

Abstract
P5A ATPases are expressed in the endoplasmic reticulum (ER) of all eukaryotic cells, and their disruption results in severe ER stress. However, the function of these ubiquitous membrane proteins, which belong to the P-type ATPase superfamily, is unknown. We purified a functional tagged version of the Saccharomyces cerevisiae P5A ATPase Spf1p and observed that the ATP hydrolytic activity of the protein is stimulated by phosphatidylinositol 4-phosphate (PI4P). Furthermore, SPF1 exhibited negative genetic interactions with SAC1, encoding a PI4P phosphatase, and with OSH1 to OSH6, encoding Osh proteins, which, when energized by a PI4P gradient, drive export of sterols and lipids from the ER. Deletion of SPF1 resulted in increased sensitivity to inhibitors of sterol production, a marked change in the ergosterol/lanosterol ratio, accumulation of sterols in the plasma membrane, and cytosolic accumulation of lipid bodies. We propose that Spf1p maintains cellular sterol homeostasis by influencing the PI4P-induced and Osh-mediated export of sterols from the ER.
INTRODUCTION
P5 ATPases belong to the large superfamily of P-type ATPases, so named because the catalytic cycle involves phosphorylation and dephosphorylation of a conserved aspartate residue (Axelsen and Palmgren, 1998; Sørensen et al., 2010). P-type ATPases are phylogenetically divided into five subfamilies, named P1 to P5, among which P1 to P3 are cation pumps and P4 ATPases are lipid flippases. Prominent examples of the P-type ATPase family are the Na+/K+-ATPase and the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA), which are P2 ATPases. In stark contrast to these pumps, which are extremely well characterized and for which more than 100 structures are deposited in the Protein Data Bank (PDB), virtually nothing is known about P5 ATPases, which have no assigned biochemical function. Sequence analysis of P5 ATPases has revealed that they are divided into two distinct subfamilies, named P5A and P5B ATPases (Sørensen et al., 2010). Whereas all P5A ATPases characterized to date localize to the endoplasmic reticulum (ER) membrane, P5B ATPases are associated with the vacuole or lysosomes (reviewed in Sørensen et al., 2015). Strikingly, whereas P5 ATPases are absent from prokaryotic genomes, all available eukaryotic genomes contain at least one P5A ATPase (Sørensen et al., 2015). This would suggest that their function is associated with some basic feature of eukaryotic cells.
The yeast Saccharomyces cerevisiae contains two P5 ATPase genes, the P5A ATPase Sensitivity to Picchia farinosa killer toxin (SPF1) and the P5B ATPase Yeast PARK9 (YPK9). One screen revealed that spf1 is insensitive to a toxin secreted by the competing yeast Picchia farinosa (Suzuki and Shimma, 1999), whereas another showed that a similar mutation causes defective degradation of 3-hydroxy-3-methylglutaryl coenzyme A reductase in the ER, which resulted in the gene being independently named Control of HMG-CoA degradation (COD1) (Cronin et al., 2000). A knockout mutation in SPF1 disrupts ER homeostasis and induces the unfolded protein response (UPR) (Jonikas et al., 2009). In the absence of Ire1p or Hac1p to initiate repair of the damage, the effect is lethal (Ng et al., 2000; Cronin et al., 2002; Vashist et al., 2002). Defects in sterol and isoprenoid biosynthesis likewise have a strong impact on the UPR (Jonikas et al., 2009). Indeed, a yeast knockout of SPF1 exhibits increased sensitivity to an inhibitor of ergosterol synthesis (Cronin et al., 2000) and an abnormal distribution of ergosterol between the ER and mitochondria (Krumpe et al., 2012). Furthermore, a mammalian homologue of Spf1p, ATP13A1, has been identified in a screen for proteins that interact directly with cholesterol (Hulce et al., 2013). Thus, a number of independent observations link P5A ATPases to sterol biosynthesis and transport.
Sterols are essential lipids in eukaryotic cells, where they influence the dynamics and functions of membranes (Mesmin and Maxfield, 2009). This effect is due to their rigid and planar structure, which reduces the flexibility of neighboring lipids, and in turn governs membrane fluidity and permeability. Sterols are synthesized and matured in the ER by a cascade of coupled enzymatic reactions (Espenshade and Hughes, 2007). Sterols are not randomly distributed in the cell; whereas the ER displays low sterol levels (around 5 mol% of lipids) (Radhakrishnan et al., 2008), sterols are abundant (up to 40 mol%) in the trans-Golgi, endosomes, and plasma membrane (PM) (Schneiter et al., 1999; Andreyev et al., 2010). Indeed, sterol accumulation in the ER is toxic (Feng et al., 2003), and sterols must therefore be rapidly transported away from their site of synthesis. Sterols leave the ER via a variety of mechanisms. For instance, sterols that are to be stored in the cytosol are esterified with long-chain fatty acids, and the resulting steryl esters accumulate in lipid droplets that bud off from the ER (Koffel et al., 2005). In contrast, those that are to be directly employed in other membranes are transported away from the ER by processes that do not seem to involve vesicular transport. These processes must be active, as sterols are transported against their concentration gradient.
The exact mechanism by which sterols are translocated from the ER to the PM is not understood (Lev, 2010; Prinz, 2010), but it has recently been proposed that lipid transfer proteins (LTPs) actively create sterol gradients by a lipid exchange mechanism (de Saint-Jean et al., 2011; Mesmin et al., 2013). According to this model, the LTP Osh4p functions as a sterol/phosphatidylinositol 4-phosphate (PI4P) exchanger that transports sterols between two membranes against their concentration gradient by dissipating the energy of the PI4P gradient (von Filseck et al., 2015). Thus, PI4P acts as the fuel that drives sterol transport and establishes active sterol concentration gradients across membrane-bound compartments. PI4P is mostly localized in the trans-Golgi and PM (Strahl and Thorner, 2007) and is virtually absent from the ER. In the trans-Golgi, PI4P is generated by the Pik1p-mediated phosphorylation of phosphatidylinositol (PI; Strahl and Thorner, 2007). In the ER, PI4P is degraded into PI by the PI4P phosphatase Sac1p (Foti et al., 2001; Faulhammer et al., 2007; Manford et al, 2010). Thus, the ATP-dependent production of PI4P in the Golgi by Pik1p and the Sac1p-mediated hydrolysis of PI4P at the ER each contribute to the establishment of a PI4P gradient between the ER and Golgi. In turn, this gradient energetically drives the creation of a sterol gradient by Osh4p (reviewed in Drin et al., 2016; Kentala et al., 2016; Mesmin and Antonny, 2016).
In this work, we attempted to elucidate the physiological role of Spf1p. Here, we present evidence that the hydrolytic activity of Spf1p is stimulated by PI4P. We show that, in the absence of SPF1, cells accumulate sterols at the plasma membrane and have disturbances in ergosterol homeostasis and that these changes affect protein function in the ER and PM. Our results suggest a possible role for Spf1p in the PI4P-regulated export of sterols from the ER.
RESULTS
P5 ATPases have a common ancestor with lipid-flipping P4 ATPases
A first step in understanding the physiological function of Spf1p is to identify the putative transported substrate or other binding ligands. One approach would be to identify the closest relatives of P5 ATPases with known functions. In early phylogenetic work, the closest relatives of P5 ATPases were suggested to be P4 ATPases, which are lipid flippases; however, statistical support for this assumption was not provided (Axelsen and Palmgren, 1998). To reinvestigate the phylogenetic placement of P5 ATPases, we made an inventory of P-type ATPase sequences from the genomes of 11 organisms from representative prokaryotic and eukaryotic supergroups and analyzed the data by maximum likelihood and Bayesian inference methods (Figure 1). We confirmed the monophyly of P5 ATPases in eukaryotes; furthermore, a common root with P4 ATPases could be identified with maximal statistical support for both methods. Although we cannot discard other possibilities, it is plausible that the common ancestor of P4 and P5 ATPases was a phospholipid-binding protein.
FIGURE 1:

P5 ATPases share a common origin with P4 ATPases. Phylogenetic tree of 171 amino acid sequences of P-type ATPases as inferred from the combined output of two independent statistical methods of measurement (Bayesian inference and maximum likelihood analysis; see Materials and Methods). Colors represent sequences from the major phyla of representative taxonomic supergroups. Black dots represent Bayesian inference values of 1.00 and black circles represent Bayesian inference values of 0.99. Numbers noted at key nodes are Bayesian inference statistical values on the left and maximum likelihood statistical values on the right of the dash (left/right). A star indicates the Spf1 protein. Question marks indicate that the transported ligand has not been identified. Scale bars represent evolutionary distance. Accession numbers of all sequences can be found in Supplemental Table S5.
Expression and purification of ATP hydrolytically active Spf1p
To analyze a possible role of Spf1p in phospholipid transport, we produced a recombinant protein and characterized it biochemically. For this purpose, we expressed the protein as a C-terminally FLAG-10xHis-tagged fusion construct in S. cerevisiae under control of the galactose-dependent GAL1 promoter. This construct complemented the spf1 mutant line with respect to its sensitivity to caffeine (Supplemental Figure 1A), demonstrating that it encodes a functional protein. Growth was optimal during glucose repression and was inhibited when SPF1 expression was boosted by galactose (Supplemental Figure 1A). The GAL1 promoter is leaky under glucose repression (Rodrigo-Brenni et al., 2004; Gohil et al., 2005), which would explain why complementation was observed on glucose medium and, furthermore, indicates that very low levels of Spf1p are optimal for cell growth. A catalytically dead version of Spf1p, in which the canonical aspartate residue required for autophosphorylation from ATP in all P-type ATPases was mutated to an asparagine (spf1pD488N), failed to complement the loss of SPF1. Tagged Spf1p was detectable by immunoblot analysis when cells were grown on glucose medium, but the signal was much stronger after 16 h on galactose medium (Supplemental Figure 1B).
We then purified the protein from the galactose-induced expression system. After induction, total membranes were isolated and solubilized with n-dodecyl-β-d-maltopyranoside (DDM), and the tagged protein was purified by Ni2+-NTA affinity chromatography in a single step. Eluted Spf1p-FLAG-10xHis appeared in both monomeric and oligomeric forms when analyzed by SDS–PAGE (Supplemental Figure 1C). Relipidation by 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC; 0.5 mg/mg protein) resulted in significant increases in both ATP hydrolysis (∼0.045 µmol Pi/min per mg protein) and phosphoenzyme formation (∼0.9 pmol 32P/μg protein), while the catalytically dead protein remained inactive (Supplemental Figure 1, E and F, purified in D). We further found the ATP hydrolytic activity of the purified enzyme to be highly sensitive to the P-type ATPase-specific inhibitor vanadate (Ki = 9.8 ± 2.4 μM; n = 3 biological replicates). Below, we refer to purified Spf1p relipidated with excess POPC (3.5 mg lipid/mg protein) as the “basal preparation.” P-type ATPases are characterized by tight coupling between ATP hydrolysis and the vectorial transport of a bound substrate (Palmgren and Nissen, 2011). As ATP hydrolysis requires binding of the transported substrate, the finding that our purified Spf1p preparation actively hydrolyzed ATP suggests that the unknown substrate for this P5 ATPase is a molecule already present in the purified preparation or the ATPase assay buffer. The purified Spf1p protein was likely associated with phospholipids, sterols, and other organic molecules present in ER membranes and/or other membrane components that copurified with the enzyme, and one or more of these, in principle, could be responsible for causing the observed basal Spf1p ATPase activity.
Spf1p activity is stimulated by phosphatidylinositol phosphates but not by cations
To identify possible lipid ligands, besides PC, we reactivated the partially delipidated Spf1p with POPC or defined phospholipid mixtures, namely, POPC supplemented with 10 mol% of single lipid components (PI, 1-palmitoyl-2-oleoyl-PS [POPS], 1-palmitoyl-2-oleoyl-PA [POPA], 1-palmitoyl-2-oleoyl-PE [POPE], ergosterol, sphingomyelin, ceramide, or PI4P), and assayed ATP hydrolytic activity (Figure 2A). None of the phospholipids typically abundant in biological membranes significantly stimulated ATPase activity above that of the basal preparation. Indeed, POPS appeared to slightly reduce ATP turnover, in contrast to previous reports (Corradi et al., 2012). Ergosterol and ceramide slightly increased the ATPase activity (less than 30% activation). A porcine brain extract of PI4P stimulated the activity by ∼75%, making it the most potent stimulator of Spf1p. The addition of PI4P resulted in a dose-responsive curve with a K0.5 value of ∼1.4 mol% for PI4P (Figure 2B). An effect at this low PI4P:POPC ratio indicates that the mixed acyl PI4P extract acts as a high-affinity activator of Spf1p.
