

Abstract
shocked (sho) is a zebrafish mutation that causes motor deficits attributable to CNS defects during the first2dof development. Mutant embryos display reduced spontaneous coiling of the trunk, diminished escape responses when touched, and an absence of swimming. A missense mutation in the slc6a9 gene that encodes a glycine transporter (GlyT1) was identified as the cause of the sho phenotype. Antisense knock-down of GlyT1 in wild-type embryos phenocopies sho, and injection of wild-type GlyT1 mRNA into mutants rescues them. A comparison of glycine-evoked inward currents in Xenopus oocytes expressing either the wild-type or mutant protein found that the missense mutation results in a nonfunctional transporter. glyt1 and the related glyt2 mRNAs are expressed in the hindbrain and spinal cord in nonoverlapping patterns. The fact that these regions are known to be required for generation of early locomotory behaviors suggests that the regulation of extracellular glycine levels in the CNS is important for proper function of neural networks. Furthermore, physiological analysis after manipulation of glycinergic activity in wild-type and sho embryos suggests that the mutant phenotype is attributable to elevated extracellular glycine within the CNS.
Keywords: behavior, zebrafish, mutation, glycine transporter, behavior, strychnine
Introduction
The zebrafish embryo is amenable to genetic analysis of neuronal circuit formation and function during development. The earliest behaviors exhibited by zebrafish embryos are stereotyped behaviors (Saint-Amant and Drapeau, 1998) that are regulated by a relatively simple hindbrain and spinal cord (Kimmel et al., 1982; Mendelson, 1985; Metcalfe et al., 1986; Bernhardt et al., 1990). The embryo is transparent, and its neurons and muscles can be assayed with both electrophysiology (Legendre and Korn, 1994; Ribera and Nusslein-Volhard, 1998; Drapeau et al., 1999; Buss and Drapeau, 2000; Saint-Amant and Drapeau, 2000) and calcium imaging (O'Malley et al., 1996; Friedrich and Korsching, 1997; Ritter et al., 2001; Higashijima et al., 2003). The nervous system develops quickly and is functional early in development, which allows the analysis of the CNS even in mutants that have compromised cardiovascular and/or muscular function. To date, several mutagenesis screens for using behavioral assays have been performed and have yielded an impressive collection of mutants (Granato et al., 1996; Neuhauss et al., 1999).
One of the behavioral mutations, shocked (sho), reacts to mechanosensory stimulation with one or two large contractions of the trunk at 48 h post-fertilization (hpf) rather than swimming away as wild-type embryos (Granato et al., 1996). Subsequently, sho embryos were found to exhibit abnormally persistent coupling between muscle fibers (Luna et al., 2004) and defective signaling in the CNS (Cui et al., 2004). Mechanosensory stimulation of 24-28 hpf wild-type embryos elicits a fast escape response consisting of two to three vigorous, alternating contractions of the trunk. This behavior is disrupted by the strongest allele, shota229g, because these embryos fail to respond or respond weakly. Because this touch-evoked response requires the caudal hindbrain and spinal cord (Saint-Amant and Drapeau, 1998), the absence of escape in 24 hpf mutants implies that sho gene activity is important for the maturation and/or function of hindbrain and/or spinal networks. Later, as the embryo matures, the escape response evolves to include swimming after 28 hpf while sho embryos respond to mechanosensory stimulation with one or two large amplitude contractions but never swimming (Cui et al., 2004). Neurophysiological analysis during this stage demonstrated a defect in the sho CNS. In wild-type embryos, sensory stimulation or application of NMDA evokes a synaptically dependent, sustained plateau depolarization that gives rise to a train of rhythmic action potentials in motor neurons to mediate swimming. In sho embryos, touch- and NMDA-induced plateau potentials were not sustained, resulting in an abbreviated burst of action potentials. Because the properties of sho motor neurons were normal, the aberrant response of motor neurons suggests that motor processing within the CNS was defective and that normal motor processing requires the sho gene product (Cui et al., 2004).
Here we report the molecular identification of the sho gene as one that encodes a glycine transporter (GlyT1). Analysis of wild-type and sho embryos in which the action of glycine within the CNS is manipulated demonstrates that regulation of extracellular glycine levels is required for normal information processing within the CNS.
Materials and Methods
Animals. Zebrafish were bred and raised according to established procedures (Westerfield, 1993), which meet the guidelines set forth by the University of Michigan animal care and use protocols. Developmental staging of the embryos was done by counting somites as described previously (Kimmel et al., 1995). The shota229g line was provided by Dr. Michael Granato (University of Pennsylvania, Philadelphia, PA). All analyses were performed using shota229g.
Behavioral observation and video recording. Embryonic and larval behaviors were observed using a Leica (Heerbrugg, Switzerland) stereomicroscope. Mechanosensory stimuli were delivered with a thin tungsten wire probe. Videos were captured using a Panasonic (Secaucas, NJ) wvbp-330 CCD camera and a Scion (Frederick, MD) LG-3 frame grabber on a G4 Mac and analyzed with NIH Image.
Mapping and cloning of slc6a9 and slc6a5. High-resolution mapping of zebrafish mutations has been described previously (Zhang et al., 1998; Talbot and Schier, 1999; Shafizadeh et al., 2002). Hybrid sho carriers were generated from crosses between the Tübingen carrier and polymorphic WIK11 wild-type grandparents. Bulk segregant analysis (Geisler, 2002) from 48 homozygous embryos and 48 wild-type siblings using a panel of agarose-scorable microsatellite markers mapped the sho locus to linkage group (LG) 2. Grandparent and parent genotypes were individually tested against additional microsatellite markers near the region of interest on LG 2, and several markers (z13281, z22526, z7632, and z8451) were found to be polymorphic and linked to the sho locus. More than 1800 homozygous embryos were collected and tested for recombination events between these markers and the sho locus. Fingerprint contigs and bacterial artificial chromosome (BAC) sequences were obtained from the Sanger Center online database (http://www.sanger.ac.uk/Projects/D_rerio/). The GENSCAN software is maintained online at http://genes.mit.edu/GENSCAN.html.
Based on the predicted cDNA sequence of slc6a9 that encodes GlyT1, the following primers were used to amplify a fragment of its cDNA: forward, 5′- AATGGAGCAGTTCCAGGAGAGCC-3′; and reverse, 5′- ATGTAAAGGTCTCCTGTCCCGCAG-3′. For 5′ rapid amplification of cDNA ends (RACE), GeneRacer kit (Invitrogen, Carlsbad, CA) was used, and the 3′ internal primer was 5′-GTTGGCGAAAGTGGCATTGGCAC-3′. Twenty individual clones from the RACE reaction were sequenced, and multiple transcripts with one of three different 5′ sequence were obtained.
To clone the slc6a5 gene that encodes GlyT2, the human GlyT2 amino acid sequence was blasted against the BAC sequences of the Sanger Institute, and clone zK7N10 was found to contain the zebrafish homolog. The following primers were used to amplify a cDNA fragment: forward, 5′- GATGAGAATAAGGCTCGGGGAAAC -3′; and reverse, 5′- ACTCACAGGGAAGGAGGAGGATTA-3′. For 5′ RACE, an internal primer (5′- CTGACCCAACGACACCTCCAACAA-3′) was used, and a single RACE product was found.
Knock-down of GlyT1 expression and mRNA rescue. An antisense slc6a9 morpholino oligonucleotides (MO) (Gene Tools, Philomath, OR) was designed against the last 10 bases of exon 2 and first 15 bases of intron 2 (5′-gataaaaacggtcacCTCCTCCATT-3′; bases in lowercase are complementary to the intronic sequence). A standard control MO of randomized sequence from Gene Tools was used as the negative control. Capped and poly(A+) tailed wild-type and G81D mRNA of GlyT1A were synthesized using the mMESSAGE mMACHINE T7 Ultra kit (Ambion, Austin, TX) and injected at a concentration of 500 μm as described previously (Hirata et al., 2004).
Expression of GlyT1 and electrophysiology in Xenopus oocytes. GlyT1 was expressed in defolliculated, stage V-VI Xenopus laevis oocytes. mRNA encoding wild-type or G81D mutant GlyT1A that contained a sequence encoding a FLAG epitope inserted before the stop codon was synthesized using the mMESSAGE mMACHINE T7 Ultra kit. Each oocyte was injected with 50 nl of RNA (500 ng/μl). After injection, oocytes were maintained in Barth's solution at 17°C for ∼48 h before electrophysiology.
