

Abstract
A healthy aorta exerts a powerful cushioning function, which limits arterial pulsatility and protects the microvasculature from potentially harmful fluctuations in pressure and blood flow. Large artery (aortic) stiffening, which occurs with aging and various pathologic states, impairs this cushioning function and has important consequences on cardiovascular health, including isolated systolic hypertension, excessive penetration of pulsatile energy into the microvasculature of target organs that operate at low vascular resistance, and abnormal ventricular-arterial interactions that promote left ventricular remodeling, dysfunction and failure. Large artery stiffness independently predicts cardiovascular risk and represents a high-priority therapeutic target to ameliorate the global burden of cardiovascular disease. We provide an overview of key physiologic and biophysical principles related to arterial stiffness, the impact of aortic stiffening on target organs, non-invasive methods for the measurement of arterial stiffness, mechanisms leading to aortic stiffening, therapeutic approaches to reduce it, and clinical applications of arterial stiffness measurements.
Keywords: Arterial stiffness, systolic hypertension, heart failure, renal disease, dementia, liver disease, pre-eclampsia, vascular calcification, MGP
Condensed Abstract:
Large artery (aortic) stiffening occurs with aging and various disease states and has profound consequences on cardiovascular health. Aortic stiffening causes isolated systolic hypertension, abnormal ventricular-arterial interactions that contribute to heart failure, and target organ microvascular damage due to excessive pulsatility. Aortic stiffness independently predicts cardiovascular risk and represents a high-priority therapeutic target to ameliorate the global burden of cardiovascular disease. We review physiologic principles related to arterial stiffness, the impact of aortic stiffening on target organs, methods measurements of arterial stiffness, mechanisms of aortic stiffening, therapeutic approaches to reduce it, and applications of arterial stiffness measurements in the clinic.
Introduction
Systemic conduit arteries normally exert a powerful cushioning function, which delivers nearly steady flow in the microvasculature despite the intermittent left ventricular (LV) ejection. This cushioning function becomes impaired with large artery stiffening (LAS), leading to multiple consequences that have a major impact on cardiovascular health. First, LAS causes isolated systolic hypertension,(1) an endemic condition responsible for a large proportion of the global burden of cardiovascular morbidity and mortality, which is characterized by increased systolic blood pressure with normal or low diastolic blood pressure (i.e., increased pulse pressure). Second, LAS reduces coronary perfusion pressure and increases LV afterload, promoting LV remodeling, dysfunction and failure, even in the absence of coronary artery disease.(2) Third, given its impact on pressure and flow pulsatility, LAS promotes increased penetration of pulsatile energy into the microvasculature of target organs, particularly those that require high blood flow and therefore must operate at low arteriolar resistance.(3) Interestingly, although arterial wall stiffening precedes isolated systolic hypertension(1) and causally contributes to target organ damage, the arterial wall itself is a target organ, which is profoundly affected by aging and various pathologic states, including diabetes, obesity, smoking, hypercholesterolemia and chronic kidney disease (CKD).(4) Therefore, LAS plays a central role in a vicious cycle of hemodynamic dysfunction characterized by excess pulsatility that ultimately contributes to heart failure (HF), impaired coronary perfusion, CKD, cerebrovascular disease, and other chronic conditions. Consistent with the central role of arterial stiffness in cardiovascular function, measures of LAS independently predict the risk of incident cardiovascular events in both clinical and community-based cohorts (5,6).
In this review, we discuss: (1) basic principles and definitions related to arterial stiffness; (2) The general hemodynamic consequences of LAS; (3) Specific consequences of LAS on various organ systems; (4) Important non-invasive methods currently available for measurements of LAS in the clinic; (5) Mechanisms of LAS and potential therapeutic targets; (6) Clinical applications of LAS measurements (Central Illustration).
Central Illustration.

Role of large artery stiffness in health and disease. In young healthy adults, a compliant aorta (left): (a) effectively buffers excess pulsatilty due to the intermittent left ventricular ejection; (b) exhibits a slow pulse wave velocity (PWV), which allows pulse wave reflections to arrive to the heart during diastole, increasing diastolic coronary perfusion pressure but not systolic ventricular load. A number of factors (aging, lifestyle, etc.) increase aortic wall stiffness, which leads to several adverse hemodynamic consequences. Aortic stiffening leads to increased aortic root characteristic impedance (Zc) and forward wave amplitude on one hand, and premature arrival of wave reflections to the heart on the other. These hemodynamic changes result in adverse patterns of pulsatile load to the left ventricle in systole and a reduced coronary perfusion pressure in diastole, ultimately promoting myocardial remodeling, dysfunction, failure and a reduced perfusion reserve (even in the absence of epicardial coronary disease). This adverse hemodynamic pattern also results in excessive pulsatility in the aorta, which is transmitted preferentially to low-resistance vascular beds (such as the kidney, placenta and brain), because in these organs, microvascular pressure is more directly coupled with aortic artery pressure fluctuations. PWV=pulse wave velocity; Zc=characteristic impedance; BP=blood pressure.
Because it would be impossible to comprehensively reference all important contributions in this rapidly growing field, a limited number of original research publications are cited in this review. More extensive references to original research contributions can be found in other recent publications (7–12).
Basic principles and definitions related to arterial stiffness
Stiffness is the resistance offered by an elastic body to deformation. Although wall stiffness cannot be measured directly in vivo, indices of arterial stiffness can be obtained via: (1) Assessment of the relation between changes in arterial pressure and changes in arterial volume, cross-sectional area or diameter; (2) Assessment of arterial pulse wave velocity (PWV), a functional parameter influenced by arterial wall stiffness.
Quantification of arterial stiffness from changes in pressure vs. volume, cross-sectional area or diameter
The overall buffer (cushioning) capacity of the arterial tree can be quantified by the total arterial compliance, which is the change in volume of the entire arterial tree for a given change in pressure (Table 1). Compliance can be approximated as the ratio of stroke volume to arterial pulse pressure (PP), or more properly estimated from the diastolic pressure decay, or by fitting windkessel models to pressure and flow waveforms.(2) It is, however, not often used in clinical research, as it requires measurement of aortic pressure and flow, and the absolute value is highly dependent on body size.
Table 1.
Commonly used Indices of arterial stiffness
| Index | Definition | Formula | Comments |
|---|---|---|---|
| Volume compliance | Change in arterial volume relative to the change in arterial pressure | Expressed in m1/mmHg (or equivalent volume/pressure units). Influenced by wall stiffness, arterial size and wall thickness (and thus also by body size). Most often used to quantify the total arterial compliance. The latter is virtually impossible to measure directly in vivo, but can be approximated as the ratio of stroke volume to pulse pressure (SV/PP) or estimated from the diastolic decay time or fitting a windkessel model to measured pressure and flow (with different models yielding slightly different values). | |
| Area compliance | Absolute change in cross-sectional area relative to the change in arterial pressure | ; mostly calculated as | Expressed in mn2/mmHg (or equivalent area/pressure units). Estimation of compliance based on cross-sectional (rather than volume) measurements. In case of availability of pressure and area waveforms as a function of time, area compliance can be calculated at any pressure level as the local tangent to the pressure-area relation (dA/dP). In practice, it is often calculated from systolic-diastolic differences in area (ΔA) and pressure (ΔP), ignoring the pressure-dependency of compliance and yielding one single value. It relates linearly to volume compliance under the assumption of a perfectly homogeneous arterial segment and no changes in segmental arterial length. The dependence on vessel caliber makes area compliance less useful for clinical studies. |
| Assuming circular cross-section, equals | |||
| Distensibility and distensibility coefficient (DC) | Fractional change in cross-sectional area relative to the change in arterial pressure | , mostly calculated as | Area compliance normalized for arterial caliper, and thus expressed in 1/mmHg or %/mmHg (relative change in lumen cross-sectional area per unit increase in pressure). Just like area compliance, the distensibility can be calculated at any given pressure level, but is most often derived using diastolic-systolic pressure and area differences, and normalized to diastolic lumen area and is known as the distensibility coefficient DC. |
| Assuming circular cross-section, equals | |||
| Pulse Wave Velocity | Speed at which the arterial pulse propagates within the arteries | Bramwell-Hill equation: | Pulse Wave Velocity (PWV) is typically expressed in m/s. For a uniform tube, PWV relates to distensibility via the Bramwell-Hill equation, demonstrating the inverse relation between both. The Bramwell-Hill equation is an elegant way to convert distensibility into PWV, which then expresses the local wave propagation speed at that specific point in the circulation. PWV is easily measured as the ratio of the distance Ax between two measuring sites, and the time delay Δx of the arterial pulse between these sites. The clinical reference method is carotid-femoral pulse wave velocity, measured from these 2 easily accessible measuring sites, but several other variants exist. |
| PWV = distance/time |
ΔP is the difference between maximum and minimum pressure. ΔV is the difference between the corresponding maximum and minimum volume. ΔA is the difference between maximum and minimum cross-sectional vessel area. Dmax is the maximum lumen diameter, Dmin is the minimum lumen diameter.
While total arterial compliance integrates the cushioning function of the entire arterial tree, the local mechanical properties of an artery can be quantified from the relation between intra-arterial pressure and cross-sectional area (A), with the tangent to the pressure-area curve defined as the area compliance (CA=dA/dP, where dA is the change in cross-sectional area associated with a change in pressure, dP; Figure-1A). When pressure is varied over a large enough pressure range, the pressure-area relation exhibits a non-linear shape, with maximal CA reached in the low-pressure range, rapidly declining at higher pressures. This non-linear pressure-dependent behavior is a consequence of the composition of arterial wall, with the load taken by elastin in the artery at low pressure (stretch), and stiffer collagen fibers being progressively recruited as pressure increases (Figure-1A). In practice, within single heartbeats, the non-linearity of the pressure-area relation is often ignored, because it is measured over a relatively narrow pressure range (i.e., between diastolic and systolic blood pressure). Thus, for area compliance computations, dP is substituted by the arterial PP, and dA is substituted by the diastolic-to-systolic increase in lumen cross-sectional area. Area compliance can be normalized to the size of the vessel, yielding the distensibility coefficient (Table 1). The distensibility coefficient is most often calculated from cyclic changes in arterial diameter, assuming a circular cross-sectional area. Of note, in all these computations, PP needs to be known at the site of arterial area measurement. This is an important consideration, given that PP varies along the arterial tree due to the phenomenon of PP amplification.(2,7,8) Key definitions, formulas and concepts for commonly used parameters of arterial stiffness are summarized in Table 1.
Figure 1. Compliance, distensibility and PWV.


