

SUMMARY
Hydrogen sulfide (H2S) is a gasotransmitter exhibiting pivotal functions in diverse biological processes, including activation of multiple cardioprotective pathways. Sulfide:quinone oxidoreductase (SQOR) is an integral membrane flavoprotein that catalyzes the first step in the mitochondrial metabolism of H2S. As such, it plays a critical role in controlling physiological levels of the gasotransmitter and has attracted keen interest as a potential drug target. We report the crystal structure of human SQOR, unraveling the molecular basis for the enzyme’s ability to catalyze sulfane sulfur transfer reactions with structurally diverse acceptors. We demonstrate that human SQOR contains unique features: an electro-positive surface depression implicated as a binding site for sulfane sulfur acceptors and postulated to funnel negatively charged substrates to a hydrophilic H2S-oxidizing active site, which is connected to a hydrophobic internal tunnel that binds coenzyme Q. These findings support a proposed model for catalysis and open the door for structure-based drug design.
Graphical Abstract
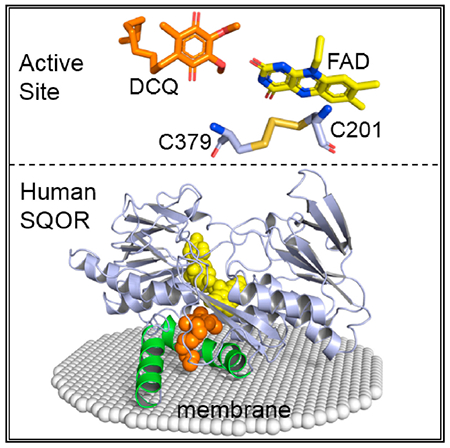
In Brief
Jackson et al. determine the structure of human SQOR, a monotopic mitochondrial membrane protein that regulates physiological levels of the gasotransmitter hydrogen sulfide. The structure uncovers the basis for the enzyme’s ability to catalyze sulfane sulfur transfer reactions with structurally diverse acceptors and opens the door for structure-based drug design.
INTRODUCTION
Human sulfide:quinone oxidoreductase (SQOR) is an integral membrane protein that catalyzes the first irreversible step in the mitochondrial metabolism of hydrogen sulfide (H2S) (Jackson et al., 2012). H2S is an important gasotransmitter exhibiting crucial functions in diverse biological processes, such as post-translational protein modification, smooth muscle relaxation, neuronal transmission and regulation of ion channels, and the mitochondrial respiratory chain (Li et al., 2011). H2S signaling is especially critical in the cardiovascular system where the gasotransmitter protects against the development of hypertension, atherosclerosis, and pathological cardiac remodeling triggered by injury that leads to heart failure (Hackfort and Mishra, 2016; Mani et al., 2013; Yang et al., 2008). Because H2S activates multiple cardioprotective pathways, increasing H2S levels is an attractive new strategy to treat heart failure. SQOR plays a major role in controlling physiological levels of H2S and sits at a key pharmacological intervention point; as such, it has attracted keen interest as a potential drug target.
SQOR catalyzes a two-electron oxidation of H2S to sulfane sulfur (S0). It uses coenzyme Q (CoQ) as electron acceptor and sulfite or glutathione as sulfane sulfur acceptor in reactions that produce thiosulfate or glutathione persulfide (GSS−), respectively (Augustyn et al., 2017; Jackson et al., 2012; Libiad et al., 2014). Human SQOR belongs to a family of flavoprotein disulfide reductases (FDR) (Miller, 2013), members of which contain a pair of redox-active cysteine residues that typically form a disulfide in the resting enzyme. The FDR family includes eukaryotic and prokaryotic SQORs as well as flavocytochrome c sulfide dehydrogenase (FCSD), a soluble bacterial H2S-oxidizing enzyme that uses cytochrome c, instead of CoQ, as electron acceptor. Crystal structures have been determined for prokaryotic SQORs and FCSDs (Brito et al., 2009; Chen et al., 1994; Cherney et al., 2010; Hirano et al., 2012; Marcia et al., 2009; Osipov et al., 2018).
Unlike human SQOR, bacterial SQORs catalyze an oxidative polymerization of H2S to produce sulfane sulfur metabolites (polysulfide chains, cyclooctasulfur rings) in reactions that do not utilize a sulfane sulfur acceptor. The active-site cysteines in bacterial SQORs are far apart (6.5–9.3 Å), a feature that provides space for the growing polysulfide chains (Brito et al., 2009; Cherney et al., 2010; Marcia et al., 2009). In contrast, FCSDs generate H2S2 as a major product of H2S oxidation (Lu et al., 2017) and their active-site cysteines are closer together, and in fact form a disulfide bridge in A. vinosum FCSD (Chen et al., 1994). Human SQOR can also convert H2S to H2S2 in the absence of an “exogenous” sulfane sulfur acceptor (Jackson et al., 2012), and we therefore reasoned that the active-site cysteines in human SQOR (Cys201, Cys379) were also likely to form a disulfide bridge. In human SQOR, Cys201 aligns with the “proximal” cysteine in FCSD, the cysteine closer to the C(4a) position of FAD. We proposed that the human SQOR reaction is initiated by nucleophilic attack of HS− at the distal cysteine, Cys379, to produce a charge-transfer (CT) complex of FAD with either Cys201S− (shown) or Cys379SS− (Scheme 1, step 1) (Jackson et al., 2012). Nucleophilic attack of Cys201S− at the C(4a) position of FAD produces a covalent flavin adduct, 4a-adduct I (step 2). Reaction of 4a-adduct I with a sulfane sulfur acceptor (N:) generates 4a-adduct II and the thiolate form of Cys379 (step 3). Nucleophilic attack of Cys379S− at the sulfur atom in the 4a-adduct produces 1,5-dihydroFAD and regenerates the disulfide bridge (step 4). The catalytic cycle is completed upon transfer of electrons from 1,5-dihydroFAD to CoQ (step 5).
Scheme 1. Catalytic Mechanism of Human SQOR.
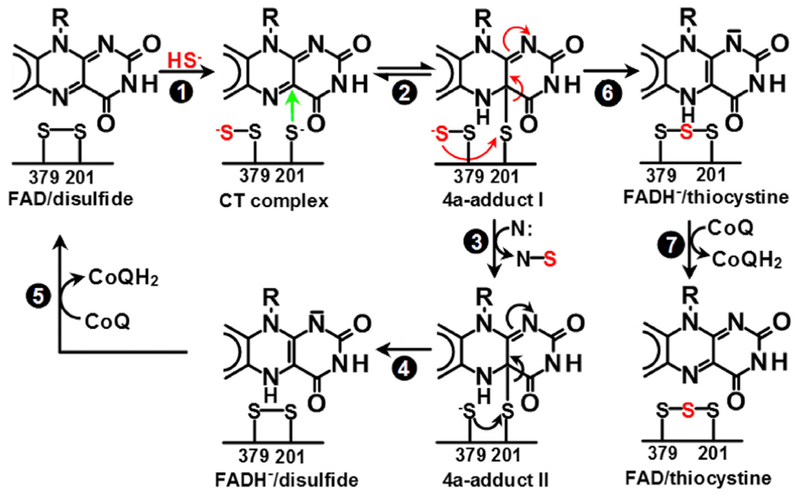
Proposed mechanism for the reaction catalyzed by human SQOR in the presence of a sulfane sulfur acceptor, denoted N: (steps 1–5) and a postulated slow conversion of 4a-adduct I to thiocystine in the absence of N: (steps 6–7) (Jackson et al., 2012).
We report here the X-ray structure of human SQOR at 2.59 Å resolution, the first of any eukaryotic SQOR. This structure supports the proposed model for catalysis and reveals a unique binding site for sulfane sulfur acceptors, unraveling the molecular basis for the enzyme’s ability to catalyze sulfane sulfur transfer reactions with structurally diverse acceptors. Other features of the eukaryotic enzyme comprise an apparent molecular hybrid of structural and functional properties observed for soluble and membrane-bound bacterial H2S-oxidizing enzymes. The structure of human SQOR opens the door for structure-based drug design.
RESULTS
Structure Determination
Crystal structures were determined for both selenomethionine (SeMet)-substituted and native human SQOR, at 2.59 and 2.99 Å resolution, respectively (Table 1). In both SeMet-substituted and native crystal forms, the asymmetric unit contains four highly similar chains (A-D) (root-mean-square deviations [RMSD] in Cα positions ≤0.18 and 0.26 Å, respectively). As expected, the SeMet and native structures are essentially identical, with Cα RMSD values ≤0.2 Å. Therefore, the following descriptions of the structure focus primarily on the SeMet-substituted enzyme, unless otherwise indicated.
Table 1.
Diffraction Data, Phasing Statistics, and Model Refinement
| SeMet Combined Dataset 1a | SeMet Combined Dataset 2b | Nativec | ||||
|---|---|---|---|---|---|---|
| Space group | P6122 | P6122 | P6122 | |||
| Cell dimensions | ||||||
| a, b, c (Å) | 119.27, 119.27, 551.68 | 119.39, 119.39, 551.86 | 119.34, 119.34, 551.98 | |||
| α, β, γ (°) | 90, 90, 120 | 90, 90, 120 | 90, 90, 120 | |||
| Mosaicity (°) | 0.44 | 0.46 | 0.49 | |||
| overall | highest shell | overall | highest shell | overall | highest shell | |
| Wavelength (Å) | 0.97918 | 0.97918 | 0.97918 | |||
| Beam/detector | 24-ID-C | Pilatus 6M-F | 24-ID-C | Pilatus 6M-F | 24-ID-E | Eiger 16M |
| Observations | 3,925,944 | 104,010 | 5,907,898 | 227,087 | 770,154 | 50,223 |
| Unique reflections | 69,991 | 4,232 | 68,911 | 2,776 | 48,607 | 4,315 |
| Resolution (Å) | 62.66–2.64 | 2.7–2.64 | 68.72–2.59 | 2.65–2.59 | 96.79–2.99 | 3.09–2.99 |
| Rmerged | 0.414 | 6.203 | 0.251 | 1.655 | 0.326 | 2.76 |
| Rmease | 0.418 | 6.334 | 0.253 | 1.666 | 0.336 | 2.89 |
| Rpimf | 0.058 | 1.27 | 0.027 | 0.18 | 0.083 | 0.83 |
| I/σ(I) | 12.5 | 1.1 | 22.9 | 5.6 | 9.6 | 1.1 |
| CC1/2 | 0.994 | 0.251 | 0.998 | 0.646 | 0.983 | 0.224 |
| Completeness | 99.7 | 95.6 | 93.7 | 60.5 | 100 | 99.9 |
| Multiplicity | 56.1 | 24.6 | 85.7 | 81.8 | 15.8 | 11.6 |
| Refinement | PDB: ID 6MO6 | PDB: ID 6MP5 | ||||
| Rwork/Rfree | 0.187/0.245 | 0.208/0.264 | ||||
| No. of atoms | ||||||
| Overall | 13,533 | 13,260 | ||||
| Protein | 13,040 | 13,020 | ||||
| Ligands | 216 | 212 | ||||
| Waters | 277 | 28 | ||||
| RMSD from ideality | ||||||
| Bonds (Å) | 0.004 | 0.003 | ||||
| Angles (°) | 0.98 | 0.605 | ||||
| B factors (Å2) | ||||||
| Mean | 39.6 | 69.2 | ||||
| Protein | 39.7 | 69.4 | ||||
| Ligands | 33.9 | 61.1 | ||||
| Waters | 35.8 | 54.1 | ||||
SeMet combined dataset 1 was produced by combining datasets collected with two crystals of SeMet-substituted enzyme, crystal A and crystal B, at a detector distance of 400 and 440 mm, respectively. Crystal A was grown using the condition 20% glycerol, 7.8% PEG 4K, 0.1 M sodium acetate trihydrate pH 4.6, 0.32 M ammonium acetate, 0.1% DHPC, 0.31 mM DCQ, and 10 mM N-ethylmaleimide. Crystal B was grown using the condition 20% glycerol, 8.2% PEG 4K, 0.1 M sodium acetate trihydrate pH 4.6, 0.32 M ammonium acetate, 0.1% DHPC, 0.31 mM DCQ, and 10 mM thiosulfate pentahydrate.
