

Summary
Currently, there is a growing need for culturing hematopoietic stem/progenitor cells (HSPCs) in vitro for various clinical applications including gene therapy. Compared to cord blood (CB) CD34+ HSPCs, it is more challenging to maintain or expand peripheral blood mobilized stem/progenitor cell (PBSC) CD34+ cells ex vivo. To fill this knowledge gap, we have systematically surveyed 466 small molecule drug compounds for their potential in cytokine-dependent expansion of human CD34+CD90+ HSPCs. We found that epigenetic modifiers, especially histone deacetylase inhibitors (HDACi), could preferentially maintain and expand these cells. Specially, treatment of CD34+ PBSCs with a single dose of HDACi Trichostatin A (TSA) at a concentration of 50nM ex vivo yielded the greatest expansion (11.7-fold) of CD34+CD90+ cells when compared to the control (DMSO plus cytokines) group. Additionally, TSA-treated PBSC CD34+ cells had a statistically significant higher engraftment rate than the control-treated group in xenotransplantation experiments. Mechanistically, TSA-treatment was associated with increased expressions of HSPC-related genes such as GATA2 and SALL4. Furthermore, TSA-mediated CD34+CD90+ expansion was reduced by down-regulation of SALL4 but not GATA2. Overall, we have developed a robust, short-term (5-day), PBSC ex vivo maintenance/expansion culture technique and demonstrated that the HDACi-TSA/SALL4 axis is important for the biological process.
Keywords: hematopoietic stem cell, CD34+CD90+, self-renewal, expansion, HDACi, SALL4
Introduction
Hematopoietic stem cells (HSCs) possess the unique capacity to self-renew and give rise to all types of mature cells within the blood and immune systems. HSC self-renewal is regulated by both intrinsic and extrinsic signals(1) (2) (3). Genes and pathways that are functionally linked to self-renewal of HSCs include CEBPα(4), Notch ligands(5, 6), Angiopoietin-like proteins(7), SALL4(8), homeobox protein B4 (HOXB4)(9) and c-MPL(10). Although self-renewal divisions of HSCs clearly occur in vivo, induction of such event ex vivo has been difficult. This is due to the fact that despite our progress in understanding the molecular factors that support self-renewal and differentiation of the hematopoietic system in vivo, not much is known about the modulation of these factors ex vivo (11).
Currently, there is a growing need for culturing PBSC in vitro for transplant-related applications such as gene therapy(12) or genome-editing via TALENs or CRISPR/Cas9(13). Furthermore, the same PBSC in vitro culture technique has the potential to be used for HSCP expansion for poor autologous mobilizations to avoid additional collections (14).
Unlike embryonic stem (ES) cells, expansion of human CD34+ HSPCs ex vivo in culture is associated with differentiation and loss of “stemness”. This is, at least in part, due to the effects of the cytokines used in the culture conditions, which induce HSPCs to proliferate and differentiate. Several approaches have been reported to modify the cytokine-based culture conditions to achieve HSPC expansion ex vivo. These include the use of Prostaglandin E2(15) (16), Pleiotrophin(17), SR1(18), UNC0638(19), Pyrimidoindole derivatives(20), and TEPA(21) (22). However, even after several decades of research, the quest for condition(s) that are able to stimulate self-renewal ex vivo still continues(23). Compared to cord blood (CB) CD34+ cells, it is more challenging to maintain and expand PBSC CD34+ cells ex vivo(24) (25).
In this study, we searched for a robust and short term ex vivo culture condition that can maintain or expand PBSCs without the loss of their “stemness”. We utilized a short term assay (5 days) that can be easily modified for use in the current clinical HSPC transplantation setting, and co-expression of CD34 and CD90 to identify compounds with potentials for PBSC expansion. After surveyed 466 compounds, including multiple chromatin modifiers, we found that a single dose of TSA treatment led to the greatest expansion of these cells. We further characterized the TSA-mediated PBSC maintenance/expansion functionally and mechanistically. Moreover, we propose a model of an HDACi-TSA/SALL4 axis in the maintenance and expansion of HSPC ex vivo.
Materials and methods
Isolation of PBSC and CB CD34+ cells and ex vivo culture
PBSC were collected after G-CSF mobilization and enriched by CD34+ immunoselection. Fresh CB collections were obtained from Cell Manipulation Core Facility in Dana-Farber Cancer Institute (DF/HCC; Boston, MA) according to guidelines established by DF/HCC Institutional Review Board. CB cells were isolated by density centrifugation on Ficoll-Paque (Stem Cell Technologies, Vancouver, BC, Canada) and enriched using the CD34 positive cell isolation kit (Stem Cell Technologies). Cells were allotted to 2 × 104 /well and incubated in IMDM containing 30% fetal bovine serum (FBS; GIBCO) supplemented with 1X CC100 cytokine mix (SCF, FL, IL3, and IL6; Stem Cell Technologies) or a serum-free expansion system (StemSpanTM SFEM II, SCF, FL, IL3, and IL6; STEMCELL Technologies) supplemented with 1X CC100 cytokine mix for 5 to 7 days without changing medium.
Engraftment of CD34+ cells in NSG mice
NSG (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ, The Jackson Laboratory, ME, USA) mice were bred and maintained in the Children’s Hospital Boston animal facility. All animal work has been approved by and done according to the guidelines of the IACUC under protocol 10-10-1832. The CB or PBSC CD34+ cells treated with TSA or DMSO were injected intravenously via the tail vein into sub-lethally irradiated (220 rads) 8 to 16-week-old NSG mice. Transplantation or IV administration was performed within 24 h after irradiation. Peripheral blood (PB) chimerism was monitored at 8 weeks post transplantation. Bone marrow (BM) chimerism was monitored at 8 and 18 weeks post transplantation. These samples were subsequently subjected to flow cytometry analysis utilizing FITC-conjugated anti-human CD45 antibody and APC-conjugated anti-mouse CD45 antibody (eBiosciences, CA, USA). The percentage of human CD45+ cells was calculated as follows: % human CD45+ cells = No. human CD45+ cells/ (No. human CD45+ cells + No. murine CD45+ cells) × 100. A threshold of 0.2% human CD45+ cells was established as a reliable predictor of positive engraftment. BM cells from primary recipients were reinfused into sub-lethally irradiated (220 rads) secondary recipient mice. Mice were sacrificed 8 weeks after transplantation and a threshold of 0.025% human CD45+ cells was established as a reliable predictor of positive engraftment. For limiting dilution analysis, a threshold of 2.8% human CD45+ cells was established as a reliable predictor of positive engraftment. The frequency of human SRCs was calculated using L-Calc software (StemCell Technologies Inc.)
Statistical analysis
Results are expressed as mean ± Standard Deviation (SD) or Standard Error (SE) when appropriate. Statistical differences were evaluated using the student t test with significance at p of 0.05 or less.
Additional materials and methods are listed supplemental material.
Results
HTS approach to identify small molecule compounds including chromatin modifiers for the maintenance/expansion of PBSC CD34+CD90+ cell ex vivo
We first reviewed HSPC assays. The xenotransplantation model provides a direct quantitative in vivo assay to measure human HSPC functional activity, and HSPCs are therefore also called severe combined immunodeficiency (SCID)-repopulating cells (SRC). Among the published studies, ex vivo cultured CD34+CD90+ cells are most well established to be SRC or have marrow-repopulating potential(26). Therefore, in our study, we use co-expression of CD34 and CD90 to identify compounds with potentials for PBSC maintenance/expansion. We developed a high throughput screening (HTS) assay based on the co-expression of these two surface markers. Primary human PBSC CD34+ cells were cultured in 96-well plates with the addition of cytokines and a drug panel for 3 to 5 days. The cells were then evaluated for expression of CD34 and CD90 by flow cytometry. Using this approach, we surveyed 446 FDA approved compounds and 20 additional small molecule drugs including a panel of chromatin modifiers (Table S1).
