

Abstract
The activity-dependent remodeling of postsynaptic structure is a fundamental process underlying learning and memory. Insulin receptor substrate p53 (IRSp53), a key player in cytoskeletal dynamics, is enriched in the postsynaptic density (PSD) fraction, but its significance in synaptic functions remains unclear. We report here that IRSp53 is accumulated rapidly at the postsynaptic sites of cultured hippocampal neurons after glutamate or NMDA stimulation in an actin cytoskeleton-dependent manner. Pharmacological profiles showed that a PKC inhibitor, but not other kinase inhibitors, specifically suppressed the synaptic translocation of IRSp53 in response to NMDA, and the selective activation of PKC with phorbol ester markedly induced the synaptic translocation. Reverse transcriptase-PCR and Western blotting showed that IRSp53-S is the major isoform expressed in cultured hippocampal neurons. The synaptic targeting of IRSp53-S was found to be mediated through N-terminal coiled-coil domain and the PDZ (PSD-95/Discs large/zona occludens-1)-binding sequence at its C-terminal end and regulated by the PKC phosphorylation of its N terminus. In electrophysiological experiments, overexpression of IRSp53-S wild type and IRSp53-S mutant that is spontaneously accumulated at the postsynaptic sites enhanced the postsynaptic function as detected by an increased miniature EPSC amplitude. These data suggest that IRSp53 is involved in NMDA receptor-linked synaptic plasticity via PKC signaling.
Keywords: actin cytoskeleton, EPSC, hippocampus, postsynaptic density, AMPA receptor, synaptic targeting
Introduction
Excitatory synapses are formed through a complex assembly process, in which glutamatergic receptors are assembled at the postsynaptic membrane of spine heads (Harris and Kater, 1994). When viewed by electron microscopy, asymmetric excitatory synapses are characterized by a thick, electron-dense structure underlying the postsynaptic membrane, termed the postsynaptic density (PSD), which is composed of glutamate receptors, the PSD scaffold and cytoskeletal proteins, and signaling molecules (Sheng and Kim, 2002; McGee and Bredt, 2003). The PSD undergoes rapid and remarkable remodeling of its structure and function in response to synaptic input (Toni et al., 1999). This structural plasticity, mediated through the molecular reorganization of the PSD constituents described above, is the fundamental molecular basis for information storage and processing in the brain (Yuste and Bonhoeffer, 2001; Ehlers, 2003).
Insulin receptor substrate p53 (IRSp53), which was originally identified as a substrate for insulin receptor tyrosine kinase, is expressed highly in the brain and is a constituent of the PSD (Yeh et al., 1996; Abbott et al., 1999). The IRSp53 protein contains several protein-protein interacting domains: an N-terminal coiled-coil/IMD domain (IRSp53/missing in metastasis homology domain), partial Cdc42/Rac interactive-binding (CRIB) domain, proline-rich domain, Src homology 3 (SH3) domain, and a tryptophan-tryptophan (WW)-binding domain (WW-BD). One of the 3′-terminal splicing variants is the IRSp53-S isoform, which contains a C-terminal PDZ (PSD-95/Discs large/zona occludens-1)-binding sequence (PDZ-BS) (Hori et al., 2003; Yamagishi et al., 2004). In fibroblasts or neuroblastoma cells, IRSp53 is activated by the Rho family small GTPases, Rac and Cdc42, and forms a complex with the actin regulators WAVE2 (Wiskott-Aldrich syndrome protein family Verprolinhomologous protein) and Mena (Miki et al., 2000; Govind et al., 2001; Krugmann et al., 2001). These complexes enhance membrane ruffling and filopodial extension, suggesting a key role for IRSp53 in cortical actin dynamics. Although the molecular characteristics of IRSp53 have been elucidated over the past few years, most functional studies of this protein have used an overexpression strategy in non-neuronal cells. The significance of IRSp53 in neurons, therefore, remains unclear.
To identify PSD constituents and their functions, we cloned several hybridoma cell lines producing PSD monoclonal antibodies (mAbs). Using these mAbs, we isolated an EVH1 (N-terminal Enabled/Vasp homology 1)- and unique leucine-zipper motif-containing PSD scaffold protein, PSD-Zip45 (Homer 1c) (Sun et al., 1998; Tadokoro et al., 1999; Okabe et al., 2001; Ebihara et al., 2003; Usui et al., 2003), and a three leucine-zipper motif- and coiled-coil domain-containing novel PSD protein, PSD-Zip70 (Konno et al., 2002). Using the mAb 149H, we identified IRSp53, which colocalized with its binding partner, mammalian LIN-seven (MALS), at cell-cell contacts in an actin cytoskeleton-dependent manner (Hori et al., 2003). Here, we found that in cultured hippocampal neurons, activation of the NMDA-type glutamate receptor induced the synaptic translocation of endogenous IRSp53 in an actin cytoskeleton-dependent manner. N-terminal phosphorylation of IRSp53 by PKC regulated its synaptic translocation. We also demonstrated that the synaptic targeting of IRSp53-S occurred via its PDZ-binding sequence and that the synaptic translocation of IRSp53-S enhanced synaptic activity as detected by increased miniature EPSCs (mEPSCs). This is a first report demonstrating the activity-dependent synaptic translocation of IRSp53 and its significance in synaptic function.
Materials and Methods
Primary antibodies. Primary antibodies used for this study include rabbit polyclonal IRSp53 antibodies (Hori et al., 2003) and an anti-bassoon mAb, which was isolated from a hybridoma cell line (336H) cloned by a previously described procedure (Sun et al., 1998). An anti-PSD-95 mAb (clone 6G6-1C9; Affinity BioReagents, Golden, CO), anti-microtubule-associated protein 2 (MAP-2) mAb (clone HM-2; Sigma, St. Louis, MO), anti-GABAA receptor mAb (Upstate Biotechnology, Lake Placid, NY), rabbit polyclonal and monoclonal (M2) anti-FLAG (Sigma), polyclonal anti-green fluorescent protein (GFP) (Molecular Probes, Eugene, OR), and anti-phosphotyrosine (clone 4G10; Upstate Biotechnology) antibodies were purchased.
Plasmid construction. All constructs were amplified by PCR and subcloned into the mammalian expression vector pcDNA3.1(+)-FLAG (modified from Invitrogen, San Diego, CA), pEF-BOS (Mizushima and Nagata, 1990), pCAGGS-FLAG modified from pCAGGS (Niwa et al., 1991), or pECFP-N1 (Clontech, Cambridge, UK) using wild-type pcDNA3.1(+)-FLAG-IRSp53-S (Hori et al., 2003) or pSP64 poly(A)-PSD-95 (Iwamoto et al., 2004) as templates. The N-terminal half of IRSp53-S (IRSp53-S-N-half; residues 1-270), N terminus (IRS-N-term; residues 1-180), central region (IRS-central; residues 180-340), and C terminus (IRS-C-term; residues 322-522) were amplified by PCR and ligated into the pcDNA3.1(+)-FLAG vector. A series of deletion mutants lacking amino-acid residues 375-438 (IRSp53-S-ΔSH3), 270-287 and 375-438 (IRSp53-S-ΔPro+ΔSH3), 375-438 and 468-472 (IRSp53-S-ΔSH3+ΔWW-BD), 375-438 and 517-522 (IRSp53-S-ΔSH3+ ΔPDZ-BS), or 250-507 (N-half+PDZ-BS), and a set of serine-to-alanine point mutations [S27A, S158A, S169A; IRSp53-S(3)A, and N-S(3)A] were generated by site-directed mutagenesis. The sequences of all the constructs were confirmed by DNA sequence analysis.
Cell culture and transfection. COS-7 and human embryonic kidney 293 (HEK293T) cells were maintained in DMEM supplemented with 10% fetal calf serum and transfected with TransIT-LT1 (Mirus, Madison, WI). Hippocampal neurons were prepared from rat brains at embryonic day 18 as described previously with some modifications (Konno et al., 2002). The dispersed neurons were plated at a density of 7500-10,000 cells/cm2 (for immunocytochemical studies) on cover glasses and at 25,000 cells/cm2 (for immunoblotting and isolation of total RNA) on 60 mm Petri dishes and maintained in glial-conditioned MEM containing 2% B27 supplement (Invitrogen). After 1 week, one-half of the medium was changed to neurobasal medium (Invitrogen) containing 2% B27 supplement and 0.5 mm l-glutamine. Plasmid DNAs (5-25 ng/μl) were microinjected through glass capillaries into the nuclei of neurons using a micromanipulator (Narushige, Tokyo, Japan). After 12-18 h, the neurons were fixed for immunocytochemistry as described below.
Western blot analysis and reverse transcriptase-PCR. The lysates of COS7 cells transfected with pEF-BOS-IRSp53-S, hippocampal neurons cultured on 60 mm dishes at 21 d in vitro (DIV), or the PSD fraction prepared from adult rat brains (Konno et al., 2002) were solubilized in SDS sample buffer and separated by SDS-PAGE. Proteins were transferred onto a nitrocellulose membrane, immunoblotted with an anti-IRSp53 antibody (1:5000), and visualized using peroxidase-conjugated secondary antibody (Amersham Biosciences, Arlington Heights, IL) followed by ECL (Amersham Biosciences). The expression levels of the mRNAs for the IRSp53-S, IRSp53-T, and IRS-58 isoforms in cultured hippocampal neurons were quantified by reverse transcriptase-PCR (RT-PCR). The total RNAs were extracted from cultured hippocampal neurons at 19 DIV using Triazol reagent (Invitrogen), and oligo-dT19-primed single-stranded cDNAs were synthesized using Super Script II (Invitrogen). Heat-denatured single-stranded cDNAs were subjected to PCR using ExTaq DNA polymerase (TaKaRa, Tokyo, Japan) and primer sets specific for the rat glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and IRSp53 isoforms. The following primers were used: the sense primer encoding the common region of all rat IRSp53 isoforms, 5′-CTCCAAGTCCAACCTGGTCA-3′; IRSp53-S antisense primer, 5′-ATCTCGAGTCACACTGTGGACACCAG-3′; IRSp53-T antisense primer, 5′-GGCTGATCTGTCATTGGTCA-3′; IRS-58 antisense primer, 5′-AGAGGGGCTGATCTGTCATT-3′; GAPDH sense primer, 5′-GTGCTGAGTATGTCGTGGAGTC-3′; and GAPDH antisense primer, 5′-GTTGTTATGGGGTCTGGGATGG-3′. The PCR products were sampled at two-cycle intervals between 20 and 28 cycles and separated by electrophoresis on 1% agarose gels. All of the PCR-positive products obtained from 40 cycle samples were isolated from the gel using GFX PCR DNA and a Gel Band Purification kit (Amersham Biosciences) and then confirmed by sequencing.