Two-electrode voltage-clamp recordings were made with an NPI Electronics (Tamm, Germany) TurboTec 3 amplifier. Experiments were performed at 22°C at a holding potential of -50 mV. The oocyte external recording solution was as follows (in mm): 90 NaCl, 1 KCl, 1.7 MgCl2, and 10 HEPES at pH 7.6. The recording pipette solution contained 3 m KCl. To apply glycine, a stock solution (50 mm) was diluted to the desired final concentration in external recording solution. Up to seven different solutions were applied to each oocyte using a BPS-8 solution switcher (ALA Scientific Instruments, Westbury, NY). Data acquisition and the switching of solutions were controlled by pClamp 8 software using a Digidata 1322A interface (Axon Instruments, Union City, CA). The initial data analysis was done in pClamp, and concentration-response relationships were fit using functions included in Sigma Plot 8.0 (SPSS, Chicago, IL).
In situ hybridization and immunohistochemistry of whole-mounted embryos. In situ hybridization and immunohistochemistry were performed as described previously (Westerfield, 1993). Briefly, embryos were anesthetized and fixed with 4% paraformaldehyde in PBS buffer. Embryos were dehydrated with methanol and rehydrated and treated with Proteinase K to increase the permeability of the riboprobes, which were digoxigenin (DIG) labeled using a RNA labeling kit (Roche Products, Welwyn Garden City, UK). The hybridization was performed at 65°C overnight. Anti-DIG antibody conjugated to alkaline phosphatase was used for the development of nitroblue-tetrazolium- chloride/5-bromo-4-chloro-indolyl-phosphate (blue color) or fast red (rhodamine fluorescence) reaction. For the GlyT1 and anti-Hu (a neuron-specific, RNA-binding protein) colabeling experiment, embryos were incubated with anti-Hu (1:100; Molecular Probes, Eugene, OR) after the in situ fast red reaction, washed, and visualized using an Alexa488-conjugated anti-mouse secondary (1:1500; Molecular Probes), and the fluorescent images were collected and processed with a confocal microscope (Zeiss, Thorn-wood, NY) and LSM 500 software.
In vivo whole-cell voltage recording from embryonic muscle cells. Methods for electrophysiological recording from muscles of zebrafish embryos have been reported previously (Drapeau et al., 1999; Buss and Drapeau, 2000). Briefly, a zebrafish embryo was anesthetized and paralyzed in recording solution [in mm: 134 NaCl, 2.9 KCl, 2.1 CaCl2, 1.2 MgCl2, 10 glucose, 10 HEPES, and 0.0075 d-tubocurarine, pH 7.8 (290 mOsm)] that contained tricaine (0.02%). The embryo was immobilized by tungsten wires (25 μm diameter) through its notochord. Before recordings, slow twitch muscles from several segments between segments 8 and 16 were exposed by peeling off the overlying skin. The embryo was constantly perfused with recording solution (2-5 ml/min). The intracellular solution consisted of the following (in mm): 116 potassium gluconate, 16 KCl, 2 MgCl2, 10 HEPES, 10 EGTA, and 4 Na3ATP, pH 7.2 (273 mOsm). Patch pipettes were pulled from borosilicate glass to yield electrodes with resistances of 6-10 MΩ. The pipette junction potential has been measured to be 6.4 mV, and all potentials were corrected for this junction potential. Recordings were made with an Axopatch 200 amplifier (Axon Instruments), low-pass filtered at 5 kHz, and sampled at 10 kHz. Data was collected with Clampex 8.2 software (Axon Instruments) and analyzed with Clampfit 9.0 software (Axon Instruments). Fictive swimming was elicited by mechanosensory stimulation via pressure ejection of bath solution delivered to the tail from a pipette with a 15-30 μm tip opening. The duration (20 ms) and pressure (20-40 psi) of the stimulation were regulated by a Picospritzer II (Parker Hannifin, Fairfield, NJ). For pharmacology experiments, glycine (from 0.1 to 1 mm) and strychnine (from 10 nm to 50 μm) were added to the bath solution when needed, and the muscle response to mechanosensory stimulation was recorded after at least 5 min of perfusion.
Drug injection into the CNS. N[3-(4′-fluorophenyl)-3-(4′-phenylphenoxy)propyl]sarcosine (NFPS) was dissolved in DMSO and stored as a 10 mm stock solution. For injection, NFPS was further diluted to 100 μm in zebrafish recording solution containing 0.1% Phenol Red. Injection micropipettes were pulled from fiber-filled borosilicate glass and backfilled. Wild-type embryos were anesthetized with tricaine (0.02%) and embedded in 1% low-melting-point agarose in extracellular recording solution. The micropipette was advanced through the agarose and through the tectum into the ventricle. The NFPS solution was pressure injected into the CNS with repeated 20 ms/20 psi pulses via a Picospritzer until the brain ventricles became visibly red. Stage-matched control, wild-type siblings were injected with the extracellular solution with only Phenol Red. All chemical reagents were purchased from Sigma-Aldrich (St. Louis, MO) unless noted otherwise.
Results
sho embryos exhibit defects in the earliest motor behavior
Because shota229g embryos exhibit defective behavior as early as 24 hpf (Cui et al., 2004), we examined the earliest motor behavior, spontaneous coiling, in these mutants. Spontaneous coiling consists of alternating, slow contractions of the trunk that are much slower than trunk contractions during the escape response and is independent of sensory stimulation (Saint-Amant and Drapeau, 1998). Time-lapse video microscopy showed that the coiling behavior first appeared in wild-type siblings of sho shortly after 17 hpf (Fig. 1A). The frequency peaked at 19 hpf (n = 28; 0.53 ± 0.15 Hz, mean ± SD) and gradually decreased as the embryos mature (Fig. 1B), as reported previously (Saint-Amant and Drapeau, 1998). However, sho embryos displayed severely reduced coiling frequencies at all stages examined. The maximum frequency observed in sho was 0.27 ± 0.09 Hz (n = 11) at 19 hpf, after which the frequency decreased and mutants stopped coiling almost completely after 21 hpf. Because lesion experiments demonstrated that the spinal cord was sufficient in generating spontaneous coiling (Saint-Amant and Drapeau, 1998), decreased coiling by mutants suggests that the sho gene product was necessary for proper function of the spinal motor circuit as early as 19 hpf.
Figure 1.

Spontaneous coiling behavior is defective in sho embryos. A, Individual frames from a time-lapse video showing two consecutive coils by a wild-type sibling embryo (22 hpf). Each coil lasted ∼500 ms and is denoted by an arrow. Time of each frame is shown at the top right corner. Scale bar, 0.5 mm. B, The frequency of coiling is reduced at all stages in sho embryos (circles) compared with wild-type siblings (wtsib; squares). *p < 0.001 by Student's t test. Error bars are SDs.
The sho gene product is a CNS glycine transporter
As the first step toward cloning the sho gene, the sho locus was determined to be located on LG 2 by bulk segregant analysis. Meiotic mapping identified several microsatellite markers that flanked the sho locus (Fig. 2A). The closest flanking markers were z22527 [0.97 centimorgans (cM), 14 recombinants in 1108 diploid embryos] and z7632 (0.26 cM, six recombinants in 1756 embryos). Using the database from the Zebrafish Genomic Fingerprinting Project (http://www.sanger.ac.uk/Projects/D_rerio/), both markers were located on a “fingerprint” contig (number 165), and six ordered, contiguous, and sequenced BAC clones that spanned the critical region between the two markers were identified. Potential open reading frames (ORFs) were identified from the BAC sequences using the GENSCAN program to be possible candidates for the sho gene. A total of 46 potential ORFs were identified, 13 of which were homologous to known proteins. Only one of these 13 ORFs encoded for a CNS-related protein. Clone zC261P7 (GenBank accession number BX537271) encoded a zebrafish homolog of a CNS glycine transporter (GlyT1) that transports glycine via a sodium- and chloride-dependent mechanism (Aragon and Lopez-Corcuera, 2003). The sho gene could encode GlyT1 because glycine is a neurotransmitter in the vertebrate CNS (for review, see Legendre, 2001) and GlyT1 knock-out mice exhibit abnormal behaviors (Gomeza et al., 2003; Tsai et al., 2004).
Figure 2.

Molecular identification of the sho gene. A, The sho locus meiotically mapped to a region on LG 2 flanked by several zebrafish microsatellite markers. The relative meiotic distance between these markers and the sho locus are shown. Six contiguous, ordered BACs (shown below the meiotic map) spanned the entire region containing the two closest markers, z22527 and z7632. BAC zC261P7 contained an ORF homologous to the mammalian gene slc6a9 that encodes GlyT1A. B, The amino acid sequence alignment of three isoforms of zebrafish GlyT1 (zGlyT1A, zGlyT1B, and zGlyT1Z) with human GlyT1A. Identical sequences are lightly shaded, and the highly conserved transmembrane domains (TM1-TM12) are darkly shaded. The position of point mutation G81D in the sho allele is denoted by an asterisk.
