

Abstract
Prion diseases are transmissible neurodegenerative disorders characterized by extensive neuronal apoptosis and accumulation of misfolded prion protein (PrPSC). Recent reports indicate that PrPSC induces neuronal apoptosis via activation of the endoplasmic reticulum (ER) stress pathway and activation of the ER resident caspase-12. Here, we investigate the relationship between prion replication and induction of ER stress during different stages of the disease in a murine scrapie model. The first alteration observed consists of the upregulation of the ER chaperone of the glucose-regulated protein Grp58, which was detected during the presymptomatic phase and followed closely the formation of PrPSC. An increase in Grp58 expression correlated with PrPSC accumulation at all stages of the disease in different brain areas, suggesting that this chaperone may play an important role in the cellular response to prion infection. Indeed, in vitro studies using N2a neuroblastoma cells demonstrated that inhibition of Grp58 expression with small interfering RNA led to a significant enhancement of PrPSC toxicity. Conversely, overexpression of Grp58 protected cells against PrPSC toxicity and decreased the rate of caspase-12 activation. Grp58 and PrP were shown to interact by coimmunoprecipitation, observing a higher interaction in cells infected with scrapie prions. Our data indicate that expression of Grp58 is an early cellular response to prion replication, acting as a neuroprotective factor against prion neurotoxicity. Our findings suggest that targeting Grp58 interaction may have applications for developing novel strategies for treatment and early diagnosis of prion diseases.
Keywords: prion, Grp58, endoplasmic reticulum stress, apoptosis, scrapie, transmissible spongiform encephalopathy, caspase-12
Introduction
Prion disorders are fatal neurodegenerative diseases characterized by spongiform degeneration of the brain, accompanied by extensive neuronal loss and accumulation of PrPSC, a misfolded and protease-resistant form of the normal prion protein (PrPC) (Prusiner, 1998). The generation of PrPSC is associated with structural changes in the protein that lead to alterations in its biochemical properties, such as insolubility in nondenaturating detergents and partial resistance to proteases.
We found previously that highly purified PrPSC from scrapie-infected mice induces endoplasmic reticulum (ER) stress and apoptosis in vitro (Hetz et al., 2003). In addition, we provided evidence for the induction of ER stress in the brain of patients affected with Creutzfeldt-Jakob disease (CJD) (Hetz et al., 2003). The ER stress response is frequently associated with cell death under pathological conditions in which misfolded proteins accumulate in the ER or when calcium homeostasis is perturbed (Ferri and Kroemer, 2001). ER stress triggers a survival pathway known as the unfolding protein response (UPR), associated with the increased expression of chaperones and folding enzymes that decrease unspecific aggregation or target misfolded proteins to proteasome-mediated degradation (Sitia and Braakman, 2003). However, when the ER homeostasis cannot be restored, a proapoptotic response is induced (Breckenridge et al., 2003), which is linked with the activation of the ER resident caspase-12 via the stress sensor Ire-1α (Yoneda et al., 2001). Other components induce the expression of the proapoptotic transcription factor GADD153/CHOP (Harding et al., 2000).
The prosurvival effects of UPR activation include an increased expression of chaperones of the glucose-regulated protein (grp) family, such as Grp78/Bip (immunoglobulin chain binding protein) and Grp94 (Reddy et al., 1999; Rao et al., 2002; Sitia and Braakman, 2003). In the terminal stage of experimental murine scrapie, the ER stress response was mainly associated with the upregulation of Grp58 [also called ERp60, ER-60, ERp58, ER-58, ERp61, and Q2 (Turano et al., 2002)], a chaperone with protein disulfide isomerase (PDI)-like activity. However, only a few reports have described a participation of Grp58 in ER stress, and the molecular mechanism is not known (Mazzarella et al., 1994; Holtz and O'Malley, 2003; Dimcheff et al., 2003b). Two structural homologs of Grp58, PDI and endothelial PDI (EndoPDI), have a neuroprotective activity against ischemia (Tanaka et al., 2000; Fischer et al., 2002; Sullivan et al., 2003). Grp58 resembles PDI in structure and activity, because both proteins catalyze thiol/disulfide exchange, including disulfide bond formation and rearrangement reactions (Turano et al., 2002).
In the present article, we investigated the relationship between prion replication and the induction of the ER stress response in a murine-scrapie model. Our results suggest that increased expression of the disulfide-isomerase Grp58 is an early event in the pathology and seems to play an important role in modulating prion neurotoxicity. These findings provide a novel target for transmissible spongiform encephalopathy (TSE) therapy and suggest a general mechanism of neuronal toxicity initiated by the accumulation of misfolded proteins, a common event in several neurodegenerative diseases (Soto, 2003).
Materials and Methods
Materials. Staurosporine, tunicamycin, brefeldin A, the calcium ionophore A23187, and thapsigargin were purchased from VWR International (Darmstadt, Germany). Cell medium, fetal calf serum, and antibiotics were obtained from Invitrogen (Carlsbad, CA). Fluo-4 and BAPTA AM were purchased from Molecular Probes (Eugene, OR).
Cell culture and viability assays. N2a cells were cultured in DMEM supplemented with 10% fetal calf serum and antibiotics (10,000 U/ml penicillin and 10 μg/ml streptomycin) at 37°C and 5% CO2. For cell viability analysis, cells were grown in collagen IV-coated 96-well plates for 24 h in cell culture medium containing 1% serum before the addition of the agonist. Cell viability was quantified using 3-(4,5-dimethylthazol-2-yl)-5-3-carboxymethoxy-phenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) and phenazine methosulfate according to the recommendations of the supplier (CellTiter 96 AQueous; Promega, Madison, WI). Purification of PrPSC and toxicity studies were performed as described previously (Hetz et al., 2003). For calcium chelation with BAPTA AM, cells were loaded for 30 min with 37 μm BAPTA AM, as described previously (Barros et al., 2001).
Plasmids and cell transfections. Vector-based small interfering RNA (siRNA) against Grp58 RNA was constructed as follows: human U6 promoter sequence was amplified from genomic DNA by nested PCR using the primers 5′-CCC GAGTCCAACACCCGTGG-3′ and 5′-GGTGTTTCGTCCTTTCCACA-3′ for the first PCR and then reamplified with 5′-ATAGAATTCCCGAGTCCAACACCCGTGGG-3′ and 5′-ATAGAATTCGGTGTTTCGTCCTTTCCACAAG-3′. The second amplicon was cloned, using EcoRI (underlined), into a shuttle vector containing a hygromycin resistance. Two different siRNAs were cloned using a forward primer specific for the vector 5′-CCG ATT TCG GCC TAT TGG T-3′ and a reverse primer containing a portion that anneals with the 3′ end of the U6 promoter and the specific hairpin sequence (Grp58-1, 5′-ATAGCGCCGCAAAAAATAGTCCCATTAGCAAAGGGAAGCTTGAACCTTTGCTAATGGGACTATCGGTGTTTCGTCCTTTCCACA-3′ and Grp58-5, 5′-ATAGCGGCCGCAAAAAGAGATACCTGAAGTCTGGAAGCTTGAACAGACTTCAGGTATCTCGGTGTTTCGTCCTTTCCACA-3′). The PCR fragments obtained were digested using BglII and NotI and cloned into the shuttle vector predigested with the same enzymes. All PCRs were performed using Pfx polymerase (Invitrogen), following the instructions of the manufacturer. As a negative control, a complete U6 promoter fused to a degenerated hairpin was used. The selected sequences of both constructs (underlined) were “blasted” against the National Center for Biotechnology Information Expressed Sequence Tag database (http://www.ncbi.nlm.nih.gov/BLAST/) and were specific for both human and mouse Grp58. The siRNA constructs were totally sequenced in both directions. Stably expressing N2a cells were produced by transfection using the SuperFect kit (Qiagen, Valencia, CA), following the instructions of the manufacturer. After 48 h of transfection, cells were selected using G418 (geneticin; 1.3 mg/ml) for 5 d, and individual clones were obtained by limiting dilution.
