Microscopy of Healthy and Pathologic Human Bone Sections. Sci. Rep. 2012, 2, 435. 10.1038/srep00435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey C.; Combes C.; Drouet C.; Glimcher M. J. Bone Mineral: Update on Chemical Composition and Structure. Osteoporosis Int. 2009, 20 (6), 1013–1021. 10.1007/s00198-009-0860-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kular J.; Tickner J.; Chim S. M.; Xu J. An Overview of the Regulation of Bone Remodelling at the Cellular Level. Clin. Biochem. 2012, 45 (12), 863–873. 10.1016/j.clinbiochem.2012.03.021. [DOI] [PubMed] [Google Scholar]
- Abdallah B. M.; Kassem M. Human Mesenchymal Stem Cells: From Basic Biology to Clinical Applications. Gene Ther. 2008, 15 (2), 109–116. 10.1038/sj.gt.3303067. [DOI] [PubMed] [Google Scholar]
- Rho J.-Y.; Kuhn-Spearing L.; Zioupos P. Mechanical Properties and the Hierarchical Structure of Bone. Medical Engineering & Physics 1998, 20 (2), 92–102. 10.1016/S1350-4533(98)00007-1. [DOI] [PubMed] [Google Scholar]
- Green M. V.; Seidel J.; Vaquero J. J.; Jagoda E.; Lee I.; Eckelman W. C. High Resolution PET, SPECT and Projection Imaging in Small Animals. Computerized Medical Imaging and Graphics 2001, 25 (2), 79–86. 10.1016/S0895-6111(00)00057-4. [DOI] [PubMed] [Google Scholar]
- Sargenti A.; Castiglioni S.; Olivi E.; Bianchi F.; Cazzaniga A.; Farruggia G.; Cappadone C.; Merolle L.; Malucelli E.; Ventura C.; et al. Magnesium Deprivation Potentiates Human Mesenchymal Stem Cell Transcriptional Remodeling. Int. J. Mol. Sci. 2018, 19 (5), 1410. 10.3390/ijms19051410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazzaniga A.; Maier J. A. M.; Castiglioni S. Impact of Simulated Microgravity on Human Bone Stem Cells: New Hints for Space Medicine. Biochem. Biophys. Res. Commun. 2016, 473 (1), 181–186. 10.1016/j.bbrc.2016.03.075. [DOI] [PubMed] [Google Scholar]
- Castiglioni S.; Romeo V.; Locatelli L.; Cazzaniga A.; Maier J. A. M. TRPM7 and MagT1 in the Osteogenic Differentiation of Human Mesenchymal Stem Cells in Vitro. Sci. Rep. 2018, 8 (1), 16195. 10.1038/s41598-018-34324-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter G. K. Role of Osteopontin in Modulation of Hydroxyapatite Formation. Calcif. Tissue Int. 2013, 93 (4), 348–354. 10.1007/s00223-013-9698-6. [DOI] [PubMed] [Google Scholar]
- Lappalainen R.; Knuuttila M.; Lammi S.; Alhava E. M.; Olkkonen H. Zn and Cu Content in Human Cancellous Bone. Acta Orthop. Scand. 1982, 53 (1), 51–55. 10.3109/17453678208992178. [DOI] [PubMed] [Google Scholar]
- Driessens F. C. M.; Verbeeck R. M. H.; Kiekens P. Mechanism of Substitution in Carbonated Apatites. Z. Anorg. Allg. Chem. 1983, 504 (9), 195–200. 10.1002/zaac.19835040925. [DOI] [Google Scholar]
- Fleet M. E.; Liu X. Calcium L2,3-Edge XANES of Carbonates, Carbonate Apatite, and Oldhamite (CaS). Am. Mineral. 2009, 94 (8–9), 1235–1241. 10.2138/am.2009.3228. [DOI] [Google Scholar]
- de Groot F. M. F.; Fuggle J. C.; Thole B. T.; Sawatzky G. A. L2,3 X-ray-absorption edges of d0 compounds: K+, Ca2+, Sc3+, and Ti4+ in Oh (octahedral) symmetry. Phys. Rev. B: Condens. Matter Mater. Phys. 1990, 41 (2), 928–937. 10.1103/PhysRevB.41.928. [DOI] [PubMed] [Google Scholar]
- Cosmidis J.; Benzerara K.; Nassif N.; Tyliszczak T.; Bourdelle F. Characterization of Ca-Phosphate Biological Materials by Scanning Transmission X-Ray Microscopy (STXM) at the Ca L2,3-, P L2,3- And C K-Edges. Acta Biomater. 2015, 12 (1), 260–269. 10.1016/j.actbio.2014.10.003. [DOI] [PubMed] [Google Scholar]
