

Abstract
The biosynthesis of antibiotics and self-protection mechanisms employed by antibiotic producers are an integral part of the growing antibiotic resistance threat. The origins of clinically relevant antibiotic resistance genes found in human pathogens have been traced to ancient microbial producers of antibiotics in natural environments. Widespread and frequent antibiotic use amplifies environmental pools of antibiotic resistance genes and increases the likelihood for the selection of a resistance event in human pathogens. This perspective will provide an overview of the origins of antibiotic resistance to highlight the crossroads of antibiotic biosynthesis and producer self-protection that result in clinically relevant resistance mechanisms. Some case studies of synergistic antibiotic combinations, adjuvants, and hybrid antibiotics will also be presented to show how native antibiotic producers manage the emergence of antibiotic resistance.
Keywords: antibiotic resistance, natural product biosynthesis, adjuvant, combination therapy, hybrid antibiotics, antibiotic inactivating enzymes, resistance-guided antibiotic discovery, genome mining, synthetic biology, directed evolution
INTRODUCTION
Antibiotics continue to be the most important resource in the global management of infectious diseases.1, 2 The increased occurrence of antibiotic resistance in human pathogens has raised global concern as antibiotics steadily lose efficacy in clinical and community settings. Despite the rise in antibiotic resistance and shifting drug discovery programs in the private sector the market for antibiotics remains strong.3, 4 In 2018, the FDA approved four new antibiotics from traditional antibiotic classes including the aminoglycoside plazomicin and tetracyclines eravacycline, omadacycline, and sarecycline.5 These new antibiotics gained approval, in part, by overcoming established clinical resistance mechanisms to meet a clinical need.6, 7 One can speculate, based on past clinical practice, that new resistance mechanisms will emerge following large-scale deployment of these new drugs (Figure 1).
Figure 1.
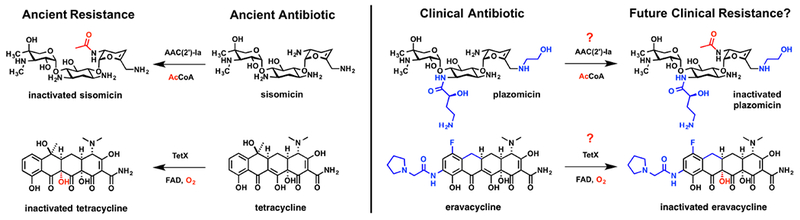
The ancient antibiotics sisomicin and tetracycline are enzymatically inactivated by aminoglycoside acetyltransferase AAC(2’)-Ia and tetracycline monooxygenase TetX, respectively. The plazomicin and eravacycline scaffolds are derived from the parent sisomicin and tetracycline scaffolds, respectively, and were FDA approved for human use in 2018. Plazomicin and eravacycline have been structurally optimized to overcome some of the established clinical resistance mechanisms for aminoglycoside and tetracycline antibiotics, respectively, and could potentially select for new clinical resistance mechanisms of ancient origin, such as enzymatic inactivation.
Recently, Wright and coworkers performed a detailed investigation of the vulnerability of plazomicin to clinically relevant aminoglycoside inactivating enzymes and ribosomal methyltransferases in a panel of isogenic E. coli strains.8 Plazomicin is a semisynthetic aminoglycoside derived from sisomicin and is designed to chemically block the sites of chemical modification by aminoglycoside inactivating enzymes, including N-acetyltransferases (AACs), O-nucleotidylyltransferases (ANTs), and O-phosphotransferases (APHs).9, 10 Indeed, plazomicin was found to maintain activity against E. coli expressing the majority of canonical AACs, ANTs, and APHs that confer broad-spectrum resistance to several subclasses of aminoglycoside antibiotics.8 However, plazomicin was covalently inactivated by AAC(2’)-Ia and APH(2”)-IVa with a reduction in growth inhibitory activity against E. coli strains expressing these enzymes (Figure 1). Due to the flexible substrate plasticity and high selective pressure for mutation under plazomicin challenge, it is likely that these enzymes could emerge as real threats after clinical deployment of plazomicin.9, 11 Furthermore, it was discovered that some plasmid-encoded 16S rRNA ribosomal methyltransferases completely abolish plazomicin antibacterial activity, although these resistance elements are still relatively rare.8 Clearly, the struggle with antibiotic resistance begins far before human use.
Similar to plazomicin, the recently FDA approved 3rd generation tetracycline antibiotic eravacycline was strategically designed to overcome traditional resistance mechanisms to tetracycline antibiotics including efflux pumps and ribosome protection proteins.6, 7 Eravacycline also holds the unique place as the only fully synthetic tetracycline to be approved for human use, although it still has the conserved tetracyclic A,B,C,D-ring core of all FDA approved natural and semisynthetic tetracyclines.12 Eravacycline was evaluated against a panel of isogenic E. coli strains expressing efflux pumps (TetK, TetA, TetB), ribosomal protection proteins (TetM), and tetracycline inactivating enzymes (TetX).7 Eravacycline maintained nearly full antibacterial activity against E. coli expressing TetK, TetA, TetB, and TetM, which represent commonly encountered efflux pumps and ribosomal protection proteins in clinical isolates. It is well known that bulky substituents on the D-ring, a motif common to all 3rd generation tetracyclines, improves ribosome affinity, blocks the binding of ribosomal protection proteins at the ribosomal A-site, and slows the rate of efflux; so, it is no surprise that eravacycline does the same.13 However, eravacycline was found to be vulnerable to resistance by TetX, a tetracycline-inactivating enzyme from the flavin monooxygenase (FMO) superfamily.7, 14 E. coli expressing TetX showed a 64-fold increase in minimal inhibitory concentration (MIC) relative to wild-type E. coli challenged with eravacycline. TetX has been shown to inactivate 1st, 2nd, and 3rd generation tetracyclines including tigecycline, the first 3rd generation tetracycline introduced to the clinic.15–17 Recently, genes encoding TetX along with homologs TetX3 and TetX4 were identified on transferable and inducible plasmids from Enterobacteriaceae and Acinetobacter in humans and animals that conferred high levels of resistance to all tetracycline antibiotics including eravacycline and omadacycline.18 Antibiotic inactivation by TetX, and presumably related homologs, occurs via formation of a C4a-hydroperoxy flavin with subsequent hydroxyl group transfer to C11a of the tetracycline substrate leading to irreversible fragmentation of the tetracycline scaffold.19
Eravacycline, omadacycline, sarecycline, and future generations of tetracycline antibiotics might fall victim to resistance by enzymatic inactivation (Figure 1). This lesson has been learned time and again for each new generation of beta-lactam antibiotics introduced to combat widespread inactivation by beta-lactamase resistance enzymes since the first report of a beta-lactamase by Abrahama and Chain in 1940.20, 21 TetX and related tetracycline inactivating enzymes known as the tetracycline destructases are widely dispersed in natural environments and hospital settings making this emerging resistance a potential clinical threat.22, 23 The risk for amplifying TetX-like tetracycline inactivating enzymes in human pathogens is a real possibility following widespread use of eravacycline. Widespread distribution of TetX amongst human pathogens could introduce pan-tetracycline resistant phenotypes that compromise the future use of the entire antibiotic class. Thus, it is imperative to develop resistance management plans proactively, including next-generation tetracyclines that overcome inactivation by TetX and adjuvants that inhibit TetX-mediated degradation of tetracycline antibiotics.24, 25 The freedom to fully control the eravacycline scaffold through total synthesis and advances in synthetic biology will greatly enhance the production of next-generation analogs and inhibitors to keep pace with emerging resistance.12, 26 Continuous evolution of beta-lactam antibiotic scaffolds through chemical and biochemical means has proved paramount for maintaining clinical efficacy of this critical drug class.27
Lessons learned from >80 years of clinical antibiotic use and development have provided a roadmap for the mechanisms, emergence, and dissemination of antibiotic resistance in hospital and community settings.28 Scientific advancements from the genomic era have been especially important for making the connections between antibiotic resistance in environmental microbes and human pathogens. The vast majority of clinical antibiotics are derived from microbial natural products.29 Thus, the vast majority of clinical antibiotic resistance originates from natural environments where antibiotic biosynthesis is prevalent.30 In fact, the aminoglycoside modifying and tetracycline inactivating enzymes discussed previously are likely to have origins in the biosynthesis of aminoglycoside and tetracycline antibiotics, respectively.31–33 This crossroads of antibiotic biosynthesis and resistance is fertile ground for microbial evolution. Antibiotic resistance has reached equilibrium in well-established natural ecosystems where antibiotic-resistant and antibiotic-sensitive organisms enjoy mutually beneficial lifestyles in the presence of diverse antibiotic cocktails.30, 34, 35 The evolutionary pressure from humans in the balance of antibiotic resistance is best appreciated in arenas where large-scale deployment of antibiotics is used for health and economic benefits; namely in hospital and agricultural settings.36 Unlike natural environments where microbes produce low concentrations of multiple antibiotics, humans tend to deploy large concentrations of single antibiotics for desired applications.37 This is best practice for the selection of highly resistant phenotypes, including multi-drug resistance (MDR) to mechanistically related clinical antibiotics such as observed for the well-known macrolide-lincosamide-streptogramin (MLS) phenotypes for Grampositive human pathogens resulting from expression of ribosome-modifying methyltransferases that perturb common antibiotic binding sites around the peptidyl transferase center.38
The exact trajectory of gene transfer for any given antibiotic resistance event is difficult to delineate.28, 30 There are known and speculated hotspots for enriched pools of antibiotic resistance genes including wastewater treatment plants, antibiotic manufacturing sites, agricultural sites employing antibiotic feedstock, and hospitals. The vectors for exchange of antibiotic resistance genes are thought to be genetically competent opportunistic human pathogens that readily grow in natural environments where antibiotics are prevalent. The individual antibiotic resistance genes representing the full spectrum of antibiotic resistance mechanisms (efflux, exclusion, target modification, sequestration, covalent inactivation) originate from selective pressure where self-protection genes that provide producers innate resistance are transferred between environmental microbes via mobile genetic elements.39 Thus, studying the biosynthetic origins of antibiotics provides insight into potential resistance mechanisms that currently exist or might emerge upon clinical use of the antibiotic or a derivative thereof. Proactive investigation of dormant and emerging resistance mechanisms is further merited because environmental microbes often provide natural strategies for overcoming resistance associated with a given antibiotic in the form of adjuvants, synergistic antibiotic combinations, and pro-drug formulations. Comprehensive reviews on the mechanisms, origins, and dissemination of antibiotic resistance are readily available.30, 40 More recently, Philmus and coworkers provided a survey with case studies on the topic of self-protection by antibiotic producing microbes.44 Here, we aim to provide a more focused perspective on the origins, mechanisms, and evolutionary trajectory of antibiotic resistance in the form of enzymatic inactivation to highlight crossroads with antibiotic biosynthesis that inform new methods for predicting and managing emerging clinical resistance mechanisms in human pathogens.
ANTIBIOTIC RESISTANCE AND PRODUCER SELF-PROTECTION
Self-protection is a pre-requisite for any antibiotic-producing microbe.42–44 It is common for antibiotic biosynthetic gene clusters (BGCs) to contain genes encoding the enzymes required for both biosynthetic assembly of the antibiotic scaffold and self-protection mechanisms.44 This co-clustering leads to co-expression of genes and ensures self-protection during production of the toxic antibiotic. Often BGCs also contain genes associated with quorum sensing regulation to ensure timely antibiotic production in mixed microbial environments where community metabolism is at play.45 Self-protection by antibiotic producers is often referred to as innate resistance while the appearance of self-protection genes in non-antibiotic producers is known as acquired resistance.
Typically, what is classified as authentic clinical resistance in human pathogens falls under the category of acquired resistance; however, human pathogens are often innately resistant to certain antibiotics. External gene acquisition is not a strict requirement for acquired resistance mechanisms. Additionally, resistance events caused by single nucleotide polymorphisms resulting in amino acid point mutations in the antibiotic target protein would fall under the category of spontaneous resistance. This type of resistance is commonly observed when human pathogens are treated with small molecule antibiotics including natural products. A serial passage experiment with increasing dose of the antibiotic can often reveal the molecular target of a given antibiotic through genome sequencing to locate the sites of resistance-conferring mutations.46
Innate resistance in the form of self-protection during antibiotic production does not always require a set of dedicated antibiotic resistance genes (ARGs). For example, Eleftheria terrae is a Gram-negative member of a new genus related to Aquabacteria that produces a highly potent lipid I–III sequestering antibiotic known as teixobactin.47, 48 This molecule rose to fame in 2015 amongst claims of no “detectable resistance”, an apparent contradiction for any antibiotic. Teixobactin is produced via non-ribosomal peptide synthetase (NRPS) enzymes in the E. terrae cytoplasm followed by export to the extracellular space via an inner membrane/outer membrane spanning RND family efflux pump encoded by genes in the teixobactin BGC. Gram-negative bacteria, including the producer E. terrae, are innately resistant to NRP antibiotics targeting lipid I-III, including teixobactin and vancomycin, that fail to permeate the outer lipid membrane that shields the lipid I-III cell membrane components localized to the inner lipid membrane.49 In Grampositive bacteria, the lipid I-III molecules are left exposed on the extracelluar surface of the lipid membrane where complex formation with teixobactin can occur to halt peptidoglycan assembly and induce a futile cycle leading to cell lysis.
Both antibiotic producers and non-producers are capable of achieving innate, acquired, and spontaneous types of antibiotic resistance. BGCs in antibiotic producing microbes are a long-term repository for ARGs.34 Genetic mobilization of these ARGs creates an on-demand supply for mixed microbiomes that can lead to dissemination into human pathogens.28, 30 The complete spectrum of antibiotic resistance mechanisms (efflux, exclusion, target modification, sequestration, enzymatic inactivation) is represented in the self-protection strategies invoked by antibiotic producing microbes.40 A classic example of this multi-pronged approach is found in Streptomyces antibioticus, a natural producer of the macrolide antibiotic oleandomycin (Figure 2).50 Oleandomycin is kept in an inactive glycosylated form in the S. antibioticus cytoplasm via two glycosyl transferases, OleI and OleD, encoded in the oleandomycin BGC.54 OleI is specific for oleandomycin, while OleD shows a broader substrate scope hinting at its role as a general macrolide inactivating resistance enzyme.52 Glycosylated oleandomycin is excreted to the extracellular space in pro-drug form via the OleB/OleC efflux pumps. An excreted glycosyl hydrolase, OleR, reveals the active protein synthesis inhibitor safely beyond the reach of the cytoplasmic ribosome target of the producer. Competing microbes that ingest oleandomycin are susceptible to ribosome inhibition allowing the producer a competitive growth advantage. These competing microbes still have opportunities for resistance gain-of-function through acquisition of efflux pumps, ribosome methyltransferases, and/or macrolide-inactivating enzymes including glycosyl transferases related to OleI and OleD.38, 53
Figure 2.

Macrolide resistance in producers and competing microbes.
Glycosylation of oleandomycin represents a model for the co-evolution of substrate specificity and plasticity into antibiotic inactivating enzymes.52 The reversible glycosylation of oleandomycin by the glycosyl transferase OleI and secreted glycosyl hydrolase OleR represents a useful self-protection strategy in S. antibioticus that preserves the antibiotic payload enabling dual use as a pro-drug strategy. Expression of OleD ensures survival in the presence of exogenous macrolides produced by competing microbes and represents a useful resistance strategy that can be passed on to human pathogens resulting in pan resistance to clinical macrolides.53 The complex interplay between producer immunity and resistance evolution through microbial competition provides the chemical breeding grounds for evolving antibiotic inactivating enzymes.
ORIGINS OF ANTIBIOTIC INACTIVATING ENZYMES
The most potent form of antibiotic resistance comes in the form of enzymatic inactivation.40, 54, 55 Unlike other resistance mechanisms, covalent modification of antibiotics depletes the total concentration of the antibiotic challenge below inhibitory levels rather than controlling local concentrations (intracellular vs extracellular) through exclusion, efflux, and sequestration or modifying the target to tolerate higher antibiotic concentrations. The flux of enzymatic antibiotic construction (biosynthesis) and destruction (self-protection/resistance) influences microbial population dynamics and drives the emergence of antibiotic inactivating enzymes.
Reversibility of Antibiotic Inactivation.
Antibiotic inactivating enzymes provide cellular immunity by modifying a portion of the antibiotic scaffold that is required for target binding. Differences arise in the types of chemical bonds formed when comparing producer self-protection and resistance in clinical pathogens. Antibiotic inactivation by self-protection enzymes tends to be reversible, while inactivation by resistance enzymes tends to be irreversible on a biologically relevant time scale. For example, consider the glutamine synthetase (GS) inhibitor tabtoxinine-beta-lactam (TBL) produced by plant pathogenic strains of Pseudomonassyringae (Figure 3).56 The alpha-carboxyl group of TBL is converted to the corresponding l-Thr dipeptide by the ATP-dependent l-Thr ligase TblF to facilitate efflux by TblR to the periplasm away from cytoplasmic GS.57 The TBL-Thr dipeptide is effluxed to the extracellular space where competing microbes can import the pro-drug via surface-displayed dipeptide permeases. Cytoplasmic dipeptidases can hydrolyze the TBL-Thr dipeptide revealing the GS inhibitor TBL in the cytoplasm where GS inhibition can be achieved.58 If intracellular TBL concentrations become too high, P. syringae can produce the Gcn5-related N-acetyltransferase (GNAT) Ttr that inactivates TBL via acetylation of the alpha-amino group.57 Both amidation of the TBL alpha-carboxylate by TblF and acetylation of the TBL alpha-amino group by Ttr result in covalent modifications that prevent GS binding. However, the amidation of TBL is reversible while acetylation appears to be irreversible. For this reason, only heterologous expression of Ttr, not TblF, in E. coli confers resistance to TBL-Thr.58 Dipeptide formation is a common pro-drug approach for non-proteinogenic amino acid antimetabolites including phosphinothricin, alaphosphin, and many more.59, 60 Phosphinothricin is a dipeptide pro-drug of the phosphinate-based GS inhibitor glufosinate and Streptomyces producer self-protection is achieved by expression of the bar gene, a GNAT related to Ttr in structure and function.61 Transgenic plants expressing the bar gene are resistant to glufosinate and form the basis of a broad-spectrum herbicide strategy marketed by Bayer under the trade name Liberty Link®.62 This is a case where irreversible antibiotic inactivation can be used to benefit society through application to agricultural chemistry.
Figure 3.

Reversible modification of tabtoxinine-beta-lactam (TBL) by amino acid ligase TblF to form the pro-drug TBL-Thr dipeptide in P. syringae. Irreversible modification of TBL by GNAT acetyltransferase Ttr confers resistance in competing microbes.
Thermodynamic and Kinetic Considerations.
