

Abstract
In vertebrates, synaptic activity exerts an important influence on the formation of neural circuits, yet our understanding of its role in directing presynaptic and postsynaptic differentiation during synaptogenesis is incomplete. This study investigates how activity influences synaptic differentiation as synapses mature during early postnatal life. Specifically, we ask what happens to presynaptic terminals when synapses develop without functional postsynaptic receptors and without fast synaptic transmission.
To address this issue, we investigated cholinergic nicotinic synapses in sympathetic ganglia of mice with a null mutation for the α3 nicotinic ACh receptor gene. Disrupting the α3 gene completely eliminates fast excitatory synaptic potentials on postganglionic sympathetic neurons, establishing a crucial role for α3-containing postsynaptic receptors in synaptic transmission. Interestingly, the preganglionic nerve terminals form morphologically normal synapses with sympathetic neurons, and these synapses persist without activity in postnatal animals. Surprisingly, when stimulating the preganglionic nerve at physiological rates, we discovered a significant decrease in ACh output from the presynaptic terminals in these α3–/– sympathetic ganglia. We show that this decrease in ACh output from the presynaptic terminals results, in part, from a lack of functional high-affinity choline transporters. We conclude the following: (1) fast synaptic transmission in mammalian SCG requires α3 expression; (2) in the absence of activity, the preganglionic nerve forms synapses that appear morphologically normal and persist for several weeks; and (3) to sustain transmitter release, developing presynaptic terminals require an activity-dependent retrograde signal.
Keywords: nicotinic receptors, cholinergic synaptic transmission, high-affinity choline transporter, muscarinic receptors, autonomic nervous system, synapse formation
Introduction
Synaptogenesis is directed by both activity-independent and activity-dependent mechanisms (Sanes and Lichtman, 1999; Tao and Poo, 2001; Cohen-Cory, 2002). Strong support for this view comes from recent studies on embryonic mice that lack proteins essential for transmitter release (Verhage et al., 2000; Misgeld et al., 2002; Washbourne et al., 2002). However, because such embryos die at birth, it has been difficult to determine the longerterm consequences of the absence of activity during synapse formation and differentiation in postnatal animals.
To address this issue, we investigate the development of synapses in sympathetic ganglia of mice because sympathetic activity is not essential for embryonic development (Crowley et al., 1994; Smeyne et al., 1994; Fagan et al., 1996). One can, therefore, investigate postnatal animals carrying mutations that disrupt ganglionic synaptic transmission and determine how synapses differentiate and mature in these ganglia without synaptic activity. Specifically, we ask the following: (1) whether the presynaptic terminals establish morphological synapses when postsynaptic receptor function is disrupted; and (2) as synapses form, whether the absence of postsynaptic activity in sympathetic neurons affects the functional properties of the presynaptic terminals.
In vertebrates, neuronal nicotinic ACh receptors (nAChRs) mediate fast excitatory synaptic transmission between cholinergic preganglionic axons and postsynaptic sympathetic neurons (Role, 1992; Sargent, 1993; McGehee and Role, 1995). Five nAChR genes are expressed by rodent sympathetic neurons: α3, α5, α7, β2, and β4 (Mandelzys et al., 1994; De Koninck and Cooper, 1995); however, the precise subunit composition of the postsynaptic receptors is unknown. In avian autonomic ganglia, both α3-containing and α7-containing receptors contribute to fast synaptic transmission (Zhang et al., 1996; Ullian et al., 1997; Chang and Berg, 1999); however, the role for α7-containing receptors in synaptic transmission in mammalian sympathetic ganglia is less well understood. We have shown previously that the developmental increase in ACh-evoked current densities on sympathetic neurons correlates well with the increase in α3 mRNA expression (Mandelzys et al., 1994; De Koninck and Cooper, 1995). Moreover, in ganglia, α3-containing receptors are located postsynaptically (Loring et al., 1988), and preganglionic neurons to the superior cervical ganglia (SCG) do not express α3 (Wada et al., 1989). Based on these observations, we reasoned that deleting the α3 gene (Xu et al., 1999) would seriously disrupt postsynaptic activity in the SCG without altering presynaptic function.
Here we show that, without α3 expression, fast synaptic transmission in sympathetic ganglia is completely absent. Remarkably, the preganglionic nerve does not require fast nicotinic transmission to establish synapses with postganglionic sympathetic neurons, and these electrophysiologically disrupted synapses persist well into postnatal life. However, we show that the output of transmitter from the preganglionic terminals is significantly impaired, in part, because these preganglionic terminals lack functional high-affinity choline transporters. Our results indicate that fast synaptic transmission in SCG requires α3 expression and that the functional maturation of presynaptic terminals in SCG requires an activity-dependent retrograde signal.
Materials and Methods
Mice. A colony of α3 neuronal nAChR-deficient mice was maintained by breeding heterozygous animals and genotyping pups by PCR (Xu et al., 1999). The survival rate of homozygote α3–/– pups in our colony was similar to that reported by Xu et al. (1999): ∼20% died within the first few postnatal days, and >60% survived until postnatal day 10 (P10) to P12, with many surviving at least 3 weeks. Most experiments were done with SCG from neonatal (P7–P10) α3–/– pups and wild-type (WT) littermates. In a few experiments, we used ganglia from P21 α3–/– and WT animals.
Neuron cultures. We cultured SCG neurons from P4–P8 mice as described previously for SCG from neonatal rats (McFarlane and Cooper, 1992). Briefly, ganglia were dissociated for 30 min at 37°C in trypsin (180–200 U/ml; Worthington, Freehold, NJ) dissolved in HBSS buffered with HEPES and adjusted to pH 7.4 with NaOH, washed with HBSS, and plated on laminin-coated coverslips in growth media. The growth media consists of L-15 media supplemented with vitamins, cofactors, penicillin–streptomycin, 5% rat serum, and NGF (25–50 ng/ml). Cultures were fed every 3 d and treated with cytosine arabinoside (10 μm; Sigma, St. Louis, MO) from days 2 to 4 to eliminate non-neuronal cells.
Extracellular recordings. Ganglia were dissected rapidly from neonatal pups, pinned down in a recording chamber (7 ml volume), and perfused continuously at 10 ml/min with oxygenated Ringer's solution at 22°C. The preganglionic nerve in the cervical sympathetic trunk was connected to an S88 stimulator and SIU5 stimulus isolation unit (Grass Instruments, Quincy, MA) with a suction electrode. The postganglionic trunk was connected to an alternating current differential amplifier (DP-301; Warner Instruments, Hamden, CT) with a suction electrode; the postganglionic compound action potentials were amplified, filtered at 100 Hz (low-pass cutoff) and 1 kHz (high-pass cutoff), digitized at 44 kHz by a pulse code modulation unit (PCM701; Sony, Tokyo, Japan), and stored on a videocassette recorder (Sony). The data were transferred to a Pentium II-based personal computer with Patchkit (Alembic Software, Montreal, Quebec, Canada) and analyzed off-line with Igor (WaveMetrics, Lake Oswego, OR). All drugs were added directly to the oxygenated Ringer's solution.
Intracellular recording. For intracellular recordings, neonatal ganglia were placed in a small recording chamber (1.5 ml volume) perfused continuously at 3–4 ml/min with oxygenated Ringer's solution at 36–37°C; the chamber was mounted on a fixed stage of dissecting microscope (SMZ-10; Nikon, Tokyo, Japan), and the ganglia were held down with minutia pins. The cervical sympathetic trunk was connected to a stimulator (4710 ORTEC dual channel; EG&G, Gaithersburg, MD) with a suction electrode, and the preparation was left undisturbed for 2–3 h while being continuously perfused with oxygenated Ringer's solution at 36–37°C. To record from ganglion cells, we used 70–120 MΩ glass microelectrodes (G150F-4; Warner Instruments) made with a DMZ universal puller (Zeitz Instruments, Munich, Germany). Stable intracellular recordings were achieved with a high inertial precision microdrive (Inchworm 8200; EXFO, Vanier, Quebec, Canada) attached to a micromanipulator (SM11; Narshige, Tokyo, Japan) that drove the electrode through the ganglion. The recording electrode was filled with 2 m KAc and connected by a thin silver chlorided wire to the head stage of an Axoclamp 2A amplifier (Axon Instruments, Union City, CA) used in current-clamp mode; depolarizing or hyperpolarizing constant current pulses were applied through the recording electrode. Membrane potentials were sampled, displayed, and stored on a Pentium II-based personal computer. Stimulation and acquisition was done with Patchkit software (Alembic Software), and the data were analyzed off-line with IGOR. The preganglionic nerve was stimulated with brief (0.1–0.3 ms) voltage pulses applied to the cervical sympathetic trunk through the suction electrode. All drugs were dissolved in oxygenated Ringer's solution: for long-term (>1 h) applications, drugs were added directly to the perfusion; for shorter (seconds to minutes) applications, drugs were added directly to the bath through a separate perfusion line. Only neurons with membrane potentials greater than –40 mV were included in this study.
For the experiments with hemicholinium (HC-3), first we stimulated the preganglionic nerve at 20 Hz for 2 s in the presence of neostigmine (10 μm) and hexamethonium (100 μm) and recorded the muscarinic response from SCG neurons intracellularly. Next, we added HC-3 (10 μm), waited ∼15 min, and then stimulated the preganglionic nerve at 20 Hz for 2 s at 1 min intervals. The first response after 15 min in HC-3 was comparable to the responses before adding HC-3; the subsequent responses in HC-3 decayed rapidly (see Results). For experiments with low-Ca2+, high-Mg2+ Ringer's solution, we lowered Ca2+ to 0.5 mm and increased Mg2+ to 5 mm.
