

Abstract
Neurexins constitute a large family of highly variable cell-surface molecules that may function in synaptic transmission and/or synapse formation. Each of the three known neurexin genes encodes two major neurexin variants, α- and β-neurexins, that are composed of distinct extracellular domains linked to identical intracellular sequences. Deletions of one, two, or all three α-neurexins in mice recently demonstrated their essential role at synapses. In multiple α-neurexin knock-outs, neurotransmitter release from excitatory and inhibitory synapses was severely reduced, primarily probably because voltage-dependent Ca2+ channels were impaired. It remained unclear, however, which neurexin variants actually influence exocytosis and Ca2+ channels, which domain of neurexins is required for this function, and which Ca2+-channel subtypes are regulated. Here, we show by electrophysiological recordings that transgenic neurexin 1α rescues the release and Ca2+-current phenotypes, whereas transgenic neurexin 1β has no effect, indicating the importance of the extracellular sequences for the function of neurexins. Because neurexin 1α rescued the knock-out phenotype independent of the α-neurexin gene deleted, these data are consistent with a redundant function among different α-neurexins. In both knock-out and transgenically rescued mice, α-neurexins selectively affected the component of neurotransmitter release that depended on activation of N- and P/Q-type Ca2+ channels, but left L-type Ca2+ channels unscathed. Our findings indicate that α-neurexins represent organizer molecules in neurotransmission that regulate N- and P/Q-type Ca2+ channels, constituting an essential role at synapses that critically involves the extracellular domains of neurexins.
Keywords: transgenic mouse, synapse, transmission, calcium channels, cell adhesion, brainstem
Introduction
Neurotransmission requires formation and differentiation of presynaptic and postsynaptic compartments, including the assembly of an extensive molecular machinery for exocytosis and signal transduction. The number, ultrastructure, and physiological properties of synapses vary considerably between individual neurons and brain regions and can change dramatically during synaptogenesis and synaptic plasticity. Neurexins were proposed as candidate molecules to mediate some of the diversity (Missler and Sudhof, 1998) because (1) their structure indicates a receptor-like transmembrane protein (Ushkaryov et al., 1992, 1994; Ushkaryov and Sudhof, 1993), (2) they bind to postsynaptic cell-surface proteins (Ichtchenko et al., 1995; Sugita et al., 2001), (3) they are receptors for α-latrotoxin, which induces neurotransmitter release (Geppert et al., 1998; Sugita et al., 1999), and (4) they are polymorphic in their extracellular sequences because of significant alternative splicing (Ichtchenko et al., 1995; Missler et al., 1998; Sugita et al., 1999, 2001). Mammalian neurexins are encoded by three genes that contain independent promoters for α- and β-neurexins (Rowen et al., 2002; Tabuchi and Sudhof, 2002). α-Neurexins contain substantially longer extracellular sequences than the much shorter β-neurexins, but share with β-neurexins the same C terminus (Missler and Sudhof, 1998). Consequently, available biochemical evidence has revealed overlapping C-terminal (intracellular) binding partners (Hata et al., 1996; Butz et al., 1998; Biederer and Sudhof, 2000), but distinct extracellular interactions for α- and β-neurexins (Ichtchenko et al., 1995; Missler et al., 1998; Sugita et al., 2001).
To determine the role of neurexins, we previously produced knock-out (KO) mice that lack one, two, or all three α-neurexins (Missler et al., 2003). Mutants with different combinations of knock-out alleles mostly died prematurely as a result of respiratory problems. Analyses of newborn mice deficient for multiple α-neurexins revealed almost no changes in brain architecture or synapse structure but uncovered a severe reduction in neurotransmitter release. As the presumptive cause of the inefficient exocytosis in mutant mice, we suggested that voltage-dependent Ca2+ channels (VDCCs) were impaired because the response pattern of synaptic transmission to specific Ca2+-channel blockers was altered, and whole-cell Ca2+ currents were reduced (Missler et al., 2003). Although no knock-out mice are available yet for β-neurexins, their putative function was addressed recently by cell culture assays: overexpression of neurexin 1β induced differentiation of postsynaptic receptors (Graf et al., 2004), and its trans-synaptic partner neuroligin stimulated de novo formation and differentiation of presynaptic terminals in vitro (Scheiffele et al., 2000; Dean et al., 2003; Sara et al., 2005). These studies established a prominent role for neurexins at synapses but also raised important questions: is the phenotype of reduced neurotransmitter release in knock-out mice specific for the deletion of α-neurexins? Are there functional differences between various neurexin genes? Is the highly variable extracellular domain of neurexins required for regulating VDCCs? Why were the β-neurexins that are still abundantly expressed in α-neurexin KO mice apparently unable to compensate for the loss of α-neurexins? Which subtypes of high voltage-activated Ca2+ channels are impaired in α-neurexin KO mice?
Here, we show that (1) a single transgenically expressed neurexin 1α splice variant rescued the impaired neurotransmission at excitatory and inhibitory synapses in all deletions of α-neurexins tested; (2) comparable transgenic expression of β-neurexins had no effect, indicating that extracellular sequences of α-neurexins are important for regulating VDCCs; and (3) α-neurexins selectively altered N- and P/Q-type Ca2+-channel function but left L-type Ca2+ channels unchanged.
Materials and Methods
Generation and breeding of transgenic mice. Transgenic vectors for neurexin 1α (αNrx::HRP) (Missler et al., 2003) and neurexin 1β (βNrx::HRP) (generated de novo for this study) were constructed using cDNA plasmids pCMVL2 and pCMVL13, respectively. Plasmid pBS-HRP served as the source of the horseradish peroxidase (a gift from Dr. H. Kraemer, University of Texas Southwestern Medical Center). Two internal restriction sites (the 5′ NotI site in pCMVL2 and the PvuI site in pBS-HRP) had to be destroyed by silent mutagenesis. A 980 bp HRP fragment was cloned into the 3′ NotI site of the modified pCMVL2 and of pCMVL13 [resulting junctional amino acid sequence: neurexin... GRQPAMQLT.HRP.NSNSGQPRGR... neurexin (italicized residues represent the HRP cDNA)]. The chimeric neurexin-HRP cDNAs were blunt-end cloned into the unique XhoI site of the mouse Thy1.2 expression vector pEX21 (a gift from Dr. M. Goedert, Medical Research Council, Cambridge, UK) to generate the transgenic vectors. The Thy1.2 promoter was chosen because of its developmentally early widespread neuronal expression (Andra et al., 1996). Transgenic mice were produced by pronucleus injection, and founder mice and progeny were genotyped using dot/Southern blotting with EagI/EagI-isolated 32P-labeled HRP cDNA probes. Because our rescue experiments required crossing mice from FVB strains (transgenics) into the mixed 129Sv × C57BL/6 background (α-neurexin KOs), each set of experiments was done on littermate mice with and without additional transgene. Genotyping a large number of newborn offspring [n = 265 pups killed at postnatal day 1 (P1)] confirmed a normal mendelian ratio for all combinations of mutated neurexin alleles (data not shown). In addition, 52% of pups (n = 96 of 183) inherited the αNrx::HRP transgene, and 51% inherited the βNrx::HRP transgene (n = 42 of 82). All analyses were performed with hemizygous transgenic mice on wild-type or α-neurexin KO backgrounds, as described below.
HRP antibodies and biochemical procedures. Expression of neurexin-HRP transgenic fusion proteins was tested in brains of hemizygous mice using antibodies against HRP epitopes. Most commercial antibodies were generated against the plant HRP and cross-react with endogenous mouse brain proteins (data not shown). Therefore, we produced a recombinant HRP-glutathione S-transferase (GST) fusion protein (from pET28-HRP) in BL21 cells to affinity purify a commercially available polyclonal serum (Nordic) using nitrocellulose membrane stripes containing HRP separated by SDS-PAGE (Olmsted, 1981). Immunoprecipitations and GST pull-downs were performed with P2 brain membrane proteins solubilized in 1% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate with affinity-purified HRP antibodies (anti-HRPaff) or recombinant calcium/calmodulin-dependent serine kinase (CASK) immobilized on protein A and glutathione agarose beads, respectively (Hata et al., 1996). To measure the enzymatic activity of transgenic neurexin-HRP fusion proteins, mouse brain membrane fractions (nonsolubilized P2 fractions) were incubated for 15 min with diaminobenzidine (1 mg/ml) and H2O2 (0.04%), and the absorbance at 595 nm was determined with reactions from nontransgenic mice serving as controls.
Morphological studies. To analyze the distribution of transgenic expression of both αNrx::HRP and βNrx::HRP proteins in the brainstem of newborn mice, brains were freshly dissected in PBS, and brainstem preparations were immersion fixed in 2% paraformaldehyde in 0.1 m phosphate buffer for 2 h, postfixed for 2 h in the same solution, and cryoprotected in 25% sucrose. For light microscopy of transgenic mice, 12 μm cryosections were thaw mounted on poly-l-lysine-coated glass slides and labeled overnight with anti-HRPaff antibodies in blocking buffer (0.1% Triton X-100, 50% normal goat serum, and PBS). Secondary goat anti-rabbit antibody and PAP antibodies (rabbit PAP) were incubated with sections in blocking solution (both antibodies were from Sternberger Monoclonals, Lutherville, MD) and visualized with diaminobenzidine (0.05% w/v) plus H2O2 (0.005% v/v) and heavy metal enhancement (NiCl2, 0.15% w/v). To compare tissues from different genotypes, sections from several mice were usually processed in parallel. Overview panels of newborn brainstem (see Fig. 2) are montages from up to 15 individual pictures captured via an AxioCam HR digital camera system mounted on an Axioscope 2 microscope (Zeiss, Oberkochen, Germany) and composed using Adobe Photoshop 6.0 software (Adobe Systems, San Jose, CA).
Figure 2.

Transgenically expressed α- and β-neurexin can be explored for their potential to rescue the knock-out phenotype. A-C, Overviews of brainstem areas from newborn αNrx::HRPtg/+ (A) or βNrx::HRPtg/+ (B) mice or from nontransgenic (non-Tg) littermates (C), assembled from sections at the indicated level (see inset). Transgenic neurexins visualized by immunohistochemistry with anti-HRPaff antibodies are present in areas used for functional analyses [PBC, part of the rostroventrolateral nucleus (RVLN), shadowed in C; and the nucleus of the hypoglossal nerve, nucleus of the 12th cranial nerve (XII), shadowed in C]. NA, Nucleus ambiguus; IO, inferior olive complex; Sp5I, spinal trigeminal nucleus interpolar part; 4V, fourth ventricle; ECu, external cuneate nucleus. Scale bar: A-C, 250 μm. D, A calorimetric enzyme assay of membrane fractions from adult nontransgenic mice and mice expressing βNrx::HRP or αNrx::HRP was used to assess the integrity of the HRP moiety. Peroxidase activity is depicted below the corresponding Western blot lanes and expressed as extinction at OD595 (means from n = 3 experiments). E, Mouse breeding strategy for electrophysiological experiments. Animals homozygous for the neurexin 2α and double heterozygous for neurexin 1α and 3α genes (Nrx1α+/- 2α-/- 3α+/-) were bred to mice of the same KO genotype but carrying an additional αNrx::HRPtg/+ or βNrx::HRPtg/+ or no transgene. All functional analyses were done on their newborn offspring (P1).
