

Abstract
The transcription factor nuclear factor erythroid 2-related factor 2 (Nrf2) coordinates expression of genes required for free radical scavenging, detoxification of xenobiotics, and maintenance of redox potential. Previously, activation of this pleiotropic response was neuroprotective in cell culture models that simulate components of stroke damage. However, the role of Nrf2 in limiting stroke damage in vivo remained unclear. We report that Nrf2 activation protects the brain from cerebral ischemia in vivo. Acute (1-3 d) intracerebroventricular or intraperitoneal pretreatment with tert-butylhydroquinone (tBHQ), an Nrf2 activity inducer, reduced cortical damage and sensorimotor deficit at 24 h and even 1 month after ischemia-reperfusion in rats. Cortical glutathione levels robustly increased with tBHQ administration to rats and Nrf2-expressing mice, but not Nrf2-/- mice. Basal and inducible activities of antioxidant/detoxification enzymes in Nrf2-/- mice were reduced when compared with Nrf2+/+ controls. Interestingly, larger infarcts were observed in Nrf2-/- mice at 7 d after stroke, but not at 24 h, suggesting that Nrf2 may play a role in shaping the penumbra well after the onset of ischemia. Neuronal death caused by a “penumbral” model of stroke, using intracortical endothelin-1 microinjection, was attenuated by tBHQ administration to Nrf2+/+, but not to Nrf2-/- mice, confirming the Nrf2-specific action of tBHQ in vivo. We conclude that Nrf2 plays a role in modulating ischemic injury in vivo. Accordingly, Nrf2 activation by small molecule inducers may be a practical preventative treatment for stroke-prone patients.
Keywords: Nrf2, NF-E2-related factor, astrocyte, tert-butylhydroquinone, antioxidant response element, NAD(P)H:quinone oxidoreductase, glutathione, oxidative stress, neuroprotection, stroke, ischemia, brain, endothelin-1
Introduction
Oxidative stress leading to ischemic cell death involves the formation of reactive oxygen species/reactive nitrogen species (ROS/RNS) through multiple injury mechanisms, such as mitochondrial inhibition, Ca2+ overload, reperfusion injury, and inflammation (Coyle and Puttfarcken, 1993; Lipton, 1999; Love, 1999; Lewen et al., 2000). Although many treatment strategies have implemented antioxidants to promote neuroprotection during ischemia, their clinical efficacy has proven disappointing (De Keyser et al., 1999; Lo et al., 2003). The Cap `n' Collar (CNC) transcription factor nuclear factor erythroid 2-related factor 2 (Nrf2) regulates an expansive set of antioxidant/detoxification genes that act in synergy to remove ROS/RNS through sequential enzymatic reactions (Li et al., 2002; Thimmulappa et al., 2002; Shih et al., 2003). As an improvement on previous neuroprotective approaches, we examined whether a coordinated cellular defense activated by Nrf2 provides effective prophylaxis for stroke.
Nrf2 gene targets, collectively known as phase 2 genes, are involved in glutathione (GSH) production and use, detoxification of ROS/RNS and xenobiotics, and NADPH production (Chan and Kwong, 2000; Ishii et al., 2000; Talalay, 2000; Thimmulappa et al., 2002). Nrf2 is normally localized to the cytoplasm, tethered to the regulatory protein, kelch-like erythroid cell-derived protein with CNC homology-associated protein 1 (Keap1) (Itoh et al., 1999). Oxidative stress, or electrophilic agents that mimic oxidative stress (Nrf2 inducers), can modify key sulfhydryl group interactions in the Keap1-Nrf2 complex, allowing dissociation and nuclear translocation of Nrf2 (Itoh et al., 1999; Dinkova-Kostova et al., 2002). When activated, Nrf2 specifically targets genes bearing an antioxidant response element (ARE) within their promoters (Venugopal and Jaiswal, 1996; Itoh et al., 1997). A wide range of natural and synthetic small molecules are potent inducers of Nrf2 activity (Fahey et al., 1997, 2001; Rushmore and Kong, 2002). These molecules have been identified from diverse chemical backgrounds including the following: isothiocyanates (abundant in cruciferous vegetables), 1,2-dithiole-3-thiones, heavy metals, and hydroperoxides (Talalay et al., 1995; Talalay, 2000). One well characterized inducer, tert-butylhydroquinone (tBHQ), a metabolite of the widely used food antioxidant butylated hydroxyanisole, is already approved for human use (National Toxicology Program, 1997; Food and Agriculture Organization of the United Nations/World Health Organization, 1999). tBHQ possesses an oxidizable 1,4 diphenolic structure that confers its potent ability to dissociate the Keap1-Nrf2 complex (De Long et al., 1987; Talalay, 1989; van Ommen et al., 1992).