SeMet combined dataset 2 was produced by combining two datasets collected from different positions within with crystal B.
Native dataset was collected at a detector distance of 500 mm from a single crystal of native enzyme grown in 10% glycerol, 14% PEG 4K, 0.1 M sodium acetate trihydrate pH 4.6, 0.5 M ammonium acetate, and 0.1% DHPC.
Rmerge = ΣhklΣi|Ihk,i − 〈Ihkl〉|/Σhkl〈Ihkl〉.
Rmeas = Σhkl[N/(−1)]½ Σi|Ii(hkl) − 〈I(hkl)〉|/Σhkl Σi Ii(hkl).
Rp.i.m. = Σhkl[1/(−1)]½ Σi|Ii(hkl) − 〈I(hkl)〉|/Σhkl Σi Ii(hkl).
Overall Structure
Human SQOR comprises two tandem Rossmann fold domains and a C-terminal domain containing two helices (denoted the C-terminal and penultimate C-terminal helices) (Figures 1A, 1B, and S1). FAD is noncovalently bound in an extended conformation and is in the oxidized state, as judged by the bright yellow color of the crystals. The first Rossmann fold starts near the N terminus and binds the ADP portion of FAD. The second Rossmann fold is closer to the isoalloxazine ring of FAD but is mostly positioned at least 10 Å away from the flavin ring with one notable exception, as will be discussed.
Figure 1. Overall Structure of Human SQOR.
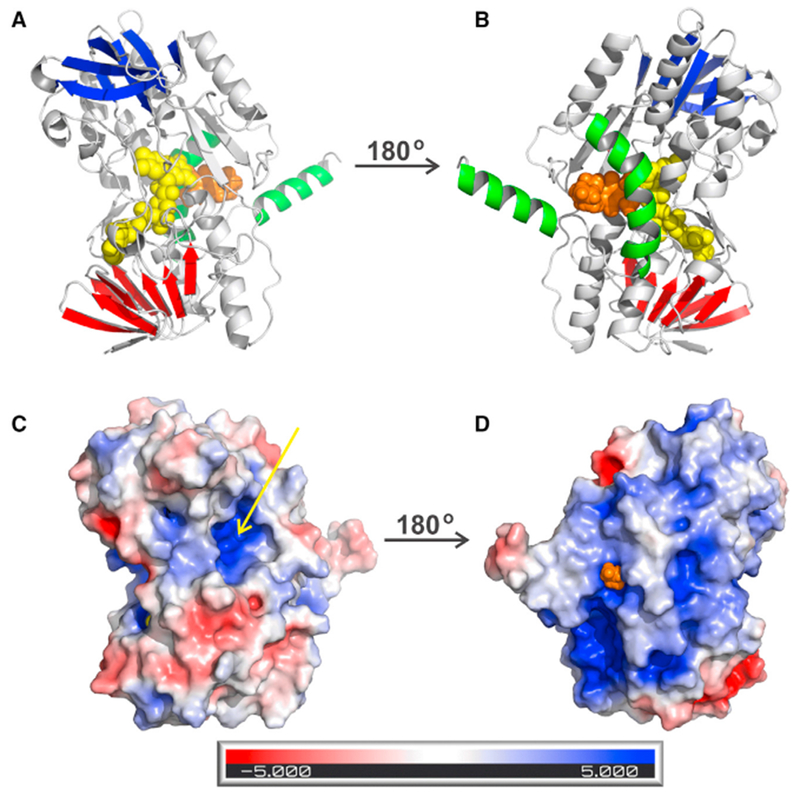
Cartoon and electrostatic surface diagrams of the matrix-facing (A and C) and membrane-facing (B and D) sides of the SQOR monomer. FAD and DCQ in (A) and (B) are shown as spheres, colored yellow and gold, respectively. β strands in the first and second Rossmann fold domains in (A) and (B) are colored red and blue, respectively; the C-terminal and penultimate C-terminal helices are colored green. The arrow in (C) points to the opening of a small channel leading to the active site. The tip of the decyl tail of DCQ in (D) is visible at the entrance to the CoQ-binding pocket. See also Figure S1.
The two active-site cysteine residues (Cys201, Cys379) lie just above the re-face of the flavin ring. In both the SeMet-substituted and native enzyme structures, these cysteine residues are linked by a bridging sulfur to form thiocystine (Cys-S-S-S-Cys) (Figures 2A and S2). These structures validate a previous proposal (Jackson et al., 2012) that a species containing thiocystine and reduced FAD is produced from 4a-adduct I when the catalytic intermediate is incubated under anaerobic conditions in the absence of a sulfane sulfur acceptor (Scheme 1, step 6). Subsequent addition of CoQ results in the rapid oxidation of the reduced flavin to produce enzyme that contains oxidized FAD and thiocystine (Scheme 1, step 7).
Figure 2. Active Site of Human SQOR.
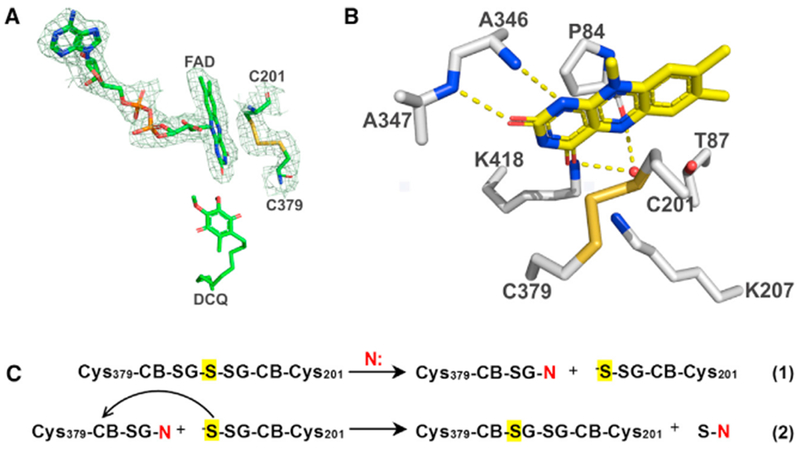
The redox-active site in SQOR (A and B) and the proposed mechanism for conversion of thiocystine-containing enzyme to a catalytically active cystine form (C). The 2Fo – Fc electron density omit map around FAD and the active-site cysteines in (A) is drawn at 1.0 σ. Hydrogen bonds in (B) are indicated by dashed yellow lines. Thiocystine-containing enzyme is converted to a catalytically active cystine form by reaction with a sulfane sulfur acceptor (N: = sulfite or glutathione) present in assay mixtures (C). The proposed activation mechanism results in the retention of the central sulfane sulfur in thiocystine (highlighted in yellow) in the active cystine form of SQOR. See also Figures S1–S3, S6, and S7.
The crystal structure of human SQOR provides clues about how the enzyme interacts with the membrane. The C-terminal helix is strongly amphipathic (Figure 3A) and protrudes from the globular body of the protein (see Figures 1A and 1B) with its hydrophobic face pointing outward (Figure 3C). Just upstream of the C-terminal helix, the penultimate helix also contributes a strongly hydrophobic outward-facing surface (Figure 3B); 16 of this helix’s 21 amino acids are nonpolar and 11 face outward (Figure 3C). The other 5 nonpolar residues (methionines and a tyrosine) point into a cavity that we suggest is the binding site for CoQ (Figure 5A). Thus, we propose that the two C-terminal helices lie in the plane of the mitochondrial membrane, with their hydrophobic faces communicating with the membrane interior. This monotopic mode of association would allow the enzyme to gain access to its hydrophobic electron acceptor, CoQ.
Figure 3. The C-Terminal Membrane-Binding Domain of SQOR.
