

Summary
Epithelial-mesenchymal interactions involve fundamental communication between tissues during organogenesis and are primarily regulated by growth factors and extracellular matrix. It is unclear whether RNA-containing exosomes are mobile genetic signals regulating epithelial-mesenchymal interactions. Here we identify that exosomes loaded with mesenchyme-specific mature miRNA contribute mobile genetic signals from mesenchyme to epithelium. The mature mesenchymal miR-133b-3p, loaded into exosomes was transported from mesenchyme to the salivary epithelium, which did not express primary miR-133b-3p. Knockdown of miR-133b-3p in culture decreased endbud morphogenesis, reduced proliferation of epithelial KIT+ progenitors and increased expression of a target gene, Disco-interacting protein 2 homolog B (Dip2b). DIP2B, which is involved in DNA methylation, was localized with 5-methylcytosine in the prophase nucleus of a subset of KIT+ progenitors during mitosis. In summary, exosomal transport of miR-133b-3p from mesenchyme to epithelium decreases DIP2B, which may function as an epigenetic regulator of genes responsible for KIT+ progenitor expansion during organogenesis.
Keywords: microRNA, exosomes, miR-133b-3p, DIP2B, progenitor cells, salivary gland, epithelial-mesenchymal interaction, KIT
Introduction
MicroRNAs (miRNAs) are small non-coding RNAs that regulate gene expression and influence many diverse cellular functions (Leonardo, et al., 2012; Bushati and Cohen, 2007; Bartel, 2004). Mature miRNAs are processed from primary transcripts (pri-miRNAs) through sequential steps that occur in the nucleus and cytoplasm (Ameres and Zamore, 2013; Carthew and Sontheimer, 2009; Kim, et al., 2009; Siomi and Siomi, 2009). Although miRNA can function within the cell that produces it, there is increasing evidence that miRNAs are used as mobile genetic signals for intercellular communication or via the systemic circulation within an organism (Mittelbrunn and Sanchez-Madrid, 2012). Cells can communicate using miRNAs within extracellular vesicles (Desrochers, et al., 2016) including exosomes (Yanez-Mo, et al., 2015; Valadi, et al., 2007), which are produced by most cells (Edgar, 2016) and found in many body fluids, such as serum and saliva. Many studies have been carried out to investigate exosome function using in vitro 2D cell culture models or systemic circulating exosomes in vivo (Yanez-Mo, et al., 2015; Mittelbrunn and Sanchez-Madrid, 2012; Kosaka and Ochiya, 2011). However, it is unclear whether specific miRNAs are transported across 3D cellular tissue boundaries, such as between epithelium and mesenchyme, during normal fetal organogenesis.
We predicted that exosomal miRNAs released from the mesenchyme are mobile genetic signals important for epithelial-mesenchymal interactions during organogenesis. We used fetal mouse submandibular glands (SMG) to isolate exosomes and to study exosomal miRNA transport between tissue types, as this model has been classically used to study epithelial-mesenchymal interactions (Patel, et al., 2006; Grobstein, 1953b; Grobstein, 1953a). The mesenchyme and epithelium can be separated, cell membranes fluorescently labeled and the tissue recombined in ex vivo culture to study, for example, exosomal transport during SMG organogenesis. Classic mesenchyme/epithelium recombination experiments using different organs, showed that the mesenchyme is inductive to the epithelium, inducing cell proliferation, differentiation and ultimately its morphogenesis (Kusakabe, et al., 1985; Sakakura, et al., 1976; Grobstein, 1953b; Grobstein, 1953a). These mobile inductive signals from the mesenchyme have been mainly attributed to secreted signals, such as growth factors and extracellular matrix. However, we previously identified that epithelial miRNAs in the SMG regulate epithelial morphogenesis (Rebustini, et al., 2012; Hayashi, et al., 2011), and thus we hypothesized that exosomal miRNAs transported between mesenchyme and epithelium could be inductive signals during organogenesis.
We first determined whether SMGs produce exosomes during fetal organogenesis at a stage of development when epithelial-mesenchymal interactions are critical. We collected conditioned media from embryo day 13 (E13) SMGs cultured for 48 h on filters, and subjected the media to sequential centrifugation to enrich for exosomes in the final pellet (Fig. 1a). We confirmed that exosomes were present using a number of methods. First, transmission electron microscopy (TEM) analysis of the pellet revealed the presence of exosome-like vesicles of the expected size (Yanez-Mo, et al., 2015; Mittelbrunn and Sanchez-Madrid, 2012; Gibbings, et al., 2009) (Fig. 1a). Secondly, Western blot analysis on the exosome pellet confirmed the presence of the exosome markers TSG101 and Alix, and absence of intracellular cytoskeletal actin (Lotvall, et al., 2014) (Fig. 1a). Thirdly, RNA electrophoresis showed that the exosome pellet contained small RNAs but not 18S- and 28S-rRNA (Fig. 1b, Fig. S1). The yield of exosomal RNA from 50 SMGs was ~2 ng and we confirmed that this was resistant to RNase A treatment, in contrast to total SMG RNA, as would be expected if the RNA were protected in exosomes (Fig. 1b). Taken together, we show that E13 mouse SMGs produce exosomes containing small RNAs.
Figure 1. Exosomes containing small RNAs are secreted into the medium of fetal SMGs cultured ex vivo.
