

Abstract
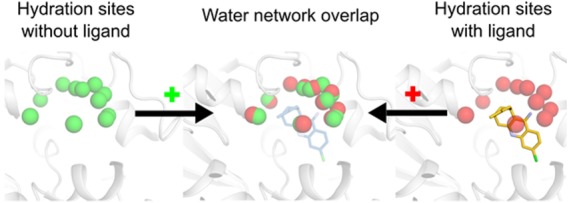
Water plays a major role in ligand binding and is attracting increasing attention in structure-based drug design. Water molecules can make large contributions to binding affinity by bridging protein–ligand interactions or by being displaced upon complex formation, but these phenomena are challenging to model at the molecular level. Herein, networks of ordered water molecules in protein binding sites were analyzed by clustering of molecular dynamics (MD) simulation trajectories. Locations of ordered waters (hydration sites) were first identified from simulations of high resolution crystal structures of 13 protein–ligand complexes. The MD-derived hydration sites reproduced 73% of the binding site water molecules observed in the crystal structures. If the simulations were repeated without the cocrystallized ligands, a majority (58%) of the crystal waters in the binding sites were still predicted. In addition, comparison of the hydration sites obtained from simulations carried out in the absence of ligands to those identified for the complexes revealed that the networks of ordered water molecules were preserved to a large extent, suggesting that the locations of waters in a protein–ligand interface are mainly dictated by the protein. Analysis of >1000 crystal structures showed that hydration sites bridged protein–ligand interactions in complexes with different ligands, and those with high MD-derived occupancies were more likely to correspond to experimentally observed ordered water molecules. The results demonstrate that ordered water molecules relevant for modeling of protein–ligand complexes can be identified from MD simulations. Our findings could contribute to development of improved methods for structure-based virtual screening and lead optimization.
1. Introduction
Water molecules in the binding sites of proteins make important contributions to molecular recognition.1,2 High-resolution crystal structures have revealed that waters frequently mediate protein–ligand interactions.3 Hydrogen bonds formed between ligands and ordered water molecules can be important for the affinity of the complex4,5 as well as for determining substrate specificity.6−8 One remarkable example is the binding site of oligopeptide-binding protein A, in which peptide ligands were found to be coordinated by a large number of ordered water molecules in a buried pocket.8,9 Ligand binding also causes displacement of waters from the binding site surface into the bulk solvent, and introduction of substituents that displace high-energy water molecules can yield remarkable increases of affinity.10−13 For example, liberation of a water molecule from the binding site of p38α MAP kinase resulted in more than a 60-fold improvement of inhibitor activity.14 Understanding the roles of water molecules in ligand binding at the molecular level is gaining increasing attention as it could contribute to more efficient structure-based drug design.2,15
Crystal structures can only provide limited information about water molecules in protein binding sites. If the resolution is too low, it may be difficult to distinguish ordered water molecules, which is clearly illustrated by the fact that 60–70% more water molecules are identified in structures with 1.0 Å resolution compared to those solved at 2.0 Å.16 Furthermore, the solvent network observed at the surface of experimentally determined protein structures may also be artifacts of the crystallization conditions.16,17 Computational techniques have become increasingly used to study water molecules in protein binding sites because such approaches can provide a more complete view of the structure, dynamics, and energetics of the solvent network. Methods for prediction of ordered water molecules in protein binding sites range from empirical and knowledge-based approaches18,19 to more rigorous techniques based on molecular dynamics (MD) or Monte Carlo (MC) simulations.13,20,21 Rapid scoring functions have been demonstrated to reproduce the positions of water molecules observed in crystal structures and identify waters that are likely to be displaced by ligands.19,22−25 Several algorithms for identification of ordered water molecules from MD or MC simulations have been developed, and applications of these have revealed large differences between the water network at the protein surface compared to the bulk solvent.20,21,26 Such methods are computationally demanding but can provide more detailed information about water networks. In addition to predicting positions of ordered waters, atomistic simulations in combination with alchemical free energy methods or inhomogeneous solvation theory27 have been used to estimate energetic contributions to affinity from displacement of water molecules from the protein surface, which provided insights into the driving forces of ligand binding.4,5,26,28,29 These techniques have also been applied in lead optimization by identifying high-energy waters that can be displaced to gain affinity by introducing substituents on a ligand11,26,30 and to characterize the druggability of binding sites.31 Simulation-based methods have mainly focused on the water molecules that are displaced by ligands, whereas those that remain in the binding site after complex formation have received less attention. The water molecules that are not displaced by the ligand reorganize at the surface of the complex or are bound in the interface between the protein and the ligand. These interfacial water molecules, in particular those that bridge hydrogen bonds, can be important for accurate atomic level modeling of protein–ligand complexes and could be utilized to improve virtual screening performance.32−34 However, as waters cannot always be identified in experimentally determined protein structures, there is a need for computational methods that can predict ordered waters in binding sites accurately.
In this work, prediction of ordered water molecules in protein binding sites and the influence of ligand binding on the solvent network were investigated using MD simulations. Particular focus was put on interfacial water molecules that interact with both the protein and ligand after complex formation. Highly populated locations for water molecules (hydration sites) were identified from simulations of experimentally determined structures of 13 protein–ligand complexes in the presence and absence of the cocrystallized ligands. The hydration sites were first used to investigate if ordered water molecules observed in crystal structures could be predicted. The sets of hydration sites derived in the presence and absence of the bound ligands were then compared to assess the influence of binding on the solvent network, which was also complemented with calculations for apo crystal structures. The use of simulation-derived water networks to improve modeling of protein–ligand complexes and applications to drug design will be discussed.
2. Methods
Selection of Proteins and Crystal Structures
A set of 13 crystal structures of protein–ligand complexes (Table 1) that had ≤2.5 Å resolution and at least two water molecules interacting via hydrogen bonds with both protein and ligand was created to study ordered water molecules in binding sites using MD simulations. In this work, this data set will be referred to as the reference crystal structures. High-resolution apo crystal structures with water molecules in the binding site were also available for eight out of 13 proteins. For the remaining five proteins, apo crystal structures were either not available, there were mutations in the binding site or no relevant crystal waters. The binding site water molecules were defined as all water oxygen atoms within 4 Å of any heavy atom of both the ligand and protein in the reference structure. For the apo form, the corresponding region was identified by aligning the apo crystal structure to the reference coordinates in PyMol.35 Electron density maps were inspected visually if these were available from the protein data bank (PDB) or the Uppsala electron density server (http://eds.bmc.uu.se/eds/) to ensure that there was experimental data supporting the binding site waters. Analyses of ordered waters for larger numbers of crystal structures were carried out by first extracting all entries with the same UniProt code and resolution ≤2.5 Å from the PDB. These were then aligned to the reference coordinates, and all structures with RMSD values <1 Å were retained for further analysis.
Table 1. Summary of Results from Clustering of MD Snapshots of the Binding Site Solvent Networks for 13 Protein–Ligand Complexes in the Presence and Absence of Ligands.
| crystal
waters |
hydration sites with
ligand |
||||
|---|---|---|---|---|---|
| protein | PDB codea | binding siteb | bridgingc | presentd | absente |
| acetylcholinesterase | 1e66 (1ea5) | 5 (5/4) | 2 | 10 | 8 (22) |
| coagulation factor VII | 1w7x (1klj) | 19 (12/10) | 4 | 22 | 16 (34) |
| fatty acid binding protein adipocyte | 2nnq (3q6l) | 7 (7/5) | 2 | 9 | 6 (22) |
| glutamate ionotropic receptor, AMPA subunit 2 | 3kgc (4o3b) | 13 (8/7) | 3 | 13 | 11 (25) |
| heat shock protein 90-alpha | 1uyg (1uyl) | 11 (8/7) | 4 | 14 | 10 (19) |
| tyrosine-protein kinase JAK2 | 3lpb | 9 (6/6) | 3 | 11 | 10 (24) |
| poly[ADP-ribose] polymerase-1 | 3l3m | 4 (4/4) | 2 | 11 | 11 (23)f |
| serine/threonine-protein kinase PLK1 | 2owb | 10 (6/5) | 2 | 8 | 6 (16)f |
| protein-tyrosine phosphatase 1B | 2azr (2cm2) | 2 (2/1) | 2 | 7 | 3 (14) |
| GAR transformylase | 1njs | 12 (10/7) | 3 | 19 | 12 (31)f |
| muscle glycogen phosphorylase | 1c8k | 6 (6/5) | 2 | 11 | 11 (21) |
| thrombin | 1ype (2uuf) | 5 (3/1) | 2 | 11 | 4 (22) |
| trypsin I | 2ayw (1s0q) | 10 (5/3) | 4 | 15 | 9 (24) |
| sum: | 113 (82/65) | 35 | 161 | 117 (297) | |
PDB codes of reference crystal structures of the holo and apo (in parentheses) forms.