FIGURE 2:

PI4P stimulates the ATP hydrolytic activity of purified Spf1p and protects against irreversible denaturation. Spf1p was relipidated with POPC or POPC plus the indicated phospholipids (10 mol% unless otherwise stated), and ATPase activity was measured after 30 min. (A) The effect of various phospholipids on Spf1p activity. POPS, palmitoyl-oleoyl-sn-phosphatidylserine; PI, phosphatidylinositol; Sph, sphingomyelin; POPA, palmitoyl-oleoyl-sn-phosphatidic acid; POPE, palmitoyl-oleoyl-sn-phosphatidylethanolamine; Erg, ergosterol; Cer, ceramide; PI4P, phosphatidylinositol 4-phosphate. (B) Dose–response effect of PI4P on Spf1p activity. (C) Effect of different phosphatidylinositol/phosphatidylinositol phosphate lipids on Spf1p activity. Extract, PI4P extracted from porcine brain lipids. diC16, synthetic dipalmitoyl phospholipids. (D) Stabilization of Spf1p by PI4P as a function of time. (E) pH dependence of Spf1p ATPase activity (black lines; Pi release) and phosphoenzyme (EP) formation (gray lines) in the presence (dashed lines) and absence (solid lines; POPC) of PI4P. (F) Temperature dependence of Spf1p ATPase activity in the presence and absence of PI4P. For all data, error bars represent SEM; n = 3 biological replicates.
To examine the specificity of Spf1p for phosphorylated phosphoinositide (PI) lipids, we tested synthetic PI lipids that all had identical acyl chains (dipalmitoyl, 16:0) but differed in the positions and number of phosphorylations present on the inositide head group. Spf1p appeared to be responsive to the placement and amount of phosphorylations on the inositol ring (Figure 2C). PI(3,4)P and PI4P caused the highest ATPase activity, whereas PI(3)P had a minor stimulatory effect (Figure 2C). Phosphorylation on the 5′ site of the inositol ring (PI(5)P, PI(3,5)P, and PI(4,5)P), full phosphorylation of the inositol ring (PI(3,4,5)P), or no phosphorylation at all (PI) resulted in loss of the stimulatory effect (Figure 2C). Even though the signaling lipid PI(3,4)P2 stimulated Spf1p to higher levels than PI4P, several studies failed to detect the presence of PI(3,4)P2 in yeast cells (Desrivières et al., 1998; Mitra et al., 2004). It therefore seemed likely that only PI4P was a physiologically relevant activator of Spf1p.
Binding of a ligand usually results in increased protein thermostability (Celej et al., 2003). Therefore, should PI4P be a true Spf1p ligand, we would expect that its binding would prevent thermal denaturation of the protein. Spf1p reactivated with POPC or POPC/PI4P was preincubated at 30°C, and the subsequent loss of activity was studied. We observed that PI4P increased the thermostability of Spf1p, suggesting that binding of this lipid to Spf1p protects the protein against heat denaturation (Figure 2D). A similar protective effect of PI4P was observed when Spf1p was exposed to alkaline pH (Figure 2E). The PI4P-induced activity of Spf1p peaked at around 40–45°C (Figure 2F).
An alternative possibility for a ligand could still be a contaminating cation present in the Spf1p preparation. To exclude this possibility, we added biologically relevant cations to a metal ion–deprived medium to measure their influence on the ATPase activity of Spf1p or its catalytic autophosphorylation. None of the tested metal ions stimulated Spf1p activity at biologically relevant concentrations (Supplemental Figure 2). Ca2+ and Mn2+, which have previously been considered ligands of Spf1p, had no stimulatory effect on the ability of Spf1p to hydrolyze ATP (Supplemental Figure 2A) or on its steady-state phosphorylation level (Supplemental Figure 2B). Likewise, addition of cation chelators or pretreatment of the assay medium with a chelating resin to remove trace metal ions had no effect on the intrinsic residual activity of Spf1p (Supplemental Figure 2C). P2 and P3 ATPases contain a conserved binding pocket for K+ in the cytoplasmic headpiece of the protein, which has a millimolar affinity for K+, and binding of K+ to this site accelerates dephosphorylation during catalysis (Sørensen et al., 2004; Buch-Pedersen et al., 2006; Schack et al., 2008). We did not observe any effect of K+ on either ATP hydrolysis or phosphoenzyme turnover for Spf1p (Supplemental Figure 2D). In accordance with this finding, residues in the regulatory K+ site are not conserved in P5A ATPases (Sørensen et al., 2010).
In conclusion, we show that Spf1p activity is stimulated by lipids but not by cations and that PI4P is the physiologically relevant lipid with the strongest positive impact on Spf1p activity.
A stimulatory effect of PI4P is associated with a shift in the conformational equilibrium of Spf1p
Specific binding of phosphorylated PIs to Spf1p could result from binding to a transport site or, alternatively, a regulatory site. As all P-type ATPases undergo large conformational movements when binding and transporting their specific ligand(s), shifts in conformational equilibrium states can be used to determine the effect of ligands on partial reactions in the turnover mechanism related to transport. One classical method of doing so involves tracing phosphoenzyme formation using radiolabeled 32P-ATP and measuring the sensitivity of the phosphoenzyme to unlabeled ADP and ATP (Buch-Pedersen et al., 2006; Schack et al., 2008; Coleman et al., 2012; Sørensen et al., 2012). Normally, only one of the two phosphorylated conformational states of the enzyme (called the E1P phosphoenzyme state) is sensitive to ADP, whereas the other (E2P state) is not.
To test the effect of PI4P on phosphoenzyme turnover, we incubated Spf1p with radiolabeled 32P-ATP and subsequently measured the effect of PI4P on the decay rate of the phosphoenzyme. The assay was carried out at 45°C, where phosphoenzyme turnover was high (Figure 3A). We observed that addition of PI4P increased the ATP-induced decay rate of the phosphoenzyme (Figure 3B) from T0.5 = 2.65 ± 0.6 s, when only POPC was present, to T0.5 = 0.95 ± 0.23 s, when PI4P was added. The ADP-induced decay rate was very high, and an effect of PI4P could not be detected (Figure 3C). To confirm that PI4P did not affect Spf1p through changes in the fluidity of the detergent/lipid micelles, we used an Arrhenius plot to calculate enzyme activity across a temperature span. We observed a linear relationship between temperature plotted as K–1 and activity (Figure 3D), which suggests that the activation energy required for ATP hydrolysis is constant at all temperatures and that PI4P does not affect Spf1p through changes in micelle fluidity. The effect of PI4P on phosphoenzyme turnover is further evidence that PI4P binds to Spf1p and stimulates its catalytic turnover by changing the conformational equilibrium of the enzyme.
FIGURE 3:

Spf1p phosphoenzyme turnover is affected by PI4P. The 32P phosphoenzyme intermediate of purified Spf1p was formed by incubation with γ-32P-labeled ATP, and phosphoenzyme decay was traced following the addition of excess amounts of unlabeled ATP or ADP to the reaction at 45°C. (A) The temperature-dependent effect of PI4P on Spf1p phosphoenzyme formation is not related to the changed fluidity of the membranes. Increasing the temperature reduced the phosphoenzyme formation of Spf1p relipidated in both POPC or POPC + mol% PI4P, with the phosphorylated state of the PI4P-activated enzyme being slightly less abundant. Steady-state conditions were ensured as all temperatures gave stable activity for up to 2 min. * indicates two-tailed t test, P = 0.0007. (B) In the presence of ATP, PI4P increases the decay rate of the Spf1p phosphoenzyme intermediate compared with that of the enzyme relipidated with POPC only. (C) In the presence of ADP, an effect of PI4P on the decay rate of the Spf1p phosphoenzyme was not observed relative to that of the enzyme relipidated with POPC only. (D) Arrhenius plot of Spf1p activity across the temperature span shows a linear relationship between temperature plotted as K–1 and specific activity, suggesting that the activation energy required for ATP hydrolysis is constant at all temperatures. For all data, error bars represent SEM; n = 3 biological replicates.
Spf1p localizes to ER domains devoid of the PI4P hydrolytic machinery
PI4P is rapidly degraded in the ER by the action of the phosphatase Sac1p (von Filseck et al., 2015); therefore, physiologically relevant PI4P stimulation of Spf1p activity did not seem plausible in the presence of Sac1p. To study the localization of Sac1p and Spf1p under physiological conditions, we genetically tagged the loci encoding both enzymes using a previously described method (Lee et al., 2013). This resulted in a yeast strain in which natively expressed Spf1p is fused to GFP and natively expressed Sac1p is fused to RFP. When analyzing the cells by fluorescence microscopy, we observed that both enzymes localize to the ER membrane (Figure 4A). However, while Sac1p is mainly found in the PM-associated part of the cortical ER, Spf1p localizes to both cortical ER-PM sites and the nuclear envelope/ER network. Notably, the cortical ER spots containing Spf1p and Sac1p do not appear to overlap, suggesting that Spf1p and Sac1p are present in distinct parts of the cortical ER network. For comparison, we conducted a similar experiment by tagging another locus encoding an ER-resident protein (SEC61) with RFP. The expression patterns of Spf1p-GFP and Sec61-RFP clearly overlapped (Figure 4B). Our results suggest that hydrolysis of PI4P in the ER is strictly separated from Spf1p localization, indicating that this phosphoinositide could be a physiologically relevant stimulator of Spf1p activity.
FIGURE 4:
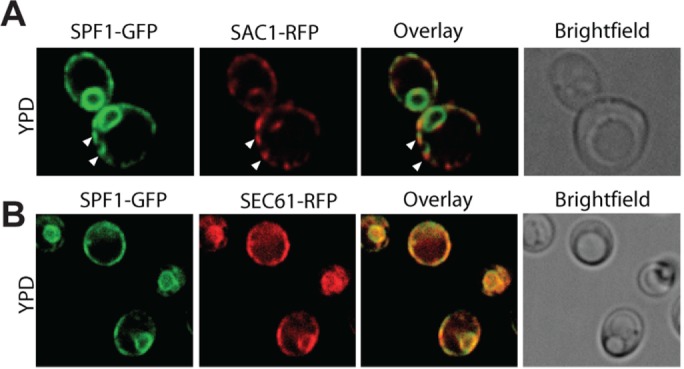
Endogenously expressed Spf1p and Sac1p localize to distinct parts of the ER membrane. SPF1 and SAC1 were genetically tagged with GFP and RFP, respectively, to allow for visualization by confocal microscopy of their intracellular localization (images representative of individual cells; n = 55). (A) Sac1p is found mainly in the cortical ER, while Spf1p localizes to both cortical ER-PM sites and the nuclear envelope/ER network. Cortical ER spots containing Spf1p and Sac1p do not appear to overlap (indicated by white arrowheads). (B) Spf1p-GFP shows a high degree of overlap with the ER marker Sec61p-RFP (image representative of individual cells, n = 23).
Loss of SPF1 is associated with perturbed cellular sterol and sphingolipid homeostasis
Having identified a role for PI4P in stimulating Spf1p activity, we aimed to determine the physiological relevance of such an effect. A key role for PI4P in cells is to control the intracellular distribution of sterols (Drin et al., 2016). If PI4P regulation of sterol homeostasis in some way involves Spf1p, we expected that loss of Spf1p would influence this equilibrium. Inhibition of sterol biosynthesis by lovastatin has already been demonstrated to affect the growth of spf1 cells (Cronin et al., 2002; Figure 5A). To further analyze any possible links between SPF1 and sterol homeostasis, we tested the effect of multiple drugs that inhibit sterol synthesis and the related sphingolipids on spf1, ypk9, and wild-type cells. We found that manumycin A, terbinafine, and fenpropimorph, which inhibit enzymes farther downstream in the mevalonate pathway likewise required for ergosterol synthesis (Supplemental Figure 3), negatively affected the growth of spf1 cells (Figure 5A). Similarly, drugs that inhibit sphingolipid synthesis at the committed steps of the pathway, that is, at the synthesis of 3-ketodihydrosphingosine (myriocin) and inositolphospho ceramides (IPCs; aureobasidin A), had a specific effect on spf1 cells (Figure 5B). Furthermore, spf1 cells showed increased sensitivity to amphotericin B and nystatin, drugs that work by inducing the formation of pores in the plasma membrane following binding to sterols (Figure 5C; Kamin´ski, 2014). This result indicates that the biological availability of sterols is increased in the plasma membrane of spf1 cells compared with that in wild-type and ypk9 cells. When stained with filipin, a dye that associates with sterol-rich compartments in membranes (Cohen et al., 2013), spf1 cells displayed increased staining at the plasma membrane (Figure 5D), consistent with the notion that the plasma membrane of spf1 cells is enriched in sterols.