To determine whether the gene slc6a9, which encodes GlyT1, is mutated in sho, full-length wild-type and mutant alleles of the glyt1 cDNA were cloned and sequenced. Analysis of products from reverse transcription (RT)-PCR reactions and comparison of the cDNA and genomic sequences revealed that the gene appears to generate three different transcripts, which have different first exons but share the ensuing 11 exons (GenBank accession numbers DQ005477, DQ005478, and DQ005479). This predicts three proteins (GlyT1A, GlyT1B, and GlyT1Z) that are 649, 653, and 644 amino acids long and differ only in the initial N-terminus sequences. The amino acid sequences are ∼74% identical with human GlyT1, with conservation of the 12 transmembrane domains especially high (Fig. 2B). A missense point mutation was identified in the sho allele of the slc6a9 gene that results in the substitution of glycine by aspartic acid at residue 81 in the second transmembrane domain, which is present in all three GlyT1 isoforms. Furthermore, the same missense mutation was found in genomic DNA encoding GlyT1 from five mutants, ruling out changes in RNA editing as the reason for the difference seen between mutant and wild-type cDNAs. The glycine residue that is mutated is highly conserved not only across species but also in other members of the 12-transmembrane, neurotransmitter transporter superfamily to which GlyT1 belongs (Melamed and Kanner, 2004). The replacement of glycine, a small nonpolar residue, by aspartic acid, a residue with a large, charged side chain, is likely to disrupt transporter function and suggests that the sho gene encodes GlyT1.
Loss of GlyT1 function mimics sho mutation
If sho encodes GlyT1, then suppressing the slc6a9 gene function in wild-type embryos should mimic the sho mutation. The expression of slc6a9 was knocked down in wild-type embryos by injecting antisense slc6a9 MOs into fertilized embryos. Because there are three transcripts of GlyT1 with different 5′ starting sequences, an antisense MO (E2I2) that is complementary to the sequence that spans the junction between exon 2 and intron 2 was used. This MO should prevent correct pre-mRNA splicing (Corey and Abrams, 2001) in all isoforms of GlyT1 because the target sequence is present in all three.
E2I2-injected wild-type morphants clearly had deficits in their motor development. At 22 hpf, nearly all E2I2 morphants exhibited a severe reduction in spontaneous coiling frequency (0.07 ± 0.12 Hz; n = 57) (Fig. 3A), whereas the frequency of the wild-type embryos injected with a standard control MO was higher (0.34 ± 0.16 Hz; n = 67) and comparable with that of uninjected embryos (0.48 ± 0.13 Hz; n = 23). Thus, an apparent knock-down of GlyT1 in wild-type embryos mimicked the decreased spontaneous coiling observed in sho embryos.
Figure 3.

GlyT1 function is required for normal spontaneous coiling behavior. A, GlyT1 antisense MO injections phenocopy sho. Histogram showing the percentage (y-axis) of uninjected wild-type (wt; top), control (ctrl) MO-injected wild-type (middle), and GlyT1 E2I2 MO-injected wild-type (bottom) embryos exhibiting spontaneous coiling at various frequencies (x-axis). Knock-down of GlyT1 function severely reduced the frequency of spontaneous coiling by wild-type embryos. B, Injection of wild-type GlyT1A mRNA but not G81D mutant mRNA rescues sho embryos. Approximately one-quarter of uninjected embryos (top) and embryos injected with G81D mRNA (middle) from a cross of sho carriers coiled with reduced frequencies, but nearly all embryos from a cross of carriers injected with wild-type mRNA coiled at frequencies within the normal range (bottom).
E2I2 morphants also displayed aberrant escape and swimming responses to mechanosensory stimulation. At 26 hpf, 100% of the wild-type siblings (n = 10) responded to tactile stimulation, with an escape response consisting of alternating contractions of the trunk (Fig. 4A), whereas 100% of sho embryos (n = 13) (Fig. 4B) responded with a weak contraction or not at all (Fig. 4B). All of the control MO-injected wild-type embryos (n = 64) exhibited normal escape responses after mechanosensory stimulation (Fig. 4Ci), but 80% of E2I2 morphants (n = 55) were unresponsive or responded weakly (Fig. 4Cii). At 36 hpf, 89% of E2I2 morphants (n = 55) displayed trunk contractions but no swimming (data not shown), whereas all control morphants (n = 64) swam away normally. Because an apparent knock-down of GlyT1 mimics the sho phenotype, the sho gene either encodes for GlyT1 or is in the same genetic pathway with GlyT1.
Figure 4.

GlyT1 function is required for normal escape responses at 26 hpf. A, A wild-type sibling (wtsib) embryo responded to tactile stimulation with fast, alternating trunk contractions. Shown is the first of two contractions. Time of each frame is noted in the top right corner. B, A sho embryo failed to respond to tactile stimulation. C, Apparent knock-down of GlyT1 phenocopied sho. A control (ctrl) MO-injected wild-type embryo responds to tactile stimulation with fast, alternating trunk contractions (top), whereas an antisense E2I2 MO-injected wild-type embryo failed to respond (bottom). D, Apparent restoration of GlyT1 activity in sho embryos rescued the mutant phenotype. A sho embryo injected with G81D mutant mRNA failed to respond to tactile stimulation (top), whereas a sho embryo injected with wild-type GlyT1 mRNA responded normally (bottom). The wild-type mRNA-injected embryo was genotyped based on its mutant response to tactile stimulation at 48 hpf (for details, see Results).
GlyT1 function rescues the sho phenotype
If the missense mutation in GlyT1 caused malfunction of the transporter and defective behaviors, then expression of the wild-type protein in sho embryos should restore normal behaviors. Because previous work demonstrated that the different N-terminus sequences appear to have no effect on transporter activity (Kim et al., 1994), any of the isoforms should be sufficient for functional restoration in sho. mRNA encoding either the wild-type or G81D GlyT1A protein was injected into embryos spawned from heterozygous parents, and their behaviors were followed over the next 2 d. If the injected mRNA rescued the mutation, then none or nearly none of the embryos from a cross between heterozygotes should display mutant behavior, as opposed to the usual 25% that one would expect for an autosomal recessive mutation.
Thirteen of 58 embryos (22%) injected with mutant G81D mRNA exhibited mutant levels of four or fewer spontaneous coils per minute at 22 hpf, whereas 8 of 40 (20%) uninjected progeny displayed four or fewer coils per minute (Fig. 3B). In contrast, all 65 embryos injected with wild-type GlyT1 mRNA displayed normal coiling frequencies, much like the uninjected wild-type embryos (n = 10). The finding that all of the progeny from a cross between heterozygous fish injected with wild-type mRNA coiled normally suggests that the homozygous sho embryos among the progeny were rescued.
Injection of wild-type GlyT1 mRNA into sho embryos also restores normal escape responses. At 26 hpf, 24% of the mutant G81D mRNA injected progeny (n = 58) from a cross between heterozygous sho carriers displayed a sho-like weak or no response to mechanosensory stimulation (Fig. 4Di), much like the 23% of the uninjected progeny (n = 40; data not shown). In contrast, only 3% of progeny injected with wild-type mRNA (n = 63) exhibited aberrant escape responses (Fig. 4Dii). Because no more than one-quarter of the mutant mRNA injected embryos displayed sho phenotypes, the G81D mutation appears not to act in a dominant-negative manner. Conversely, the finding that nearly all embryos injected with wild-type GlyT1A mRNA spawned from a cross of sho carriers exhibited normal behavior suggests that mutant embryos were rescued by wild-type mRNA. Interestingly, nine of these putative “rescued” embryos, representing 16% of injected embryos, responded aberrantly to tactile stimulation by failing to swim at 48 hpf (data not shown). Thus, the rescue appears to be transient, as one might expect because of degradation of injected mRNA. In fact, increasing the amount of injected wild-type mRNA can extend the rescue into the third day (data not shown). The return of these transiently rescued embryos to sho phenotypes further strengthens the conclusion that mutants were indeed rescued by the wild-type GlyT1 mRNA. Mutant phenocopy by knock-down of GlyT1 in wild-type embryos and mutant rescue by wild-type GlyT1A mRNA in sho embryos strongly suggest that the sho gene encodes for GlyT1.