Electrophysiological recordings. All electrophysiological analyses were performed on brainstem neurons of mice whose genotype was unknown to the experimenter, essentially as described previously (Zhang et al., 1999; Missler et al., 2003). Acute slices containing the pre-Botzinger complex (PBC) and hypoglossal motor nucleus from newborn (P1) littermate mice were used for whole-cell recordings. The bath solution in all experiments consisted of the following (in mm): 118 NaCl, 3 KCl, 1.5 CaCl2, 1 MgCl2, 25 NaHCO3, 1 NaH2PO4, and 20 glucose, pH 7.4 (aerated with 95% O2 and 5% CO2 and kept at 28-30°C). Evoked EPSCs (eEPSCs) and evoked IPSCs (eIPSCs) were recorded from hypoglossal motor neurons in the presence of 1 μm strychnine and 1 μm bicuculline or from PBC neurons in the presence of 10 μm CNQX, respectively. EPSCs were evoked by 0.1 Hz field stimulations of axons of interneurons close to the PBC using a custom-built bipolar platinum electrode. An isolation unit, IsoFlex (A.M.P.I., Jerusalem, Israel), with a custom-built power supply, was used to apply currents of supramaximal stimulation strength (∼1 mA actual current near the slice). During the long time course of investigations represented in this study (extending over 3 years), different stimulation setups and types of electrodes had to be used, preventing the direct comparison of absolute EPSC amplitudes between the αNrx::HRPtg/+ and βNrx::HRPtg/+ experiments. This, however, does not affect our conclusions regarding their respective rescue potential. The pipette solution for eEPSC and eIPSC measurements contained the following (in mm): 140 K-gluconate (eEPSCs) or 140 KCl (eIPSCs), 1 CaCl2, 10 EGTA, 2 MgCl2, 4 Na3ATP, 0.5 Na3GTP, and 10 HEPES, pH 7.3. Peak amplitudes were averaged from 25 consecutive responses. To monitor changes in input resistance, current responses to a -10 mV voltage step (20 ms) from a holding potential of -70 mV were recorded before every fifth stimulus. In all experiments, the distance between the stimulation and recording electrodes was similar between slices of different genotypes, and we could not find any significant differences in latency of evoked responses between the genotypes. The supramaximal stimulation protocol using constant current amplitudes (see above) differs slightly from the one applied in our previous paper for the KO analysis (Missler et al., 2003), in which we adjusted the stimulation strength according to the failure rate obtained. The current approach was chosen to emphasize differences between mice that are homozygous for the same α-neurexin mutation and that either lack or contain the neurexin transgene. Using the supramaximal stimulation strength in all cases produced a higher resolution of rescue effect even in the case of multiple α-neurexin KOs. However, using supramaximal stimulation strength obscured comparison of absolute responses between the different genotypes in these experiments, explaining why the differences between KO combinations are less pronounced than in our previous study.
Miniature postsynaptic current analysis. Spontaneous miniature IPSCs (mIPSCs) and miniature EPSCs (mEPSCs) were recorded from neurons of the PBC and hypoglossal nucleus at a Cl- reversal potential of ∼0 mV in 10 μm CNQX or 1 μm strychnine and 1 μm bicuculline, respectively; signals with amplitudes of at least 2.5 times above background noise were selected, and statistical significance was tested in each experiment. Based on our selection criteria, miniature postsynaptic current (mini) events close to the detection level could be missed; however, the noise levels and amplitudes were similar between genotypes. Voltage-activated Ca2+ currents were measured from neurons of the PBC with electrodes containing (in mm) 110 CsCl2, 30 TEA-Cl, 1 CaCl2, 10 EGTA, 2 MgCl2, 4 Na3ATP, 0.5 Na3GTP, and 10 HEPES, pH 7.3, and with 0.5 μm tetrodotoxin in the bath solution. Serial and membrane resistances were estimated from current transients induced by 20 mV hyperpolarization voltage commands from a holding potential of -70 mV. The serial resistance was compensated by 80%, and patches with a serial resistance of >20 MΩ, a membrane resistance of <0.8 GΩ, or leak currents of >150 pA were excluded. The membrane currents were filtered by a four-pole Bessel filter at a corner frequency of 2 kHz and digitized at a sampling rate of 5 kHz using the DigiData 1200B interface (Axon Instruments, Union City, CA). Currents were recorded in response to voltage step changes from the -70 mV holding potential to test potentials between -80 and +30 mV and quantified as peak currents in response to voltage steps from -70 to 0 mV. Ca2+-current measurements were corrected using the P/4 protocol that subtracts leak currents measured during four leak-subtraction prepulses applied immediately before each voltage step. Blocking experiments using specific VDCC inhibitors were always performed in the same sequential order, applying 10 μm nifedipine for L-type channel contributions, 0.5 μm ω-conotoxin MVIIA for N-type channel contributions, 0.2 μm agatoxin IVA for P/Q-type channel contributions, and 100 μm Cd2+ to probe for residual Ca2+ channel activities. Data acquisition and analysis were performed using commercially available software [pClamp 6.0 and AxoGraph 4.6 (Axon Instruments); Prism 3 software (GraphPad Software, San Diego, CA)].
Miscellaneous. Primary antibodies to neurexins (A473), neuroligin (4C12), and synaptotagmin 1 (W855) were characterized previously (Ushkaryov et al., 1992; Butz et al., 1998; Song et al., 1999) or obtained commercially: Sy38, anti-synaptophysin (DakoCytomation, Hamburg, Germany), anti-P/Q Ca2+-channel subunit and anti-NMDA R1 (Chemicon, Temecula, CA), and heat shock protein 70 (Affinity BioReagents, Golden, CO). Statistical significance was tested with a two-tailed Student's t test in Excel (Microsoft, Redmond, WA) spreadsheets or with InStat (GraphPad Software) and, in case of mini events, with the Kolmogorov-Smirnov test integrated in MiniAnalysis (Synaptosoft, Decatur, GA).
Results
Transgenic mice expressing neurexins that differ solely in their extracellular sequences
To elucidate the molecular determinants that link neurexins to Ca2+ channels, we used a comparative transgenic rescue analysis. This analysis allows us to explore the role of structurally distinct α- and β-neurexins in synaptic transmission. In our initial characterization of α-neurexin KO mice, we showed that the decrease in whole-cell Ca2+ currents caused by deletion of α-neurexins can be partially reversed by transgenic neurexin 1α expressed as a fusion protein with HRP under control of the Thy1 promoter (Missler et al., 2003). This finding raised questions as to whether the reduced neurotransmitter release was also reversed by the transgene and whether the partial rescue was a specific consequence of transgenic neurexin 1α expression (or could be extended to β-neurexins). To address these questions, here we have compared the physiological effects mediated by two sets of transgenic mice that express two different neurexins (Fig. 1A): (1) the original neurexin 1α transgene (αNrx::HRP) (Missler et al., 2003) and (2) a newly generated neurexin 1β transgene (βNrx::HRP).
Figure 1.

Transgenic mice expressing two distinct neurexins under control of a heterologous promoter. A, Domain model of transgenically introduced αNrx::HRP and βNrx::HRP proteins with an internal HRP epitope to monitor expression. The transgenic α- and β-neurexins under control of the Thy1.2 promoter differ exclusively in their extracellular sequences. Gray sheet, transmembrane domain (Missler and Sudhof, 1998). B, Comparative immunoblot analysis of several transgenic (Tg) mouse lines expressing βNrx::HRP. Brain membrane proteins (20 μg/lane) were probed with anti-HRPaff. Loading control blots were probed with antibodies against the NMDA receptor subunit R1 (anti-NR1). C, Bound material from pull-down experiments with the protein CASK was analyzed by immunoblotting with affinity-purified HRP antibodies (top) or pan-neurexin antibodies (middle). Both transgenically expressed (arrows) and endogenous (asterisks) neurexins interact with their intracellular ligand CASK. Equivalent amounts of recombinantly expressed GST-CASK were used for the pull-down procedures (bottom; Coomassie stain). D, Immunoprecipitation (IP) of brain membrane proteins from transgenic mice expressing αNrx::HRP and βNrx::HRP and from control mice (non-Tg). Transgenic neurexin-HRP fusion proteins were precipitated with anti-HRPaff and analyzed by immunoblotting with antibodies to neurexins (top) or neuroligins (middle; bottom shows control samples before immunoprecipitation). Only βNrx::HRP binds to neuroligin, confirming that isoform-specific interactions are intact in brains of transgenic animals.
Both sets of transgenic mice were produced with identical vectors in which an HRP cDNA was inserted at the same position into neurexin 1α or 1β to monitor expression (Fig. 1A). HRP was chosen as an internal epitope tag, because studies in Drosophila demonstrated that HRP did not interfere with membrane traffic when inserted into the extracellular sequences of a cell-surface protein (Sunio et al., 1999; Dubois et al., 2001). The major difference between the two sets of transgenic mice arises from variations in neurexin 1α and 1β sequences, which are identical apart from their extracellular domains, the former containing a large extracellular sequence consisting of multiple domains and the latter consisting of a shorter extracellular region (Ushkaryov et al., 1992; Missler and Sudhof, 1998) (Fig. 1A). To ensure that the differences observed between the activity of these transgenes were not attributable to insertion effects, we generated multiple lines of both transgenes, which, as described below, produced similar transgene protein levels and expression patterns and thus allowed a direct comparison of the functions of the two types of neurexins.
Heterozygous (αNrx::HRPtg/+ and βNrx::HRPtg/+) and homozygous (αNrx::HRPtg/tg and βNrx::HRPtg/tg) transgenic mice were viable and fertile (data not shown), indicating that the transgenic neurexins did not strongly interfere with normal neurexin function or the role of an unidentified essential gene close to the insertion site of the transgenes. To examine protein expression, we affinity-purified commercially available HRP antibodies on immobilized recombinant HRP and used anti-HRPaff for immunoblotting. The anti-HRPaff reacted with a single protein on immunoblots from adult transgenic mice and detected no cross-reactive bands in nontransgenic littermates (Fig. 1B). Comparable immunoblots of α-neurexin transgenic mice (data not shown) indicated that expression levels of the transgenic βNrx::HRP were approximately the same as those for transgenic αNrx::HRP (for a direct comparison of their expression in newborn mice, see Fig. 2). We next investigated whether the HRP-tagged neurexins expressed in transgenic mice were capable of normal protein-protein interactions typical for endogenous neurexins. Pull-down experiments using a GST fusion protein containing the postsynaptic density-95/Discs large/zona occludens-1 (PDZ) domain of CASK, a protein that binds to the C terminus of neurexins (Hata et al., 1996), revealed that both transgenic neurexins bound CASK efficiently (Fig. 1C). This observation suggested that endogenous (Fig. 1C, asterisks) and transgenic (Fig. 1C, arrows) neurexins engage in similar intracellular binding activities, as predicted from their identical C terminus. To explore the integrity of specific extracellular interactions, we performed immunoprecipitations of brain proteins from transgenic mice with anti-HRPaff antibodies. These experiments showed that the βNrx::HRP fusion protein is present in the brain in a complex with neuroligin 1, as predicted for this particular splice variant (Fig. 1D) (Ichtchenko et al., 1995). In contrast, no neuroligin 1 coimmunoprecipitated with αNrx::HRP was detectable.