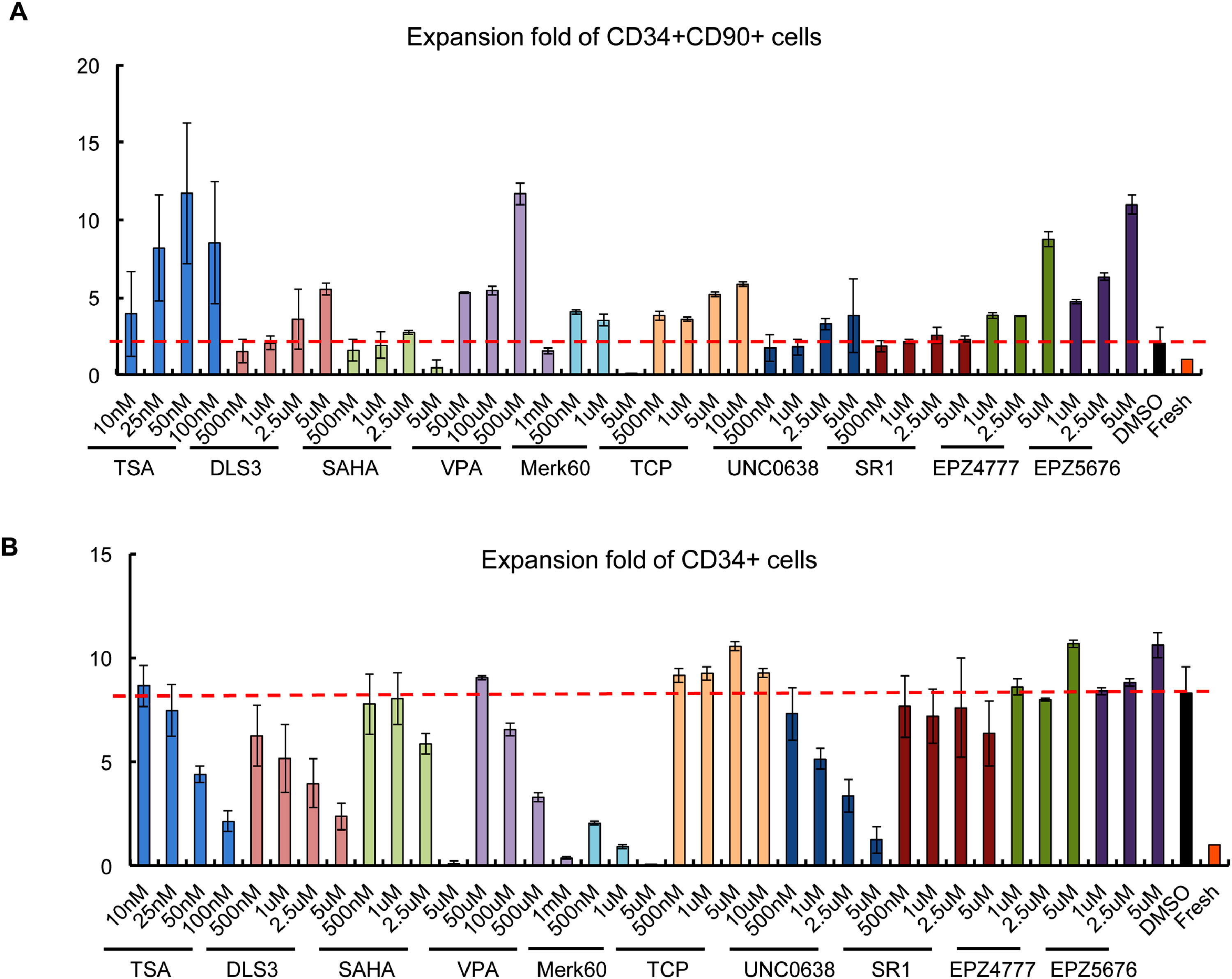
Ten compounds were considered to be positive hits based on the increased percentage of CD34+CD90+. Among them, five were histone deacetylase inhibitors, namely trichostatin A (TSA), DLS3, Valproic Acid (VPA), SAHA, and Merk60. The others were H3K9me2 methyltransferase inhibitor UNC0638, Dot1 inhibitors EPZ4777 and EPZ5676 (Figure 1A). A mild increase in the CD34+CD90+ percentage was also observed with treatment of antagonist of the aryl hydrocarbon receptor (SR1) and Lysine-Specific Demethylase 1 (LSD1) Inhibitor Tranylcypromine (TCP) (Figure 1A). None of the 446 FDA-approved drug compounds were positive in the screen. In addition, epigenetic modifiers such as BET inhibitor JQ1-S et al were all negative.
Figure 1. Screening for compounds which increase CD34+CD90+ cells from PBSC.

(A) Effect of the compounds on percentage of CD34+CD90+ expression following 5 days of culture. (B) Absolute number of CD34+CD90+cells after 5 days of culture with the compounds. Each value represents mean ± SE of two or three independent experiments.
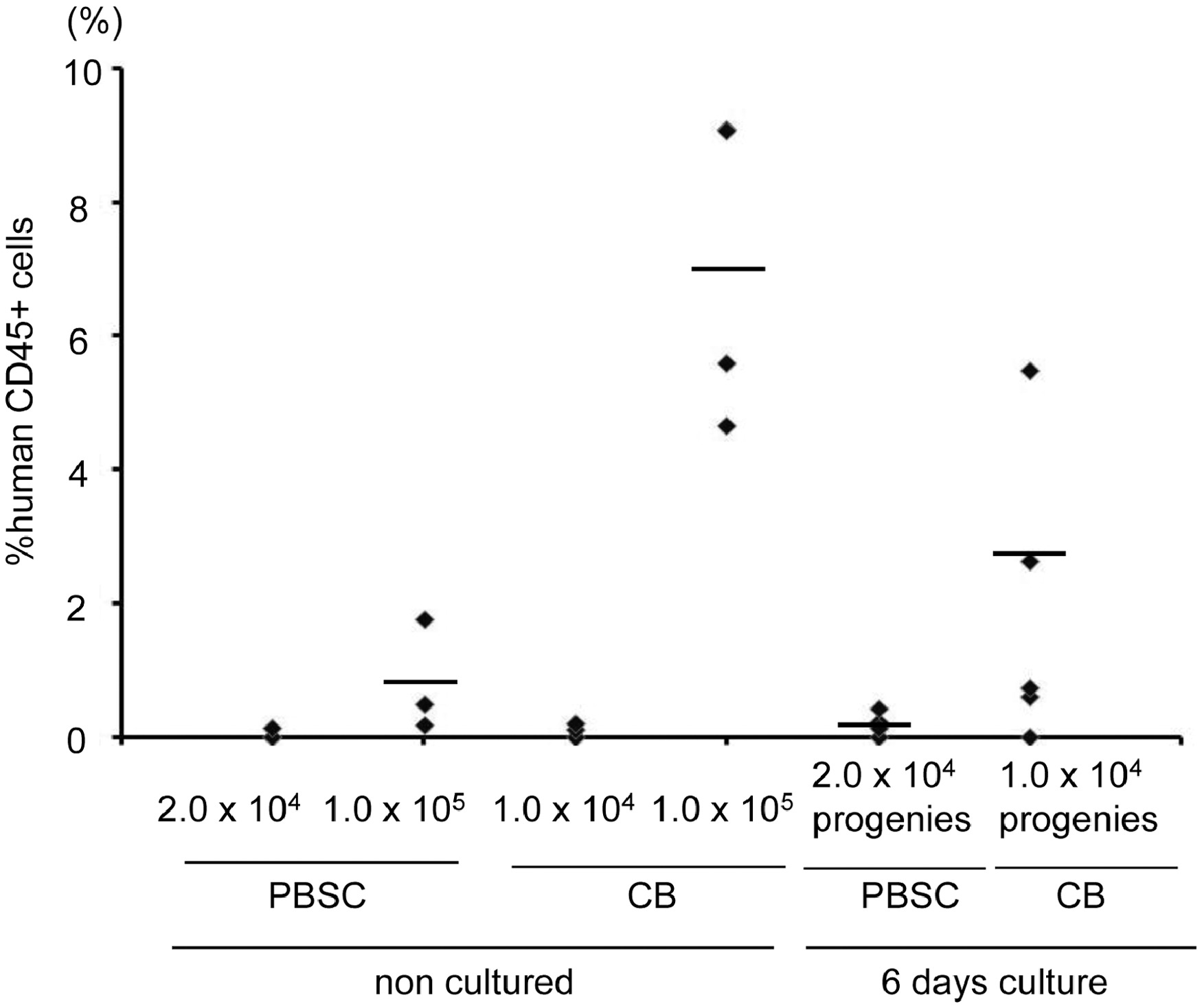
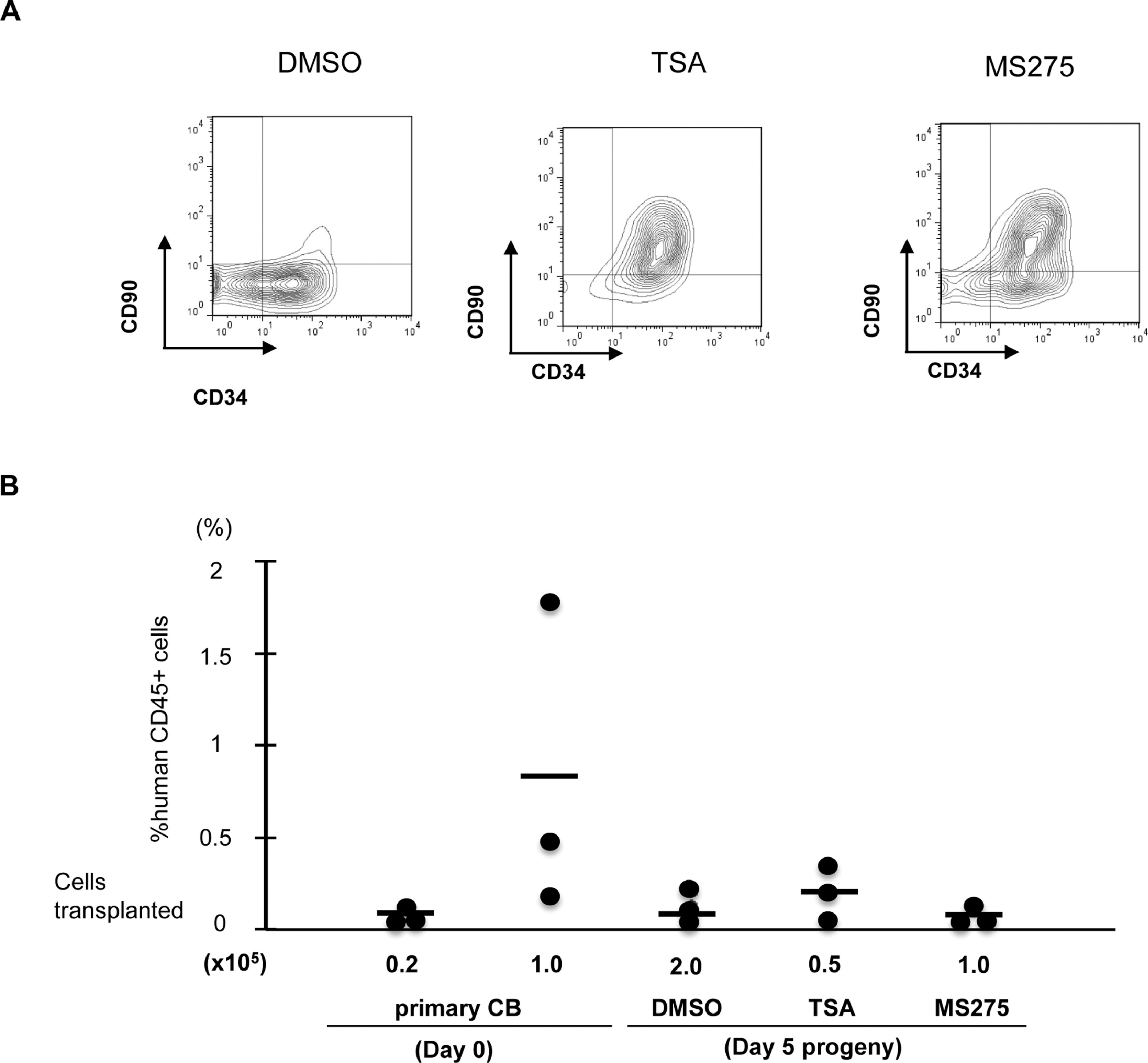
By measuring the absolute CD34+CD90+ cell number, we noticed that while most of the positive hits could expand the CD34+CD90+ population compared to control (DMSO plus cytokines) treated CD34+ cells, there was a range of ability. Among the ten positive hits, TSA, VPA, EPZ4777 and EPZ5676 treatment demonstrated the greatest (11.7-fold, 11.6-fold, 8.7-fold, 10.9-fold) expansion of CD34+CD90+ cells compared to control based on absolute cell number (Figure 1B) and expansion fold (Figure S1A). These compounds did not markedly expand CD34+ and CD34+CD90− cells in 5 days (Figure S1B). To further enhance expansion, we combined the treatment of SR1 with TSA (Figure S2A). However, this combination treatment only mildly improved the expansion of CD34+CD90+ cells. In addition, we observed that HDACi such as TSA and MS275 can also expand the CD34+CD90+ population from cord blood (Figure S3).