FIGURE 5:
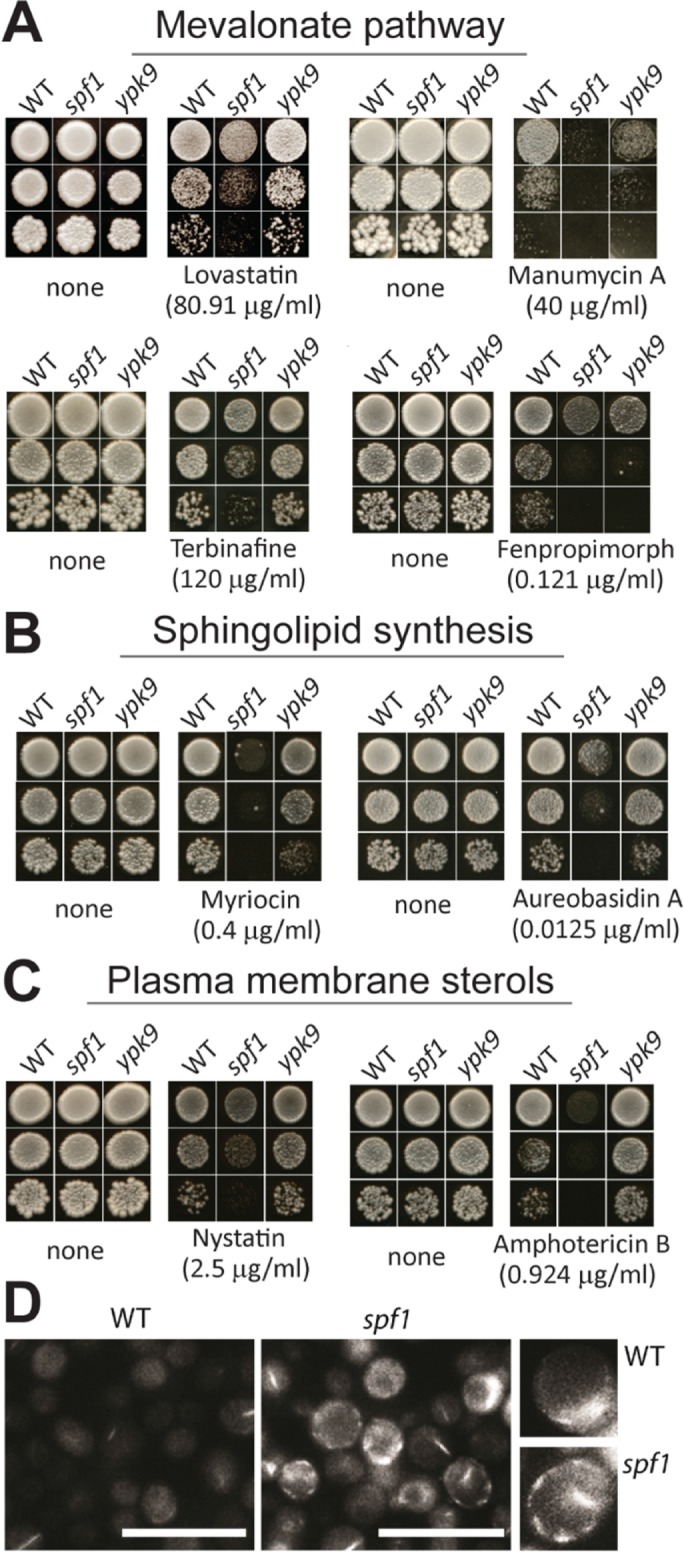
Functional sterol and sphingolipid synthesis pathways are needed to withstand loss of SPF1 in S. cerevisiae. (A, B) Wild-type (WT), spf1, and ypk9 yeast cells grown on rich media supplemented with inhibitors and antibiotics against lipid biosynthesis pathways, as summarized in Supplemental Figure S3. Inhibitors against enzymes in the mevalonate pathway (A) and sphingolipid biosynthesis pathways (B) negatively affect growth of spf1 cells to different extents, whereas ypk9 cells are insensitive to these inhibitors. (C, D) The plasma membrane of spf1 cells is enriched in sterols. (C) Cytolytic drugs that impact cells with a high plasma membrane sterol content negatively affect growth of spf1 cells to different extents, whereas ypk9 cells are insensitive. (D) spf1 cells show increased plasma membrane staining by filipin compared with wild-type cells. Scale bar, 10 μm. All inhibitors were screened by titration until knockdown of growth for all cell types, and only data related to the biggest observable difference between wild-type and spf1 cells are shown.
Taken together, spf1 cells showed signs of both sterol deficiency in the ER (i.e., increased sensitivity to inhibitors of sterol biosynthesis) and sterol surplus (i.e., accumulation at the plasma membrane). Notably, these effects were not observed in the ypk9 mutant. These data indicate that a specific loss of spf1 results in sterol dyshomeostasis.
Next, total sterol content was measured by mass spectrometry (Silvestro et al., 2013), and phospholipids and sphingolipids were labeled metabolically by adding inorganic 32P and 3H-inositol, respectively, to the growth medium, followed by total lipid extraction and analysis on thin-layer chromatography (TLC) plates. We identified a significant increase in the total level of ergosterol in spf1 cells over wild-type cells (3.0 µg ergosterol compared with 2.1 µg ergosterol per OD600 in spf1 and wild-type cells, respectively; p < 0.0001; n = 3), which coincided with a significant decrease in total lanosterol as measured by mass spectrometry (Figure 6A). Consequently, the ergosterol-to-lanosterol ratio was ∼4-fold greater in spf1 knockout cells than in wild-type cells (Figure 6B). This indicates a shift in the sterol synthesis pathway toward the end product. In contrast, we observed only minor changes in the phospholipid profile between wild-type and spf1 cells (Supplemental Figure 4). Although the combined total level of sphingolipids seemed to markedly increase in spf1 cells (Figure 6C), we found little change in the ratio between the three major sphingolipid classes found in yeast, IPCs, mannosylinositol phosphoceramide (MIPC), and mannosyl-di-(inositolphosphoryl) ceramide (M(IP)2C) (Figure 6D).
FIGURE 6:
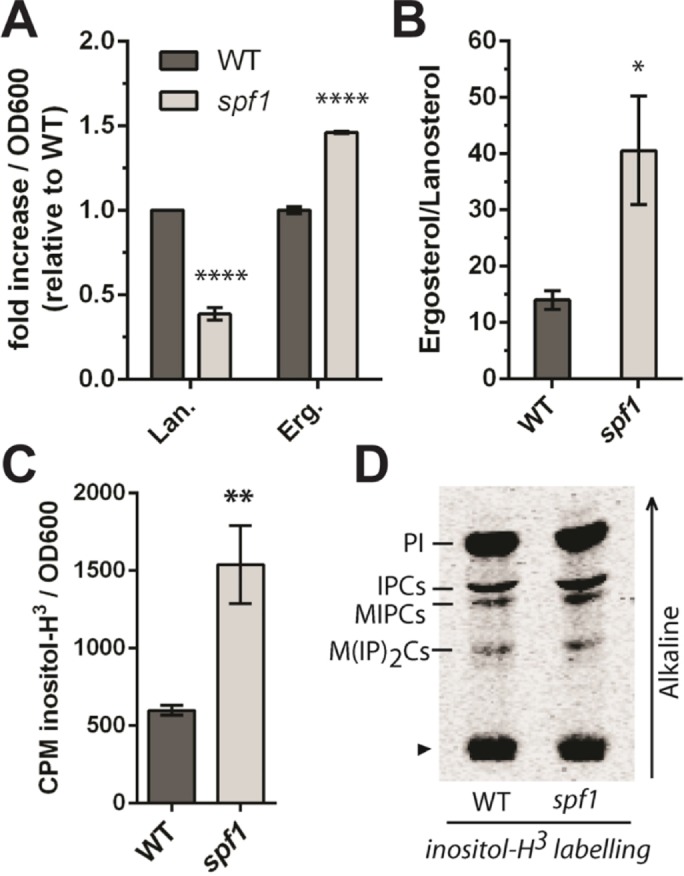
Deletion of SPF1 results in alterations of cellular sterol composition and an increase in the total sphingolipid content. (A) Lanosterol (Lan) is decreased, whereas ergosterol (Erg) is increased in spf1 cells. Data represent three biological replicates. ****p < 0.0001, according to a two-tailed t test. Actual specific values measured represent ergosterol, 3.0 μg (spf1)/2.1 μg (WT), and lanosterol, 0.04 μg (spf1)/0.11 μg (WT) per OD600. (B) The total ergosterol-to-lanosterol ratio is ∼4 times greater in spf1 cells than in wild-type cells (n = 6 biological replicates, two-tailed t test, *p = 0.0303). (C) Sphingolipids are more abundant in spf1 cells. Total lipid extracts from a similar number of wild-type and spf1 cells were labeled with 3H-inositol and measured by scintillation counting (two-tailed t test, **P = 0.0030, n = 3 biological replicates). Error bars in A–C represent SEM. (D) Comparison of sphingolipid classes in wild-type and spf1 cells. Lipid extracts containing an equal amount of 3H label (based on CPM) were separated on TLC plates. An arrowhead indicates the loading spot. The arrow to the right indicates the migration of alkaline solvent. PI, phosphatidylinositol; IPC, inositolphosphoryl ceramide; MIPC, mannosyl-inositolphosphoryl ceramide; M(IP)2C, mannosyl-di-(inositolphosphoryl) ceramide.
Lipid bodies with altered sterol content accumulate in the spf1 mutant background
Lipid bodies are a major reservoir for sterols in cells (Thiam et al., 2013). The sterol C-24 methyltransferase Erg6p is present in both lipid bodies and cortical ER sites associated with the plasma membrane (Athenstaedt et al., 1999; Pichler et al., 2001). When an Erg6p-GFP reporter construct was expressed in yeast cells, the signal was strong in internal structures of wild-type cells (Figure 7A, white arrowheads), whereas it was not detected in spf1 cells (Figure 7A, broken arrow marks the plasma membrane). Overall, Erg6p expression was strongly down-regulated in spf1 cells (Figure 7B). The spf1 mutant cells reacted strongly with Nile red, a dye that binds to neutral lipids and accumulates in lipid bodies (Greenspan et al., 1985; Figure 7, C and D). Measurement of the total energy content of wild-type and spf1 cells by calorimetry also revealed that spf1 cells contained more energy stored in chemical bonds than did wild-type cells (Supplemental Table S1). This is in accordance with the finding that spf1 cells accumulate lipid bodies.
FIGURE 7:

Disruption of SPF1 induces formation of lipid bodies with altered sterol content. (A) Deletion of spf1 results in depletion of an Erg6-GFP reporter from internal structures (white arrowheads; the broken arrow marks the plasma membrane) imaged by fluorescence microscopy. (B) Erg6-GFP expression levels quantified by fluorescence intensity of GFP (n = 100 examined cells each of WT and spf1; two-tailed t test; ****p < 0.0001). (C) Nile red staining of lipids in wild-type (WT) and spf1 cells imaged by fluorescence microscopy. White arrowheads indicate lipid bodies (LBs). (D) Box plot of Nile red fluorescence within LBs (n = 20) from wild-type (WT) and spf1 cells (two-tailed t test, ****p < 0.0001). (E) Depletion of Spf1p is associated with a shift in the ergosterol-to-phosphatidylcholine ratio in LBs. Lipids were extracted from cellular fractions carrying LBs from spf1 cells and spf1 cells overexpressing the Spf1p construct. Total ergosterol content was determined by TLC analysis, whereas total PC content was determined by an enzymatic assay. (F) Emission spectra for Nile red–stained regions within the yeast cells were derived from lambda scans ranging from 540 to 660 nm using a 514-nm laser line and 4-nm steps. Four peaks were detected at 576, 595, 615, and 645 nm (n = 6, two-tailed t test; *p < 0.04).