The G81D mutation results in a nonfunctional glycine transporter
To further test the hypothesis that the G81D mutation causes the sho phenotype, wild-type and G81D GlyT1A mRNAs were injected into Xenopus oocytes, and two-electrode voltage-clamp recordings from these oocytes were performed to assay transporter function (Fig. 5A). GlyT1 couples glycine uptake with ionic gradients by transporting one glycine molecule for every two Na+ and one Cl- ions into the cell (Aragon and Lopez-Corcuera, 2003). As a consequence, a net inward current is carried by GlyT1 in the presence of extracellular glycine (Roux and Supplisson, 2000). Both the untagged and FLAG-tagged versions of the wild-type and G81D mutant proteins were analyzed. The addition of a FLAG epitope at the C terminus of GlyT1 did not appear to affect the transporter activity, and the result for the FLAG-tagged proteins are reported here. Wild-type GlyT1A mRNA-injected oocytes exhibited inward currents in response to applied glycine at concentrations from 2 to 600 μm, with an EC50 at 63 ± 14 μm (n = 10) (Fig. 5B) and peak inward current of 621 ± 158 nA at 600 μm glycine (n = 6) (Fig. 5C), which was significantly greater than uninjected control oocytes (4 ± 26 nA; n = 6; p < 0.001). However, oocytes injected with G81D mutant mRNA did not respond significantly to exogenous glycine at concentrations ranging from 2 to 600 μm, with peak inward currents to 600 μm glycine (3 ± 23 nA; n = 6), comparable with that of uninjected oocytes (p > 0.95). By adding a FLAG epitope to the C terminus of each protein, we were able to examine whether the lack of current in mutant mRNA-injected oocytes was attributable to impaired protein synthesis. Western blots of total protein extract from oocytes injected with either wild-type or mutant mRNAs probed with anti-FLAG antibody showed that there were comparable amounts of protein synthesized (data not shown). Thus, the physiological analysis in oocytes demonstrated that the G81D mutant GlyT1 was nonfunctional and confirmed that the sho phenotype was attributable to a loss-of-function mutation in GlyT1.
Figure 5.

The G81D mutation in GlyT1 results in a nonfunctional glycine transporter. A, Current responses in Xenopus oocytes injected with wild-type zebrafish GlyT1A mRNA, G81D mutant, and control oocytes, to bath application of glycine. The shaded bars at the top represent different concentrations of glycine in the bath (in order: 2, 5, 10, 50, 100, 300, and 600 μm). B, A plot of the average relative current responses of oocytes expressing wild-type GlyT1A to different concentrations of glycine (n = 10). The current measurements were normalized to the maximum response of each oocyte and expressed as percentage of the maximum current. C, The G81D mutant GlyT1 does not generate a glycine-dependent inward current. Shown are the average maximum inward currents induced by 600 μm glycine in wild-type GlyT1 mRNA-injected, G81D mRNA-injected, and uninjected control oocytes. Error bars represent SEs.
Glycine transporters are expressed in the CNS
In situ hybridization, using a probe specific for the coding sequence shared by all three isoforms, showed that GlyT1 is strongly expressed in the hindbrain and spinal cord during development (Fig. 6). GlyT1 is expressed in the rostral spinal cord by 18 hpf, the earliest time point examined (data not shown). At 24 hpf, the transporter is expressed in a segmental manner as five dorsally located stripes in the hindbrain (Fig. 6B). In the caudal hindbrain and spinal cord, GlyT1-positive cells are located at the dorsal midline and are not labeled by the neuronal marker anti-Hu, suggesting that the GlyT1-positive cells are non-neuronal (Fig. 6C,D). Because neurons are not found in the dorsal hindbrain at 24 hpf, the GlyT1-positive cells in the dorsal hindbrain stripes are likely non-neuronal as well. By 48 hpf, the transporter is expressed by cells along the dorsal midline of the midbrain and hindbrain in addition to the dorsal hindbrain stripes (Fig. 6E) and by the hindbrain floor plate cells (data not shown). Because there are no neurons along the dorsal midline of these regions (Bernhardt et al., 1990), these GlyT1-positive cells are likely non-neuronal as well. GlyT1 is also expressed in the retina, as reported for mammals (Jones, 1995; Zafra et al., 1995b; Pow and Hendrickson, 2000). In the spinal cord, the midline floor plate cells express GlyT1 at 48 hpf (Fig. 6F), in addition to the dorsal midline cells. By 72 hpf, GlyT1 expression in the hindbrain is more diffuse but remains primarily dorsal and superficial, with the strongest signal associated with cells lining the ventricle (Fig. 6G). However, in the spinal cord, the expression pattern of the transporter changes dramatically so that GlyT1 is now expressed in a pattern that appears to be complementary to that at 48 hpf, with the medial cells between the floor plate and dorsal midline cells becoming GlyT1 positive (Fig. 6H). Expression of GlyT1 by the early hindbrain and spinal cord is consistent with the finding that these regions are sufficient for those early behaviors that are deficient in sho mutants. Furthermore, GlyT1 is expressed primarily by non-neuronal cells in the early zebrafish CNS, consistent with expression of GlyT1 predominantly by glial cells in the mammalian CNS (Guastella et al., 1992; Smith et al., 1992; Borowsky et al., 1993; Adams et al., 1995; Zafra et al., 1995a,b; Jursky and Nelson, 1996).
Figure 6.
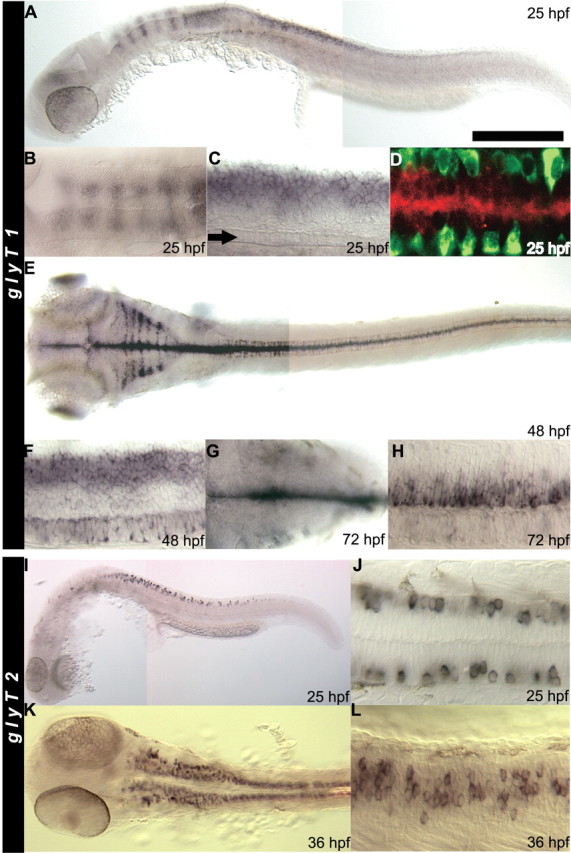
glyT1 and glyT2 are predominantly expressed by the hindbrain and spinal cord. In all panels, anterior is to the left, and, in the lateral views, dorsal is up. A, Lateral view of a whole-mounted embryo showing that glyT1 is expressed in hindbrain stripes, dorsal spinal cord, and the tectum at 25 hpf. B, Dorsal view showing the stripes of glyT1-positive cells in the dorsal hindbrain at 25 hpf. C, Lateral view focused at the midline focal plane showing that glyT1 is expressed by cells in the medial dorsal spinal cord. The row of midline floor-plate cells can be seen at the ventral midline (arrow). D, A confocal image of the 25 hpf spinal cord seen in a dorsal view demonstrating that glyT1-positive dorsal cells (red) at the midline are not postmitotic neurons. Neurons are labeled with anti-Hu (green) and located more laterally. E, Dorsal view of a whole-mounted 48 hpf embryo showing that glyT1 is expressed at the midline of the CNS, stripes in the hindbrain, and scattered cells in the lateral spinal cord. F, Lateral view of the 48 hpf spinal cord showing that glyT1 is expressed in longitudinal dorsal and ventral stripes in the spinal cord but not in between. G, Dorsal view of the 72 hpf hindbrain showing expression of glyT1 by cells at the superficial surface near the ventricle. H, Lateral view of the 72 hpf spinal cord showing that glyT1 is now expressed by a longitudinal stripe of cells that occupies the intermediate region that was glyT1 negative at 48 hpf. glyT1 is no longer expressed in the dorsal and ventral stripes. I, Lateral view of a whole-mounted 25 hpf embryo showing that glyT2 is expressed by apparent neurons in the spinal cord. J, Dorsal view of the 25 hpf spinal cord showing that the glyT2-positive cells are located at the margin of the spinal cord and are likely to be neurons. K, Dorsal view of a 36 hpf embryo showing an increased number of glyT2-positive cells in the spinal cord and many glyT2-positive cells in the hindbrain. L, Lateral view of the spinal cord showing that the glyT2-positive cells are located in the position occupied by commissural neurons at 36 hpf. Scale bar: A, E, K, 300 μm; B, 210 μm; C, F, G, J, L, 60 μm; D, 70 μm; H, 160 μm; I, 450 μm.