Whole-cell recording. Whole-cell patch-clamp recordings were made with VE-2 amplifier (Alembic Software) at room temperature. The patch electrodes had resistances of 2–5 MΩ. Membrane currents were filtered at 1.5 kHz with a four-pole Bessel filter, sampled at 2.5–5 kHz, displayed, and stored on-line. Patchkit software was used for stimulation and data acquisition. The patch electrodes were filled with the following (in mm): 65 KF, 55 KAc, 5 NaCl, 0.2 CaCl2, 1 MgCl2, 10 EGTA, and 10 HEPES, pH adjusted to 7.3 with KOH. The neurons were perfused continuously throughout the recording at 1 ml/min with perfusion media consisting of the following: 140 mm NaCl, 5.4 mm KCl, 0.33 mm NaH2PO4, 0.44 mm KH2PO4, 2.8 mm CaCl2, 0.18 mm MgCl2, 10 mm HEPES, 5.6 mm glucose, 2 mm glutamine, 5 μg/ml phenol red, 2 μm tetrodotoxin, and 1–2 μm atropine, pH adjusted to 7.4 with NaOH. ACh was dissolved in perfusion media and applied by pressure ejection from pipettes with tip diameters of 5–10 μm positioned 20–30 μm from the soma (Mandelzys et al., 1994).
Calcium imaging. Ratiometric fluorescence imaging with fura-2 AM (Molecular Probes, Eugene, OR) was used to measure ACh-induced changes in intracellular free Ca2+ concentration. Neurons were incubated at 37°C for 30 min to 1 h with L-15 CO2 (Sigma) containing fura-2 AM (10 μm), washed once with perfusion medium (see above, Whole-cell recording), and left for 20–30 min at room temperature. The cultures were then placed on the stage of an inverted microscope (Axiovert 200 M; Zeiss, Oberkochen, Germany) and viewed through a 40×, 1.3 numerical aperture Plan Neofluor oil-immersion objective (Zeiss). The cells were continuously perfused throughout the recording at 1 ml/min with perfusion media at 37°C. ACh (100 μm) was dissolved in perfusion media and applied to neurons by pressure ejection from pipettes with tip diameters of 5–10 μm positioned 20–30 μm from the soma. As a control, we applied 40 mm K+, which produced a clear increase in intracellular Ca2+ from both WT and α3–/– neurons. Fluorescent images were obtained by exciting the cultures with either 340 or 380 nm wavelength for 100–200 ms from a 150 W xenon arc lamp (LAMBDA DG-4; Sutter Instruments, Novato, CA) and collecting 510 nm emissions with a cooled CCD camera (CoolSnap HQ; PhotoMetrics, Tucson, AZ), all controlled by Metafluor software (Universal Imaging Corporation, West Chester, PA). Images were acquired with Metafluor at 1/s during the ACh application and 0.2/s between applications and analyzed off-line with Metafluor and Igor. The 340/380 ratios were converted to calcium concentration using a fura-2 calcium calibration kit (Molecular Probes).
Adenovirus. Full-length α3 neuronal nAChR subunit cDNA was ligated into pAdTrack-cytomegalovirus (CMV), and replication-deficient viral vectors were made according to He et al. (1998). All viruses were titered in duplicate with the cytopathic effect method (Nyberg-Hoffman et al., 1997). SCG cultures were infected at day 2 with 50 infectious particles/μl overnight, rinsed twice with virus-free medium, and stimulated with 40 mm K+ for 12–24 h to obtain high expression from the CMV promoter (Wheeler and Cooper, 2001).
Ultrastructural studies. Ganglia were place directly into 2% paraformaldehyde–2% glutaraldehyde in phosphate buffer (PB) 0.1 m at room temperature on a shaker for 30 min and then cut in half and fixed for an additional 60–90 min. Next, the tissue was rinsed three times, 10 min each, in 0.1 m PB at room temperature and postfixed in 1% OsO4 plus 1.5% potassium ferricyanide in H2O on a shaker for 1 h at room temperature. The tissue was rinsed briefly with distilled H20, dehydrated in graded series of ethanol concentrations up to 100%, placed in 100% propylene oxide for 10–15 min for two times, and then in a 1:1 mixture of propylene oxide and Embed812 on a shaker for 1 h, in a 1:2 mixture on a shaker for 2 h, and then 100% Embed812 on a shaker overnight at room temperature. The next day, the tissue was embedded in Embed812 and polymerized (24 h in an oven at 60°C). Thin sections of ganglia were cut on an ultramicrotome (UltracutE; Reichert-Jung, Nussloch, Germany), stained with 2% aqueous uranyl acetate and 3% lead citrate, and viewed with a Philips (Aachen, Germany) CM10 electron microscope.
Immunocytochemistry. Freshly dissected ganglia were placed in 0.5% paraformaldehyde in 0.1 m PB for 1 h, overnight in 40% sucrose in 0.1 m PB, and then embedded in OCT compound and frozen immediately by immersion in 2-methylbutane cooled in liquid nitrogen. Frozen sections (30 μm) from these ganglia were cut with a cryostat and placed onto Probe-on Plus slides (Fisher Scientific, Houston, TX); the sections were rinsed with PBS for 30 min, blocked for 1 h in 10% normal donkey serum in PBS and 0.5% Triton X-100 at room temperature for 1 h, and then overnight in primary antibodies (polyclonal goat anti-VAChT (1:750; Chemicon, Temecula, CA), polyclonal goat anti-choline acetyltransferase (ChAT) (1:50; Chemicon), monoclonal mouse anti-neurofilament (1:800; Sternberger Monoclonals, Lutherville, MD), polyclonal goat anti-vesicular ACh transporter (VAChT) and rabbit anti-synaptobrevin (1:400; Synaptic Systems, Göttingen, Germany), rabbit anti-synaptotagmin (1:200; Synaptic Systems), or anti-syntaxin 1a (1: 400; Synaptic Systems) in PBS-containing 10% normal donkey serum at 4°C. The sections were rinsed three times with PBS and then placed in secondary antibodies [donkey anti-goat FITC and/or donkey anti-mouse rhodamine (Jackson ImmunoResearch, West Grove, PA)] in PBS-containing 10% normal donkey serum for 1 h at room temperature, then rinsed three times with PBS, and mounted with Vectashield (Vector Laboratories, Burlingame, CA). Nonspecific staining, judged by processing sections without the primary antibody, was very low.
A series of z-stacks (0.3 μm/plane) were obtained with a confocal microscope (LSM 510; Zeiss) and a 63×, 1.4 numerical aperture Plan Neofluor oil-immersion objective. Images were quantified off-line with MetaMorph (Universal Imaging Corporation). All z-stacks were thresholded at twice the background pixel intensity obtained from a line scan of the image. For quantification of VAChT-positive terminals and for colocalization, we took running averages of three consecutive planes through the entire z-stack, and only images present on three consecutive planes and 1–3 μm in diameter were counted.
Results
ACh-evoked currents are absent on α3–/– neurons and restored by overexpressing α3 cDNA
Peripheral autonomic neurons express two different subtypes of nAChRs: α3-containing receptors and α7-containing receptors (McGehee and Role, 1995; Rosenberg et al., 2002). We have shown previously that α3 is highly expressed in rodent sympathetic neurons and that the developmental increase in ACh-evoked current densities correlates well with the increase in α3 mRNA expression (Mandelzys et al., 1994; De Koninck and Cooper, 1995). To determine whether deleting the α3 gene would abolish ACh-evoked currents, we recorded from freshly isolated sympathetic neurons with whole-cell patch-clamp techniques while applying ACh exogenously. Brief ACh (100 μm) application onto freshly dissociated neurons from a WT neonatal SCG evoked a large inward current (Fig. 1A); the average ACh-evoked current density on neurons from WT SCG was 62.3 pA/pF (n = 25) (Fig. 1B). Conversely, ACh (100 μm) applied to dissociated neurons from α3–/– ganglia evoked no change in membrane current (Fig. 1A,B). These results suggest that sympathetic neurons require the expression of α3 subunit for the appearance of ACh-evoked inward currents.
Figure 1.
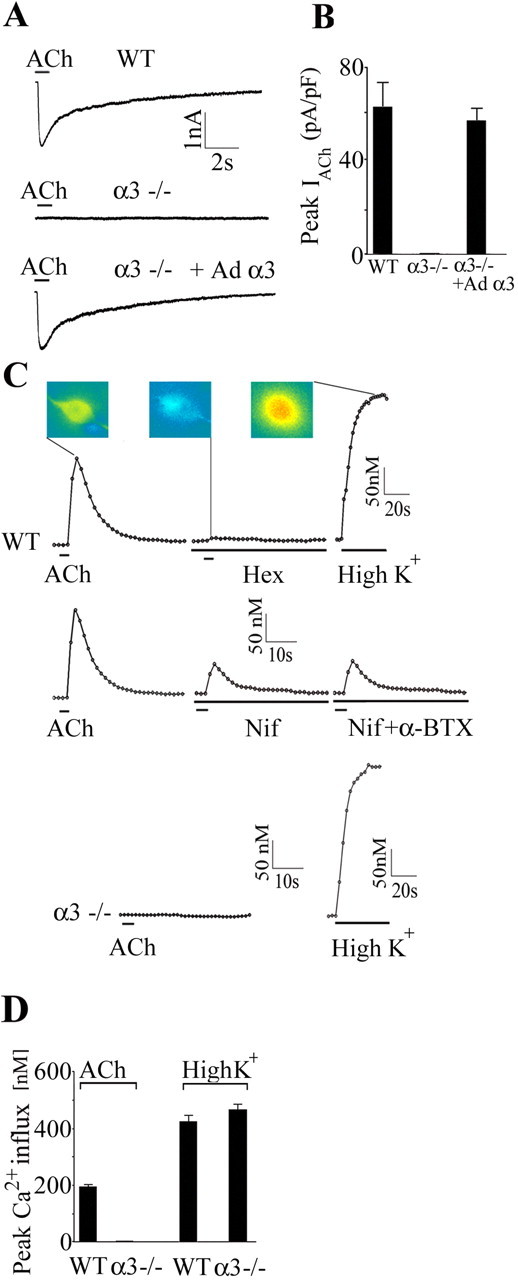
Sympathetic neurons in α3–/– SCG lack ACh-evoked currents and nAChR-induced changes in intracellular calcium. A, Whole-cell recordings from P7 WT SCG neuron in culture for 2 d (top), P8 α3–/– SCG neuron in culture for 2 d (middle), and P8 α3–/– neuron in culture for 5 d and infected with Ad α3 at day 2 in culture (bottom). A brief application of ACh (100 μm) produced a large, rapid inward current on the WT SCG neuron and on the α3–/– neuron infected with Ad α3 but produced no detectable inward current from the α3–/– SCG neuron. B, Mean ACh-evoked current densities on the three types of neurons shown in A. Each column represents the mean ± SEM; n = 25–27. None of the 25 neurons from α3–/– ganglia had detectable ACh-evoked inward currents; however, infecting α3–/– neurons with α3 cDNA restores ACh-evoked current densities to WT levels. C, P6 WT and α3–/– neurons in culture for 1 d were loaded with fura-2 AM and imaged with 340 and 380 nm excitation. The 340/380 ratios were converted to intracellular Ca2+ concentrations and plotted over time. A brief application of ACh (100 μm) produced a large, rapid increase in intracellular Ca2+ concentration in the WT SCG neuron, which was reversibly blocked by hexamethonium (100 μm). The middle three traces in C show that a large fraction of the ACh-induced increase in intracellular Ca2+ was blocked by nifedipine (Nif) (5 μm) and unaffected by αBgt (0.5–1 μm). The bottom two traces in C show that ACh (100μm) produced no detectable change in intracellular Ca2+ concentration in the α3–/– SCG neuron. Depolarizing neurons with 40 mm K+ produced large increases in intracellular Ca2+ in WT and α3–/– neurons. The 340 and 380 images were obtained at 1 s intervals during ACh applications and 5 s intervals during high K+ at 37°C. The inset shows the actual 340/380 images from a WT neuron at the peak of the ACh response, in the presence of hexamethonium (Hex), and at the peak of the high K+ response. D, Mean change in calcium influx induced by ACh (100 μm) and high K+ (40 mm) for WT and α3–/– neurons. ACh did not induce detectable changes in intracellular Ca2+ in α3–/– neurons; however, all α3–/– neurons responded to high K+. The values are the mean ± SEM; n = 32–47.