Lipid bodies from spf1 cells and cells overexpressing SPF1 were isolated and their lipid content analyzed. We observed that the ergosterol-to-phosphatidylcholine ratio was higher in cells overexpressing SPF1 than in the spf1 mutant (Figure 7E). An emission scan of the Nile red–stained particles in wild-type and spf1 cells revealed a significant shift in the relative distribution of two peaks associated with a red-shifted population of Nile red molecules in spf1 cells (Figure 7F). As Nile red undergoes changes in spectral properties depending on solvent polarity (Greenspan et al., 1985), the data confirm that the lipid bodies of spf1 cells have a different composition than those of wild-type cells.
Vital sterol-sensitive processes are compromised in the spf1 background
Next, we tested whether correct functioning of sterol-sensitive processes was affected in spf1 cells. Sec61p is a sterol-sensitive ER translocon protein that mediates secretion of the α-mating factor (Nilsson et al., 2001, Plath et al., 2004, Yamamoto et al., 2012). To test Sec61p function, we studied the secretion ability of a GFP protein fused to the pre-pro sequence of the α-mating factor signal peptide (sec-yEGFP) in spf1 and wild-type cells. We found that removal of spf1 results in increased GFP retention in the cytosol, which indicates a decreased capacity to secrete this peptide (Figure 8, A and B). This suggests that the Sec61p translocon function is hampered in the spf1 background, resulting in decreased uptake of the reporter protein into the ER.
FIGURE 8:

Deletion of SPF1 inhibits sterol-sensitive processes in the ER and PM. (A, B) Processing of pre-pro-α-mating factor signal peptide is defective in spf1 cells and the peptide accumulates in the cytoplasm. The peptide was fused to the yEGFP reporter (sec-yEGFP), and intracellular fluorescence was quantified by whole-cell flow cytometry (A) and visualized in intact cells by confocal imaging (B). Cyto-yEGFP, free yEGFP. Data are reported as the average of three independent experiments (two-tailed t test; *p = 0.0127; ns. = not significant; error bars represent SEM). (C, D) Deletion of SPF1 results in failure to activate the plasma membrane proton ATPase. (C) Growth of spf1 cells is inhibited at alkaline pH, where Pma1p action is required to establish a proton gradient. (D) Pma1p cannot be glucose-activated in spf1 cells. In the basal state, the maximal specific activity (Vmax) and ATP affinity (Km) were comparable between Pma1p in wild-type and spf1 yeast. After glucose activation, Pma1p from spf1 exhibited a lower Vmax and higher Km than did that from wild-type yeast. The dashed lines indicate glucose-activated Pma1p (n = 3 biological replicates; error bars represent SEM).
The Pma1p PM H+-ATPase proton pump is excluded from detergent-insoluble sterol-rich domains of the PM (Grossmann et al., 2007) and would therefore be expected to be affected by changes in sterol composition of the PM. We observed that, in contrast to wild-type cells, mutant spf1 cells hardly grew at alkaline external pH (pH 7.6), whereas growth was unchanged at pH 7.0 (Figure 8C). This suggests a failure to maintain the proton gradient at limiting conditions when the extracellular proton concentration is low. A common way to test the functionality of Pma1p in isolated membranes is to study ATP hydrolysis after glucose activation of the protein (Serrano, 1983). Indeed, whereas Pma1p in wild-type cells showed high activity following glucose activation of isolated membrane fractions (from Vmax = 0.6 ± 0.1 to 1.4 ± 0.1), Pma1p in spf1 cells responded only marginally to glucose treatment (from Vmax = 0.6 ± 0.1 to 0.8 ± 0.2; Figure 8D). Likewise, the Km for ATP decreased in wild-type Pma1p following glucose activation but not in the spf1 background (Figure 8D). These data suggest that the formation of the PM proton gradient is hampered in the spf1 background as a result of decreased Pma1p activity.
Our results show that vital membrane proteins in both the ER and PM have altered properties in the spf1 background, which could be explained by dyshomeostasis of sterol content in these membranes. As these proteins maintain basal functions in cation balance and protein folding and processing, their impaired activity would affect a multitude of downstream processes. This, along with similar negative effects on other sterol-dependent enzymes, explains the pleiotropy of phenotypes described for spf1 cells (reviewed in Sørensen et al., 2015).
SPF1 exhibits a strong negative genetic interaction with OSH genes
The nonvesicular export of ergosterol from its site of synthesis in the ER is carried out by Osh proteins that are energized by the dissipation of the PI4P gradient between the ER and Golgi (Drin et al., 2016; Kentala et al., 2016; Mesmin and Antonny, 2016). Therefore, PI4P and sterol homeostasis are directly linked in the cell. As Spf1p is stimulated by PI4P and affects sterol accumulation at the PM, we next examined a possible connection between SPF1 and OSH genes by attempting to generate double knockout strains. In the spf1 background, we failed to generate stable knockouts of six (OSH1 to OSH6) of the seven OSH genes found in S. cerevisiae. Generation of the double knockout spf1/osh7 was not attempted, as OSH7 is a paralogue of OSH6 and was suggested to have redundant functions with this gene (Tian et al., 2018). To confirm this result, we introduced a plasmid carrying SPF1 and a URA3 marker that confers sensitivity to 5-fluoroorotic acid (5-FOA) into individual cell lines carrying knockouts of OSH1 to OSH6. Subsequently, we knocked out the chromosomal copy of SPF1 in all lines prior to treating the cells with 5-FOA to select against the plasmid. None of the resulting strains were viable (Figure 9). This demonstrates that deletion of SPF1 in any of the osh1–6 backgrounds results in synthetic lethality, indicating a strong negative genetic interaction between ER-related Osh proteins and Spf1p.
FIGURE 9:
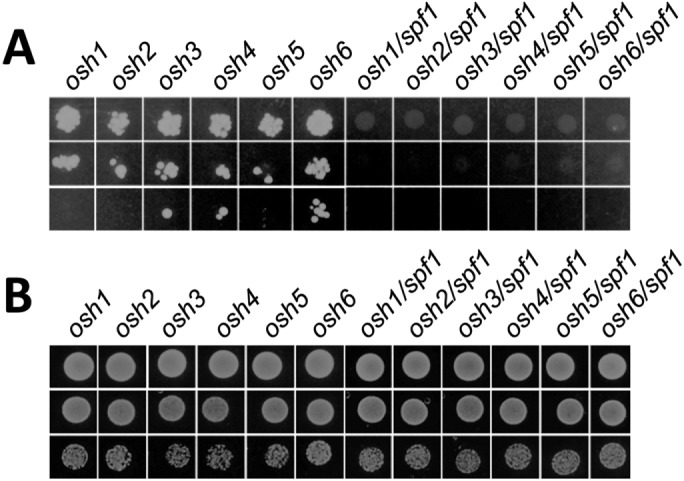
SPF1 knockouts are not viable in a background carrying a deletion of an OSH gene. Individual cell lines carrying knockouts of OSH1 to OSH6 were transformed with a plasmid carrying SPF1 and a URA3 marker that confers sensitivity to 5-FOA. In a parallel series, the chromosomal copy of SPF1 was knocked out in all lines. (A) Growth of cells during 5-FOA selection. Transformed cells were grown on selective minimal medium with uracil and treated with 5-FOA to select against the plasmid. (B) Growth of cells in the absence of 5-FOA selection. Transformed osh and double knockout osh/spf1 cells were grown on selective minimal media without uracil.
SPF1 interacts functionally and genetically with SAC1 in maintaining plasma membrane sterol contents
Next, we tested whether Spf1p has an effect on PI4P cellular distribution that may affect the sterol transport to the PM by Osh proteins. To test this, we expressed a fluorescent PI4P-binding reporter (GFP-2xPHOsh) in wild-type cells and studied PI4P distribution under conditions where Spf1p activity is essential for cell survival. We observed that PI4P levels decreased in intracellular membranes and increased at the PM in response to drug-induced depletion of sterols and sphingolipids, and at increased temperatures (Figure 10, A and B). Deletion of SAC1 affected PI4P distribution in a manner analogous to treatment with drug inhibitors (Figure 10C), corroborating previous findings (Jesch et al., 2010). Moreover, deletion of SPF1 in wild-type or sac1 cells did not result in any discernible changes in PI4P localization, indicating that PI4P redistribution in response to sterol depletion is linked to removal of Sac1p, but not to changes in Spf1p activity.
FIGURE 10:

Redistribution of PI4P in response to sterol dyshomeostasis is a function of SAC1 but not SPF1. Intracellular localization of PI4P was detected by the fluorescent lipid-associated GFP-2XPHOsh2 PI4P reporter construct. (A) Inhibition of sterol and sphingolipid biosynthesis by fenpropimorph (Fenprop.; 0.121 μg/ml) and myriocin (0.4 μg/ml), respectively, results in accumulation of PI4P in the PM. In the control, DMSO was added. (B) Increasing the temperature from 30 to 37°C results in accumulation of PI4P in the PM. (C) Deletion of SAC1, but not SPF1, results in accumulation of PI4P in the PM. Filled arrows, PI4P in intracellular spots most likely representing Golgi membranes. Broken arrows, PM. All images are representative of individual cells: A, B, n = 20; C, WT, n = 36; sac1, n = 25; spf1, n = 25; sac1/spf1, n = 29.
Next, we compared the growth of a wild-type strain with that of single and double knockout strains carrying genetic lesions in either SPF1 or SAC1, or in both genes concurrently. Whereas the single mutants were not affected in growth on rich medium, growth of the spf1/sac1 double knockout was inhibited, particularly in the early growth phase (Figure 11A). The genetic interaction between SPF1 and SAC1 was enhanced under rich media conditions by caffeine (Figure 11B), which interferes with phosphoinositide homeostasis (Saiardi et al., 2005), and by fenpropimorph (Figure 11C), which suppresses sterol biosynthesis (Kelly et al., 1994). Compared with wild-type cells, the plasma membrane of sac1 cells displayed a lower sterol content, as judged by decreased sensitivity to nystatin (Figure 11D) and decreased staining with filipin (Figure 11E). In contrast, three individual spf1/sac1 double knockout cell lines resembled wild-type cells with regard to nystatin sensitivity and filipin staining (Figure 11, D and E). This would suggest that Spf1p and Sac1p work in opposing directions to maintain plasma membrane sterol levels. Our results demonstrate a genetic interaction between SPF1 and SAC1, which may be related to disruption of phosphoinositide and sterol homeostasis.
FIGURE 11:

SPF1 interacts functionally and genetically with SAC1 in maintaining plasma membrane sterol content. (A) Growth of the spf1/sac1 double mutant is slower than that of the single mutants. Three individual spf1/sac1 mutants were tested. Growth was recorded after 1 d at 30°C. (B) Growth of the spf1/sac1 double mutant is more sensitive to caffeine than the combined effect of the single mutants. Overexpression of SPF1 functionally complements the phenotype. (C) Growth of the spf1/sac1 double mutant is impaired compared with the single mutants when sterol synthesis is inhibited. Fenpropimorph (Fenprop.) was added at 0.0605 μg/ml. (D) Deletion of sac1 partially rescues the accumulation of PM sterols in the spf1 mutant. Nystatin was added at 1.25 μg/ml. (E) Deletion of sac1 partially rescues the accumulation of PM sterols in the spf1 mutant. Three individual spf1/sac1 mutants were tested. Mean intensities of filipin staining were quantified (box plot, two-tailed Welch t test, ****p < 0.0001).
DISCUSSION
P5 ATPases constitute a eukaryotic subfamily of P-type ATPases ubiquitous in the endoplasmic reticulum, where they appear to fulfill a vital function. Although the transport function, if any, is unknown, we show that they share a common ancestor with P4 ATPases, which also are specific for eukaryotes and transport phospholipids. As both subfamilies evolved before the appearance of the major eukaryotic supergroups, P5 ATPases might have functional roles related to lipid homeostasis.
Ligand dependency of Spf1p activity
The spf1 mutant is sensitive to divalent metal cations (Cronin et al., 2002; Heuck et al., 2010), and the addition of extracellular Ca2+ suppresses a subset of spf1 phenotypes (Cronin et al., 2000). Therefore, it was previously proposed that the transported substrate of Spf1p could be Ca2+ (Cronin et al., 2000; Vashist et al., 2002) or Mn2+ (Cohen et al., 2013). However, our results dispute the notion that cations, including K+, are the transport substrates of Spf1p. These results are in accordance with previous studies showing that metal cations have no or only a minimal stimulatory effect on Spf1p activity at physiological concentrations and, indeed, seem to inhibit enzyme function at higher concentrations (Cronin et al., 2002; Corradi et al., 2012). The metal ion sensitivity of spf1 cells is therefore likely to be a secondary effect not related to the catalytic properties of Spf1p. Indeed, similar metal sensitivities are observed in mutant strains of P4 lipid flippases (Pomorski et al., 2003), which share a common evolutionary origin with P5 ATPases (Figure 1).
