

Abstract
Histone deacetylase 4 (HDAC4) undergoes signal-dependent shuttling between the cytoplasm and nucleus, which is regulated in part by calcium/calmodulin-dependent kinase (CaMK)-mediated phosphorylation. Here, we report that HDAC4 intracellular trafficking is important in regulating neuronal cell death. HDAC4 is normally localized to the cytoplasm in brain tissue and cultured cerebellar granule neurons (CGNs). However, in response to low-potassium or excitotoxic glutamate conditions that induce neuronal cell death, HDAC4 rapidly translocates into the nucleus of cultured CGNs. Treatment with the neuronal survival factor BDNF suppresses HDAC4 nuclear translocation, whereas a proapoptotic CaMK inhibitor stimulates HDAC4 nuclear accumulation. Moreover, ectopic expression of nuclear-localized HDAC4 promotes neuronal apoptosis and represses the transcriptional activities of myocyte enhancer factor 2 and cAMP response element-binding protein, survival factors in neurons. In contrast, inactivation of HDAC4 by small interfering RNA or HDAC inhibitors suppresses neuronal cell death. Finally, an increase of nuclear HDAC4 in granule neurons is also observed in weaver mice, which harbor a mutation that promotes CGN apoptosis. Our data identify HDAC4 and its intracellular trafficking as key effectors of multiple pathways that regulate neuronal cell death.
Keywords: apoptosis, cell death, deacetylase, HDAC4, MEF2, neuron
Introduction
Neuronal cell death is a tightly regulated process. It is involved intimately in both normal development of the nervous system and neurological disorders (Contestabile, 2002). Developmentally regulated neuron death eliminates the large excess of neurons that are produced, thereby preserving only those neurons needed for proper synaptic connections (Benn and Woolf, 2004). In contrast, neuron death in the adult brain is common in a variety of disorders including those associated with cerebral ischemia and neurodegenerative diseases (Contestabile, 2002). Deciphering the machinery that controls neuronal cell death can thus provide both great insight into nervous system development and new therapeutic targets to prevent disease-induced neuron loss.
Many signaling events have been shown to regulate neuron death. Calcium signaling has been identified as a major pathway that protects neurons from cell death (Hack et al., 1993; Contestabile, 2002). A key target of calcium signaling is the transcription factor myocyte enhancer factor 2 (MEF2). Although initially characterized as regulators of muscle differentiation, MEF2 members have now emerged as important neuronal survival factors as well (Mao et al., 1999; Gaudilliere et al., 2002). Calcium signaling regulates MEF2 through multiple mechanisms. A critical one involves a subfamily of the histone deacetylases (HDACs): the class IIA HDACs.
HDACs have been characterized primarily as transcriptional corepressors that catalyze local histone deacetylation (Nagy et al., 1997; Strahl and Allis, 2000). The class IIA histone deacetylases, which include HDAC4, 5, 7, and 9, share several unique features (for review, see Verdin et al., 2003). They all bind MEF2 and repress its activity, and they all undergo intracellular trafficking between the cytoplasm and nucleus regulated by signal-induced phosphorylation (Miska et al., 1999; Bertos et al., 2001). For example, HDAC4 can be phosphorylated by calcium/calmodulin-dependent kinase IV (CaMKIV) (Zhao et al., 2001). Phosphorylation recruits the phospho-binding protein 14-3-3, and the resulting complex is exported efficiently from the nucleus (McKinsey et al., 2001; Wang and Yang, 2001). HDAC4 can subsequently reenter the nucleus after dephosphorylation and dissociation from 14-3-3 (Grozinger and Schreiber, 2000). Importantly, active shuttling of HDAC4 can be observed under specific conditions, such as the differentiation of C2C12 myoblasts, although the physiological significance of this is not completely understood (Zhao et al., 2001).
Although the focus of HDAC4 research has centered on muscle and chondrocyte development (Lu et al., 2000; Vega et al., 2004), HDAC4 mRNA is highly abundant in the brain (Grozinger et al., 1999), suggesting it has a function there. In this study, we report that HDAC4 is a critical regulator for neuronal cell death. We show that HDAC4 accumulates in the nucleus in response to cell-death conditions in cultured cerebellar granule neurons (CGNs) and that this translocation can be modulated by other survival factors. Nuclear accumulation of HDAC4 promotes CGN death and represses MEF2- and cAMP response element (CRE)-binding protein (CREB)-dependent transcription, whereas inactivation of HDAC4 by small interfering RNA (siRNA) or pharmacological inhibitors inhibits cell death. Finally, we observe increased nuclear HDAC4 in vivo in the external granule layer (EGL) of mice with increased CGN apoptosis. Together, these data identify HDAC4 and its intracellular trafficking as important regulatory elements in neuronal cell death.
Materials and Methods
Reagents and antibodies. The α-HDAC4 polyclonal antibody ab186 was used as described previously (Zhao et al., 2001). Monoclonal antibodies against green fluorescent protein (GFP) and actin were from Roche (Indianapolis, IN) and Sigma (St. Louis, MO), respectively. The α-active-caspase-3 antibody (polyclonal) was from Promega (Madison, WI). Secondary antibodies used were from Jackson ImmunoResearch (West Grove, PA) and included goat anti-rabbit indocarbocyanine (Cy 3), goat anti-rabbit RedX, goat anti-mouse Cy3, and goat anti-mouse Cy2. Leptomycin B, trichostatin A (TSA), and trapoxin A (TPX) were gifts from M. Yoshida (University of Tokyo, Tokyo, Japan). 2-[N-(2-hydroxyethyl)]-N-(4-methoxybenzenesulfonyl)]amino-N-(4-chloro-cinnamyl-N-methylbenzylamine) (KN-93) was a gift from A. R. Means (Duke University). Other reagents were from Sigma unless otherwise noted.
Plasmids. Vectors for siRNA experiments (pSuper) were obtained from OligoEngine (Seattle, WA). The oligo used for HDAC4 knock-down was against murine HDAC4, which was inferred from sequence comparison to human HDAC4, starting with the predicted murine HDAC4 gene in the Ensembl database. The sequence used was gcggcagatactcattgcaga (5′-3′), to which was appended a hairpin loop structure. pEGFP-N3 plasmids (Invitrogen, Carlsbad, CA) were used to construct to express GFP, human HDAC4, and the 3SA mutant. The NT-3(-1117)-luciferase fusion reporter used in luciferase assays was obtained from the Bonni laboratory (Harvard University, Boston, MA). pEGFP-C2 plasmids containing HDAC4-Δ118 and HDAC4-Δ118-3SA mutants were obtained from the Wang laboratory (McGill University, Montreal, Quebec, Canada).
Tissue blot. Organs were obtained from adult male C57BL/6 mice (∼20 weeks of age). Organs were homogenized in 25 mm HEPES buffer with 1 mm EGTA, 1 mm EDTA, 10% glycerol, and protease inhibitors. Fifty micrograms from each sample were used for SDS-PAGE and Western blotting with ab186. Data were confirmed with independent samples from three different mice.
Genotyping. Heterozygous weaver mice (obtained from The Jackson Laboratory, Bar Harbor, ME) were bred to produce the mice used in the weaver experiments. To genotype weaver mutant mice, toe or tail clippings from mouse pups were digested in 10 mm Tris buffer, pH 8.0, with 100 mm NaCl, 25 mm EDTA, 0.5% SDS, and 10 mg/ml Proteinase K. Genomic DNA was isolated by phenol extraction and ethanol precipitation, then subjected to PCR of the region surrounding the weaver mutation in the Kcnj6 (potassium inwardly rectifying channel, subfamily J, member 6) gene. The PCR primers used were gattccatggaccaggatgtg and catgaaggcgttgacaatgga (5′-3′). PCR products were gel purified and sequenced using a third primer (cgatacctgacggacatcttc), ∼200 bp upstream from the weaver mutation site. The chromotograms were then analyzed to determine whether the pup was wild type, heterozygous, or homozygous for the weaver mutation.
Immunohistochemistry. Neonatal mouse cerebellums were dissected, fixed in paraformaldehyde, saturated with 25% sucrose solution, then frozen in Tissue-Tek OCT media (VWR, West Chester, PA). Frozen sections were then sliced by cryostat by members of the Duke Department of Pathology. Frozen sections from adult rat cortex were obtained from the Schwartz-Bloom laboratory (Duke University). For immunofluorescent staining, all frozen sections were thawed, fixed in paraformaldehyde, washed, blocked in 5% normal goat serum, and incubated with primary antibody overnight at 4°C. Sections were then washed, blocked again, incubated with secondary antibody, washed, stained with Hoechst dye, and mounted with Fluoromount-G (Southern Biotechnologies, Birmingham, AL).
