

Abstract
The contribution of intracellular stores to axonal Ca2+ overload during chemical ischemia in vitro was examined by confocal microscopy. Ca2+ accumulation was measured by fluo-4 dextran (low-affinity dye, KD ≈ 4 μm) or by Oregon Green 488 BAPTA-1 dextran (highaffinity dye, KD ≈ 450 nm). Axonal Na+ was measured using CoroNa Green. Ischemia in CSF containing 2 mm Ca2+ caused an ∼3.5-fold increase in fluo-4 emission after 30 min, indicating a large axonal Ca2+ rise well into the micromolar range. Axonal Na+ accumulation was enhanced by veratridine and reduced, but not abolished, by TTX. Ischemia in Ca2+-free (plus BAPTA) perfusate resulted in a smaller but consistent Ca2+ increase monitored by Oregon Green 488 BAPTA-1, indicating release from intracellular sources. This release was eliminated in large part when Na+ influx was reduced by replacement with N-methyl-d-glucamine (NMDG+; even in depolarizing high K+ perfusate), Li+, or by the application of TTX and significantly increased by veratridine. Intracellular release also was reduced significantly by neomycin or 1-(6-[(17β-methoxyestra-1,3,5 [10]-trien-17-yl) amino] hexyl)-1H-pyrrole-2,5-dione ( U73122) (phospholipase C inhibitors), heparin [inositol trisphosphate (IP3) receptor blocker], or 7-chloro-5-(2-chlorophenyl)-1,5-dihydro-4,1-benzothiazepin-2(3H)-one (CGP37157; mitochondrial Na+/Ca2+ exchange inhibitor) as well as ryanodine. Combining CGP37157 with U73122 or heparin decreased the response more than either agent alone and significantly improved electrophysiological recovery. Our conclusion is that intra-axonal Ca2+ release during ischemia in rat optic nerve is mainly dependent on Na+ influx. This Na+ accumulation stimulates three distinct intra-axonal sources of Ca2+: (1) the mitochondrial Na+/Ca2+ exchanger driven in the Na+ import/Ca2+ export mode, (2) positive modulation of ryanodine receptors, and (3) promotion of IP3 generation by phospholipase C.
Keywords: axon, Na/Ca exchanger, IP3, ryanodine, endoplasmic reticulum, mitochondria, phospholipase C
Introduction
Ischemia in CNS white matter causes axonal Na+ influx through noninactivating Na+ channels. Accumulation of axoplasmic Na+ is critical for promoting cellular injury; coupled with severe K+ depletion that results in large membrane depolarization, high Na+i stimulates reverse Na+/Ca2+ exchange and axonal Ca2+ overload (Stys et al., 1992; Stys and Lopachin, 1998). Ca2+ overloading in turn triggers processes that are detrimental to affected cells (Schanne et al., 1979; Kostyuk et al., 2000; Sattler and Tymianski, 2000; Rizzuto, 2001). In addition to an extracellular source of Ca2+, many excitable cells contain enough bound or sequestered ion to increase [Ca2+]i markedly (Stys et al., 1997; Kristian and Siesjo, 1998) if these intracellular pools of Ca2+ [e.g., endoplasmic reticulum (ER), mitochondria] are released. Results obtained in our laboratory indicate that intracellular Ca2+ overload because of anoxia in rat optic nerve is attributable primarily to Ca2+ influx from the extracellular space (Stys et al., 1992; Stys and Lopachin, 1998; Stys, 2004). During in vitro ischemia, however, removal of external Ca2+ failed to protect spinal cord dorsal columns; instead, under these conditions, Ca2+ was released from the ER in amounts sufficient to cause severe damage via a mechanism similar to “excitation-contraction coupling” in skeletal muscle (Ouardouz et al., 2003). Given the potentially differing responses of central white matter tracts to injury in different regions of the CNS [e.g., optic nerve vs dorsal column (Jiang et al., 2002)] or among species [e.g., mouse vs rat (Tekkok et al., 2003)], we investigated the contribution of intracellular Ca2+ stores to axoplasmic Ca2+ rise during in vitro ischemia in rat optic nerve with the use of high-resolution confocal laser-scanning microscopy. Our results indicate that a significant portion of ischemic axoplasmic Ca2+ increase originates from the ER and from mitochondria; unexpectedly, this Ca2+ release is strongly dependent on axonal Na+ influx, similar to the dependence of extracellular Ca2+ entry on Na+ (for review, see Stys, 2004).
Materials and Methods
Adult Long-Evans male rats were anesthetized with 80% CO2/20% O2 and decapitated. Optic nerves were dissected out and immersed in Ca2+-free artificial CSF (aCSF) at 4°C. Nerves were placed in an interface perfusion chamber and loaded with fluorescent dyes. For Ca2+ and Na+ imaging, the nerves were placed in a custom-built perfusion chamber, mounted on an upright Nikon (Tokyo, Japan) C1 confocal laserscanning microscope. Imaging was performed at 36°C with a 60 or 40× immersion objective maintained at the same temperature to avoid cooling of the sample. Fluorescence changes were normalized to average basal levels and reported as a ratio of the signal collected from ion-sensitive and ion-insensitive fluorophores plotted against time. Electrophysiological measurements of compound action potentials (CAPs) were performed by using suction electrodes as described previously (Stys et al., 1991).