Number of waters in the binding site of the crystal structure, i.e. within 4 Å of both the protein and ligand. The number of binding site crystal waters that were reproduced by hydration sites derived from simulations carried out with the ligand present/absent, respectively, is shown in parentheses.
Number of crystal waters that bridge protein–ligand interactions, i.e. within 3.3 Å of a polar atom (N or O) of both the ligand and protein.
Number of hydration sites within 4 Å of the protein and ligand from MD simulations carried out in the presence of the ligand.
Number of hydration sites within 4 Å of the protein and ligand from MD simulations carried out in the absence of the ligand after removing sites that overlap with the ligand. The number of hydration sites prior to removing the hydration sites that overlapped with the ligand is shown in parentheses.
This protein was excluded from the analysis of frequently observed ordered waters (Table S2), which resulted in a total of 227 hydration sites.
Molecular Dynamics Simulations
Up to three sets of MD simulations were carried out for each protein. For the 13 holo crystal structures, simulations were performed in the presence and absence of the cocrystallized ligand. An additional set of simulations was also carried out for apo crystal structures of eight proteins. MD simulations were performed in the program Q36 using the OPLS all atom (OPLSAA) force field.37 Force field parameters (OPLSAA_2005) for the ligands were derived using the program hetgrp_ffgen (Schrödinger, LLC, New York, NY, 2014). Prior to the simulations, all nonprotein atoms were removed from the system. The simulations were carried out using spherical boundary conditions. A sphere with an 18 Å radius was centered on the binding site, and the volume not occupied by solute atoms was filled with water molecules. For the simulations carried out in the absence of the ligand, the structures were solvated using a grid of TIP3P water molecules.38 In the simulations carried out with the cocrystallized ligand present, all the structures were solvated using the last snapshot of the simulations performed without the ligand after removing waters that were overlapping with solute atoms. During the simulation, waters at the sphere surface were subjected to radial and polarization restraints according to the SCAAS model.36,39 Nonbonded interactions were truncated at a 10 Å cutoff, and long-range interactions beyond this radius were treated with the local reaction field (LRF) method.40 The SHAKE algorithm was applied to constrain solvent bonds and angles.41 The ionizable residues Asp, Glu, Arg, and Lys within the sphere were assigned protonation states based on their most probable state at pH 7 except in the case of histidines, which were protonated based on the hydrogen bonding network (Table S1). A time step of 1 fs was used, and nonbonded pair lists were updated every 25 steps. Each system was equilibrated for 360 ps, and the temperature was gradually increased to 300 K. A simulation at 300 K was then performed for an additional 8 ns, and 8000 snapshots of the system were saved. The solute heavy atoms were tightly restrained using a harmonic restraint of 100 kcal mol–1 Å–2 to their initial coordinates in order to characterize the solvent network in the binding site.
Identification of Hydration Sites
The clustering method by Young et al.20,26 was used to identify locations of ordered binding site waters based on 8000 snapshots from the MD simulations for each system. The MD snapshots were first aligned based on the (rigid) protein coordinates, and then all water molecules within 1 Å of each water were identified based on the oxygens. The water molecule with the highest number of neighbors was then defined as a “hydration site”, and all water molecules belonging to this site were excluded in the following iteration. The occupancy of a hydration site was defined as the fraction of MD simulation snapshots that contributed to it. This procedure was then repeated until the remaining hydration sites had occupancies <0.3. The convergence of the protocol was assessed by clustering two sets of independent MD simulation trajectories for three proteins (heat shock protein 90-alpha, acetylcholinesterase, and fatty acid binding protein adipocyte), which resulted in 97% overlapping hydration sites. The results obtained by clustering were compared to random placement of hydration sites, which was estimated based on three independent sets of binding site water molecules generated using the grid-based solvation algorithm in the program Q.36
3. Results
Prediction of Ordered Water Molecules Based on MD Simulations of Protein–Ligand Complexes
We first investigated if hydration sites derived from MD simulations could be used to predict positions of water molecules present in crystal structures of 13 protein–ligand complexes (Table 1). All selected proteins were drug targets, and, in each case, there was a cocrystallized ligand and several water molecules in the binding pocket. The binding site water molecules, i.e. those interacting with both the protein and ligand, were defined as all waters with oxygens within 4 Å of both the cocrystallized ligand and the protein. This resulted in 113 experimentally observed binding site waters that were used to benchmark predictions based on explicit solvent MD simulations.
All atom MD simulations were carried out with the heavy atoms of the proteins and ligands held rigid by using restraints to the crystal structure coordinates. The binding site water molecules were clustered based on snapshots from an 8 ns MD simulation for each complex using the method proposed by Young et al.20,26 The number of identified hydration sites for the protein–ligand complexes is summarized in Table 1. A total of 161 hydration sites were obtained in the binding site region, which was 42% more than the number of crystal waters. The hydration sites were then compared to the crystal waters (Tables 1 and 2). In order to avoid boundary effects, the 223 hydration sites within 5 Å of the complex were included in these calculations. A crystal water was considered to be correctly predicted if it was within 1 Å of a hydration site. This criterion led to 82 correctly predicted binding site crystal waters, corresponding to 73% of those observed experimentally. This result can be compared to that obtained by random placement of hydration sites, which yielded an average of 9% reproduced crystal waters. The percentage of predicted crystal waters ranged from 50% to 100% for the 13 proteins. Analysis of the protein structures suggested that a smaller fraction of the crystal waters was reproduced for flat binding sites with solvent exposed regions, whereas the results were typically better for proteins that had defined binding pockets with waters occupying deep cavities. The simulation of Trypsin I (PDB code: 2ayw(42)) had the smallest fraction of reproduced crystal waters. Visual inspection of the binding site revealed that two identical ligands had been cocrystallized in this case, which may be an artifact of crystallization. The MD simulations were repeated for a second high-resolution structure (PDB code: 1c1t(43)), resulting in a slightly improved percentage of reproduced crystal waters (63%). Examples of predicted hydration sites for three different protein–ligand complexes are shown in Figure 1. All crystal waters present in the binding site were correctly predicted for five of the complexes, e.g. the muscle glycogen phosphorylase (Figure 1A). For heat shock protein 90-alpha (Figure 1B) and glutamate ionotropic receptor AMPA subunit 2 (Figure 1C) the hydration sites reproduced 8 out of 11 and 13 crystal waters, respectively.
Table 2. Percent of the Binding Site and Bridging Crystal Waters That Were Reproduced by Hydration Sites Derived from the Holo and Apo Crystal Structures.
| cocrystallized ligand |
||
|---|---|---|
| crystal waters | presenta | absentb |
| binding site (holo)c | 73% | 58% |
| bridging (holo)d | 91% | 77% |
| binding site (apo)e | 72% | |
The cocrystallized ligand was present in MD simulation of the holo binding site.
The cocrystallized ligand was absent in MD simulation of the holo binding site.
Hydration sites were compared to crystal waters within 4 Å of both the protein and ligand.
Hydration sites were compared to crystal waters within 3.3 Å of a polar atom (N and O atoms) of both the ligand and protein.
