

Abstract
Mutations in the intracellular metalloenzyme superoxide dismutase 1 (SOD1) are linked to neurotoxicity in familial amyotrophic lateral sclerosis (ALS) by an unclear mechanism. Golgi fragmentation and endoplasmic reticulum stress are early hallmarks of spinal motor neuron pathology in transgenic mice overexpressing mutant SOD1, suggesting that dysfunction of the neuronal secretory pathway may contribute to ALS pathogenesis. We therefore proposed that mutant SOD1 directly engages and modulates the secretory pathway based on recent evidence of SOD1 secretion in diverse human cell lines. Here, we demonstrate that a fraction of active endogenous SOD1 is secreted by NSC-34 motor neuron-like cells via a brefeldin-A (BFA)-sensitive pathway. Expression of enhanced green fluorescent protein-tagged mutant human SOD1 (hSOD1-EGFP) in NSC-34 cells induced frequent cytoplasmic inclusions and protein insolubility that correlated with toxicity. In contrast, transfection of non-neuronal COS-7 cells resulted in mutant hSOD1-EGFP cytoplasmic inclusions, oligomerization, and fragmentation without detectable toxicity. Importantly, impaired secretion of hSOD1-EGFP was common to all 10 SOD1 mutants tested relative to wild-type protein in NSC-34 cells. Treatment with BFA inhibited hSOD1-EGFP secretion with pronounced BFA-induced toxicity in mutant cells. Extracellular targeting of mutant hSOD1-EGFP via SOD3 signal peptide fusion attenuated cytoplasmic inclusion formation and toxicity. The effect of elevated extracellular SOD1 was then evaluated in a transgenic rat model of ALS. Chronic intraspinal infusion of exogenous wild-type hSOD1 significantly delayed disease progression and endpoint in transgenic SOD1G93A rats. Collectively, these results suggest novel extracellular roles for SOD1 in ALS and support a causal relationship between mutant SOD1 secretion and intraneuronal toxicity.
Keywords: amyotrophic lateral sclerosis, CSF, neuronal inclusion, NSC-34 cell, protein aggregation, secretion, superoxide dismutase 1, transgenic rat
Introduction
Amyotrophic lateral sclerosis (ALS) is a progressive and fatal adult-onset neurodegenerative disorder characterized by premature loss of spinal, cranial, and cortical motor neurons (Rowland and Shneider, 2001). Cytoplasmic Cu,Zn-superoxide dismutase 1 (SOD1) mutations are associated with 20% of familial ALS cases (Rosen et al., 1993) and induce motor neuron disease when overexpressed in transgenic mice or rats (Gurney et al., 1994; Howland et al., 2002). Mutant SOD1-mediated toxicity is primarily linked to pro-oxidative chemistry and/or protein aggregates, provoking neuroinflammatory, excitotoxic, cytoskeletal, and mitochondrial damage to motor neurons by a non-cell-autonomous mechanism (Bendotti and Carri, 2004; Bruijn et al., 2004). However, the selective lethality of ubiquitously expressed SOD1 mutations in motor neuron degeneration remains unknown.
Increasing evidence suggests early disturbances in the neuronal secretory apparatus and pathway in ALS. Fragmentation of the Golgi apparatus is a consistent neuropathologic feature of lower or upper motor neurons in SOD1-linked familial and sporadic ALS (Mourelatos et al., 1990; Fujita et al., 1999, 2000) and Guamanian ALS-parkinsonism-dementia complex (Mourelatos et al., 1994). Golgi pathology was also detected in spinal motor neurons of presymptomatic transgenic SOD1G93A mice (Mourelatos et al., 1996), increasing with disease progression but primarily independent of inclusion and aggregate burden (Stieber et al., 2000a,b,c). Furthermore, fragmented Golgi were not characteristic of transgenic neurofilament heavy subunit mice overexpressing neurofilament protein (Stieber et al., 2000b), suggesting that mutant SOD1 may directly target and disrupt the Golgi apparatus early in ALS pathogenesis. Endoplasmic reticulum (ER) stress is also evident in ALS rodent models, with induction of the local chaperone glucose-regulated protein 78 reported in spinal motor neurons of preclinical transgenic SOD1L84V mice (Tobisawa et al., 2003). Activation of ER-resident caspase-12 was also demonstrated early in spinal cords of transgenic SOD1G93A mice (Wootz et al., 2004). Together, these data suggest a progressive dysfunction of neuronal ER-Golgi transport and secretion in SOD1-mediated ALS, although the secreted proteins involved are yet to be identified.
SOD1 is traditionally regarded as a cytosolic protein, with separate mitochondrial (SOD2) and extracellular (SOD3) targeted enzymes found in other cell compartments (Zelko et al., 2002). However, SOD1 was localized to the intermembrane space of yeast mitochondria, suggesting a wider protective role in oxidant stress (Sturtz et al., 2001). Mutant SOD1 was subsequently detected within spinal mitochondria of transgenic SOD1G93A mice, implying novel toxic mechanisms (Jaarsma et al., 2001). Recently, evidence for extracellular SOD1 and paracrine activities distinct from SOD3 was demonstrated in vitro. SOD1 secretion into the culture medium from human hepatocyte, fibroblast, neuroblastoma, and thymic-derived cell lines was shown (Mondola et al., 1996, 1998; Cimini et al., 2002). SOD1 export was linked to an ATP-dependent and brefeldin-A (BFA)-sensitive secretory pathway, implicating microvesicular transport from the ER-Golgi system (Mondola et al., 2003).
Based on the evidence of secretory pathway dysfunction in ALS, we hypothesized that mutant SOD1 secretion is compromised and linked to toxicity. To address this, endogenous, wild-type (WT) and mutant SOD1 secretion was characterized in a motor neuron cell culture model of mutant SOD1-mediated ALS. Here, we report that selective impairment of mutant SOD1 secretion is associated with toxic inclusions and is reversed by extracellular mutant protein targeting. Accordingly, elevation of extracellular SOD1 in transgenic SOD1G93A rats improved survival outcomes, suggesting novel extracellular roles for SOD1 in ALS pathogenesis and therapy.
Materials and Methods
Expression vectors. The pEF-BOS vectors containing human SOD1WT, SOD1A4V, SOD1G37R, SOD1G85R, SOD1G93C, and SOD1FS126 cDNAs were provided by Dr. David Borchelt (Johns Hopkins University, Baltimore, MD). Constructs were used as templates for PCR amplification using the following primers: 5′-GCGCGCGTCGACAAGCATGGC-3′ (forward) and 5′-GCGCGCGTCGACGCTTGGGCGATCCCAAT-3′ (reverse). Primers were designed to introduce a SalI site to allow subcloning into pEGFP-N1 (Clontech, Palo Alto, CA) and to remove the SOD1 translation stop codon. Additional mutant SOD1H43R, SOD1H46R, SOD1H48Q, SOD1D90A, and SOD1G93A plasmids were generated via site-directed mutagenesis of the SOD1WT template using the Quik Change kit (Stratagene, La Jolla, CA). For mitochondrial targeting constructs, a 72 bp sequence encoding the N-terminal 24 amino acid leader peptide of human SOD2 was inserted upstream of the SOD1WT and SOD1G93A sequences in pEGFP-N1 vectors. For extracellular targeting constructs, a 54 bp sequence encoding the N-terminal 18 amino acid signal peptide of human SOD3 was inserted upstream of the SOD1WT and SOD1G93A sequence in pEGFP-N1 vectors. Both sets of constructs were prepared by site-directed mutagenesis using the following primers: mitochondrial, 5′-CAGTCGACAAGCATGTTGAGCCGGGCAGTGTGCGGCACCAGC-AGGCAGCTGGCTCCGGCTTTGGGGTATCTGGGCTCCAGGCAGG-CGACGAAGGC-3′ (forward) and 5′-CGGCCTTCGTCGCCTGCCTG-GAGCCCAGATACCCCAAAGCCGGAGCCAGCTGCCTGCT GGTGC-CGCACACTGCCCGGCTCAACATGCTTGTCGACTG-3′ (reverse); extracellular, 5′-CAGTCGACAAGCATGCTGGCGCTACTGTGTT-CCTGCCTGCTCCTGGCAGCCGGTGCCTCGGACGCGACGAAGG-CCG-3′ (forward) and 5′-CGGCCTTCGTCGCGTCCGAGGCACCG-GCTGCCAGGAGCAGGCAGGAACACAGTAGCGC CAGCATGCTT-GTCGACT-3′ (reverse).
The sequence of the PCR-amplified or mutagenized region was verified in each case using an automated fluorescent dye-terminator cycle DNA sequencer (PerkinElmer Life Sciences, Emeryville, CA).