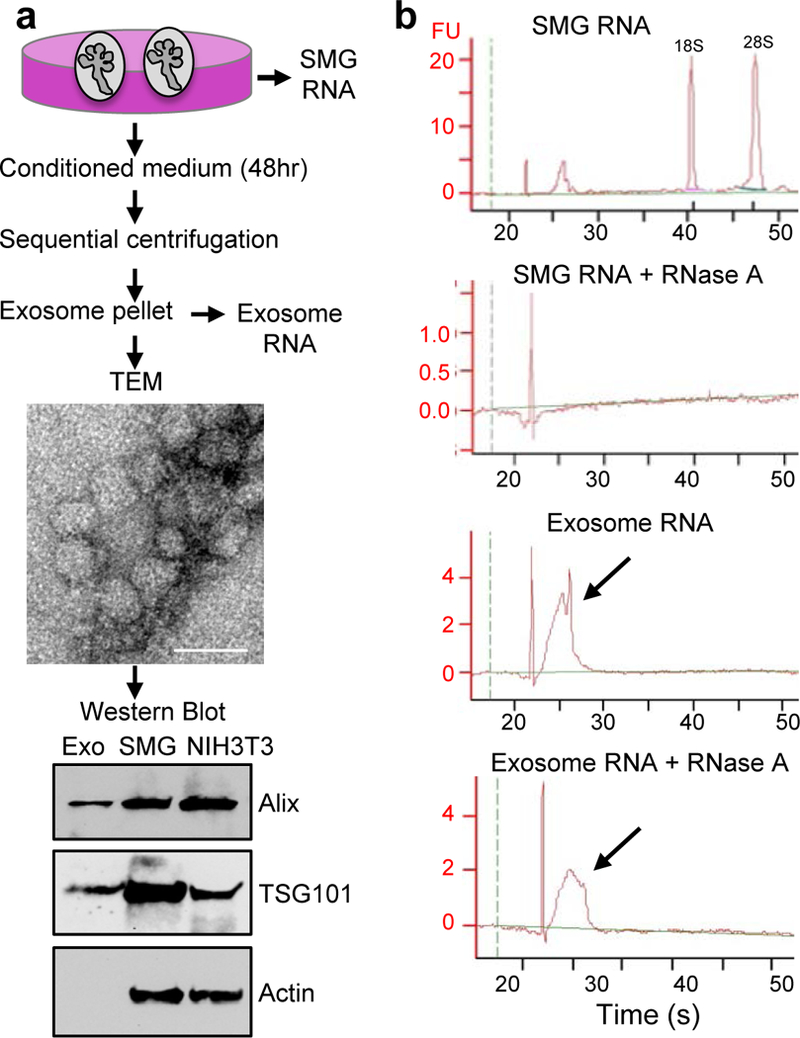
(a) Exosomes (Exo) isolated by sequential centrifugation were analyzed by transmission electron microscopy (TEM, Scale bar = 50 nm) and Western blot for the exosome markers Alix and TSG101. Intact SMG and NIH3T3 cell lysates are positive controls. (b) Bioanalyzer analysis shows that small RNAs in exosomes were resistant to RNase A degradation. Arrows indicate the peak of small RNA before and after RNase A treatment. FU; fluorescence units. N = 3, graph is a representative experiment.
To investigate whether the transfer of exosomes occurs between epithelium and mesenchyme, or vice-versa, we used E13 SMGs in tissue recombination experiments. The cell membranes of either epithelium or mesenchyme were labeled with a fluorescent BODIPY-ceramide dye, which labels cell membranes as well as exosome membranes (Kosaka, et al., 2012; Trajkovic, et al., 2008) (Fig. 2a). We first confirmed that BODIPY ceramide also labeled exosomes isolated from SMGs, by repeating the exosome preparation as described above, but using labeled SMGs and measuring the emission spectrum of the isolated exosome fraction to confirm that it contained BOPIDY-ceramide, i.e. 540–680nm light emission (data not shown). Further, when labeled mesenchyme was recombined with unlabeled epithelium and cultured for 12 h, ceramide-labeled vesicles were detected in the unlabeled epithelium (Fig. 2a, upper panel). Since the vesicles were detected by confocal microscopy it suggests they are accumulations of labeled exosomes or include other labeled vesicles, as the size of an individual exosome would not be resolved at this magnification. In the converse recombination experiment, ceramide-labeled vesicles from labeled epithelium were not detected in the unlabeled mesenchyme by confocal analysis (Fig. 2a, lower panel) although we cannot rule out the possibility that low levels of epithelial exosomes are present. However, we focused on the abundant labeled vesicles, which included exosomes, that were transported from the mesenchyme to the epithelium. In addition, we observed labeled-vesicle uptake in isolated SMG epithelium physically separated from labeled mesenchyme by a laminin extracellular matrix (ECM). The labeled mesenchyme was placed ~100 μm away on top of the ECM (Fig. 2b). After 8 h of incubation, ceramide-labeled vesicles were observed in the epithelial endbuds (Fig. 2b). To confirm that ceramide-labeled vesicles released by the mesenchyme contained exosomes, we co-stained the mesenchyme sitting on top of the ECM with antibodies to TSG101, an exosome marker (Fig. S2). The TSG101 staining was punctate and often associated with larger ceramide labeled vesicles. We also confirmed that the ceramide label released from the mesenchyme was in vesicles produced by the labeled cells by both increasing and reducing vesicle release with the ionophore monensin, which increases exosome release (Savina, et al., 2003) and brefeldin-A, which decreases exosome release (Mittelbrunn, et al., 2011). We treated ceramide-labeled mesenchyme recombined with non-labeled epithelium with each chemical for 6 h. With monensin we observed an increase in the ceramide-labeled vesicles in the epithelium of ~1.5 fold (Fig. 2c). With brefeldin-A treatment there was ~65 % reduction in the number of ceramide-labeled vesicles in the epithelium (Fig. 2c). Taken together, these data provide evidence that fluorescently labeled vesicles, including TSG101-labeled exosomes, are released from the mesenchyme and that they can diffuse through an ECM to the epithelium.
Figure 2. Fluorescently-labeled exosomes are transported from the SMG mesenchyme to the epithelium.
