

Abstract
Activated microglia may be detrimental to neuronal survival in a number of neurodegenerative diseases. Thus, strategies that reduce microglial neurotoxicity may have therapeutic benefit. Stimulation of group II metabotropic glutamate (mGlu) receptors on rat primary microglia with the specific group II agonist 2S,2′R,3′R-2-(2′,3′-dicarboxy-cyclopropyl)glycine for 24 h induced microglial activation and resulted in a neurotoxic microglial phenotype. These effects were attributable to preferential mGlu2 stimulation, because N-acetyl-l-aspartyl-l-glutamate, a specific mGlu3 agonist, did not induce microglial activation or neurotoxicity. Stimulation of microglial mGlu2 but not mGlu3 induced caspase-3 activation in cerebellar granule neurons in culture, using microglial-conditioned media as well as cocultures. Stimulation of microglial mGlu2 induced tumor necrosis factor-α (TNFα) release, which contributed to microglial neurotoxicity mediated via neuronal TNF receptor 1 and caspase-3 activation. Stimulation of microglial group I or III mGlu receptors did not induce TNFα release. TNFα was only neurotoxic in the presence of microglia or microglial-conditioned medium. The toxicity of TNFα could be prevented by coexposure of neurons to conditioned medium from microglia stimulated by the specific group III agonist l-2-amino-4-phosphono-butyric acid. The neurotoxicity of TNFα derived from mGlu2-stimulated microglia was potentiated by microglial-derived Fas ligand (FasL), the death receptor ligand. FasL was constitutively expressed in microglia and shed after mGlu2 stimulation. Our data suggest that selective and inverse modulation of microglial mGlu2 and mGlu3 may prove a therapeutic target in neuroinflammatory diseases such as Alzheimer's disease and multiple sclerosis.
Keywords: metabotropic glutamate receptor, neurodegenerative disease, microglia, neuroprotection, TNFα, FasL
Introduction
Microglia are increasingly implicated in the signaling cascades leading to neuronal death in numerous neurodegenerative diseases (Cagnin et al., 2001; Gebicke-Haerter, 2001; Liu and Hong, 2003) through their excessive release of neurotoxins (Piani et al., 1991; Kingham et al., 1999; Kingham and Pocock, 2001; Hanisch, 2002). Strategies that reduce sustained microglial activation may provide therapeutic benefit. Our recent data suggest that modulation of microglial metabotropic glutamate (mGlu) receptors may constitute a means to control microglial activation (Taylor et al., 2002, 2003).
At least eight subtypes of mGlu receptors exist, and microglia express mGlu receptor subtypes belonging to group I (Biber et al., 1999), group II (mGlu 2/3; Taylor et al., 2002), and group III (mGlu4,6,8) (Taylor et al., 2003). Previously, we have shown that microglial group II and group III mGlu receptors are both negatively coupled with adenylate cyclase but that the consequences for neurons after stimulation of each group are very different (Taylor et al., 2002, 2003). Stimulation of microglial group II mGlu receptors induces microglial activation, mitochondrial depolarization, and apoptosis as well as inducing a neurotoxic phenotype (Taylor et al., 2002). In contrast, activation of microglial group III mGlu receptors protects neurons against microglial neurotoxicity (Taylor et al., 2003).
Substances that are both released by activated microglia and implicated in neurotoxicity include the cytokine tumor necrosis factor α (TNFα) (Lee et al., 1993; Hanisch, 2002; Wang et al., 2003). TNFα is a member of the superfamily of cytokines that also includes Fas ligand (FasL). TNFα is implicated as an important inflammatory mediator in many neurodegenerative diseases including Alzheimer's disease and multiple sclerosis (Mattson et al., 1997; Gebicke-Haerter, 2001). FasL and its receptor Fas show upregulated expression in acute and chronic neurological conditions (Nishimura et al., 1995; Dowling et al., 1996; D'Souza et al., 1996; Felderhoff-Mueser et al., 2000). FasL is constitutively expressed in microglia in vitro and in normal brain (D'Souza et al., 1996; Frigerio et al., 2000). The upregulation and shedding of FasL in microglia after stimulation (Badie et al., 2000; Terrazzino et al., 2002) and in neurodegenerative diseases (D'Souza et al., 1996) may contribute to neurotoxicity by promoting apoptosis in target cells (Griffith et al., 1995; Nagata, 1999).
In this study, we wanted to determine the mechanism by which stimulation of microglial group II mGlu receptors induces neurotoxicity. We show that activation of microglial group II mGlu receptors, specifically mGlu2 and not mGlu3, induced microglial activation and substantial TNFα release. Stimulation of microglial group I or group III mGlu receptors did not evoke TNFα release. TNFα was a component of mGlu2-induced microglial neurotoxicity, inducing neuronal apoptosis through activation of TNF receptor 1 (TNFR1) and promoting the activation of caspase-3. FasL released by microglia was a crucial cofactor for TNFα neurotoxicity after mGlu2 stimulation. These findings suggest that specific modulation of microglial mGlu2 receptors could provide therapeutic benefit in Alzheimer's disease and other neurodegenerative diseases in which microglial reactivity and neuroinflammation are implicated.
Materials and Methods
Materials. Wistar rats were bred and reared in house from stock animals originally purchased from Charles River UK (Kent, UK). Fetal calf serum and minimum essential medium with Earle's salts (MEM) were obtained from Invitrogen (Paisley, UK). Tissue culture plasticware and coverslips were obtained from Scientific Laboratory Supplies (Nottingham, UK). PBS powder was purchased from ICN Biomedicals (Maidenhead, UK). All mGlu receptor agonists were from Tocris Cookson (Bristol, UK), except for N-acetyl-l-aspartyl-l-glutamic acid (NAAG), which was purchased from Sigma (Poole, UK). 5,5′,6,6′-Tetrachloro-1,1′,3,3′-tetraethylbenzimidazole carboncyanine iodide (JC-1) was from Molecular Probes Europe (Leiden, The Netherlands). Fas-Fc (recombinant) set was purchased from Q Biogen Alexis (Nottingham, UK). Anti-TNFα (L-19), anti-FasL (N-20), anti-Fas (M-20), anti-TNFR1 (H-271), anti-TNFR2 (H-202), goat anti-rabbit IgG horseradish peroxidase (HRP), goat anti-mouse IgG HRP, donkey anti-goat IgG HRP, protein A/G-agarose and TNFR1 (BA-162), and TNFR2 (BA-174) purified proteins were from Autogen Bioclear (Calne, UK). Anti-inducible nitric oxide synthase (iNOS) and iNOS lysate standard were from Pharmingen (Oxford, UK). ED1 antibody was from Serotec (Oxford, UK). Rat recombinant TNFα and Quantikine M TNFα ELISA were from R & D Systems (Abingdon, UK). Biotinylated secondary antibodies, the avidinbiotin complex kit, and aqueous mountant were from Vector Laboratories (Peterborough, UK). Precast gels were from Bio-Rad (Hemel Hempstead, UK). CaspaTag caspase-3 (DEVD) activity kit was purchased from Intergen (Purchase, NY) and Affiniti Research Products (Exeter, UK). All other chemicals were purchased from Sigma.
Preparation of microglial cell cultures. Microglial cells were isolated from 3- to 6-d-old Wistar rat pups as described previously (Kingham and Pocock, 2000). Briefly, animals were killed by cervical dislocation and decapitation in accordance with the United Kingdom Animals (Scientific Procedures) Act, 1986. Brains were removed into ice-cold PBS (in mm: 140 NaCl, 5 KCl, 25 Na2HPO4, 2.9 NaH2PO4.2H2O, and 11 glucose, pH 7.4) and homogenized. The homogenate was centrifuged at 500 × g for 10 min, and the pellet was resuspended in 70% isotonic Percoll (stock with 1% PBS) diluted with PBS. This was overlaid with 35% Percoll and PBS. The Percoll gradient was centrifuged at 1250 × g for 45 min, and microglial cells were collected from the 35/70% interface. After washing, the cells were plated at a density of 9 × 104 per 13 mm coverslip. Cultures were maintained at 37°C in 5% CO2 in MEM supplemented with 25 mm KCl, 30 mm glucose, 25 mm NaHCO3, 1 mm glutamine, 10% heat-inactivated fetal calf serum, 50 U/ml penicillin, and 50 μg/ml streptomycin. This medium was used to culture the microglia because this was the same medium in which cultured cerebellar granule neurons were grown and allows us to add conditioned medium from the microglia to the neurons. In particular, adding conditioned medium with a reduced KCl concentration would be detrimental to the survival of cerebellar granule neurons. Microglial cells were routinely used 1 d after plating. The microglial cultures contained 99.05 ± 0.93% microglia as determined by staining with the microglial marker isolectin B4. Cells were counted on a minimum of three separate fields on four coverslips, and results are expressed as a percentage of the total cell number (4′,6-diamidino-2-phenylindole-stained cells) ± SEM. This is a similar level of purity as we have observed previously (Taylor et al., 2002, 2003; Morgan et al., 2004). We confirmed our previous findings that after 1 d in vitro, microglial cells exhibit a resting-like, ramified morphology with only a small number of cells staining positive for ED-1 (a marker for activated microglia) (data not shown) (Taylor et al., 2002, 2003; Morgan et al., 2004).
Isolation and culture of cerebellar granule neurons. Primary cultures of cerebellar granule cells (CGCs) were isolated from 3- to 6-d-old rat pups and prepared as described previously (Evans and Pocock, 1999). Cells were plated on 13 mm poly-d-lysine-coated glass coverslips at a density of 6.5 × 106 per coverslip and maintained in MEM-supplemented medium (as above). After 24 h in vitro, cytosine furanoarabinoside (10 μm) was added to prevent proliferation of non-neuronal cells, and after 3 d in vitro, 100 μl of sterile distilled water was added to reduce medium concentration by evaporation. The cultures were maintained at 37°C in 5% CO2 and used at 7 d in vitro.
Growth and maintenance of the N9 microglial cell line. The N9 microglial cell line was a kind gift from Dr. Paola Ricciardi Castagnoli (CNR Cellular and Molecular Pharmacology Centre, Milan, Italy). The cell line was derived originally from embryonic day 13 mouse microglial cultures (Corradin et al., 1993), and responses from these cells are similar to those from primary cultures of rat microglia (Kingham and Pocock, 2000). N9 microglial cells were maintained in culture medium (DMEM supplemented with 4.4 mm NaHCO3, 50 μm β-mercaptoethanol, 5% newborn calf serum, 50 U/L penicillin, and 50 μg/ml streptomycin) at 37°C in 5% CO2. N9 cells were plated at a density of 1 × 104 cell/well on 13 mm glass coverslips and were used at 2 d in vitro.
Treatment of microglial cell cultures. Primary rat microglial cells were exposed to the specific mGlu receptor agonist: 250 μm trans-azetidine-2,4,-dicarboxylic acid (t-ADA; group I), 500 nm 2S,2′R,3′R-2-(2′,3′-dicarboxy-cyclopropyl)glycine (DCGIV; group II), 50 μm NAAG (specific mGlu3 agonist), or 100 μm l-2-amino-4-phosphono-butyric acid (l-AP-4; group III). In some experiments, cells were exposed to the specific mGlu receptor antagonist: 250 μm 1-aminoindan-1,5-dicarboxylic acid (AIDA; group I) or 500 μm 2S,1′S,2′S-2-methyl-2-(2′carboxycyclopropyl)glycine (MCCG; group II). In some cases, microglia were activated with lipopolysaccharide (LPS; 1 μg/ml). At defined times, the tissue culture medium was collected, and the cells were evaluated for mitochondrial depolarization with the fluorescent probe JC-1 and apoptosis by Hoechst 33342 staining, fixed for immunocytochemical detection of FasL and TNFα expression, or lysed for Western blotting. N9 microglial cells were exposed to DCGIV (500 nm), NAAG (50 μm), l-AP-4 (100 μm), or LPS (1 μg/ml) for 24 h, after which the cell culture medium was retained and the cells were fixed for immunocytochemical detection of FasL expression or lysed for Western blotting.