Antibiotic resistance by enzymatic inactivation evolves with chemistry. The chemical reactions catalyzed by antibiotic inactivating enzymes tend to exploit the weakest link in the antibiotic scaffold. Consider beta-lactam antibiotics: all contain the strained 2-azetidonine heterocycle (a.k.a. beta-lactam) as the source of antibacterial activity.63 Beta-lactams are kinetically stable, but thermodynamically labile and are susceptible to irreversible hydrolysis of the beta-lactam ring (Figure 4a). Beta-lactamase enzymes catalyze the irreversible hydrolysis of 2-azetidinones with fast kinetics that can approach diffusion-controlled efficiency for some beta-lactam subclasses.21, 64 Each structural class of antibiotic possesses unique reactivity exploited by inactivating enzymes. For example, tetracycline antibiotics are not readily susceptible to hydrolysis like beta-lactams but the electron rich extended pi-conjugation of the tetracyclic core is naturally susceptible to photooxidation and chemical oxidation.14 It follows that a class of flavin monooxygenase (FMO) enzymes have emerged as tetracycline inactivating enzymes.22 These tetracycline inactivating FMOs catalyze the irreversible oxidation of tetracycline antibiotics via formation of a reactive C4a-flavin peroxy intermediate (Figure 4b).19
Figure 4.
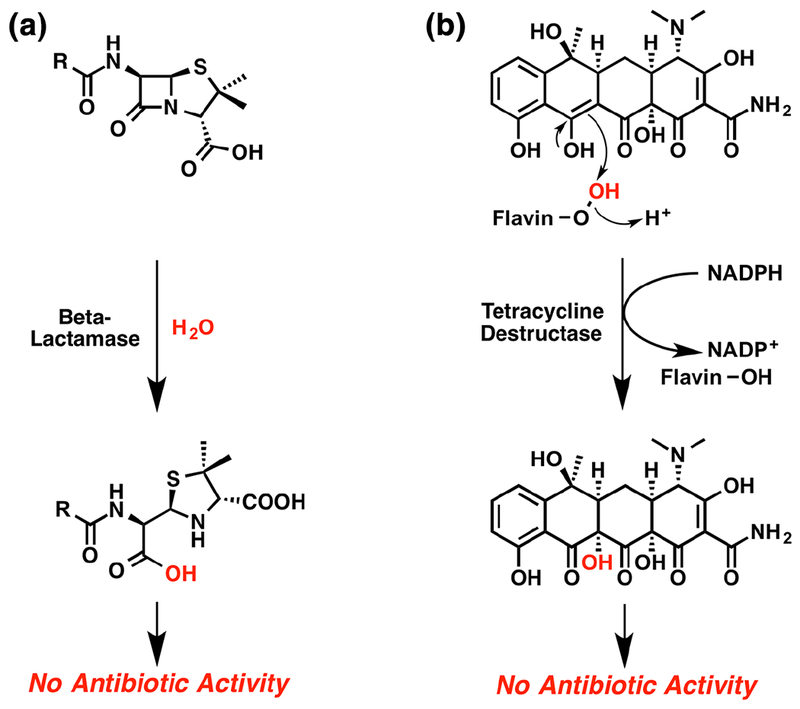
Resistance evolves with chemistry. (a) Hydrolysis of strained beta-lactam ring in penicillin by a beta-lactamase. (b) Oxidation of electron rich C11a-enol in tetracycline by a tetracycline destructase C4a-flavin peroxy intermediate.
Evolution from the Biological Target.
The emergence of antibiotic inactivating enzymes can follow distinct evolutionary paths. In the case of beta-lactamases, at least 4 distinct evolutionary classes have emerged (A–D).65 The most common class A beta-lactamases, including TEM-1, are serine hydrolases evolved directly from the target d,d-transpeptidase domain of the bacterial penicillin-binding proteins (PBPs) (Figure 5a).66 The primary differences between beta-lactamases and PBPs arise in the kinetics of the active site serine deacylation step, which in comparison is fast for beta-lactamses and slow for PBPs.21, 67 The emergence of extended spectrum beta-lactamases (ESBLs) followed shortly after widespread distribution of plasmid-encoded TEM and SHV beta-lactamases through gain-of-function mutations of the parent antibiotic resistance gene.64 ESBLs benefit from an expanded substrate tolerance and improved catalytic efficiencies. Such enhancements in substrate plasticity can be recreated in the laboratory setting through directed evolution experiments, as discussed in a later section.68
Figure 5.
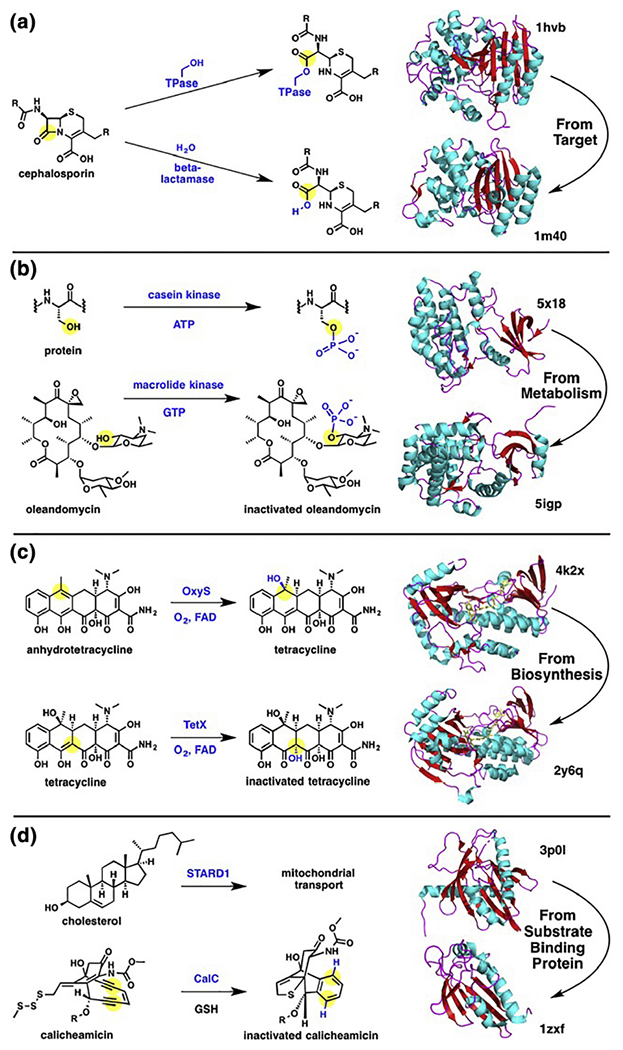
Evolution of antibiotic inactivating enzymes. (a) Evolution of beta-lactamases from target d,d-transpeptidases. (b) Evolution of protein kinase into a macrolide inactivating enzyme. (c) Evolution of a tetracycline inactivating enzyme (“antibiotic destructase”) from a biosynthetic flavin monooxygenase (“antibiotic constructase”). (d) Evolution of an endiyne self-sacrificing protein from a substrate binding protein.
Evolution from General Metabolism.
Mutation of the target enzyme into an antibiotic destructase seems plausible for cases where suicide inhibition (inhibition of the enzyme at the expense of the inhibitor scaffold) is at play; as is true for the beta-lactam antibiotics. For antibiotics where target inhibition is non-covalent, it is possible for enzymes from general metabolism to emerge as antibiotic destructases. Both housekeeping metabolic enzymes and antibiotic inactivating enzymes covalently modify substrates, but there are a few noteworthy differences (Figure 6) 52, 69, 70 Housekeeping enzymes tend to have broad substrate plasticity, exhibit slow kinetics, and are expressed constitutively. In some cases antibiotic inactivating enzymes are highly selective for a specific structural class of antibiotic substrates with broad substrate plasticity within the antibiotic class, exhibit fast kinetics, and are inducible by the antibiotic challenge. Furthermore, antibiotic resistance genes can be plasmid-encoded reflecting genetic mobility under selective pressure applied by antibiotic exposure while housekeeping genes are carried on the chromosome. It should be noted that housekeeping and antibiotic resistance genes are not necessarily mutually exclusive as many bacteria harbor species-specific chromosomal beta-lactamases.21
Figure 6.
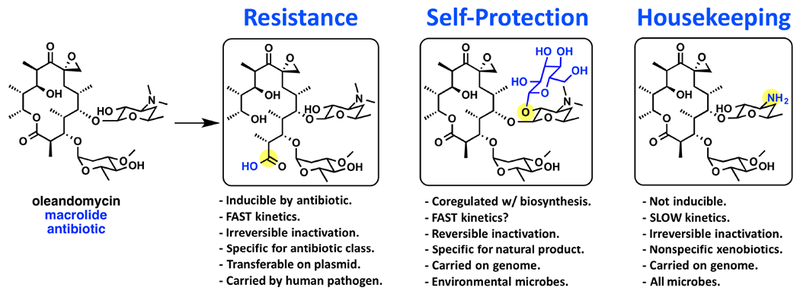
Comparison of macrolide inactivating enzymes used for resistance (macrolide esterase), self-protection (macrolide glycosyltransferase), and general metabolic housekeeping (demethylase) in microbes. Chemical modifications of the macrolide erythromycin are highlighted in blue with the site of functional group modification highlighted by a yellow circle.
Wright and coworkers recently demonstrated how substrate specificity emerges in a family of GTP-dependent macrolide inactivating kinases (Mph) using ancestral sequence reconstruction combined with directed evolution and functional selections.74 The Mph kinases phosphoiylate the conserved 2’-OH group of the dimethylamino sugar to prevent ribosome binding and confer resistance to Mph producers.72, 73 Similar kinases have emerged as resistance enzymes against aminoglycoside and tuberactinomycin antibiotics and all seem to have a structural relationship to Eukaryotic protein kinases supported by the retention of residual, albeit weak, Ser-protein kinase activity (Figure 5b).74, 75 Narrow spectrum Mphs were found to be common in Bacillales with catalytic phosphorylation rates against clinical macrolides being too low to confer resistance.74 In the Wright study, four residues were logically connected to expanding substrate accommodation in Mphs, while several less obvious mutations were shown to improve catalytic efficiency to provide gain-of-function resistance when expressed in a heterologous host. This approach for ancestral sequence reconstruction will likely be generally applicable to antibiotic inactivating enzymes that are amenable to gain-of-function via directed evolution including beta-lactamases68, tetracycline FMOs45, and aminoglycoside phosphotransferases.76, 77
Evolution from a Biosynthetic Enzyme.
While macrolide phosphorylation does appear to be a sufficiently irreversible modification to confer resistance, a more effective resistance strategy would seem to be the destructive hydrolysis of the macrolactone ring common to the entire antibiotic class. In fact, plasmid encoded macrolide esterases, including erythromycin esterase EreA, have been discovered in clinical pathogens (Figure 6).53, 69 To date, no structure of a macrolide esterase has been solved. Sequence comparison and homology modeling suggests that macrolide esterases (EreA, EreA2, EreB, EreC, and EreD) are metalloenzymes predicted to be structurally related to esterases involved in succinoglycan biosynthesis (BcR135 and BcR136). It is intriguing that macrolides are products of type I polyketide synthases where product release from the enzymatic assembly line is facilitated by a C-terminal hydrolase domain that catalyzes macrolactonization as opposed to hydrolysis of the intermediate acyl enzyme oxoester intermediate.78 It is conceivable that these macrolactonization catalytic domains could also evolve into autonomous macrolide esterases through accommodation of a nucleophilic water in the active site. Such conversion of an “antibiotic constructase” to an “antibiotic destructase” might be favorable since the ancestral biosynthetic enzyme has already evolved the substrate-binding pocket and catalyzes the necessary chemistry for antibiotic inactivation (lactonization vs hydrolysis in the case of macrolides). Based on the ancestral sequence reconstruction and directed evolution approach applied towards other classes of antibiotic inactivating enzymes the acquisition of gain-of-function mutations in ancestral biosynthetic enzymes that confer resistance through antibiotic inactivation seems plausible.71
Another case for emergence of an “antibiotic destructase” from an “antibiotic constructase” might be found in the tetracycline FMO family of tetracycline inactivating enzymes (Figure 5C).22 Oxidation of the anhydrotetracycline C-ring at carbon-6 by a flavin monooxygenase, OxyS, provides the mature tetracycline scaffold during biosynthetic assembly.32, 33 The tetracycline FMOs are more versatile and oxidize tetracyclines at a variety of positions including enol Cl la and ketone C12 leading to oxidized degradation products via hydroxylation or Baeyer-Villiger oxygen insertion, respectively, that lack antibacterial activity.49 Some tetracycline FMOs, including TetX, can also oxidize anhydrotetracycline and show broad substrate plasticity within the tetracycline antibiotic class that rivals clinically prevalent antibiotic inactivating enzymes including macrolide and aminoglycoside kinases.14, 24, 25 Similar to macrolide kinases, TetX readily accepts gain-of-function mutations via directed evolutionary pressure that enhance catalytic efficiency towards the oxidative inactivation of 3rd generation tetracyclines including tigecycline, eravacylcine, and omadacycline.15, 18 The evolutionary trajectory from OxyS or related flavin monooxygenases to tetracycline destructases has not been fully explored.
Evolution from a Substrate-Binding Protein.
Yet another evolutionary line of antibiotic inactivation has emerged from substrate-binding proteins (SBPs) (Figure 5d). SBPs are typically considered non-catalytic proteins that reversibly bind small molecules with high affinity.79 A somewhat odd case has emerged where an SBP encoded in BGCs for enediyne natural products can act either as a sequestering agent to prevent association with target DNA80 or a single turnover catalyst to quench reactive para-benzyne radicals generated from protein sequestered parent enediyne through C-H abstraction from a conserved glycine residue leading to protein fragmentation.81, 82 BGCs for anthraquinone-fused 9-membered enediynes such as dynemicin seem to be rich in SBPs such as TnmSl, TnmS2, and TnmS3 that non-covalently sequester the DNA-damaging enediynes from DNA in the cytoplasm to ensure self-protection 80 Recently, the X-ray structure of the DNA gyrase inhibitor albicidin bound to a BGC-encoded sequestering protein AlbA was solved showing how high-affinity cytoplasmic sequestration can be an effective self-protection strategy.83 The calicheamicin BGC, and related 10-membered enediynes, are also DNA-damaging agents with mechanism of action similar to the 9-membered enediynes. Calicheamicin is a prodrug where activation is triggered via disulfide reduction leading to formation of a reactive para-benzyne radical capable of DNA cleavage chemistry. Calicheamicin producers encode self-sacrificing SBPs such as CalC that act as single turnover catalysts to inactivate cytoplasmic pools of activated eneidynes by quenching reactive benzyne radicals through C-H abstraction from a glycine residue in the active site binding calyx resulting in CalC cleavage.81, 82 It is unclear if antibiotic sequestration by reversible or self-sacrificing SBPs represent a clinical antibiotic resistance threat given that the large doses of antibiotics used in clinical settings are likely to outcompete sequestration as a viable resistance strategy.
No matter the evolutionary trajectory for emergence of antibiotic inactivating enzymes, ultimately, the antibiotic scaffold determines the modification site and type of enzymatic inactivation that will be effective for a given antibiotic class (Table 1) 11, 14, 17, 22, 31, 50–53, 57, 59, 60, 65, 69, 71, 72, 74, 76, 80–82, 84–130 Highly conserved functional groups and reactive “soft spots” in the scaffold (e.g. strained beta-lactam rings) are prime targets for inactivating resistance enzymes. Resistance can only be achieved if the chemical modification is irreversible on a biological time scale and if the modification blocks access and/or binding to the target. The most dangerous antibiotic inactivating enzymes completely “destroy” the antibiotic scaffold (e.g. beta-lactamases and tetracycline destructases), possess broad substrate coverage across the entire antibiotic class, and are expressed in response to the antibiotic challenge. Presumably, no antibiotic, even the famed teixobactin claimed to be free of “detectable resistance” (see previous discussion), can fully evade emergence of inactivating enzymes.47 Recently, Qian and coworkers discovered a d-stereospecific peptidase associated with NRPS BGCs that can confer pan resistance to antibacterial NRPs containing d-amino acids.85 Specifically, two d-stereospecific peptidases, BogQ and TriF, from the bogorol and tridecaptin BGCs, respectively, were shown to confer self-protection to the producing microbes via enzymatic hydrolysis of the corresponding NRPs in vitro. Surprisingly, BogQ was also able to hydrolyze the nonnative substrate bacitracin and confer resistance to bacitracin in strains expressing BogQ. While a truncated teixobactin peptide was not a very good substrate for BogQ, this type of peptidase could emerge as a teixobactin-inactivating enzyme. Directed evolution of BogQ with functional selections against teixobactin might be a good way to anticipate gain-of-function mutations in d-stereospecific peptidases leading to teixobactin resistance.
Table 1.