- Beniash E.; Metzler R. A.; Lam R. S.; Gilbert P. U. Transient Amorphous Calcium Phosphate in Forming Enamel. J. Struct. Biol. 2009, 166 (2), 133–143. 10.1016/j.jsb.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Politi Y.; Metzler R. A.; Abrecht M.; Gilbert B.; Wilt F. H.; Sagi I.; Addadi L.; Weiner S.; Gilbert P. U. P. A. Transformation Mechanism of Amorphous Calcium Carbonate into Calcite in the Sea Urchin Larval Spicule. Proc. Natl. Acad. Sci. U. S. A. 2008, 105 (45), 17362–17366. 10.1073/pnas.0806604105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sviben S.; Gal A.; Hood M. A.; Bertinetti L.; Politi Y.; Bennet M.; Krishnamoorthy P.; Schertel A.; Wirth R.; Sorrentino A.; et al. A Vacuole-like Compartment Concentrates a Disordered Calcium Phase in a Key Coccolithophorid Alga. Nat. Commun. 2016, 7, 11228. 10.1038/ncomms11228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zougrou I. M.; Katsikini M.; Brzhezinskaya M.; Pinakidou F.; Papadopoulou L.; Tsoukala E.; Paloura E. C. Ca L2,3-Edge XANES and Sr K-Edge EXAFS Study of Hydroxyapatite and Fossil Bone Apatite. Naturwissenschaften 2016, 103 (7), 60. 10.1007/s00114-016-1383-y. [DOI] [PubMed] [Google Scholar]
- Wang X.; Yang J.; Andrei C. M.; Soleymani L.; Grandfield K. Biomineralization of Calcium phate Revealed by in Situ Liquid-Phase Electron Microscopy. Communications Chemistry 2018, 1 (1), 80. 10.1038/s42004-018-0081-4. [DOI] [Google Scholar]
- Boonrungsiman S.; Gentleman E.; Carzaniga R.; Evans N. D.; McComb D. W.; Porter A. E.; Stevens M. M. The Role of Intracellular Calcium Phosphate in Osteoblast-Mediated Bone Apatite Formation. Proc. Natl. Acad. Sci. U. S. A. 2012, 109 (35), 14170–14175. 10.1073/pnas.1208916109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannini C.; Fischer A.; Lange C.; Ploog K.; Tapfer L. Heavy Carbon Doping of GaAs Grown by Solid-source Molecular-beam Epitaxy. Appl. Phys. Lett. 1992, 61 (2), 183–185. 10.1063/1.108212. [DOI] [Google Scholar]
- Boanini E.; Gazzano M.; Bigi A. Ionic Substitutions in Calcium Phosphates Synthesized at Low Temperature. Acta Biomater. 2010, 6 (6), 1882–1894. 10.1016/j.actbio.2009.12.041. [DOI] [PubMed] [Google Scholar]
- Lappalainen R.; Knuuttila M. Atomic Absorption Spectrometric Evidence of Relationships between Some Cationic Elements in Human Dentine. Arch. Oral Biol. 1982, 27 (10), 827–830. 10.1016/0003-9969(82)90037-1. [DOI] [PubMed] [Google Scholar]
- Malucelli E.; Fratini M.; Notargiacomo A.; Gianoncelli A.; Merolle L.; Sargenti A.; Cappadone C.; Farruggia G.; Lagomarsino S.; Iotti S. Where Is It and How Much? Mapping and Quantifying Elements in Single Cells. Analyst 2016, 141 (18), 5221–5235. 10.1039/C6AN01091A. [DOI] [PubMed] [Google Scholar]
- de Jonge M. D.; Holzner C.; Baines S. B.; Twining B. S.; Ignatyev K.; Diaz J.; Howard D. L.; Legnini D.; Miceli A.; McNulty I.; et al. Quantitative 3D Elemental Microtomography of Cyclotella Meneghiniana at 400-Nm Resolution. Proc. Natl. Acad. Sci. U. S. A. 2010, 107 (36), 15676–15680. 10.1073/pnas.1001469107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urist M. R. Bone: Formation by Autoinduction. Science 1965, 150 (3698), 893–899. 10.1126/science.150.3698.893. [DOI] [PubMed] [Google Scholar]
- Ripamonti U. Soluble and Insoluble Signals Sculpt Osteogenesis in Angiogenesis. World J. Biol. Chem. 2010, 1 (5), 109–132. 10.4331/wjbc.v1.i5.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solé V. A.; Papillon E.; Cotte M.; Walter Ph; Susini J. A Multiplatform Code for the Analysis of Energy-Dispersive X-Ray Fluorescence Spectra. Spectrochim. Acta, Part B 2007, 62 (1), 63–68. 10.1016/j.sab.2006.12.002. [DOI] [Google Scholar]