Overall, our findings suggest that epigenetic modifiers, especially HDAC inhibitors have the potential to expand the CD34+CD90+ cells in our short-term culture condition.
TSA-treatment of PBSC CD34+ cells ex vivo leads to preferential expansion of CD34+CD90+ cells
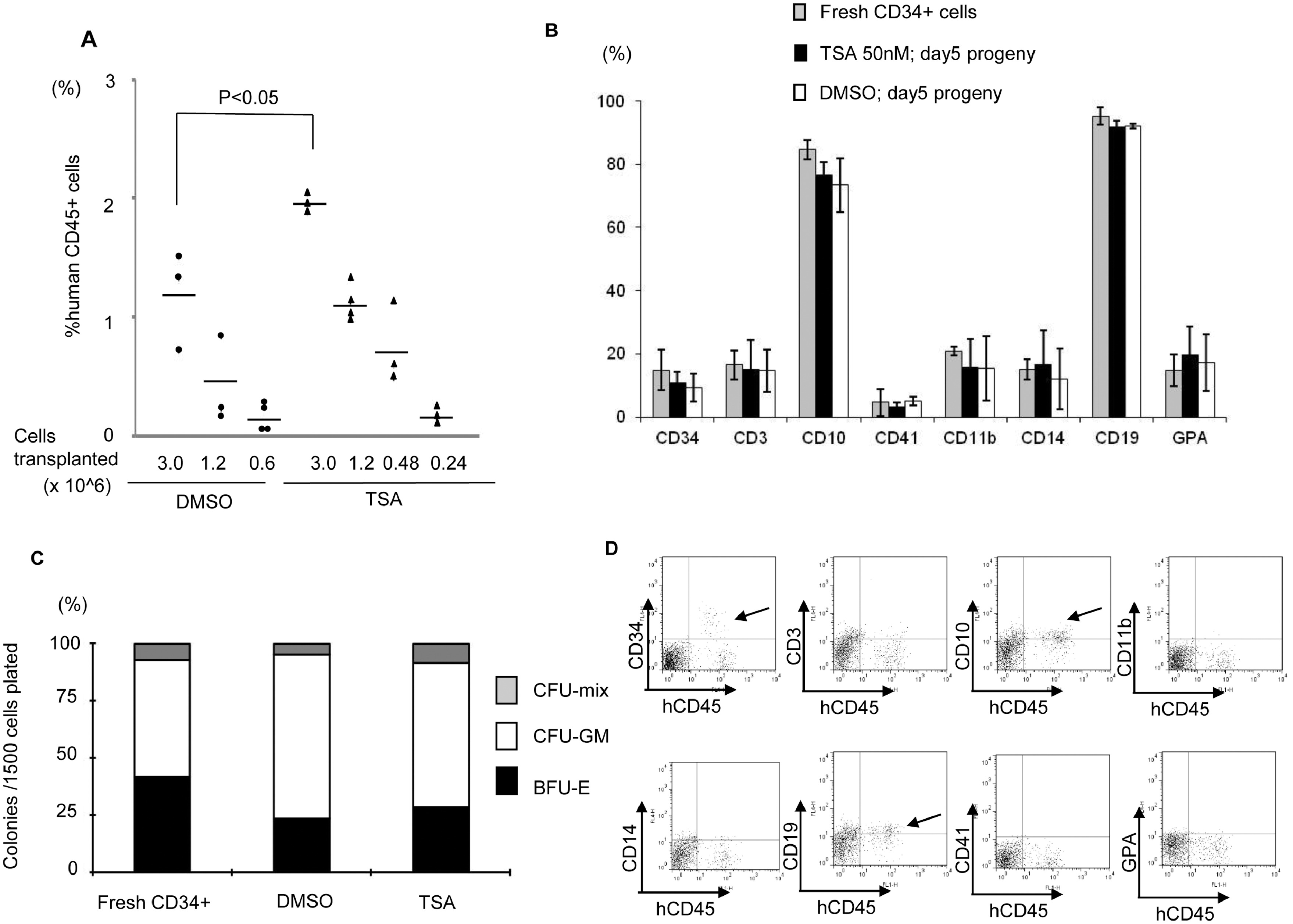
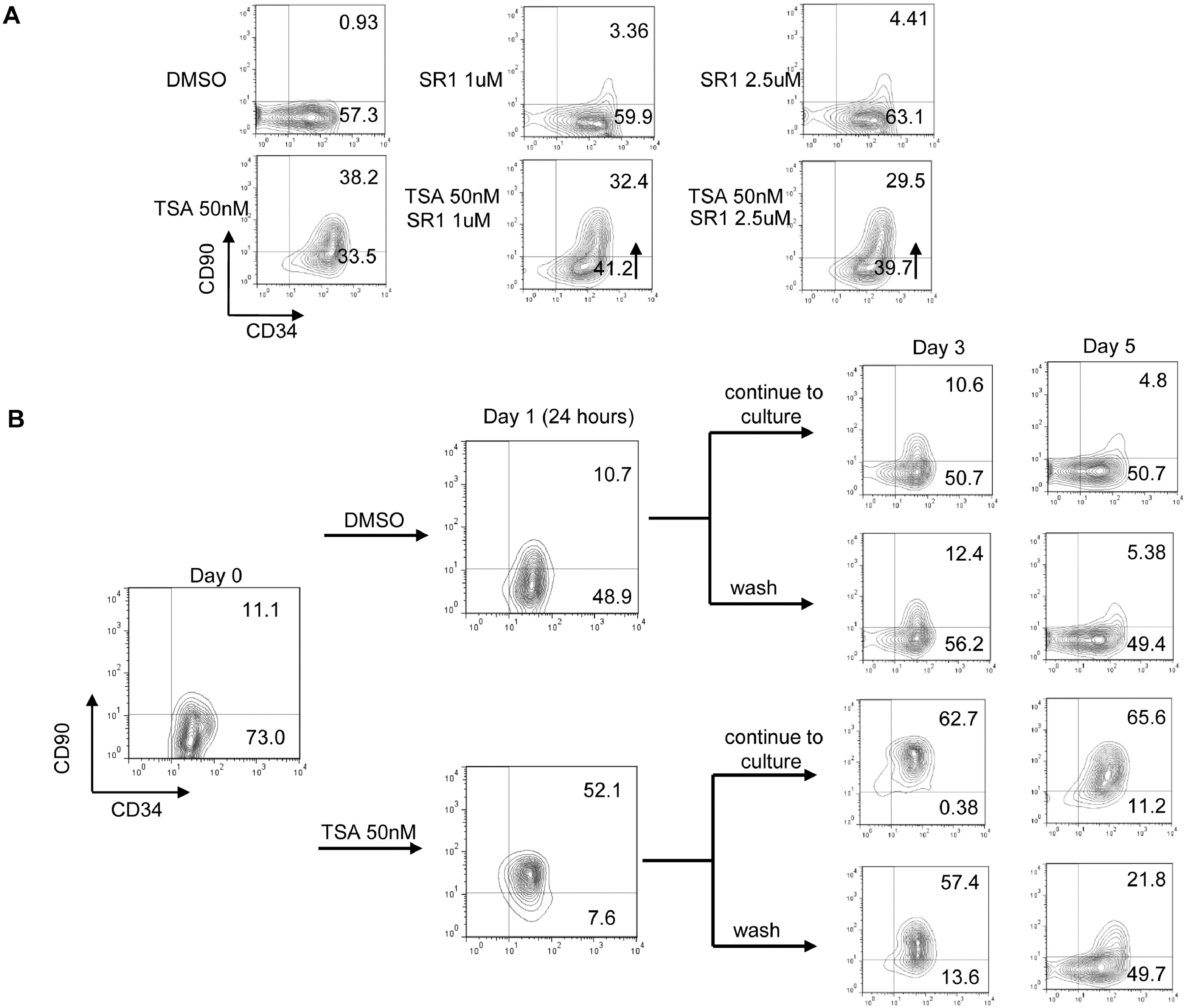
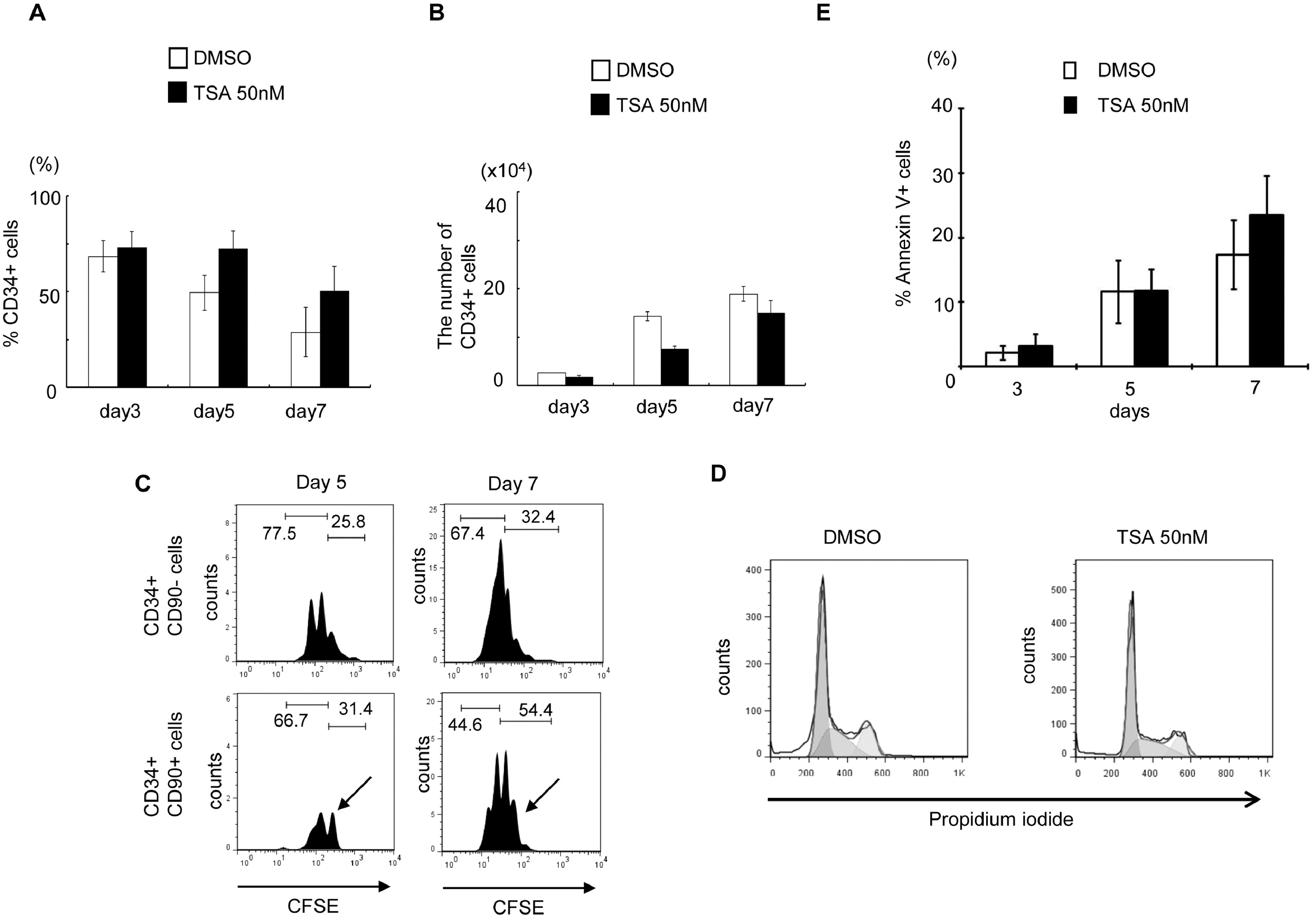
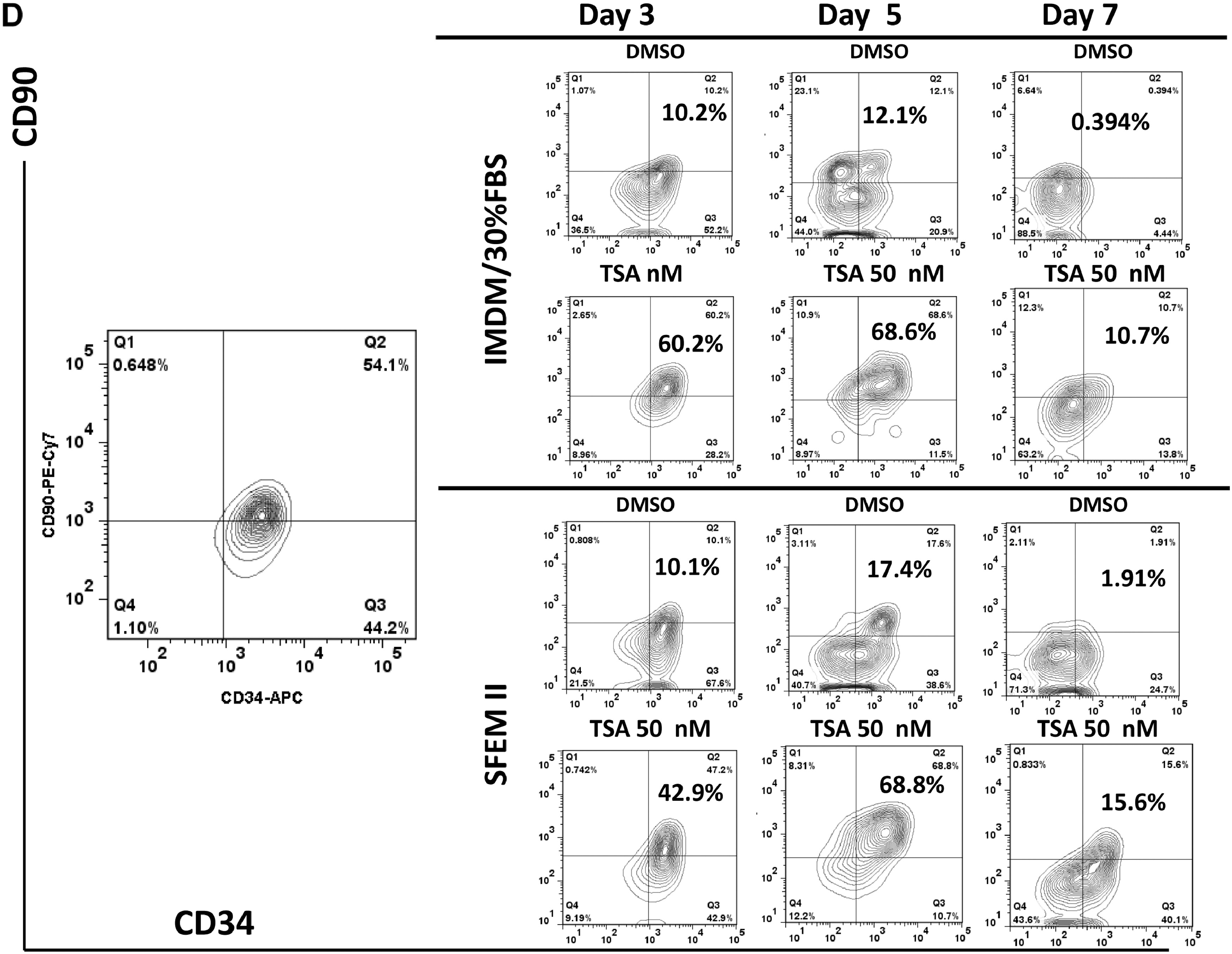
From our screening, TSA demonstrated the highest CD34+CD90+ ex vivo expansion potential at a single dose with the lowest functional concentration of 50nM since higher dosage is toxic to cells (Figure 1, & Figure S1A). We then focused on TSA to investigate its role in PBSC maintenance/expansion in greater detail. Serial time-point studies were carried out. We observed that TSA treatment led to a preferential expansion of CD34+CD90+ cells on day 3, 5, and 7 compared to control treated cells (Figure 2B&2C; p<0.05). There was a significant increase in CD34+CD90+ cells 24 hours post TSA treatment (Figure S2B). To examine whether TSA is required for further expansion of this population during the subsequent culture period, we performed the following experiments. Since the half-life of TSA in culture is 3 days, washing out TSA after 24 hours of treatment was performed, which resulted in decreased CD34+CD90+ cells (Figure S3B). This suggests that the TSA mediated expansion of CD34+CD90+ cells is reversible if TSA treatment is terminated prematurely. We next examined the effects of TSA treatment on cell growth. We found that the overall cell growth