The aCSF contained the following (in mm): 126 NaCl, 26 NaHCO3, 3.0 KCl, 1.25 NaH2PO4, 2.0 MgSO4, 2.0 CaCl2, and 10 glucose. Solutions were bubbled continuously with 95% O2/5% CO2, pH ≈7.4. As Ca2+ indicators, two fluorescent probes were used: fluo-4 dextran (KD ≈ 4 μm) or Oregon Green 488 BAPTA-1 dextran (KD ≈ 450 nm). Na+ changes were measured by CoroNa Green. For visualization of axonal profiles, the nerves were coloaded with red Alexa Fluor 594 dextran. The dextran-conjugated dyes were restricted to the axoplasmic compartment and loaded optic nerve fibers to >1 mm from the cut end after 1.5 h, likely by axoplasmic transport (Ren et al., 2000; Verbny et al., 2002; Ouardouz et al., 2003). The red ion-independent fluorescence was used to outline regions of interest (ROIs) from which green Ca2+- or Na+-dependent fluorescence was measured. Because larger axons were more easily distinguishable, ROIs represent mainly larger (>1.5 μm in diameter) fibers. The loading buffer contained a low Na+ concentration [NaCl was replaced by 126 mm of N-methyl-d-glucamine (NMDG)] and a low Ca2+ concentration (CaCl2 was omitted) to mimic intra-axonal concentrations. Dye concentrations in the loading pipette included the following: 150 μm for fluo-4 dextran, 75 μm for Oregon Green 488 BAPTA-1 dextran, 1.4 mm for CoroNa Green, and 100-200 μm Alexa Fluor 594 dextran. Because loading efficiency was estimated to be only ∼1% (Verbny et al., 2002), the final concentration of Ca2+ dyes in the axon was probably only several micromolar. The imaging was performed at a relatively constant distance (∼1.2 mm) from the cut end to minimize the effects of spatial dye gradients. Chemical ischemia was induced by using NaN3 (2 mm), which causes rapid inhibition of mitochondrial complex IV (Petersen, 1977; Malek et al., 2005), and 0 glucose (replaced with 10 mm sucrose) in the perfusate. At the conclusion of every 0 Ca2+ experiment, chemical ischemia was induced in the presence of high Ca2+ (2 mm) CSF to confirm that axonal Ca2+ increased; this control was deemed important to ensure that the absence of a Ca2+ rise as a result of manipulations was real and not a technical failure (e.g., inadequate dye loading, etc.). Minimally, three nerves were studied per treatment group, with approximately three ROIs chosen per nerve and typically six axons per ROI. All fluorescent dyes were purchased from Invitrogen (San Diego, CA). BAPTA (tetrapotassium salt, with KCl concentration adjusted accordingly) was purchased from A.G. Scientific (San Diego, CA). Neomycin, heparin (Sigma, St. Louis, MO), and an oxygen-bridged dinuclear ruthenium amine complex (Ru360; Calbiochem, La Jolla, CA) were dissolved directly into aCSF or loading solution. Next, 1-(6-[(17β-methoxyestra-1,3,5 [10]-trien-17-yl) amino] hexyl)- 1H-pyrrole-2,5-dione ( U73122), ryanodine, and 7-chloro-5-(2-chlorophenyl)-1,5-dihydro-4,1-benzothiazepin-2(3H)-one (CGP37157) (Tocris Bioscience, Ellisville, MO) were dissolved first in DMSO and then added to aCSF. Cyclosporin A (Sigma) was dissolved in ethanol. All drugs were applied 15 min before the onset of 0 Ca2+ perfusate (see below) and continued throughout ischemia.
Statistics. All data are expressed as the means ± SD. Statistical differences were calculated by ANOVA with Bonferroni's correction for multiple comparisons or ANOVA with Dunnett's test for multiple comparisons with a common control group. Student's t test was used for single comparisons between two groups. Reported n values represent the number of individually analyzed axons.
Results
Axonal Ca2+ changes during ischemia in normal and Ca2+-free CSF
To assess the extent of Ca2+ rise during in vitro ischemia in optic axons, we initially used the low-affinity Ca2+ indicator fluo-4 dextran. Images were taken during perfusion in normal aCSF (containing 2 mm Ca2+) to determine the basal level of fluorescence and then during ischemia (0 glucose plus 2 mm NaN3) at 1 min intervals.
Figure 1 shows representative images of optic axons before and after ischemia. Red fluorescence represents Ca2+-insensitive dextran dye loaded to outline axon cylinders from which ROIs were derived for analysis of Ca2+- or Na+-dependent fluorescence. Panels A and B show a substantial increase in green Ca2+ signal after 30 min of chemical ischemia (C and D show the Ca2+ signal in pseudocolor). The middle panels show ischemic axonal Ca2+ rise in 0 Ca2+ perfusate with the use of the higher-affinity Oregon Green 488 BAPTA-1 indicator. The bottom panels show axonal Na+ increase (green signal with CoroNa Green indicator) as a function of ischemia (for more details, see the legend to Fig. 1). The fluorescence changes were normalized to the average basal fluorescence before the application of the ischemic buffer, and the change in fluorescence (F/F0) was plotted against time (Fig. 2). There was a substantial increase in Ca2+-dependent fluorescence, which began after an average lag time of 5-6 min after ischemia was introduced and increased to ∼3.5 times control after 30 min (Fig. 2A).
Figure 1.
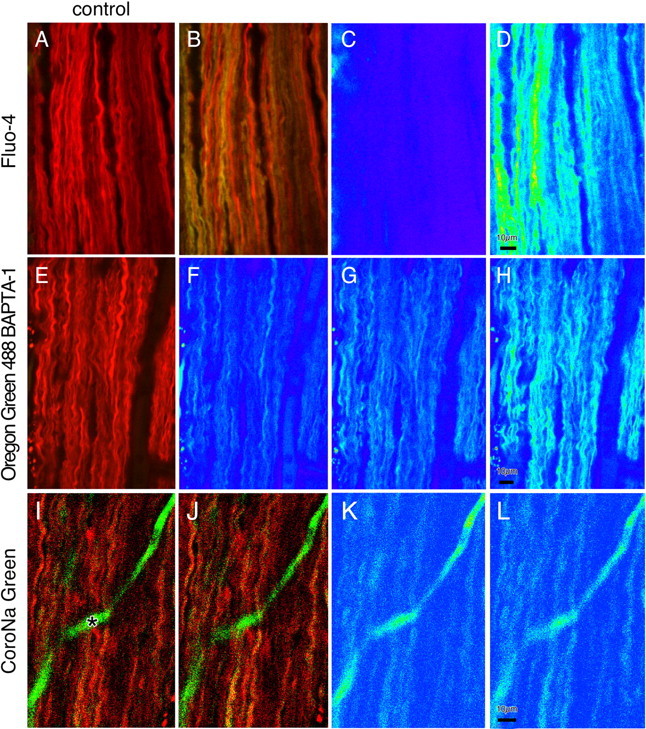
Confocal images of live optic nerve before and during ischemia. Axons were coloaded with red ion-insensitive Alexa Fluor 594 dextran for visualization of axonal profiles and with either low-affinity fluo-4 dextran (KD, 4 μm; top row) to demonstrate larger increases in axonal Ca2+ (A, before ischemia; B, 30 min in ischemic buffer; C, D, Ca2+-sensitive fluorescence in pseudocolor) or high-affinity Oregon Green 488 BAPTA-1 dextran (KD, 500 nm; middle row) to demonstrate contributions of intracellular Ca2+ stores in 0 Ca2+ (plus BAPTA) conditions (E, pre-ischemia image; F, same image in pseudocolor; G, after 7 min in 0 Ca/BAPTA ischemic buffer; H, after 15 min in Ca2+-replete ischemic buffer). The bottom row demonstrates an Na+-sensitive CoroNa Green fluorescence increase in ischemia (I, before ischemia; J, 30 min in ischemic buffer; K, L, Na+-dependent fluorescence in pseudocolor). The solid green structure (asterisk) is a dye-filled capillary.
Figure 2.

Time course of normalized axonal green/red fluorescence ratio showing a 3.5-fold Ca2+-dependent fluorescence increase over baseline after 30 min of ischemia in 2 mm Ca2+ with fluo-4 as the Ca2+ indicator (A). In 0 Ca2+ (plus BAPTA) perfusate, the ischemia evoked a 19% increase with Oregon Green 488 BAPTA-1 dextran as the Ca2+ indicator (B), indicating a release of this ion from intra-axonal compartments. The inset demonstrates a fluorescence decrease in control experiments without ischemia, indicating that perfusion with the 0 Ca2+/BAPTA solution gradually reduced axoplasmic [Ca2+].