Cell culture and transfection. Mouse motor neuron-like NSC-34 cells were supplied by Dr. Neil Cashman (University of Toronto, Toronto, Ontario, Canada), and simian COS-7 cells were provided by George Christopoulos (Howard Florey Institute, Parkville, Victoria, Australia). Cell lines were maintained in DMEM with 10% (v/v) fetal calf serum and 1% (v/v) penicillin-streptomycin (Invitrogen, Mt. Waverley, Australia). Cells were subcultured in 24-well plates (1 × 105 cells per well) and were transfected transiently with plasmids (1 μg DNA per well) using a 1:1 ratio of Transfast lipofection reagent (Promega, Annandale, Australia) to DNA. Cells were cotreated with 0.1 μg/ml BFA (Sigma, Seven Hills, Australia) at this time point. Cells were visualized at 72 hr after transfection with an inverted fluorescence microscope (Olympus, Melville, NY). For inclusion quantification, aggregate-positive cells from a minimum of three wells were counted as a percentage of total enhanced green fluorescent protein (EGFP)-positive cell transfectants.
Protein extraction. Culture medium was removed from wells and was clarified by centrifugation at 5000 × g for 5 min. Medium fractions were assayed for cytolysis using a lactate dehydrogenase (LDH) activity kit (Roche, Castle Hill, Australia). Cells were lysed in Tris-NaCl (TN) buffer (50 mm Tris-HCl, pH 7.5, 150 mm NaCl) with 0.1% (v/v) NP-40, 0.1% (w/v) SDS, and 1% (v/v) protease inhibitor mixture (Sigma) for 10 min on ice. Lysates were clarified by centrifugation at 15,800 × g for 10 min. Resulting pellets were resuspended in TN buffer with 1% (w/v) SDS and ultrasonicated for 15 sec.
Mitochondrial fractions were prepared as described previously with modifications (Epperly et al., 2002). Cells were resuspended in hypotonic buffer [5 mm Tris-HCl, pH 7.5, 5 mm KCl, 1.5 mm MgCl2, 0.1 mm EDTA, 1 mm DTT, and 1% (v/v) protease inhibitor mixture] and incubated on ice for 30 min. Cells were disrupted with 20 strokes in a Dounce homogenizer and centrifuged at 500 × g for 5 min. Resulting supernatants were centrifuged at 10,000 × g for 30 min to clarify cytosolic fractions. Mitochondrial pellets were solubilized in TN buffer with 1% (w/v) SDS and ultrasonicated for 15 sec. Proteins were quantified using the BCA assay kit (Promega).
Immunoprecipitation. Medium, cell lysate, and subcellular fractions were incubated with SOD1 (1:750; Calbiochem, La Jolla, CA) or GFP (1:750; Chemicon, Boronia, Australia) antibodies and 30 μl of 50% (w/v) protein A-Sepharose CL-4B (Amersham Biosciences, Castle Hill, Australia) in Tris buffer [50 mm Tris-HCl, pH 7.5, 0.02% (w/v) NaN3] on a rotating wheel overnight at 4°C. Samples were centrifuged at 15,800 × g for 1 min, and Sepharose pellets were washed twice in Tris buffer for 10 min each. For immunoblotting, immunoprecipitates were liberated by boiling in 2% (w/v) SDS reducing loading buffer. For zymography, immunoprecipitates were eluted in 0.1 m glycine, pH 3.0, for 15 sec and neutralized with 1 m Tris-HCl, pH 9.0, before mixing with native loading buffer.
Immunoblotting and zymography. Samples were electrophoresed through 12.5% SDS-polyacrylamide gels and transferred to nitrocellulose membranes (Millipore, North Ryde, Australia). Membranes were blocked with 5% (w/v) nonfat milk and 0.1% (v/v) Tween 20 in Tris-buffered saline, pH 8.0, for 1 hr and incubated with SOD1 (1:1000) or p38 mitogen-activated protein kinase (p38MAPK) (1:1000; Cell Signaling, Beverly, MA) overnight at 4°C. Blots were probed with HRP-conjugated antibodies (1:2500; Chemicon) and visualized with enhanced chemiluminescence reagents (Roche). For zymography, samples were separated using 7.5% nondenaturing polyacrylamide gels and stained for dismutase activity as described previously (Beauchamp and Fridovich, 1971).
Cell survival assay. Cells were subcultured in 96-well plates (1 × 104 cells per well) and transfected (0.2 μg DNA per well). Cell viability at 72 hr was assessed by reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (Sigma). Cells were treated with MTT (0.5 mg/ml) for 1 hr at 37°C, medium was aspirated, and cells were solubilized in DMSO for absorbance measurements at 530 nm.
Animal drug treatment. Transgenic SOD1G93A rats derived from the SD-TgN(SOD1G93A)L26H line (Taconic, Germantown, NY) were bred on a Sprague Dawley (SD) background and were PCR genotyped using tail genomic DNA. At postnatal day 100 (P100), rats were anesthetized by intraperitoneal injection of Nembutal (Lyppard, Cheltenham, Australia) (0.7 ml/kg) and atropine (0.5 ml/kg) in 0.9% (w/v) saline. Alzet model 2004 mini-osmotic pumps (Durect Corporation, Cupertine, CA) filled with 50 μm erythrocyte hSOD1WT (Sigma) or PBS vehicle solution, pH 7.4, were preincubated at 37°C for 48 hr and then inserted intraspinally at vertebrae L4-5 and implanted subcutaneously between the hindlimbs. Eight rats were used per group, consisting of equal male and female numbers. Pumps supply 33 d of continuous infusion (0.3 nmol/d), and accuracy was determined by aspirating pumps postmortem. Aliquots of hSOD1WT solution were also incubated at 37°C and periodically tested with zymography to verify stability at physiologic temperature.
Locomotor function was assessed three times per week using a rat rotarod (6.4 cm axial diameter; Scientific Concepts, Glen Waverley, Australia), commencing at P90. Rats were trained once by two ramping sessions of 2.7-27 rpm over 5 min and one constant session of 16.2 rpm for 5 min, with 10 min rest periods between sessions. Rats were tested at a constant speed of 16.2 rpm for a maximum of 3 min. Disease onset was defined by the age of initial rotarod performance deficit. Disease progression was scored as follows: stage I, onset of hindlimb paresis; stage II, severe hindlimb paresis and abnormal gait; stage III, hindlimb paralysis usually with unilateral forelimb involvement. Rats were killed at stage III onset by lethal intraperitoneal injection (150 mg/kg, sodium pentobarbitone), thus defining this time point as the age of death. The time interval between disease onset and stage III was considered as disease duration. A minority of rats from both groups developed moribund forelimb paralysis without posterior involvement and were excluded from this study. These experiments were approved by the Howard Florey Institute Animal Ethics Committee (permit number 03-101).
Immunohistochemistry and histology. Rats at disease endpoint were transcardially perfused with PBS followed by 4% (w/v) paraformaldehyde in PBS. Lumbar spinal cords were dissected, postfixed, cryoprotected overnight in 30% (w/v) sucrose in PBS, embedded, and transverse frozen 15 μm cryostat sections were cut. Sections were permeabilized in 0.3% (v/v) Triton X-100 in PBS for 15 min, blocked in 5% (w/v) nonfat milk in PBS for 1 hr, and incubated overnight at 4°C with SOD1 antibodies (1:500) in blocking solution. Sections were incubated with anti-sheep biotinylated antibodies (1:200; Vector Laboratories, Burlingame, CA) for 1 hr and fluorescein-streptavidin (1:200) for 2 hr before visualizing. Adjacent sections were counterstained with 0.1% (w/v) cresyl fast violet using standard protocols, dehydrated, and coverslipped.
Fluid and tissue extraction. A separate cohort of transgenic and wild-type SD rats were killed at P60, P90 (preclinical), and P120 (symptomatic). CSF was tapped from the cisterna magna and clarified by centrifugation at 8000 × g. Lumbar spinal cords were dissected and homogenized with liquid nitrogen-chilled mortars in radioimmunoprecipitation assay lysis buffer [50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 0.1% (w/v) SDS, 1% (w/v) sodium deoxycholate, 1% (v/v) Triton X-100, and 1% (v/v) protease inhibitor mixture]. Homogenates were stored on ice for 20 min and clarified by centrifugation at 15,800 × g for 20 min for immunoblotting, as described above.
Statistical analysis. Cell-culture data were analyzed using one-way ANOVA with Tukey's post hoc test. Disease onset and duration data were compared using an unpaired t test. Survival data were compared using Kaplan-Meier survival analysis with the log-rank test. All statistical tests were performed with Graph-Pad Prism software (version 3.02; GraphPad Software, San Diego, CA).