We evaluated the effect of multiple lipids on Spf1p-mediated ATP hydrolysis. Among the phospholipids tested, PI4P and PI3,4P were by far the most potent species in stimulating basal ATP hydrolysis by Spf1p. As PI4P increases the stability of Spf1p without influencing micelle fluidity, it is likely that the effect of PI4P on Spf1p reflects direct binding to the protein. However, it remains unclear whether PI4P binds to a transport site or to a site that merely serves a regulatory function. The P4 lipid flippase Drs2p is strongly activated by binding of PI4P to regulatory sequences in the C-terminal region distant from the transport site (Azouaoui et al., 2014), and the human P5B ATPase ATP13A2 is regulated by binding of phosphatidylinositol bisphosphate (PI3,5P) to its N-terminal region (Holemans et al., 2015). The N- and C-termini of Spf1p are predicted to be short (less than 25 amino acid residues each) and do not contain lipid-binding motifs as described for P4 and P5B ATPases.
Role of Spf1p in maintaining sterol homeostasis
The sensitivity of spf1 cells to inhibitors affecting lipid metabolism suggests that fully functional lipid biosynthesis pathways are needed to withstand the loss of SPF1. Furthermore, in the spf1 mutant, 1) lipid bodies accumulate and have an altered morphology, most likely due to a decrease in sterol content, 2) ergosterol accumulates at the expense of lanosterol, and 3) sterols accumulate in lipid aggregates at the plasma membrane. That Spf1p influences sterol homeostasis is compatible with earlier observations in the literature (Cronin et al., 2000; Jonikas et al., 2009; Krumpe et al., 2012). Most notably, SPF1 interacts strongly with HMG2 (Cronin et al., 2000), which encodes an HMG-CoA reductase catalyzing the first committed step in the mevalonate pathway, which is responsible for ergosterol production. In addition, several negative genetic interactions between SPF1 and genes involved in lipid synthesis and trafficking have been reported (references given in Supplemental Table S2).
We have previously argued that malfunction of proper sterol transport and distribution within the cell could explain the pleiotropy of phenotypes observed in P5A ATPase knockout cells (Sørensen et al., 2015). We now show that two sterol-sensitive membrane proteins, Sec61 in the ER and Pma1p in the PM, have altered properties in the spf1 background. As these proteins are closely connected to basic functions in protein secretion and cellular cation balance, their malfunction would be expected to influence numerous other processes in the cell. Misdistribution of sterols further explains other key features observed in spf1 cells related to proper insertion of membrane proteins and glycolipid-anchored proteins (Sørensen et al., 2015). Hence, the results presented in this work confirm that changes in overall sterol homeostasis and distribution are likely responsible for the pleiotropic phenotypes of spf1 cells.
In yeast, newly synthesized sterols leave the ER through a process mediated by the PI4P-hydrolyzing phosphatase Sac1p (Drin et al., 2016). In this process, Sac1p works with members of the Osh class of proteins to drive sterols from their biogenesis compartment in the ER to receiving membranes. The Osh proteins act as the actual lipid/PI4P exchangers between opposing membrane leaflets, and Sac1p drives dissipation of the PI4P gradient in the donor membranes (Drin et al., 2016; Kentala et al., 2016; Mesmin and Antonny, 2016). The S. cerevisiae genome encodes seven Osh proteins, several of which have been shown to act as translocators of sterols, sphingolipids, and phospholipids. When testing the genetic relationship between SPF1 and members of the OSH gene family, we identified a synthetic lethality between spf1 and osh1, osh2, osh3, osh4, osh5, and osh6. This demonstrates that both ER- and PM-associated Osh functions are required for survival in the spf1 background.
Deletion of SAC1 results in a decrease in the sterol content at the PM (Figure 11, D and E), which is in line with a role for Sac1p in driving the forward motion of sterols out of the ER through depletion of the PI4P gradient. Notably, we observed that deletion of SPF1 results in a significant increase in PM sterols. Three independently derived spf1/sac1 cell lines further showed PM sterol levels similar to those of wild-type cells (Figure 11E). This, together with an increase in lipid bodies, suggests a putative involvement of Spf1p in regulating sterol fluxes between different cellular membranes. Whether this effect is due to direct Spf1p action or whether this is merely another aspect of a pleiotropic phenotype independent of Spf1p remains to be determined. However, as most, if not all, phenotypes of the spf1 mutant seem to be explained by a downstream dysfunctional sterol homeostasis, the impact of Spf1p on cellular sterol distribution must be upstream and possibly a direct effect related to assisting in the PI4P-induced export of sterols from the ER.
Sac1p and Osh proteins work in the forward transfer of sterols from the ER to PM. The findings reported in this study suggest that Spf1p works in the opposite direction in a PI4P-stimulated manner to restore the balance between PM and ER sterols. How Spf1p function slots into the known sterol trafficking machinery of the cell is unclear. However, given its close evolutionary relationship with P4 lipid flippases, the stimulatory effect of PI4P, and the absence of a cation stimulus on the purified enzyme, Spf1p might act as a PI4P-stimulated flippase in the cellular trafficking of sterols.
MATERIALS AND METHODS
Materials and yeast growth media
Yeast strains and plasmids used in this study are listed in Supplemental Tables S3 and S4, respectively. Lipids were purchased from Avanti Polar Lipids, except for all diC16 phosphatidylinositols/phosphatidylinositol phosphates (Echelon Biosciences). All other chemicals were purchased from Sigma-Aldrich, except for glass beads (0.5 mm; BioSpec Products); Pierce 660 nm Protein Assay (Thermo Scientific); n-dodecyl β-d-maltoside (DDM) and octyl β-d-glucopyranoside (OG) (Glycon Biochem); ATP (PanReac AppliChem); BSA-free RGS-His Antibody (Qiagen); agar, yeast extract, and peptone (Becton, Dickinson and Co.); and γ-32P-ATP, 32P-orthophosphate, and myo-inositol-[3H] (Perkin-Elmer).
Yeast cells were grown in YPD (1% wt/vol yeast extract, 2% wt/vol bactopeptone, 2% wt/vol glucose) or YPG (1% wt/vol yeast extract, 2% wt/vol bactopeptone, 2% wt/vol galactose) medium. Selection was performed on a synthetic defined minimal medium (0.7% wt/vol yeast nitrogen base, 0.02 mg/ml amino acid, 50 mM succinic acid-Tris, pH 5.5) containing the appropriate dropout nutritional supplements (Sigma) and 2% wt/vol glucose (SD) or galactose (SG). For plates, 2% wt/vol agar was added. Unless otherwise stated, yeast was transformed using the Li-Ac method (Gietz et al., 1995).
Plasmid generation for expression of Spf1p
The SPF1 open reading frame was amplified by PCR from genomic DNA from wild-type S. cerevisiae BY4741 cells and a FLAG-tag followed by a 10xHis tag was added at the 3′ end. The fragment was ligated into the EcoRI and SalI digestion sites of a 2µ vector carrying an HIS3 selection marker, under the control of the galactose-inducible GAL1 promoter, to generate pRS423:pGAL1-SPF1-FLAG/RGS10xhis. From this construct, overlap extension PCR (Ho et al., 1989) was employed to generate the D488N mutation (pRS423:pGAL1-SPF1-D488N-FLAG/RGS10xhis). Correct sequences were confirmed by sequencing. spf1 and sac1 knockout strains of S. cerevisiae BY4741 were transformed with the indicated constructs using the Li-Ac method (Gietz et al., 1995), and positive transformants were selected on selective SD plates. To generate double spf1/osh mutants, the BamHI/SalI fragment of pRS423:pGAL1-SPF1-FLAG/RGS10xhis containing the GAL1 promoter and the SPF1 gene was ligated into pRS426-GAL bearing a URA3 marker. For drop tests, wild-type BY4741 yeast and transformed spf1 cells were grown overnight on YPD and selective SD plates, respectively, suspended in sterile ultrapure H2O, and diluted to OD600 = 1. A dilution series of OD600 = 1, 0.1, and 0.01 was made with sterile H2O, and 5 µl of each dilution was dropped onto YPD or YPG plates with or without inhibitors at the indicated concentrations. Spotted cells were grown for 2 d at 30°C.
Microscopy
Wild-type and spf1 cells were grown in YPD media and harvested at the beginning of the stationary phase. Cells were used immediately for microscopy (see below) or harvested by centrifugation at 4000 × g for 5 min at 4°C before being frozen in liquid N2 for storage at −80°C. Frozen samples were used for sterol extraction and calorimetry.
For staining with Nile red, a modification of the protocol from Greenspan et al. (1985) was used. Equal volumes of live cell suspension diluted to 0.1 OD600 units and 0.1 µg/ml Nile red (from a 10-mg/ml stock solution in dimethyl sulfoxide [DMSO]) were mixed in an Eppendorf tube and incubated for 10 min in darkness at room temperature. A 10-µl drop was placed on a microscope slide and covered with an 18 × 32–mm coverslip, and cells were imaged with a Leica SP5x confocal microscope (Leica Microsystems Mannheim, Germany) using a 40×/1.4 numerical aperture (NA) water objective. Nile red was excited with the 514-nm laser line, and the emission was recorded from 538 to 600 nm. Emission spectra for stained lipid bodies were recorded from 530 to 690 nm using 4-nm steps. A region of interest was drawn around one lipid body within each of 20 cells from wild-type and spf1 populations, and the intensity of the Nile red signal was analyzed using Leica LAS AF Lite software.
For staining with filipin, cells were incubated at room temperature for 1 min with 20 µg/ml dye (freshly made from a 10-mM DMSO stock) and imaged immediately with a Leica inverted DMI 4000B fluorescence microscope using a 63×/1.2 NA objective, with excitation at 340–380 nm and emission at 425 nm (long-pass filter). Similar samples were imaged with a Leica SP5x confocal microscope with 355-nm UV laser excitation/400–475–nm emission, using a 63×/1.2 NA water objective. Regions of interest were drawn around cells in the focal plane within each of 10 images for wild-type (total, n = 38 cells) and spf1 (total, n = 53 cells) cells and analyzed using Leica LAS AF Lite software.
For PI4P localization, the indicated yeast cells were transformed with a GFP-2xPHOsh reporter construct (pRS424GFP-2xPH(Osh2); Addgene plasmid 36095; kind gift of Scott Emr) and visualized as described (Stefan et al., 2011), using a Leica SP5x confocal microscope (Leica Microsystems Mannheim).
For localization of Spf1p, Sac1p, and Sec61p, the encoding genes were chromosomally tagged at the 3′ end with GFP (SPF1) or RFP (SAC1 and SEC61), as described (Lee et al., 2013). Correct tagging was verified by PCR, and the isolated strains were inoculated onto YPD media plates and grown overnight at 150 rpm, 30°C. Cells were scraped and diluted 10-fold into 1 ml of the indicated liquid media containing the indicated final concentrations of inhibitors. After 4 h of growth at the indicated temperatures, cells were harvested in a tabletop microfuge, washed in phosphate-buffered saline (PBS; 130 mM NaCl, 2.6 mM KCl, 7 mM Na2HPO4, 1.2 mM KH2PO4, pH 7.2), and resuspended in 100–200 µl PBS. Yeast cells were then mixed with one volume of a melted 1% wt/vol agarose solution (kept at 45°C) containing the indicated final concentration of inhibitors and spotted onto a 10-well Teflon glass slide, and the glass slide was immediately covered with a glass slip. Cells were imaged using an Andor Revolution XD spinning-disk confocal system with excitation at 488 mm and a band-pass filter of 500–550 nm (GFP), or excitation at 561 mm and an emission long-pass filter of 568 nm (RFP). The exposure time for RFP was 4000 ms with the EM gain set to 100, and the exposure time for GFP was 2000 ms with the EM gain set to 100.
To visualize retained intracellular GFP in the secretion assays, confocal microscopy images were obtained using a Leica TCS SP5-X MP UV confocal laser scanning microscope with a 100×/1.40 NA oil immersion objective.
Calorimetry
Frozen wild-type and spf1 cells prepared as above were dehydrated by freeze drying. The dehydrated cell material was pressed into pellets and weighed on an analytical scale, and the total energy content was determined as joules per gram of cell material in a bomb calorimeter (IKA C4000 adiabatic calorimeter).