with TSA treatment was about 2.5 times lower than that with control (Figure 2A; TSA 10.7× 104 ± 1.12 /well vs TSA 28.3 × 104 ± 0.49 /well; p<0.05). We further quantitated the absolute cell number of CD34+ (Figure S4A&4B), and CD34+CD90+ subpopulations (Figure 2D). We observed that the absolute number of CD34+CD90+ cells was significantly increased by TSA treatment especially from day 5 to day 7.
Figure 2. TSA-treated culture leads to preferential expansion of slowly dividing PBSC CD34+CD90+ cells.

(A) Cell growth of PBSC CD34+ cells cultured in the presence of cytokines with TSA or DMSO at various concentration of TSA after 5 days of culture. The data shown are the mean of three independent experiments. Error bars indicate SD. (B) Time course experiment from day 0 to 7 days showing that TSA increased in the number of CD34+CD90+ cells. The data shown are the mean of three independent experiments. (C) Percentage of CD34+CD90+ cells at 3, 5 and 7 days of culture in the presence or the absence of 50nM TSA. Error bars indicate SD; * indicates p<0.05. (D) Absolute number of CD34+CD90+ cells at 3, 5 and 7 days of culture in the presence or the absence of 50nM TSA. Error bars indicate SD; * indicates p<0.05. (E) CSFE-labeled CD34+ cells were cultured in the presence of cytokines with TSA or DMSO treatment for 7 days. The panel shows a representative (1 of 3 experiment) flow cytometric profile of CFSE fluorescence intensity after 5 and 7 days of culture. The arrow indicates the fraction of cells that have undergone fewer cell divisions when compared to CD34+CD90− cells.