This Ca2+ accumulation may originate from two sources: (1) influx of extracellular Ca2+ and (2) release of Ca2+ from intracellular stores. To determine the contribution of the latter and anticipating a smaller [Ca2+] increase when bath Ca2+ was removed, we loaded optic nerves with the higher-affinity Ca2+ indicator Oregon Green 488 BAPTA-1 dextran instead. The nerves were pretreated with 0 Ca2+ (plus 0.75 mm BAPTA) for 5 min to remove any residual external Ca2+ and then perfused in an ischemic 0 Ca2+ (plus BAPTA) buffer. We needed to ensure that for all experiments in 0 Ca2+ perfusate there was minimal extracellular Ca2+ remaining that could contribute to any observed intracellular Ca2+ increases. To simulate the extracellular environment near the surface of optic nerves from which images were acquired, we dissolved Ca2+-sensitive and Ca2+-insensitive dextran dyes in a bead of 2% agar and imaged them 20 μm below the surface (a depth typically used for optic nerve imaging) as the perfusate was switched from normal (2 mm) Ca2+ to 0 Ca2+/EGTA. Ca fluorescence decreased to ∼50% after ∼4 min of perfusion with the 0 Ca2+/EGTA solution. If we assume that at 2 mm the dye was saturated, the half-maximal fluorescence implies that the free [Ca2+] in the agar bead dropped to ≈ KD level, which is 400-500 nm. Therefore, we are confident that our standard paradigm of preapplying 0 Ca2+/BAPTA solution for 5 min before ischemia effectively eliminated virtually all contribution of Ca2+ influx from the extracellular space. We switched to BAPTA as the Ca2+ chelator to avoid the possibility of reduced Ca2+ binding capacity with EGTA in an acidified extracellular environment during ischemia, known to occur in optic nerve (Ransom et al., 1992). Representative confocal images are shown in Figure 1 (middle row). In Ca2+-free conditions, the green fluorescence began to rise within 1-2 min of ischemia onset (Fig. 1G) and reached its maximum (19 ± 6% increase) after ∼7 min. When Ca2+ was reintroduced, the fluorescence rose further, indicating there was an additional dynamic range of the dye, which could report additional Ca2+ increases from influx across the axolemma. It should be noted that in control experiments without ischemia, Ca2+-dependent fluorescence decreased by ∼9% below baseline (Fig. 2B, inset). For this reason, the reported 19% increase in the above experiment and in all subsequent studies performed in 0 Ca2+-BAPTA perfusate probably underestimated the true Ca2+ rise.
Axonal Na+ accumulation during ischemia
To explore the hypothesis that Na+ accumulation may modulate Ca2+ release, we studied the temporal profiles of axonal Na+ changes as a function of the same manipulations that modulated Ca2+ release from internal compartments (see below). Axonal Na+ measurements were performed under identical experimental conditions as Ca2+ measurements except that the axons were loaded with the Na+-sensitive dye CoroNa Green along with red dextran-conjugated Alexa Fluor 594. Unfortunately, there is no commercially available Na+ indicator conjugated to dextran. As a result, in contrast to dextran-conjugated dyes, the intensity of axonal CoroNa Green fluorescence decreased with time, likely attributable to loss of this low-molecular-weight (MW) dye (MW 586 vs 10 kDa dextrans) by export and/or leakage. The rate of CoroNa Green fluorescence decay was determined first in control experiments without ischemia and was found to be approximately linear over the time required for our measurements (∼50 min), decreasing by ∼1.5%/min (Fig. 3A). All experiments therefore included a 20 min baseline from which a linear regression was determined, allowing for extrapolation of expected fluorescence loss for the duration of the study. Raw data (Fig. 3B) thus were adjusted, yielding fluorescence changes approximately corrected for dye loss (Fig. 3C).
Figure 3.

Time course of normalized green/red (Na+-sensitive CoroNa Green/Na+-insensitive) fluorescence ratio. A, Approximately linear decrease in axoplasmic fluorescence in nonischemic conditions (each point represents the mean from ∼30 axons). B, Reversal of the fluorescence decline during chemical ischemia. C, Fluorescence changes adjusted for loss of the Na+-sensitive dye, demonstrating the net rise of axoplasmic [Na+] during ischemia (traces represent the means from 60-90 axons). Activating Na+ channels with veratridine further exacerbated axonal Na+ loading during ischemia, whereas blocking these channels with TTX reduced, but did not abolish completely, axonal Na+ accumulation. Dashed lines indicate baseline fluorescence.
Na+-dependent fluorescence began to increase within 3-4 min of ischemia onset and continued for the 30 min duration of the experiment, reaching 58 ± 10% of control (Fig. 3C). Increasing Na+ channel permeability with veratridine (50 μm), an alkaloid that inhibits voltage-dependent Na+ channel inactivation (Catterall, 1980), accelerated and amplified axonal ischemic Na+ accumulation (95 ± 8%). Conversely, blocking Na+ channels with TTX (1 μm) significantly reduced (25 ± 5 vs 58 ± 10% after 30 min of ischemia), but did not eliminate completely, the axonal Na+ rise, indicating that voltage-gated TTX-sensitive Na+ channels were the major, but not the only, source of axonal Na+ influx during ischemia.
Intracellular Ca2+ release is Na+ dependent
Axonal Na+ accumulation during anoxia/ischemia may drive a number of pathological Ca2+-dependent cascades (e.g., promotion of mitochondrial Ca2+ release via reversal of its Na+/Ca2+ exchanger, neurotransmitter release via reversal of Na+-dependent transporters and potential stimulation of metabotropic receptors leading to Ca2+ release, and promotion of depolarization-induced release from Ca2+ stores). To examine the role of Na+ in mediating Ca2+ release from intraaxonal compartments, we reduced Na+ influx by substituting this cation with impermeable NMDG+ or permeable Li+ or by blocking voltage-gated Na+ channels with 1 μm TTX. Ischemia then was applied in 0 Ca2+/BAPTA perfusate. The replacement of Na+ with NMDG+/choline or blocking voltage-gated Na+ channels with TTX [treatments that both significantly reduce the degree of ischemic depolarization in optic nerve (Malek et al., 2003)] prevented axonal Ca2+ rise almost completely (Fig. 4), with an average fluorescence increase of 1 ± 4 and 4 ± 5% over baseline, respectively. This suggests that Na+ influx and/or membrane depolarizations promote intracellular Ca2+ release in optic nerve axons during ischemia. However, the replacement of bath Na+ with Li+, which permeates Na+ channels (Fontana et al., 1995) and allows for axonal depolarization (Leppanen and Stys, 1997), also abolished ischemic Ca2+ increase (1 ± 4%) (Fig. 4). Moreover, so that axonal depolarization during Na+-depleted conditions could be ensured, perfusion of optic nerve in 40 mm K+ buffer (Na+ replaced with NMDG+ and Cl- partially replaced with impermeable gluconate, maintaining a constant K+ × Cl- product to reduce Donnan-mediated volume changes) did not induce any increase in Ca2+ fluorescence during ischemia in 0 Ca2+/BAPTA perfusate (Fig. 4). Together, these observations suggested that, in the optic nerve, Ca2+ release from intracellular stores was dependent not on ischemic axonal depolarization per se but on the influx of Na+ ions. The role of Na+ entry during 0 Ca2+ ischemia was investigated additionally by using veratridine to promote axonal Na+ accumulation. Application of 50 μm veratridine during 0 Ca2+ ischemia caused a marked increase in axonal Ca2+ fluorescence (50 ± 8%) (Fig. 4), which parallels the exaggerated axonal overload with Na+ (Fig. 3), further supporting the notion that intracellular Ca2+ release is proportional to ischemic Na+ influx.