Previous studies have shown that increasing Nrf2 activity in mixed neuronal/glial cultures is highly neuroprotective during in vitro models that simulate components of stroke damage, such as oxidative glutamate toxicity (a model involving GSH depletion), H2O2 exposure, metabolic inhibition by rotenone, and Ca2+ overload (Murphy et al., 1991; Duffy et al., 1998; Lee et al., 2003b; Shih et al., 2003; Kraft et al., 2004). However, the protective role of Nrf2 activity during in vivo stroke has not been examined. Here, we report that administration of tBHQ before stroke significantly improves sensorimotor and histological outcome after ischemia-reperfusion. Conversely, mice lacking Nrf2 have increased sensitivity to permanent focal ischemia and fail to respond to Nrf2 inducers. Our data suggest that Nrf2 function is important for controlling stroke damage, and application of Nrf2 inducers may be an effective prophylactic treatment for those at risk for stroke.
Materials and Methods
Reagents. All reagents were obtained from Sigma (St. Louis, MO) unless otherwise stated.
Astrocyte-enriched primary cultures. Enriched cortical astrocyte cultures were prepared from 0- to 2-d-old postnatal rat or Nrf2-/- colony mice pups, as described previously (Shih et al., 2003). The conditions used primarily result in a population of type I and II astrocytes, as assessed by anti-glial fibrillary acid protein staining.
Plasmids, adenoviruses, and transfections. The ARE sequence was obtained from the rat NAD(P)H:quinone oxidoreductase gene promotor and was used to make the human placental alkaline phosphatase (hPAP) reporter construct (rQR51wt) and mutant (rQR51mut), as described previously (Murphy et al., 2001; Shih et al., 2003). Astrocytes seeded (2 × 105 cell/ml) in 24-well plates were transfected with reporter constructs using Lipofectamine 2000 (Invitrogen, Carlsbad, CA), according to the protocol of the manufacturer, except that 1 μg of DNA and 1 μl of Lipofectamine were used for each well, and transfections were terminated after 6 h. The transfection efficiency of astrocytes was typically ∼20% as assessed by β-galactosidase staining. Mammalian expression plasmids carrying cDNA-encoding mouse Nrf2 and a dominant-negative version of Nrf2 (Nrf2DN) were a generous gift from Dr. Jawed Alam (Alton Ochsner Medical Foundation, New Orleans, LA) (Alam et al., 1999). These cDNAs were used to construct recombinant adenoviral vectors using the Cre-lox system (Canadian Stroke Network Core Facility, University of Ottawa, Ottawa, Ontario, Canada) (Hardy et al., 1997) and used as described previously (Shih et al., 2003).
Human placental alkaline phosphatase assay. To assess ARE-mediated gene expression, hPAP activity was measured as described by previous studies (Henthorn et al., 1988). Briefly, astrocyte cultures were collected in lysis buffer consisting of 10 mm Tris-HCl, pH 8.0, 1 mm MgCl2, and 0.1% Triton X-100. One-half of the sample was saved for determination of protein concentration and β-galactosidase activity (for normalization of transfection efficiency). The remaining sample was heated to 65°C for 30 min to inactivate endogenous phosphatase activity. The assay was initiated by mixing ∼15 μg of protein with diethanolamine buffer (0.73 m diethanolamine with 0.36 mm MgCl2, pH 9.8, final concentration) and 13.6 mm p-nitrophenyl phosphate (final volume, 150 μl) The reaction was monitored at 405 nm. hPAP activity from rQR51mut transfected astrocytes was subtracted as ARE-independent background.
Enzyme assays for astrocyte cell culture and brain tissue. Astrocyte-enriched cultures were rinsed with 10 mm PBS, pH 7.4, scraped into ice-cold PBS, and sonicated for 10 s. These lysates were centrifuged at 15,000 × g for 10 min at 4°C. Supernatants were collected and promptly assayed for enzyme activity. Brain and liver tissue was dissected in ice-cold PBS and homogenized with 10 strokes of a dounce homogenizer in tissue buffer containing 25 mm Tris, pH 7.4, and 250 mm sucrose. Crude homogenates were centrifuged at 15,000 × g for 10 min at 4°C. Tissue homogenate supernatants were collected and promptly assayed for enzyme activity. NAD(P)H:quinone oxidoreductase (NQO1) enzyme activity was determined by calculating the dicumarol-sensitive fraction of 2,6-dichlorophenolindophenol (DCPIP) reduction (Benson et al., 1980). Reactions consisting of 25 mm Tris-HCl buffer, pH 7.4, with 0.7 mg/ml BSA, 5 μm flavin adenine dinucleotide, 200 μm nicotinamide adenine dinucleotide (NADH), and 30 μg/ml protein, with and without 20 μm dicumarol, were preincubated for 10 min at 25°C (final concentrations in 200 μl reaction volume). DCPIP was then added to a final concentration of 36 μm (20 μl volume), and the reaction was monitored at 540 nm. The extinction coefficient for DCPIP was 2.1 × 104 m/cm. The glutathione S-transferase (GST) assay consisted of 1 mm 1-chloro-2,4-dinitrobenzene (CDNB), 1 mm GSH, and 100 μg/ml protein at 37°C in 100 mm potassium phosphate buffer, pH 6.5 (final concentration in 150 μl reaction volume) (Kelly et al., 2000). The GST reaction was monitored at 340 nm, and the spontaneous nonenzymatic slope was subtracted from the total observed slope. The extinction coefficient for CDNB was 9600 m/cm. The lactate dehydrogenase (LDH) assay consisted of 3.3 mm pyruvate, 0.34 mm NADH, and 100 μg/ml protein in PBS at 37°C (final concentrations in 150 μl reaction volume) (Everse et al., 1970). The LDH reaction was monitored at 340 nm, and the spontaneous nonenzymatic slope was subtracted from the total observed slope. Purified LDH enzyme standards were used to ensure that values were obtained within the linear of the assay. The extinction coefficient for NADH was 6219 m/cm. Protein concentration was determined using the bicinchronic acid method according to the protocol of the manufacturer (Pierce, Rockford, IL).
