

Abstract
An increasing body of evidence suggests that mitochondrial dysfunction plays an important role in the pathogenesis of familial amyotrophic lateral sclerosis associated with “gain of function” mutations in Cu/Zn superoxide dismutase 1 (SOD1). SOD1 is mostly a cytosolic protein, but a portion of SOD1 is localized in mitochondria of patients with familial amyotrophic lateral sclerosis and transgenic mouse models of the disease. Despite the finding that mutant SOD1 localizes in mitochondria, the pathogenic significance of the mitochondrial mutant SOD1 remains to be elucidated. Here, we demonstrate that both wild-type and mutant human SOD1 accumulate in brain mitochondria of transgenic mice and that SOD1 displays a very complex intramitochondrial compartmentalization. For the first time, we show that, in addition to being in the mitochondrial outer membrane and intermembrane space, SOD1 is also localized in the mitochondrial matrix. Importantly, we show that aberrant SOD1 macromolecular aggregates are formed in the matrix of brain mitochondria. This suggests that mutant SOD1 in the brain mitochondrial matrix is misfolded and prone to aggregation, which may contribute to selective neuronal degeneration.
Keywords: ALS, amyotrophic lateral sclerosis, matrix, mitochondria, aggregation, SOD1, brain
Introduction
Amyotrophic lateral sclerosis (ALS) is a progressive paralytic disease characterized by selective degeneration of the upper and lower motor neurons. Approximately 10% of ALS cases are familial (fALS), ∼20% of which are associated with mutations in the Cu/Zn superoxide dismutase 1 gene SOD1 (Rosen et al., 1993). The fALS-associated SOD1 mutations are suggested to cause a “toxic gain of function” because (1) many of the fALS-associated SOD1 mutants retain dismutase activity (Borchelt et al., 1994), and (2) transgenic mice harboring mutant SOD1 genes display an ALS phenotype (Bruijn et al., 1997), whereas SOD1 knock-out mice exhibit no symptoms of ALS (Reaume et al., 1996).
Among other pathological features of transgenic mouse models of fALS is the presence of membrane-bound vacuoles derived from massive mitochondrial degeneration in the motor neurons of mice expressing mutant human SOD1 (hSOD1) mutations at early disease stages (Wong et al., 1995; Jaarsma et al., 2000; Higgins et al., 2003). We and others have shown that mitochondrial oxidative phosphorylation is dysfunctional in isolated brain and spinal cord mitochondria from symptomatic G93A transgenic mice (Jung et al., 2002; Mattiazzi et al., 2002). These findings suggest that mitochondrial alterations may represent a triggering factor in the onset of fALS.
Originally found to be a cytosolic enzyme (Weisiger and Fridovich, 1973), SOD1 has also been detected in the mitochondrial intermembrane space (IMS) in yeast (Sturtz et al., 2001; Field et al., 2003) and rat liver (Okado-Matsumoto and Fridovich, 2001). Immunochemical and ultrastructural studies have confirmed that hSOD1 proteins expressed in transgenic mouse models of fALS are localized in mitochondria (Jaarsma et al., 2001; Higgins et al., 2002; Mattiazzi et al., 2002). The mechanisms of mutant SOD1 toxicity and its tissue specificity have yet to be determined, but one proposed toxic gain of function of mutant SOD1 is protein aggregation (Durham et al., 1997; Bruijn et al., 1998; Koide et al., 1998; Johnston et al., 2000; Watanabe et al., 2001; Wang et al., 2002). Recently, it was suggested that mutant hSOD1 aggregates preferentially in mitochondria from spinal cord in both humans and mouse models of fALS (Liu et al., 2004), and large SOD1-immunoreactive structures associated with mitochondrial vacuolar membranes have been identified by electron microscopy (Higgins et al., 2003). Mutant hSOD1 was also found to interact with the antiapoptotic protein Bcl-2 in mitochondria of transgenic mice and human patients (Pasinelli et al., 2004). Although the physiological and the pathological meaning of mitochondrial localization of SOD1 have yet to be clarified, these findings suggest that mutant hSOD1 localized in mitochondria may play a role in mitochondrial dysfunction and motor neuron degeneration in fALS.
Here, we show that a portion of mitochondrial hSOD1 localizes in the mitochondrial matrix of transgenic fALS mice. In contrast with the hSOD1 in the IMS, we find that the majority of hSOD1 in the matrix is enzymatically inactive, suggesting that it is improperly folded and/or improperly metallated. We also find that mutant, but not wild-type (Wt), hSOD1 forms large molecular weight aggregates in the matrix of brain mitochondria. SOD1 aggregates in the matrix may be linked to fALS pathology, because they are found in the matrix of brain mitochondria, which are affected in fALS but not in liver that is spared by the disease.
Materials and Methods
Transgenic mice. Transgenic mice expressing wild-type, G93A mutant (Gurney et al., 1994), and G85R mutant (Wong et al., 1995; Bruijn et al., 1997) hSOD1 were from The Jackson Laboratory (Bar Harbor, ME).
Antibodies. Western blotting analysis was performed using the following antibodies: SOD1 (Calbiochem, San Diego, CA), protein disulfide isomerase (PDI) (Stressgen Biotechnologies, San Diego, CA), cathepsin D (Athens Research and Technology, Athens, GA), synapsin I (Sigma, St. Louis, MO), syntaxin 6 (Stressgen Biotechnologies), manganese SOD (MnSOD) (Stressgen Biotechnologies), voltage-dependent anion channel (VDAC) (Calbiochem) and Akt kinase (Santa Cruz Biotechnology, Santa Cruz, CA), cytochrome c (BD Biosciences Pharmingen, San Diego, CA), cytochrome c oxidase subunit I (COX I) (Molecular Probes, Eugene, OR), Bcl-2 (BD Biosciences, San Jose, CA), and heat shock protein 60 (Hsp60) (Stressgen Biotechnologies).
Mitochondrial isolation and alkali extraction. All of the procedures were approved by the Animal Care and Use Committee of the Weill Medical College of Cornell University. Mitochondria were purified as described previously (Pallotti and Lenaz, 2001), with some modifications. Briefly, tissue were excised and homogenized (glass-Teflon Potter-Elvehjem homogenizer) in buffer H, consisting of 10 mm Tris, pH 7.4, 320 mm sucrose, 1 mm EDTA, 1 mm DTT, and 1 mg/ml fatty-acid-free bovine serum albumin (BSA) (Sigma). The homogenate was centrifuged at 1500 × g for 5 min at 4°C. The pellet was resuspended in one-half of the original volume of buffer H and centrifuged again under the same conditions. The supernatants were combined and centrifuged at 15,000 × g for 20 min at 4°C. The crude mitochondrial pellet was resuspended in 1 ml of buffer H without BSA and then layered on top of a chilled, 7.5/10% (w/v) discontinuous Ficoll gradient (made in buffer H without BSA), using 5 ml for each layer. The samples were centrifuged at 24,000 rpm for 24 min at 4°C in a Beckman Coulter (Fullerton, CA) Optima LE-80K ultracentrifuge using a SW 41 Ti swing-out rotor. The purified mitochondrial pellet was resuspended in 200 μl of chilled buffer H. Sample aliquots were snap frozen in liquid nitrogen and kept at -80°C. Protein concentration was determined by a colorimetric assay kit (Bio-Rad, Hercules, CA).