Grp58 cDNA (clone BI153534; Invitrogen) was amplified by PCR using specific primers (5′-GCGGATCCGCCACCATGCGCTTCAGCTGCCTAG-3′ and 5′-GCTCTAGATTAGAGGTCCTCTTGTG-3′. The amplicon was digested with XhoI and BamHI (underlined) and cloned into pcDNA4 vector (Invitrogen). Murine PrP was cloned by reverse transcription-PCR from N2a RNA and cloned into pcDNA3 using the HindIII and EcoRI restriction sites. Green fluorescent protein (GFP) was fusioned to the N-terminal of PrP at codon 25.
SDS-PAGE and Western blot analysis. Brain tissue was homogenized on ice in radioimmunoprecipitation assay (RIPA) buffer (20 mm Tris, pH 8.0, 150 mm NaCl, 0.1% SDS, 0.5% deoxycholic acid, and 0.5% Triton X-100) containing a protease inhibitor mixture (F. Hoffmann-La Roche, Basel, Switzerland). Protein concentration was determined by micro-BCA assay (Pierce, Rockford, IL). The equivalent of 30-50 μg of total protein was generally loaded onto 10% SDS-polyacrylamide minigels (NuPAGE Novex; Invitrogen, Basel, Switzerland) and analyzed by Western blotting as described previously (Hetz et al., 2003). The following antibodies and dilutions were used: 6H4 anti-PrP, 1:10,000 (Prionics, Schlieren, Switzerland); anti-caspase-12, 1:2000 (Exalpha Biologicals, Watertown, MA); anti-Grp78/Bip, anti-Hsp60 (heat shock protein 60), anti-Hsp70, anti-calnexin, anti-Grp58, and anti-Grp94, 1:2000 (Stress-Gene, San Diego, CA); and anti-actin or tubulin, 1:2000 (Santa Cruz Biotechnology, Santa Cruz, CA). After incubation with the primary antibody, membranes were incubated for 1 h at room temperature with horseradish peroxidase-coupled secondary antibodies diluted 1:10,000 in washing buffer. After being washed, specifically bound antibodies were detected by enhanced chemiluminescence assay (Amersham Biosciences, Cardiff, UK).
PrP subcellular localization studies. To assess the subcellular localization of PrPC, N2a cell clones stably expressing Grp58, Grp58siRNA, or empty vector were transiently transfected with a PrP-GFP expression vector, and, after 48 h, the GFP fluorescence emission was analyzed. In parallel, cells were fixed and stained with an ER immunofluorescent marker, as described previously (Leyton et al., 2001). In brief, cells were fixed for 30 min in PBS containing 4% paraformaldehyde and permeabilized with cold methanol at -20°C for 10 min. After blocking for 1 h with 5% gelatin, cells were subsequently incubated with anti-calnexin antibodies for 1 h at 37°C, washed three times in PBS, and incubated with a rhodamine-conjugated secondary antibody for analysis on an epifluorescence microscope. Nucleus was stained with Hoechst.
Deglycosylation studies. For analysis of PrP glycosylation status, N2a cell clones stably expressing Grp58, Grp58siRNA, or empty vector were transiently transfected with a mouse PrP expression vector. After 48 h, cell extracts were prepared in RIPA buffer, and PrP was deglycosylated by treating the extracts with PNGase F (peptide N-glycosidase F) (New England BioLabs, Beverly, MA) as described previously (Russelakis-Carneiro et al., 2002).
Coimmunoprecipitation studies. Cells were infected with Rocky Mountain Laboratory (RML) prions, as described previously (Hetz et al., 2003) and treated with 10 μm epoxomycin or left untreated, and cell extracts were prepared in RIPA buffer. Immunoprecipitation was performed with the anti-PrP antibody 6H4 and protein A/G-Sepharose beads, following the recommendations of the manufacturer (Amersham Biosciences). The presence of PrP and Grp58 was evaluated by Western blots, as described above.
Animal samples. C57BL/6J mice were injected stereotaxically in the hippocampus with 1 μl of a 10% brain homogenate taken from mice infected with 139A scrapie at a terminally ill stage. Onset of the clinical disease was measured weekly by determining body-weight loss and decrease in muscle strength using a grid system, as described previously (Soto et al., 2000). Scrapie incubation times were defined from the date of injection to the time when the clinical symptoms persisted for 3 consecutive weeks, which, for the conditions used in this study, corresponded to 16 weeks after injection. For Western blot analysis, animals were killed at different times after infection, and different brain areas were dissected and analyzed separately.
Immunohistochemistry. Coronal sections (20 μm) were taken from dorsal hippocampus of mice. Sections were fixed overnight in a solution containing 4% paraformaldehyde and transferred to a 20% sucrose solution. Immunostaining was performed on free-floating sections. Anti-neuronal-specific nuclear protein (NeuN) antibody was used to specifically stain neurons and was detected with avidin-biotin peroxidase using reagents from Vector Laboratories (Vectastain Elite ABC; Burlingame, CA) and diaminobenzidine as the chromogen. For the analysis of the expression of Grp58 and a neuronal marker, the brain slices were coincubated with anti-Grp58 polyclonal antibody and the monoclonal antibody NeuN and developed with secondary antibody conjugated with FITC or rhodamine.
Calcium signaling. The changes in intracellular calcium levels resulting from stimulation of ER calcium release were measured using the fluorometric imaging plate reader 1 (FLIPR1) machine (Molecular Devices, Sunnyvale, CA) by the use of the fluorescent dye Fluo-4, as described previously (Hetz et al., 2003). In brief, cells were grown on 96-well black plates coated with collagen IV and loaded with Fluo-4 at a final concentration of 10 μg/ml. After 120 min of incubation at 37°C, cells were stimulated with different calcium agonists in FLIPR buffer (in mm: 150 NaCl, 5 KCl, 1 MgCl2, 10 HEPES, and 10 glucose) in the absence of extracellular calcium. Fluorescence emission was quantified every 5 s for 30 min.
Statistical analysis. Data were analyzed by parametric t test (two-tailed), and significance was expressed as follows: *p < 0.05; **p < 0.01 (see Figs. 4, 6). When more than two groups were analyzed, an ANOVA test was also used to estimate statistical significance.
Figure 4.

Grp78 and Grp94 are transiently induced by 139A scrapie infection. A, B, Grp78 (A) and Grp94 (B) expression levels at 12, 16, and 20 wpi were determined by Western blotting, and the band intensities from three different infected animals were quantified by densitometry. Top, A representative Western blot analysis at 16 and 20 wpi. Bottom, Averaged intensities were compared with the corresponding values from three control animals for both Grp78 and Grp94. Analysis by two-way ANOVA, using brain regions and weeks postinjection as the variables, showed that Grp78 and Grp94 expression levels were not significantly different. t test analysis of individual values showed some statistically significant differences (*p < 0.05; **p < 0.01). Cx, Cortex; Hip, hippocampus; Thal, thalamus; BS, brainstem. C, Calnexin, Hasp70, Hsp60, and GADD153/CHOP levels were analyzed in different brain areas at 20 wpi by Western blotting (left) and quantified as described in A and B (right).
Figure 6.

