

Abstract
Brain-derived neurotrophic factor (BDNF) has been implicated in regulating neuronal survival, differentiation, and synaptic plasticity. Reduced expression of BDNF within the substantia nigra accompanies the deterioration of dopaminergic neurons in Parkinson's disease (PD) patients. Analysis of the effects of long-term BDNF absence from the CNS has been difficult because of the early postnatal lethality of BDNF-/- mice. Mice with a floxed BDNF allele were bred with Wnt1-Cre mice to generate Wnt-BDNFKO mice that lack BDNF from the midbrain-hindbrain (MHB). These mice are viable but exhibit hindlimb clutching and poor rotarod performance. Tyrosine hydroxylase (TH)-positive neuron numbers in the substantia nigra pars compacta (SNC) were estimated using stereological methods, revealing a persistent ∼23% reduction of these cells at postnatal day 21 (P21) in Wnt-BDNFKO mice compared with controls. The diminishment of TH-expressing neurons was present at birth and continued through P120. This deficit appears selective for the dopaminergic population, because at P21, total neuron number within the SNC, defined as neuronal nuclei protein-positive cells, was not significantly reduced. Interestingly, and similar to observations in PD patients, SNC neuron subpopulations are not equally affected. Calbindin- and calretinin-expressing SNC populations show no significant difference between Wnt-BDNFKO mice and controls. Thus, BDNF depletion from the MHB selectively leads to reduced TH expression in a subpopulation of neurons, but it remains unclear whether these cells are lost.
Keywords: BDNF, substantia nigra pars compacta, Parkinson's disease, mouse, Cre-lox, stereology, neurotrophin
Introduction
Parkinson's disease (PD) is the most common neurodegenerative disorder affecting the basal ganglia. In ∼10% of cases, the disease is associated with a genetic defect (Goldman and Tanner, 1998) in genes, including α-synuclein, parkin, or DJ-1 (Feany and Bender, 2000; Shimura et al., 2000; Lotharius and Brundin, 2002). In the majority of cases, the appearance of PD is sporadic, and its etiology is unknown. The common feature of PD is a progressive loss of dopaminergic (DA) neurons from the substantia nigra pars compacta (SNC). Identifying the mechanisms that regulate the generation and survival of nigral DA neurons, both during development and in the adult, should enhance our ability to treat PD through either pharmacological or transplant approaches.
Brain-derived neurotrophic factor (BDNF) is a member of the neurotrophin family of secreted growth factors vital for trophic support of neurons within both the peripheral nervous system and the CNS. BDNF signals through the tyrosine kinase receptor B (TrkB) and through the p75 receptor (Huang and Reichardt, 2001). Both BDNF and TrkB are expressed by DA neurons (Okazawa et al., 1992; Nishio et al., 1998; Numan and Seroogy, 1999; Venero et al., 2000). Postmortem studies of PD patients using in situ hybridization, immunohistochemistry, and Western blot analysis demonstrate that reduced levels of BDNF within the SNC accompany the disease condition (Mogi et al., 1999; Parain et al., 1999; Howells et al., 2000; Chauhan et al., 2001). In culture experiments, BDNF has been found to promote the survival and differentiation of mesencephalic DA neurons (Hyman et al., 1991; Feng et al., 1999). In vitro and in vivo, BDNF protects DA neurons from the toxic effects of low levels of the neurotoxins 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and 6′-OHDA (Levivier et al., 1995; Shults et al., 1995; Hung and Lee, 1996; for review, see Murer et al., 2001). BDNF stimulates dopamine activity and turnover in vitro and in vivo and is hypothesized to play a role in the mechanisms that allow the surviving DA neurons in early stage PD patients to compensate for cell losses (Blochl and Sirrenberg, 1996; Murer et al., 2001). This evidence suggests a critical role for BDNF in supporting the survival and differentiation of midbrain DA neurons in both normal and PD individuals. These functions are likely to complement and overlap with those of other secreted factors, including glial cell line-derived neurotrophic factor (GDNF) and bone morphogenetic proteins (BMPs), which are also known to enhance differentiation and survival of DA neurons (Feng et al., 1999; Gratacos et al., 2001b; Brederlau et al., 2002; Zuch et al., 2004).
Although BDNF has been studied intensely, the uncertainty about the importance of the role of BDNF in the early development and long-term survival of nigral DA neurons remains. The poor postnatal health, retarded growth, and subsequent premature death (by ∼21 d of age) of BDNF-/- mice complicates genetic analyses of CNS development (Jones et al., 1994; Ernfors et al., 1995; Conover and Yancopoulos, 1997; Liebl et al., 1997). To circumvent these complications, we use the bacteriophage P1 Cre/loxP system to analyze BDNF requirements in the SNC. We used a mouse line with a floxed BDNF allele (Gorski et al., 2003) and a transgenic line expressing Cre recombinase in the midbrain-hindbrain (MHB) under control of the Wnt-1 promoter (Danielian et al., 1998) to generate mice, Wnt-BDNFKO, lacking BDNF in this region. We report here that BDNF plays a role in the generation, differentiation, and/or survival of a specific subpopulation of DA neurons within the SNC.
Materials and Methods
Generation of midbrain/hindbrain-restricted BDNF mutant mice. All animal procedures were conducted in accordance with Public Health Service guidelines and with the approval of the University of Colorado Institutional Animal Care and Use Committee. BDNFLacZ (Bennett et al., 1999) and R26R Cre reporter (Soriano, 1999) transgenic lines have been described previously. Wnt1-Cre induces efficient MHB-specific Cre-mediated loxP recombination (Danielian et al., 1998). Wnt1-Cre;BDNFneo/+ mice, in which BDNFneo is a null allele with a neomycin cassette in place of the BDNF coding exon (Jones et al., 1994), and BDNFlox/+ mice were bred; offspring containing all three mutated alleles (Wnt1-Cre;BDNFneo/lox) are referred to as Wnt-BDNFKO, and BDNFneo/lox mice are referred to as heterozygous. The mice used here were backcrossed at least three to four generations onto the C57BL/6 strain. Both BDNF+/+ and BDNFlox/+ were pooled with wild type in data collection, because BDNFlox/+ appears to function as near wild type (Gorski et al., 2003). PCR analysis was used to determine genotype. BDNF protein ELISA was performed as described previously (Baquet et al., 2004).
Immunocytochemistry and histochemistry. Postnatal mice younger than 8 d of age were anesthetized by hypothermia on wet ice. Older mice were heavily sedated by intraperitoneal injection of 2.5% avertin (20 μl/g) in PBS, pH 7.4 (100% avertin is 10 g of tribromoethanol and 10 ml of tert-amyl alcohol), and then treated in the same manner as described previously (Vigers et al., 2000). Primary antibodies included mouse monoclonal anti-neuronal nuclei protein (NeuN) (1:1000; Chemicon, Temecula, CA), a rabbit anti-β-galactosidase (β-gal) antibody (1:1000; ICN Pharmaceuticals, Auroria, OH), mouse monoclonal (1:2500; Sigma, St. Louis, MO) and rabbit polyclonal (1:2000; Chemicon) tyrosine hydroxylase (TH) antibodies, calbindin (CB) (1:200; Sigma), and calretinin (CR) (1:1000; Chemicon). The secondary antibodies used were biotinylated goat anti-rabbit or biotinylated goat anti-mouse antibody (Jackson ImmunoResearch, West Grove, PA) diluted 1:250 in blocking solution. Visualization was done by diaminobenzidine reaction as described by Vigers et al. (2000). Slides were counterstained when appropriate with cresyl violet. β-Gal expression in CNS tissues was visualized histochemically by staining with 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-gal) as described previously (Vigers et al., 2000) or via immunofluorescence using the β-galactosidase antibody.