For alkali extraction, purified whole mitochondria were incubated for 30 min on ice in the presence of 20 vol of one of the following: buffer H, freshly prepared 0.1 m sodium carbonate, pH ∼11.5, or buffer H plus 0.5% Triton X-100 (TX-100) (Sigma). The samples were then centrifuged at 91,000 × g for 25 min at 4°C. The supernatants were precipitated with ice-cold 12% trichloroacetic acid (TCA) and centrifuged at 18,000 × g for 15 min at 4°C, followed by an ice-cold acetone wash using the same centrifugation conditions.
Preparation and sonication of brain mitoplasts. For mitoplast (MP) preparation, purified mitochondria (5 mg/ml) were incubated with 5 vol of cold hypotonic buffer (in mm: 10 Tris, pH 7.4, 1 EDTA, and 1 DTT) for 10 min on ice, followed by incubation in the presence of 150 mm KCl or 150 mm NaCl for 2 min. The samples were centrifuged at 18,000 × g for 20 min at 4°C to obtain postmitoplast supernatants (PMSs) and MP pellets.
MP fractions were sonicated with a microtip (Branson sonifier; Branson, Danbury, CT) three times at 2 min intervals on ice with 10 s pulses using a 50% duty cycle with a power output of 3. The samples were then centrifuged at 91,000 × g for 60 min at 4°C. The pellets were resuspended in 200 μl of 1 m KCl for 5 min on ice and then centrifuged again under the same conditions.
All of the PMS fractions, TCA precipitated as described above, and MP fractions were analyzed by 12% SDS-PAGE and Western blotting.
Immunoprecipitation of brain mitoplasts. MP fractions were resuspended in 600 μl of buffer H containing 150 mm NaCl and 5 mm MgCl2 (IP buffer). Protein G-Sepharose beads (Zymed Laboratories, South San Francisco, CA) were washed in IP buffer before incubation with 20 μl of SOD1 antibody for 1 h on a rocking platform at room temperature. After being washed in IP buffer, the bead/antibody complex was incubated with either intact or sonicated MP fractions overnight at 4°C on a rocker platform. The immune complexes were recovered by centrifugation at 600 × g for 30 s and then washed extensively with IP buffer. Volume equivalents of the pellets and the supernatants were analyzed by 12% SDS-PAGE and Western blotting.
Proteinase K protection assay. Mitochondria (25 μg) swelled in 5 vol of hypotonic buffer were incubated with or without 20 μg/ml proteinase K (PK) (Sigma) in the presence of 0.3% BSA for 20 min on ice, followed by an additional 10 min incubation with 2 mm phenylmethylsulfonyl fluoride (PMSF) to terminate proteolysis. In replicate samples, swelled mitochondria were incubated with 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS) (Sigma) at a final concentration of 10 mm for 10 min on ice or sonicated as described above before the addition of PK.
SOD1 gel activity assay and PK treatments. Twenty microliters of a 10 mg/ml solution of purified mitochondria were subfractionated as mentioned above, removing a 20 μl aliquot before centrifugation. MP fractions were resuspended in a volume of buffer SN (in mm: 10 Tris, pH 7.4, 50 sucrose, 150 NaCl, 1 EDTA, and 1 DTT) equivalent to that of the PMS. After incubating all of the samples with 0.5% Triton X-100 on ice for 15 min, 20 μl of each fraction was electrophoresed in a 10% native polyacrylamide gel (Invitrogen, Carlsbad, CA). SOD activity was determined using an activity gel assay as described previously by Beauchamp and Fridovich (1971). One unit of purified hSOD1 (Sigma) was used as a positive control. Activities were quantified by densitometry of band signal intensity using a Fluor-S MultiImager equipped with Quantity One software (Bio-Rad) on inverted gel images.
To assess SOD1 sensitivity to proteolysis, the remaining MP and PMS fractions were incubated in the presence of 200 μg/ml PK at room temperature for 3 h. After incubation with PK, aliquots were removed and mixed with equal volumes of 2× 4% SDS sample buffer containing 1 mm PMSF. Densitometric measurements of the Western blots were performed as described above.
Filter-trap assay. Brain and liver PMS and MP fractions were prepared as described above and resuspended in buffer SN. Both MP and PMS fractions were incubated with 0.5% TX-100 for 15 min on ice and then filtered onto a prewetted 0.22 μm cellulose acetate membrane (GE Osmonics, Minnetonka, MN) using a 96-well dot-blot apparatus (Schleicher & Schuell BioScience, Dassel, Germany) (Wang et al., 2002). After washing each well with 200 μl of PBS, the membrane was blocked with 5% milk in PBS/0.4% Tween 20 and then immunoprobed with the appropriate antibodies.
Results
hSOD1 accumulated in brain mitochondria does not behave as an integral membrane protein
To study the mitochondrial localization of SOD1, we purified brain and liver mitochondria from transgenic human Wt or mutant (G93A and G85R) mice. Volume equivalents of postnuclear supernatants, postmitochondrial supernatants, and purified mitochondria were analyzed by Western blot. The majority of SOD1 was associated with the postmitochondrial supernatant, but a portion of SOD1 was associated with the mitochondrial fraction in both Wt and mutant mice (Fig. 1A, lanes 3, 6, 9). Because of its lower expression levels, a longer exposure of the Western blot was necessary to visualize G85R hSOD1 associated with the mitochondrial fraction (Fig. 1A, bottom, lane 9). The mitochondrial matrix enzyme MnSOD and VDAC, a protein of the mitochondrial outer membrane (OM), were used as mitochondrial markers. Various organellar marker proteins were used to exclude contamination of the mitochondria with other cellular components (Fig. 1A, lanes 3, 6, 9). Akt kinase, a soluble cytosolic enzyme, was used to exclude contamination by cytosolic proteins.
Figure 1.