There is a second glycine transporter, GlyT2, that is expressed by glycinergic neurons in mammals and zebrafish (Jursky and Nelson, 1995; Higashijima et al., 2004a,b). To assess whether the loss of GlyT1 function in sho embryos could lead to compensatory changes in the expression of other related genes, the status of GlyT2 was investigated in those embryos. Full-length cDNA for zebrafish GlyT2 was cloned, and the expression of GlyT2 was assayed. At 24 hpf, GlyT2-positive cells were located in the marginal zone of the spinal cord in which neurons reside, suggesting that the GlyT2-positive cells were neurons (Fig. 6I,J). Occasionally, a few faintly GlyT2-positive cells were observed in the hindbrain as well (data not shown). By 36 hpf, many more cells in spinal cord and hindbrain were GlyT2 positive (Fig. 6K,L). With further development to 72 hpf, there was an increase in presumptive GlyT2-positive neurons, but they were restricted to the hindbrain and spinal cord as described previously (Higashijima et al., 2004a,b). At all stages examined, there were no differences in the expression pattern of glyT2 in sho compared with wild-type embryos (data not shown). Thus, no compensatory change in GlyT2 expression resulted from the absence of functional GlyT1.
High extracellular glycine can account for much of the sho phenotype
The loss of GlyT1 function in sho embryos should lead to increased extracellular glycine in the CNS and increased signaling via glycine receptors. If the decrease in spontaneous coiling and lack of swimming in sho embryos is attributable to the increased activity of glycine receptors, then blocking the effect of excessive glycine with the glycine receptor blocker strychnine should restore normal behavior in mutants. At 21 hpf, sho embryos rarely coil (Fig. 1B), but, after 5 min of exposure to 200 μm strychnine, mutant embryos (21 hpf; n = 7) coil with a frequency of 0.37 ± 0.10 Hz. This is not significantly different from two age-matched control groups: wild-type siblings in strychnine (n = 12) that coil with a frequency of 0.37 ± 0.09 Hz (t test, p > 0.95) and wild-type siblings not exposed to strychnine (n = 12) that coil with a frequency of 0.36 ± 0.13 Hz (t test, p > 0.95). Thus, blocking glycinergic synaptic transmission restores normal spontaneous coiling in sho embryos.
Whether strychnine could restore swimming in older embryos was also examined. Slow twitch muscle fibers from midtrunk segments in wild-type embryos (40-46 hpf; n = 5) responded to mechanosensory stimulation with a large, nonrhythmic depolarization in the presence of 1-10 μm strychnine (n = 20) (Fig. 7Aii) rather than the rhythmic swimming depolarizations seen in the absence of strychnine (Fig. 7Ai). In sho embryos, midtrunk muscles respond to mechanosensory stimulation with a nonrhythmic depolarization without strychnine (Cui et al., 2004) (Fig. 7Bi). However, in the presence of 1-10 μm strychnine, sho muscles responded to mechanosensory stimulation with an initial large, nonrhythmic depolarization immediately followed by rhythmic depolarizations in three of four embryos (Fig. 7Bii). Thus, it appears that a reduction of strychnine-sensitive glycinergic transmission in sho embryos restores some swimming as well as spontaneous coiling.
Figure 7.

Partial block of glycinergic inhibitory synaptic transmission can restore swimming in sho embryos, but the response to mechanosensory stimulation is still aberrant. A, A series of muscle voltage recordings from a wild-type embryo in response to mechanosensory stimulation (arrow) after exposure to different concentrations of strychnine. i, Rhythmic depolarizations are elicited in the absence of strychnine. ii, An irregular, large depolarization is elicited in 10 μm strychnine. iii, An irregular, large depolarization is elicited in 50 μm strychnine. B, A series of muscle recordings from a sho embryo in response to mechanosensory stimulation (arrow) after exposure to different concentrations of strychnine. i, A typical sho-like depolarization is elicited. ii, An irregular, large depolarization followed by rhythmic depolarizations (bracket) similar to swimming is elicited in 10 μm strychnine. iii, Only an irregular, large depolarization is elicited in 50 μm strychnine. wtsib, Wild-type sibling.
When strychnine was >10 μm, the rhythmic activity disappeared in sho embryos, leaving only the large, nonrhythmic depolarization (n = 16) (Fig. 7Biii). Strychnine at 10 and 50 μm appeared to be a saturating concentration in, respectively, wild-type and sho embryos because there were no additional changes in the muscle response with higher concentrations. Interestingly, the maximum amplitude and duration of the large, nonrhythmic depolarization of sho muscles seen in saturating levels of strychnine were greater than that for wild-type muscles (16.1 ± 0.9 mV in sho vs 11.0 ± 0.7 mV in wild type, p < 0.001; 378 ± 22 ms in sho vs 259 ± 18 ms in wild type, p < 0.001). Duration was measured as the amount of time the depolarization was at or greater than half the peak amplitude. Thus, pharmacologically blocking glycinergic transmission appears to partially restore normal activity in mutant embryos.
To further explore whether the putative, increased extracellular glycine contributes to the mutant phenotype, NFPS, which is a persistent antagonist specific for GlyT1 but not GlyT2 (Aubrey and Vandenberg, 2001), was applied to wild-type embryos to see whether it would phenocopy the sho mutation. We first demonstrated that NFPS effectively blocked zebrafish GlyT1 expressed in Xenopus oocytes. Application of 50 μm glycine to oocytes injected with wild-type GlyT1A mRNA induced a 140 ± 25 nA current that was inhibited by 98% within 60 s after the addition of 500 nm NFPS (n = 8; data not shown). Next, NFPS (100 μm) was injected into the CNS of anesthetized wild-type embryos (32 hpf). Drug-injected embryos quickly recovered from anesthesia and were morphologically normal and initially displayed normal escape responses and swimming in response to mechanosensory stimulation (data not shown). After 3 h, however, 73% of the NFPS-injected embryos (n = 45) responded to touch with a hypercontraction of the trunk and no swimming (data not shown), much like sho embryos, whereas all of the control-injected embryos escaped and swam normally (n = 10). At 48 hpf, only 31% of the NFPS-injected embryos (n = 42) responded with trunk hypercontractions and no swimming (data not shown), suggesting that the effect of the drug was transient. Voltage recordings from muscles of control embryos and NFPS-injected embryos confirmed the behavioral results. Muscles from NFPS-injected embryos (three at 36 hpf and two at 48 hpf) exhibited one or two large, nonrhythmic depolarizations in response to mechanosensory stimulation reminiscent of the sho response (Fig. 8B). Muscles from control-injected embryos (two at 36 hpf) displayed normal rhythmic depolarizations typical of the fictive swimming activity (Fig. 8A). Thus, pharmacologically blocking GlyT1 phenocopies the sho mutation. The delayed appearance of sho-like responses in NFPS-injected embryos is consistent with the hypothesis that the abnormal behaviors are attributable to the accumulation of extracellular glycine in the CNS resulting from the inhibition of GlyT1.
Figure 8.

The sho response to mechanosensory stimulation is phenocopied by pharmacologically blocking GlyT1 in wild-type embryos, and putative dilution of high extracellular glycine in sho embryos restores swimming responses. A, Muscle voltage recording from a wild-type (wt) embryo (36hpf) 3 h after a control injection showed a normal fictive swimming in response to mechanosensory stimulation (arrow). B, Muscle recording from a wild-type embryo (36 hpf) 3 h after a NFPS injection showing showed a large, nonrhythmic depolarization resembling the sho in response to touch. C, Muscle recording from a sho embryo (42 hpf) after the hindbrain was exposed and perfused for 30 min with glycine-free solution showed rhythmic depolarizations similar to fictive swimming in response to touch. D, Muscle recording from the same embryo as in C after switching from perfusion with glycine-free solution to perfusion with solution containing 200 μm glycine showed the large, nonrhythmic depolarization characteristic of sho in response to touch.