To test this idea directly, we infected dissociated neurons from α3–/– ganglia with adenoviruses constructed to express α3 cDNA (Ad α3) and the reporter gene green fluorescent protein (GFP) (Wheeler and Cooper, 2001) and then recorded from infected neurons 48–72 h later. α3–/– neurons infected with Ad α3 had ACh-evoked inward currents that were similar in magnitude and time course to those on WT neurons (Fig. 1A); ACh-evoked current densities on α3–/– neurons infected with Ad α3 were not significantly different from those on WT neurons (Fig. 1B), whereas α3–/– neurons infected with control viruses expressing GFP alone had no detectable ACh-evoked currents (n = 27) (data not shown). The results in Figure 1, A and B, indicate that expressing α3 cDNAs in neurons from α3–/– SCG is sufficient to restore ACh-evoked currents to wild-type levels on these neurons.
We did not observe α7-type currents on freshly dissociated neurons from α3–/– ganglia. One possibility is that such α7-containing receptors produce their effects by mobilizing Ca2+ from internal stores (Sharma and Vijayaraghavan, 2003). To test this, we blocked muscarinic receptors with atropine (2 μm) and monitored changes in cytoplasmic Ca2+ with the calcium indicator fura 2 in response to brief ACh (100 μm) applications. In neurons dissociated from P7 WT SCG, ACh (100 μm) applications produced a large increase in cytoplasmic Ca2+ that was reversibly blocked by hexamethonium (100 μm), an nAChR antagonist (Fig. 1C,D). Most of the ACh-induced increase in Ca2+ was attributable to depolarization-induced Ca2+ influx through L-type Ca2+ channels, and a fraction was attributable to an α-bungarotoxin (αBgt)-insensitive Ca2+ influx through nAChRs (n = 31) (Fig. 1C). Conversely, ACh (100 μm) produced no change in cytoplasmic Ca2+ in dissociated neurons from α3–/– SCG, although all α3–/– SCG neurons responded to depolarization with a large increase in cytoplasmic Ca2+ when exposed to high K+ (40 mm) (Fig. 1C,D).
SCG from α3–/– mice lack synaptic transmission
Because isolated α3–/– SCG neurons do not have functional nAChRs (Fig. 1), we anticipated that synaptic transmission in α3–/– ganglia would be severely disrupted. To test this, we stimulated the preganglionic nerve and recorded from the postganglionic trunk of the SCG from neonatal α3–/– mice and their WT littermates.
Suprathreshold stimuli applied to the preganglionic nerve of SCG from neonatal WT mice produced large compound action potentials (Fig. 2A); these compound action potentials were reversibly blocked by hexamethonium (100 μm). In addition, because we recorded close to the ganglion, we could also detect the preganglionic afferent volley preceding the postganglionic compound action potential. Results similar to those in Figure 2A were obtained from all (18 of 18) WT SCG.
Figure 2.
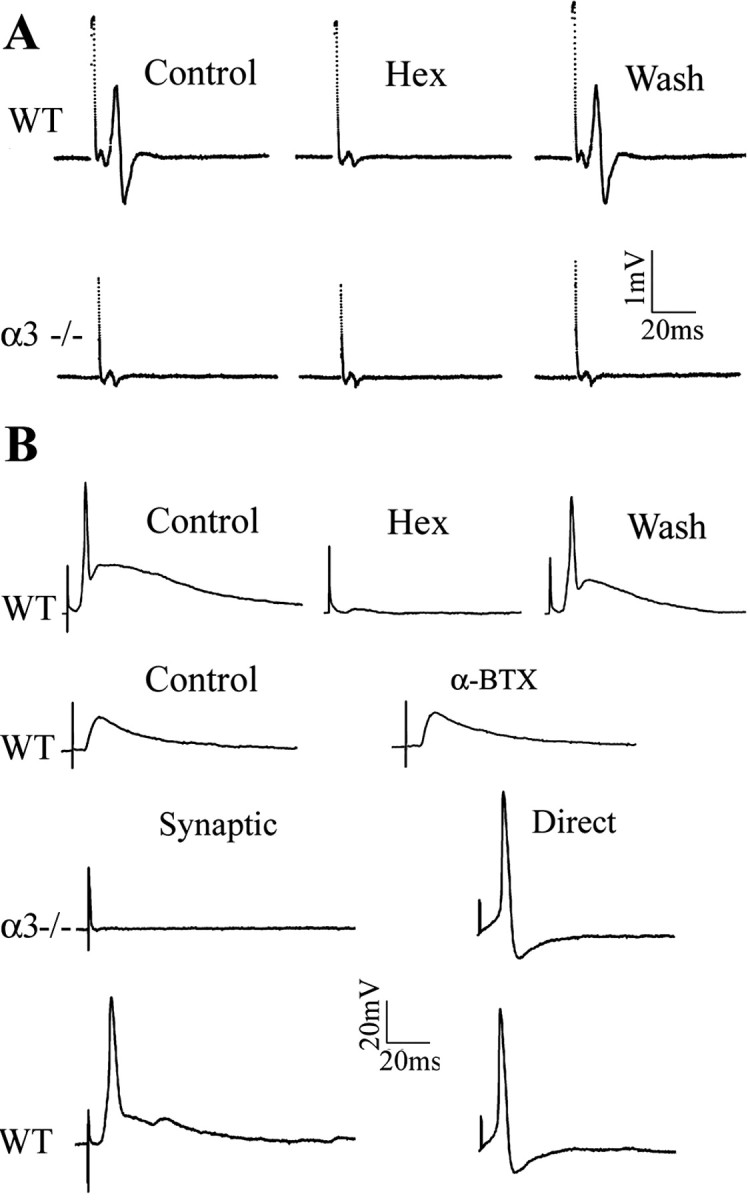
No postganglionic compound action potential or fast EPSPs on sympathetic neurons in α3–/– superior cervical ganglion. A shows postganglionic compound action potentials and preganglionic potentials recorded from the sympathetic trunk of a P7WT SCG (top traces) in response to suprathreshold stimuli to the preganglionic nerve. In WT ganglia, hexamethonium (Hex) (100 μm) reversibly blocked the compound action potential but had no effect on the preganglionic potential. In P7 α3–/– ganglia (bottom trace), suprathreshold stimuli to the preganglionic nerve failed to evoke postganglionic compound action potentials, whereas the preganglionic potentials were unchanged. B, Stimulating the preganglionic nerve evoked large suprathreshold EPSPs from a P8 WT sympathetic neuron recorded intracellularly; the EPSPs were reversibly blocked by hexamethonium (100μm) but unaffected byαBgt (0.5μm). Stimulating the preganglionic nerve failed to produce any change in membrane potential on a P8 α3–/– sympathetic neuron, demonstrating that fast synaptic EPSPs are absent inα3–/– ganglia. Direct intracellular current injection, conversely, evoked fast, overshooting action potentials (right) on both WT and α3–/– neurons, indicating that α3–/– neurons do not require fast synaptic transmission to express voltage-gated currents that underlie the action potential.
In contrast, suprathreshold stimuli applied to the preganglionic nerve of SCG from α3–/– littermates failed to evoke detectable compound action potentials in all (25 of 25) ganglia examined, although the preganglionic afferent volleys were clearly present (Fig. 2A). These results in Figure 2A indicate that the preganglionic nerve does not evoke suprathreshold EPSPs on sympathetic neurons in α3–/– ganglia.
Sympathetic neurons in α3–/– SCG lack fast EPSPs
To test whether the preganglionic nerve evoked subthreshold EPSPs, we recorded intracellularly from neurons in neonatal SCG. Stimulating the preganglionic nerve in a P8 WT ganglia produced large, fast suprathreshold EPSPs on SCG neurons lasting tens of milliseconds (Fig. 2B). These EPSPs were reversibly blocked by hexamethonium (100 μm), indicating that the synapses were cholinergic and mediated by nAChRs (Fig. 2B). We observed similar results from all neurons (27 of 27) in neonatal WT SCG. α-Bgt (0.5 μm) had no effect on these EPSPs (n = 5) (Fig. 2B).
Conversely, stimulating the preganglionic nerve in P8 α3–/– ganglia failed to produce any detectable change in membrane potential (Fig. 2B). Similar results were obtained from all neurons (53 of 53) in neonatal α3–/– ganglia. These results demonstrate that fast synaptic transmission requires α3-containing nAChRs. Moreover, our results indicate that, in α3–/– ganglia, (1) no other nAChR α subunit substitutes for the missing α3 to restore fast synaptic transmission, and (2) no other transmitter system compensates for the absence of cholinergic-nicotinic transmission at these synapses.