Functional activity and evolutionary origins of representative antibiotic modifying enzymes.
| Antibiotic destructases | Susceptible functional groups | Relevant antibiotic classes | Representative antibiotics | Natural role | References | |
|---|---|---|---|---|---|---|
| Hydrolases | Beta-lactamases | Beta-lactam ring, beta-lactone ring | Beta-lactams, beta-lactones | Amoxicillin, cephamycin, thienamycin, aztreonam, obafluorin | Self-protection | [65, 84] |
| Esterases | Esters, macrolactones | Macrolides | Erythromycin, oleandomycin | Unknown | [69] | |
| d,d-Peptidases | Peptide bonds | Non-ribosomal peptides | Bogorol, bacitracin, zwittermicin, colibactin, xenocoumacin | Self-protection/pro-drug release | [85–88] | |
| Cyclopropane hydrolase | Cyclopropane ring | Cyclopropane antibiotics | Colibactin | Self-protection | [89] | |
| Epoxide hydrolase | Epoxides | Epoxide antibiotics | Fosfomycin | Unknown | [90] | |
| Di-/tripeptidase | Peptide bond | Di- and tripeptides | Phosphinothricin, tabtoxin, bacilysin | Prodrug release | [91] | |
| Glycosidase | Glycosidic linkage | Macrolides | Oleandomycin | Prodrug release | [50–52] | |
| Phosphatase | Phosphoester | Aminoglycosides | Streptomycin | Prodrug release | [31, 92] | |
| Amidase | Amide | Amphenicols, beta-lactams | Chloramphenicol, penicillin | Self-protection, antibiotic catabolism | [93–95] | |
| Transferases | Thioltransferases | Epoxides, ketones, aldehydes, michael acceptors | Fosfomycin, abyssomicin | Fosfomycin, abyssomicin | Xenobiotic detoxification | [96–98] |
| Phosphotransferases | Hydroxyl groups | Aminoglycosides, macrolides, amphenicols, fosfomycin, ansamycins | Kanamycin, erythromicin, chloramphenicol, fosfomycin, rifampin | Self-protection/pro-drug formation | [11, 53, 71, 72, 74, 76, 99–103] | |
| Acetyltransferases | Hydroxyl groups, amino groups | Streptogramins, antimetabolites, amphenicols, aminoglycosides, fluoroquinolones | Virginiamycin, TBL, azetidine-2-carboxylate, chloramphenicol, kanamycin, ciprofloxacin | Self-protection, antibiotic catabolism | [57, 61, 104–113] | |
| Glycosyltransferases | Hydroxyl groups | Macrolides, ansamycins | Oleandomycin, erythromicin, rifamycin | Self-protection/pro-drug formation | [52, 53, 114] | |
| Methyltransferases | Sulfhydryl, amino, and hydroxyl groups | Dithiolpyrrolones, dithioldikemiddleiperazines | Holomycin, gliotoxin | Self-protection | [115, 116] | |
| Nucleotidylyltransferases | Hydroxyl groups | Aminoglycosides, lincosamides | Kanamycin, clindamycin | Self-protection | [11, 76, 117–120] | |
| ADP-ribosyltransferases | Hydroxyl groups | Ansamycins | Rifamycin | Self-protection | [121, 122] | |
| Amino acid ligase | Amino acids | Amino acid antimetabolites | Phosphinothricin, tabtoxin, alaphosphin | Prodrug formation | [57, 59, 60] | |
| Asparagine acyl transferase | Asparagine amino group | Non-ribosomal peptides | Zwittermicin, colibactin, xenocoumacin | Prodrug formation | [86–88] | |
| Lyases | Streptogramin lyase | Esters | Streptogramins | Virginiamycin, quinupristin | Self-protection | [123, 124] |
| Oxido-reductases | Flavin monooxygenases | Electron-rich olefins, electron-deficient ketones | Tetracyclines, ansamycins | Tigecycline, rifamycin | Biosynthesis/self-protection | [14, 17, 22, 125, 126] |
| Quinone reductase | Quinones | Mitomycins | Mitomycin C | Self-protection/pro-drug | [127] | |
| Nitro reductase | Nitro groups | Amphenicols | Chloramphenicol | Xenobiotic detoxification | [128, 129] | |
| Ketoreductase | Ketone | Streptogramins | Virginiamycin M1 | Self-protection | [130] | |
| Self-sacrificing proteins | Substrate-binding proteins | Chemical sequestration | Enediynes, albicidin | Dynemicin, albicidin | Self-protection | [81–83] |
| Glycyl cleavage proteins | Enediyne | Enediynes | Calicheamicin | Self-protection | [80] | |
PROSPECTING FOR RESISTANCE
The emergence of environmental antibiotic resistance mechanisms in human pathogens through horizontal gene transfer is now recognized as a major route for dissemination of ARGs in the community and clinical settings (Figure 7) 28, 30 Monitoring for antibiotic resistance using functional screens (e.g. antibacterial susceptibility assays on clinical isolates) in hospitals is a longstanding clinical practice.131 New advancements in next-generation DNA sequencing and functional metagenomics have expanded the capacity to prospect for resistance in environments where microbial cultivation is challenging.28, 132 Functional metagenomics has proved useful in revealing new members and new families of ARGs providing a refined view of sequence diversity and environmental distribution of a given ARG class.22 Historically, resistance among antibiotic producers and heterologous hosts has been used advantageously by pharmaceutical companies to screen for new antimicrobial agents in natural product extracts at the beginning of the antibiotic discovery process. Leveraging modern advancements in genetics and analytical instrumentation revisiting these fruitful discovery paradigms are turning up new molecules and revealing associated biosynthesis and resistance pathways.
Figure 7.
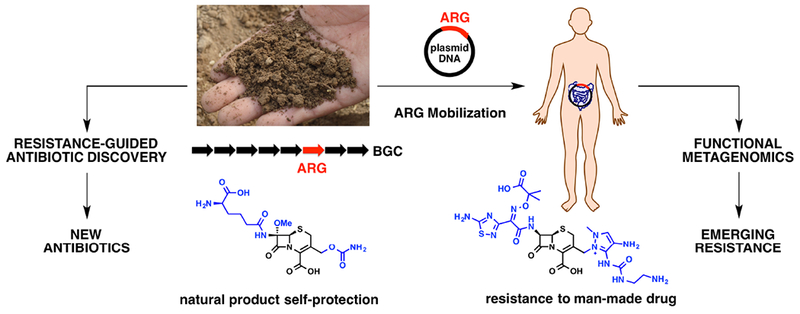
Dissemination of ARGs from soil to the human gut. Ancient antibiotic resistance to the natural cephalosporin can be a source of resistance to the man-made cephalosporin used in hospitals when ARGs from a BGC are mobilized on plasmids. This natural phenomenon can be leveraged for resistance-guided antibiotic discovery (left) to prospect for new antibiotics and functional metagenomic screens (right) to prospect for emerging resistance. The image of soil was obtained from pixabay.com and is free for use in the public domain.
Functional Metagenomic Screens for ARGs.
In a seminal study, Wright and coworkers demonstrated that the equilibrium pool of ARGs in soil Actinomycetes is rich.39, 133 It was no surprise to discover that environmental resistance to natural product-derived antibiotics such as macrolides is more common than resistance to fully synthetic antibiotics such as linezolid. Pioneering work by Jo Handelsman and coworkers revealed the diversity, distribution, and origins of beta-lactamase resistance genes in Alaskan soil and validated soil environments as a reservoir of resistance even in the absence of anthropogenic activity.134 The Dantas lab expanded on this principle in the search for environmental ARGs by broadly applying functional metagenomics using environmental DNA (eDNA) samples from diverse environments to screen for antibiotic resistance against all major classes of antibiotics in an E. coli heterologous host.135 By carefully screening soil and human gut metagenomes the connection between ARGs in soil microbes, human commensals, and pathogens was solidified. Functional screens can reveal ARGs associated with all mechanistic types of antibiotic resistance, including enzymatic inactivation.
In the case of antibiotic inactivating enzymes, functional metagenomics combined with in vitro reconstitution has played a key role in validating enzyme function connected to antibiotic resistance.28, 132 The flavin-dependent tetracycline inactivating FMOs highlighted throughout this review were discovered during functional metagenomic analysis of soil and human gut microbiomes (Figure 4).22, 24 Homologous FMOs that inactivate rifamycin antibiotics via a similar oxidative degradation mechanisms have been the focus of recent study by Wright, Tanner, and Sobrado.125, 126, 136 The pool of rifamycin FMOs could presumably be expanded using functional metagenomic screens as done for the related tetracycline FMOs. Functional screens have also led to the discovery of previously overlooked antibiotic inactivating enzymes such as chloramphenicol nitro reductases.128, 129 Related approaches have even turned up novel antibiotic degradation mechanisms including “antibiotic eater” strains that provide an expanded view of the natural roles for antibiotics beyond bacterial growth inhibition.95, 108 Antibiotic catabolism in environmental microbes could turn out to be a rich source of antibiotic inactivating enzymes.137 Expanding known ARGs and enriching the sequence pool for a given ARG class is the first step in gaining predictive capacity for resistance based solely on genome sequencing. Building predictive sequence similarity networks for ARGs will be an important tool for identifying emerging clinically relevant ARGs in human pathogens. Even with increasingly diverse genomic and metagenomic sequence databases, it is still difficult to predict functional ARGs from sequence alone. A recent study by Ruppe and coworkers revealed that coupling functional metagenomics with three dimensional protein structure prediction through pairwise comparative modeling, so called “homology comparative modeling”, can refine the pool of predicted ARGs and provide new candidate antibiotic inactivating enzymes that would otherwise remain overlooked by direct sequence comparison.138
Computational Approaches to Predicting Resistance.
New innovations in computational methods to predict function based on sequence and structural identity will play an important role alongside functional screens in future campaigns to expand ARG predictive capacity.25, 132, 138 Ancestral reconstruction of ARGs present in the clinic will connect gain-of-function mutants in natural environments and clinical settings where the evolutionary connection is not necessarily mutually exclusive.71, 135 Purely computational approaches for predicting protein evolution are hampered by molecular ensembles and statistical thermodynamics.139 The use of Markov state models is gaining traction to explore cryptic functional states of proteins, including TEM beta-lactamases, that can provide positive and negative allosteric modulators when coupled to small molecule screens.140, 141 Directed evolution and deep mutational scanning has proven to recreate clinically relevant gain-of-function mutations in TEM beta-lactamases. In a seminal study by Stevens and coworkers, TEM-1 was subjected to directed evolution in hypermutator E. coli, which recreated three mutations (E104K/M182T/G238S) found in the clinical beta-lactamase TEM-52 that improve stability and impart activity against 3rd generation cephalosporins such as cephotaxime.142 Similar approaches have been applied to aminoglycoside and macrolide inactivating enzymes suggesting that computational methods will need to be supplemented with functional screens to provide useful information on the catalytic efficiency, cellular abundance, stability, and evolutionary trajectory of antibiotic resistance enzymes in a given pathogen.71, 77
Resistance-Guided Antibiotic Discovery.
The link between antibiotic biosynthesis and resistance is playing an increasingly important role in prospecting for antibiotic resistance and next-generation antibiotics. Microbial antibiotic BGCs are a long-term repository for ARGs in diverse environments.34, 39, 41, 133, 135 Resistance-guided BGC mining has turned up several new antibiotics in recent years and has helped to expand the structural pool of known antibiotic classes. Wright and coworkers reported the first successful demonstration of this method in 2013.143 By screening a small library of 1,000 actinomyces against vancomcyin a 4% resistance hit rate was achieved that correlated with expression of the canonical vanHAX operon for glycopeptide resistance. Genome sequencing guided by co-clustering of the vanHAX operon with glycopeptide BGCs led to the discovery of pekiskomycin, a scaffold novel glycopeptide. Resistance-guided screening has proved even more useful in recent years where the number of genome-sequenced strains of antibiotic producers is on the rise.144 Advancements in computational methods and smart screens that combine structure/resistance prediction with functional screens can greatly enhance the rate of antibiotic dereplication to reveal novel antibiotic structures and adjuvants.145, 146 Co-clustering of a duplicate DNA sliding clamp variant (GriR) with a BGC in Streptomycesgriseus led to the discovery of a new NRP antibiotic, griselimycin, and confirmation of the DNA polymerase sliding clamp domain (DnaN) as the cellular target.147 Heterologous expression of GriR in Streptomyces coelicolor conferred griselimycin resistance. Co-crystallization of griselmycin with the M. smegmatis DNA sliding clamp dimer revealed a novel binding mode in adjacent hydrophobic pockets created by an interdomain protein-protein interaction that is stabilized by the macrocyclic NRP core and proline-valine tail of griselimycin, respectively. Griselimycin and the DNA clamp represent a novel antibiotic/target pair with promising anti-TB activity. Future resistance-guided screens against griselimycin paired with detailed analysis of the biosynthetic assembly will likely turn up novel structural analogs.148 More recently, Shen and coworkers have connected three proteins, TnmSl-S3, with anthraquinone-fused enediyne self-resistance in producers. The TnmSl-S3 proteins are beta-barrel homodimers that bind the enediynes with nanomolar affinity providing resistance via sequestration. The genes encoding these resistance proteins are conserved in producer BGCs and widespread in diverse environments including the human microbiome80. Enrichment of the human microbiome with ARGs creates a selection-ready environment for resistance upon clinical antibiotic use.
Philmus and coworkers recently highlighted a nice selection of ARG-BGC genome mining efforts including blasticidin (BlsJ; efflux pump)149, andrimid (AdmT; beta-subunit of the target acetyl coA carboxylase)150, mycophenolic acid (MpaF; resistant homolog of the target inosine-5’-monophosphate dehydrogenase)151, platensimycin/platencin (PtmP3; resistant homolog of the target beta-ketoacyl-acyl carrier protein synthase III)152, indolmycin (IndO; resistant homolog of the target Trp-tRNA synthetase)153, 154 and geldanamycin (HtpG; resistant homolog of the target heat shock protein 90)155, 156 New examples of ARG-BGC mining continue to emerge including the recent discovery by Nair and Metcalf of a resistant threonine synthase homolog ThrC in Bacillus subtilis ATCC 6633, a natural producer of the potent phosphono-oligopeptide threonine synthase inhibitor rhizocticin.157 In this case, in vitro reconstitution of sensitive and resistant ThrC homologs from B. subtilis provided mechanistic insight into the mode of threonine synthase inhibition that can guide the design of improved phosphono-oligopeptide inhibitors that regain activity against current insensitive ThrC targets. Resistance-guided genome mining combined with advancements in high-throughput microbial genome sequencing and mass spectrometry provides new opportunities in natural product discovery.145, 146, 158 The targeted mining for ARGs embedded in BGCs will help to distinguish antibacterial agents from natural products with alternate functions. Furthermore, heterologous expression of BGC-derived ARGs can be used to screen for new producers and identify structural relatives to expand the pool of a given natural product class. Utilization of isogenic panels of bacterial strains expressing clinical ARGs has already proven useful as an antibiotic discovery strategy and a way to optimize antibiotic structures to evade resistance.8, 159
STAYING AHEAD OF RESISTANCE
Antibiotic resistance is something that should be considered at the earliest stages of antibiotic development. Resistance should be managed, not avoided, through pharmaceutical optimization and responsible stewardship following deployment into medical practice.160A pro-active resistance-management plan should be incorporated into the development of all new antibiotics to extend the clinically useful lifetime and allow for sustainable introduction of future scaffold generations. While nature is the origin of antibiotic resistance it also holds the solutions. Nature has provided a plethora of novel antibiotics acting on unexploited biological targets that still await pre-clinical development.29 These molecules represent great starting points for new antibiotic development programs where resistance has not saturated the target and scaffold iteration has not saturated the market. Rediscovery of new variants of established clinical antibiotics reveals scaffold modifications that might improve potency, evade established resistance mechanisms, or provide a useful prodrug delivery strategy. Additionally, genome mining is proving to reveal that antibiotic BGCs are often grouped into “super clusters” that enable co-production of synergistic antibiotics or hybrid antibiotics with enhanced bioactivity.
Synergistic Antibiotic Combinations.
The first documented example of a biosynthetic “super cluster” is the cephamycin-clavulanate BGC in Streptomyces clavuligerus and related Streptomycetes.161, 162 The cephamycin and clavulanate operons are co-regulated by the ccaR gene in the cephamycin BGC.163 CcaR is a Streptomyces antibiotic regulatory protein (SARP) that acts as a positive regulator of the cephamycin-clavulanate “super cluster”. CcaR over-expression leads to increased production of cephamycin and clavulanate.164 This co-production of two beta-lactams, cephamycin and clavulanate, is beneficial given the specific bioactivity of each molecule (Figure 8a). Cephamycin is a cephalosporin antibiotic that inhibits bacterial penicillin binding proteins and is susceptible to some classes of beta-lactamase enzymes. Clavulanic acid is a potent inhibitor of many beta-lactamases and rescues beta-lactam antibiotic activity against competing microbes expressing beta-lactamase enzymes.165 It should also be noted that Streptomyces clavuligerus secretes a beta-lactamase inhibitory protein (BLIP) in addition to clavulanic acid.166, 167 Structural details of proteinaceous BLIPs in complex with inhibited beta-lactamases might offer a starting point for designing new small molecule inhibitors.168–170 Clavulanic acid has been developed as a combination product with clinical beta-lactam antibiotics, including the penem antibiotic amoxicillin forming a combination antibiotic therapy known as Augmentin™.171
Figure 8.
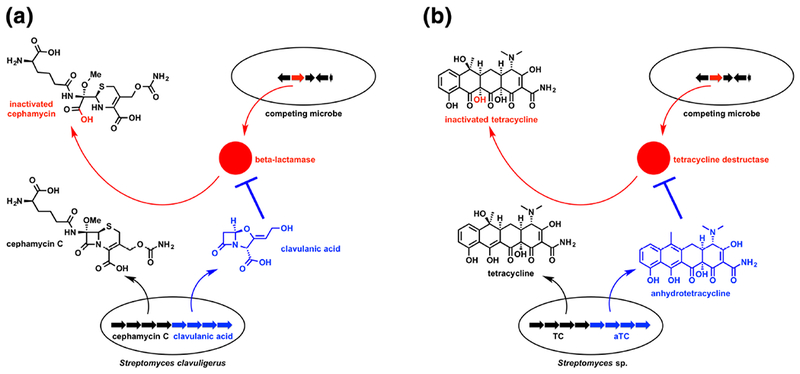
Co-production of antibiotic (black) and adjuvant (blue) to overcome resistance during microbial competition. (a) Transcription of the beta-lactam “super” BGC in S. clavuligerus results in co-production of cephamycin C and clavulanic acid, a beta-lactam antibiotic/beta-lactamase inhibitor combination that rescues beta-lactam antibacterial activity against competing microbes expressing beta-lactamase resistance enzymes. (b) Similarly, co-production of anhydrotetracycline (aTC), a tetracycline destructase inhibitor, and tetracycline (TC), a ribosome inhibitor, might provide a competitive advantage against microbes expressing tetracycline destructase resistance enzymes.
There is a key lesson that may apply to other natural products where co-expression of multiple biosynthetic products provides synergistic bioactivity. For example, anhydrotetracycline was recently reported to be a potent inhibitor of tetracycline destructase resistance enzymes isolated from environmental soil microbes.24, 25 Anhydrotetracycline is also the biosynthetic precursor to tetracycline with both metabolites accumulating in the extracellular space of the producing microbe (Figure 5b).32, 33 It could be that co-production of anhydrotetracycline and tetracycline is “intentional” and provides a growth advantage against competing microbes expressing tetracycline destructase enzymes (Figure 8b).14 Genome mining and smart screens can also reveal antibiotic adjuvants whether or not coclustering is at play.145 Recently, Wright and coworkers used such a method to identify aspergillomarasmine A, a potent inhibitor of metallo-beta-lactamases including NDM-1 for which there is currently no clinically useful inhibitor.172 Revisiting natural product discovery with an angle towards antibiotic combination discovery is needed to enrich the pool of antibiotic potentiating agents to validate combination therapies as a sustainable option for managing resistance.173
A more recent example of a biosynthetic “super cluster” encoding coproduction of sulfazecin and bulgecin was discovered in Paraburkholderia acidophila ATCC 31363 and Burkholderia ubonensis ATCC 31433 (Figure 9a) 174 Sulfazecin is an N-sulfated monobactam inhibitor of the transpeptidase domain of bacterial penicillin binding proteins, while bulgecin is a 2-(hydroxymethyl)-3-(O-(N-acetyl-d-glucosamine-4-sulfate))-pyrrolidine inhibitor of lytic transglycosylases MltD, MltG, and Slt.175 Consequently, co-production of sulfazecin and bulgecin results in simultaneous inhibition of peptidoglycan peptidyl crosslinking and glycan chain hydrolysis, respectively, worsening the effects of the cell wall repair futile cycle and leading to rapid cell lysis.176 The bactericidal effects of sulfazecin and bulgecin are synergistic and bulgecin has been shown to potentiate the antibacterial activity of clinical beta-lactam antibiotics including third generation cephalosporin ceftazidime and meropenem. Further investigation of bulgecin and beta-lactams as combination therapeutics is merited. The bulgecin/sulfazecin “super cluster” is a reminder that stand alone antibiotic BGCs should be revisited to see if flanking genomic operons harbor BGCs for synergistic molecules. Recently it was realized that all producers of rapamycin, a well-known immunosuppressant with antifungal activity, also produce a group of actinoplanic acids that provide a synergistic antifungal effect via inhibition of farnesyltransferases.177 The rapamycin and actinoplanic acid BGCs are organized as “super clusters” in producing microbes. Surprisingly, combinations of rapamycin and farnesyltransferase inhibitors have been found to have synergistic effects against several types of mammalian cancers and are currently under clinical investigation.178
Figure 9.