Glutathione assay. Total GSH was quantified by the method of Tietze (1969). Briefly, the acid-soluble fraction was obtained from astrocyte cell lysates and tissue homogenates by adding perchloric acid to a final concentration of 3%, followed by centrifugation at 14,000 × g for 10 min. The acid-soluble fraction was neutralized to pH 7 with 0.5 m KOH/50 mm Tris. After the removal of precipitate (potassium perchlorate) by a second centrifugation, 50 μl aliquots of sample were combined with 100 μl of reaction mixture consisting of 2.5 ml of 1 mm 5,5′-dithio-bis-(2-nitrobenzoic acid) (DTNB), 2.5 ml of 5 mm NADPH, 2.5 ml of phosphate buffer solution (100 mm NaPO4, pH 7.5, 1 mm EDTA), and glutathione reductase (5 U/ml final volume). GSH-mediated reduction of DTNB was measured at 412 nm at 30 s intervals over 30 min. GSH content was normalized to protein.
Oxidative glutamate toxicity and assay for neuronal viability. The oxidative glutamate toxicity paradigm was used as described previously (Shih et al., 2003). Briefly, immature cortical neurons [1-4 d in vitro (DIV)] were pretreated with 20 μm tBHQ or DMSO vehicle for 24 h, followed by washout and exposure to 1 mm glutamate for an additional 24 h. Neuronal viability was evaluated using the MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2 H-tetrazolium bromide] cell viability assay.
Animals and experimental treatments. Our experiments were approved by the Canadian Council for Animal Care and the University of British Columbia Animal Care Committee. All experiments were conducted in strict accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Wistar [University of British Columbia (UBC) animal facility] and Sprague Dawley rats (Charles River Laboratories, Wilmington, MA) weighing 250-350 g at the time of stroke were housed in pairs and maintained in a 12 h light/dark cycle, with food and water ad libitum. All rats and mice were normally fed the LabDiet 5001 formulation (PMI Nutrition International, Richmond, IN), which contains no tBHQ but trace amounts of BHA for preserving animal fats (2 ppm). Osmotic pump studies were performed using Wistar rats, but because of a high incidence of subarachnoid hemorrhage during stroke and higher poststroke morbidity, later experiments (intraperitoneal injections) were performed using Sprague Dawley rats. For intracerebroventricular osmotic mini-pump delivery, tBHQ was prepared at a final concentration of 1 mm in vehicle (1% DMSO in PBS). tBHQ was first dissolved in pure DMSO and PBS was added to final volume. The mixture was rapidly and vigorously vortexed to redissolve any precipitates. We found that tBHQ prepared in this manner was stable for over 1 week when maintained at 37°C, retaining its ability to induce phase 2 enzymes and protect immature cortical neurons from oxidative glutamate toxicity (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). A rust color developed with time, although this transition did not affect its neuroprotective ability. Fresh tBHQ solution was sterile filtered and loaded into pumps according to the protocol of the manufacturer [Alzet (Cupertino, CA) model 1003D, designed to deliver 1 μl/h for 4 d]. Each pump was then attached via a 2 cm length of tubing to a cannula (Plastics One, Roanoke, VA) placed at bregma +1.5 mm mediolateral (ML), -0.9 mm anteroposterior (AP), and 4.5 mm dorsoventral (DV), targeting the right lateral ventricle. tBHQ was infused continuously for 72 h, at which time focal ischemia was initiated. Proper cannula placement was verified by eye during brain sectioning at the time the animals were killed. For intraperitoneal delivery, a more concentrated tBHQ solution was prepared at 5 mg/ml in vehicle (1% DMSO in PBS) using the same method described above. The tBHQ solution was injected three times at 3.33 or 16.7 mg/kg separated by 8 h intervals. The volume of each injection was set at 3.33 ml/kg. The concentrated tBHQ stock solution was prepared fresh for each injection.
Nrf2-/- mice (C57B/SV129 background) originating from the laboratory of Dr. Yuet Wai Kan (Cardiovascular Research Institute and Department of Laboratory Medicine, University of California-San Francisco, San Francisco, CA) were maintained in a 12 h dark/light cycle with food and water ad libitum (Chan et al., 1996). Male mice were used between the ages of 10 and 16 weeks. All animals in this study were derived from breeding Nrf2+/- heterozygotes, because it was conceivable that some selection for mice with reduced sensitivity to oxidative stress may occur if the knock-out animals were allowed to inbreed over several generations.
For tBHQ feeding of mice, food pellets (LabDiet 5001) were powdered in a coffee grinder and dry mixed with 1% tBHQ (w/w). Distilled water was added to the powder (equal v/w), thoroughly mixed, and reshaped into food pellets. The pellets were then baked at 60°C for 3 h. Control food was processed in the same manner without the addition of tBHQ.