Grp58 protects N2a cells against PrPSC toxicity. A, N2a clones transfected with Grp58siRNA, mock siRNA, or full-length Grp58 were treated with different concentrations of purified PrPSC, and cell viability was determined after 48 h by MTS analysis. Data were analyzed by Student's t test to compare the differences between mock-transfected cells and those overexpressing Grp58 or transfected with Grp58siRNA. *p < 0.05; **p < 0.01. B, In parallel, cells were treated with PrPSC, and caspase-12 activation was measured by Western blot analysis. Actin levels are shown as an internal control of protein loading. C, N2a clones transfected with Grp58siRNA, mock siRNA, or full-length Grp58 (OE) were treated with 50 nm PrPSC or 800 ng/ml tunicamycin for 48 h, and GADD153/CHOP and actin levels were determined by Western blot analysis.
Results
Scrapie prion infection induces upregulation of Grp58 during the presymptomatic phase of the disease
To assess the contribution of ER stress to the process of neuronal loss during the development of prion diseases, we performed a time course experiment using the murine scrapie strain 139A as a model. To establish the kinetics of disease progression, animals were injected into the hippocampus with 1 μl of 139A scrapie brain homogenate, and the time to onset of clinical signs was determined. Muscle capacity was measured using an inverted grill setup, and the first signs of muscle weakness were observed at 16 weeks after injection (Fig. 1A) (n = 8 per group). This was accompanied by evident ruffling fur, hunched posture, and reduced mobility (data not shown). Animals were killed after 23 weeks because of difficulties with food ingestion. Based on these observations, the period up to 12 weeks postinjection (wpi) was considered the presymptomatic stage, whereas 16, 20, and 23 weeks postinjection were considered representative of the early, late, and terminal stages of the disease, respectively.
Figure 1.
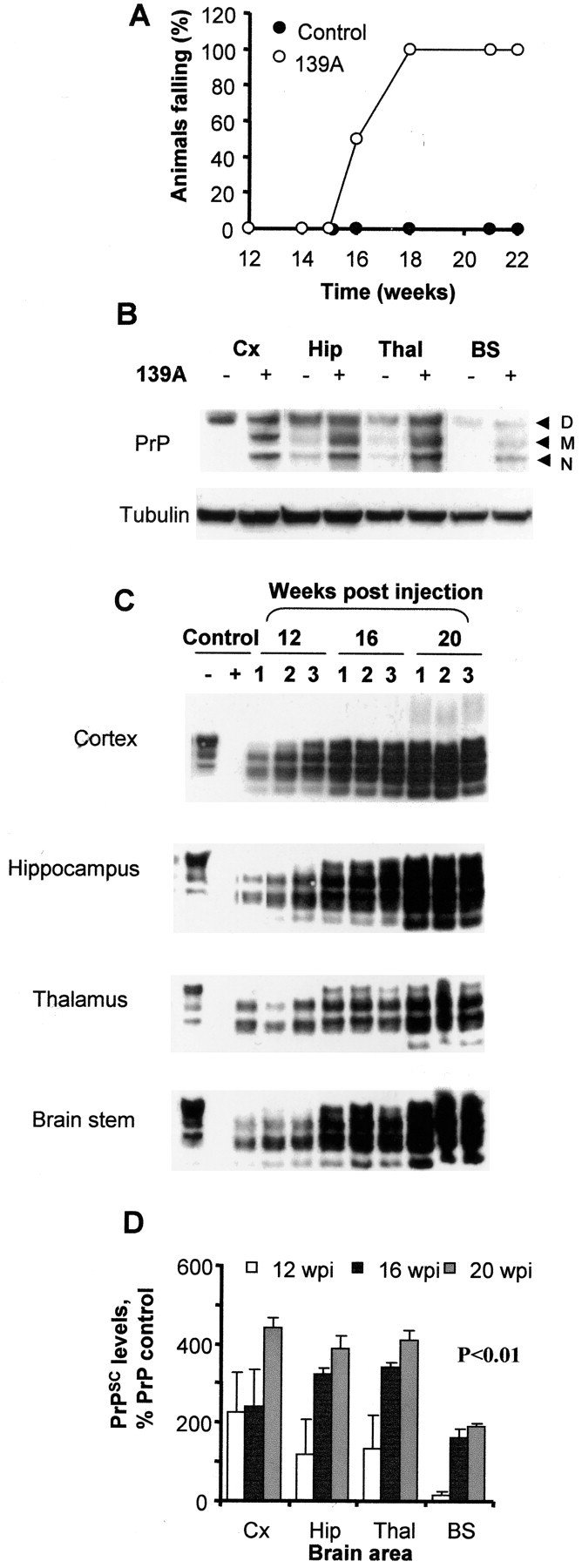
Disease progression and prion propagation in a 139A murine scrapie model. A, Eight animals per group were injected in the hippocampus with 1 μl of 10% brain homogenate from normal (controls) or 139A scrapie-infected mice. Muscle strength was determined by measuring the time during which animals were able to cling to an inverted grill. Values represent the percentage of animals in each group that fell within 1 min after inversion. B, Western blot analysis of total PrP levels in control and infected brains in different brain areas after 16 wpi. Tubulin levels were used as an internal control for protein loading. Cx, Cortex; Hip, hippocampus; Thal, thalamus; BS, brainstem. D, M, and N represent the diglycosylated, monoglycosylated, and nonglycosylated forms of PrP, respectively. C, Western blot analysis of PrPSC in PK-digested brain homogenates from infected animals at 12, 16, and 20 wpi. Results from three different animals per group are shown. As controls, normal brain homogenates were treated with PK or left untreated. D, Quantification of PrPSC levels in different brain areas of scrapie-infected animals as a percentage of total PrPC in controls animals. Differences in PrPSC accumulation were statistically significant (p < 0.001), as analyzed by two-way ANOVA using brain regions and weeks postinjection as the variables.
Four groups of infected and control animals, corresponding to 12, 16, 20, and 23 wpi, were killed, and the biochemical and histological analysis of the brain was performed on each individual animal. The brain was dissected into hippocampus, cortex, brainstem, and thalamus. Western blot analysis of total PrP levels revealed an accumulation over time compared with control animals (Fig. 1B and data not shown). Accumulation of PrP was associated with increased levels of the monoglycosylated and nonglycosylated forms of PrP (Fig. 1B), which is a common feature of this scrapie strain, as described previously (Russelakis-Carneiro et al., 2002). The level of PrPSC was determined by Western blot analysis of brain homogenates after proteinase K (PK) treatment (Fig. 1C). During the presymptomatic phase of the disease, at 12 wpi, PrPSC was detectable in all brain areas, with higher levels present near the site of injection (hippocampus and thalamus). With continued disease progression, PrPSC spread throughout the whole brain with highly reproducible kinetics (Fig. 1D).
Neuronal loss was assessed by histological analysis using the NeuN neuronal marker. The first signs of neuronal loss were detected in the thalamus at 20 wpi, with a higher decrease at the terminal stage of the disease (data not shown). In the hippocampus and other brain areas, neuronal loss was apparent only at the terminal stage of the disease, as described previously (Hetz et al., 2003). These data indicate that the onset of clinical signs of scrapie is not attributable to neuronal loss but rather to either neuronal dysfunction or synaptic alterations.