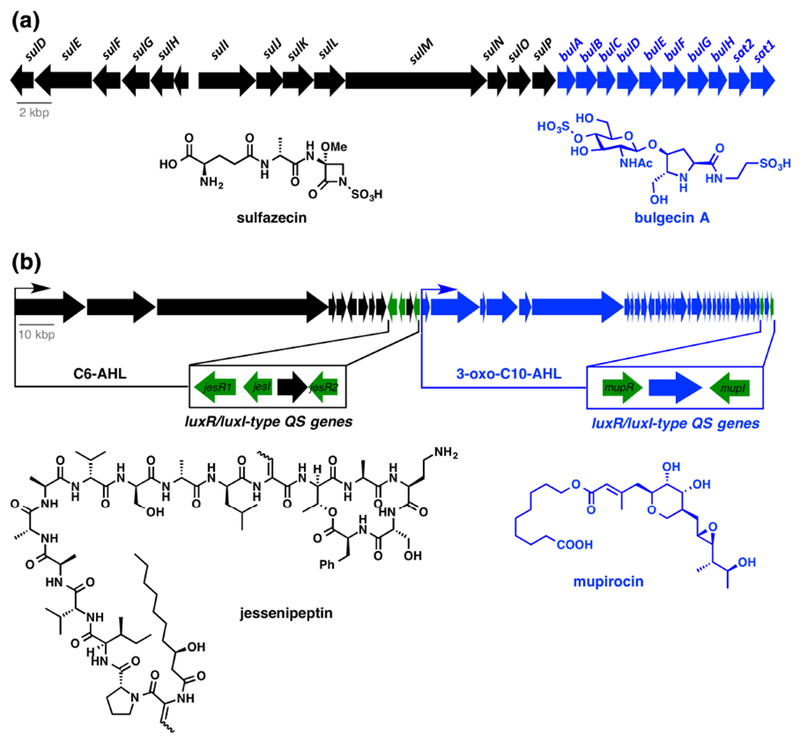
Co-production of synergistic antibiotic combinations to improve potency and limit resistance development. (a) Sulfazecin and bulgecin BGCs in Paraburkholderia acidophila ATCC 31363 are arranged as a “super cluster”. (b) Mupirocin and jessenpeptin BGCs in amoeba-associated Pseudomonas sp. QS1027 are co-regulated by LuxR/LuxI-type quorum sensing (QS) systems. Genes are color coded blue or black to match biosynthetic products (green genes are regulatory).
A second example of co-antibiotic production was recently discovered in Pseudomonas sp. QS1027, an amoeba associated strain isolated from the fruiting bodies of Dictyostelium discoideum in soild179 Pseudomonas sp. QS1027 was found to produce the polyketide antibiotic mupirocin, a clinically useful inhibitor of Ile-tRNA synthetase marketed as Bactroban®, along with a cooperatively excreted cyclic lipopeptide, jessenipeptin, produced via an NRPS pathway (Figure 9b). The jessenipeptin and mupirocin BGCs are organized in a “super cluster” and both sub-BGCs contain a dedicated luxR/luxl-type pair of quorum sensing (QS) genes. Production of mupirocin and jessenipeptin is stimulated by unique acyl homoserine lactones (AHLs) (C6-AHL activates transcription of the jessenipeptin BGC; 3-oxo-C10-AHL activates transcription of the mupricon BGC) indicating a role for quorum sensing in the cooperative biological activity of the antibiotic pair. Co-production of mupirocin and jessenipeptin did not play a role in the immunity of strain QS1027 to D. discoideum grazing although jessenipeptin, not mupirocin, did show potent amoebicidal activity (IC50 = 4 μg/mL). However, the antibiotics were synergistic in the killing of Gram-positive bacteria including B. subtilis and methicillin-resistant S. aureus (MRSA). The target of jessenipeptin is unknown, but the synergistic relationship with mupirocin, a Ile-tRNA synthetase inhibitor, suggests membrane depolarization as a potential mechanism of action that would enhance cell permeability of mupirocin allowing for greater cytoplasmic accumulation. Mupirocin-jessenipeptin synergy points to a role for co-biosynthetic regulation of the antibiotic pair by unique quorum sensing agents that might contribute to the adaptation of certain Pseudomonads to coexist with predatory amoebas. In the broader sense, this example should encourage the study of microbial interactions in ecology to better understand the plethora of “silent” antibiotic BGCs found in microbial producers that reserve antibiotic deployment for highly specialized scenarios.180
Hybrid Antibiotics.
An alternative evolutionary approach to antibiotic “super clusters” resulting in co-production of distinct antibiotic entities is the merging of BGCs through biosynthetic enzymes that catalyze the covalent ligation of multiple antibiotic scaffolds creating multi-functional hybrid antibiotics.181–183 Here the term “hybrid antibiotic” is used to define the joining of two different antibiotic moieties via a biosynthetic pathway present in a single organism; as opposed to the traditional use of the term “hybrid” in plant and animal breeding where the product results from the combination of genes from two disparate strains or species. There are however some parallels between the two uses of the term since hybrid antibiotic biosynthetic operons in a single microbe might arise from a horizontal gene transfer event between two disparate microbes each harboring biosynthetic operons for individual antibiotic moieties that end up in the hybrid scaffold. Hybrid antibiotics can take advantage of expanded target engagement and leverage access to membrane transport pathways provided by the merging of two antibiotic scaffolds.
Classic examples of hybrid antibiotics are the naturally occurring sideromycins (siderophore-antibiotic conjugates), that achieve energy-dependent receptor-mediated import across the cell envelope through iron transport pathways to access periplasmic and cytoplasmic targets of the antibiotic attached to the siderophore delivery vector (Figure 10a).184 In a sense, the sideromycins are similar to dipeptide prodrugs such as tabtoxin that were discussed earlier (Figure 3), where the siderophore component facilitates cell entry in a manner similar to the dipeptide prodrug.57, 58, 91 Since siderophore transport is driven by ATP hydrolysis, intracellular concentrations of the antibiotic can reach levels that well exceed extracellular concentrations leading to enhanced potency, often in the nanomolar range.184 The location and nature of the biological target determines if antibiotic release from the siderophore is required for target engagement.185–187 In the case of albomycin, a thionucleoside Ser-tRNA synthetase inhibitor is linked to a ferrichrome-like trihydroxamate siderophore through a peptide bond that is cleaved by the cytoplasmic peptidease PepN found in many Gram-negative bacteria (Figure 10a).188 PepN deletion mutants are resistant to albomycin reflecting that antibiotic release from the siderophore is critical for antibacterial activity.189 Resistance can also be achieved via mutations or deletion of transport proteins indicating that targeted uptake pathways must be required for pathogen virulence.190, 191 Albomycin serves as a model template for hybrid antibiotic biosynthesis where the ferrichrome siderophore is assembled via a dedicated NRPS assembly line and the thionucleoside is produced via an independent series of enzymes encoded in the same BGC.192‘ 193 An ATP-dependent ligase, AbmC, is predicted to join the siderophore and thionucleoside via a serine linker. A similar convergent strategy was used to assemble the albomycin scaffold via total synthesis opening the opportunity for the production of new analogs for pharmaceutical optimization.194 It is noteworthy that the albomycin BGC also contains a duplicate copy of Ser-tRNA synthetase, AbmK, that serves a role in self-protection against poisoning of the canonical Ser-tRNA synthetase encoded outside the albomycin BGC192. The sideromycin drug delivery approach has gained wide traction in academic and industrial settings as a method for developing pathogen-targeted narrow spectrum antibacterial agents and platforms for pathogen diagnostics, although it should be noted that pre-clinical toxicity and resistance development have hindered clinical study of sideromycins.184, 191 Cefiderocol is the lone example of a siderophore cephalosporin that has shown promise through phase 2 clinical trials and is being pursued as a treatment for MDR Gram-negative pathogens such as Pseudomonas aeruginosa.195
Figure 10.

Biosynthesis and activation of hybrid antibiotics to improve potency, engage multiple biological targets, and limit resistance development. (a) Albomycin BGC from Streptomyces sp. ATCC 700974 encodes an amide synthetase AbmC that presumably ligates the siderophore and Ser-tRNA synthetase inhibitor components to form the hybrid sideromycin antibiotic. (b) Thiomarinol BGC from Pseudoalteromonas spp. SANK73390 encodes for a CoA-ligase TmlU and acyl transferase HolE that ligates the mupirocin and holomycin components to form the hybrid thiomarinol antibiotic. Genes are color coded to match biosynthetic products.
A more recent example of biosynthetic hybridity is the antibiotic thiomarinol C produced by Pseudomonas spp. SANK73390.196, 197 Thiomarinol C is composed of pseudomonic acid C, a component of the FDA-approved antibiotic mupirocin, joined to the dithiolopyrrolone antibiotic holomycin (Figure 10b).198 Pseudomonic acid C is an Ile-tRNA synthetase inhibitor while holomycin has been shown to influence Zn-dependent metabolic pathways and inhibit some Zn-dependent metalloenzymes.199, 200 Thiomarinol C is at least equally as potent an inhibitor of Ile-tRNA synthetase as the parent pseudomonic acid C and benefits from improved cell permeation, improved spectrum of activity, and increased stability.201 It is unclear if holomycin is cleaved from mupirocin in the cytoplasm, but this does not seem to be required since the intact thiomarinol scaffold remains inhibitory towards the target Ile-tRNA synthetase. Furthermore, the holomycin moiety might potentiate the inhibition of protein synthesis through Ile-tRNA synthetase inhibition via disruption of RNA replication and other Zn-dependent metabolic pathways.199, 200 Thiomarinol biosynthesis is analogous to albomycin biosynthesis in that each antibiotic component is produced separately by genes contained in the same biosynthetic “super cluster” with a final ligation step of the two antibiotic components.196, 202, 203 The free carboxylate of pseudomonic acid C is converted to the corresponding coenzyme A thioester by the ATP-dependent ligase TmlU. Acyltransferase HolE catalyzes nucleophilic attack of the thioester carbonyl by the free amino group of holomycin to give free conenzyme A and the amide linked holomycin-pseudomonic acid C conjugate, thiomarinol C, following nucleophilic acyl substitution. The reconstitution of TmlU/HolE could enable the chemoenzymatic production of new thiomarinol inspired hybrid structures to complement existing methods based on chemical synthesis and mutasynthesis.201, 203–205 Hybrid antibiotics offer a potential advantage if one component faces modification by an inactivating enzymes since the second antibiotic component might remain functionally active. Mining microbial BGCs for hybrid antibiotics is needed to complement current synthetic strategies aimed at synthesizing multifunctional hybrid antibiotics that overcome established resistance mechanisms.181–183 advancements in synthetic biology206, 207, protein engineering208, chemoenzymatic synthesis209, and heterologous natural product expression210 are needed to advance the study and production of new hybrid antibiotics in engineered microbes.
Biosynthetic promiscuity.
According to the screening hypothesis, promiscuity in natural product biosynthesis provides an evolutionary advantage.244 That is, organisms that produce a larger number of antibiotics, at the potential cost of less potency, have a greater chance of making beneficial compounds for a more diverse set of evolutionary scenarios. Biosynthetic promiscuity can be obtained in several ways including enzyme substrate plasticity212, post-biosynthetic tailoring reactions213, release of shunt biosynthetic products214–216, and even non-enzymatic post-biosynthetic scaffold rearrangements217–220. Macrolides represent a classic example in biosynthetic diversity where control of macrocycle ring size, degree of framework oxidation, and post-polyketide synthase glycodiversificiation results in production of a structurally diverse pool of macrolides (Figure 11a).78, 216 The pikromycin polyketide synthase (PKS) assembly line in Streptomyces venezuelae provides at least six structurally distinct macrolide antibiotics. The PKS is composed of 6 modules (PikAI-IV) and a stand-alone type II thioesterase (PikAV) that acts in trans to release and cyclize the penultimate thioester precursor to the macrolide cores. Remarkably, it appears as though PikAV can cleave and cyclize thioesters from both module 5 and module 6 resulting in the formation of 12- and 14- membered macro lactones, respectively, that serve as substrates for a downstream P450 oxidase PikC and a glycosyltransferase DesVII that generate the final cocktail of pikromycin protein synthesis inhibitors with a high degree of substrate plasticity. In the laboratory, the substrate plasticity of the macrolide post-PKS modification enzymes have proved useful in the chemoenzymatic diversification of macrolides through late stage enzymatic oxidations and glycorandomization.213, 221, 222 While directed evolution approaches for tailoring enzyme optimization typically turn up highly specific enzymes, natural evolution seems to favor tailoring enzymes with a high degree of substrate plasticity.223 Presumably, producing a pool of macrolides increases the odds of overcoming resistance by the armament of environmental macrolide inactivating enzymes by supplying decoy substrates, non-substrates, and possibly inhibitors.
Figure 11.
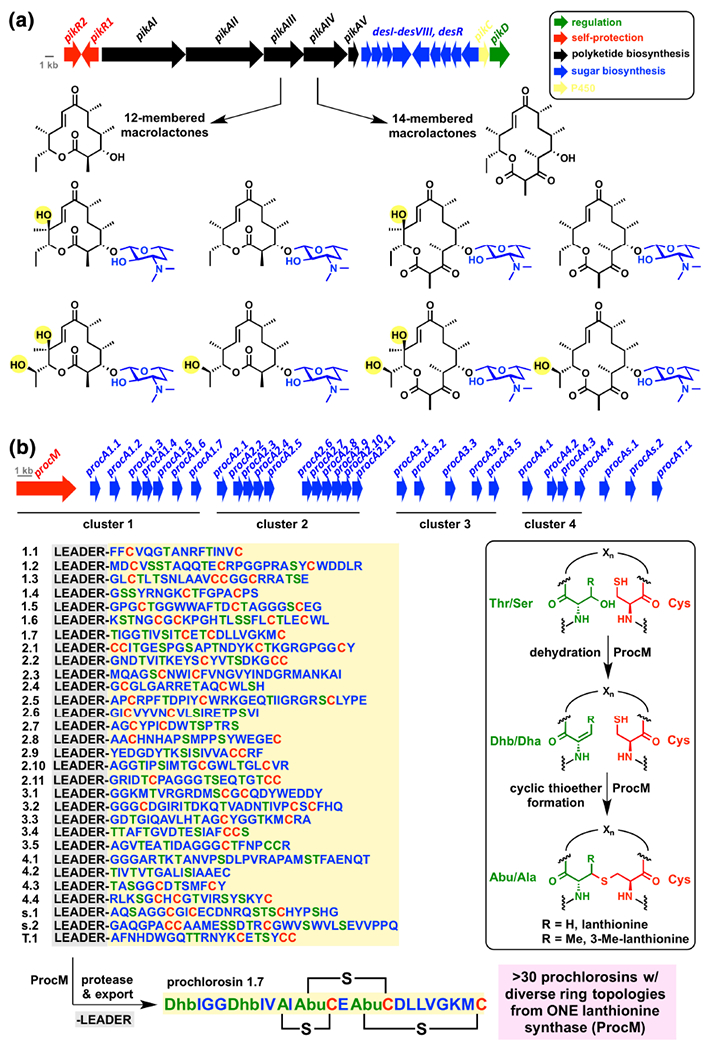
Biosynthetic promiscuity as a source of antibiotic cocktails. (a) Pikromycin biosynthesis in Streptomyces venezuelae produces a pool of 12- and 14-membered macrolides with varying degrees of side chain oxidation. Genes are color coded as defined in the legend where the color of biosynthetic genes match the structural fragment derived from catalysis by enzyme gene products. (b) Prochlorosin biosynthesis in Prochlorococcus MIT9313 generates a library of structurally diverse lantipeptides with unique ring topologies from genetically encoded precursor peptides and a single lanthionine synthetase ProcM with low substrate selectivity. The procM gene is shown in red while all genes encoding product precursor peptides are shown in blue.