Stroke models. Anesthesia was induced with 4% isofluorane (Baxter International, Deerfield, IL) and maintained with 2% isofluorane in 30% O2 and 70% N2O. Body temperature was maintained at 37°C with a feedback-controlled rectal probe-heat blanket system (FHC, Bowdoinham, ME) and an overhead incandescent lamp. Ischemia-reperfusion was induced in rats using the intraluminal suture method as described previously (Longa et al., 1989; Kawamura et al., 1991). Briefly, a ventral neck incision was made and neck muscles were pulled aside with ligatures and the right common carotid artery (CCA) was carefully dissected away from the vagus nerve. The external carotid artery (ECA) was ligated and cauterized. A 3 cm 3-0 nylon suture (Ethilon, Tilburg, The Netherlands) (tip blunted with a soldering iron) was inserted through the ECA and pushed into the internal carotid artery (ICA) until a gentle resistance was felt, leaving ∼7-10 mm of the suture remaining outside the ECA/ICA bifurcation. The suture was clamped in place with a micro-aneurysm clip (Harvard Apparatus, Holliston, MA), and the animal was then sutured and awoken from anesthesia. After 1.5 h of ischemia, the animal was then reanesthetized and the suture was retracted. Although the ECA was permanently ligated, blood flowing from the CCA could still pass through the ICA allowing full reperfusion. Animals were tested for stroke-induced sensorimotor deficits (described below) just before termination of stroke to ensure proper placement of the suture. Blood samples were collected from a PE-10 femoral artery catheter (Intramedic; BD Diagnostic Systems, Sparks, MD) and blood-gas measurements (Rapidlab 348; Bayer, Etobicoke, Ontario, Canada) were made before, during, and 15 min after the ischemic period. Animals were included in the ischemia-reperfusion study based on a number of criteria: (1) must show all signs of stroke behavior during ischemia (behavioral score of 10 out of 12; see scoring scheme below). (2) No subarachnoid hemorrhage (assessed during removal of the brain from the skull). (3) Must have a visible infarct, regardless of size (lack of infarct is likely attributable to poor suture placement). (4) Must not be hypothermic at any point during or 30 min after the stroke surgery (i.e., <36°C core temperature).
For Nrf2-/- mice, cortical ischemia was induced by permanently occluding the right distal middle cerebral artery (MCA) using a procedure modified from previous studies (Cechetto et al., 1989; Majid et al., 2000). Anesthesia and body temperature regulation were same as focal ischemia with rats. The mice were placed in a stereotaxic frame (David Kopf Instruments, Tujunga, CA) for surgery. An incision was made in the skin between the right eye and ear. The temporal muscle was cut and pulled aside, and the MCA was accessed by drilling a small hole in the skull using an air-powered dental drill. The artery was carefully cauterized using either a fine-tip hand-held heat cautery (Fine Science Tools, Foster City, CA) or a bipolar electrocoagulator (ME102; Martin Elektrotom MC, Tuttlingen, Germany). Any animals that developed subarachnoid hemorrhage from this procedure were immediately killed. The successful occlusion of the MCA was guided with a laser Doppler [Perimed (Piscataway, NJ) PF3]. The Doppler probe was placed above the temporal ridge, but not over an artery, to ensure measurement of cerebral microcirculation. Heat cauterization itself produced negligible tissue damage in sham surgeries, in which no change in regional cerebral blood flow was observed.
Intracortical endothelin-1 (American Peptide Company, Sunnyvale, CA) microinjections were performed as described previously (Zhang et al., 2005), with some modifications. Briefly, a hole was drilled in the skull at +1.5 mm ML, -1.0 mm AP, and -0.8 mm DV. Glass pipettes (60-90 μm tip diameter), linked by PE-10 tubing to a 10 μl Hamilton (Reno, NV) syringe, were used to inject 0.5 μl of endothelin-1 (1 μg/μl) at a rate of 6 μl/h using a syringe pump (World Precision Instruments, Sarasota, FL). Mice were killed 7 d after endothelin-1 injection.
Semiqualitative neurological scoring for rat ischemia-reperfusion. Sensorimotor performance was scored based on a previous description, with some modifications (Reglodi et al., 2003). Briefly, the scoring system involved the evaluation of six tests, each with a three-tiered scoring scale (0, 1, or 2), with 0 being normal behavior and 2 being a severe deficit of contralateral side. Tests included the following: (1) contralateral thorax twisting when elevated by tail, (2) contralateral forelimb flexion, (3) resistance to lateral push, (4) forelimb bracing against table surface, (5) forelimb placement on table edge, and (6) forelimb and hindlimb table edge sensation. Each animal received a cumulative score out of 12 for each trial. Behavior was assessed just before termination of ischemia (1.5 h) and again after 22.5 h of reperfusion. In long-term survival studies, neurological scoring was performed weekly. Only animals that received at least a score of 10 out of 12 during neurological testing at the time of occlusion were included in the study. From our experience, a score <10 (at the time of occlusion) was most likely attributable to improper placement of the suture and incomplete ischemia. An experimenter blinded to the animal treatment groups performed behavioral assessments and surgeries.