For double-label analysis of cells, immunofluorescence was used. To examine the effectiveness of the Wnt1-Cre mouse line at driving MHB loxP recombination, free-floating cryostat sections (40 μm) were cut and then washed in PBS three times for 15 min each. Sections were placed in block [Tris-buffered saline (TBS), 0.4% Triton X-100, 1% normal goat serum, 0.5% glycine, 0.5% bovine serum albumin] for 1 h and then in primary antibody(s) for 48 h at 4°C. Afterward, sections were washed three times in TBST (Tris-buffered saline, 0.4% Triton X-100) for 15 min each and then incubated in either Alexa 488 goat anti-mouse IgG antibody (1:200; Molecular Probes, Eugene, OR) or Alexa 546 goat anti-rabbit IgG antibody (1:200; Molecular Probes, Eugene, OR) for 2 h in the dark at room temperature. Sections were washed twice in TBST, once in TBS, and once in 0.5 m Tris, pH 7.5, before mounting on Superfrost slides (Fisher Scientific, Pittsburgh, PA) and coverslipping with Fluoromount (Fisher Scientific). Cryostat sections (10 μm) were mounted directly onto slides and were allowed to air dry for 1 h before the blocking step. Images were captured using a Zeiss (Oberkochen, Germany) Axioplan deconvolution microscope equipped with a Hamamatsu (Bridgewater, NJ) C4742-95 digital camera and OpenLab software (Improvision, Coventry, UK).
Behavioral analysis. Wild-type, heterozygous, and Wnt-BDNFKO mice at various ages were analyzed for limb clasping by suspending them from their tails at least one foot above a surface for 1 min. A clasping event was defined by the retraction of either or both hindlimbs into the body and toward the midline. Mice were scored on a simple “yes” or “no” basis with the investigator blind to genotype. Rotarod analysis was performed with 4- to 5-week-old mice as described previously (Baquet et al., 2004).
Protein blot analysis of TH. Postnatal day 35 (P35) Wnt-BDNFKO mice and controls were killed by cervical dislocation, and the brains were removed. Coronal sections (800 μm) were cut, and the striatum was dissected out. Striatal tissue was placed in cold homogenization buffer (1% SDS, 1 mm EDTA, 10 mm Tris, pH 8), minced, and sonicated. Total protein concentrations were determined using a modified BCA assay (Pierce, Rockford, IL), and 10 μg of each sample was electrophoresed on 12% SDS-PAGE by a standard procedure (Bio-Rad, Hercules, CA). Gels were transferred to nitrocellulose membranes, which were then blocked in 1:1 solution of Licor blocking solution (LI-COR Biosciences, Lincoln, NE) and PBS. Membranes were incubated with agitation in 1:1 Licor blocking solution:PBS with 0.1% Tween 20 and anti-tyrosine hydroxylase mouse monoclonal (1:8000; Sigma) at 4°C overnight. After washing, membranes were incubated at room temperature for 2 h in the same buffer with Alexa Fluor 680 goat anti-mouse (1:1000; Molecular Probes) and then washed again in 0.1% Tween 20 in PBS. A final wash was done in PBS, and the blot was visualized using an Odyssey infrared-imaging system (LI-COR Biosciences). Samples were corrected for background and quantified using Odyssey software. All samples were normalized to wild type for the purposes of comparison (n = 4 for Wnt-BDNFKO and wild type; n = 3 for heterozygotes).
Cell counting. The total number of DA neurons in both hemispheres of the SNC and the ventral tegmental area (VTA) was estimated using a fractionator-sampling design (Gundersen et al., 1988; West et al., 1991; Finkelstein et al., 2000; Parish et al., 2001). Staining with TH antibody delineated the mediodorsal boundary of the SNC in each 40 μm cryostat section. The ventrolateral boundary was defined by the dorsal portion of the SN pars reticulata; the anterior medial boundary was defined by the ventral tegmental area and by size and orientation of stained cells. DA neurons of the SNC are larger than ventral tegmental area DA neurons, and SNC DA neurons orient along the long axis of the SNC. The posterior medial portion of the SNC was defined by the medial lemniscus. In each of the sections sampled, TH-positive neuron somas were used as the counting unit according to optical dissector rules (Gundersen et al., 1988). Labeled profiles were counted only if the first recognizable profile of the cell soma came into focus within the counting frame (West et al., 1991). Using every third coronal section, the analysis was performed starting with the first appearance of TH-positive neurons, extending to the most caudal parts of the SNC and including both hemispheres. Using StereoInvestigator, a morphometry and stereology software package (version 5.0; MicroBrightField, Colchester, VT), cell counts were made at regular predetermined intervals (x = 250 μm; y = 130 μm) within an unbiased counting frame of known area (65 × 65 μm = 4225 μm2) superimposed on the image. Sections were viewed under a 60×, numerical aperture 1.40, oil immersion objective on a Nikon (Tokyo, Japan) Eclipse E600 photomicroscope having a motorized Biopoint XYZ axis computer-controlled stage (Ludl Electronic, Hawthorne, NY) and a 3CCD MTI video camera (Dage-MTI, Michigan City, IN). The counting frame within the SNC was positioned randomly by the StereoInvestigator software, thereby creating a systematic random sample of the area. Twenty-eight micrometers was defined for the z-dimension of the counting brick with a 2 μm guard on either side. StereoInvestigator was used to make outlines of both hemispheres of the SNC as defined by TH antibody staining, and these outlines were used on adjacent sections stained for NeuN (Mullen et al., 1992), calbindin, or calretinin. Stained cells were counted within the outlines defined by TH expression, and total estimates were obtained. Counting parameters for NeuN and calbindin differed slightly from those set for TH counts. For NeuN, every sixth section was counted, and the counting frame was reduced to (50 × 50 μm = 2500 μm2) to prevent oversampling. For calbindin, every second section was counted, and the counting frame was (100 × 100 μm = 10000 μm2). Each experiment was conducted with the investigator blind to genotype. The numbers of mice used for each experiment are as follows: TH counts (for P0 and ∼P120, n = 3 for each genotype; P21, n = 9 for wild type and Wnt-BDNFKO; n = 8 for heterozygous), NeuN counts at P21 (n = 4 for Wnt-BDNFKO and heterozygous; n = 3 for wild type), and calbindin and calretinin (n = 3 for each genotype at P21). TH-positive cell counts in the VTA were performed using a grid of x = 200 μm by y = 200 μm and a frame of 50 × 50 μm = 2500 μm2. TH stain was used to define the outline of the VTA with the medial lemniscus as the lateral border. Counts represent combined totals of both hemispheres. VTA and NeuN counts were done using an Axioskop2 Plus (Zeiss) photomicroscope with 63× oil immersion objective (numerical aperture, 1.40) and an Optronics CCD camera (Optronics, Galeta, CA).
An estimate of the percentages of TH-positive neurons having undergone recombination in Wnt1-Cre;R26R mice was obtained from 40 μm coronal cryostat sections. The sections were immunofluorescently labeled for TH and β-galactosidase (marker of recombination). Montage images (FITC and rhodamine) of the SNC (every third section, entire extent of the SNC in both hemispheres) were created with OpenLab using a 40× objective, and the total number of TH-positive neurons and total number of TH-positive, β-gal-positive neurons were counted for each mouse. The number of double-labeled neurons divided by the total number of TH-positive neurons was the percentage of recombination. P7, P21, and P28 were examined.
Results
Expression of BDNF in the SNC
We used the BDNFLacZ mouse strain (Bennett et al., 1999) to characterize the expression of BDNF within the SNC. X-gal staining was apparent as early as embryonic day 16.5 (E16.5) (Fig. 1) in a subset of cells and continuing through the postnatal period, consistent with BDNF having a role in both substantia nigra development and maintenance. To further identify the subset of BDNF-expressing cells, we performed double-labeling experiments with anti-β-gal and anti-tyrosine hydroxylase antibodies. Expression of TH, a marker for differentiated DA neurons, was apparent in a subset of β-gal-expressing cells. Representative images from P21 BDNFLacZ/+ mice brains (Fig. 2B) reveal numerous cells that are positive for both TH and β-gal as well as cells that express one or the other marker.
Figure 1.