A) Typical relationship between intra-arterial pressure and lumen area when varying the pressure over a sufficiently large range. The area compliance (red line) is the slope to the pressure-area relationship, which can be calculated at any pressure level. At low pressure, the load is mainly taken by elastin, and the artery has a high compliance. As pressure increases, the load is progressively shifted to stiffer collagen fibers, leading to a functionally lower compliance. Vascular smooth muscle tone also affects compliance, particularly in distal aortic segments and intermediate-sized arteries. Normalizing area compliance to the local radius yields the distensibility coefficient (see Table 1). B) For a homogenous tube, the distensibility coefficient (Dist coeff) is theoretically linked to the PWV via the Bramwell-Hill equation (where ρ is the density of the blood) in an inverse, non-linear fashion; an increase in PWV by a factor 2 (which is about the change observed in humans from the age of 20 to the age of 70) implies a decrease in distensibility by a factor 4. An alternative formulation is the Moens-Korteweg equation, linking PWV to the stiffness of the wall material (incremental elastic modulus, Einc), the wall thickness (h) and lumen diameter (D).
Quantification of arterial wall stiffness from the speed of wave travel
Because of the distensibility of the arteries, the pulse generated by the ejecting heart propagates as a wave through the arterial wall, with a given speed, the PWV. For a uniform and infinitely long tube, the Bramwell-Hill equation provides the theoretical link between the distensibility of the tube and the propagation speed of the pulse (Figure-1B). Similarly, as demonstrated by the Moens-Korteweg equation, PWV is a mathematical function of the stiffness (elastic modulus) of the arterial wall material (Figure-1B). As such, PWV provides a straightforward and tangible way to quantify arterial stiffness in vivo: the stiffer the artery, the higher PWV.
PWV (usually expressed in m/s) is in principle calculated as the ratio of the distance (Δx) between two arterial sites, and the time delay (Δt) of the pulse between these sites, as shown in Figure 1B. The timing of the pulse in key arterial locations can be inferred from the timing of the pressure upstroke (measured with, for instance, non-invasive arterial tonometry) or from the timing of the flow upstroke (measured with, for instance, Doppler ultrasound).
The primary target for clinically-relevant arterial stiffness measurements is the aorta, because it is the largest and most distensible artery in the body, and also the most prone to abnormal stiffening in response to the cumulative exposure to hemodynamic loading and risk factors. Accordingly, LAS, but not medium-sized (i.e., muscular artery) stiffness is a strong independent predictor of cardiovascular risk (7).
Hemodynamic Consequences of LAS
LAS has complex hemodynamic effects, mediated via: (1) Effects on the early aortic systolic pressure rise; (2) The speed at which the cardiac pulse travels in the arterial wall;(2,13) (3) Effects on wave reflections at first-order bifurcations.
Effect on the early-systolic aortic pulse pressure rise
At the beginning of each cardiac cycle, the heart generates a forward-traveling pulse that increases pressure and flow in the proximal aorta. The amount of pressure increase required to push the pulse flow through the aortic root in early systole depends on the local impedance offered by the proximal aorta. This local impedance, called the characteristic impedance (Zc), increases due to aortic root wall stiffness, a small aortic caliber, or both.(2) Interestingly, Zc is much more sensitive to size than to stiffness. Therefore, aortic dilation can partially overcome the hemodynamic consequences of aortic root stiffening, and conversely, wall stiffening in the absence of eccentric remodeling of the root can have particularly pronounced effects on pulse pressure. However, excessive aortic dilation with aneurysm formation is clearly deleterious and associated with a risk of aortic rupture.
For general reference, it is worth clarifying the difference between arterial characteristic impedance and resistance. Resistance (the ratio of mean pressure to mean flow) occurs due to the friction that blood flow experiences from shear near the vascular wall and is mainly determined by vessel size: resistance is negligible in vessels larger than ~0.3–0.4 mm and is thus a microvascular property. In contrast, characteristic impedance pertains to pulsatile pressure and flow and is a macrovascular property.
Effect on forward and backward wave speed (Figure 2)
Figure 2. Consequences of increased aortic stiffness on the central pressure waveform.

The left side represents what occurs with a highly compliant aorta, as occurs in young healthy adults; the right side represents a very stiff aorta, as occurs in the elderly and/or in the presence of various disease states. A stiff aorta is associated with a high-ampitude forward wave and a faster PWV, determining a faster forward and backward (reflected) wave speeds. This determines an earlier arrival of wave reflections to the aorta, with progressive loss of diastolic pressure augmentation and increased late systolic augmentation. The figure illustrates 2 rather extreme situations, with most people falling “in between”. Moreover, it should be emphasized that the apparent distance to reflection sites is highly variable between individuals and may become a dominant determinant of the timing of wave reflections in populations that exhibit stiff aortas or in the setting of hypertension. The term “apparent” is used because reflection sites can modify the morphology of reflected waves making it appear “shifted” in time (i.e., a shift to the right appears as if a reflection site is further away from the heart, for any given PWV). FW=forward wave. BW=backward wave.
The energy wave generated by the LV (incident wave) is transmitted by conduit vessels and partially reflected at innumerable sites of impedance mismatch throughout the arterial tree, including points of change in wall diameter, change in material properties along the artery wall and points of branching. A myriad of scattered tiny reflections from individual reflection sites are conducted back to the proximal aorta and merge into a net reflected wave, which appears discrete (and is often mistakenly interpreted as arising from a single reflection site at the “effective” length of the arterial tree). As will be discussed further in this review, given the highly distributed nature of wave reflections, individual reflection sites have little importance relative to the bulk of reflections originating elsewhere (except for strong reflection sites in specific conditions, such as aortic coarctation or complete distal aortic occlusion).
The timing of arrival of reflected waves to the proximal aorta is highly dependent on the PWV of conduit vessels, particularly the aorta, which transmits both forward and backward traveling waves. Stiffer aortas conduct waves at greater PWV, and therefore promote an earlier arrival of the reflected waves for any given distance to reflection sites.
In the presence of a slow PWV, as occurs in young, healthy adults, wave reflections arrive at the aorta predominantly during diastole, augmenting diastolic pressure. When PWV increases due to wall stiffening, however, reflections arrive at the aorta in mid-to-late systole, producing systolic pressure augmentation, with loss of diastolic pressure augmentation and widening of the aortic PP (Figure 2). This phenomenon may be totally missed or underestimated by PP measurements in the brachial artery, due to 2 reasons: (1) the reflected wave tends to augment central systolic pressure to a greater degree than brachial pressure; (2) The reflected wave not only increases central peak pressure, but is has a predominant effect on the morphology of the pressure waveform, with a pronounced mid-to-late systolic component relative to early systolic and diastolic pressure. For any given absolute blood pressure level, this time pattern is a key determinant of abnormal ventricular-vascular interactions,(8,14,15) as will be discussed further in this review. It is noteworthy that individuals with identical central PP values can have markedly different central pressure profiles.
Interestingly, just like the amplitude of the reflected wave is partially dependent on the amplitude of the forward wave, wave reflections can re-reflect at the LV, becoming rectified and thus contributing to the forward wave.(16) In the presence of very high PWV, as occurs with advanced age and/or disease, multiple reflections and re-reflections occur in a single cardiac cycle, and forward and backward waves are highly dependent on each other. Finally, it is important to note that wave reflections and arterial stiffness do not necessarily vary in the same direction. In general, arterial stiffness modulates the effects of wave reflections on the central pressure profile by affecting the timing of their arrival to the proximal aorta. However, wave reflections can vary independently of arterial stiffness, due to modulation of muscular artery (17) and microvascular tone. Finally, although the timing of wave reflections is correlated with LAS, there is also great variability in the distance to reflection sites, such that this relationship is not perfect. Moreover, the variability in the distance to reflection sites can become the dominant determinant of the timing of wave reflections within patient populations that in general exhibit stiff aortas (such as older, diseased populations) or in the setting of hypertension.
Effect of aortic stiffness on wave reflections in first-order bifurcations?
In young adults, aortic PWV is much lower than the PWV of intermediate-sized muscular arteries. With aging, aortic PWV increases and matches the PWV of these more distal segments. This has led to the proposition that increased PWV with aging produces impedance matching with the stiffer muscular arterial segments, reducing wave reflections at first order bifurcations (18). This principle has been extended to a controversial proposition of a protective reflection at first-order branches of target organs such as the brain and the kidney. This theory has been met with relatively little critical analysis and has often been misinterpreted in the literature. Several aspects of this model are inconsistent with physiologic considerations. First, wave reflections at the inlet of target organs amplify the PP, and thus increase it in daughter branches to a much greater degree than they prevent energy penetration. This is because PP amplification into the branches is linearly related to the local reflection coefficient, whereas prevention of energy penetration is related to its square root; for example, a local reflection coefficient at the aortic-carotid interface of 0.10 would prevent only 1% of energy penetration but would increase carotid pulse pressure by 10%. Second, impedance matching does not equal PWV matching between parent and daughter vessels, but largely depends on vessel size (specifically, the ratio of the area of the parent vessels to the sum of the areas of the daughter vessels). This is due to the fact that, as discussed earlier in this review, the characteristic impedance of a vessel is much more dependent on its caliber than on its stiffness. The geometry of vessels at first-order bifurcations tends to be already optimized for forward energy transmission (19), such that impedances are relatively well-matched even before the aorta stiffens with age. Third, due to this matched geometry, reflections at single bifurcations are rather small relative to the composite reflected wave arising from the sum of innumerable tiny reflections elsewhere. For example, reflection coefficients as small as 0–0.15 have been reported for the aorta-carotid interface,(18) which would prevent only up to ~2.25% of energy transmission into the carotid artery. This contrasts with systemic reflection coefficients of ~0.5 or higher, which in sum return as much as ~25% of the energy contained in the pulse. Fourth, the bulk of reflections originating outside the target organs (such as the brain and kidney) and returning to the aorta, penetrate these organs as forward waves. Furthermore, arteries upstream of the carotid appear to be more effective at attenuating high-frequency flow pulsations (such as those produced by the initial cardiac contraction) than low-frequency oscillations (contained in the reflected wave from the lower body),(20) suggesting that the latter wave more effectively penetrates the cerebral microvasculature. Therefore, a protective effect of wave reflections at the inlet of target organs is not substantiated from a theoretical standpoint, not demonstrated from a clinical standpoint, and its quantitative importance remains to be determined by rigorous studies that account for the complexity of whole-system hemodynamics. What is unquestionable is that increased pulsatile energy in central arteries can penetrate and damage the microvasculature, particularly when local arteriolar tone/resistance (which protects more distal microvessels) is low.
LAS and Target Organ Damage
LAS increases markedly with aging (particularly after age 50) and in the presence of various cardiovascular risk factors such as diabetes (7). LAS mediates increased central arterial pressure and flow pulsatility, which has ill-effects in various organs, including the left heart (which directly interacts with the pulsatile load imposed by aorta and downstream wave reflections) and organs that require “torrential” blood flow, and thus must operate at low levels of local microvascular resistance.
Systemic organs that exhibit richly-perfused low-resistance vascular beds include the kidney, the placenta, the brain, and to a lesser extent, the liver and testicles (Figure 3). Both increased pulsatile pressure (barotrauma) and pulsatile flow (with excessive shear forces from increased pulsatile flow velocity) can result in microvascular damage in these organs. Myocardial vessels also exhibit low resistance, but are protected during systole by the myocardial contraction, which extrinsically compresses intra-myocardial blood vessels. Therefore, it is useful to discuss the cardiac consequences of arterial stiffening separately from consequences in other target organs. A list of potential consequences of arterial stiffening on various target organs is shown in Figure 4.
Figure 3. Arterial blood flow per unit of tissue mass in various organs.