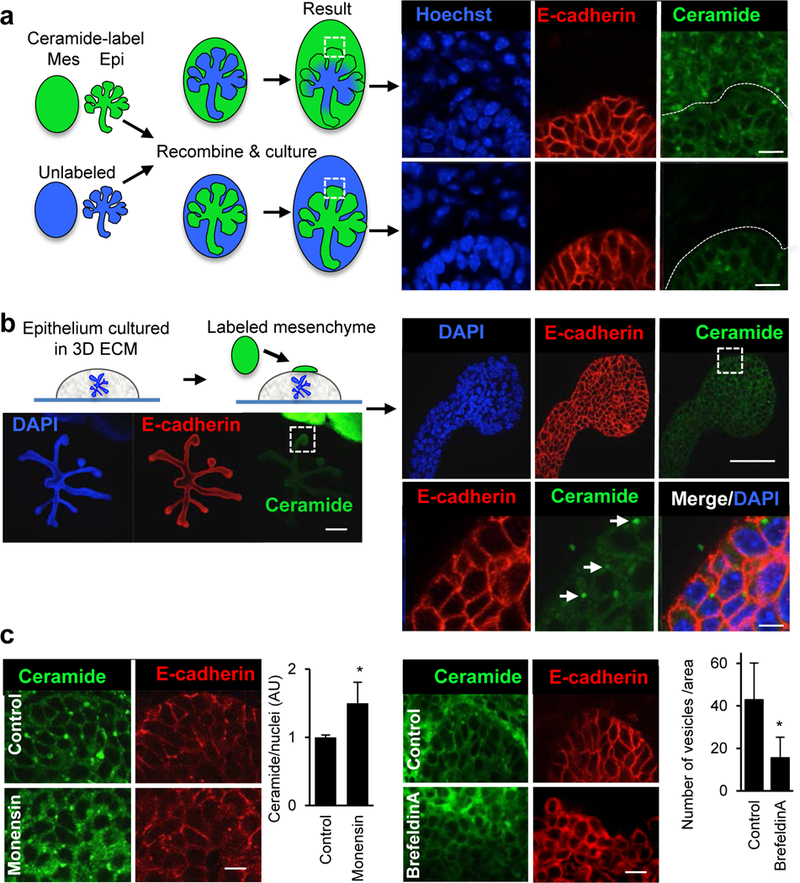
(a) SMG epithelium (Epi) separated from mesenchyme (Mes) were both labeled with fluorescent ceramide and recombined with unlabeled epithelium or mesenchyme. After 12 h of recombination they were analyzed by confocal microscopy. The ceramide label was transported from the mesenchyme to the epithelium but not the other way around. Scale bar = 10 μm. (b) Isolated epithelium in 3D laminin extracellular matrix was cultured with labeled mesenchyme. Labeled vesicles were detected in the epithelium after 8 h (white arrows). Scale bar = 200 μm (left), 50 μm (right upper), 5 μm (right lower). (c) Monensin (1 μM) increases ceramide-labeled vesicles in the epithelium, whereas brefeldin-A (1 μg/ml) decreases the number of vesicles in the epithelium. Scale bars = 10 μm. Student’s t-test, * P< 0.05.
Next we identified which miRNAs were present in exosomes isolated from SMG conditioned media using real-time PCR (qPCR) miRNA arrays and compared these to miRNAs isolated directly from intact SMGs. We detected 81 exosomal miRNAs, which were a subset of the 153 miRNAs identified in intact SMGs (Fig. 3a, Supplemental Table 1). Three of the exosomal miRNAs have sequence motifs previously identified as being important for miRNA loading into exosomes (CCCU) (Villarroya-Beltri, et al., 2013). These three miRNAs were significantly enriched in the exosome pellet isolated from the culture media as compared to the RNA isolated from intact SMGs in culture, miR-133a-3p (14.8-fold), miR-133b-3p (6.6-fold), and miR-409–3p (2.7-fold) (Fig. 3b). On the other hand, miR-200c-3p, a miRNA previously reported to be abundant in SMG epithelium (Rebustini, et al., 2012), was less abundant in exosomes (0.07-fold) than in intact SMGs. These data suggest that exosomes contain a subset of miRNAs produced by the SMG, and that some of these may be selectively packaged in exosomes.
Figure 3. miR-133b-3p is enriched in exosomes but its primary transcript is only expressed in the mesenchyme while the mature miRNA is detected in both epithelium and mesenchyme.
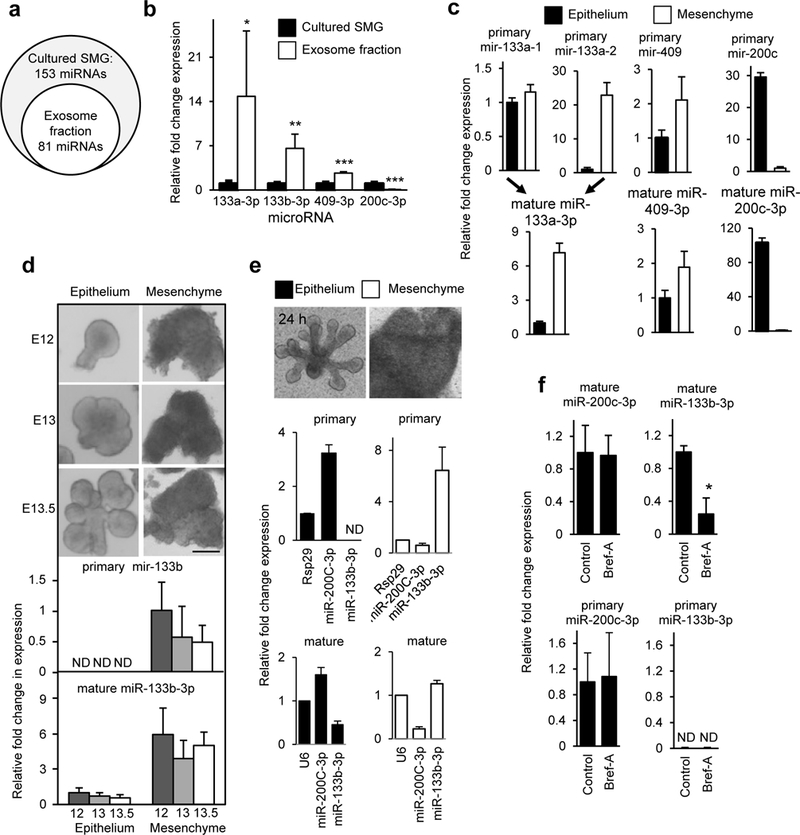
(a) Exosomal miRNAs are a subset of SMG miRNAs. N = 3. (b) miR-133a-3p, miR-133b-3p, and miR-409–3p were relatively more abundant in the RNA isolated from the exosome fraction than SMGs. miR-200c-3p is an epithelial-related miRNA. Student’s t-test, * P< 0.05, ** P< 0.01, *** P< 0.001. N = 3. (c) TaqMan qPCR of the primary and mature forms of exosomal miRNAs (miR-133a-3p, miR-409–3p) and epithelial-expressed miR-200c-3p shows that primary and mature transcripts were detected in both epithelium and mesenchyme. N = 3. (d) Expression analysis by TaqMan qPCR of primary and mature forms of miR-133b-3p in isolated epithelium and mesenchyme. Images are epithelium and mesenchyme at embryo day 12, 13, and 13.5. The primary transcript of miR-133b-3p was not detected (ND) in epithelium, although the mature form was found in both epithelium and mesenchyme. (e) Primary miR-133–3p is not detected in cultured epithelium but its mesenchymal expression increases. Analysis of primary and mature miRNA expression, normalized to time 0 (dotted line), in isolated epithelium and mesenchyme cultured separately for 24 h. (f) Brefeldin-A (1ng/ml) treatment of SMGs for 6 h, which reduces exosome secretion, decreases mature miR-133b-3p expression in the epithelium by ~80 %. N = 3. All graphs are means ± s.d.