Phosphorylation of IRSp53 in vitro. COS-7 cells were transfected with FLAG-tagged IRSp53-S wild type, IRS-N-term, IRS-central, or IRS-C-term and harvested in lysis buffer containing 50 mm Tris-Cl, pH 7.5, 150 mm NaCl, 1 mm EDTA, 1% Triton X-100, 10 mm NaF, 1 mm PMSF, 10 μg/ml pepstatin, and 10 μg/ml leupeptin. IRSp53 was immunoprecipitated using anti-FLAG mAb-coupled protein-G-Sepharose beads (Amersham Biosciences) for 2 h at 4°C. The beads were then washed four times with the lysis buffer containing 300 mm NaCl and 0.1% sodium deoxycholate (DOC) and resuspended in 50 μl of kinase reaction mixture [20 mm HEPES, pH 7.4, 1.3 mm CaCl2, 10 mm MgCl2, 1 mm DTT, 200 μg/ml phosphatidylserine, 20 μg/ml diacylglycerol, 1 μm PKI (a specific PKA inhibitor peptide; Sigma), 0.5 U/ml PKCs purified from rat brain (Calbiochem, La Jolla, CA), 0.25 mm ATP, and 10 μCi [γ-32P]ATP]. The reaction was performed at 30°C for 30 min with or without PKC inhibitor [bisindolylmaleimide-I (Bis-I); 1 μm] and terminated by adding SDS sample buffer. One-half of each sample was loaded onto a gel, separated by SDS-PAGE, and subjected to autoradiography. For the quantification of precipitated IRSp53, the other half of each sample was analyzed by immunoblotting using polyclonal anti-FLAG (1:5000) as the primary antibody.
Phosphorylation of IRSp53 in cells. HEK293T cells transiently transfected with or without FLAG-IRSp53-S were grown for 24 h and starved in serum-free DMEM overnight. After being starved for 4 h in serumfree, phosphate-free DMEM (Invitrogen), the cells were incubated with 0.1 μCi/ml [32P]orthophosphate (Amersham Biosciences) for 2 h before stimulation with 100 nm 12-O-tetradecanoylphorbol-13-acetate (TPA) plus 100 nm okadaic acid (Calbiochem) or 0.2 μm insulin (Sigma) plus 0.5 mm vanadate for 30 min. After stimulation, the plates were washed with ice-cold PBS, and the cells were harvested in lysis buffer containing phosphatase inhibitor mixture (Sigma). IRSp53 was immunoprecipitated with anti-FLAG mAb-coupled protein-G-Sepharose beads, and the immunoprecipitates were dissolved in SDS sample buffer. The radioactivity and amount of IRSp53 in the immunoprecipitates were determined as described above. Phosphorylation of tyrosine residues was detected using the anti-phosphotyrosine antibody 4G10 (1:3000).
Pharmacological treatment. Neurons were preincubated in 1 μm TTX (Sankyo, Tokyo, Japan) for 30 min and then stimulated with 100 μm glutamate plus 10 μm glycine, 50 μm NMDA (Tocris Cookson, Bristol, UK), or 100 nm TPA (Sigma) for the indicated time, as described in Results and figure legends. The other pharmacological agents were used at the following concentrations (in μm): 100 AP-5 (Tocris Cookson), 50 CNQX (Tocris Cookson), 20 AP-3 (Tocris Cookson), 5 latrunculin A (Molecular Probes), 50 genistein (Calbiochem), 10 lavendustin A (Calbiochem), 10 1-[N, O-bis(5-isoquinolinesulfonyl)-N-methyl-l-tyrosyl]-4-phenylpiperazine (KN-62) (Calbiochem), 10 4-amino-5-(4-chlorophenyl) -7(t-butyl)pyrazol(3,4-d)pyramide (PP2) (Calbiochem), 5 nifedipine (Tocris Cookson), 50 picrotoxin (Tocris Cookson), and 1 bisindolylmaleimide-I (Calbiochem). With the exception of latrunculin A, the pharmacological agents were applied to the culture medium for 30-120 min before stimulation.
Detergent extraction. For immunocytochemistry, hippocampal neurons grown on cover glasses were washed with Ca2+- and Mg2+-containing PBS and extracted with Triton X-100 buffer (50 mm NaCl, 10 mm PIPES, pH 6.8, 3 mm MgCl2, 0.5% Triton X-100, and 300 mm sucrose) on ice for 10 min before fixation. Immunocytochemical procedures were as described below. For biochemical study, hippocampal neurons grown on 60 mm Petri dishes were untreated or treated with 50 μm NMDA for 10 min, and the cells were solubilized in SDS sample buffer (total lysate). To prepare the Triton X-100-insoluble fraction, stimulated cells were subsequently incubated with Triton X-100 buffer on ice for 10 min. After extraction, the cells were washed with Ca2+- and Mg2+-containing PBS and collected in the same volume of SDS sample buffer (insoluble fraction). Equal amounts of sample solutions were then subjected to SDS-PAGE and transferred to nitrocellulose membranes. An anti-IRSp53 antibody was used as the primary antibody and visualized using a peroxidase-conjugated secondary antibody followed by ECL. The measured IRSp53 level in the insoluble fraction was normalized to the total amount of IRSp53 protein in the whole-cell lysate and quantified by NIH Image software.
Immunoprecipitation. HEK293T cells transiently cotransfected with FLAG-IRSp53 wild type or S(3)A mutant and PSD-95-cyan fluorescent protein (CFP) were grown in serum-free DMEM for 24 h. After stimulation with 100 nm TPA or vehicle for 30 min, the cells were washed with ice-cold PBS and then harvested in radioimmunoprecipitation assay (RIPA) buffer containing 50 mm Tris-Cl, pH 8.0, 150 mm NaCl, 5 mm EDTA, 1% NP-40, 0.5% DOC, 0.05% SDS, 10 mm NaF, 1 mm PMSF, 10 mg/ml pepstatin, 10 mg/ml leupeptin, and phosphatase inhibitor mixture. IRSp53 was immunoprecipitated using anti-FLAG mAb-coupled protein-G-Sepharose beads for 3 h at 4°C. After washing with RIPA buffer, all precipitates were solubilized with SDS sample buffer and detected by immunoblotting using anti-FLAG and anti-GFP polyclonal antibodies, respectively.
Immunocytochemistry and image analysis. All analyses were performed at 18-24 DIV. Immunostaining was performed as described previously (Usui et al., 2003). For all immunocytochemical experiments except F-actin staining with phalloidin, the neurons were fixed in 4% formaldehyde/4% sucrose in PBS for 40 min on ice. For the staining of MAP-2/F-actin shown in Figure 3, the neurons were fixed in 4% formaldehyde/4% sucrose in PBS for 20 min at room temperature. Neurons were immunolabeled overnight using the following primary antibodies: anti-IRSp53 (1:1000), anti-PSD-95 (1:2000), anti-bassoon (1:500), anti-GABAA receptor (1:500), and polyclonal anti-FLAG (1:5000) antibodies. For F-actin staining, the neurons were incubated with Alexa Fluor 568-conjugated phalloidin (2 μg/ml; Molecular Probes) in 3% BSA for 30 min at room temperature. Fluorescence images were acquired using a cooled CCD camera (Roper Scientific, Tucson, AZ) mounted on an Olympus (Melville, NY) IX-70 microscope. Morphometric studies were performed using MetaMorph imaging software (Universal Imaging Corporation, West Chester, PA). To compare the expression levels of exogenous and endogenous IRSp53, transfected cells were labeled immunofluorescently with an anti-IRSp53 antibody, and the total pixel intensity of transfected versus untransfected cells was compared. The synapse/dendrite fluorescence intensities (see Figs. 1C, 4 B) were analyzed as the ratios of the average fluorescence intensities of IRSp53 in the synapses colocalized with the PSD-95 immunoclusters to those in the adjacent dendrites. Statistical significance was determined using Student's t test.
Figure 3.
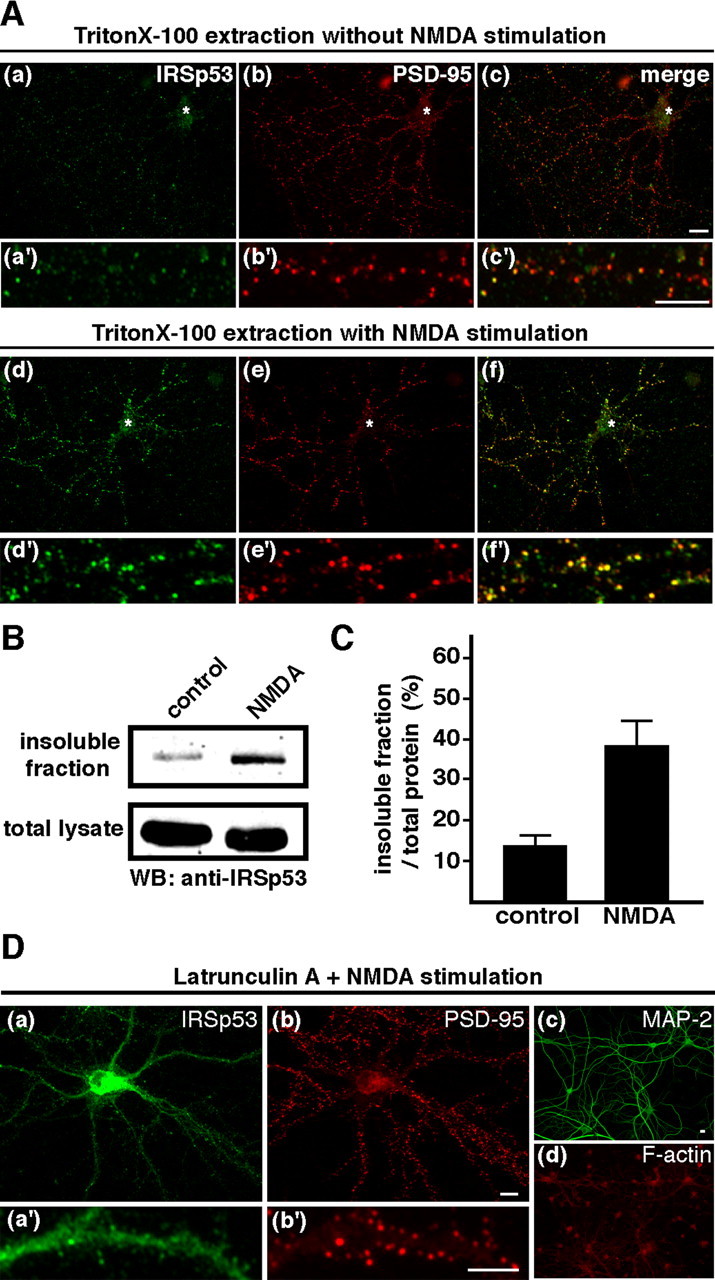
NMDA-induced change in IRSp53 distribution. A, Neurons treated with (Ad-Af, Ad′-Af′) or with out (Aa-Ac, Aa′-Ac′) 50 μm NMDA for 10 min were treated with 0.5% Triton X-100 before fixation. These cells were subsequently double labeled for IRSp53 (green) and PSD-95 (red). Merged images are shown in the right panels. Asterisks indicate the soma. B, Western blot (WB) analysis of untreated (control) and NMDA-treated (NMDA) neurons. Neurons were extracted with Triton X-100, and equivalent volumes of the lysate (bottom) and the insoluble fraction (top) were immunoblotted using an anti-IRSp53 antibody. C, The amount of IRSp53 in the insoluble fraction of hippocampal neurons with (NMDA) or without (control) 50 μm NMDA stimulation normalized to the total amount of protein in the whole-cell lysate, quantified by densitometry. Error bars represent SEM. Scale bar, 10 μm. D, Requirement of F-actin for the synaptic translocation of IRSp53. Neurons were treated with 5 μm latrunculin A for 16 h to depolymerize F-actin before the application of NMDA. After stimulation with 50 μm NMDA for 10 min, neurons were double labeled for IRSp53 (Da, Da′) and PSD-95 (Db, Db′), MAP-2 (Dc), or F-actin (Dd). Scale bars, 10 μm.