Infarct measurement. The extent of infarction was measured with 2,3,5-triphenyl-tetrazolium chloride (TTC) for 24 h survival times (Bederson et al., 1986) or cresyl violet (CV), which is more appropriate for assessing infarct area at longer time points because TTC vital staining will not differentiate between gliosis and viable neuronal tissue. For TTC staining, animals were deeply anesthetized with euthanyl (Bimeda, Cambridge, Ontario, Canada) and the brains removed, washed in room temperature (RT) PBS, and sliced into 2 mm sections for rats and into 1 mm sections for mice with the aid of brain matrices (Plastics One). The sections were carefully placed into 3% TTC in PBS and incubated at RT for 10 min. The TTC solution was then removed and replaced with 4% paraformaldehyde (PFA) for overnight fixation at 4°C. For CV staining, animals were anesthetized with euthanyl and transcardially perfused with RT PBS followed with ice-cold 4% PFA (for each solution, 100 ml for rats and 15 ml for mice). The brains were removed and postfixed for 24 h, and then cryoprotected in 30% sucrose in PBS with 0.02% sodium azide for an additional 48 h. Coronal sections were collected with a cryostat and mounted on Superfrost slides (Fisher Scientific, Houston, TX). CV staining was performed on 40 μm sections using standard protocols. All stained sections were scanned at 600 dpi on a desktop scanner [Epson (Toronto, Ontario, Canada) 1660]. Infarct area was measured using NIH ImageJ version 1.30. Given that hemispheric volume could change as a result of the ischemia, we normalized the infarct area of each section to ipsilateral hemisphere area (or cortex area for permanent MCA occlusion in mice) and reported data as percentage infarct area of ischemic hemisphere or cortex. Similar results were obtained when infarct area was expressed as percentage of contralateral hemisphere/cortex. Infarct volume was calculated according to the principle of Cavalieri (volume = s1d1 + s2d2 + s3d3 + s4d4), where s is infarct surface area, and d is distance between two sections. Infarct quantification was performed by a researcher blinded to the experimental conditions.
FluoroJade staining used to quantify endothelin-1-induced infarcts was performed on 20 μm mouse brain sections as described previously (Schmued et al., 1997). FluoroJade images were collected using Northern Eclipse software (version 6.0) and a wide-field fluorescence microscope (Axiophot; Zeiss, Oberkochen, Germany) equipped with a Retiga EXi CCD camera (QImaging, Burnaby, British Columbia, Canada). Infarct area was measured using ImageJ. Because endothelin-1 (ET-1) injection does not always produce a contiguous volume of tissue damage, infarct size was expressed as summed FluoroJade-positive area measured over six brain sections taken at 600 μm intervals (bregma +2.0 to -2.0 mm) rather than as a calculated brain volume. Because a 7 d time point was used to assess FluoroJade staining, it was also not always possible to view all individually degenerating neurons and count them. Therefore, we assessed FluoroJade staining by outlining an infarcted region and measuring its area.
Statistical analyses. All data are shown as means ± SEM. Statistical analyses were performed using the two-tailed Student's t test, one-way ANOVA with Bonferroni's post hoc test, or two-way ANOVA in GraphPad Prism (version 2.01; GraphPad Software, San Diego, CA). *p < 0.05, **p < 0.01, and ***p < 0.001.
Results
Induction of multiple antioxidant systems in astrocyte cultures by inducers of Nrf2-dependent transcription
In cultured astrocytes, Nrf2-mediated induction of antioxidant enzymes can be achieved by treatment with low molecular-weight electrophilic compounds such as tBHQ (Ahlgren-Beckendorf et al., 1999; Eftekharpour et al., 2000; Murphy et al., 2001; Lee et al., 2003b; Shih et al., 2003). Based on previous work, three established markers of Nrf2 activation were evaluated throughout this study: total GSH content, as well as GST and NQO1 activity (Chan and Kwong, 2000; Hayes et al., 2000; Ishii et al., 2000; McMahon et al., 2001; Ramos-Gomez et al., 2001; Chanas et al., 2002; Shih et al., 2003). Consistent with other reports, all three markers were induced in astrocytes within 10-24 h in response to tBHQ treatment (Fig. 1A) (Ahlgren-Beckendorf et al., 1999; Lee et al., 2003a; Kraft et al., 2004). As a positive control, adenoviral-mediated overexpression of Nrf2 (Ad-Nrf2) was sufficient to increase all three markers, compared with GFP control (Ad-GFP), whereas Ad-Nrf2DN produced no change (Fig. 1B) (Shih et al., 2003). Importantly, these effects were specific, because no increase in cellular LDH activity (not regulated by Nrf2) was observed with either tBHQ treatment or overexpression of Nrf2 (Fig. 1A,B).
Figure 1.