Immunodepletion of microglial conditioned medium. Microglial conditioned medium (MGCM) was immunodepleted of either FasL or TNFα. Briefly, 600 μl sterile aliquots of either control MGCM or MGCM from treated microglia were incubated with FasL or TNFα antibody at 4°C with constant rotation. After 4 h, 20 μl of protein A/G-agarose was added and left for an additional 16 h at 4°C with constant rotation. The immunocomplex was removed by centrifugation at 10,000 rpm for 1 min, and the supernatant was collected and transferred to CGC cultures. Samples of the supernatants that had been immunodepleted were subjected to Western blotting or ELISA to ensure the removal of FasL or TNFα. The FasL immunocomplex was retained and subjected to Western blotting for the assessment of FasL release from control and treated microglia.
Treatment of neuronal cultures. Conditioned medium from control or treated microglia was subsequently transferred to CGC cultures, which were then incubated for an additional 24 or 48 h before cells were fixed with 4% formaldehyde (in PBS). In TNFα toxicity experiments, the appropriate concentration of TNFα was added to the neuronal cultures at the same time as the MGCM. In some experiments, antibodies for TNFα, Fas, TNFR1, or TNFR2 were added 30 min before the addition of the MGCM. For the Fas-Fc experiments, 5 μg/ml Fas-Fc and 1 μg/ml enhancer was added to the CGC cultures 30 min before the addition of DCGIV-MGCM. After fixing, cells were either stained with Hoechst 33342 for the assessment of neuronal apoptosis or used for immunocytochemical detection of Fas or TNFR1/2 expression.
Cocultures. Primary rat microglial cells (two coverslips) were placed in a 35 mm Petri dish in 2 ml of medium, and mGlu receptor agonists were added as described above. After 24 h, CGCs (two coverslips) were added to each dish, and the preparation was maintained at 37°C in 5% CO2 for an additional 24 or 48 h. Cells were then fixed with 4% formaldehyde, and apoptosis was assessed with Hoechst 33342 staining. Controls consisted of neurons cultured alone (basal) or microglia cultured together with neurons (control).
Measurement of microglial mitochondrial membrane polarization. Microglial mitochondrial membrane polarization (ΔΨm) was assessed with the fluorescent probe JC-1. At 490 nm, cells with depolarized mitochondria contained JC-1 predominantly in the monomeric form and fluoresced green. Cells with polarized mitochondria predominantly contain JC-1 in aggregate form, and the mitochondria fluoresce red/orange (Salvioli et al., 1997). We have shown previously that the state of mitochondrial depolarization as determined with JC-1 staining is the same as results obtained after staining with tetramethylrhodamine ethyl ester, another mitochondrial membrane potential-sensitive fluorescence dye, and that the fluorescence associated with either stain subsequently decreased in the microglial mitochondria after the addition of the mitochondrial uncoupler carbonyl cyanide p-(trifluoromethoxy) phenylhydrazone (Kingham and Pocock, 2000). Microglia were incubated with JC-1 (5 μm) in basic medium (in mm: 153 NaCl, 3.5 KCl, 0.4 KH2PO4, 20 N-tris(hydroxymethyl)methyl-2-aminoethanesulphonic acid, 5 NaHCO3, 5 glucose, 1.2 Na2SO4, and 1.3 CaCl2, pH 7.4) at 37°C for 10 min, washed, and placed on a thermostat stage at 37°C. Coverslips were viewed using an Olympus (Tokyo, Japan) IX70 inverted fluorescence microscope with excitation at 490 nm and emission at >520 nm. Microglia with polarized mitochondria were seen with distinct mitochondria fluorescing red/orange, and in microglia with depolarized mitochondria, the cell cytoplasm and mitochondria appeared green (Kingham and Pocock, 2000). The numbers of cells with mitochondria stained red/orange or green were counted, and the degree of mitochondrial depolarization was expressed as the percentage of green cells per field. A minimum of 6 fields per coverslip were counted, and between 6 and 10 coverslips from at least three independent experiments were assessed for each variable.
Assessment of apoptosis. After measurement of mitochondrial polarization, the same cells were fixed with 4% formaldehyde in PBS (4°C) for 10 min and incubated with Hoechst 33342 (5 μg/ml) for 10 min (Yan et al., 1994; Kingham et al., 1999). Nuclear morphology was viewed using a fluorescence microscope with excitation at 360 nm and emission >490 nm. Apoptotic cells were identified as those cells containing brightly stained pyknotic nuclei compared with healthy cells in which the nuclei appeared less bright and less condensed. A minimum of 6 fields per coverslip were counted, and between 6 and 10 coverslips were assessed for each variable.
Assessment of caspase-3 expression. The expression of active caspase-3 was assessed in live cells using the CaspaTag Caspase-3 (DEVD) activity kit. Cells were counterstained with propidium iodide (PI; 5 μg/ml) to indicate dead cells. Briefly, 2.5 μl of CaspaTag solution was added to the cells in culture in 250 μl of their normal culture medium and placed back in the incubator at 37°C for 30 min. Coverslips were removed from the wells and placed in warm (37°C) basic medium and immediately observed in a fluorescence microscope (Zeiss, Oberkochen, Germany). CaspaTag fluorescence was captured with an FITC filter set and manual exposure of 4000 ms. PI fluorescence was measured with a rhodamine filter set. A phase contrast image of the same field was also captured.
Immunocytochemical detection of ED1, TNFα, Fas, FasL, and TNFR1/2. Cells were fixed in 4% formaldehyde in PBS at 4°C and in 100% methanol (-20°C) for 20 min. After washing with PBS, cells were preincubated with PBS containing 2.5% normal goat serum (or normal horse serum for ED1 staining) for 30 min at room temperature. Cells were then incubated with primary antibody overnight at 4°C (1:250 anti-Fas or anti-FasL; 1:500 anti-ED1, anti-TNFα, anti-TNFR1, or anti-TNFR2 in PBS). After washing, cells were exposed to goat anti-mouse biotinylated antibody for ED1 (1:1000), donkey anti-goat biotinylated antibody for TNFα (1:500), or goat anti-rabbit biotinylated antibody (1:500 in PBS) for 2 h at room temperature and avidinbiotin complex for 1 h at room temperature. After three final washes, cells were exposed to DAB. Negative controls were treated in the same way, but the primary antibody was omitted. For each primary antibody, an isotype control was also performed.
Western blotting. Cells were harvested into lysis buffer (20 mm Tris-acetate, 1 mm EDTA, 1 mm EGTA, 10 mm sodium-β-glycerophosphate, 1 mm sodium orthovanadate, 5% glycerol, 1% Triton X-100, 0.27 m sucrose, 1 mm benzamidine, 4 μg/ml leupeptin, and 0.1%β-mercaptoethanol, pH 7.4) and incubated on ice for 10 min. Cell lysates were then centrifuged to remove cell debris, and the protein concentration in the supernatant was determined using bovine serum albumin as a standard. Cell proteins or FasL immunocomplexes prepared by immunoprecipitation were resolved using SDS-PAGE and transferred electrophoretically onto Immobilon-P-polyvinylidene difluoride membranes. Membranes were washed with Tween 20-Trisbuffered saline (T-TBS; 10 mm Tris-HCl, 150 mm NaCl, and 0.5% Tween 20, pH 7.4) and blocked for 2 h at room temperature in blocking buffer (T-TBS plus 5% nonfat dried milk). Membranes were incubated with the primary antibody overnight at 4°C [anti-FasL (1:500), anti-Fas (1:500), anti-TNFR1 (1:1000), anti-TNFR2 (1:1000), anti-iNOS (1:5000), or anti-β-actin (1:500) in 5% nonfat milk]. After repeated washes in T-TBS, the membranes were incubated with HRP-conjugated secondary antibody [goat anti-rabbit (1:1000) for anti-FasL, anti-Fas, anti-TNFR1, anti-TNFR2, and anti-iNOS, and goat anti-mouse (1:1000) for anti-β-actin in 5% nonfat milk] for 2 h at room temperature. Blots were developed by enhanced chemiluminescence detection.
ELISA determination of TNFα. For the quantitative determination of TNFα concentration in microglial cell culture supernatants, Quantikine M Rat TNFα Immunoassay kits were used according to the manufacturer's instructions. Briefly, MGCM was removed from the cultures and centrifuged at 10,000 rpm for 1 min. To 96-well plates precoated with monoclonal antibody for TNFα, 50 μl of supernatant or standard was added in duplicate and incubated at room temperature for 2 h. After washing, a 100 μl rat TNFα conjugate was added to each well for 2 h at room temperature before washing again. Then 100 μl of substrate solution was added for 30 min at room temperature, followed by stop solution. Optical density was determined using a microplate reader set to 450 nm with a correction filter at 540 nm. TNFα concentrations in the cell supernatants were determined using a standard curve of known TNFα concentrations.
Statistical analysis. Significant differences were estimated using Welch's two-sided t test with levels of significance at ***p < 0.0005, **p < 0.005, and *p < 0.05; p > 0.05 was not significant.
Results
Stimulation of mGlu2 induces microglial reactivity and apoptosis
Microglial apoptosis as a consequence of activation is mediated by upstream mitochondrial depolarization (Kingham and Pocock, 2000). Stimulation of group II mGlu receptors (mGlu2 and mGlu3) on microglia with the specific agonist DCGIV (500 nm) induced microglial mitochondrial depolarization and apoptosis (Fig. 1A), in line with our previous findings (Taylor et al., 2002). DCGIV at 1-100 nm induced 6.4 ± 0.89% to 10.65 ± 1.05% microglial apoptosis, respectively, with 500 nm inducing 33.14 ± 1.89% and 1 μm inducing 34.1 ± 1.061 microglial apoptosis. There was no significant difference between the level of apoptosis induced with 500 nm DCGIV compared with 1 μm DCGIV, as assessed by Student's t test, suggesting saturation of the response (p = 0.7345). A concentration of 500 nm DCGIV was used in all subsequent experiments because we showed previously that this induced ∼50% microglial activation as determined by ED1 staining (Taylor et al., 2002).
Figure 1.
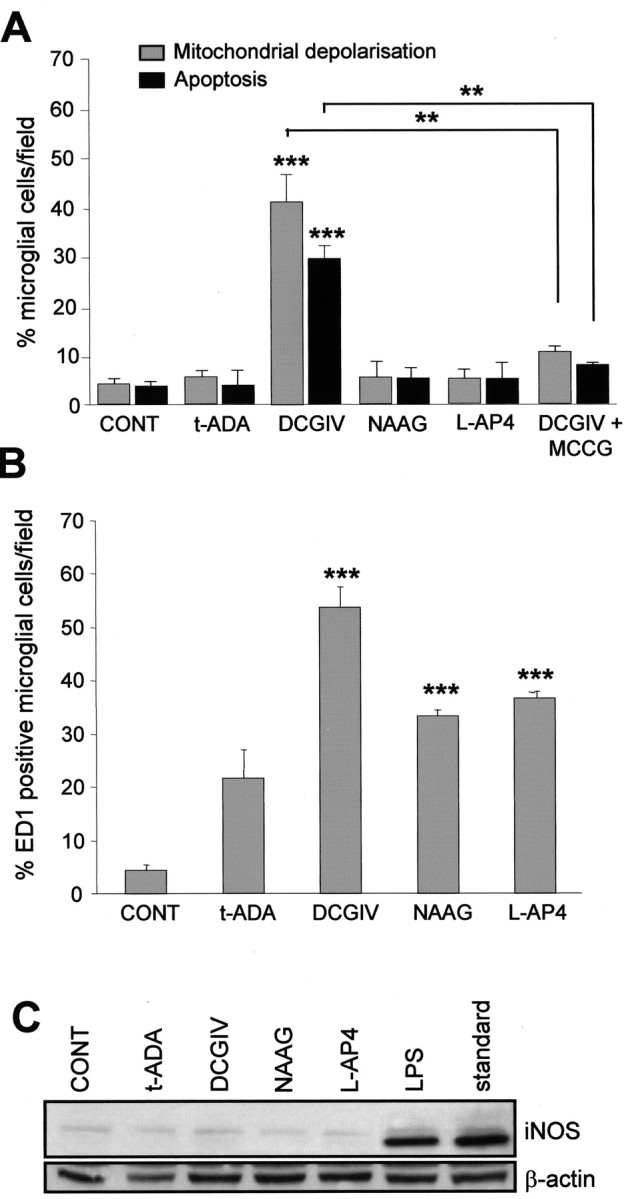
Activation of mGlu2 induces microglial mitochondrial depolarization, apoptosis, and ED1 expression. Primary rat microglia were exposed to specific mGlu receptor agonists: t-ADA (group I; 250 μm), DCGIV [group II; 500 nm; with or without the group II antagonist MCCG (500 μm)], NAAG (mGlu3; 50 μm), and l-AP-4 (group III; 100 μm). After 24 h, the number of cells with depolarized mitochondria (determined by JC-1 staining), those exhibiting apoptotic nuclei (as indicated by Hoechst 33342 staining), were counted (A), as were those positive for ED1 staining (with antibody to ED1, 1:500, counterstained with hematoxylin), a marker for activated microglia (B). Data are mean ± SEM of at least six determinations. C, Microglial mGlu receptor stimulation does not induce iNOS expression. Primary microglial cultures were treated for 24 h with the agonists, as above, or with LPS (1 μg/ml) for 24 h. Cells were lysed, and cell proteins were resolved by 10% SDS-PAGE and immunoblotted with anti-iNOS antibody (1:5000). Standard, Commercially available iNOS lysate used as a positive control (1 μg/ml). The blot was then reprobed for β-actin (1:500) to confirm equal loading. Significance levels are shown in all graphs: **p < 0.005; ***p < 0.0005 compared with control unstimulated microglia. CONT, Control.