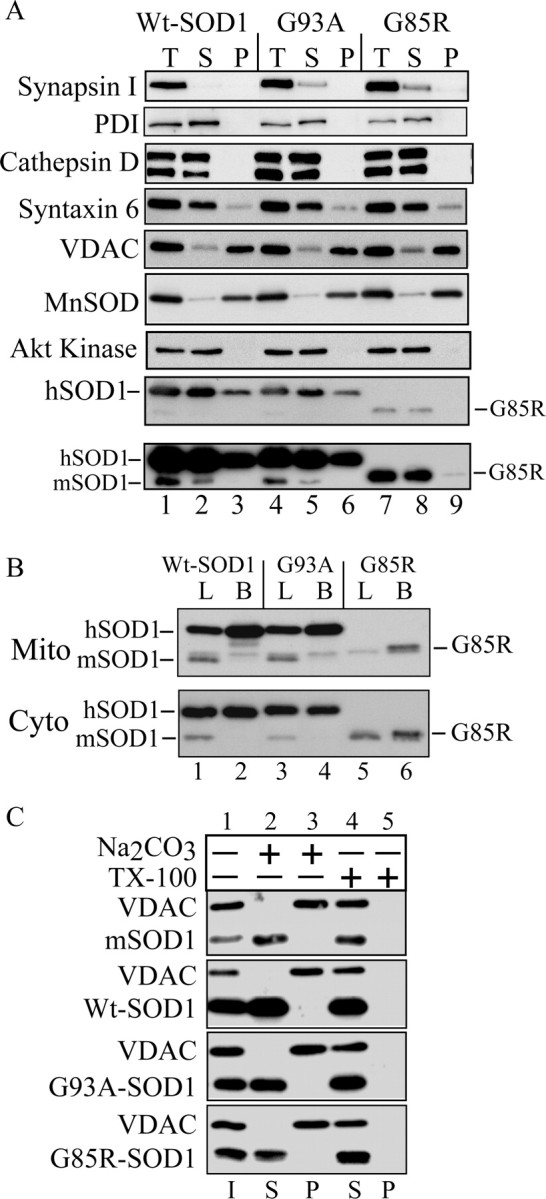
SOD1 in purified mitochondria from fALS transgenic mice. A, Immunoblotting of volume equivalents of postnuclear supernatant (T), postmitochondrial supernatant (S), and purified mitochondria (P). Mitochondrial content and structural integrity were analyzed using antibodies against the mitochondrial markers MnSOD and VDAC. Contamination was assessed with antibodies against synapsin I (synaptic vesicles), PDI (endoplasmic reticulum) and cathepsin D (lysosomes), Akt kinase (cytosol), and syntaxin 6 (trans-Golgi network). SOD1 was detected with antibodies that react with both the mouse SOD1 (mSOD1) and the transgenic hSOD1. The position of the faster-migrating G85R mutant hSOD1 is indicated. The bottom panel is a prolonged exposure of the above SOD1 blot to detect small amounts of G85R hSOD1 in the mitochondrial pellet. B, Comparison of the relative SOD1 content in purified mitochondria (Mito) and cytosol (Cyto) from brain and liver of hSOD1 transgenic mice. B, Brain; L, liver. C, Alkali treatment of mitochondrial membranes. Purified brain mitochondria were incubated in the presence of 0.1 m sodium carbonate, pH ∼11.5, or buffer H plus 0.5% TX-100. After centrifugation, supernatants (S) and pellets (P) were analyzed by SDS-PAGE and immunoblotting for SOD1 and the mitochondrial outer membrane protein VDAC. I, Input.
Because mitochondrial dysfunction in fALS mice is limited to the neural tissue (Mattiazzi et al., 2002) despite the ubiquitous expression of mutant SOD1, we compared the concentrations of transgenic hSOD1 in mitochondria from brain, an affected tissue, and liver, a nonaffected tissue. In both Wt and mutant mice, the content of hSOD1 was higher in brain compared with liver mitochondria (Fig. 1B, top). Compared with standards of purified hSOD1 (10 and 50 ng) (data not shown), in brain, hSOD1 was ∼3% of total mitochondrial protein. In liver mitochondria, hSOD1 was <1% of total protein. The hSOD1 content was independent from the expression levels of the protein, because in both Wt and G93A mutant hSOD1 mice, the amounts of cytosolic hSOD1 in liver and brain were virtually identical (Fig. 1B, bottom).
To determine whether SOD1 behaves like an integral membrane protein, we performed an alkaline extraction on whole-brain mitochondria from Wt and mutant mice. Alkaline extraction removes soluble and peripheral proteins, but not integral proteins, from membranes (Fujiki et al., 1982). After alkaline extraction, the majority of hSOD1 was released into the supernatant (Fig. 1C, lane 2). In contrast, there was no release of VDAC, a mitochondrial integral membrane protein, from the membrane pellet into the supernatant (Fig. 1C, lane 3). No difference was observed in the behavior of Wt and mutant hSOD1 in alkaline conditions or in the presence of detergents. These results indicate that the majority of hSOD1 in mitochondria does not behave like an integral membrane protein.
hSOD1 localizes in the matrix space of brain mitochondria
To investigate the submitochondrial localization of hSOD1, we fractionated brain mitochondria, generating MPs by hypoosmotic lysis of the OM. After hypoosmotic lysis, samples were incubated in the presence of 150 mm KCl to disrupt ionic protein interactions. Under these conditions, the majority of cytochrome c, a protein of the IMS, was released into the PMS (Fig. 2A, lanes 1, 3, 5), confirming the disruption of the OM. Conversely, the majority of MnSOD remained in the pellet, indicating that intact mitoplasts were recovered (Fig. 2A, lanes 2, 4, 6). Approximately 50% of the total mitochondrial hSOD1 was found in the PMS (Fig. 2A, lanes 1, 3, 5). Surprisingly, the remaining 50% of hSOD1 remained associated with the MPs (Fig. 2A, lanes 2, 4, 6). Solubilization of MPs with detergents (CHAPS or Triton X-100) resulted in the release of SOD1 from the MPs into the PMSs (supplemental Fig. S1, available at www.jneurosci.org as supplemental material).
Figure 2.
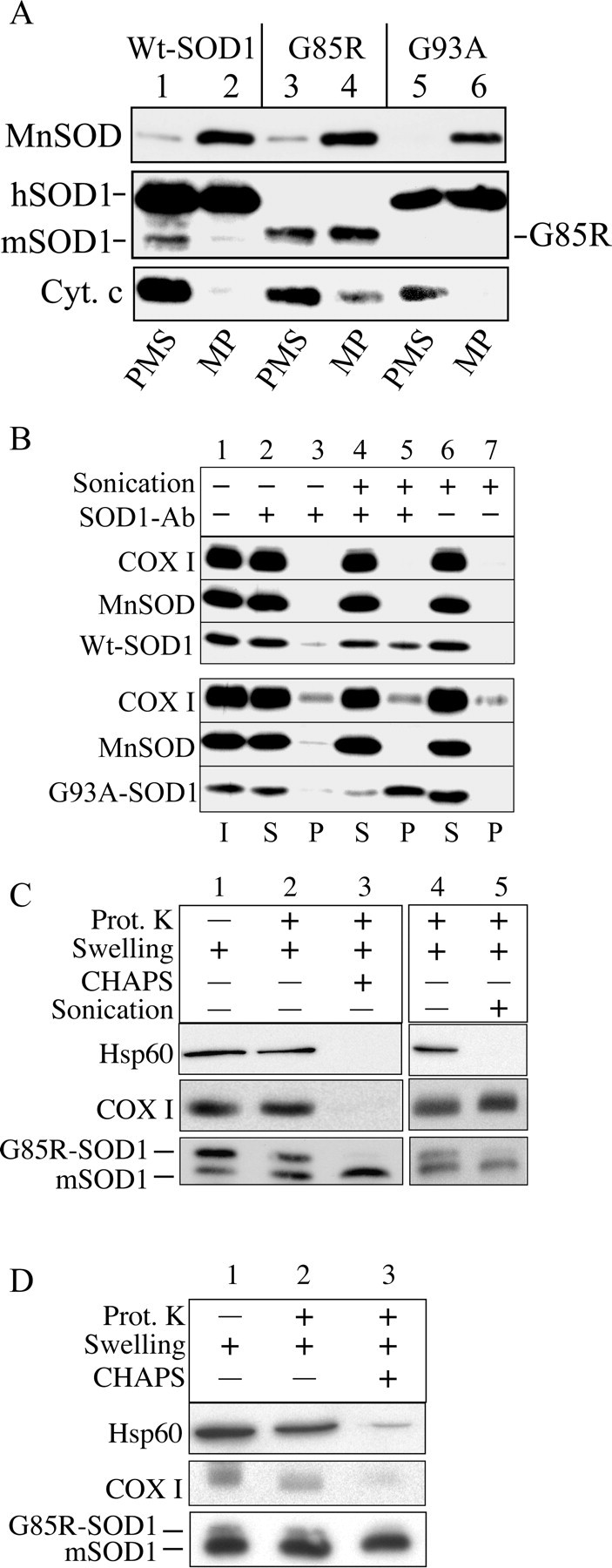
hSOD1 resides in the mitochondrial matrix. A, hSOD1 is associated with mitoplasts in a high salt-resistant manner. Mitochondria were subfractionated into MPs and PMSs, and MPs were subsequently incubated in the presence of 150 mm KCl. After centrifugation, MP and PMS fractions were immunoblotted for SOD1, the intermembrane component cytochrome, as well as the matrix component MnSOD. Cyt. c, Cytochrome c. B, Disruption of the mitochondrial IM is required to immunoprecipitate hSOD1 associated with mitoplasts. Intact or sonicated MPs were incubated with SOD1 antibody (SOD1 Ab)-protein G-Sepharose and then centrifuged at 600 × g for 30 s. Samples were immunoprobed for SOD1. MnSOD and COX I antibodies were used as markers of a soluble and an IM component of the mitoplasts, respectively. Lanes 6 and 7 represent a negative control without the addition of SOD1 antibody. I, Mitoplast input; S, supernatant; P, immunoprecipitated pellet. C, Swelled mitochondria from G85R brain were incubated in the absence or presence of 20 μg/ml PK (Prot. K) for 20 min on ice, with and without the addition of 10 mm CHAPS (lanes 2, 3) or with and without sonication (lanes 4, 5). The mouse SOD1 (mSOD1) was resistant to proteolysis in contrast to the human G85R-SOD1. Hsp60, a matrix component, and the mitochondrial IM protein COX I were used as markers to assess IM integrity. Lane 1 contains swelled mitochondria without treatment with PK, detergents, or sonication. D, Liver mitochondria from a 10-month-old G85R mouse treated as in C.