Hydration sites were compared to crystal waters within 4 Å of both the apo protein and the ligand from the holo structure, which was placed into the binding site by aligning the two structures.
Figure 1.

Examples of hydration sites derived from MD simulations and comparison to crystal waters for three protein–ligand complexes. Hydration sites derived in the presence (A-C) and absence (D-F) of the ligands are shown for muscle glycogen phosphorylase (A, D), heat shock protein 90-alpha (B, E), and glutamate receptor ionotropic, AMPA 2 (C, F). In (D-F), the cocrystallized ligands are shown, but they were not included in the MD simulations. Hydration sites and crystal waters are shown as transparent green and red spheres, respectively. The proteins are shown as gray cartoons, and the cocrystallized ligands are depicted in sticks.
In the next step, the occupancies of the hydration sites were analyzed. The occupancies of the hydration sites ranged from 0.3 to 1.0, and the average over all complexes was 0.71. We then examined if the probability that hydration sites overlapped with crystal waters was correlated with occupancy. Whereas the preceding paragraph focused on the percentage of the crystal waters that were reproduced by hydration sites (73%), we instead calculated the percentage of the hydration sites that corresponded to crystal waters. The hydration sites were divided into five groups representing different occupancy intervals between 0.3 and 1.0. For each group, the predictive rate (i.e., the percentage of the hydration sites that were present in the crystal structures) was calculated (Figure 2A). The lowest percentage of verified hydration sites (26%) was obtained in the interval with the lowest occupancy (<0.44). The highest percentage was found for the group of hydration sites with the highest occupancies (≥0.86), in which 51 out of the 68 (75%) corresponded to the same positions as crystal waters. Overall, there was a strong linear correlation between the occupancies of the hydration sites and the percentage of the hydration sites that corresponded to crystal waters (R2 = 0.85). The overall predictive rate could hence be improved by increasing the occupancy cutoff. For example, with a cutoff of ≥0.5, the number of hydration sites was reduced from 161 to 114, which was close to the total number of crystal waters (113). The number of reproduced crystal waters was then reduced from 82 to 73, but the predictive rate increased from 51% to 64%.
Figure 2.

(A) The percentage of the hydration sites that reproduced crystal waters for different levels of hydration site occupancy from simulations carried out with the ligand present (red bars) and absent (blue bars), respectively. (B) The percentage of the hydration sites that reproduced frequently observed crystal waters for different levels of hydration site occupancy from simulations carried out with the ligand absent. The number of crystal structures per protein is shown in Table S2.
Prediction of Ordered Water Molecules in Protein–Ligand Complexes Based on MD Simulations Carried out with the Ligand Absent
MD simulations of the 13 crystal structures were also carried out with the ligand removed from the binding site. These calculations were performed to investigate if crystal water positions could be predicted by hydration sites derived in the absence of the ligand and, more generally, to assess the influence of the ligand on the solvent network. A total of 297 hydration sites were identified from clustering of MD snapshots (Table 1). An additional 136 hydration sites were hence obtained compared to the simulations carried out for the complexes, and a majority of these were located in the volume occupied by the ligand. The number of hydration sites that did not overlap with the cocrystallized ligand (defined as 2.2 Å within its heavy atoms) was 117, which can be compared to the 161 hydration sites identified with the ligand present in the simulations (Table 1 and Figure 3). Analogous to the analysis made for the simulations carried with ligands bound to the proteins, the hydration sites obtained in the absence of the ligand were compared to the experimental structures of the complexes, and 65 of the 113 crystal waters (58%) were reproduced, which was only 17 less than for the simulations carried out in the presence of the ligand (Tables 1 and 2). This can be compared to the result obtained from random placement of hydration sites, which yielded an average of 9% reproduced crystal waters. Interestingly, 63 of the 65 reproduced crystal waters were also identified from the simulations carried out in the presence of the ligand. Examples of predicted hydration sites for three different proteins are shown in Figure 1D-F. Five out of six crystal waters were reproduced by hydration sites in the muscle glycogen phosphorylase structure (Figure 1D). Seven crystal waters were correctly predicted both for the crystal structures of heat shock protein 90-alpha (Figure 1E) and glutamate ionotropic receptor AMPA subunit 2 (Figure 1F) complexes of a total of 11 and 13, respectively. In all three cases, one less crystal water was predicted compared to the set of hydration sites derived from simulations of the complexes (Figure 1A-C).
Figure 3.
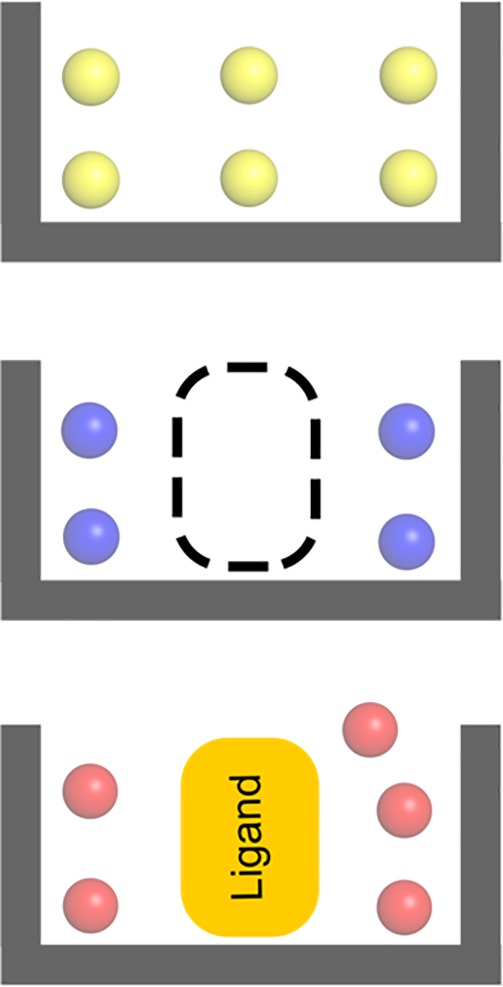
Illustration of hydration sites derived from different simulation sets. Hydration sites obtained from simulations carried out in the absence of the cocrystallized ligand are depicted as yellow spheres. The subset of blue spheres corresponds to hydration sites that are not overlapping with the cocrystallized ligand. The black dashed lines represent the volume occupied by the cocrystallized ligand. Hydration sites derived from simulations with the cocrystallized ligand present are depicted as red spheres.
The analysis of hydration site occupancy was also repeated for the simulations carried out in the absence of the ligand. In order to allow a comparison to the simulations carried out in the presence of the ligand, only the 117 hydration sites that did not overlap with the ligand were included. The average occupancy for this subset of hydration sites was 0.62, which was lower than for the corresponding simulations of the complexes (0.71). There was a strong correlation between the MD derived occupancies of the hydration sites and the predictive rates (R2 = 0.95, Figure 2A). The correlation was slightly stronger than for the simulations carried out with the ligands present, but the number of hydration sites in the interval with the highest occupancies decreased from 86 to 25. Of 25 hydration sites with occupancies ≥0.86, 88% overlapped with crystal waters.
Crystal structures of the same protein often contain varying numbers of water molecules depending on resolution, bound ligands, and experimental conditions.3,16 To investigate if the hydration sites were reoccurring, we extended the analysis for proteins with >20 available structures with resolution ≤2.5 Å and an RMSD to the initially selected reference structure of <1.0 Å. The total number of crystal structures for the ten proteins that fulfilled these criteria ranged from 25 to 371 (Table S2). The hydration sites derived from simulations carried out with the ligand absent were used in this case as water molecules could occupy any part of the binding site. In order to identify frequently observed ordered waters, a hydration site was required to overlap with a crystal water in at least 10% of the structures. Of the 227 hydration sites identified in the absence of the ligand for the ten proteins (Table 1 and Table S2), 81 predicted reoccurring crystal waters. The correlation between the occupancies of the hydration sites and the predictive rates (R2 = 0.55, Figure 2B) was weaker compared to that obtained for the reference crystal structures (Figure 2A). An increase in the percentage of correctly predicted crystal waters compared to the other intervals was found for the sites with occupancies ≥0.86. In this group, 27 out of the 41 hydration sites (66%) were verified by frequently observed crystal waters. Nine of the 24 hydration sites for Trypsin I were found in at least 37 other structures, and one of these was even present in 363 cases. The hydration site with the highest occupancy was present in 134 crystal structures and was involved in hydrogen bonds to several different ligands.