Cerebellar granule neuron isolation and culture. Cultures were obtained essentially as described previously (Oliver et al., 2003). Briefly, hind-brains were obtained from postnatal day 7 (P7) to P9 mice from C57BL/6 background. Cells were dissociated through papain treatment, trituration with ovomucoid (50 mg/ml BSA and 50 mg/ml trypsin inhibitor; Roche), and 70 μm cell strainer (BD Biosciences, San Jose, CA). Samples were then run over a Percoll step gradient (0, 35, 65%), and granule cells at the 35/65 interface were recovered and plated onto poly-d-lysinecoated coverslips (Fisher, Pittsburgh, PA) and plates (BD Biosciences). Unless otherwise noted, cells were generally cultured in full media (FM), which consisted of Neurobasal media with 5% FBS, B27 supplement, sodium pyruvate, l-glutamine, penicillin/streptomycin, 0.6 mg/ml glucose, and 25 mm KCl. After 1 d in culture, 10 μm cytosine arabinoside was added to cells to inhibit non-neuronal cells. When cultured longer than 2 d, media was changed every 2-3 d by removing one-half of the conditioned media and then replacing it with fresh FM media.
CGN treatment conditions. For low-potassium (LK) conditions, media consisted of FM with only 5 mm KCl and without serum and B27. For high-potassium (HK) media used in glutamate excitotoxicity experiments, media consisted of FM without serum and B27. When cells were switched from FM to LK or HK, media were aspirated from cells, they were washed in plain Neurobasal, and new media were added. Cells were then incubated for various times in the altered media. When used, leptomycin B, BDNF, KN-93, and glutamic acid were added either directly to cells already in FM media or to cells in LK or HK media immediately after the media were changed. For the HDAC inhibitor experiments, cells were first pretreated with TSA and TPX for 5 h. The media were changed for LK samples as above, and TSA and TPX were readded to the media. All samples were then incubated an additional 12 h before harvest.
Transfection of CGNs. Cells were transfected immediately after isolation using the Mouse Neuron Nucleofector kit (Amaxa, Gaithersburg, MD). Briefly, 3-5 × 106 cells were centrifuged and resuspended in transfection buffer. DNA was added, and samples were transferred to electroporation cuvettes. Program O-05 was then run on the samples in the Amaxa Nucleofector device. Samples were then diluted with DMEM with 10% calf serum before being split onto four to six wells of 24-well plates or one or two 6 cm plates. Cells were allowed to settle onto coverslips and plates for 2-3 h, then the cell media were replaced with FM media. Cells were then cultured for 2 d before harvesting.
Immunofluorescent staining of CGNs. Samples were stained for immunofluorescence essentially as described previously (Zhao et al., 2001). When samples were harvested, the poly-d-lysine-coated coverslips were rinsed with PBS and fixed with 4% paraformaldehyde. They were washed, then permeabilized with 0.2% Triton X-100. Samples were blocked with 5% NGS and incubated with primary antibody overnight at 4°C. Sections were then washed, incubated with secondary antibody, washed again, stained with Hoechst dye, and mounted with Fluoromount-G.
CGN cell sorting. For showing the reduction of HDAC4 expression by siRNA, CGNs were first transfected as above with either pEGFP and empty pSuper vector or pEGFP and pSuper-m4HD4, and they were then cultured for 2 d in vitro. Cells were harvested by incubation with papain and ovomucoid solutions as in CGN isolation. Cells were then sorted by the Duke Department of Pharmacology fluorescence-activated cell sorting (FACS) facility for GFP positives. Cells from control and HDAC4 siRNA transfections were lysed with SDS sample buffer and boiled. Equal cell numbers were run on SDS-PAGE and immunoblotted using ab186 and α-actin. Relative band intensity was calculated using NIH Image software. In the image shown, lysates corresponding to 71,000 GFP-positive cells were used.
Luciferase assays. For luciferase assays, CGNs were transfected as above with an NT-3(-1117)-luciferase fusion reporter (Shalizi et al., 2003) and GFP, HDAC4, or HDAC4-3SA. After 2 d in culture, three wells of GFP cells were shifted to LK media for 3 h. Luciferase assays were then performed in triplicate essentially as described previously (Zhao et al., 2001). A measuring time of 30 s was used to show sufficient luciferase activity. The assay results were normalized to GFP in FM media for fold activation. Three independent experiments showed similar trends.
CGN lysates. For preparation of lysates, cultured CGNs were collected and pelleted, then lysed in buffer containing 20 mm HEPES, 0.1% Triton X-100, 50 mm NaCl, 1 mm EGTA, and inhibitors (aprotinin, leupeptin, PMSF, DTT, Na2SO4, and NaF). Samples were sonicated briefly, then rocked for 30 min at 4°C. Samples were spun at 6000 × g for 5 min to remove cell debris. Protein concentration was determined by Bradford assay (Bio-Rad, Hercules, CA).
Microscopy. All images shown were taken with a Zeiss (Thornwood, NY) LSM 410 confocal microscope with a 63× objective lens, except for Figure 1 B and supplemental Figure 1 (available at www.jneurosci.org as supplemental material), which were taken with a Zeiss Axioskop compound microscope with a 20× objective (adult cortex) or with the LSM 410 with a 40× objective (neonatal cerebellum). Images were pseudocolored with green for GFP or HDAC4 staining, blue for nuclei, and red for active caspase-3. For quantitation of nuclear fluorescence, the same microscope-lens combination was used. For analyses of localization and cell death in cultured CGNs, samples were examined with the Zeiss Axioskop with a 63× objective. For the analysis of cerebellar sections for in vivo localization, photographs were taken using the LSM 410 with a 40× objective.
Figure 1.
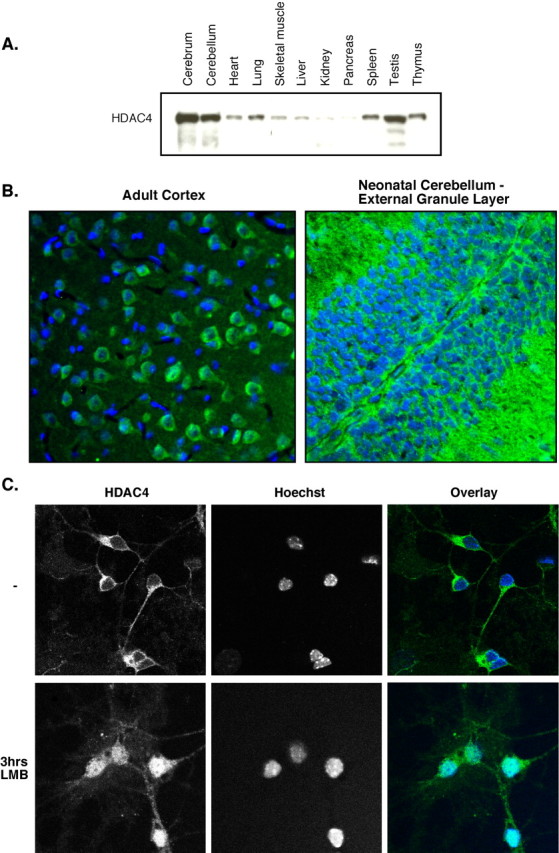
HDAC4 is expressed highly in the brain and is primarily cytoplasmic in neurons. A, C57BL/6 adult male mice were dissected, and organs were homogenized. Fifty micrograms of each sample were run on SDS-PAGE and blotted with ab186 (HDAC4). B, Adult rat neocortex (left) and neonatal mouse cerebellum (right; EGL pictured) were immunofluorescently stained with ab186 (green) and Hoechst (blue). C, CGNs were isolated and cultured for 6 d in vitro in full media. Leptomycin B (LMB) was then added to the cells or not, and cells were stained after 3 h of treatment with ab186 and Hoechst dye. Representative images are shown.