The kidney, placenta-fetal unit, heart, brain, liver and testicle are shown. Although liver blood flow is much higher than shown, most of it is from the portal vein (not accounted for in the graph, which represents only arterial flow).
Figure 4. Potential consequences of LAS on target organs.

LAS can impact the heart through effects on afterload (particularly, modulation of the timing of wave reflections) and may damage low-resistance organs via excess arterial pulsatility. Importantly, not all these potential consequences are similarly well-established. In general, there is a large body of evidence supporting deleterious effects on the kidney and heart, moderate evidence for the brain and the placenta, and early/limited evidence for the liver and testicles (see text for details). ASH=Non-alcoholic steatohepatitis.
Arterial stiffness and the Heart
The adverse consequences of LAS and abnormal pulsatile hemodynamics on the heart have been recently reviewed (8) The aorta affects ventricular-arterial interactions via: (1) Its impact on the early systolic pressure rise, which is a key determinant of peak myocardial wall stress; (2) Its impact on the timing of arrival of wave reflections to the LV, which affects LV remodeling, fibrosis, diastolic dysfunction and coronary perfusion pressure (Figure 5).
Figure 5. Cardiac Consequences of Arterial Stiffening.

Earlier arrival of wave reflections favor a loss of coronary perfusion pressure on one hand, and increased mid-to-late systolic load on the other, due to a shift of wave reflection arrival from diastole to systole. In populations with stiff aortas, muscular artery function may become a key determinant of the apparent distance to reflection sites and thus the effects of wave reflections on the central aorta, for any given PWV. PWV=pulse wave velocity. FW=forward wave. BW=backward wave.
The aortic property that governs the aortic and LV pressure rise associated during early systolic ejection is the characteristic impedance of the proximal aorta, which as stated previously, is dependent on aortic root stiffness and caliber. Peak myocardial wall stress occurs in early systole, when systolic pressure coexists with quasi-diastolic LV geometry (thinner wall and larger cavity), consistent with Laplace’s law (21). In general, in the absence of ischemia, the LV myocardium seems to be able to adapt relatively well to increased load during this early period of contraction (22). However, peak myocardial wall stress is a key determinant of myocardial oxygen consumption (23), implying that a high aortic root characteristic impedance increases myocardial oxygen demand.
As stated previously, when PWV increases, reflected waves arrive at the heart during mid-to-late systole rather than diastole, which has deleterious effects on the myocardium, including: (a) An increase in mid-to-late systolic load (relative to early systolic load); (b) A decrease in the area under the pressure curve in diastole, which is a key determinant of coronary blood flow (Figures 2 and 5).
A large body of evidence now indicates that pulsatile afterload during mid- and late-systole has adverse consequences on LV remodeling and function (8,24,25). Experimental studies in animal models demonstrate that increased late systolic load leads to LV hypertrophy and fibrosis (24), and these findings are supported by human studies (26,27) Experimental studies also demonstrate that late systolic load impairs LV relaxation and systolic-diastolic coupling (28), and in support of these findings, late systolic load is independently associated with diastolic dysfunction in clinical and community-based cohorts (29–31) and strongly predicts HF in the general population.(32,33) Late systolic load is also associated with atrial dysfunction(34) and independently predicted the risk of atrial fibrillation in the Framingham Heart Study.(35) Interestingly, direct measures of LAS (carotid-femoral PWV) independently predicted incident heart failure in some studies (6,36), but this finding has been inconsistent.(37) Augmentation index (a rough measure of late systolic hypertension) was a more robust predictor of new-onset atrial fibrillation than CF-PWV in the Framingham Heart Study (35). These findings are consistent with our understanding of the effect of LAS on arterial load, which critically depend on its modulation of the timing of wave reflection, which is variable between individuals due to differences in the apparent distance to reflection sites. Therefore, direct measurements of LV pulsatile afterload and late systolic load (which can be accomplished with analyses of time-resolved aortic pressure and flow waveforms),(8) rather than measurements of LAS per se, appear to most informative for the prediction of HF and atrial fibrillation risk, and for the phenotypic characterization of patients with established HF.
Finally, a high PWV with premature arrival of wave reflections to the proximal aorta determines a loss of diastolic pressure augmentation by wave reflections (Figures 2 and 5). This, in addition to the reduction in aortic compliance, determines a steep diastolic pressure decay, reducing the area under the diastolic pressure curve, which is a key determinant of coronary perfusion.(38) Given that LAS increases systolic load and reduces coronary perfusion, it contributes to a reduction in the myocardial supply/demand ratio, and may partially exhaust the coronary perfusion reserve at rest, particularly in women,(39) who exhibit shorter distances to reflection sites and earlier arrival of wave reflections to the proximal aorta.
Arterial stiffness and the kidney
In the hypertension literature, the kidney has often been described as a villain and a victim, given its ability to both influence blood pressure regulation (villain) and its vulnerability to damage from hypertension over time (victim)(40). With respect to arterial stiffness, it is likely that the same labels apply, since declines in kidney function are associated with the factors known to increase LAS, which in turn promotes further kidney function loss.
The kidney exhibits the highest flow rate and lowest vascular resistance of any large organ. This low resistance/high blood flow state renders the kidneys highly susceptible to trauma from pulsatile pressure and blood flow, which can damage the glomeruli, leading to albuminuria and a reduced glomerular filtration rate (41).
Several studies demonstrate that in established CKD, carotid-femoral PWV increases, particularly as renal function declines, and much more so in age-matched diabetics with CKD compared to non-diabetics (42,43).
CKD enhances LAS due to several mechanisms (Figure 6). Impaired kidney function results in dysregulation of bone and mineral metabolism. Serum concentrations of osteoprotegerin and fibroblast growth factor-23, which increase in CKD, are associated with greater LAS (44,45). Vascular calcification is more prevalent in patients with CKD and end-stage kidney disease, and is strongly associated with increased LAS.(46) Serum levels of inflammatory biomarkers such as transforming growth factor-beta, hsCRP, tumor necrosis factor-alpha, and interleukin-6, are elevated and CKD, and are significantly associated, in a cross-sectional fashion, with LAS.(47) CKD is also characterized by a reduced ability to handle a sodium load (48), detrimental increases in the renin-angiotensin-aldosterone axis,(49) and sympathetic hyperactivity,(50) all of which also affect adversely LAS.
Figure 6. Impact of LAS on the kidney.

The kidney and the arterial wall exert important influences on each other and this interaction may lead to a vicious circle of arterial stiffening and the appearance/progression of chronic kidney disease and its complications. RAAS=renin-angiotensin-aldosterone system.
LAS independently predicts adverse cardiovascular outcomes in CKD. In the chronic renal insufficiency cohort (CRIC) study, a large longitudinal study of CKD patients in the USA, participants with higher carotid-femoral PWV were more likely to develop incident hospitalized HF,(6) progression to end-stage kidney disease, and all-cause death, independent of office blood pressure.(51) In a French study, patients with end-stage kidney disease on hemodialysis were far less likely to survive when their PWV remained elevated on antihypertensive treatment (52).
Arterial stiffness and the brain
The brain accounts for <2% of the average adult body weight, but utilizes ~25% of the body’s glucose and 20% of the body’s oxygen in order to meet metabolic demands.(53) The brain is densely vascularized and requires high-flow, low-resistance arterial hemodynamics, and is thus susceptible to pulsatile microvascular trauma on one hand, and associated regional hypoperfusion on the other.(3) The white matter is far less vascularized and receives less perfusion than the gray matter, making it more sensitive to hypoperfusion. Maintaining consistent and well-regulated cerebral perfusion is essential for proper cognitive and physical function, and abnormalities in regional perfusion may be key for the progression of cognitive impairment and dementia.
Normally, cerebral blood flow (CBF) is remarkably constant and is affected by cardiac output, LAS, pressure gradients along arteries, and cerebral autoregulation. Atherothrombotic arterial disease is a well-established determinant of CBF and the risk for stroke. Emerging data suggest that arterial stiffness also increases the risk for subclinical brain infarction and incident stroke (5).
As LAS increases, higher aortic pulsatility occurs, which is transmitted into the cerebral microcirculation, which may promote brain microvascular remodeling and impaired oxygen delivery. Excess pulsatile energy delivered to the cerebral microvasculature may also impair cerebral autoregulation, a multifactorial process of the cerebral circulation which regulates vascular resistance and perfusion through myogenic, autonomic, and metabolic mechanisms.(54) While cerebral autoregulation remains incompletely understood in humans, it is often modeled by CBF (at rest) and its response to a functional challenge (such as exercise, hypercapnia or orthostatic changes), which is generally termed cerebrovascular reactivity (CVR). CVR is lower among individuals with hypertension and greater LAS.(55) Studies of CVR and cognitive impairment are inconsistent, but favor reduced CVR among individuals with mild cognitive impairment, the prodromal phase of dementia.(55,56)
LAS, measured by carotid-femoral PWV, has been consistently associated with cognitive impairment, cognitive decline and incident dementia.(11,57,58) and with imaging evidence of regional brain atrophy and cerebral small vessel disease, including white matter hyperintensities, lacunar infarcts, cerebral microbleeds, and enlarged perivascular spaces (Figure 7).(59,60) LAS is also associated with subtle changes in the content of the white matter microstructure indicative of hypoperfusion and the future development of white matter hyperintensities.(61)
Figure 7. Arterial stiffness and age-related changes in the brain commonly seen in Alzheimer’s disease and related dementias.