Because of this association of hSOD1 with brain MPs, we hypothesized that the portion of hSOD1 associated with MPs resided in the mitochondrial matrix. To test this hypothesis, we followed two experimental approaches. First, we used a protein G-SOD1 antibody complex to immunoprecipitate hSOD1 from brain MPs. We expected that, after physical disruption of the inner membrane (IM) by sonication, hSOD1 would be released from MPs and become accessible to the protein G-SOD1 anti-body complex, whereas without sonication, hSOD1 residing on the inside of an intact IM would not be accessible for immunoprecipitation. A portion of both Wt and G93A hSOD1 could be immunoprecipitated from sonicated MPs (Fig. 2B, lane 5), whereas only a negligible amount of hSOD1 sedimented from intact mitoplasts (Fig. 2B, lane 3). The absence of hSOD1 in the pellet fraction of the negative control (protein G only) excluded that hSOD1 bound protein G-Sepharose nonspecifically (Fig. 2B, lane 7). These data indicate that the disruption of the IM is necessary to render matrix hSOD1 accessible to the protein G-SOD1 antibody complex.
Second, we took advantage of the fact that G85R mutant hSOD1 is completely PK sensitive (Ratovitski et al., 1999). We subjected G85R brain mitochondria to PK treatment after OM disruption by hypoosmotic swelling. We tested various protease concentrations and conditions and found optimal conditions that preserved the integrity of the mitochondrial IM. Samples were treated with 20 μg/ml PK for 20 min on ice, followed by inactivation of the protease. A portion of G85R hSOD1 was protected from proteolysis in swelled mitochondria (Fig. 2C, lane 2). The soluble matrix protein Hsp60 and the IM integral protein COX I were protected from proteolysis in swelled mitochondria, indicating that the IM was preserved. After CHAPS solubilization, G85R hSOD1 as well as Hsp60 and COX I were digested (Fig. 2C, lane 3), indicating that all of these proteins were sensitive to PK degradation. To exclude that G85R hSOD1 digestion was attributable to enhanced PK activity after detergent treatment, we replicated the experiment on sonicated swelled mitochondria without the addition of CHAPS. As expected, sonication made both G85R hSOD1 and Hsp60 available to protease digestion, whereas in the absence of detergent, the transmembrane protein COX I was still essentially protected (Fig. 2C, lane 5). The wild-type murine SOD1 was resistant to PK treatment (Ratovitski et al., 1999; Tiwari and Hayward, 2003). For comparison with a nonaffected tissue, we performed a PK protection assay on swelled mitochondria from G85R mouse liver. Protease-protected mutant hSOD1 was also localized in liver mitochondrial matrix, but, relative to endogenous mouse SOD1, it was much less abundant than in brain (Fig. 2D). This result was consistent with the finding that transgenic hSOD1 accumulates more in brain than in liver mitochondria (Fig. 1B). Together, these data indicate that a significant portion of mitochondrial hSOD1 localizes in the matrix.
SOD1 associated with brain mitoplasts is enzymatically impaired
We examined the distribution of SOD1 enzymatic activity in mitochondrial fractions of Wt and G93A brain. Mitochondria, mitoplasts, and PMSs were analyzed by gel activity assays. In both Wt and G93A mice, the majority of the mitochondrial SOD1 activity was found in the PMS fractions (Fig. 3A, lanes 3, 6), whereas a smaller amount was detected in the MP fractions (Fig. 3A, lanes 4, 7). The SOD1 enzymatic activities in the submitochondrial fractions did not seem to correlate with the relative distribution of hSOD1 protein. The in-gel SOD1 activity was normalized by the amount of SOD1 detected by Western blot in brain mitochondrial fractions from Wt mice. The normalized SOD1 activity in the MP fraction was on average 2.7-fold lower than in the PMS (Fig. 3B). These results suggest that hSOD1 associated with MPs has reduced enzymatic activity.
Figure 3.

hSOD1 associated with brain mitoplasts is enzymatically impaired. A, Distribution of SOD1 enzymatic activity in brain mitochondria from Wt and mutant G93A hSOD1 transgenic mice. Purified mitochondria (MT) were subfractionated into PMS and MP fractions and were analyzed using an activity gel assay, as described in Materials and Methods. Lane 1, Purified hSOD1 was used as a positive control. Numbers in parentheses are band signal intensities of SOD1 activity [in optical density units (ODU) per square millimeter]. B, Quantification of the relative SOD1 activity in brain MT, PMSs, and MPs from two Wt mice. SOD1 activity was quantified in-gel as in A. Lane 1, Purified hSOD1 was used as a positive control. SOD1 activities in PMSs and MPs were normalized by the density of SOD1 bands detected by Western blot in aliquots from the same samples (optical density ratios). Numbers in parentheses are optical density ratios (OD Ratio; in arbitrary units). C, PK (Prot. K) treatment of subfractionated mitochondria. The same PMS (lanes 1, 2) and MP (lanes 3, 4) fractions shown in A were treated with or without 200 μg/ml PK for 180 min at room temperature, followed by protease inactivation with 1 mm PMSF and 4% SDS sample buffer. Western blots were immunoprobed for SOD1. Endogenous murine SOD1 (mSOD1) was not PK sensitive.