Figure 4.

Effects of Na+ influx on Ca2+-dependent fluorescence changes during ischemia in 0 Ca2+ (plus BAPTA) perfusate. Combining 0 Ca2+ with 0 Na+ (126 mm of NaCl replaced with the impermeant NMDG+ ion and 26 mm of NaHCO3 with choline bicarbonate) in the perfusate or the application of 1 μm TTX in large part prevented any Ca2+ increase. Preapplication of Li+-substituted 0 Na+ perfusate (126 mm of NaCl replaced with LiCl and 26 mm of NaHCO3 with choline bicarbonate) in 0 Ca2+ (plus BAPTA), which allows axonal depolarization to occur (in contrast to the impermeable NMDG ion), also prevented ischemic Ca2+ rise. Veratridine (which increased Na+ influx) (Fig. 3) markedly increased the ischemic Ca2+ response. Forcing depolarization with 40 mm K+ in the absence of Na+ did not promote ischemic Ca2+ rise, indicating that it is mainly the Na+ influx, and not axonal depolarization, that triggers the release of Ca2+ from intracellular compartments. The dashed line indicates baseline fluorescence.
Inositol trisphosphate and ryanodine receptors and the mitochondrial Na+/Ca2+ exchanger are major sources of intracellular Ca2+ during ischemia
The primary intracellular Ca2+ storage/release organelle in most cells is the ER, possessing two major families of Ca2+ release channels: the inositol trisphosphate (IP3) receptor and ryanodine receptor (RyR) (Berridge, 1998; Rizzuto, 2001). The IP3 receptor is activated by inositol 1,4,5-trisphosphate, which is generated from phosphatidylinositol 4,5-bisphosphate by phospholipase C (PLC). We investigated the role of the IP3 receptor in Ca2+ release by using three agents acting at distinct points in the IP3 signaling pathway: U73122, an irreversible antagonist of PLC (Jin et al., 1994); neomycin, which complexes phosphoinositide lipids to render them unavailable as PLC substrates (Schacht, 1976, 1978); and heparin, which inhibits IP3-induced Ca2+ release by binding to IP3-binding sites (Worley et al., 1987; Supattapone et al., 1988). U73122 and neomycin were bath-applied. Because heparin is a large polysaccharide and is therefore incapable of crossing cell membranes, it was loaded intra-axonally along with the dextran-conjugated dyes. Each experiment was performed in 0 external Ca2+ (plus BAPTA), as explained in Materials and Methods. As shown in Figure 5, each of these three antagonists reduced the peak Ca2+ response by ∼50% compared with drugfree controls (p < 0.01), suggesting that ischemia-induced Ca2+ release is mediated in part by the IP3 signaling pathway. There remained a substantial component of Ca2+ release, however, that was independent of IP3 receptors.
Figure 5.

Bar graph summarizing the effects of ER and mitochondrial Ca2+ release blockers (measured at the point of maximum Ca2+-dependent fluorescence increase, which occurred within 5-10 min after chemical ischemia was applied). Neomycin (300-500 μm), U73122 (20 μm), heparin (85 mg/ml), ryanodine (60 μm), and CGP37157 (20 μm) all significantly suppressed ischemic Ca2+ increases (all in 0 Ca2+ plus BAPTA perfusate; *p < 0.01 compared with drug-free control). Combining U73122 or heparin with CGP37157 reduced the Ca2+ rise even more. White numbers within the bars represent the number of individual axons analyzed for each treatment. Error bars indicate SD.
Another important ER intracellular Ca2+ release channel is the RyR, activated either by Ca2+ itself or by depolarization via coupling with voltage-gated Ca2+ channels (Berridge, 1998; Rizzuto, 2001). At high concentrations (≥50 μm), ryanodine blocks RyRs (Solovyova et al., 2002). In our experiments, the application of ryanodine (60 μm) resulted in a significant decrease in the ischemic Ca2+ response in 0 external Ca2+ perfusate (13 ± 5 vs 19 ± 6% without ryanodine; p < 0.01).
Mitochondria are also capable of storage and release of intracellular Ca2+ (Bernardi, 1999; Rizzuto et al., 1999). Under physiological conditions, Ca2+ efflux from mitochondria is mediated primarily by the mitochondrial Na+/Ca2+ exchanger. In addition, under certain conditions, the Ca2+ uniporter and the permeability transition pore may serve as conduits of Ca2+ release (Gunter et al., 2000; Montero et al., 2001). To determine the specific route through which Ca2+ could exit from mitochondria in the optic nerve, we tested three blockers: CGP37157, an inhibitor of mitochondrial Na+/Ca2+ exchange (Chiesi et al., 1988); Ru360, an inhibitor of the mitochondrial Ca2+ uniporter (Matlib et al., 1998); and cyclosporin A, a permeability transition pore blocker (Broekemeier et al., 1989; Crompton et al., 1999). Pretreatment with CGP37157 reduced the Ca2+ fluorescence increase by ∼50% from 19 ± 6 to 8 ± 4% (p < 0.01). Cyclosporin A and Ru360 did not reduce the peak Ca2+ signal significantly: 15 ± 7 and 16 ± 7%, respectively (p > 0.05). Together, these results indicate that the mitochondrial Na+/Ca2+ exchanger accounts for the majority of mitochondrial Ca2+ release during ischemia.
If IP3 receptors and the mitochondrial Na+/Ca2+ exchanger account for almost one-half of the Ca2+ rise, blocking both should reduce the Ca2+ rise even further if the two pathways are independent. Indeed, a combination of U73122 or heparin with CGP37157 reduced the ischemic axonal Ca2+ increase to 5 ± 4% (p < 0.05; U73122 vs U73122 plus CGP37157; ANOVA with Bonferroni's correction for multiple comparisons) and 6 ± 5% (p < 0.01; heparin vs heparin plus CGP37157), respectively (Fig. 5), confirming that these two sources are the major contributors to intracellular release during ischemia and that they appear to operate independently. To assess whether this restraint of Ca2+ rise has functional implications, we performed electrophysiological CAP recordings on optic nerves treated with U73122 plus CGP37157 (both 20 μm) during a 1 h exposure to in vitro chemical ischemia (95% N2/5% CO2 atmosphere plus glucose replaced with sucrose), followed by reperfusion. Because Ca2+-free bath readily removes Ca2+ from axoplasm in optic nerve (see Discussion) and is itself therefore highly neuroprotective (Stys et al., 1990), this experiment was performed in normal Ca2+-replete CSF. Despite the large source of available extracellular Ca2+, the combination of U73122 plus CGP37157 improved electrophysiological recovery from 5 ± 3% (n = 8 nerves) of control CAP area in ischemia/DMSO to 17 ± 8% (n = 12) in the drug combination (p < 0.001).