More recently, the explosion of research in the area of ribosomally synthesized post-translationally modified peptide (RiPP) family of antibiotics has revealed some of the biosynthetic secrets that lead to immense structural diversification of small peptides derived from proteinogenic amino acid building blocks.224 Surprisingly, the enzymes that perform the post-translational modifications (PTMs) following ribosomal generation of RiPP precursor peptides display a large degree of substrate plasticity that is tamed by inclusion of a traceless leader peptide that guides substrate peptides to enzymes responsible for installing PTMs. One PTM common to the lanthipeptide subclass of RiPPs is the conversion of a Ser or Thr to the corresponding dehydroalanine (Dha) or dehydrobutyrine (Dhb) residue, respectively, via O-glutamylation or O-phosphoiylation followed by dehydration with loss of glutamate or phosphate, respectively (Figure 11b). Nearby Cys residues can add as nucleophiles in a 1,4-conjugate addition (Michael reaction) to the newly formed Dha or Dhb residues followed by α-protonation of the resulting enolate to give a cyclic thioether known as a lanthionine or 3-methylanthionine residue, respectively. Bifunctional lanthipeptide synthetases (LanM) that catalyze tandem dehydrative conjugate additions to form lanthionine residues in lantipeptide antibiotics show great substrate plasticity and result in the formation of structurally diverse cocktails of lanthipeptides with varying degrees of macrocyclizations and dehydrations.225 In nisin biosynthesis, NisC catalyzes the formation of all five cyclic ethers in the lipid-II sequestering lantipeptide scaffold 226 The prochlorosin lantipeptides produced by the cyanobacterium Prochlorococcus MIT9313 represent a more extreme example of combinatorial diversity-generating biosynthesis (Figure 11b) 227 The genome of Prochlorococcus MIT9313 encodes at least 30 ProcA lantipeptide pre-peptides and only one bifunctional lantipeptide synthase, ProcM.228 The ProcA peptides (1.1-1.7; 2.1-2.11; 3.1-3.5; 4.1-4.4, s.l, s.2, T.l) share >60% sequence similarity in the leader peptide and <30% sequence similarity in the antibiotic pre-peptide ranging form 12-32 amino acids.229 Nearly all possible Thr/Ser dehydrations and Cys cyclic thioether formations of the peptide cocktail are formed enzymatically by ProcM resulting in a staggering array of ring topologies for the kinetically controlled pool of >30 lantipeptides excreted by Prochlorococcus MIT9313.230, 231 This level of biosynthetic flexibility has inspired several approaches to building DNA-encoded libraries of RiPP antibiotics using codon randomization and even phage display in yeast.232–234 While cell-based approaches to RiPP diversification will presumably eliminate production of compounds that are toxic to the host, the ease of producing large compound libraries via these systems should enable sufficient chemical diversity to provide hits in a wide range of therapeutic areas including “undruggable” protein-protein interactions.235 Advancements in the in vitro reconstitution of RiPP biosynthetic enzymes have also enabled chemoenzymatic approaches to certain members of the RiPP antibiotic family including pyridine-containing thiopeptides and lantipeptides.209 RiPP antibiotics show a wide range of biological activity and many show promise as antimicrobial and anticancer agents.236 Perhaps pursuing defined cocktails of RiPPs for desired therapeutic applications will provide a higher hit rate for lead optimization and outpace resistance development. The topic of “diversitygenerating biosynthesis” was recently taken up by Schmidt237 and the argument is made that enzyme “promiscuity” seems to be the rule rather than the exception for certain pharmacophores, including lanthionine units, where scaffold diversification provides a competitive advantage against resistance development consistent with the natural product “screening hypothesis” first proposed by Firn and Jones.211
FINAL THOUGHTS
Turning to nature’s therapeutic logic and resistance management plans is a smart way to supplement and inspire new directions to overcome antibiotic resistance in the clinic.29, 238 This philosophy has proved fruitful in providing the first wave of antibiotic scaffolds that has sustained 80 years of clinical success. Today new antibiotic drugs, including multiple iterations of beta-lactam antibiotic and beta-lactamase inhibitor combinations, derived from natural products continue to provide essential cures for bacterial infections. The slow pace of new antibiotic classes entering the clinic and increasing rates of resistance against new and established antibiotic drugs demands constant nurturing of the antibiotic discovery pipeline.239 Scaffold iterations are no longer keeping pace with antibiotic resistance.240 There are many lessons still to learn from the balance of antibiotic resistance and susceptibility in natural environments that can inspire new therapeutic strategies and methods for managing antibiotic resistance. In natural environments, resistance is at equilibrium and microbial populations welcome all phenotypes including antibiotic producers, sacrificial lambs, cheaters, and police.241, 242 While resistance has reached equilibrium in natural environments, resistance in hospitals has tipped in favor of MDR pathogens. While humans did not create antibiotic resistance, we have amplified resistance in certain settings by our practice of antibiotic consumption that is now trickling into the general population.36, 37
Chemical synthesis243, traditional medicinal chemistry236 and advances in drug delivery244 will always serve as a pillar of antibiotic development. It is time again to turn to natural products in the genomics and metabolomics era to provide the next wave of molecules that will sustain decades of infectious disease treatment. Synergistic combination therapies will play an important role in the future of antibiotic therapeutics to both overcome established resistance and delay the emergence of new resistance mechanisms. Mining microbial genomes for “super” and “hybrid” BGCs encoding the production of synergistic antibiotics promises to deliver new lead antibiotic combinations and fuel the development of non-traditional therapeutic strategies. As some molecules are synergistic and others antagonistic, the timing of biosynthetic production is key with transcriptional regulation controlling the molecular pool in response to external signals (e.g. quorum sensing). An improved understanding of this antibiotic regulation in mixed microbial populations will help guide the development and use of antibiotics in the era of harnessing the human microbiome.245, 246 New drug delivery strategies and modification of existing antibiotic scaffolds to favor intracellular accumulation in target bacteria merit continued investigation inspired by natural compounds that efficiently cross the bacterial cell envelope.49 Advancements in measuring cellular accumulation of antibiotics and exploration of nontraditional antibiotic targets including virulence factors are welcomed additions to antibiotic discovery programs. New smart screens including chemical genetic assays for synthetically lethal gene targets and resistance-guided antibiotic discovery have promise to reveal new antibacterial natural products.159, 247 Recent efforts to improve microbial cultivation248, activate silent antibiotic BGCs245, 246, and expand metagenomic capture of BGCs expressed in heterologous hosts have turned up new antibacterial leads.249
The connection between environmental and clinical resistance now seems undeniable. It remains challenging to distinguish between producer self-protection and authentic antibiotic resistance. It is even more difficult to determine if emerging resistance mechanisms from natural environments will have clinical relevance. Based on the short history of antibiotic use the appearance of environmental resistance in human pathogens is a safe bet if the associated natural antibiotic is widely deployed. Prospecting for emerging resistance and anticipating clinically relevant ARGs using computational methods and functional screens is important to develop resistance management plans for established antibiotics and new lead compounds. Proactive development of new diagnostic agents for specific resistance mechanisms and MDR pathogens along with development of adjuvants for overcoming potential resistance enzymes should be implemented early in the preclinical development of antibiotics.
In the long run, antibiotic stewardship will probably impact the dissemination of resistance more than any other factor.160 Our use of antibiotics in medical, agricultural, and related venues should be reevaluated to assess the longterm effects of resistance development on human health, national security, and the economy. For example, the current standard of care for pre-mature infants is administration of antibiotics in neonatal intensive care units.250 The lack of statistical correlation to increased survival rate combined with concerns about disrupting the developing infant microbiome with an oversized pool of ARGs suggests that long-term consequences of antibiotic use in infants should be investigated.251 This could be a scenario where prophylactic antibiotic use does more harm than good. A second example worth mentioning is the recent approval by the US EPA to allow the spraying of orange orchards in the state of Florida with the antibiotics streptomycin and oxytetracycline to control citrus greening, a bacterial disease common to Florida citrus trees.252 This practice could result in the release of 440,000 kilograms of the antibiotics into the soil – an unprecedented scale. Kishony and coworkers have shown that tetracycline and associated degradation products can dramatically influence microbial population dynamics.35, 253, 254 It is unclear how large-scale deployment of oxytetracycline and streptomycin will influence the soil and plant microbiomes in the exposure zone although it is reasonable to hypothesize that tetracycline and aminoglycoside ARGs should increase under selective pressure. This enriched pools of ARGs could compromise the efficacy of newly approved life-saving aminoglycoside and tetracycline drugs including plazomicin and eravacycline, respectively (Figure 1).5, 6, 9 The short-sided economic benefits of agricultural antibiotic use are clear, but the potential for antibiotic resistance dissemination might have unintended economic costs related to infectious disease control. Additionally, it is unclear if extended large-scale antibiotic use on crops will provide sustainable management of crop disease given that target pathogens will eventually become resistant. This will be a unique opportunity to explore the crossroads of antibiotic resistance and biosynthesis in soil on relevant geographical and time scales.
RESEARCH HIGHLIGHTS.
Antibiotic resistance is balanced by antibiotic biosynthesis in natural environments.
Resistance in human pathogens originates from self-protection mechanisms in antibiotic producers.
Antibiotic inactivating enzymes evolve to modify conserved scaffold motifs required for target binding.
Hybrid antibiotics, adjuvants, and synergistic combinations are natural platforms for overcoming resistance.
Proactive study of resistance can be leveraged to guide next-generation antibiotic discovery.
ACKNOWLEGEMENTS
Research in Prof. Timothy Wencewicz’s laboratory related to antibiotics and resistance is supported by the National Institute of Allergy and Infectious Diseases, National Institutes of Health (grant R01 123394), the National Science Foundation through a CAREER Award (grant 1654611), the Children’s Discovery Institute at St. Louis Children’s Hospital (grant MI-PD-II-2018-748), the Research Corporation for Science Advancement through a Cottrell Scholar award, and the Sloan Foundation through a Sloan Fellowship award.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Antibiotic resistance threats in the United States, 2013; Center for Disease Control and Prevention, U.S. Department of Health and Human Services: Washington DC, Published Online, 2013. [Google Scholar]
- [2].O’Neill J Tackling drug-resistant infections globally: Final report and recommendations, 2016; Review on Antimicrobial Resistance, Wellcome Trust: London, Published Online, May, 2016. [Google Scholar]
- [3].Hamad B (2010) The antibiotics market. Nature Rev. Drug Discov 9, 675–676. [DOI] [PubMed] [Google Scholar]
- [4].Simpkin VL, Renwick MJ, Kelly R, & Mossialos E (2017) Incentivising innovation in antibiotic drug discovery and development: progress, challenges and next steps. J. Antibiot 70,1087–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Mullard A (2019) 2018 FDA drug approvals. Nature Rev. Drug Discov 18, 85–89. [DOI] [PubMed] [Google Scholar]
- [6].Zhanel GG, Cheung D, Adam H, Zelenitsky S, Golden A, Schweizer F, Gorityala B, Lagace-Wiens PR, Walkty A, Gin AS, Hoban DJ, & Karlowsky JA (2016) Review of eravacycline, a novel fluorocycline antibacterial agen. Drugs 76, 567–588. [DOI] [PubMed] [Google Scholar]
- [7].Grossman TH, Starosta AL, Fyfe C, O’Brien W, Rothstein DM, Mikolajka A, Wilson DN, & Sutcliffe JA (2012) Target- and resistance-based mechanistic studies with TP-434, a novel fluorocycline antibiotic. Antimicrob. Agents Chemother 56, 2559–2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Cox G, Ejim L, Stogios PJ, Koteva K, Bordeleau E, Evdokimova E, Sieron AO, Savchenko A, Serio AW, Krause KM, & Wright GD (2018) Plazomicin retains antibiotic activity against most aminoglycoside modifying enzymes. ACS Infect. Dis 4, 980–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Wagenlehner FME, Cloutier DJ, Komirenko AS, Cebrik DS, Krause KM, Keepers TR, Connolly LE, Miller LG, Friedland I, & Dwyer JP (2019) Once-daily plazomicin for complicated urinary tract infections. N. Engl. J. Med 380, 729–740. [DOI] [PubMed] [Google Scholar]
- [10].Armstrong ES, & Miller GH (2010) Combating evolution with intelligent design: the neoglycoside ACHN-490. Curr. Opin. Microbiol 13, 565–573. [DOI] [PubMed] [Google Scholar]
- [11].Garneau-Tsodikova S, & Labby KJ (2016) Mechanisms of resistance to aminoglycoside antibiotics: Overview and perspectives. MedChemComm 7, 11–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Sun C, & Xiao X-Y (2018) Fully synthetic tetracyclines: Increasing chemical diversity to combat multidrug-resistant bacterial infections In Antibacterials: Volume II (Fisher JF, Mobasheiy S, and Miller MJ, Eds.), pp 55–95, Springer International Publishing, Cham. [Google Scholar]
- [13].Jenner L, Starosta AL, Terry DS, Mikolajka A, Filonava L, Yusupov M, Blanchard SC, Wilson DN, & Yusupova G (2013) Structural basis for potent inhibitory activity of the antibiotic tigecycline during protein synthesis. Proc. Natl. Acad. Sci. U. S. A 110, 3812–3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Markley JL, & Wencewicz TA (2018) Tetracycline-inactivating enzymes. Front. Microbiol 9,1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Linkevicius M, Sandegren L, & Andersson DI (2016) Potential of tetracycline resistance proteins to evolve tigecycline resistance. Antimicrob. Agents Chemother 60, 789–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Volkers G, Damas JM, Palm GJ, Panjikar S, Soares CM, & Hinrichs W (2013) Putative dioxygen-binding sites and recognition of tigecycline and minocycline in the tetracycline-degrading monooxygenase TetX. Acta Crystallogr D Biol Crystallogr. 69,1758–1767. [DOI] [PubMed] [Google Scholar]
- [17].Moore IF, Hughes DW, & Wright GD (2005) Tigecycline is modified by the flavin-dependent monooxygenase TetX. Biochemistry 44,11829–11835. [DOI] [PubMed] [Google Scholar]
- [18].He T, Wang R, Liu D, Walsh TR, Zhang R, Lv Y, Ke Y, Ji Q, Wei R, Liu Z, Shen Y, Wang G, Sun L, Lei L, Lv Z, Li Y, Pang M, Wang L, Sun Q, Fu Y, Song H, Hao Y, Shen Z, Wang S, Chen G, Wu C, Shen J, and Wang Y (2019) Emergence of plasmid-mediated high-level tigecycline resistance genes in animals and humans. Nat. Microbiol Doi: 10.1038/s41564-019-0445-2. [DOI] [PubMed] [Google Scholar]
- [19].Yang W, Moore IF, Koteva KP, Bareich DC, Hughes DW, & Wright GD (2004) TetX is a flavin-dependent monooxygenase conferring resistance to tetracycline antibiotics. J. Biol. Chem 279, 52346–52352. [DOI] [PubMed] [Google Scholar]
- [20].Abraham EP, & Chain E (1940) An enzyme from bacteria able to destroy penicillin. Nature. 146, 837–837. [PubMed] [Google Scholar]
- [21].Bush K (2018) Past and present perspectives on β-lactamases. Antimicrob. Agents Chemother 62, e01076–01018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Forsberg KJ, Patel S, Wencewicz TA, & Dantas G (2015) The tetracycline destructases: A novel family of tetracycline-inactivating enzymes. Chem. Biol 22, 888–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Leski TA, Bangura U, Jimmy DH, Ansumana R, Lizewski SE, Stenger DA, Taitt CR, & Vora GJ (2013) Multidrug-resistant tet(X)-containing hospital isolates in Sierra Leone. Int. J. Antimicrob. Agents 42, 83–86. [DOI] [PubMed] [Google Scholar]
- [24].Park J, Gasparrini AJ, Reck MR, Symister CT, Elliott JL, Vogel JP, Wencewicz TA, Dantas G, & Tolia NH (2017) Plasticity, dynamics, and inhibition of emerging tetracycline resistance enzymes. Nat. Chem. Biol 13, 730–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Markley JL, Fang L, Gasparrini AJ, Symister CT, Kumar H, Tolia NH, Dantas G, & Wencewicz TA (2019) Semisynthetic analogues of anhydrotetracycline as inhibitors of tetracycline destructase enzymes. ACS Infect. Dis 5, 618–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Sun C, Wang Q, Brubaker JD, Wright PM, Lerner CD, Noson K, Charest M, Siegel DR, Wang YM, & Myers AG (2008) A robust platform for the synthesis of new tetracycline antibiotics. J. Am. Chem. Soc 130, 17913–17927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Scholar EM, Scholar EM, and Pratt WB (2000) The Antimicrobial Drugs, Oxford University Press. [Google Scholar]