We showed previously, in both humans affected with CJD and murine scrapie models, that prion replication in brain is accompanied by increased expression of Grp58 in the terminal stage of the disease (Yoo et al., 2002; Hetz et al., 2003). However, it was not clear whether this alteration was an early or late event during the disease progression. To define more precisely the contribution of ER stress during the propagation of PrPSC, Western blot analysis of Grp58 expression levels was performed on different brain areas during the course of scrapie infection. Figure 2, A and B, shows that a 1.5-fold to twofold induction of Grp58 can be detected as early as 12 wpi, 4 weeks before the appearance of the first clinical signs. Expression levels of Grp58 increased over time, with high Grp58 levels in the hippocampus (the site of injection), cortex, and thalamus, and intermediate levels in the brainstem, compared with control animals (Fig. 2B). Interestingly, the level of Grp58 expression correlated closely with the accumulation of misfolded prion protein, as observed when PrPSC levels are plotted with the corresponding data for Grp58 expression levels in different brain areas and at distinct times during the disease (Fig. 2C; data from 12, 16, and 20 wpi). During the terminal stage of the disease (23 wpi), a decrease in the expression levels of Grp58 were observed in hippocampus (until 1.5-fold induction) and thalamus (returning to basal levels). However, in cortex, the expression levels of Grp58 continued increasing up to a ninefold induction (Fig. 2B). The downregulation of Grp58 at the terminal stage of the disease correlated with the occurrence of neuronal loss in this scrapie model when different brain areas were compared, because, in thalamus and hippocampus, massive neuronal loss was observed (data not shown) (Hetz et al., 2003).
Figure 2.
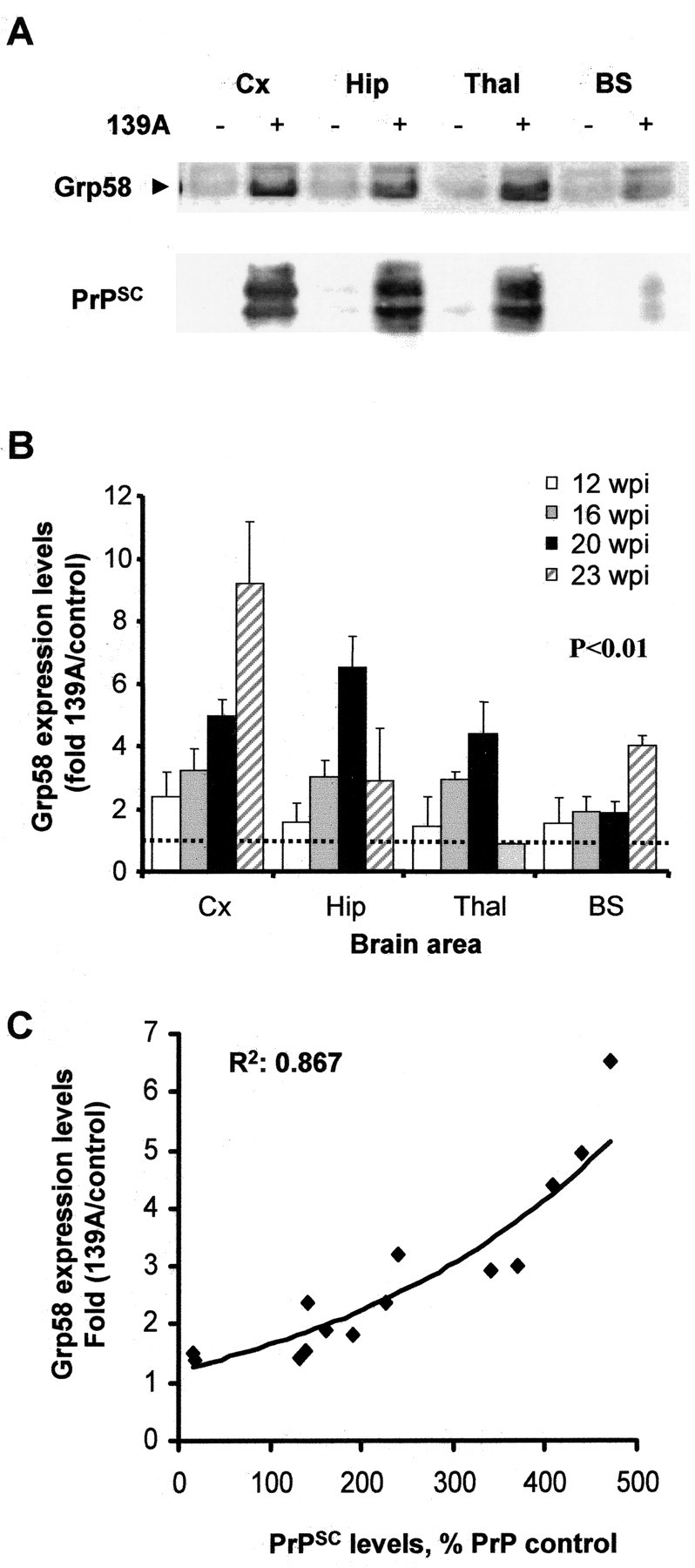
Grp58 is upregulated in the brain of 139A-infected animals during early stages of the disease and correlates with PrPSC levels. A, Grp58 and PrPSC levels were analyzed by Western blot in different brain areas at 16 wpi. B, Grp58 expression levels were determined by quantitative Western blot analysis of brain homogenates taken at different stages of the disease, and the results from three animals were averaged. The ratio of the Grp58 signal from infected and normal animals was determined for different brain areas at various times postinjection. Results represent the average ± SD of three different animals. Differences in Grp58 expression levels were statistically significant (p < 0.01), as analyzed by two-way ANOVA using brain regions and weeks postinjection as the variables. Cx, Cortex; Hip, hippocampus; Thal, thalamus; BS, brainstem. C, The levels of PrPSC and Grp58 were compared from all brain homogenates described in B and Figure 1 E.
As an alternative approach to determine the expression levels of Grp58 and to analyze the cellular types in which Grp58 is expressed, we performed immunofluorescence of brain sections from control and scrapie-infected animals at 20 wpi. Scrapie-infected animals presented a higher staining with anti-Grp58 antibodies compared with control animals (Fig. 3). Interestingly, when the sections were double stained with a specific neuronal marker (NeuN), a coexpression of Grp58 and the neuronal marker was observed, indicating that the induction of Grp58 is a neuronal response to prion replication (Fig. 3A, B shows magnification). The pattern of Grp58 staining reveals a discrete intracellular localization, consisting of the typical staining of ER-associated proteins.
Figure 3.

Neuronal expression of Grp58. A, Thalamus brain section from control and scrapie-infected animals were coimmunostained with anti-Grp58 (red fluorescence) and anti-NeuN (green fluorescence). Right panels indicate superposition of NeuN and Grp58 staining. B, Magnification of two pictures obtained from scrapie-infected animals, as described in A. C, Control experiments performed in the absence of primary antibodies. Phase-contrast picture is shown as control.
Scrapie prion infection triggers a transient induction of Grp78 and Grp94
Small changes in the expression levels of Grp78 and Grp94, the most common proteins used as ER stress markers, were observed after infection, but without a clear increase during the course of the disease. Only transient significant changes in the expression levels of Grp78 and Grp94 were observed in thalamus and brainstem after 16 weeks of infection, at the beginning of the symptomatic phase of the disease (Fig. 4A,B). No correlation between Grp78/Grp94 levels and PrPSC accumulation was observed (compare Figs. 4A,B and 1E). However, no significant upregulation of other chaperones, such as calnexin, Hsp60, or Hsp70, was observed in any of the samples analyzed (Fig. 4C and data not shown). In addition, the levels of the ER stress-inducible transcription factor GADD153/CHOP were undetectable in any of the samples analyzed (Fig. 4C) (see Fig. 6C for positive control for antibody), nor were changes in the expression levels of its target Bcl-2 observed (data not shown). Together, these data suggest that prion replication triggers a nonclassical ER stress response, in which the major change observed is the specific induction of the disulfide isomerase Grp58.