Data analysis. For localization studies, 200 or more cells were examined in random fields in at least three experiments. Non-neuronal cell contaminants in the culture were not counted. A cell was counted as “cytoplasmic” if the HDAC4 staining in the cytoplasm was visibly stronger than in the nucleus. Cells were counted as “high nuclear” if the intensity of nuclear staining equaled or exceeded the cytoplasmic staining. Therefore, cytoplasmic plus high nuclear always equaled 100% for each sample. For overexpression studies, > 100 cells were counted in at least five experiments. First, cells were identified as transfected by staining for GFP or ab186, then the nuclei of transfected cells were examined. If the nucleus of a cell was condensed, fragmented, or perforated, the cell was counted as “dead/dying.” For the caspase-3 experiment, 200 or more cells were counted for each condition in three independent experiments. GFP-positive cells were identified and then assessed for active-caspase-3 staining. In HDAC inhibitor studies, > 400 cells were counted in at least three experiments. Nuclei were examined as above in random fields. Nuclei that appeared morphologically distinct from CGN nuclei were excluded. For nuclear fluorescence quantitation, images were obtained of ∼120 cells in both FM and LK media in each of two independent experiments. Hoechst staining was used to determine the boundaries of the nuclei, and Zeiss software was used to measure the average fluorescence of HDAC4 staining within that area. Values were expressed in relative units with a theoretical range of 0-255. To correct for differences in staining intensity, machine settings, etc. between experiments, the average nuclear fluorescence for each cell in both conditions was normalized to the overall mean for FM media. A histogram was then plotted for each condition (FM and 3 h in LK media) using bins of 15 (all of the cells with a fluorescence of 0-15 in one group, 15-30 in the next, etc.). The histogram values were corrected for the small difference in number of cells between conditions by conversion into frequencies per 100 cells. For localization analysis in vivo, three or four mice were used for each genotype, and seven or more fields of EGL were photographed for each section. Localization was determined using the same criteria as in cell cultures. Five hundred or more cells were counted for each animal. For all experiments, data were tallied, analyzed, and graphed using Microsoft (Redmond, WA) Excel. Error bars represent 1 SD above and below the mean for each sample. Significance was ascertained by Student's t test.
Results
HDAC4 has been characterized previously in muscle and chondrocyte differentiation (Vega et al., 2004). However, immunoblotting of tissue homogenates from a variety of mouse organs using an antibody specific for HDAC4 revealed that HDAC4 protein is expressed highly in both the cerebrum and cerebellum (Fig. 1A). Immunohistochemical staining of mouse brain tissue confirms the abundant presence of HDAC4. Interestingly, HDAC4 is primarily localized to the cytoplasm in both adult and neonatal neurons (Fig. 1B) while being primarily excluded from astrocytes (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). A portion of HDAC4 may also be present in the molecular layer, observed both in tissue sections and in the processes of cultured neurons (data not shown). Consistent with these observations, HDAC4 is a cytoplasmic protein in primary CGNs isolated from neonatal mice (Fig. 1C). Importantly, treatment with the nuclear export inhibitor leptomycin B caused accumulation of HDAC4 in nuclei, revealing that HDAC4 is subject to active nuclear export and dynamically cycles between the nucleus and cytoplasm.
We next wished to explore whether HDAC4 intracellular trafficking is regulated in neurons. Because MEF2, a major target of HDAC4, is a prosurvival factor in neurons (Mao et al., 1999), we reasoned that HDAC4 might be involved in neuronal survival. Primary CGNs are responsive to a number of different neurotrophic and neurotoxic stimuli (Vaudry et al., 2003). Cultured CGNs are protected from spontaneous cell death by the presence of depolarizing levels of potassium in the medium, which is believed to simulate synaptic activity that occurs in vivo. If the potassium concentration is reduced in the CGN media, the cells undergo apoptotic cell death. This effect has been used to study many factors influencing neuron survival (Contestabile, 2002).
To determine whether HDAC4 is involved in neuronal cell death, we incubated CGNs in FM or LK media and then examined the subcellular localization of HDAC4 by immunostaining. We found that after only 20 min in LK media, HDAC4 began to translocate from the cytoplasm to the nucleus (Fig. 2A,B). After 3 h in LK media, > 75% of the cells displayed significant nuclear accumulation of HDAC4. This result was further confirmed by quantifying the average nuclear fluorescence of HDAC4 in cells maintained in FM and in LK media for 3 h (Fig. 2C). A histogram of the normalized nuclear HDAC4 fluorescence demonstrates that the cell population in LK media (mean of 120) is clearly right shifted compared with the FM population (mean of 64). This is not a result of a change in expression of HDAC4 because HDAC4 levels do not change significantly during the treatment times (Fig. 2D). To further confirm HDAC4 translocation, we transfected CGNs with wild-type HDAC4 and a phosphorylation-deficient mutant, HDAC4-3SA, that has been shown to be resistant to nuclear export in other cell types (Zhao et al., 2001). We again examined the subcellular distribution of the transfected cells in FM and LK media (Fig. 2E). As expected, wild-type HDAC4 accumulated in the nucleus in a majority of transfected cells after 3 h of LK treatment, whereas the HDAC4-3SA mutant was present in the nucleus in high amounts under all conditions.
Figure 2.
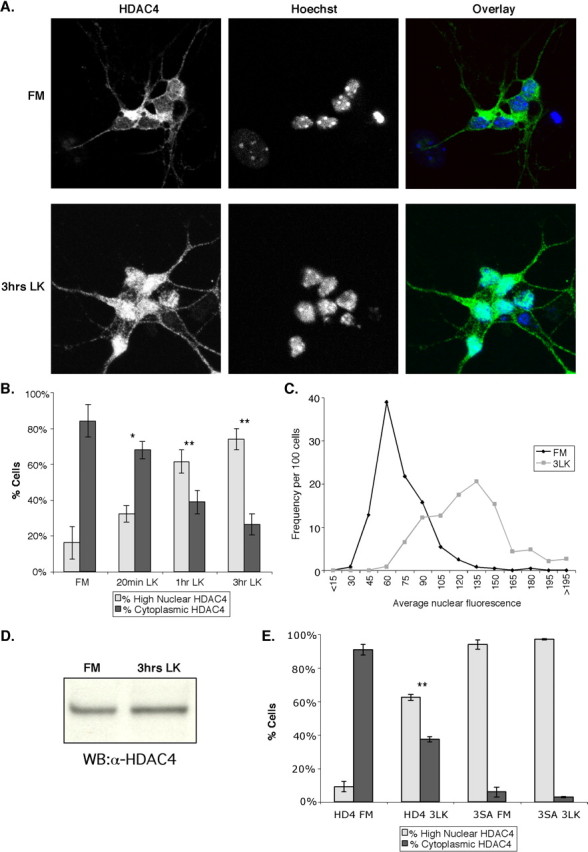
HDAC4 translocates to the nucleus under conditions that induce cell death. After 5-7 d invitro, CGNs were left in FM or replaced with LK media. Cells were stained with ab186 and Hoechst dye. Representative images are shown in A. B, A quantification of the data is shown. Cells were considered cytoplasmic if cytoplasmic staining was brighter than nuclear staining. If the nuclear staining was equal to or greater than the cytoplasmic staining, the cell was counted as high-nuclear HDAC4. *p < 0.05; **p < 0.005 compared with FM. Three or more experiments were performed for each condition, and > 200 cells were counted in each experiment. C, After 5 d in vitro, CGNs were left in FM media or replaced with LK media for 3 h. Cells were then stained with ab186 and Hoechst. Images were taken of ∼120 cells in both FM and LK cells in each of two independent experiments, and average nuclear fluorescence of each cell was obtained and normalized. The histogram was then plotted of nuclear fluorescence versus number of cells for the FM and 3 h in LK conditions (3LK). The x-axis groups the cells by average nuclear fluorescence; for example, the “45” group represents all the cells with nuclear fluorescences between 30 and 45 units (on a 0-255 scale). The y-axis gives the number of cells in each group per 100 cells counted. D, CGN lysates from cells in FM and in LK media for 3 h were run on SDS-PAGE and immunoblotted with ab186. WB, Western blot. E, CGNs were transfected with GFP, HDAC4 (HD4), or HDAC4-3SA (3SA) immediately after isolation. After 2 d in vitro, the localization of HDAC4- and HD4-3SA-transfected CGNs was assessed in FM media and LK media after 3 h (3LK). Cells were stained with ab186 and Hoechst dye. **p < 0.005 versus HDAC4 in FM. Three or more experiments were performed for each condition, and > 100 cells were counted in each experiment. Error bars represent one SD above and below the mean.