Figure 1.

Translocation of IRSp53 to spines mediated through NMDA receptor activation. A, Rat hippocampal neurons cultured for 3 weeks with (Ad-Af, Ad′-Af′) or without (Aa-Ac, Aa′-Ac′) 100 μm glutamate stimulation for 10 min were double labeled with anti-IRSp53 (green) and anti-PSD-95 (red) antibodies. The merged images are shown in the right panels (Ac, Ac′, Af, Af′). B, Hippocampal neurons in culture were pretreated with 100 μm AP-5 (Bg-Bi), 50 μm CNQX (Bj-Bl), or 20 μm AP-3 (Bm-Bo) before 100 μm glutamate (Glu) stimulation. After stimulation for 10 min, the cells were labeled for IRSp53 (green) and PSD-95 (red). C, Summary of the spine-targeting intensities of IRSp53. The targeting intensities of IRSp53 are expressed as spine/dendrite fluorescence intensities as shown in A and B. Three to six independent experiments were performed for each condition, and a total of 300 spines from 8 to 10 neurons were selected randomly. Error bars represent SEM. p < 0.001; asterisks indicate Student's t test values against control. Scale bar, 10 μm.
Figure 4.
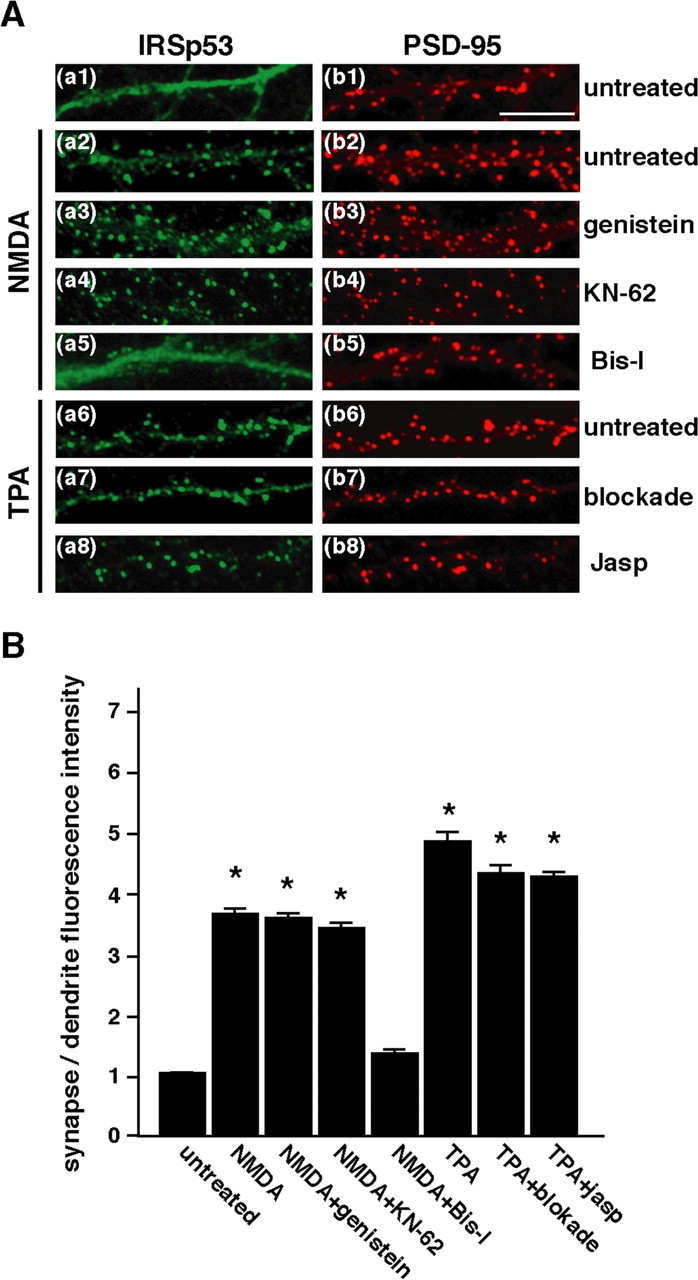
Involvement of PKC activation in the synaptic translocation of IRSp53. A, Neurons were left untreated (Aa1, Ab1), treated with 50 μm NMDA (Aa2-Aa5, Ab2-Ab5) for 10 min, or treated with 100 nm TPA (Aa6-Aa8, Ab6-Ab8) for 30 min and double labeled for IRSp53 (left) and PSD-95 (right). Where indicated, pretreatment with pharmacological agents was performed before stimulation. Blockade, Blocking mixture containing AP-5/CNQX/AP-3/picrotoxin/nifedipine/TTX; Jasp, jasplakinolide. B, The pharmacological effects on the synaptic targeting of IRSp53 are shown in A. The synaptic targeting intensities of IRSp53 are expressed as shown in Figure 1C. Four to six independent experiments were performed for each condition, and a total of 400 synapses from 10 to 12 neurons were randomly selected. Error bars represent SEM. p < 0.001; asterisks indicate Student's t test values against control. Scale bar, 10 μm.
Electrophysiology. To analyze mEPSCs in transfected neurons, wholecell recordings were made as described previously (Yasuda et al., 2003). Briefly, neurons from the same culture preparation were transfected with GFP, GFP and FLAG-IRSp53-S wild type, GFP and FLAG-IRSp53-S-ΔSH3, or GFP and FLAG-IRSp53-S-ΔSH3+ΔPDZ-BS. After 24 h, these cells were continuously superfused with an external solution containing 150 mm NaCl, 2.5 mm KCl, 2 mm MgCl2, 2 mm CaCl2, 10 mm HEPES, 10 mm glucose, 0.1 mm picrotoxin, and 0.5 μm TTX, pH 7.4. Where indicated, the IRSp53-S wild-type-transfected neurons were treated with 100 nm TPA for 1 h before recording. Whole-cell recording pipettes (5-7 MΩ) were filled with an internal solution containing the following (in mm): 130 Cs-MeSO4, 10 HEPES, 0.2 EGTA, 8 NaCl, 4 Mg-ATP, 0.3 Na3-GTP, and 5 N-ethyl bromide quaternary salt Cl, pH 7.2 with CsOH, osmolarity adjusted to 280-290 mOsm. GFP-positive cells were voltage clamped at -70 mV, and the input and series resistances were monitored continuously on-line. If the series resistance was >25 MΩ, the cell was discarded. The mEPSCs were recorded at 2 kHz with Igor Pro software (WaveMetrics, Lake Oswego, OR). Usually >300 consecutive events were collected and then analyzed with Mini Analysis software (Synaptosoft, Decatur, GA). All mEPSCs above a threshold value (4.5 pA) were included in the data analysis, and each mEPSC was verified visually. The mean mEPSC amplitudes and frequencies were calculated and statistically analyzed with one-way ANOVA followed by Fisher's test.
Results
Synaptic translocation of IRSp53 mediated through the NMDA receptor
It has been reported that IRSp53 is enriched in the PSD fraction (Abbott et al., 1999). The localization of IRSp53 in cultured hippocampal neurons is, however, complicated (Abbott et al., 1999; Bockmann et al., 2002; Soltau et al., 2004). This may be because of a usage of different culture conditions. To investigate the function of IRSp53, we immunolabeled cultured hippocampal neurons (21 DIV) with anti-IRSp53 and anti-PSD-95 antibodies. Although punctates of IRSp53 immunolabeling colocalized with PSD-95 immunoclusters were observed in a minor population of neurons, likely as reported previously (Bockmann et al., 2002; Soltau et al., 2004), most of IRSp53 immunolabels were distributed through dendrites as well as spines in a major population of neurons (Fig. 1Aa,Aa′). We next examined the synaptic activity-dependent localization of IRSp53. After glutamate stimulation for 10 min, IRSp53 was localized predominantly to the dendritic spines, with PSD-95s present (Fig. 1Ad-Af,Ad′-Af′). To characterize the glutamate receptor-mediated IRSp53 accumulation, neurons were pretreated with glutamate receptor antagonists before stimulation. Treatment with the NMDA receptor antagonist AP-5 markedly suppressed the accumulation of IRSp53 in spines (Fig 1Bg-Bi), whereas neither the AMPA receptor antagonist CNQX nor the metabotropic glutamate receptor antagonist AP-3 did (Fig. 1Bj-Bo), suggesting NMDA receptor activation was required for the IRSp53 accumulation in spines.
We next analyzed the NMDA receptor-mediated IRSp53 accumulation in spines as a function of time. Within 3 min after NMDA stimulation, IRSp53 rapidly accumulated in the spines. This accumulation was maximal at 10 min and then decreased thereafter (Fig. 2A). A similar time-dependent accumulation was observed when the neurons were treated with glutamate (data not shown). To further characterize the accumulation of IRSp53 in spines, neurons were double labeled with anti-IRSp53 and antisynaptic marker antibodies. When neurons were stimulated with NMDA, the immunoclusters of IRSp53 along the dendrites completely overlapped with those of PSD-95, a postsynaptic marker (Fig. 2Ba-Bc,Ba′-Bc′), and partially overlapped with those of bassoon, a presynaptic maker (Fig. 2Bd-Bf). IRSp53 did not, however, overlap with the GABAA receptor, an inhibitory postsynaptic marker (Fig. 2Bg-Bi). These results suggest that IRSp53 specifically translocates to the postsynaptic sites of excitatory synapses in response to NMDA receptor activation.