To test whether hSOD1 is misfolded, brain MPs and PMSs were solubilized and subjected to a harsh PK treatment (200 μg/ml for 3 h at room temperature). We expected that properly folded hSOD1 would be PK insensitive (Ratovitski et al., 1999; Tiwari and Hayward, 2003). We found that Wt SOD1 was unaffected by PK in both the MPs and the PMSs (Fig. 3C). Likewise, the enzymatically active mutant G93A-SOD1 was PK resistant in the PMS (Fig. 3C, lane 2). However, ∼30% of the mutant G93A-SOD1 in the MP fraction was digested by PK as determined by Western blot densitometry (Fig. 3C, lane 4). This data suggest that a portion of G93A mutant hSOD1 in the matrix may acquire an altered conformation. The enzymatically inactive G85R mutant hSOD1 was completely PK sensitive in both MPs and PMSs (Fig. 3C, lanes 2, 4).
Mutant hSOD1 forms macromolecular aggregates in brain mitoplasts
We performed a size-exclusion filter assay on subfractionated brain and liver mitochondria (Wang et al., 2002). Both MP and PMS fractions were solubilized in 0.5% TX-100, vacuum filtered onto a cellulose acetate membrane, and immunoprobed with SOD1 antibodies. In liver MPs and PMSs from nontransgenic, Wt, and SOD1 mutant mice, we found negligible amounts of trapped SOD1 cross-reacting material (Fig. 4A, PMS, L). In brain PMS of both G93A and G85R mutant SOD1 mice, there was a small amount of trapped SOD1 (Fig. 4A, PMS, B). However, in brain MPs from mutant mice, we found abundant amounts of trapped SOD1 compared with nontransgenic and Wt MPs (Fig. 4A, MP, B). We tested whether these nonfilterable aggregates contained other matrix proteins and found that MnSOD was also trapped on the membrane in brain MPs from G85R mice. This suggests that matrix proteins can be involved with SOD1 in forming high molecular weight aggregates. A titration of the G85R MP sample excluded that the retention of MnSOD on the membrane was attributable to saturation of the membrane with nonfilterable mutant G85R-hSOD1 aggregates (Fig. 4B).
Figure 4.
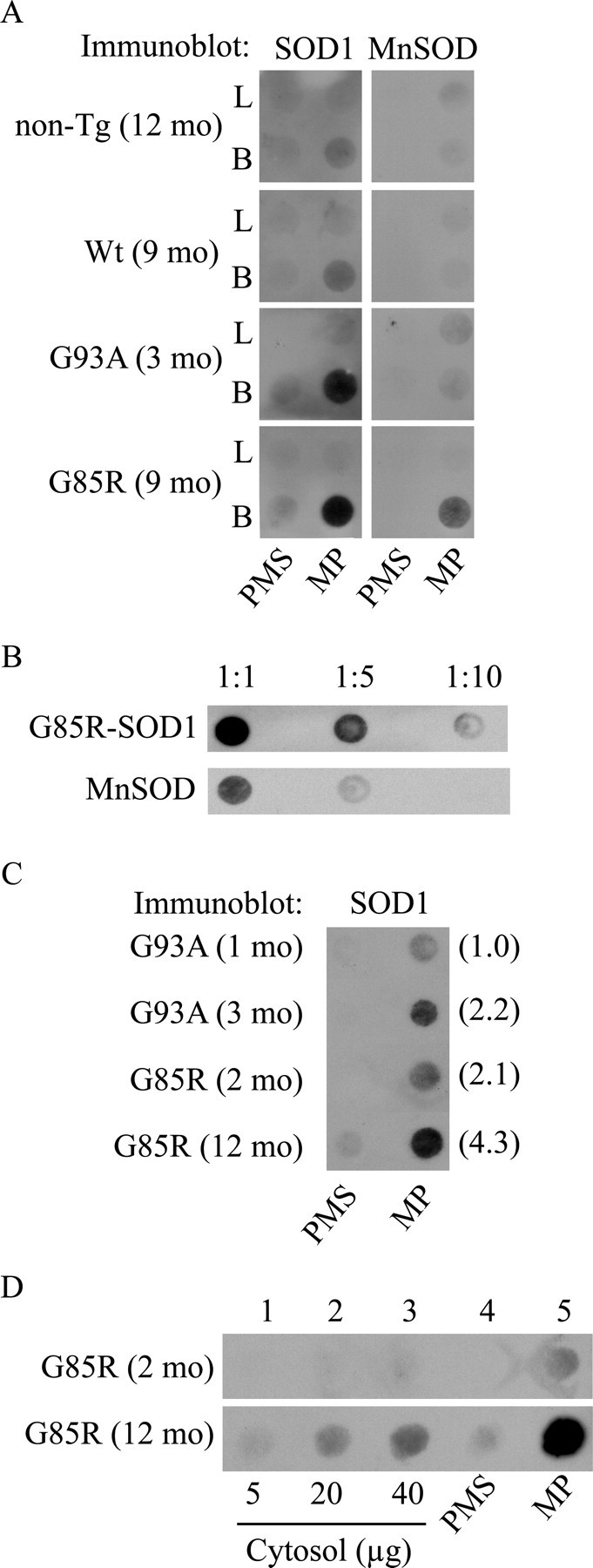
Mutant hSOD1 forms macromolecular aggregates in mitoplasts from brain mitochondria of fALS transgenic mice. A, MP and PMS fractions were prepared from brain (B) and liver (L) of nontransgenic (non-Tg) and transgenic hSOD1 mice. For each sample, 40 μg of total mitochondrial protein was subjected to fractionation in MPs and PMSs. The fractions were incubated in the presence of 0.5% TX-100, vacuum filtered onto a 0.22 μm cellulose acetate membrane using a 96-well dot-blot apparatus, and then immunoprobed for SOD1 and the matrix protein MnSOD. B, Dilutions of the G85R-hSOD1 MP samples prepared as in A. C, Age-dependent accumulation of mutant hSOD1 aggregates. Filter-trap assay of MP and PMS fractions from brain mitochondria of hSOD1 mutant mice at different disease stages. Samples were prepared as in A. G93A mice at 1 month of age and G85R mice at 2 months of age were presymptomatic, whereas G93A mice at 3 months of age and G85R mice at 12 months of age were symptomatic. In parentheses, densitometric intensities of SOD1 immunoreactivity in arbitrary optical density units. D, Comparison of mutant hSOD1 aggregate content in brain MP and PMS fractions (prepared as in A) with their corresponding cytosolic fractions from G85R mice at 2 and 12 months of age. mo, Month.