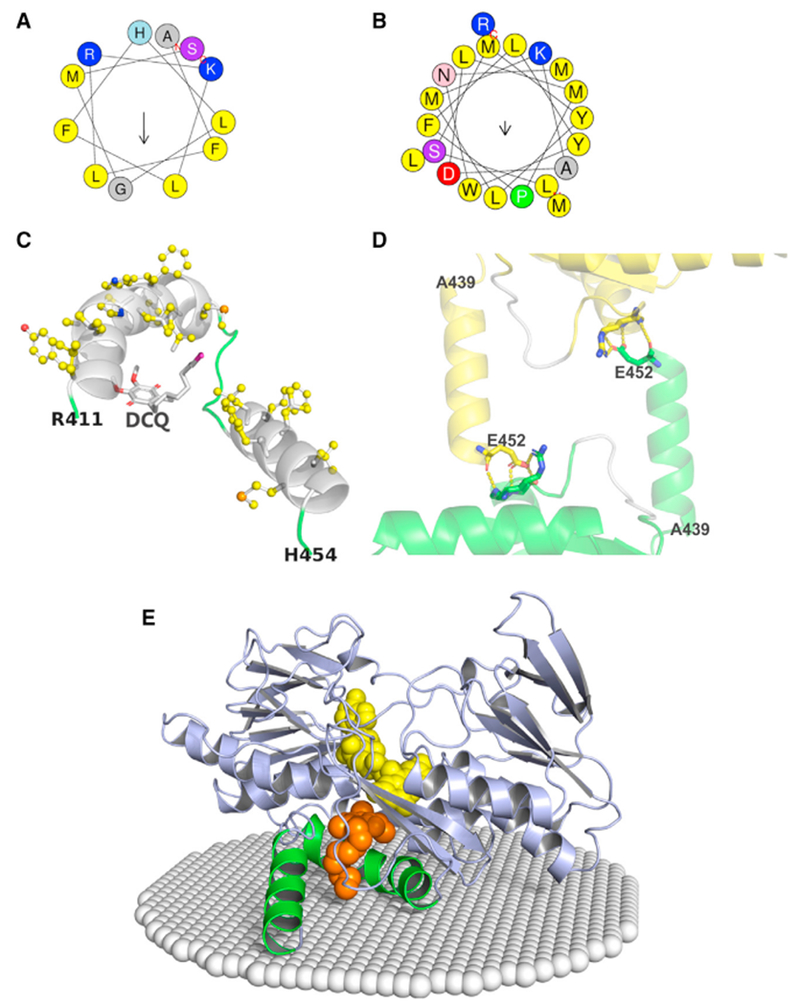
(A and B) Helical wheels of the C-terminal (A) and penultimate C-terminal (B) helices. Residues are color coded according to the type of amino acid: nonpolar (yellow); polar (purple or pink); positively charged (blue); negatively charged (red). Arrows represent hydrophobic moments. Analysis was performed using heliQuest (http://heliquest.ipmc.cnrs.fr/) (Gautier et al., 2008).
(C) The cartoon diagram provides a view from the membrane interior of the C-terminal and penultimate C-terminal helices (white), adjacent loop regions (green), and DCQ (sticks); nonpolar residues are shown as balls and sticks with carbons colored yellow. The decyl tail of DCQ points toward the membrane-facing surface; the tip of the tail (colored magenta) protrudes from the CoQ binding cavity.
(D) Crystallographic packing constraints on the C-terminal helix (Ala439-Glu452). Chain A from one asymmetric unit and an adjacent symmetry-related chain D are shown as cartoons colored green and yellow, respectively, except for a portion of a loop (400-405), which is colored white in both chains. The C-terminal helix touches loop 400-405, but makes no specific contacts. Glu452, the Arg411- Ser413 peptide backbone, and the side chains of Arg411 and Ser413 are shown as sticks. Hydrogen bonds are indicated by the yellow dotted lines.
(E) The cartoon diagram shows the spatial position of SQOR in the inner mitochondrial membrane (white spheres) that was calculated using the PPM server (http://opm.phar.umich.edu/server.php) (Lomize et al., 2011). The C-terminal and penultimate C-terminal helices are colored green; all other regions are colored blue. FAD and DCQ are shown as spheres, colored yellow and gold, respectively.
Figure 5. View of the CoQ-Binding Pocket and the Proposed Mechanism for Electron Transfer from Reduced FAD to DCQ.
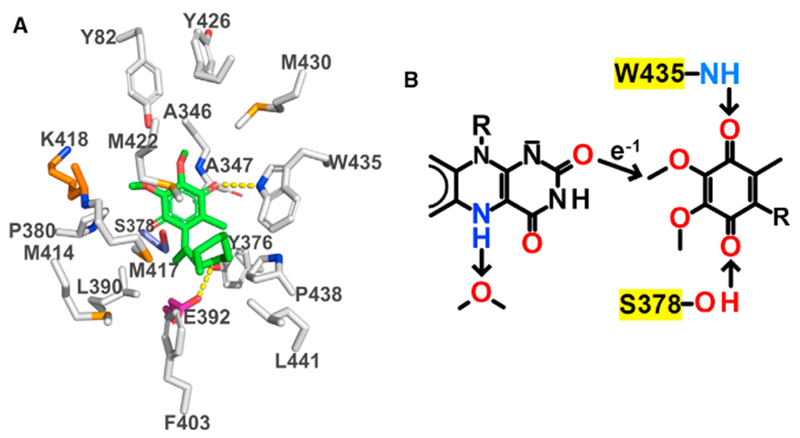
(A) Carbons in residues in the CoQ-binding pocket are colored according to residue type: nonpolar, white; negatively charged, magenta; positively charged, orange; polar, blue; carbons in DCQ are colored green. Hydrogen bonds are indicated by dashed yellow lines.
(B) The arrow shows the single through-space jump from FAD:O2 to DCQ:C3M predicted by the program HARLEM. Trp435:NH1, Ser378:O, and a water molecule are proposed to act as proton donors or acceptors, as discussed in the text.
See also Figures 2A and 2B.
The crystal structure deviates from the model described above, in that the C-terminal and penultimate helices are not quite coplanar (Figure 3C). However, the position of the C-terminal helix is likely perturbed by a crystal contact with the penultimate helix in a symmetry-related molecule. Thus, the last residue in the C-terminal helix, Glu452, forms hydrogen bonds with residues (Arg411, Leu412, Ser413) in the penultimate helix of the adjacent chain (Figure 3D). In the absence of this crystal-packing constraint, the C-terminal helix, being only loosely tethered to the remainder of the protein, can likely move so as to become coplanar with the penultimate helix, forming the membrane-binding surface. The proposed model for the insertion of human SQOR into the mitochondrial inner membrane is supported by a model calculated using the PPM server (http://opm.phar.umich.edu/server.php) (Lomize et al., 2011) which shows the penultimate helix bound to the membrane. The C-terminal helix is also in contact with the membrane at its C terminus but the N terminus is ~8 Å above the membrane surface (Figure 3E). The latter is consistent with the modest movement needed to achieve coplanarity of the two helices.
The matrix-facing (membrane-distal) surface of SQOR is located above the re-face of the flavin ring and features a small opening that provides access to an active-site cysteine (Figures 1A and 1C). The membrane-facing surface lies on the opposite side of the protein, above the si-face of the flavin ring (Figures 1B and 1D) and contains the entrance to a hydrophobic pocket, which we predict is responsible for binding CoQ. Electron density consistent with an alkyl tail is observed in this pocket, regardless of whether or not the enzyme was co-crystallized with decyl-CoQ (DCQ), suggesting that the site is occupied by some unknown detergent or lipid species that competes with DCQ; this species may have been carried through the purification or introduced during crystallization. However, DCQ could be convincingly modeled into this pocket using either Flare 1.0 (Cresset) or dockingserver.com (Figure S3), suggesting that this pocket indeed serves as the CoQ-binding site. The docked DCQ structure (Flare 1.0) is included for reference in some of this paper’s figures. The DCQ molecule lies just below the si-face of the flavin ring (Figure 2A) with the methyl carbon of its quinone ring, C3M, lying 3.7 Å away from the O2 position in the isoalloxazine ring of FAD.
The membrane-facing and matrix-facing surfaces of human SQOR have profoundly different electrostatic characters. The membrane-facing surface is characterized by hydrophobic patches and the hydrophobic opening to the CoQ-binding cavity (Figure 1D). This surface also contains large regions of positive charge, which may interact with negatively charged surface of the phospholipid bilayer. In contrast, the matrix-facing surface exhibits significant negatively charged regions, one of which is associated with the N-terminal Rossmann fold domain. The negatively charged regions cluster around a centrally located bowl-shaped surface depression that is highly electropositive. In the center of this surface depression lies a small opening that provides access to one of the redox-active cysteine residues (Figure 1C).
SQOR Quaternary Structure
The four SQOR chains in the crystal asymmetric unit form a roughly square structure (Figure 4). We questioned whether any of the packing interactions seen in SQOR crystals might reflect physiologically relevant interactions. Given that SQOR is a monotopic membrane protein, complex formation would require that the membrane-facing surfaces of all protomers are on the same face of the complex and coplanar. For example, the asymmetric unit of A. aeolicus SQOR contains two trimers; within each, the membrane-binding surfaces of the three monomers are on the same face of the trimer and coplanar (Figure S4), suggesting that the membrane-bound enzyme is a trimer (Marcia et al., 2009). This feature is not, however, observed for any pair of adjacent monomers in the asymmetric unit of human SQOR. Potential complexes can also be evaluated using such criteria as buried surface area (Krissinel and Henrick, 2007). Thus, to investigate candidate biological assemblies involving crystallographic symmetry operations, all possible dimeric assemblies of human SQOR monomers were analyzed based on the orientations of the membrane-binding surfaces, as well as buried surface area and interaction energies, using the PISA application (qtPISA) in the CCP4 Software Suite (Krissinel, 2015). No dimeric assemblies were found with the membrane-binding surfaces on the same face of the dimer and coplanar, nor were any dimers inferred from calculations of interaction energies.
Figure 4. Arrangement of the Four SQOR Monomers in the Crystallographic Asymmetric Unit.
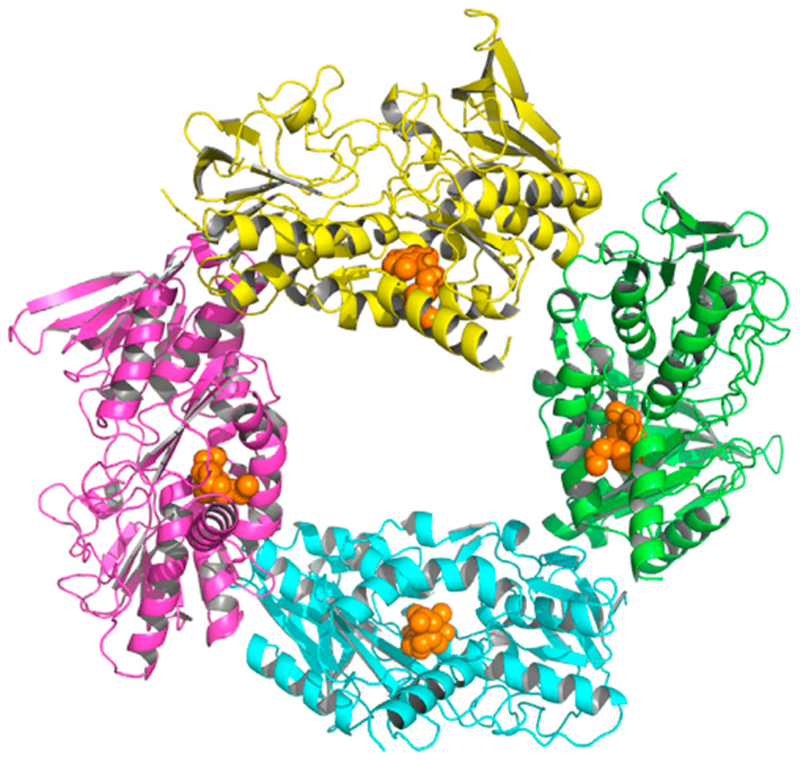
Chains A, B, C, and D are shown as cartoons colored green, cyan, magenta, and yellow, respectively. DCQ is shown as spheres, colored gold. See also Figures S4 and S5.