- [28].Crofts TS, Gasparrini AJ, & Dantas G (2017) Next-generation approaches to understand and combat the antibiotic resistome. Nat. Rev. Microbiol 15, 422–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wencewicz TA (2016) New antibiotics from nature’s chemical inventory. Bioorg. Med. Chem 24, 6227–6252. [DOI] [PubMed] [Google Scholar]
- [30].Davies J, & Davies D (2010) Origins and evolution of antibiotic resistance. Microbiol. Mol. Biol. Rev 74, 417–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Piwowarski JM, & Shaw PD (1979) Streptomycin resistance in a streptomycin-producing microorganism. Antimicrob. Agents Chemother 16, 176–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wang P, Bashiri G, Gao X, Sawaya MR, & Tang Y (2013) Uncovering the enzymes that catalyze the final steps in oxytetracycline biosynthesis. J. Am. Chem. Soc 135, 7138–7141. [DOI] [PubMed] [Google Scholar]
- [33].Pickens LB, & Tang Y (2010) Oxytetracycline biosynthesis. J. Biol. Chem 285, 27509–27515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].D’Costa VM, King CE, Kalan L, Morar M, Sung WW, Schwarz C, Froese D, Zazula G, Calmels F, Debruyne R, Golding GB, Poinar HN, & Wright GD (2011) Antibiotic resistance is ancient. Nature 477, 457–461. [DOI] [PubMed] [Google Scholar]
- [35].Chait R, Vetsigian K, & Kishony R (2011) What counters antibiotic resistance in nature? Nat. Chem. Biol 8, 2–5. [DOI] [PubMed] [Google Scholar]
- [36].Palumbi SR (2001) Humans as the world’s greatest evolutionary force. Science 293, 1786–1790. [DOI] [PubMed] [Google Scholar]
- [37].Klein EY, Van Boeckel TP, Martinez EM, Pant S, Gandra S, Levin SA, Goossens H, & Laxminarayan R (2018) Global increase and geographic convergence in antibiotic consumption between 2000 and 2015. Proc. Natl. Acad. Sci. U. S. A 115, E3463–e3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Leclercq R (2002) Mechanisms of resistance to macrolides and lincosamides: nature of the resistance elements and their clinical implications. Clin. Infect. Dis 34, 482–492. [DOI] [PubMed] [Google Scholar]
- [39].Wright GD (2007) The antibiotic resistome: the nexus of chemical and genetic diversity. Nat. Rev. Microbiol 5, 175–186. [DOI] [PubMed] [Google Scholar]
- [40].Blair JM, Webber MA, Baylay AJ, Ogbolu DO, & Piddock LJ (2015) Molecular mechanisms of antibiotic resistance. Nat. Rev. Microbiol 13, 42–51. [DOI] [PubMed] [Google Scholar]
- [41].Almabruk KH, Dinh LK, & Philmus B (2018) Self-resistance of natural product producers: Past, present, and future focusing on self-resistant protein variants. ACS Chem. Biol 13, 1426–1437. [DOI] [PubMed] [Google Scholar]
- [42].Cundliffe E (1992) Self-protection mechanisms in antibiotic producers. Ciba Foundation Symposium 171, 199–208; discussion 208–114. [DOI] [PubMed] [Google Scholar]
- [43].Hopwood DA (2007) How do antibiotic-producing bacteria ensure their self resistance before antibiotic biosynthesis incapacitates them? Mol. Microbiol 63, 937–940. [DOI] [PubMed] [Google Scholar]
- [44].Cundliffe E (1989) How antibiotic-producing organisms avoid suicide. Ann. Rev. Microbiol 43, 207–233. [DOI] [PubMed] [Google Scholar]
- [45].Duerkop BA, Varga J, Chandler JR, Peterson SB, Herman JP, Churchill ME, Parsek MR, Nierman WC, & Greenberg EP (2009) Quorumsensing control of antibiotic synthesis in Burkholderia thailandensis. J. Bacteriol 191, 3909–3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Mehta HH, Prater AG, & Shamoo Y (2018) Using experimental evolution to identify druggable targets that could inhibit the evolution of antimicrobial resistance. J. Antibiot 71, 279–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Ling LL, Schneider T, Peoples AJ, Spoering AL, Engels I, Conlon BP, Mueller A, Schaberle TF, Hughes DE, Epstein S, Jones M, Lazarides L, Steadman VA, Cohen DR, Felix CR, Fetterman KA, Millett WP, Nitti AG, Zullo AM, Chen C, & Lewis K (2015) A new antibiotic kills pathogens without detectable resistance. Nature 517, 455–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Guo C, Mandalapu D, Ji X, Gao J, & Zhang Q (2018) Chemistry and biology of teixobactin. Chem. Eur. J 24, 5406–5422. [DOI] [PubMed] [Google Scholar]
- [49].Acosta-Gutierrez S, Ferrara L, Pathania M, Masi M, Wang J, Bodrenko I, Zahn M, Winterhalter M, Stavenger RA, Pages JM, Naismith JH, van den Berg B, Page MGP, & Ceccarelli M (2018) Getting drugs into Gram-negative bacteria: Rational rules for permeation through general porins. ACS Infect. Dis 4, 1487–1498. [DOI] [PubMed] [Google Scholar]
- [50].Quiros LM, Aguirrezabalaga I, Olano C, Mendez C, & Salas JA (1998) Two glycosyltransferases and a glycosidase are involved in oleandomycin modification during its biosynthesis by Streptomyces antibioticus. Mol. Microbiol 28, 1177–1185. [DOI] [PubMed] [Google Scholar]
- [51].Aguirrezabalaga I, Olano C, Allende N, Rodriguez L, Brana AF, Mendez C, & Salas JA (2000) Identification and expression of genes involved in biosynthesis of L-oleandrose and its intermediate L-olivose in the oleandomycin producer Streptomyces antibioticus. Antimicrob. Agents Chemother 44, 1266–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Bolam DN, Roberts S, Proctor MR, Turkenburg JP, Dodson EJ, Martinez-Fleites C, Yang M, Davis BG, Davies GJ, & Gilbert HJ (2007) The crystal structure of two macrolide glycosyltransferases provides a blueprint for host cell antibiotic immunity. Proc. Natl. Acad. Sci. U. S. A 104, 5336–5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Golkar T, Zielinski M, and Berghuis AM (2018) Look and outlook on enzyme-mediated macrolide resistance. Front. Microbiol 9,1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Wright GD (2005) Bacterial resistance to antibiotics: enzymatic degradation and modification. Adv. Drug Deliv. Rev 57,1451–1470. [DOI] [PubMed] [Google Scholar]
- [55].De Pascale G, & Wright GD (2010) Antibiotic resistance by enzyme inactivation: from mechanisms to solutions. Chembiochem. 11,1325–1334. [DOI] [PubMed] [Google Scholar]
- [56].Patrick GJ, Fang L, Schaefer J, Singh S, Bowman GR, & Wencewicz TA (2018) Mechanistic basis for ATP-dependent inhibition of glutamine synthetase by tabtoxinine-beta-lactam. Biochemistry 57,117–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Wencewicz TA, & Walsh CT (2012) Pseudomonas syringae self-protection from tabtoxinine-beta-lactam by ligase TblF and acetylase Ttr. Biochemistry 51, 7712–7725. [DOI] [PubMed] [Google Scholar]
- [58].Hart KM, Reck M, Bowman GR, & Wencewicz TA (2016) Tabtoxrnrne-β-lactam is a “stealth” β-lactam antibiotic that evades β-lactamase-medrated antibiotic resistance. MedChemComm 7,118–127. [Google Scholar]
- [59].Blodgett JA, Zhang JK, Yu X, & Metcalf WW (2016) Conserved biosynthetic pathways for phosalacine, bialaphos and newly discovered phosphonic acid natural products. J. Antibiot 69,15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Bougioukou DJ, Ting CP, Peck SC, Mukherjee S, & van der Donk WA (2019) Use of the dehydrophos biosynthetic enzymes to prepare antimicrobial analogs of alaphosphin. Org. Biomol. Chem 17, 822–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].VanDrisse CM, Hentchel KL, & Escalante-Semerena JC (2016) Phosphinothricin acetyltransferases identified using in vivo, in vitro, and bioinformatic analyses. Appl. Environ. Microbiol 82, 7041–7051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Carbonari CA, Latorre DO, Gomes GL, Velini ED, Owens DK, Pan Z, & Dayan FE (2016) Resistance to glufosinate is proportional to phosphinothricin acetyltransferase expression and activity in LibertyLink® and WideStrike® cotton. Planta 243, 925–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Hamed RB, Gomez-Castellanos JR, Henry L, Ducho C, McDonough MA, & Schofield CJ (2013) The enzymes of beta-lactam biosynthesis. Nat. Prod. Rep 30, 21–107. [DOI] [PubMed] [Google Scholar]
- [64].Palzkill T (2018) Structural and mechanistic basis for extended-spectrum drug-resistance mutations in altering the specificity of TEM, CTX-M, and KPC beta-lactamases. Front. Mol. Biosci 5,16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Bush K (2013) Proliferation and significance of clinically relevant beta-lactamases. Ann. N. Y. Acad. Sci 1277, 84–90. [DOI] [PubMed] [Google Scholar]
- [66].Meroueh SO, Minasov G, Lee W, Shoichet BK, & Mobashery S (2003) Structural aspects for evolution of beta-lactamases from penicillin-binding proteins. J. Am. Chem. Soc 125, 9612–9618. [DOI] [PubMed] [Google Scholar]
- [67].Lu WP, Kincaid E, Sun Y, & Bauer MD (2001) Kinetics of beta-lactam interactions with penicillin-susceptible and -resistant penicillin-binding protein 2x proteins from Streptococcus pneumoniae. Involvement of acylation and deacylation in beta-lactam resistance. J. Biol. Chem 276, 31494–31501. [DOI] [PubMed] [Google Scholar]
- [68].Salverda ML, De Visser JA, & Barlow M (2010) Natural evolution of TEM-1 beta-lactamase: experimental reconstruction and clinical relevance. FEMS Microbiol. Rev 34, 1015–1036. [DOI] [PubMed] [Google Scholar]
- [69].Nakamura A, Nakazawa K, Miyakozawa I, Mizukoshi S, Tsurubuchi K, Nakagawa M, O’Hara K, & Sawai T (2000) Macrolide esterase-producing Escherichia coli clinically isolated in Japan. J. Antibiot 53, 516–524. [DOI] [PubMed] [Google Scholar]
- [70].Nolin TD, Appiah K, Kendrick SA, Le P, McMonagle E, & Himmelfarb J (2006) Hemodialysis acutely improves hepatic CYP3A4 metabolic activity. J. Am. Soc. Nephrol 17, 2363–2367. [DOI] [PubMed] [Google Scholar]
- [71].Pawlowski AC, Stogios PJ, Koteva K, Skarina T, Evdokimova E, Savchenko A, & Wright GD (2018) The evolution of substrate discrimination in macrolide antibiotic resistance enzymes. Nat. Comm 9, 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Bhullar K, Waglechner N, Pawlowski A, Koteva K, Banks ED, Johnston MD, Barton HA, & Wright GD (2012) Antibiotic resistance is prevalent in an isolated cave microbiome. PLoS One 7, e34953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].O’Hara K, Kanda T, & Kono M (1988) Structure of a phosphorylated derivative of oleandomycin, obtained by reaction of oleandomycin with an extract of an erythromycin-resistant strain of Escherichia coli. J. Antibiot 41, 823–827. [DOI] [PubMed] [Google Scholar]
- [74].Hon WC, McKay GA, Thompson PR, Sweet RM, Yang DS, Wright GD, & Berghuis AM (1997) Structure of an enzyme required for aminoglycoside antibiotic resistance reveals homology to eukaryotic protein kinases. Cell 89, 887–895. [DOI] [PubMed] [Google Scholar]
- [75].Skinner RH, & Cundliffe E (1980) Resistance to the antibiotics viomycin and capreomycin in the Streptomyces species which produce them. J. Gen. Microbiol 120, 95–104. [DOI] [PubMed] [Google Scholar]
- [76].Magnet S, & Blanchard JS (2005) Molecular insights into aminoglycoside action and resistance. Chem. Rev 105, 477–498. [DOI] [PubMed] [Google Scholar]
- [77].Kramer JR, & Matsumura I (2013) Directed evolution of aminoglycoside phosphotransferase (3’) type IIIa variants that inactivate amikacin but impose significant fitness costs. PLoS One 8, e76687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Xue Y, & Sherman DH (2001) Biosynthesis and combinatorial biosynthesis of pikromycin-related macrolides in Streptomyces venezuelae. Metab. Eng 3, 15–26. [DOI] [PubMed] [Google Scholar]
- [79].Berntsson RPA, Smits SHJ, Schmitt L, Slotboom D-J, & Poolman B (2010) A structural classification of substrate-binding proteins. FEBS Lett. 584, 2606–2617. [DOI] [PubMed] [Google Scholar]
- [80].Chang CY, Yan X, Crnovcic I, Annaval T, Chang C, Nocek B, Rudolf JD, Yang D, Hindra, Babnigg G, Joachimiak A, Phillips GN Jr., & Shen B (2018) Resistance to enediyne antitumor antibiotics by sequestration. Cell Chem. Biol 25, 1075–1085. e1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Biggins JB, Onwueme KC, & Thorson JS (2003) Resistance to enediyne antitumor antibiotics by CalC self-sacrifice. Science 301, 1537–1541. [DOI] [PubMed] [Google Scholar]
- [82].Elshahawi SI, Ramelot TA, Seetharaman J, Chen J, Singh S, Yang Y, Pederson K, Kharel MK, Xiao R, Lew S, Yennamalli RM, Miller MD, Wang F, Tong L, Montelione GT, Kennedy MA, Bingman CA, Zhu H, Phillips GN Jr., & Thorson JS (2014) Structure-guided functional characterization of enediyne self-sacrifice resistance proteins, CalU16 and CalU19. ACS Chem. Biol 9, 2347–2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Rostock L, Driller R, Gratz S, Kerwat D, von Eckardstein L, Petras D, Kunert M, Alings C, Schmitt FJ, Friedrich T, Wahl MC, Loll B, Mainz A, & Sussmuth RD (2018) Molecular insights into antibiotic resistance - how a binding protein traps albicidin. Nat. Comm 9, 3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Schaffer JE, Reck MR, Prasad NK, & Wencewicz TA (2017) beta-Lactone formation during product release from a nonribosomal peptide synthetase. Nat. Chem. Biol 13, 737–744. [DOI] [PubMed] [Google Scholar]
- [85].Li YX, Zhong Z, Hou P, Zhang WP, and Qian PY (2018) Resistance to nonribosomal peptide antibiotics mediated by D-stereospecific peptidases. Nat. Chem. Biol 14, 381–387. [DOI] [PubMed] [Google Scholar]
- [86].Brotherton CA, & Balskus EP (2013) A prodrug resistance mechanism is involved in colibactin biosynthesis and cytotoxicity. J. Am. Chem. Soc 135, 3359–3362. [DOI] [PubMed] [Google Scholar]
- [87].Kevany BM, Rasko DA, & Thomas MG (2009) Characterization of the complete zwittermicin A biosynthesis gene cluster from Bacillus cereus. Appl. Environ. Microbiol 75, 1144–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Reimer D, Pos KM, Thines M, Grun P, & Bode HB (2011) A natural prodrug activation mechanism in nonribosomal peptide synthesis. Nat. Chem. Biol 7, 888–890. [DOI] [PubMed] [Google Scholar]
- [89].Tripathi P, Shine EE, Healy AR, Kim CS, Herzon SB, Bruner SD, & Crawford JM (2017) ClbS is a cyclopropane hydrolase that confers colibactin resistance. J. Am. Chem. Soc 139, 17719–17722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Fillgrove KL, Pakhomova S, Schaab MR, Newcomer ME, & Armstrong RN (2007) Structure and mechanism of the genomically encoded fosfomycin resistance protein, FosX, from Listeria monocytogenes. Biochemistry 46, 8110–8120. [DOI] [PubMed] [Google Scholar]
- [91].Circello BT, Miller CG, Lee JH, van der Donk WA, & Metcalf WW (2011) The antibiotic dehydrophos is converted to a toxic pyruvate analog by peptide bond cleavage in Salmonella enterica. Antimicrob. Agents Chemother 55, 3357–3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Sugiyama M, Mochizuki H, Nimi O, & Nomi R (1981) Mechanism of protection of protein synthesis against streptomycin inhibition in a producing strain. J. Antibiot 34, 1183–1188. [DOI] [PubMed] [Google Scholar]
- [93].Kim SH, Kang PA, Han K, Lee SW, & Rhee S (2019) Crystal structure of chloramphenicol-metabolizing enzyme EstDL136 from a metagenome. PLoS One 14, e0210298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Tao W, Lee MH, Wu J, Kim NH, Kim JC, Chung E, Hwang EC, & Lee SW (2012) Inactivation of chloramphenicol and florfenicol by a novel chloramphenicol hydrolase. Appl. Environ. Microbiol 78, 6295–6301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Crofts TS, Wang B, Spivak A, Gianoulis TA, Forsberg KJ, Gibson MK, Johnsky LA, Broomall SM, Rosenzweig CN, Skowronski EW, Gibbons HS, Sommer MOA, & Dantas G (2018) Shared strategies for beta-lactam catabolism in the soil microbiome. Nat. Chem. Biol 14, 556–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Pakhomova S, Rife CL, Armstrong RN, & Newcomer ME (2004) Structure of fosfomycin resistance protein FosA from transposon Tn2921. Protein Sci. 13, 1260–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Klontz EH, Tomich AD, Gunther S, Lemkul JA, Deredge D, Silverstein Z, Shaw JF, McElheny C, Doi Y, Wintrode PL, MacKerell AD Jr., Sluis-Cremer N, & Sundberg EJ (2017) Structure and dynamics of FosA-mediated fosfomycin resistance in Klebsiella pneumoniae and Escherichia coli. Antimicrob. Agents Chemother 61, e01572–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Rigsby RE, Fillgrove KL, Beihoffer LA, & Armstrong RN (2005) Fosfomycin resistance proteins: a nexus of glutathione transferases and epoxide hydrolases in a metalloenzyme superfamily. Methods Enzymol. 401, 367–379. [DOI] [PubMed] [Google Scholar]
- [99].Izard T (2001) Structural basis for chloramphenicol tolerance in Streptomyces venezuelae by chloramphenicol phosphotransferase activity. Protein Sci. 10, 1508–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Izard T, & Ellis J (2000) The crystal structures of chloramphenicol phosphotransferase reveal a novel inactivation mechanism. EMBO J. 19, 2690–2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Pakhomova S, Bartlett SG, Augustus A, Kuzuyama T, & Newcomer ME (2008) Crystal structure of fosfomycin resistance kinase FomA from Streptomyces wedmorensis. J. Biol. Chem 283, 28518–28526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Kobayashi S, Kuzuyama T, & Seto H (2000) Characterization of the fomA and fomB gene products from Streptomyces wedmorensis, which confer fosfomycin resistance on Escherichia coli. Antimicrob. Agents Chemother 44, 647–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Stogios PJ, Cox G, Spanogiannopoulos P, Pillon MC, Waglechner N, Skarina T, Koteva K, Guarne A, Savchenko A, & Wright GD (2016) Rifampin phosphotransferase is an unusual antibiotic resistance kinase. Nat. Comm 7, 11343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Favrot L, Blanchard JS, & Vergnolle O (2016) Bacterial GCN5-related N-acetyltransferases: From resistance to regulation. Biochemistry 55, 989–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Sugantino M, & Roderick SL (2002) Crystal structure of Vat(D): an acetyltransferase that inactivates streptogramin group A antibiotics. Biochemistry 41, 2209–2216. [DOI] [PubMed] [Google Scholar]
- [106].He H, Ding Y, Bartlam M, Sun F, Le Y, Qin X, Tang H, Zhang R, Joachimiak A, Liu J, Zhao N, & Rao Z (2003) Crystal structure of tabtoxin resistance protein complexed with acetyl coenzyme A reveals the mechanism for beta-lactam acetylation. J. Mol. Biol 325, 1019–1030. [DOI] [PubMed] [Google Scholar]