To establish whether HDAC4 translocation is limited to the low-potassium paradigm of cell death, we induced cell death in CGNs via glutamate excitotoxicity. Importantly, a similar nuclear translocation of HDAC4 was observed in these excitotoxic conditions (Fig. 3A). This observation indicates that the HDAC4 translocation is not limited to the low-potassium state but rather seems to be a more general response to neuronal cell death. Addition or withdrawal of neurotrophic factors is another method of affecting neuronal cell death. Neurotrophins such as BDNF have been shown to prevent cell death of neurons, including CGNs under LK conditions (Segal et al., 1992; Kubo et al., 1995). To further establish that HDAC4 nuclear translocation is a specific response to neuronal cell-death signaling, we examined whether BDNF treatment affected HDAC4 localization in response to LK medium. As expected, BDNF had no effect on HDAC4 localization in FM media (data not shown). In contrast, BDNF mostly prevented HDAC4 nuclear translocation in LK media, with the majority (65%) of cells still displaying cytoplasmic HDAC4 staining (Fig. 3B). These results support the notion that HDAC4 nuclear translocation is a bona fide response to cell-death conditions.
Figure 3.

Glutamate excitotoxicity, BDNF, and CaMK can regulate HDAC4 translocation in CGNs. A, After 6-7 d in vitro, CGNs were left in FM or replaced with no-serum HK media (3HK). One hundred micromolar l-glutamic acid (Glut) was then added to some HK samples to induce an excitotoxic response, and all cells were incubated for 3 h. Cells were then stained with ab186 and Hoechst dye. A quantification of the data are shown. **p < 0.001 compared with untreated HK. Three or more experiments were performed for each condition, and > 200 cells were counted in each experiment. B, After 2 d in vitro, CGNs were left in FM or replaced with LK media. Some cells (FM and LK) were treated with 100 ng/ml BDNF and/or 8 μm KN-93 at the same time as potassium deprivation (3 h). Cells were stained with ab186 and Hoechst dye. A quantification of the data is shown. ++p < 0.001 compared with untreated LK for 3 h; ** p < 0.001 compared with untreated FM. Three or more experiments were performed for each condition, and > 200 cells were counted in each experiment. Error bars represent one SD above and below the mean.
CaMKs also act to promote neuronal survival (Hack et al., 1993). Notably, CaMKs have been shown to directly phosphorylate HDAC4 and promote its nuclear export (Zhao et al., 2001). We therefore tested whether inhibition of CaMKs affects HDAC4 subcellular localization. As shown in Figure 3B, even in FM media, treatment with a CaMK inhibitor, KN-93, induced HDAC4 nuclear translocation in > 70% of cells. Interestingly, HDAC4 translocation induced by KN-93 is not reversed by BDNF (Fig. 3B), suggesting that CaMK may be a downstream effector of BDNF in a signaling pathway that regulates HDAC4 intracellular trafficking. Together, these observations confirm that CaMK can regulate neuronal HDAC4 localization and provide an additional link between neuronal cell death and HDAC4 nuclear accumulation.
Our results so far established a correlation between HDAC4 translocation and neuronal cell death. To determine whether HDAC4 nuclear accumulation is important for neuronal cell death, wild-type HDAC4 and the phosphorylation-deficient mutant of HDAC4 (HDAC4-3SA) were transfected into CGNs. Overexpressed wild-type HDAC4 is found primarily in the cytoplasm of untreated CGNs (Figs. 2E, 4A). In contrast, high-nuclear staining is observed for the HDAC4-3SA mutant in both FM and LK media. The expression levels of these constructs were similar (Fig. 4B). We then assessed cell death in the transfected CGNs under different potassium conditions. Two days after transfection, <3% of control cells transfected with GFP displayed nuclear morphology indicative of cell death (Fig. 4C). In contrast, > 11% of neurons expressing HDAC4-3SA were undergoing cell death. Similarly, CGNs transfected with the HDAC4-3SA mutant display a much higher rate of cell death under LK conditions (nearly 40% after 6 h of LK) than either control cells or cells transfected with wild-type HDAC4. Overexpression of wild-type HDAC4 also showed a moderate effect in inducing cell death; however, it was consistently less potent than the HDAC4-3SA mutant (Fig. 4C). To confirm that the effect of HDAC4 on CGN death is caused by the increased nuclear localization of HDAC4, CGNs were transfected with two mutants of HDAC4 (HDAC4-Δ118 and HDAC4-Δ118-3SA) that have been shown previously to be primarily restricted to the cytoplasm (Wang and Yang, 2001). Cells transfected with these mutants did not have increased cell death compared with GFP controls, demonstrating that nuclear localization of HDAC4 is necessary for its effect on neuronal cell death (supplemental Fig. 2A, available at www.jneurosci.org as supplemental material). The increase in cell death is also dependent on the enzymatic activity of HDAC4, because transfection of an enzymatically dead mutant of HDAC4 (H803A) (Miska et al., 1999) does not promote cell death effectively (data not shown). The effect of HDAC4-3SA indicates that nuclear accumulation of HDAC4 promotes CGN cell death.
Figure 4.
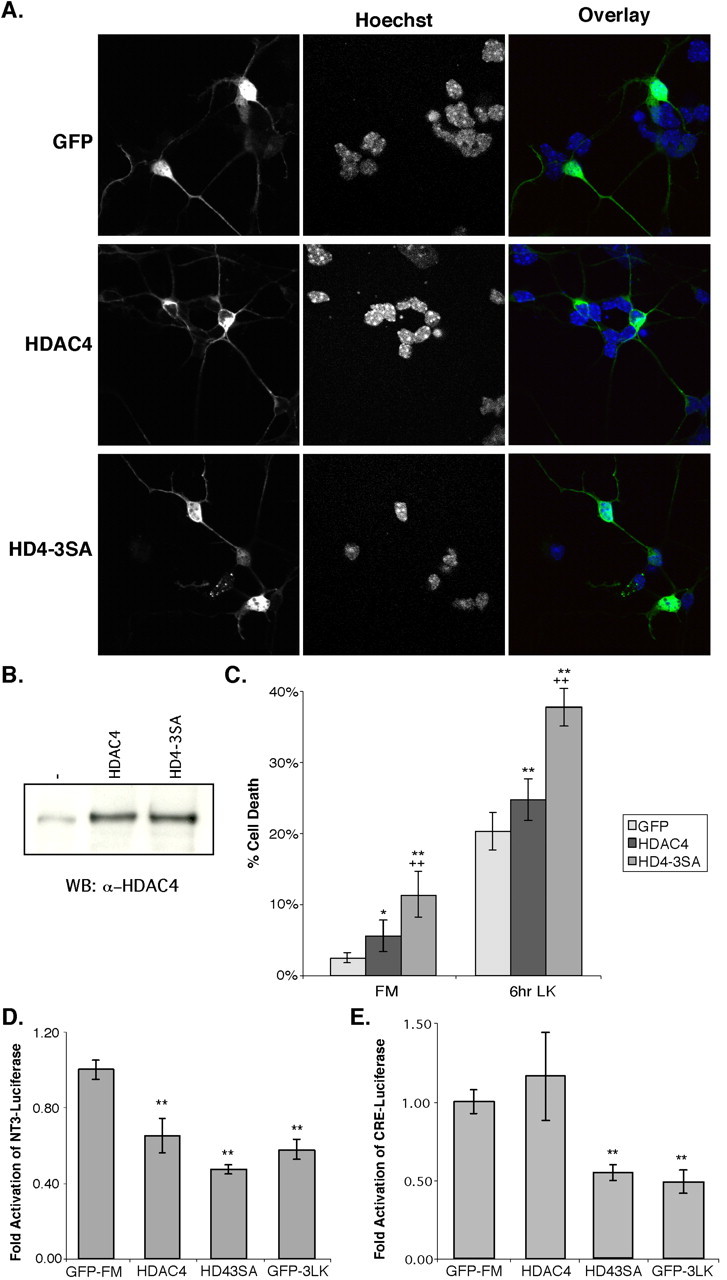
Overexpression of HDAC4 causes increased sensitivity to cell death and represses MEF2- and CREB-dependent activity. Immediately after isolation, CGNs were transfected with GFP, HDAC4, or HDAC4-3SA. After 2 d in vitro, the cells were left in FM or replaced with LK media for either 3 or 6 h. Cells were stained with Hoechst dye and either ab186 or αGFP. A, Representative images of cells in FM media are shown. B, Untransfected, HDAC4-transfected, or HD4-3SA-transfected CGNs were harvested and immunoblotted using ab186. WB, Western blot. C, Transfected cells were assessed for their viability by observing nuclei. Cells with nuclei that appeared condensed, fragmented, or perforated were considered dead/dying. *p < 0.05, **p < 0.01 compared with GFP control at each condition; ++p < 0.01 compared with wild-type HDAC4 at each condition. Five or more experiments were performed for each condition, and > 100 cells were counted in each experiment. D, CGNs were transfected with an NT-3(-1117)-luciferase fusion reporter (Shalizi et al., 2003) and GFP as a control, HDAC4, or HDAC4-3SA. After 2 d in vitro, some GFP cells were shifted to LK media for 3 h (GFP-3LK). Luciferase assays were then performed in triplicate, and the results were normalized to GFP in FM media for fold activation. A representative experiment is shown. **p < 0.01 compared with GFP control in FM. E, CGNs were transfected with a CRE-containing luciferase reporter (from the somatostatinpromoter) and GFP as a control, HDAC4, or HDAC4-3SA. After 2 d invitro, some GFP cells were shifted to LK media for 3 h (GFP-3LK). Luciferase assays were then performed in triplicate, and the results were normalized to GFP in FM media for fold activation. **p <0.001 versus CRE-luciferasein FM. A representative experiment is shown. HD4-3SA, HDAC4-3SA; 3 LK, 3 h in LK conditions. Error bars represent one SD above and below the mean.