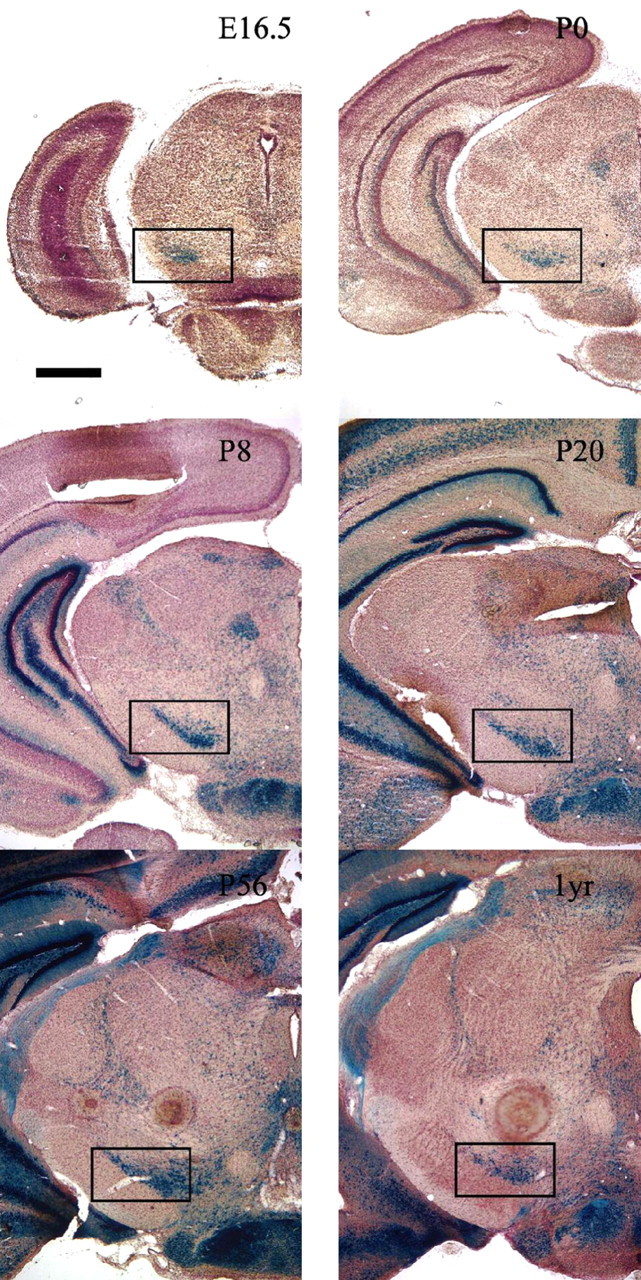
BDNFLacZ expression in the substantia nigra. BDNFLacZ/+ embryos, neonates, and postnatal mice demonstrate the presence of BDNF within the substantia nigra pars compacta. The black box indicates the location of the SNC. Coronal sections (40 μm) of different ages, starting at E16.5 and ending at 1 year (1yr) (indicated in the top right portion of each panel), were stained with X-gal, counterstained with Neutral Red, and then photographed at 20× magnification. Scale bar, 1 mm.
Figure 2.
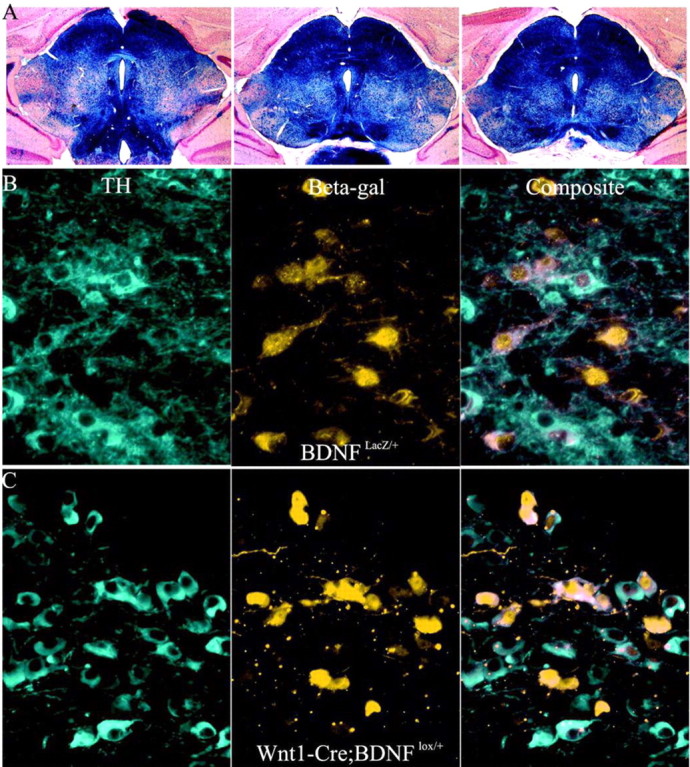
Expression of Cre in midbrain BDNF-expressing neurons. A, X-gal stain of 40 μm coronal sections (counterstained with Neutral Red) from the midbrain-hindbrain of P21 Wnt1-Cre;R26R mice. The dark blue stain indicates areas of Cre activity. Outside the MHB, there are only scattered CNS cells that undergo recombination. B, Immunofluorescent double labeling using antibodies to tyrosine hydroxylase and β-gal of 10 μm cryostat sections from the anterior portion of the SNC of a BDNFLacZ/+ mouse documents expression in multiple SNC cells. Images were photographed at 400× magnification. Lateral is to the left of the image, and the midline is to the right of the image. C, Effectiveness of Wnt1-Cre at excising BDNF from DA neurons. Images of sections are processed as in B of the SNC of a Wnt1-Cre; BDNFlox/+ mouse.
Wnt1-Cre is effective at driving recombination in the SNC
The Wnt1-Cre transgenic mouse line has been used previously to recombine floxed alleles in the MHB region (Chai et al., 2000; Rico et al., 2002). Mating a Wnt1-Cre mouse to a R26R (Soriano, 1999) mouse produces offspring in which MHB-Cre activity is seen as early as E9.5 (Danielian et al., 1998). In the region of the SNC, ∼80-90% of neurons that were TH positive were also β-gal-positive (Fig. 2A), which demonstrates that Wnt1-Cre can drive extensive lox recombination within the midbrain DA neurons.
To further assay the extent of Wnt1-Cre-mediated recombination, we bred these mice to the BDNFlox/+ strain to produce Wnt1-Cre:BDNFlox/+ mice. If Wnt1-Cre is effective at the excision of a floxed BDNF gene, MHB β-gal-expressing Wnt1-Cre;BDNFlox/+ mice should mimic the expression seen in BDNFLacZ/+. The SNCs of P21 Wnt1-Cre;BDNFlox/+ and BDNFLacZ/+ mice double labeled for β-gal and TH were compared. Similar TH/β-gal expression patterns were apparent (Fig. 2B,C) in which TH and β-gal sometimes colocalize as well as TH-positive β-gal-negative and TH-negative β-gal-positive cells. These observations relating TH and BDNF expression also are consistent with the pattern of BDNF mRNA expression described in the SNC of rats (Seroogy et al., 1994).
To examine the consequences of Wnt1-Cre-mediated BDNF deletion on MHB BDNF concentration, we bred Wnt-BDNFKO mice. ELISA of MHB tissue revealed a significant reduction of 95% for P7/P9 (p < 0.001) in levels of BDNF protein in mutants compared with wild-type controls (Fig. 3A).
Figure 3.

Reduced BDNF protein in MHB leads to motor deficits and reduced striatal TH in Wnt-BDNFKO mice. A, BDNF protein was quantified by ELISA for MHB and expressed as nanograms of BDNF protein per gram of wet tissue. These extracts were obtained from P7 and P9 mice and pooled to increase the n (for P7/P9, n = 5 for Wnt-BDNFKO and wild-type; n = 4 for heterozygous; **p < 0.01; ***p < 0.001; one-way ANOVA with a Newman-Keuls post hoc test). B, Performance of 4- to 5-week-old mice on an accelerating rotarod. Each mouse had three trials each day, which were averaged for the day (Wnt-BDNFKO, n = 7; heterozygous, n = 16; wild type, n = 6; **p < 0.01 heterozygous vs wild type for day 1 only; ***p < 0.001 Wnt-BDNFKO vs heterozygous and wild type all 3 d). C, Wnt-BDNFKO, heterozygous, and wild-type mice at 1, 2, and 4 months of age were suspended by their tails for 1 min. Clasping was defined as the balling up of one or both of the hindlimb paws, accompanied by pulling them into the body and movement of the limbs toward the midline. Four-month-old wild-type, heterozygous, and Wnt-BDNFKO mice undergoing tail suspension are shown. D, Western blot analysis of TH levels in total striatal protein from P35 Wnt-BDNFKO and control mice (n = 4 for Wnt-BDNFKO and wild type; n = 3 for heterozygous; *p < 0.05; one-way ANOVA with a Newman-Keuls post hoc test). White bar, Wnt-BDNFKO; gray bar, heterozygous; black bar, wild type.