- [107].Shichiri M, Hoshikawa C, Nakamori S, & Takagi H (2001) A novel acetyltransferase found in Saccharomyces cerevisiae Sigma1278b that detoxifies a proline analogue, azetidine-2-carboxylic acid. J. Biol. Chem 276, 41998–42002. [DOI] [PubMed] [Google Scholar]
- [108].Gross C, Felsheim R, & Wackett LP (2008) Genes and enzymes of azetidine-2-carboxylate metabolism: detoxification and assimilation of an antibiotic. J. Bacteriol 190, 4859–4864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Shaw WV (1983) Chloramphenicol acetyltransferase: enzymology and molecular biology. CRC Crit. Rev. Biochem 14, 1–46. [DOI] [PubMed] [Google Scholar]
- [110].Huang L, Yuan H, Liu M-F, Zhao X-X, Wang M-S, Jia R-Y, Chen S, Sun K-F, Yang Q, Wu Y, Chen X-Y, Cheng A-C, & Zhu D-K (2017) Type B chloramphenicol acetyltransferases are responsible for chloramphenicol resistance in Riemerella anatipestifer, China. Front. Microbiol 8, 297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Shaw WV, Packman LC, Burleigh BD, Dell A, Morris HR, & Hartley BS (1979) Primary structure of a chloramphenicol acetyltransferase specified by R plasmids. Nature 282, 870–872. [DOI] [PubMed] [Google Scholar]
- [112].Leslie AG (1990) Refined crystal structure of type III chloramphenicol acetyltransferase at 1.75 A resolution. J. Mol. Biol 213, 167–186. [DOI] [PubMed] [Google Scholar]
- [113].Robicsek A, Strahilevitz J, Jacoby GA, Macielag M, Abbanat D, Park CH, Bush K, & Hooper DC (2006) Fluoroquinolone-modifying enzyme: a new adaptation of a common aminoglycoside acetyltransferase. Nat. Med 12, 83–88. [DOI] [PubMed] [Google Scholar]
- [114].Spanogiannopoulos P, Thaker M, Koteva K, Waglechner N, & Wright GD (2012) Characterization of a rifampin-inactivating glycosyltransferase from a screen of environmental actinomycetes. Antimicrob. Agents Chemother 56, 5061–5069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Li B, Forseth RR, Bowers AA, Schroeder FC, & Walsh CT (2012) A backup plan for self-protection: S-methylation of holomycin biosynthetic intermediates in Streptomyces clavuligerus. Chembiochem. 13, 2521–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Duell ER, Glaser M, Le Chapelain C, Antes I, Groll M, & Huber EM (2016) Sequential inactivation of gliotoxin by the S-methyltransferase TmtA. ACS Chem. Biol 11, 1082–1089. [DOI] [PubMed] [Google Scholar]
- [117].Ramirez MS, & Tolmasky ME (2010) Aminoglycoside modifying enzymes. Drug Resist. Updat 13, 151–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Cox G, Stogios PJ, Savchenko A, & Wright GD (2015) Structural and molecular basis for resistance to aminoglycoside antibiotics by the adenylyltransferase ANT(2”)-Ia. mBio 6, e02180–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Levings RS, Hall RM, Lightfoot D, & Djordjevic SP (2006) linG, a new integron-associated gene cassette encoding a lincosamide nucleotidyltransferase. Antimicrob. Agents. Chemother 50, 3514–3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Brisson-Noel A, Delrieu P, Samain D, & Courvalin P (1988) Inactivation of lincosaminide antibiotics in Staphylococcus. Identification of lincosaminide O-nucleotidyltransferases and comparison of the corresponding resistance genes. J. Biol. Chem 263, 15880–15887. [PubMed] [Google Scholar]
- [121].Baysarowich J, Koteva K, Hughes DW, Ejim L, Griffiths E, Zhang K, Junop M, & Wright GD (2008) Rifamycin antibiotic resistance by ADP-ribosylation: Structure and diversity of Arr. Proc. Natl. Acad. Sci. U. S. A 105, 4886–4891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Quan S, Imai T, Mikami Y, Yazawa K, Dabbs ER, Morisaki N, Iwasaki S, Hashimoto Y, & Furihata K (1999) ADP-Ribosylation as an intermediate step in inactivation of rifampin by a mycobacterial gene. Antimicrob. Agents Chemother 43,181–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Mukhtar TA, Koteva KP, Hughes DW, & Wright GD (2001) Vgb from Staphylococcus aureus inactivates streptogramin B antibiotics by an elimination mechanism not hydrolysis. Biochemistry 40, 8877–8886. [DOI] [PubMed] [Google Scholar]
- [124].Korczynska M, Mukhtar TA, Wright GD, & Berghuis AM (2007) Structural basis for streptogramin B resistance in Staphylococcus aureus by virginiamycin B lyase. Proc. Natl. Acad. Sci. U. S. A 104,10388–10393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Liu LK, Dai Y, Abdelwahab H, Sobrado P, & Tanner JJ (2018) Structural evidence for rifampicin monooxygenase inactivating rifampicin by cleaving its ansa-bridge. Biochemistry 57, 2065–2068. [DOI] [PubMed] [Google Scholar]
- [126].Koteva K, Cox G, Kelso JK, Surette MD, Zubyk HL, E]im L, Stogios P, Savchenko A, Sorensen D, & Wright GD (2018) Rox, a rifamycin resistance enzyme with an unprecedented mechanism of action. Cell Chem. Biol 25, 403–412. [DOI] [PubMed] [Google Scholar]
- [127].He M, Sheldon PJ, & Sherman DH (2001) Characterization of a quinone reductase activity for the mitomycin C binding protein (MRD): Functional switching from a drug-activating enzyme to a drug-binding protein. Proc. Natl. Acad. Sci. U. S. A 98, 926–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Crofts TS, Sontha P, King A0, Wang B, Biddy BA, Zanolli N, Gaumnitz J, & Dantas G (2019) Discovery and characterization of a nitroreductase capable of conferring bacterial resistance to chloramphenicol. Cell Chem. Biol 26, 559–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Green KD, Fosso MY, Mayhoub AS, & Garneau-Tsodikova S (2019) Investigating the promiscuity of the chloramphenicol nitroreductase from Haemophilus influenzae towards the reduction of 4-nitrobenzene derivatives. Bioorg. Med. Chem. Lett 29,1127–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Suzuki N, Lee CK, Nihira T, & Yamada Y (1998) Purification and characterization of virginiamycin Ml reductase from Streptomyces virginiae. Antimicrob. Agents Chemother 42, 2985–2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Johnson AP (2015) Surveillance of antibiotic resistance. Philos. Trans. R. Soc. Lond. B Biol. Sci 370, 20140080–20140080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Boolchandani M, D’Souza AW, & Dantas G (2019) Sequencing-based methods and resources to study antimicrobial resistance. Nat. Rev. Genet Published Online, 18 March 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].D’Costa VM, McGrann KM, Hughes DW, & Wright GD (2006) Sampling the antibiotic resistome. Science 311, 374–377. [DOI] [PubMed] [Google Scholar]
- [134].Allen ΗK, Moe LA, Rodbumrer J, Gaarder A, & Handelsman J (2009) Functional metagenomics reveals diverse beta-lactamases in a remote Alaskan soil. ISME J 3, 243–251. [DOI] [PubMed] [Google Scholar]
- [135].Forsberg KJ, Reyes A, Wang B, Selleck EM, Sommer MO, & Dantas G (2012) The shared antibiotic resistome of soil bacteria and human pathogens. Science 337,1107–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Liu LK, Abdelwahab H, Martin Del Campo JS, Mehra-Chaudhary R, Sobrado P, & Tanner JJ (2016) The structure of the antibiotic deactivating, N-hydroxylating rifampicin monooxygenase. J. Biol. Chem 291, 21553–21562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Dantas G, Sommer MO, Oluwasegun RD, & Church GM (2008) Bacteria subsisting on antibiotics. Science 320, 100–103. [DOI] [PubMed] [Google Scholar]
- [138].Ruppe E, Ghozlane A, Tap J, Pons N, Alvarez AS, Maziers N, Cuesta T, Hernando-Amado S, Clares I, Martinez JL, Coque TM, Baquero F, Lanza VF, Maiz L, Goulenok T, de Lastours V, Amor N, Fantin B, Wieder I, Andremont A, van Schaik W, Rogers M, Zhang X, Willems RJL, de Brevern AG, Batto JM, Blottiere HM, Leonard P, Lejard V, Letur A, Levenez F, Weiszer K, Haimet F, Dore J, Kennedy SP, & Ehrlich SD (2019) Prediction of the intestinal resistome by a three-dimensional structure-based method. Nat. Microbiol 4, 112–123. [DOI] [PubMed] [Google Scholar]
- [139].Sailer ZR, & Harms MJ (2017) Molecular ensembles make evolution unpredictable. Proc. Natl. Acad. Sci. U. S. A 114, 11938–11943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Zimmerman MI, Hart KM, Sibbald CA, Frederick TE, Jimah JR, Knoverek CR, Tolia NH, & Bowman GR (2017) Prediction of new stabilizing mutations based on mechanistic insights from Markov state models. ACS Cent. Sci 3, 1311–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Hart KM, Moeder KE, Ho CMW, Zimmerman MI, Frederick TE, & Bowman GR (2017) Designing small molecules to target cryptic pockets yields both positive and negative allosteric modulators. PLoS One 12, e0178678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Orencia MC, Yoon JS, Ness JE, Stemmer WP, & Stevens RC (2001) Predicting the emergence of antibiotic resistance by directed evolution and structural analysis. Nat. Struct. Biol 8, 238–242. [DOI] [PubMed] [Google Scholar]
- [143].Thaker MN, Wang W, Spanogiannopoulos P, Waglechner N, King AM, Medina R, & Wright GD (2013) Identifying producers of antibacterial compounds by screening for antibiotic resistance. Nat. Biotechnol 31, 922–927. [DOI] [PubMed] [Google Scholar]
- [144].Doroghazi JR, & Metcalf WW (2013) Comparative genomics of actinomycetes with a focus on natural product biosynthetic genes. BMC Genomics. 14, 611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Cox G, Sieron A, King AM, De Pascale G, Pawlowski AC, Koteva K, & Wright GD (2017) A common platform for antibiotic dereplication and adjuvant discovery. Cell Chem. Biol 24, 98–109. [DOI] [PubMed] [Google Scholar]
- [146].Dejong CA, Chen GM, Li H, Johnston CW, Edwards MR, Rees PN, Skinnider MA, Webster AL, & Magarvey NA (2016) Polyketide and nonribosomal peptide retro-biosynthesis and global gene cluster matching. Nat. Chem. Biol 12, 1007–1014. [DOI] [PubMed] [Google Scholar]
- [147].Kling A, Lukat P, Almeida DV, Bauer A, Fontaine E, Sordello S, Zaburannyi N, Herrmann J, Wenzel SC, Konig C, Ammerman NC, Barrio MB, Borchers K, Bordon-Pallier F, Bronstrup M, Courtemanche G, Gerlitz M, Geslin M, Hammann P, Heinz DW, Hoffmann H, Klieber S, Kohlmann M, Kurz M, Lair C, Matter H, Nuermberger E, Tyagi S, Fraisse L, Grosset JH, Lagrange S, & Muller R (2015) Antibiotics. Targeting DnaN for tuberculosis therapy using novel griselimycins. Science 348, 1106–1112. [DOI] [PubMed] [Google Scholar]
- [148].Lukat P, Katsuyama Y, Wenzel S, Binz T, Konig C, Blankenfeldt W, Bronstrup M, & Muller R (2017) Biosynthesis of methyl-proline containing griselimycins, natural products with anti-tuberculosis activity. Chem. Sci 8, 7521–7527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Cone MC, Petrich AK, Gould SJ, & Zabriskie TM (1998) Cloning and heterologous expression of blasticidin S biosynthetic genes from Streptomyces griseochromogenes. J. Antibiot 51, 570–578. [DOI] [PubMed] [Google Scholar]
- [150].Liu X, Fortin PD, & Walsh CT (2008) Andrimid producers encode an acetyl-CoA carboxyltransferase subunit resistant to the action of the antibiotic. Proc. Natl. Acad. Sci. U. S. A 105, 13321–13326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Hansen BG, Genee HJ, Kaas CS, Nielsen JB, Regueira TB, Mortensen UH, Frisvad JC, & Patil KR (2011) A new class of IMP dehydrogenase with a role in self-resistance of mycophenolic acid producing fungi. BMC Microbiol. 11, 202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Peterson RM, Huang T, Rudolf JD, Smanski MJ, & Shen B (2014) Mechanisms of self-resistance in the platensimycin- and platencin-producing Streptomyces platensis MA7327 and MA7339 strains. Chem. Biol 21, 389–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Vecchione JJ, & Sello JK (2009) A novel tryptophanyl-tRNA synthetase gene confers high-level resistance to indolmycin. Antimicrob. Agents Chemother 53, 3972–3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Kitabatake M, Ali K, Demain A, Sakamoto K, Yokoyama S, & Soll D (2002) Indolmycin resistance of Streptomyces coelicolor A3(2) by induced expression of one of its two tryptophanyl-tRNA synthetases. J. Biol. Chem 277, 23882–23887. [DOI] [PubMed] [Google Scholar]
- [155].Millson SH, Chua CS, Roe SM, Polier S, Solovieva S, Pearl LH, Sim TS, Prodromou C, & Piper PW (2011) Features of the Streptomyces hygroscopicus HtpG reveal how partial geldanamycin resistance can arise with mutation to the ATP binding pocket of a eukaryotic Hsp90. FASEB J. 25, 3828–3837. [DOI] [PubMed] [Google Scholar]
- [156].Prodromou C, Nuttall JM, Millson SH, Roe SM, Sim TS, Tan D, Workman P, Pearl LH, & Piper PW (2009) Structural basis of the radicicol resistance displayed by a fungal hsp90. ACS Chem. Biol 4, 289–297. [DOI] [PubMed] [Google Scholar]
- [157].Petronikolou N, Ortega MA, Borisova SA, Nair SK, & Metcalf WW (2019) Molecular basis of Bacillus subtilis ATCC 6633 self-resistance to the phosphono-oligopeptide antibiotic rhizocticin. ACS Chem. Biol 14, 742–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Luzzatto-Knaan T, Melnik AV, & Dorrestein PC (2019) Mass spectrometry uncovers the role of surfactin as an interspecies recruitment factor. ACS Chem. Biol 14, 459–467. [DOI] [PubMed] [Google Scholar]
- [159].Gehrke SS, Kumar G, Yokubynas NA, Cote JP, Wang W, French S, MacNair CR, Wright GD, & Brown ED (2017) Exploiting the sensitivity of nutrient transporter deletion strains in discovery of natural product antimetabolites. ACS Infect. Dis 3, 955–965. [DOI] [PubMed] [Google Scholar]
- [160].Charani E, & Holmes A (2019) Antibiotic stewardship-Twenty years in the making. Antibiotics 8, E7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Ward JM, & Hodgson JE (1993) The biosynthetic genes for clavulanic acid and cephamycin production occur as a ‘super-cluster’ in three Streptomyces. FEMS Microbiol. Lett 110, 239–242. [DOI] [PubMed] [Google Scholar]
- [162].Mellado E, Lorenzana LM, Rodriguez-Saiz M, Diez B, Liras P, & Barredo JL (2002) The clavulanic acid biosynthetic cluster of Streptomyces clavuligerus: genetic organization of the region upstream of the car gene. Microbiol. 148, 1427–1438. [DOI] [PubMed] [Google Scholar]
- [163].Perez-Llarena FJ, Liras P, Rodriguez-Garcia A, & Martin JF (1997) A regulatory gene (ccaR) required for cephamycin and clavulanic acid production in Streptomyces clavuligerus: amplification results in overproduction of both beta-lactam compounds. J. Bacteriol 179, 2053–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Santamarta I, Lopez-Garcia MT, Kurt A, Nardiz N, Alvarez-Alvarez R, Perez-Redondo R, Martin JF, & Liras P (2011) Characterization of DNA-binding sequences for CcaR in the cephamycin-clavulanic acid supercluster of Streptomyces clavuligerus. Mol. Microbiol 81, 968–981. [DOI] [PubMed] [Google Scholar]
- [165].Drawz SM, & Bonomo RA (2010) Three decades of beta-lactamase inhibitors. Clin. Microbiol. Rev 23, 160–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Hata T, Omura S, Iwai Y, Ono H, & Takeshima H (1972) Studies on penicillinase inhibitors produced by microorganisms. J. Antibiot 25, 473–474. [DOI] [PubMed] [Google Scholar]
- [167].Doran JL, Leskiw BK, Aippersbach S, & Jensen SE (1990) Isolation and characterization of a beta-lactamase-inhibitory protein from Streptomyces clavuligerus and cloning and analysis of the corresponding gene. J. Bacteriol 172, 4909–4918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Strynadka NC, Jensen SE, Alzari PM, & James MN (1996) A potent new mode of beta-lactamase inhibition revealed by the 1.7 A X-ray crystallographic structure of the TEM-1-BLIP complex. Nat. Struct. Biol 3, 290–297. [DOI] [PubMed] [Google Scholar]
- [169].Lim D, Park HU, De Castro L, Kang SG, Lee HS, Jensen S, Lee KJ, & Strynadka NC (2001) Crystal structure and kinetic analysis of beta-lactamase inhibitor protein-II in complex with TEM-1 beta-lactamase. Nat. Struct. Biol 8, 848–852. [DOI] [PubMed] [Google Scholar]
- [170].Eiamphungporn W, Schaduangrat N, Malik AA, & Nantasenamat C (2018) Tackling the antibiotic resistance caused by class A beta-lactamases through the use of beta-lactamase inhibitory protein. Int. J. Mol. Sci 19, E2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Bush K (2018) Game changers: New beta-lactamase inhibitor combinations targeting antibiotic resistance in Gram-negative bacteria. ACS Infect. Dis 4, 84–87. [DOI] [PubMed] [Google Scholar]
- [172].King AM, Reid-Yu SA, Wang W, King DT, De Pascale G, Strynadka NC, Walsh TR, Coombes BK, & Wright GD (2014) Aspergillomarasmine A overcomes metallo-beta-lactamase antibiotic resistance. Nature 510, 503–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Melander RJ, & Melander C (2017) The challenge of overcoming antibiotic resistance: An adjuvant approach? ACS Infec. Dis 3, 559–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Horsman ME, Marous DR, Li R, Oliver RA, Byun B, Emrich SJ, Boggess B, Townsend CA, & Mobashery S (2017) Whole-genome shotgun sequencing of two beta-proteobacterial species in search of the bulgecin biosynthetic cluster. ACS Chem. Biol 12, 2552–2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Dik DA, Madukoma CS, Tomoshige S, Kim C, Lastochkin E, Boggess WC, Fisher JF, Shrout JD, & Mobashery S (2019) Slt, MltD, and MltG of Pseudomonas aeruginosa as targets of bulgecin A in potentiation of beta-lactam antibiotics. ACS Chem. Biol 14, 296–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Tomoshige S, Dik DA, Akabane-Nakata M, Madukoma CS, Fisher JF, Shrout JD, & Mobashery S (2018) Total syntheses of bulgecins A, B, and C and their bactericidal potentiation of the beta-lactam antibiotics. ACS Infect. Dis 4, 860–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Mrak P, Krastel P, Pivk Lukancic P, Tao J, Pistorius D, & Moore CM (2018) Discovery of the actinoplanic acid pathway in Streptomyces rapamycinicus reveals a genetically conserved synergism with rapamycin. J. Biol. Chem 293, 19982–19995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Bikkul MU, Clements CS, Godwin LS, Goldberg MW, Kill IR, & Bridger JM (2018) Farnesyltransferase inhibitor and rapamycin correct aberrant genome organisation and decrease DNA damage respectively, in Hutchinson-Gilford progeria syndrome fibroblasts. Biogerontology 19, 579–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Arp J, Gotze S, Mukherji R, Mattern DJ, Garcia-Altares M, Klapper M, Brock DA, Brakhage AA, Strassmann JE, Queller DC, Bardl B, Willing K , Peschel G, & Stallforth P (2018) Synergistic activity of cosecreted natural products from amoebae-associated bacteria. Proc. Natl. Acad. Sci. U. S. A 115, 3758–3763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Helfrich EJN, Vogel CM, Ueoka R, Schafer M, Ryffel F, Muller DB, Probst S, Kreuzer M, Piel J, & Vorholt JA (2018) Bipartite interactions, antibiotic production and biosynthetic potential of the Arabidopsis leaf microbiome. Nat. Microbiol 3, 909–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Domalaon R, Idowu T, Zhanel GG, & Schweizer F (2018) Antibiotic hybrids: The next generation of agents and adjuvants against Gram-negative pathogens? Clin. Microbiol. Rev 31, e00077–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Klahn P, & Bronstrup M (2017) Bifunctional antimicrobial conjugates and hybrid antimicrobials. Nat. Prod. Rep 34, 832–885. [DOI] [PubMed] [Google Scholar]