Although fast excitatory synaptic transmission is absent from sympathetic neurons in α3–/– neonatal SCG, direct electrical stimulation of these neurons evoked overshooting action potentials that were similar in magnitude and time course to those on neurons in WT SCG (Fig. 2B). This indicates that α3–/– neurons continue to differentiate electrically and do not require fast synaptic activity to express voltage-gated currents that underlie the action potential.
Muscarinic responses on α3–/– and WT neurons are similar
Stimulating the preganglionic nerve in P7 WT ganglia at 20 Hz for 2 s while blocking the nAChRs with hexamethonium (100 μm) produced a 5–7 mV slow EPSP that was blocked by atropine (Fig. 3A). Deleting the α3 nAChR subunit gene abolishes fast nicotinic EPSPs, but we expected that these slow muscarinic EPSPs in α3–/– ganglia would be unaffected; however, this was not the case. In P7 α3–/– ganglia, we observed no detectable slow EPSPs when we applied similar stimulations to the preganglionic nerve (Fig. 3A). This unanticipated absence of slow EPSPs in α3–/– ganglia raised several questions. Are the postsynaptic muscarinic responses normal? Do the preganglionic nerves innervate sympathetic neurons in α3–/– ganglia? If they innervate the SCG neurons and form synapses, can they release normal amounts of ACh?
Figure 3.

The absence of slow EPSPs but normal muscarinic responses in α3–/– ganglia. A, Stimulating the preganglionic nerve with a 20 Hz train for 2 s in the presence of hexamethonium (100 μm) produced a slow depolarization in a P7 WT neuron (top). A similar 20 Hz train stimulation produced no detectable change in membrane potential in a P7 α3–/– neuron (bottom). The stimulus artifacts were removed for clarity. Experiments were done at 37°C. B, Depolarization of a P7 WT SCG neuron (left) and a P8α3–/– SCG neuron (right) induced by three ACh concentrations (0.1×, 1×, and 5×) applied directly to each ganglia. [1× represents the concentration of ACh that produced a similar depolarization to that after preganglionic nerve stimulation (2 s at 20 Hz in neostigmine) and was usually 100μm added to the perfusion solution.] Below shows response to 1× ACh with and without neostigmine (Neo). C, ACh dose–response relationship for 11 WT (•) and 9 α3–/– (▪) neurons. The values are the means ± SEM; n = 11 for WT and 9 for α3–/–. There is no significant difference in the muscarinic response between neurons in WT and α3–/– ganglia. All experiments were done in the presence of hexamethonium (100 μm) and at 37°C.
To address whether the muscarinic responses are normal, we directly applied ACh to the ganglia. Muscarinic receptors on rat sympathetic neurons are not focally concentrated at synapses but are widely distributed over their somatic and dendritic surfaces, with higher densities on the soma (Ramcharan and Matthews, 1996). For acetylcholine to reach muscarinic receptors during synaptic transmission, it must diffuse from the synapses (Brown and Selyanko, 1985).
We applied increasing concentrations of ACh to the ganglia while recording from SCG neurons intracellularly to measure the muscarinic response. We adjusted the concentration of ACh (typically ∼100 μm) to give us responses that mimicked the nerve-evoked response in WT ganglia without acetylcholinesterase (see below). We refer to this concentration as 1× and measured the response of neonatal WT and α3–/– SCG neurons to four ACh concentrations: 0.01×, 0.1×, 1×, and 5× (Fig. 3B,C). We observed no significant difference in muscarinic responses among WT and α3–/– neurons (Fig. 3B,C); moreover, the responses were unaltered by acetylcholinesterase inhibitors. These results show that the appearance of functional muscarinic receptors on SCG neurons does not require fast synaptic activity and that the muscarinic response on WT and α3–/– neurons is not different.
Morphological synapses form on neurons without synaptic activity
The absence of nerve-evoked slow EPSPs in α3–/– SCG is not attributable to the lack of postsynaptic muscarinic responsiveness but is more likely attributable to the low amount of ACh released from the presynaptic terminals. Therefore, we determined whether the density of cholinergic varicosities in P7 α3–/– ganglia was altered. As a marker for cholinergic varicosities, we immunostained for VAChT (Fig. 4A). We found no significant differences in either the number of cholinergic varicosities (Fig. 4B) or the mean VAChT fluorescence (data not shown) in α3–/– compared with WT ganglia.
Figure 4.
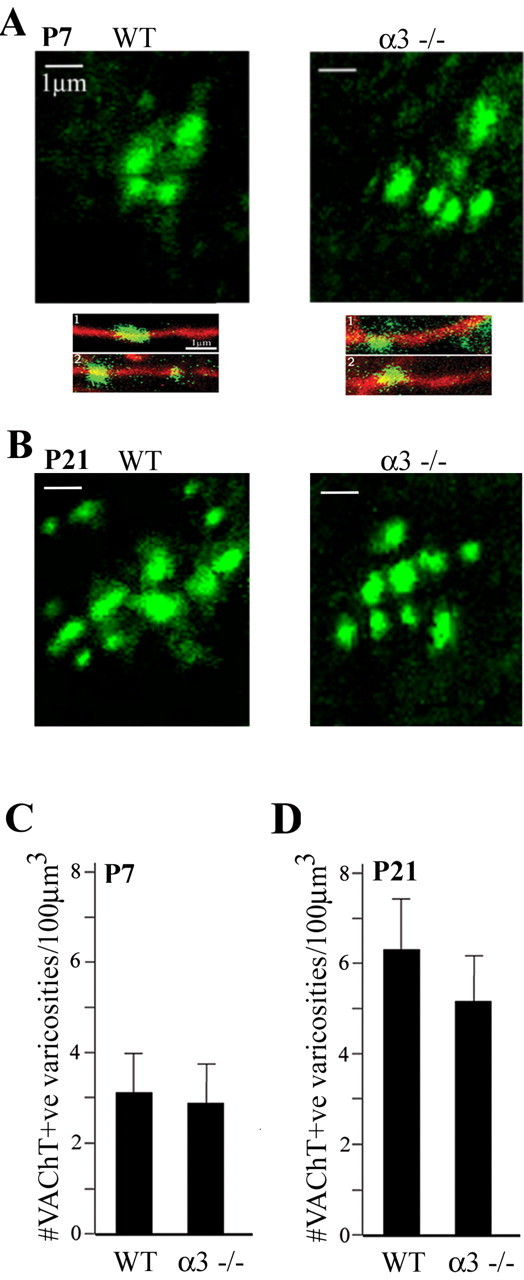
The densities of preganglionic varicosities in α3–/– ganglia are similar to those in WT ganglia. A, Confocal images of VAChT-positive varicosities in P7 WT andα3–/– ganglia. The bottom panels show higher magnification images of VAChT-positive varicosities (green) located on neurofilament-positive preganglionic axons (red). Scale bars, 1 μm. B, Average number of VAChT-positive varicosities in P7 WT and α3–/– ganglia. More than 450 varicosities were counted from confocal images in WT andα3–/– ganglia. The number of varicosities in each ganglion was normalized to a constant volume, and the values are the mean ± SEM; n = 5. C, Confocal images of VAChT-positive varicosities in P21 WT and α3–/– ganglia. D, Average number of VAChT-positive varicosities in P21 WT andα3–/– ganglia. More than 270 varicosities were counted from confocal images in WT and α3–/– ganglia. The number of varicosities in each ganglion was normalized to a constant volume, and the values are the mean ± SEM; n = 3.
The density of cholinergic varicosities in P21 ganglia was approximately twofold greater than in P7 for both α3–/– and WT ganglia (Fig. 4C,D). At P21, there was no significant difference in the density of cholinergic varicosities between α3–/– and WT ganglia.
In addition, we immunostained for vesicle-associated membrane protein (VAMP), synaptotagmin, syntaxin 1a, or ChAT in >670 varicosities from 25 ganglia (Fig. 5). More than 90% of these proteins colocalized with VAChT in presynaptic varicosities; ChAT also localized to preganglionic axons. We found no significant differences in immunostaining for these proteins between WT and α3–/– ganglia.
Figure 5.
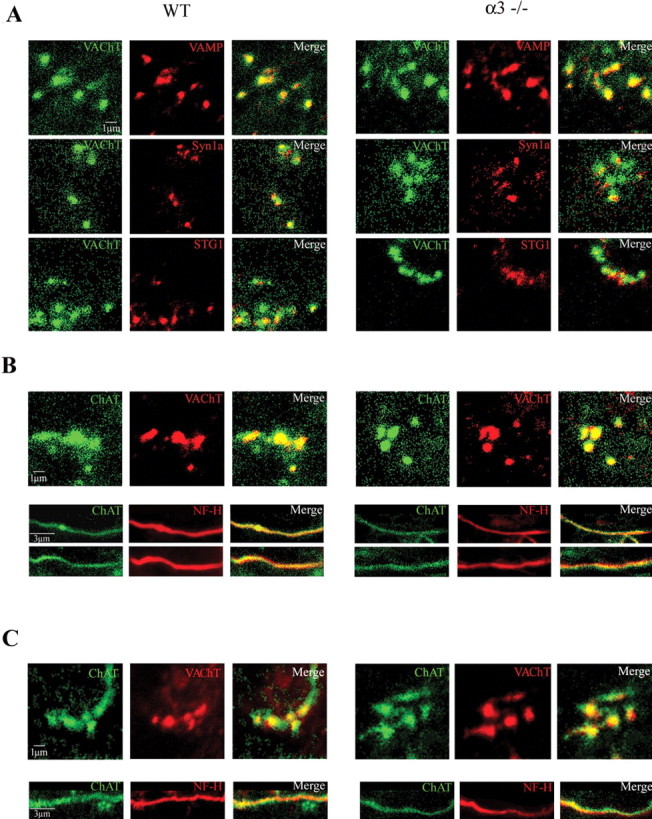
The preganglionic varicosities in α3–/– ganglia contain ChAT, VAChT, and SNARE (soluble N-ethylmaleimide-sensitive factor attachment protein receptor) proteins and are similar to those in WT ganglia. Immunostaining of varicosities in WT ganglia (left set) and α3–/– ganglia (right set) for VAChT, synaptobrevin (VAMP), syntaxin 1a (Syn1a), synaptotagmin (STG1), and ChAT. In A, the left columns show immunostaining for VAChT (green), the middle columns show immunostaining for VAMP (synaptobrevin) (top, red), syntaxin 1a (middle, red), and synaptotagmin (bottom, red), and the right columns are the merged images of the left and middle columns. The top three sets of panels show that VAMP, syntaxin 1a, and synaptotagmin colocalize with VAChT in presynaptic varicosities in P7WT and α3–/– ganglia. Scale bar, 1 μm. In B, the left columns show immunostaining for ChAT (green), the middle columns show immunostaining for VAChT (red) in presynaptic varicosities (top panels) and neurofilament (Nf-H, red) along preganglionic axons (bottom 2 panels). The right columns are the merged images of the left and middle columns and show that ChAT is present in preganglionic axons and in presynaptic varicosities in P7 WT and α3–/– ganglia. C, Similar to B but from P21 WT and α3–/– ganglia. Scale bars (in B, C): 1 μm for the varicosities; 3 μm for axons.