Results
Active endogenous mouse SOD1 is secreted by NSC-34 cells
To establish whether SOD1 secretion is a relevant phenomenon in the NSC-34 motor neuron cell line, medium fractions and cell lysates were collected from cultures at various time points for immunoprecipitation experiments. Endogenous mouse SOD1 (mSOD1) was first detected in medium fractions at 48 hr and increased at 72 hr (Fig. 1a), representing ∼20% of intracellular mSOD1 by immunoblot analysis. Assay of medium fractions by zymography also demonstrated weak activity at 72 hr (Fig. 1a), suggesting that extracellular mSOD1 is homodimeric and active. These results were not attributable to cytolysis, because LDH activity in the media was not significantly elevated during this time period (Fig. 1a), and cytoplasmic p38MAPK was not present in medium fractions (Fig. 1b). Treatment of cells with BFA for 72 hr inhibited mSOD1 release into the medium (Fig. 1c), suggesting that ER-Golgi-dependent secretory transport may underlie extracellular mSOD1.
Figure 1.

Endogenous mSOD1 secretion in NSC-34 cells is time dependent and BFA sensitive. a, Anti-SOD1 immunoblot analysis (top) and dismutase activity gel (bottom) of culture medium fractions and cell extracts immunoprecipitated for SOD1 at various time points. Control incubations with culture medium alone (-) and 0.1% (v/v) Triton X-100 lysed cells (+) are shown. LDH activity was quantified in culture media as an index of cytolysis. Data are presented as mean ± SEM; n = 4. b, Anti-p38MAPK immunoblot analysis of culture medium fractions. c, Anti-SOD1 immunoblot analysis of cells treated with BFA for 72 hr reveals inhibition of mSOD1 secretion.
Mutant hSOD1-EGFP cytoplasmic inclusions correlate with toxicity in NSC-34 cell transfectants
NSC-34 cells were transfected transiently with WT and ALS-linked mutant hSOD1-EGFP. Ten mutants were selected for study, involving WT-like active (A4V, G37R, H43R, D90A, G93A, G93C), copperbinding site (H46R, H48Q, G85R), and truncation (FS126) mutations. Transient expression of vector alone (Vec) and WT plasmids induced widespread and diffuse fluorescence in cells (Fig. 2a). Furthermore, these cells maintained a characteristic neuronal morphology with neurite outgrowth. Expression of eight mutant hSOD1-EGFP constructs induced large, prominent, and multiple cytoplasmic inclusions in cells (Fig. 2a). Cells bearing aggregates appeared dystrophic and devoid of neurites. Inclusions were not evident in two mutants, H48Q and D90A, which instead resembled WT transfectants with uniformly distributed fluorescence and preserved neurites. The frequency of inclusion-positive cells was quantified (Fig. 2b), revealing significant hSOD1-EGFP aggregation in A4V, G85R (p < 0.01), and FS126 (p < 0.05) transfectants. Cell survival data correlated inversely with inclusion burden, because viability was significantly reduced by A4V, G85R (p < 0.01), G93A, and G93C (p < 0.05) expression (Fig. 2c). Thus, increased cell death was associated with most mutants exhibiting a high aggregation propensity. Immunoblots of cell supernatant fractions demonstrated an ∼50 kDa band representing hSOD1-EGFP on a background of 16 kDa mSOD1 in transfectants (Fig. 2d). Increased mobility of G85R, D90A, and FS126 variants was also evident. Analysis of pellet fractions revealed accumulation of all 10 mutants but not WT nor mSOD1, suggesting mutant-specific enhanced protein insolubility. Importantly, high molecular weight or oligomeric mutant hSOD1-EGFP species were not detected in either NSC-34 cell fraction. Transfection with original pEF-BOS plasmids also failed to produce oligomeric hSOD1, suggesting that EGFP fusion does not impede aggregation (data not shown). Nevertheless, hSOD1-EGFP activity was detected using zymography (Fig. 2e), implying homodimer formation under native conditions. Activity was absent for H46R, H48Q, G85R, and FS126 mutants, whereas slightly increased native mobility was evident for G93A and G93C variants, and the A4V mutant exhibited the greatest mobility shift (Fig. 2e).
Figure 2.
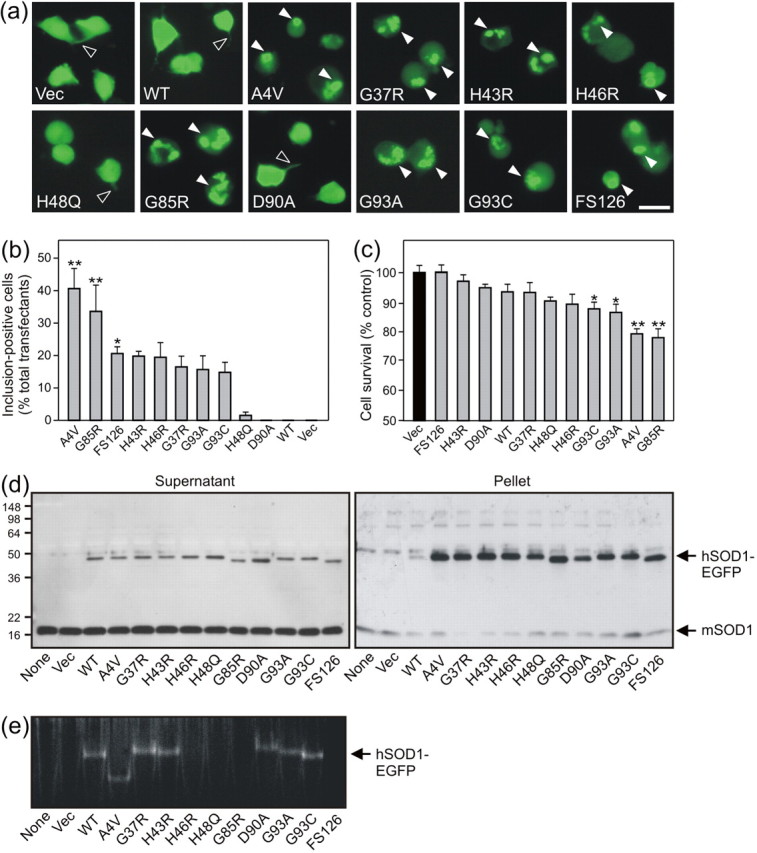
Mutant hSOD1-EGFP expression induces abundant cytoplasmic aggregates, protein insolubility without oligomerization, and toxicity in transfected NSC-34 cells. a, Fluorescence microscopy of cells 72 hr after transfection reveals global hSOD1-EGFP distribution and preserved neurite outgrowth (open arrowhead) in Vec and WT cells, whereas inclusion formation (arrowhead) is promoted by most mutant hSOD1-EGFPs. Scale bar, 20 μm. b, The frequency of aggregate-positive cells is quantified. c, Cell viability of cultures determined by MTT reduction; mean ± SEM; n = 4; *p < 0.05; **p <0.01 versus Vec cell transfectants by one-way ANOVA with Tukey's post hoc test. d, Anti-SOD1 immunoblot analysis of supernatant and pellet fractions from cell lysates (10 μg per lane) reveals hSOD1-EGFP monomers and mutant protein-enriched pellets. e, Dismutase activity gel analysis of cell lysate supernatants (50 μg per lane) shows differential hSOD1-EGFP activities and native mobilities.
COS-7 cells are resistant to mutant hSOD1-EGFP-induced toxicity despite an intracellular aggregate burden
To test the selective toxicity of SOD1 mutants, hSOD1-EGFP plasmids were expressed transiently in non-neuronal COS-7 cells. Uniformly distributed fluorescence was again observed in Vec and WT cell transfectants, whereas the majority of mutants formed large cytoplasmic inclusions (Fig. 3a). Interestingly, organelles presumed to be vacuoles or endosomes were refractory to hSOD1-EGFP localization. Quantification of cells containing inclusions revealed significant hSOD1-EGFP aggregation in A4V, G85R (p < 0.01), G93C, and G37R (p < 0.05) transfectants (Fig. 3b); however, inclusion frequency was reduced approximately fourfold overall compared with NSC-34 cells. Furthermore, cell viability was not significantly impaired by expression of any mutants examined in COS-7 cells (Fig. 3c). Western blots of cell supernatant fractions indicated robust expression of two major bands at ∼50 and ∼27 kDa, representing hSOD1-EGFP and cleaved EGFP, respectively, on a background of 16 kDa endogenous mSOD1 (Fig. 3d). Importantly, analysis of pellet fractions revealed insoluble high molecular weight material and protein fragments associated with most mutants. hSOD1-EGFP oligomers and fragments were not observed in H48Q and D90A mutant pellets. Finally, enzyme activity assay of transfected cell lysates produced a similar profile to NSC-34 cell transfectants (Fig. 3e).