We now show that the effect of DCGIV on microglia was attributable to its stimulation of mGlu2, because NAAG (50 μm), a specific agonist of mGlu3 (Wroblewska et al., 1997), did not induce microglial mitochondrial depolarization or apoptosis after 24 h (Fig. 1A) or 48 h (data not shown). NAAG has been shown to be a highly selective mGlu3 agonist with a potency that rivaled glutamate in cells transfected with mGlu3 cDNA (Wroblewska et al., 1993), and concentrations of NAAG up to 300 μm did not activate mGlu2 in transfected cell lines (Wroblewska et al., 1997; Hess et al., 1999). Therefore, the effects of NAAG (50 μm) seen is this study are likely attributable to its specific stimulation of mGlu3. The addition of the group II antagonist MCCG (500 μm), together with DCGIV, reduced both the DCGIV-induced changes in mitochondrial polarization and apoptosis to control levels (Fig. 1A). Stimulation of microglial group I mGlu receptors with the specific agonist t-ADA (250 μm) (Klein et al., 1997) or group III mGlu receptors with l-AP-4 (100 μm) (Okamoto et al., 1994; Schoepp et al., 1999) did not induce microglial mitochondrial depolarization or apoptosis at 24 h (Fig. 1A) or after 48 h (data not shown).
Control untreated microglial cultures contained very few activated (ED1-positive) cells (Fig. 1B). Microglia exposed to DCGIV for 24 h became immunoreactive for ED1 (53.6 ± 3.9 cells/field); this increased to >80% cells per field after 48 h (data not shown). Although the other mGlu receptor agonists increased the number of ED1-positive cells after 24 h (Fig. 1B), this was not associated with a change in mitochondrial polarization or apoptosis (Fig. 1A), and no further increase in ED1-positive cells was seen after 48 h (data not shown).
To further assess the reactive state of the microglia, we measured the expression of iNOS, which is upregulated in microglia activated with chromogranin A or LPS (Kingham et al., 1999; Morgan et al., 2004). Control microglia did not express iNOS (Fig. 1C), which confirmed ED1 data, suggesting that control cultures were in a resting-like state. Exposing microglia to the different mGlu receptor agonists for 24 h did not induce an increase in iNOS expression (Fig. 1C). In contrast, exposing microglia to LPS (1 μg/ml), a known microglial activator for 24 h, induced a strong upregulation of iNOS expression. This suggested that microglial activation after mGlu receptor stimulation, and any subsequent neurotoxicity, involved an iNOS-independent pathway.
Stimulation of microglial mGlu2 but not mGlu3 activates microglia to a neurotoxic phenotype
Medium from activated microglia is toxic to neurons (Kingham et al., 1999; Taylor et al., 2002, 2003). Exposing neuronal cultures to MGCM from DCGIV-treated microglia (DCGIV-MGCM) induced an increase in the number of neurons displaying pyknotic nuclei as determined by Hoechst staining (Fig. 2A). Medium from microglia exposed to 1-100 nm DCGIV induced neuronal apoptosis from 8.75 ± 0.35 to 21.7 ± 0.09%, respectively, whereas 500 nm DCGIV-MGCM lead to 45 ± 0.25% and 1 μm DCGIV-MGCM lead to 46.57 ± 2.25% apoptosis. Student's t test revealed no significant difference between the level of apoptosis induced with conditioned medium from 500 nm DCGIV-stimulated microglia compared with the level of apoptosis induced with conditioned medium from 1 μm DCGIV-stimulated microglia (p = 0.5809).
Figure 2.
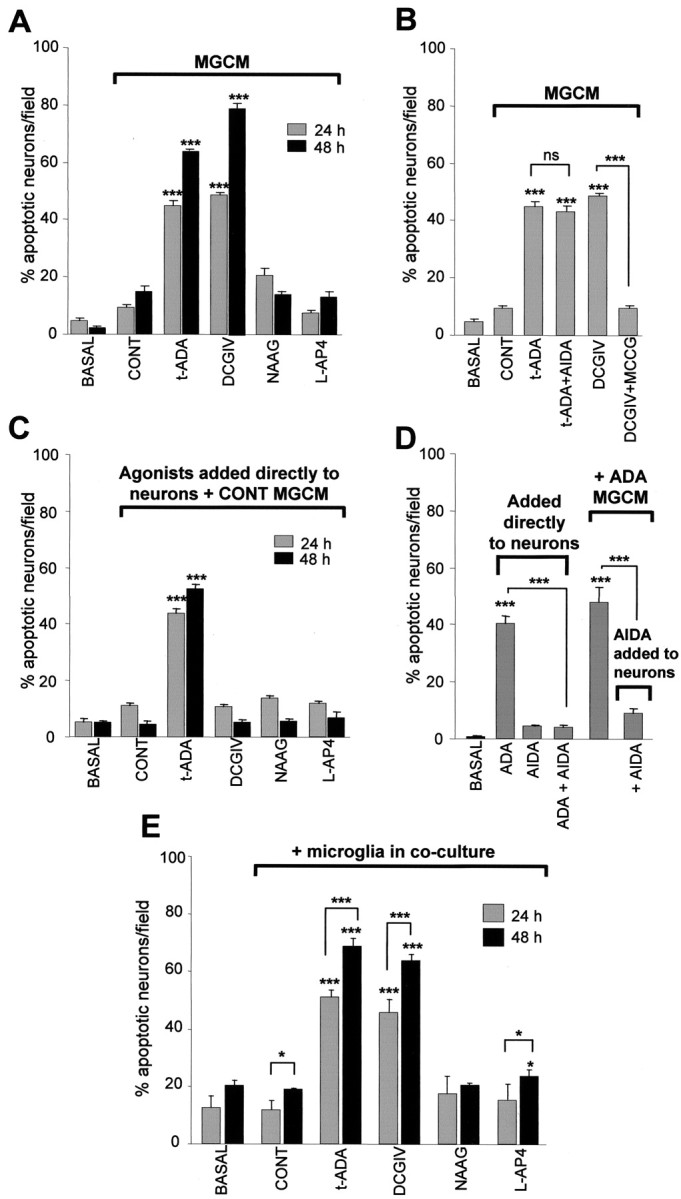
Stimulation of microglial mGlu2 induces a neurotoxic microglial phenotype. Cerebellar granule neurons were untreated (basal), incubated with MGCM from control unstimulated microglia (CONT), or exposed to MGCM from microglia exposed to different mGlu receptor agonists for 24 or 48 h, where indicated: t-ADA (250 μm) with or without the antagonist AIDA (250 μm), DCGIV (500 nm) with or without the antagonist MCCG (500 μm), NAAG (50 μm), and l-AP-4 (100 μm). Neuronal apoptosis was measured by Hoechst 33342 staining. A, A cerebellar granule neuron exposed to MGCM from t-ADA (group I)-treated microglia or DCGIV (group II)-treated microglia becomes apoptotic. MGCM from microglia treated with the group II mGlu3 agonist NAAG and the group III mGlu agonist l-AP-4 does not evoke CGC neuronal apoptosis. B, The neurotoxicity of MGCM from t-ADA-treated microglia is not reduced when the microglia are coexposed to the group I antagonist AIDA. The MGCM from DCGIV-treated microglia is significantly reduced when the microglia are coexposed to the group II antagonist MCCG. C, The direct addition of the mGlu receptor agonists to neuronal cultures in the presence of MGCM from control unstimulated microglia revealed that t-ADA is directly neurotoxic. D, The direct neurotoxicity of t-ADA can be abated by the addition of the group I antagonist AIDA to the neuronal cultures. E, Activation of microglial mGlu2 induces the release of soluble, stable neurotoxins. Untreated microglia in coculture with neurons (CONT) did not induce apoptosis above that in primary cultures of CGC neurons alone (basal). Neurons were added to the microglial cultures 24 h after the addition of the mGlu receptor agonist t-ADA, DCGIV, NAAG, or l-AP-4. In each case, the number of apoptotic neurons per field was counted in six separate fields on atleast four coverslips per condition and is expressed as a percentage of total cells (mean ± SEM). Significance values are shown compared with control MGCM-treated neuronal cultures in A-D and control cocultures (CONT; neurons cultured with microglia) in E, unless indicated otherwise (***p < 0.0005).
MGCM from microglia exposed to NAAG was not toxic to CGC neurons at 24 or 48 h, suggesting that activation of microglial mGlu2 receptors by DCGIV induced microglial neurotoxicity, whereas stimulation of mGlu3 does not. Conditioned medium from microglia exposed to DCGIV, together with the group II mGlu receptor antagonist MCCG, significantly reduced DCGIV-induced microglial neurotoxicity (Fig. 2B), suggesting that the release of toxins from microglia was a specific response to mGlu2 stimulation. We confirmed that the neurotoxicity was not attributable to a direct toxic effect of DCGIV carried over in the MGCM, because exposing neuronal cultures to 500 nm DCGIV in the presence of control MGCM (Fig. 2C) (Taylor et al., 2002) or alone (data not shown) did not induced neuronal apoptosis above control MGCM alone after 24 or 48 h.
Stimulation of group III receptors on microglia with l-AP-4 did not induce a neurotoxic phenotype (Fig. 2A) (Taylor et al., 2003). Medium from microglia exposed to the group I agonist t-ADA was neurotoxic after 24 h, with an additional increase in neuronal apoptosis observed after a 48 h exposure (Fig. 2A). However, the direct addition of t-ADA to neuronal cultures together with MGCM (Fig. 2C) or alone (Fig. 2D) caused a significant increase in neuronal death. This implied that the neurotoxic effect seen with MGCM from t-ADA-treated microglia was more likely because of the carry-over of t-ADA to the neurons via the MGCM rather than to the release of microglial toxins induced by t-ADA. This neurotoxicity could not be reduced by exposing the microglia to t-ADA together with the group I antagonist AIDA (250 μm) (Fig. 2B) but was prevented when the antagonist AIDA was added directly to the neurons (Fig. 2D). Because the levels of neuronal death induced by 24 h t-ADA-MGCM were similar to t-ADA added directly to the neuronal cultures, this also suggested that no additional toxins were released when group I mGlu receptors on microglia are stimulated.
The addition of MGCM to neurons allows the assessment of released stable components of microglial neurotoxicity. Therefore, to determine the involvement of nonstable microglial-released toxins, microglia were cocultured with neurons in the presence of the mGlu receptor agonists. Similar results for neuronal apoptosis after mGlu receptor modulation were obtained when microglia and neurons were grown in coculture (Fig. 2E), indicating that soluble, stable toxins are released from microglia and that the presence of neurons does not modulate the microglial response. In coculture experiments, basal (neurons alone) and control (neurons with unstimulated microglia) cultures have a slightly higher background level of apoptosis than MCGM experiments, because the neurons are incubated with fresh medium. It is known that fresh medium containing 10% serum is toxic to cells by an additional mechanism of excitotoxicity induced by the high amounts of glutamate present in the serum (Schramm et al., 1990).
Exposing neuronal cultures for 48 h to MGCM from DCGIV-stimulated microglia induced a high level of apoptosis (Fig. 2A), and the cells started to loose adherence and float off the coverslips. Because this made accurate counting of neuronal cells more difficult, in all subsequent experiments, neuronal cultures were exposed to MGCM for only 24 h.