Figure 2.
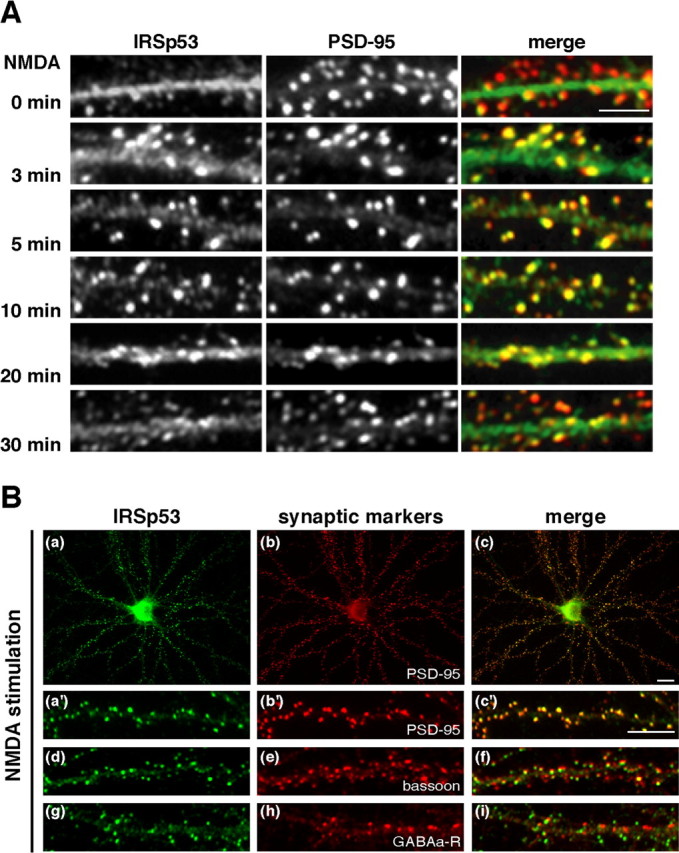
NMDA-induced synaptic translocation of IRSp53 in hippocampal neurons. A, Time course of the synaptic translocation of IRSp53 after NMDA stimulation. Neurons were stimulated with 50 μm NMDA for the indicated times, fixed, and then immunolabeled for IRSp53 (left) and PSD-95 (middle). Merged images are shown in the right panels. B, After stimulation with 50 μm NMDA for 10 min, neurons were double labeled for IRSp53 (Ba, Ba′, Bd, Bg) and PSD-95 (Bb, Bb′), bassoon (Be), or GABAA receptor (GABAa-R; Bh). Merged images are shown in the right panels. Scale bars: A, 5 μm; B, 10 μm.
Involvement of the actin cytoskeleton in the NMDA receptor-mediated synaptic translocation of IRSp53
To investigate the importance of the cytoskeleton in the synaptic translocation of IRSp53, we performed in situ extraction with Triton X-100. The detergent extraction of living cells has been used to assess molecular interactions at the PSD in cultured hippocampal neurons (Allison et al., 1998) and the membrane cytoskeleton at cell-cell contacts of non-neuronal cells (Hori et al., 2003). In untreated neurons, most of the cellular IRSp53 was extracted by Triton X-100, leaving only a small amount of it at synaptic sites (Fig. 3Aa-Ac,Aa′-Ac′). After NMDA stimulation, intensely labeled IRSp53 immunoclusters appeared at synaptic sites that were resistant to Triton X-100 extraction (Fig. 3Ad-Af,Ad′-Af′). We then determined the amount of IRSp53 protein in the Triton X-100-insoluble fraction of cultured hippocampal neurons with or without NMDA stimulation. Western blotting showed that, consistent with the immunocytochemistry results (Fig. 3A), the amount of IRSp53 in the Triton X-100-insoluble fraction increased after NMDA stimulation (Fig. 3B,C). We also examined the involvement of the actin cytoskeleton in the synaptic translocation of IRSp53. Treatment with latrunculin A for ∼16 h almost completely disrupted the F-actin (Fig. 3Dd) but showed no changes in microtubule organization (Fig. 3Dc). NMDA stimulation did not induce the synaptic translocation of IRSp53 in neurons pretreated with latrunculin A (Fig. 3Da,Da′). Together, these results suggest that the actin cytoskeleton is involved in the NMDA receptor-mediated synaptic translocation of IRSp53.
NMDA-induced synaptic translocation of IRSp53 via PKC signaling
Using a pharmacological approach, we screened for signaling molecules that could potentially be involved in the NMDA receptor-mediated synaptic translocation of IRSp53. It has been reported that IRSp53 is tyrosine phosphorylated by the insulin (or its related) receptor tyrosine kinases and by Src family tyrosine kinases. We verified that the tyrosine phosphorylation of IRSp53 was involved in its synaptic targeting. Treatment with genistein, which is a broad inhibitor of tyrosine kinases, showed no significant effect on the NMDA receptor-mediated synaptic translocation of IRSp53 (Fig. 4Aa3). Furthermore, neither lavendustin A, another kind of tyrosine kinase inhibitor, nor PP2, a specific Src family tyrosine kinase inhibitor, had an effect (data not shown). We next examined the possible role of other kinases that have been implicated in NMDA receptor signaling. Treatment with the calcium/calmodulin-dependent protein kinase II (CaMKII) inhibitor KN-62 did not inhibit the synaptic translocation of IRSp53 (Fig. 4Aa4). Notably, a specific PKC inhibitor, Bis-I, almost completely blocked the NMDA-induced IRSp53 translocation (Fig. 4Aa5). Treatment with a PKC agonist, TPA, induced a more remarkable synaptic translocation of IRSp53 than did glutamate or NMDA treatment (Fig. 4Aa6,B). The TPA-induced synaptic translocation of IRSp53 was sustained for up to 2 h (data not shown).
In addition to postsynaptic PKC activation, phorbol esters have been reported to enhance vesicle fusion at presynaptic sites (Malenka et al., 1986; Parfitt and Madison, 1993). To block the presynaptic and postsynaptic activities, we pretreated neurons with a mixture of inhibitors containing TTX (for voltage-gated sodium channels), nifedipine (for voltage-gated L-type calcium channels), picrotoxin (for GABA receptors), and AP-5, CNQX, and AP-3 (for glutamate receptors) before TPA application. There were no significant effects of these pharmacological agents on the TPA-induced synaptic translocation of IRSp53 (Fig. 4Aa7). Fong et al. (2002) reported that F-actin accumulates in spines after TPA-induced PKC activation. Consistent with this, we demonstrated that the synaptic localization of IRSp53 depends on the actin cytoskeleton (Fig. 3). We then used jasplakinolide, which binds to and stabilizes F-actin (Halpain at al., 1998), to prevent rearrangements of the actin cytoskeleton. No effect of this agent was found on the synaptic translocation of IRSp53 (Fig. 4Aa8). Together, these results indicate that the NMDA receptor-mediated synaptic translocation of IRSp53 specifically depends on the activation of postsynaptic PKC and not on tyrosine kinase, CaMKII activity, or F-actin turnover.
Requirement of the N-terminal half and PDZ-binding sequence of IRSp53 for synaptic targeting
To determine the key domain for the activity-dependent synaptic targeting, we first identified the IRSp53 isoform(s) expressed in cultured hippocampal neurons. Four alternatively spliced isoforms of IRSp53 that vary in the 3′-terminal exon [IRSp53-L, -S (-A), -T, and IRS-58] have been reported (Okamura-Oho et al., 2001). In addition, there is a fifth alternatively spliced isoform, IRSp53-B, which has an identical primary structure to IRSp53-S but lacks 40 amino acids abutting the CRIB/SH3-binding domain (Alvarez et al., 2002). Western blotting using anti-IRSp53 polyclonal antibodies that recognize a common region of all IRSp53 isoforms revealed a single band with 58 kDa in the COS7 lysate expressing recombinant IRSp53-S and a 58/53 kDa doublet in the PSD fraction and the lysate of cultured hippocampal neurons. In hippocampal neurons, there was much less of the 53 kDa isoform than the 58 kDa one. Thus, IRSp53-S, -T, and IRS-58 might be expressed in hippocampal neurons, but IRSp53-L, with a calculated molecular weight of ∼74 kDa, is not (Fig. 5A). We also examined the expression of these three isoforms by RT-PCR using rat cDNA obtained from cultured hippocampal neurons with pairs of oligonucleotides specific for each isoform. Using RT-PCR analysis, all these isoforms were detected at 40 cycles (Fig. 5B). To compare the expression levels, the PCR products of these isoforms were sampled at two-cycle intervals between 20 and 28 cycles. Among these isoforms, IRSp53-S and IRSp53-T were detected, but the expression level of the latter was much lower than that of IRSp53-S. IRS-58 was not detected at these cycles (Fig. 5B). These results indicate that IRSp53-S is the major and unique isoform in hippocampal neurons.
Figure 5.
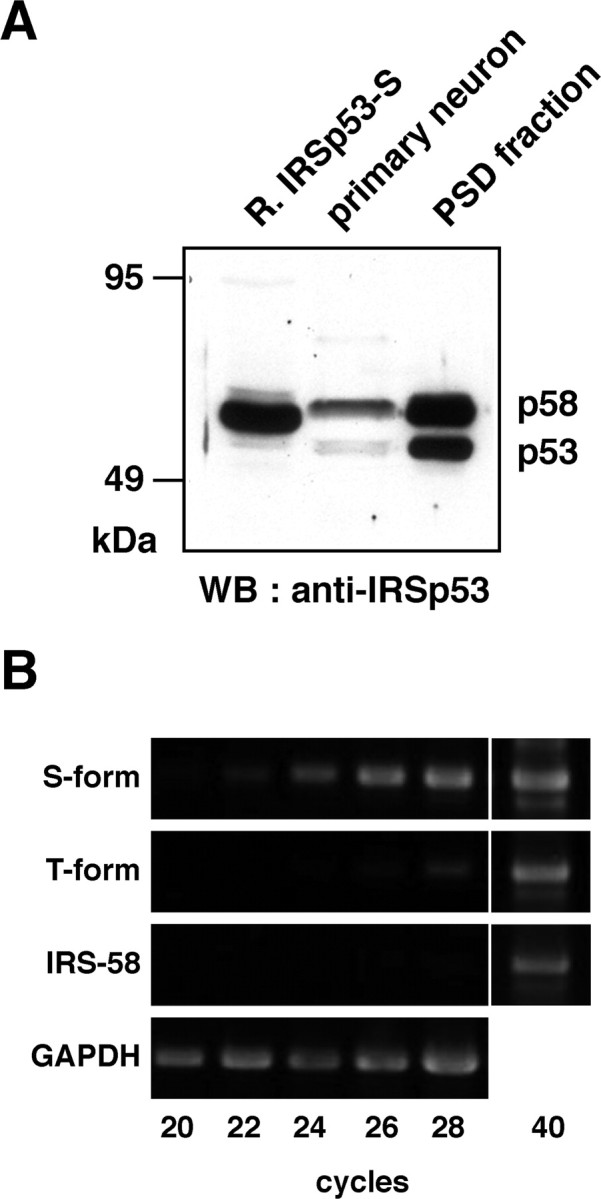
Expression of IRSp53 isoforms in cultured hippocampal neurons. A, The lysates of COS7 cells expressing IRSp53-S (left lane), the whole-cell lysates of cultured hippocampal neurons at 21 DIV (middle lane), and the PSD fraction (right lane) were labeled with an anti-IRSp53 antibody. WB, Western blot. B, Expression of the S-form, T-form, and IRS-58 isoforms in rat hippocampal neurons was analyzed by RT-PCR. Representative results for the RT-PCR products from three separate experiments are shown.