Time course and Nrf2 dependency of tBHQ-mediated phase 2 enzyme induction in astrocyte-enriched cultures. A, Astrocyte cultures (7-10DIV) prepared from postnatal rats (postnatal day 0-2) were treated with 20 μm tBHQ or DMSO vehicle for various times before harvesting for GSH, GST, NQO1, and LDH assays. Induction of GSH, GST, and NQO1 was observed within 10-24 h, whereas LDH activity (negative control) was unchanged. B, As a positive control, Ad-Nrf2 was sufficient to increase GSH content, as well as NQO1 and GST activity, compared with GFP control (Ad-GFP), where as Ad-Nrf2DN produced nochange (Shih et al., 2003). C, Nrf2+/+ and Nrf2-/- cultures were transiently transfected with an hPAP reporter construct regulated by an ARE-bearing promotor and treated with 20 μm tBHQ for 24 h. Increased hPAP activity was detected in Nrf2+/+ but not Nrf2-/- cultures. D, Basal activities of GST and NQO1, as well as GSH content, were suppressed in astrocyte cultures derived from Nrf2-/- mice, compared with Nrf2+/+ control cultures. E, tBHQ-mediated induction of GSH, GST, and NQO1 by tBHQ was abrogated in Nrf2-/- cultures but could be rescued with adenoviral Nrf2 overexpression (Lee et al., 2003b; Shih et al., 2003). Data represent the mean ± SEM collected from at least three independent experiments. *p < 0.05, **p < 0.01, and ***p < 0.001; two-tailed t test.
Previous studies have shown that tBHQ does not activate the phase 2 response in Nrf2-/- cultures, confirming its specificity for Nrf2 (Lee et al., 2003a). Consistent with this report, we find that astrocytes prepared from Nrf2-/- mice have reduced basal ARE-mediated gene expression (23.8 ± 4.3% of Nrf2+/+ control; n = 3), which fails to be induced by tBHQ, as assessed by reporter assay (Fig. 1C). As a result, basal and inducible GSH, GST, and NQO1 levels were also suppressed in Nrf2-/- astrocytes (Fig. 1 D, E). Importantly, induction of these markers could be rescued by Nrf2 overexpression (Fig. 1 E). Previous in vitro experiments by Kraft et al. (2004) indicate that the neuroprotective effects of tBHQ are lost when using Nrf2-/- astrocytes. These results verify that the tBHQ-stimulated Nrf2 pathway, not induction of another pathway, plays a causal role in neuroprotection.
The three markers tested (GSH, GST, and NQO1) do not represent the full scope of Nrf2 gene targets but nonetheless indicate that the phase 2 response is activated. For a more comprehensive description of Nrf2-regulated genes in vitro, see recent microarray studies (Li et al., 2002; Thimmulappa et al., 2002; Lee et al., 2003a; Shih et al., 2003; Kraft et al., 2004). In this in vitro paradigm, the inductive effect of tBHQ requires 10-24 h to be manifested because new transcription and protein translation must precede its neuroprotective function. Accordingly, in our in vivo studies, we examined the effects of tBHQ pretreatment on transient forebrain ischemia in rats.
Intracerebroventricular infusion of tBHQ is protective in a rat model of ischemia-reperfusion
As one strategy to induce phase 2 enzymes in the brain in vivo, we implanted osmotic mini-pumps, which continuously delivered a small volume (1 μl/h) of 1 mm tBHQ solution (prepared in 1% DMSO vehicle) for 4 d into the right lateral ventricle (intracerebroventricularly), bypassing the blood-brain barrier. The tBHQ solution was active for the entire duration of in vivo administration because it was capable of protecting cultured neurons from oxidative glutamate toxicity even when extracted from the pump after 4 d at 37°C (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
After 3 d of continuous tBHQ delivery, rats were subjected to 1.5 h of focal ischemia-reperfusion using the intraluminal suture model of MCA occlusion (MCAO) (Fig. 2E) (Longa et al., 1989; Kawamura et al., 1991). In animals treated with vehicle, we observed a subcortical (i.e., striatum) and cortical infarct 24 h after stroke using vital staining with TTC (Fig. 2A-C). However, in animals treated with tBHQ, we observed a significant reduction of infarction in the cortex but not of the stroke core within the subcortex (volume of cortical infarct expressed as percentage of total ischemic hemisphere: vehicle, 9.2 ± 2.9; tBHQ, 1.1 ± 0.7; **p = 0.008; two-tailed t test) (Fig. 2A-C). For both vehicle and tBHQ-treated animals, we observed no significant hypothermia or apparent differences in core temperature between the groups (Table 1). Both vehicle and tBHQ-treated animals had a similar level of contralateral sensorimotor deficits during occlusion, suggesting that both groups were subjected to similar levels of ischemia (Fig. 2D). However, 24 h after stroke, we found a significant reduction in the neurological scores in the tBHQ-treated group (indicating a more mild deficit) when compared with vehicle-treated controls, consistent with infarct volume measurements (Fig. 2D).
Figure 2.