Next we measured the tissue distribution of the primary and mature forms of the exosomal-enriched miRNA using TaqMan qPCR. We predicted that the mature miRNAs would be detected in both epithelium and mesenchyme due to exosomal transport. However, if the primary miRNA were detected only in mesenchyme but not epithelium, it would suggest that the miRNA was transcribed in the mesenchyme, processed into its mature form, and then transported to the epithelium in exosomes. We measured the primary and mature forms of the exosome-enriched miR-133a-3p, miR-133b-3p, and miR-409–3p in E13 SMG epithelium and mesenchyme. We also used the epithelial-enriched miR-200c-3p as a control, since both its primary and mature forms were detected in E13 epithelium (Fig. 3c). Likewise, both primary and mature forms of miR-409–3p were detected in both mesenchyme and epithelium. Similarly, mature miR-133a-3p and its two different primary forms, primary miR-133a-1 and primary miR-133a-2 were detected in both mesenchyme and epithelium (Fig. 3c). In contrast, the primary form of miR-133b-3p was only detected in the mesenchyme but not in the epithelium, although the mature form miR-133b-3p was detected in both tissues (Fig. 3d, Fig. S3). To confirm this expression pattern, we also separated epithelium from mesenchyme at two other early stages of SMG development, E12 (single epithelial endbud), and E13.5 (5–8 endbuds) and found that primary miR-133b-3p was only detectable in the mesenchyme, whereas the mature form was detected in both epithelium and mesenchyme (Fig. 3d). We also cultured isolated E13.5 epithelium and mesenchyme separately for 24 h and measured both primary and mature miR-133b-3p and miR-200c with culture (Fig. 3e). We could not detect primary miR-133b-3p in cultured epithelia and the level of mature miR-133b-3p decreased with culture, as expected as it was not being made by the epithelium. In contrast, both primary and mature miR-200c increased during epithelial culture (Fig. 3e). Also, as expected both primary and mature forms of miR-133b-3p increased in mesenchymal culture. Finally, we treated intact SMGs with brefeldin-A for 6 h and then isolated the epithelium from the mesenchyme. Brefeldin-A reduced exosome release, resulting in less mature miR-133b-3p being detected in the epithelium compared to control treatment (Fig. 3f). Taken together with the previous experiments with fluorescent-labeled mesenchyme, these data suggest that miR-133b-3p is transcribed in the mesenchyme, processed to its mature form and transported to the epithelium in exosomes during SMG organogenesis. In contrast, the mature forms of other exosomal miRNAs are expressed in both epithelium and mesenchyme.
To identify potential direct targets of miR-133b-3p we combined results from microarray analysis with bioinformatic prediction. We treated isolated epithelium with an antagomir to miR-133b-3p (Anta-133b-3p) for 20 h, a time during which no change in morphogenesis was apparent. In this loss-of-function assay, we identified 88 upregulated messenger RNAs (mRNAs) by microarray analysis. Based on three bioinformatic miRNA target prediction programs (Supplemental Table 2), we focused on eight candidate mRNAs that were potential direct targets of miR-133b-3p and upregulated in the microarray by the antagomir. Using qPCR analysis, we confirmed that one of these, the disco-interacting protein 2 homolog b (Dip2b) was significantly increased in epithelia after 20 h of Anta-133b-3p treatment (Fig. S4a). One possible explanation for the apparent modest changes in gene expression was that only a subpopulation of cells was being targeted. Dip2b is known to contain a binding site for the transcriptional regulator DNA methyltransferase 1 associated protein 1 (DMAP1), and is involved in DNA methylation to epigenetically regulate cell proliferation and differentiation (Winnepenninckx, et al., 2007). In humans, DIP2B has also been identified as a potential susceptibility gene associated with colorectal cancer (Closa, et al., 2014). Interestingly, DNA methylation has also been associated with aberrant miR-133b expression in colorectal cancer (Lv, et al., 2015).