Next, we used electron microscopic analysis to determine whether these preganglionic terminals formed morphological synapses on sympathetic neurons in the absence of activity. We quantified 29 synapses from nine different ganglia in neonatal α3–/– mice and 36 synapses from six different WT littermates and found no significant difference in morphology of synapses in SCG from WT and α3–/– mice (Fig. 6A–F). The synapses have the characteristic features of accumulations of synaptic vesicles adjacent to the presynaptic surface membrane, enhanced postsynaptic density, a parallel arrangement and thickening of the presynaptic and postsynaptic membranes, and a widened synaptic cleft. We measured the number of docked vesicles at the active zone, synaptic cleft width, length of specialized presynaptic and postsynaptic membrane, number of multivesicular bodies, and diameter of small clear synaptic vesicles and of large densecore vesicles. We found no significant differences in any of these parameters in WT versus α3–/– SCG (Table 1). These results indicate that morphologically normal synapses are established in ganglia in the absence of fast synaptic activity.
Figure 6.

The ultrastructure of synapses in α3–/– ganglia are similar to those in WT ganglia. A–F, Electron micrographs of synapses in P7 in α3–/– (A, C, E) and WT (B, D, F) SCG. The synapses have the characteristic morphological features: accumulations of synaptic vesicles adjacent to the presynaptic membrane, enhanced postsynaptic density, a parallel arrangement and thickening of the presynaptic and postsynaptic membranes, and a widened synaptic cleft. Scale bars, 300 nm.
Table 1.
Quantification of synaptic parameters in the SCG of α3 null-mutant and control littermates at neonatal P6-P7
|
|
α3-/- |
Control |
|---|---|---|
| Small clear synaptic vesicles (nm, diameter) | 30 ± 1.5 | 33 ± 1.2 |
| Large dense core vesicles (nm, diameter) | 71 ± 3.3 | 71 ± 5.6 |
| Docked vesicles (# per synapse) | 0.9 ± 0.3 | 1.1 ± 0.2 |
| Multivesicular bodies (# per synapse) | 0.7 ± 0.2 | 0.5 ± 0.3 |
| Synaptic cleft width (nm, diameter) | 25 ± 1.2 | 24 ± 0.7 |
| Active zone length (nm, diameter) | 261 ± 30 | 263 ± 22 |
| PSD length (nm, diameter) |
265 ± 19 |
273 ± 29 |
Data represent means ± SEMs; n = 29 α3-/- synapses and 36 control synapses from 9 α3-/- and 6 control animals, respectively. Docked vesicles per synapse are defined as those that are located less than one vesicle diameter from the presynaptic plasma membrane. PSD, Postsynaptic density.
ACh release from preganglionic varicosities requires fast synaptic activity
Our results indicate that the postsynaptic muscarinic responsiveness in α3–/– neurons is normal, that the density of cholinergic varicosities in α3–/– ganglia is similar to those in WT ganglia, and that the preganglionic nerve in α3–/– ganglia forms morphologically normal synapses. This suggests that the absence of slow EPSPs in α3–/– ganglia results from a presynaptic malfunction in transmitter output.
If ACh released from cholinergic varicosities in α3–/– ganglia was lower than that released from varicosities in WT ganglia, most of the ACh might be hydrolyzed by acetylcholinesterase before it could diffuse to the muscarinic receptors and is therefore incapable of producing slow EPSPs. To test this, we inhibited the acetylcholinesterase with neostigmine. In control solution, a brief (2 s) 20 Hz train in a P6 WT ganglion produced a small 5–7 mV slow depolarization, whereas in the presence of neostigmine (10 μm), a similar stimulation produced a large (∼25 mV) slow EPSP lasting 1–2 min that was reversibly blocked by atropine (10 μm) (Fig. 7A). In neonatal α3–/– SCG in control solution, a 2 s, 20 Hz train applied to the preganglionic nerve produced no detectable response; however, after adding neostigmine (10 μm), the 20 Hz train produced a small (5–7 mV) atropine-sensitive depolarization that lasted only 10–20 s (Fig. 7A). These nerve-evoked depolarizations in neostigmine were reversibly blocked by perfusing the ganglia with low-Ca2+, high-Mg2+ Ringer's solution (n = 3) (data not shown). We obtained similar results from P21 ganglia (Fig. 7B). These results suggest that cholinergic varicosities in α3–/– ganglia can release ACh but in significantly smaller amounts than varicosities in WT ganglia.
Figure 7.
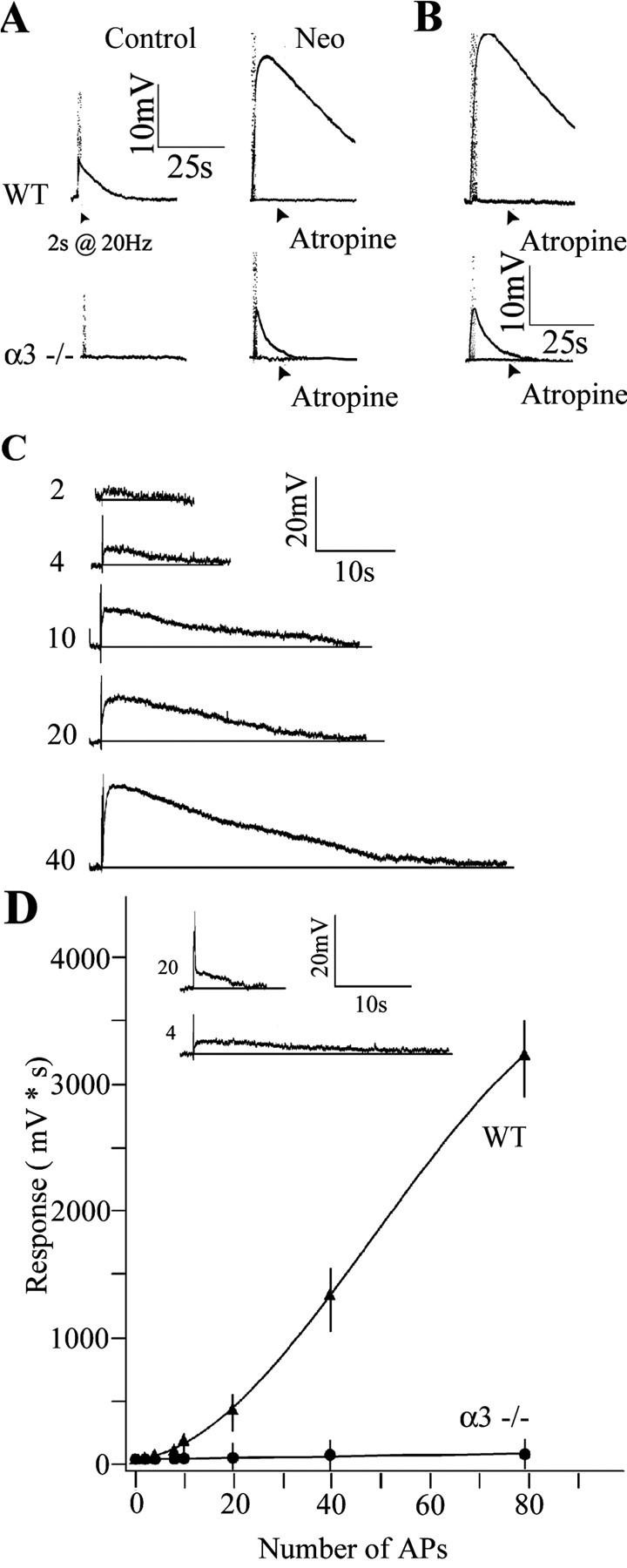
Reduced ACh output from preganglionic varicosities in α3–/– SCG. A, Top traces, Stimulating the preganglionic nerve at 20 Hz for 2 s produced a small, slow muscarinic depolarization on a P6 WT SCG neuron in control solution with hexamethonium (100 μm) (left); the response increased ∼4.5-fold after adding neostigmine (10 μm) (Neo) to inhibit acetylcholinesterase (right) and was blocked by adding atropine (3 μm). Bottom traces, Stimulating the preganglionic nerve at 20 Hz for 2 s produced no detectable response from a P8 α3–/– SCG neuron (left); however, after inhibiting cholinesterase with neostigmine (10 μm) (right), preganglionic nerve stimulation produced a small muscarinic response that was blocked by atropine (3 μm). B, Response from a P21 WT (top) and α3–/– (bottom) SCG neuron after inhibiting cholinesterase with neostigmine (10 μm). C, Muscarinic responses on a P8WT neuron increased with the number of stimuli applied to the preganglionic nerve. The preganglionic nerve was stimulated at 20 Hz; the number of stimuli delivered to the preganglionic nerve is indicated at the left of each trace. D, Integrated muscarinic responses from neurons in WT (▴) and α3–/– (•) ganglia were plotted against the number of applied stimuli to the preganglionic nerve. The solid line represents a fourth-order polynomial fit to the data. The inset compares the response of a P7 WT neuron after four stimuli to the response of a P7α3–/– neuron to 20 stimuli. The values are the means ± SEM (n = 8 for WT and n = 7 for α3–/– neurons). All experiments were done with neostigmine (10 μm) and hexamethonium (100 μm) added to the perfusion fluid at 37°C. AP, Action potential.