To begin to address the mechanism by which HDAC4 promotes neuronal cell death, we investigated whether HDAC4 represses transcription in CGNs. As discussed above, MEF2, a known HDAC4 target, promotes CGN survival by activating the transcription of neurotrophin-3 (NT-3) (Shalizi et al., 2003). Therefore, we examined whether HDAC4 could repress activity of the NT-3 promoter in CGNs. As shown in Figure 4D, HDAC4-3SA repressed transcription of a luciferase reporter driven by the NT-3 promoter to a level similar to the reduction in control cells after 3 h in LK media. Wild-type HDAC4 repressed NT3-luciferase to an intermediate level (Fig. 4D). Thus, the repression of MEF2-mediated NT-3 promoter activity by HDAC4 is consistent with the increase in cell death induced by HDAC4-3SA and low-potassium conditions. We also considered whether HDAC4 might have other targets in addition to MEF2. CREB, another transcription factor regulated by calcium pathways, also promotes neuron survival (Bonni et al., 1995). Therefore, we tested whether HDAC4 could repress transcription directed by CREB by transfecting a CRE-dependent luciferase reporter into CGNs along with HDAC4 constructs. The HDAC4-3SA mutant was able to repress transcription from this reporter, again reducing activity to a similar extent as 3 h of LK treatment (Fig. 4E). Using this reporter, wild-type HDAC4 did not have a consistent effect. These results indicate that HDAC4 promotes neuron death by repressing multiple prosurvival transcription factors, including MEF2 and CREB.
If nuclear accumulation of HDAC4 is important for neuronal cell death, then CGNs lacking HDAC4 should be more resistant to cell death-inducing agents such as low potassium. To test this, CGNs were cotransfected with an HDAC4-specific siRNA (m4HD4) plasmid and GFP. Immunoblotting demonstrated that HDAC4 protein levels were reduced between 60 and 70% in the siRNA-transfected CGNs relative to vector controls after FACS sorting for GFP-positive cells (Fig. 5A). Unsorted control and HDAC4 knock-down CGNs were subjected to potassium withdrawal, and cell death was assessed in GFP-positive cells with two different apoptotic markers, nuclear morphology and the active form of caspase-3. Although only a few cells displayed abnormal nuclear morphology in FM media, 22% of vector control cells had nuclear morphology indicative of cell death after 6 h in LK media (Fig. 5B). Strikingly, only 10% of cells transfected with HDAC4 siRNA were dead or dying after 6 h of LK. To confirm these results, transfected cells were stained for active caspase-3. As expected, very few cells displayed positive staining for active caspase-3 in FM media (Fig. 5Cb,Cf). In contrast, after 6 h of LK treatment, nearly 30% of vector control cells were stained positively for active caspase-3 (Fig. 5Ci-Cl, arrows, D). In contrast, only 11% of cells transfected with the m4HD4 siRNA were positive for active caspase-3 (Fig. 5Cm-Cp, arrows, D), whereas untransfected cells in these samples underwent normal cell death in response to LK conditions (Fig. 5Cj,Cn, arrowheads). Transfection of a second siRNA targeted to a different region of HDAC4 showed similar results, further strengthening these observations (supplemental Fig. 3A, available at www.jneurosci.org as supplemental material). These findings strongly suggest that HDAC4 is required for efficient neuronal cell death.
Figure 5.
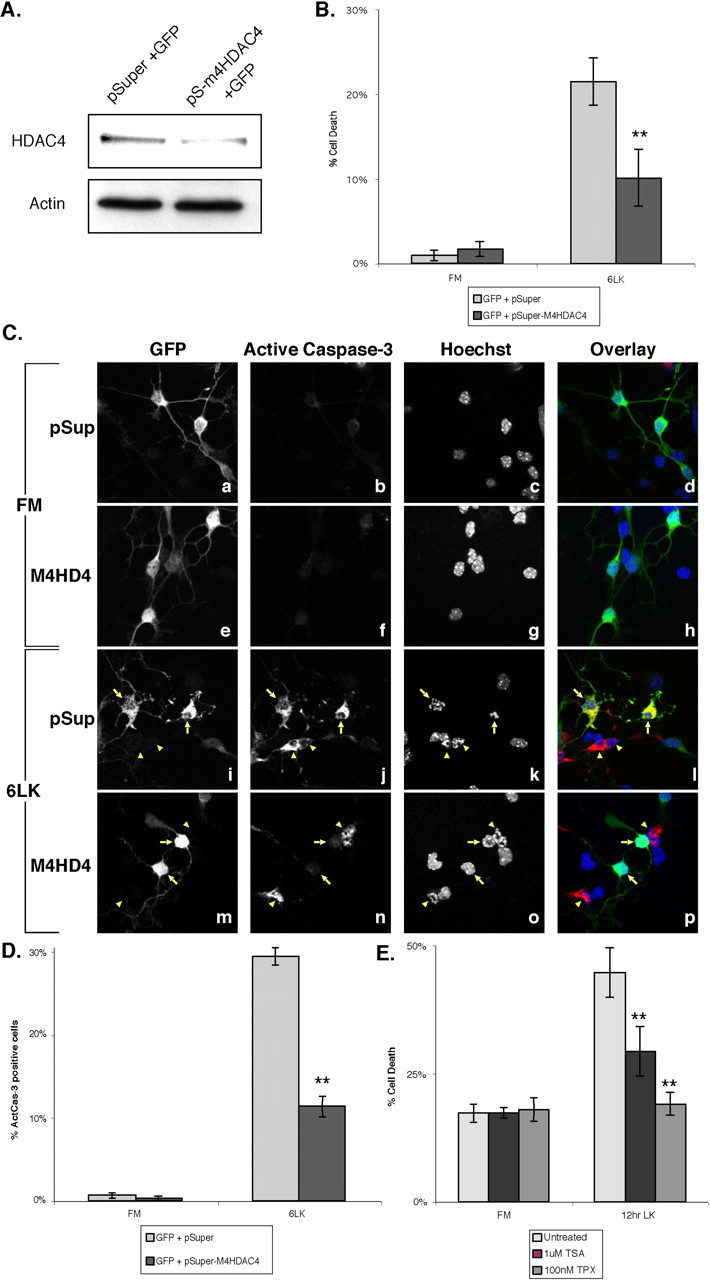
Inhibition of HDAC4 by siRNA or pharmacological inhibitors reduces cell death of CGNs. Immediately after isolation, CGNs were cotransfected with GFP and either pSuper (pSup) or pSuper-m4HD4 (pS-m4HD4). A, After 2 d in vitro, cells were sorted for GFP positives by FACS. Lysates were equalized for cell number, then blotted for HDAC4 and actin. Band intensity was quantitated with NIH Image. B-D, Transfected cells were left in FM media (Ca-Ch) or switched to LK media for 6 h (Ci-Cp). The cells were stained with αGFP, Hoechst dye, and α-active-caspase-3 (C, D only). B, Nuclear morphology of GFP-positive cells was observed, and cells with nuclei that appeared condensed, fragmented, or perforated were considered dead/dying. **p <0.002 compared with control at 6LK. Four or more experiments were performed for each condition, and 100 or more cells were counted in each experiment. C, D, GFP-positive cells were assessed for the presence of positive staining for active caspase-3. Representative images are shown in C. Ci-Cp, Arrows point to transfected cells, and arrowheads to apoptotic untransfected cells. The data are quantitated in D. **p < 0.002 compared with control at 6LK. Three experiments were performed for eachcondition, and 200 or more cells were counted in each experiment. E, Untransfected CGNs after 6-8 d in vitro were pretreated with 1 μm TSA or 100 nm Trapoxin-A for 5 h or left untreated. Samples were then left in FM or replaced with LK media with TSA or TPX added for an additional 12 h. Cells were stained with Hoechst dye, and cell death was evaluated by examination of nuclei as above. **p < 0.002 compared with untreated LK media for 12 h (12 hr LK). Three or more experiments were performed for each condition, and > 400 cells were counted in each experiment. 6LK, 6 h in LK media. Error bars represent one SD above and below the mean.