- [183].Parkes AL, & Yule IA (2016) Hybrid antibiotics - clinical progress and novel designs. Expert Opin. Drug Discov 11, 665–680. [DOI] [PubMed] [Google Scholar]
- [184].Wencewicz TA, & Miller MJ (2018) Sideromycins as pathogen-targeted antibiotics, In Antibacterials. Topics in Medicinal Chemistry (Fisher JF, Mobashery S, and Miller MJ, Eds.), 26, 151–183, Springer, Cham [Google Scholar]
- [185].Wencewicz TA, Mollmann U, Long TE, & Miller MJ (2009) Is drug release necessary for antimicrobial activity of siderophore-drug conjugates? Syntheses and biological studies of the naturally occurring salmycin “Trojan Horse” antibiotics and synthetic desferridanoxamine-antibiotic conjugates, Biometals. 22, 633–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Wencewicz TA, Long TE, Mollmann U, & Miller MJ (2013) Trihydroxamate siderophore-fluoroquinolone conjugates are selective sideromycin antibiotics that target Staphylococcus aureus. Bioconjug. Chem 24, 473–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Wencewicz TA, & Miller MJ (2013) Biscatecholate-monohydroxamate mixed ligand siderophore-carbacephalosporin conjugates are selective sideromycin antibiotics that target Acinetobacter baumannii. J. Med. Chem 56, 4044–4052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Braun V, Pramanik A, Gwinner T, Koberle M, & Bohn E (2009) Sideromycins: tools and antibiotics. Biometals. 22, 3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Braun V, Gunthner K, Hantke K, & Zimmermann L (1983) Intracellular activation of albomycin in Escherichia coli and Salmonella typhimurium. J. Bacteriol 156, 308–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Pramanik A, & Braun V (2006) Albomycin uptake via a ferric hydroxamate transport system of Streptococcus pneumoniae R6. J. Bacteriol 188, 3878–3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Kim A, Kutschke A, Ehmann DE, Patey SA, Crandon JL, Gorseth E, Miller AA, McLaughlin RE, Blinn CM, Chen A, Nayar AS, Dangel B, Tsai AS, Rooney MT, Murphy-Benenato KE, Eakin AE, & Nicolau DP (2015) Pharmacodynamic profiling of a siderophore-conjugated monocarbam in Pseudomonas aeruginosa: Assessing the risk for resistance and attenuated efficacy. Antimicrob. Agents Chemother 59, 7743–7752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Zeng Y, Kulkarni A, Yang Z, Patil PB, Zhou W, Chi X, Van Lanen S, and Chen S (2012) Biosynthesis of albomycin delta(2) provides a template for assembling siderophore and aminoacyl-tRNA synthetase inhibitor conjugates. ACS Chem. Biol 7, 1565–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Ushimaru R, & Liu HW (2019) Biosynthetic origin of the atypical stereochemistry in the thioheptose core of albomycin nucleoside antibiotics, J. Am. Chem. Soc 141, 2211–2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Lin Z, Xu X, Zhao S, Yang X, Guo J, Zhang Q, Jing C, Chen S, & He Y (2018) Total synthesis and antimicrobial evaluation of natural albomycins against clinical pathogens. Nat. Comm 9, 3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Zhanel GG, Golden AR, Zelenitsky S, Wiebe K, Lawrence CK, Adam HJ, Idowu T, Domalaon R, Schweizer F, Zhanel MA, Lagace-Wiens PRS, Walkty AJ, Noreddin A, Lynch Iii JP, and Karlowsky JA (2019) Cefiderocol: A Siderophore Cephalosporin with Activity Against Carbapenem-Resistant and Multidrug-Resistant Gram-Negative Bacilli, Drugs 79, 271–289. [DOI] [PubMed] [Google Scholar]
- [196].Dunn ZD, Wever WJ, Economou NJ, Bowers AA, & Li B (2015) Enzymatic basis of “hybridity” in thiomarinol biosynthesis. Angew. Chem. Int. Ed. Engl 54, 5137–5141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Shiozawa H, Kagasaki T, Kinoshita T, Haruyama H, Domon H, Utsui Y, Kodama K, & Takahashi S (1993) Thiomarinol, a new hybrid antimicrobial antibiotic produced by a marine bacterium. Fermentation, isolation, structure, and antimicrobial activity. J. Antibiot 46, 1834–1842. [DOI] [PubMed] [Google Scholar]
- [198].Li B, Wever WJ, Walsh CT, & Bowers AA (2014) Dithiolopyrrolones: biosynthesis, synthesis, and activity of a unique class of disulfide-containing antibiotics. Nat. Prod. Rep 31, 905–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Chan AN, Shiver AL, Wever WJ, Razvi SZA, Traxler MF, & Li B (2017) Role for dithiolopyrrolones in disrupting bacterial metal homeostasis. Proc. Natl. Acad. Sci. U. S. A 114, 2717–2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Lauinger L, Li J, Shostak A, Cemel IA, Ha N, Zhang Y, Merkl PE, Obermeyer S, Stankovic-Valentin N, Schafmeier T, Wever WJ, Bowers AA, Carter KP, Palmer AE, Tschochner H, Melchior F, Deshaies RJ, Brunner M, & Diernfellner A (2017) Thiolutin is a zinc chelator that inhibits the Rpn11 and other JAMM metalloproteases. Nat. Chem. Biol 13, 709–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [201].Marion O, Gao X, Marcus S, & Hall DG (2009) Synthesis and preliminary antibacterial evaluation of simplified thiomarinol analogs. Bioorg. Med. Chem 17, 1006–1017. [DOI] [PubMed] [Google Scholar]
- [202].Fukuda D, Haines AS, Song Z, Murphy AC, Hothersall J, Stephens ER, Gurney R, Cox RJ, Crosby J, Willis CL, Simpson TJ, & Thomas CM (2011) A natural plasmid uniquely encodes two biosynthetic pathways creating a potent anti-MRSA antibiotic. PLoS One 6, e18031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Murphy AC, Fukuda D, Song Z, Hothersall J, Cox RJ, Willis CL, Thomas CM, & Simpson TJ (2011) Engineered thiomarinol antibiotics active against MRSA are generated by mutagenesis and mutasynthesis of Pseudoalteromonas SANK73390. Angew. Chem. Int. Ed. Engl 50, 3271–3274. [DOI] [PubMed] [Google Scholar]
- [204].Gao X, & Hall DG (2005) Catalytic asymmetric synthesis of a potent thiomarinol antibiotic. J. Am. Chem. Soc 127, 1628–1629. [DOI] [PubMed] [Google Scholar]
- [205].Gao SS, Wang L, Song Z, Hothersall J, Stevens ER, Connolly J, Winn PJ, Cox RJ, Crump MP, Race PR, Thomas CM, Simpson TJ, & Willis CL (2017) Selected mutations reveal new intermediates in the biosynthesis of mupirocin and the thiomarinol antibiotics. Angew. Chem. Int. Ed. Engl 56, 3930–3934. [DOI] [PubMed] [Google Scholar]
- [206].Qi Y, Ding E, & Blodgett JAV (2018) Native and engineered clifednamide biosynthesis in multiple streptomyces spp. ACS Syn. Biol 7, 357–362. [DOI] [PubMed] [Google Scholar]
- [207].Kim E, Moore BS, & Yoon YJ (2015) Reinvigorating natural product combinatorial biosynthesis with synthetic biology. Nat. Chem. Biol 11, 649–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Awakawa T, Fujioka T, Zhang L, Hoshino S, Hu Z, Hashimoto J, Kozone I, Ikeda H, Shin-Ya K, Liu W, & Abe I (2018) Reprogramming of the antimycin NRPS-PKS assembly lines inspired by gene evolution. Nat. Comm 9, 3534–3534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [209].Fleming SR, Bartges TE, Vinogradov AA, Kirkpatrick CL, Goto Y, Suga H, Hicks LM, & Bowers AA (2019) Flexizyme-enabled benchtop biosynthesis of thiopeptides. J. Am. Chem. Soc 141, 758–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [210].Huo L, Hug JJ, Fu C, Bian X, Zhang Y, & Muller R (2019) Heterologous expression of bacterial natural product biosynthetic pathways. Nat. Prod. Rep Published Online, Jan. 8. [DOI] [PubMed] [Google Scholar]
- [211].Firn RD, & Jones CG (2003) Natural products--a simple model to explain chemical diversity. Nat. Prod. Rep 20, 382–391. [DOI] [PubMed] [Google Scholar]
- [212].Shrestha SK, & Garneau-Tsodikova S (2016) Expanding substrate promiscuity by engineering a novel adenylating-methylating NRPS bifunctional enzyme. Chembiochem. 17, 1328–1332. [DOI] [PubMed] [Google Scholar]
- [213].Narayan AR, Jimenez-Oses G, Liu P, Negretti S, Zhao W, Gilbert MM, Ramabhadran RO, Yang YF, Furan LR, Li Z, Podust LM, Montgomery J, Houk KN, & Sherman DH (2015) Enzymatic hydroxylation of an unactivated methylene C-H bond guided by molecular dynamics simulations. Nat. Chem 7, 653–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [214].Bohac TJ, Fang L, Giblin DE, & Wencewicz TA (2019) Fimsbactin and acinetobactin compete for the periplasmic siderophore binding protein BauB in pathogenic Acinetobacter baumannii. ACS Chem. Biol 14, 674–687. [DOI] [PubMed] [Google Scholar]
- [215].Proschak A, Lubuta P, Grun P, Lohr F, Wilharm G, De Berardinis V, & Bode HB (2013) Structure and biosynthesis of fimsbactins A-F, siderophores from Acinetobacter baumannii and Acinetobacter baylyi. Chembiochem. 14, 633–638. [DOI] [PubMed] [Google Scholar]
- [216].Kittendorf JD, & Sherman DH (2009) The methymycin/pikromycin pathway: a model for metabolic diversity in natural product biosynthesis. Bioorg. Med. Chem 17, 2137–2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].Sattely ES, & Walsh CT (2008) A latent oxazoline electrophile for N-O-C bond formation in pseudomonine biosynthesis. J. Am. Chem. Soc 130, 12282–12284. [DOI] [PubMed] [Google Scholar]
- [218].Wuest WM, Sattely ES, & Walsh CT (2009) Three siderophores from one bacterial enzymatic assembly line. J. Am. Chem. Soc 131, 5056–5057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [219].Shapiro JA, & Wencewicz TA (2016) Acinetobactin isomerization enables adaptive iron acquisition in Acinetobacter baumannii through pH-triggered siderophore swapping. ACS Infect. Dis 2, 157–168. [DOI] [PubMed] [Google Scholar]
- [220].Lin Z, Koch M, Abdel Aziz MH, Galindo-Murillo R, Tianero MD, Cheatham TE, Barrows LR, Reilly CA, & Schmidt EW (2014) Oxazinin A, a pseudodimeric natural product of mixed biosynthetic origin from a filamentous fungus. Org. Lett 16, 4774–4777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [221].Gantt RW, Peltier-Pain P, & Thorson JS (2011) Enzymatic methods for glyco(diversification/randomization) of drugs and small molecules. Nat. Prod. Rep 28, 1811–1853. [DOI] [PubMed] [Google Scholar]
- [222].Zhang G, Li Y, Fang L, & Pfeifer BA (2015) Tailoring pathway modularity in the biosynthesis of erythromycin analogs heterologously engineered in E. coli. Sci. Adv 1, e1500077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [223].Arnold FH (2018) Directed evolution: Bringing new chemistry to life. Angew. Chem. Int. Ed 57, 4143–4148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [224].Hudson GA, & Mitchell DA (2018) RiPP antibiotics: biosynthesis and engineering potential. Curr. Opin. Microbiol 45, 61–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Zhang Q, Yu Y, Velasquez JE, & van der Donk WA (2012) Evolution of lanthipeptide synthetases. Proc. Natl. Acad. Sci. U. S. A 109, 18361–18366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [226].Li B, Yu JP, Brunzelle JS, Moll GN, van der Donk WA, & Nair SK (2006) Structure and mechanism of the lantibiotic cyclase involved in nisin biosynthesis. Science 311, 1464–1467. [DOI] [PubMed] [Google Scholar]
- [227].Li B, Sher D, Kelly L, Shi Y, Huang K, Knerr PJ, Joewono I, Rusch D, Chisholm SW, & van der Donk WA (2010) Catalytic promiscuity in the biosynthesis of cyclic peptide secondary metabolites in planktonic marine cyanobacteria. Proc. Natl. Acad. Sci. U. S. A 107, 10430–10435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [228].Zhang Q, Yang X, Wang H, & van der Donk WA (2014) High divergence of the precursor peptides in combinatorial lanthipeptide biosynthesis. ACS Chem. Biol 9, 2686–2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [229].Tang W, & van der Donk WA (2012) Structural characterization of four prochlorosins: a novel class of lantipeptides produced by planktonic marine cyanobacteria. Biochemistry 51, 4271–4279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [230].Yu Y, Mukherjee S, & van der Donk WA (2015) Product formation by the promiscuous lanthipeptide synthetase ProcM is under kinetic control. J. Am. Chem. Soc 137, 5140–5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [231].Bobeica SC, & van der Donk WA (2018) The enzymology of prochlorosin biosynthesis. Methods Enzymol. 604, 165–203. [DOI] [PubMed] [Google Scholar]
- [232].Hetrick KJ, Walker MC, & van der Donk WA (2018) Development and application of yeast and phage display of diverse lanthipeptides. ACS Cent. Sci 4, 458–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [233].Young TS, Dorrestein PC, & Walsh CT (2012) Codon randomization for rapid exploration of chemical space in thiopeptide antibiotic variants. Chem. Biol 19, 1600–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [234].Tran HL, Lexa KW, Julien O, Young TS, Walsh CT, Jacobson MP, & Wells JA (2017) Structure-activity relationship and molecular mechanics reveal the importance of ring entropy in the biosynthesis and activity of a natural product. J. Am. Chem. Soc 139, 2541–2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [235].Yang X, Lennard KR, He C, Walker MC, Ball AT, Doigneaux C, Tavassoli A, & van der Donk WA (2018) A lanthipeptide library used to identify a protein-protein interaction inhibitor. Nat. Chem. Biol 14, 375–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [236].LaMarche MJ, Leeds JA, Amaral A, Brewer JT, Bushell SM, Deng G, Dewhurst JM, Ding J, Dzink-Fox J, Gamber G, Jain A, Lee K, Lee L, Lister T, McKenney D, Mullin S, Osborne C, Palestrant D, Patane MA, Rann EM, Sachdeva M, Shao J, Tiamfook S, Trzasko A, Whitehead L, Yifru A, Yu D, Yan W, & Zhu Q (2012) Discovery of LFF571: an investigational agent for Clostridium difficile infection. J. Med. Chem 55, 2376–2387. [DOI] [PubMed] [Google Scholar]
- [237].Gu W, & Schmidt EW (2017) Three principles of diversity-generating biosynthesis. Acc. Chem. Res 50, 2569–2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [238].Walsh C, & Wencewicz T (2016) Antibiotics: Challenges, Mechanisms, Opportunities, ASM Press. [Google Scholar]
- [239].Brown ED, & Wright GD (2016) Antibacterial drug discovery in the resistance era. Nature 529, 336–343. [DOI] [PubMed] [Google Scholar]
- [240].Walsh CT, & Wencewicz TA (2014) Prospects for new antibiotics: a molecule-centered perspective. J. Antibiot 67, 7–22. [DOI] [PubMed] [Google Scholar]
- [241].Snoussi M, Talledo JP, Del Rosario NA, Mohammadi S, Ha BY, Kosmrlj A, & Taheri-Araghi S (2018) Heterogeneous absorption of antimicrobial peptide LL37 in Escherichia coli cells enhances population survivability. eLife 7, e38174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [242].Chen R, Deziel E, Groleau MC, Schaefer AL, & Greenberg EP (2019) Social cheating in a Pseudomonas aeruginosa quorum-sensing variant. Proc. Natl. Acad. Sci. U. S. A 116, 7021–7026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [243].Rossiter SE, Fletcher MH, & Wuest WM (2017) Natural products as platforms to overcome antibiotic resistance. Chem. Rev 117, 12415–12474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [244].Cheng AV, & Wuest WM (2019) Signed, sealed, delivered: Conjugate and prodrug strategies as targeted delivery vectors for antibiotics. ACS Infect. Dis Published Online, Apr. 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [245].Xu F, Wu Y, Zhang C, Davis KM, Moon K, Bushin LB, & Seyedsayamdost MR (2019) A genetics-free method for high-throughput discovery of cryptic microbial metabolites. Nat. Chem. Biol 15, 161–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [246].Moon K, Xu F, Zhang C, & Seyedsayamdost MR (2019) Bioactivity-HiTES unveils cryptic antibiotics encoded in Actinomycete bacteria. ACS Chem. Biol 14, 767–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [247].Huber J, Donald RG, Lee SH, Jarantow LW, Salvatore MJ, Meng X, Painter R, Onishi RH, Occi J, Dorso K, Young K, Park YW, Skwish S, Szymonifka MJ, Waddell TS, Miesel L, Phillips JW, & Roemer T (2009) Chemical genetic identification of peptidoglycan inhibitors potentiating carbapenem activity against methicillin-resistant Staphylococcus aureus. Chem. Biol 16, 837–848. [DOI] [PubMed] [Google Scholar]
- [248].Sherpa RT, Reese CJ, & Montazeri Aliabadi H (2015) Application of iChip to grow “uncultivable” microorganisms and its impact on antibiotic discovery. J. Pharm. Pharm. Sci 18, 303–315. [DOI] [PubMed] [Google Scholar]
- [249].Hover BM, Kim SH, Katz M, Charlop-Powers Z, Owen JG, Ternei MA, Maniko J, Estrela AB, Molina H, Park S, Perlin DS, & Brady SF (2018) Culture-independent discovery of the malacidins as calcium-dependent antibiotics with activity against multidrug-resistant Gram-positive pathogens. Nat. Microbiol 3, 415–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [250].Gasparrini AJ, Crofts TS, Gibson MK, Tarr PI, Warner BB, & Dantas G (2016) Antibiotic perturbation of the preterm infant gut microbiome and resistome. Gut Microbes. 7, 443–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [251].Jernberg C, Lofmark S, Edlund C, & Jansson JK (2010) Long-term impacts of antibiotic exposure on the human intestinal microbiota. Microbiol. 156, 3216–3223. [DOI] [PubMed] [Google Scholar]
- [252].McKenna M (2019) Antibiotics set to flood Florida’s troubled orange orchards. Nature 567, 302–303. [DOI] [PubMed] [Google Scholar]
- [253].Palmer AC, Angelino E, & Kishony R (2010) Chemical decay of an antibiotic inverts selection for resistance. Nat. Chem. Biol 6, 105–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [254].Stone LK, Baym M, Lieberman TD, Chait R, Clardy J, & Kishony R (2016) Compounds that select against the tetracycline-resistance efflux pump. Nat. Chem. Biol 12, 902–904. [DOI] [PMC free article] [PubMed] [Google Scholar]