To ensure a meaningful comparison of the effects of the transgenic neurexins (see below), we confirmed by immunohistochemistry that transgenic neurexin 1α and 1β are expressed in similar patterns in the brainstem of newborn mice in areas used for the electrophysiological rescue analysis (Fig. 2A-C, RVLN, XII). Although activity measurements revealed that the HRP moiety of both transgenic proteins was enzymatically active (Fig. 2D), the peroxidase activity conferred by HRP could not be used for these morphological studies because of the low sensitivity of the technique (data not shown). For functional analyses, we bred mice that are homozygous for the neurexin 2α KO allele (Nrx2α-/-) and heterozygous for the two other neurexin KO alleles (Nrx1α+/- and Nrx3α+/-) and, in addition, either lack or contain a hemizygous transgene (nontransgenic, αNrx::HRPtg/+, or βNrx::HRPtg/+) (Fig. 2E). Mating of this parent generation resulted in littermate homozygous neurexin 2α KO offspring that additionally carried various combinations of the neurexin 1α and 3α alleles and also (in 50% of the cases) a single copy of the respective transgene.
Transgenic neurexin 1α enhances synaptic transmission and Ca2+ currents in α-neurexin KO mice
To explore the potential of one α-neurexin isoform to rescue the synaptic phenotype of multiple α-neurexin KO mice, we first asked whether αNrx::HRPtg/+ was able to alter the amplitude of evoked synaptic responses. Because α-neurexin KO mice homozygous for two or three mutant alleles die early postnatally, we analyzed all mutant mice immediately after birth (P1) in acute brainstem slices, because the synaptic connections in this area mature relatively early and are involved in breathing control [normal breathing was disrupted in α-neurexin KO mice (Missler et al., 2003)]. Synaptic responses were monitored in hypoglossal motor neurons after afferent fiber stimulation near the PBC by measuring eEPSCs in littermate α-neurexin mutant mice with and without the αNrx::HRP transgene.
Representative EPSC traces in Figure 3A and their quantification in Figure 3B show that transgenic expression of αNrx::HRP caused a dramatic increase in evoked synaptic responses in α-neurexin KO mice (Fig. 3B). In single neurexin 2α (SKO2) and double neurexin 2α/3α (DKO2/3) or 1α/2α KO (DKO1/2) mice, αNrx::HRP doubled the mean EPSC to ∼198-223% compared with their nontransgenic littermates. Higher absolute EPSC amplitudes in SKO mice expressing αNrx::HRP, compared with DKO and triple KO (TKO) mice containing αNrx::HRP, suggested that a single transgene was not sufficient to overcome deletion of multiple α-neurexins (Fig. 3B). Only a limited number of TKO mice could be analyzed in these experiments because TKO mice are difficult to obtain by breeding (the probability of obtaining a TKO plus αNrx::HRPtg/+ was 3.125%). Consistent with the increased amplitude of eEPSCs, the αNrx::HRPtg/+ also decreased the failure rate in all genotypes (from 19.2 to 7.2% in SKO2 mice when the αNrx::HRP transgene was expressed and from 30.0 to 7.3% in DKO1/2 mice; data not shown). To test whether this rescue also extended to evoked responses at inhibitory terminals, eIPSCs were recorded from PBC neurons after field stimulations of axons close to the PBC in the presence of 10 μm CNQX. For these experiments, because of the difficulty of breeding a sufficient number of mice with the correct genotype, quantifications were restricted to littermate mice of one α-neurexin genotype (DKO2/3) with and without αNrx::HRPtg/+. As for excitatory synapses, eIPSCs increased significantly when an α-neurexin transgene was present (DKO2/3, 165.4 ± 38.11 pA, n = 6; DKO2/3 and αNrx::HRPtg/+, 347.7 ± 68.55 pA, n = 5; p < 0.05).
Figure 3.
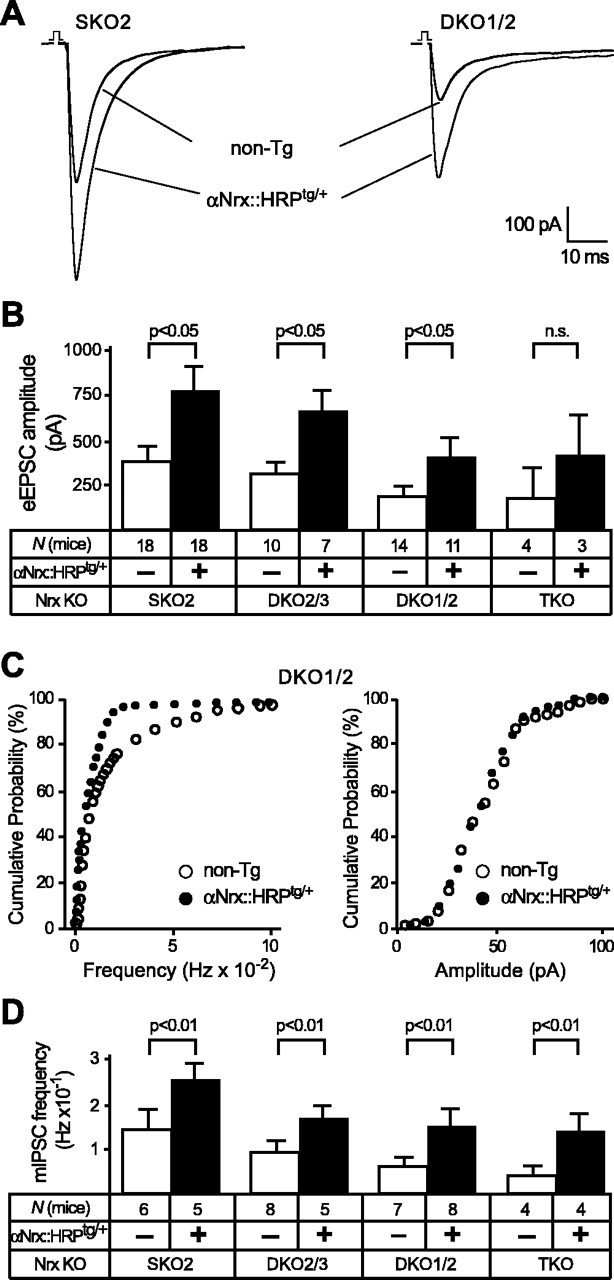
Transgenically expressed αNrx::HRP increases evoked and spontaneous neurotransmitter release in neurons of KO mice. A, Sample traces of eEPSCs, representing an average from 25 consecutive responses to field stimulations recorded under 1 μm bicuculline and 1 μm strychnine. Recordings were obtained in hypoglossal motor neurons from newborn SKO2 and DKO1/2 mice without [nontransgenic (non-Tg)] and with an additional αNrx::HRPtg/+ transgene. B, Corresponding mean EPSC amplitudes of multiple α-neurexin KO littermate mice (SKO2, DKO2/3, DKO1/2, and TKO) with and without the αNrx::HRPtg/+ transgene. All recordings were performed on newborn mice (P1) before genotyping in this and all subsequent experiments. n.s., Not significant. C, Cumulative histogram of miniature frequency (left) and amplitudes (right) of spontaneous inhibitory neurotransmitter release (mIPSCs) in brainstem neurons, recorded from double KO (DKO1/2) neurons with (filled symbols) and without (open symbols) an additional αNrx::HRPtg/+ transgene. D, Corresponding mean frequency of mIPSCs in brainstem neurons recorded in the presence of TTX and CNQX from multiple α-neurexin KO littermate mice (SKO2, DKO2/3, DKO1/2, and TKO) without (open bars) and with (filled bars) the αNrx::HRPtg/+ transgene. In all graphs and panels, statistical significance is indicated above the bars. Data shown are means ± SEM. Error bars represent SEM.
To analyze whether the increase in evoked synaptic responses by the neurexin 1α transgene reflects a general improvement of vesicle release, we next measured spontaneous frequencies and amplitudes of minis in PBC neurons (Fig. 3C,D). Confirming the results from the evoked release experiments, we observed that the αNrx::HRPtg/+ transgene caused a significant enhancement of the spontaneous neurotransmitter release in all α-neurexin KO combinations studied. Mini frequencies at inhibitory synapses increased to ∼160% in SKO2 and αNrx::HRPtg/+ neurons, to ∼174% in DKO2/3 and αNrx::HRPtg/+ neurons, to ∼233% in DKO1/2 and αNrx::HRPtg/+ neurons, and to ∼274% in TKO and αNrx::HRPtg/+ neurons compared with their respective nontransgenic littermate controls (Fig. 3D). Although this proportional increase in mini frequency by the αNrx::HRP transgene appears stronger the more α-neurexin alleles are missing, the lower absolute mIPSC frequencies in rescued DKO and TKO versus SKO2 neurons are consistent with our previous notion that a single transgene is not enough to replace multiple α-neurexins (see above). In contrast to the frequency, no changes in mini amplitudes were observed in rescued neurons (Fig. 3C and data not shown), supporting the idea that neurexins primarily function presynaptically. To test whether the rescue of mini release also extends to excitatory synapses, we measured minis in hypoglossal motor neurons in the presence of 1 μm strychnine and 1 μm bicuculline, again by restricting quantitative analysis to littermate mice of one particular α-neurexin genotype (DKO2/3) with and without αNrx::HRPtg/+ Frequencies of mEPSCs increased from 0.7 ± 0.013 × 10-1 Hz (DKO2/3; n = 4) to 3.9 ± 0.24 × 10-1 Hz (DKO2/3 and αNrx::HRPtg/+; n = 4), confirming that transgenic α-neurexin potently enhances spontaneous and evoked synaptic neurotransmitter release in both excitatory and inhibitory synapses of knock-out mice.