Labeling of cells and lipid analysis
To label phospholipids and sphingolipids, cells were grown as above in YPD medium supplemented with either [32P]-orthophosphate or [3H]-myo-inositol. Total phospholipid extracts were purified from equal OD600 units of cells using the Bligh and Dyer protocol (Bligh and Dyer, 1959). The organic solvent was evaporated under N2 gas, and lipids were stored in closed glass tubes at −20°C and were only handled using glass pipettes. Total 32P label was determined by liquid scintillation counting, and an equal amount (10 nCi) was spotted onto TLC plates (silica gel 60; Merck) and separated in two dimensions using first an alkaline solvent (CHCl3/MeOH/NH4OH; 65/25/4 vol/vol) and then an acidic solvent (CHCl3/MeOH/acetone/acetic acid/H2O; 50/10/20/10/4 vol/vol). Plates were analyzed with a phosphor imager (Storm 860; GE Healthcare). Labeled spots were quantified from three independent experiments, each compared with a standard dilution of [32P]. For control plates, a lipid mix of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoinositol (POPI), 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-l-serine (POPS), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE), and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphate (POPA) was spotted onto TLC plates, separated in a similar manner before being sprayed with primuline (0.005% in acetone/water; 8/2 vol/vol), and visualized using UV detection at 375 nm. Omission of individual lipids allowed assignment of individual spots onto the control plate.
Total sphingolipid extracts were obtained by the method of Hanson and Lester (1980). Briefly, wild-type and spf1 cells were suspended in 100 μl of ethanol/water/diethylether/pyridine/ammonia (15/15/5/1/0.018 vol/vol) and incubated at 60°C for 15 min. The residue was centrifuged at 1500 × g for 5 min at room temperature and extracted once more in the same manner. The resulting supernatants were dried under N2 gas and resuspended in chloroform, and equal amounts based on [3H] CPM measurements were spotted onto TLC plates and separated in one dimension using chloroform/methanol/4.2 M ammonia (9/7/2 vol/vol) as solvent. Equal amounts based on [3H] CPM measurements were spotted onto TLC plates and separated in one dimension. Plates were exposed to a [3H]-sensitive screen for 5 d and scanned by a phosphor imager (Storm 860; GE Healthcare).
Total sterol extracts were obtained from 10 OD600 units of wild-type or spf1 cells as previously described (Silvestro et al., 2013; Marek et al., 2014). Briefly, cells were resuspended in 50 ml of 6% wt/vol KOH in MeOH in a Falcon tube by vortexing for 30 s. Complete saponification was performed at 80°C for 2 h. Then one volume of H2O and two volumes of hexane were added and the samples were vortexed and then centrifuged at 500 × g for 5 min at room temperature. The hexane fraction was collected, and the water phase was reextracted with another 20 ml of hexane. The two fractions were pooled and dried completely in a rotavapor. Lipids were resuspended in toluene and derivatized by acetylation in acetic anhydride and pyridine for 1 h at 80°C. When dried, the samples were resuspended in hexane, analyzed by GC-MS (Agilent 6890 gas chromatograph and 5973 mass analyzer), and quantified by GC-FID (Varian 8400 gas chromatograph). β-Sitosterol was used both as an internal and as an external standard in individual experiments, and sample peaks were obtained from ergosterol and lanosterol.
Total lipid was extracted from fractions containing lipid droplets as described by Bligh and Dyer (1959). Phosphatidylcholine content was measured by enzymatic assay (Phosphatidylcholine Assay Kit STA-600; Cell Biolabs). The ergosterol content was calculated based on an external ergosterol standard and related to the PC content. The solvent used was hexane/diethylether/acetic acid (80/15/1), and samples were stained with primuline (0.005% in acetone/water; 8/2 vol/vol) and quantified by UV detection at 375 nm.
Analysis of chemical inhibitors and genetic interactors in yeast lipid metabolism
Growth assays were performed for wild-type, spf1, and ypk9 cells. The ypk9 knockout was a generous gift from the laboratory of Susan Lindquist, Whitehead Institute for Biomedical Research, Cambridge, MA (Gitler et al., 2009). Briefly, cells in the exponential phase were diluted in sterilized MQ water, and a dilution series was spotted onto YPD plates containing the indicated concentration of inhibitor prepared in either ethanol or DMSO as carrier. For each individual inhibitor, a control plate was prepared using the same volume of carrier but without inhibitor. Plates were incubated at 30°C for 2–3 d before scanning. Genetic interaction data used in Supplemental Figure S4 were retrieved from the Saccharomyces Genome Database (www.yeastgenome.org/).
Pre-pro-α secretion assay
The yeast centromeric plasmid encoding a secreted version of a yeast-enhanced yEGFP under the control of a galactose-inducible promoter (pRS316:sec-yEGFP) was a generous gift from Eric Shusta, Department of Chemical and Biological Engineering, University of Wisconsin–Madison (Huang and Shusta, 2005). Homologous recombination was used to remove the pre-pro signal peptide and yield a cytoplasmic yEGFP under the control of a galactose-inducible promoter (pRS316:cyto-yEGFP) in ZHY709 yeast cells. For this, a PCR fragment containing yEGFP without the signal peptide was transformed into yeast together with the pRS316:sec-yEGFP plasmid linearized with BamHI. Selection was carried out in SD selective media, and plasmid DNA extraction was carried out using the GenElute Plasmid Miniprep Kit (Sigma-Aldrich), followed by amplification in E. coli and sequencing (Costa et al., 2016).
For cytometry analysis, wild-type and spf1 cells harboring sec-yEGFP or cyto-yEGFP, respectively, were grown in selective minimal SD medium before induction in selective SG media for 24 h. Flow cytometry measurements were carried out using a Becton Dickinson FACS Calibur cytometer equipped with a 488-nm argon laser and Cell Quest software (BD Biosciences). Prior to analysis, cells were labeled with 1 µl of 1 mg/ml propidium iodide for staining of nonviable cells. For yEGFP and propidium iodide detection, fluorescence emission was measured with a 530/30-nm band-pass filter and a 670-nm long-pass filter, respectively. Twenty thousand cells were analyzed without gating during the acquisition. Data were analyzed using FlowJo software (FlowJo). Live yeast cells were selected based on forward/side-scatter gating and PI exclusion. The yEGFP fluorescence of living cells was plotted on a histogram, and the geometric mean fluorescence intensity was used for further statistical analysis. Confocal microscopy images were obtained using a Leica TCS SP5-X MP UV confocal laser scanning microscope with a 100×/1.40 NA oil immersion objective (Leica Microsystems). yEGFP was excited at 488 nm, and the emitting light was measured in the interval 505–539 nm. Fluorescence images were processed using Leica software (Leica Microsystems).
Glucose activation of Pma1p in wild-type and spf1 cells
Wild-type or spf1 cells were incubated in 100 ml YPD for 48 h at 30°C. Then 25 ml of saturated yeast solution was inoculated into 250 ml of YPD medium and grown at 30°C for 20 h. Cells were harvested by centrifugation (2000 × g, 5 min at 4°C) and washed with cold ultrapure H2O. Cells were then incubated for 1 h in cold H2O and pelleted again by centrifugation. Samples were split in two, and then half was incubated in H2O and the other in a 4% wt/vol glucose solution in ultrapure H2O for 30 min. Cells were subsequently collected by centrifugation (2000 × g, 5 min at 4°C), frozen in liquid N2, and stored at −80°C. ATPase activity was measured on isolated total membranes.
Generation of spf1/sac1 and spf1/osh double knockout strains
Double spf1/sac1 knockouts were generated by crossing and tetrad dissection. First, BY4742 spf1::HIS3 knockout cells were crossed with BY4741 sac1::KanMX4 cells on YPD media. The resulting cells were diluted in sterile H2O, and single colonies of diploid sac1/spf1 cells were confirmed to be resistant to G418 and to have lost histidine auxotrophy. Tetrad formation was induced by growth on sporulation media, and tetrads were dissected using a MSM400 Singer dissection microscope. Tetrad spores were tested for segregation of selection markers on SD media by selection marker screening, and true tetrads, which showed proper segregation of markers, were selected for further analysis. This approach allowed the generation of three haploid spf1/sac1 double knockout lines. The resulting genotype was confirmed by PCR and by growth of the cells on selective media.
To generate double spf1/osh yeast mutants, the ohs1-to-osh6 strains were first transformed with a plasmid bearing a URA3 marker and the SPF1 gene. Single colonies were selected on uracil-selective media, and transformants were further transformed with a PCR product containing the two fragments corresponding to the 5′- and 3′-ends of the SPF1 gene separated by a HIS3 marker. Single colonies were selected on uracil- and histidine-selective media, and the knockout of genomic SPF1 was confirmed using PCR with specific primers annealing to the knockout cassette and to the SPF1 promoter or terminator regions in the genome. To test the viability of the double knockout, the positive transformants were grown overnight in SD liquid media lacking histidine and uracil, suspended in ultrapure H2O, and diluted to OD600 1, 0.1, and 0.01; then 5 µl of each dilution was dropped on minimal media plates containing 1 g/l 5-FOA for uracil counterselection.
Subsequently, BY4741 and deletion strains lacking OSH genes were transformed with a PCR product containing two fragments corresponding to the 5′- and 3′-ends of SPF1 (705 and 1010 nucleotides, respectively) separated by a URA3 marker. First, the original yeast plasmid containing SPF1 was digested with BamHI/PstI and HindIII/ApaI (3′-end) to obtain the 5′- and 3′-ends of SPF1, respectively. Both fragments were sequentially cloned into pBluescript II KS (+) vector digested with the same enzymes. PCR was employed to generate a fragment containing the URA3 selection marker surrounded by EcoRI and SmaI sites, which was inserted between the two SPF1 fragments by cutting the plasmid with EcoRI and EcoRV. Finally, the cassette containing the 5′- and 3′-ends of SPF1 separated by the URA3 selection marker was amplified by PCR. BY4741 and deletion strains lacking Osh genes were transformed with the whole PCR reaction volume and selected on plates lacking uracil. Eight different individual colonies from each successful transformation were tested for deletion of SPF1 by PCR, using two pairs of specific primers amplifying the deleted SPF1 fragment and the deletion construct respectively.
Purification of Spf1p
To prepare total membrane fractions, cell pellets from rich media were thawed in ice water, resuspended in 2 ml of ice-cold resuspension buffer (50 mM Tris-HCl, 1 mM EDTA, 0.1 M KCl, 0.6 M sorbitol, pH 7.4) per gram wet weight, incubated on an oscillating table for 5–15 min at 4°C, and then centrifuged (2800 × g, 5–10 min, 4°C). The supernatant was discarded, and the cell pellet was resuspended in 1 ml of ice-cold lysis buffer (50 mM Tris-HCl, 1 mM EDTA, 0.6 M sorbitol, 250 µM phenylmethylsulfonyl fluoride [PMSF], 1 mM dithiothreitol [DTT], and 2 µg/µl pepstatin A, pH 7.4) per gram wet weight. Glass beads (0.5 mm) at a ratio of 1 g per gram of wet weight were added, and the cells were lysed by eight to ten 30-s vortex cycles, with 30-s breaks on ice between cycles. The cell lysis mixture was transferred to 50-ml Falcon tubes and centrifuged (1000 × g, 15–20 min, 4°C), and the resulting supernatant was extracted and further centrifuged (160,000 × g, 1 h, 4°C). The supernatant was discarded and the pelleted membranes were resuspended in ice-cold lysis buffer, homogenized with a Potter–Elvehjem glass tissue homogenizer, and centrifuged again (160,000 × g, 1 h, 4°C) before resuspension in solubilization buffer (20 mM MOPS, 20% glycerol, 130 mM KCl, 1 mM MgCl2, 5 mM imidazole, 250 µM PMSF, 1 mM DTT, and 2 µg/µl Pepstatin A, pH 7.4, adjusted with N-methyl d-glucamine) in a total volume of 10 ml. Protein concentration was determined using a NanoDrop 1000 (Thermo-Fisher Scientific), and the preparation was covered in N2 gas, frozen in liquid N2, and stored at −80°C.