Intracerebroventricular infusion of tBHQ reduced sensorimotor deficit and infarct size 24 h after ischemia-reperfusion. Intracerebroventricular delivery of 1 mm tBHQ (1 μl/h) for 72 h before focal ischemia reduced cortical infarction compared with vehicle-treated controls. A, Animals were killed 24 h after the initiation of transient MCAO (1.5 h), and infarcted brain regions were visualized using TTC staining (lighter colored tissue is infarcted). Representative examples are shown from each treatment group. B, Cortical infarction was attenuated with tBHQ treatment. Infarct areas are shown between sections 1 (anterior) and 6 (posterior). Section 2 approximately corresponds to bregma = 0.0. C, Subcortical infarction was not attenuated. D, Acute motor deficits assessed during the stroke (1.5 h) were similar, suggesting that animals from both groups received the same degree of ischemia. However, sensorimotor deficit was significantly lower in the tBHQ-treated group when evaluated 24 h after reperfusion. E, A schematic diagram summarizing the pump implant and stroke timeline. Intracerebroventricular infusion was continuous from pump implant to killing. Data represent mean ± SEM collected from n = 9 animals for tBHQ and n = 7 for vehicle treatment groups. *p < 0.05, two-tailed t test or one-way ANOVA with Bonferroni's post hoc test.
Table 1.
Summary of physiological parameters for rat and mouse stroke studies
|
|
Vehicle |
tBHQ |
|---|---|---|
| Intracerebroventricular osmotic pump study | ||
| Number of animals | 9 | 7 |
| Core temp. before occlusion (°C) | 37.4 ± 0.3 | 36.7 ± 0.3 |
| Core temp. during occlusion (°C) | 38.0 ± 0.2 | 37.8 ± 0.5 |
| Core temp. 30 min after occlusion (°C) | 36.9 ± 0.5 | 36.9 ± 0.5 |
| Weight before pump implant (g) | 267 ± 15 | 274 ± 17 |
| Weight before stroke (g) | 272 ± 13 | 275 ± 13 |
| Weight 24 h after stroke (g) | 243 ± 15 | 253 ± 20 |
| Intraperitoneal delivery: 24 h survival | ||
| Number of animals | 11 | 11 |
| Core temp. before occlusion (°C) | 36.6 ± 0.2 | 36.9 ± 0.2 |
| Core temp. during occlusion (°C) | 39.1 ± 0.1 | 38.9 ± 0.1 |
| Core temp. 30 min after occlusion (°C) | 37.6 ± 0.3 | 37.6 ± 0.2 |
| Weight before intraperitoneal injection (g) | 309 ± 7 | 315 ± 7 |
| Weight before stroke (g) | 292 ± 7 | 294 ± 7 |
| Weight 24 h after stroke (g) | 259 ± 6 | 273 ± 7 |
| Intraperitoneal delivery: 1 month survival | ||
| Number of animals | 5 | 6 |
| Core temp. before occlusion (°C) | 37.1 ± 0.1 | 36.9 ± 0.1 |
| Core temp. during occlusion (°C) | 38.7 ± 0.3 | 38.7 ± 0.3 |
| Core temp. 30 min after occlusion (°C) | 37.5 ± 0.2 | 37.2 ± 0.2 |
| Weight before intraperitoneal injection (g) | 330 ± 12 | 316 ± 15 |
| Weight before stroke (g) | 299 ± 10 | 291 ± 14 |
| Weight 24 h after stroke (g) | 263 ± 8 | 258 ± 13 |
| Weight 1 month after stroke (g) | 429 ± 10 | 420 ± 18 |
| Blood gas analysis (before) | ||
| pH | 7.31 ± 0.05 | 7.32 ± 0.05 |
| pCO2 (mmHg) | 39.1 ± 2.7 | 40.5 ± 3.1 |
| pO2 (mmHg) | 111.7 ± 7.1 | 103 ± 4.5 |
| Cl− (mmol/L) | 102.5 ± 0.6 | 103.6 ± 1.1 |
| Hematocrit (%) | 41.2 ± 1.2 | 40.6 ± 1.0 |
| Blood gas analysis (during) | ||
| pH | 7.32 ± 0.07 | 7.39 ± 0.09 |
| pCO2 (mmHg) | 30.4 ± 7.8 | 38.5 ± 1.7 |
| pO2 (mmHg) | 124.9 ± 5.9 | 116.0 ± 6.8 |
| Cl− (mmol/L) | 101.4 ± 1.8 | 101.5 ± 1.0 |
| Hematocrit (%) | 37.3 ± 0.3 | 37.0 ± 2.0 |
| Blood gas analysis (15 min after) | ||
| pH | 7.50 ± 0.01 | 7.47 ± 0.05 |
| pCO2 (mmHg) | 37.7 ± 1.4 | 36.8 ± 1.4 |
| pO2 (mmHg) | 109.5 ± 2.4 | 111.0 ± 7.8 |
| Cl− (mmol/L) | 101.7 ± 1.2 | 102 ± 0.6 |
| Hematocrit (%) | 37.3 ± 0.3 | 38.5 ± 2.1 |
|
|
Nrf2+/+ |
Nrf2+/− |
Nrf2−/− |
|---|---|---|---|
| Adult mouse focal (24 h survival) | |||
| Number of animals | 8 | 5 | |
| Core temp. 30 min after cautery (°C) | 36.6 ± 0.2 | 36.1 ± 0.7 | |
| Weight before stroke (g) | 26.4 ± 1.1 | 25.9 ± 0.7 | |
| Weight at killing (g) | 24.8 ± 1.0 | 25.2 ± 0.9 | |
| Adult mouse focal (7 d survival) | |||
| Number of animals | 9 | 3 | 8 |
| Core temp. before cautery (°C) | 37.2 ± 0.2 | 37.4 ± 0.1 | 37.2 ± 0.2 |
| Core temp. 30 min after cautery (°C) | 37.1 ± 0.2 | 37.7 ± 0.2 | 36.7 ± 0.2 |
| Weight before stroke (g) | 27.0 ± 0.8 | 32.0 ± 0.3 | 29.2 ± 0.6 |