Figure 3.
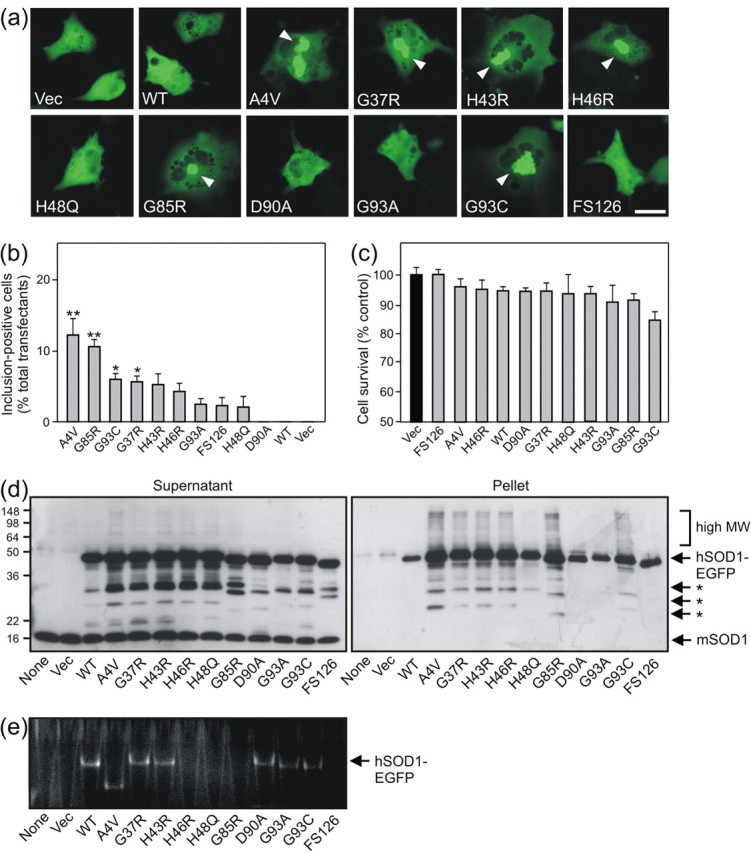
Mutant hSOD1-EGFP expression induces cytoplasmic aggregates, protein insolubility, oligomerization, and fragmentation without toxicity in transfected COS-7 cells. a, Fluorescence microscopy of cells 72 hr after transfection reveals diffuse hSOD1-EGFP localization in Vec and WT cells, whereas inclusion formation (arrowhead) is induced by most mutant hSOD1-EGFPs. Scale bar, 20 μm. b, The frequency of aggregate-positive cells is quantified. c, Cell viability of cultures determined by MTT reduction assay; mean ± SEM; n = 4; *p < 0.05; **p < 0.01 versus Vec cells by one-way ANOVA with Tukey's post hoc test. d, Anti-SOD1 immunoblot analysis of supernatant and pellet fractions from cell lysates (10 μg per lane) reveals monomeric, high molecular weight (MW; bracket), and fragmented (asterisk) hSOD1-EGFP species. e, Dismutase activity gel analysis of cell lysate supernatants (50 μg per lane) reveals different hSOD1-EGFP activities and native mobilities.
Impaired secretion is a common property of mutant hSOD1-EGFP in transfected NSC-34 cells
The secretion of hSOD1-EGFP was next investigated in the NSC-34 cell culture model of mutant SOD1-mediated ALS. A high proportion of WT protein was detected in the medium fraction 72 hr after transfection (Fig. 4a). The extracellular secretion of all 10 mutants, however, was diminished relative to extracellular WT protein. This was independent of expression level, and indirectly transfection efficiency, as suggested by variable intracellular mutant expression (Fig. 4a). Furthermore, culture media were completely deficient in hSOD1-EGFP from A4V, G85R, and FS126 cell transfectants. Conversely, secretion of H48Q and D90A proteins was highest among the mutants tested. Treatment of transfected cells with BFA for 72 hr reduced both WT and mutant hSOD1-EGFP levels in the medium (Fig. 4b), suggesting a common secretory pathway to endogenous mSOD1. When visualized, BFA-treated cells displayed rounded morphology suggestive of toxicity (Fig. 4c). The intracellular distribution of hSOD1-EGFP was not altered in WT cells exposed to BFA; however, mutant cells adopted compacted and fine inclusion material after treatment. Cell viability was accordingly reduced by BFA treatment in WT cells (p < 0.01); however, mutant hSOD1-EGFP-expressing cells displayed enhanced sensitivity to BFA-induced toxicity (p < 0.001) (Fig. 4d).
Figure 4.
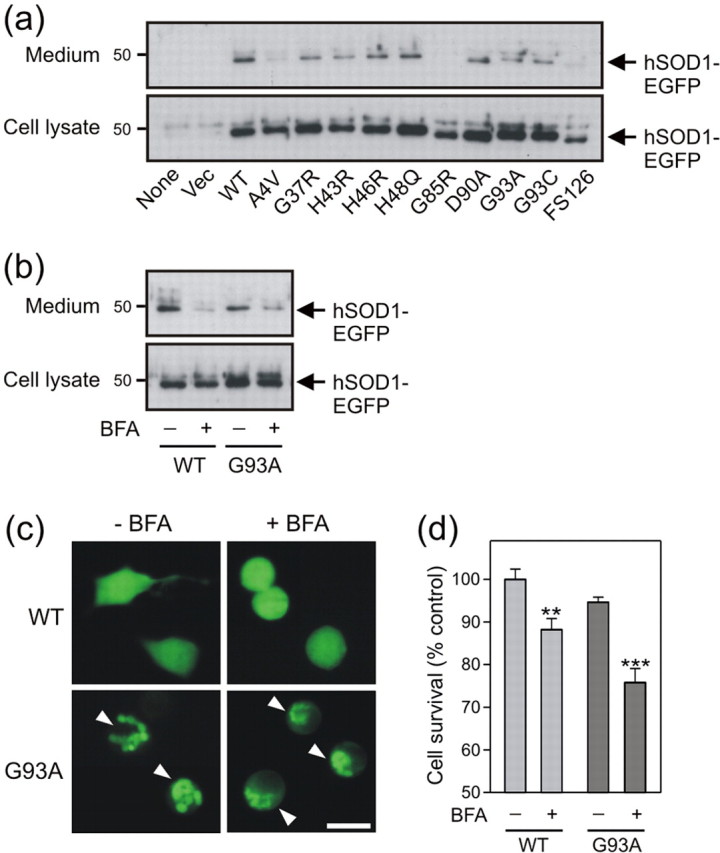
Mutant hSOD1-EGFP secretion is impaired and toxicity potentiated by BFA treatment in transfected NSC-34 cells. a, Anti-SOD1 immunoblot analysis of culture medium fractions and cell extracts immunoprecipitated for GFP reveals deficient extracellular accumulation of mutant hSOD1-EGFP. b, Anti-SOD1 immunoblot analysis of cells treated with BFA for 72 hr demonstrates inhibition of hSOD1-EGFP secretion. c, Fluorescence microscopy of mutant cells treated with BFA for 72 hr reveals fine and granular cytoplasmic inclusions (arrowhead). Scale bar, 20 μm. d, Cell viability of cultures determined by MTT reduction is reduced by BFA exposure with pronounced toxicity in mutant cells. Data are presented as mean ± SEM; n = 4; **p < 0.01; ***p < 0.001 versus untreated genotype-matched cells by one-way ANOVA with Tukey's post hoc test.
SOD3 signal peptide-mediated extracellular targeting reduces mutant hSOD1-EGFP toxic inclusions in transfected NSC-34 cells
To modulate the hSOD1-EGFP secretory pathway in NSC-34 cells, the human SOD2 mitochondrial import sequence or SOD3 secretion signal peptide was fused in frame to the N terminus of hSOD1-EGFP, as shown in Figure 5a. Expression of hSOD2-targeted hSOD1-EGFP in cells resulted in heterogeneous fluorescence, with inclusions still evident in mutant transfectants (Fig. 5b). Expression of hSOD3-targeted hSOD1-EGFP induced weak intracellular fluorescence in transfectants with little nuclear localization (Fig. 5b). Importantly, extracellular targeting of mutant hSOD1-EGFP reduced and confined inclusion formation to the cell periphery. Consistent with these observations, crude mitochondrial preparations were enriched with hSOD2-targeted hSOD1-EGFP compared with nontargeted protein (Fig. 5c). Furthermore, medium fractions contained greater levels of hSOD3-targeted hSOD1-EGFP than nontargeted protein. No relative differences between WT and mutant protein import or export were evident, whether targeted or not. A mobility shift of targeted hSOD1-EGFP was also noted in cytosolic and cell lysate fractions, most likely attributable to intact precursor proteins. Importantly, the viability of mutant cells was significantly rescued by extracellular targeting of mutant hSOD1-EGFP (p < 0.01) (Fig. 5d).