It has been reported that prolonged ER stress leads to apoptotic cell death, mediated by caspase-12, an ER resident caspase, which, after activation by ER stress, induces caspase-3-executed apoptosis (Mehmet, 2000). Caspase-12 is activated by N-terminal cleavage to generate fragments of 42 and 35 kDa. Western blot analysis of caspase-12 activation over time revealed that this protease is activated only during terminal stages of the disease and in the brain regions showing extensive neuronal loss (Hetz et al., 2003). A weak signal corresponding to the active fragments was detected at 20 wpi, and a more pronounced activation was detected at 23 wpi (data not shown).
Grp58 expression is a survival factor that protects cells against ER stress and PrPSC toxicity
An increase in the expression levels of Grp58 was observed in different brain areas until the late stage of the disease. However, at the terminal stage of the disease, in the brain areas that presented neuronal death and caspase-12 activation, a downregulation of Grp58 was observed, suggesting that Grp58 could act as a neuroprotective factor in response to prion replication. Grp58 has been shown to be induced under stress conditions, triggered by the pharmacological targeting of ER function (Mazzarella et al., 1994); however, its participation in the ER stress response in not known. To analyze the relationship between Grp58 and prion replication, N2a cells were stably transfected with vector-based siRNA targeted against Grp58 mRNA. Those cells were selected as a model because they reproduce several aspects associated with TSEs, such as sensitivity to PrPSC toxicity (Hetz et al., 2003), they express PrPC and, they are able to sustain prion replication in vitro (Butler et al., 1988). Several cell lines were obtained in which Grp58 expression was completely abolished (Fig. 5A). No alteration in the expression levels of Grp78, Grp94, or calnexin were observed in these cells, suggesting that targeting Grp58 RNA was specific (Fig. 5A,B). This result also indicates that the downregulation of Grp58 does not affect the expression of other related chaperones, such as PDI, suggesting that no evident compensatory changes are observed after blocking Grp58 expression. Even after the induction of ER stress by treatment with tunicamycin (an inhibitor of N-linked glycosylation) or brefeldin A (an inhibitor of ER-Golgi transport), no detection of Grp58 was observed in Grp58siRNA-transfected cells (Fig. 5B). No alteration on the endogenous levels of PrP expression was observed in cells expressing Grp58siRNA (data not shown).
Figure 5.

Blocking the expression of Grp58 increases the susceptibility of N2a cells to some forms of ER stress. A, Different N2a subclones were obtained, which were stably transfected with a vector-based siRNA against Grp58 RNA (RNAi), an irrelevant siRNA (mock), or a Grp58 expression plasmid (OE). The expression levels of Grp58, Grp94, PDI, calnexin, and actin were analyzed in several different clones by Western blot. B, The induction levels of Grp58, Grp78, and Grp94 (top) were determined after treatment with tunicamycin (T) or brefeldin A (B). In parallel, activation of caspase-12 was determined in the same samples (bottom). Actin levels were measured as a control for proper protein loading. C, Three different clones transfected with Grp58siRNA or with mock siRNA were treated with different concentrations of tunicamycin, and cell viability was determined after 48 h by MTS analysis. The effect of tunicamycin on cell death of Grp58siRNA was found highly significantly different (p < 0.0001) compared with the effect on mock-transfected cells as analyzed by one-way ANOVA. D, One representative clone transfected with Grp58siRNA or mock siRNA was treated with different concentrations of brefeldin A. Analysis by one-way ANOVA found the effect significantly different (p < 0.01) compared with mock-transfected cells. E, The same clone was treated with 5 or 20 nm A23187 (A), thapsigargin (Thap; 10 μm), or staurosporine (Sta; 80 nm). In these experiments, the differences between Grp58siRNA cells were not considered significantly different from mock-transfected cells, as evaluated by Student's t tests. In all of these studies, data show the mean and SD of at least two different experiments performed in triplicate.
Previous studies have shown that the expression of Grp78 and Grp94 has a protective activity against ER stress-mediated apoptosis (Liu et al., 1997; Reddy et al., 1999; Rao et al., 2002). Grp58 has no sequence or functional similarity with these other members of the Grp family. Indeed, no direct relationship between Grp58 and apoptosis has been described, although other disulfide isomerases with sequence homology to Grp58, such as PDI and EndoPDI, have been shown to protect against ischemia (Tanaka et al., 2000; Fischer et al., 2002; Sullivan et al., 2003). Therefore, we decided to test the susceptibility of Grp58siRNA cells to ER stress induced by several different mechanisms. Tunicamycin leads to ER stress via the accumulation of misfolded proteins in the ER (Breckenridge et al., 2003). As shown in Figure 5C, three different stably transfected clones were more susceptible to tunicamycin treatment compared with mock-transfected cells. Similar results were observed after treatment with brefeldin A (Fig. 5D). On the other hand, ER stress induced by reagents that affect calcium homeostasis, such as thapsigargin (an inhibitor on the ER-calcium ATPase) or the calcium ionophore A23187, is not exacerbated by loss of Grp58 expression (Fig. 5E). Furthermore, no difference between mock-transfected and Grp58siRNA-transfected cells was observed after treatment with staurosporine, which is known to induce the mitochondrial apoptosis pathway (Fig. 5E). Thus, Grp58 seems to be specifically involved in apoptosis protection within the ER stress pathway. In this respect, it is interesting that the increased toxicity of brefeldin A and tunicamycin in Grp58siRNA cells is associated with the activation of caspase-12, as evidenced by the decrease in the level of the inactive proform (Fig. 5B). Together, these data suggest that the neuroprotective activity of Grp58 is upstream of caspase-12 activation and is triggered specifically by ER stress events involving accumulation of misfolded proteins in this organelle.
We then tested the susceptibility of Grp58siRNA cells or Grp58-overexpressing cells to PrPSC toxicity. As shown in Figure 6A, reduced expression of Grp58 in Grp58siRNA-expressing cells leads to increased toxicity of PrPSC, whereas overexpression of Grp58 confers resistance to PrPSC. For example, at 50 nm PrPSC, although no significant neuronal death was observed in cells overexpressing Grp58, >70% cell death was obtained in Grp58 null neurons. The protective activity of Grp58 is associated with a decrease in caspase-12 activation (Fig. 6B). No induction of GADD153/CHOP levels was observed after PrPSC treatment of control, Grp58siRNA, or Grp58-overexpressing cells (Fig. 6C). However, induction of this proapoptotic transcription factor was detected in cells treated with tunicamycin or brefeldin A, which was negatively regulated by Grp58 expression levels, reinforcing the notion that Grp58 has a neuroprotective activity (Fig. 6C and data not shown). Thus, our data suggest that PrPSC triggers an atypical form of ER stress that can be differentiated from the effect of typical ER stressors, such as tunicamycin and brefeldin A.
Grp58 interacts with PrP but does not affect its maturation process
To study the possible interaction between PrP and Grp58, we performed coimmunoprecipitation experiments. Immunoprecipitation of endogenous PrP revealed that Grp58 coprecipitated with PrP in N2a cells (Fig. 7A). In addition, low levels of Grp78 were detected after PrP immunoprecipitation. Interestingly, when N2a cells were infected with RML scrapie prions, a larger amount of Grp58 was detected in association with PrP (Fig. 7A). Because the amount of PrP immunoprecipitated was similar in infected and noninfected cells, we conclude that Grp58 might be expressed in larger quantities in infected cells or have a higher affinity for PrPSC. The interaction between PrP and Grp78 was not affected by RML scrapie infection. Similar results were observed when accumulation of misfolded PrP was induced by treatment of wild-type N2a cells with the proteasome inhibitor epoxomycin (Fig. 7A). This treatment induces the accumulation of abnormally folded PrP in the ER.