Stimulation of mGlu2 induces TNFα release from microglia
Because activated microglia can release the cytokine TNFα, a potential neurotoxin that induces neuronal apoptosis (Hanisch, 2002; Yang et al., 2002), we investigated whether TNFα was released from microglia after mGlu receptor stimulation. Low levels of TNFα were detected in the medium from untreated microglial (32.9 ± 6.8 pg/ml), indicating that microglia in culture constitutively release small amounts of this cytokine (Fig. 3A), in line with the findings of others (Ciesielski-Treska et al., 1998; Kim et al., 2004; Weydt et al., 2004). Stimulation of microglial mGlu2 and mGlu3 with 500 nm DCGIV for 24 h induced significant TNFα release to 128.7 ± 29.7 pg/ml (Fig. 3A), whereas specific activation of microglial mGlu3 with NAAG (50 μm) failed to evoke release of TNFα above basal, suggesting that stimulation of mGlu2 triggers TNFα release. DCGIV-induced TNFα release was reduced by the specific group II antagonist MCCG (500 μm). Interestingly, MGCM from microglia exposed to MCCG alone had significantly lower TNFα levels than MGCM from control untreated cultures, suggesting that slight activation of these cells through stimulation of mGlu2 occurs in unstimulated cultures (Fig. 3A). Agonists of group I or III mGlu receptors did not evoke microglial TNFα release compared with control cultures after 24 h (Fig. 3A).
Figure 3.
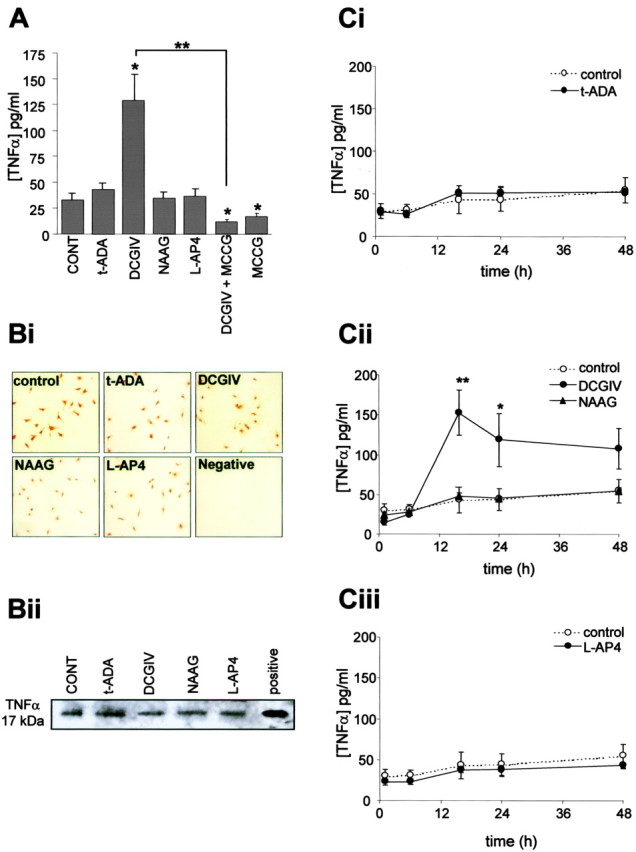
Activation of mGlu2 induces TNFα release from microglia. A, Microglial cells were exposed to the specific mGlu receptor agonist t-ADA (group I; 250 μm), DCGIV [group II; 500 nm; with or without group II antagonist MCCG (500 μm)], NAAG (mGlu3; 50 μm), l-AP-4 (group III; 100 μm), or MCCG (500 μm). After 24 h, the TNFα concentration in the culture medium was measured by ELISA. Data are the mean ± SEM from four to five separate experiments (defined as different cell preparations) per condition in which the replicates per experiment were three to four (defined as the number of separate coverslips for each condition). Bi, Immunolocalization of TNFα expression in control microglia and microglia exposed to mGlu receptor agonists, as above, for 24 h. Fixed cells were stained with a specific antibody to TNFα (1:500). Negative controls were treated in the same way, but the primary antibody was omitted. Bii, Cell lysates (45 μg of protein) from microglia exposed to mGlu receptor agonists for 24 h were subjected to immunoprecipitation for TNFα, and the immunocomplexes were resolved by 10% SDS-PAGE. Positive, Rat recombinant TNFα (1 μg/ml). C, Time course of TNFα release from microglia. Cells were exposed to the specific mGlu receptor agonists t-ADA (Ci; 250 μm), DCGIV (Cii; 500 nm), NAAG (Cii; 50 μm), and l-AP-4 (Ciii; 100 μm). At 1, 6, 16, 24, and 48 h after exposure, the culture medium was removed and the TNFα concentration was measured by ELISA. For each time point, values are mean ± SEM from four to five separate batches of cells with replicates of three to four separate coverslips. Significance values in all graphs are compared with control untreated MGCM (*p < 0.05; **p < 0.005), unless indicated otherwise.
Immunocytochemical determination of TNFα expression on microglia showed that control untreated microglia expressed TNFα (Fig. 3Bi). This staining was specific because no positive staining was observed in the negative control in which primary antibody was omitted (Fig. 3Bi, Negative) or with an isotype control (data not shown). Microglia exposed to different mGlu receptor agonists for 24 h had a similar level of TNFα expression to that seen in untreated cells. Western blotting of microglial cell lysates showed that microglia expressed TNFα as a 17 kDa protein, and stimulation of different subtypes of microglial mGlu receptors did not alter TNFα expression (Fig. 3Bii). These data suggested that although microglia constitutively express TNFα, it is only when microglia become activated after mGlu2 stimulation that TNFα is released into the tissue culture medium (Fig. 3A).
DCGIV-induced TNFα release increased between 6 and 16 h, at which time it peaked but remained elevated 48 h after the addition of the agonist (Fig. 3Cii). The increase in TNFα levels up to 16 h was attributable to release from microglia rather than cell lysis, because before this time no dead (PI-positive) or apoptotic microglial cells were observed (data not shown). No alteration in TNFα release compared with control untreated microglia was observed with NAAG, t-ADA, or l-AP-4 (Fig. 3Ci-iii). These findings also suggested that t-ADA neurotoxicity seen at 24 and 48 h was not attributable to an earlier release of TNFα.
TNFα released after mGlu2 stimulation contributes to DCGIV-induced microglial neurotoxicity
We investigated whether the TNFα released after microglial mGlu2 stimulation was responsible for the neurotoxic effect of MGCM from DCGIV-stimulated microglia (Fig. 4A). When DCGIV-MGCM was immunodepleted of TNFα before its addition to the neuronal cultures, a significant reduction in neuronal apoptosis was observed (to 25.0 ± 1.7 cells/field; p < 0.0005 compared with apoptosis in neurons exposed to DCGIV-MGCM at 24 h) (Fig. 4A). These results suggested that TNFα released by microglia after mGlu2 stimulation was a contributing factor to mGlu2-induced microglial neurotoxicity. Other factors may also be involved because neuronal death after TNFα immunodepletion of DCGIV-MGCM still remained significantly higher than that observed with control MGCM. We confirmed that the TNFα antibody (at the concentrations used for immunodepletion) did not induce neuronal apoptosis when added directly to neuronal cultures together with control MGCM (Fig. 4A).
Figure 4.
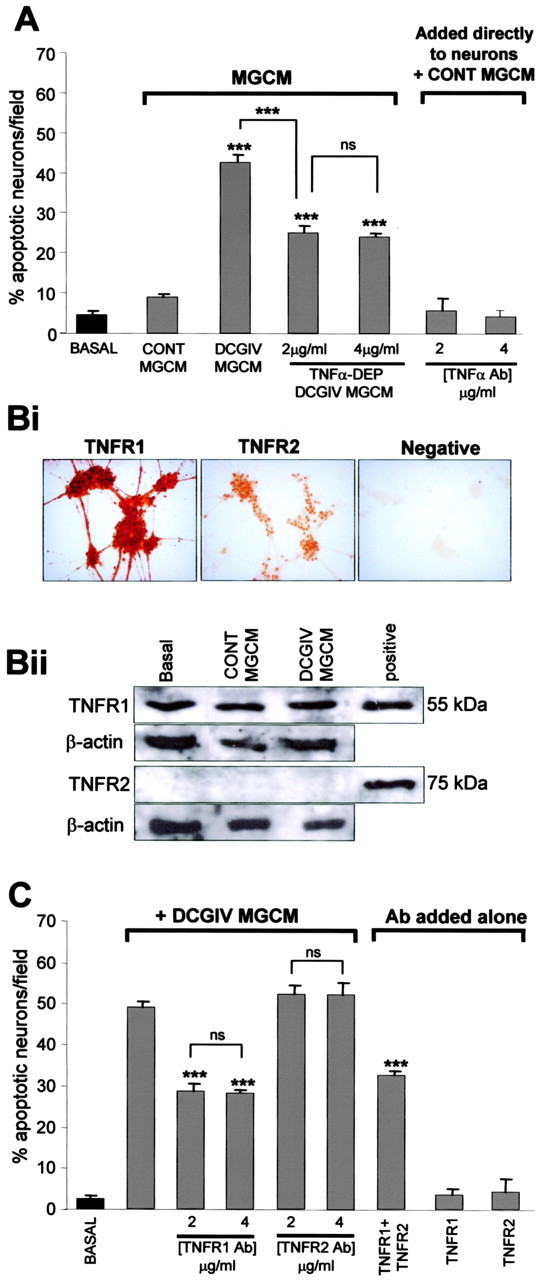
TNFα contributes to microglial mGlu2-induced neurotoxicity through stimulation of neuronal TNFR1. A, Cerebellar granule neurons were incubated with DCGIV-MGCM or DCGIV-MGCM that was immunodepleted of TNFα. Neuronal apoptosis was assessed with Hoechst 33342 staining 24 h after the addition of the MGCM. Immunodepleting DCGIV-MGCM of TNFα reduced the neurotoxicity compared with normal DCGIV-MGCM. The direct addition of the TNFα antibody at the concentrations used for immunodepletion (2 and 4 μg/ml) in the presence of control MGCM did not affect neuronal survival. Data are the mean ± SEM of at least four determinations. Bi, Immunolocalization of TNFR1 and TNFR2 on CGC neurons. Cells were fixed and stained with specific antibodies for either TNFR1 or TNFR2 (1:500). The negative control was treated in the same way, but the primary antibody was omitted. Bii, Western blots of neuronal cell lysates (45 μg of protein) from untreated neurons (basal) and those exposed to control MGCM or DCGIV-MGCM for 24 h. Blots were stained for TNFR1 or TNFR2 (1:1000) and reprobed for β-actin (1:500) to ensure equal protein loading. Positive, Commercial standard of TNFR1 or TNFR2 protein (1 μg/ml). C, Neuronal cultures were exposed to antibodies for TNFR1 or TNFR2 (2 or 4 μg/ml) for 30 min before the addition of DCGIV-MGCM. Blocking neuronal TNFR1 significantly reduced DCGIV-induced neuronal death at 24 h. The addition of the antibodies alone (4 μg/ml) had no effect on neuronal survival. Data are the mean ± SEM of at least four determinations. Significance values in both graphs are shown compared with DCGIV-MGCM, unless indicated otherwise: ***p < 0.0005; p > 0.05 was not significant (ns).
TNFα contributes to mGlu2-induced microglial neurotoxicity through stimulation of neuronal TNFR1
TNFα binds to the TNF receptors TNFR1 (p55) and TNFR2 (p75), both of which have distinct signaling pathways; TNFα signaling through TNFR1 can induce apoptosis, whereas signaling through TNFR2 promotes protection (Yang et al., 2002). There was a very strong expression of TNFR1 localized on cell bodies and neurites of CGC neurons, in contrast to the staining observed with TNFR2 (Fig. 4Bi). Western blotting of neuronal cell lysates revealed a strong band at 55 kDa, the expected molecular weight of TNFR1 in untreated neurons (Fig. 4Bii, Basal). TNFR1 expression was not altered when neuronal cultures were incubated with either control MGCM or DCGIV-MGCM for 24 h. No band at 75 kDa was detected when blots from CGC neurons were stained for TNFR2 in either basal- or MGCM-treated neurons, suggesting that the amount of TNFR2 protein expressed (as shown in Fig. 4Bi) is below the resolution for Western blotting.