Next, we examined the synaptic targeting domain of IRSp53-S by expressing a series of FLAG-tagged IRSp53-S deletion mutants in hippocampal neurons (Fig. 6). It has been reported that the overexpression of IRSp53 induces drastic changes in cell shape (Govind et al., 2001; Yamagishi et al., 2004). We also confirmed the observation of similar morphological changes at the spines and dendrites of cultured hippocampal neurons by the overexpression of IRSp53 (data not shown). To avoid changing the shape of spines and exceeding the capacity of IRSp53 at postsynapses, only a small amount (5-10 ng/μl) of the expression vector was microinjected into the nuclei of cultured neurons. The expression levels of exogenous IRSp53-S or its deletion mutants were up to ∼2.5 times the level of endogenous IRSp53, as confirmed by anti-IRSp53 antibody (see Materials and Methods). The synaptic targeting of the IRSp53-S variants was defined by double-labeling with PSD-95. Consistent with the endogenous IRSp53, exogenous FLAG-IRSp53-S was targeted specifically to synapses only when neurons were stimulated with NMDA (Fig. 6B, 1-C and 1-S). The N-terminal half of IRSp53-S, which contains the binding domain of the Rho family small GTPases, was not targeted to synapses, with or without NMDA stimulation (Fig. 6B, 2-C and 2-S). Notably, the SH3-deleted mutant (ΔSH3) was specifically targeted to synapses without NMDA stimulation (Fig. 6B, 3-C and 3-S), suggesting that SH3 inhibits the synaptic targeting of IRSp53 in the basal state. Deletion of the WW-binding (ΔSH3+ΔWW-BD) (Fig. 6B, 4-C and 4-S) and prolinerich (ΔPro+ΔSH3) (Fig. 6B, 5-C and 5-S) domains in addition to the SH3 domain did not impair the activity-independent synaptic targeting of the IRSp53-S ΔSH3-deleted mutant, whereas deletion of the PDZ-binding sequence from the SH3-deleted mutant (ΔSH3+ΔPDZ-BS) (Fig. 6B, 6-C and 6-S) resulted in the loss of synaptic targeting; this mutant was rather diffusely distributed within the dendrites and did not respond to NMDA stimulation. In addition, the PDZ-binding-sequence-deleted IRSp53-S mutant was not targeted to synapses in response to the stimulation (ΔPDZ-BS) (Fig. 6B, 7-C and 7-S). To examine whether the PDZ-binding sequence itself is sufficient for synaptic targeting, the C-terminal half of IRSp53 was expressed in neurons. With or without NMDA stimulation, this fragment was diffusely distributed throughout dendrites and spines. A weak accumulation of this fragment at synapses was, however, detected (Fig. 6B, 8-C and 8-S). This result suggests the requirement of an additional domain of IRSp53 for synaptic targeting. Yamagishi et al. (2004) reported that the predicted N-terminal helical stretch (250 amino acids) of IRSp53 binds to F-actin in vitro. We tested another construct that comprises N-terminal half of IRSp53-S and its PDZ-binding sequence (N-half + PDZ-BS). This construct localized efficiently to synapses (Fig. 6B, 9-C and 9-S). We confirmed that the actin cytoskeleton was required for synaptic localization of this construct because pretreatment with latrunculin A inhibited the synaptic targeting of this construct (data not shown). Together, our present results suggest that both PDZ-binding sequence and N-terminal half of coiled-coil domain, which interacts with the actin cytoskeleton, are critical for synaptic targeting.
Figure 6.
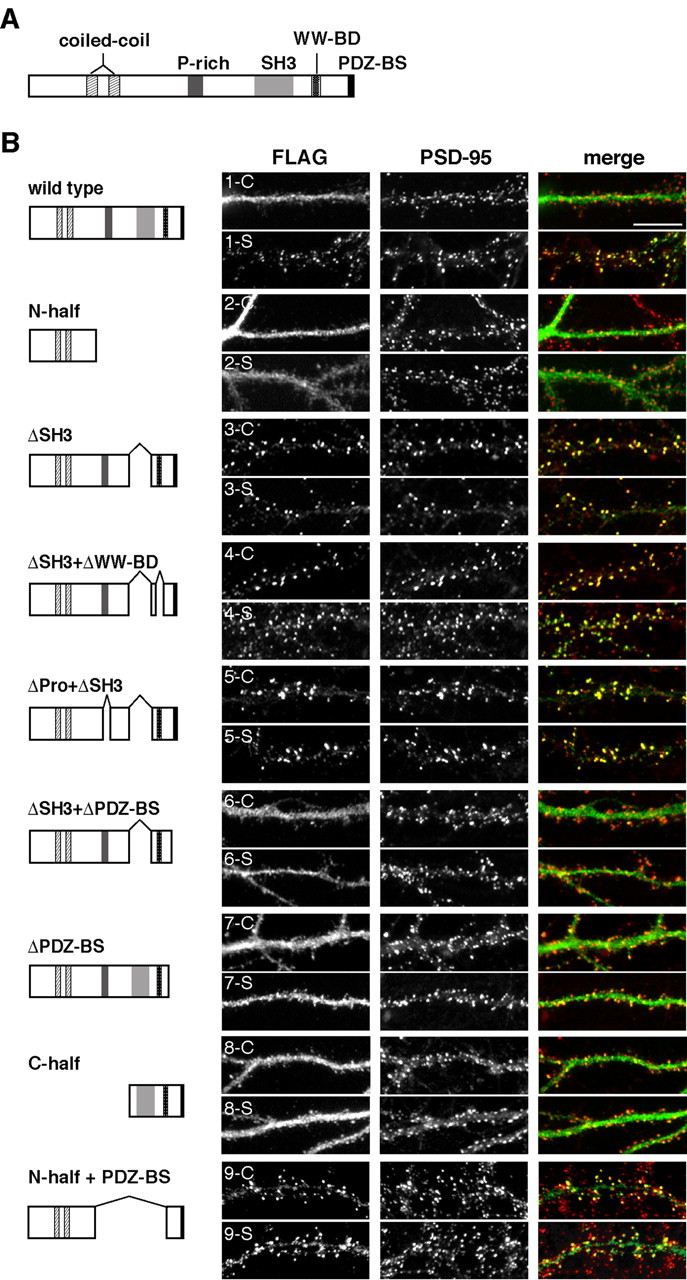
Functional domains of IRSp53-S required for synaptic targeting. A, Schematic diagram of the IRSp53-S domain structure. IRSp53 contains several protein-protein-interacting domains: from the N terminus, a coiled-coil domain, partial CRIB domain, proline-rich (P-rich) domain, SH3 domain, WW-binding domain, and a PDZ-binding sequence (BS) at the C-terminal end. B, Immunofluorescence micrographs show the distribution of transfected FLAG-tagged wild-type IRSp53-S and its deletion mutants in hippocampal neurons (19-22 DIV). The domain structures of the full length and deletions of IRSp53-S are shown to the left of each image. Transfected neurons were fixed in the basal state (control; 1-9-C) or treated with 50 μm NMDA (stimulated; 1-9-S) for 10 min before fixation and double labeled with anti-FLAG (left) and anti-PSD-95 (middle) antibodies. Merged images are shown in the right panels. Scale bar, 10 μm.
Regulation of synaptic targeting of IRSp53 by PKC phosphorylation
To elucidate the molecular mechanism underlying the synaptic targeting of IRSp53, we analyzed the role of IRSp53 phosphorylation by PKC. To verify the phosphorylation of IRSp53 in vivo, we labeled HEK293T cells with [32P]orthophosphate. Under these conditions, FLAG-IRSp53 expressed in HEK293T cells was phosphorylated at a basal level. TPA increased the phosphorylation of FLAG-IRSp53 (Fig. 7Aa1). Western blotting using an anti-phosphotyrosine antibody (4G10) showed that IRSp53 was tyrosine phosphorylated by insulin stimulation but not under the above experimental conditions (Fig. 7Aa3). IRSp53 has nine candidate phosphorylation sites for PKC according to the canonical pattern (S/T)X(R/K), where X is any residue. We performed an in vitro kinase assay using rat brain PKCs and FLAG-tagged IRSp53 immunoprecipitates obtained from IRSp53-transfected COS7 cells in the presence of [γ-32P]ATP. Phosphorylated IRSp53 was detected clearly only when PKC was added, whereas preincubation with bisindolylmaleimide-I suppressed the phosphorylation of IRSp53, indicating that IRSp53 is phosphorylated by PKC (Fig. 7Bb1). To further define the phosphorylation sites for PKC, IRSp53 was divided into three regions, N-terminal (1-180 aa), central (180-340 aa), and C-terminal (320-522 aa) fragments. Although the central fragment does not contain a PKC phosphorylation site, the N- and C-terminal fragments have three and six putative PKC phosphorylation sites, respectively (Ser-27, Ser-158, and Ser-169 in the N-terminal fragment and Thr-349, Ser-396, Ser-419, Ser-441, Ser-475, and Thr-486 in the C-terminal fragment). As shown in Figure 7B, phosphorylation of the N-terminal fragment, but not of the central or C-terminal fragment, was detected (Fig. 7Bb3, left). We also confirmed that the substitution of the three potential residues for PKC phosphorylation (Ser-27, 158, and 169) with alanine [N-S(3)A] significantly reduced the phosphorylation levels (Fig. 7Bb3, right).
Figure 7.