To obtain additional evidence of the role of HDAC4 in neuronal cell death, we treated neurons with pharmacological inhibitors of HDAC4. Mature CGNs were pretreated for 5 h with the HDAC inhibitors TSA or TPX, then switched to LK media containing the inhibitors for 12 h. Treatment with either inhibitor for 17 h in FM media did not affect cell death significantly (Fig. 5E). In untreated cells, cell death was ∼45% after 12 h in low potassium. Intriguingly, TPX treatment completely reversed cell death caused by low potassium, reducing it to the level seen in FM media (Fig. 5E). TSA also inhibited cell death, although not to the same extent as TPX. These results show that HDAC inhibitors are capable of suppressing neuronal cell death and support the idea that HDAC4 plays an important role in this process.
Finally, we sought evidence of nuclear HDAC4 accumulation after neuron death in vivo during mouse development. We chose to study the weaver mutant mouse strain because it has a mutation in a potassium channel (Kcnj6/G-protein-gated inwardly rectifying K+ channel 2) that causes substantial death of cerebellar granule cells (Rakic and Sidman, 1973; Patil et al., 1995). If HDAC4 nuclear accumulation were involved in granule neuron cell death, we expected to observe an increase of nuclear HDAC4 in neurons in weaver mutant mice. To this end, cerebellar sections from wild-type and weaver mutant mice were prepared, and the subcellular localization of HDAC4 in the EGL was determined by immunostaining. The analysis of HDAC4 subcellular localization revealed a marked increase in the number of cells with nuclear HDAC4 in EGL sections from weaver mutant mice (Fig. 6A). In wild-type mice, 12.2% of the CGNs in the EGL displayed nuclear HDAC4 (Fig. 6B). However, this percentage was approximately doubled (22.4%) in the weaver mutants. This result shows that HDAC4 nuclear translocation also occurs in vivo in conditions associated with neuronal cell death.
Figure 6.
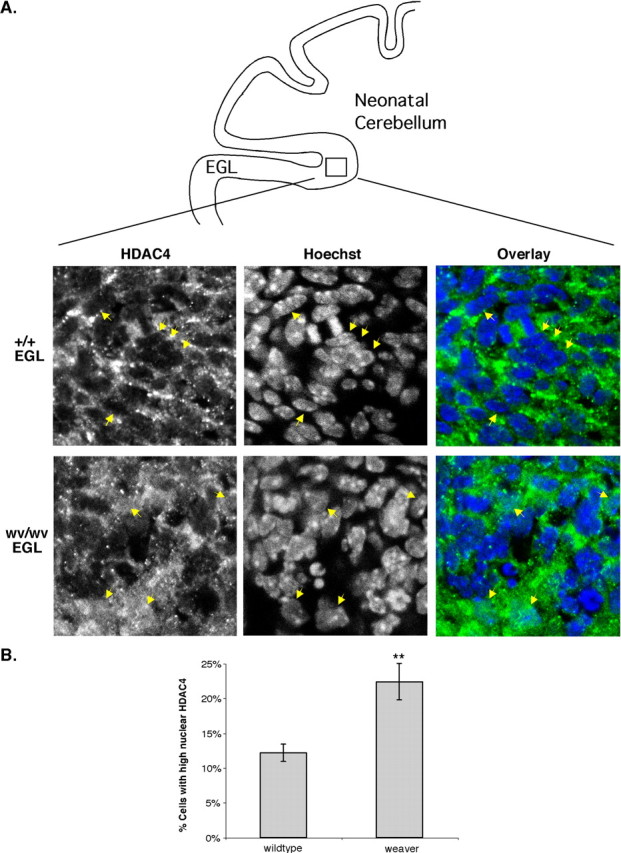
Increased nuclear accumulation of HDAC4 is observed in weaver mutant mice. Weaver and wild-type cerebella from P7-P8 mice were sectioned and then stained with ab186 and Hoechst dye. A, Examples of the EGL of wild-type (+/+) and weaver homozygous (wv/wv) sections are shown. A schematic of a cross section of neonatal mouse cerebellum is included (top) to help orient the reader. In the wild-type section, arrows point to nuclear exclusion of HDAC4. In the weaver section, arrows point to nuclear accumulations of HDAC4. B, A quantification of HDAC4 localization in the sections is shown. Only cells in the EGL of the cerebellum were counted. If the nuclear staining was equal to or greater than the cytoplasmic staining, the cell was counted as high-nuclear HDAC4. Three or four animals were used of each genotype, and > 500 cells were counted per animal. **p < 0.001 compared with wild type. Error bars represent one SD above and below the mean.
Discussion
In this study, we have identified HDAC4 and its subcellular localization as novel regulatory elements important for the execution of cerebellar granule neuron cell death. We found a tight correlation between neuronal cell death signaling and HDAC4 nuclear translocation. Furthermore, we provide evidence that nuclear accumulation of HDAC4 promotes neuronal cell death, whereas inactivation of HDAC4 by specific siRNA and pharmacological inhibitors prevents cell death. These data connect signal-dependent intracellular trafficking of HDAC4 to the regulation of neuronal cell death.
We found that although HDAC4 is dynamically transported between the nucleus and cytoplasm, HDAC4 is localized predominantly to the cytoplasm of neurons in culture or in brain tissues under normal conditions. This is consistent with a previous report showing that HDAC4 is normally cytoplasmic in cultured hippocampal neurons (Chawla et al., 2003). Importantly, HDAC4 undergoes nuclear accumulation in response to conditions that induce cell death in these neurons. The nuclear accumulation of HDAC4 is inhibited by BDNF, a neurotrophin that suppresses CGN cell death. Conversely, a CaMK inhibitor induces neuronal cell death and causes HDAC4 nuclear accumulation. Increased nuclear localization of HDAC4 in the EGL of weaver mice provides additional evidence for the involvement of HDAC4 in neuronal cell death and shows that this function is biologically relevant in vivo. Supporting the functional importance of HDAC4 nuclear translocation in neuronal cell death, introduction of a nuclear-localized HDAC4 (HDAC4-3SA) can significantly promote CGN cell death. Most importantly, knock-down of HDAC4 by siRNA gives the neurons a substantially higher resistance to cell death, showing that the endogenous protein promotes neuronal cell death. These results strongly indicate that HDAC4 is a key target of multiple signaling events that control neuronal cell death.
The mechanism of action for HDAC4 in promoting cell death is not fully understood; however, it most likely involves the repression of transcription factors that promote neuron survival. MEF2 is an important prosurvival factor in neurons, including CGNs (Mao et al., 1999). Given the known ability of HDAC4 to bind and repress MEF2 activity, HDAC4 could promote apoptosis, at least in part, by inhibiting the transcriptional activity of MEF2. Indeed, we showed that nuclear HDAC4, which promotes neuronal cell death, can also efficiently repress activation of neurotrophin-3 (Fig. 4D), which likely mediates the neuroprotective activity of MEF2 (Shalizi et al., 2003). This is also consistent with the reduction of MEF2 activity after low-potassium treatment in CGNs (Fig. 4D) (Shalizi et al., 2003) and excitotoxic insults in cortical neuron cultures (Okamoto et al., 2002; Gong et al., 2003), conditions in which HDAC4 accumulates in the nucleus (Figs. 2A,3A). It seemed likely that HDAC4 also has additional targets. CREB, another prosurvival transcription factor that is regulated by calcium signaling, was a second potential target (Bonni et al., 1995). Indeed, as we show in Figure 4E, the HDAC4-3SA mutant can repress transcription from a CRE-dependent luciferase reporter in CGNs. This is consistent with results from heterologous cells also showing that HDAC4 can repress CREB (T. A. Bolger and T.-P. Yao, unpublished observation). A full elucidation of the transcriptional targets of HDAC4 in neuronal cell death pathways will be important for understanding how HDAC4 controls this highly regulated process.