Discussion
Most studies indicate that ischemic intracellular Ca2+ overload in neurons is attributable primarily to Ca2+ influx from the extracellular space (Lipton, 1999; Arundine and Tymianski, 2003; Paschen, 2003). Although the role of external Ca2+ is well established, less is known about the sources and release mechanisms of intracellular stores of Ca2+ during ischemia. Nevertheless, intracellular Ca2+ stores are known to contribute to cytosolic Ca2+ rise in ischemic neurons (Mitani et al., 1993; Zhang and Lipton, 1999), and inhibition of this release may be protective (Yano et al., 2001; Wang et al., 2002). Very little is known about such mechanisms in myelinated axons.
In the present study, ischemic Ca2+ accumulation was measured by a low-affinity dye when extracellular Ca2+ was maintained in the physiological range (∼2 mm) to estimate the degree of axonal [Ca2+] rise in comparison with that induced in 0 external Ca2+ solution. In agreement with studies in ischemic neurons (Lipton, 1999; Sattler and Tymianski, 2000; Erecinska and Silver, 2001; Arundine and Tymianski, 2003; Paschen, 2003), we show that ischemic central axons also suffer large increases in axoplasmic Ca2+ concentrations. Although we could not perform absolute Ca2+ measurements, the large increase in fluorescence of the low-affinity fluo-4 dextran indicator suggests that free [Ca2+] increased well into the micromolar range. Under conditions when bath Ca2+ was removed, a higher-affinity indicator was preferred to show the smaller but still substantial ischemic Ca2+ rise originating from internal compartments. Our measurements probably underestimated the contribution of Ca2+ release from internal stores because of potential continuous Ca2+ washout in 0 Ca2+/BAPTA buffer. This decrease might be related to the proximity of portions of the ER to the axolemma (Berridge, 1998; Ouardouz et al., 2003). In fact, some ER cisternas, including those of axoplasmic reticulum, might approach the plasma membrane to within 20 nm (Lindsey and Ellisman, 1985; Berridge, 1998) and may be depleted in the face of a strongly reversed gradient favoring efflux. Measurements of total axoplasmic Ca2+ (which included regions of ER) revealed that exposure of optic nerve to 0 Ca2+ perfusate reduced Ca2+ to undetectable levels (Stys and Lopachin, 1998), suggesting a robust exchange of the majority of axonal Ca2+ with the extracellular space in optic fibers. The 0 Ca2+ perfusion was necessary experimentally to dissect out the two components of Ca2+ accumulation, but clearly such conditions would not apply during normal physiological or pathophysiological signaling. Curiously, dorsal column fibers are able to maintain levels of internally released Ca2+ much more readily than optic fibers under conditions of perfusion with 0 Ca2+ solution (Ouardouz et al., 2003). The reasons for this disparity are unclear but may point to differences in how Ca2+ handling machinery is organized in different fiber tracts.
We demonstrated previously that in spinal cord dorsal columns, intracellular Ca2+ stores caused a robust Ca2+ rise in ischemic axons even in Ca2+-free perfusate (Ouardouz et al., 2003). In this tissue, this Ca2+ release was triggered to a large extent by axonal depolarization and was reduced significantly by ryanodine. The present study suggests that in optic axons the relative contribution of various intracellular Ca2+ stores may be somewhat different, with IP3-dependent stores and mitochondria being the more important sources, whereas ryanodine receptor-dependent stores play a smaller role. Even in dorsal columns, however, ryanodine reduced, but did not abolish, axoplasmic Ca2+ increase caused by ischemia in 0 external Ca2+, suggesting additional parallel sources as in optic nerve. In the present study, CAP recordings indicate that, although extracellular Ca2+ plays a major role as a source of deleterious Ca2+ into ischemic axons, release of this ion from IP3-sensitive stores and mitochondria also significantly contributes to functional injury of optic nerve axons.
Na+ dependence of internal Ca2+ release
Na+ entry is characteristic of ischemic neurons and has been shown to be important in overall cellular injury (Lipton, 1999; Taylor et al., 1999; Erecinska and Silver, 2001; LoPachin et al., 2001; Varadarajan et al., 2001; Tanonaka and Takeo, 2003; Banasiak et al., 2004). This is also true of injured axons, which accumulate Na+ through TTX-sensitive Na+ channels, leading to Ca2+ influx via reverse Na+/Ca2+ exchange (Stys et al., 1992) and voltage-gated Ca2+ channels (Fern et al., 1995).
The ER plays a prominent role in maintaining Ca2+ homeostasis in neurons (Berridge, 2002; Verkhratsky and Toescu, 2003). Alterations in Ca2+ homeostasis in the ER contribute to neuronal apoptosis and excitotoxicity (Mattson, 1989; Frandsen and Schousboe, 1991; Mody and MacDonald, 1995; Mattson et al., 2000). In white matter, the Ca2+ released from ER adds to the injurious effects of Ca2+ influx during traumatic compressive injury (Thorell et al., 2002). The unexpected finding in this study was the almost complete dependence of Ca2+ release from intracellular sources on Na+ influx in ischemic optic axons. Although it is known that Na+i can promote the release of Ca2+ from stores by coupling via the Na+/Ca2+ exchanger, resulting in more Ca2+ entry and enhanced Ca2+-induced Ca2+ release (Han et al., 2002; Pogwizd and Bers, 2002; Ritter et al., 2003), this would not apply in our experiments because of a lack of external Ca2+; therefore, a direct role for Na+ is suggested. The link between Na+ influx and activation of the PLC/IP3 pathway was described almost 20 years ago (Gusovsky et al., 1986). However, it still remains unclear how Na+ influences ER Ca2+ release other than via the plasmalemmal Na+/Ca2+ exchanger as mentioned above.
Our results suggest that there exist three distinct intracellular sources of Ca2+ release in ischemic optic axons: IP3 receptors (IP3Rs), RyRs, and mitochondria, with all three being dependent on Na+ influx. Zhang and Lipton (1999) reported that in ischemic rat hippocampal slices, in addition to influx via NMDA receptors, a substantial proportion of Ca2+ originated from mitochondria, being released via the mitochondrial Na+/Ca2+ exchanger. The present study demonstrates that in the ischemic optic nerve mitochondria account for ∼50% of the Ca2+ rise from intracellular sources. In agreement with Zhang and Lipton (1999), we also found that the increase is Na+ dependent and is reduced significantly by the application of CGP37157, a blocker of the mitochondrial Na+/Ca2+ exchanger (Chiesi et al., 1988) that mediates most of the efflux of Ca2+ from mitochondria in exchange for the influx of Na+ (Rizzuto, 2003). Blocking the Ca2+ uniporter or the permeability transition pore resulted in a statistically insignificant reduction in ischemic Ca2+ release; whether this indicates that these two pathways truly do not contribute to Ca2+ release from mitochondria in our paradigm or whether our sampling was insufficient to demonstrate a significant effect is unknown and would require additional studies.