Figure 7.
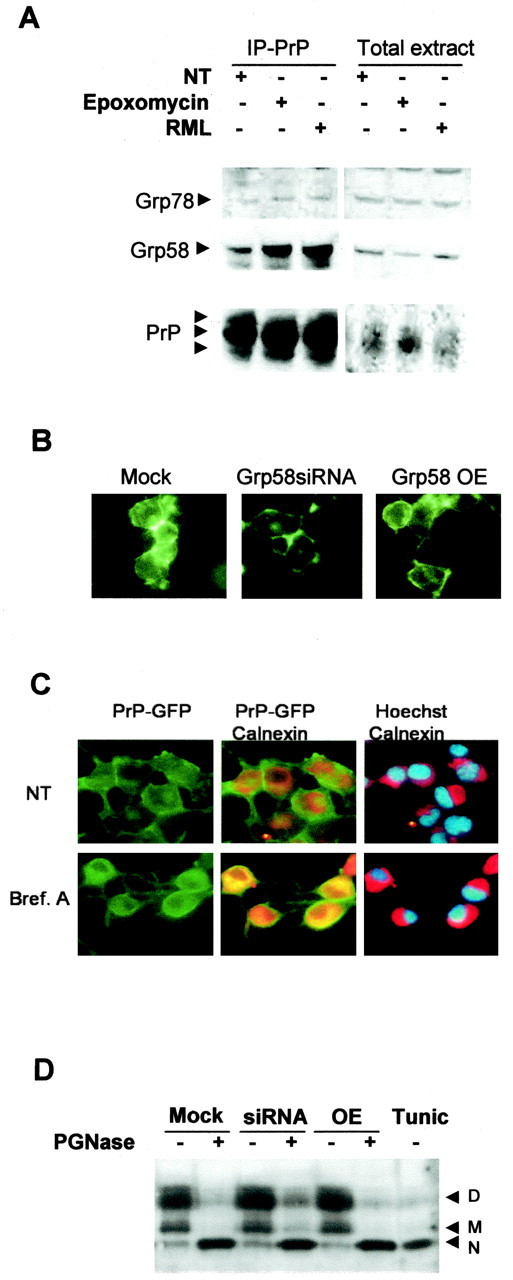
Grp58 interacts with PrP but does not alter its maturation process. A, N2a cells untreated (NT), infected with RML scrapie prions, or treated with 5 μm epoxomycin were lysed and immunoprecipitated (IP) with the anti-PrP monoclonal antibody 6H4. The levels of Grp58 and Grp78 were analyzed by Western blot. As control, the levels of these proteins were analyzed in the total extracts before immunoprecipitation of PrP. B, Prp-GFP fusion protein was transiently expressed in N2a clones transfected with Grp58siRNA, mock siRNA, or full-length Grp58 (Grp58 OE), and the GFP fluorescence distribution was analyzed. C, N2a cells transfected with PrP-GFP were treated with 50 μm brefeldin A (Bref. A) or left untreated, and PrP distribution was analyzed (green fluorescence). As an intracellular marker, stained calnexin was analyzed (red fluorescence). Nuclear was stained with Hoechst (blue fluorescence). Superposition of GFP and calnexin or calnexin and Hoechst staining are shown. D, Murine PrP was overexpressed, cell extracts were treated with PGNase F or left untreated, and the PrP pattern was analyzed by Western blot. As a positive control, wild-type cells were treated with 5 μg/ml tunicamycin (Tunic) for 12 h. D, Diglycosylated; M, monoglycosylated; N, nonglycosylated.
Because PrPC has an intracellular disulfide bond, the possible effects of changes in Grp58 expression on PrP maturation and cell-sorting processes were assessed. The expression of PrPC in the cell surface was analyzed by expressing a PrP-GFP fusion protein in Grp58siRNA, Grp58-overexpressing, or mock-transfected cells. PrP targeting to the plasma membrane was not affected by modification of Grp58 expression levels, because the majority of PrP was detected at the cell surface (Fig. 7B). In most of the cells analyzed, a minor fraction of PrP was detected in intracellular compartments with a perinuclear distribution, probably reflecting an ER-Golgi localization of the protein being synthesized and sorted to the plasma membrane. As a control, wild-type N2a cells were treated with brefeldin A to induce an intracellular accumulation of PrP. As shown in Figure 7C, brefeldin A triggered a drastic change in the subcellular localization of PrP, observing a perinuclear distribution (compare with Hoechst staining) and a partial colocalization with the ER marker calnexin (Fig. 7C).
In addition, the glycosylation state of PrP was investigated in Grp58siRNA, Grp58-overexpressing, or mock-transfected cells. Cells were transiently transfected with a murine PrPC expression vector, and N-deglycosylation of PrP was performed by treatment of the protein extracts with PGNase. As a positive control, cells were treated with tunicamycin to detect the endogenous unglycosylated form of PrPC. The results indicate that the addition of mature glycosylations on PrPC was unaffected by alterations in Grp58 expression (Fig. 7D), suggesting that PrP passed the ER-Golgi protein quality control.
Discussion
Alteration of ER homeostasis by several forms of stress, such as heat shock, overexpression of mutant proteins, or alteration on proteasome activity, can cause the accumulation of misfolded proteins in the ER. These alterations trigger the specific activation of a protective cellular response, the so-called UPR, which leads to the upregulation of ER chaperones and a general decrease in protein synthesis (Liu et al., 1997; Reddy et al., 1999; Ferri and Kroemer, 2001; Rao et al., 2002). Sustained ER stress generates an irreversible cellular damage that can lead to cell death by apoptosis or necrosis, depending on the type and intensity of the stimulus. Caspase-12 activation is one of the molecular effectors of ER-dependent apoptosis (Nakagawa et al., 2000). Interestingly, caspase-12 knock-out mice are viable and do not show abnormalities in programmed cell death during development, suggesting that this pathway may be mainly associated with cellular death resulting from pathological conditions.
In the present article, we show that, during the progression of murine scrapie, the first signs of ER stress are associated with the upregulation of Grp chaperones, particularly Grp58 and, to a lesser extent, Grp78 and Grp94. Grp58 induction directly correlated with the kinetics of prion replication in different brain areas, suggesting that Grp58 upregulation is a specific cellular response induced by prion replication in vivo. The activation of caspase-12 and neuronal loss was detected only at late and terminal stages of the disease. These observations establish for the first time a spatial/temporal relationship between prion replication and ER stress-mediated apoptosis. Caspase-12 activation and the upregulation of Grps have been described previously in the brain of individuals affected by sporadic and variant CJD and in murine scrapie during terminal stages of the disease (Yoo et al., 2002; Hetz et al., 2003). Our data rule out the possibility that the ER stress response described in individuals affected with prion diseases is an epiphenomenon associated with massive neuronal loss during the late stages of disease progression. Furthermore, our findings suggest that ER stress is a primary response to prion replication in vivo. Similar observations have been made recently in an experimental system of retroviral-induced spongiform degeneration, a model that is neuropathologically related to TSE (Dimcheff et al., 2003a). However, the ER stress response observed in our scrapie model seems to be particular to this type of diseases, because not all of the common features of ER stress induction are observed, such as GADD153/CHOP, heat shock proteins, and calnexin induction.