Next, we asked whether the lower total nucleated cell number observed in TSA treatment was due to a delay in cell proliferation. We performed a cell-division-monitoring assay, using the carboxyfluorescein succinimidyl ester (CFSE) fluorescent dye, on CD34+ cells during the culture period. While 66.0 % of control treated CD34+ cells divided more than 4 times by day 5, only 9.02% of TSA treated cells went through a similar number of cell divisions (Figure 2E). Comparable results were observed on day 7. In addition, there was no significant difference in cell cycle progression between TSA and control treated cells demonstrated by propidium iodide (PI) staining (Figure S4C). To investigate whether the lower total nucleated cell number observed after TSA treatment was due to increased apoptosis, we performed annexin V and PI staining with TSA- or control-treated cells on day 3, 5 and 7. No difference in apoptosis was observed between the two treatment groups (Figure S4D). We then asked whether TSA treatment could lead to increased CD34+CD90+ cells through proliferation. Using the CFSE assay, we observed that CD34+CD90+ cells divided upon TSA treatment. Since the CD34+CD90+ is the dominant population in TSA treatment, and these cells divide slower than the CD34+CD90− cells (Figure S4E), these observations could explain why there are fewer cell divisions in the TSA treated group (Figure 2E).
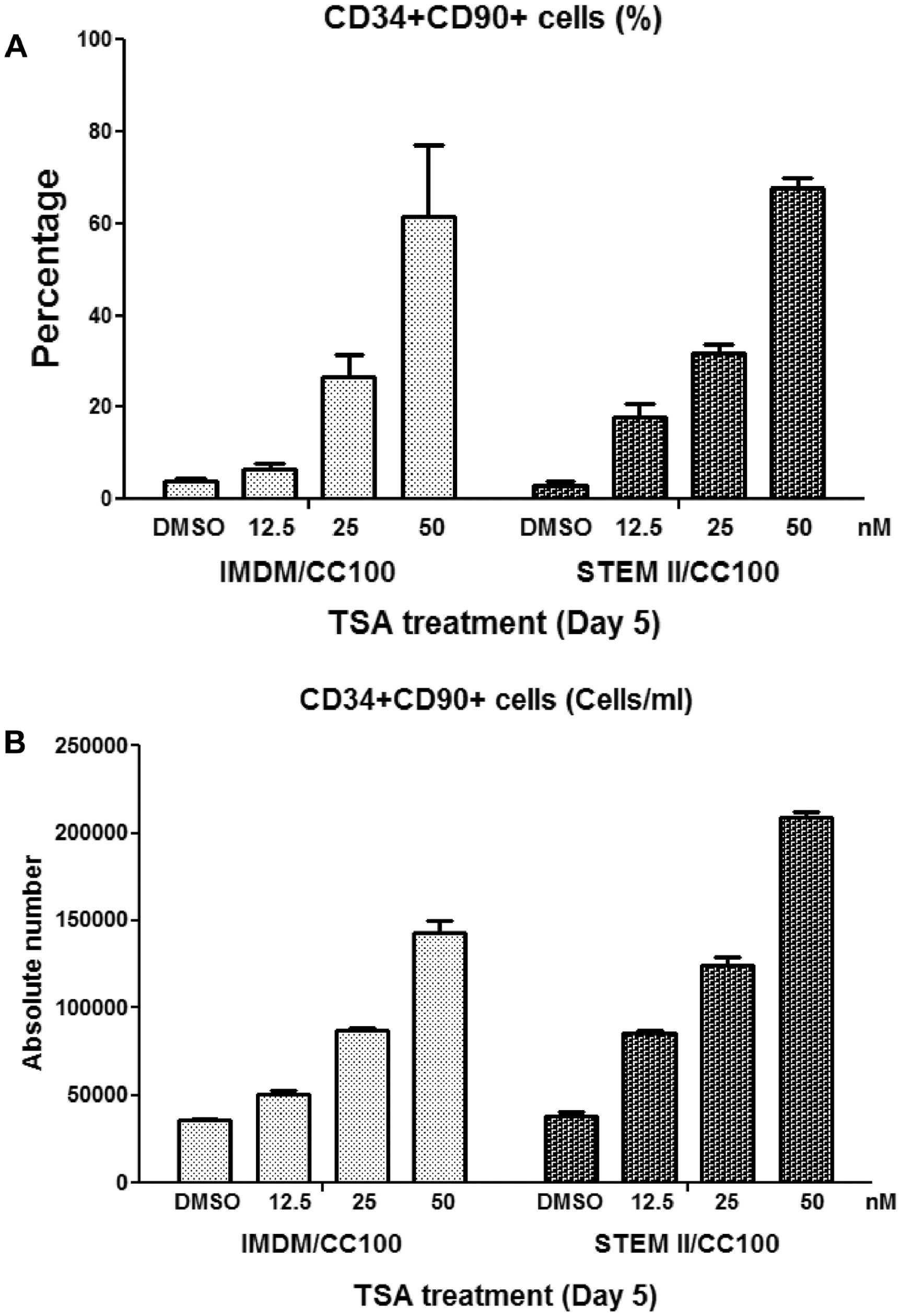
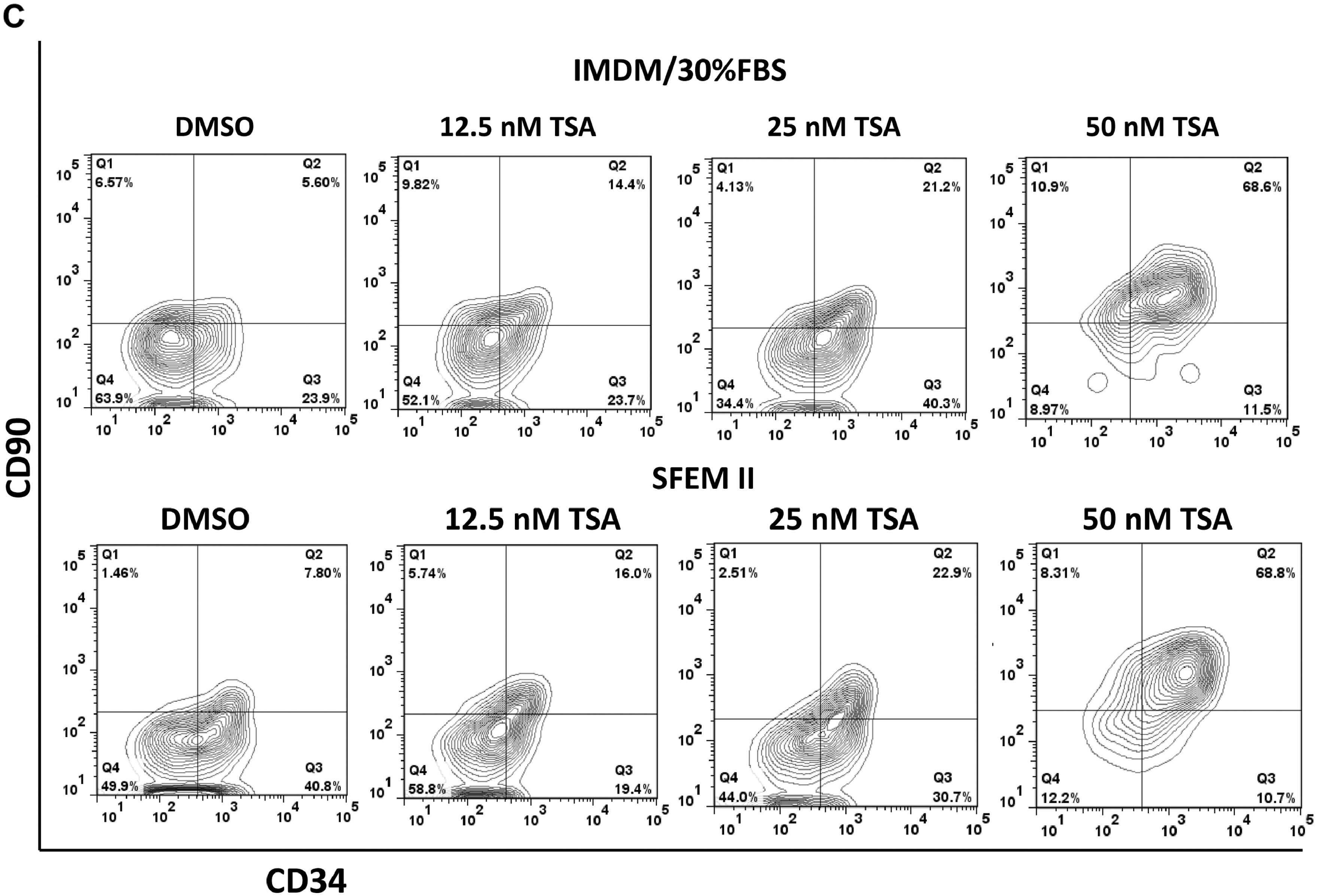
In addition, we have tested the TSA-mediated protocol in a serum-free StemSpan SFEM II Medium, we observed a similar result in expansion of CD34+CD90+ population (Figure S5).
In summary, the lower expansion of TSA-treated CD34 cells was due to a slower cell division rather than apoptosis.