Cherney et al. (2010) previously identified a potential biological dimer of A. ferrooxidans SQOR by PISA analysis using the PDBePISA server (Figure S5A). The only interaction holding the postulated dimer together is an edge-on association of two β sheets, and the buried surface area is not large. More importantly, although the membrane-binding surfaces of the protomers are on the same face of the dimer, they are not coplanar. The latter is readily apparent in the model calculated for the interaction of the proposed biological dimer with the membrane using the PPM server (http://opm.phar.umich.edu/server.php) Lomize et al., 2011) which shows limited contact of only one monomer with the membrane surface (Figure S5B).
PISA analysis using qtPISA identified a dimeric assembly of human SQOR monomers with a similar anti-parallel β sheet at the dimer interface, formed by equivalent β strands from each protomer. The membrane-binding surfaces in the hypothetical human dimer are not coplanar. Thus, the model calculated by the PPM server shows interaction of only one monomer in this dimer with the membrane surface (Figure S5C), similar to the model calculated for monomeric human SQOR (Figure 3C).
In contrast to results obtained for the A. ferrooxidans and human dimers, the model calculated by the PPM server for the proposed A. aeolicus biological assembly shows that the membrane-binding surfaces of all protomers in the proposed biological trimer are in contact with the membrane (Figure S4B).
The FAD Binding Site
FAD is bound in an extended conformation. Its interactions with the protein include 12 hydrogen bonds (Figure S6) and electrostatic interactions with two helix dipoles. Two basic residues, Lys207 and Lys418, lie above the flavin’s re- and si-face, respectively (Figure 2B) with their respective ε-amino groups 3.2 and 3.3 Å from a carbonyl oxygen in the isoalloxazine ring (O4). Lys207 and Lys418 are part of a cluster of four lysine residues near FAD; the other two lysines, Lys200 and Lys344, are located near the ribityl chain of FAD (Figure 6A). FAD forms hydrogen bonds between its flavin ring atoms N1 and O2 and main chain atoms of Ala346 and Ala347, respectively, while FAD:O4 hydrogen bonds to a water molecule that is also hydrogen bonded to Pro84:O (Figure 2B). This water is close to FAD:N5 and is likely mechanistically significant (see Discussion). The N terminus of helix α11 points toward the N(1)-O(2) position of the FAD ring (Figure S7), so that its helix dipole will help to stabilize the electrophilic character of the flavin ring and the anionic form of the flavin hydroquinone. Another helix dipole interaction occurs with helix α1, the N terminus of which points toward the pyrophosphate group of FAD and partially neutralizes its charge. Helices α11 and α1 form part of the N-terminal Rossmann fold (Figure S1).
Figure 6. Substrate Access to the H2S-Oxidizing Active Site.
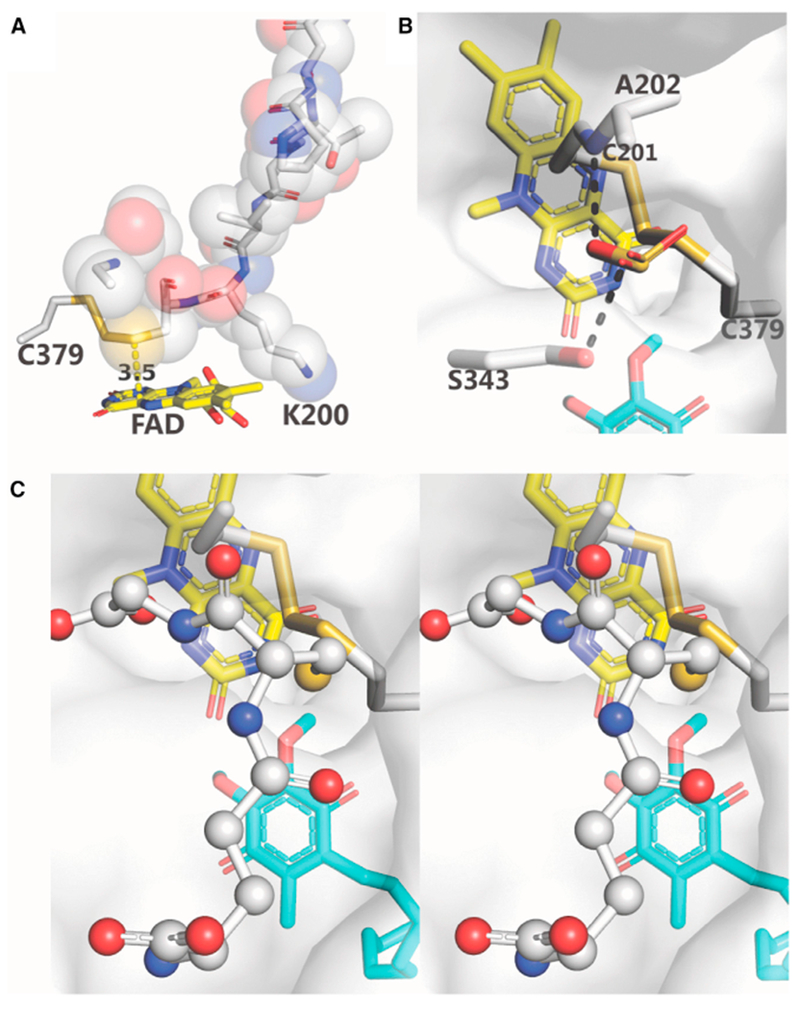
(A) The loop, P195-G203, is shown in sticks and spheres; Cys379 and FAD are shown in sticks. The distance between Cys201:SG and FAD:C4a (3.5 Å) is indicated. Lys344 (not shown) is behind V199-K200.
(B and C) Mono and wall-eye stereoviews of the docking of sulfite (B) and glutathione (C), respectively, to a proposed binding site. The semitransparent protein surface is colored white. Hydrogen bonds are indicated by dashed black lines. Sulfite, FAD, DCQ, and amino acid residues are shown as sticks. Glutathione is shown as ball and sticks.
See also Figures S1 and S8.
The CoQ-Binding Pocket
The entrance to the CoQ-binding pocket is located on the membrane-facing surface of human SQOR (Figure 1D). Within the pocket, the docked molecule of DCQ is surrounded by hydrophobic residues, including five from the penultimate C-terminal helix, along with ten other hydrophobic residues (Figure 5A). Two charged residues are found in the vicinity of DCQ: Lys318, whose side chain points away from the CoQ-binding pocket toward the flavin ring, and Glu392, which forms a hydrogen bond with Tyr376:O. The polar quinone ring of DCQ is inserted deep into the pocket and is close to the isoalloxazine ring of FAD (Figure 2A). The O2 carbonyl oxygen in the quinone ring of DCQ is hydrogen bonded to Trp345:NE1; the other carbonyl oxygen, O5, is close to Ser378:OG (2.8 Å) but not properly oriented for hydrogen bond formation. The hydrophobic decyl tail of DCQ threads through the pocket opening toward the outside of the protein.
Substrate Access to the Active-Site Redox Centers
Solvent access to the re-face of the flavin ring is obstructed by a nine-residue loop (195-203) from the second Rossmann fold (Figures 6A and S1). The loop includes the proximal active-site cysteine (Cys201) and serves to align Cys201:SG directly above and 3.5 Å from FAD:C4a. Importantly, these two atoms are positioned appropriately to form a 4a-thiolate flavin adduct (4a-adduct I), a postulated catalytic intermediate (Scheme 1, step 2). The distal active-site cysteine (Cys379) communicates with the matrix-facing surface of the protein via a small channel (Figures 6B and 6C), which is well-positioned to provide substrate access required for the initiating nucleophilic attack of HS− at Cys379:SG (Scheme 1, step 1). In addition, this channel affords access to sulfane sulfur acceptors (sulfite, glutathione), which must contact the distal cysteine in 4a-adduct I to mediate the postulated sulfur transfer step (Scheme 1, step 3). Intriguingly, the channel opening is located within a bowl-shaped electropositive surface depression (see Figure 1C). Since sulfite and glutathione are both negatively charged at physiological pH, this electropositive region might serve as a binding site for sulfite or glutathione and promote access to Cys379.
To evaluate this hypothesis, we used Flare 1.0 (Cresset) to dock sulfite to a candidate binding site centered within 6 Å of the opening of the small channel. Figure 6B shows the top docking pose. The docked sulfite molecule is bound within the channel opening, just below the protein surface, and forms hydrogen bonds to Ala202:N and Ser343:O. Very similar results were obtained for the top 6 sulfite poses, which differ only by rotation of the docked ligand about the central sulfur atom.
We next used Glide (Schrödinger Release, 2018-2) in XP- or peptide-mode to dock glutathione, a flexible peptide with nine rotatable bonds. The two docking modes resulted in very similar predictions and Glide gscores (−5.82 and −5.39 kcal/mol, respectively). Remarkably, in the top pose obtained with either docking mode, the SH group of glutathione is positioned within the opening of the small channel leading to the redox-active site (Figure 6C). The remainder of the glutathione molecule forms multiple hydrogen bonds with the protein and water molecules (Figure S8).