Because TNFα released from microglia after mGlu2 stimulation was neurotoxic and CGC neurons express TNF receptors, we determined whether either of the TNF receptors were involved in neuronal death after microglial mGlu2 stimulation. Blocking neuronal TNFR1 with a specific TNFR1 antibody (2-4 μg/ml) before the addition of DCGIV-MGCM to the cultures significantly reduced DCGIV-MGCM-induced neuronal death (Fig. 4C). Inhibition of TNFR2 with a specific TNFR2 antibody did not modulate neuronal apoptosis after the addition of DCGIV-MGCM. In addition, blockade of TNFR1 and TNFR2 together did not significantly alter neuronal apoptosis compared with blocking TNFR1 alone. These data suggested that TNFR1 alone plays a significant role in neuronal apoptosis induced by DCGIV-MGCM. Inhibition of TNFR1 or TNFR2 in control untreated neurons did not effect neuronal apoptosis (Fig. 4C), indicating that activation of these receptors is not involved in the maintenance of neuronal survival in these cultures.
TNFα requires microglia for neurotoxicity
Although we found that TNFα contributed to neurotoxicity induced by DCGIV-MGCM (Fig. 4A), exposing neuronal cultures to TNFα at concentrations within and above the maximum amount measured in DCGIV-MGCM by ELISA did not induce neuronal apoptosis (Fig. 5A). Thus, the direct addition of rat recombinant TNFα alone at 0.1 and 0.25 ng/ml for 24 h did not increase neuronal death above that in controls (Fig. 5A). Furthermore, higher concentrations of TNFα (up to 100 ng/ml) did not induce neuronal apoptosis at 24 h or after up to 6 d (data not shown). These observations, which are in line with those from other groups (Barone et al., 1997; Ciesielski-Treska et al., 1998), suggested that TNFα by itself was unable to induce neuronal apoptosis.
Figure 5.
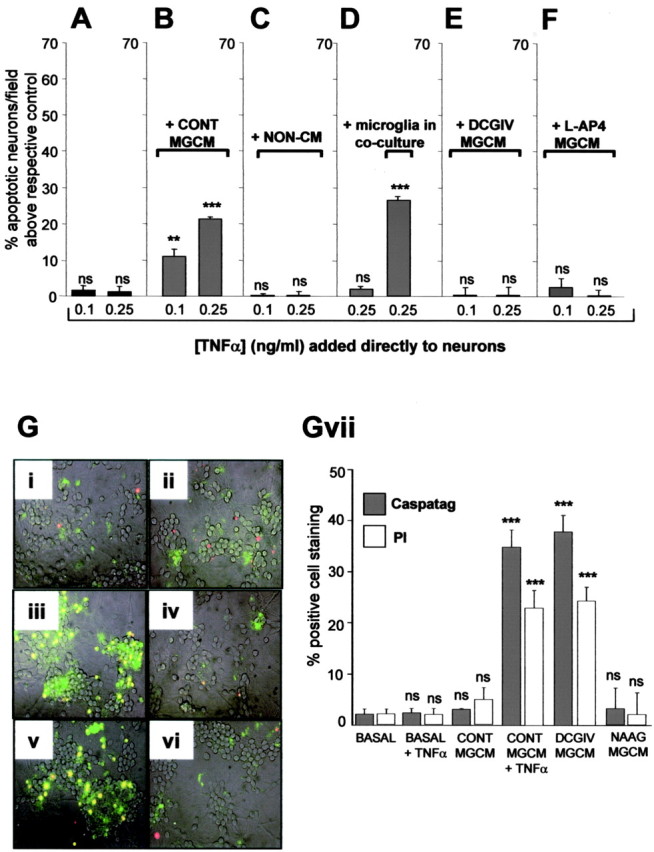
TNFα requires microglia to induce neurotoxicity. Cerebellar neuronal cultures were exposed directly to TNFα (0.1 or 0.25 ng/ml) alone (A) or together with control MGCM (B), nonconditioned medium (C), microglia in coculture (D), DCGIV-MGCM (E), or l-AP-4-MGCM (F). Neuronal apoptosis was assessed with Hoechst 33342 staining after 24 h. TNFα was only neurotoxic in the presence of MGCM or microglia in coculture. Data (mean ± SEM) are shown as the percentage of apoptotic neurons per field above respective control (untreated neurons; A, C, D), control MGCM (B), DCGIV-MGCM (E), or l-AP-4-MGCM (F). Significance values are shown compared with respective control: *p < 0.05; **p < 0.005; ***p < 0.0005; p > 0.05 is nonsignificant (ns). G, Caspase-3 activity (CaspaTag; green) and PI staining (red) were assessed in untreated neuronal cultures (i) and those exposed for 24 h to control MGCM (ii), DCGIV-MGCM (iii), 0.25 ng/ml TNFα alone (iv), 0.25 ng/ml TNFα with control MGCM (v), or NAAG-MGCM (vi). Cells were incubated with CaspaTag and PI for 30 min, and staining was viewed using a fluorescence microscope. Where CaspaTag staining and PI staining overlap, cells appear yellow. Gvii, The number of caspase-3- or PI-positive cells were counted and expressed as a percentage of the total cell number per field in at least three fields per coverslip from at least three separate coverslips. Data are expressed as mean ± SEM. Significance values are shown compared with respective CaspaTag or PI controls in CONT MGCM: *p < 0.05; **p < 0.005; ***p < 0.0005; p > 0.05 is nonsignificant (ns).
The addition of TNFα to CGC neurons, together with MGCM from control unstimulated microglia, induced a concentration-dependent increase in neuronal apoptosis (Fig. 5B). This effect was not attributable to a component of the medium used for microglial cell culture, because the addition of TNFα in the presence of nonconditioned medium (medium maintained in the incubator without microglial cells) did not induce neuronal apoptosis (Fig. 5C). In addition, TNFα (0.25 ng/ml) only became toxic to neurons cocultured with microglia (Fig. 5D). These findings hinted at the possibility that a cofactor present in MGCM was required for TNFα to become neurotoxic. To determine whether the TNFα present in DCGIV-MGCM was at its maximal concentration for neurotoxicity, we added DCGIV-MGCM to neurons with additional TNFα. Neither supplemented TNFα at 0.1 nor 0.25 ng/ml induced additional neuronal apoptosis above that induced with DCGIV-MGCM alone, suggesting that neurotoxicity is not limited by the availability of TNFα (Fig. 5E). Because we have shown that stimulation of group III mGlu receptors on microglia does not induce a neurotoxic phenotype and can be neuroprotective against different paradigms of microglial activation (Taylor et al., 2003), we tested whether activation of this mGlu receptor group on microglia modulated the neurotoxicity of TNFα added in the presence of control MGCM. Stimulation of microglial group III mGlu receptors with l-AP-4 (100 μm) completely prevented the neurotoxicity of TNFα (at 0.1 or 0.25 ng/ml) when added to the neurons in the presence of control MCGM (Fig. 5F).
Because activation of microglial mGlu2 induced neuronal apoptosis, as did TNFα in the presence of microglia or MGCM, we wanted to determine whether this involved the activation of caspase-3. Although TNFα induces apoptotic pathways involving the activation of caspase-3 in other cell types (MacEwan, 2002; Dempsey et al., 2003; Alikhani et al., 2004), exposing CGC neurons directly to 0.25 ng/ml TNFα did not induce caspase-3 activity above that seen in basal, nonstimulated neuronal cultures (Fig. 5Gi, iv). Although the addition of control MGCM did not significantly increase the level of caspase-3-expressing neurons (Fig. 5Gii), the addition of control MGCM together with 0.25 ng/ml TNFα significantly increased the number of caspase-3-positive neurons (Fig. 5Gv). This was also the case for neurons exposed to DCGIV-MGCM (Fig. 5Giii), whereas exposure of neurons to NAAG-MGCM failed to evoke an elevation of caspase-3-expressing neurons above that found with control MGCM (Fig. 5Gvi), again suggesting that stimulation of mGlu2 but not mGlu3 induces neurotoxicity. The direct addition of FasL (at 0.01 or 0.1 μg/ml) induced caspase-3 activation in these neurons (data not shown). The numbers of caspase-3-positive neurons in each condition (Fig. 5Gvii) was similar to the number of apoptotic neurons determined by Hoechst 33342 staining (Figs. 2A, 5A, B) but was slightly less than the number of dead cells identified by PI (Fig. 5Gvii), because this dye will report dead and late apoptotic cells.
FasL may act as a cofactor in TNFα-induced microglial neurotoxicity
Our findings above suggest that DCGIV-MGCM evokes neurotoxicity because it contains TNFα (Fig. 4A) but that TNFα cannot induce the neurotoxicity without the presence of some other factor released by microglia (Fig. 5B, D). We thus investigated whether this other factor could be FasL. Microglia have been shown to constitutively express and shed FasL (Badie et al., 2000; Terrazzino et al., 2002), and soluble FasL may contribute to neurotoxicity by promoting apoptosis in target cells (Griffith et al., 1995; Nagata, 1999). TNFα (0.25 ng/ml) was added to neuronal cultures together with either control MGCM or control MGCM that had been immunodepleted of FasL. Neuronal apoptosis induced by TNFα in the presence of FasL-depleted control MGCM was significantly less than in cultures exposed to TNFα with control MGCM (15.9 ± 1.9 and 32.3 ± 1.8, respectively; p < 0.0005) (Fig. 6A). This indicated that FasL was a cofactor in TNFα-induced microglial neurotoxicity. Other factors may also be involved because neuronal death induced by TNFα in the presence of FasL-depleted MGCM remained significantly higher than in cultures exposed to control MGCM alone. No further decrease in neuronal apoptosis was observed when the FasL antibody concentration used for immunodepletion was increased to 4 μg/ml (data not shown). The presence of FasL in MGCM from untreated microglia in itself did not exert a neurotoxic effect, because there was no significant difference in the levels of neuronal apoptosis after exposure to nondepleted control MGCM compared with FasL-depleted control MGCM (Fig. 6A). This suggests that either FasL is present in control MGCM below a toxic threshold or that the presence of a certain threshold level of TNFα is required for FasL toxicity. We find that FasL can be directly neurotoxic but requires a concentration of at least 0.01 μg/ml to induce neuronal apoptosis above that induced by control MGCM (Fig. 6B).
Figure 6.
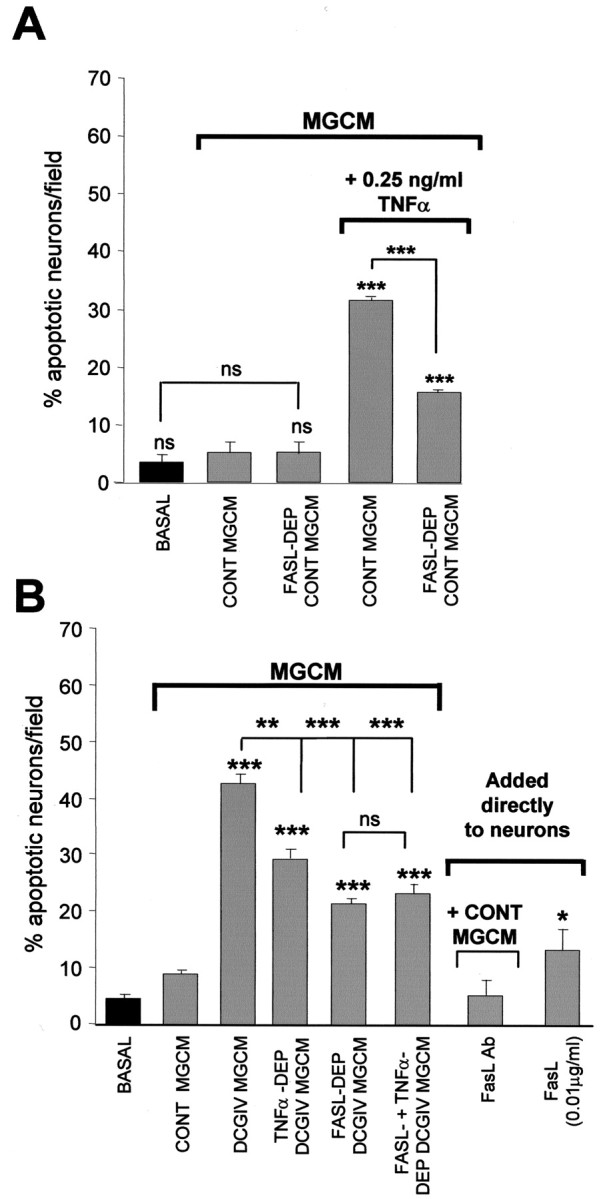
FasL may act as a cofactor for TNFα-induced microglial neurotoxicity. A, Neuronal cultures were incubated with 0.25 ng/ml TNFα together with control MGCM (CONT MGCM) or FasL-immunodepleted control MGCM (FASL-DEP CONT MGCM). After 24 h, the number of apoptotic neurons were counted after Hoechst staining. Immunodepletion of FasL in MGCM decreased TNFα toxicity seen with nondepleted MGCM. Data are mean ± SEM of at least four determinations. B, Incubating CGC neurons with FasL-immunodepleted DCGIV-MGCM for 24 h significantly reduced DCGIV-induced neurotoxicity, as did immunodepleting DCGIV-MGCM of TNFα. Direct application of FasL (0.1 μg/ml) increased the number of apoptotic neurons after 24 h, as determined by Hoechst staining. Data are mean ± SEM of at least four determinations. Significance values in both graphs are compared with control, unless indicated otherwise: *p < 0.05; **p < 0.005; ***p < 0.0005; p > 0.05 is nonsignificant (ns).