Hydration Sites and Crystal Waters That Bridge Protein–Ligand Interactions
As water molecules that bridge receptor–ligand interactions are of particular interest in drug design, we analyzed if such crystal waters were predicted by hydration sites. A bridging crystal water was defined as an oxygen atom within 3.3 Å of a polar atom (N or O) of the ligand and the protein, which resulted in a total of 35 waters for the 13 studied complexes. The results for the hydration sites with occupancies >0.3 from simulations with the ligand present and absent are summarized in Figure 4. Almost all bridging crystal waters, 32 out of 35 (91%), were captured by the hydration sites derived in the presence of the ligand, whereas 27 out of 35 (77%) were reproduced by hydration sites obtained with the ligand absent. In both cases, the percentages of reproduced bridging crystal waters were higher than for all crystal waters (Table 2). Interestingly, the average occupancies for the predicted bridging waters were also higher than the average for all hydration sites. The average occupancies for the hydration sites representing bridging crystal waters were 0.91 and 0.78 for the simulations carried out in the presence and absence of the ligand, respectively, which can be compared to 0.71 and 0.62 for all hydration sites. Finally, we analyzed if hydration sites that were not observed in the crystal structures bridged protein–ligand interactions. Among the nine hydration sites that fulfilled these criteria, two were in close vicinity of a crystal water (1.2 and 1.4 Å, respectively). We visually inspected the electron density maps for the remaining seven cases, but there was no clear evidence for ordered waters in the same positions as the hydration sites.
Figure 4.
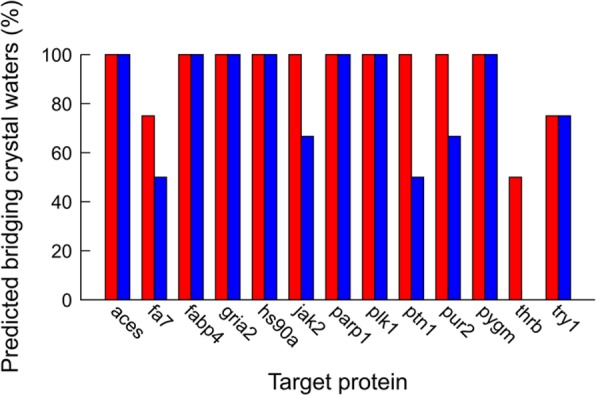
Percent correctly predicted crystal waters that bridge protein–ligand interactions using hydration sites derived from simulations with the ligand present (red) and absent (blue). Abbreviations for proteins: aces, acetylcholinesterase; fa7, coagulation factor VII; fabp4, fatty acid binding protein adipocyte; gria2, glutamate ionotropic receptor AMPA subunit 2; hs90a, heat shock protein alpha-90; jak2, tyrosine protein kinase JAK2; parp1, poly[ADP-ribose] polymerase-1; plk1, serine/threonine-protein kinase PLK1; ptn1, protein-tyrosine phosphatase 1B; pur2, GAR transformylase; pygm, muscle glycogen phosphorylase; thrb, thrombin; try1, trypsin I.
As the analysis of the water network in the binding site was based on a single structure for each protein, it was possible that the hydration sites could act as bridging waters in other crystal structures. For this reason, we extended our analysis to 1312 high-resolution crystal structures from the PDB (Table S2). Bridging crystal waters in these structures were compared to the hydration sites obtained from the simulations carried out with the ligand absent. Two observations were made from this analysis. First, crystal water molecules reproduced by hydration sites were found to stabilize different ligand scaffolds in several cases (Figure 5). Second, hydration sites that did not overlap with bridging waters in the reference structure were found to hydrogen bond to both the protein and ligand in other crystal structures. Six examples of hydration sites derived from MD simulations of the reference crystal structure that were found to bridge protein–ligand interactions in the extended set of structures are shown in Figure 6. In all these cases, the hydration sites overlapped with the cocrystallized ligand in the reference structure and could hence only be identified from the simulations carried out with the ligand absent.
Figure 5.

Examples of hydration sites derived from the reference crystal structures (Table 1) that reproduced crystal waters forming hydrogen bonds with diverse ligands in other experimental structures. Two crystal structures for three different proteins are shown: (A, B) Fatty acid binding protein adipocyte (PDB codes: 3fr4 and 3p6h); (C, D) Heat shock protein 90-alpha (PDB codes: 1uyh and 3b28); (E, F) Muscle glycogen phosphorylase (PDB codes: 1uzu and 3ebo). Hydration sites and crystal waters are shown as transparent green and red spheres, respectively. The proteins are shown as gray cartoons with selected residues and the cocrystallized ligands in sticks. Hydrogen bonds are depicted as black dashed lines.
Figure 6.

Examples of hydration sites derived from the reference crystal structure that bridged hydrogen bonds between the protein and ligand in other experimental structures. In each case, the hydration site did not bridge hydrogen bonds between the protein and ligand in the reference crystal structure. Two crystal structures for three different proteins are shown: (A, B) Tyrosine-protein kinase JAK2 (PDB codes: 4d0w and 4zim); (C, D) Thrombin (PDB codes: 2hj6 and 3p17); (E, F) Trypsin I (PDB codes: 1c1t and 3a7y). Hydration sites and crystal waters are shown as transparent green and red spheres, respectively. The proteins are shown as gray cartoons with selected residues in sticks. The cocrystallized ligands are depicted in sticks with either blue (reference crystal structure) or yellow (other crystal structure) carbon atoms. Hydrogen bonds are depicted as black dashed lines.
Comparisons of Binding Site Hydration Networks in the Presence and Absence of the Ligand
The initial results showed that there was an overlap between hydration sites derived from simulations of the holo structures in the absence and presence of the cocrystallized ligands. In order to further quantify similarities and perturbations of the water network upon ligand binding, the hydration sites obtained from simulations of these two states and apo structures were compared.
The hydration sites obtained within 4 Å of both the protein and ligand from simulations of the holo structure in the absence of the ligand were first compared to those identified within 5 Å of the complex. A larger radius in the latter case was used because a pair of hydration sites was considered to be overlapping if they were <1 Å from each other, which makes inclusion of sites within 5 Å necessary to avoid boundary effects. For this reason, the degree of overlap between the two sets of hydration sites differed depending on the selection of reference point. Of the 297 hydration sites with occupancies >0.3 derived in the absence of the ligand, 94 overlapped with the sites obtained with the ligand present, which corresponded to 80% of the 117 sites that were not overlapping with the ligand. Conversely, if the hydration sites derived in the presence of the ligand were compared to the set obtained with the ligand absent, 100 out of 161 hydration sites (62%) overlapped. The fraction of overlapping hydration sites further increased if only those with high occupancies were considered. For the 25 hydration sites with occupancies ≥0.86 that were derived with the ligand absent, all overlapped with hydration sites determined with the ligand present. Of the 68 hydration sites with occupancies ≥0.86 from the simulations with the ligand present, 56 (82%) were found among the hydration sites derived from simulations with the ligand absent. Remarkably, all of the hydration sites obtained with the ligand present were also present in the absence of the ligand in the case of acetylcholinesterase (Figure 7).
Figure 7.