To first confirm that miR-133b-3p directly targets Dip2b in a sequence-dependent manner, we performed luciferase reporter assays with NIH3T3 cells using plasmids with the Dip2b 3’ untranslated region (3’ UTR) containing a miR-133b-3p binding site (Dip2b-wt) (Fig. S4b). An RNAHybrid program predicted a secondary structure between miR-133b-3p and the potential binding site of Dip2b 3’UTR, suggesting they can make an RNA/RNA duplex with a minimum free energy of −29.5 kcal/mol (Rehmsmeier, et al., 2004). We mutated the target site sequence by substituting three nucleotides of the base-pairing seed region in miR-133b-3p (Dip2b-mutation). A miR-133b-3p mimic (Mimic-133b-3p) reduced luciferase levels in the Dip2b-wild type but not with the Dip2b-mutation, indicating that miR-133b-3p targets Dip2b in a ‘seed region-dependent’ manner. As expected, treating SMG epithelium with Mimic-133b-3p downregulated Dip2b expression (Fig. S4c). Taken together, these results suggest that miR-133b-3p directly targets Dip2b in a sequence-dependent manner to downregulate its mRNA.
Next, we treated isolated SMG epithelium with Anta-133b-3p for 45 h to measure changes in epithelial morphogenesis and mature miR-133b-3p gene expression (Fig. 4a). Treatment with Anta-133b-3p not only reduced expression of miR-133b-3p but also epithelial morphogenesis, as measured by a morphogenic index. We confirmed that these changes were associated with an increase in Dip2b expression (Fig. 4b), and ~ 2-fold increase in DIP2B staining in the epithelium (Fig. 4c and Fig. 4d), which was more highly expressed in certain individual cells that appeared to be proliferating, based on their nuclear morphology. In control epithelia, DIP2B was also apparent in the nuclei of a subpopulation of endbud cells, but with less staining intensity than Anta-133b-3p-treated epithelium (Fig. 4d, arrows). In addition, there was also a striking reduction of SMG endbud expansion with antagomir treatment. Therefore, we measured the expression of the endbud progenitor marker Kit, which we previously identified to mark proliferative endbud progenitors in SMG epithelium (Lombaert, et al., 2013). Knockdown of miR-133b-3p significantly reduced Kit expression (Fig. 4b), and there was a relative increase in expression of the ductal marker Krt19 as well as Krt5 and Krt14, which label both endbud and ductal cells. Fgfr2b, which is essential for SMG epithelial proliferation, was also upregulated (Fig. 4b). While combined KIT and FGFR2b signaling regulates endbud expansion (Lombaert, et al., 2013), Fgfr2b is also expressed throughout the duct (Patel, et al., 2006). The increased Fgfr2b is likely due to the proportional increase in the remaining ductal cells. We then measured proliferation by immunostaining antagomir-treated epithelium for CCND1 (Fig. 4e) and counting the number of proliferating cells in the endbud epithelium. There was ~76% reduction in the number of CCND1+ cells in Anta-133b-3p-treated epithelium compared to control. Together, these data suggest that miR-133b-3p specifically targets proliferating KIT-expressing (KIT+) endbud progenitors and consequently reduces endbud morphogenesis. In order to define whether miR133b-3p regulates KIT+ progenitor cell expansion via the epigenetic modulator DIP2B, we evaluated the expression of DIP2B in SMG KIT+ cells. Our previous microarray analysis of SMG development shows that Dip2b is expressed from ontogenesis until adulthood, and is present in both E13 epithelium and mesenchyme (sgmap.nidcr.nih.gov). Immunostaining confirmed broad cytoplasmic staining for DIP2B in both epithelium and mesenchyme. However, there was a subpopulation of individual cells within the SMG endbud that showed nuclear localization of DIP2B, and the protein expression further increased after Anta-133b-3p treatment (Fig. 4c). These cells also expressed KIT and E-Cadherin (Ecad). Further analysis of DIP2B protein in the SMG endbud revealed that during epithelial mitosis DIP2B condensed in the prophase nucleus in a discrete cell population (Fig. 4d, Fig. 4f and Figs. S4d and S4e). Moreover, co-staining with anti-5-methylcytosine (5mC) to label methylated DNA, an epigenetic modification due to DNA methyltransferase activity, suggested an association of DIP2B with 5mC in heterochromatin (Fig. 4f and Fig. S4f). However, at other stages of mitosis DIP2B staining was dissociated from mitotic chromatin (Fig. S4d). 5mC is an important repressor of gene expression in the genome, and DNA methylation is maintained through mitosis to the formation of daughter cells. Yet, certain transcription factors and chromatin binding proteins are excluded from chromatin during certain stages of mitosis (Ma, et al., 2015; Zaret, 2014; Egli, et al., 2008). Taken together, these data suggest that in a subset of KIT+ endbud progenitors miR-133b-3p suppresses DIP2B expression, and we speculate that this may be associated with DNA methylation during prophase mitosis.
Figure 4. Knockdown of miR-133b-3p reduces epithelial morphogenesis and endbud proliferation by increasing DIP2B in the nucleus of a KIT+ epithelial progenitor subpopulation.