To determine whether mitochondrial matrix macromolecular aggregates accumulate with disease progression, we compared equal volumes of brain PMS and MP fractions from presymptomatic and symptomatic G93A and G85R mice. In both mutants, there was a clear age-dependent increase in the amount of aggregates retained on the filter (Fig. 4C). Furthermore, we compared the distribution of macromolecular aggregates between the cytosolic and the mitochondrial PMS and MP fractions from G85R brain. We filtered increasing amounts of cytosol up to a protein amount equivalent to that of PMS and MP fractions (40 μg of total protein). Interestingly, in the presymptomatic mouse (2 months of age), aggregates were detected only in the MP fraction but not in the PMS or cytosolic fractions. In the end-stage disease mouse (12 months of age), aggregates were detected in all of the fractions; however, they were most abundant in the MP fraction (Fig. 4D), suggesting that these aggregates form preferentially in the mitochondrial matrix.
Discussion
In this study, we find that, in both cytosol and mitochondria, hSOD1 represents ∼1-3% of total proteins. This is consistent with previous reports in rat (Okado-Matsumoto and Fridovich, 2001), mouse (Higgins et al., 2002; Mattiazzi et al., 2002), and yeast (Sturtz et al., 2001). Our findings also show that hSOD1 is at least threefold more concentrated in brain mitochondria than liver mitochondria. It is unlikely that the difference in mitochondrial hSOD1 content between brain and liver is attributable to faster liver mitochondrial turnover, because the adult mammalian liver has a low cellular (Fausto, 1990) and mitochondrial turnover. Instead, there is an accumulation of hSOD1 in mitochondria of neural cells, perhaps in response to increased oxidative stress in postmitotic tissues that have a higher oxidative metabolism. The finding of increased mitochondrial targeting of mutant hSOD1 in neural tissue may provide some clues for the specificity of the neurodegenerative process in fALS.
Previously, it has been proposed that mammalian mitochondrial SOD1 localizes in the IMS (Okado-Matsumoto and Fridovich, 2001; Mattiazzi et al., 2002). Although detailed ultrastructural studies of spinal cord mitochondria revealed SOD1 on both the surface and inside mitochondria (Jaarsma et al., 2001; Higgins et al., 2002), these studies could not resolve the precise intramitochondrial distribution of the protein. Here, we present three lines of evidence in support of the novel concept that mitochondrial hSOD1 is associated with the mitochondrial matrix: hSOD1 (1) can only be released from MPs if the IM is solubilized with detergents or disrupted by sonication, (2) can only be immunoprecipitated with SOD1 antibodies if the IM is disrupted by sonication, and (3) can only be digested by PK (G85R mutant hSOD1) if the IM is solubilized by detergents or sonicated. In regard to the last point, we had observed previously that both Wt and G93A mitochondria from spinal cord and brain contained a portion of hSOD1 that was associated with MPs (Mattiazzi et al., 2002). At the time, we thought that this portion of mitochondrial hSOD1 was bound to the outer side of the IM, as a result of the disappearance of hSOD1 from MPs after PK treatment. However, because both Wt and G93A hSOD1 are insensitive to PK digestion, the disappearance of hSOD1 from MPs in our previous experiments was presumably attributable to the proteolytic disruption of the IM by a harsh PK treatment. We have since worked out appropriate conditions for a PK treatment that preserve the integrity of the IM (Fig. 2C).
A previous study did not report the presence of SOD1 in rat liver mitochondrial matrix (Okado-Matsumoto and Fridovich, 2001). In this study, the intracellular distribution of rat endogenous SOD1 in liver was clearly different from the one we found in brain from transgenic mice expressing hSOD1. We show that, when equal protein amounts from the cytosolic and the mitochondrial fractions are compared, hSOD1 content is almost equal in the two fractions, whereas in the study by Okado-Matsumoto and Fridovich (2001), rat SOD1 was vastly more abundant in the cytosolic than in the mitochondrial fraction. These differences in intracellular SOD1 distribution may explain why SOD1 was not found previously in the rat liver mitochondrial matrix, supporting the concept that hSOD1 accumulates preferentially in the mitochondrial matrix of affected tissues.
The mechanisms underlying mitochondrial importation of hSOD1 are still unclear. Typically, proteins imported into mitochondria contain either a canonical N-terminal mitochondrial targeting signal (MTS) or an internal noncleavable MTS (Pfanner et al., 1987; Millar and Shore, 1994). However, it is known that some soluble proteins with dual subcellular localization can be targeted to the mitochondrial matrix without the aid of a canonical MTS, such as 2-methylacyl-CoA racemase (Amery et al., 2000). Furthermore, mitochondrial proteins that are associated with the matrix side of the mitochondrial IM, such as the accessory subunit of complex I, MWFE, are imported without an MTS (Zhuchenko et al., 1996). Although primary sequence analysis of SOD1 reveals no obvious MTS, SOD1 may contain a cryptic importation signal, and it may follow mitochondrial importation pathways similar to those of these proteins. Clearly, additional studies are needed to define the mechanism underlying SOD1 import into mitochondria and its implications in mitochondrial dysfunction.
Recently, it was reported that only mutant hSOD1, and not Wt hSOD1, associates with mitochondria from spinal cord and brain of transgenic mice (Liu et al., 2004). Here, we show that both Wt and mutant hSOD1 localize in mitochondria. These findings are in agreement with previous biochemical (Okado-Matsumoto and Fridovich, 2001; Mattiazzi et al., 2002) and ultrastructural (Jaarsma et al., 2001; Higgins et al., 2002) evidence. In addition, we find that Wt hSOD1 also localizes in spinal cord mitochondria (data not shown).
When we replicated the same mitochondrial purification scheme of Liu et al. (2004), we found that the mitochondrial markers MnSOD1, cytochrome c, and COX I are not only contained in the 25-30% interphase but also in several other fractions of the Nycodenz (Nycomed Pharma, Oslo, Norway) gradient. Because mutant and Wt SOD1 mitochondria may have different shape and size (Higgins et al., 2002), they may partition in different layers of the gradient. Perhaps, in the work by Liu et al. (2004), the analysis of the 25-30% interphase fraction alone might have led to an underestimation of mitochondrial Wt hSOD1. Conversely, in our Ficoll purification gradient, we recover most brain mitochondria in the pellet fraction, as indicated by the mitochondrial markers MnSOD1 and VDAC. This pellet fraction contains purified mitochondria, as shown by a lack of contamination with other organelles (Fig. 1A). We also show that the majority of hSOD1 associating with brain mitochondria does not behave as an integral membrane protein nor is it bound to peripheral membrane proteins. Again, our findings are different from those of Liu et al. (2004), who suggested that the majority of mutant hSOD1 in spinal cord mitochondria is associated with integral membrane proteins. It is possible that mutant hSOD1 establishes more stable bonds with mitochondrial membrane proteins in spinal cord than in brain, although the biological mechanisms underlying these differences remain to be understood. Finally, based on PK protection assays performed on whole mitochondria, Liu et al. (2004) suggested that mutant hSOD1 is primarily associated with the cytosolic side of the OM. Fine-tuning of the conditions for PK digestion of MP membranes demonstrates that the distribution of Wt and mutant hSOD1 inside mitochondria is more complex, involving partitioning in multiple compartments.