Motor abnormalities and reduced TH in Wnt-BDNFKO mice
PD patients are afflicted with characteristic movement disorders, including tremor at rest, limb rigidity, reduction of spontaneous movements, and difficulty in performing complex motor movements (Fahn, 2003). If BDNF plays a pivotal role in SNC development and/or function, its absence from the MHB might translate into movement disorders. Thus, qualitative and quantitative analyses of the motor behavior of Wnt-BDNFKO and controls were performed. Wnt-BDNFKO mice, observed in their home environment, displayed no obvious movement abnormalities such as reduced locomotion, rotation, or alteration of stereo-typed behaviors that distinguish them from controls (personal observation). However, rotarod analysis of 4- to 5-week-old Wnt-BDNFKO mice showed a statistically significant impairment of motor skills when compared with controls (Fig. 3B). Wnt-BDNFKO mice also exhibit an early onset clutching phenotype that is seen in many neurodegenerative mouse models. At 1 month, 78% of Wnt-BDNFKO mice clutch compared with 28% for heterozygous and 8% for wild type. By 4 months, all Wnt-BDNFKO mice clutch compared with 67% for heterozygous and 16% for wild type (Fig. 3C) (Lalonde, 1987b; Hamilton et al., 1996; van den Akker et al., 1999; Auerbach et al., 2001; Guidetti et al., 2001; van Dellen et al., 2001). Western blot analysis of total striatal protein revealed a reduction of 42% in the amount of tyrosine hydroxylase in Wnt-BDNFKO mice compared with wild type and a 30% reduction compared with heterozygous (Fig. 3D), and TH fibers appeared less dense in the dorsal striatum (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). These results led us to more deeply examine TH cells within the SNC and VTA.
A selective reduction in the number of dopaminergic neurons
Future rodent DA neurons undergo their final mitoses between E10 and E13 (Bayer et al., 1995) and have migrated to their final position by ∼E16 (Gerfen et al., 1987). TH is the rate-limiting enzyme in the production of dopamine, and it can be used to mark DA neurons as early as E12.5. The number of TH-positive neurons in the rodent MHB is low at birth but reaches a peak by P14 and then decreases to adult levels by P21. This numerical increase is the result of postmitotic cells assuming a DA neuronal fate (Lieb et al., 1996; Jackson-Lewis et al., 2000). Based on TH immunoreactivity, Jackson-Lewis et al. (2000) concluded that the mouse SNC achieved its adult appearance by ∼P14. Inspection of the SNCs of P21 Wnt-BDNFKO mice revealed that TH staining is neither as intense nor as expansive as that found in controls (Fig. 4A). This is true in both anterior and posterior portions of the SNC. TH-positive neurons and fibers also appear disorganized in Wnt-BDNFKO SNC compared with controls. The total number of TH neurons in both hemispheres of the SNC of wild-type, heterozygous, and Wnt-BDNFKO mice were compared. At P0, P21, and approximately P120 (Fig. 4B), stereological counts demonstrated reductions in the number of DA neurons of 27, 23, and 23%, respectively, in the SNC of Wnt-BDNFKO mice compared with controls. Unlike the SNC, counts of total VTA DA neurons of both hemispheres revealed no significant difference between Wnt-BDNFKO mice and controls (Fig. 4C).
Figure 4.

Wnt-BDNFKO mice have reduced TH expression in the SNC but not in the VTA. A, The extent of the SNC appears reduced in P21Wnt-BDNFKO mice compared with controls, and the number of TH fibers also seems to be reduced. The most anterior section is at the top, and the most posterior section is at the bottom. Coronal cryostat sections (40 μm) taken at 240 μm intervals stained for TH and counterstained with cresyl violet are shown. B, Optical fractionator estimates of the number of DA neurons in both hemispheres of the SNC of Wnt-BDNFKO mice and controls at P0, P21, and P120 (P0, n = 3/genotype; P21, n = 9, 8, and 9 for Wnt-BDNFKO, heterozygous, and wild type, respectively; P120, n = 3/genotype; *p < 0.05; **p < 0.01; one-way ANOVA with a Newman-Keuls posthoc test). C, There is no significant reduction in the total number of TH-positive cells of both hemispheres of the VTA of Wnt-BDNFKO mice compared with controls (P21, n = 11, 7, and 8 for Wnt-BDNFKO, heterozygous, and wild type, respectively, p = 0.6454; P120, n = 3/genotype, p = 0.8170). White bars, Wnt-BDNFKO; gray bars, heterozygous; black bars, wild type.
No significant decrease in the number of NeuN-positive cells in the SNC
The reduced TH-positive neuron numbers in Wnt-BDNFKO SNC could be attributable to the failure of neurons fated to be dopaminergic to express TH or the absence of such neurons. To further investigate these possibilities, alternating coronal serial sections of Wnt-BDNFKO and control brains were stained for TH or NeuN. Outlines of the SNC, as defined by TH expression, were used to designate an area for stereological counts on adjacent NeuN-stained sections. These counts revealed a 7% decrease in total NeuN number in both Wnt-BDNFKO mice and heterozygous controls compared with wild-type mice, but this was not significant by one-way ANOVA (Fig. 5A).
Figure 5.

NeuN-positive neuron loss in the SNC of Wnt-BDNFKO mice was not detected. A, NeuN-positive cells were counted in the SNC, defined by outlining the TH-stained population of neurons on adjacent sections, and estimates were obtained for total numbers of neurons in the SNC at P21 (n = 4 for Wnt-BDNFKO and heterozygous; n = 3 for wild type; p = 0.5093). B, Stereological estimates of calbindin-positive cell population in the SNC. C, Estimates of calretinin-positive cell population in the SNC. For both experiments, n = 3/genotype. No significant difference was found between genotypes by one-way ANOVA. White bars, Wnt-BDNFKO; gray bars, heterozygous; black bars, wild type.
Calbindin and calretinin populations are not reduced in Wnt-BDNFKO mice
Distinct subpopulations of DA neurons in the SNC can be recognized based on their expression of various markers. Notably, TH-expressing neurons that also express CB and CR (Liang et al., 1996a; Nemoto et al., 1999; Gonzalez-Hernandez and Rodriguez, 2000) are known to be more resistant to 6′-OHDA and MPTP toxicity and are less affected in Parkinson's disease (Yamada et al., 1990; Liang et al., 1996b; Isaacs et al., 1997; McMahon et al., 1998; Damier et al., 1999a; Kim et al., 2000; Tsuboi et al., 2000) than DA neurons lacking expression of these Ca2+-binding proteins. CB and CR expression within the SNC of Wnt-BDNFKO mutant and control mice were analyzed. In coronal sections, the extent of the SNC appears reduced in Wnt-BDNFKO mice, but the density of CB profiles appears increased compared with controls (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). CR expression patterns within mutants and controls also appear similar (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). To quantify CB- and CR-expressing populations within the SNC, CB- and CR-positive cells were counted in the same manner as described for the NeuN analysis. The results indicate that there is no significant reduction of either the CB or the CR populations within the SNC (Fig. 5 B, C) (p = 0.89 and p = 0.96, respectively; n = 3 mice/genotype).
Discussion
β-galactosidase expression in BDNFLacZ/+ embryos indicates that BDNF is present during differentiation of DA neurons from approximately E16 onwards (Fig. 1), but it is not present during the phase of DA neuron precursor proliferation (from E10 to E13) (Bayer et al., 1995). This suggests that BDNF could act as a survival factor, a differentiation factor, or both for DA neurons. It is interesting to note the presence of a large group of faintly staining β-gal-positive cells that are also TH positive (Fig. 2B,C). The existence of these cells suggest that BDNF expression is more widespread among DA neurons than detected using in situ hybridization (Seroogy et al., 1994; Howells et al., 2000). To study BDNF requirements in SNC development, we used the Wnt1-Cre mouse line to achieve MHB-specific deletion of BDNF. Wnt1-Cre excised the floxed BDNF allele in a MHB-specific manner with >80% efficiency in SNC DA neurons. ELISA analysis of the MHB of Wnt-BDNFKO mice confirms near-complete elimination of BDNF protein.