Figure 5.

Extracellular targeting of mutant hSOD1-EGFP attenuates cytoplasmic aggregates and toxicity in transfected NSC-34 cells. a, Schematic diagram of hSOD1-EGFP constructs with hSOD2 or hSOD3 signal peptide preceding cDNAs. hCMV, Human cytomegalovirus. b, Fluorescence microscopy of cells 72 hr after transfection reveals distinct subcellular and extracellular localizations of SOD2- and SOD3-targeted hSOD1-EGFP, respectively. Inclusion material (arrowhead) in mutant cells is reduced by extracellular targeting. Scale bar, 20 μm. c, Anti-SOD1 immunoblot analysis of subcellular, intracellular, and extracellular accumulation of SOD2- and SOD3-targeted hSOD1-EGFP immunoprecipitated for GFP. Mito, Mitochondrial; Cyto, cytosolic. d, Cell viability of cultures determined by MTT reduction assay is rescued by SOD3 targeting in mutant cells. Data are presented as mean ± SEM; n = 4; **p < 0.01 versus G93A cells by one-way ANOVA with Tukey's post hoc test.
Chronic intrathecal treatment with hSOD1WT delays disease progression and endpoint in transgenic ALS rats
To support these observations in culture, we performed analogous experiments in vivo. SOD1 was next detected in an extracellular compartment of a transgenic rat model of ALS. Endogenous rat SOD1 (rSOD1) was observed in CSF of nontransgenic animals (Fig. 6). Mutant hSOD1 was detected in CSF of preclinical and symptomatic transgenic SOD1G93A rats, exceeding rSOD1 levels with an age-dependent increase. Extracellular SOD1 again represented a fraction of intracellular protein in spinal cord, without apparent oligomer formation in CSF (Fig. 6).
Figure 6.

Presence of extracellular endogenous and mutant SOD1 in CSF of transgenic ALS rats. Anti-SOD1 immunoblot analysis of CSF and lumbar spinal cord lysates (10 μg per lane) extracted from WT and transgenic SOD1G93A rats at various postnatal day ages. Open arrowheads indicate putative high molecular weight species.
Next, the effect of elevated extracellular SOD1 on disease progression was tested in transgenic ALS rats. Exogenous hSOD1WT was intraspinally delivered at an intracellular physiological concentration to presymptomatic rats. Animal weights did not vary significantly between the different treatment groups during the infusion period (Fig. 7a). The onset of locomotor impairment as measured by rotarod analysis was not significantly delayed by hSOD1WT treatment at 115 ± 6 d (mean ± SEM) compared with the vehicle group at 115 ± 5 d (Fig. 7b). However, hSOD1WT therapy significantly prolonged survival in rats to 149 ± 8 d compared with saline infused animals at 124 ± 4 d (p < 0.05) (Fig. 7c). Disease duration was also significantly extended by hSOD1WT treatment (p < 0.05) (Table 1), suggesting that extracellular elevation of hSOD1WT alters disease outcome but not onset in transgenic ALS rats. To verify that implanted hSOD1WT maintains antioxidative activity and stability, the enzyme was incubated at physiologic temperature for 1 month. Remarkably, dismutase activity persisted up to 4 weeks incubation (Fig. 7d), validating this intrathecal method of delivery.
Figure 7.
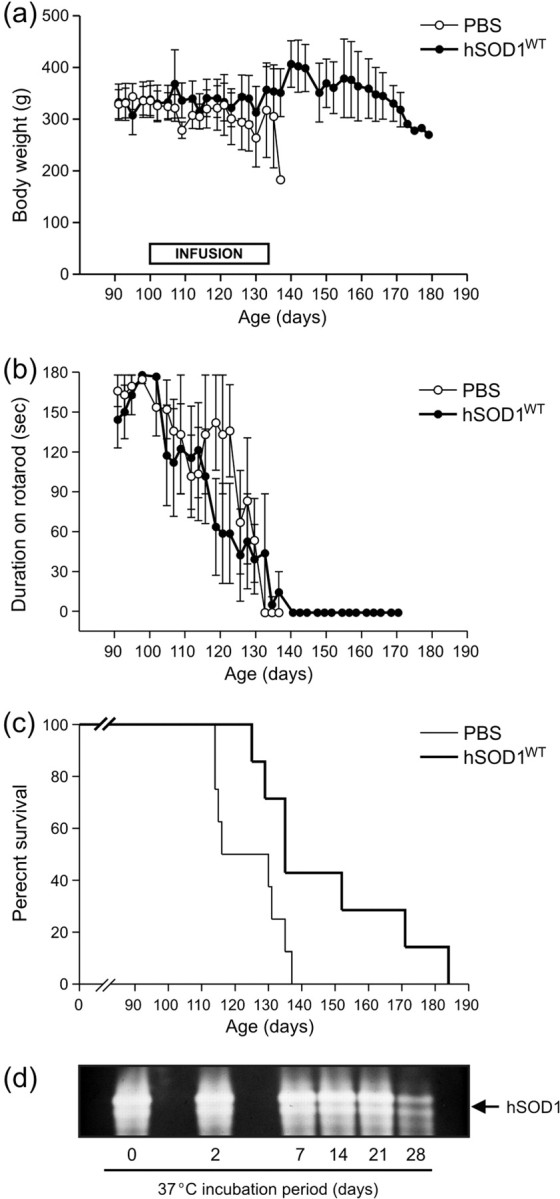
Chronic intraspinal infusion of extracellular hSOD1WT delays disease progression and endpoint in transgenic SOD1G93A rats. Animals were implanted with intrathecal pumps containing hSOD1WT or PBS vehicle solutions at P100; n = 8 per group. a, Animal weights as a function of treatment. b, Onset of locomotor impairment as assessed by rotarod analysis was not significantly altered by treatment. Error bars represent SEM. c, Kaplan-Meier survival analysis reveals a significant increase in survival of hSOD1WT infused rats versus PBS-treated animals (p < 0.05). d, Dismutase gel assay establishes that hSOD1WT (1 μg per lane) maintains antioxidative activity over 4 week period of incubation at physiologic temperature.
Table 1.
Comparison of disease parameters in transgenic SOD1G93A rats intrathecally administered hSOD1WT or vehicle solution
|
Parameter |
hSOD1WT |
PBS |
|---|---|---|
| Disease onset (rotarod) | 115.3 ± 6.4 | 115.3 ± 4.9 |
| Disease duration | 26.5 ± 7.3* | 9.0 ± 2.1 |
| Disease endpoint |
148.7 ± 8.0**
|
124.0 ± 3.6 |
Values represent mean ± SEM (days). *p < 0.05, statistical difference from PBS group by unpaired t test; **p < 0.05, statistical difference from PBS group by log-rank test.
Consistent with these survival data, increased SOD1 immunoreactivity was detected in both gray and white matter of spinal cords from hSOD1WT-treated rats (Fig. 8a). Immunoreactive deposits radiating in the white matter were consistent with glial cell profiles as determined by Nissl staining (Fig. 8b). Furthermore, vacuolar pathology in the spinal cord was markedly reduced by hSOD1WT treatment compared with vehicle controls (Fig. 8b).
Figure 8.

Extracellular hSOD1WT infusion results in spinal SOD1 accumulation and less vacuolar pathology in transgenic SOD1G93A rats. a, Analysis of lumbar spinal cord ventral horns reveals increased focal SOD1 immunoreactivity (arrowheads) in hSOD1WT-treated rats compared with saline infusion. b, Vacuolation is also less evident in spinal cords from hSOD1WT-treated rats as shown by cresyl violet staining. Motor neurons are indicated (open arrowhead). Scale bars, 100 μm; inset, 50 μm.