These changes include declines in cerebral perfusion to the gray matter, evidence of multiple forms of cerebral small vessel disease [lacunar infarcts (white circles), white matter hyperintensities (white streaks), cerebral microbleed (red triangles), enlarged perivascular (Virchow-Robin) spaces], loss of brain volume, and ß-amyloid deposition in the brain (brown plaques). Such changes are suspected to affect the integrity of the white matter and neurovascular unit.
There are emerging relationships between vascular risk factors and pathologic hallmarks of Alzheimer’s disease, such as protein aggregation of fibrillar β-amyloid (Aβ) and phosphorylated tau that precede brain atrophy and cognitive impairment.(62) Aβ proteins begin to aggregate and deposit as fibrillar Aβ plaques in the neocortex, subcortex and vasculature. Tau protein deposits in the entorhinal region and progresses towards the cortex and more closely related to atrophy and cognitive impairment.(62) Arterial stiffness is associated with the extent and progression of cerebral Aβ plaque burden quantified by positron-emission tomography imaging among non-demented older adults.(63,64) Studies assessing the relationship between arterial stiffness and tau aggregation have not yet been performed.
Although LAS is associated with multiple aspects of pathology in Alzheimer’s disease and related dementias, it is important to note that such pathology rarely occurs in isolation. Neuropathology studies suggest that a majority of dementia cases are “mixed”, namely exhibiting vascular and Alzheimer’s disease features. LAS is associated with greater odds of concomitant cerebral small vessel disease and Aβ pathology in older adults.(63) Further, cerebral Aβ may contribute to impaired cerebrovascular functional responses. Aggregation of Aβ in the vessel wall, described clinically as cerebral amyloid angiopathy often co-occurs with classical parenchymal Aβ plaques in Alzheimer’s disease patients.(65) CAA is emerging as an important marker of risk for Alzheimer’s disease, microinfarction, brain microbleeds and macro-hemorrhage, and vascular cognitive impairment.
In conclusion, LAS is a key determinant of pulsatile hemodynamics and appears to lead to microvascular damage, CBF and hypoperfusion of the brain. Moreover, emerging data demonstrate associations between LAS and pathologic changes associated with Alzheimer’s disease and related dementias. Interventions targeting LAS could have the potential to prevent vascular dementia and pathologic changes associated with AD and related dementias.
Arterial Stiffness and the placental circulation
Given the fetal metabolic needs, the placenta must operate at very high-flow/low vascular resistance, and is second only to the kidney in regard to blood flow rates per unit of tissue mass (Figure 3). Pregnancy is associated with a pronounced increase in stroke volume, which must be met with low LAS to prevent large increases in PP. Indeed, normal pregnancy is associated with a decrease in LAS throughout most of the gestational period, which increases towards the end of pregnancy.(66)
When compared with their healthy pregnant counterparts, women with established preeclampsia (a late pregnancy complication characterized by hypertension and proteinuria, sometimes also associated with damage to other organs such as the liver) exhibit increased LAS.(66) Interestingly, several studies now indicate that differences in LAS and pulsatile hemodynamics precede the development of late pregnancy complications. It has long been known that a high PP early in pregnancy is predictive of subsequent pre-eclampsia, but not of uncomplicated gestational hypertension.(67) More recent studies assessed the relationship between LAS and subsequent obstetric complications. A recent meta-analysis of such studies(68) demonstrated that carotid-femoral PWV is increased in women who subsequently develop preeclampsia or intrauterine growth retardation (a condition in which the fetus is smaller than expected for gestational age). It remains to be determined whether LAS causally contributes to late pregnancy complications (thus representing a target for treatment) and whether LAS measurements can be used as predictive tests for these complications to facilitate early interventions.
LAS, the Liver and Metabolic disease
The liver receives as much as ~25% of the cardiac output, and the hepatic artery provides approximately one-third of the liver blood flow (with the remainder being provided by the portal vein). The hepatic artery therefore exhibits high-flow (Figure 3) and Doppler characteristics of a low-resistance vascular bed. The microvasculature downstream of the hepatic artery constantly regulates arterial flow to maintain total blood flow to the liver constant, regardless of changes in portal vein flow. This regulatory process, termed the hepatic arterial buffer response, is recognized as an important mechanism to maintain metabolic homeostasis.(69)
Non-alcoholic fatty liver disease (NAFLD) is the most prevalent chronic liver disease, affecting ~20% of the population worldwide. NAFLD is strongly associated with the metabolic syndrome and diabetes, and can progress to non-alcoholic steatohepatitis (NASH), which is characterized by liver fibrosis.
Accumulating evidence demonstrates complex interrelationships between diabetes, NAFLD and arterial stiffness. First, diabetic individuals exhibit much higher carotid-femoral PWV than their non-diabetic counterparts,(7) even in the setting of established CKD (42,43) or HFpEF.(70) This has been assumed to be due to effects of diabetes on the arterial wall. However, recent data suggest that LAS precedes diabetes and predicts its future development. Among 2,450 older adults without diabetes at baseline enrolled in a large cohort study in southern Sweden, carotid-femoral PWV was recently reported to predict the future development of diabetes over ~4.4 years, with roughly a doubling and tripling of risk in the mid and highest PWV tertiles, respectively, compared to the lowest tertile. This association was independent of potential confounders. This study confirms a previous large study that demonstrated that PP independently predicted new-onset diabetes among 2,685 hypertensive patients enrolled in the Candesartan Antihypertensive Survival Evaluation in Japan (CASE-J) trial.
Multiple studies have demonstrated an association between NAFLD and LAS.(71) Furthermore, among patients with NAFLD, LAS may be associated with liver fibrosis. In a recent longitudinal study among 291 diabetics with NAFLD, serial carotid-femoral-PWV measurements and liver fibrosis assessments via transient elastography were performed over ~7 years. In this study, a high LAS and a steeper increase in LAS over time were associated with an increased likelihood of advanced liver fibrosis. These findings are consistent with a smaller study that examined histologic liver fibrosis among patients with NAFLD.(72)
Given the important intersection of hemodynamic arterial function and metabolic function in the liver, it is possible that LAS contributes to the progression of NAFLD and hepatic insulin resistance, but further studies are required to assess whether a causal relationship between LAS and liver fibrosis exists.
Arterial Stiffness and Testicular dysfunction
Several considerations also suggest a bidirectional relationship between testicular function and LAS. Androgen receptors can modulate vascular function and conversely, LAS with excess pulsatility may impact testicular microvascular function. Lower androgen levels are independently associated with greater LAS in older men.(73) The testicles exhibit Doppler features of a low-resistance vascular bed, suggesting that increased pulsatile energy in conduit vessels may damage the testicular microcirculation. An intact testicular microvascular network is essential to maintain oxygen delivery and uptake by testicular tissue.(74) Abnormal vasoactive responses and structural degeneration of the testicular microvasculature occurs with aging, which is thought to play an active role in promoting aging of the testis. In animal models, a reduced capacity to regulate testicular blood flow is sufficient to impact testosterone secretion, even in the presence of intact Leydig cell function (75). Although hemodynamic considerations and limited observational data suggest that LAS may contribute to microvascular damage in the testicles, further mechanistic research is required in this area.
Mechanisms of Arterial Stiffening and Therapeutic Approaches
The stiffness of an artery, and how stiffness changes with loading conditions (intraluminal pressure), is determined by the 3-dimensional architecture and interconnections of its major constituents. Large arteries have 3 main layers (Figure 8), and various processes in these layers can affect LAS. The medial layer is the main determinant of LAS, via its various constituents: the extracellular matrix proteins elastin and collagen, vascular smooth muscle cells (VSMCs), cell-matrix connections, and matrix constituents such as glycosaminoglycans interconnecting the extracellular matrix. In large arteries, these elements are organized in concentric elastin sheets, intertwined by smooth muscle cells and matrix material, along with collagen fibers running both in the axial and circumferential directions, providing mechanical strength to the artery. With progressive distance from the heart, arteries evolve from elastic (rich in elastin) to muscular (rich in VSMCs). The abdominal aorta in particular, exhibits transitional ultrastructure, with a significant amount of both elastic elements and VSMCs.
Figure 8. General Mechanisms of Arterial Stiffness.

Various mechanisms can increase the stiffness of the intima, media and adventitia. The media is however, the most important layer in large arteries. See text for details. NO=nitric oxide. AGE=advanced glycation end-products. RAAS=renin-angiotensin-aldosterone system. ERP=elastin-related peptides.
Multiple pathologic processes related to elastin fragmentation/degradation, collagen deposition, collagen and elastin cross-linking by advanced glycation end-products, VSMC stiffening, medial calcification, endothelial dysfunction and inflammation, can impact LAS, as summarized in Figure 8 and Table 2. It should be noted that these processes do not occur in isolation, but often interact to promote LAS. For instance, elastin fragmentation is often followed by collagen accumulation, and the generation of elastin degradation products (elastin-related peptides) may activate signaling pathways that promote VSMC osteogenic differentiation and vascular calcification.(76) Similarly, inflammatory pathways can lead to endothelial cell dysfunction and activation, and to the release of bone morphogenetic protein-2 by endothelial cells, which in turn promotes VSMC osteogenic differentiation and vascular calcification. Given the complex interrelationships in various processes listed in Figure 8 and Table 2, it is not surprising that pleiotropic mediators, such as various micro-RNAs (77) and Klotho (9) appear to exert a coordinated regulation of various processes involved in LAS, providing opportunities for therapeutic interventions.
Table 2.
General Mechanisms of Large Artery Stiffening
| Mechanism | General Description | Key Molecular Mediators |
|---|---|---|
Elastin Fragmentation
|
Elastin deposition in the arterial is limited to the fetal development period and infancy and is subsequently turned off; therefore, in adulthood, elastin fiber damage is essentially irreparable. Progressive elastin fragmentation/loss occurs due to: (a) Mechanical fatigue over the lifetime (cyclic stress); (b) Elastase-mediated proteolysis. |
|
Collagen deposition
|
Collagen deposits occur at the arterial wall (predominantly the media), including sites of elastin breakdown |
|
AGE-mediated collagen and elastin cross-linking
|
Glucose cross-links develop and AGEs form between proteins with a long half-life (such as collagen). Cross-linked collagen is more resistant to enzymatic proteolysis. Glycated elastin is more susceptible to degradation. Hyperglycemia leads to AGE formation, whereas renal dysfunction leads to reduced AGE elimination |
|
VSMC Stiffening/tone
|
Alterations in the VSMC cytoskeleton and integrin interactions with the extracellular matrix. Likely to be more important in distal aortic segments, which are richer in VSMCs. |
|
Calcification
|
Medial calcification is predominantly mediated by the osteochondrogenic differentiation of VSMCs. This process is different from the intimal calcification associated with atherosclerosis, which is predominantly mediated by chondrocyte-like cells of bone marrow origin. Adventitial calcification may also occur, which involves myofibroblasts and/or microvascular pericytes. |
|
|
Endothelial dysfunction |
Reduced NO production by the endothelium may play a role in regulating VSMC stiffness/tone in distal aortic segments like the abdominal aorta. Increased stiffness of the endothelial cell cytoskeleton that is in close to the membrane (cortical cytoskeleton) may also play a role in modulating stiffness |
|
Inflammation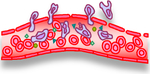
|
There are interconnections between inflammatory pathways and various mechanisms of large artery stiffening (elastin degradation, medial calcification, calcification, endothelial dysfunction) |
|
VSMC=vascular smooth muscle cell; MMP=matrix metalloproteinase; MGP=matrix GIa protein; FGF-23=fibroblast growth factor 23; BMP-2=Bone morphogenetic protein 2; Msx2=Homeobox Protein Hox-8; Wnt=willingness Int; PiT-1=phosphate transporter 1; ALP=alkaline phosphatase; AGEs=advanced glycation end-products; RAGE=receptor of AGEs; NO=nitric oxide.
Although a detailed description of potential therapeutic approaches to LAS is outside of the scope of this review, approaches under investigation include: (1) Anti-inflammatory therapies; (2) Direct inhibitors of vascular calcification (such as SNF472, a hydroxyapatite crystal growth inhibitor currently being tested to reduce vascular calcification in end-stage renal disease); (3) Agonists or activators of endogenous calcification inhibitory pathways, such as the Matrix Gla-Protein (MGP) pathway, discussed in more detail below; (4) Collagen cross-linking inhibitors or breakdown promoters; (5) Mineralocorticoid receptor antagonism (which has been shown to reduce carotid-femoral PWV independent of mean blood pressure in patients with CKD)(78); (6) Elastase inhibition; (7) Elastin-related peptide-signaling inhibition; (8) NO donors; (9) Micro-RNA therapy.
The MGP pathway represents a particularly attractive and clinically relevant therapeutic target. MGP is a small secretory protein produced by chondrocytes and VSMCs.(79) The inactive form of MGP (dephospho-uncarboxylated MGP, dp-ucMGP) undergoes serial post-translational carboxylation and phosphorylation to form active MGP (Figure 9). The active form of MGP is a potent inhibitor of arterial calcification by inhibiting bone morphogenetic protein-2-induced osteogenic differentiation of VSMCs, and via calcium phosphate scavenging. Carboxylation of MGP is dependent upon the availability of vitamin K in the vascular wall.(79) Interestingly, vascular vitamin K deficiency appears to occur at lower thresholds of severity than those leading to coagulopathy and can be completely unapparent on standard clinical testing. Since dp-uc-MGP (inactive MGP) is secreted into the circulation, it represents an interesting circulating biomarker of vascular vitamin K availability. An increase in circulating dp-uc-MGP has been shown to be independently associated with increased LAS in various populations, including the general population,(80,81) adults with diabetes,(82) hypertension,(83) chronic kidney disease(84), and HF with preserved or reduced ejection fraction.(85) Interestingly, warfarin, a vitamin K antagonist, inhibits MGP carboxylation/activation and is a potent promoter of vascular calcification in animal models. Warfarin use has been associated with LAS in HF(85) and with vascular and aortic valve calcification in various populations. Limited data suggest that vitamin K2 supplementation can ameliorate or reduce age-related arterial stiffening,(86) and this intervention is currently being tested to reduce aortic valvular calcification in a randomized trial. Further research is required before this approach can be recommended to reduce age-related LAS.
Figure 9. Vitamin K-dependent Matrix Gla protein (MGP) maturation.