Total membranes from YPG-induced yeast cultures expressing Spf1p or spf1p D488N were solubilized in solubilization buffer with DDM at 4°C, at a protein:DDM ratio of 1:2 for 50–60 min before centrifugation at 100,000 × g for 1 h at 4°C. Affinity purification of Spf1p was performed by transferring the supernatant to a column with equilibrated Ni-NTA resin, followed by a 1.5- to 4-h incubation at 4°C. The column was washed twice with three column volumes of 10 mM wash buffer (solubilization buffer with 10 mM imidazole, 0.05% wt/vol DDM), and two times with two volumes of 20 mM wash buffer (solubilization buffer with 20 mM imidazole, 0.05% DDM), and then eluted in seven fractions (in a total of 4–5 ml) of elution buffer (solubilization buffer with 300 mM imidazole, 0.005% DDM). Fractions containing Spf1p were pooled, divided into 200-µl aliquots, covered with N2 gas, frozen in liquid N2, and stored at −80°C. The protein concentration was quantified by the Bradford assay using γ-globulin as a standard. Typically, 1 l of yeast culture yielded 2–4 mg of purified protein at a concentration of 0.6–1.4 mg/ml after pooling of elution fractions.
Relipidation and ATPase assays
Individual lipids were dissolved in chloroform or other appropriate solvents according to the supplier, transferred to a clean glass tube, and dried under a stream of N2 gas. One 5-mm glass bead was added to each glass tube, and reactivation buffer (50 mM Tris-HCl, 50 mM NaCl, 0.5% wt/vol octyl β-d-glucopyranoside, pH 7.2) was added to the lipids to give a final concentration of 4.086 mg lipid/ml. The lipid–buffer mixture was covered in N2 gas and shaken gently at room temperature followed by sonication (Branson 1510 bath sonicator) until the mixture appeared clear. The lipid–buffer solution was frozen in liquid N2 and stored at −20°C.
ATPase activity was assayed using a modified Baginski assay (Baginski et al., 1967; Regenberg et al., 1995). In all assays, unless specified, 5 µg of purified protein was preincubated on ice with the indicated lipids at 3.552 µg lipid/µg protein, added to 300 µl of ATPase buffer (20 mM MOPS, 8 mM MgSO4, 50 mM KNO3, 5 mM NaN3, 0.25 mM Na2MoO4, 3 mM ATP, pH 7.4, adjusted with N-methyl d-glucamine), and then incubated at 30°C for 30 min before the reaction was stopped as described (Regenberg et al., 1995). Activity was calculated by subtracting the concentration of inorganic phosphate in ATPase buffer without protein. For POPC titration experiments, the lipids were titrated to protein samples by preincubation on ice with increasing µg lipid/µg protein ratios, from 0 to 3.552, for 30 min. Dissolved lipids were thawed and diluted with ice-cold reactivation buffer without OG to the indicated concentrations (up to 3.552 µg lipid/µg protein) to keep the volume constant, and placed on ice. Phosphatidylinositides were added at a final concentration of 10 mol% to POPC.
For ion titration experiments, different concentrations of CaCl2, MnCl2, ZnCl2, or CuCl2 were added to ATPase buffer treated with Chelex resin containing 100 µM EDTA and 100 µM ethylene glycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA). Free metal ion concentrations were calculated using the MaxChelator Webmaxc Extended calculator (available at https://somapp.ucdmc.ucdavis.edu/pharmacology/bers/maxchelator/downloads.htm).
Data points in all experiments represent averages from at least three individual experiments, each using protein from an individual purification.
Phosphorylation of Spf1p by 32P-ATP
Phosphorylation and subsequent dephosphorylation of Spf1p were performed essentially as described by Sørensen et al. (2012). Briefly, 2.5 µg of enzyme relipidated as above was added to 95 µl EP reaction buffer (20 mM MOPS, 8 mM MgSO4, 50 mM KCl, pH 7.4, adjusted with N-methyl d-glucamine) on ice with a stirring magnet, and phosphorylation was initiated by the addition of 5 µl of 32P-ATP mix (3.75 µCi γ-32P-ATP/5 µM cold ATP) at the indicated temperature for 40 s. Steady-state conditions were confirmed under all experimental conditions. Reactions were stopped by quenching with stop buffer (20% wt/vol trichloroacetic acid, 1 mM phosphoric acid). Dephosphorylation experiments were performed by the addition of 1 mM ADP or 1 mM ATP (final concentrations) to radiolabeled phosphoenzyme at steady state (following 40 s of phosphorylation) and then stopped by the addition of stop buffer at the indicated time points. Then 50 µl of 10 mg/ml BSA was added to all samples before centrifugation (16,000 × g, 15 min) and washing. The radioactivity of the labeled samples was measured by scintillation counting. For all measurements, the 32P-ATP mix used for phosphorylation was serially diluted and used as an internal standard for quantification in each data set.
Phylogenetic analysis
Amino acid sequences with significant similarity (expected value <10–30) to the Homo sapiens P5A-ATPase ATP13A1 were first identified using the Basic Local Alignment Search Tool (BLAST) for annotated sequences in completed genomes in the KEGG database (www.genome.jp/tools-bin/search_sequence?prog=blast&db=cmy&seqid=aa). This data set was extended by similar BLAST searches using the National Center for Biotechnology Information (NCBI) nonredundant protein sequence database (http://blast.ncbi.nlm.nih.gov/) against the species Methanococcoides methylutens and Nannochloropsis gaditana. Intact P-type ATPase sequences were identified by the presence of signature motifs characteristic of P-type ATPases. The resulting data set comprised 171 sequences from 11 genomes, which represented different stages in evolution from eubacteria to mammals. Accession numbers of the protein sequences are listed in Supplemental Table S5.
The phylogenetic analysis was carried out as described by Sørensen et al. (2018). All positions containing gaps or ambiguous data were eliminated, leaving a total of 999 positions in the final data set. The average SD of split frequencies at termination of the Bayesian inference analysis, after 2,220,000 generations, was 0.009991 for the tree in Figure 1. In the RAxML analysis, clade robustness was assessed with 100 rapid bootstrap inferences followed by thorough analysis of maximum likelihood. Bootstrap values were inferred from 1000 replicates to obtain statistical support for the placement of nodes.
Supplementary Material
Acknowledgments
We acknowledge Steen Holmberg and Thi Ngoc Hoa Phan for their help in establishing yeast double knockout strains. We acknowledge Eric Shusta for providing the pRS316:s-yEGFP plasmid and Aaron Gitler for providing the ypk9 knockout cells. This work was funded by a grant from the Danish Natural Science Research Council to M.P. (4002-00207B). D.S. acknowledges funding from “Biomass for the 21st Century,” the Danish National Advanced Technology Foundation (now Innovation Found Denmark; 001-2011-4). T.G.P. and R.L.L. acknowledge funding from Villium Fonden (Grants 022868 and 13234, respectively). All bioimaging was carried out at the Center for Advanced Bioimaging Denmark (CAB).
Abbreviations used:
- DMSO
dimethyl sulfoxide
- ER
endoplasmic reticulum
- PI4P
phosphatidylinositol 4-phosphate
- Spf1p
sensitivity to Picchia farinosa 1 protein
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E18-06-0365) on February 20, 2019.
REFERENCES
- Andreyev AY, Fahy E, Guan Z, Kelly S, Li X, McDonald JG, Milne S, Myers D, Park H, Ryan A, et al (2010). Subcellular organelle lipidomics in TLR-4-activated macrophages. J Lipid Res , 2785–2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Athenstaedt K, Zweytick D, Jandrositz A, Kohlwein SD, Daum G. (1999). Identification and characterization of major lipid particle proteins of the yeast Saccharomyces cerevisiae. J Bacteriol , 6441–6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelsen KB, Palmgren MG. (1998). Evolution of substrate specificities in the P-type ATPase superfamily. J Mol Evol , 84–101. [DOI] [PubMed] [Google Scholar]
- Azouaoui H, Montigny C, Ash MR, Fijalkowski F, Jacquot A, Grønberg C, López-Marqués RL, Palmgren MG, Garrigos M, le Maire M, et al (2014). A high-yield co-expression system for the purification of an intact Drs2p-Cdc50p lipid flippase complex, critically dependent on and stabilized by phosphatidylinositol-4-phosphate. PLoS One , e112176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baginski ES, Foa PP, Zak B. (1967). Microdetermination of inorganic phosphate, phospholipids, and total phosphate in biologic materials. Clin Chem , 326–332. [PubMed] [Google Scholar]
- Bligh EG, Dyer WJ. (1959). A rapid method of total lipid extraction and purification. Can J Biochem Physiol , 911–917. [DOI] [PubMed] [Google Scholar]
- Buch-Pedersen MJ, Rudashevskaya EL, Berner TS, Venema K, Palmgren MG. (2006). Potassium as an intrinsic uncoupler of the plasma membrane H+-ATPase. J Biol Chem , 38285–38292. [DOI] [PubMed] [Google Scholar]
- Celej MS, Montich GG, Fidelio GD. (2003). Protein stability induced by ligand binding correlates with changes in protein flexibility. Protein Sci , 1496–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen Y, Megyeri M, Chen OC, Condomitti G, Riezman I, Loizides-Mangold U, Abdul-Sada A, Rimon N, Riezman H, Platt FM, et al (2013). The yeast p5 type ATPase, spf1, regulates manganese transport into the endoplasmic reticulum. PLoS One , e85519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman JA, Vestergaard AL, Molday RS, Vilsen B, Andersen JP. (2012). Critical role of a transmembrane lysine in aminophospholipid transport by mammalian photoreceptor P4-ATPase ATP8A2. Proc Natl Acad Sci USA , 1449–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corradi GR, de Tezanos Pinto F, Mazzitelli LR, Adamo HP. (2012). Shadows of an absent partner: ATP hydrolysis and phosphoenzyme turnover of the Spf1 (sensitivity to Pichia farinosa killer toxin) P5-ATPase. J Biol Chem , 30477–30484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa SR, Marek M, Axelsen KB, Theorin L, Pomorski TG, López-Marqués RL. (2016). Role of posttranslational modifications at the β-subunit ectodomain in complex association with a promiscuous plant P4-ATPase. Biochem J , 1605–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronin SR, Khoury A, Ferry DK, Hampton RY. (2000). Regulation of HMG-CoA reductase degradation requires the P-type ATPase Cod1p/Spf1p. J Cell Biol , 915–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronin SR, Rao R, Hampton RY. (2002). Cod1p/Spf1p is a P-type ATPase involved in ER function and Ca2+ homeostasis. J Cell Biol , 1017–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Saint-Jean M, Delfosse V, Douguet D, Chicanne G, Payrastre B, Bourguet W, Antonny B, Drin G. (2011). Osh4p exchanges sterols for phosphatidylinositol 4-phosphate between lipid bilayers. J Cell Biol , 965–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desrivières S, Cooke FT, Parker PJ, Hall MN. (1998). MSS4, a phosphatidylinositol-4-phosphate 5-kinase required for organization of the actin cytoskeleton in Saccharomyces cerevisiae. J Biol Chem , 15787–15793. [DOI] [PubMed] [Google Scholar]
- Drin G, von Filseck JM, Cˇopicˇ A. (2016). New molecular mechanisms of inter-organelle lipid transport. Biochem Soc Trans , 486–492. [DOI] [PubMed] [Google Scholar]
- Espenshade PJ, Hughes AL. (2007). Regulation of sterol synthesis in eukaryotes. Annu Rev Genet , 401–427. [DOI] [PubMed] [Google Scholar]
- Faulhammer F, Kanjilal-Kolar S, Knödler A, Lo J, Lee Y, Konrad G, Mayinger P. (2007). Growth control of Golgi phosphoinositides by reciprocal localization of Sac1 lipid phosphatase and Pik1 4-kinase. Traffic , 1554–1567. [DOI] [PubMed] [Google Scholar]
- Feng B, Yao PM, Li Y, Devlin CM, Zhang D, Harding HP, Sweeney M, Rong JX, Kuriakose G, Fisher EA, et al (2003). The endoplasmic reticulum is the site of cholesterol-induced cytotoxicity in macrophages. Nat Cell Biol , 781–792. [DOI] [PubMed] [Google Scholar]
- Foti M, Audhya A, Emr SD. (2001). Sac1 lipid phosphatase and Stt4 phosphatidylinositol 4-kinase regulate a pool of phosphatidylinositol 4-phosphate that functions in the control of the actin cytoskeleton and vacuole morphology. Mol Biol Cell , 2396–2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietz RD, Schiestl RH, Willems AR, Woods RA. (1995). Studies on the transformation of intact yeast cells by the LiAc/SS-DNA/PEG procedure. Yeast , 355–360. [DOI] [PubMed] [Google Scholar]