DISCUSSION
Human SQOR exhibits structural features, such as two tandem Rossmann fold domains, that are characteristic of the FDR family of proteins (Miller, 2013). The overall fold is most similar to that of prokaryotic H2S-oxidizing enzymes, as judged by RMSD values observed upon superimposition of the human SQOR structure with those of bacterial SQORs and FCSDs (RMSD ≈2.1–2.6 Å) (Table 2). FAD is bound noncovalently to human SQOR in an extended conformation, as seen in many FDR family members, with the notable exception of most prokaryotic H2S-oxidizing enzymes where the flavin is covalently attached to the protein via a thioether link to a cysteine residue (Table 2). The matrix-facing surface of human SQOR contains negatively charged regions that cluster around a central electropositive “bowl,” which we propose serves to funnel negatively charged substrates (HS− sulfite, glutathione) to the hydrophilic H2S-oxidizing active site. On the opposite, membrane-facing surface of the protein, large positively charged regions and hydrophobic patches can be found, framing the entrance to a hydrophobic CoQ-binding pocket. Human SQOR is a basic protein (isoelectric point [pI] = 9.02) and exhibits a net positive charge at physiological pH, unlike bacterial H2S-oxidizing enzymes that are weakly acidic proteins (pI ≈ 5.3–6.5) and negatively charged at pH 7.4 (Table 2). Nevertheless, bacterial SQORs exhibit pronounced differences in electrostatic potential between their membrane-facing and solvent-facing surfaces (Cherney et al., 2010; Marcia et al., 2009), similar to human SQOR.
Table 2.
Comparison of Human SQOR with Bacterial H2S-Oxidizing Enzymes
| Structural Comparison |
Electrostatics |
Distance (Å) |
Active-Site
Accessibility |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Protein (pdb code) | RMSD (Å) | No. of Cα Matches | FAD Attachment | Net Charge (pl)a | Membrane- vs Solvent-Facing Surfacesb | CysP:SG to CysD:SGc | FAD:C4a to |
Loop Above re-face of FADd | re-face of FAD | Active-Site Cysteines | |
| CysP:SG | CysD:SG | ||||||||||
| Human SQOR (PDB: 6MO6) | – | – | noncovalent | positive (9.02) | polarized | 3.4 | 3.5 | P195-G203 | obstructed | CysD: close to the surface, accessible via a short channel; CysP: shielded | |
| A. vinosum FCSD (PDB: 1fcd) | 2.60 | 245 | covalente | negative (5.28) | NA | 2.1 | 3.3 | P154-P162 | obstructed | on the surface, solvent exposed | |
| T. tepidum FCSD (PDB: 3vrd) | 2.61 | 241 | covalente | negative (5.55) | NA | 4.9 | 3.5 | P154-P162 | obstructed | on the surface, solvent exposed | |
| T. paradoxus FCSD (PDB: 5n1t) | 2.58 | 239 | covalente | negative (5.53) | NA | 4.7 | 3.5 | P186-P194 | obstructed | on the surface, solvent exposed | |
| A. aeolicus SQOR (PDB: 3hyw) | 2.25 | 212 | covalente | negative (6.53) | polarized | 8.1, 8.3, 9.0, 9.3f | 6.1, 6.2f | 5.7, 6.9f | I151-C156 | accessible cavity, accommodates sulfur polymerization products | accessible via channels |
| A. ferrooxidans SQOR (PDB: 3t31) | 2.91 | 248 | noncovalent | negative (6.48) | polarized | 6.7, 6.8g | 6.0 | 4.7, 5.2g | M155-C160 | partially shielded, but solvent accessible | accessible via channels |
| A. ambivalens SQOR (PDB: 3h8l) | 2.11 | 221 | covalente | negative (6.33) | ND | 6.5, 7.1g | 4.9 | 7.4, 7.6g | G154-E179 | partially shielded, but solvent accessible | accessible via a channel |
Net charge at pH 7.4 estimated based on the calculated isoelectric point (pI).
Solvent refers to the inner mitochondrial matrix (human SQOR) or the periplasmic space (bacterial SQORs), respectively. The property is not applicable (NA) for FCSDs, which are soluble proteins. Electrostatic surface properties were not determined (ND) for A. ambivalens SQOR because no interpretable electron density was observed for the C-terminal membrane-binding domain.
The proximal active-site cysteine (CysP) is closer to the N terminus of the protein than the distal cysteine (CysD). CysP:SG is also closer to FAD:C4a in human SQOR, FCSD and some bacterial SQORs. (CysP) = Cys201 (human); Cys161 (A. vinosum FCSD); Cys161 (T. tepidum FCSD); Cys193 (T. paradoxus FCSD); Cys156 (A. aeolicus SQOR); Cys160 (A. ferrooxidans SQOR); Cys178 (A. ambivalens SQOR). CysD = Cys379 (human); Cys337 (A. vinosum FCSD); Cys337 (T. tepidum FCSD); Cys364 (T. paradoxus FCSD); Cys347 (A. aeolicus SQOR); Cys356 (A. ferrooxidans SQOR); Cys350 (A. ambivalens SQOR).
The loop from the second Rossmann fold in human SQOR, FCSDs and bacterial SQOR’s includes CysP.
Thioether link between FAD:C8M and a cysteine residue: Cys42, A. vinosum FCSD; Cys42, T. tepidum FCSD; Cys73, T. paradoxus FCSD; Cys124, A. aeolicus SQOR; Cys129, A. ambivalens SQOR.
Two alternate conformations are observed for both CysP and CysD.
Two alternate conformations are observed for CysD.
Active-Site Architecture
Certain features of human SQOR indicate that the eukaryotic enzyme is an apparent molecular hybrid of structural and functional properties observed for soluble and membrane-bound bacterial H2S-oxidizing enzymes. Thus, the architecture of the hydrophilic H2S-oxidizing active site in human SQOR resembles that of soluble prokaryotic FCSDs. Specifically, the spacing between active-site cysteines, the positions of the cysteines relative to the flavin and the protein surface, and solvent accessibility in these enzymes are strikingly dissimilar from bacterial SQORs, which can be rationalized in terms of the different substrates, products, and catalytic mechanisms.
The spacing between active-site cysteines varies widely in H2S-oxidizing enzymes and is also sensitive to changes in the redox state of the cysteines (Table 2). In A. vinosum FCSD, which oxidizes H2S to the small product H2S2, the proximal and distal cysteines (CysP and CysD) are close and form a disulfide bridge (Chen et al., 1994), which likely represents the resting state in bacterial FCSDs. SG-SG distances are longer in otherwise highly similar FCSDs from T. tepidum and T. paradoxus (RMSD ≤0.76 Å) because the cysteines are linked by two bridging sulfane sulfurs or present as a cysteine/cysteine persulfide pair (Hirano et al., 2012; Osipov et al., 2018). In human SQOR, an intermediate SG-SG distance is seen with the cysteines being linked by a single bridging sulfur to form thiocystine. This close separation is consistent with the similar catalytic properties of human SQOR and FCSDs and our proposal that the disulfide bridge corresponds to the catalytically active form of human SQOR (Jackson et al., 2012). In contrast, large SG-SG separations (>6.5 Å) are observed in bacterial SQORs (Table 2) and provide space for sulfur polymerization products that are produced only by these enzymes.
The positions of CysP and CysD in human SQOR and FCSDs relative to the flavin and the protein surface, respectively, exhibit mechanistically significant differences from those observed in bacterial SQORs. Thus, in human SQOR, CysP:SG is located directly above FAD:C4a, at a distance of 3.5 Å, and is optimally positioned to form a 4a-thiolate flavin adduct, a postulated catalytic intermediate (see Scheme 1); a similar positioning is observed in FCSDs. In contrast, the corresponding distances in bacterial SQORs are much longer (4.7–7.6 Å), and apparently incompatible with 4a-thiolate adduct formation (Table 2). CysD is close to the matrix-facing surface in human SQOR, with CysD:SG only a few angstroms from a short channel that mediates both substrate entry from and product release into the mitochondrial matrix. Similarly, CysD is on the protein surface in FCSDs and directly exposed to solvent. In contrast, CysD:SG is much further from the surface in bacterial SQORs (10–12 Å), and the proposed substrate access channels are considerably longer than in human SQOR (Brito et al., 2009; Cherney et al., 2010; Marcia et al., 2009). Furthermore, separate channels in bacterial SQOR’s have been postulated for H2S entry from the periplasmic space and release of the hydrophobic polysulfide product into the bacterial inner membrane (Cherney et al., 2010; Marcia et al., 2009).
Differences in the solvent accessibility of the H2S-oxidizing active center in human SQOR and FCSDs versus bacterial SQORs also correlates with differences in substrate/product preference. Thus, access to the active site in both human SQOR and FCSDs is obstructed by a nine-residue loop that includes the proximal active site cysteine. Although corresponding loops are also found in bacterial SQORs (Table 2), they only partially shield the re-face of FAD in A. ferrooxidans and A. ambivalens SQOR (Brito et al., 2009; Cherney et al., 2010) while the re-face of the flavin in A. aeolicus SQOR is completely unobstructed and faces a 15-Å-long solvent-accessible cavity that provides space for sulfur polymerization products (Marcia et al., 2009). It is worth noting that the re-face of the flavin in most other members of the FDR family typically faces an open and freely accessible binding site for pyridine nucleotide substrates (Miller, 2013).
The observed similarity in the architecture of the H2S-oxidizing active site in human SQOR and prokaryotic FCSDs is consistent with predictions made by Marcia et al. (2010) based on sequence alignments and phylogenetic tree computation on representative sequences of SQORs (eukaryotic and prokaryotic) and bacterial FCSDs.
Proposed Binding Site for Sulfane Sulfur Acceptors
Unlike bacterial H2S-oxidizing enzymes, human SQOR requires a sulfane sulfur acceptor. We demonstrate that human SQOR contains unique structural features: a bowl-shaped electropositive hollow that serves as a binding site for sulfite and glutathione and postulated to funnel negatively charged substrates to its hydrophilic H2S-oxidizing active site. This site is connected to Cys379 (CysD) by a narrow channel (Figures 6B and 6C), which allows direct access for small substrates such as sulfite, but limits active-site access to only the thiol group for the larger glutathione substrate. Being just large enough to allow access for the actual sulfane sulfur acceptor, this narrow channel opening may shield the active site and protect reactive sulfur intermediates from undesirable reactions with other metabolites.
The modeled complexes of human SQOR with sulfite or glutathione resemble catalytically competent enzyme·substrate complexes but must differ from the actual complexes formed during turnover in at least two ways. Firstly, the modeled complexes contain oxidized FAD instead of a 4a-thiolate flavin adduct with Cys201 (Scheme 1). Secondly (and more importantly), Cys379 is present as a persulfide (Cys379SS−) in 4a-adduct I, but in the modeled complexes forms thiocystine with Cys201 via a bridging sulfane sulfur. The modeled complexes place the nucleophilic sulfur atom of sulfite or glutathione too far away from the sulfane sulfur in thiocystine for efficient transfer (4.7 or 6.7 Å, respectively). However, thiocystine formation likely causes Cys379 to move away from the channel opening; in the persulfide form of this residue found in 4a-adduct 1 we expect the sulfane sulfur of Cys379SS− to be ~1–3 Å closer to the sulfane sulfur acceptor, and thus suitably positioned for transfer.