Investigation of α-neurexin KO mice had prompted the hypothesis that the impairment in neurotransmission is essentially a result of an impairment of Ca2+-channel function (Missler et al., 2003). Here, we performed whole-cell recordings of total high voltage-activated Ca2+ currents in KO neurons of the PBC with and without the transgenically expressed αNrx::HRP. Consistent with our previous results, we found that Ca2+ currents were low in α-neurexin KO mice, but transgenic expression of α-neurexin (αNrx::HRPtg/+) robustly increased peak amplitudes (Fig. 4A). To compare the current data with our previous study, we quantified the whole-cell Ca2+ current as current densities (Fig. 4B), which confirmed and extended our previous observation that the αNrx::HRP transgene can increase Ca2+-current densities approximately two-fold in all α-neurexin KO combinations. Although we noted that the increase in whole-cell Ca2+ currents paralleled the increase in the evoked and spontaneous responses (shown above), it remains an open question to what extent these presumably somatodendritic currents reflect the activity of presynaptic Ca2+ channels. Even the best rescue by αNrx::HRP in single and double KO mice (to 17-18 pA/pF) (Fig. 4B), however, did not completely reach the Ca2+-current densities observed in corresponding neurons of genetically matching wild-type mice (∼20 pA/pF) (Missler et al., 2003).
Figure 4.
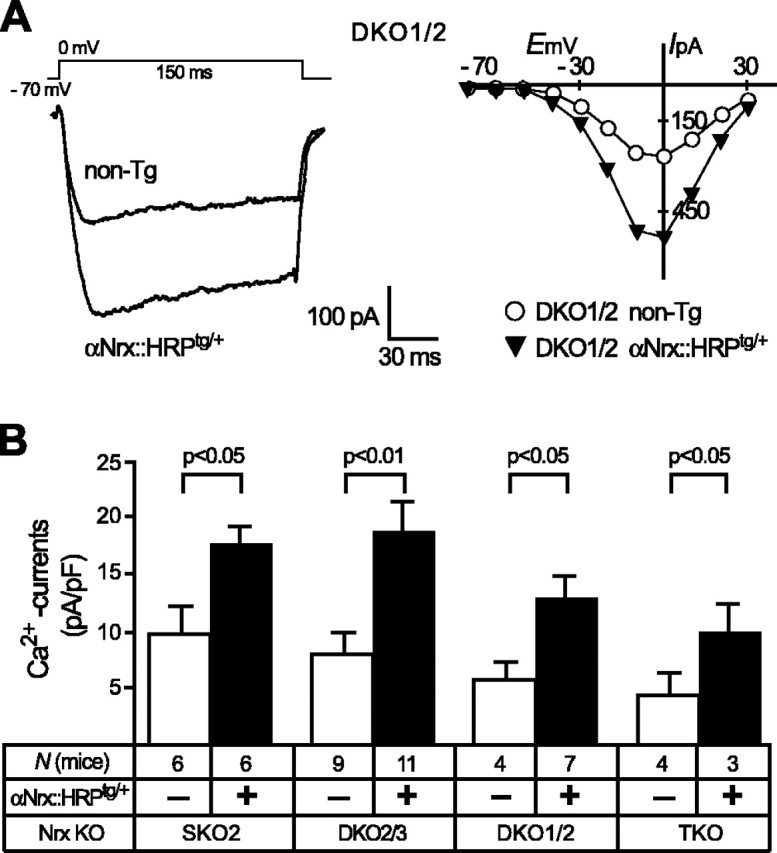
The αNrx::HRPtg/+ transgene improves Ca2+ currents in all combinations of α-neurexin KO mice. A, Individual sample traces (left) and current-voltage relationships (right) of whole-cell Ca2+-current recordings from PBC neurons in DKO1/2 mice without [nontransgenic (non-Tg)] and with additional αNrx::HRPtg/+ transgene. High voltage-activated Ca2+ currents were evoked by 150-ms-long voltage steps (10 mV each) from a holding potential of -70 mV to various test potentials ranging from -70 to +30 mV. B, The mean current densities show quantification of maximal depolarization steps to 0 mV (as shown in A, left). Whole-cell Ca2+ current increases significantly in neurons of all combinations of α-neurexin KO mice when an αNrx::HRPtg/+ transgene is present (filled bars). Data shown are means ± SEM. Statistical significance is indicated above the bars. Error bars represent SEM.
Transgenic neurexin 1β has no effect on the α-neurexin KO phenotype
We next examined whether the neurexin 1β transgene (βNrx::HRPtg/+) caused rescue activity similar to that seen for the neurexin 1α transgene. A successful rescue effect with βNrx::HRPtg/+ would suggest that the difference in extracellular sequences between neurexin 1α and 1β is unimportant with respect to synaptic transmission and that the strong phenotype in α-neurexin KO mice in the continued presence of β-neurexins is attributable to the differential expression or localization of α- and β-neurexins. Conversely, if the neurexin 1β transgene is unable to rescue the phenotype despite similar expression levels and patterns, an essential function of the α-neurexin-specific extracellular domains in affecting presynaptic neurotransmitter release seems likely.
We found that the βNrx::HRPtg/+ transgene had no effect on the size of evoked synaptic responses or on the failure rate in any of the α-neurexin KO mice tested (Fig. 5A,B and data not shown). In these experiments, we compared littermates that carry the same KO combinations with or without an additional βNrx::HRPtg/+ transgene and measured the amplitudes in hypoglossal motor neurons after field stimulation of their inputs from the PBC. However, no significant differences in mean currents were observed (Fig. 5B). To test whether βNrx::HRP had a detectable influence on spontaneous release at inhibitory synapses, we measured inhibitory mini events in PBC neurons with and without a βNrx::HRPtg/+ transgene. However, mini frequencies were unchanged between all KO mice with or without the βNrx::HRPtg/+ transgene present (Fig. 5C). In addition, transgenically expressed β-neurexin caused no alterations of postsynaptic receptor function, as evidenced by unchanged mini amplitudes in all KO combinations tested (SKO, 57.3 ± 5.98 pA, n = 7; SKO and βNrx::HRPtg/+, 55.6 ± 11.1 pA, n = 6; DKO2/3, 52.1 ± 10.9 pA, n = 4; DKO2/3 and βNrx::HRPtg/+, 49.7 ± 8.8 pA, n = 4; DKO1/2, 48.1 ± 6.9, n = 7; DKO1/2 and βNrx::HRPtg/+, 45.9 ± 4.2 pA, n = 6; TKO, 47.2 ± 12.9 pA, n = 3; TKO and βNrx::HRPtg/+, 48.3 ± 11.2 pA, n = 4). Finally, to exclude the possibility that a potential rescue effect by the extracellularly short β-neurexin was restricted to increasing the VDCC activity without significantly improving presynaptic function, we also measured whole-cell Ca2+ currents (Fig. 5D,E). No difference in traces or current-voltage curves, however, was apparent from littermate mice with or without the βNrx::HRPtg/+ transgene (Fig. 5D), and the respective current densities were unchanged (Fig. 5E). The experiments with βNrx::HRP established that extracellular sequences of α-neurexins are required for their role in neurotransmission. It should be noted, however, that the failure of βNrx::HRP to improve any of the KO parameters does not exclude an important function of the C-terminal domains of neurexins but demonstrates that they are not sufficient to regulate Ca2+ channels.
Figure 5.
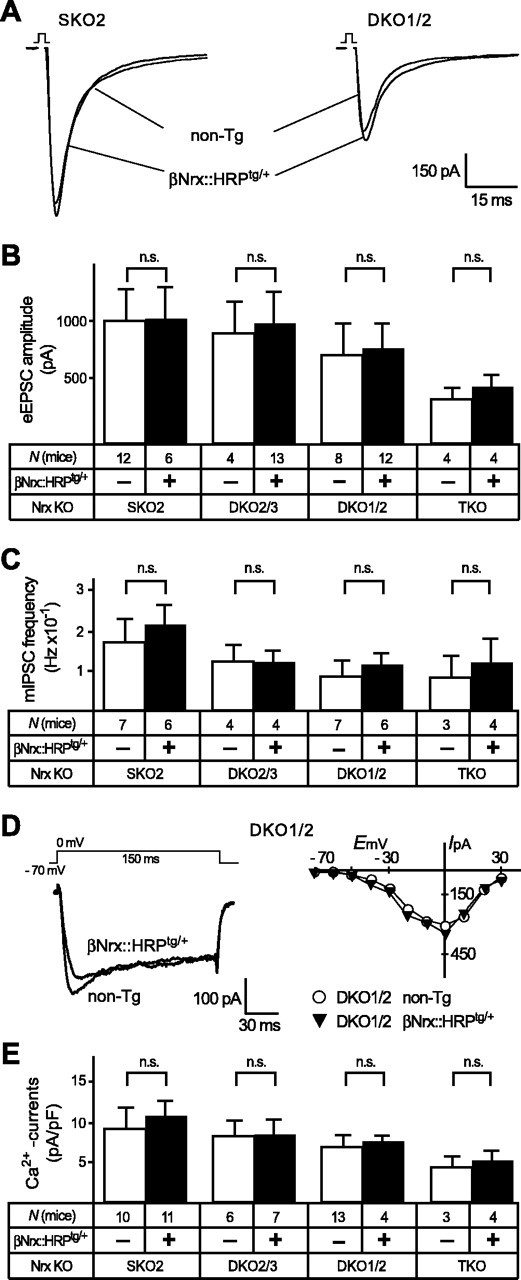
Transgenically expressed βNrx::HRP is not sufficient to improve synaptic function and Ca2+ currents. Experiments similar to those described in Figures 3 and 4, with the exception that a different neurexin variant (β-neurexin) with distinct extracellular but identical C-terminal sequences was transgenically expressed and tested for its rescue potential (βNrx::HRP; see Fig. 1A). A-E, Evoked EPSC amplitudes (A, B), spontaneous IPSC frequencies (C), and high voltage-activated Ca2+ currents (D, E) are unchanged in KO mice (SKO2, DKO2/3, DKO1/2, and TKO) without [nontransgenic (non-Tg); open bars] and with (filled bars) the βNrx::HRPtg/+ transgene present. In all graphs and panels, statistical significance is indicated above the bars. Data shown are means ± SEM. Error bars represent SEM.
The robust rescue mediated by transgenic αNrx::HRP raises the question of whether the αNrx::HRP acts as a general enhancer of synaptic transmission that would also increase neurotransmitter release within a wild-type genetic background. We therefore investigated whether transgenic neurexins potentiate evoked postsynaptic responses or Ca2+-channel function even in wild-type neurons that express endogenous α-neurexins normally. For this purpose, we measured eEPSCs in neurons from wild-type littermate mice that carried or lacked a transgenic αNrx::HRP but found no differences in release (Fig. 6A,B). We also tested whether the normal Ca2+-channel activity could be facilitated (αNrx::HRP) or inhibited (βNrx::HRP) by recording high voltage-activated Ca2+ currents in neurons from wild-type littermate mice that carried or lacked an αNrx::HRPtg/+ or βNrx::HRPtg/+ transgene (Fig. 6C,D). However, current traces and quantitations revealed no changes (Fig. 6C,D), consistent with observations that expression levels of VDCC pore-forming subunits were neither reduced in α-neurexin KO mice (Missler et al., 2003) nor increased in the transgenic mice shown here (Fig. 6E). Thus, expression of additional transgenic neurexins by itself does not produce a gain-of-function or a dominant-negative phenotype.
Figure 6.