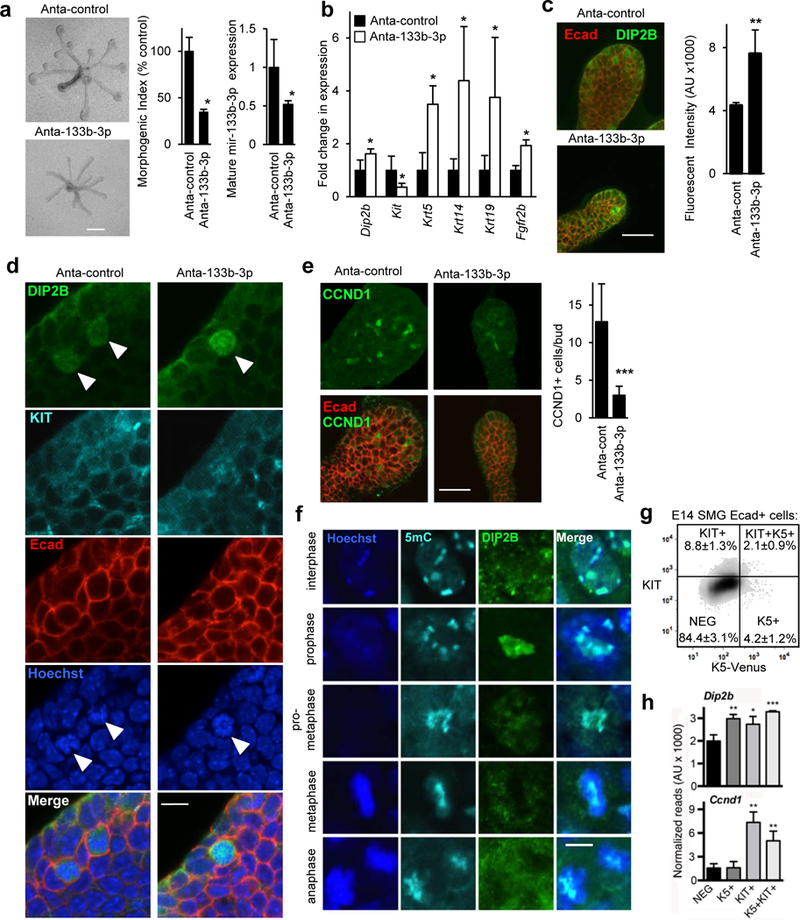
(a) miR-133b-3p loss-of-function with an antagomir (Anta-133b-3p) decreases epithelial morphogenesis and mature miR-133b-3p expression. Morphogenic index = number of endbuds x width of endbuds x length of ducts in arbitrary units, normalized to epithelia treated with control antagomir (Anta-control). N = 3. Means ± s.e.m. (b) Anta-133b-3p increased expression of Dip2b, a predicted target gene, as well as Krt5, Krt14, Krt19, and Fgfr2b and decreased expression of Kit. Expression was normalized to Rps29 and compared to epithelia treated with Anta-control. Student’s t-test, * P < 0.05. N = 3. Means ± s.e.m. (c) Anta-133b-3p treatment increased DIP2B staining. Scale bar 50 μm. N = 3. (d) Anta-133b-3p treatment increased nuclear localization of DIP2B protein in certain KIT+ epithelial (Ecad+) endbud cells. Scale bar 10 μm. N = 3. (e) Anta-133b-3p treatment reduced endbud proliferation and the number of CCND1+ cells in the endbuds decreased. Scale bar 50 μm. N = 3. (f) DIP2B associates with methylated-cytosine (5mc) in mitotic epithelial cells during prophase. DIP2B staining condenses in the nucleus of epithelial cells in prophase and was disassociated from mitotic chromatin in other phases. DIP2B partially co-localizes with methylated-cytosine (5mc) in heterochromatin. Scale bar 5 μm. Images are 1μm optical sections. (g) E14 SMG progenitors were sorted by FACS using K5-Venus and KIT expression. Percentage values are means ± s.e.m. of each cell population; graph is a representative experiment. (h) Dip2b and Ccnd1 were detected by RNAseq (N = 3) in epithelial Ecad+ cells that express KIT with or without K5.
To identify the subset of KIT+ progenitors expressing DIP2B, we isolated four epithelial Ecad+ cell populations by fluorescence-activated cell sorting (FACS) from E14 SMGs based on KIT and KRT5-Venus expression (Lombaert, et al., 2013; Knox, et al., 2010): KIT+K5+, KIT+K5-, KIT-K5+, and KIT-K5- (NEG) (Fig. 4g). RNA-sequencing (RNAseq) analysis showed that DIP2B transcripts were present in all cell types but significantly enriched in KIT+K5-, K5+KIT- and KIT+K5+ cells, when compared to KIT-K5- cells (Fig. 4h, GEO accession: GSE89896). In addition, Ccnd1 expression was also significantly enriched in the KIT+K5- and KIT+K5+ cells, suggesting they are proliferating (Fig. 4h). This was also supported by CCND1 staining as shown in Fig. 4e. The RNAseq data also shows Dip2b expression in K5+KIT- cells, which are not proliferating, and these may include the basal cells showing cytoplasmic staining of DIP2B (Fig. 4c). The function of cytoplasmic DIP2B in non-proliferating cells remains to be determined. Together, these data suggest that miR133–3p downregulates DIP2B in a subset of proliferating KIT+ progenitors i.e., the KIT+K5- cells, to allow their expansion.
In summary, it is well established that the mesenchyme provides soluble external signals to instruct epithelial development during organogenesis. While the most widely studied soluble cues are growth factors, we propose that exosomal transport of miRNAs is a distinct mechanism to regulate epithelial-mesenchymal interactions during organogenesis. Although an ECM-rich basement membrane separates the epithelium from mesenchyme, it was shown that micro-perforations in the basement membrane occur in multiple embryonic organs including lung, kidney, and SMG (Harunaga, et al., 2014). The reported holes (1.6 μm2 in average size) are potentially large enough for exosomes (20–100 nm in diameter) or cellular processes that secrete exosomes to pass through.
We propose that miR-133b-3p downregulates DIP2B, which participates in epigenetic repression of genes responsible for expansion of a subset of KIT+K5-progenitors. Specific exosome transport of miR-133b-3p from the mesenchyme to the epithelium reduces Dip2b expression to allow expansion of specific KIT+K5- progenitors, potentially by reducing DNA methylation. This is feasible as DIP2B has a binding domain to DNA methyltransferase 1 associated protein (DMAP1) (Winnepenninckx, et al., 2007) and is associated with methylcytosine in heterochromatin. The DMAP1-binding domain is highly conserved throughout eukaryotes, suggesting it has an important function. DMAP1 is a member of the TIP60-p400 histone acetyltransferase complex, which contributes to self-renewal and differentiation of embryonic stem cells (Chen, et al., 2013; Koizumi, et al., 2010; Fazzio, et al., 2008; Sapountzi, et al., 2006).