- MessaoudiI C.; Boudier T.; Sorzano C. O. S.; Marco S. TomoJ: Tomography Software for Three-Dimensional Reconstruction in Transmission Electron Microscopy. BMC Bioinf. 2007, 8 (1), 288. 10.1186/1471-2105-8-288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueden C. T.; Schindelin J.; Hiner M. C.; DeZonia B. E.; Walter A. E.; Arena E. T.; Eliceiri K. W. ImageJ2: ImageJ for the next Generation of Scientific Image Data. BMC Bioinf. 2017, 18 (1), 529. 10.1186/s12859-017-1934-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. A visualization system for exploratory research and analysis - Pettersen. J. Comput. Chem. 2004, 25, 1605. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Mirone A.; Brun E.; Gouillart E.; Tafforeau P.; Kieffer J. The pyhst2 hybrid distributed code for high speed tomographic reconstruction with iterative reconstruction and a priori knowledge capabilities. Nucl. Instrum. Methods Phys. Res., Sect. B 2014, 324, 41–48. 10.1016/j.nimb.2013.09.030. [DOI] [Google Scholar]
- Sorrentino A.; Nicolás J.; Valcárcel R.; Chichón F. J.; Rosanes M.; Avila J.; Tkachuk A.; Irwin J.; Ferrer S.; Pereiro E. MISTRAL: A Transmission Soft X-Ray Microscopy Beamline for Cryo Nano-Tomography of Biological Samples and Magnetic Domains Imaging. J. Synchrotron Radiat. 2015, 22 (4), 1112–1117. 10.1107/S1600577515008632. [DOI] [PubMed] [Google Scholar]
- Pereiro E.; Nicolás J.; Ferrer S.; Howells M. R. A Soft X-Ray Beamline for Transmission X-Ray Microscopy at ALBA. J. Synchrotron Radiat. 2009, 16 (4), 505–512. 10.1107/S0909049509019396. [DOI] [PubMed] [Google Scholar]
- Schneider G.; Guttmann P.; Heim S.; Rehbein S.; Mueller F.; Nagashima K.; Heymann J. B.; Müller W. G.; McNally J. G. Three-Dimensional Cellular Ultrastructure Resolved by X-Ray Microscopy. Nat. Methods 2010, 7 (12), 985–987. 10.1038/nmeth.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Meirer F.; Williams P. A.; Wang J.; Andrews J. C.; Pianetta P. TXM-Wizard: A Program for Advanced Data Collection and Evaluation in Full-Field Transmission X-Ray Microscopy. J. Synchrotron Radiat. 2012, 19 (2), 281–287. 10.1107/S0909049511049144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer J. R.; Mastronarde D. N.; McIntosh J. R. Computer Visualization of Three-Dimensional Image Data Using IMOD. J. Struct. Biol. 1996, 116 (1), 71–76. 10.1006/jsbi.1996.0013. [DOI] [PubMed] [Google Scholar]
- Altamura D.; Lassandro R.; Vittoria F. A.; De Caro L.; Siliqi D.; Ladisa M.; Giannini C. X-Ray Microimaging Laboratory (XMI-LAB). J. Appl. Crystallogr. 2012, 45 (4), 869–873. 10.1107/S0021889812025733. [DOI] [Google Scholar]
- Sibillano T.; De Caro L.; Scattarella F.; Scarcelli G.; Siliqi D.; Altamura D.; Liebi M.; Ladisa M.; Bunk O.; Giannini C. Interfibrillar Packing of Bovine Cornea by Table-Top and Synchrotron Scanning SAXS Microscopy. J. Appl. Crystallogr. 2016, 49 (4), 1231–1239. 10.1107/S1600576716010396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siliqi D.; De Caro L.; Ladisa M.; Scattarella F.; Mazzone A.; Altamura D.; Sibillano T.; Giannini C. SUNBIM: A Package for X-Ray Imaging of Nano- and Biomaterials Using SAXS, WAXS, GISAXS and GIWAXS Techniques. J. Appl. Crystallogr. 2016, 49 (3), 1107–1114. 10.1107/S1600576716006932. [DOI] [Google Scholar]
- Rodríguez-Carvajal J. Recent Advances in Magnetic Structure Determination by Neutron Powder Diffraction. Phys. B 1993, 192 (1), 55–69. 10.1016/0921-4526(93)90108-I. [DOI] [Google Scholar]
- Denton A. R.; Ashcroft N. W. Vegard’s Law. Phys. Rev. A: At., Mol., Opt. Phys. 1991, 43 (6), 3161–3164. 10.1103/PhysRevA.43.3161. [DOI] [PubMed] [Google Scholar]
- Yang Y.; Fus F.; Pacureanu A.; da Silva J. C.; De Nolf W.; Biot C.; Bohic S.; Cloetens P. Three-Dimensional Correlative Imaging of a Malaria-Infected Cell with a Hard X-ray Nanoprobe. Anal. Chem. 2019, 91 (10), 6549–6554. 10.1021/acs.analchem.8b05957. [DOI] [PubMed] [Google Scholar]
- Mokso R.; Cloetens P.; Maire E.; Ludwig W.; Buffiere J.-Y. Nanoscale zoom tomography with hard X-rays using Kirkpatrick-Baez optics. Appl. Phys. Lett. 2007, 90, 144104. 10.1063/1.2719653. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.