Na+-dependent mitochondrial Ca2+ release during ischemia could contribute significantly to free axoplasmic Ca2+ because resting mitochondria contain substantial amounts of this cation (Stys et al., 1997; Rizzuto et al., 1999; Yang et al., 2003), mostly in a complex with phosphate, which nevertheless readily can absorb and release Ca2+ ions (David, 1999; Yang et al., 2003). Moreover, release of mitochondrial Ca2+ also can be damaging because of compromised ATP synthesis; depletion of mitochondrial Ca2+ may suppress Ca2+-sensitive matrix dehydrogenases that catalyze key Krebs' cycle reactions (McCormack et al., 1990; Bernardi, 1999). It is curious that previous reports on anoxic axonal mitochondria indicated that these organelles accumulate Ca2+ during injury (LoPachin and Stys, 1995), yet the present results suggest the opposite. It is possible that during anoxia, these organelles are able to maintain some electrochemical potential across their inner membrane, likely by hydrolysis of glycolytically derived ATP (Nicholls and Budd, 2000). The residual polarization could be sufficient to support a modest Ca2+ accumulation into the matrix, whereas in the face of a profound energy deficit during ischemia the mitochondria would be depolarized more strongly and would thus be driven to release matrix Ca2+.
The Na+ dependence of IP3R- and RyR-mediated Ca release is more puzzling. The effects of this ion may be direct on these receptors or indirect, acting at upstream signaling points. In the case of IP3Rs, there is no convincing evidence of a direct effect of Na+ ions. Instead, we favor the indirect explanation of ischemic Na+ influx promoting neurotransmitter release, which may in turn stimulate metabotropic receptors, activate PLC, and generate IP3. Several observations support this scenario. We had shown that anoxia causes glutamate release from central axons via reverse Na+-dependent glutamate transport (Li et al., 1999). We also have evidence for the involvement of group I metabotropic glutamate receptors in mediating Ca2+ overload in ischemic spinal axons (Stys and Ouardouz, 2002). Moreover, several neurotransmitter uptake mechanisms are Na+ dependent, and their metabotropic receptors are coupled to PLC (e.g., serotonin, dopamine, norepinephrine); therefore, glutamate may be only one of several neurotransmitters released from ischemic axons that then stimulate metabotropic receptors to generate IP3. Finally, if Na+ exerted a direct effect on IP3Rs, PLC inhibition would not reduce ischemic Ca2+ release, yet both U73122 and neomycin reduced the Ca2+ response. In the case of RyRs, there is evidence of direct positive modulation by Na+ ions, which increases the release of Ca2+ from the sarcoplasmic reticulum in skeletal muscle even in the absence of Na+/Ca2+ exchange activity (Allard and Rougier, 1992; Hu et al., 2003).
Conclusion
During optic nerve ischemia, axonal intracellular Ca2+ stores release Ca2+ in a Na+-dependent manner via IP3Rs and RyRs from the ER and via the mitochondrial Na+/Ca2+ exchanger (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). This additional contribution to axoplasmic Ca2+ increase further exacerbates the deleterious effects of Ca2+ overload originating from the extracellular space in the genesis of ischemic and traumatic injury to central myelinated axons.
Footnotes
This work was supported by operating grants from the National Multiple Sclerosis Society and Canadian Stroke Network and an equipment grant from the Canadian Institutes of Health Research (MMA-48299). P.K.S. is supported by a Career Investigator Award from the Heart and Stroke Foundation of Ontario.
Correspondence should be addressed to Dr. Peter K. Stys, Division of Neuroscience, Ottawa Health Research Institute, 725 Parkdale Avenue, Ottawa, Ontario, Canada K1Y 4K9. E-mail: pstys@ohri.ca.
Copyright © 2005 Society for Neuroscience 0270-6474/05/259960-08$15.00/0
References
- Allard B, Rougier O (1992) Reappraisal of the role of sodium ions in excitation-contraction coupling in frog twitch muscle. J Muscle Res Cell Motil 13: 117-125. [DOI] [PubMed] [Google Scholar]
- Arundine M, Tymianski M (2003) Molecular mechanisms of calcium-dependent neurodegeneration in excitotoxicity. Cell Calcium 34: 325-337. [DOI] [PubMed] [Google Scholar]
- Banasiak KJ, Burenkova O, Haddad GG (2004) Activation of voltage-sensitive sodium channels during oxygen deprivation leads to apoptotic neuronal death. Neuroscience 126: 31-44. [DOI] [PubMed] [Google Scholar]
- Bernardi P (1999) Mitochondrial transport of cations: channels, exchangers, and permeability transition. Physiol Rev 79: 1127-1155. [DOI] [PubMed] [Google Scholar]
- Berridge MJ (1998) Neuronal calcium signaling. Neuron 21: 13-26. [DOI] [PubMed] [Google Scholar]
- Berridge MJ (2002) The endoplasmic reticulum: a multifunctional signaling organelle. Cell Calcium 32: 235-249. [DOI] [PubMed] [Google Scholar]
- Broekemeier KM, Dempsey ME, Pfeiffer DR (1989) Cyclosporin A is a potent inhibitor of the inner membrane permeability transition in liver mitochondria. J Biol Chem 264: 7826-7830. [PubMed] [Google Scholar]
- Catterall WA (1980) Neurotoxins that act on voltage-sensitive sodium channels in excitable membranes. Annu Rev Pharmacol Toxicol 20: 15-43. [DOI] [PubMed] [Google Scholar]
- Chiesi M, Schwaller R, Eichenberger K (1988) Structural dependency of the inhibitory action of benzodiazepines and related compounds on the mitochondrial Na+-Ca2+ exchanger. Biochem Pharmacol 37: 4399-4403. [DOI] [PubMed] [Google Scholar]
- Crompton M, Virji S, Doyle V, Johnson N, Ward JM (1999) The mitochondrial permeability transition pore. Biochem Soc Symp 66: 167-179. [DOI] [PubMed] [Google Scholar]
- David G (1999) Mitochondrial clearance of cytosolic Ca2+ in stimulated lizard motor nerve terminals proceeds without progressive elevation of mitochondrial matrix [Ca2+]. J Neurosci 19: 7495-7506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erecinska M, Silver IA (2001) Tissue oxygen tension and brain sensitivity to hypoxia. Respir Physiol 128: 263-276. [DOI] [PubMed] [Google Scholar]
- Fern R, Ransom BR, Waxman SG (1995) Voltage-gated calcium channels in CNS white matter: role in anoxic injury. J Neurophysiol 74: 369-377. [DOI] [PubMed] [Google Scholar]