Origin of Thiocystine and Proposed Conversion to Cystine during Turnover
Despite the similarity between the catalytic reactions and the H2S-oxidizing active sites of human SQOR and FCSDs (Jackson et al., 2012; Lu et al., 2017), the active-site cysteines in human SQOR are covalently linked by a bridging sulfur to form thiocystine, instead of being directly linked to form cystine, as observed with A. vinosum FCSD. We propose here a mechanism to explain the presence of thiocystine. We anticipate that expression of SQOR in E. coli initially generates recombinant enzyme containing oxidized FAD and cystine. This species is converted to 4a-adduct I upon reaction with H2S (Scheme 1, steps 1 and 2). Rearrangement of 4a-adduct I generates thiocystine plus reduced FAD (Scheme 1, step 6) which reacts with CoQ to yield enzyme containing oxidized FAD and thiocystine (Scheme 1, step 6), the species observed in SQOR crystals. The H2S required to initiate this cycle is likely produced endogenously by E. coli. The active-site cysteines in the crystal structure of recombinant A. ferrooxidans SQOR are covalently linked by five bridging sulfur atoms that form a branched pentasulfane. This active-site cysteine modification has also been attributed to reaction with endogenous H2S produced during expression (Cherney et al., 2010).
The thiocystine species can be rapidly converted back to an active form of the enzyme, consistent with the lack of an initial lag in catalytic assays that monitor the reduction of CoQ1 in the presence of H2S and a sulfane sulfur acceptor (sulfite or glutathione) (Augustyn et al., 2017; Jackson et al., 2012). We propose that the catalytically active cystine form of human SQOR is rapidly produced under assay conditions by reaction of thiocystine-containing enzyme with the sulfane sulfur acceptor (N) (Figure 2C). Nucleophilic attack of N: at the sterically accessible SG position of Cys379 results in the formation of Cys379SN, accompanied by the expulsion of Cys201SS−, a good leaving group (Figure 2C, step 1). Nucleophilic attack of Cys201SS− at the CB position in Cys379SN produces cystine plus thiosulfate or glutathione persulfide, depending on whether N: corresponds to sulfite or glutathione, respectively (Figure 2C, step 2).
Mechanism of Electron Transfer between FAD and CoQ
Analysis using the program HARLEM (Kurnikov, 2018) predicts that electron flow between FAD and DCQ involves a single through-space jump from FAD:O2 to DCQ:C3M (Figure 5B), the point of closest approach (3.7 Å) between the isoalloxazine and quinone rings. We propose that the reaction is initiated by 1-electron transfer from the anionic form of reduced FAD to produce a neutral flavin radical (FADH− ⇒ FADH⋅ + e−1). Consistent with this idea, the pKa for ionization at N(1) in free reduced 1,5-dihydroflavins is 6.7, making the anionic species readily accessible at physiological pH, and in fact only the anionic reduced flavin has been detected in 15NMR studies with various reduced flavoproteins (Muller, 1992). The α11 helix dipole points toward the N(1)-O(2) position of the FAD ring (Figure S7), stabilizing the anionic flavin hydroquinone, and additional stabilization may be provided by nearby lysine residues (Figure 2B).
We observe a water molecule 3.0 Å from FAD:N5, and propose that it serves as a proton acceptor in the second electron transfer step, which requires loss of a proton from the N(5) position of the flavin radical (FADH⋅ ⇒ FAD + H+ + e−1). We also propose that two residues in the CoQ-binding pocket serve as immediate proton donors to the carbonyl oxygens during reduction of DCQ (Figure 5B); specifically, Trp345:NE1 is 3.1 Å from DCQ:O2 and Ser378:OG is 2.8 Å from DCQ:O5. These reactions will require concomitant proton transfer from nearby water molecules to reprotonate the protein side chains; no water molecules are seen in the CoQ-binding pocket, but this is not surprising, given the modest resolution of this structure. However, waters are found in the CoQ-binding pocket in the higher-resolution bacterial SQOR structures (PDB: 3hyw and 3t31).
The Biological Assembly of Human SQOR in the Inner Mitochondrial Membrane
SQOR catalysis couples H2S oxidation to CoQ reduction. Oxidized CoQ10 gains access to the hydrophilic H2S-oxidizing active site via a 13 Å long hydrophobic tunnel, and, once reduced, returns to the inner mitochondrial membrane. This cyclic process requires that SQOR is stably associated with the membrane. Membrane association might conceivably be enhanced by cooperative binding of several SQOR monomers to form a complex on the membrane surface; any such complex would require the coplanar orientation of the membrane-binding surfaces of each protomer. However, no such complex is observed in the crystals of human SQOR or in assemblies generated by applying symmetry operations to the monomers in the asymmetric unit, and we therefore conclude that human SQOR exists as a monomer in cells. This differs from a previous proposal that SQOR exists as a dimer, based on a molecular mass estimated by gel filtration (Mishanina et al., 2015); however, this result fails to account for bound detergent, which will lead to an inflated estimate of the protein’s Stokes radius (le Maire et al., 2008). It is also possible that artifactual dimers can arise in detergent solutions via nonspecific association of the protein’s hydrophobic membrane-binding surfaces, as observed for the dimer in the asymmetric unit of A. ferrooxidans SQOR (PDB: 3T2Z) (Cherney et al., 2010). It is worth noting that the PDBePISA server predicts that the crystallographic dimer in PDB: 3T2Z is stable in solution.
Concluding Remarks
SQOR catalyzes the crucial first step in the mitochondrial metabolism of H2S, and we present here the first crystal structure for any eukaryotic SQOR. This structure supports the proposed model for catalysis and reveals a unique binding site for sulfane sulfur acceptors, unraveling the molecular basis for the enzyme’s ability to catalyze sulfane sulfur transfer reactions with structurally diverse acceptors: sulfite and glutathione. Other features of the eukaryotic enzyme comprise an apparent molecular hybrid of structural and functional properties observed for prokaryotic soluble (FCSDs) and membrane-bound (SQORs) H2S-oxidizing enzymes. The structure of human SQOR opens the door for structure-based drug design.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Marilyn S. Jorns (mj27@drexel.edu).
METHOD DETAILS
Expression and Purification of Human SQOR
Native SQOR was expressed as previously described (Jackson et al., 2012). Briefly, E. coli BL21(DE3) cells harboring a pET23a-based plasmid with a synthetic version of the gene (sqrdl) encoding human SQOR (pET23a_MATopzSQOR) and a plasmid with genes (cpn10, cpn60) for cold-adapted chaperonins from Oleispira antarctica (pCPN10/60) were grown in Terrific Broth (TB) media at 15°C until the A595 reached 1.1. Native SQOR expression was induced with isopropyl β-D-thiogalactopyranoside (0.5 mM). Cells were harvested 20 h after induction. The cell pellets (~100 g from 9 L of culture) were stored at −80°C. SeMet-substituted enzyme was expressed by growing E. coli BL21 (DE3)/pET23a_MATopzSQOR/pCPN10/60 cells in auto-induction media (PASM-5052) (Doublie, 2007; Studier, 2005). Cells were grown with shaking at 37°C until A595 had reached 1.3 (24-36 h). The temperature was then lowered to 15°C. Cells were harvested after 91 h of growth at 15°C. The cell pellets (40 - 60 g from 12 L of culture) were stored at −80°C.
SQOR was purified essentially as previously described (Jackson et al., 2012), except as noted. All steps of the purification were conducted at 4°C. Cell pellets (~50 g) were suspended in Tris-acetate buffer, pH 7.6 containing 0.5 M sucrose and 0.1 mM EDTA (75 mL), and then mixed with lysozyme (0.5 mg/mL) plus a cocktail of nucleases and protease inhibitors (DNAase, 20 μg/mL; RNAase, 20 μg/mL; 5 mM magnesium sulfate; soybean trypsin inhibitor, 12.6 μg/mL; aprotinin, 2 μg/mL; phenylmethanesulfonylfluoride, 25 μg/mL; and tosyllysine chloromethylketone, 3 μg/mL). The suspension was incubated with stirring for 20 min and then sonicated (Branson Model 350, power setting = 6, duty cycle = 40%) for a total of 450 s in 30-s intervals, separated by 30-s cooling periods. Cell debris was removed by centrifugation at low speed (10 min at 10,000g).
Membrane-bound SQOR is found in the low speed supernatant which contains membrane fragments generated during sonication. Modifications have been introduced in the protocol for enzyme solubilization and subsequent chromatography steps, except as otherwise noted. The undiluted low-speed supernatant was modified to contain 50 mM potassium phosphate buffer, pH 6.8, 300 mM NaCl and 1% (w/v) DM. The sample was stirred for 2 h at 4°C to solubilize SQOR and then centrifuged at high-speed after adjusting the pH to 8. The high-speed supernatant was loaded onto two tandem 5 mL HiTrap IMAC columns (GE Healthcare), previously equilibrated with 50 mM Tris-HCl (pH 8) containing 0.05% (w/v) DHPC, 300 mM NaCl, 10% (w/v) glycerol, 20 mM imidazole and 0.01% sodium azide (buffer A). Buffer B contained 0.03% DHPC and 600 mM imidazole but was otherwise identical to buffer A. The column was washed with 5 column volumes of a linear gradient from 0 to 6-7% buffer B. SQOR was eluted with 2 column volumes of a linear gradient from 7 to 100% buffer B and stored at −80°C.
The IMAC column eluate was dialyzed using a 2K MWCO membrane (SpectraPor®) versus 50 mM potassium carbonate/bicarbonate buffer (pH 9.6) containing 2% (w/v) glycerol, 30 mM NaCl and 5% (w/v) PEG 3350. The pH of the eluate was adjusted to pH 9.4 (if necessary) and the sample was then centrifuged (10 min at 30,000g). The eluate was purified using a 50 mL HiLoad 26/10 Q Sepharose High Performance anion exchange column (GE Healthcare), as previously described (Jackson et al., 2012), except for the following changes. Buffer A contained 0.01% sodium azide and 0.03% (w/v) DHPC. Buffer B contained 0.03% (w/v) DHPC. The column was washed with 1 column volume of buffer A (50 mM Tris-HCl buffer, pH 8.0, containing 2% glycerol, 100 mM sodium chloride, 0.01% sodium azide, and 0.03% DHPC) and then with 5 column volumes of a linear gradient to 100% buffer B (50 mM Tris-HCl buffer, pH 8.0, containing 2% glycerol, 1.0 M sodium chloride, and 0.03% DHPC). Larger volumes of purified SQOR (>40 mL) were first concentrated using 2K MWCO dialysis tubing over a bed of solid PEG 3350. Additional concentration to achieve a desired final concentration (9-15 mg/mL) was performed using a 10 K Macrosep Advance Centrifugal Device (Pall Life Sciences), as previously described (Jackson et al., 2012). The purified enzyme was stored in 50 mM Tris-HCl (pH 8), 0.03% (w/v) DHPC, 2% (w/v) glycerol, ~0.002% (w/v) sodium azide and 250-300 mM NaCl at −80°C. This method yields 0.5-0.7 mg of purified human SQOR per g of cell pellet.