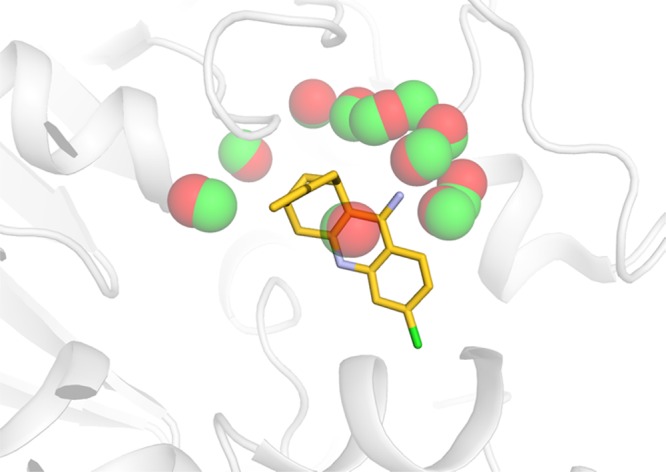
Overlap between hydration networks from simulations carried out in the presence (red) and absence (green) of the ligand for acetylcholinesterase. The hydration sites determined with the ligand present and absent are shown as red and green spheres, respectively. The protein is shown as a gray cartoon, and the cocrystallized ligand is shown in sticks.
Histograms describing how the hydration sites were distributed over different occupancy intervals were generated to investigate how ligand binding influenced the binding site water networks (Figure 8). The hydration sites obtained with the ligand absent had the largest number of occurrences in lowest occupancy interval and were almost evenly distributed over the other. For the subset of (117) sites that did not overlap with the ligand, the largest number of sites was found in the interval with the lowest occupancies, whereas the number of sites in the other intervals was relatively similar. In the presence of the ligand, the number of hydration sites with occupancies <0.86 decreased compared to the set obtained in the absence of the ligand, whereas it was slightly higher for the highest interval (0.86–1.00). If the subsets of hydration sites in the region not occupied by the ligand were compared, the only marked difference was found in the 0.86–1.00 interval, in which there was almost 3-fold as many hydration sites from the simulations carried out with the ligand present. In summary, hydration sites derived in the absence of the ligand were to some extent preserved in the presence of the ligand, but complex formation also led to an increase of the occupancies and an additional number of ordered water molecules.
Figure 8.
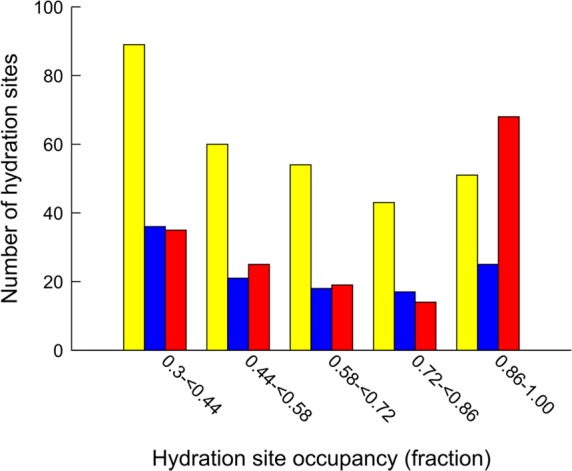
Distribution of the number of hydration sites over different occupancy intervals from simulations with the ligand absent and present. Yellow bars represent all hydration sites obtained with the ligands absent, whereas the blue bars show the distribution after removing sites that overlapped with the ligands. Red bars represent hydration sites obtained with the ligands present.
Apo crystal structures for the 13 studied proteins were also analyzed if such were available. There were high-resolution crystal structures with ≤2.5 Å resolution for eight out of the 13 proteins in the PDB. MD simulations of the eight apo structures identified a total of 187 hydration sites in the region occupied by the ligand in the holo structure. Of the 108 crystal waters, 78 (72%) were reproduced by a hydration site (Table S3), which was the same number as obtained for the simulations carried out for the corresponding eight complexes. The average occupancy was 0.61, which can be compared to 0.71 that was obtained using the holo structure with the ligand present. To investigate if the hydration sites derived for the apo structures in the absence of the ligand were captured by the simulation of the holo form, these two sets of hydration sites were compared. Of the 187 hydration sites derived from simulations of the apo structure, 99 (53%) were also present among the sites derived from the holo structure in the absence of the ligand. For hydration sites with high occupancy (≥0.86), 76% overlapped if the sets from the apo and holo structures were compared. As expected, large differences in the solvent network were observed where conformational reorganization led to changes in the shape of the binding site. For example, in the case of heat shock protein 90-alpha (Figure 9A) a local conformational change upon ligand binding led to reorganization of the water network in one subpocket, whereas the hydration sites were conserved in the rest of the binding pocket. For protein-tyrosine phosphatase 1B (Figure 9B) reorganization of the loop regions led to changes in the hydration site network in the entire binding site.
Figure 9.

Examples of hydration sites derived from MD simulations of apo and holo crystal structures for two proteins. (A) Hydration sites derived for heat shock protein 90-alpha and (B) protein-tyrosine phosphatase 1B. The protein structures of the apo and holo structures are depicted as gray cartoons, and the regions undergoing major conformational changes are colored red. Hydration sites that were present in simulations of both apo and holo structures are shown as red spheres. Hydration sites that were unique for the apo and holo structures are represented as green and blue spheres, respectively.
4. Discussion
The aims of this work were to assess if ordered water molecules in protein–ligand complexes could be predicted based on MD simulations and to investigate the impact of ligands on binding site solvent networks. An important first step toward understanding the roles of water in ligand binding is to accurately predict ordered water molecules in experimentally determined structures. We demonstrated that hydration sites derived from simulations of 21 high resolution crystal structures, representing both apo and holo forms of 13 proteins, reproduced 73% of the experimentally observed waters. Our results are similar to those previously obtained based on clustering of MD snapshots13 and empirical scoring functions such as AcquaAlta18 and WaterDock,19 which identified between 62% to 81% of the crystal waters. A compelling result, which was made possible by the use of MD simulations, was the strong correlation between the occupancy of the hydration sites and the probability of observing these in crystal structures. The vast majority of the hydration sites (75%) with the highest occupancies was also present in the experimentally determined structures. These results are in agreement with the work of Sun et al., which in addition demonstrated a correlation between the B-factors of crystal waters and the probability that hydration sites reproduced these.13 Locations where binding site water molecules are frequently present in MD simulations are hence likely to be observed experimentally, and, conversely, there is a high probability that crystal waters with low B-factors are predicted by hydration sites. Two caveats of the method used in this work should be noted. First, it may be challenging to obtain accurate models of the water network if the solvent molecules cannot diffuse in and out of the binding site in the simulations, as demonstrated by Maurer et al. for the oligopeptide-binding protein A.44 Exchange with the bulk solvent may be limited for enclosed binding sites and could also be influenced by the tight restraints on the protein heavy atoms used in the MD simulations. Second, a large number of hydration sites did not correspond to crystal waters. This discrepancy can partly be explained by the resolution of the structures, which strongly influences the number of identified waters.16 In addition, hydration sites were considered even if they had occupancies as low as 0.3, which may not be detected as ordered waters by crystallography. In agreement with this hypothesis, there was a consistent increase of the percentage of verified hydration sites if only subsets with high occupancies were considered. In prospective applications, the fraction of hydration sites that will not overlap with crystal waters could be reduced by simply increasing the occupancy cutoff but at the expense of reducing the total number of reproduced crystal waters. Overall, our results demonstrate that hydration sites derived from MD simulations can predict ordered water molecules in protein binding sites with high accuracy.
Previous studies have demonstrated that including ordered waters in molecular docking calculations can enhance virtual screening performance. Improvements of binding mode predictions and ligand enrichment have been observed using several different algorithms, but performance is still target dependent.32−34,45−48 Although relatively short MD simulations were required to obtain maps of the solvent network, high-throughput analysis of complexes would be computationally expensive. We hypothesized that waters interacting with the protein and ligand in complexes would in some cases be tightly bound even if the ligand was not present. If this was the case, MD simulations carried out in the absence of the ligand could be used to identify ordered waters relevant for modeling of complexes. Although the percentage of crystal waters that were reproduced by hydration sites was lower than for the simulations of the complexes, it was encouraging to find that a majority (58%) could still be identified. The hydration sites with high occupancies were very likely to be present in the crystal structures of the complexes, which is remarkable considering that the ligand was not present in the simulation. In order for identified hydration sites to be useful for prospective ligand discovery, the water molecules at these locations should also be present in complexes with different ligand scaffolds. Interestingly, analysis of up to hundreds of crystal complexes per protein showed that an average of eight hydration sites per structure were frequently observed.