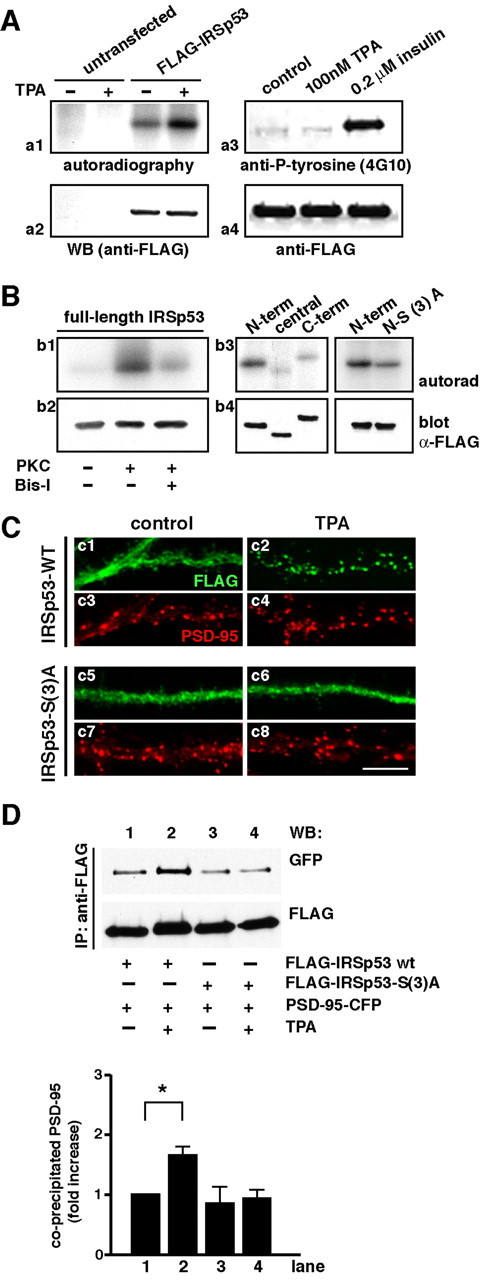
IRSp53 phosphorylation by PKC regulates the synaptic targeting of IRSp53. A, HEK293T cells transfected with FLAG-IRSp53 were labeled with [32P]orthophosphate and treated with 100 nm TPA or vehicle for 30 min. FLAG-IRSp53 was immunoprecipitated from the cell lysates, followed by the detection of [32P]orthophosphate incorporation by autoradiography (Aa1). The amount of IRSp53 protein was determined by immunoblotting with an anti-FLAG antibody (Aa2). WB, Western blot. Under the same conditions as in Aa1 and Aa2, IRSp53 was not tyrosine phosphorylated, as detected with an anti-phosphotyrosine antibody (4G10) (Aa3, Aa4). B, FLAG-tagged full-length IRSp53 (Bb1, Bb2) or IRSp53 fragments [Bb3, Bb4; left, IRS-N-term (1-180 aa), IRS-central (180-322 aa), IRS-C-term (320-522 aa); right, IRS-N-term and the N-term mutant for which the 27th, 158th, and 169th serine residues are substituted with alanine] in the immunoprecipitates were used as a substrate for purified PKC. Where indicated, the phosphorylation assay was performed with bisindolylmaleimide-I, a PKC inhibitor. Protein phosphorylation was visualized by autoradiography (autorad) (Bb1, Bb3). The amount of IRSp53 protein was determined by immunoblotting with an anti-FLAG antibody (Bb2, Bb4). C, Immunofluorescence micrographs show the distribution of transfected FLAG-tagged wild-type (WT) IRSp53-S and IRSp53-S with three point mutations [S27A, S158A, S169A; IRSp53-S(3)A] in hippocampal neurons. Transfected neurons were fixed in the basal state (control; Cc1, Cc3, Cc5, Cc7) or treated with 100 nm TPA for 10 min before fixation (TPA; Cc2, Cc4, Cc6, Cc8) and double labeled with anti-FLAG (green) and anti-PSD-95 (red) antibodies. Scale bar, 10 μm. D, HEK293T cells were cotransfected with PSD-95-CFP and FLAG-IRSp53-S wild type (wt) (lanes 1 and 2) or FLAG-IRSp53-S(3)A (lanes 3 and 4). After treatment with 100 nm TPA (lanes 2 and 4) or vehicle (lanes 1 and 3) for 30 min, IRSp53 was precipitated with anti-FLAG antibody from the cell lysates. Immunoprecipitates (IP) were analyzed by Western blotting (WB) using anti-GFP (PSD-95-CFP; top) and anti-FLAG (FLAG-IRSp53; bottom) antibodies. Representative results from three separate experiments are shown. E, The graph shows the amount of coprecipitated PSD-95-CFP proteins normalized to the amount of IRSp53 immunoprecipitates in the same lanes. Values represent the relative amount of PSD-95 precipitate and are expressed as means ± SD of three separate experiments. p < 0.05; the asterisk indicates the Student's t test value against lane 1.
To investigate the role of this phosphorylation in TPA-dependent IRSp53 synaptic accumulation, we examined the intracellular distribution of FLAG-IRSp53 carrying the three point mutations [IRSp53-S(3)A] in hippocampal neurons. Both wild-type IRSp53-S and its mutant were distributed throughout the dendrites and spines in the basal state (Fig. 7Cc1-Cc4). After TPA stimulation for 30 min, wild-type IRSp53-S was markedly translocated to synapses (Fig. 7Cc5), consistent with the result described above (Fig. 4A). The IRSp53-S(3)A mutant, however, did not respond to TPA stimulation (Fig. 7Cc7). To examine whether the interaction between IRSp53-S and one of its binding partners, PSD-95, is affected by PKC phosphorylation, we cotransfected PSD-95-CFP and FLAG-IRSp53 wild type or S(3)A mutant in HEK293T cells with or without TPA stimulation. By immunoprecipitating IRSp53 with anti-FLAG mAb, PSD-95 was specifically precipitated with IRSp53 wild type and S(3)A mutant, respectively (Fig. 7D, lanes 1 and 3). TPA stimulation increased in the amount of precipitated PSD-95 in the cells cotransfected with IRSp53 wild type but not in the cells cotransfected with IRSp53-S(3)A mutant (Fig. 7D, lanes 2 and 4). These results suggest that PKC phosphorylation of IRSp53 is critically involved in the synaptic targeting of this protein by regulating the interaction with its binding partners such as PSD-95.
Effect of IRSp53 on synaptic function
To explore the functional role of IRSp53 in synapses, we analyzed the peak amplitude and frequency of mEPSCs in the IRSp53-S variant-transfected neurons. The coexpression of GFP and IRSp53-S wild type in neurons enhanced the mEPSC amplitude but had no effect on the mEPSC frequency compared with neurons expressing GFP alone (Fig. 8A,B) (n = 7 for GFP+IRSp53-S wild type, 13 for GFP control; p < 0.05). In agreement with previous reports (Malenka et al., 1986; Parfitt and Madison, 1993), TPA stimulation markedly increased in the frequency of mEPSC recorded from neurons coexpressing GFP and IRSp53-S wild type, but an additional increase in mEPSC amplitude was hardly detected (Fig. 8A,B) (n = 9 for GFP+IRSp53-S wild type with TPA). It is likely that IRSp53 localized at synapses may be saturated by overexpression of IRSp53-S before TPA stimulation. To confirm this possibility, we also examined the synaptic currents recorded from neurons expressing IRSp53-S-ΔSH3, which spontaneously targets to synapses without stimulation. In IRSp53-S-ΔSH3-transfected neurons, the enhanced mEPSC amplitude but not frequency was detected. However, there was no significant effect on either mEPSC amplitude or frequency recorded from neurons coexpressing GFP and IRSp53-S-ΔSH3-ΔPDZ-BS (Fig. 8A,B) (n = 17 for GFP+IRSp53-S-ΔSH3, 12 for GFP+IRSp53-S-ΔSH3-ΔPDZ-BS; p < 0.05). Together, these results suggest that the synaptic translocation of IRSp53 enhances the postsynaptic activity.
Figure 8.
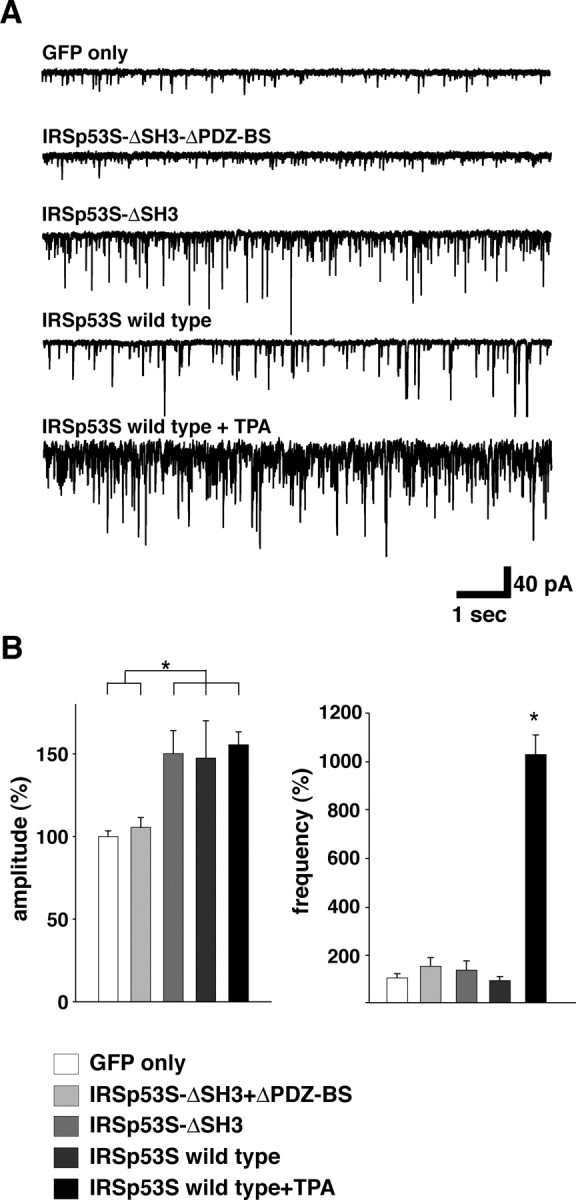
Synaptic expression of IRSp53 enhances the synaptic function. A, Examples of miniature EPSCs recorded from GFP-, GFP+IRSp53p53S-ΔSH3-ΔPDZ-BS-, GFP+IRSp53p53S-ΔSH3-, GFP+IRSp53-S wild-type-, and TPA-stimulated GFP+IRSp53-S wild-type-transfected sister neurons. B, Summary of the averaged amplitude and frequency of miniature EPSCs. Error bars represent SD (GFP only, n = 13; IRSp53-S-ΔSH3-ΔPDZ-BS, n = 12; GFP+IRSp53-S-ΔSH3, n = 17; GFP+IRSp53-S wild type, n = 7; GFP+IRSp53-S wild type with TPA, n = 9; *p < 0.05). The recorded values are normalized to the mean amplitude and frequency of the GFP-transfected sister neurons (12.09 ± 2.30 pA and 7.39 ± 4.74 Hz; n = 13).