The muscarinic responses recorded from WT sympathetic neurons varied directly with the number of stimuli applied to the preganglionic nerve (Fig. 7C) (Brown and Selyanko, 1985). The integrated muscarinic responses on WT sympathetic neurons increased dramatically with increasing number of stimuli to the preganglionic nerve (Fig. 7C,D). In sharp contrast, the muscarinic responses from sympathetic neurons in α3–/– ganglia were difficult to detect unless the preganglionic nerve was stimulated for at least 1 s at 20 Hz (Fig. 7C, inset). Moreover, increasing numbers of stimuli to the preganglionic nerve in α3–/– ganglia had little effect on the magnitude of the response (Fig. 7C). These results indicate that ACh output from preganglionic varicosities is dramatically reduced in α3–/– ganglia.
In WT ganglia, the muscarinic responses to successive stimuli (2 s at 20 Hz) decreased slowly (Fig. 8A,B); the second application delivered after a 60 s rest was ∼80% of the initial response and eventually leveled off to ∼70% of initial response after five to six intervals (Fig. 8B). In contrast, the muscarinic responses in α3–/– ganglia decreased sharply as if the stores of ACh became rapidly depleted (Fig. 8A,B); the second response was ∼25% of the initial response (Fig. 8B) and became undetectable after the third or fourth application (Fig. 8B). This rapid depletion suggests that, when cholinergic nicotinic synapses develop without functional postsynaptic nAChRs and fast synaptic activity, the preganglionic varicosities are abnormal in their ability to synthesize and/or store ACh.
Figure 8.
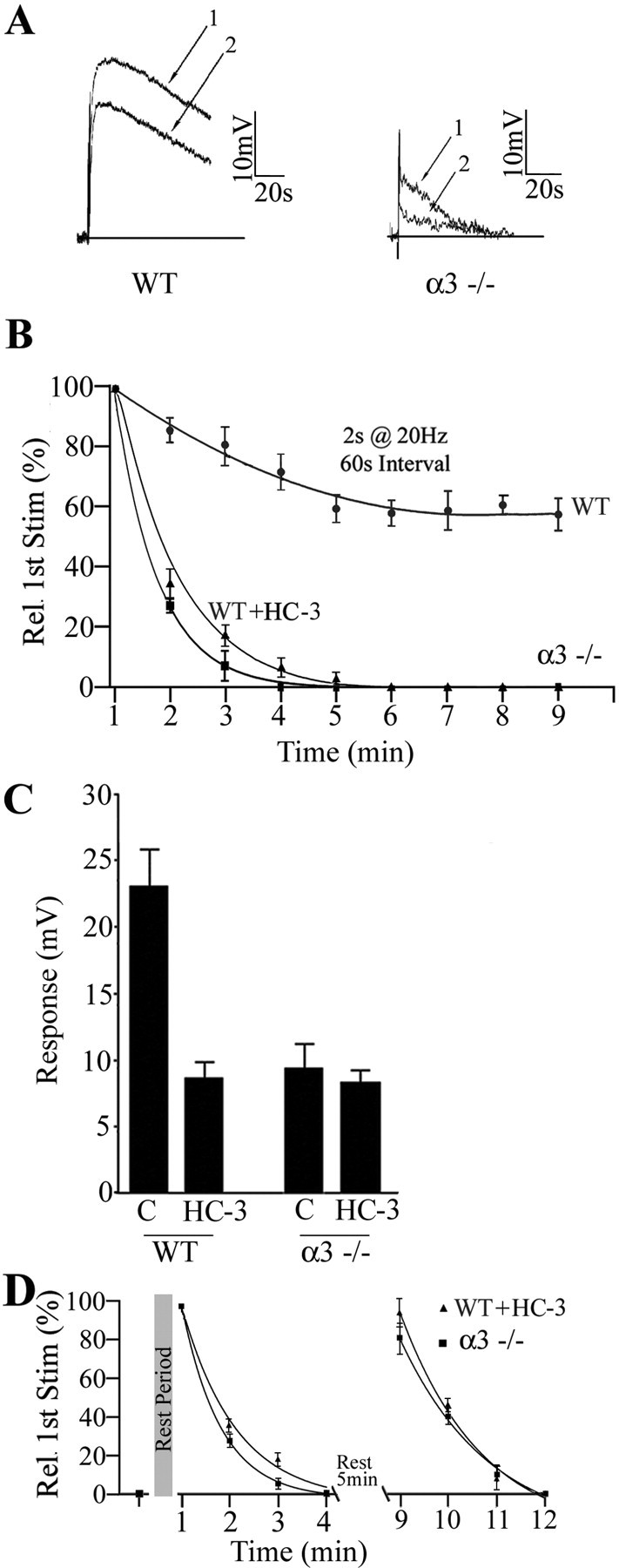
ACh output from α3–/– varicosities after burst stimulation depletes rapidly, recovers slowly, and is independent of the high-affinity choline transporter. A, Muscarinic depolarizations on a P7 neuron in a WT ganglion (left) and P7 neuron in an α3–/– ganglion (right) in response to paired stimuli to the preganglionic nerve (2 s at 20 Hz) delivered 40 s apart. B, The response to a series of stimuli (2 s at 20 Hz) delivered at 60 s intervals and normalized to the first response in the series. In WT ganglia, the response declined gradually to ∼70% of the initial response and leveled off after five to six trials. In α3–/– ganglia, conversely, the response declined rapidly and was not detectable after the third stimulus. In WT ganglia treated with HC-3 (10 μm), the response declined similarly and was not detectable after the fourth interval. The solid lines are single-exponential fits to the data. C, The bar graph shows that blocking the high-affinity choline transporter in WT ganglia with HC-3 (20 μm) decreased transmitter output to ∼30% of the control level, whereas HC-3 (20 μm) had little effect on responses in α3–/– ganglia. D shows the peak muscarinic responses from P7 WT neurons treated with HC-3 (▴) and from P7 α3–/– neurons (▪) when stimulated with two consecutive series of four stimuli (2 s at 20 Hz every 60 s) with 5 min rest intervals between each series; the response recovered almost completely after each rest interval and then rapidly became depleted. All experiments were done with neostigmine (10μm) and hexamethonium (100μm) added to the perfusion fluid at 37°C. The values are the means ± SEM; n = 5 for neurons in WT ganglia and n = 6 for neurons in α3–/– ganglia.
Preganglionic terminals in α3–/– ganglia lack functional high-affinity choline transporters
Synthesis of ACh in preganglionic terminals depends predominantly on the acute uptake of extracellular choline by the Na+-dependent, HC-3-sensitive, high-affinity choline transporter, particularly during high-frequency firing (Birks and MacIntosh, 1961; Okuda et al., 2000; Ferguson et al., 2004). If the deficit in ACh output from preganglionic varicosities in α3–/– ganglia results from impaired high-affinity choline transporters then (1) HC-3 should have little effect on ACh output from α3–/– varicosities, and (2) blocking the high-affinity choline transporter in WT ganglia should lead to ACh depletion whose kinetics mimic those of terminals in α3–/– ganglia.
To test the first prediction, we inhibited the high-affinity choline transporter with HC-3 (10 μm) in α3–/– ganglia and stimulated the preganglionic nerve at 20 Hz for 2 s in neostigmine-containing solution. Inhibiting the high-affinity choline transporter had no effect on the muscarinic response in α3–/– ganglia, indicating that α3–/– terminals lack functional high-affinity choline transporters (Fig. 8C).
To test the second prediction, we stimulated the preganglionic nerve at 20 Hz for 2 s at 1 min intervals while perfusing with HC-3 (10 μm) (see Materials and Methods). After blocking the high-affinity choline transporter, the response decayed rapidly and was no longer detectable after the fourth interval, similar to what we observed from terminals in α3–/– ganglia (Fig. 8B).
In addition, when we rested the terminals in α3–/– ganglia for 5 min, the response to 20 Hz, 2 s stimulation recovered (Fig. 8D). Similarly, in WT ganglia treated with HC-3, after a 5 min rest, the muscarinic response recovered to approximately one-third of control, likely using the low-affinity choline transporter to resynthesize ACh (Fig. 8C). This ACh output from WT terminals in HC-3, however, decayed quickly and was undetectable after the third or fourth interval, mimicking the kinetics for depletion and recovery observed in α3–/– terminals (Fig. 8D).
The lack of effect of HC-3 on terminals in α3–/– ganglia and the rapid depletion of ACh from WT terminals treated with HC-3 together indicate that preganglionic terminals in α3–/– ganglia lack functional high-affinity choline transporters.
Discussion
Our main findings in this study are as follows: (1) sympathetic neurons must express the α3 nAChR gene to acquire fast nicotinic-type ACh-evoked inward currents; (2) morphologically normal synapses are established on neurons that lack functional postsynaptic receptors; (3) when synapses in SCG develop without fast synaptic activity, the output of transmitter from the presynaptic terminals is impaired; and (4) the decrease in transmitter output results from a lack of functional high-affinity choline transporters on presynaptic terminals. We discuss each of these findings in more detail below.
Functional nAChRs on postsynaptic SCG neurons require α3 subunits
Deleting the α3 gene completely disrupts fast synaptic transmission between presynaptic and postsynaptic neurons. Interestingly, we observed no compensatory upregulation of other nAChR subunit genes in the SCG of α3 nAChR null mice, nor did we observe the upregulation of any other transmitter system to restore fast synaptic transmission in SCG in the absence of α3-containing receptors.
This lack of compensatory upregulation to restore synaptic transmission was unexpected. Recent work has demonstrated that, when activity is perturbed, synapses dynamically adjust their strengths to ensure that firing rates are maintained within some functional range (Turrigiano and Nelson, 2004). These important homeostatic mechanisms have a clear role in the activity-dependent refinement of connectivity. Our observation of the lack of compensatory mechanisms does not necessarily contradict this principle of synaptic homeostasis but indicates that, if synaptic transmission in sympathetic ganglia is drastically perturbed during embryogenesis, then homeostatic mechanisms cannot restore synaptic transmission. It may become impossible to trigger such homeostatic mechanisms when the average level of postsynaptic depolarization at developing synapses is absent for long periods of time.