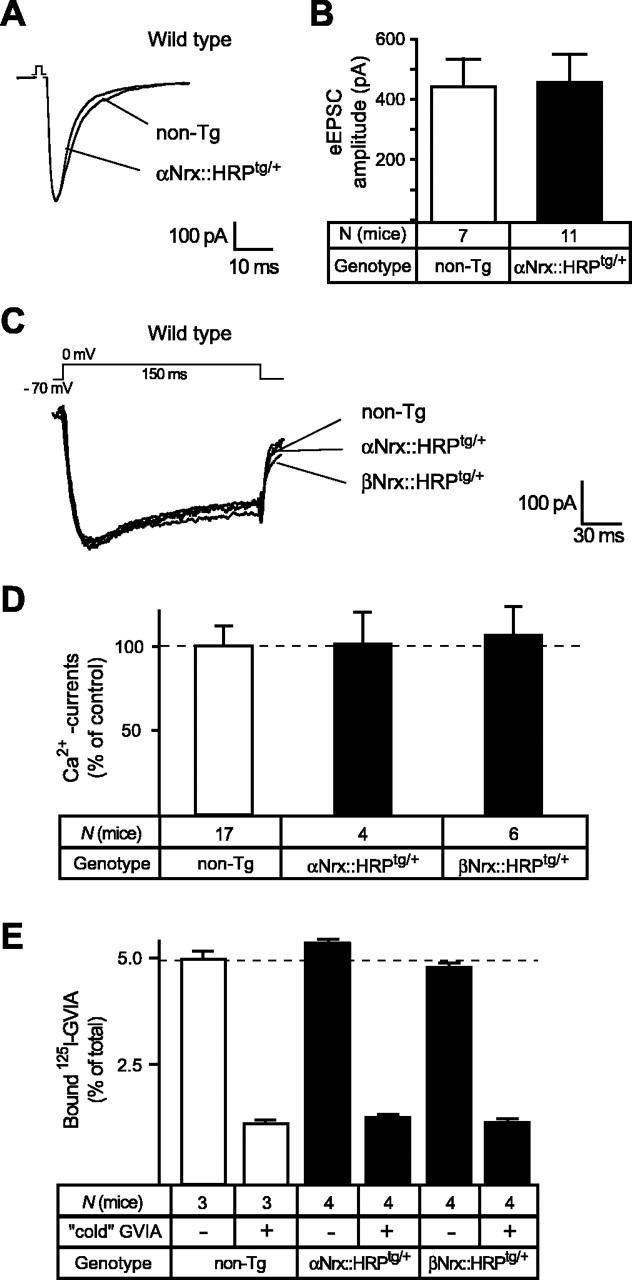
Transgenic α- and β-neurexins do not induce a general gain-of-function in wild-type neurons. A, B, Sample traces (A) and averaged evoked excitatory postsynaptic responses (B) from wild-type neurons (carrying no neurexin deletions) without [nontransgenic (non-Tg); open bar] and with (filled bar) an additional αNrx::HRPtg/+ transgene, exhibiting no effect of transgenically expressed α-neurexin on wild-type levels of synaptic transmission. C, Representative traces of high voltage-activated Ca2+ currents from wild-type neurons without (non-Tg) and with an additional αNrx::HRPtg/+ or βNrx::HRPtg/+ transgene. The depolarization protocol was identical to the one used in Figures 4A and 5D. D, Quantitative comparison of current densities in littermate wild-type mice without (non-Tg, 100%) and with an additional αNrx::HRPtg/+ or βNrx::HRPtg/+ transgene, indicating that the expression of neurexin transgenes did not increase Ca2+ currents beyond wild-type levels. Data shown are means ± SEM. E, Binding of 125I-labeled ω-conotoxin GVIA at 30 pm to brain membranes from wild-type control (non-Tg) and transgenic mice expressing αNrx::HRPtg/+ or βNrx::HRPtg/+. All experiments were done with and without a 100-fold excess of unlabeled (“cold”) toxin to account for unspecific binding. Data shown are means ± SEM. Error bars represent SEM.
α-Neurexins specifically affect non-L-type Ca2+ channels
Although transgenic neurexin 1α increased synaptic transmission in all KO combinations, evoked postsynaptic responses are not completely abolished in KO neurons, even in neurons lacking all three α-neurexins (Figs. 3B, 5B). The residual synaptic transmission that is independent of α-neurexins could be explained by at least two hypotheses: that α-neurexins only partially modulate the activity of all VDCCs or that α-neurexins modulate only specific subtypes of VDCCs. To test which Ca2+ channels are affected by α-neurexins, we first analyzed the contribution of different VDCC subtypes to evoked neurotransmitter release in hypoglossal motor neurons from wild-type mice by sequentially blocking specific channels pharmacologically (Fig. 7A,B). Addition of nifedipine, ω-conotoxin, and agatoxin (blockers of L-, N-, and P/Q-type Ca2+ channels, respectively) reduced the EPSC to almost zero (Fig. 5A). The small residual EPSC after these additions was blocked by Cd2+, suggesting that it most likely was mediated by R-type Ca2+ channels (Fig. 5A). Therefore, we focused our quantitative analysis on L-, N-, and P/Q-type VDCCs that appear to be dominant at this synapse.
Figure 7.
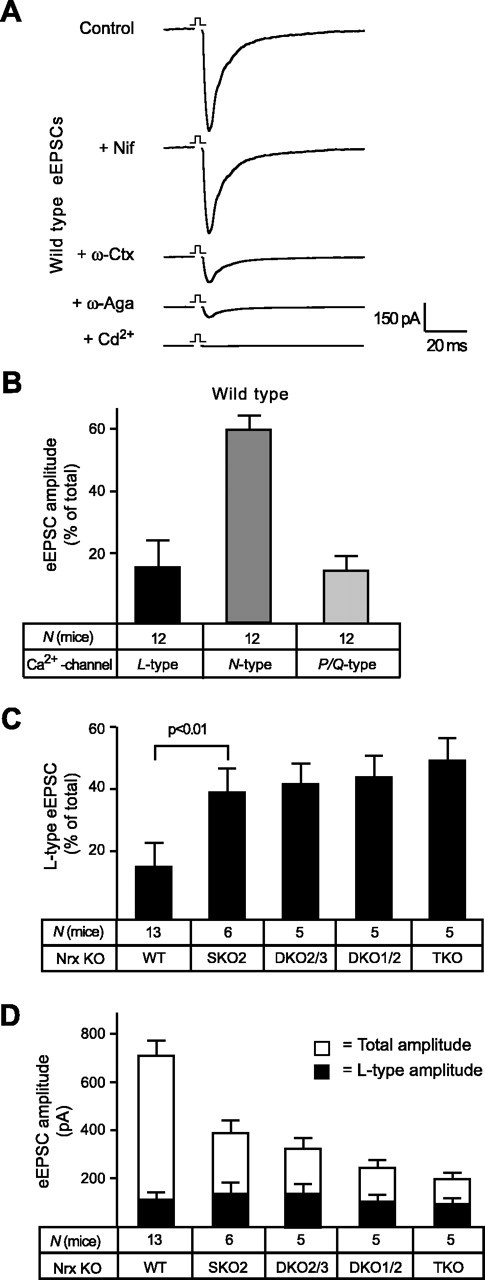
L-type Ca2+-channel-mediated EPSC components are unaffected by α-neurexins. A, Averaged traces of glutamatergic EPSCs monitored in wild-type hypoglossal motor neurons before (control) and after sequential application of 10 μm nifedipine (Nif) for L-type channel contributions, 0.5 μm ω-conotoxin MVIIA (ω-Ctx) for N-type channel contributions, 0.2 μm agatoxin IVA (ω-Aga) for P/Q-type channel contributions, and 100 μm Cd2+ to probe for residual Ca2+-channel activities (presumably mediated by R-type). B, Relative contributions of different VDCC subtypes to total EPSC amplitudes calculated from blocking experiments, as shown in A. The evoked synaptic response in the neonatal brainstem preparation depends mostly on N-type Ca2+ channels but also has significant contributions from L- and P/Q-type VDCCs. C, Comparison of L-type-mediated EPSC components in wild-type (WT) and multiple α-neurexin KO mice, uncovering a significantly increased proportion of the L-type VDCC component in mutant neurons, which is already evident in single KO (SKO2) mutant mice. D, Absolute currents of evoked postsynaptic responses (EPSCs; in picoamperes) during field stimulations of hypoglossal neurons. Total (open bars) and L-type-mediated (filled bars) amplitudes were analyzed in wild-type controls and in multiple α-neurexin KO mice. Statistical significance is indicated above the bars. Data shown are means ± SEM. Error bars represent SEM.
The amplitude of EPSCs in wild-type neurons depended >60% on an N-type Ca2+ channel-dependent component, supplemented by smaller but approximately equal contributions from L-type and P/Q-type Ca2+ channels (Fig. 7B). The contributions of these three VDCC subtypes to neurotransmission make the neonatal brainstem particularly suitable to study regulatory preferences. Control experiments in wild-type neurons using different mixtures of blockers instead of adding them sequentially resulted in a similar contribution of Ca2+-channel subtypes (data not shown). Because the participation of L-type channels (>15% of the total amplitude) (Fig. 7B) reported here is larger than the L-type-dependent component observed in other central synapses (Wu et al., 1999), we tested how this L-type dependence was affected in α-neurexin KO neurons. Analyzing the relative proportion of L-type-mediated EPSCs to the total postsynaptic response revealed that the L-type component increases from ∼15% in wild-type neurons to >50% in TKO neurons (Fig. 7C). In contrast to most other parameters studied thus far in α-neurexin KO mice (survival rates, ventilation activity, evoked and spontaneous release, and Ca2+ currents) (Missler et al., 2003), the relative L-type contribution increases dramatically in SKO2 mice (Fig. 7C).
The augmented L-type dependence of EPSCs in KO neurons could reflect a true compensation caused by upregulated activity of L-type Ca2+ channels or merely constitute a relative increase caused by the dramatically reduced total EPSC. Evaluation of absolute amplitudes demonstrated that the L-type-mediated EPSC amplitudes did not differ significantly between α-neurexin genotypes (Fig. 7D, filled bars). This result suggests that the activity of L-type VDCCs is not dependent on α-neurexins, indicating that the impairment of N- and P/Q-type Ca2+ channels is primarily responsible for the phenotype.
To obtain direct evidence for a positive regulation of non-L-type Ca2+ channels by α-neurexins, we applied the sequential blocking protocol described above to two different α-neurexin KO combinations (SKO2 and DKO1/2) with and without the αNrx::HRPtg/+ transgene (Fig. 8A,B). The presence of αNrx::HRPtg/+ increased the contributions of N- and P/Q-type VDCCs to the total EPSC twofold to threefold (Fig. 8C,D). Consistent with the unchanged L-type-mediated EPSC in KO neurons (Fig. 7D), expression of an additional αNrx::HRPtg/+ transgene did not change the size of L-type-dependent amplitudes in rescued neurons (Fig. 8C,D).
Figure 8.