The demonstration that BDNF and CaMK can regulate HDAC4 localization (Fig. 3B) provides an initial clue to unraveling the signaling pathways leading to HDAC4 in neurons. It has been shown previously that tropomyosin-related kinase B, the BDNF receptor, can bind and activate phospholipase Cγ (PLCγ) (Huang and Reichardt, 2003), and PLCγ can then produce a rise in intracellular calcium through inositol triphosphate production. This allows activation of CaMKs, which have been demonstrated to be responsive to BDNF (Finkbeiner et al., 1997). As demonstrated extensively in other cell types previously, CaMK can directly phosphorylate HDAC4, inducing 14-3-3 binding and HDAC4 nuclear export (Zhao et al., 2001). This would shift the equilibrium of HDAC4 localization to the cytoplasm, resulting in increased neuronal survival. Supporting this model in which CaMK is one of the downstream effectors of BDNF, BDNF treatment cannot prevent HDAC4 nuclear translocation induced by CaMK inhibition, although it can effectively suppress HDAC4 nuclear accumulation caused by low potassium (Fig. 3B). Interestingly, BDNF is still able to repress cell death in CGNs transfected with HDAC4-3SA, suggesting that BDNF targets multiple factors that may be downstream of HDAC4 (supplemental Fig. 2B, available at www.jneurosci.org as supplemental material). Two recent reports indicated the possibility of further HDAC4 regulation by caspase-dependent cleavage in response to DNA damage (Liu et al., 2004; Paroni et al., 2004). In CGN cell death, however, no cleavage product of HDAC4 was seen in immunoblots of cell lysates from LK media (data not shown). Furthermore, HDAC4 nuclear translocation is not affected by the caspase inhibitor N-benzyloxycarbonyl-Val-Ala-Asp-fluoromethyl ketone (Z-VAD-fmk) (data not shown). These results provide additional evidence that nuclear accumulation of HDAC4 is upstream of caspase activation and is an early event in neuronal cell-death signaling.
Using cultured granule neurons as a model, our results strongly indicate an important role for HDAC4 in the regulation of cell death. The observation of increased HDAC4 nuclear accumulation in weaver mutant mice further supports this argument (Fig. 6). Mice lacking functional HDAC4 have been described recently (Vega et al., 2004). The dominant phenotype associated with HDAC4 inactivation was found in skeletal development. Interestingly, it was also reported that a subset of HDAC4 knock-out mice appear to have enlarged brains and exencephaly (McKinsey and Olson, 2004; Vega et al., 2004). Although it remains to be determined whether HDAC4-deficient mice have reduced neuronal cell death, the observed defects are consistent with a possible neuronal phenotype. We note that a related HDAC, HDAC5, was reported to undergo similar nuclear translocation in response to low potassium (Linseman et al., 2003). However, unlike HDAC4 knock-out mice, HDAC5 knock-out mice are viable and without obvious brain defects (Chang et al., 2004). Although it is possible that HDAC5 or other related class IIA members might be involved, the fact that reduction of HDAC4 by siRNA effectively suppresses CGN cell death indicates that HDAC4 may be the dominant deacetylase in the regulation of neuronal cell death (Fig. 5).
The inhibition of CGN cell death by pharmacological inhibitors of HDAC4 (Fig. 5E) is potentially significant in that it may offer a potential therapeutic avenue for preventing neuron death. This observation is initially somewhat surprising because HDAC inhibitors, including TSA and TPX, are known for their activity in inducing cell-cycle arrest and apoptosis in a number of cell lines (for review, see Marks et al., 2004). The reasons for the different response between cancer cell lines and primary neurons are not clear. However, it is of great interest to note that HDAC inhibitors have been shown previously to reduce neurodegeneration caused by expanded polyglutamine proteins in Drosophila and mouse models (Steffan et al., 2001; Ferrante et al., 2003; Hockly et al., 2003; Minamiyama et al., 2004). HDAC inhibitors have also been shown to reduce cell death in at least one cell culture model of polyglutamine disease (McCampbell et al., 2001). Although TSA and TPX also affect other members of HDAC family (Furumai et al., 2001), our study clearly establishes an important role for HDAC4 in the execution of cell death in cultured primary neurons. Therefore, it is reasonable to speculate that inhibition of HDAC4 activity may be at least partly responsible for the effects of HDAC inhibitors seen in the polyglutamine disease models. Indeed, we have observed HDAC4 nuclear translocation in primary neurons in response to the expression of disease-associated polyglutamine proteins (Bolger and Yao, unpublished observation). These observations imply that specific pharmacological inhibition of HDAC4 might offer a more targeted therapeutic approach to prevent neuronal cell death induced by various pathological conditions. We note that others have found that TSA can have toxic effects on cultured CGNs with extended treatment times (Boutillier et al., 2003). We also observe increased cell death in TSA- or TPX-treated CGNs compared with untreated cells in FM media after > 24 h; however, TSA and TPX treatments still reduce low-potassium-induced cell death (supplemental Fig. 3B, available at www.jneurosci.org as supplemental material, and data not shown). Inhibition of other HDACs is likely to be the cause of the toxicity, which further underscores the need for a more targeted approach.
We propose that HDAC4 is a mediator of neuronal cell death and that a tightly regulated equilibrium exists between nuclear HDAC4, which promotes neuronal cell death, and cytoplasmic HDAC4, which permits cell survival. The conditions and factors affecting this equilibrium have all been shown previously to influence neuronal survival, supporting the importance of HDAC4 subcellular localization in cellular survival decisions. It will be of great interest to determine whether other factors implicated in neuronal survival and death can also affect HDAC4 localization, particularly those associated with pathological conditions that cause neuron death. Our demonstration that HDAC inhibitors can reduce cell death in primary neuron culture may provide a molecular basis for the use of HDAC inhibitors in treating neuronal cell death associated with various diseases and points to the potential utility of developing new, more specific HDAC4 inhibitors in the future.
Footnotes
T.A.B. is supported by a National Science Foundation Graduate Research Fellowship, and T.-P.Y. is a Leukemia and Lymphoma Society Scholar. We thank Drs. Azad Bonni and Xiang-Jiao Yang for the gifts of plasmids, Drs. Rochelle Schwartz-Bloom and Anthony Means for reagents, Drs. Robert Wechsler-Reya and Michael Ehlers for their advice, and Jeffrey Kovacs and Todd Cohen for critical reading of this manuscript.
Correspondence should be addressed to Tso-Pang Yao, Box 3813, Duke University Medical Center, Durham, NC 27710. E-mail: yao00001@mc.duke.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/259544-10$15.00/0
References
- Benn SC, Woolf CJ (2004) Adult neuron survival strategies-slamming on the brakes. Nat Rev Neurosci 5: 686-700. [DOI] [PubMed] [Google Scholar]
- Bertos NR, Wang AH, Yang XJ (2001) Class II histone deacetylases: structure, function, and regulation. Biochem Cell Biol 79: 243-252. [PubMed] [Google Scholar]
- Bonni A, Ginty DD, Dudek H, Greenberg ME (1995) Serine 133-phosphorylated CREB induces transcription via a cooperative mechanism that may confer specificity to neurotrophin signals. Mol Cell Neurosci 6: 168-183. [DOI] [PubMed] [Google Scholar]
- Boutillier AL, Trinh E, Loeffler JP (2003) Selective E2F-dependent gene transcription is controlled by histone deacetylase activity during neuronal apoptosis. J Neurochem 84: 814-828. [DOI] [PubMed] [Google Scholar]
- Chang S, McKinsey TA, Zhang CL, Richardson JA, Hill JA, Olson EN (2004) Histone deacetylases 5 and 9 govern responsiveness of the heart to a subset of stress signals and play redundant roles in heart development. Mol Cell Biol 24: 8467-8476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chawla S, Vanhoutte P, Arnold FJ, Huang CL, Bading H (2003) Neuronal activity-dependent nucleocytoplasmic shuttling of HDAC4 and HDAC5. J Neurochem 85: 151-159. [DOI] [PubMed] [Google Scholar]