From work done in avian ciliary ganglia (Zhang et al., 1996; Ullian et al., 1997; Chang and Berg, 1999; Rosenberg et al., 2002), we had anticipated that SCG neurons in mice would have α7 responses. However, we did not detect any α7 depolarizations in neurons in intact α3–/– ganglia; in fact, we did not detect any ACh-induced whole-cell currents in isolated α3–/– neurons, nor did we detect any changes in intracellular calcium. Because α7 mRNA levels are regulated by activity (De Koninck and Cooper, 1995; Brumwell et al., 2002), the absence of synaptic activity on α3–/– sympathetic neurons may have caused a downregulation of α7-containing receptors. Alternatively, rodent sympathetic neurons may not express conventional α7 receptors on their cell bodies (Helekar et al., 1994; Cuevas and Berg, 1998). Importantly, by overexpressing α3 in α3–/– neurons with adenoviruses, we restored the ACh-evoked current densities to levels found on WT neurons. Therefore, α3 expression is critical for the appearance of functional nAChRs on sympathetic neurons.
Fast synaptic transmission is not required for the development of morphological synapses
Previous observations indicate that morphological synapses can form on neurons in vivo in the absence of presynaptic function. Our results demonstrate that electrophysiologically silent synapses are established on α3–/– SCG neurons and are consistent with observations that the initial events in synapse formation proceed without activity (Verhage et al., 2000; Misgeld et al., 2002; Washbourne et al., 2002). In rodent SCG, synaptogenesis starts at embryonic day 13–14 (Rubin, 1985), although most synapses form during the early postnatal period. If neurons in embryonic α3–/– ganglia lack synaptic activity like those in postnatal α3–/– ganglia, then it is likely that some of the synapses that we observed in P7 α3–/– ganglia have been maintained without activity for ∼2 weeks. Morphologically, the presynaptic terminals at these electrophysiologically silent synapses appear normal; we observed no significant difference in the vesicle population in the terminals of α3–/– ganglia compared with those of WTs, nor was there any significant difference in the number of docked vesicles at the active zones. In addition, we did not detect any differences in the presence of choline acetyltransferase, VAChT, VAMP, syntaxin 1a, or synaptotagmin between terminals in α3–/– ganglia and those of WTs. Unexpectedly, however, we discovered that these morphologically normal terminals release significantly less ACh than those in WT ganglia.
Presynaptic output in α3–/– SCG is impaired
We used the muscarinic response to quantify differences in ACh release from the preganglionic nerve in WT versus α3–/– ganglia. Muscarinic receptors on rat sympathetic neurons are widely distributed over their somatic and dendritic surfaces and are not focally concentrated at synapses, as demonstrated in a definitive study by Ramcharan and Matthews (1996) using light and electron microscopic autoradiography.
In WT ganglia, when we stimulated the preganglionic nerve at 20 Hz in control solutions, we recorded slow, small (∼5 mV) muscarinic depolarizations on sympathetic neurons that became significantly larger and more prolonged when we inhibited cholinesterase activity. Conversely, stimulating the preganglionic nerve under identical conditions in α3–/– ganglia produced markedly smaller muscarinic responses. In fact, unlike WT ganglia, we could not detect a muscarinic response in α3–/– ganglia unless we inhibited the esterase.
We observed no significant difference in postsynaptic muscarinic responsiveness in WT versus α3–/– SCG neurons. Moreover, we observed no difference in the density of preganglionic cholinergic varicosities between WT and α3–/– ganglia. Therefore, the difference in response to preganglionic stimulation in WT and α3–/– ganglia must result from differences in presynaptic release of ACh.
Because preganglionic neurons in rodents do not appear to express the α3 gene (Wada et al., 1989), it seems unlikely that the absence of α3 can account for the low ACh output from terminals in α3–/– ganglia. Moreover, our experiments on ACh output were done in blocking concentrations of hexamethonium, thereby eliminating any possible contribution of presynaptic nAChRs to the difference in transmitter output between varicosities in α3–/– and WT ganglia (Liang and Vizi, 1997; Rogers and Sargent, 2003).
High-affinity choline transporter function
Instead, our results suggest that the low ACh output from varicosities in α3–/– ganglia result from malfunction, dysregulation, and/or low expression of the high-affinity HC-3-sensitive, choline transporter CHT (Okuda et al., 2000; Ferguson et al., 2004). Choline uptake through CHT is thought to be the rate-limiting determinant of ACh synthesis, particularly during activity-induced increases in release (Birks and MacIntosh, 1961). We show that repetitive stimulation depletes transmitter rapidly from terminals in α3–/– ganglia, unlike those in WT, and that α3–/– terminals recover slowly from depletion with a 5–10 min rest; these results are consistent with a malfunction/misexpression of the high-affinity choline transporter. Relevantly, we show that HC-3 significantly reduced ACh output from WT preganglionic terminals when stimulated with a 20 Hz train but had no effect on ACh output from terminals in α3–/– ganglia; these results indicate that functional high-affinity choline transporters are absent from terminals in α3–/– ganglia.
Retrograde control of transmitter output at developing synapses
Our results suggest that the absence of functional CHT and the low ACh output from presynaptic terminals in α3–/– ganglia result from a lack of an activity-dependent retrograde signal. Recently, it has been demonstrated that CHT resides on a subset of vesicles in cholinergic terminals (Ferguson et al., 2003); moreover, it has been proposed that this subpopulation of CHT-containing vesicles are specifically recruited for release during prolonged periods of activity, delivering CHT to the membrane of the terminal, enabling high-affinity choline uptake for continued ACh synthesis (Ferguson et al., 2003). Therefore, if this recruitment depends on an activity-dependent retrograde signal, then the absence of such a signal could account for the reduced amount of ACh released from terminals in α3–/– ganglia and fewer functional CHT on the membrane. Alternatively, it has been shown that the expression of CHT in motor neurons is downregulated when the axon is axotomized and subsequently upregulated during reinnervation of muscle (Oshima et al., 2004). This suggests that motor axons receive an activity-dependent retrograde signal from muscle that regulates the expression of CHT.
In two paralytic zebrafish mutants, Sofa potato and Nic, the postsynaptic muscle is not sensitive to ACh, and, although the NMJs in these mutants are morphologically normal, the junctions have no spontaneous or evoked endplate potentials (Westerfield et al., 1990; Li et al., 2003). Nonetheless, vesicle recycling in the presynaptic terminals at these inactive neuromuscular junctions (NMJs) proceeds normally, as revealed by optical measures of exocytosis with FM1-43 [N-(3-triethylammoniumpropyl)-4-(4-(dibutylamino)styryl) pyridinium dibromide] (Li et al., 2003). If autonomic synapses in mice develop similarly to NMJs in zebrafish, then it is unlikely that the difference in ACh output that we observed between varicosities in α3–/– and WT ganglia is attributable to a difference in vesicular release or recycling; instead, it suggests that an activity-dependent retrograde signal influences the output of transmitter by regulating some other process.
Two good examples of activity-dependent retrograde signaling that affect presynaptic transmitter output at developing synapses are the studies on NMJ in Drosophila (Petersen et al., 1997; Davis et al., 1998; DiAntonio et al., 1999; Haghighi et al., 2003) and the studies on Xenopus nerve–muscle junctions in culture (Tao and Poo, 2001). Extrapolating from the studies in Drosophila, we anticipate that depolarization, calcium influx, and the subsequent activation of CaM kinase II in the postsynaptic cell are crucial steps in regulating retrograde signals that influence transmitter output from presynaptic terminals (Haghighi et al., 2003). The studies on Xenopus nerve–muscle cocultures suggest that neurotrophins act as an activity-dependent retrograde signal to boost transmitter release (Tao and Poo, 2001). Because BDNF plays an important role in the developmental innervation of sympathetic ganglia (Causing et al., 1997), it will be interesting to discover whether BDNF is involved in activity-dependent retro-grade signaling that regulates transmitter output from preganglionic terminals.
Footnotes
This work was supported by the National Institutes of Health (M.H.J.) and the Canadian Institutes of Health Research (E.C.). We thank A. L. Beaudet for the α3 KO mice, C. Bourque for his advice with the intracellular recordings, and Pejmun Haghighi and Linda Cooper for comments on this manuscript.
Correspondence should be addressed to Dr. Ellis Cooper, Department of Physiology, McGill University, McIntyre Medical Science Building, 3655 Promenade Sir William Osler, Montreal, Quebec, Canada H3G 1Y6. E-mail: ellis.cooper@mcgill.ca.
DOI:10.1523/JNEUROSCI.1983-05.2005
Copyright © 2005 Society for Neuroscience 0270-6474/05/258555-12$15.00/0
S.R. and A.K. contributed equally to this work.