Treatment of PBSC CD34+ cells with TSA enhances marrow-repopulating potential in vivo
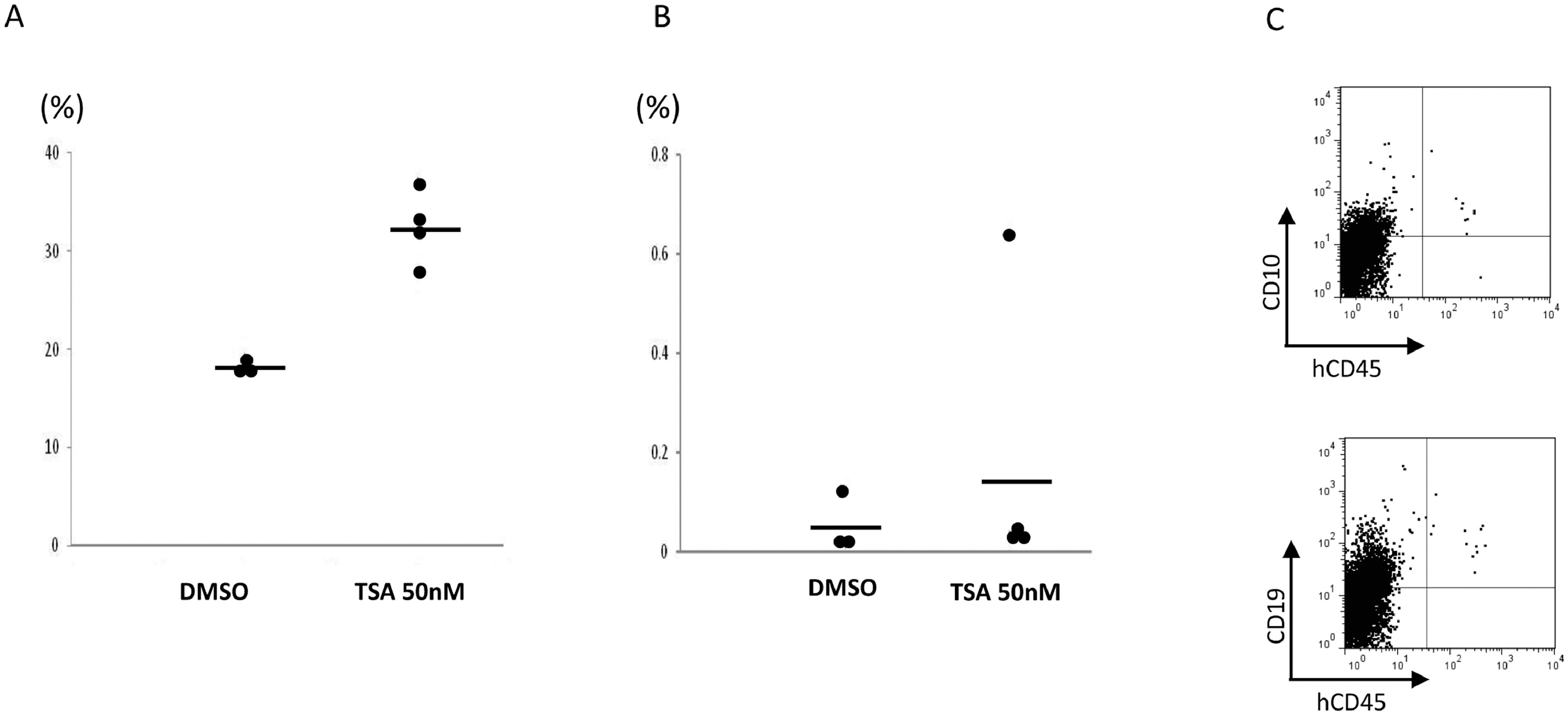
Compared to CB, PBSC CD34+ cells have reduced marrow-repopulation potential (Figure S6)(24) (25). To validate whether TSA-treated PBSC CD34+ cells have enhanced function in vivo, we performed xenotransplantation experiments. During the ex vivo course of culturing PBSC CD34+ cells, we noticed that on day 5, the majority of control-treated cells were CD34+CD90−, while a large portion of TSA-treated cells were still CD34+CD90+ (Figure 2B). To compare the marrow-repopulation potential between control and TSA treatment, we transplanted cells after culture for 5 days with control or TSA into NSG mice as a measurement for HSPC function. Eight weeks after transplantation, the mice were sacrificed and their peripheral blood (PB) and bone marrow (BM) samples analyzed (Figure S7A & Figure 3A). Mice transplanted with 3×106 TSA-treated day 5 progeny demonstrated a higher level of human hematopoietic cell engraftment (average engraftment of 32.74%), while transplantation of the same number of control-treated cells resulted in a lower level of BM engraftment (average engraftment of 18.43%; p<0.005).
Figure 3. Treatment of PBSCs with TSA enhanced the marrow-repopulating potential.

(A) A scatter plot showing the levels of human CD45+ cell engraftment in the bone marrow (BM) of NSG mice 8 weeks after transplantation with CD34+ cells cultured with TSA or DMSO. (B) Limiting dilution analysis to compare the frequency of SRCs in the progeny from TSA-treated, control-treated culture conditions and those from fresh uncultured CD34+ cells. (C) Representative BM flow cytometric analysis of 8 weeks multilineage hematopoietic differentiation potential of engrafted human hematopoietic cells treated with TSA in NSG mice. (D) A scatter plot showing the levels of human CD45+ cell engraftment in the BM of NSG mice 18 weeks after transplantation with their progeny after culture with TSA or DMSO.
To assess the degree of HSPC maintenance/expansion, we used limiting dilution analysis to compare the frequency of SRCs in the progeny from TSA-treated and control-treated culture conditions. Poisson distribution analysis revealed a SRC frequency of 1 in 247,567 (95% CI: 1 in 367,071 to 1 in 166,969) in TSA-treated cells, and 1 in 1,164,807 (95% CI: 1 in 1,784,517 to 1 in 760,304) in control-treated cells indicating the effective expansion of SRC number (about 4.7 fold) by TSA treatment (Figure 3A). The SRC frequency of fresh untreated CD34+ cells was 1 in 355,285 (95% CI: 1 in 555,478 to 1 in 227,241) (Figure 3B).
We next asked whether cells treated with TSA have the ability to give rise to mature hematopoietic cells. We observed that TSA-treated cells capable of differentiating into multiple lineages following transplantation in vivo (Figure 3C). The overall differentiation properties were similar between TSA- and control-treated cells (Figure S7B). We also assessed their functional capacity by colony forming cell assays, and found TSA- and control-treated cells had similar differentiation abilities (Figure S7C). To explore the long-term reconstitution capacity of the transplanted cells, the engraftment rates in the BM were evaluated at 18 weeks post-transplantation (Figure 3D). Mice transplanted with 6×105 TSA-treated day 5 progeny demonstrated a higher level of human hematopoietic cell engraftment (average engraftment of 13.5%), while transplantation of the same number of control-treated cells resulted in a lower level of BM engraftment (average engraftment 0.68%; p<0.05). Multi-lineage engraftment in the BM was also observed at 18 weeks post-transplant with TSA-treatment in vivo (Figure S7D).
In addition, to evaluate whether TSA treated cells still retain their self-renewal capacity after primary transplantation, bone marrow (BM) cells from the primary NSG (NOD-scid gamma null) recipients were harvested at 8 weeks and were reinfused into sub-lethally irradiated (220 rads) secondary NSG recipients (Figure S8 & Table S3). We have observed a trend toward increased engraftment in TSA-treated cells. This result is in agreement with our conclusion from the long-term (18 weeks post xenotransplantation) and limiting dilution analysis, both suggesting that TSA treatment can enhance PBSC engraftment.
In summary, these results indicate that our TSA-mediated CD34+ cell ex vivo culture technique can successfully expand SRCs with long-term and multilineage hematopoietic differentiation potential.
SALL4 contributes to TSA-mediated expansion of CD34+CD90+ cells
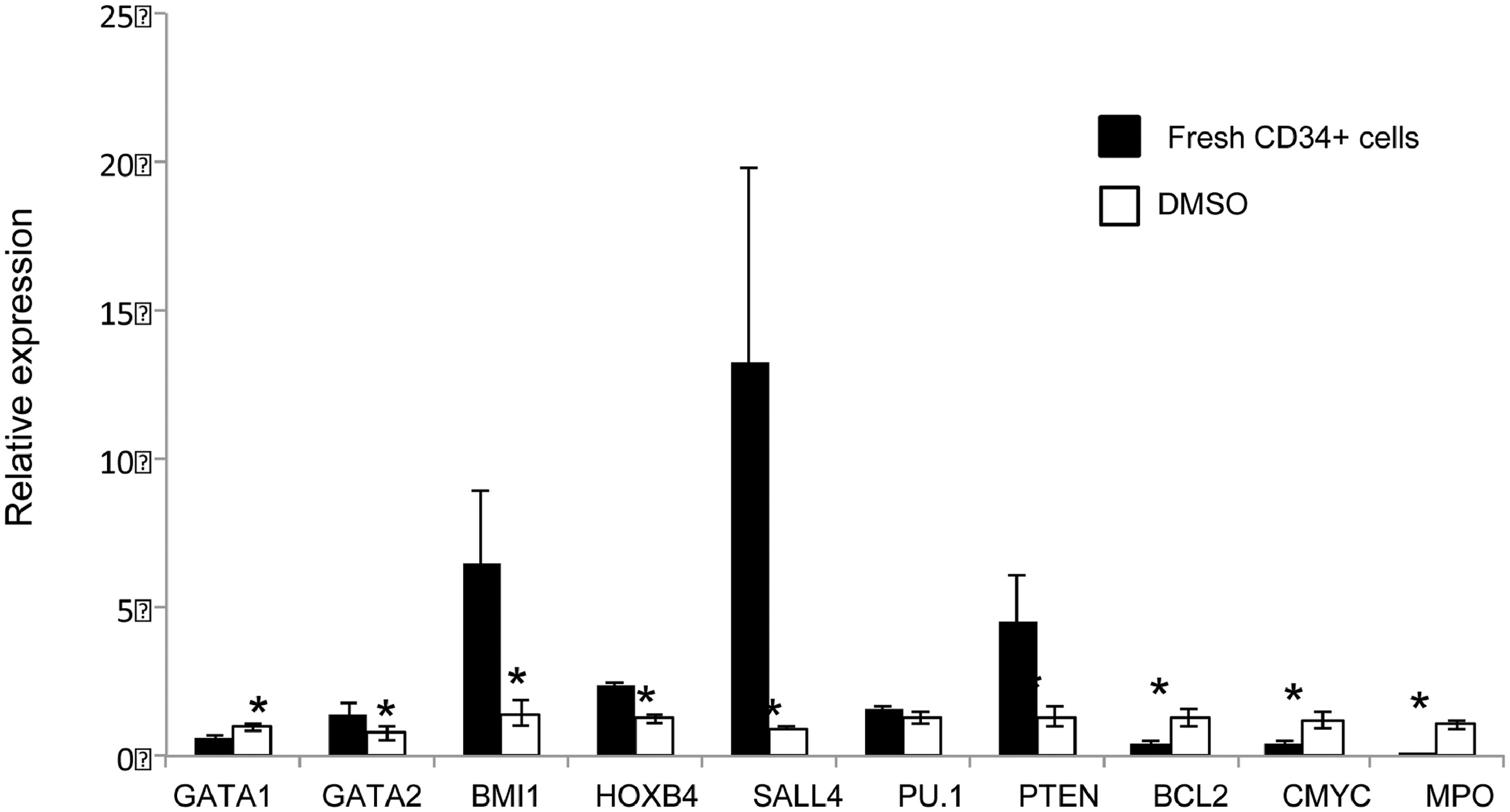
Next, we investigate the molecular mechanism(s) responsible for the expansion of functional HSPCs following TSA treatment. We hypothesized that cytokine-alone based HSPC culture conditions stimulate CD34+ cells to proliferate and differentiate, along with down-regulation of HSPC-related genes. Therefore, we examined the expression levels of a panel of genes known to be involved in self-renewal or differentiation of HSCs by qRT- PCR. When compared to fresh uncultured CD34+ cells, we observed a decrease in the transcript levels of GATA2, BMI1, HOXB4, SALL4, and PTEN after cytokine-mediated ex vivo culture (Figure S9). We theorized that by adding TSA to the cytokine-based culture condition, we could limit the down-regulation of these genes. Therefore, we compared the expression profile of these genes with or without TSA treatment during the ex vivo culture period. We observed higher levels of transcripts for GATA1, GATA2, HOXB4, and SALL4 in cells treated with TSA (Figure 4A; p<0.05). To assess the potential contribution(s) of these four genes to the CD34+CD90+ expansion, TSA treated CD34+CD90− and CD34+CD90+ cells were sorted and analyzed. The expression levels of GATA2 and SALL4 were increased in CD34+CD90+ cells when compared to those in CD34+CD90− cells (Figure 4B), while the expression levels of GATA1 and HOXB4 were not significantly different between the two cell populations. These data suggest that GATA2 and SALL4 may contribute to the TSA-mediated CD34+CD90+ expansion.