At early ages, Wnt-BDNFKO mice display a clutching phenotype similar to that seen in other mouse models of neurodegeneration (Lalonde, 1987a; Hamilton et al., 1996; van den Akker et al., 1999; Auerbach et al., 2001; Guidetti et al., 2001). Wnt-BDNFKO mice also display significant deficits in motor ability as assayed by rotarod, yet their improvement in successive trials demonstrates that they are capable of motor learning. Comparison of the slope of the lines, a measure of motor skill acquisition (Crawley, 1999; Tarantino et al., 2000), indicates that mutant and wild-type mice learn at similar rates. We suggest that the rotarod deficiency observed is more indicative of impairment of motor abilities such as coordination and balance, which are less dependent on basal ganglia function than is motor learning. Importantly, the cellular basis of these Wnt-BDNFKO mouse motor abnormalities is somewhat ambiguous, because the BDNF gene is excised from the SN as well as other MHB structures important in motor function, including the cerebellum and pons as well as portions of the PNS, notably including the dorsal root ganglia (S. Tamowski and K. R. Jones, unpublished observation) (Danielian et al., 1998; Rico et al., 2002). Importantly, in PD patients, the appearance of motor pathologies requires a ≥60% loss of DA neurons (Bernheimer et al., 1973), also consistent with the idea that the motor deficits in Wnt-BDNFKO mice are unlikely to be simply nigral in origin.
Loss of BDNF from the MHB adversely affects the amount of TH present in the striatum and establishment of the proper number of DA neurons within the SNC. TH levels, as quantified by Western blot analysis, are significantly reduced in Wnt-BDNFKO mice, and this parallels what has been reported for dopamine levels in BDNF-/- mice (Dluzen et al., 1999). At birth, there is a 27% reduction of the number of DA neurons in the SNC of Wnt-BDNFKO mice; this magnitude of deficit is maintained at later ages, suggesting that a specific subpopulation of DA neurons is affected by the absence of BDNF. BDNF is known to promote the differentiation and survival of DA neurons (Hyman et al., 1994; Studer et al., 1995; Hagg, 1998; Feng et al., 1999; Son et al., 1999). DA neuronal differentiation and cellular apoptosis occur concurrently during the initial development of the SNC. Starting at approximately E12, increasing numbers of neurons in the SNC assume the DA fate as marked by TH expression (Bayer et al., 1995; Kawano et al., 1995). The majority of DA neurons appear between birth and 3 weeks of age in rodents (Lieb et al., 1996; Jackson-Lewis et al., 2000). Apoptosis, in contrast, occurs in two peaks with one at P2 and the other at P10 (Jackson-Lewis et al., 2000).
Because proliferation of DA precursors has ceased before observable BDNF expression within the SNC, it is doubtful that BDNF plays a role in the initial generation of DA neurons. It is more likely that BDNF is responsible for DA neuron differentiation, survival, or both in conjunction with other trophic factors such as GDNF and BMPs (Lin et al., 1993; Tomac et al., 1995; Jordan et al., 1997; Reiriz et al., 1999; Oo et al., 2003; Kholodilov et al., 2004). Synergistic effects between BDNF and other trophic factors might further SNC development. Combined application of BDNF and BMPs to cultured cortical and striatal neurons and stem cells are known to enhance neuronal differentiation (Gratacos et al., 2001a; Chang et al., 2003) and could have similar effects in the nigra.
If BDNF is only involved in the differentiation of DA neurons, its absence might result in fewer DA neurons but would not necessarily have a significant impact on total SNC neuronal number. Although we found a small decrease in the number of NeuN-positive cells in the SNC of Wnt-BDNFKO mice (∼7% compared with wild-type mice), a comparable reduction was found in heterozygotes. Because the reduction in NeuN number is present in both Wnt-BDNFKO and heterozygotes, but the TH reduction is present only in the Wnt-BDNFKO mice, we suggest that this is indicative of a loss of TH expression and not necessarily of neurons from the SNC. It is nonetheless possible that, given TH neurons are a subset of the SNC, a loss of cells might have occurred, perhaps suggested by the NeuN reduction.
Contrary to our results, Baker et al. (2005) found no loss of TH cells in BDNF null mutant mice at P14 and P21. Such differences in results might be related to methodology, or small changes might be missed or detected depending on how restrictively or permissively the boundaries of the SNC are defined.
Porritt et al. (2005) injected antisense BDNF oligonucleotide unilaterally into adult rat SNC and observed a reduction of TH-positive cells on both the ipsilateral and contralateral sides. Counts of Nissl-stained sections indicated neuronal loss, although not as dramatic as the TH losses, especially on the contralateral side. In Wnt-BDNFKO mice, we observed TH losses and minor neuronal losses but not nearly to the extent of Porritt et al. (2005). Perhaps their observations suggest enhanced BDNF dependence at a later stage, developmental compensation in our mice, or a difference between mouse and rat.
We detected a similar, although not quite as severe, phenotype as that found in TrkB hypomorphic mice (Zaman et al., 2004). The greater loss of TH expression in TrkB mutant mice suggests that the widespread expression of TrkB within the SNC (Numan and Seroogy, 1999) may confer responsiveness to more than BDNF alone. Notably, both neurotrophin (NT)-4 and NT-3 can activate TrkB (Patapoutian and Reichardt, 2001), and NT-4 has been shown in some instances to be more effective than BDNF at protecting SNC neurons from degeneration and death (Hagg, 1998). Upregulation of NT-3 within the forebrain caused by loss of BDNF occurred in another mouse we studied (Gorski et al., 2003) and could mitigate adverse effects. Because Zaman et al. (2004) examined older mice (3 months of age), it would be interesting to determine whether this loss occurs earlier and is accompanied by reduction in neuronal number.
To determine whether other SNC subpopulations might be BDNF dependent, we assessed the number of CB- and CR-positive cells. We found that CB- and CR-positive population sizes were comparable across genotypes. Perhaps these calcium-binding proteins help protect SNC neurons from the adverse effects of BDNF absence. Observations by others that ∼20% of the SNC is comprised of TH-positive cells that are negative for CB and CR suggest an intriguing possibility that this subpopulation loses TH expression in Wnt-BDNFKO mice. Notably, CB- and CR-positive cells are more resistant to damage caused by the ravages of PD or toxic insult than other nigral cells (Yamada et al., 1990; German et al., 1992; Mouatt-Prigent et al., 1994; Kim et al., 2000).
Although not displaying the more drastic aspects of PD, Wnt-BDNFKO mice may nevertheless be a useful model for the study of aspects of this disease. The noncalbindin populations of DA neurons are some of the first to succumb to the pathologies of PD (Damier et al., 1999b). This is similar to the losses we observed in Wnt-BDNFKO mice, in which calbindin-expressing cells in the SNC are spared. Elimination of BDNF-expressing cells from the SNC in early stages of PD could accelerate later losses by leaving the remaining non-CB/non-CR-expressing DA neurons especially vulnerable to insult. A recent report suggests that wild-type α-synuclein, mutations of which are known to result in PD (Polymeropoulos et al., 1997), plays a role in regulating BDNF expression and that mutant forms of α-synuclein lead to reduced levels of BDNF (Kohno et al., 2004). This is reminiscent of Huntington's disease, in which wild-type huntingtin protein is a positive regulator of BDNF transcription (Zuccato et al., 2003). Downregulation of BDNF expression in the SNC might be one of the earlier steps in PD, and the resulting sensitization of neurons could be a common feature of many neurodegenerative diseases.
The early onset of the Wnt-BDNFKO phenotype could suggest a mechanism for juvenile PD. Cases of PD can be categorized depending on the age of onset of the patient as either juvenile (10-20 years of age), young (20-40 years of age), or late (≥40 years of age). Juvenile PD cases are usually the result of hereditary factors, which might prove to modulate BDNF expression (Hattori et al., 2000; Giasson and Lee, 2001; West et al., 2002; West and Maidment, 2004). Future studies directed toward better understanding the interplay between BDNF and other trophic factors in regulating DA differentiation and survival of specific subpopulations of these neurons promises to lead to new approaches for treating PD.
Footnotes
This work was supported initially by a Howard Hughes Medical Institute fellowship (Z.C.B.) and the Burroughs-Wellcome New Investigator in Pharmacology Award and recently by National Institutes of Health Grant RO1 EY014998 (K.R.J.). We thank Andrew P. McMahon for the generous gift of the Wnt1-Cre transgenic line. We thank Min Han for the use of a microscope and OpenLab software, Brad Olwin for the use of his cryostat, Jeanne Wehner for the use of her rotarod, Edward Dudek for the use of his stereology setup, and Richard Blaskovich, Tiffany Farnum, Geoff Greene, and Wendy Pouliot for technical assistance. We thank Alison Vigers, Jessica Gorski, Edoardo Marcora, and Micheal Klymkowsky for helpful suggestions on this manuscript and Jason Carnes for mouse photography.