Discussion
This study provides evidence for extracellular secretion of SOD1 in a motor neuron cell line, extending a growing list of neural and non-neural cells capable of this function (Mondola et al., 1996, 1998; Cimini et al., 2002). SOD1 secretion involved BFA-sensitive transport in NSC-34 cells, suggesting an ER-Golgi transduction pathway from the cytosol. SOD1 export also bears resemblance to nonconventional protein secretion such as interleukin-1β, because SOD1 lacks both a conventional N-terminal secretion signal peptide and posttranslational modifications typical of ER-Golgi processing (Nickel, 2003). We also detected extracellular dismutase activity concomitant with SOD1 secretion, suggesting a potential paracrine antioxidant and neuroprotective role. Interestingly, an extracellular function was recently assigned to intracellular zinc-binding metallothionein (MT) proteins in cortical wound regeneration (Chung and West, 2004). Thus, SOD1, MT, and additional metalloproteins may be actively recruited beyond the cytoplasm as a result of oxidant stress and injury in the CNS.
In contrast to endogenous and wild-type SOD1, a selective impairment of ALS-linked mutant SOD1 secretion was demonstrated in NSC-34 cells. These data are in accordance with recent reports of diminished CD4 receptor and glutamate transporter-1 surface expression in mutant SOD1-transfected cell lines (Stieber et al., 2004; Vanoni et al., 2004), suggesting that mutant SOD1 expression has an adverse effect on protein secretion and intracellular trafficking. Fragmentation of the motor neuronal Golgi apparatus is consistently documented in humans and transgenic mice with ALS (Mourelatos et al., 1990, 1996; Fujita et al., 1999). Furthermore, endoplasmic reticulum, microvesicular, and vacuolar neuropathology are characteristic of certain transgenic ALS mouse lines before clinical disease (Dal Canto and Gurney, 1994; Tobisawa et al., 2003; Wootz et al., 2004). Hence, it is possible that mutant SOD1-induced structural damage to secretory organelles compromises not only fast axoplasmic transport in ALS (Mourelatos et al., 1996; Zhang et al., 1997) but also proteins destined for transynaptic or extracellular compartments. Blockade of mutant SOD1 secretion may be incidental but injurious, contributing to intracellular protein accumulation, aggregation, and toxicity. The SOD3 signal peptide-mediated extracellular targeting experiments further support this conclusion. Impaired secretion of SOD1 as a pathogenic factor may also offer one explanation for the relationship between SOD1 and other unrelated gene products mutated in familial ALS, including endosomal ALS2/alsin (Hadano et al., 2001; Yang et al., 2001) and recently, vesicle-trafficking protein VAPB (Nishimura et al., 2004). These proteins are all implicated in intracellular protein transport, mutations in which may disrupt secretion, leading to motor neuron dysfunction.
BFA treatment inhibited mutant SOD1 secretion and promoted toxicity in NSC-34 cells. BFA is a widely used secretion inhibitor, inducing ER stress and Golgi dispersion (Dinter and Berger, 1998). Exposure of cultured spinal neurons to BFA induces preferential motor neuron death, recapitulating key features of ALS (Kikuchi et al., 2003). In fact, BFA and mutant SOD1 share common neurotoxic mechanisms, including Golgi fragmentation, abolished neurite outgrowth, and induction of apoptosis (Jareb and Banker, 1997; Kikuchi et al., 2003), further implicating the secretory pathway in mutant SOD1 toxicity. In contrast, artificial extracellular secretion of mutant SOD1 via SOD3 signal peptide fusion attenuated inclusions and cell death, implicating an intracellular deleterious role of mutant SOD1 in toxicity.
Impaired mutant SOD1 secretion was associated with toxic protein inclusion formation in NSC-34 cells. Abnormal intracytoplasmic SOD1-immunoreactive inclusions are a common feature of motor neurons, astrocytes, and oligodendrocytes in transgenic ALS mice (Bruijn et al., 1998; Stieber et al., 2000c). Preceding these inclusions are possibly high molecular weight SOD1 oligomers concentrated in the spinal cords of predisease mice (Johnston et al., 2000; Wang et al., 2002). Mutant SOD1 inclusions, but not oligomeric pathology, were identified in NSC-34 cells. Moreover, the increased aggregation propensity of select mutants, A4V and G85R, correlated with enhanced cell death, supporting previous studies relating mutant SOD1 aggregate burden to cultured neuronal death (Durham et al., 1997; Takeuchi et al., 2002). Intriguingly, two mutants, H48Q and D90A, were devoid of toxic aggregates, perhaps reflecting the benign disease course and homozygous expression required for ALS in the latter mutation (Andersen et al., 1995). Regardless of aggregation and toxicity, however, all mutants examined here demonstrated enhanced detergent insolubility. Although implicated in mutant SOD1-mediated disease (Shinder et al., 2001), protein insolubility alone did not predispose to toxicity in this study. By comparison, transfected COS-7 cells were impervious to mutant SOD1-mediated death, suggesting an inherent susceptibility of NSC-34 cells based on motor neuron physiology. Previously, COS-7 cells expressing mutant SOD1 exhibited granular cytoplasmic inclusions without evidence for apoptosis (Koide et al., 1998). Increased protein turnover and proteasomal activity in COS-7 cells may account for these differences, as suggested by the presence of SOD1 peptide fragments in this study and others (Wang et al., 2003).
Based on the evidence in NSC-34 cells, we proposed that extracellular confined SOD1 may be therapeutic in a transgenic ALS rat model also exhibiting motor neuronal inclusions (Howland et al., 2002). To circumvent the potential mutant SOD1-mediated impairment of protein secretion in vivo, exogenous hSOD1WT was delivered directly into the CNS. Chronic intraspinal infusion of hSOD1WT significantly prolonged survival in transgenic ALS rats, compatible with the reported benefits of antioxidant therapy in transgenic ALS mice (Gurney et al., 1997; Reinholz et al., 1999; Andreassen et al., 2000; Jung et al., 2001). In particular, injection of SOD1-catalase mimetics delayed mortality but not disease onset in transgenic SOD1G93A mice (Jung et al., 2001), matching these results with hSOD1WT infusion. Extracellular SOD1 may exert both antioxidant and antiexcitotoxic protection in ALS, because cell surface-bound SOD1 modulates intracellular calcium-dependent signaling pathways (Mondola et al., 2004). In contrast, disease in double transgenic mice overexpressing hSOD1WT was either unaltered in SOD1G85R mice (Bruijn et al., 1998) or accelerated in SOD1G93A mice (Jaarsma et al., 2000; Fukada et al., 2001). We suggest that intrinsic elevation of hSOD1WT by these transgenic cross-breeding methods may burden the secretory pathway already targeted by mutant SOD1 overexpression, even potentiating toxicity in the latter mutant. Indeed, wild-type cell contribution in chimeric transgenic ALS mice substantially delays disease (Clement et al., 2003), perhaps mediated by astrocytic extracellular release of neuroprotective hSOD1WT. Finally, it is also noteworthy that intrathecal administration of recombinant hSOD1WT was trialed in sporadic and familial ALS patients (Cudkowicz et al., 1997). Although no positive effects were obtained because of limited sample size and late intervention, extracellular hSOD1WT therapy may provide benefit if commenced preclinically or early, as suggested by this study in transgenic ALS rats.
In conclusion, these experiments link deficient extracellular accumulation of mutant SOD1 to intracellular aggregates and toxicity in NSC-34 cells. Extracellular targeting of mutant SOD1 ameliorated toxic inclusions, suggesting that extracellular SOD1 may modify disease. Accordingly, extracellular delivery of hSOD1WT improved clinical disease in transgenic ALS rats. These data support novel extracellular roles for mutant and wild-type SOD1 in ALS pathogenesis and therapy, respectively.
Footnotes
This work was supported by National Health and Medical Research Council Program Grant 236805 and the R. D. Wright Fellowship to A.F.H. and by the Motor Neuron Disease Research Institute of Australia (J.D.A.). We thank Dr. David Borchelt and David Fromholt for provision of the pEF-BOS SOD1 plasmids, Dr. Neil Cashman for supplying the NSC-34 cell line, George Christopoulos for donation of the COS-7 cells, and Dr. Jennifer Callaway for use of the rat rotarod.
Correspondence should be addressed to Bradley J. Turner, Motor Neuron Disease Research Laboratory, Brain Injury and Repair Group, Howard Florey Institute, University of Melbourne, Victoria 3010, Australia. E-mail: b.turner@hfi.unimelb.edu.au.