FasL contributes to mGlu2-induced microglial neurotoxicity
Because we found that FasL may be a cofactor in TNFα-induced neurotoxicity (Fig. 6A) and that TNFα contributes to mGlu2-induced microglial toxicity (Fig. 4A), we next investigated the role FasL in DCGIV-MGCM-induced neurotoxicity. Exposing neuronal cultures to DCGIV-MGCM that was immunodepleted of FasL significantly reduced neuronal death after 24 h compared with DCGIV-MGCM (21.5 ± 1.9 and 45.5 ± 2.4% apoptotic neurons per field; p < 0.0005) (Fig. 6B). No further reduction in neuronal cell death was observed when neurons were exposed to DCGIV-MGCM that had been immunodepleted of both FasL and TNFα. The direct addition of FasL antibody with control MGCM did not alter neuronal death above that with control MGCM (Fig. 6B).
Thus, data from these experiments imply that control unstimulated cultures of microglia released FasL (Fig. 6) but that this is not neurotoxic because unstimulated microglia do not release significant TNFα (Fig. 3C). TNFα released from microglia after mGlu2 stimulation significantly contributed to microglial neurotoxicity by acting in concert with this FasL released from the microglia (Fig. 6B). Therefore, we determined directly whether microglial cells expressed and/or released FasL and whether this was altered by mGlu2 stimulation. Immunolocalization confirmed that both control and DCGIV-treated primary microglia expressed FasL (Fig. 7Ai) as did microglia activated with LPS (1 μg/ml) for 24 h. Western blotting of microglial cell lysates confirmed that control microglia expressed FasL protein, with a strong band observed at 38 kDa, the expected molecular weight of FasL (Fig. 7Aii). In addition, a strong band with a molecular weight of ∼100 kDa was evident in all cell lysates. This higher band may represent an aggregated form of FasL (Schneider et al., 1988). Exposing microglia to DCGIV or LPS reduced FasL expression, with a decrease observed in both the 38 and 100 kDa FasL bands compared with β-actin expression (Fig. 7Aiv).
Figure 7.
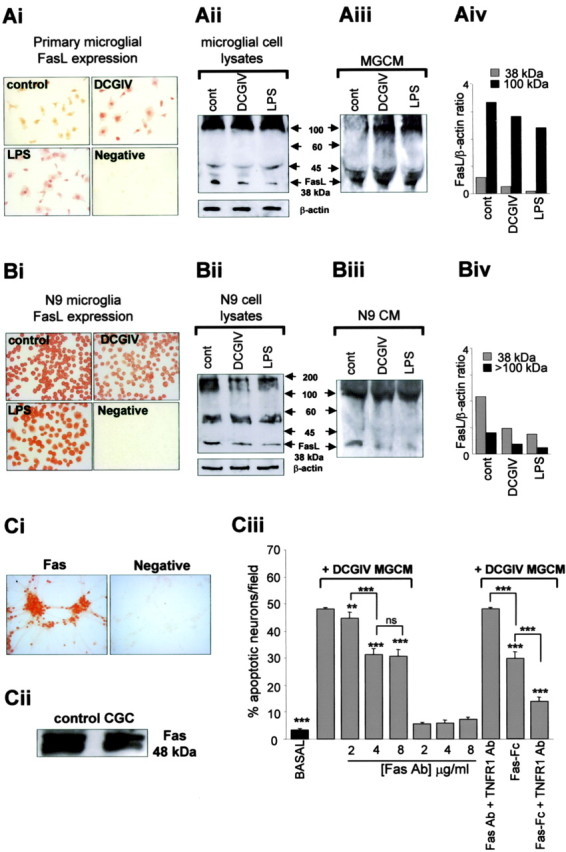
Microglia express and release FasL, which induces Fas-mediated neuronal death. Ai, Immunolocalization of FasL on unstimulated primary microglia (control) and microglia exposed to DCGIV (500 nm) or LPS (1 μg/ml) for 24 h. Cells were fixed and stained with a specific antibody to FasL (1:500). Primary antibody was omitted in the negative control. Western blot immunoblotted with anti-FasL antibody (1:500) of microglial cell lysates (60 μg of protein) (Aii) or MGCM (FasL immunocomplex from immunodepleted medium) (Aiii) from unstimulated primary microglia (cont) and microglia treated with DCGIV (500 nm) or LPS (1 μg/ml) for 24 h is shown. The blot for microglial cell lysates was reprobed for β-actin to ensure equal loading. Aiv, Densitometry data expressed as a ratio of FasL to β-actin from microglial cell lysate blot (Ai) showed a reduction in the 38 and <100 kDa FasL bands after activation with DCGIV or LPS. Bi, Immunolocalization of FasL on unstimulated N9 microglia (control) and N9 microglia exposed to DCGIV (500 nm) or LPS (1 μg/ml) for 24 h. Cells were fixed and stained with anti-FasL (1:500). Negative control was treated in the same way, but the primary antibody was omitted. Western blot immunoblotted with anti-FasL antibody (1:500) of N9 microglial cell lysates (60 μg protein) (Bii) or N9 conditioned medium (CM) (FasL immunocomplex from immunodepleted medium) (Biii) from unstimulated N9 microglia (cont) and N9 microglia treated with DCGIV (500 nm) or LPS (1 μg/ml) for 24 h is shown. The blot for microglial cell lysates was reprobed for β-actin. Biv, Densitometry data of FasL to β-actin ratio from N9 microglial cell lysate blot (Bi) showed a reduction in the 38 and <100 kDa FasL bands after activation with DCGIV or LPS. Ci, Immunolocalization of Fas on CGC neurons. Cells were fixed and stained with a specific antibody to Fas (1:500). The primary antibody was omitted in the negative control. Cii, Western blot of neuronal cellular protein. Cell lysates (45 μg of protein) were resolved by 10% SDS-PAGE and immunoblotted with anti-Fas antibody (1:500). Ciii, Neuronal cultures were incubated with an antibody to Fas (2-8 μg/ml), Fas antibody (Ab) (4 μg/ml) plus TNFR1 Ab (4 μg/ml), Fas-Fc (5 μg/ml), or Fas-Fc (5 μg/ml) plus TNFR1 Ab (4 μg/ml) 30 min before the addition of DCGIV-MGCM. Neuronal apoptosis was assessed using Hoechst 33342 staining. Data are expressed the percentage of apoptotic cells per field mean ± SEM of at least four determinations. Significance values are shown compared with DCGIV-MGCM-treated neuronal cultures, unless indicated otherwise: **p < 0.005; ***p < 0.0005.
To assess whether primary microglia released FasL into the culture medium, FasL was immunoprecipitated from the different MGCM, and the immunocomplexes were subjected to Western blotting. A FasL band at 38 kDa was present in control, DCGIV, and LPS-MGCM (Fig. 7Aiii). There was a strong band at ∼100 kDa in DCGIV and LPS-MGCM samples but not in control MGCM. These findings suggest that control microglia express FasL (as a 38 and 100 kDa protein) and release FasL (predominantly as a 38 kDa protein), although the cells are not in an activated state as determined by ED1 staining, mitochondrial polarization, iNOS expression, and apoptosis (Fig. 1). In contrast, after activation with DCGIV or LPS, microglia released FasL as a 100 kDa protein and as a 38 kDa protein (Fig. 7Aiii).
We obtained similar results for FasL expression and release in the murine N9 microglial cell line. Positive FasL staining was observed in unstimulated N9 microglia with decreasing intensity when the cells were exposed to DCGIV or LPS for 24 h (Fig. 7Bi). FasL protein expression (38 kDa band) in N9 cell lysates was decreased in DCGIV- and LPS-treated cells (Fig. 7Bii, iv). N9 microglia also expressed FasL as a higher molecular weight protein, with a strong band closer to 200 kDa rather than at 100 kDa as observed in primary microglia. As with primary microglia, N9 microglia also released FasL as a 38 and 100 kDa protein (Fig. 7Biii).
Microglial FasL induced neuronal death by binding to Fas
Endogenous FasL initiates apoptosis in target cells by binding to its receptor Fas (Griffith et al., 1995). Because we have shown that FasL in DCGIV-MGCM is a factor for neurotoxicity, we determined whether activation of Fas on CGC neurons was involved. Immunolocalization confirmed that control cultures of CGC neurons expressed Fas, in line with previous findings (Castiglione et al., 2004), and that the expression was localized to the neuronal cell bodies (Fig. 7Ci). In addition, Western blotting of neuronal cell lysates showed a strong expression of Fas protein (band at 48 kDa) in these cultures (Fig. 7Cii). Blocking neuronal Fas with a specific antibody (2 μg/ml) before the addition of DCGIV-MGCM for 24 h significantly reduced neuronal apoptosis compared with DCGIV-MGCM alone (Fig. 7Ciii). Increasing the antibody concentration further decreased neuronal apoptosis with a maximal reduction seen with an antibody concentration of 4 μg/ml (to 31.0 ± 1.4% apoptotic neurons/field compared with 45.3 ± 0.6% with DCGIV-MGCM; p < 0.0005). Surprisingly, blocking neuronal Fas and TNFR1 completely prevented any protection from apoptosis induced by DCGIV-MGCM. However, use of Fas-Fc, which inhibits the activity of soluble FasL, significantly reduced DCGIV-MGCM-induced neuronal apoptosis; this was further reduced in the presence of TNFR1 antibody (Fig. 7Ciii).
Discussion
Here, we investigated the mechanisms by which activation of mGlu receptors on microglia results in neurotoxicity. Stimulation of microglial group II mGlu receptors, specifically mGlu2, induced microglial TNFα release, which contributed to neurotoxicity after microglial mGlu2 stimulation. Microglial-derived TNFα was only neurotoxic in the presence of microglial-derived FasL, and neurotoxicity did not involve microglial iNOS because it was not expressed. Stimulation of microglial mGlu2 lead to neuronal caspase-3 activation and apoptosis. Stimulation of group I or group III microglial mGlu receptors did not evoke TNFα release or microglial-driven neurotoxicity.
We have shown previously that stimulation of group II mGlu receptors (mGlu2 and mGlu3) on microglia with the specific agonist DCGIV (Brabet et al., 1998) induces microglial activation and neurotoxicity (Taylor et al., 2002). Here, the effects of DCGIV are attributable to activation of microglial mGlu2, because stimulation of mGlu3 with the specific mGlu3 agonist NAAG (Wroblewska et al., 1997) did not induce microglial TNFα release or neurotoxicity. The consequences of microglial mGlu2 stimulation were prevented by MCCG, a selective antagonist of group II mGlu receptors (Jane et al., 1994; Knöpfel et al., 1995). We have shown that 0.5-1 μm DCGIV induced microglial neurotoxicity. Cloned group II mGlu receptors have EC50 values for glutamate of 12 μm (mGlu2) and 4 μm (mGlu3) (Pin and Duvoisin, 1995), and basal levels of glutamate can stimulate group II mGlu receptors on pyramidal neurons in the rat sensorimotor cortex (Bandrowski et al., 2003). We do not currently know what levels of glutamate are required to activate microglial group II mGlu receptors, but if these receptors are similar to the cloned receptors, it is likely that any sustained elevation in extracellular glutamate [from the reported baseline level in interstitial glutamate of 0.5-2 μm (Meldrum, 2000; Bandrowski et al., 2003) to the level of 2-5 μm, which can trigger excitotoxic damage in neurons (Lipton and Rosenberg, 1994)] may well activate microglial group II mGlu receptors in vivo. Sustained elevation of glutamate and thus sustained activation of microglia via group II mGlu receptors will be further influenced by any failure in the clearance of intercellular glutamate by compromised neurons and glia.