MALDI Mass Spectrometry
Purified samples of SeMet-substituted and native SQOR were analyzed using MALDI mass spectrometry at the Wistar Proteomics Facility. Samples of SeMet-substituted enzyme exhibited >98.8% incorporation of the expected amount of selenomethionine.
Crystallization of SQOR
Native SQOR was initially crystallized using reagent numbers 57 and 58 from the Hampton Research sparse reagent kit (MembFac HT™ HR2-137). Crystallization trials were conducted using MRC Maxi 48-well sitting drop plates (Hampton Research) that contained 100 μL of reagent in each reservoir. A 1 μL aliquot of 0.1% (w/v) DM in H2O was added to the drop well, followed by 1 μl of protein stock (~ 10mg/mL) and then 1 μL of reagent, with no mixing. Plates were sealed with tape, protected from light, and incubated at room temperature (17 - 28°C). Obelisk-shaped crystals were observed after ~2 months, with dimensions of 0.015 × 0.015 × 0.05 mm3.
Refinement and optimization of the crystallization of native and SeMet-substituted SQOR were conducted using the hanging drop vapor diffusion method in VDX™ plates (Hampton Research). Solutions of monoolein were prepared as a 10.66% (w/v) stock in ethanol and stored at −35°C. Stock solutions of DCQ (31 mM) were prepared in 10% DHPC solution. The final reservoir solution consisted of 500 μL of 20% (w/v) glycerol, 0.1 M sodium acetate trihydrate pH 4.6, 0.3-0.5 M ammonium acetate, 7-10% (w/v) PEG 4K, 0.1% (w/v) DHPC. The precipitant solution for SeMet-substituted enzyme also contained 310 μM DCQ and 10 mM sodium thiosulfate. A 1 μL aliquot of monoolein stock solution was added to the coverslip and air dried for 3 minutes, followed by the sequential addition (with no mixing) of 1 μL of native (9-10 mg/mL) or SeMet-substituted (11-13 mg/mL) enzyme, and 1 μL of the reservoir condition. Diffraction-quality obelisk-shaped crystals were obtained after ~1-2 weeks of incubation at room temperature. The dimensions of crystals obtained with SeMet-substituted enzyme were 0.08 × 0.08 × 0.5 mm3. The linear dimensions of crystals obtained with native enzyme were typically 50-70% smaller.
Data Collection, Phases and Refinement
X-ray diffraction data were collected at a temperature of 100 K and a wavelength of 0.97918 Å at beamlines 24-ID-E and 24-ID-C of the Advanced Photon Source (APS). Datasets for each individual crystal were initially processed using iMOSFLM (Battye et al., 2011) and purposely indexed as P3. Combinations of datasets were then evaluated using the CCP4 program BLEND (Foadi et al., 2013; Winn et al., 2011). Eventually, two paired datasets were chosen for the SeMet-substituted enzyme, re-indexed using Pointless, and then scaled using Aimless into the space group P6122 (Evans, 2011; Evans and Murshudov, 2013).
During data collection, crystals were aligned with the long unit-cell axis (c) approximately parallel to the rotation axis. One of the best diffracting crystals, however, happened to lie with its c-axis essentially coincident with the rotation axis, leading to a sizable missing cone of data (Dauter, 1999). To overcome this problem, these data were combined with data from a second, more weakly diffracting crystal, yielding SeMet combined dataset 1, which was used for phasing (Table 1). Two data sets measured from different positions on the strong crystal were combined to give SeMet combined dataset 2, which was used for refinement of the SeMet enzyme structure.
Selenium positions were found and refined using SHELX with a resolution search range of 15 to 4 Å (Sheldrick, 2008); 11 of the 13 SeMet residues per chain were found. PHASER was then used to refine the phases, followed by solvent flattening using the Parrot density modification algorithm in CCP4 (McCoy et al., 2007). The resulting phases were applied to SeMet combined dataset 2 and refined, yielding a map that was clearly interpretable for initial chain tracing using BUCANEER (part of CCP4i). Approximately 98% of the residues in the four molecules of SQOR in the asymmetric unit were built. Initial iterative refinement and manual model correction was then done using REFMAC5 and Coot (Emsley and Cowtan, 2004; Vagin et al., 2004). Final refinement stages were performed with PHENIX (Adams et al., 2010), including PHENIX.autobuild (Zwart et al., 2008) and PHENIX.refine (Afonine et al., 2012). A single dataset was collected for a crystal of the native enzyme (Table 1), and its structure determined by molecular replacement (MR) using PHASER. During refinement, 5% of all reflections were flagged as “free” (Brünger, 1992). The stereochemical quality of the refined models was assessed with MolProbity (Chen et al., 2010) and the wwPDB Validation server http://wwpdb-validation.wwpdb.org. Statistics generated from X-ray crystallography data processing, refinement, and structure validation are displayed in Table 1.
All structural figures were prepared using Pymol 2.0.6. Hydrogen bonds involving ligands and/or water molecules were assigned using Maestro 11.5 (Schrödinger). The 2Fo - Fc electron density omit maps are drawn at 1.0 σ; the electron density was carved using a 2 Å carving radius. Polder omit maps (Liebschner et al., 2017) were calculated in Phenix. Maps are drawn at 3.0 σ; the electron density was carved using a 2 Å carving radius. Electrostatic surface potentials were calculated using the APBS electrostatic plugin to Pymol 2.0. A topology diagram was generated using TopDraw, a program within the CCP4 suite.
Docking Studies
Docking studies with DCQ and sulfite were performed using Flare 1.0 (Cresset). Glutathione was docked using Schrödinger Release 2018-2: Glide (Schrödinger, LLC, New York) in XP-mode or peptide-mode. Structure data files (Ideal SDF files), used in docking studies with DCQ and glutathione, were downloaded from the Protein Data Bank website. Docking studies with DCQ were also performed using dockingserver.com and a .smi file for DCQ that was generated from a SMILES string using EXCEL (Office 365).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistics generated from X-ray crystallography data processing, refinement, and structure validation are displayed in Table 1.
DATA AND SOFTWARE AVAILABILITY
Coordinates and structure factors have been deposited in the Protein Data Bank for SeMet-substituted SQOR and native SQOR under ID codes PDB: 6MO6 and PDB: 6MP5, respectively.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| E. coli BL21(DE3) | Merck-Novagen | Catalog # 69450 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Sulfide:quinone oxidoreductase (SQOR) | (Jackson et al., 2012)This paper | N/A |
| Monoolein (1-oleoyl-rac-glycerol) | Sigma-Aldrich | Catalog # M7765 |
| n-decyl-β-D-maltopyranoside (DM) | Anatrace | Catalog # D322S |
| 1,2-diheptanoyl-sn-glycero-3-phosphocholine (DHPC) | Anatrace | Catalog # D607 |
| Decylubiquinone (DCQ) | Enzo Life Sciences, Inc. | Catalog # BML-CM115 |
| HiTrap IMAC HP column | GE Healthcare | Catalog # 17092005 |
| Q Sepharose High Performance column | GE Healthcare | Catalog # 17101401 |
| L-selenomethionine | Tokyo Chemical Industry | Catalog # S0442 |
| Critical Commercial Assays | ||
| MembFac HT™ | Hampton Research | HR2-137 |
| Deposited Data | ||
| Crystal structure of Selenomethioine-substituted SQOR | This paper | PDB: 6MO6 |
| Crystal structure of Native SQOR | This paper | PDB: 6MP5 |
| Crystal structure of A. aeolicus SQOR | Marcia et al., 2009 | PDB: 3HYW |
| Crystal structure of A. ferrooxidans SQOR | Cherney et al., 2010 | PDB: 3T31 |
| Crystal structure of A. ferrooxidans SQOR | Cherney et al., 2010 | PDB: 3T2Z |
| Crystal structure of A. ambivalens SQOR | Brito et al., 2009 | PDB: 3H8L |
| Crystal structure of A. vinosum FCSD | Chen et al., 1994 | PDB: 1FCD |
| Crystal structure of T. tepidum FCSD | Hirano et al., 2012 | PDB: 3VRD |
| Crystal structure of T. paradoxus FCSD | Osipov et al., 2018 | PDB: 5N1T |
| Recombinant DNA | ||
| pET23a_MATopzSQOR | (Jackson et al., 2012) | N/A |
| Software and Algorithms | ||
| CCP4i | (Winn et al., 2011) | http://www.ccp4.ac.uk/ |
| iMOSFLM | (Battye et al., 2011) | http://www.ccp4.ac.uk/html/mosflm.html |
| BLEND | (Foadi et al., 2013) | http://www.ccp4.ac.uk/html/blend.html |
| REFMAC5 | (Vagin et al., 2004) | http://www.ccp4.ac.uk/html/refmac5.html |
| PHASER | (McCoy et al., 2007) | http://www.ccp4.ac.uk/html/phaser/html |
| ShelXC/D/E | (Sheldrick, 2008) | http://shelx.uni-goettingen.de/ |
| COOT | (Emsley and Cowtan, 2004) | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| PyMOL | Schrödinger | https://pymol.org |
| Phenix | (Adams et al., 2010) | https://www.phenix-online.org/ |
| Phenix.autobuild | (Zwart et al., 2008) | https://www.phenix-online.org/ |
| Phenix.refine | (Afonine et al., 2012) | https://www.phenix-online.org/ |
| Maestro 11.5 | Schrödinger | https://www.schrodinger.com/maestro |
| Flare 1.0 | Cresset | https://www.cresset-group.com/tag/structure-based/ |
| Glide (Release 2018-2) | Schrödinger | https://www.schrodinger.com/glide |
| PPM server | (Lomize et al., 2011) | http://opm.phar.umich.edu/server.php |
| qtPISA | (Krissinel, 2015) | http://www.ccp4.ac.uk/ |
| HARLEM | (Kurnikov, 2018) | https://crete.chem.cmu.edu/index.php/software/harlem-software |
| heliQuest | (Gautier et al., 2008) | http://heliquest.ipmc.cnrs.fr/ |
| dockingserver.com | (Bikadi and Hazai, 2009) | https://dockingserver.com/web/ |
Highlights.