Copyright © 2005 Society for Neuroscience 0270-6474/05/250108-10$15.00/0
References
- Andersen PM, Nilsson P, Ala-Hurula V, Keranen ML, Tarvainen I, Haltia T, Nilsson L, Binzer M, Forsgren L, Marklund SL (1995) Amyotrophic lateral sclerosis associated with homozygosity for an Asp90Ala mutation in CuZn-superoxide dismutase. Nat Genet 10: 61-66. [DOI] [PubMed] [Google Scholar]
- Andreassen OA, Dedeoglu A, Klivenyi P, Beal MF, Bush AI (2000) N-acetyl-l-cysteine improves survival and preserves motor performance in an animal model of familial amyotrophic lateral sclerosis. NeuroReport 11: 2491-2493. [DOI] [PubMed] [Google Scholar]
- Beauchamp C, Fridovich I (1971) Superoxide dismutase: improved assays and an assay applicable to acrylamide gels. Anal Biochem 44: 276-287. [DOI] [PubMed] [Google Scholar]
- Bendotti C, Carri MT (2004) Lessons from models of SOD1-linked familial ALS. Trends Mol Med 10: 393-400. [DOI] [PubMed] [Google Scholar]
- Bruijn LI, Houseweart MK, Kato S, Anderson KL, Anderson SD, Ohama E, Reaume AG, Scott RW, Cleveland DW (1998) Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science 281: 1851-1854. [DOI] [PubMed] [Google Scholar]
- Bruijn LI, Miller TM, Cleveland DW (2004) Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu Rev Neurosci 27: 723-749. [DOI] [PubMed] [Google Scholar]
- Chung RA, West AK (2004) A role for extracellular metallothioneins in CNS injury and repair. Neuroscience 123: 595-599. [DOI] [PubMed] [Google Scholar]
- Cimini V, Ruggiero G, Buonomo T, Seru R, Sciorio S, Zanzi C, Santangelo F, Mondola P (2002) CuZn-superoxide dismutase in human thymus: immunocytochemical localisation and secretion in thymus-derived epithelial and fibroblast cell lines. Histochem Cell Biol 118: 163-169. [DOI] [PubMed] [Google Scholar]
- Clement AM, Nguyen MD, Roberts EA, Garcia ML, Boillee S, Rule M, McMahon AP, Doucette W, Siwek D, Ferrante RJ, Brown Jr RH, Julien JP, Goldstein LS, Cleveland DW (2003) Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science 302: 113-117. [DOI] [PubMed] [Google Scholar]
- Cudkowicz ME, Warren L, Francis JW, Lloyd KJ, Friedlander RM, Borges LF, Kassem N, Munsta TL, Brown Jr RH (1997) Intrathecal administration of recombinant human superoxide dismutase 1 in amyotrophic lateral sclerosis: a preliminary safety and pharmacokinetic study. Neurology 49: 213-222. [DOI] [PubMed] [Google Scholar]
- Dal Canto MC, Gurney ME (1994) Development of central nervous system pathology in a murine transgenic model of human amyotrophic lateral sclerosis. Am J Pathol 145: 1271-1279. [PMC free article] [PubMed] [Google Scholar]
- Dinter A, Berger EG (1998) Golgi-disturbing agents. Histochem Cell Biol 109: 571-590. [DOI] [PubMed] [Google Scholar]
- Durham HD, Roy J, Dong L, Figlewicz DA (1997) Aggregation of mutant Cu/Zn superoxide dismutase proteins in a culture model of ALS. J Neuropathol Exp Neurol 56: 523-530. [DOI] [PubMed] [Google Scholar]
- Epperly MW, Sikora CA, DeFilippi SJ, Gretton JA, Zhan Q, Kufe DW, Greenberger JS (2002) Manganese superoxide dismutase (SOD2) inhibits radiation-induced apoptosis by stabilization of the mitochondrial membrane. Radiat Res 157: 568-577. [DOI] [PubMed] [Google Scholar]
- Fujita Y, Okamoto K, Sakurai A, Amari M, Nakazato Y, Gonatas NK (1999) Fragmentation of the Golgi apparatus of Betz cells in patients with amyotrophic lateral sclerosis. J Neurol Sci 163: 81-85. [DOI] [PubMed] [Google Scholar]
- Fujita Y, Okamoto K, Sakurai A, Gonatas NK, Hirano A (2000) Fragmentations of the Golgi apparatus of the anterior horn cells in patients with familial amyotrophic lateral sclerosis with SOD1 mutations and posterior column involvement. J Neurol Sci 174: 137-140. [DOI] [PubMed] [Google Scholar]
- Fukada K, Nagano S, Satoh M, Tohyama C, Nakanishi T, Shimizu A, Yanagihara T, Sakoda S (2001) Stabilization of mutant Cu/Zn superoxide dismutase (SOD1) protein by coexpressed wild SOD1 protein accelerates the disease progression in familial amyotrophic lateral sclerosis mice. Eur J Neurosci 14: 2032-2036. [DOI] [PubMed] [Google Scholar]
- Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX, Chen W, Zhai P, Sufit RL, Siddique T (1994) Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 264: 1772-1775. [DOI] [PubMed] [Google Scholar]
- Gurney ME, Cutting FB, Zhai P (1996) Benefit of vitamin E, riluzole, and gabapentin in a transgenic model of familial amyotrophic lateral sclerosis. Ann Neurol 39: 147-157. [DOI] [PubMed] [Google Scholar]
- Hadano S, Hand CK, Osuga H, Yanagisawa Y, Otomo A, Devon RS, Miyamoto N, Showguchi-Miyata J, Okada Y, Singaraja R, Figlewicz DA, Kwiatkowski T, Hosler BA, Sagie T, Skaug J, Nasir J, Brown Jr RH, Scherer SW, Rouleau GA, Hayden MR, Ikeda JE (2001) A gene encoding a putative GTPase regulator is mutated in familial amyotrophic lateral sclerosis 2. Nat Genet 29: 166-173. [DOI] [PubMed] [Google Scholar]
- Howland DS, Liu J, She Y, Goad B, Maragakis NJ, Kim B, Erickson J, Kulik J, DeVito L, Psaltis G, DeGennaro LJ, Cleveland DW, Rothstein JD (2002) Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis. Proc Natl Acad Sci USA 99: 1604-1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaarsma D, Haasdijk ED, Grashorn JA, Hawkins R, van Duijn W, Verspaget HW, London J, Holstege JC (2000) Human Cu/Zn superoxide dismutase (SOD1) overexpression in mice causes mitochondrial vacuolization, axonal degeneration, and premature motoneuron death and accelerates motoneuron disease in mice expressing a familial amyotrophic lateral sclerosis mutant SOD1. Neurobiol Dis 7: 623-643. [DOI] [PubMed] [Google Scholar]
- Jaarsma D, Rognoni F, van Duijn W, Verspaget HW, Haasdijk ED, Holstege JC (2001) CuZn superoxide dismutase (SOD1) accumulates in vacuolated mitochondria in transgenic mice expressing amyotrophic lateral sclerosis-linked SOD1 mutations. Acta Neuropathol 102: 293-305. [DOI] [PubMed] [Google Scholar]
- Jareb M, Banker G (1997) Inhibition of axonal growth by brefeldin A in hippocampal neurons in culture. J Neurosci 17: 8955-8963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston JA, Dalton MJ, Gurney ME, Kopito RR (2000) Formation of high molecular weight complexes of mutant Cu,Zn-superoxide dismutase in a mouse model for familial amyotrophic lateral sclerosis. Proc Natl Acad Sci 97: 12571-12576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung C, Rong Y, Doctrow S, Baudry, Malfroy B, Xu ZS (2001) Synthetic superoxide dismutase/catalase mimetics reduce oxidative stress and prolong survival in a mouse amyotrophic lateral sclerosis model. Neurosci Lett 304: 157-160. [DOI] [PubMed] [Google Scholar]
- Kikuchi S, Shinpo, Tsuji S, Yabe I, Niino M, Tashiro K (2003) Brefeldin A-induced neurotoxicity in cultured spinal cord neurons. J Neurosci Res 71: 591-599. [DOI] [PubMed] [Google Scholar]
- Koide T, Igarashi S, Kikugawa K, Nakano R, Inuzuka T, Yamada M, Takahashi H, Tsuji S (1998) Formation of granular cytoplasmic aggregates in COS7 cells expressing mutant Cu/Zn superoxide dismutase associated with familial amyotrophic lateral sclerosis. Neurosci Lett 257: 29-32. [DOI] [PubMed] [Google Scholar]
- Mondola P, Annella T, Santillo M, Santangelo F (1996) Evidence for secretion of cytosolic CuZn superoxide dismutase by Hep G2 cells and human fibroblasts. Int J Biochem Cell Biol 28: 677-681. [DOI] [PubMed] [Google Scholar]