Figure 4. Treatment of PBSC CD34+ cells with TSA modulates expression of stem cell related genes.

(A) Effects of TSA treatment on the relative transcript level of genes (GATA1, GATA2, NOTCH1, BMI1, HOXB4, SALL4, PU.1, PU.1, PTEN, BCL2, c-MYC, and MPO) were measured by real-time quantitative PCR. Total RNA was extracted from cells obtained after 3 days of culture in the presence of cytokines with TSA or DMSO. GAPDH was used as internal calibrator (control gene). Measurements were obtained in duplicate using at least 2 independent samples. (B) TSA treated CD34+CD90+ cells and CD34+CD90− cell were sorted and the expression levels of GATA1, GATA2, HOXB4, and SALL4 were analyzed. Measurements were obtained in duplicate using at least 2 independent samples. Error bars indicate SE; * indicates p<0.05.
Both GATA2 and SALL4 are known transcription factors involved in HSC function. Enforcing GATA2 expression can increase the quiescence of CB CD34+ cells, reduce proliferation and cell performance in long term culture-initiating cell and colony forming cell (CFC) assays (27). We previously demonstrated that SALL4 is a key regulator in normal human hematopoiesis (28). Overexpression of SALL4 led to rapid and efficient expansion of CD34+ cells with enhanced engraftment and long-term repopulation capacity in vivo (8).
To investigate whether SALL4 and GATA2 may play a role in TSA-mediated CD34+CD90+ expansion, we down-regulated their expression by shRNA (Figure 5A). Transfection efficiency was evaluated using a vector expressing GFP only (Figure 5B). ShRNA-mediated knockdown led to markedly reduced expression of GATA2 or SALL4 transcripts (Figure 5C&D). We observed a significant reduction in the percentage of CD34+CD90+ cells after treatment only with SALL4 shRNA (Figure 5E), but not GATA2 (Figure 5F). These data suggest that SALL4 is involved, at least partially, in TSA-mediated expansion of CD34+CD90+ cells.
Figure 5. SALL4 silencing decreased TSA-mediated expansion of CD34+CD90+ cells.

(A) The strategy of knockdown of SALL4 and GATA2. (B) Transduction efficacy was evaluated by using lentiviral vectors pLL3.7 and pLKO.3G expressing GFP. (C) SALL4 mRNA expression after shRNA targeting in PBSC CD34+ cells (D) GATA2 mRNA expression after shRNA targeting. (E) The percentage of TSA-mediated CD34+CD90+ cells after SALL4 silencing. (F) The percentage of TSA-mediated CD34+CD90+ cells after GATA2 silencing. (N=3, Error bars indicate SD)
Discussion
Compared to cord blood, PBSC CD34+ cells have reduced marrow-repopulation potential (Supplementary Figure 4)(24) (25), and there is a lack of systematic efforts in searching for method(s) that can maintain and expand HSPC from a PBSC source. However, there is growing need for expanding PBSCs for transplant-related practices such as gene therapy or genome-editing via TALENs or CRISPR/Cas9. Developing a technology, which allows for HSPC ex vivo expansion is a key step towards these applications. We have set up a HTS assay to screen for small molecule compounds for this purpose. Previously, TSA, in combination with 5-aza-2’-deoxycytidine (5azaD) or valproic acid (VPA), had been reported in the expansion of CD34+CD90+ population (29) (30) (31) (32). We have further simplified and optimized the TSA -only culture period to 5 days to better fit a clinical transplant setting, in which a shorter culture period is associated with less cost.
HSPC fate is governed by transcription factors, which are down-regulated during ex vivo expansion. The combination of cytokines and chromatin modifiers could expand HSPC and favor self-renewal during cell division. This phenomenon can be attributed to, at least in part, the maintained expression of key transcription factors. Based on this hypothesis, in further searching for potential mechanism(s) of TSA-mediated PBSC expansion, we examined gene expression profiles with a focus on transcription factors. The expression of several key HSPC function-related genes, such as SALL4 and GATA2, in PBSC CD34+ cells is affected by the treatment of TSA. Both genes are also enriched in the CD34+CD90+ population when compared to the CD34+CD90− cells. To evaluate which of these two genes is important for the TSA-mediated expansion of CD34+CD90+ population, we performed loss-of-function studies on these two genes. Knocking down SALL4, but not GATA2, can block TSA-mediated expansion of CD34+CD90+ cells. SALL4 is known to be important in self-renewal and differentiation of HSPCs and ES cells (8, 28, 33–38), but it has never been invested in HDACi-mediated CD34+ cell expansion/maintenance.
Altogether, we propose the following model for our ex vivo PBSC expansion technology (Figure 6). The cytokine-based HSPC culture condition stimulates CD34+ cells to proliferate ad differentiate; as a result, the majority population after culture will be CD34−, CD34+CD90−, CD34+CD90+ cells. This could be due to the decreased expression of HSPC-related transcription factors such as SALL4 during this process (Figure S9). With the addition of TSA to the cytokine-based culture condition, the expression level of some of these HSPC-related transcription factors such as SALL4 is maintained/increased (Figure 4A), and our TSA-mediated HSPC expansion method can induce the CD34+ cells to divide, and result in the expansion of the CD34+CD90+ population. The mechanism of HDACi TSA-medicated expansion of CD34+CD90+ cells is, at least in part, by maintaining the expression of SALL4 during the ex vivo culture period. This model is further supported by down-regulation of SALL4 can reduce TSA-mediated CD34+CD90+ cells expansion (Figure 5E).
Figure 6. A schematic model of ex vivo CD34+CD90+ expansion from human PBSC HSPCs.

We propose the following model: Under culture conditions with stimulating cytokines, CD34+ HSPCs tend to differentiate and become CD34− or CD34+CD90− cells. Addition of epigenetic modifiers, such as the HDACi TSA to the culture media, results in expansion of CD34+CD90+ cells. The TSA-mediated HSPC expansion functions, at least in part, through transcription factor SALL4 (TF SALL4).
Conclusion
In summary, we report a robust, 5-day, TSA-mediated ex vivo culture method to expand the CD34+CD90+ HSPC population obtained from a PBSC source, which has the potential to be used in PBSC-related transplants such as gene therapy. We further demonstrate that for the first time, SALL4, a transcription factor known to be important in self-renewal and differentiation of HSPCs, is important for this TSA-mediated ex vivo CD34+CD90+ HSPC expansion process.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Highlight.
HTS approach to identify compounds for the maintenance of PBSC CD34+CD90+ cells
TSA treated CD34+ cells ex vivo leads to preferential expansion of CD34+CD90+ cells
Treatment of PBSC CD34+ cells with TSA enhances marrow-repopulating potential in vivo
SALL4 contributes to TSA-mediated expansion of CD34+CD90+ cells
Acknowledgment
This work was supported in part through NIH grant PO1HL095489, research funds from Leukemia and Lymphoma Society and V foundation (to LC), and by the Singapore Ministry of Health’s National Medical Research Council under its Singapore Translational Research (STaR) Investigator Award and the National Research Foundation Singapore and the Singapore Ministry of Education under its Research Centres of Excellence initiative (to DGT). We want to thank Kol Jia Young, Justin Tan, Thalia Chai-Zhang and Alicia Stein for their help in editing the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict-of-interest disclosure:
The authors have no competing financial interests.