In general, the induction of ER chaperones is implicated in the protection against cell death after stress (Liu et al., 1997; Reddy et al., 1999; Tanaka et al., 2000; Fischer et al., 2002; Rao et al., 2002; Sullivan et al., 2003). In this respect, our data suggest that PrPSC is recognized by the cell as a stress factor. ER chaperones are likely to be implicated in the removal of PrPSC by either correcting the misfolded protein or promoting its translocation into the cytoplasm for proteasome degradation. This mechanism has been described extensively for wild-type and mutant PrP molecules during synthesis and maturation via ER-Golgi (for review, see Dimcheff et al., 2003a); however, it has not been proposed in relation to the infectious forms of the disease.
To analyze the involvement of Grp58 in response to prion infection, we studied the effect of different expression levels of Grp58 in PrPSC-induced toxicity in vitro. siRNA-mediated knock-out of Grp58 increased the susceptibility of N2a cells to treatment with PrPSC, whereas overexpression of the protein significantly protected cells against PrPSC toxicity. Furthermore, in Grp58siRNA cells, increased activation of caspase-12 was observed after exposure to PrPSC, suggesting that Grp58 is a key survival factor in protecting neurons against PrPSC toxicity. Similar observations were made after treatment of Grp58siRNA cells with other reagents that promote the accumulation of misfolded proteins in the ER. On the other hand, ER stress induced by perturbation in calcium homeostasis was insensitive to the level of Grp58 expression.
The high capacity and low affinity for calcium of Grp78/Bip and Grp94 has led to the suggestion that these chaperones act as a “calcium buffer” and are thus protective against alteration in calcium homeostasis (Michalak et al., 2002). Analysis of the primary structure of Grp58 revealed that its C-terminal region does not contain the putative calcium-binding domain, which is observed in other disulfide isomerases, such as PDI and EndoPDI, and which is thought to protect against calcium-mediated injuries such as ischemia [for alignments, see Sullivan et al. (2003)]. A recent article indicates that Grp58 negatively modulates the calcium oscillation activity mediated by the ER-calcium ATPase in Xenopus oocytes (Li and Camacho, 2004). It was suggested previously that the induction of ER stress by PrPSC treatment could be mediated via signaling, depending on the release of calcium from this organelle (Hetz et al., 2003). However, we observed that, in neuroblastoma cells, the stimulation of calcium release from the ER with thapsigargin, A23187, or histamine is not perturbed in Grp58siRNA or Grp58-overexpressing cells (data not shown). In addition, PrPSC toxicity was not prevented by the intracellular calcium chelation with BAPTA AM (data not shown). Therefore, these data indicate that the mechanism by which PrPSC induces ER stress is more complex. We could speculate on a model in which PrPSC might interact with a cell-surface receptor and be transported to the ER and by itself induce ER stress. This mechanism has been described for bacterial toxins, such as cholera toxin, which bind to lipid raft structures in the plasma membrane and are then internalized by endocytosis toward the ER (Sandvig and van Deurs, 2002). Interestingly, PDI has been implicated in the regulation of the pathogenic effects of cholera toxin (Tsai and Rapoport, 2002). Moreover, it was shown that, in neuroblastoma cells infected with scrapie, modification of intracellular trafficking between Golgi-ER or endosome-Golgi increases PrPSC formation (Beranger et al., 2002), opening up the possibility that PrPSC can reach the ER and be recognized by Grp58. In this sense, we observed that PrP and Grp58 interact, as shown by immunoprecipitation experiments. Strikingly, this interaction was increased in cells infected with RML prions or in cells treated with a proteasome inhibitor. These data suggest that Grp58 may bind preferentially to abnormally folded PrP. The subcellular localization of this interaction remains to be determined. Grp58 is one of the few chaperones that have been shown to be active in the plasma membrane, in which it is located in lipid rafts and binds to N-glycosylated proteins (Turano et al., 2002). Interestingly, PrP is an N-glycosylated protein that contains one intramolecular disulfide bridge and is also located mainly in lipid raft structures (Vey et al., 1996).
Grp58 is a member of the Grp family based on its ability to be induced by glucose deprivation but not because of a structural/functional relationship with other members, such as Grp78 or Grp94, which are not related to PDI activity. Grp58 activity has been extensively characterized in terms of its function in major histocompatibility complex class I assembly and in signal transduction regulation via the STAT (signal transducer and activator of transcription) pathway (Turano et al., 2002), but its function in the ER stress response has so far been speculative. Our findings represent the first time in which a protective role of Grp58 against apoptosis has been described. Grp58 is a chaperone with disulfide isomerase activity. Conditions that affect the formation of disulfide bridges, such as DTT treatment, favor the formation of un-glycosylated PrP (Capellari et al., 1999), a form that has been shown to be more susceptible to conversion into PrPSC. In addition, DTT treatment induces PrP to adopt some PrPSC-like properties, such as proteinase K resistance and insolubility in nondenaturating detergents (Ma and Lindquist, 1999). These data suggest that factors affecting the stability of the disulfide bridge could have an impact on prion replication. Additional support for this hypothesis comes from experiments showing that in vitro amplification of PrPSC is dependent on the presence of free sulfhydryl groups (Lucassen et al., 2003). Finally, reshuffling of cysteine bridges from intramolecular to intermolecular forms has been proposed to play a role on PrP conversion and on the stabilization of the misfolded protein aggregates (Tompa et al., 2002). These findings suggest that the protective activity of Grp58 against PrPSC neurotoxicity may be mediated by a direct interaction between the two proteins, resulting in a reduction of PrPSC misfolding. We are currently generating knock-out and transgenic animals overexpressing Grp58 to analyze the progression of PrPSC replication and neurotoxicity in vivo. Our findings indicate that the induction of Grp58 is directly related to the cellular response triggered by prion replication and suggest that the pharmacological induction of this protein may have clinical benefits in prion-related diseases in which protein misfolding leads to ER stress and cell death.
Footnotes
S.W. was supported by a postdoctoral fellowship from Federation of European Biochemical Societies and the Fond National Suisse de la Recherche Scientifique, and C.H. was supported by a postdoctoral fellowship from Damon Runyon Cancer Research Foundation. We dedicate this work to the memory of Alfonso Alicari Flores, grandfather of Claudio Hetz, who died from brain stroke during the course of this work.