MGP maturation requires vitamin K, and leads to the formation of pc-MGP (phosphorylated, carboxylated MGP), which is a strong inhibitor of vascular calcification via inhibition of Bone morphogenetic protein 2 (BMP-2) signaling and VSMC osteogenic-differentiation. Vitamin K deficiency in the vascular wall leads to the formation of uncarboxylated (uc-)MGP (which accumulates in the vessel wall) and dephosphorylated, uncarboxylated (dpuc-)MGP which can be measured in serum.
Finally, it is worth noting that with the likely exception of renin-angiotensin-aldosterone system blockers, standard antihypertensive medications do not change the intrinsic material properties of the arterial wall and largely act through reductions in vascular resistance or cardiac output. Nevertheless, blood pressure reduction by standard antihypertensive medications can lead to a reduction in operating stiffness, given the non-linear arterial compliance curve (Figure 1A). Therapies to safely improve the material properties of the arterial wall represent the “next frontier” in cardiovascular prevention, and could lead to marked reductions in the global burden of age-related chronic conditions that will occur even in the absence of atherosclerotic cardiovascular disease.
Methods to measure LAS
Practical considerations and the relative ease of measurement in large populations have made carotid-femoral PWV the current reference method for LAS. Carotid-femoral PWV takes advantage of the superficial location of the common carotid and femoral arteries, which arise from 2 distant locations in the aorta, and can be interrogated with pressure sensors (such as applanation tonometers) or Doppler probes applied to the body surface (Figure 10A–B). Carotid and femoral signals can be simultaneously or consecutively acquired, in the latter case with an electrocardiographic tracing in which the R wave serves as a reference point to compute the time delay of the pulse between these locations. The arterial path length (distance traveled by the pulse) is usually computed as 80% of the directly measured distance between the carotid and femoral arteries or by subtracting the sternal notch-to-carotid distance from the notch-to-femoral distance.(7,87) For clinical purposes, the former method is recommended (preferably on the right side of the body), since it provides more accurate path length estimations,(88) and was the method utilized to derive normative data in a recent large study.(87) Measurements made with other methods can be transformed to the standard value (i.e., value corresponding to 80% of the direct carotid-femoral distance) using specific formulas and/or an online calculator, as recently described in detail.(88) Recent data strongly suggest that equations based on standard clinical and anthropometric variables can be used to estimate carotid-femoral distance without the need for direct carotid-femoral distance measurements,(89) but this approach requires further study.
Figure 10. Methods of measurement of carotid-femoral PWV (A–C), CAVI (D) and ba-PWV (E) by various devices.

Sensors and examples of recorded signals are shown. C=carotid signal; F=femoral signal; P=phonocardiographic signal; B=brachial signal; A=ankle signal. 1: FDA-approved; 2: the vicorder uses a neck cuff, rather than a tonometry sensor.
Important drawbacks of carotid-femoral PWV include: (1) an unequivocal travel path does not really exist (while the pulse travels up into the carotids, it simultaneously travels down the descending aorta towards the femoral site); (2) the measurement bypasses the ascending aorta and part of the arch, neglecting the most distensible segment of the aorta, which also exhibits the earliest changes with aging; (3) the distance between measurement sites needs to be derived from surface measurements and thus represents only an approximation; (4) The acquisition of carotid and femoral pulse waveforms can be technically difficult with pressure sensors, particularly in obese individuals. More recent devices have replaced the common femoral artery tonometer with a blood pressure cuff applied to the proximal lower extremity (Figure 10C); further research is required to determine to what extent this modification impacts the accuracy and predictive ability of LAS assessments.
Another commonly used method is the acquisition of phonocardiographic data along with brachial and ankle cuff waveforms (Figure 10D), as done with the VaSera device. In this method, the heart-to-arm pulse transit time is first computed as the time elapsed between the 2nd heart sound (aortic valve closure) and the dicrotic notch (which marks end-systole) in the brachial waveform. Next, the time of pulse onset at the heart is computed via subtraction of the heart-to-arm transit time from the time of the upstroke in the brachial waveform. Finally, the heart-to-ankle transit time is computed as the time elapsed between the pulse onset at the heart and the upstroke of the ankle waveform. This method can estimate the heart-to-ankle PWV or a related parameter, the cardiac-ankle vascular index (CAVI), which is less blood pressure-dependent than PWV. CAVI has been shown to predict incident cardiovascular events in studies performed predominantly in Asian populations,(90) and has recently been incorporated into a large cohort study in the Multiethnic Study of Atherosclerosis (MESA). A potential disadvantage relative to carotid-femoral PWV is the inclusion of a long muscular arterial segment (femoral to ankle), which may confound LAS measurements. Therefore, the value of an alternative method using femoral cuffs instead of ankle cuffs is also being investigated in the MESA study.
Finally, brachial and ankle cuff waveforms can be utilized to compute the brachial-ankle PWV (baPWV). In this method, the delay between the brachial and ankle upstrokes is computed directly (Figure 10E), and the distance from the brachium to the ankle is calculated from body height. Although the simplicity of this method is attractive, there is not a true transit path between the brachial and the ankle locations, because the pulse simultaneously travels down to the arm as is it traveling down the arch and thoracic aorta. Similar to CAVI, the inclusion of long muscular arterial segments in the upper and lower extremities also represents a potential disadvantage of baPWV relative to carotid-femoral PWV. A recent meta-analysis that included 14,673 Japanese participants without cardiovascular disease at baseline (91) demonstrated an independent association between baPWV and incident of cardiovascular events.
Finally, it is worth mentioning some parameters that should not be considered reliable indicators of LAS. These include: (1) The effective arterial elastance (ratio of end-systolic pressure to stroke volume), which is not significantly influenced by LAS, but almost exclusively a function of heart rate and peripheral vascular resistance (a microvascular property)(92); (2) carotid-femoral PWV values estimated from pulse wave analysis of a single site (such as oscillometric brachial cuffs or finger sensors), which are highly confounded and not recommended by current guidelines(7). In particular, recent studies indicate that “PWV” values provided by 2 devices that utilize single-site brachial pressure waveforms are almost entirely a function of age and measured systolic blood pressure, rather than representing an incremental measure of LAS.(93,94) Whereas pulse wave analysis of a single arterial sites provides useful data for various hemodynamic analyses,(2,7,8,10,13) we strongly recommend against the use of such methods for the purposes of LAS estimations.
Finally, methods that utilize cardiac imaging modalities commonly used in clinical practice may allow more widespread assessment of LAS by clinicians. A promising method is the echocardiographic assessment of thoracic aortic PWV. With echocardiography, the timing of the pulse at the LV outflow tract (aortic inlet), the proximal descending aorta and the proximal abdominal aorta are easily interrogated, and such interrogations are already part of comprehensive echocardiographic clinical scans. A straightforward method can be applied to standard echocardiographic DICOM images to compute the time delay between these 3 locations (Figure-11). Methods to estimate the path length between these locations using anthropometric and echocardiographic parameters could be developed, opening the possibility to quantify aortic PWV with every clinical echocardiogram. Another method that is increasingly utilized is through-plane phase-contrast MRI, in which ascending and descending thoracic aortic flow can be simultaneously interrogated in cross-section, and the path length can be precisely measured from a 3D-reconstruction of the aorta (Figure-12). The main limitation of this method is its limited temporal resolution, which compromises its precision over the short path length interrogated, and is likely insufficient to adequately resolve fast PWVs in individuals with stiff aortas.
Figure 11. Echocardiographic Assessment of Thoracic Aortic transit time.

ECG signals and digital flow traces are extracted from DICOM images of pulsed-wave Doppler interrogations from the left ventricular outflow tract (LVOT, 1), descending aorta (DA, 2) and proximal abdominal aorta (AA, 3). Beats at each location relate consistently to the QRS complex, which can be used as a time-reference. The top left panel shows the signal averaged pulses at the 3 locations, clearly demonstrating the transit time. The development of methods to estimate aortic path length (distance) between 2 points using anthropometric and echocardiographic imaging data is under development, and could facilitate a broad application of PWV measurements in clinical practice.
Figure 12. Assessment of Thoracic Aortic PWV by through-plane phase-contrast MRI.