A hydrophilic H2S-oxidizing site is connected to a hydrophobic tunnel that binds CoQ
Cys201 is optimally positioned to form a postulated 4a-thiolate flavin intermediate
A unique electropositive depression serves as a binding site for sulfane acceptors
This hollow may also funnel negatively charged substrates to the H2S-oxidizing site
ACKNOWLEDGMENTS
Research reported in this publication was supported by the National Institutes of Health General Medical Sciences under Award R01 GM107389 (M.S.J.) and by the NIH National Heart, Lung, and Blood Institute under Award R41 HL134435 (to M.S.J.). This work is based on research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences from the NIH (P41 GM103403). The Pilatus 6M detector on 24-ID-C beam line is funded by a NIH-ORIP HEI grant (S10 RR029205). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated forthe DOE Office ofScience byArgonne National Laboratory under contract no. DE-AC02-06CH11357. Work reported here was run on hardware supported by Drexel’s University Research Computing Facility. We thank Cresset (UK; http://www.cresset-group.com/) for access to their software via an academic license. WethankAdel Ahmed for performing docking studies with glutathione.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.str.2019.03.002.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, and Grosse-Kunstleve RW (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, and Adams PD (2012). Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr 68, 352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustyn KDC, Jackson MR, and Jorns MS (2017). Use of tissue metabolite analysis and enzyme kinetics to discriminate between alternate pathways for hydrogen sulfide metabolism. Biochemistry 56, 986–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battye TGG, Kontogiannis L, Johnson O, Powell HR, and Leslie AGW (2011). iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D Biol. Crystallogr 67, 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bikadi Z, and Hazai E (2009). Application of the PM6 semi-empirical method to modeling proteins enhances docking accuracy of AutoDock. J. Cheminform 1, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brito JA, Sousa FL, Stelter M, Bandeiras TM, Vonrhein C, Teixeira M, Pereira MM, and Archer M (2009). Structural and functional insights into sulfide:quinone oxidoreductase. Biochemistry 48, 5613–5622. [DOI] [PubMed] [Google Scholar]
- Brünger AT (1992). Free R-Value - a novel statistical quantity for assessing the accuracy of crystal structures. Nature 355, 472–475. [DOI] [PubMed] [Google Scholar]
- Chen ZW, Koh M, Vandriessche G, Vanbeeumen JJ, Bartsch RG, Meyer TE, Cusanovich MA, and Mathews FS (1994). The structure of flavocytochrome c sulfide dehydrogenase from a purple phototrophic bacterium. Science 266, 430–432. [DOI] [PubMed] [Google Scholar]
- Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, and Richardson DC (2010). MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherney MM, Zhang YF, Solomonson M, Weiner JH, and James MNG (2010). Crystal structure of sulfide:quinone oxidoreductase from Acidithiobacillus ferrooxidans: insights into sulfidotrophic respiration and detoxification. J. Mol. Biol 398, 292–305. [DOI] [PubMed] [Google Scholar]
- Dauter Z (1999). Data-collection strategies. Acta Crystallogr. D Biol. Crystallogr 55, 1703–1717. [DOI] [PubMed] [Google Scholar]
- Doublie S (2007). Production of selenomethionyl proteins in prokaryotic and eukaryotic expression systems. Methods Mol. Biol 363, 91–108. [DOI] [PubMed] [Google Scholar]
- Emsley P, and Cowtan K (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Evans PR (2011). An introduction to data reduction: space-group determination, scaling and intensity statistics. Acta Crystallogr. D Biol. Crystallogr 67, 282–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans PR, and Murshudov GN (2013). How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr 69, 1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foadi J, Aller P, Alguel Y, Cameron A, Axford D, Owen RL, Armour W, Waterman DG, Iwata S, and Evans G (2013). Clustering procedures for the optimal selection of data sets from multiple crystals in macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr 69, 1617–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier R, Douguet D, Antonny B, and Drin G (2008). HELIQUEST: a web server to screen sequences with specific a-helical properties. Bioinformatics 24, 2101–2102. [DOI] [PubMed] [Google Scholar]
- Hackfort BT, and Mishra PK (2016). Emerging role of hydrogen sulfide-microRNA crosstalk in cardiovascular diseases. Am. J. Physiol. Heart Circ. Physiol 310, H802–H812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano Y, Kimura Y, Suzuki H, Miki K, and Wang ZY (2012). Structure analysis and comparative characterization of the cytochrome c’ and flavocytochrome c from thermophilic purple photosynthetic bacterium Thermochromatium tepidum. Biochemistry 51, 6556–6567. [DOI] [PubMed] [Google Scholar]
- Jackson MR, Melideo SL, and Jorns MS (2012). Human sulfide:quinone oxidoreductase catalyzes the first step in hydrogen sulfide metabolism and produces a sulfane sulfur metabolite. Biochemistry 51, 6804–6815. [DOI] [PubMed] [Google Scholar]
- Krissinel E (2015). Stock-based detection of protein oligomeric states in jsPISA. Nucleic Acids Res. 43, W314–W319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E, and Henrick K (2007). Inference of macromolecular assemblies from crystalline state. J. Mol. Biol 372, 774–797. [DOI] [PubMed] [Google Scholar]
- Kurnikov IV (2018). HARLEM computer program https://crete.chem.cmu.edu/index.php/software/harlem-software.
- le Maire M, Arnou B, Olesen C, Georgin D, Ebel C, and Møller JV (2008). Gel chromatography and analytical ultracentrifugation to determine the extent of detergent binding and aggregation, and Stokes radius of membrane proteins using sarcoplasmic reticulum Ca2+-ATPase as an example. Nat. Protoc 3, 1782–1795. [DOI] [PubMed] [Google Scholar]
- Li L, Rose P, and Moore PK (2011). Hydrogen sulfide and cell signaling. Annu. Rev. Pharmacol. Toxicol 51, 169–187. [DOI] [PubMed] [Google Scholar]
- Libiad M, Yadav PK, Vitvitsky V, Martinov M, and Banerjee R (2014). Organization of the human mitochondrial H2S oxidation pathway. J. Biol. Chem 289, 30901–30910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebschner D, Afonine PV, Moriarty NW, Poon BK, Sobolev OV, Terwilliger TC, and Adams PD (2017). Polder maps: improving OMIT maps by excluding bulk solvent. Acta Crystallogr. D Struct. Biol 73, 148–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomize MA, Pogozheva ID, Joo H, Mosberg HI, and Lomize AL (2011). OPM database and PPM web server: resources for positioning of proteins in membranes. Nucleic Acids Res. 40, D370–D376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu CJ, Xia YZ, Liu DX, Zhao R, Gao R, Liu HL, and Xun LY (2017). Cupriavidus necator H16 uses flavocytochrome c sulfide dehydrogenase to oxidize self-produced and added sulfide. Appl. Environ. Microbiol 83, 10.1128/AEM.01610-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani S, Li HZ, Untereiner A, Wu LY, Yang GD, Austin RC, Dickhout JG, Lhotak S, Meng QH, and Wang R (2013). Decreased endogenous production of hydrogen sulfide accelerates atherosclerosis. Circulation 127, 2523–2534. [DOI] [PubMed] [Google Scholar]
- Marcia M, Ermler U, Peng G, and Michel H (2009). The structure of Aquifex aeolicus sulfide:quinone oxidoreductase, a basis to understand sulfide detoxification and respiration. Proc. Natl. Acad. Sci. U S A 106, 9625–9630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcia M, Ermler U, Peng G, and Michel H (2010). A new structure-based classification of sulfide:quinone oxidoreductases. Proteins 78, 1073–1083. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007). Phaser crystallographic software. J. Appl. Crystallogr 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller SM (2013). Flavoprotein disulfide reductases and structurally related flavoprotein thiol/disulfide-linked oxidoreductases. In Handbook of Flavoproteins, Hille R, Miller SM, and Palfey BA, eds. (De Gruyter; ), pp.165–201. [Google Scholar]
- Mishanina TV, Yadav PK, Ballou DP, and Banerjee R (2015). Transient kinetic analysis of hydrogen sulfide oxidation catalyzed by human sulfide quinone oxidoreductase. J. Biol. Chem 290, 25072–25080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller F (1992). Nuclear magnetic resonance studies on flavoproteins In Chemistry and Biochemistry of Flavoenzymes 3, Muller F, ed. (CRC Press; ), pp. 557–595. [Google Scholar]
- Osipov EM, Lilina AV, Tsallagov SI, Safonova TN, Sorokin DY, Tikhonova TV, and Popov VO (2018). Structure of the flavocytochrome c sulfide dehydrogenase associated with the copper-binding protein CopC from the haloalkaliphilic sulfur-oxidizing bacterium Thioalkalivibrio paradoxus ARh 1. Acta Crystallogr. D Struct. Biol 74, 632–642. [DOI] [PubMed] [Google Scholar]
- Sheldrick GM (2008). A short history of SHELX. Acta Crystallogr. A Found. Adv 64, 112–122. [DOI] [PubMed] [Google Scholar]
- Studier FW (2005). Protein production by auto-induction in high-density shaking cultures. Protein Expr. Purif 41, 207–234. [DOI] [PubMed] [Google Scholar]
- Vagin AA, Steiner RA, Lebedev AA, Potterton L, McNicholas S, Long F, and Murshudov GN (2004). REFMAC5 dictionary: organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr. D Struct. Biol 60, 2184–2195. [DOI] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, and McCoy AJ (2011). Overview of the CCP4 suite and current developments. Acta Crystallogr. D Struct. Biol 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Wu L, Jiang B, Yang W, Qi J, Cao K, Meng Q, Mustafa AK, Mu W, Zhang S, et al. (2008). H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine γ-lyase. Science 322, 587–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwart PH, Afonine PV, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, McKee E, Moriarty NW, Read RJ, Sacchettini JC, et al. (2008). Automated structure solution with the PHENIX suite In Structural Proteomics. Methods in Molecular Biology™, 426, Kobe B, Guss M, and Huber T, eds. (Humana Press; ), pp. 419–435. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Coordinates and structure factors have been deposited in the Protein Data Bank for SeMet-substituted SQOR and native SQOR under ID codes PDB: 6MO6 and PDB: 6MP5, respectively.