If the sho defects are attributable to an accumulation of CNS glycine, then the removal of extracellular glycine may alleviate this problem. Because hindbrain/brainstem is known to be important for the generation of swimming patterns in other species (Grillner, 2003), we hypothesized that the lack of swimming in sho may be attributable to the persistent inhibition of the hindbrain network by high extracellular glycine. To test this, we sought to dilute out the putative high extracellular glycine in the hindbrain of sho embryos by removing the skin overlying the fourth ventricle to expose the hindbrain to the glycine-free recording solution. Indeed, after 30 min of perfusion, sho embryos responded to mechanosensory stimulation with rhythmic depolarizations characteristic of swimming (Fig. 8C). The duration of the fictive swimming episodes were not distinguishable between the hindbrain-exposed wild-type (1324 ± 69 ms; n = 7) and sho (1287 ± 76 ms; n = 11; p > 0.7) embryos, although the frequency of the rhythmic depolarizations were significantly higher in wild-type (24.0 ± 0.6 Hz) compared with sho (16.8 ± 0.4 Hz; p < 0.001) embryos. Furthermore, we found that, after a switch of the perfusion solution from the glycine-free solution to one containing 200 μm glycine, mechanosensory stimulation again elicited the large, nonrhythmic depolarizations in sho embryos (n = 3) characteristic of the mutant response (Fig. 8D). The finding that swimming can be restored in sho embryos by the putative removal of excess glycine from the hindbrain and that the mutant phenotype can be reinduced by raising the glycine level corroborate the partial rescue of swimming in mutants by strychnine and suggests that the behavioral abnormality in sho is attributable to the loss of the capacity to remove extracellular glycine in the CNS.
Discussion
We identified the zebrafish sho gene as one encoding for the GlyT1 glycine transporter. GlyT1 is expressed by non-neuronal cells in the CNS, whereas a second glycine transporter, GlyT2, is expressed by neurons during the first3dof embryonic development. Because shota229g embryos exhibit aberrant behaviors attributable to a CNS defect (Cui et al., 2004), glycine uptake by GlyT1 on non-neuronal cells in the CNS appears to be required for normal CNS function.
sho mutants also exhibit a muscle phenotype. Muscle fibers in zebrafish embryos are initially electrically coupled, with coupling decreasing as development progresses (Buss and Drapeau, 2000). Embryos of a different allele of sho, shote301, exhibit persistent electrical coupling between muscle fibers (Luna et al., 2004). The electrical coupling leads to prolonged depolarization of muscles and subsequent hypercontraction exhibited by mutants. Thus, persistent muscle coupling in mutants is likely to reinforce noncyclical output by motor neurons attributable to the sho CNS defect (Cui et al., 2004). Because GlyT1 is not expressed in muscles, the muscle defect is secondary to the CNS defect, suggesting that input from the CNS to muscles can regulate coupling between muscle fibers. shote301 embryos were also reported to exhibit patterned output by motor axons from extracellular ventral root recordings (Luna et al., 2004), unlike the irregular, noncyclical motor output seen in shota229g (Cui et al., 2004). This difference is likely attributable to the fact that shota229g is the strongest allele, whereas shote301 is a weaker allele (Granato et al., 1996). Thus, it is possible that the CNS phenotype in shote301 embryos is more subtle and perhaps not detectable by ventral root recordings.
We found a missense mutation, G81D, in GlyT1 that is the cause of abnormal motor behaviors in sho. The point mutation occurs in the highly conserved second transmembrane domain, and the affected residue is conserved across species as well as in transporters for GABA, serotonin, taurine, and dopamine that also belong to the 12-transmembrane neurotransmitter transporter superfamily (Melamed and Kanner, 2004). Not much is known about the role of this transmembrane domain in GlyT1 function. Because the G81D mutation replaces the simplest amino acid with a larger, acidic residue, the mutation is likely to affect function. We confirmed that the G81D mutation severely compromises the activity of GlyT1 by voltage-clamp analysis of wild-type and mutant RNA-injected Xenopus oocytes, providing evidence for an important role of the second transmembrane domain for GlyT1 function. Previous mutagenesis analysis in which the corresponding glycine residue in the serotonin transporter was replaced by cysteine found no effect on the activity of the transporter or its membrane localization (Sato et al., 2004). However, changes to the side-chain size of the amino acid immediately adjacent to the glycine residue in the GABA transporter GAT-4 affected the substrate specificity and interaction of the transporter to its antagonist, taurine (Melamed and Kanner, 2004). Thus, it is possible that the G81D mutation may affect the substrate specificity of GlyT1. Another possibility is that changes in the second transmembrane domain attributable to the G81D mutation may disrupt the function of the adjacent first extracellular loop that is thought to undergo conformational changes during transport (Lopez-Corcuera et al., 2001; Roux et al., 2001).
Glycine has three known or potential physiological roles within the CNS. First, glycine is a major neurotransmitter in the hindbrain and spinal cord and opens Cl--selective channels (Legendre, 2001). In very young animals, the Cl- gradient results in a modest depolarization when channels open, whereas in older animals, neurons hyperpolarize to glycine (Rivera et al., 1999; Ben-Ari, 2001). Second, glycine is a coagonist with glutamate for synaptic transmission via NMDA-gated, excitatory glutamatergic receptors (Mayer et al., 1989). Third, glycine without glutamate can serve as an excitatory neurotransmitter acting via NR1/NR3 receptors in Xenopus oocytes (Chatterton et al., 2002). Because GlyT1 activity can regulate the concentration of glycine in the extracellular fluid layer adjacent to the membrane of Xenopus oocytes expressing GlyT1 in vitro (Supplisson and Bergman, 1997), it is hypothesized to regulate levels of glycine within the synaptic regions of the CNS. Thus, the loss of GlyT1 function should lead to increased levels of extracellular glycine in the CNS and increased and/or chronic inhibition at glycinergic synapses and increased excitation at NMDA synapses in GlyT1 mutants. In fact, homozygous GlyT1 knock-out mice exhibit physiological defects in respiratory neural circuits that are accountable by chronic inhibition attributable to increased extracellular glycine (Gomeza et al., 2003). Strychnine-sensitive, spontaneous glycinergic synaptic noise recorded in hypoglossal motor neurons is higher in GlyT1-/- mice than in controls, and the change in steady-state holding currents after application of strychnine to hypoglossal motor neurons are greater, suggesting chronic inhibition of these neurons. Rhythmic respiratory activity in brainstem slices of GlyT1-/- mice is disrupted but was restored by decreasing glycinergic inhibition with strychnine. Furthermore, addition of high glycine or sarcosine to wild-type brainstem slices phenocopies GlyT1-/- mice. Thus, the defects in respiratory circuits of knock-outs can be accounted for as a straightforward physiological consequence of high extracellular glycine.
Increased extracellular glycine induced by interference with GlyT1 can also enhance NMDA receptor (NMDAR) synaptic transmission and a variety of behaviors, including learning and memory. Pharmacological blockade of GlyT1 increases NMDAR-mediated synaptic transmission in the hippocampus, prefrontal cortex, and brainstem, including those onto hypoglossal motor neurons, and also enhances prepulse inhibition of the acoustic startle response (Bergeron et al., 1998; Chen et al., 2003; Kinney et al., 2003; Lim et al., 2004; Martina et al., 2004). Similarly, glycine transport is reduced by 50% in GlyT1+/- mice, and there is an increase in the ratio of NMDAR current to AMPA receptor current in hippocampal slices and enhanced performance in prepulse inhibition and spatial learning tasks (Tsai et al., 2004). As with the defects in the respiratory circuits after elimination of GlyT1 activity, the enhancement of NMDAR-mediated synaptic transmission and the resultant behavior can also be explained as a simple physiological consequence of increased extracellular glycine.
Many features of sho embryos can also be accounted for by a higher level of extracellular glycine within the CNS. sho embryos exhibit a decrease in the frequency of spontaneous coiling compared with wild-type embryos, and the application of strychnine to mutants appears to restore normal levels of spontaneous coiling. Thus, increased or chronic glycinergic activity in mutants could be responsible for the decrease in coiling. Saint-Amant and Drapeau (2000, 2001) demonstrated two types of network-driven activity in zebrafish embryos before 24 hpf when spontaneous coiling is most evident: periodic depolarizations that generate action potentials and bursts of glycinergic synaptic potentials. The latter are sensitive to strychnine, suggesting that glycine receptors are active in early neurons within the neural circuit for spontaneous coiling. Despite the existence of glycine receptors at these early stages, glycinergic synaptic input is not required for normal coiling because strychnine does not affect coiling in wild-type embryos. This is consistent with the finding that coiling is unaffected after block of chemical synaptic transmission, suggesting that coiling is driven primarily by electrical synapses within the neural circuit (Saint-Amant and Drapeau, 2001). However, in sho mutants, defective transport of glycine could lead to high extracellular glycine, chronic inhibition of the coiling circuit, and decreased spontaneous coiling.