Correspondence should be addressed to Dr. Claudio Soto, Department of Neurology, University of Texas Medical Branch, 301 University Boulevard, Galveston, TX 77555. E-mail: clsoto@utmb.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/252793-10$15.00/0
References
- Barros LF, Stutzin A, Calixto A, Catalan M, Castro J, Hetz C, Hermosilla T (2001) Nonselective cation channels as effectors of free radical-induced rat liver cell necrosis. Hepatology 33: 114-122. [DOI] [PubMed] [Google Scholar]
- Beranger F, Mange A, Goud B, Lehmann S (2002) Stimulation of PrP(C) retrograde transport toward the endoplasmic reticulum increases accumulation of PrP(Sc) in prion-infected cells. J Biol Chem 277: 38972-38977. [DOI] [PubMed] [Google Scholar]
- Breckenridge DG, Germain M, Mathai JP, Nguyen M, Shore GC (2003) Regulation of apoptosis by endoplasmic reticulum pathways. Oncogene 22: 8608-8618. [DOI] [PubMed] [Google Scholar]
- Butler DA, Scott MR, Bockman JM, Borchelt DR, Taraboulos A, Hsiao KK, Kingsbury DT, Prusiner SB (1988) Scrapie-infected murine neuroblastoma cells produce protease-resistant prion proteins. J Virol 62: 1558-1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capellari S, Zaidi SI, Urig CB, Perry G, Smith MA, Petersen RB (1999) Prion protein glycosylation is sensitive to redox change. J Biol Chem 274: 34846-34850. [DOI] [PubMed] [Google Scholar]
- Dimcheff DE, Portis JL, Caughey B (2003a) Prion proteins meet protein quality control. Trends Cell Biol 13: 337-340. [DOI] [PubMed] [Google Scholar]
- Dimcheff DE, Askovic S, Baker AH, Johnson-Fowler C, Portis JL (2003b) Endoplasmic reticulum stress is a determinant of retrovirus-induced spongiform neurodegeneration. J Virol 77: 12617-12629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferri KF, Kroemer G (2001) Organelle-specific initiation of cell death pathways. Nat Cell Biol 3: E255-E263. [DOI] [PubMed] [Google Scholar]
- Fischer H, Koenig U, Eckhart L, Tschachler E (2002) Human caspase 12 has acquired deleterious mutations. Biochem Biophys Res Commun 293: 722-726. [DOI] [PubMed] [Google Scholar]
- Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D (2000) Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell 6: 1099-1108. [DOI] [PubMed] [Google Scholar]
- Hetz C, Russelakis-Carneiro M, Maundrell K, Castilla J, Soto C (2003) Caspase-12 and endoplasmic reticulum stress mediate neurotoxicity of pathological prion protein. EMBO J 22: 5435-5445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtz WA, O'Malley KL (2003) Parkinsonian mimetics induce aspects of unfolded protein response in death of dopaminergic neurons. J Biol Chem 278: 19367-19377. [DOI] [PubMed] [Google Scholar]
- Leyton L, Schneider P, Labra CV, Ruegg C, Hetz CA, Quest AF, Bron C (2001) Thy-1 binds to integrin beta(3) on astrocytes and triggers formation of focal contact sites. Curr Biol 11: 1028-1038. [DOI] [PubMed] [Google Scholar]
- Li Y, Camacho P (2004) Ca2+-dependent redox modulation of SERCA 2b by ERp57. J Cell Biol 164: 35-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Bowes III RC, van de Water B, Sillence C, Nagelkerke JF, Stevens JL (1997) Endoplasmic reticulum chaperones GRP78 and calreticulin prevent oxidative stress, Ca2+ disturbances, and cell death in renal epithelial cells. J Biol Chem 272: 21751-21759. [DOI] [PubMed] [Google Scholar]
- Lucassen R, Nishina K, Supattapone S (2003) In vitro amplification of protease-resistant prion protein requires free sulfhydryl groups. Biochemistry 42: 4127-4135. [DOI] [PubMed] [Google Scholar]
- Ma J, Lindquist S (1999) De novo generation of a PrPSc-like conformation in living cells. Nat Cell Biol 1: 358-361. [DOI] [PubMed] [Google Scholar]
- Mazzarella RA, Marcus N, Haugejorden SM, Balcarek JM, Baldassare JJ, Roy B, Li LJ, Lee AS, Green M (1994) Erp61 is GRP58, a stress-inducible luminal endoplasmic reticulum protein, but is devoid of phosphatidylinositide-specific phospholipase C activity. Arch Biochem Biophys 308: 454-460. [DOI] [PubMed] [Google Scholar]
- Mehmet H (2000) Caspases find a new place to hide. Nature 403: 29-30. [DOI] [PubMed] [Google Scholar]
- Michalak M, Robert Parker JM, Opas M (2002) Ca2+ signaling and calcium binding chaperones of the endoplasmic reticulum. Cell Calcium 32: 269-278. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, Yuan J (2000) Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature 403: 98-103. [DOI] [PubMed] [Google Scholar]
- Prusiner SB (1998) Prions. Proc Natl Acad Sci USA 95: 13363-13383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao RV, Peel A, Logvinova A, del Rio G, Hermel E, Yokota T, Goldsmith PC, Ellerby LM, Ellerby HM, Bredesen DE (2002) Coupling endoplasmic reticulum stress to the cell death program: role of the ER chaperone GRP78. FEBS Lett 514: 122-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy RK, Lu J, Lee AS (1999) The endoplasmic reticulum chaperone glycoprotein GRP94 with Ca2+-binding and antiapoptotic properties is a novel proteolytic target of calpain during etoposide-induced apoptosis. J Biol Chem 274: 28476-28483. [DOI] [PubMed] [Google Scholar]
- Russelakis-Carneiro M, Saborio GP, Anderes L, Soto C (2002) Changes in the glycosylation pattern of prion protein in murine scrapie. Implications for the mechanism of neurodegeneration in prion diseases. J Biol Chem 277: 36872-36877. [DOI] [PubMed] [Google Scholar]
- Sandvig K, van Deurs B (2002) Transport of protein toxins into cells: pathways used by ricin, cholera toxin and Shiga toxin. FEBS Lett 529: 49-53. [DOI] [PubMed] [Google Scholar]
- Sitia R, Braakman I (2003) Quality control in the endoplasmic reticulum protein factory. Nature 426: 891-894. [DOI] [PubMed] [Google Scholar]
- Soto C (2003) Unfolding the role of protein misfolding in neurodegenerative diseases. Nat Rev Neurosci 4: 49-60. [DOI] [PubMed] [Google Scholar]
- Soto C, Kascsak RJ, Saborio GP, Aucouturier P, Wisniewski T, Prelli F, Kascsak R, Mendez E, Harris DA, Ironside J, Tagliavini F, Carp RI, Frangione B (2000) Reversion of prion protein conformational changes by synthetic beta-sheet breaker peptides. Lancet 355: 192-197. [DOI] [PubMed] [Google Scholar]
- Sullivan DC, Huminiecki L, Moore JW, Boyle JJ, Poulsom R, Creamer D, Barker J, Bicknell R (2003) EndoPDI, a novel protein-disulfide isomerase-like protein that is preferentially expressed in endothelial cells acts as a stress survival factor. J Biol Chem 278: 47079-47088. [DOI] [PubMed] [Google Scholar]
- Tanaka S, Uehara T, Nomura Y (2000) Up-regulation of protein-disulfide isomerase in response to hypoxia/brain ischemia and its protective effect against apoptotic cell death. J Biol Chem 275: 10388-10393. [DOI] [PubMed] [Google Scholar]
- Tompa P, Tusnady GE, Friedrich P, Simon I (2002) The role of dimerization in prion replication. Biophys J 82: 1711-1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai B, Rapoport TA (2002) Unfolded cholera toxin is transferred to the ER membrane and released from protein disulfide isomerase upon oxidation by Ero1. J Cell Biol 159: 207-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turano C, Coppari S, Altieri F, Ferraro A (2002) Proteins of the PDI family: unpredicted non-ER locations and functions. J Cell Physiol 193: 154-163. [DOI] [PubMed] [Google Scholar]
- Vey M, Pilkuhn S, Wille H, Nixon R, DeArmond SJ, Smart EJ, Anderson RG, Taraboulos A, Prusiner SB (1996) Subcellular colocalization of the cellular and scrapie prion proteins in caveolae-like membranous domains. Proc Natl Acad Sci USA 93: 14945-14949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneda T, Imaizumi K, Oono K, Yui D, Gomi F, Katayama T, Tohyama M (2001) Activation of caspase-12, an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the ER stress. J Biol Chem 276: 13935-13940. [DOI] [PubMed] [Google Scholar]
- Yoo BC, Krapfenbauer K, Cairns N, Belay G, Bajo M, Lubec G (2002) Overexpressed protein disulfide isomerase in brains of patients with sporadic Creutzfeldt-Jakob disease. Neurosci Lett 334: 196-200. [DOI] [PubMed] [Google Scholar]