Stimulation of microglial group I mGlu receptors did not induce microglial activation, whereas neurotoxicity was attributable to a direct effect of t-ADA rather than one mediated by microglia. Furthermore, stimulation of microglial group I mGlu receptors did not induce iNOS expression or TNFα release, suggesting any microglial toxicity is unlikely to involve iNOS or TNFα. Activation of group III mGlu receptors (mGlu4, mGlu6, and mGlu8) on microglia with the specific agonist l-AP-4 (Okamoto et al., 1994; Schoepp et al., 1999) did not induce iNOS expression, TNFα release, or transform the microglia to a neurotoxic phenotype, the latter in line with previous findings (Taylor et al., 2003).
We found that microglia expressed TNFα as the soluble 17 kDa form. Although both cell surface and soluble TNFα are biologically active, released soluble TNFα appears to be primarily detrimental (Schneider et al., 1998). In accordance with this, control unstimulated microglia, group III stimulated microglia, and mGlu3-stimulated microglia, which were not toxic to neurons, expressed TNFα, (as determined by immunolocalization and Western blotting) but released low amounts of this cytokine (<40 pg/ml measured by ELISA of MGCM). Stimulation of microglial mGlu2 triggered the release of significant amounts of soluble TNFα, and depletion of TNFα from DCGIV-MGCM afforded significant neuroprotection. The toxicity of TNFα to the CGC neurons is likely a result of its interaction with the TNFα receptor TNFR1 (p55), rather than TNFR2 (p75) (Yang et al., 2002), because antibody block of TNFR2 on the CGC neurons did not prevent the neurotoxicity of DCGIV-MGCM, whereas block of TNFR1 did. Furthermore, the expression of TNFR2 on these cells was very low compared with that of TNFR1. These findings are in line with data demonstrating the role of these two different TNF receptors in triggering distinct signaling pathways. Thus, TNFα signaling through TNFR1 induces apoptosis, whereas signaling through TNFR2 promotes protection (Yang et al., 2002; Micheau and Tschopp, 2003). Furthermore, TNFR1 is the primary receptor for soluble TNFα, whereas TNFR2 is the main receptor for membrane TNFα (Grell et al., 1995; Varfolomeev and Ashkenazi, 2004).
Although TNFα released after mGlu2 stimulation contributed to neuronal death after exposure to DCGIV-MGCM, exposure of neurons to TNFα at concentrations similar to those found by ELISA of DCGIV-MGCM was not directly neurotoxic, in line with previous findings (Chao et al., 1995; Barone et al., 1997; Ciesielski-Treska et al., 1998). However, TNFα became neurotoxic when added in the presence of control MGCM (which did not contain significant amounts of TNFα) or control microglia (which did not release significant amounts of TNFα). This suggests that microglia may also release cofactors that work with TNFα to induce neurotoxicity. Furthermore, the ability of TNFα to become neurotoxic may also reflect changes in the ratio of shed soluble TNFR to TNFα, which may explain why the basal levels of TNFα secreted by the resting-state microglia are not neurotoxic but added TNFα is. It has been suggested that microglia can shed TNFR2 and may thus control the bioavailability of TNFα (Hanisch et al., 2001).
Stimulation of group III mGlu receptors on microglia is not neurotoxic and protects neurons against microglial neurotoxicity (Taylor et al., 2003). Here, conditioned medium from microglia treated with a specific agonist of group III was neuroprotective when TNFα was added to the neurons at concentrations that induced apoptosis when it was added with control MGCM. This suggests that stimulation of group III mGlu receptors on microglia may trigger the release of a neuroprotective factor or reduce toxin release.
To determine the nature of the cofactor acting with TNFα to induce the neurotoxicity of DCGIV-MGCM, we investigated the role of FasL. Microglia constitutively express FasL at low levels in normal human white matter (Dowling et al., 1996; Bechmann et al., 1999), and microglial activation results in increased expression and release of FasL (Spanaus et al., 1998; Badie et al., 2000; Niinobu et al., 2000; Ciesielski-Treska et al., 2001; Terrazzino et al., 2002). Accordingly, the N9 microglial cell line and primary cultured microglia were found to express FasL. Immunodepleting DCGIV-MGCM of FasL significantly reduced neuronal apoptosis, suggesting, as in other cells (Nagata 1999), that FasL was able to trigger apoptosis in CGC neurons. Furthermore, immunodepleting control MGCM of FasL significantly reduced the neurotoxicity of added TNFα, confirming that FasL acts in concert with TNFα to induce neurotoxicity. These data suggest that microglia shed FasL in culture. We found that control microglia released 38 kDa FasL, which was enhanced (with a concomitant fall in the cellular expression) after stimulation with DCGIV or LPS, together with a prominent release of FasL at 100 kDa. This is likely to be an aggregated form of FasL (molecular weight, >100 kDa), which is more efficient at inducing apoptosis in Fasbearing cells compared with unaggregated FasL (molecular weight, ∼38 kDa) (Schneider et al., 1988; Tanaka et al., 1995; Spanaus et al., 1998; Huang et al., 1999). In accordance with this, FasL released from control microglia did not induce neuronal death (in the absence of additional TNFα), most likely because it was not in the aggregated form seen to be released after stimulation with DCGIV or LPS. Alternatively, it is possible that the amount of FasL released in control MGCM is below the threshold to trigger apoptosis alone, because we have shown that the addition of soluble FasL can trigger neuronal apoptosis.
It is likely that TNFα and FasL act here through convergent pathways to trigger neuronal apoptosis, because depletion of DCGIV-MGCM of both FasL and TNFα did not reduce apoptosis below that with medium depleted of only one of these substances. Furthermore, neuronal apoptosis triggered by the direct addition of FasL was not further increased by the presence of TNFα. The point of convergence of these pathways is certainly at, or above, the level of caspase-3 activation, because DCGIV-MGCM, control MGCM with added TNFα, or the direct addition of FasL all induced neuronal caspase-3 activation. Activation of Fas triggers apoptosis by signaling via caspase-8, leading to caspase-3-mediated apoptosis (Houston and O'Connell, 2004), and activation of TNFR1 can trigger caspase-3-mediated apoptosis (Dempsey et al., 2003). We also showed previously that CGC neurons exposed to conditioned medium from activated microglia undergo caspase-3-mediated apoptosis (Kingham et al., 1999).
In conclusion, the present results provide evidence that stimulation of microglial mGlu2 receptors can evoke the release of TNFα, which in concert with microglial released FasL can trigger neuronal apoptosis. Although TNFα may be released from microglia via stimulation of other pathways (Hanisch 2002), its implication in the neuroinflammatory cascades underlying many neurodegenerative diseases makes it an important target for modulation. Bezzi et al. (2001) report that TNFα can trigger the release of glutamate, which is directly neurotoxic. However, this glutamate may also lead to additional microglial activation via stimulation of mGlu2 and additional TNFα release. Thus, modulation of microglial mGlu2 receptors may provide therapeutic benefit.
Footnotes
This work was supported by the Wellcome Trust UK.
Correspondence should be addressed to Dr. J. M. Pocock, Cell Signalling Laboratory, Department of Neuroinflammation, Institute of Neurology, University College London, 1 Wakefield Street, London WC1N 1PJ, UK. E-mail: j.pocock@ion.ucl.ac.uk.
Copyright © 2005 Society for Neuroscience 0270-6474/05/252952-13$15.00/0
References
- Alikhani M, Alikhani Z, Raptis M, Graves DT (2004) TNFα in vivo stimulates apoptosis in fibroblasts through caspase-8 activation and modulates the expression of pro-apoptotic genes. J Cell Physiol 201: 341-348. [DOI] [PubMed] [Google Scholar]
- Badie B, Schartner J, Vorpahl J, Preston K (2000) Interferon-gamma induces apoptosis and augments the expression of Fas and Fas ligand by microglia in vitro. Exp Neurol 162: 290-296. [DOI] [PubMed] [Google Scholar]
- Bandrowski AE, Huguenard JR, Prince DA (2003) Baseline glutamate levels affect group I and II mGluRs in layer V pyramidal neurons of rat sensorimotor cortex. J Neurophysiol 1308-1316. [DOI] [PMC free article] [PubMed]
- Barone FC, Arvin B, White RF, Miller A, Webb CL, Willette RN, Lysko PG, Feuerstein GZ (1997) Tumor necrosis factor-alpha. A mediator of focal ischemic brain injury. Stroke 28: 1233-1244. [DOI] [PubMed] [Google Scholar]
- Bechmann I, Mor G, Nilsen J, Eliza M, Nitsch R, Naftolin F (1999) FasL (CD95L, Apo1L) is expressed in the normal rat and human brain: evidence for the existence of an immunological brain barrier. Glia 27: 62-74. [DOI] [PubMed] [Google Scholar]
- Bezzi P, Domercq M, Brambilla L, Galli R, Schols D, De Clercq E, Vescovi A, Bagetta G, Kollias G, Meldolesi J, Volterra A (2001) CXCR4-activated astrocyte glutamate release via TNFalpha: amplification by microglia triggers neurotoxicity. Nat Neurosci 4: 702-710. [DOI] [PubMed] [Google Scholar]
- Biber K, Laurie DJ, Berthele A, Sommer B, Tolle TR, Gebicke-Harter PJ, van Calker D, Boddeke HW (1999) Expression and signaling of group I metabotropic glutamate receptors in astrocytes and microglia. J Neurochem 72: 1671-1680. [DOI] [PubMed] [Google Scholar]
- Brabet I, Parmentier M-L, De Colle C, Blockaert J, Acher F, Pin J-P (1998) Comparative effect of L-CCG-I, DCG-IV and γ-carbocy-l-glutamate on all cloned metabotropic glutamate receptor subtypes. Neuropharmacology 37: 1043-1051. [DOI] [PubMed] [Google Scholar]
- Cagnin A, Brooks DJ, Kennedy AM, Gunn RN, Myers R, Turkheimer FE (2001) In-vivo measurement of activated microglia in dementia. Lancet 358: 461-467. [DOI] [PubMed] [Google Scholar]
- Castiglione M, Spinsanti P, Iacovelli L, Lenti L, Martini F, Gradini R, Di Giorgi Gerevini V, Caricasole A, Caruso A, De Maria R, Nicoletti F, Melchiorri D (2004) Activation of Fas receptor is required for the increased formation of the disialoganglioside GD3 in cultured cerebellar granule cells committed to apoptotic death. Neuroscience 126: 889-898. [DOI] [PubMed] [Google Scholar]
- Chao CC, Hu S, Ehrlich L, Peterson PK (1995) Interleukin-1 and tumor necrosis factor-α synergistically mediate neurotoxicity: involvement of nitric oxide and of N-methyl-d-aspartate receptors. Brain Behav Immunol 9: 355-365. [DOI] [PubMed] [Google Scholar]
- Ciesielski-Treska J, Ulrich G, Taupenot L, Chasserot-Golaz S, Corti A, Aunis D, Bader M-F (1998) Chromogranin A induces a neurotoxic phenotype in brain microglial cells. J Biol Chem 273: 14339-14346. [DOI] [PubMed] [Google Scholar]
- Ciesielski-Treska J, Ulrich G, Chasserot-Golaz S, Zwiller J, Revel M-O, Aunis D, Bader M-F (2001) Mechanisms underlying neuronal death induced by chromogranin A-activated microglia. J Biol Chem 276: 13113-13120. [DOI] [PubMed] [Google Scholar]
- Corradin SB, Mauel J, Donimi SD, Quattrocchi E, Ricciardi-Castagnoli P (1993) Inducible nitric oxide synthase activity of cloned murine microglial cells. Glia 7: 255-262. [DOI] [PubMed] [Google Scholar]