References
- Birks RI, MacIntosh FC (1961) Acetylcholine metabolism of a sympathetic ganglion. Can J Biochem Physiol 39: 787–827. [Google Scholar]
- Brown DA, Selyanko AA (1985) Membrane currents underlying the cholinergic slow excitatory post-synaptic potential in the rat sympathetic ganglion. J Physiol (Lond) 365: 365–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brumwell CL, Johnson JL, Jacob MH (2002) Extrasynaptic α7-nicotinic acetylcholine receptor expression in developing neurons is regulated by inputs, targets, and activity. J Neurosci 22: 8101–8109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Causing CG, Gloster A, Aloyz R, Bamji SX, Chang E, Fawcett J, Kuchel G, Miller FD (1997) Synaptic innervation density is regulated by neuron-derived BDNF. Neuron 18: 257–267. [DOI] [PubMed] [Google Scholar]
- Chang KT, Berg DK (1999) Nicotinic acetylcholine receptors containing α7 subunits are required for reliable synaptic transmission in situ J Neurosci 19: 3701–3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen-Cory S (2002) The developing synapse: construction and modulation of synaptic structures and circuits. Science 298: 770–776. [DOI] [PubMed] [Google Scholar]
- Crowley C, Spencer SD, Nishimura MC, Chen KS, Pitts-Meek S, Armanini MP, Ling LH, McMahon SB, Shelton DL, Levinson AD, Phillips HS (1994) Mice lacking nerve growth factor display perinatal loss of sensory and sympathetic neurons yet develop basal forebrain cholinergic neurons. Cell 76: 1–20. [DOI] [PubMed] [Google Scholar]
- Cuevas J, Berg DK (1998) Mammalian nicotinic receptors with α7 subunits that slowly desensitize and rapidly recover from α-bungarotoxin blockade. J Neurosci 18: 10335–10344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis GW, DiAntonio A, Petersen SA, Goodman CS (1998) Postsynaptic PKA controls quantal size and reveals a retro-grade signal that regulates presynaptic transmitter release in Drosophila. Neuron 20: 305–315. [DOI] [PubMed] [Google Scholar]
- De Koninck P, Cooper E (1995) Differential regulation of neuronal nicotinic ACh receptor subunit genes in cultured neonatal rat sympathetic neurons: specific induction of α7 by membrane depolarization through a Ca2+/calmodulin-dependent kinase pathway. J Neurosci 15: 7966–7978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiAntonio A, Petersen SA, Heckmann M, Goodman CS (1999) Glutamate receptor expression regulates quantal size and quantal content at the Drosophila neuromuscular junction. J Neurosci 19: 3023–3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagan AM, Zhang H, Landis S, Richard J, Smeyne RJ, Silos-Santiago I, Barbacid M (1996) TrkA, but not TrkC, receptors are essential for survival of sympathetic neurons in vivo. J Neurosci 16: 6208–6218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson SM, Savchenko V, Apparsundaram S, Zwick M, Wright J, Heilman CJ, Yi H, Levey AI, Blakely RD (2003) Vesicular localization and activity-dependent trafficking of presynaptic choline transporters. J Neurosci 23: 9697–9709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson SM, Bazalakova M, Savchenko V, Tapia JC, Wright J, Blakely RD (2004) Lethal impairment of cholinergic neurotransmission in hemicholinium-3-sensitive choline transporter knockout mice. Proc Natl Acad Sci USA 101: 8762–8767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghighi AP, McCabe BD, Fetter RD, Palmer JE, Hom S, Goodman CS (2003) Retrograde control of synaptic transmission by postsynaptic CaMKII at the Drosophila neuromuscular junction. Neuron 39: 255–267. [DOI] [PubMed] [Google Scholar]
- He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B (1998) A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci USA 95: 2509–2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helekar SA, Char D, Neff S, Patrick J (1994) Prolyl isomerase requirement for the expression of functional homo-oligomeric ligand-gated ion channels. Neuron 12: 179–189. [DOI] [PubMed] [Google Scholar]
- Li W, Ono F, Brehm P (2003) Optical measurements of presynaptic release in mutant zebrafish lacking postsynaptic receptors. J Neurosci 23: 10467–10474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang S-D, Vizi ES (1997) Positive feedback modulation of acetylcholine release from isolated rat superior cervical ganglion. J Pharmacol Exp Ther 280: 650–655. [PubMed] [Google Scholar]
- Loring RH, Sah DW, Landis SC, Zigmond RE (1988) The ultrastructural distribution of putative nicotinic receptors on cultured neurons from the rat superior cervical ganglion. Neuroscience 24: 1071–1080. [DOI] [PubMed] [Google Scholar]
- Mandelzys A, Pie B, Deneris ES, Cooper E (1994) The developmental increase in ACh current densities on rat sympathetic neurons correlates with changes in nicotinic ACh receptor α-subunit gene expression and occurs independent of innervation. J Neurosci 14: 2357–2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarlane S, Cooper E (1992) Postnatal development of voltage-gated K currents on rat sympathetic neurons. J Neurophysiol 67: 1291–1300. [DOI] [PubMed] [Google Scholar]
- McGehee DS, Role LW (1995) Physiological diversity of nicotinic acetylcholine receptors expressed by vertebrate neurons. Annu Rev Physiol 57: 521–546. [DOI] [PubMed] [Google Scholar]
- Misgeld T, Burgess RW, Lewis RM, Cunningham JM, Lichtman JW, Sanes JR (2002) Roles of neurotransmitter in synapse formation: development of neuromuscular junctions lacking choline acetyltransferase. Neuron 36: 635–648. [DOI] [PubMed] [Google Scholar]
- Nyberg-Hoffman C, Shabram P, Li W, Giroux D, Aguilar-Cordova E (1997) Sensitivity and reproducibility in adenoviral infectious titer determination. Nat Med 3: 808–811. [DOI] [PubMed] [Google Scholar]
- Okuda T, Haga T, Kanai Y, Endou H, Ishihara T, Katsura I (2000) Identification and characterization of the high-affinity choline transporter. Nat Neurosci 3: 120–125. [DOI] [PubMed] [Google Scholar]
- Oshima S, Yamadab K, Shirakawa T, Watanabe M (2004) Changes of high-affinity choline transporter CHT1 mRNA expression during degeneration and regeneration of hypoglossal nerves in mice. Neurosci Lett 365: 97–101. [DOI] [PubMed] [Google Scholar]
- Petersen SA, Fetter RD, Noordermeer JN, Goodman CS, DiAntonio A (1997) Genetic analysis of glutamate receptors in Drosophila reveals a retrograde signal regulating presynaptic transmitter release. Neuron 19: 1237–1248. [DOI] [PubMed] [Google Scholar]
- Ramcharan EJ, Matthews MR (1996) Autoradiographic localization of functional muscarinic receptors in the rat superior cervical sympathetic ganglion reveals an extensive distribution over non-synaptic surfaces of neuronal somata, dendrites and nerve endings. Neuroscience 71: 797–832. [DOI] [PubMed] [Google Scholar]
- Rogers M, Sargent PB (2003) Rapid activation of presynaptic nicotinic acetylcholine receptors by nerve-released transmitter. Eur J Neurosci 18: 2946–2956. [DOI] [PubMed] [Google Scholar]
- Role LW (1992) Diversity in primary structure and function of neuronal nicotinic acetylcholine receptor channels. Curr Opin Neurobiol 2: 254–262. [DOI] [PubMed] [Google Scholar]
- Rosenberg MM, Blitzblau RC, Olsen DP, Jacob MH (2002) Regulatory mechanisms that govern nicotinic synapse formation in neurons. J Neurobiol 53: 542–555. [DOI] [PubMed] [Google Scholar]
- Rubin E (1985) Development of the rat superior cervical ganglion: initial stages of synapse formation. J Neurosci 5: 697–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanes JR, Lichtman JW (1999) Development of the vertebrate neuromuscular junction. Annu Rev Neurosci 22: 389–442. [DOI] [PubMed] [Google Scholar]
- Sargent PB (1993) The diversity of neuronal nicotinic acetylcholine receptors. Annu Rev Neurosci 16: 403–443. [DOI] [PubMed] [Google Scholar]
- Sharma G, Vijayaraghavan S (2003) Modulation of presynaptic store calcium induces release of glutamate and postsynaptic firing. Neuron 38: 929–939. [DOI] [PubMed] [Google Scholar]
- Smeyne RJ, Klein R, Schnapp A, Long LK, Bryant S, Lewin A, Lira S, Barbacid M (1994) Severe sensory and sympathetic neuropathies in mice carrying a disrupted Trk/NGF receptor gene. Nature 368: 246–249. [DOI] [PubMed] [Google Scholar]
- Tao HW, Poo M (2001) Retrograde signaling at central synapses. Proc Natl Acad Sci USA 98: 11009–11015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG, Nelson SB (2004) Homeostatic plasticity in the developing nervous system. Nat Rev Neurosci 5: 97–107. [DOI] [PubMed] [Google Scholar]
- Ullian EM, McIntosh JM, Sargent PB (1997) Rapid synaptic transmission in the avian ciliary ganglion is mediated by two distinct classes of nicotinic receptors. J Neurosci 17: 7210–7219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhage M, Maia AS, Plomp JJ, Brussaard AB, Heeroma JH, Vermeer H, Toonen RF, Hammer RE, van den Berg TK, Missler M, Geuze HJ, Thomas C, Sudhof TC (2000) Synaptic assembly of the brain in the absence of neurotransmitter secretion. Science 287: 864–869. [DOI] [PubMed] [Google Scholar]
- Wada E, Wada K, Boulter J, Deneris E, Heinemann S, Patrick J, Swanson LW (1989) Distribution of alpha 2, alpha 3, alpha 4, and beta 2 neuronal nicotinic receptor subunit mRNAs in the central nervous system: a hybridization histochemical study in the rat. J Comp Neurol 284: 314–335. [DOI] [PubMed] [Google Scholar]
- Washbourne P, Thompson PM, Carta M, Costa ET, Mathews JR, López-Bendito G, Molnár Z, Becher MW, Valenzuela CF, Partridge LD, Wilson MC (2002) Genetic ablation of the t-SNARE SNAP-25 distinguishes mechanisms of neuroexocytosis. Nat Neurosci 5: 19–26. [DOI] [PubMed] [Google Scholar]
- Westerfield M, Liu DW, Kimmel CB, Walker C (1990) Pathfinding and synapse formation in a zebrafish mutant lacking functional acetylcholine receptors. Neuron 4: 867–874. [DOI] [PubMed] [Google Scholar]
- Wheeler DG, Cooper E (2001) Depolarization strongly induces human cytomegalovirus major immediate-early promoter/enhancer activity in neurons. J Biol Chem 276: 31978–31985. [DOI] [PubMed] [Google Scholar]
- Xu XW, Gelber S, Orr-Urtreger A, Armstrong D, Lewis RA, Ou C, Patrick J, Role L, De Biasi M, Beaudet AL (1999) Megacystis, mydriasis, and ion channel defect in mice lacking the α3 neuronal nicotinic acetylcholine receptor. Proc Natl Acad Sci USA 96: 5746–5751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Coggan J, Berg DK (1996) Synaptic currents generated by neuronal acetylcholine receptors sensitive to alpha-bungarotoxin. Neuron 17: 1231–1240. [DOI] [PubMed] [Google Scholar]