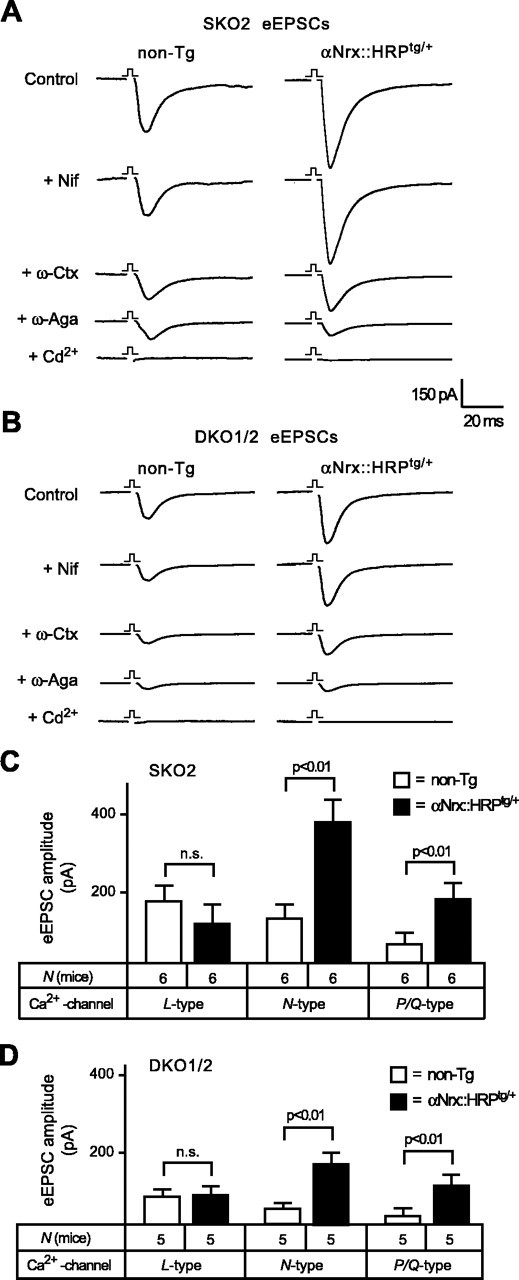
Transgenic α-neurexin increases evoked responses by selectively activating N- and P/Q-type Ca2+ channels. A, B, Averaged traces representing glutamatergic EPSCs from SKO2 and DKO1/2 mice without [nontransgenic (non-Tg)] and with additional αNrx::HRPtg/+ transgene. Evoked responses were recorded before (control) and after sequential application of specific VDCC blockers (same protocol as in Fig. 7A). Nif, Nifedipine; ω-Ctx, ω-conotoxin; ω-Aga, agatoxin. C, D, Quantitative comparison of the contributions from different VDCC subtypes to EPSCs in single KO and double KO mice without (open bars) and with (filled bars) additional αNrx::HRPtg/+ transgene shows an increased amplitude mediated by N- and P/Q-type calcium channels when a transgenic α-neurexin is present. The proportion of L-type-dependent current components is unchanged in rescued neurons. n.s., Not significant. In all graphs and panels, statistical significance is indicated above the bars. Data shown are means ± SEM. Error bars represent SEM.
Our finding of a specific regulation of N- and P/Q-type VDCC activity by α-neurexins documents the molecular specificity of the neurexin-Ca2+-channel link. To further validate this result, we performed experiments that tested the effect of Ca2+-channel inhibitors on the frequency of mIPSCs. Recording mini frequencies in PBC neurons to study Ca2+-channel activity is meaningful, because in these neurons approximately one-half of the mini events depend on VDCCs and can be blocked by Cd2+ (45-55%) (Missler et al., 2003) (data not shown). We first recorded minis mediated by different VDCC subtypes in wild-type neurons (Fig. 9A) after a sequential blocking protocol described above (Figs. 7, 8). Consistent with the data on evoked responses, we found that N-type Ca2+ channels mediated most of the Ca2+ channel-dependent minis (>60%). However, the P/Q-type-dependent component of minis was slightly higher (26%) than the L-type-dependent component (6%) (Fig. 9B). Despite the small contribution of L-type VDCCs to wild-type minis, their relative proportion increased greatly in the different α-neurexin KO mice, reaching proportions similar to the EPSC data shown above (Fig. 9C). Corresponding to the EPSC analysis, however, this apparent compensation was not attributable to an actual increase in L-type-dependent minis, because the overall mini frequency is reduced in KO neurons (Missler et al., 2003) (Figs. 3D, 5C), supporting the notion that the function of L-type VDCCs is mostly unaffected by the absence of α-neurexins. We then determined how the VDCC subtype contributions to mini frequencies changed when an αNrx::HRPtg/+ transgene was crossed into the same α-neurexin KO combinations used above (SKO2 and DKO1/2) (Fig. 10A,B). Consistent with the results from evoked release, the presence of the αNrx::HRPtg/+ transgene significantly increased the N- and P/Q-type-mediated mini frequencies, whereas L-type-dependent mIPSC events were unchanged (Fig. 10C,D), indicating that L-type channels are also excluded from α-neurexin regulation at inhibitory synapses.
Figure 9.
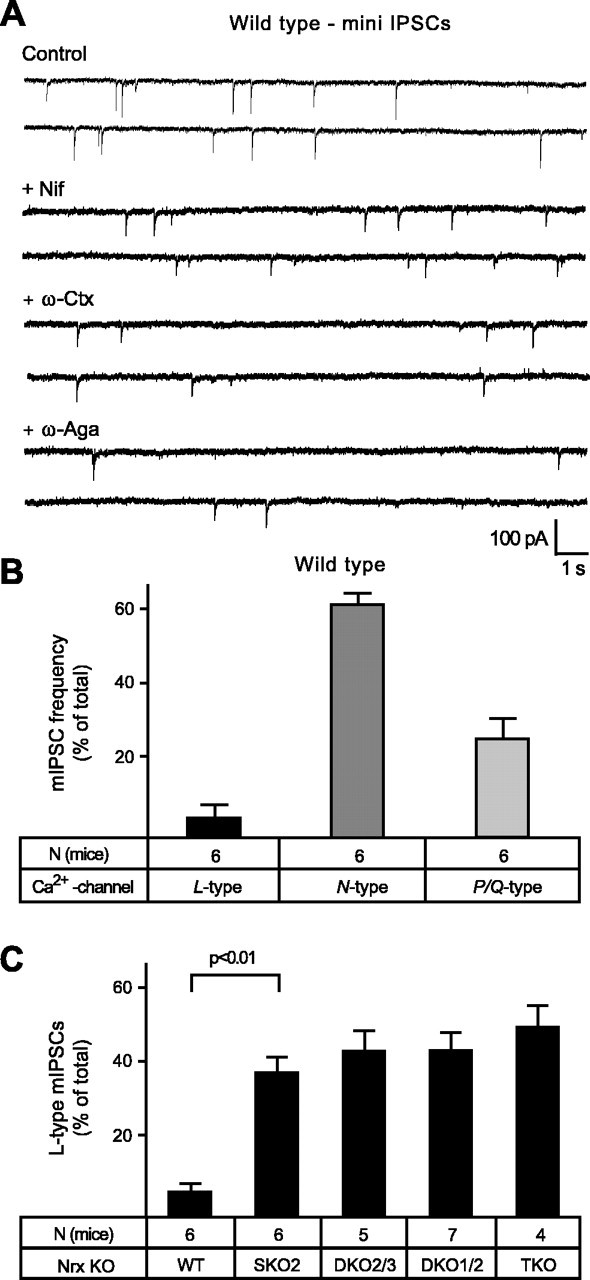
Ca2+-dependent minis at inhibitory synapses of wild-type and α-neurexin KO mice. A, Representative traces of spontaneous release events (mIPSCs) recorded under TTX and CNQX from neonatal PBC neurons of wild-type mice before (control) and after the sequential blocking of Ca2+ channels, as described in Figures 7 and 8 for evoked release. Nif, Nifedipine; ω-Ctx, ω-conotoxin; ω-Aga, agatoxin. B, Dependence of wild-type mIPSCs on different VDCC subtypes is expressed as a percentage of the respective Ca2+-dependent mini frequency, quantitated from blocking experiments, as shown in A. In wild-type neurons of this brainstem area, approximately one-half of all minis depend on Ca2+ influx (data not shown). C, In knock-out mice lacking one, two, or all three α-neurexins (SKO2, DKO2/3, DKO1/2, and TKO), the proportion of L-type-mediated minis appears significantly larger because of the strongly reduced overall mIPSC frequencies (compare with Figs. 3D and 5C). WT, Wild type. Statistical significance is indicated above the bars. Data shown are means ± SEM. Error bars represent SEM.
Figure 10.
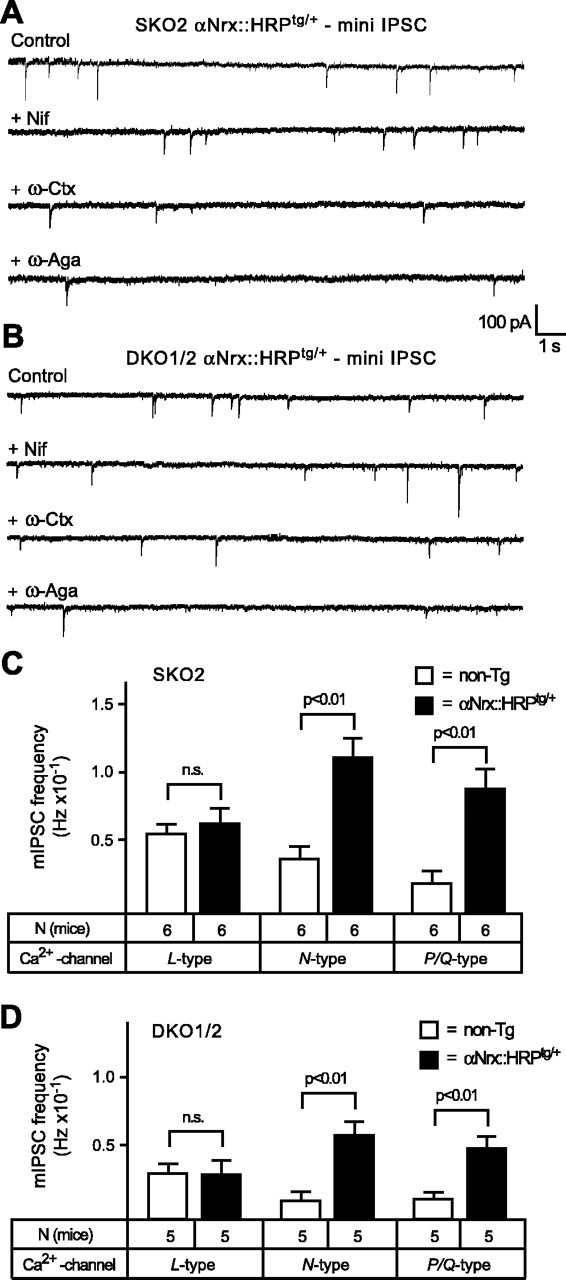
Transgenically expressed α-neurexin specifically increases N- and P/Q-type-mediated mini frequencies. A, B, Sample traces of mini events (mIPSCs) recorded under TTX and CNQX from SKO2 and DKO1/2 mice, both with an additional αNrx::HRPtg/+ transgene present. Spontaneous postsynaptic responses were recorded before (control) and after sequential application of Ca2+-channel inhibitors (same as in Fig. 9A). To show representative changes in mini events after drug treatment, recordings with above-average frequencies were chosen for all conditions (compare with absolute frequencies in C, D). Nif, Nifedipine; ω-Ctx, ω-conotoxin; ω-Aga, agatoxin; n.s., not significant. C, D, Because transgenic α-neurexin rescued mini frequencies in all α-neurexin KO combinations studied (Fig. 3D), the contributions of different VDCC subtypes were compared in single KO (C) and double KO (D) mice without [nontransgenic (non-Tg); open bars] and with (filled bars) additional αNrx::HRPtg/+ transgene. Increased mini frequencies reveal an augmented activity of N- and P/Q-type VDCCs but not of L-type channels. In all graphs and panels, statistical significance is indicated above the bars. Data shown are means ± SEM. Error bars represent SEM.