Mutant G93A hSOD1, which is PK resistant elsewhere, exhibits partial PK sensitivity in the matrix. This suggests that mutant, but not Wt, SOD1 acquires a conformation more susceptible to proteolysis in the matrix, denoting a biochemical difference between Wt and mutant hSOD1 in this mitochondrial compartment. Consistent with abnormal folding and/or metallation, we also show that both Wt and mutant G93A hSOD1 associated with MPs are partially inactive. Although copper metallation is a prerequisite for SOD1 activity, presumably it is not required to form the folded, soluble SOD1 protein (Hayward et al., 2002). Although the SOD1 holoenzyme cannot be imported into mitochondria, a completely demetallated SOD1 is transported into the mitochondrial IMS and activated in the presence of the copper chaperone for SOD1 (CCS) (Sturtz et al., 2001; Okado-Matsumoto and Fridovich, 2002; Field et al., 2003). This is consistent with our findings of normally active wild-type and G93A mutant hSOD1 in the PMS fractions of brain mitochondria. However, there was some residual SOD1 activity in the MP fractions. We do not know whether CCS is also present in the matrix. However, there are potential alternative pathways for copper loading of SOD1 in the matrix. For example, CCS-/- mice retain some SOD1 activity (Subramaniam et al., 2002), because SOD1 can be loaded with copper in a CCS-independent manner via a pathway involving the reduced form of glutathione (Carroll et al., 2004), which is abundant in mitochondria.
We showed previously that G93A mitochondria suffer from impaired oxidative phosphorylation. This impairment is markedly worse in brain and spinal cord than in liver mitochondria (Mattiazzi et al., 2002). Likewise, we detect mutant hSOD1 macromolecular aggregates associated with brain MPs but not liver MPs. The aggregates correlate with disease progression and are more abundant in G85R than in G93A MPs (Fig. 4C), whereas the mitochondrial content of G85R is lower than G93A hSOD1 (Fig. 1). This may reflect an increased propensity of the G85R mutant to misfold and aggregate in the matrix. In addition, in MPs of G85R mutant mice, the trapped macromolecular aggregates also contain MnSOD, suggesting that proteins other than hSOD1 may participate in the formation of these aggregates. Strikingly, brain mutant hSOD1 macromolecular aggregates are more abundant in the mitochondrial matrix than in the cytosol, suggesting that the matrix environment is conducive for mutant SOD1 aggregate formation.
In summary, our results show that both Wt and mutant hSOD1 associate with various mitochondrial compartments in the brain of transgenic mice, including the IM, the IMS, and the matrix. In addition, we observed the age-dependent formation of abundant hSOD1 macromolecular aggregates in the matrix. What remains to be determined is whether these aggregates are the cause or the consequence of mitochondrial dysfunction associated with mutant SOD1. The role of the complex compartmentalization and aggregation of mitochondrial hSOD1 in pathogenesis of fALS is a promising field of future investigation.
Footnotes
This work was supported by National Institutes of Health-National Institute of Neurological Disorders and Stroke, the Robert Packard Amyotrophic Lateral Sclerosis Research Center at Johns Hopkins University, and the Amyotrophic Lateral Sclerosis Association. We thank Dr. Khatuna Kipiani for assistance with the fALS transgenic mice, Dr. Anatoly Starkov for discussion and invaluable advice, and Dr. Erik Falck-Pedersen for support.
Correspondence should be addressed to Giovanni Manfredi, Weill Medical College of Cornell University, 525 East 68th Street, A-505, New York, NY 10021. E-mail: gim2004@mail.med.cornell.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/252463-08$15.00/0
References
- Amery L, Fransen M, De Nys K, Mannaerts GP, Van Veldhoven PP (2000) Mitochondrial and peroxisomal targeting of 2-methylacyl-CoA racemase in humans. J Lipid Res 41: 1752-1759. [PubMed] [Google Scholar]
- Beauchamp C, Fridovich I (1971) Superoxide dismutase: improved assays and an assay applicable to acrylamide gels. Anal Biochem 44: 276-287. [DOI] [PubMed] [Google Scholar]
- Borchelt DR, Lee MK, Slunt HS, Guarnieri M, Xu ZS, Wong PC, Brown Jr RH, Price DL, Sisodia SS, Cleveland DW (1994) Superoxide dismutase 1 with mutations linked to familial amyotrophic lateral sclerosis possesses significant activity. Proc Natl Acad Sci USA 91: 8292-8296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruijn LI, Becher MW, Lee MK, Anderson KL, Jenkins NA, Copeland NG, Sisodia SS, Rothstein JD, Borchelt DR, Price DL, Cleveland DW (1997) ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron 18: 327-338. [DOI] [PubMed] [Google Scholar]
- Bruijn LI, Houseweart MK, Kato S, Anderson KL, Anderson SD, Ohama E, Reaume AG, Scott RW, Cleveland DW (1998) Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science 281: 1851-1854. [DOI] [PubMed] [Google Scholar]
- Carroll MC, Girouard JB, Ulloa JL, Subramaniam JR, Wong PC, Valentine JS, Culotta VC (2004) Mechanisms for activating Cu- and Zn-containing superoxide dismutase in the absence of the CCS Cu chaperone. Proc Natl Acad Sci USA 101: 5964-5969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durham HD, Roy J, Dong L, Figlewicz DA (1997) Aggregation of mutant Cu/Zn superoxide dismutase proteins in a culture model of ALS. J Neuropathol Exp Neurol 56: 523-530. [DOI] [PubMed] [Google Scholar]
- Fausto N (1990) Hepatocyte differentiation and liver progenitor cells. Curr Opin Cell Biol 2: 1036-1042. [DOI] [PubMed] [Google Scholar]
- Field LS, Furukawa Y, O'Halloran TV, Culotta VC (2003) Factors controlling the uptake of yeast copper/zinc superoxide dismutase into mitochondria. J Biol Chem 278: 28052-28059. [DOI] [PubMed] [Google Scholar]
- Fujiki Y, Hubbard AL, Fowler S, Lazarow PB (1982) Isolation of intracellular membranes by means of sodium carbonate treatment: application to endoplasmic reticulum. J Cell Biol 93: 97-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX (1994) Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation Science 264: 1772-1775. [ [DOI] [PubMed] [Google Scholar]; Erratum (1995) 269: 149] [Google Scholar]
- Hayward LJ, Rodriguez JA, Kim JW, Tiwari A, Goto JJ, Cabelli DE, Valentine JS, Brown Jr RH (2002) Decreased metallation and activity in subsets of mutant superoxide dismutases associated with familial amyotrophic lateral sclerosis. J Biol Chem 277: 15923-15931. [DOI] [PubMed] [Google Scholar]