Discussion
Regulation of the synaptic translocation of IRSp53
It has been well documented that NMDA receptor signaling is critical for several forms of synaptic plasticity. In response to NMDA receptor activation, postsynaptic proteins are reorganized via translocation and degradation (Shen and Meyer, 1999; Colledge et al., 2003; Hering and Sheng, 2003). In this study, we observed the synaptic activity-dependent translocation of IRSp53 to postsynaptic sites and analyzed its molecular mechanism. A majority of IRSp53 was diffusely distributed through dendrites as well as spines under nonstimulated conditions, whereas the NMDA receptor stimulation markedly induced the translocation of IRSp53 from dendrites to excitatory postsynaptic sites. Only a limited number of synapses (<5%) was filled with IRSp53 even under nonstimulated conditions (data not shown). The other groups, however, reported the synaptic localization of IRSp53 without stimulation (Abbott et al., 1999; Bockmann et al., 2002; Soltau et al., 2004). This inconsistency may be attributable to different basal levels of the synaptic activity under different culture conditions. In support to this, our pharmacological study showed that there was no synaptic localization of IRSp53 in neurons pretreated with TTX. A PKC inhibitor, but not other kinase inhibitors, specifically suppressed the NMDA-dependent translocation of IRSp53 to synapses, and a potent PKC activator, TPA, markedly induced a sustained synaptic translocation of IRSp53 without NMDA receptor activation, suggesting the NMDA-dependent synaptic translocation of IRSp53 via PKC signaling. We also demonstrated that the PKC phosphorylation of IRSp53 at its N terminus positively regulates the synaptic targeting of IRSp53.
Wu et al. (2003) reported phosphorylation of the calmodulin-binding protein neurogranin by transient NMDA-triggered PKC activation in mouse hippocampal slices. In our study, NMDA receptor activation resulted in the rapid translocation of IRSp53 to synapses within 10 min and its redistribution to dendrites after 30 min, with dynamics similar to the time course of PKC activation/deactivation by NMDA stimulation. We hypothesize that the intracellular translocation of IRSp53 between dendrites and postsynapses may be regulated by its phosphorylation/dephosphorylation state. Another possibility is that the translocation is caused by the actin dynamics. We showed by in situ detergent extraction and latrunculin A treatment that the actin cytoskeleton is required for anchoring the IRSp53 at synapses. A similar result for IRSp53 localization at the cell-cell contacts of epithelial cells was reported previously (Hori et al., 2003). In neurons, glutamate stimulation leads to the disassembly of actin filaments (Halpain et al., 1998) and a reduction in the actin turnover rate in spines (Star et al., 2002). We also observed that, similar to the synaptic IRSp53 dynamics, which change in a time scale of minutes, actin filaments stained with fluorescence-conjugated phalloidin remained in the synapses at 10 min but disappeared after 30 min of NMDA treatment (data not shown). This observation is consistent with the results reported by Okamoto et al. (2004). Together, our results suggest that the redistribution of IRSp53 from the spines to the dendrites may be secondary to the actin depolymerization.
We also demonstrated that the PDZ-binding sequence in the C terminus of IRSp53 is involved in anchoring it to synapses. Our immunoprecipitation experiments using heterologous cells further support this finding that PKC phosphorylation of IRSp53 at its N terminus positively regulates the interaction with one of its binding partners, PSD-95. Soltau et al. (2002) reported that a proline-rich region of shank1 binds to the SH3 domain of IRSp53 in a cdc42-dependent manner. In our findings, however, IRSp53 lacking the SH3 domain was targeted spontaneously to synapses without NMDA receptor and PKC activation, suggesting that shank1 is not involved in the synaptic localization of IRSp53. It has been proposed that IRSp53 itself binds intramolecularly at the basal state, but the binding of active Rac or Cdc42 to the N-terminal region or the partial CRIB motif of IRSp53, respectively, liberates the SH3 domain in the C terminus from its N terminus, thereby allowing IRSp53 to associate with its binding partners (Miki and Takenawa, 2002; Soltau et al., 2002). Taking these findings into consideration, our results suggest that the SH3 domain of IRSp53 serves as an inhibitory domain for the synaptic translocation of IRSp53 at the basal state and that PKC phosphorylation of IRSp53 at its N terminus may cause a conformational change, followed by an association of the PDZ-binding sequence at its C-terminal end with PDZ-containing PSD scaffold proteins, such as MALS, synapse-associated protein-97 (SAP-97), PSD-95, or SAP-102 (Hori et al., 2003; Soltau et al., 2004). In addition, the N-terminal half of IRSp53 interacted with the actin cytoskeleton is also necessary for the efficient synaptic targeting. In fact, we demonstrated that the actin cytoskeleton is required for the synaptic localization of IRSp53. Thus, our results suggest that the candidate domain for interaction with the actin cytoskeleton is the N-terminal coiled-coil/IMD (Yamagishi et al., 2004).
Importance of the synaptic translocation of IRSp53
Several studies have demonstrated the involvement of PKC mediated through the NMDA receptor in the plastic properties associated with the long-term changes of synaptic activity (Angenstein and Staak, 1997; Van der Zee and Douma, 1997). Loading a constitutively active form of the PKC catalytic fragment or treatment with phorbol esters causes the augmentation of the mEPSC amplitude in cultured hippocampal neurons (Wang et al., 1994; Carroll et al., 1998; Goldin and Segal, 2003). The phosphorylation of several PKC substrates in the long-term potentiation mediated by NMDA receptor activation has also been reported (Chen et al., 1997; Ramakers et al., 1999). It is, however, unclear which of the many processes activated by PKC is the fundamental component of synaptic plasticity.
Electrophysiological data in this study showed that the overexpression of IRSp53-S wild type or ΔSH3 mutant enhanced the synaptic function as measured by increased mEPSC amplitude without affecting frequency, suggesting a postsynaptic effect of the expression of these proteins. As a control, the expression of another IRSp53-S mutant lacking the C-terminal PDZ-binding sequence had no effect on the synaptic activity. These results support the above findings that changes in postsynaptic activity can be attributed to an increase in the IRSp53 accumulation at postsynapses. It has been reported previously that the IRSp53-S-ΔSH3 serves as a dominant-negative form on the regulation of actin dynamics (Miki et al., 2000; Govind et al., 2001). In this study, an increase in mEPSC amplitude was detected in neurons overexpressing IRSp53-S wild type, indicating that IRSp53 positively regulates the synaptic function via other domain(s) except for SH3 domain. Neurons after 14 DIV exhibit a mature type of synaptic transmission consisting of AMPA receptor-mediated responses (Renger et al., 2001). It is likely that the augmentation of the synaptic activity by IRSp53 translocation is caused by the stabilization/increase of AMPA receptor number in the postsynaptic membrane or by positive modification of the single channel conductance. AMPA receptors are recycled rapidly at the synaptic membrane (Bredt and Nicoll, 2003), and the delivery of AMPA receptors to the postsynaptic membrane could be a major mechanism underlying NMDA receptor-dependent long-term potentiation (Bredt and Nicoll, 2003). El-Husseini et al. (2000) reported that postsynaptic overexpression of PSD-95, which is a potential IRSp53 binding partner (Soltau et al., 2004), enhances the postsynaptic clustering and activity of AMPA receptors by binding with stargazin, an AMPA receptor-binding tetraspanning membrane protein (Chen et al., 2000; El-Husseini et al., 2000). Furthermore, it has been reported that SAP-97, another membrane-associated guanylate kinase family protein (MAGUK), interacts with and delivers the GluR1 subunit of the AMPA receptor to synapses in a CaMKII-dependent manner (Mauceri et al., 2004). IRSp53 may act together with these proteins in the synaptic clustering or delivery of AMPA receptor. However, additional study is required to delineate the detailed molecular mechanisms with which this occurs.
In conclusion, we demonstrated a novel mechanism underlying the regulatory role of IRSp53, in which PKC phosphorylation via NMDA signaling controls the translocation of IRSp53 from dendrites to synapses in cultured neurons, and the synapsetargeted IRSp53 is involved in the maturation of excitatory synapses. It would be interesting to investigate whether and how the molecular complex of IRSp53 and its binding partners, such as the MAGUK proteins, regulate the function of glutamate receptors in excitatory synapses.
Footnotes
This work was supported by Grant-in-Aid for Scientific Research 15GS0312 from the Ministry of Education, Science, Sports and Culture of Japan (K.S.).
Correspondence should be addressed to Kenji Sobue, Department of Neuroscience (D13), Osaka University Graduate School of Medicine, 2-2 Yamadaoka, Suita City, Osaka 565-0871, Japan. E-mail: sobue@nbiochem.med.osaka-u.ac.jp.