Magnitude and phase (top right panels) images of an aortic cross section are used to extract time-resolved flow curves. The path length can be directly measured from aortic images.
LAS, Cardiovascular Risk and Clinical Applications of LAS Measurements
As mentioned previously, a large body of evidence has demonstrated the prognostic value of LAS for the prediction of cardiovascular events, (5–7,90,91) particularly of carotid-femoral PWV, which is currently considered the reference method. It is interesting to note that although LAS and atherosclerosis are distinct processes from a mechanistic and pathologic standpoint, atherosclerotic risk factors and biologic processes overlap with those leading to LAS, such that LAS predicts multiple endpoints, including atherosclerotic and non-atherosclerotic cardiovascular events.
A recent participant-level meta-analysis of prospective studies including 17,635 participants among whom 1,785 experienced a cardiovascular event, found that each standard-deviation change in carotid-femoral PWV is associated with a 35%, 54% and 45% increase in the risk of coronary heart disease, stroke and overall cardiovascular disease, respectively. Carotid-femoral-PWV was an independent predictor of risk across multiple subpopulations (Figure 13) although the effect was more pronounced among participants aged 60 or younger, among whom there was a doubling of risk for every standard deviation increase.
Figure 13. Carotid-femoral PWV as a predictor of cardiovascular events in a recent meta-analysis of prospective studies.

Carotid-femoral-PWV was an independent predictor of risk across multiple subpopulations although the effect was more pronounced among participants aged 60 or younger. Reproduced from (5)
Age-specific reference values of carotid-femoral PWV have been well-established in several populations, with the largest sample provided by the European Reference Values for Arterial Stiffness’ Collaboration.(87) In this large sample, there was a marked increase in carotid-femoral PWV with aging, and with blood pressure categories at any particular age (Figure 14A). It should be noted that normative data in this study were generated using 80% of the direct carotid-femoral distance for path length estimations, and should be strictly applied using this method. Among subjects with optimal or normal BP values and no additional cardiovascular risk factors, 90th percentile values for carotid-femoral PWV were as follows: <30 years: 7.1 m/s; 30–39 years: 8 m/s; 40–49 years: 8.6 m/s; 50–59 years: 10 m/s; 60–69 years: 13.1 m/s and ≥70 years: 14.6 m/s. These values can be practically used to assess values of carotid-femoral PWV across the adult lifespan and in turn, enhance cardiovascular risk assessments. Furthermore, to the degree that LAS is a lifelong marker of vascular aging that precedes and predicts the development of hypertension,(1) it represents a simple, inexpensive and non-invasive marker that can be monitored across the lifespan. Age-specific values, which have been well defined, are best understood in the context of the lifetime course of the health-disease continuum, a concept emphasized in the report of the Lancet Commission on Hypertension.(95) In this model, the development of elevated blood pressure represents an avoidable threshold, which is preceded and subsequently accompanied by premature vascular aging.
Figure 14. Normal values of carotid-femoral PWV by age and blood pressure status established by the European Arterial Stiffness Collaboration (A) and concept of life-course of vascular aging.