Water molecules frequently bridge protein–ligand interactions and the design of ligands to form such hydrogen bonds has successfully been utilized in structure-based drug discovery.2 For example, HIV-1 protease inhibitors were found to interact with the same active site water molecule, which was demonstrated to contribute favorably to binding by MD free energy calculations.5 In a prospective study, Powers et al. took advantage of ordered waters in a docking screen against the binding site of AmpC β-lactamase, leading to the discovery of an inhibitor that was confirmed to interact with the included water molecules in a subsequently solved crystal structure.49 In this work, ∼2.7 bridging waters per crystal structure were identified, which agrees well with the average of three obtained by Lu et al. for a larger set of protein–ligand complexes.3 Almost all bridging crystal waters (91%) were reproduced by hydration sites derived from MD simulations of the complexes, which was an improvement over the results obtained for the full set of crystal waters (73%). A similar increase of accuracy for bridging crystal waters was obtained with the empirical method AquaAlta.18 This result may be explained by the fact that bridging waters will form two or more hydrogen bonds to the protein–ligand complex. Analyses of crystal structures have revealed that waters with several hydrogen bonds to the complex have low B-factors, indicating low mobility.3,50 In agreement with this observation, hydration sites that bridged hydrogen bond interactions between the protein and ligand had high occupancies, which we in turn found to increase the probability of predicting crystal waters. A remarkable finding was that a high percentage of the bridging crystal waters (77%) was also reproduced if hydration sites were derived in the absence of the ligand. As many of these hydration sites maintained high occupancies and reoccurred in multiple experimental structures, our results show that a large fraction of the bridging waters is relatively ordered even if the ligand is not present. This result is in agreement with the analysis of apo and holo crystal structures by Garcia-Sosa et al., which showed that waters with low B-factors and many protein interactions are unlikely to be displaced.24 In lead optimization, it may hence be preferential to design ligands that interact with ordered waters that form multiple hydrogen bonds rather than attempting to displace them with a substituent.
Protein–ligand association involves displacement of water molecules from the binding site and leads to reorganization of the water network on the surface created by the complex. MD simulation with explicit solvent is currently the most rigorous approach to model the influence of water on ligand binding and can, in combination with free energy calculations, be used to estimate the associated energetic contributions to affinity.4,12,51−53 However, calculation of binding free energies by alchemical transformations is still computationally demanding and complex to carry out efficiently. More rapid and approximate methods have been limited to implicit solvent models that cannot capture the effects of interactions with ordered water molecules. The clustering algorithm by Young et al. reduces an essentially infinite number of possible solvent configurations in a binding site to a map of the most ordered waters.20,26 In this work, MD-derived hydration sites were used to quantify the influence of ligands on the binding site solvent network. For the 13 studied proteins, the number of hydration sites per complex was reduced from an average of 23 to 12 upon ligand binding. Liberation of ordered water molecules into the bulk solvent is considered to be one of the major driving forces of ligand binding, and hydration site displacement energies, e.g. calculated from inhomogeneous solvation theory, have been shown to reproduce structure–activity relationships for congeneric ligand series.26 Approaches for inclusion of receptor desolvation energies based on MD-derived water networks were recently implemented in the WSCORE and DOCK scoring functions.54,55 Sun et al.13 and Balius et al.54 incorporated hydration site displacement energies in the molecular docking program DOCK. Some improvements of pose prediction were obtained, but there was only a moderate enhancement of virtual screening performance.13,54 This lack of improvement could in part be due to the fact that none of the two scoring functions took energetic contributions from interactions with hydration sites at the interface between the protein and the ligand into account. As demonstrated by Krimmer et al., networks of ordered water molecules on the surface of the complex can play a major role in determining ligand binding thermodynamics.56 In this region, we identified an average of 12 hydration sites per binding site. More than half of these interfacial water molecules were located at the same positions both in the absence and presence of the ligand, suggesting that the protein to a large extent will dictate the locations of ordered waters. It should be noted that there was also an average of five hydration sites interacting with both the protein and ligand that were only present in the simulations carried out for the complex. Furthermore, a large increase of the average occupancies for the interfacial waters was observed in the MD simulations, which will likely counteract the gain of free energy from displacing water molecules from the pocket. In order to obtain a more accurate description of solvent effects on ligand binding in scoring functions, both displacement and hydrogen bonding to discrete waters as well as a description of the contributions from reorganization of the solvent network on the surface of the complex should be taken into account.
The results presented in this work illustrate the importance of ordered waters in molecular recognition. Our results support that hydration sites derived from a single simulation carried out in the absence of the bound ligand can be used to identify ordered waters involved in ligand recognition, and this approach can be applied to a broad range of therapeutic targets. We envision that information extracted from MD-derived binding site solvent networks can be used to improve atomic resolution modeling of protein–ligand complexes. For example, the hydration sites could be utilized to solvate complexes of a protein with different compounds prior to running MD simulations. A better initial description of the binding site hydration network may improve the accuracy of structure refinement and free energy calculations. Hydration sites can also be included in molecular docking algorithms to enhance virtual screening performance or guide lead optimization. Our work can thereby contribute to the development of rapid and more accurate methods for structure-based drug discovery.
Acknowledgments
This project has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement no. 715052). This work was also supported by the Swedish e-Science Research Center (SeRC), the Swedish Research Council (2013-5708), the Göran Gustafsson Foundation, and the Science for Life Laboratory. Computational resources were provided by the Swedish National Infrastructure for Computing (SNIC).
Glossary
ABBREVIATIONS
- MD
molecular dynamics
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jcim.7b00520.