In conclusion, microRNA transport in exosomes is a mechanism to mobilize genetic signals from mesenchyme to epithelium and specifically influence progenitor cell expansion during organogenesis. It remains to be determined whether exosomes are somehow specifically targeted to epithelial KIT+K5- progenitors, whether all epithelial cells ultimately take up exosomes and/or the uptake of exosomes in KIT+K5- cells is enhanced due to their high proliferative status, and whether only a subset of progenitors respond to specific miRNAs. In the future microRNA-mediated mechanisms may be useful as targets for inducing proliferative regeneration of damaged adult organs.
Experimental Procedures
SMG culture
Fetal SMG explants and isolated SMG epithelium were cultured as previously described (Rebustini, et al., 2009).
Mouse lines
Embryos were obtained from ICR timed pregnant females (Harlan). K5-Venus mice were produced as previously described (Knox, et al., 2010). All experiments were approved by the Animal Care and Use Committee at NIDCR, NIH.
Exosome preparation
Exosomes were prepared from conditioned medium of E13 SMG after 48 h of culture and processed for TEM. Conditioned medium was centrifuged to remove cells and cellular debris. The supernatant was ultracentrifuged to obtain an exosome pellet.
Ceramide labeling
SMG cell membranes were labelled with BODIPY-TR-ceramide (Molecular Probes). Labeled tissue was then recombined with unlabeled tissue in culture or placed on top of 3D laminin-111 (Trevigen) to observe ceramide transfer to unlabeled epithelium.
Analysis of microRNA and mRNA
RNA was prepared using mirVana miRNA isolation and analyzed using the Megaplex Pools protocol and reagents (Applied Biosystems) to detect miRNAs as previously described (Rebustini, et al., 2012). For mature miRNA PCR analysis, TaqMan microRNA assays were performed (Applied Biosystems). qPCR of mRNAs were performed as previously described (Rebustini, et al., 2012).
Transfection assays
E13 epithelia were transfected with antagomirs (Exiqon) or mimics (Ambion) using RNAiFect (Qiagen) reagents as previously described (Rebustini, et al., 2012).
Immunofluorescence and FACS
Immunofluorescence was performed as previously described (Rebustini, et al., 2009) and FACS analysis as previously described (Knox, et al., 2010).
Supplementary Material
Acknowledgments
The authors would like to thank the NIDCR Combined Technical Research Core for FACs analysis, Daniel Martin for bioinformatics analysis, Kelly Ten Hagen for critical reading of the manuscript and Masanori Kashimata at Asahi University for technical support. This research was supported by the Intramural Research Program of the National Institute of Dental and Craniofacial Research at the National Institutes of Health and by JSPS KAKENHI Grant Numbers JP15K11060, JP15K20370, and by Grant for Basic Science Research Projects from The Sumitomo Foundation.
References
- Ameres SL, and Zamore PD (2013). Diversifying microRNA sequence and function. Nat Rev Mol Cell Biol 14, 475–88. [DOI] [PubMed] [Google Scholar]
- Bartel DP (2004). MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116, 281–97. [DOI] [PubMed] [Google Scholar]
- Bushati N, and Cohen SM (2007). microRNA functions. Annu Rev Cell Dev Biol 23, 175–205. [DOI] [PubMed] [Google Scholar]
- Carthew RW, and Sontheimer EJ (2009). Origins and Mechanisms of miRNAs and siRNAs. Cell 136, 642–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen PB, Hung JH, Hickman TL, Coles AH, Carey JF, Weng Z, Chu F, and Fazzio TG (2013). Hdac6 regulates Tip60-p400 function in stem cells. Elife 2, e01557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Closa A, Cordero D, Sanz-Pamplona R, Sole X, Crous-Bou M, Pare-Brunet L, Berenguer A, Guino E, Lopez-Doriga A, Guardiola J, et al. (2014). Identification of candidate susceptibility genes for colorectal cancer through eQTL analysis. Carcinogenesis 35, 2039–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desrochers LM, Antonyak MA, and Cerione RA (2016). Extracellular Vesicles: Satellites of Information Transfer in Cancer and Stem Cell Biology. Developmental cell 37, 301–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar JR (2016). Q&A: What are exosomes, exactly? BMC Biol 14, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egli D, Birkhoff G, and Eggan K (2008). Mediators of reprogramming: transcription factors and transitions through mitosis. Nat Rev Mol Cell Biol 9, 505–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazzio TG, Huff JT, and Panning B (2008). An RNAi screen of chromatin proteins identifies Tip60-p400 as a regulator of embryonic stem cell identity. Cell 134, 162–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbings DJ, Ciaudo C, Erhardt M, and Voinnet O (2009). Multivesicular bodies associate with components of miRNA effector complexes and modulate miRNA activity. Nat Cell Biol 11, 1143–9. [DOI] [PubMed] [Google Scholar]
- Grobstein C (1953a). Inductive epitheliomesenchymal interaction in cultured organ rudiments of the mouse. Science (New York, N.Y.) 118, 52–5. [DOI] [PubMed] [Google Scholar]
- Grobstein C (1953b). Morphogenetic interaction between embryonic mouse tissues separated by a membrane filter. Nature 172, 869–70. [DOI] [PubMed] [Google Scholar]
- Harunaga JS, Doyle AD, and Yamada KM (2014). Local and global dynamics of the basement membrane during branching morphogenesis require protease activity and actomyosin contractility. Developmental biology 394, 197–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, Koyama N, Azuma Y, and Kashimata M (2011). Mesenchymal miR-21 regulates branching morphogenesis in murine submandibular gland in vitro. Developmental biology 352, 299–307. [DOI] [PubMed] [Google Scholar]
- Kim VN, Han J, and Siomi MC (2009). Biogenesis of small RNAs in animals. Nat Rev Mol Cell Biol 10, 126–39. [DOI] [PubMed] [Google Scholar]