- Gitler AD, Chesi A, Geddie ML, Strathearn KE, Hamamichi S, Hill KJ, Caldwell KA, Caldwell GA, Cooper AA, Rochet JC, Lindquist S. (2009). Alpha-synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity. Nat Genet , 308–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gohil VM, Thompson MN, Greenberg ML. (2005). Synthetic lethal interaction of the mitochondrial phosphatidylethanolamine and cardiolipin biosynthetic pathways in Saccharomyces cerevisiae. J Biol Chem , 35410–35416. [DOI] [PubMed] [Google Scholar]
- Greenspan P, Mayer EP, Fowler SD. (1985). Nile red: a selective fluorescent stain for intracellular lipid droplets. J Cell Biol , 965–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossmann G, Opekarová M, Malinsky J, Weig-Meckl I, Tanner W. (2007). Membrane potential governs lateral segregation of plasma membrane proteins and lipids in yeast. EMBO J , 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson BA, Lester RL. (1980). The extraction of inositol-containing phospholipids and phosphatidylcholine from Saccharomyces cerevisiae and Neurospora crassa. J Lipid Res , 309–315. [PubMed] [Google Scholar]
- Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. (1989). Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene , 51–59. [DOI] [PubMed] [Google Scholar]
- Holemans T, Sørensen DM, van Veen S, Martin S, Hermans D, Kemmer GC, Van den Haute C, Baekelandt V, Günther Pomorski T, Agostinis P, et al (2015). A lipid switch unlocks Parkinson’s disease-associated ATP13A2. Proc Natl Acad Sci USA , 9040–9045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D, Shusta EV. (2005). Secretion and surface display of green fluorescent protein using the yeast Saccharomyces cerevisiae. Biotechnol Prog , 349–357. [DOI] [PubMed] [Google Scholar]
- Heuck S, Gerstmann UC, Michalke B, Kanter U. (2010). Genome-wide analysis of caesium and strontium accumulation in Saccharomyces cerevisiae. Yeast , 817–835. [DOI] [PubMed] [Google Scholar]
- Hulce JJ, Cognetta AB, Niphakis MJ, Tully SE, Cravatt BF. (2013). Proteome-wide mapping of cholesterol-interacting proteins in mammalian cells. Nat Methods , 259–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jesch SA, Gaspar ML, Stefan CJ, Aregullin MA, Henry SA. (2010). Interruption of inositol sphingolipid synthesis triggers Stt4p-dependent protein kinase C signaling. J Biol Chem , 41947–41960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonikas MC, Collins SR, Denic V, Oh E, Quan EM, Schmid V, Weibezahn J, Schwappach B, Walter P, Weissman JS, Schuldiner M. (2009). Comprehensive characterization of genes required for protein folding in the endoplasmic reticulum. Science , 1693–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamin´ski DM. (2014). Recent progress in the study of the interactions of amphotericin B with cholesterol and ergosterol in lipid environments. Eur Biophys J , 453–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly DE, Rose ME, Kelly SL. (1994). Investigation of the role of sterol Δ 8→7-isomerase in the sensitivity of Saccharomyces cerevisiae to fenpropimorph. FEMS Microbiol Lett , 223–226. [DOI] [PubMed] [Google Scholar]
- Kentala H, Weber-Boyvat M, Olkkonen VM. (2016). OSBP-related protein family: Mediators of lipid transport and signaling at membrane contact sites. Int Rev Cell Mol Biol , 299–340. [DOI] [PubMed] [Google Scholar]
- Koffel R, Tiwari R, Falquet L, Schneiter R. (2005). The Saccharomyces cerevisiae YLL012/YEH1, YLR020/YEH2, and TGL1 genes encode a novel family of membrane-anchored lipases that are required for steryl ester hydrolysis. Mol Cell Biol , 1655–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krumpe K, Frumkin I, Herzig Y, Rimon N, Özbalci C, Brügger B, Rapaport D, Schuldiner M. (2012). Ergosterol content specifies targeting of tail-anchored proteins to mitochondrial outer membranes. Mol Biol Cell , 3927–3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Lim WA, Thorn KS. (2013). Improved blue, green, and red fluorescent protein tagging vectors for S. cerevisiae. PLoS One , e67902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lev S. (2010). Non-vesicular lipid transport by lipid-transfer proteins and beyond. Nat Rev Mol Cell Biol , 739–750. [DOI] [PubMed] [Google Scholar]
- Manford A, Xia T, Saxena AK, Stefan C, Hu F, Emr SD, Mao Y. (2010). Crystal structure of the yeast Sac1: implications for its phosphoinositide phosphatase function. EMBO J , 1489–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marek M, Silvestro D, Fredslund MD, Andersen TG, Günther-Pomorski T. (2014). Serum albumin promotes ATP-binding cassette transporter-dependent sterol uptake in yeast. FEMS Yeast Res , 1223–1233. [DOI] [PubMed] [Google Scholar]
- Mesmin B, Antonny B. (2016). The counterflow transport of sterols and PI4P. Biochim Biophys Acta , 940–951. [DOI] [PubMed] [Google Scholar]
- Mesmin B, Bigay J, Moser von Filseck J, Lacas-Gervais S, Drin G, Antonny B. (2013). A four-step cycle driven by PI4P hydrolysis directs sterol/PI4P exchange by the ER-Golgi tether OSBP. Cell , 830–843. [DOI] [PubMed] [Google Scholar]
- Mesmin B, Maxfield FR. (2009). Intracellular sterol dynamics. Biochim Biophys Acta , 636–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra P, Zhang Y, Rameh LE, Ivshina MP, McCollum D, Nunnari JJ, Hendricks GM, Kerr ML, Field SJ, Cantley LC, et al (2004). A novel phosphatidylinositol(3,4,5)P 3 pathway in fission yeast. J Cell Biol , 205–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng DT, Spear ED, Walter P. (2000). The unfolded protein response regulates multiple aspects of secretory and membrane protein biogenesis and endoplasmic reticulum quality control. J Cell Biol , 77–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson I, Ohvo-Rekilä H, Slotte JP, Johnson AE, von Heijne G. (2001). Inhibition of protein translocation across the endoplasmic reticulum membrane by sterols. J Biol Chem , 41748–41754. [DOI] [PubMed] [Google Scholar]
- Palmgren MG, Nissen P. (2011). P-type ATPases. Annu Rev Biophys , 243–266. [DOI] [PubMed] [Google Scholar]
- Pichler H, Gaigg B, Hrastnik C, Achleitner G, Kohlwein SD, Zellnig G, Perktold A, Daum G. (2001). A subfraction of the yeast endoplasmic reticulum associates with the plasma membrane and has a high capacity to synthesize lipids. Eur J Biochem , 2351–2361. [DOI] [PubMed] [Google Scholar]
- Plath K, Wilkinson BM, Stirling CJ, Rapoport TA. (2004). Interactions between Sec complex and prepro-alpha-factor during posttranslational protein transport into the endoplasmic reticulum. Mol Biol Cell , 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomorski T, Lombardi R, Riezman H, Devauz PF, van Meer G, Holthuis JC. (2003). Drs2p-related P-type ATPases Dnf1p and Dnf2p are required for phospholipid translocation across the yeast plasma membrane and serve a role in endocytosis. Mol Biol Cell , 1240–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz WA. (2010). Lipid trafficking sans vesicles: where, why, how? Cell , 870–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radhakrishnan A, Goldstein JL, McDonald JG, Brown MS. (2008). Switch-like control of SREBP-2 transport triggered by small changes in ER cholesterol: a delicate balance. Cell Metab , 512–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regenberg B, Villalba JM, Lanfermeijer FC, Palmgren MG. (1995). C-terminal deletion analysis of plant plasma membrane H+-ATPase: yeast as a model system for solute transport across the plant plasma membrane. Plant Cell , 1655–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigo-Brenni MC, Thomas S, Bouck DC, Kaplan KB. (2004). Sgt1p and Skp1p modulate the assembly and turnover of CBF3 complexes required for proper kinetochore function. Mol Biol Cell , 3366–3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saiardi A, Resnick AC, Snowman AM, Wendland B, Snyder SH. (2005). Inositol pyrophosphates regulate cell death and telomere length through phosphoinositide 3-kinase-related protein kinases. Proc Natl Acad Sci USA , 1911–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schack VR, Morth JP, Toustrup-Jensen MS, Anthonisen AN, Nissen P, Andersen JP, Vilsen B. (2008). Identification and function of a cytoplasmic K+ site of the Na+, K+-ATPase. J Biol Chem , 27982–27990. [DOI] [PubMed] [Google Scholar]
- Schneiter R, Brügger B, Sandhoff R, Zellnig G, Leber A, Lampl M, Athenstaedt K, Hrastnik C, Eder S, Daum G, et al (1999). Electrospray ionization tandem mass spectrometry (ESI-MS/MS) analysis of the lipid molecular species composition of yeast subcellular membranes reveals acyl chain-based sorting/remodeling of distinct molecular species en route to the plasma membrane. J Cell Biol , 741–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano R. (1983). In vivo glucose activation of the yeast plasma membrane ATPase. FEBS Lett , 11–14. [DOI] [PubMed] [Google Scholar]
- Silvestro D, Andersen TG, Schaller H, Jensen PE. (2013). Plant sterol metabolism. Δ7-sterol-C5-desaturase (STE1/DWARF7), Δ5,7-sterol-Δ7-reductase (DWARF5) and Δ24-sterol-Δ24-reductase (DIMINUTO/DWARF1) show multiple subcellular localizations in Arabidopsis thaliana (Heynh) L. PLoS One , e56429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sørensen DM, Buch-Pedersen MJ, Palmgren MG. (2010). Structural divergence between the two subgroups of P5 ATPases. Biochim Biophys Acta , 846–855. [DOI] [PubMed] [Google Scholar]
- Sørensen TL, Clausen JD, Jensen AM, Vilsen B, Møller JV, Andersen JP, Nissen P. (2004). Localization of a K+-binding site involved in dephosphorylation of the sarcoplasmic reticulum Ca2+-ATPase. J Biol Chem , 46355–46358. [DOI] [PubMed] [Google Scholar]
- Sørensen DM, Holemans T, van Veen S, Martin S, Arslan T, Haagendahl IW, Holen HW, Hamouda N, Eggermont J, Palmgren M, Vangheluwe P. (2018). Parkinson disease related ATP13A2 evolved early in animal evolution. PLoS One , e0193228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sørensen DM, Holen HW, Holemans T, Vangheluwe P, Palmgren MG. (2015). Towards defining the substrate of orphan P5A-ATPases. Biochim Biophys Acta , 524–535. [DOI] [PubMed] [Google Scholar]
- Sørensen DM, Møller AB, Jakobsen MK, Jensen MK, Vangheluwe P, Buch-Pedersen MJ, Palmgren MG. (2012). Ca2+ induces spontaneous dephosphorylation of a novel P5A-type ATPase. J Biol Chem , 28336–28348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefan CJ, Manford AG, Baird D, Yamada-Hanff J, Mao Y, Emr SD. (2011). Osh proteins regulate phosphoinositide metabolism at ER-plasma membrane contact sites. Cell , 389–401. [DOI] [PubMed] [Google Scholar]
- Strahl T, Thorner J. (2007). Synthesis and function of membrane phosphoinositides in budding yeast, Saccharomyces cerevisiae. Biochim Biophys Acta , 353–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki C, Shimma YI. (1999). P-type ATPase Spf1 mutants show a novel resistance mechanism for the killer toxin SMKT. Mol Microbiol , 813–823 [DOI] [PubMed] [Google Scholar]
- Thiam AR, Farese RV, Walther TC. (2013). The biophysics and cell biology of lipid droplets. Nat Rev Mol Cell Biol , 775–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian S, Ohta A, Horiuchi H, Fukuda R. (2018). Oxysterol-binding protein homologs mediate sterol transport from the endoplasmic reticulum to mitochondria in yeast. J Biol Chem , 5636–5648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vashist S, Frank CG, Jakob CA, Ng DT. (2002). Two distinctly localized p-type ATPases collaborate to maintain organelle homeostasis required for glycoprotein processing and quality control. Mol Biol Cell , 3955–3966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Filseck JM, Vanni S, Mesmin B, Antonny B, Drin G. (2015). A phosphatidylinositol-4-phosphate powered exchange mechanism to create a lipid gradient between membranes. Nat Commun , 6671. [DOI] [PubMed] [Google Scholar]
- Yamamoto H, Fujita H, Kida Y, Sakaguchi M. (2012). Pleiotropic effects of membrane cholesterol upon translocation of protein across the endoplasmic reticulum membrane. Biochemistry , 3596–3605. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.