- Dempsey PW, Doyle SE, He JQ, Chen G (2003) The signaling adaptors and pathways activated by TNF superfamily. Cytokine Growth Factor Rev 14: 193-209. [DOI] [PubMed] [Google Scholar]
- Dowling P, Shang G, Raval S, Menonna J, Cook S, Husar W (1996) Involvement of the CD95 (APO-1/Fas) receptor/ligand system in multiple sclerosis brain. J Exp Med 184: 1513-1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Souza SD, Bonetti B, Balasingam V, Cashman NR, Barker PA, Troutt AB, Raine CS, Antel JP (1996) Multiple sclerosis: Fas signaling in oligodendrocyte cell death. J Exp Med 184: 2361-2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans GJO, Pocock JM (1999) Modulation of neurotransmitter release by dihydropyridine-sensitive calcium channels involves tyrosine phosphorylation. Eur J Neurosci 11: 279-292. [DOI] [PubMed] [Google Scholar]
- Felderhoff-Mueser U, Taylor DL, Greenwood K, Kozma M, Stibenz D, Joashi JC, Edwards AD, Mehmet H (2000) Fas/CD95/APO-1 can function as a death receptor for neuronal cells in vitro and in vivo and is upregulated following cerebral hypoxic-ischemic injury to the developing rat brain. Brain Pathol 10: 17-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frigerio S, Silei V, Ciusani E, Massa G, Laura GM, Salmaggi A (2000) Modulation of Fas-ligand (Fas-L) on human microglial cells: an in vitro study. J Neuroimmunol 105: 109-114. [DOI] [PubMed] [Google Scholar]
- Gebicke-Haerter PJ (2001) Microglia in neurodegeneration: molecular aspects. Mol Res Tech 54: 47-58. [DOI] [PubMed] [Google Scholar]
- Grell M, Douni E, Wajant H, Lohden M Clauss M, Maxeiner B, Georgopoulos S, Less W, Kollias G, Pfizenmaier K (1995) The transmembrane from of tumor necrosis factor is the prime activating ligand of the 80 kDa tumor necrosis factor receptor. Cell 83: 793-802. [DOI] [PubMed] [Google Scholar]
- Griffith TS, Brunner T, Fletcher SM, Green DR, Ferguson TA (1995) Fas ligand-induced apoptosis as a mechanism of immune privilege. Science 270: 1189-1192. [DOI] [PubMed] [Google Scholar]
- Hanisch U-W (2002) Microglia as a source and target of cytokines. Glia 40: 140-155. [DOI] [PubMed] [Google Scholar]
- Hanisch UK, Prinz M, Angstwurm K, Hausler KG, Kann O, Kettenmann H, Weber JR (2001) The protein tyrosine kinase inhibitor AG126 prevents the massive microglial cytokine induction by pneumococcal cell walls. Eur J Immunol 31: 2104-2115. [DOI] [PubMed] [Google Scholar]
- Hess SD, Pasienczny R, Rao SP, Jachec C, Varney MV, Johnson EC (1999) Activity of N-acectylaspartylglutamate at human recombinant glutamate receptors. Soc Neurosci Abstr 25: 975. [Google Scholar]
- Houston A, O'Connell J (2004) The Fas signalling pathway and its role in the pathogenesis of cancer. Curr Opin Pharmacol 4: 321-326. [DOI] [PubMed] [Google Scholar]
- Huang DC, Hahne M, Schroeter M, Frei K, Fontana A, Villunger A, Newton K, Tschopp J, Strasser A (1999) Activation of Fas by FasL induced aopotosis by a mechanism that cannot be blocked by Bcl-2 or Bclx (L). Proc Natl Acad Sci USA 96: 14871-14876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jane DE, Hones PL, Pook PC, Tse HW, Watkins JC (1994) Actions of two new sub-type selective metabotropic glutamate receptor antagonists in the neonatal rat spinal cord. Br J Pharmacol 112: 809-816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim W-K, Hwang S-Y, Oh E-S, Piao HZ, Kim K-W, Han I-O (2004) TGF-β1 represses activation and resultant death of microglia via inhibition of phosphatidylinositol 3-kinase activity. J Immunol 172: 7015-7023. [DOI] [PubMed] [Google Scholar]
- Kingham PJ, Pocock JM (2000) Microglial apoptosis induced by chromogranin A is mediated by mitochondrial depolarization and the permeability transition but not by cytochrome c release. J Neurochem 74: 1452-1462. [DOI] [PubMed] [Google Scholar]
- Kingham PJ, Pocock JM (2001) Microglial secreted cathepsin B induces neuronal apoptosis. J Neurochem 76: 1475-1484. [DOI] [PubMed] [Google Scholar]
- Kingham PJ, Cuzner ML, Pocock JM (1999) Apoptotic pathways mobilized in microglia and neurones as a consequence of chromogranin A-induced microglial activation. J Neurochem 73: 538-547. [DOI] [PubMed] [Google Scholar]
- Klein J, Reymann KG, Riedel (1997) Activation of phospholipases C and D by the novel metabotropic glutamate receptor agonist t-ADA. Neuropharmacology 36: 261-263. [DOI] [PubMed] [Google Scholar]
- Knöpfol T, Lukic S, Leonardt T, Flor PJ, Kuhn R, Gasparini F (1995) Pharmacological characterization of MCCG and MAP4 at mGluR1b, mGluR2 and mGluR4a human metabotropic glutamate receptor subtypes. Neuropharmacology 34: 1099-1102. [DOI] [PubMed] [Google Scholar]
- Lee SC, LiuW, Dickson DW, Brosnan CF, German JW (1993) Cytokine production by human fetal microglia and astrocytes. Differential induction by lipopolysaccharide and IL-1beta. J Immunol 150: 2659-2667. [PubMed] [Google Scholar]
- Lipton SA, Rosenberg PA (1994) Excitatory amino acids as a final common pathway for neurologic disorders. N Engl J Med 330: 613-622. [DOI] [PubMed] [Google Scholar]
- Liu B, Hong J-S (2003) Role of microglia in inflammation-mediated neurodegenerative diseases; mechanisms and strategies for therapeutic intervention. J Pharmacol Exp Ther 304: 1-7. [DOI] [PubMed] [Google Scholar]
- MacEwan DJ (2002) TNF receptor subtype signalling: differences and cellular consequences. Cell Signal 14: 477-492. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Barger SW, Furukawa K, Bruce AJ, Wyss-Coray T, Mark RJ, Mucke L (1997) Cellular signaling roles of TGFβ, TNFα and βAPP in brain injury responses and Alzheimer's disease. Brain Res Rev 23: 47-61. [DOI] [PubMed] [Google Scholar]
- Meldrum BS (2000) Glutamate as a neurotransmitter in the brain: review of physiology and pathology. J Nutr 130: 1007S-1015S. [DOI] [PubMed] [Google Scholar]
- Micheau O, Tschopp J (2003) Induction of TNF receptor I-mediated apoptosis via two sequential signalling complexes. Cell 114: 181-190. [DOI] [PubMed] [Google Scholar]
- Morgan SC, Taylor DL, Pocock JM (2004) Microglia release activators of neuronal proliferation mediated by activation of mitogen-activated protein kinase, phosphatidylinositol-3-kinase/Akt and delta-Notch signalling cascades. J Neurochem 90: 89-101. [DOI] [PubMed] [Google Scholar]
- Nagata S (1999) Fas ligand-induced apoptosis. Annu Rev Genet 33: 29-55. [DOI] [PubMed] [Google Scholar]
- Niinobu T, Fukuo K, Yasuda O, Tsubakimoto M, Mogi M, Nishimaki H, Morimoto S, Ogiharrra T (2000) Negative feedback regulation of activated macrophages via Fas-mediated apoptosis. Am J Physiol Cell Physiol 279: C504-C509. [DOI] [PubMed] [Google Scholar]
- Nishimura T, Akiyama H, Yonehara S, Kondo H, Ikeda K, Kato M, Iseki E, Kosaka K (1995) Fas antigen expression in brains of patients with Alzheimer-type dementia. Brain Res 695: 137-145. [DOI] [PubMed] [Google Scholar]
- Okamoto N, Hori S, Akazawa C, Hayashi Y, Shigemoto R, Mizuno N, Nakanishi S (1994) Molecular characterization of a new metabotropic glutamate receptor mGluR7 coupled to inhibitory cyclic AMP signal transduction. J Biol Chem 269: 1231-1236. [PubMed] [Google Scholar]
- Piani D, Frei K, Do KQ, Cuenod M, Fontana A (1991) Murine brain macrophages induced NMDA receptor mediated neurotoxicity in vitro by secreting glutamate. Neurosci Lett 133: 159-162. [DOI] [PubMed] [Google Scholar]
- Pin J-P, Duvoisin R (1995) The metabotropic glutamate receptors: structure and functions. Neuropharmacology 34: 1-26. [DOI] [PubMed] [Google Scholar]
- Salvioli S, Ardizzoni A, Franceschi C, Cossarizza A (1997) JC-1, but not DiOC6(3) or rhodamine 123, is a reliable fluorescent probe to assess ΔΨm changes in intact cells: implications for studies on mitochondrial functionality during apoptosis. FEBS Lett 411: 77-82. [DOI] [PubMed] [Google Scholar]
- Schneider P, Holler N, Bodmer J-L, Hahne M, Frei K, Fontana A, Tschopp J (1988) Conversion of membrane-bound Fas (CD95) ligand to its soluble form is associated with downregulation of its proapototic activity and loss of liver toxicity. J Exp Med 187: 1205-1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoepp DD, Jane DE, Monn JA (1999) Pharmacological agents acting at subtypes of metabotropic glutamate receptors. Neuropharmacology 38: 1431-1476. [DOI] [PubMed] [Google Scholar]
- Schramm M, Eimerl S, Costa E (1990) Serum and depolarising agents cause acute neurotoxicity in cultured cerebellar granule cells: role of glutamate receptors responsive to N-methyl-d-aspartate. Proc Natl Acad Sci USA 87: 1193-1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spanaus KS, Schlapbach R, Fontana A (1998) TNF-α and IFN-γ render microglia sensitive to Fas ligand-induced apoptosis by induction of Fas expression and down-regulation of Bcl-2 and Bcl-xl. Eur J Immunol 28: 4398-4408. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Suda T, Takahashi T, Nagata S (1995) Expression of the functional soluble form of human fas ligand in activated lymphocytes. EMBO J 14: 1129-1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor DL, Diemel LT, Cuzner ML, Pocock JM (2002) Activation of group II metabotropic glutamate receptors underlies microglial reactivity and neurotoxicity following stimulation with peptides upregulated in Alzheimer's disease. J Neurochem 82: 1179-1191. [DOI] [PubMed] [Google Scholar]
- Taylor DL, Diemel LT, Pocock JM (2003) Activation of microglial group III metabotropic glutamate receptors protects neurons against microglial neurotoxicity. J Neurosci 23: 2150-2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terrazzino S, Bauleo A, Baldan A, Leon A (2002) Peripheral LPS administrations up-regulate Fas and FasL on brain microglial cells; a brain protective or pathogenic event? J Immunol 124: 45-53. [DOI] [PubMed] [Google Scholar]
- Varfolomeev EE, Ashkenazi A (2004) Tumor necrosis factor: an apoptosis JuNKie? Cell 116: 491-497. [DOI] [PubMed] [Google Scholar]
- Wang W, Ping J, Dow KE (2003) Corticotropin-releasing hormone induces proliferation and TNFα release in cultured rat microglia via MAP kinase signalling pathways. J Neurochem 84: 189-195. [DOI] [PubMed] [Google Scholar]
- Weydt PY, Yuen EC, Ransom BR, Möller T (2004) Increased cytotoxic potential of microglia from ALS-transgenic mice. Glia 48: 179-182. [DOI] [PubMed] [Google Scholar]
- Wroblewska B, Wroblewski JT, Saab O, Neal JH (1993) N-acetylaspartyl-glutamate inhibits forskolin-stimulated cyclic AMP levels via a metabotropic glutamate receptor in cultured cerebellar granule cells. J Neurochem 61: 943-948. [DOI] [PubMed] [Google Scholar]
- Wroblewska B, Wroblewski JT, Pshenichkin S, Surin A, Sullivan SE, Neal JH (1997) N-acetylaspartylglutamate selectively activates mGluR3 receptors in transfected cells. J Neurochem 69: 174-181. [DOI] [PubMed] [Google Scholar]
- Yan G-M, Ni BH, Weller M, Wood KA, Paul SM (1994) Depolarization or glutamate-receptor activation blocks apoptotic cell death of cultured cerebellar granule neurons. Brain Res 656: 43-51. [DOI] [PubMed] [Google Scholar]
- Yang L, Lindholm K, Konishi Y, Li R, Shen Y (2002) Target depletion of distinct tumor necrosis factor receptor subtypes reveals hippocampal neuron death and survival through different signal transduction pathways. J Neurosci 22: 3025-3032. [DOI] [PMC free article] [PubMed] [Google Scholar]