A physiological response to increased extracellular glycine may also be responsible for the swimming defect of sho embryos. First, mechanosensory stimulation of sho embryos elicits a large, nonrhythmic depolarization in trunk muscles rather than the normal rhythmic depolarizations that underlie swimming (Cui et al., 2004). However, partial blockade of glycinergic transmission by intermediate levels of strychnine often restored rhythmic depolarizations after the initial, large irregular depolarization in mutant muscles. Second, the larger and longer irregular depolarization seen after saturating levels of strychnine in sho embryos compared with wild-type embryos could be attributable to increased synaptic transmission via NMDAR one might predict because of the higher levels of extracellular glycine. Third, pharmacological inhibition of GlyT1 in wild-type embryos induced both sho-like behavior and muscle voltage responses to mechanosensory stimulation. Fourth, putative washout of high extracellular glycine from the hindbrain of sho embryos restored fictive swimming, and apparent reintroduction of high glycine reinduced the mutant response to mechanosensory stimulation. These finding suggests that swimming circuits are made dysfunctional by excessive glycine in mutants and that they can generate a swimming pattern when the effect of elevated glycine is removed.
sho embryos may be useful for further investigating the effects of high glycine on the CNS by a combination of electrophysiology, in vivo imaging of fluorescently tagged proteins, antisense knock-down of gene products, and targeted misexpression of relevant genes. This may provide insights for the effects of high CNS glycine and the resultant motor dysfunction that occur in inherited human diseases such as glycine encephalopathy and nonketotic hyperglycemia (Applegarth and Toone, 2004).
Footnotes
This work was supported in part by Center for Organogenesis Training Grant 5-T32-HD007505 (W.W.C.), a Long-Term Fellowship from the Human Frontier Science Program (H.H.), National Science Foundation Grant IBN-0077634 (S.E.L.), National Institutes of Health Grant NS039196 (R.I.H.), German Human Genome Project Grant 01 KW 9919 (R.G.), and National Institute of Neurological Disorders and Stroke Grant NS36587 (J.Y.K.). We thank members of the Kuwada laboratory for helpful discussions, Shawn Sprague for fish care, and J. Power and N. Nagaya for help with the Western analysis.
Correspondence should be addressed to Dr. John Y. Kuwada, Department of Molecular, Cellular, and Developmental Biology, University of Michigan, Ann Arbor, MI 48109-1048. E-mail: kuwada@umich.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/256610-11$15.00/0
References
- Adams RH, Sato K, Shimada S, Tohyama M, Puschel AW, Betz H (1995) Gene structure and glial expression of the glycine transporter GlyT1 in embryonic and adult rodents. J Neurosci 15: 2524-2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Applegarth DA, Toone JR (2004) Glycine encephalopathy (nonketotic hyperglycinaemia): review and update. J Inherit Metab Dis 27: 417-422. [DOI] [PubMed] [Google Scholar]
- Aragon C, Lopez-Corcuera B (2003) Structure, function and regulation of glycine neurotransporters. Eur J Pharmacol 479: 249-262. [DOI] [PubMed] [Google Scholar]
- Aubrey KR, Vandenberg RJ (2001) N[3-(4′-fluorophenyl)-3-(4′-phenyl-phenoxy)propyl]sarcosine (NFPS) is a selective persistent inhibitor of glycine transport. Br J Pharmacol 134: 1429-1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ari Y (2001) Developing networks play a similar melody. Trends Neurosci 24: 353-360. [DOI] [PubMed] [Google Scholar]
- Bergeron R, Meyer TM, Coyle JT, Greene RW (1998) Modulation of N-methyl-d-aspartate receptor function by glycine transport. Proc Natl Acad Sci USA 95: 15730-15734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhardt R, Chitnis A, Lindamer L, Kuwada J (1990) Identification of spinal neurons in the embryonic and larval zebrafish. J Comp Neurol 302: 603-616. [DOI] [PubMed] [Google Scholar]
- Borowsky B, Mezey E, Hoffman BJ (1993) Two glycine transporter variants with distinct localization in the CNS and peripheral tissues are encoded by a common gene. Neuron 10: 851-863. [DOI] [PubMed] [Google Scholar]
- Buss RR, Drapeau P (2000) Physiological properties of zebrafish embryonic red and white muscle fibers during early development. J Neurophysiol 84: 1545-1557. [DOI] [PubMed] [Google Scholar]
- Chatterton JE, Awobuluyi M, Premkumar LS, Takahashi H, Talantova M, Shin Y, Cui J, Tu S, Sevarino KA, Nakanishi N, Tong G, Lipton SA, Zhang D (2002) Excitatory glycine receptors containing the NR3 family of NMDA receptor subunits. Nature 415: 793-798. [DOI] [PubMed] [Google Scholar]
- Chen L, Muhlhauser M, Yang CR (2003) Glycine tranporter-1 blockade potentiates NMDA-mediated responses in rat prefrontal cortical neurons in vitro and in vivo. J Neurophysiol 89: 691-703. [DOI] [PubMed] [Google Scholar]
- Corey DR, Abrams JM (2001) Morpholino antisense oligonucleotides: tools for investigating vertebrate development. Genome Biol 2: REVIEWS1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui WW, Saint-Amant L, Kuwada JY (2004) shocked gene is required for the function of a premotor network in the zebrafish CNS. J Neurophysiol 92: 2898-2908. [DOI] [PubMed] [Google Scholar]
- Drapeau P, Ali DW, Buss RR, Saint-Amant L (1999) In vivo recording from identifiable neurons of the locomotor network in the developing zebrafish. J Neurosci Methods 88: 1-13. [DOI] [PubMed] [Google Scholar]
- Friedrich RW, Korsching SI (1997) Combinatorial and chemotopic odorant coding in the zebrafish olfactory bulb visualized by optical imaging. Neuron 18: 737-752. [DOI] [PubMed] [Google Scholar]
- Geisler R (2002) Mapping and cloning. In: Zebrafish, a practical approach. Oxford: Oxford UP.
- Gomeza J, Hulsmann S, Ohno K, Eulenburg V, Szoke K, Richter D, Betz H (2003) Inactivation of the glycine transporter 1 gene discloses vital role of glial glycine uptake in glycinergic inhibition. Neuron 40: 785-796. [DOI] [PubMed] [Google Scholar]
- Granato M, van Eeden F, Schach U, Trowe T, Brand M, Furutani-Seiki M, Haffter P, Hammerschmidt M, Heisenberg C, Jiang Y, Kane D, Kelsh R, Mullins M, Odenthal J, Nusslein-Volhard C (1996) Genes controlling and mediating locomotion behavior of the zebrafish embryo and larva. Development 123: 399-413. [DOI] [PubMed] [Google Scholar]
- Grillner S (2003) The motor infrastructure: from ion channels to neuronal networks. Nat Rev Neurosci 4: 573-586. [DOI] [PubMed] [Google Scholar]
- Guastella J, Brecha N, Weigmann C, Lester HA, Davidson N (1992) Cloning, expression, and localization of a rat brain high-affinity glycine transporter. Proc Natl Acad Sci USA 89: 7189-7193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashijima S, Masino MA, Mandel G, Fetcho JR (2003) Imaging neuronal activity during zebrafish behavior with a genetically encoded calcium indicator. J Neurophysiol 90: 3986-3997. [DOI] [PubMed] [Google Scholar]
- Higashijima SI, Mandel G, Fetcho JR (2004a) Distribution of prospective glutamatergic, glycinergic, and GABAergic neurons in embryonic and larval zebrafish. J Comp Neurol 480: 1-18. [DOI] [PubMed] [Google Scholar]
- Higashijima SI, Schaefer M, Fetcho JR (2004b) Neurotransmitter properties of spinal interneurons in embryonic and larval zebrafish. J Comp Neurol 480: 19-37. [DOI] [PubMed] [Google Scholar]
- Hirata H, Saint-Amant L, Waterbury J, Cui WW, Zhou W, Li Q, Goldman D, Granato M, Kuwada JY (2004) accordion, a zebrafish behavioral mutant, has a muscle relaxation defect due to a mutation in the ATPase Ca2+ pump SERCA1. Development 131: 5457-5468. [DOI] [PubMed] [Google Scholar]
- Jones EM (1995) Na+- and Cl--dependent neurotransmitter transporters in bovine retina: identification and localization by in situ hybridization histochemistry. Vis Neurosci 12: 1135-1142. [DOI] [PubMed] [Google Scholar]