Discussion
Neurexins are among several neuronal cell-adhesion molecules that were proposed to contribute to synaptic neurotransmitter release and/or synapse formation (for review, see Sudhof, 2001; Missler, 2003; Scheiffele, 2003). For many of these candidate cell-adhesion molecules, studies in knock-out mice have produced illuminative phenotypes, although their suggested functions are often only indirectly related to synapses (Tomasiewicz et al., 1993; Cremer et al., 1994; Evers et al., 2002; Togashi et al., 2002; Wang et al., 2002). The α-neurexin KO phenotype, in contrast, strongly points toward a specifically synaptic function, because their deletion severely impairs both excitatory and inhibitory neurotransmitter release, presumably as a result of impaired Ca2+-channel function, but does not affect brain architecture and synaptic ultrastructure (Missler et al., 2003). Despite the present inavailability of β-neurexin knock-out mice, recent in vitro work on the β-neurexin/neuroligin complex has suggested that it may have a role in promoting synapse formation and differentiation (Scheiffele et al., 2000; Dean et al., 2003; Graf et al., 2004).
We have now directly compared the putative roles of α- and β-neurexins and investigated the molecular specificity of the neurexin-Ca2+-channel regulation using a comparative rescue strategy in transgenic mice. Such a strategy is rarely used because of the amount of work involved, although a similar approach has been followed, for example, to analyze the functional redundancy of the closely related growth cone proteins GAP43 and cytoskeleton-associated protein 23 (Frey et al., 2000). For questions relating to the normal regulation of Ca2+ channels by a cell-adhesion molecule in an intact brain, a transgenic strategy may be advantageous, because cell-adhesion events are best studied in acute slices of an intact neuronal network. The brainstem system was chosen because multiple α-neurexin KO mutants die early postnatally as a result of respiratory problems arising from brainstem defects, and data from the rescue analysis could be tested for consistency compared with the previous KO study. Furthermore, the background-free detection of transgenic neurexin::HRP proteins allows an unbiased estimation of expression levels and distribution patterns. In addition, choosing transgenic lines that express neurexin::HRP fusion proteins within the range of endogenous neurexins reduces the problems of dominant-negative effects and high variability, frequently associated, for example, with overexpression approaches in vitro. Our current study extends the previous analysis of neurexins by reporting three novel findings.
First, transgenic neurexin 1α, but not transgenic neurexin 1β, partly rescued the well characterized properties of the α-neurexin KO phenotype. Because transgenic neurexin 1α and 1β differ solely in their extracellular domains, but contain identical transmembrane regions and cytoplasmic sequences, we conclude that α- and β-neurexins have nonredundant functions that involve their distinct N-terminal domains. However, if the extracellular sequences of α-neurexin define their role as organizer molecules of presynaptic release, what then is the respective role of β-neurexins? The work by the Scheiffele and Craig laboratories suggests, as originally proposed when neurexins were first identified (Ushkaryov et al., 1992), that neurexins can promote the formation of de novo synapses and differentiation of postsynaptic receptors (Scheiffele et al., 2000; Dean et al., 2003; Graf et al., 2004). Because these reports were confined to the in vitro activities of β-neurexins, whereas we study the effects of transgenes in vivo, it is at present difficult to compare their results with ours. However, we did not detect an effect of transgenic expression of neurexin 1β on mini amplitudes or evoked responses (as would be expected if postsynaptic receptors were recruited or activated) and did not find that transgenic neurexin 1β augmented mini frequencies (as would be expected from a synaptogenetic activity). A possible interpretation of this discrepancy could be that the synapse formation and differentiation observed by overexpression in cultures uncovers an additional (rather than essential) functional activity of neurexins that is redundant with other molecules and thus is not present in intact tissue. Alternatively, it is possible that a synaptogenetic function for neurexins is developmentally compensated, although this raises the question of why the essential function for α-neurexins in executing neurotransmitter release, as documented here and previously (Missler et al., 2003), is not compensated.
Second, the neurexin 1α transgene ameliorated all facets of the α-neurexin KO phenotype, independent of which neurexin genes were mutated, demonstrating that the various α-neurexins do not exhibit fundamental functional differences. The fact that a single transgene with only one set of splice variants was active in reversing the various phenotypes suggests that α-neurexins mediate a single integrated activity. The increase in neurotransmitter release and the regulation of VDCCs mediated by α-neurexin were not simple gain-of-function activities, because the transgenic proteins did not further facilitate their activity in wild-type mice. This result suggests that neurexins may regulate VDCC activity only within their normal range but not beyond.
Third, our data now show that deletion of α-neurexins selectively impairs neurotransmitter release that is dependent on non-L-type Ca2+ channels. Other classes of extracellular molecules such as the matrix component tenascin-C may modulate L-type-dependent synaptic plasticity (Evers et al., 2002), but L-type-mediated EPSC amplitudes and mini frequencies in brainstem synapses were unaffected by α-neurexins, as shown here. The fact that a preference of α-neurexins for N- and P/Q-type Ca2+ channels exists is interesting, because the VDCCs involved in presynaptic Ca2+ entry are usually of the N- and P/Q-type (Dunlap et al., 1995; Reid et al., 1998; Wu et al., 1999), but R- and L-type channels may be involved at particular synapses as well (Wu et al., 1998; Brandt et al., 2003). Although all presynaptic terminals contain VDCCs, the release probability and Ca2+ dynamics of presynaptic terminals vary significantly (Hessler et al., 1993; Rosenmund et al., 1993; Koester and Sakmann, 2000), even at synaptic terminals of the same neuron (Maccaferri et al., 1998; Markram et al., 1998; Reyes et al., 1998; Scanziani et al., 1998). Because of the high Ca2+ cooperativity and Ca2+ sensitivity of neurotransmitter release (Bollmann et al., 2000; Schneggenburger and Neher, 2000), regulating VDCCs efficiently controls synaptic strength, and small changes in Ca2+ influx lead to magnified effects on synaptic release (Felmy et al., 2003). It comes as no surprise, therefore, that VDCCs are extensively regulated. In addition to the intracellular free calcium concentration itself, several helix-loop-helix Ca2+-binding motifs containing proteins called EF-hand proteins (e.g., calmodulin, normal calf serum, calcium-binding protein) (for review, see Weiss and Burgoyne, 2002), G-protein-coupled receptor pathways (such as GABAB or 5-HT) (for review, see Catterall, 2000; Zamponi, 2001), components of the exocytotic machinery (e.g., syntaxin 1 or synaptosomal-associated protein of 25 kDa) (for review, see Catterall, 1999; Atlas, 2001), or even signaling events induced by lipid bilayer components (Wu et al., 2002) facilitate or inhibit VDCCs.
Our findings on α-neurexins now put the first synaptic cell-adhesion molecule on the list of important VDCC regulators. These results, however, raise additional questions. Why are there multiple α-neurexin genes with hundreds of splice variants if a single isoform, expressed in a single splice variant, is at least partly able to rescue different phenotypic effects produced by the deletions of multiple α-neurexin genes? What are the extracellular interactions that mediate the essential functions of α-neurexins, and how are these extracellular interactions coupled to intracellular effector pathways? What is the actual role of β-neurexins, including their splice variants that do not bind to neuroligin in a trans-synaptic complex? Addressing these questions will significantly promote our understanding of the organization of presynaptic terminals but will require new experimental approaches.
Footnotes
This work was supported by the Deutsche Forschungsgemeinschaft (Sonderforschungsbereich 406 and Center for Molecular Physiology of the Brain grants to M.M. and W.Z.) and by the National Institutes of Health (T.C.S.). We thank R. W. Tsien and R. Schneggenburger for discussion, M. Goedert (Medical Research Council, Cambridge, UK) and H. Kramer (University of Texas Southwestern Medical Center) for plasmids, and S. Gerke for excellent technical assistance.
Correspondence should be addressed to Markus Missler, Center for Physiology and Pathophysiology, Georg-August University, Humboldtallee 23, D-37073 Göttingen, Germany. E-mail: mmissle1@gwdg.de.
Copyright © 2005 Society for Neuroscience 0270-6474/05/254330-13$15.00/0
W.Z. and A.R. contributed equally to this work.