Correspondence should be addressed to Kevin R. Jones, Department of Molecular, Cellular, and Developmental Biology, University of Colorado, Campus Box 347, Boulder, CO 80309. E-mail: krjones@stripe.colorado.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/256251-09$15.00/0
References
- Auerbach W, Hurlbert MS, Hilditch-Maguire P, Wadghiri YZ, Wheeler VC, Cohen SI, Joyner AL, MacDonald ME, Turnbull DH (2001) The HD mutation causes progressive lethal neurological disease in mice expressing reduced levels of huntingtin. Hum Mol Genet 10: 2515-2523. [DOI] [PubMed] [Google Scholar]
- Baker SA, Stanford LE, Brown RE, Hagg T (2005) Maturation but not survival of dopaminergic nigrostriatal neurons is affected in developing and aging BDNF-deficient mice. Brain Res 1039: 177-188. [DOI] [PubMed] [Google Scholar]
- Baquet ZC, Gorski JA, Jones KR (2004) Early striatal dendrite deficits followed by neuron loss with advanced age in the absence of anterograde cortical brain-derived neurotrophic factor. J Neurosci 24: 4250-4258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer SA, Wills KV, Triarhou LC, Ghetti B (1995) Time of neuron origin and gradients of neurogenesis in midbrain dopaminergic neurons in the mouse. Exp Brain Res 105: 191-199. [DOI] [PubMed] [Google Scholar]
- Bennett JL, Zeiler SR, Jones KR (1999) Patterned expression of BDNF and NT-3 in the retina and anterior segment of the developing mammalian eye. Invest Ophthalmol Vis Sci 40: 2996-3005. [PubMed] [Google Scholar]
- Bernheimer H, Birkmayer W, Hornykiewicz O, Jellinger K, Seitelberger F (1973) Brain dopamine and the syndromes of Parkinson and Huntington. Clinical, morphological and neurochemical correlations. J Neurol Sci 20: 415-455. [DOI] [PubMed] [Google Scholar]
- Blochl A, Sirrenberg C (1996) Neurotrophins stimulate the release of dopamine from rat mesencephalic neurons via Trk and p75Lntr receptors. J Biol Chem 271: 21100-21107. [DOI] [PubMed] [Google Scholar]
- Brederlau A, Faigle R, Kaplan P, Odin P, Funa K (2002) Bone morphogenetic proteins but not growth differentiation factors induce dopaminergic differentiation in mesencephalic precursors. Mol Cell Neurosci 21: 367-378. [DOI] [PubMed] [Google Scholar]
- Chai Y, Jiang X, Ito Y, Bringas Jr P, Han J, Rowitch DH, Soriano P, McMahon AP, Sucov HM (2000) Fate of the mammalian cranial neural crest during tooth and mandibular morphogenesis. Development 127: 1671-1679. [DOI] [PubMed] [Google Scholar]
- Chang MY, Son H, Lee YS, Lee SH (2003) Neurons and astrocytes secrete factors that cause stem cells to differentiate into neurons and astrocytes, respectively. Mol Cell Neurosci 23: 414-426. [DOI] [PubMed] [Google Scholar]
- Chauhan NB, Siegel GJ, Lee JM (2001) Depletion of glial cell line-derived neurotrophic factor in substantia nigra neurons of Parkinson's disease brain. J Chem Neuroanat 21: 277-288. [DOI] [PubMed] [Google Scholar]
- Conover JC, Yancopoulos GD (1997) Neurotrophin regulation of the developing nervous system: analyses of knockout mice. Rev Neurosci 8: 13-27. [DOI] [PubMed] [Google Scholar]
- Crawley JN (1999) Behavioral phenotyping of transgenic and knockout mice: experimental design and evaluation of general health, sensory functions, motor abilities, and specific behavioral tests. Brain Res 835: 18-26. [DOI] [PubMed] [Google Scholar]
- Damier P, Hirsch EC, Agid Y, Graybiel AM (1999a) The substantia nigra of the human brain. I. Nigrosomes and the nigral matrix, a compartmental organization based on calbindin D(28K) immunohistochemistry. Brain 122: 1421-1436. [DOI] [PubMed] [Google Scholar]
- Damier P, Hirsch EC, Agid Y, Graybiel AM (1999b) The substantia nigra of the human brain. II. Patterns of loss of dopamine-containing neurons in Parkinson's disease. Brain 122: 1437-1448. [DOI] [PubMed] [Google Scholar]
- Danielian PS, Muccino D, Rowitch DH, Michael SK, McMahon AP (1998) Modification of gene activity in mouse embryos in utero by a tamoxifen-inducible form of Cre recombinase. Curr Biol 8: 1323-1326. [DOI] [PubMed] [Google Scholar]
- Dluzen DE, Story GM, Xu K, Kucera J, Walro JM (1999) Alterations in nigrostriatal dopaminergic function within BDNF mutant mice. Exp Neurol 160: 500-507. [DOI] [PubMed] [Google Scholar]
- Ernfors P, Kucera J, Lee KF, Loring J, Jaenisch R (1995) Studies on the physiological role of brain-derived neurotrophic factor and neurotrophin-3 in knockout mice. Int J Dev Biol 39: 799-807. [PubMed] [Google Scholar]
- Fahn S (2003) Description of Parkinson's disease as a clinical syndrome. Ann NY Acad Sci 991: 1-14. [DOI] [PubMed] [Google Scholar]
- Feany MB, Bender WW (2000) A Drosophila model of Parkinson's disease. Nature 404: 394-398. [DOI] [PubMed] [Google Scholar]
- Feng L, Wang CY, Jiang H, Oho C, Mizuno K, Dugich-Djordjevic M, Lu B (1999) Differential effects of GDNF and BDNF on cultured ventral mesencephalic neurons. Brain Res Mol Brain Res 66: 62-70. [DOI] [PubMed] [Google Scholar]
- Finkelstein DI, Stanic D, Parish CL, Tomas D, Dickson K, Horne MK (2000) Axonal sprouting following lesions of the rat substantia nigra. Neuroscience 97: 99-112. [DOI] [PubMed] [Google Scholar]
- Gerfen CR, Baimbridge KG, Thibault J (1987) The neostriatal mosaic. III. Biochemical and developmental dissociation of patch-matrix mesostriatal systems. J Neurosci 7: 3935-3944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- German DC, Manaye KF, Sonsalla PK, Brooks BA (1992) Midbrain dopaminergic cell loss in Parkinson's disease and MPTP-induced parkinsonism: sparing of calbindin-D28k-containing cells. Ann NY Acad Sci 648: 42-62. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Lee VM (2001) Parkin and the molecular pathways of Parkinson's disease. Neuron 31: 885-888. [DOI] [PubMed] [Google Scholar]
- Goldman SM, Tanner C (1998) Etiology of Parkinson's disease. In: Parkinson's disease and movement disorder (Jankovic J, Tolosa E, eds), pp 1333-1358. Baltimore: Williams and Wilkins.