- Jursky F, Nelson N (1995) Localization of glycine neurotransmitter transporter (GLYT2) reveals correlation with the distribution of glycine receptor. J Neurochem 64: 1026-1033. [DOI] [PubMed] [Google Scholar]
- Jursky F, Nelson N (1996) Developmental expression of the glycine transporters GLYT1 and GLYT2 in mouse brain. J Neurochem 67: 336-344. [DOI] [PubMed] [Google Scholar]
- Kim KM, Kingsmore SF, Han H, Yang-Feng TL, Godinot N, Seldin MF, Caron MG, Giros B (1994) Cloning of the human glycine transporter type 1: molecular and pharmacological characterization of novel isoform variants and chromosomal localization of the gene in the human and mouse genomes. Mol Pharmacol 45: 608-617. [PubMed] [Google Scholar]
- Kimmel C, Powell S, Metcalfe W (1982) Brain neurons which project to the spinal cord in young larvae of the zebrafish. J Comp Neurol 205: 112-127. [DOI] [PubMed] [Google Scholar]
- Kimmel C, Ballard W, Kimmel S, Ullmann B, Schilling T (1995) Stages of embryonic development of the zebrafish. Dev Dyn 203: 253-310. [DOI] [PubMed] [Google Scholar]
- Kinney GG, Sur C, Burno M, Mallorga PJ, Williams JB, Figueroa DJ, Wittmann M, Lemaire W, Conn PJ (2003) The glycine transporter type 1 inhibitor N-[3-(4′-fluorophenyl)-3-(4′-phenylphenoxy)propyl]sarcosine potentiates NMDA receptor-mediated responses in vivo and produces an antipsychotic profile in rodent behavior. J Neurosci 23: 7586-7591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legendre P (2001) The glycinergic inhibitory synapse. Cell Mol Life Sci 58: 760-793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legendre P, Korn H (1994) Glycinergic inhibitory synaptic currents and related receptor channels in the zebrafish brain. Eur J Neurosci 6: 1544-1557. [DOI] [PubMed] [Google Scholar]
- Lim R, Hoang P, Berger AJ (2004) Blockade of glycine transporter-1 (GLYT-1) potentiates NMDA receptor-mediated synaptic transmission in hypoglossal motorneurons. J Neurophysiol 92: 2530-2537. [DOI] [PubMed] [Google Scholar]
- Lopez-Corcuera B, Nunez E, Martinez-Maza R, Geerlings A, Aragon C (2001) Substrate-induced conformational changes of extracellular loop 1 in the glycine transporter GLYT2. J Biol Chem 276: 43463-43470. [DOI] [PubMed] [Google Scholar]
- Luna VM, Wang M, Ono F, Gleason MR, Dallman JE, Mandel G, Brehm P (2004) Persistent electrical coupling and locomotory dysfunction in the zebrafish mutant shocked. J Neurophysiol 92: 2003-2009. [DOI] [PubMed] [Google Scholar]
- Martina M, Gorfinkel Y, Halman S, Lowe JA, Periyalwar P, Schmidt CJ, Bergeron R (2004) Glycine transporter type 1 blockade changes NMDA receptor-mediated responses and LTP in hippocampal CA1 pyramidal cells by altering extracellular glycine levels. J Physiol (Lond) 557: 489-500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer ML, Vyklicky Jr L, Clements J (1989) Regulation of NMDA receptor desensitization in mouse hippocampal neurons by glycine. Nature 338: 425-427. [DOI] [PubMed] [Google Scholar]
- Melamed N, Kanner BI (2004) Transmembrane domains I and II of the gamma-aminobutyric acid transporter GAT-4 contain molecular determinants of substrate specificity. Mol Pharmacol 65: 1452-1461. [DOI] [PubMed] [Google Scholar]
- Mendelson B (1985) Soma position is correlated with time of development in three types of identified reticulospinal neurons. Dev Biol 112: 489-493. [DOI] [PubMed] [Google Scholar]
- Metcalfe W, Mendelson B, Kimmel C (1986) Segmental homologies among reticulospinal neurons in the hindbrain of the zebrafish larva. J Comp Neurol 251: 147-159. [DOI] [PubMed] [Google Scholar]
- Neuhauss SC, Biehlmaier O, Seeliger MW, Das T, Kohler K, Harris WA, Baier H (1999) Genetic disorders of vision revealed by a behavioral screen of 400 essential loci in zebrafish. J Neurosci 19: 8603-8615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Malley D, Kao Y, Fetcho JR (1996) Imaging the functional organization of zebrafish hindbrain segments during escape behaviors. Neuron 17: 1145-1155. [DOI] [PubMed] [Google Scholar]
- Pow DV, Hendrickson AE (2000) Expression of glycine and the glycine transporter glyt-1 in the developing rat retina. Vis Neurosci 17: 1-9. [DOI] [PubMed] [Google Scholar]
- Ribera AB, Nusslein-Volhard C (1998) Zebrafish touch-insensitive mutants reveal an essential role for the developmental regulation of sodium current. J Neurosci 18: 9181-9191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritter DA, Bhatt DH, Fetcho JR (2001) In vivo imaging of zebrafish reveals differences in the spinal networks for escape and swimming movements. J Neurosci 21: 8956-8965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera C, Voipio J, Payne JA, Ruusuvuori E, Lahtinen H, Lamsa K, Pirvola U, Saarma M, Kaila K (1999) The K+/Cl- co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature 397: 251-255. [DOI] [PubMed] [Google Scholar]
- Roux MJ, Supplisson S (2000) Neuronal and glial glycine transporters have different stoichiometries. Neuron 25: 373-383. [DOI] [PubMed] [Google Scholar]
- Roux MJ, Martinez-Maza R, Le Goff A, Lopez-Corcuera B, Aragon C, Supplisson S (2001) The glial and the neuronal glycine transporters differ in their reactivity to sulfhydryl reagents. J Biol Chem 276: 17699-17705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saint-Amant L, Drapeau P (1998) Time course of the development of motor behaviors in the zebrafish embryo. J Neurobiol 37: 622-632. [DOI] [PubMed] [Google Scholar]
- Saint-Amant L, Drapeau P (2000) Motoneuron activity patterns related to the earliest behavior of the zebrafish embryo. J Neurosci 20: 3964-3972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saint-Amant L, Drapeau P (2001) Synchronization of an embryonic network of identified spinal interneurons solely by electrical coupling. Neuron 31: 1035-1046. [DOI] [PubMed] [Google Scholar]
- Sato Y, Zhang Y-W, Androutsellis-Theotokis A, Rudnick G (2004) Analysis of transmembrane domain 2 of rat serotonin transporter by cysteine scanning mutagenesis. J Biol Chem 279: 22926-22933. [DOI] [PubMed] [Google Scholar]
- Shafizadeh E, Paw BH, Foott H, Liao EC, Barut BA, Cope JJ, Zon LI, Lin S (2002) Characterization of zebrafish merlot/chablis as non-mammalian vertebrate models for severe congenital anemia due to protein 4.1 deficiency. Development 129: 4359-4370. [DOI] [PubMed] [Google Scholar]
- Smith KE, Borden LA, Hartig PR, Branchek T, Weinshank RL (1992) Cloning and expression of a glycine transporter reveal colocalization with NMDA receptors. Neuron 8: 927-935. [DOI] [PubMed] [Google Scholar]
- Supplisson S, Bergman C (1997) Control of NMDA receptor activation by a glycine transporter coexpressed in Xenopus oocytes. J Neurosci 17: 4580-4590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbot WS, Schier AF (1999) Positional cloning of mutated zebrafish genes. Methods Cell Biol 60: 259-286. [DOI] [PubMed] [Google Scholar]
- Tsai G, Ralph-Williams RJ, Martina M, Bergeron R, Berger-Sweeney J, Dunham KS, Jiang Z, Caine SB, Coyle JT (2004) Gene knockout of glycine transporter 1: characterization of the behavioral phenotype. Proc Natl Acad Sci USA 101: 8485-8490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerfield M (1993) The zebrafish book, Ed 3. Eugene, OR: University of Oregon.
- Zafra F, Gomeza J, Olivares L, Aragon C, Gimenez C (1995a) Regional distribution and developmental variation of the glycine transporters GLYT1 and GLYT2 in the rat CNS. Eur J Neurosci 7: 1342-1352. [DOI] [PubMed] [Google Scholar]
- Zafra F, Aragon C, Olivares L, Danbolt NC, Gimenez C, Storm-Mathisen J (1995b) Glycine transporters are differentially expressed among CNS cells. J Neurosci 15: 3952-3969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Talbot WS, Schier AF (1998) Positional cloning identifies zebrafish one-eyed pinhead as a permissive EGF-related ligand required during gastrulation. Cell 92: 241-251. [DOI] [PubMed] [Google Scholar]