PWV increases with age and BP category (A, reproduced with permission from (87)); note that blood pressure categories correspond to European guidelines, rather than to recent AHA/ACC categories. Panel B demonstrates the concept of life-course of vascular aging and preventable thresholds for cardiovascular disease across the health-disease continuum (Partially reproduced from (95)). QOL=quality of life.
To the degree that measurements of LAS predict cardiovascular risk independently of traditional risk factors, they can be used to enhance cardiovascular risk assessments in various clinical scenarios and can thus provide useful information for primary and primordial prevention strategies. Some suggested clinical applications are summarized in Table 3. In general, these include clinical scenarios in which a refinement of cardiovascular risk can guide clinician-patient discussions and/or enhance decision-making for intensification of lifestyle interventions, therapy initiation (such as statins or antihypertensive therapy) or frequency of cardiovascular risk assessments (particularly in younger individuals). LAS may also provide relevant information to evaluate younger individuals with a family history of isolated systolic hypertension, since a substantial proportion of the variability in LAS is inheritable (9). Finally, LAS measurements may be advantageous in special populations in which pulled cohort equations are unreliable; for instance, it is well know that equations derived from US populations can provide highly mis-calibrated absolute risk estimates other populations, particular those at earlier stages of the epidemiologic transition. To the degree that LAS measurements directly characterize the accumulation of subclinical vascular damage over many years, they should in principle be more robust than pulled cohort equations across populations.
Table 3.
Suggested clinical Applications of Measurements of LAS in primordial and primary prevention of cardiovascular disease
| Clinical Scenario | Rationale | Impact on Clinical Management | |
|---|---|---|---|
| Hypertension | ACC/AHA Stage 1 Hypertension (130–139/80–89 mmHg) with PCE-calculated 10-year ASCVD risk ~10% without diabetes or CKD | LAS can be useful to refine risk stratification when PCE-calculated 10-year ASCVD risk is close to the threshold for treatment, after an informed clinician-patient discussion. |
|
| Stage 2 isolated systolic hypertension (>140 mmHg) in very young adults with paucity of other cardiovascular risk factors | The combination of high pulse pressure amplification (with normal central systolic pressure) and low or normal LAS for age support a low CV risk |
|
|
| Non-hypertensive adults <40 years of age with a family history of ISH | LAS is partially heritable. LAS precedes and predicts the development of ISH, a potentially avoidable threshold in the life course of cardiovascular disease (see Figure 14B). A high PWV for age is consistent with early vascular aging. |
|
|
| Other CV Risk Assessment Scenarios | Refinement of cardiovascular risk assessment in non-diabetic adults 40–75 years of age at intermediate PCE-calculated 10-year ASCVD risk | In this group of patients, risk-based decisions for preventive interventions may be uncertain and LAS measurements can be utilized to refine risk assessment (particularly if various “risk enhancing” clinical parameters do not clearly favor a specific course of action). |
|
| Refinement of cardiovascular risk assessment in middle-aged non-diabetic adults at borderline PCE-calculated 10-year risk of ASCVD (5% to <7.5%) who also have other factors that increase their ASCVD risk (“risk enhancers”) | In this group of patients, LAS measurements may be useful to improve risk-based decisions as an alternative or as a “gate-keeper” for coronary calcium score testing, particularly when concerns about radiation exposure (younger age, overweight/obesity) or about cost are present |
|
|
| Assessment of CV risk in special populations | PCE-calculated 10-year risk estimations can provide notoriously mis-calibrated estimates in non-US populations, particularly those at earlier stages of the epidemiologic transition. This may also apply to immigrants from those populations in the US. |
|
ASCVD= atherosclerotic cardiovascular disease; CV=cardiovascular; PCE=pulled cohort equations.
Other potential clinical application of LAS and/or pulsatile pulsatile hemodynamics measurements that require further research include: (1) The identification of individuals with favorable responses to specific interventions, such as specific first-line antihypertensive regimens; (2) The prediction of late pregnancy complications (such as pre-eclampsia), which could facilitate more aggressive clinical monitoring/management; (3) The prediction of thoracic aneurysm growth (early data suggests that LAS is related to the rate of growth of aortic aneurysms) (96); (4) The assessment of optimal timing of interventions for aortic valve stenosis, since arterial pulsatile load and valvular load are additive, and the former may enhance the impact of the latter on the heart (97).
Conclusions
LAS has profound consequences for cardiovascular health and is likely responsible for a large proportion of the global chronic cardiovascular disease burden. LAS can be readily measured in clinical practice and can enhance cardiovascular risk prediction. Given considerations regarding the hemodynamic role of LAS and excess pulsatility on target organs, a better understanding of molecular processes leading to LAS should be aggressively pursued, in order to design effective therapies. LAS represents an unmet, high-priority therapeutic target to ameliorate the global burden of cardiovascular disease. Such LAS-related disease burden will remain (and likely grow) as atherosclerotic disease and cancer-related mortality is reduced, and represents the next frontier in cardiovascular disease prevention.
Highlights.
Large artery stiffness (LAS) is an important determinant of CV health and target organ damage.
LAS is an independent predictor of CV risk and may aid in clinical decision-making in various clinical scenarios.
Multiple mechanisms can lead to LAS, but further research is required to identify key molecular determinants of LAS in humans.
LAS represents a high-priority therapeutic target to ameliorate the global burden of CV disease.
Funding:
JAC is supported by NIH grants R01 HL 121510-01A1, R61-HL-146390, R01-AG058969, 1R01HL104106, P01HL094307 and R56 HL136730.
Abbreviations:
- LV
Left Ventricle
- LAS
Large Artery Stiffening
- CKD
Chronic kidney disease
- PWV
Pulse wave velocity
- PP
Pulse Pressure
- HF
Heart failure
- HFpEF
Heart failure with preserved ejection fraction
- Zc
Characteristic Impedance
- CBF
Cerebral blood flow
- CAVI
Cardiac-ankle vascular index
- baPWV
brachial-ankle pulse wave velocity
- CF-PWV
carotid-femoral pulse wave velocity
- VSMC
vascular smooth muscle cells
- CBF
cerebral blood flow
- CVR
Cerebrovascular reactivity
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: J.A.C. has consulted for Sanifit, Bayer, Bristol-Myers Squibb, OPKO Healthcare, Ironwood, Akros Pharma, Merck, Pfizer, Edwards Lifesciences, Microsoft and Fukuda-Denshi. He received research grants from National Institutes of Health, American College of Radiology Network, Fukuda-Denshi, Bristol-Myers Squibb and Microsoft. J.A.C. is named as inventor in University of Pennsylvania patent applications for the use of inorganic nitrates/nitrites for the treatment of Heart Failure and Preserved Ejection Fraction, novel methods of pulse wave analysis and neoepitope-based collagen biomarkers of tissue fibrosis in heart failure. Other authors report no disclosures.
References
- 1.Kaess BM, Rong J, Larson MG et al. Aortic stiffness, blood pressure progression, and incident hypertension. JAMA 2012;308:875–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chirinos JA, Segers P. Noninvasive evaluation of left ventricular afterload: part 2: arterial pressure-flow and pressure-volume relations in humans. Hypertension 2010;56:563–70. [DOI] [PubMed] [Google Scholar]
- 3.O’Rourke MF, Safar ME. Relationship between aortic stiffening and microvascular disease in brain and kidney: cause and logic of therapy. Hypertension 2005;46:200–4. [DOI] [PubMed] [Google Scholar]
- 4.Payne RA, Wilkinson IB, Webb DJ. Arterial stiffness and hypertension: emerging concepts. Hypertension 2010;55:9–14. [DOI] [PubMed] [Google Scholar]
- 5.Ben-Shlomo Y, Spears M, Boustred C et al. Aortic pulse wave velocity improves cardiovascular event prediction: an individual participant meta-analysis of prospective observational data from 17,635 subjects. J Am Coll Cardiol 2014;63:636–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chirinos JA, Khan A, Bansal N et al. Arterial stiffness, central pressures, and incident hospitalized heart failure in the chronic renal insufficiency cohort study. Circ Heart Fail 2014;7:709–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Townsend RR, Wilkinson IB, Schiffrin EL et al. Recommendations for Improving and Standardizing Vascular Research on Arterial Stiffness: A Scientific Statement From the American Heart Association. Hypertension 2015;66:698–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weber T, Chirinos JA. Pulsatile arterial haemodynamics in heart failure. Eur Heart J 2018;39:3847–3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pierce GL. Mechanisms and Subclinical Consequences of Aortic Stiffness. Hypertension 2017;70:848–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nichols WW, O’Rourke MF, Vlachopoulos C. McDonald’s Blood Flow in Arteries: Theoretical, Experimental and Clinical Principles, 6th Edition. Hodder Arnold; 2011. [Google Scholar]
- 11.Rabkin SW. Arterial stiffness: detection and consequences in cognitive impairment and dementia of the elderly. J Alzheimers Dis 2012;32:541–9. [DOI] [PubMed] [Google Scholar]
- 12.Hughes TM, Craft S, Lopez OL. Review of ‘the potential role of arterial stiffness in the pathogenesis of Alzheimer’s disease’. Neurodegener Dis Manag 2015;5:121–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chirinos JA, Segers P. Noninvasive evaluation of left ventricular afterload: part 1: pressure and flow measurements and basic principles of wave conduction and reflection. Hypertension 2010;56:555–62. [DOI] [PubMed] [Google Scholar]
- 14.Chirinos JA. Deep Phenotyping of Systemic Arterial Hemodynamics in HFpEF (Part 2): Clinical and Therapeutic Considerations. J Cardiovasc Transl Res 2017;10:261–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chirinos JA, Segers P, Gillebert TC et al. Arterial properties as determinants of time-varying myocardial stress in humans. Hypertension 2012;60:64–70. [DOI] [PubMed] [Google Scholar]
- 16.Phan TS, Li JK, Segers P, Chirinos JA. Misinterpretation of the Determinants of Elevated Forward Wave Amplitude Inflates the Role of the Proximal Aorta. J Am Heart Assoc 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chirinos JA, Londono-Hoyos F, Zamani P et al. Effects of organic and inorganic nitrate on aortic and carotid haemodynamics in heart failure with preserved ejection fraction. Eur J Heart Fail 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mitchell GF, van Buchem MA, Sigurdsson S et al. Arterial stiffness, pressure and flow pulsatility and brain structure and function: the Age, Gene/Environment Susceptibility--Reykjavik study. Brain 2011;134:3398–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li JK. Dominance of geometric over elastic factors in pulse transmission through arterial branching. Bull Math Biol 1986;48:97–103. [DOI] [PubMed] [Google Scholar]
- 20.Hirata K, Yaginuma T, O’Rourke MF, Kawakami M. Age-related changes in carotid artery flow and pressure pulses: possible implications for cerebral microvascular disease. Stroke 2006;37:2552–6. [DOI] [PubMed] [Google Scholar]
- 21.Chirinos JA, Segers P, Gupta AK et al. Time-varying myocardial stress and systolic pressure-stress relationship: role in myocardial-arterial coupling in hypertension. Circulation 2009;119:2798–807. [DOI] [PubMed] [Google Scholar]
- 22.Chirinos JA. Deciphering Systolic-Diastolic Coupling in the Intact Heart. Hypertension 2017;69:575–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Strauer BE. Myocardial oxygen consumption in chronic heart disease: role of wall stress, hypertrophy and coronary reserve. Am J Cardiol 1979;44:730–40. [DOI] [PubMed] [Google Scholar]
- 24.Kobayashi S, Yano M, Kohno M et al. Influence of aortic impedance on the development of pressure-overload left ventricular hypertrophy in rats. Circulation 1996;94:3362–8. [DOI] [PubMed] [Google Scholar]
- 25.Gillebert TC, Lew WY. Influence of systolic pressure profile on rate of left ventricular pressure fall. Am J Physiol 1991;261:H805–13. [DOI] [PubMed] [Google Scholar]
- 26.Zamani P, Bluemke DA, Jacobs DR Jr., et al. Resistive and Pulsatile Arterial Load as Predictors of Left Ventricular Mass and Geometry: The Multi-Ethnic Study of Atherosclerosis. Hypertension 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hashimoto J, Westerhof BE, Westerhof N, Imai Y, O’Rourke MF. Different role of wave reflection magnitude and timing on left ventricular mass reduction during antihypertensive treatment. J Hypertens 2008;26:1017–24. [DOI] [PubMed] [Google Scholar]
- 28.Gillebert TC, Lew WY. Influence of systolic pressure profile on rate of left ventricular pressure fall. Am J Physiol 1991;261:H805–13. [DOI] [PubMed] [Google Scholar]
- 29.Fukuta H, Ohte N, Wakami K et al. Impact of arterial load on left ventricular diastolic function in patients undergoing cardiac catheterization for coronary artery disease. Circulation journal : official journal of the Japanese Circulation Society 2010;74:1900–5. [DOI] [PubMed] [Google Scholar]
- 30.Weber T, O’Rourke MF, Ammer M, Kvas E, Punzengruber C, Eber B. Arterial stiffness and arterial wave reflections are associated with systolic and diastolic function in patients with normal ejection fraction. Am J Hypertens 2008;21:1194–202. [DOI] [PubMed] [Google Scholar]
- 31.Chirinos JA, Segers P, Rietzschel ER et al. Early and Late Systolic Wall Stress Differentially Relate to Myocardial Contraction and Relaxation in Middle-Aged Adults: The Asklepios Study. Hypertension 2013;61:296–303. [DOI] [PubMed] [Google Scholar]