- Gonzalez-Hernandez T, Rodriguez M (2000) Compartmental organization and chemical profile of dopaminergic and GABAergic neurons in the substantia nigra of the rat. J Comp Neurol 421: 107-135. [DOI] [PubMed] [Google Scholar]
- Gorski JA, Zeiler SR, Tamowski S, Jones KR (2003) Brain-derived neurotrophic factor is required for the maintenance of cortical dendrites. J Neurosci 23: 6856-6865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gratacos E, Checa N, Perez-Navarro E, Alberch J (2001a) Brain-derived neurotrophic factor (BDNF) mediates bone morphogenetic protein-2 (BMP-2) effects on cultured striatal neurones. J Neurochem 79: 747-755. [DOI] [PubMed] [Google Scholar]
- Gratacos E, Perez-Navarro E, Tolosa E, Arenas E, Alberch J (2001b) Neuroprotection of striatal neurons against kainate excitotoxicity by neurotrophins and GDNF family members. J Neurochem 78: 1287-1296. [DOI] [PubMed] [Google Scholar]
- Guidetti P, Charles V, Chen EY, Reddy PH, Kordower JH, Whetsell Jr WO, Schwarcz R, Tagle DA (2001) Early degenerative changes in transgenic mice expressing mutant huntingtin involve dendritic abnormalities but no impairment of mitochondrial energy production. Exp Neurol 169: 340-350. [DOI] [PubMed] [Google Scholar]
- Gundersen HJ, Bendtsen TF, Korbo L, Marcussen N, Moller A, Nielsen K, Nyengaard JR, Pakkenberg B, Sorensen FB, Vesterby A, West MJ (1988) Some new, simple and efficient stereological methods and their use in pathological research and diagnosis. Apmis 96: 379-394. [DOI] [PubMed] [Google Scholar]
- Hagg T (1998) Neurotrophins prevent death and differentially affect tyrosine hydroxylase of adult rat nigrostriatal neurons in vivo. Exp Neurol 149: 183-192. [DOI] [PubMed] [Google Scholar]
- Hamilton BA, Frankel WN, Kerrebrock AW, Hawkins TL, FitzHugh W, Kusumi K, Russell LB, Mueller KL, van Berkel V, Birren BW, Kruglyak L, Lander ES (1996) Disruption of the nuclear hormone receptor RORalpha in staggerer mice. Nature 379: 736-739. [DOI] [PubMed] [Google Scholar]
- Hattori N, Shimura H, Kubo S, Kitada T, Wang M, Asakawa S, Minashima S, Shimizu N, Suzuki T, Tanaka K, Mizuno Y (2000) Autosomal recessive juvenile parkinsonism: a key to understanding nigral degeneration in sporadic Parkinson's disease. Neuropathology 20 [Suppl]: S85-S90. [DOI] [PubMed] [Google Scholar]
- Howells DW, Porritt MJ, Wong JY, Batchelor PE, Kalnins R, Hughes AJ, Donnan GA (2000) Reduced BDNF mRNA expression in the Parkinson's disease substantia nigra. Exp Neurol 166: 127-135. [DOI] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF (2001) Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci 24: 677-736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung HC, Lee EH (1996) The mesolimbic dopaminergic pathway is more resistant than the nigrostriatal dopaminergic pathway to MPTP and MPP+ toxicity: role of BDNF gene expression. Brain Res Mol Brain Res 41: 14-26. [DOI] [PubMed] [Google Scholar]
- Hyman C, Hofer M, Barde YA, Juhasz M, Yancopoulos GD, Squinto SP, Lindsay RM (1991) BDNF is a neurotrophic factor for dopaminergic neurons of the substantia nigra. Nature 350: 230-232. [DOI] [PubMed] [Google Scholar]
- Hyman C, Juhasz M, Jackson C, Wright P, Ip NY, Lindsay RM (1994) Overlapping and distinct actions of the neurotrophins BDNF, NT-3, and NT-4/5 on cultured dopaminergic and GABAergic neurons of the ventral mesencephalon. J Neurosci 14: 335-347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacs KR, Wolpoe ME, Jacobowitz DM (1997) Calretinin-immunoreactive dopaminergic neurons from embryonic rat mesencephalon are resistant to levodopa-induced neurotoxicity. Exp Neurol 146: 25-32. [DOI] [PubMed] [Google Scholar]
- Jackson-Lewis V, Vila M, Djaldetti R, Guegan C, Liberatore G, Liu J, O'Malley KL, Burke RE, Przedborski S (2000) Developmental cell death in dopaminergic neurons of the substantia nigra of mice. J Comp Neurol 424: 476-488. [DOI] [PubMed] [Google Scholar]
- Jones KR, Farinas I, Backus C, Reichardt LF (1994) Targeted disruption of the BDNF gene perturbs brain and sensory neuron development but not motor neuron development. Cell 76: 989-999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan J, Bottner M, Schluesener HJ, Unsicker K, Krieglstein K (1997) Bone morphogenetic proteins: neurotrophic roles for midbrain dopaminergic neurons and implications of astroglial cells. Eur J Neurosci 9: 1699-1709. [DOI] [PubMed] [Google Scholar]
- Kawano H, Ohyama K, Kawamura K, Nagatsu I (1995) Migration of dopaminergic neurons in the embryonic mesencephalon of mice. Brain Res Dev Brain Res 86: 101-113. [DOI] [PubMed] [Google Scholar]
- Kholodilov N, Yarygina O, Oo TF, Zhang H, Sulzer D, Dauer W, Burke RE (2004) Regulation of the development of mesencephalic dopaminergic systems by the selective expression of glial cell line-derived neurotrophic factor in their targets. J Neurosci 24: 3136-3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim BG, Shin DH, Jeon GS, Seo JH, Kim YW, Jeon BS, Cho SS (2000) Relative sparing of calretinin containing neurons in the substantia nigra of 6-OHDA treated rat Parkinsonian model. Brain Res 855: 162-165. [DOI] [PubMed] [Google Scholar]
- Kohno R, Sawada H, Kawamoto Y, Uemura K, Shibasaki H, Shimohama S (2004) BDNF is induced by wild-type alpha-synuclein but not by the two mutants, A30P or A53T, in glioma cell line. Biochem Biophys Res Commun 318: 113-118. [DOI] [PubMed] [Google Scholar]
- Lalonde R (1987a) Motor abnormalities in weaver mutant mice. Exp Brain Res 65: 479-481. [DOI] [PubMed] [Google Scholar]
- Lalonde R (1987b) Motor abnormalities in staggerer mutant mice. Exp Brain Res 68: 417-420. [DOI] [PubMed] [Google Scholar]
- Levivier M, Przedborski S, Bencsics C, Kang UJ (1995) Intrastriatal implantation of fibroblasts genetically engineered to produce brain-derived neurotrophic factor prevents degeneration of dopaminergic neurons in a rat model of Parkinson's disease. J Neurosci 15: 7810-7820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang CL, Sinton CM, German DC (1996a) Midbrain dopaminergic neurons in the mouse: co-localization with calbindin-D28K and calretinin. Neuroscience 75: 523-533. [DOI] [PubMed] [Google Scholar]
- Liang CL, Sinton CM, Sonsalla PK, German DC (1996b) Midbrain dopaminergic neurons in the mouse that contain calbindin-D28k exhibit reduced vulnerability to MPTP-induced neurodegeneration. Neurodegeneration 5: 313-318. [DOI] [PubMed] [Google Scholar]
- Lieb K, Andersen C, Lazarov N, Zienecker R, Urban I, Reisert I, Pilgrim C (1996) Pre- and postnatal development of dopaminergic neuron numbers in the male and female mouse midbrain. Brain Res Dev Brain Res 94: 37-43. [DOI] [PubMed] [Google Scholar]
- Liebl DJ, Tessarollo L, Palko ME, Parada LF (1997) Absence of sensory neurons before target innervation in brain-derived neurotrophic factor-, neurotrophin 3-, and TrkC-deficient embryonic mice. J Neurosci 17: 9113-9121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin LF, Doherty DH, Lile JD, Bektesh S, Collins F (1993) GDNF: a glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science 260: 1130-1132. [DOI] [PubMed] [Google Scholar]
- Lotharius J, Brundin P (2002) Pathogenesis of Parkinson's disease: dopamine, vesicles and alpha-synuclein. Nat Rev Neurosci 3: 932-942. [DOI] [PubMed] [Google Scholar]
- McMahon A, Wong BS, Iacopino AM, Ng MC, Chi S, German DC (1998) Calbindin-D28k buffers intracellular calcium and promotes resistance to degeneration in PC12 cells. Brain Res Mol Brain Res 54: 56-63. [DOI] [PubMed] [Google Scholar]