- Contestabile A (2002) Cerebellar granule cells as a model to study mechanisms of neuronal apoptosis or survival in vivo and in vitro. Cerebellum 1: 41-55. [DOI] [PubMed] [Google Scholar]
- Ferrante RJ, Kubilus JK, Lee J, Ryu H, Beesen A, Zucker B, Smith K, Kowall NW, Ratan RR, Luthi-Carter R, Hersch SM (2003) Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington's disease mice. J Neurosci 23: 9418-9427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkbeiner S, Tavazoie SF, Maloratsky A, Jacobs KM, Harris KM, Greenberg ME (1997) CREB: a major mediator of neuronal neurotrophin responses. Neuron 19: 1031-1047. [DOI] [PubMed] [Google Scholar]
- Furumai R, Komatsu Y, Nishino N, Khochbin S, Yoshida M, Horinouchi S (2001) Potent histone deacetylase inhibitors built from trichostatin A and cyclic tetrapeptide antibiotics including trapoxin. Proc Natl Acad Sci USA 98: 87-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudilliere B, Shi Y, Bonni A (2002) RNA interference reveals a requirement for myocyte enhancer factor 2A in activity-dependent neuronal survival. J Biol Chem 277: 46442-46446. [DOI] [PubMed] [Google Scholar]
- Gong X, Tang X, Wiedmann M, Wang X, Peng J, Zheng D, Blair LA, Marshall J, Mao Z (2003) Cdk5-mediated inhibition of the protective effects of transcription factor MEF2 in neurotoxicity-induced apoptosis. Neuron 38: 33-46. [DOI] [PubMed] [Google Scholar]
- Grozinger CM, Schreiber SL (2000) Regulation of histone deacetylase 4 and 5 and transcriptional activity by 14-3-3-dependent cellular localization. Proc Natl Acad Sci USA 97: 7835-7840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grozinger CM, Hassig CA, Schreiber SL (1999) Three proteins define a class of human histone deacetylases related to yeast Hda1p. Proc Natl Acad Sci USA 96: 4868-4873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hack N, Hidaka H, Wakefield MJ, Balazs R (1993) Promotion of granule cell survival by high K+ or excitatory amino acid treatment and Ca2+/calmodulin-dependent protein kinase activity. Neuroscience 57: 9-20. [DOI] [PubMed] [Google Scholar]
- Hockly E, Richon VM, Woodman B, Smith DL, Zhou X, Rosa E, Sathasivam K, Ghazi-Noori S, Mahal A, Lowden PA, Steffan JS, Marsh JL, Thompson LM, Lewis CM, Marks PA, Bates GP (2003) Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington's disease. Proc Natl Acad Sci USA 100: 2041-2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF (2003) Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem 72: 609-642. [DOI] [PubMed] [Google Scholar]
- Kubo T, Nonomura T, Enokido Y, Hatanaka H (1995) Brain-derived neurotrophic factor (BDNF) can prevent apoptosis of rat cerebellar granule neurons in culture. Brain Res Dev Brain Res 85: 249-258. [DOI] [PubMed] [Google Scholar]
- Linseman DA, Bartley CM, Le SS, Laessig TA, Bouchard RJ, Meintzer MK, Li M, Heidenreich KA (2003) Inactivation of the myocyte enhancer factor-2 repressor histone deacetylase-5 by endogenous Ca(2+)//calmodulin-dependent kinase II promotes depolarization-mediated cerebellar granule neuron survival. J Biol Chem 278: 41472-41481. [DOI] [PubMed] [Google Scholar]
- Liu F, Dowling M, Yang XJ, Kao GD (2004) Caspase-mediated specific cleavage of human histone deacetylase 4. J Biol Chem 279: 34537-34546. [DOI] [PubMed] [Google Scholar]
- Lu J, McKinsey TA, Zhang CL, Olson EN (2000) Regulation of skeletal myogenesis by association of the MEF2 transcription factor with class II histone deacetylases. Mol Cell 6: 233-244. [DOI] [PubMed] [Google Scholar]
- Mao Z, Bonni A, Xia F, Nadal-Vicens M, Greenberg ME (1999) Neuronal activity-dependent cell survival mediated by transcription factor MEF2. Science 286: 785-790. [DOI] [PubMed] [Google Scholar]
- Marks PA, Richon VM, Miller T, Kelly WK (2004) Histone deacetylase inhibitors. Adv Cancer Res 91: 137-168. [DOI] [PubMed] [Google Scholar]
- McCampbell A, Taye AA, Whitty L, Penney E, Steffan JS, Fischbeck KH (2001) Histone deacetylase inhibitors reduce polyglutamine toxicity. Proc Natl Acad Sci USA 98: 15179-15184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinsey TA, Olson EN (2004) Dual roles of histone deacetylases in the control of cardiac growth. Novartis Found Symp 259: 132-141; discussion 141-135, 163-139. [PubMed] [Google Scholar]
- McKinsey TA, Zhang CL, Olson EN (2001) Identification of a signal-responsive nuclear export sequence in class II histone deacetylases. Mol Cell Biol 21: 6312-6321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minamiyama M, Katsuno M, Adachi H, Waza M, Sang C, Kobayashi Y, Tanaka F, Doyu M, Inukai A, Sobue G (2004) Sodium butyrate ameliorates phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Hum Mol Genet 13: 1183-1192. [DOI] [PubMed] [Google Scholar]
- Miska EA, Karlsson C, Langley E, Nielsen SJ, Pines J, Kouzarides T (1999) HDAC4 deacetylase associates with and represses the MEF2 transcription factor. EMBO J 18: 5099-5107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy L, Kao HY, Chakravarti D, Lin RJ, Hassig CA, Ayer DE, Schreiber SL, Evans RM (1997) Nuclear receptor repression mediated by a complex containing SMRT, mSin3A, and histone deacetylase. Cell 89: 373-380. [DOI] [PubMed] [Google Scholar]
- Okamoto S, Li Z, Ju C, Scholzke MN, Mathews E, Cui J, Salvesen GS, Bossy-Wetzel E, Lipton SA (2002) Dominant-interfering forms of MEF2 generated by caspase cleavage contribute to NMDA-induced neuronal apoptosis. Proc Natl Acad Sci USA 99: 3974-3979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver TG, Grasfeder LL, Carroll AL, Kaiser C, Gillingham CL, Lin SM, Wickramasinghe R, Scott MP, Wechsler-Reya RJ (2003) Transcriptional profiling of the Sonic hedgehog response: a critical role for N-myc in proliferation of neuronal precursors. Proc Natl Acad Sci USA 100: 7331-7336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paroni G, Mizzau M, Henderson C, Del Sal G, Schneider C, Brancolini C (2004) Caspase-dependent regulation of histone deacetylase 4 nuclear-cytoplasmic shuttling promotes apoptosis. Mol Biol Cell 15: 2804-2818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil N, Cox DR, Bhat D, Faham M, Myers RM, Peterson AS (1995) A potassium channel mutation in weaver mice implicates membrane excitability in granule cell differentiation. Nat Genet 11: 126-129. [DOI] [PubMed] [Google Scholar]
- Rakic P, Sidman RL (1973) Sequence of developmental abnormalities leading to granule cell deficit in cerebellar cortex of weaver mutant mice. J Comp Neurol 152: 103-132. [DOI] [PubMed] [Google Scholar]
- Segal RA, Takahashi H, McKay RD (1992) Changes in neurotrophin responsiveness during the development of cerebellar granule neurons. Neuron 9: 1041-1052. [DOI] [PubMed] [Google Scholar]
- Shalizi A, Lehtinen M, Gaudilliere B, Donovan N, Han J, Konishi Y, Bonni A (2003) Characterization of a neurotrophin signaling mechanism that mediates neuron survival in a temporally specific pattern. J Neurosci 23: 7326-7336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffan JS, Bodai L, Pallos J, Poelman M, McCampbell A, Apostol BL, Kazantsev A, Schmidt E, Zhu YZ, Greenwald M, Kurokawa R, Housman DE, Jackson GR, Marsh JL, Thompson LM (2001) Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila Nature 413: 739-743. [DOI] [PubMed] [Google Scholar]
- Strahl BD, Allis CD (2000) The language of covalent histone modifications. Nature 403: 41-45. [DOI] [PubMed] [Google Scholar]
- Vaudry D, Falluel-Morel A, Leuillet S, Vaudry H, Gonzalez BJ (2003) Regulators of cerebellar granule cell development act through specific signaling pathways. Science 300: 1532-1534. [DOI] [PubMed] [Google Scholar]
- Vega RB, Matsuda K, Oh J, Barbosa AC, Yang X, Meadows E, McAnally J, Pomajzl C, Shelton JM, Richardson JA, Karsenty G, Olson EN (2004) Histone deacetylase 4 controls chondrocyte hypertrophy during skeletogenesis. Cell 119: 555-566. [DOI] [PubMed] [Google Scholar]
- Verdin E, Dequiedt F, Kasler HG (2003) Class II histone deacetylases: versatile regulators. Trends Genet 19: 286-293. [DOI] [PubMed] [Google Scholar]
- Wang AH, Yang XJ (2001) Histone deacetylase 4 possesses intrinsic nuclear import and export signals. Mol Cell Biol 21: 5992-6005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Ito A, Kane CD, Liao TS, Bolger TA, Lemrow SM, Means AR, Yao TP (2001) The modular nature of histone deacetylase HDAC4 confers phosphorylation-dependent intracellular trafficking. J Biol Chem 276: 35042-35048. [DOI] [PubMed] [Google Scholar]