- Fontana G, Rogowski RS, Blaustein MP (1995) Kinetic properties of the sodiumcalcium exchanger in rat brain synaptosomes. J Physiol (Lond) 485: 349-364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frandsen A, Schousboe A (1991) Dantrolene prevents glutamate cytotoxicity and Ca2+ release from intracellular stores in cultured cerebral cortical neurons. J Neurochem 56: 1075-1078. [DOI] [PubMed] [Google Scholar]
- Gunter TE, Buntinas L, Sparagna G, Eliseev R, Gunter K (2000) Mitochondrial calcium transport: mechanisms and functions. Cell Calcium 28: 285-296. [DOI] [PubMed] [Google Scholar]
- Gusovsky F, Hollingsworth EB, Daly JW (1986) Regulation of phosphatidylinositol turnover in brain synaptoneurosomes: stimulatory effects of agents that enhance influx of sodium ions. Proc Natl Acad Sci USA 83: 3003-3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han C, Tavi P, Weckstrom M (2002) Role of the Na+-Ca2+ exchanger as an alternative trigger of CICR in mammalian cardiac myocytes. Biophys J 82: 1483-1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu XF, Chen KY, Xia R, Xu YH, Sun JL, Hu J, Zhu PH (2003) Modulation of the interactions of isolated ryanodine receptors of rabbit skeletal muscle by Na+ and K+. Biochemistry 42: 5515-5521. [DOI] [PubMed] [Google Scholar]
- Jiang Q, Malek S, Ouardouz M, Stys PK (2002) Differential response of rat optic nerve and spinal cord dorsal column to in vitro ischemia. Soc Neurosci Abstr 28: 299.5. [Google Scholar]
- Jin W, Lo TM, Loh HH, Thayer SA (1994) U73122 inhibits phospholipase C-dependent calcium mobilization in neuronal cells. Brain Res 642: 237-243. [DOI] [PubMed] [Google Scholar]
- Kostyuk PG, Shmigol AV, Voitenko NV, Svichar NV, Kostyuk EP (2000) The endoplasmic reticulum and mitochondria as elements of the mechanism of intracellular signaling in the nerve cell. Neurosci Behav Physiol 30: 15-18. [DOI] [PubMed] [Google Scholar]
- Kristian T, Siesjo BK (1998) Calcium in ischemic cell death. Stroke 29: 705-718. [DOI] [PubMed] [Google Scholar]
- Leppanen L, Stys PK (1997) Ion transport and membrane potential in CNS myelinated axons. I. Normoxic conditions. J Neurophysiol 78: 2086-2094. [DOI] [PubMed] [Google Scholar]
- Li S, Mealing GA, Morley P, Stys PK (1999) Novel injury mechanism in anoxia and trauma of spinal cord white matter: glutamate release via reverse Na+-dependent glutamate transport. J Neurosci 19: RC16(1-9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsey JD, Ellisman MH (1985) The neuronal endomembrane system. III. The origins of the axoplasmic reticulum and discrete axonal cisternae at the axon hillock. J Neurosci 5: 3135-3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton P (1999) Ischemic cell death in brain neurons. Physiol Rev 79: 1431-1568. [DOI] [PubMed] [Google Scholar]
- LoPachin Jr RM, Stys PK (1995) Elemental composition and water content of rat optic nerve myelinated axons and glial cells: effects of in vitro anoxia and reoxygenation. J Neurosci 15: 6735-6746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LoPachin RM, Gaughan CL, Lehning EJ, Weber ML, Taylor CP (2001) Effects of ion channel blockade on the distribution of Na, K, Ca, and other elements in oxygenglucose deprived CA1 hippocampal neurons. Neuroscience 103: 971-983. [DOI] [PubMed] [Google Scholar]
- Malek SA, Coderre E, Stys PK (2003) Aberrant chloride transport contributes to anoxic/ischemic white matter injury. J Neurosci 23: 3826-3836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malek SA, Adorante JS, Stys PK (2005) Differential effects of Na-K-ATPase pump inhibition, chemical anoxia, and glycolytic blockade on membrane potential of rat optic nerve. Brain Res 1037: 171-179. [DOI] [PubMed] [Google Scholar]
- Matlib MA, Zhou Z, Knight S, Ahmed S, Choi KM, Krause-Bauer J, Phillips R, Altschuld R, Katsube Y, Sperelakis N, Bers DM (1998) Oxygen-bridged dinuclear ruthenium amine complex specifically inhibits Ca2+ uptake into mitochondria in vitro and in situ in single cardiac myocytes. J Biol Chem 273: 10223-10231. [DOI] [PubMed] [Google Scholar]
- Mattson MP (1989) Acetylcholine potentiates glutamate-induced neurodegeneration in cultured hippocampal neurons. Brain Res 497: 402-406. [DOI] [PubMed] [Google Scholar]
- Mattson MP, LaFerla FM, Chan SL, Leissring MA, Shepel PN, Geiger JD (2000) Calcium signaling in the ER: its role in neuronal plasticity and neurodegenerative disorders. Trends Neurosci 23: 222-229. [DOI] [PubMed] [Google Scholar]
- McCormack JG, Halestrap AP, Denton RM (1990) Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol Rev 70: 391-425. [DOI] [PubMed] [Google Scholar]
- Mitani A, Yanase H, Sakai K, Wake Y, Kataoka K (1993) Origin of intracellular Ca2+ elevation induced by in vitro ischemia-like condition in hippocampal slices. Brain Res 601: 103-110. [DOI] [PubMed] [Google Scholar]
- Mody I, MacDonald JF (1995) NMDA receptor-dependent excitotoxicity: the role of intracellular Ca2+ release. Trends Pharmacol Sci 16: 356-359. [DOI] [PubMed] [Google Scholar]
- Montero M, Alonso MT, Albillos A, Garcia-Sancho J, Alvarez J (2001) Mitochondrial Ca2+-induced Ca2+ release mediated by the Ca2+ uniporter. Mol Biol Cell 12: 63-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls DG, Budd SL (2000) Mitochondria and neuronal survival. Physiol Rev 80: 315-360. [DOI] [PubMed] [Google Scholar]
- Ouardouz M, Nikolaeva MA, Coderre E, Zamponi GW, McRory JE, Trapp BD, Yin X, Wang W, Woulfe J, Stys PK (2003) Depolarization-induced Ca2+ release in ischemic spinal cord white matter involves L-type Ca2+ channel activation of ryanodine receptors. Neuron 40: 53-63. [DOI] [PubMed] [Google Scholar]
- Paschen W (2003) Mechanisms of neuronal cell death: diverse roles of calcium in the various subcellular compartments. Cell Calcium 34: 305-310. [DOI] [PubMed] [Google Scholar]
- Petersen LC (1977) The effect of inhibitors on the oxygen kinetics of cytochrome c oxidase. Biochim Biophys Acta 460: 299-307. [DOI] [PubMed] [Google Scholar]
- Pogwizd SM, Bers DM (2002) Na/Ca exchange in heart failure: contractile dysfunction and arrhythmogenesis. Ann NY Acad Sci 976: 454-465. [DOI] [PubMed] [Google Scholar]