- Higgins CM, Jung C, Ding H, Xu Z (2002) Mutant Cu, Zn superoxide dismutase that causes motoneuron degeneration is present in mitochondria in the CNS. J Neurosci 22: RC215(1-6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins CM, Jung C, Xu Z (2003) ALS-associated mutant SOD1G93A causes mitochondrial vacuolation by expansion of the intermembrane space and by involvement of SOD1 aggregation and peroxisomes. BMC Neurosci 4: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaarsma D, Haasdijk ED, Grashorn JA, Hawkins R, van Duijn W, Verspaget HW, London J, Holstege JC (2000) Human Cu/Zn superoxide dismutase (SOD1) overexpression in mice causes mitochondrial vacuolization, axonal degeneration, and premature motoneuron death and accelerates motoneuron disease in mice expressing a familial amyotrophic lateral sclerosis mutant SOD1. Neurobiol Dis 7: 623-643. [DOI] [PubMed] [Google Scholar]
- Jaarsma D, Rognoni F, van Duijn W, Verspaget HW, Haasdijk ED, Holstege JC (2001) CuZn superoxide dismutase (SOD1) accumulates in vacuolated mitochondria in transgenic mice expressing amyotrophic lateral sclerosis-linked SOD1 mutations. Acta Neuropathol (Berl) 102: 293-305. [DOI] [PubMed] [Google Scholar]
- Johnston JA, Dalton MJ, Gurney ME, Kopito RR (2000) Formation of high molecular weight complexes of mutant Cu, Zn-superoxide dismutase in a mouse model for familial amyotrophic lateral sclerosis. Proc Natl Acad Sci USA 97: 12571-12576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung C, Higgins CM, Xu Z (2002) A quantitative histochemical assay for activities of mitochondrial electron transport chain complexes in mouse spinal cord sections. J Neurosci Methods 114: 165-172. [DOI] [PubMed] [Google Scholar]
- Koide T, Igarashi S, Kikugawa K, Nakano R, Inuzuka T, Yamada M, Takahashi H, Tsuji S (1998) Formation of granular cytoplasmic aggregates in COS7 cells expressing mutant Cu/Zn superoxide dismutase associated with familial amyotrophic lateral sclerosis. Neurosci Lett 257: 29-32. [DOI] [PubMed] [Google Scholar]
- Liu J, Lillo C, Jonsson PA, Velde CV, Ward CM, Miller TM, Subramaniam JR, Rothstein JD, Marklund S, Andersen PM, Brannstrom T, Gredal O, Wong PC, Williams DS, Cleveland DW (2004) Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron 43: 5-17. [DOI] [PubMed] [Google Scholar]
- Mattiazzi M, D'Aurelio M, Gajewski CD, Martushova K, Kiaei M, Beal MF, Manfredi G (2002) Mutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice. J Biol Chem 277: 29626-29633. [DOI] [PubMed] [Google Scholar]
- Millar DG, Shore GC (1994) Mitochondrial Mas70p signal anchor sequence. Mutations in the transmembrane domain that disrupt dimerization but not targeting or membrane insertion. J Biol Chem 269: 12229-12232. [PubMed] [Google Scholar]
- Okado-Matsumoto A, Fridovich I (2001) Subcellular distribution of superoxide dismutases (SOD) in rat liver: Cu,Zn-SOD in mitochondria. J Biol Chem 276: 38388-38393. [DOI] [PubMed] [Google Scholar]
- Okado-Matsumoto A, Fridovich I (2002) Amyotrophic lateral sclerosis: a proposed mechanism. Proc Natl Acad Sci USA 99: 9010-9014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallotti F, Lenaz G (2001) Isolation and subfractionation of mitochondria from animal cells and tissue culture lines. Methods Cell Biol 65: 1-35. [DOI] [PubMed] [Google Scholar]
- Pasinelli P, Belford ME, Lennon N, Bacskai BJ, Hyman BT, Trotti D, Brown Jr RH (2004) Amyotrophic lateral sclerosis-associated SOD1 mutant proteins bind and aggregate with Bcl-2 in spinal cord mitochondria. Neuron 43: 19-30. [DOI] [PubMed] [Google Scholar]
- Pfanner N, Hoeben P, Tropschug M, Neupert W (1987) The carboxyl-terminal two-thirds of the ADP/ATP carrier polypeptide contains sufficient information to direct translocation into mitochondria. J Biol Chem 262: 14851-14854. [PubMed] [Google Scholar]
- Ratovitski T, Corson LB, Strain J, Wong P, Cleveland DW, Culotta VC, Borchelt DR (1999) Variation in the biochemical/biophysical properties of mutant superoxide dismutase 1 enzymes and the rate of disease progression in familial amyotrophic lateral sclerosis kindreds. Hum Mol Genet 8: 1451-1460. [DOI] [PubMed] [Google Scholar]
- Reaume AG, Elliott JL, Hoffman EK, Kowall NW, Ferrante RJ, Siwek DF, Wilcox HM, Flood DG, Beal MF, Brown Jr RH, Scott RW, Snider WD (1996) Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat Genet 13: 43-47. [DOI] [PubMed] [Google Scholar]
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan JP, Deng HX (1993) Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis Nature 362: 59-62. [ [DOI] [PubMed] [Google Scholar]; Erratum (1993) 364: 362] [Google Scholar]
- Sturtz LA, Diekert K, Jensen LT, Lill R, Culotta VC (2001) A fraction of yeast Cu,Zn-superoxide dismutase and its metallochaperone, CCS, localize to the intermembrane space of mitochondria. A physiological role for SOD1 in guarding against mitochondrial oxidative damage. J Biol Chem 276: 38084-38089. [DOI] [PubMed] [Google Scholar]
- Subramaniam JR, Lyons WE, Liu J, Bartnikas TB, Rothstein J, Price DL, Cleveland DW, Gitlin JD, Wong PC (2002) Mutant SOD1 causes motor neuron disease independent of copper chaperone-mediated copper loading. Nat Neurosci 5: 301-307. [DOI] [PubMed] [Google Scholar]
- Tiwari A, Hayward LJ (2003) Familial amyotrophic lateral sclerosis mutants of copper/zinc superoxide dismutase are susceptible to disulfide reduction. J Biol Chem 278: 5984-5992. [DOI] [PubMed] [Google Scholar]
- Wang J, Xu G, Borchelt DR (2002) High molecular weight complexes of mutant superoxide dismutase 1: age-dependent and tissue-specific accumulation. Neurobiol Dis 9: 139-148. [DOI] [PubMed] [Google Scholar]
- Watanabe M, Dykes-Hoberg M, Culotta VC, Price DL, Wong PC, Rothstein JD (2001) Histological evidence of protein aggregation in mutant SOD1 transgenic mice and in amyotrophic lateral sclerosis neural tissues. Neurobiol Dis 8: 933-941. [DOI] [PubMed] [Google Scholar]
- Weisiger RA, Fridovich I (1973) Superoxide dismutase. Organelle specificity. J Biol Chem 248: 3582-3592. [PubMed] [Google Scholar]
- Wong PC, Pardo CA, Borchelt DR, Lee MK, Copeland NG, Jenkins NA, Sisodia SS, Cleveland DW, Price DL (1995) An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron 14: 1105-1116. [DOI] [PubMed] [Google Scholar]
- Zhuchenko O, Wehnert M, Bailey J, Sun ZS, Lee CC (1996) Isolation, mapping, and genomic structure of an X-linked gene for a subunit of human mitochondrial complex I. Genomics 37: 281-288. [DOI] [PubMed] [Google Scholar]