- 32.Chirinos JA, Segers P, Duprez DA et al. Late systolic central hypertension as a predictor of incident heart failure: the Multi-ethnic Study of Atherosclerosis. J Am Heart Assoc 2015;4:e001335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chirinos JA, Kips JG, Jacobs DR Jr., et al. Arterial wave reflections and incident cardiovascular events and heart failure: MESA (Multiethnic Study of Atherosclerosis). J Am Coll Cardiol 2012;60:2170–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chirinos JA, Phan TS, Syed AA et al. Late Systolic Myocardial Loading Is Associated With Left Atrial Dysfunction in Hypertension. Circ Cardiovasc Imaging 2017;10:e006023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shaikh AY, Wang N, Yin X et al. Relations of Arterial Stiffness and Brachial Flow-Mediated Dilation With New-Onset Atrial Fibrillation: The Framingham Heart Study. Hypertension 2016;68:590–6. [DOI] [PubMed] [Google Scholar]
- 36.Tsao CW, Lyass A, Larson MG et al. Relation of Central Arterial Stiffness to Incident Heart Failure in the Community. J Am Heart Assoc 2015;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pandey A, Khan H, Newman AB et al. Arterial Stiffness and Risk of Overall Heart Failure, Heart Failure With Preserved Ejection Fraction, and Heart Failure With Reduced Ejection Fraction: The Health ABC Study (Health, Aging, and Body Composition). Hypertension 2017;69:267–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hoffman JI, Buckberg GD. The myocardial oxygen supply:demand index revisited. J Am Heart Assoc 2014;3:e000285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Coutinho T, Mielniczuk LM, Srivaratharajah K, deKemp R, Wells GA, Beanlands RS. Coronary artery microvascular dysfunction: Role of sex and arterial load. Int J Cardiol 2018;270:42–47. [DOI] [PubMed] [Google Scholar]
- 40.Klahr S The kidney in hypertension--villain and victim. NEnglJMed 1989;320:731–733. [DOI] [PubMed] [Google Scholar]
- 41.Hashimoto J, Ito S. Central pulse pressure and aortic stiffness determine renal hemodynamics: pathophysiological implication for microalbuminuria in hypertension. Hypertension 2011;58:839–846. [DOI] [PubMed] [Google Scholar]
- 42.Townsend RR, Wimmer NJ, Chirinos JA et al. Aortic PWV in chronic kidney disease: a CRIC ancillary study. AmJHypertens 2010;23:282–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Briet M, Boutouyrie P, Laurent S, London GM. Arterial stiffness and pulse pressure in CKD and ESRD. Kidney Int 2012;82:388–400. [DOI] [PubMed] [Google Scholar]
- 44.Scialla JJ, Leonard MB, Townsend RR et al. Correlates of Osteoprotegerin and Association with Aortic Pulse Wave Velocity in Patients with Chronic Kidney Disease. ClinJAmSocNephrol 2011;6:2612–2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Coban M, Inci A, Yilmaz U, Asilturk E. The Association of Fibroblast Growth Factor 23 with Arterial Stiffness and Atherosclerosis in Patients with Autosomal Dominant Polycystic Kidney Disease. Kidney Blood Press Res 2018;43:1160–1173. [DOI] [PubMed] [Google Scholar]
- 46.Raggi P, Bellasi A, Ferramosca E, Islam T, Muntner P, Block GA. Association of pulse wave velocity with vascular and valvular calcification in hemodialysis patients. Kidney Int 2007;71:802–807. [DOI] [PubMed] [Google Scholar]
- 47.Peyster E, Chen J, Feldman HI et al. Inflammation and Arterial Stiffness in Chronic Kidney Disease: Findings From the CRIC Study. Am J Hypertens 2017;30:400–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sanders PW. Effect of salt intake on progression of chronic kidney disease. CurrOpinNephrolHypertens 2006;15:54–60. [DOI] [PubMed] [Google Scholar]
- 49.Mehdi UF, Adams-Huet B, Raskin P, Vega GL, Toto RD. Addition of angiotensin receptor blockade or mineralocorticoid antagonism to maximal angiotensin-converting enzyme inhibition in diabetic nephropathy. Journal of the American Society of Nephrology 2009;20:2641–2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Converse RL Jr., Jacobsen TL, Toto RD et al. Sympathetic overactivity in patients with chronic renal failure. NEnglJMed 1992;327:1912–1918. [DOI] [PubMed] [Google Scholar]
- 51.Townsend RR, Anderson AH, Chirinos JA et al. Association of Pulse Wave Velocity With Chronic Kidney Disease Progression and Mortality: Findings From the CRIC Study (Chronic Renal Insufficiency Cohort). Hypertension 2018;71:1101–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Guerin AP, Blacher J, Pannier B, Marchais SJ, Safar ME, London GM. Impact of aortic stiffness attenuation on survival of patients in end-stage renal failure. Circulation 2001;103:987–992. [DOI] [PubMed] [Google Scholar]
- 53.Attwell D, Laughlin SB. An energy budget for signaling in the grey matter of the brain. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism 2001;21:1133–45. [DOI] [PubMed] [Google Scholar]
- 54.Yang SH, Liu R. Chapter 10 - Cerebral Autoregulation In: Caplan LR, Biller J, Leary MC, et al. , editors. Primer on Cerebrovascular Diseases (Second Edition). San Diego: Academic Press, 2017:57–60. [Google Scholar]
- 55.Jefferson AL, Cambronero FE, Liu D et al. Higher Aortic Stiffness Is Related to Lower Cerebral Blood Flow and Preserved Cerebrovascular Reactivity in Older Adults. Circulation 2018;138:1951–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Beishon L, Haunton VJ, Panerai RB, Robinson TG. Cerebral Hemodynamics in Mild Cognitive Impairment: A Systematic Review. Journal of Alzheimer’s disease : JAD 2017;59:369–385. [DOI] [PubMed] [Google Scholar]
- 57.Meyer ML, Palta P, Tanaka H et al. Association of Central Arterial Stiffness and Pressure Pulsatility with Mild Cognitive Impairment and Dementia: The Atherosclerosis Risk in Communities Study-Neurocognitive Study (ARIC-NCS). Journal of Alzheimer’s disease : JAD 2017;57:195–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pase MP, Beiser A, Himali JJ et al. Aortic Stiffness and the Risk of Incident Mild Cognitive Impairment and Dementia. Stroke; a journal of cerebral circulation 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Henskens LH, Kroon AA, van Oostenbrugge RJ et al. Increased aortic pulse wave velocity is associated with silent cerebral small-vessel disease in hypertensive patients. Hypertension 2008;52:1120–6. [DOI] [PubMed] [Google Scholar]
- 60.Poels MM, Zaccai K, Verwoert GC et al. Arterial stiffness and cerebral small vessel disease: the Rotterdam Scan Study. Stroke; a journal of cerebral circulation 2012;43:2637–42. [DOI] [PubMed] [Google Scholar]
- 61.Maillard P, Carmichael O, Fletcher E, Reed B, Mungas D, DeCarli C. Coevolution of white matter hyperintensities and cognition in the elderly. Neurology 2012;79:442–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jack CR Jr., Bennett DA, Blennow K et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s & dementia : the journal of the Alzheimer’s Association 2018;14:535–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hughes T, Wagenknecht L, Craft S et al. Arterial Stiffness and Dementia Pathology: Atherosclerosis Risk in Communities (ARIC)-PET Study. Neurology 2018;90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hughes TM, Kuller LH, Barinas-Mitchell EJ et al. Arterial stiffness and beta-amyloid progression in nondemented elderly adults. JAMA Neurol 2014;71:562–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bourassa P, Tremblay C, Schneider JA, Bennett DA, Calon F. Beta-amyloid pathology in human brain microvessel extracts from the parietal cortex: relation with cerebral amyloid angiopathy and Alzheimer’s disease. Acta neuropathologica 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tan I, Butlin M, Avolio A. Does increase in arterial stiffness and wave reflection precede development of placental-mediated complications in pregnancy? J Hypertens 2018;36:1029–1031. [DOI] [PubMed] [Google Scholar]
- 67.Thadhani R, Ecker JL, Kettyle E, Sandler L, Frigoletto FD. Pulse pressure and risk of preeclampsia: a prospective study. Obstet Gynecol 2001;97:515–20. [DOI] [PubMed] [Google Scholar]
- 68.Osman MW, Nath M, Breslin E et al. Association between arterial stiffness and wave reflection with subsequent development of placental-mediated diseases during pregnancy: findings of a systematic review and meta-analysis. J Hypertens 2018;36:1005–1014. [DOI] [PubMed] [Google Scholar]
- 69.Eipel C, Abshagen K, Vollmar B. Regulation of hepatic blood flow: the hepatic arterial buffer response revisited. World J Gastroenterol 2010;16:6046–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chirinos JA, Bhattacharya P, Kumar A et al. Impact of Diabetes Mellitus on Ventricular Structure, Arterial Stiffness, and Pulsatile Hemodynamics in Heart Failure With Preserved Ejection Fraction. J Am Heart Assoc 2019;8:e011457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Villela-Nogueira CA, Leite NC, Cardoso CR, Salles GF. NAFLD and Increased Aortic Stiffness: Parallel or Common Physiopathological Mechanisms? Int J Mol Sci 2016;17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sunbul M, Agirbasli M, Durmus E et al. Arterial stiffness in patients with non-alcoholic fatty liver disease is related to fibrosis stage and epicardial adipose tissue thickness. Atherosclerosis 2014;237:490–3. [DOI] [PubMed] [Google Scholar]
- 73.Dockery F, Bulpitt CJ, Donaldson M, Fernandez S, Rajkumar C. The relationship between androgens and arterial stiffness in older men. J Am Geriatr Soc 2003;51:1627–32. [DOI] [PubMed] [Google Scholar]
- 74.Dominguez JM 2nd, Davis RT 3rd, McCullough DJ, Stabley JN, Behnke BJ. Aging and exercise training reduce testes microvascular PO2 and alter vasoconstrictor responsiveness in testicular arterioles. Am J Physiol Regul Integr Comp Physiol 2011;301:R801–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Damber JE, Bergh A, Daehlin L. Testicular blood flow, vascular permeability, and testosterone production after stimulation of unilaterally cryptorchid adult rats with human chorionic gonadotropin. Endocrinology 1985;117:1906–13. [DOI] [PubMed] [Google Scholar]
- 76.Duca L, Blaise S, Romier B et al. Matrix ageing and vascular impacts: focus on elastin fragmentation. Cardiovasc Res 2016;110:298–308. [DOI] [PubMed] [Google Scholar]
- 77.Nanoudis S, Pikilidou M, Yavropoulou M, Zebekakis P. The Role of MicroRNAs in Arterial Stiffness and Arterial Calcification. An Update and Review of the Literature. Front Genet 2017;8:209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang RN, Green J, Wang Z et al. Bone Morphogenetic Protein (BMP) signaling in development and human diseases. Genes Dis 2014;1:87–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schurgers LJ, Uitto J, Reutelingsperger CP. Vitamin K-dependent carboxylation of matrix Gla-protein: a crucial switch to control ectopic mineralization. Trends Mol Med 2013;19:217–26. [DOI] [PubMed] [Google Scholar]
- 80.Pivin E, Ponte B, Pruijm M et al. Inactive Matrix Gla-Protein Is Associated With Arterial Stiffness in an Adult Population-Based Study. Hypertension 2015;66:85–92. [DOI] [PubMed] [Google Scholar]
- 81.Mayer O Jr., Seidlerova J, Wohlfahrt P et al. Desphospho-uncarboxylated matrix Gla protein is associated with increased aortic stiffness in a general population. J Hum Hypertens 2016;30:418–23. [DOI] [PubMed] [Google Scholar]
- 82.Sardana M, Vasim I, Varakantam S et al. Inactive Matrix Gla-Protein and Arterial Stiffness in Type 2 Diabetes Mellitus. Am J Hypertens 2017;30:196–201. [DOI] [PubMed] [Google Scholar]
- 83.Chirinos JA, Sardana M, Syed AA et al. Aldosterone, inactive matrix gla-protein, and large artery stiffness in hypertension. J Am Soc Hypertens 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Puzantian H, Akers SR, Oldland G et al. Circulating Dephospho-Uncarboxylated Matrix Gla-Protein Is Associated With Kidney Dysfunction and Arterial Stiffness. Am J Hypertens 2018;31:988–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hashmath Z, Lee J, Gaddam S et al. Vitamin K Status, Warfarin Use, and Arterial Stiffness in Heart Failure. Hypertension 2019;73:364–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Knapen MH, Braam LA, Drummen NE, Bekers O, Hoeks AP, Vermeer C. Menaquinone-7 supplementation improves arterial stiffness in healthy postmenopausal women. A double-blind randomised clinical trial. Thromb Haemost 2015;113:1135–44. [DOI] [PubMed] [Google Scholar]
- 87.Reference Values for Arterial Stiffness C. Determinants of pulse wave velocity in healthy people and in the presence of cardiovascular risk factors: ‘establishing normal and reference values’. Eur Heart J 2010;31:2338–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Van Bortel LM, De Backer T, Segers P. Standardization of Arterial Stiffness Measurements Make Them Ready for Use in Clinical Practice. Am J Hypertens 2016;29:1234–1236. [DOI] [PubMed] [Google Scholar]
- 89.Weir-McCall JR, Brown L, Summersgill J et al. Development and Validation of a Path Length Calculation for Carotid-Femoral Pulse Wave Velocity Measurement: A TASCFORCE, SUMMIT, and Caerphilly Collaborative Venture. Hypertension 2018;71:937–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Matsushita K, Ding N, Kim ED et al. Cardio-ankle vascular index and cardiovascular disease: Systematic review and meta-analysis of prospective and cross-sectional studies. J Clin Hypertens (Greenwich) 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ohkuma T, Ninomiya T, Tomiyama H et al. Brachial-Ankle Pulse Wave Velocity and the Risk Prediction of Cardiovascular Disease: An Individual Participant Data Meta-Analysis. Hypertension 2017;69:1045–1052. [DOI] [PubMed] [Google Scholar]
- 92.Chirinos JA, Rietzschel ER, Shiva-Kumar P et al. Effective arterial elastance is insensitive to pulsatile arterial load. Hypertension 2014;64:1022–31. [DOI] [PubMed] [Google Scholar]
- 93.Salvi P, Scalise F, Rovina M et al. Noninvasive Estimation of Aortic Stiffness Through Different Approaches. Hypertension 2019;74:117–129. [DOI] [PubMed] [Google Scholar]
- 94.Schwartz JE, Feig PU, Izzo JL Jr., Pulse Wave Velocities Derived From Cuff Ambulatory Pulse Wave Analysis. Hypertension 2019;74:111–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Olsen MH, Angell SY, Asma S et al. A call to action and a lifecourse strategy to address the global burden of raised blood pressure on current and future generations: the Lancet Commission on hypertension. Lancet 2016;388:2665–2712. [DOI] [PubMed] [Google Scholar]
- 96.Rooprai J, Boodhwani M, Beauchesne L et al. Thoracic Aortic Aneurysm Growth in Bicuspid Aortic Valve Patients: Role of Aortic Stiffness and Pulsatile Hemodynamics. J Am Heart Assoc 2019;8:e010885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chirinos JA, Akers SR, Schelbert E et al. Arterial Properties as Determinants of Left Ventricular Mass and Fibrosis in Severe Aortic Stenosis: Findings From ACRIN PA 4008. J Am Heart Assoc 2019;8:e03742. [DOI] [PMC free article] [PubMed] [Google Scholar]