- Ransom BR, Walz W, Davis PK, Carlini WG (1992) Anoxia-induced changes in extracellular K+ and pH in mammalian central white matter. J Cereb Blood Flow Metab 12: 593-602. [DOI] [PubMed] [Google Scholar]
- Ren Y, Ridsdale A, Coderre E, Stys PK (2000) Calcium imaging in live rat optic nerve myelinated axons in vitro using confocal laser microscopy. J Neurosci Methods 102: 165-176. [DOI] [PubMed] [Google Scholar]
- Ritter M, Sui Z, Philipson KD, Li F, Spitzer KW, Ishida H, Barry WH (2003) Ca2+ sparks induced by Na/Ca exchange. Cell Calcium 34: 11-17. [DOI] [PubMed] [Google Scholar]
- Rizzuto R (2001) Intracellular Ca2+ pools in neuronal signaling. Curr Opin Neurobiol 11: 306-311. [DOI] [PubMed] [Google Scholar]
- Rizzuto R (2003) Calcium mobilization from mitochondria in synaptic transmitter release. J Cell Biol 163: 441-443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzuto R, Pinton P, Brini M, Chiesa A, Filippin L, Pozzan T (1999) Mitochondria as biosensors of calcium microdomains. Cell Calcium 26: 193-199. [DOI] [PubMed] [Google Scholar]
- Sattler R, Tymianski M (2000) Molecular mechanisms of calcium-dependent excitotoxicity. J Mol Med 78: 3-13. [DOI] [PubMed] [Google Scholar]
- Schacht J (1976) Inhibition by neomycin of polyphosphoinositide turnover in subcellular fractions of guinea-pig cerebral cortex in vitro. J Neurochem 27: 1119-1124. [DOI] [PubMed] [Google Scholar]
- Schacht J (1978) Purification of polyphosphoinositides by chromatography on immobilized neomycin. J Lipid Res 19: 1063-1067. [PubMed] [Google Scholar]
- Schanne FA, Kane AB, Young EE, Farber JL (1979) Calcium dependence of toxic cell death: a final common pathway. Science 206: 700-702. [DOI] [PubMed] [Google Scholar]
- Solovyova N, Veselovsky N, Toescu EC, Verkhratsky A (2002) Ca2+ dynamics in the lumen of the endoplasmic reticulum in sensory neurons: direct visualization of Ca2+-induced Ca2+ release triggered by physiological Ca2+ entry. EMBO J 21: 622-630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stys PK (2004) White matter injury mechanisms. Curr Mol Med 4: 109-126. [DOI] [PubMed] [Google Scholar]
- Stys PK, Lopachin RM (1998) Mechanisms of calcium and sodium fluxes in anoxic myelinated central nervous system axons. Neuroscience 82: 21-32. [DOI] [PubMed] [Google Scholar]
- Stys PK, Ouardouz M (2002) Role of glutamate receptors in spinal cord dorsal column ischemia. Soc Neurosci Abstr 28: 298.8. [Google Scholar]
- Stys PK, Ransom BR, Waxman SG, Davis PK (1990) Role of extracellular calcium in anoxic injury of mammalian central white matter. Proc Natl Acad Sci USA 87: 4212-4216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stys PK, Ransom BR, Waxman SG (1991) Compound action potential of nerve recorded by suction electrode: a theoretical and experimental analysis. Brain Res 546: 18-32. [DOI] [PubMed] [Google Scholar]
- Stys PK, Waxman SG, Ransom BR (1992) Ionic mechanisms of anoxic injury in mammalian CNS white matter: role of Na+ channels and Na+-Ca2+ exchanger. J Neurosci 12: 430-439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stys PK, Lehning E, Saubermann AJ, LoPachin Jr RM (1997) Intracellular concentrations of major ions in rat myelinated axons and glia: calculations based on electron probe x-ray microanalyses. J Neurochem 68: 1920-1928. [DOI] [PubMed] [Google Scholar]
- Supattapone S, Worley PF, Baraban JM, Snyder SH (1988) Solubilization, purification, and characterization of an inositol trisphosphate receptor. J Biol Chem 263: 1530-1534. [PubMed] [Google Scholar]
- Tanonaka K, Takeo S (2003) Na+ overload-induced mitochondrial dysfunction in myocardial ischemia/reperfusion injury (in Japanese). Nippon Yakurigaku Zasshi 121: 339-348. [DOI] [PubMed] [Google Scholar]
- Taylor CP, Weber ML, Gaughan CL, Lehning EJ, LoPachin RM (1999) Oxygen/glucose deprivation in hippocampal slices: altered intraneuronal elemental composition predicts structural and functional damage. J Neurosci 19: 619-629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tekkok SB, Brown AM, Ransom BR (2003) Axon function persists during anoxia in mammalian white matter. J Cereb Blood Flow Metab 23: 1340-1347. [DOI] [PubMed] [Google Scholar]
- Thorell WE, Leibrock LG, Agrawal SK (2002) Role of RyRs and IP3 receptors after traumatic injury to spinal cord white matter. J Neurotrauma 19: 335-342. [DOI] [PubMed] [Google Scholar]
- Varadarajan SG, An J, Novalija E, Smart SC, Stowe DF (2001) Changes in [Na+]i, compartmental [Ca2+], and NADH with dysfunction after global ischemia in intact hearts. Am J Physiol Heart Circ Physiol 280: H280-H293. [DOI] [PubMed] [Google Scholar]
- Verbny Y, Zhang CL, Chiu SY (2002) Coupling of calcium homeostasis to axonal sodium in axons of mouse optic nerve. J Neurophysiol 88: 802-816. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A, Toescu EC (2003) Endoplasmic reticulum Ca2+ homeostasis and neuronal death. J Cell Mol Med 7: 351-361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Nguyen HN, Maguire JL, Perry DC (2002) Role of intracellular calcium stores in cell death from oxygenglucose deprivation in a neuronal cell line. J Cereb Blood Flow Metab 22: 206-214. [DOI] [PubMed] [Google Scholar]
- Worley PF, Baraban JM, Supattapone S, Wilson VS, Snyder SH (1987) Characterization of inositol trisphosphate receptor binding in brain. Regulation by pH and calcium. J Biol Chem 262: 12132-12136. [PubMed] [Google Scholar]
- Yang F, He XP, Russell J, Lu B (2003) Ca2+ influx-independent synaptic potentiation mediated by mitochondrial Na+-Ca2+ exchanger and protein kinase C. J Cell Biol 163: 511-523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano T, Nakayama R, Imaizumi T, Terasaki H, Ushijima K (2001) Dantrolene ameliorates delayed cell death and concomitant DNA fragmentation in the rat hippocampal CA1 neurons subjected to mild ischemia. Resuscitation 50: 117-125. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Lipton P (1999) Cytosolic Ca2+ changes during in vitro ischemia in rat hippocampal slices: major roles for glutamate and Na+-dependent Ca2+ release from mitochondria. J Neurosci 19: 3307-3315. [DOI] [PMC free article] [PubMed] [Google Scholar]