References
- Andra K, Abramowski D, Duke M, Probst A, Wiederhold KH, Burki K, Goedert M, Sommer B, Staufenbiel M (1996) Expression of APP in transgenic mice: a comparison of neuron-specific promoters. Neurobiol Aging 17: 183-190. [DOI] [PubMed] [Google Scholar]
- Atlas D (2001) Functional and physical coupling of voltage-sensitive calcium channels with exocytotic proteins: ramifications for the secretion mechanism. J Neurochem 77: 972-985. [DOI] [PubMed] [Google Scholar]
- Biederer T, Sudhof TC (2000) Mints as adaptors. Direct binding to neurexins and recruitment of munc18. J Biol Chem 275: 39803-39806. [DOI] [PubMed] [Google Scholar]
- Bollmann JH, Sakmann B, Borst JG (2000) Calcium sensitivity of glutamate release in a calyx-type terminal. Science 289: 953-957. [DOI] [PubMed] [Google Scholar]
- Brandt A, Striessnig J, Moser T (2003) CaV1.3 channels are essential for development and presynaptic activity of cochlear inner hair cells. J Neurosci 23: 10832-10840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butz S, Okamoto M, Sudhof TC (1998) A tripartite protein complex with the potential to couple synaptic vesicle exocytosis to cell adhesion in brain. Cell 94: 773-782. [DOI] [PubMed] [Google Scholar]
- Catterall WA (1999) Interactions of presynaptic Ca2+ channels and snare proteins in neurotransmitter release. Ann NY Acad Sci 868: 144-159. [DOI] [PubMed] [Google Scholar]
- Catterall WA (2000) Structure and regulation of voltage-gated Ca2+ channels. Annu Rev Cell Dev Biol 16: 521-555. [DOI] [PubMed] [Google Scholar]
- Cremer H, Lange R, Christoph A, Plomann M, Vopper G, Roes J, Brown R, Baldwin S, Kraemer P, Scheff S (1994) Inactivation of the N-CAM gene in mice results in size reduction of the olfactory bulb and deficits in spatial learning. Nature 367: 455-459. [DOI] [PubMed] [Google Scholar]
- Dean C, Scholl FG, Choih J, DeMaria S, Berger J, Isacoff E, Scheiffele P (2003) Neurexin mediates the assembly of presynaptic terminals. Nat Neurosci 6: 708-716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois L, Lecourtois M, Alexandre C, Hirst E, Vincent JP (2001) Regulated endocytic routing modulates wingless signaling in Drosophila embryos. Cell 105: 613-624. [DOI] [PubMed] [Google Scholar]
- Dunlap K, Luebke JI, Turner TJ (1995) Exocytotic Ca2+ channels in mammalian central neurons. Trends Neurosci 18: 89-98. [PubMed] [Google Scholar]
- Evers MR, Salmen B, Bukalo O, Rollenhagen A, Bosl MR, Morellini F, Bartsch U, Dityatev A, Schachner M (2002) Impairment of L-type Ca2+ channel-dependent forms of hippocampal synaptic plasticity in mice deficient in the extracellular matrix glycoprotein tenascin-C. J Neurosci 22: 7177-7194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felmy F, Neher E, Schneggenburger R (2003) Probing the intracellular calcium sensitivity of transmitter release during synaptic facilitation. Neuron 37: 801-811. [DOI] [PubMed] [Google Scholar]
- Frey D, Laux T, Xu L, Schneider C, Caroni P (2000) Shared and unique roles of CAP23 and GAP43 in actin regulation, neurite outgrowth, and anatomical plasticity. J Cell Biol 149: 1443-1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geppert M, Khvotchev M, Krasnoperov V, Goda Y, Missler M, Hammer RE, Ichtchenko K, Petrenko AG, Sudhof TC (1998) Neurexin I alpha is a major alpha-latrotoxin receptor that cooperates in alpha-latrotoxin action. J Biol Chem 273: 1705-1710. [DOI] [PubMed] [Google Scholar]
- Graf ER, Zhang X, Jin SX, Linhoff MW, Craig AM (2004) Neurexins induce differentiation of GABA and glutamate postsynaptic specializations via neuroligins. Cell 119: 1013-1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hata Y, Butz S, Sudhof TC (1996) CASK: a novel dlg/PSD95 homolog with an N-terminal calmodulin-dependent protein kinase domain identified by interaction with neurexins. J Neurosci 16: 2488-2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hessler NA, Shirke AM, Malinow R (1993) The probability of transmitter release at a mammalian central synapse. Nature 366: 569-572. [DOI] [PubMed] [Google Scholar]
- Ichtchenko K, Hata Y, Nguyen T, Ullrich B, Missler M, Moomaw C, Sudhof TC (1995) Neuroligin 1: a splice site-specific ligand for beta-neurexins. Cell 81: 435-443. [DOI] [PubMed] [Google Scholar]
- Koester HJ, Sakmann B (2000) Calcium dynamics associated with action potentials in single nerve terminals of pyramidal cells in layer 2/3 of the young rat neocortex. J Physiol (Lond) 529: 625-646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maccaferri G, Toth K, McBain CJ (1998) Target-specific expression of presynaptic mossy fiber plasticity. Science 279: 1368-1370. [DOI] [PubMed] [Google Scholar]
- Markram H, Wang Y, Tsodyks M (1998) Differential signaling via the same axon of neocortical pyramidal neurons. Proc Natl Acad Sci USA 95: 5323-5328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Missler M (2003) Synaptic cell adhesion goes functional. Trends Neurosci 26: 176-178. [DOI] [PubMed] [Google Scholar]
- Missler M, Sudhof TC (1998) Neurexins: three genes and 1001 products. Trends Genet 14: 20-26. [DOI] [PubMed] [Google Scholar]
- Missler M, Hammer RE, Sudhof TC (1998) Neurexophilin binding to alpha-neurexins. A single lns domain functions as an independently folding ligand-binding unit. J Biol Chem 273: 34716-34723. [DOI] [PubMed] [Google Scholar]
- Missler M, Zhang W, Rohlmann A, Kattenstroth G, Hammer RE, Gottmann K, Sudhof TC (2003) Alpha-neurexins couple Ca2+ channels to synaptic vesicle exocytosis. Nature 424: 939-948. [DOI] [PubMed] [Google Scholar]
- Olmsted JB (1981) Affinity purification of antibodies from diazotized paper blots of heterogeneous protein samples. J Biol Chem 256: 11955-11957. [PubMed] [Google Scholar]
- Reid CA, Bekkers JM, Clements JD (1998) N- and P/Q-type Ca2+ channels mediate transmitter release with a similar cooperativity at rat hippocampal autapses. J Neurosci 18: 2849-2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes A, Lujan R, Rozov A, Burnashev N, Somogyi P, Sakmann B (1998) Target-cell-specific facilitation and depression in neocortical circuits. Nat Neurosci 1: 279-285. [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Clements JD, Westbrook GL (1993) Nonuniform probability of glutamate release at a hippocampal synapse. Science 262: 754-757. [DOI] [PubMed] [Google Scholar]
- Rowen L, Young J, Birditt B, Kaur A, Madan A, Philipps DL, Qin S, Minx P, Wilson RK, Hood L, Graveley BR (2002) Analysis of the human neurexin genes: alternative splicing and the generation of protein diversity. Genomics 79: 587-597. [DOI] [PubMed] [Google Scholar]
- Sara Y, Biederer T, Atasoy D, Chubykin A, Mozhayeva MG, Sudhof TC, Kavalali ET (2005) Selective capability of SynCAM and neuroligin for functional synapse assembly. J Neurosci 25: 260-270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scanziani M, Gahwiler BH, Charpak S (1998) Target cell-specific modulation of transmitter release at terminals from a single axon. Proc Natl Acad Sci USA 95: 12004-12009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheiffele P (2003) Cell-cell signaling during synapse formation in the CNS. Annu Rev Neurosci 26: 485-508. [DOI] [PubMed] [Google Scholar]
- Scheiffele P, Fan J, Choih J, Fetter R, Serafini T (2000) Neuroligin expressed in nonneuronal cells triggers presynaptic development in contacting axons. Cell 101: 657-669. [DOI] [PubMed] [Google Scholar]
- Schneggenburger R, Neher E (2000) Intracellular calcium dependence of transmitter release rates at a fast central synapse. Nature 406: 889-893. [DOI] [PubMed] [Google Scholar]
- Song JY, Ichtchenko K, Sudhof TC, Brose N (1999) Neuroligin 1 is a postsynaptic cell-adhesion molecule of excitatory synapses. Proc Natl Acad Sci USA 96: 1100-1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugita S, Khvochtev M, Sudhof TC (1999) Neurexins are functional alpha-latrotoxin receptors. Neuron 22: 489-496. [DOI] [PubMed] [Google Scholar]
- Sugita S, Saito F, Tang J, Satz J, Campbell K, Sudhof TC (2001) A stoichiometric complex of neurexins and dystroglycan in brain. J Cell Biol 154: 435-445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunio A, Metcalf AB, Kramer H (1999) Genetic dissection of endocytic trafficking in Drosophila using a horseradish peroxidase-bride of sevenless chimera: hook is required for normal maturation of multivesicular endosomes. Mol Biol Cell 10: 847-859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof TC (2001) Alpha-latrotoxin and its receptors: neurexins and CIRL/latrophilins. Annu Rev Neurosci 24: 933-962. [DOI] [PubMed] [Google Scholar]
- Tabuchi K, Sudhof TC (2002) Structure and evolution of neurexin genes: insight into the mechanism of alternative splicing. Genomics 79: 849-859. [DOI] [PubMed] [Google Scholar]
- Togashi H, Abe K, Mizoguchi A, Takaoka K, Chisaka O, Takeichi M (2002) Cadherin regulates dendritic spine morphogenesis. Neuron 35: 77-89. [DOI] [PubMed] [Google Scholar]
- Tomasiewicz H, Ono K, Yee D, Thompson C, Goridis C, Rutishauser U, Magnuson T (1993) Genetic deletion of a neural cell adhesion molecule variant (N-CAM-180) produces distinct defects in the central nervous system. Neuron 11: 1163-1174. [DOI] [PubMed] [Google Scholar]
- Ushkaryov YA, Sudhof TC (1993) Neurexin III alpha: extensive alternative splicing generates membrane-bound and soluble forms. Proc Natl Acad Sci USA 90: 6410-6414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ushkaryov YA, Petrenko AG, Geppert M, Sudhof TC (1992) Neurexins: synaptic cell surface proteins related to the alpha-latrotoxin receptor and laminin. Science 257: 50-56. [DOI] [PubMed] [Google Scholar]
- Ushkaryov YA, Hata Y, Ichtchenko K, Moomaw C, Afendis S, Slaughter CA, Sudhof TC (1994) Conserved domain structure of beta-neurexins. Unusual cleaved signal sequences in receptor-like neuronal cell-surface proteins. J Biol Chem 269: 11987-11992. [PubMed] [Google Scholar]
- Wang X, Weiner JA, Levi S, Craig AM, Bradley A, Sanes JR (2002) Gamma protocadherins are required for survival of spinal interneurons. Neuron 36: 843-854. [DOI] [PubMed] [Google Scholar]
- Weiss JL, Burgoyne RD (2002) Sense and sensibility in the regulation of voltage-gated Ca2+ channels. Trends Neurosci 25: 489-491. [DOI] [PubMed] [Google Scholar]
- Wu L, Bauer CS, Zhen XG, Xie C, Yang J (2002) Dual regulation of voltage-gated calcium channels by PtdIns(4,5)P2. Nature 419: 947-952. [DOI] [PubMed] [Google Scholar]
- Wu LG, Borst JG, Sakmann B (1998) R-type Ca2+ currents evoke transmitter release at a rat central synapse. Proc Natl Acad Sci USA 95: 4720-4725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu LG, Westenbroek RE, Borst JG, Catterall WA, Sakmann B (1999) Calcium channel types with distinct presynaptic localization couple differentially to transmitter release in single calyx-type synapses. J Neurosci 19: 726-736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamponi GW (2001) Determinants of G protein inhibition of presynaptic calcium channels. Cell Biochem Biophys 34: 79-94. [DOI] [PubMed] [Google Scholar]
- Zhang W, Elsen F, Barnbrock A, Richter DW (1999) Postnatal development of GABAB receptor-mediated modulation of voltage-activated Ca2+ currents in mouse brain-stem neurons. Eur J Neurosci 11: 2332-2342. [DOI] [PubMed] [Google Scholar]