- Mondola P, Annella T, Seru R, Santangelo F, Iossa S, Gioielli A, Santillo M (1998) Secretion and increase of intracellular CuZn superoxide dismutase content in human neuroblastoma SK-N-BE cells subjected to oxidative stress. Brain Res Bull 45: 517-520. [DOI] [PubMed] [Google Scholar]
- Mondola P, Ruggiero G, Seru R, Damiano S, Grimaldi S, Garbi C, Monda M, Greco D, Santillo M (2003) The Cu,Zn superoxide dismutase in neuroblastoma SK-N-BE cells is exported by a microvesicles dependent pathway. Mol Brain Res 110: 45-51. [DOI] [PubMed] [Google Scholar]
- Mondola P, Santillo M, Seru R, Damiano S, Alvino C, Ruggiero G, Formisano P, Terrazzano G, Secondo A, Annunziato L (2004) Cu,Zn superoxide dismutase increases intracellular calcium levels via a phospholipase C-protein kinase C pathway in SK-N-BE neuroblastoma cells. Biochem Biophys Res Commun 324: 887-892. [DOI] [PubMed] [Google Scholar]
- Mourelatos Z, Adler H, Hirano A, Donnenfeld H, Gonatas JO, Gonatas NK (1990) Fragmentation of the Golgi apparatus of motor neurons in amyotrophic lateral sclerosis revealed by organelle-specific antibodies. Proc Natl Acad Sci USA 87: 4393-4395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourelatos Z, Hirano A, Rosenquist AC, Gonatas NK (1994) Fragmentation of the Golgi apparatus of motor neurons in amyotrophic lateral sclerosis (ALS). Clinical studies in ALS of Guam and experimental studies in deafferented neurons and in β,β′-imunodipropionitrile axonopathy. Am J Pathol 144: 1288-1300. [PMC free article] [PubMed] [Google Scholar]
- Mourelatos Z, Gonatas NK, Stieber A, Gurney ME, Dal Canto MC (1996) The Golgi apparatus of spinal cord motor neurons in transgenic mice expressing mutant Cu,Zn superoxide dismutase becomes fragmented in early, preclinical stages of the disease. Proc Natl Acad Sci USA 93: 5472-5477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickel W (2003) The mystery of nonclassical protein secretion. A current view on cargo proteins and potential export routes. Eur J Biochem 270: 2109-2119. [DOI] [PubMed] [Google Scholar]
- Nishimura AL, Mitne-Neto M, Silva HC, Richieri-Costa A, Middleton S, Cascio D, Kok F, Oliveira JR, Gillingwater T, Webb J, Skehel P, Zatz M (2004) A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am J Hum Genet 75: 822-831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinholz MM, Merkle CM, Poduslo JF (1999) Therapeutic benefits of putrescine-modified catalase in a transgenic mouse model of familial amyotrophic lateral sclerosis. Exp Neurol 159: 204-216. [DOI] [PubMed] [Google Scholar]
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan JP, Deng HX, Rahmani Z, Krizus A, McKenna-Yasek D, Cayabyab A, Gaston SM, Berger R, Tanzi RE, Halperin JJ, Hertzfeldt B, Van den Bergh R, Hung WY, Bird T, Deng G, Mulder DW, Smyth C, Laing NG, Soriano E, Pericak-Vance MA, Haines J, Rouleau GA, Gusella JS, Horvitz HR, Brown Jr RH (1993) Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362: 59-62. [DOI] [PubMed] [Google Scholar]
- Rowland LP, Shneider MD (2001) Amyotrophic lateral sclerosis. N Engl J Med 344: 1688-1700. [DOI] [PubMed] [Google Scholar]
- Shinder GA, Lacourse MC, Minotti S, Durham HD (2001) Mutant Cu/Zn-superoxide dismutase proteins have altered solubility and interact with heat shock/stress proteins in models of amyotrophic lateral sclerosis. J Biol Chem 276: 12791-12796. [DOI] [PubMed] [Google Scholar]
- Stieber A, Gonatas JO, Gonatas NK (2000a) Aggregation of ubiquitin and a mutant ALS-linked SOD1 protein correlate with disease progression and fragmentation of the Golgi apparatus. J Neurol Sci 173: 53-62. [DOI] [PubMed] [Google Scholar]
- Stieber A, Gonatas JO, Collard JF, Meier J, Julien JP, Schweitzer P, Gonatas NK (2000b) The neuronal Golgi apparatus is fragmented in transgenic mice expressing a mutant human SOD1, but not in mice expressing the human NF-H gene. J Neurol Sci 173: 63-72. [DOI] [PubMed] [Google Scholar]
- Stieber A, Gonatas JO, Gonatas NK (2000c) Aggregates of mutant protein appear progressively in dendrites, in periaxonal processes of oligodendrocytes, and in neuronal and astrocytic perikarya of mice expressing the SOD1G93A mutation of familial amyotrophic lateral sclerosis. J Neurol Sci 177: 114-123. [DOI] [PubMed] [Google Scholar]
- Stieber A, Gonatas JO, Moore JS, Bantly A, Kim HS, Tim MB, Gonatas NK (2004) Disruption of the structure of the Golgi apparatus and the function of the secretory pathway by mutants G93A and G85R of Cu, Zn superoxide dismutase (SOD1) of familial amyotrophic lateral sclerosis. J Neurol Sci 219: 45-53. [DOI] [PubMed] [Google Scholar]
- Sturtz LA, Diekert K, Jensen LT, Lill R, Culotta VC (2001) A fraction of yeast Cu,Zn superoxide dismutase and its metallochaperone, CCS, localize to the intermembrane space of mitochondria: a physiologic role for SOD1 in guarding against mitochondrial oxidative damage. J Biol Chem 276: 38084-38090. [DOI] [PubMed] [Google Scholar]
- Takeuchi H, Kobayashi Y, Yoshihara T, Niwa J, Doyu M, Ohtsuka K, Sobue G (2002) Hsp70 and Hsp40 improve neurite outgrowth and suppress intracytoplasmic aggregate formation in cultured neuronal cells expressing mutant SOD1. Brain Res 949: 11-22. [DOI] [PubMed] [Google Scholar]
- Tobisawa S, Hozumi Y, Arawaka S, Koyama S, Wada M, Nagai M, Aoki M, Itoyama Y, Goto K, Kato T (2003) Mutant SOD1 linked to familial amyotrophic lateral sclerosis, but not wild-type SOD1, induces ER stress in COS7 cells and transgenic mice. Biochem Biophys Res Commun 303: 496-503. [DOI] [PubMed] [Google Scholar]
- Vanoni C, Massari S, Losa M, Carrega P, Perego C, Conforti L, Pietrini G (2004) Increased internalisation and degradation of GLT-1 glial glutamate transporter in a cell model for familial amyotrophic lateral sclerosis. J Cell Sci 117: 5417-5426. [DOI] [PubMed] [Google Scholar]
- Wang J, Xu G, Borchelt DR (2002) High molecular weight complexes of mutant superoxide dismutase 1: age-dependent and tissue-specific accumulation. Neurobiol Dis 9: 139-148. [DOI] [PubMed] [Google Scholar]
- Wang J, Slunt H, Gonzales V, Fromholt D, Coonfield M, Copeland NG, Jenkins NA, Borchelt DR (2003) Copper-binding-site-null SOD1 causes ALS in transgenic mice: aggregates on non-native SOD1 delineate a common feature. Hum Mol Genet 12: 2753-2764. [DOI] [PubMed] [Google Scholar]
- Wootz H, Hansson I, Korhonen L, Napankangas U, Lindholm D (2004) Caspase-12 cleavage and increased oxidative stress during motoneuron degeneration in transgenic mouse model of ALS. Biochem Biophys Res Commun 322: 281-286. [DOI] [PubMed] [Google Scholar]
- Yang Y, Hentati A, Deng HX, Dabbagh O, Sasaki T, Hirano M, Hung WY, Ouahchi K, Yan J, Azim AC, Cole N, Gascon G, Yagmour A, Ben-Hamida M, Pericak-Vance M, Hentati F, Siddique T (2001) The gene encoding alsin, a protein with three guanine-nucleotide exchange factor domains, is mutated in a form of recessive amyotrophic lateral sclerosis. Nat Genet 29: 160-165. [DOI] [PubMed] [Google Scholar]
- Zelko IN, Mariani TJ, Folz RJ (2002) Superoxide dismutase multigene family: a comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structure, evolution and expression. Free Radic Biol Med 33: 337-349. [DOI] [PubMed] [Google Scholar]
- Zhang B, Tu P, Abtahian F, Trojanowski JQ, Lee VM (1997) Neurofilaments and orthograde transport are reduced in ventral root axons of transgenic mice that express human SOD1 with a G93A mutation. J Cell Biol 139: 1307-1315. [DOI] [PMC free article] [PubMed] [Google Scholar]