- Mogi M, Togari A, Kondo T, Mizuno Y, Komure O, Kuno S, Ichinose H, Nagatsu T (1999) Brain-derived growth factor and nerve growth factor concentrations are decreased in the substantia nigra in Parkinson's disease. Neurosci Lett 270: 45-48. [DOI] [PubMed] [Google Scholar]
- Mouatt-Prigent A, Agid Y, Hirsch EC (1994) Does the calcium binding protein calretinin protect dopaminergic neurons against degeneration in Parkinson's disease? Brain Res 668: 62-70. [DOI] [PubMed] [Google Scholar]
- Mullen RJ, Buck CR, Smith AM (1992) NeuN, a neuronal specific nuclear protein in vertebrates. Development 116: 201-211. [DOI] [PubMed] [Google Scholar]
- Murer MG, Yan Q, Raisman-Vozari R (2001) Brain-derived neurotrophic factor in the control human brain, and in Alzheimer's disease and Parkinson's disease. Prog Neurobiol 63: 71-124. [DOI] [PubMed] [Google Scholar]
- Nemoto C, Hida T, Arai R (1999) Calretinin and calbindin-D28k in dopaminergic neurons of the rat midbrain: a triple-labeling immunohistochemical study. Brain Res 846: 129-136. [DOI] [PubMed] [Google Scholar]
- Nishio T, Furukawa S, Akiguchi I, Sunohara N (1998) Medial nigral dopamine neurons have rich neurotrophin support in humans. NeuroReport 9: 2847-2851. [DOI] [PubMed] [Google Scholar]
- Numan S, Seroogy KB (1999) Expression of trkB and trkC mRNAs by adult midbrain dopamine neurons: a double-label in situ hybridization study. J Comp Neurol 403: 295-308. [DOI] [PubMed] [Google Scholar]
- Okazawa H, Murata M, Watanabe M, Kamei M, Kanazawa I (1992) Dopaminergic stimulation up-regulates the in vivo expression of brain-derived neurotrophic factor (BDNF) in the striatum. FEBS Lett 313: 138-142. [DOI] [PubMed] [Google Scholar]
- Oo TF, Kholodilov N, Burke RE (2003) Regulation of natural cell death in dopaminergic neurons of the substantia nigra by striatal glial cell line-derived neurotrophic factor in vivo J Neurosci 23: 5141-5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parain K, Murer MG, Yan Q, Faucheux B, Agid Y, Hirsch E, Raisman-Vozari R (1999) Reduced expression of brain-derived neurotrophic factor protein in Parkinson's disease substantia nigra. NeuroReport 10: 557-561. [DOI] [PubMed] [Google Scholar]
- Parish CL, Finkelstein DI, Drago J, Borrelli E, Horne MK (2001) The role of dopamine receptors in regulating the size of axonal arbors. J Neurosci 21: 5147-5157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patapoutian A, Reichardt LF (2001) Trk receptors: mediators of neurotrophin action. Curr Opin Neurobiol 11: 272-280. [DOI] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL (1997) Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science 276: 2045-2047. [DOI] [PubMed] [Google Scholar]
- Porritt MJ, Batchelor PE, Howells DW (2005) Inhibiting BDNF expression by antisense oligonucleotide infusion causes loss of nigral dopaminergic neurons. Exp Neurol 192: 226-234. [DOI] [PubMed] [Google Scholar]
- Reiriz J, Espejo M, Ventura F, Ambrosio S, Alberch J (1999) Bone morphogenetic protein-2 promotes dissociated effects on the number and differentiation of cultured ventral mesencephalic dopaminergic neurons. J Neurobiol 38: 161-170. [PubMed] [Google Scholar]
- Rico B, Xu B, Reichardt LF (2002) TrkB receptor signaling is required for establishment of GABAergic synapses in the cerebellum. Nat Neurosci 5: 225-233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seroogy KB, Lundgren KH, Tran TM, Guthrie KM, Isackson PJ, Gall CM (1994) Dopaminergic neurons in rat ventral midbrain express brain-derived neurotrophic factor and neurotrophin-3 mRNAs. J Comp Neurol 342: 321-334. [DOI] [PubMed] [Google Scholar]
- Shimura H, Hattori N, Kubo S, Mizuno Y, Asakawa S, Minoshima S, Shimizu N, Iwai K, Chiba T, Tanaka K, Suzuki T (2000) Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet 25: 302-305. [DOI] [PubMed] [Google Scholar]
- Shults CW, Kimber T, Altar CA (1995) BDNF attenuates the effects of intrastriatal injection of 6-hydroxydopamine. NeuroReport 6: 1109-1112. [DOI] [PubMed] [Google Scholar]
- Son JH, Chun HS, Joh TH, Cho S, Conti B, Lee JW (1999) Neuroprotection and neuronal differentiation studies using substantia nigra dopaminergic cells derived from transgenic mouse embryos. J Neurosci 19: 10-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano P (1999) Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet 21: 70-71. [DOI] [PubMed] [Google Scholar]
- Studer L, Spenger C, Seiler RW, Altar CA, Lindsay RM, Hyman C (1995) Comparison of the effects of the neurotrophins on the morphological structure of dopaminergic neurons in cultures of rat substantia nigra. Eur J Neurosci 7: 223-233. [DOI] [PubMed] [Google Scholar]
- Tarantino LM, Gould TJ, Druhan JP, Bucan M (2000) Behavior and mutagenesis screens: the importance of baseline analysis of inbred strains. Mamm Genome 11: 555-564. [DOI] [PubMed] [Google Scholar]
- Tomac A, Lindqvist E, Lin LF, Ogren SO, Young D, Hoffer BJ, Olson L (1995) Protection and repair of the nigrostriatal dopaminergic system by GDNF in vivo. Nature 373: 335-339. [DOI] [PubMed] [Google Scholar]
- Tsuboi K, Kimber TA, Shults CW (2000) Calretinin-containing axons and neurons are resistant to an intrastriatal 6-hydroxydopamine lesion. Brain Res 866: 55-64. [DOI] [PubMed] [Google Scholar]
- van Dellen A, Deacon R, York D, Blakemore C, Hannan AJ (2001) Anterior cingulate cortical transplantation in transgenic Huntington's disease mice. Brain Res Bull 56: 313-318. [DOI] [PubMed] [Google Scholar]
- van den Akker E, Reijnen M, Korving J, Brouwer A, Meijlink F, Deschamps J (1999) Targeted inactivation of Hoxb8 affects survival of a spinal ganglion and causes aberrant limb reflexes. Mech Dev 89: 103-114. [DOI] [PubMed] [Google Scholar]
- Venero JL, Vizuete ML, Revuelta M, Vargas C, Cano J, Machado A (2000) Upregulation of BDNF mRNA and trkB mRNA in the nigrostriatal system and in the lesion site following unilateral transection of the medial forebrain bundle. Exp Neurol 161: 38-48. [DOI] [PubMed] [Google Scholar]
- Vigers AJ, Baquet ZC, Jones KR (2000) Expression of neurotrophin-3 in the mouse forebrain: insights from a targeted LacZ reporter. J Comp Neurol 416: 398-415. [DOI] [PubMed] [Google Scholar]
- West A, Periquet M, Lincoln S, Lucking CB, Nicholl D, Bonifati V, Rawal N, Gasser T, Lohmann E, Deleuze JF, Maraganore D, Levey A, Wood N, Durr A, Hardy J, Brice A, Farrer M (2002) Complex relationship between Parkin mutations and Parkinson disease. Am J Med Genet 114: 584-591. [DOI] [PubMed] [Google Scholar]
- West AB, Maidment NT (2004) Genetics of parkin-linked disease. Hum Genet 114: 327-336. [DOI] [PubMed] [Google Scholar]
- West MJ, Slomianka L, Gundersen HJ (1991) Unbiased stereological estimation of the total number of neurons in the subdivisions of the rat hippocampus using the optical fractionator. Anat Rec 231: 482-497. [DOI] [PubMed] [Google Scholar]
- Yamada T, McGeer PL, Baimbridge KG, McGeer EG (1990) Relative sparing in Parkinson's disease of substantia nigra dopamine neurons containing calbindin-D28K. Brain Res 526: 303-307. [DOI] [PubMed] [Google Scholar]
- Zaman V, Nelson ME, Gerhardt GA, Rohrer B (2004) Neurodegenerative alterations in the nigrostriatal system of trkB hypomorphic mice. Exp Neurol 190: 337-346. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Tartari M, Crotti A, Goffredo D, Valenza M, Conti L, Cataudella T, Leavitt BR, Hayden MR, Timmusk T, Rigamonti D, Cattaneo E (2003) Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nat Genet 35: 76-83. [DOI] [PubMed] [Google Scholar]
- Zuch CL, David D, Ujhelyi L, Hudson JL, Gerhardt GA, Kaplan PL, Bickford PC (2004) Beneficial effects of intraventricularly administered BMP-7 following a striatal 6-hydroxydopamine lesion. Brain Res 1010: 10-16. [DOI] [PubMed] [Google Scholar]