Table S1, protonation states of histidine residues; Table S2, summary of number of crystal structures per protein; Table S3, summary of results for eight apo crystal structures (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Ladbury J. E. Just Add Water! The Effect of Water on the Specificity of Protein-Ligand Binding Sites and Its Potential Application to Drug Design. Chem. Biol. 1996, 3, 973–980. 10.1016/S1074-5521(96)90164-7. [DOI] [PubMed] [Google Scholar]
- Spyrakis F.; Ahmed M. H.; Bayden A. S.; Cozzini P.; Mozzarelli A.; Kellogg G. E. The Roles of Water in the Protein Matrix: A Largely Untapped Resource for Drug Discovery. J. Med. Chem. 2017, 60, 6781–6827. 10.1021/acs.jmedchem.7b00057. [DOI] [PubMed] [Google Scholar]
- Lu Y. P.; Wang R. X.; Yang C. Y.; Wang S. M. Analysis of Ligand-Bound Water Molecules in High-Resolution Crystal Structures of Protein-Ligand Complexes. J. Chem. Inf. Model. 2007, 47, 668–675. 10.1021/ci6003527. [DOI] [PubMed] [Google Scholar]
- Lu Y. P.; Yang C. Y.; Wang S. M. Binding Free Energy Contributions of Interfacial Waters in HIV-1 Protease/Inhibitor Complexes. J. Am. Chem. Soc. 2006, 128, 11830–11839. 10.1021/ja058042g. [DOI] [PubMed] [Google Scholar]
- Li Z.; Lazaridis T. Thermodynamic Contributions of the Ordered Water Molecule in HIV-1 Protease. J. Am. Chem. Soc. 2003, 125, 6636–6637. 10.1021/ja0299203. [DOI] [PubMed] [Google Scholar]
- Levinson N. M.; Boxer S. G. A Conserved Water-Mediated Hydrogen Bond Network Defines Bosutinib’s Kinase Selectivity. Nat. Chem. Biol. 2014, 10, 127–132. 10.1038/nchembio.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nissink J. W. M.; Bista M.; Breed J.; Carter N.; Embrey K.; Read J.; Winter-Holt J. J. MTH1 Substrate Recognition-an Example of Specific Promiscuity. PLoS One 2016, 11, e0151154. 10.1371/journal.pone.0151154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tame J. R. H.; Murshudov G. N.; Dodson E. J.; Neil T. K.; Dodson G. G.; Higgins C. F.; Wilkinson A. J. The Structural Basis of Sequence-Independent Peptide Binding by OppA Protein. Science 1994, 264, 1578–1581. 10.1126/science.8202710. [DOI] [PubMed] [Google Scholar]
- Tame J. R. H.; Sleigh S. H.; Wilkinson A. J.; Ladbury J. E. The Role of Water in Sequence-Independent Ligand Binding by an Oligopeptide Transporter Protein. Nat. Struct. Biol. 1996, 3, 998–1001. 10.1038/nsb1296-998. [DOI] [PubMed] [Google Scholar]
- Lam P. Y. S.; Jadhav P. K.; Eyermann C. J.; Hodge C. N.; Ru Y.; Bacheler L. T.; Meek J. L.; Otto M. J.; Rayner M. M.; Wong Y. N.; Chang C. H.; Weber P. C.; Jackson D. A.; Sharpe T. R.; Ericksonviitanen S. Rational Design of Potent, Bioavailable, Nonpeptide Cyclic Ureas as HIV Protease Inhibitors. Science 1994, 263, 380–384. 10.1126/science.8278812. [DOI] [PubMed] [Google Scholar]
- Mason J. S.; Bortolato A.; Weiss D. R.; Deflorian F.; Tehan B.; Marshall F. H. High End GPCR Design: Crafted Ligand Design and Druggability Analysis Using Protein Structure, Lipophilic Hotspots and Explicit Water Networks. In Silico Pharmacol. 2013, 1, 23. 10.1186/2193-9616-1-23. [DOI] [Google Scholar]
- Michel J.; Tirado-Rives J.; Jorgensen W. L. Energetics of Displacing Water Molecules from Protein Binding Sites: Consequences for Ligand Optimization. J. Am. Chem. Soc. 2009, 131, 15403–15411. 10.1021/ja906058w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H. Z.; Zhao L. F.; Peng S. M.; Huang N. Incorporating Replacement Free Energy of Binding-Site Waters in Molecular Docking. Proteins: Struct., Funct., Genet. 2014, 82, 1765–1776. 10.1002/prot.24530. [DOI] [PubMed] [Google Scholar]
- Liu C. J.; Wrobleski S. T.; Lin J.; Ahmed G.; Metzger A.; Wityak J.; Gillooly K. M.; Shuster D. J.; McIntyre K. W.; Pitt S.; Shen D. R.; Zhang R. F.; Zhang H. J.; Doweyko A. M.; Diller D.; Henderson I.; Barrish J. C.; Dodd J. H.; Schieven G. L.; Leftheris K. 5-Cyanopyrimidine Derivatives as a Novel Class of Potent, Selective, and Orally Active Inhibitors of P38 Alpha Map Kinase. J. Med. Chem. 2005, 48, 6261–6270. 10.1021/jm0503594. [DOI] [PubMed] [Google Scholar]
- de Beer S. B.; Vermeulen N. P.; Oostenbrink C. The Role of Water Molecules in Computational Drug Design. Curr. Top. Med. Chem. 2010, 10, 55–66. 10.2174/156802610790232288. [DOI] [PubMed] [Google Scholar]
- Carugo O.; Bordo D. How Many Water Molecules Can Be Detected by Protein Crystallography?. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1999, 55, 479–483. 10.1107/S0907444998012086. [DOI] [PubMed] [Google Scholar]
- Sondergaard C. R.; Garrett A. E.; Carstensen T.; Pollastri G.; Nielsen J. E. Structural Artifacts in Protein-Ligand X-Ray Structures: Implications for the Development of Docking Scoring Functions. J. Med. Chem. 2009, 52, 5673–5684. 10.1021/jm8016464. [DOI] [PubMed] [Google Scholar]
- Rossato G.; Ernst B.; Vedani A.; Smiesko M. AcquAalta: A Directional Approach to the Solvation of Ligand-Protein Complexes. J. Chem. Inf. Model. 2011, 51, 1867–1881. 10.1021/ci200150p. [DOI] [PubMed] [Google Scholar]
- Ross G. A.; Morris G. M.; Biggin P. C. Rapid and Accurate Prediction and Scoring of Water Molecules in Protein Binding Sites. PLoS One 2012, 7, e32036. 10.1371/journal.pone.0032036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young T.; Abel R.; Kim B.; Berne B. J.; Friesner R. A. Motifs for Molecular Recognition Exploiting Hydrophobic Enclosure in Protein-Ligand Binding. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 808–813. 10.1073/pnas.0610202104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel J.; Tirado-Rives J.; Jorgensen W. L. Prediction of the Water Content in Protein Binding Sites. J. Phys. Chem. B 2009, 113, 13337–13346. 10.1021/jp9047456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymer M. L.; Sanschagrin P. C.; Punch W. F.; Venkataraman S.; Goodman E. D.; Kuhn L. A. Predicting Conserved Water-Mediated and Polar Ligand Interactions in Proteins Using a K-Nearest-Neighbors Genetic Algorithm. J. Mol. Biol. 1997, 265, 445–464. 10.1006/jmbi.1996.0746. [DOI] [PubMed] [Google Scholar]
- Amadasi A.; Surface J. A.; Spyrakis F.; Cozzini P.; Mozzarelli A.; Kellogg G. E. Robust Classification of ″Relevant″ Water Molecules in Putative Protein Binding Sites. J. Med. Chem. 2008, 51, 1063–1067. 10.1021/jm701023h. [DOI] [PubMed] [Google Scholar]
- Garcia-Sosa A. T.; Mancera R. L.; Dean P. M. Waterscore: A Novel Method for Distinguishing between Bound and Displaceable Water Molecules in the Crystal Structure of the Binding Site of Protein-Ligand Complexes. J. Mol. Model. 2003, 9, 172–182. 10.1007/s00894-003-0129-x. [DOI] [PubMed] [Google Scholar]
- Sridhar A.; Ross G. A.; Biggin P. C. Waterdock 2.0: Water Placement Prediction for Holo-Structures with a Pymol Plugin. PLoS One 2017, 12, e0172743. 10.1371/journal.pone.0172743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abel R.; Young T.; Farid R.; Berne B. J.; Friesner R. A. Role of the Active-Site Solvent in the Thermodynamics of Factor Xa Ligand Binding. J. Am. Chem. Soc. 2008, 130, 2817–2831. 10.1021/ja0771033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazaridis T. Inhomogeneous Fluid Approach to Solvation Thermodynamics. 1. Theory. J. Phys. Chem. B 1998, 102, 3531–3541. 10.1021/jp9723574. [DOI] [Google Scholar]
- Barillari C.; Taylor J.; Viner R.; Essex J. W. Classification of Water Molecules in Protein Binding Sites. J. Am. Chem. Soc. 2007, 129, 2577–2587. 10.1021/ja066980q. [DOI] [PubMed] [Google Scholar]
- Nguyen C. N.; Cruz A.; Gilson M. K.; Kurtzman T. Thermodynamics of Water in an Enzyme Active Site: Grid-Based Hydration Analysis of Coagulation Factor Xa. J. Chem. Theory Comput. 2014, 10, 2769–2780. 10.1021/ct401110x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matricon P.; Ranganathan A.; Warnick E.; Gao Z. G.; Rudling A.; Lambertucci C.; Marucci G.; Ezzati A.; Jaiteh M.; Dal Ben D.; Jacobson K. A.; Carlsson J. Fragment Optimization for GPCRs by Molecular Dynamics Free Energy Calculations: Probing Druggable Subpockets of the A2A Adenosine Receptor Binding Site. Sci. Rep. 2017, 7, 6398. 10.1038/s41598-017-04905-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beuming T.; Che Y.; Abel R.; Kim B.; Shanmugasundaram V.; Sherman W. Thermodynamic Analysis of Water Molecules at the Surface of Proteins and Applications to Binding Site Prediction and Characterization. Proteins: Struct., Funct., Genet. 2012, 80, 871–883. 10.1002/prot.23244. [DOI] [PubMed] [Google Scholar]
- Huang N.; Shoichet B. K. Exploiting Ordered Waters in Molecular Docking. J. Med. Chem. 2008, 51, 4862–4865. 10.1021/jm8006239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forli S.; Olson A. J. A Force Field with Discrete Displaceable Waters and Desolvation Entropy for Hydrated Ligand Docking. J. Med. Chem. 2012, 55, 623–638. 10.1021/jm2005145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts B. C.; Mancera R. L. Ligand-Protein Docking with Water Molecules. J. Chem. Inf. Model. 2008, 48, 397–408. 10.1021/ci700285e. [DOI] [PubMed] [Google Scholar]
- DeLano W. L.The Pymol Molecular Graphics System, Version 1.4.1; Schrödinger, LLC: 2011.