Copyright © 2005 Society for Neuroscience 0270-6474/05/252670-12$15.00/0
References
- Abbott MA, Wells DG, Fallon JR (1999) The insulin receptor tyrosine kinase substrate p85/53 and the insulin receptor are components of CNS synapses. J Neurosci 19: 7300-7308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allison DW, Gelfand VI, Spector I, Craig AM (1998) Role of actin in anchoring postsynaptic receptors in cultured hippocampal neurons: differential attachment of NMDA versus AMPA receptors. J Neurosci 18: 2423-2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez CE, Sutcliffe JG, Thomas EA (2002) Novel isoform of insulin receptor substrate p58/53 is generated by alternative splicing in the CRIB/SH3-binding region. J Biol Chem 277: 24728-24734. [DOI] [PubMed] [Google Scholar]
- Angenstein F, Staak S (1997) Receptor-mediated activation of protein kinase C in hippocampal long-term potentiation: facts, problems and implications. Prog Neuropsychopharmacol Biol Psychiatry 21: 427-454. [DOI] [PubMed] [Google Scholar]
- Bockmann J, Kreutz MR, Gundelfinger ED, Bockers TM (2002) ProSAP/Shank postsynaptic density proteins interact with insulin receptor tyrosine kinase substrate IRSp53. J Neurochem 83: 1013-1017. [DOI] [PubMed] [Google Scholar]
- Bredt DS, Nicoll RA (2003) AMPA receptor trafficking at excitatory synapses. Neuron 40: 361-379. [DOI] [PubMed] [Google Scholar]
- Carroll RC, Nicoll RA, Malenka RC (1998) Effects of PKA and PKC on miniature excitatory postsynaptic currents in CA1 pyramidal cells. J Neurophysiol 80: 2797-2800. [DOI] [PubMed] [Google Scholar]
- Chen L, Chetkovich DM, Petralia RS, Sweeney NT, Kawasaki Y, Wenthold RJ, Bredt DS, Nicoll RA (2000) Stargazin regulates synaptic targeting of AMPA receptors by two distinct mechanisms. Nature 408: 936-943. [DOI] [PubMed] [Google Scholar]
- Chen SJ, Sweatt JD, Klann E (1997) Enhanced phosphorylation of the postsynaptic protein kinase C substrate RC3/neurogranin during longterm potentiation. Brain Res 749: 181-187. [DOI] [PubMed] [Google Scholar]
- Colledge M, Snyder EM, Crozier RA, Soderling JA, Jin Y, Langeberg LK, Lu H, Bear MF, Scott JD (2003) Ubiquitination regulates PSD-95 degradation and AMPA receptor surface expression. Neuron 40: 595-607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebihara T, Kawabata I, Usui S, Sobue K, Okabe S (2003) Synchronized formation and remodeling of postsynaptic densities: long-term visualization of hippocampal neurons expressing postsynaptic density proteins tagged with green fluorescent protein. J Neurosci 23: 2170-2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers MD (2003) Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nat Neurosci 6: 231-242. [DOI] [PubMed] [Google Scholar]
- El-Husseini AE, Schnell E, Chetkovich DM, Nicoll RA, Bredt DS (2000) PSD-95 involvement in maturation of excitatory synapses. Science 290: 1364-1368. [PubMed] [Google Scholar]
- Fong DK, Rao A, Crump FT, Craig AM (2002) Rapid synaptic remodeling by protein kinase C: reciprocal translocation of NMDA receptors and calcium/calmodulin-dependent kinase II. J Neurosci 22: 2153-2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldin M, Segal M (2003) Protein kinase C and ERK involvement in dendritic spine plasticity in cultured rodent hippocampal neurons. Eur J Neurosci 17: 2529-2539. [DOI] [PubMed] [Google Scholar]
- Govind S, Kozma R, Monfries C, Lim L, Ahmed S (2001) Cdc42Hs facilitates cytoskeletal reorganization and neurite outgrowth by localizing the 58-kD insulin receptor substrate to filamentous actin. J Cell Biol 152: 579-594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halpain S, Hipolito A, Saffer L (1998) Regulation of F-actin stability in dendritic spines by glutamate receptors and calcineurin. J Neurosci 18: 9835-9844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris KM, Kater SB (1994) Dendritic spines: cellular specializations imparting both stability and flexibility to synaptic function. Annu Rev Neurosci 17: 341-371. [DOI] [PubMed] [Google Scholar]
- Hering H, Sheng M (2003) Activity-dependent redistribution and essential role of cortactin in dendritic spine morphogenesis. J Neurosci 23: 11759-11769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori K, Konno D, Maruoka H, Sobue K (2003) MALS is a binding partner of IRSp53 at cell-cell contacts. FEBS Lett 554: 30-34. [DOI] [PubMed] [Google Scholar]
- Iwamoto T, Yamada Y, Hori K, Watanabe Y, Sobue K, Inui M (2004) Differential modulation of NR1-NR2A and NR1-NR2B subtypes of NMDA receptor by PDZ domain-containing proteins. J Neurochem 89: 100-108. [DOI] [PubMed] [Google Scholar]
- Konno D, Ko JA, Usui S, Hori K, Maruoka H, Inui M, Fujikado T, Tano Y, Suzuki T, Tohyama K, Sobue K (2002) The postsynaptic density and dendritic raft localization of PSD-Zip70, which contains an N-myristoylation sequence and leucine-zipper motifs. J Cell Sci 115: 4695-4706. [DOI] [PubMed] [Google Scholar]
- Krugmann S, Jordens I, Gevaert K, Driessens M, Vandekerckhove J, Hall A (2001) Cdc42 induces filopodia by promoting the formation of an IRSp53:Mena complex. Curr Biol 11: 1645-1655. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Madison DV, Nicoll RA (1986) Potentiation of synaptic transmission in the hippocampus by phorbol esters. Nature 321: 175-177. [DOI] [PubMed] [Google Scholar]
- Mauceri D, Cattabeni F, Di Luca M, Gardoni F (2004) Calcium/calmodulin-dependent kinase II phosphorylation derives synapse-associated protein 97 into spines. J Biol Chem 279: 23813-23821. [DOI] [PubMed] [Google Scholar]
- McGee AW, Bredt DS (2003) Assembly and plasticity of the glutamatergic postsynaptic specialization. Curr Opin Neurobiol 13: 111-118. [DOI] [PubMed] [Google Scholar]
- Miki H, Takenawa T (2002) WAVE2 serves a functional partner of IRSp53 by regulating its interaction with Rac. Biochem Biophys Res Commun 293: 93-99. [DOI] [PubMed] [Google Scholar]
- Miki H, Yamaguchi H, Suetsugu S, Takenawa T (2000) IRSp53 is an essential intermediate between Rac and WAVE in the regulation of membrane ruffling. Nature 408: 732-735. [DOI] [PubMed] [Google Scholar]
- Mizushima S, Nagata S (1990) pEF-BOS, a powerful mammalian expression vector. Nucleic Acids Res 18: 5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa H, Yamamura K, Miyazaki J (1991) Efficient selection for highexpression transfectants with a novel eukaryotic vector. Gene 108: 193-199. [DOI] [PubMed] [Google Scholar]
- Okabe S, Urushido T, Konno D, Okado H, Sobue K (2001) Rapid redistribution of the postsynaptic density protein PSD-Zip45 (Homer 1c) and its differential regulation by NMDA receptors and calcium channels. J Neurosci 21: 9561-9571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto K, Nagai T, Miyawaki A, Hayashi Y (2004) Rapid and persistent modulation of actin dynamics regulates postsynaptic reorganization underlying bidirectional plasticity. Nat Neurosci 7: 1104-1112. [DOI] [PubMed] [Google Scholar]
- Okamura-Oho Y, Miyashita T, Yamada M (2001) Distinctive tissue distribution and phosphorylation of IRSp53 isoforms. Biochem Biophys Res Commun 289: 957-960. [DOI] [PubMed] [Google Scholar]
- Parfitt KD, Madison DV (1993) Phorbol esters enhance synaptic transmission by a presynaptic, calcium-dependent mechanism in rat hippocampus. J Physiol (Lond) 471: 245-268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakers GM, McNamara RK, Lenox RH, De Graan PN (1999) Differential changes in the phosphorylation of the protein kinase C substrates myristoylated alanine-rich C kinase substrate and growth-associated protein-43/B-50 following Schaffer collateral long-term potentiation and longterm depression. J Neurochem 73: 2175-2183. [PubMed] [Google Scholar]
- Renger JJ, Egles C, Liu G (2001) A developmental switch in neurotransmitter flux enhances synaptic efficacy by affecting AMPA receptor activation. Neuron 29: 469-484. [DOI] [PubMed] [Google Scholar]
- Shen K, Meyer T (1999) Dynamic control of CaMKII translocation and localization in hippocampal neurons by NMDA receptor stimulation. Science 284: 162-166. [DOI] [PubMed] [Google Scholar]
- Sheng M, Kim MJ (2002) Postsynaptic signaling and plasticity mechanisms. Science 298: 776-780. [DOI] [PubMed] [Google Scholar]
- Soltau M, Richter D, Kreienkamp HJ (2002) The insulin receptor substrate IRSp53 links postsynaptic shank1 to the small G-protein cdc42. Mol Cell Neurosci 21: 575-583. [DOI] [PubMed] [Google Scholar]
- Soltau M, Berhörster K, Kindler S, Buck F, Richter D, Kreienkamp HJ (2004) Insulin receptor substrate of 53 kDa links postsynaptic shank to PSD-95. J Neurochem 90: 659-665. [DOI] [PubMed] [Google Scholar]
- Star EN, Kwiatkowski DJ, Murthy VN (2002) Rapid turnover of actin in dendritic spines and its regulation by activity. Nat Neurosci 5: 239-246. [DOI] [PubMed] [Google Scholar]
- Sun J, Tadokoro S, Imanaka T, Murakami SD, Nakamura M, Kashiwada K, Ko J, Nishida W, Sobue K (1998) Isolation of PSD-Zip45, a novel Homer/vesl family protein containing leucine zipper motifs, from rat brain. FEBS Lett 437: 304-308. [DOI] [PubMed] [Google Scholar]
- Tadokoro S, Tachibana T, Imanaka T, Nishida W, Sobue K (1999) Involvement of unique leucine-zipper motif of PSD-Zip45 (Homer 1c/vesl-1L) in group 1 metabotropic glutamate receptor clustering. Proc Natl Acad Sci USA 96: 13801-13806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toni N, Buchs PA, Nikonenko I, Bron CR, Muller D (1999) LTP promotes formation of multiple spine synapses between a single axon terminal and dendrite. Nature 402: 421-425. [DOI] [PubMed] [Google Scholar]
- Usui S, Konno D, Hori K, Maruoka H, Okabe S, Fujikado T, Tano Y, Sobue K (2003) Synaptic targeting of PSD-Zip45 (Homer 1c) and its involvement in the synaptic accumulation of F-actin. J Biol Chem 278: 10619-10628. [DOI] [PubMed] [Google Scholar]
- Van der Zee EA, Douma BR (1997) Historical review of research on protein kinase C in learning and memory. Prog Neuropsychopharmacol Biol Psychiatry 21: 379-406. [DOI] [PubMed] [Google Scholar]
- Wang LY, Dudek EM, Browning MD, MacDonald JF (1994) Modulation of AMPA/kainate receptors in cultured murine hippocampal neurons by protein kinase C. J Physiol (Lond) 475: 431-437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Huang KP, Huang FL (2003) Participation of NMDA-mediated phosphorylation and oxidation of neurogranin in the regulation of Ca2+- and Ca2+/calmodulin-dependent neuronal signaling in the hippocampus. J Neurochem 86: 1524-1533. [DOI] [PubMed] [Google Scholar]
- Yamagishi A, Masuda M, Ohki T, Onishi H, Mochizuki N (2004) A novel actin bundling/filopodium-forming domain conserved in insulin receptor tyrosine kinase substrate p53 and missing in metastasis protein. J Biol Chem 279: 14929-14936. [DOI] [PubMed] [Google Scholar]
- Yasuda H, Barth AL, Stellwagen D, Malenka RC (2003) A developmental switch in the signaling cascades for LTP induction. Nat Neurosci 6: 15-16. [DOI] [PubMed] [Google Scholar]
- Yeh TC, Ogawa W, Danielsen AG, Roth RA (1996) Characterization and cloning of a 58/53-kDa substrate of the insulin receptor tyrosine kinase. J Biol Chem 271: 2921-2928. [DOI] [PubMed] [Google Scholar]
- Yuste R, Bonhoeffer T (2001) Morphological changes in dendritic spines associated with long-term synaptic plasticity. Annu Rev Neurosci 24: 1071-1089. [DOI] [PubMed] [Google Scholar]