References
- 1.Orkin SH, and Zon LI. SnapShot: hematopoiesis. Cell. 2008;132(4):712. [DOI] [PubMed] [Google Scholar]
- 2.Zon LI. Intrinsic and extrinsic control of haematopoietic stem-cell self-renewal. Nature. 2008;453(7193):306–13. [DOI] [PubMed] [Google Scholar]
- 3.Kiel MJ, and Morrison SJ. Uncertainty in the niches that maintain haematopoietic stem cells. Nat Rev Immunol. 2008;8(4):290–301. [DOI] [PubMed] [Google Scholar]
- 4.Zhang P, Iwasaki-Arai J, Iwasaki H, Fenyus ML, Dayaram T, Owens BM, Shigematsu H, Levantini E, Huettner CS, Lekstrom-Himes JA, et al. Enhancement of hematopoietic stem cell repopulating capacity and self-renewal in the absence of the transcription factor C/EBP alpha. Immunity. 2004;21(6):853–63. [DOI] [PubMed] [Google Scholar]
- 5.Delaney C, Varnum-Finney B, Aoyama K, Brashem-Stein C, and Bernstein ID. Dose-dependent effects of the Notch ligand Delta1 on ex vivo differentiation and in vivo marrow repopulating ability of cord blood cells. Blood. 2005;106(8):2693–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Delaney C, Heimfeld S, Brashem-Stein C, Voorhies H, Manger RL, and Bernstein ID. Notch-mediated expansion of human cord blood progenitor cells capable of rapid myeloid reconstitution. Nat Med. 2010;16(2):232–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang CC, Kaba M, Ge G, Xie K, Tong W, Hug C, and Lodish HF. Angiopoietin-like proteins stimulate ex vivo expansion of hematopoietic stem cells. Nat Med. 2006;12(2):240–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aguila JR, Liao W, Yang J, Avila C, Hagag N, Senzel L, and Ma Y. SALL4 is a robust stimulator for the expansion of hematopoietic stem cells. Blood. 2011;118(3):576–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Amsellem S, Pflumio F, Bardinet D, Izac B, Charneau P, Romeo PH, Dubart-Kupperschmitt A, and Fichelson S. Ex vivo expansion of human hematopoietic stem cells by direct delivery of the HOXB4 homeoprotein. Nat Med. 2003;9(11):1423–7. [DOI] [PubMed] [Google Scholar]
- 10.Nishino T, Miyaji K, Ishiwata N, Arai K, Yui M, Asai Y, Nakauchi H, and Iwama A. Ex vivo expansion of human hematopoietic stem cells by a small-molecule agonist of c-MPL. Exp Hematol. 2009;37(11):1364–77 e4. [DOI] [PubMed] [Google Scholar]
- 11.Dahlberg A, Delaney C, and Bernstein ID. Ex vivo expansion of human hematopoietic stem and progenitor cells. Blood. 2011;117(23):6083–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Naldini L. Ex vivo gene transfer and correction for cell-based therapies. Nat Rev Genet. 2011;12(5):301–15. [DOI] [PubMed] [Google Scholar]
- 13.Hsu PD, Lander ES, and Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014;157(6):1262–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Olivieri A, Marchetti M, Lemoli R, Tarella C, Iacone A, Lanza F, Rambaldi A, Bosi A, and Italian Group for Stem Cell T. Proposed definition of ‘poor mobilizer’ in lymphoma and multiple myeloma: an analytic hierarchy process by ad hoc working group Gruppo ItalianoTrapianto di Midollo Osseo. Bone Marrow Transplant. 2012;47(3):342–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goessling W, Allen RS, Guan X, Jin P, Uchida N, Dovey M, Harris JM, Metzger ME, Bonifacino AC, Stroncek D, et al. Prostaglandin E2 enhances human cord blood stem cell xenotransplants and shows long-term safety in preclinical nonhuman primate transplant models. Cell Stem Cell. 2011;8(4):445–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cutler C, Multani P, Robbins D, Kim HT, Le T, Hoggatt J, Pelus LM, Desponts C, Chen YB, Rezner B, et al. Prostaglandin-modulated umbilical cord blood hematopoietic stem cell transplantation. Blood. 2013;122(17):3074–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Himburg HA, Muramoto GG, Daher P, Meadows SK, Russell JL, Doan P, Chi JT, Salter AB, Lento WE, Reya T, et al. Pleiotrophin regulates the expansion and regeneration of hematopoietic stem cells. Nat Med. 2010;16(4):475–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boitano AE, Wang J, Romeo R, Bouchez LC, Parker AE, Sutton SE, Walker JR, Flaveny CA, Perdew GH, Denison MS, et al. Aryl hydrocarbon receptor antagonists promote the expansion of human hematopoietic stem cells. Science. 2010;329(5997):1345–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen X, Skutt-Kakaria K, Davison J, Ou YL, Choi E, Malik P, Loeb K, Wood B, Georges G, Torok-Storb B, et al. G9a/GLP-dependent histone H3K9me2 patterning during human hematopoietic stem cell lineage commitment. Genes Dev. 2012;26(22):2499–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fares I, Chagraoui J, Gareau Y, Gingras S, Ruel R, Mayotte N, Csaszar E, Knapp DJ, Miller P, Ngom M, et al. Cord blood expansion. Pyrimidoindole derivatives are agonists of human hematopoietic stem cell self-renewal. Science. 2014;345(6203):1509–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peled T, Mandel J, Goudsmid RN, Landor C, Hasson N, Harati D, Austin M, Hasson A, Fibach E, Shpall EJ, et al. Pre-clinical development of cord blood-derived progenitor cell graft expanded ex vivo with cytokines and the polyamine copper chelator tetraethylenepentamine. Cytotherapy. 2004;6(4):344–55. [DOI] [PubMed] [Google Scholar]
- 22.de Lima M, McMannis J, Gee A, Komanduri K, Couriel D, Andersson BS, Hosing C, Khouri I, Jones R, Champlin R, et al. Transplantation of ex vivo expanded cord blood cells using the copper chelator tetraethylenepentamine: a phase I/II clinical trial. Bone Marrow Transplant. 2008;41(9):771–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Walasek MA, van Os R, and de Haan G. Hematopoietic stem cell expansion: challenges and opportunities. Ann N Y Acad Sci. 2012;1266(138–50. [DOI] [PubMed] [Google Scholar]
- 24.Leung W, Ramirez M, and Civin CI. Quantity and quality of engrafting cells in cord blood and autologous mobilized peripheral blood. Biol Blood Marrow Transplant. 1999;5(2):69–76. [DOI] [PubMed] [Google Scholar]
- 25.Tanavde VM, Malehorn MT, Lumkul R, Gao Z, Wingard J, Garrett ES, and Civin CI. Human stem-progenitor cells from neonatal cord blood have greater hematopoietic expansion capacity than those from mobilized adult blood. Exp Hematol. 2002;30(7):816–23. [DOI] [PubMed] [Google Scholar]
- 26.Dorrell C, Gan OI, Pereira DS, Hawley RG, and Dick JE. Expansion of human cord blood CD34(+)CD38(−) cells in ex vivo culture during retroviral transduction without a corresponding increase in SCID repopulating cell (SRC) frequency: dissociation of SRC phenotype and function. Blood. 2000;95(1):102–10. [PubMed] [Google Scholar]
- 27.Tipping AJ, Pina C, Castor A, Hong D, Rodrigues NP, Lazzari L, May GE, Jacobsen SE, and Enver T. High GATA-2 expression inhibits human hematopoietic stem and progenitor cell function by effects on cell cycle. Blood. 2009;113(12):2661–72. [DOI] [PubMed] [Google Scholar]
- 28.Gao C, Kong NR, Li A, Tatetu H, Ueno S, Yang Y, He J, Yang J, Ma Y, Kao GS, et al. SALL4 is a key transcription regulator in normal human hematopoiesis. Transfusion. 2013;53(5):1037–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Araki H, Yoshinaga K, Boccuni P, Zhao Y, Hoffman R, and Mahmud N. Chromatin-modifying agents permit human hematopoietic stem cells to undergo multiple cell divisions while retaining their repopulating potential. Blood. 2007;109(8):3570–8. [DOI] [PubMed] [Google Scholar]
- 30.Mahmud N, Petro B, Baluchamy S, Li X, Taioli S, Lavelle D, Quigley JG, Suphangul M, and Araki H. Differential effects of epigenetic modifiers on the expansion and maintenance of human cord blood stem/progenitor cells. Biol Blood Marrow Transplant. 2014;20(4):480–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Saraf S, Araki H, Petro B, Park Y, Taioli S, Yoshinaga KG, Koca E, Rondelli D, and Mahmud N. Ex vivo expansion of human mobilized peripheral blood stem cells using epigenetic modifiers. Transfusion. 2015;55(4):864–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Papa L, Zimran E, Djedaini M, Ge Y, Ozbek U, Sebra R, Sealfon SC, and Hoffman R. Ex vivo human HSC expansion requires coordination of cellular reprogramming with mitochondrial remodeling and p53 activation. Blood Adv. 2018;2(20):2766–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chai L. The role of HSAL (SALL) genes in proliferation and differentiation in normal hematopoiesis and leukemogenesis. Transfusion. 2011;51 Suppl 4(87S–93S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tatetsu H, Kong NR, Chong G, Amabile G, Tenen DG, and Chai L. SALL4, the missing link between stem cells, development and cancer. Gene. 2016;584(2):111–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang J, Chai L, Fowles TC, Alipio Z, Xu D, Fink LM, Ward DC, and Ma Y. Genome-wide analysis reveals Sall4 to be a major regulator of pluripotency in murine-embryonic stem cells. Proc Natl Acad Sci U S A. 2008;105(50):19756–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang J, Gao C, Chai L, and Ma Y. A novel SALL4/OCT4 transcriptional feedback network for pluripotency of embryonic stem cells. PLoS One. 2010;5(5):e10766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang J, Tam WL, Tong GQ, Wu Q, Chan HY, Soh BS, Lou Y, Yang J, Ma Y, Chai L, et al. Sall4 modulates embryonic stem cell pluripotency and early embryonic development by the transcriptional regulation of Pou5f1. Nat Cell Biol. 2006;8(10):1114–23. [DOI] [PubMed] [Google Scholar]
- 38.Yuri S, Fujimura S, Nimura K, Takeda N, Toyooka Y, Fujimura Y, Aburatani H, Ura K, Koseki H, Niwa H, et al. Sall4 is essential for stabilization, but not for pluripotency, of embryonic stem cells by repressing aberrant trophectoderm gene expression. Stem Cells. 2009;27(4):796–805. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.