- Knox SM, Lombaert IM, Reed X, Vitale-Cross L, Gutkind JS, and Hoffman MP (2010). Parasympathetic innervation maintains epithelial progenitor cells during salivary organogenesis. Science (New York, N.Y.) 329, 1645–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koizumi T, Negishi M, Nakamura S, Oguro H, Satoh K, Ichinose M, and Iwama A (2010). Depletion of Dnmt1-associated protein 1 triggers DNA damage and compromises the proliferative capacity of hematopoietic stem cells. Int J Hematol 91, 611–9. [DOI] [PubMed] [Google Scholar]
- Kosaka N, Iguchi H, Yoshioka Y, Hagiwara K, Takeshita F, and Ochiya T (2012). Competitive interactions of cancer cells and normal cells via secretory microRNAs. The Journal of biological chemistry 287, 1397–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosaka N, and Ochiya T (2011). Unraveling the Mystery of Cancer by Secretory microRNA: Horizontal microRNA Transfer between Living Cells. Front Genet 2, 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusakabe M, Sakakura T, Sano M, and Nishizuka Y (1985). A pituitary-salivary mixed gland induced by tissue recombination of embryonic pituitary epithelium and embryonic submandibular gland mesenchyme in mice. Developmental biology 110, 382–91. [DOI] [PubMed] [Google Scholar]
- Leonardo TR, Schultheisz HL, Loring JF, and Laurent LC (2012). The functions of microRNAs in pluripotency and reprogramming. Nat Cell Biol 14, 1114–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombaert IM, Abrams SR, Li L, Eswarakumar VP, Sethi AJ, Witt RL, and Hoffman MP (2013). Combined Kit and Fgfr2b signaling regulates epithelial progenitor expansion during organogenesis. Stem Cell Reports 1, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotvall J, Hill AF, Hochberg F, Buzas EI, Di Vizio D, Gardiner C, Gho YS, Kurochkin IV, Mathivanan S, Quesenberry P, et al. (2014). Minimal experimental requirements for definition of extracellular vesicles and their functions: a position statement from the International Society for Extracellular Vesicles. J Extracell Vesicles 3, 26913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv LV, Zhou J, Lin C, Hu G, Yi LU, Du J, Gao K, and Li X (2015). DNA methylation is involved in the aberrant expression of miR-133b in colorectal cancer cells. Oncol Lett 10, 907–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Kanakousaki K, and Buttitta L (2015). How the cell cycle impacts chromatin architecture and influences cell fate. Front Genet 6, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittelbrunn M, Gutierrez-Vazquez C, Villarroya-Beltri C, Gonzalez S, Sanchez-Cabo F, Gonzalez MA, Bernad A, and Sanchez-Madrid F (2011). Unidirectional transfer of microRNA-loaded exosomes from T cells to antigen-presenting cells. Nat Commun 2, 282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittelbrunn M, and Sanchez-Madrid F (2012). Intercellular communication: diverse structures for exchange of genetic information. Nat Rev Mol Cell Biol 13, 328–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel VN, Rebustini IT, and Hoffman MP (2006). Salivary gland branching morphogenesis. Differentiation 74, 349–64. [DOI] [PubMed] [Google Scholar]
- Rebustini IT, Hayashi T, Reynolds AD, Dillard ML, Carpenter EM, and Hoffman MP (2012). miR-200c regulates FGFR-dependent epithelial proliferation via Vldlr during submandibular gland branching morphogenesis. Development 139, 191–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebustini IT, Myers C, Lassiter KS, Surmak A, Szabova L, Holmbeck K, Pedchenko V, Hudson BG, and Hoffman MP (2009). MT2-MMP-dependent release of collagen IV NC1 domains regulates submandibular gland branching morphogenesis. Developmental cell 17, 482–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehmsmeier M, Steffen P, Hochsmann M, and Giegerich R (2004). Fast and effective prediction of microRNA/target duplexes. RNA 10, 1507–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakakura T, Nishizuka Y, and Dawe CJ (1976). Mesenchyme-dependent morphogenesis and epithelium-specific cytodifferentiation in mouse mammary gland. Science (New York, N.Y.) 194, 1439–41. [DOI] [PubMed] [Google Scholar]
- Sapountzi V, Logan IR, and Robson CN (2006). Cellular functions of TIP60. Int J Biochem Cell Biol 38, 1496–509. [DOI] [PubMed] [Google Scholar]
- Savina A, Furlan M, Vidal M, and Colombo MI (2003). Exosome release is regulated by a calcium-dependent mechanism in K562 cells. The Journal of biological chemistry 278, 20083–90. [DOI] [PubMed] [Google Scholar]
- Siomi H, and Siomi MC (2009). On the road to reading the RNA-interference code. Nature 457, 396–404. [DOI] [PubMed] [Google Scholar]
- Trajkovic K, Hsu C, Chiantia S, Rajendran L, Wenzel D, Wieland F, Schwille P, Brugger B, and Simons M (2008). Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science (New York, N.Y.) 319, 1244–7. [DOI] [PubMed] [Google Scholar]
- Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, and Lotvall JO (2007). Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol 9, 654–9. [DOI] [PubMed] [Google Scholar]
- Villarroya-Beltri C, Gutierrez-Vazquez C, Sanchez-Cabo F, Perez-Hernandez D, Vazquez J, Martin-Cofreces N, Martinez-Herrera DJ, Pascual-Montano A, Mittelbrunn M, and Sanchez-Madrid F (2013). Sumoylated hnRNPA2B1 controls the sorting of miRNAs into exosomes through binding to specific motifs. Nat Commun 4, 2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winnepenninckx B, Debacker K, Ramsay J, Smeets D, Smits A, FitzPatrick DR, and Kooy RF (2007). CGG-repeat expansion in the DIP2B gene is associated with the fragile site FRA12A on chromosome 12q13.1. Am J Hum Genet 80, 221–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanez-Mo M, Siljander PR, Andreu Z, Zavec AB, Borras FE, Buzas EI, Buzas K, Casal E, Cappello F, Carvalho J, et al. (2015). Biological properties of extracellular vesicles and their physiological functions. J Extracell Vesicles 4, 27066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaret KS (2014). Genome reactivation after the silence in mitosis: recapitulating mechanisms of development? Developmental cell 29, 132–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.