- Marelius J.; Kolmodin K.; Feierberg I.; Aqvist J. Q: A Molecular Dynamics Program for Free Energy Calculations and Empirical Valence Bond Simulations in Biomolecular Systems. J. Mol. Graphics Modell. 1998, 16, 213–225. 10.1016/S1093-3263(98)80006-5. [DOI] [PubMed] [Google Scholar]
- Jorgensen W. L.; Maxwell D. S.; Tirado-Rives J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. 10.1021/ja9621760. [DOI] [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. 10.1063/1.445869. [DOI] [Google Scholar]
- King G.; Warshel A. A Surface Constrained All-Atom Solvent Model for Effective Simulations of Polar Solutions. J. Chem. Phys. 1989, 91, 3647–3661. 10.1063/1.456845. [DOI] [Google Scholar]
- Lee F. S.; Warshel A. A Local Reaction Field Method for Fast Evaluation of Long-Range Electrostatic Interactions in Molecular Simulations. J. Chem. Phys. 1992, 97, 3100–3107. 10.1063/1.462997. [DOI] [Google Scholar]
- Ryckaert J. P.; Ciccotti G.; Berendsen H. J. C. Numerical-Integration of Cartesian Equations of Motion of a System with Constraints - Molecular-Dynamics of N-Alkanes. J. Comput. Phys. 1977, 23, 327–341. 10.1016/0021-9991(77)90098-5. [DOI] [Google Scholar]
- Sherawat M.; Kaur P.; Perbandt M.; Betzel C.; Slusarchyk W. A.; Bisacchi G. S.; Chang C.; Jacobson B. L.; Einspahr H. M.; Singh T. P. Structure of the Complex of Trypsin with a Highly Potent Synthetic Inhibitor at 0.97 Å Resolution. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2007, 63, 500–507. 10.1107/S090744490700697X. [DOI] [PubMed] [Google Scholar]
- Katz B. A.; Clark J. M.; Finer-Moore J. S.; Jenkins T. E.; Johnson C. R.; Ross M. J.; Luong C.; Moore W. R.; Stroud R. M. Design of Potent Selective Zinc-Mediated Serine Protease Inhibitors. Nature 1998, 391, 608–612. 10.1038/35422. [DOI] [PubMed] [Google Scholar]
- Maurer M.; de Beer S. B.; Oostenbrink C. Calculation of Relative Binding Free Energy in the Water-Filled Active Site of Oligopeptide-Binding Protein A. Molecules 2016, 21, 499. 10.3390/molecules21040499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenselink E. B.; Beuming T.; Sherman W.; van Vlijmen H. W. T.; IJzerman A. P. Selecting an Optimal Number of Binding Site Waters to Improve Virtual Screening Enrichments against the Adenosine A2A Receptor. J. Chem. Inf. Model. 2014, 54, 1737–1746. 10.1021/ci5000455. [DOI] [PubMed] [Google Scholar]
- Verdonk M. L.; Chessari G.; Cole J. C.; Hartshorn M. J.; Murray C. W.; Nissink J. W. M.; Taylor R. D.; Taylor R. Modeling Water Molecules in Protein-Ligand Docking Using GOLD. J. Med. Chem. 2005, 48, 6504–6515. 10.1021/jm050543p. [DOI] [PubMed] [Google Scholar]
- Rarey M.; Kramer B.; Lengauer T. The Particle Concept: Placing Discrete Water Molecules During Protein-Ligand Docking Predictions. Proteins: Struct., Funct., Genet. 1999, 34, 17–28. 10.1002/(SICI)1097-0134(19990101)34:1<17::AID-PROT3>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Santos R.; Hritz J.; Oostenbrink C. Role of Water in Molecular Docking Simulations of Cytochrome P450 2D6. J. Chem. Inf. Model. 2010, 50, 146–154. 10.1021/ci900293e. [DOI] [PubMed] [Google Scholar]
- Powers R. A.; Morandi F.; Shoichet B. K. Structure-Based Discovery of a Novel, Noncovalent Inhibitor of AmpC Beta-Lactamase. Structure 2002, 10, 1013–1023. 10.1016/S0969-2126(02)00799-2. [DOI] [PubMed] [Google Scholar]
- Poornima C. S.; Dean P. M. Hydration in Drug Design 1. Multiple Hydrogen-Bonding Features of Water Molecules in Mediating Protein-Ligand Interactions. J. Comput.-Aided Mol. Des. 1995, 9, 500–512. 10.1007/BF00124321. [DOI] [PubMed] [Google Scholar]
- Hamelberg D.; McCammon J. A. Standard Free Energy of Releasing a Localized Water Molecule from the Binding Pockets of Proteins: Double-Decoupling Method. J. Am. Chem. Soc. 2004, 126, 7683–7689. 10.1021/ja0377908. [DOI] [PubMed] [Google Scholar]
- Li Z.; Lazaridis T. The Effect of Water Displacement on Binding Thermodynamics: Concanavalin A. J. Phys. Chem. B 2005, 109, 662–670. 10.1021/jp0477912. [DOI] [PubMed] [Google Scholar]
- Wang L.; Wu Y. J.; Deng Y. Q.; Kim B.; Pierce L.; Krilov G.; Lupyan D.; Robinson S.; Dahlgren M. K.; Greenwood J.; Romero D. L.; Masse C.; Knight J. L.; Steinbrecher T.; Beuming T.; Damm W.; Harder E.; Sherman W.; Brewer M.; Wester R.; Murcko M.; Frye L.; Farid R.; Lin T.; Mobley D. L.; Jorgensen W. L.; Berne B. J.; Friesner R. A.; Abel R. Accurate and Reliable Prediction of Relative Ligand Binding Potency in Prospective Drug Discovery by Way of a Modern Free-Energy Calculation Protocol and Force Field. J. Am. Chem. Soc. 2015, 137, 2695–2703. 10.1021/ja512751q. [DOI] [PubMed] [Google Scholar]
- Balius T. E.; Fischer M.; Stein R. M.; Adler T. B.; Nguyen C. N.; Cruz A.; Gilson M. K.; Kurtzman T.; Shoichet B. K. Testing Inhomogeneous Solvation Theory in Structure-Based Ligand Discovery. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, E6839–E6846. 10.1073/pnas.1703287114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy R. B.; Repasky M. P.; Greenwood J. R.; Tubert-Brohman I.; Jerome S.; Annabhimoju R.; Boyles N. A.; Schmitz C. D.; Abel R.; Farid R.; Friesner R. A. WScore: A Flexible and Accurate Treatment of Explicit Water Molecules in Ligand-Receptor Docking. J. Med. Chem. 2016, 59, 4364–4384. 10.1021/acs.jmedchem.6b00131. [DOI] [PubMed] [Google Scholar]
- Krimmer S. G.; Betz M.; Heine A.; Klebe G. Methyl, Ethyl, Propyl, Butyl: Futile but Not for Water, as the Correlation of Structure and Thermodynamic Signature Shows in a Congeneric Series of Thermolysin Inhibitors. ChemMedChem 2014, 9, 833–846. 10.1002/cmdc.201400013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.