| Weight at 24 h after stroke (g) | 27.0 ± 1.0 | 32.1 ± 0.9 | 28.2 ± 1.0 |
| Weight at sacrifice (g) | 27.0 ± 0.9 | 29.5 ± 0.2 | 28.8 ± 0.7 |
|
|
Nrf2+/+ |
|
Nrf2−/− |
|
|---|---|---|---|---|
| Adult mouse ET-1 (7 d survival) | ||||
| Diet type | Control | 1% tBHQ | Control | 1% tBHQ |
| Number of animals | 6 | 6 | 5 | 6 |
| Core temp. after microinjection (°C) | 37.2 ± 0.1 | 37.4 ± 0.3 | 37.1 ± 0.2 | 36.9 ± 0.3 |
| Weight before tBHQ feeding (g) | 31.4 ± 1.7 | 31.8 ± 1.2 | 25.8 ± 1.0 | 27.9 ± 0.6 |
| Weight before stroke (g) | 30.7 ± 1.4 | 30.0 ± 1.2 | 25.7 ± 1.0 | 21.3 ± 0.3* |
| Weight at sacrifice (g) |
30.3 ± 2.0 |
29.4 ± 1.4 |
26.2 ± 1.0 |
19.5 ± 0.5*
|
All values are the mean ± SE. For rat studies, there were no significant differences among groups for physiological variables, Student's t test, or one-way ANOVA. For mouse studies, only a significant reduction of body weight in tBHQ-fed Nrf2−/− mice was observed, compared with Nrf2−/− mice fed the control diet. *p<0.05 versus control diet for the same genotype. temp., Temperature.
Induction of cortical glutathione levels in vivo after tBHQ treatment
Given that intracerebroventricular delivery of tBHQ is not necessarily a viable strategy for clinical prophylactic treatment, we also tested whether intraperitoneal injection of tBHQ could protect against focal ischemia-reperfusion in rats. We first optimized intraperitoneal injection of tBHQ by monitoring cortical GSH content, as well as GST and NQO1 activities. Using a protocol consisting of three 16.7 mg/kg injections, each spaced 8 h apart, we could robustly and consistently increase GSH levels at 24 h after the first injection (Fig. 3). The increase in GSH was apparently attributable to local brain synthesis, as opposed to being delivered to the brain after synthesis in peripheral organs (such as the liver), because GSH levels in the serum and CSF were unaltered with tBHQ treatment [serum GSH plus oxidized GSH (GSSG) levels 24 h after intraperitoneal injection (in μm): vehicle, 14.5 ± 2.9; tBHQ, 14.7 ± 2.8; CSF GSH plus GSSG levels (in μm): vehicle, 3.1 ± 0.3; tBHQ, 3.2 ± 0.4]. Using this treatment, we were not able to resolve significant elevation of GST and NQO1 enzyme activity in the cortex, perhaps because longer time points were needed for the induction of these enzymes. It is also possible that the site of induction for these enzymes is restricted to a subpopulation of cells in the total tissue sample (i.e., specific astrocyte types or the meninges) leading to dilution of the enzyme activity (Murphy et al., 1998). The 16.7 mg/kg dose (total 50 mg/kg injected) was used for subsequent stroke experiments because this treatment produced a significant increase in cortical GSH, whereas a dose of 3.3 mg/kg was unable to produce this induction (Fig. 3). With higher doses (33.3 mg/kg), animals displayed adverse behavior with ataxia of the hindlimbs and trunk, as observed in previous studies (Food and Agriculture Organization of the United Nations/World Health Organization, 1975). The 16.7 mg/kg dose sometimes produced a mild and transient ataxia observed within the first 5 min of injection, which lasted for <10 min. However, this effect is unlikely to be related to Nrf2 because of its rapid and transient nature and may be attributable to discomfort from the injected solution.
Figure 3.

Induction of glutathione in the cortex after intraperitoneal injection of tBHQ. Sprague Dawley rats were injected with tBHQ intraperitoneally (vehicle, 3.3 mg/kg, or 16.7 mg/kg) three times daily (8 h apart), and GSH content was evaluated 24 h after the first injection. Homogenized cortical tissue was protein precipitated, and the acid-soluble GSH was immediately assayed using the glutathione reductase method (Tietze, 1969). The 16.7 mg/kg dose, but not the 3.3 mg/kg dose, significantly increased cortical GSH levels. Symbols represent paired animals in each experiment. Data represent means ± SEM from n = 6 animals per treatment group. **p = 0.007, two-tailed t test.
Prophylactic intraperitoneal injection of tBHQ confers protection both 24 h and 1 month after ischemia-reperfusion
Comparable with intracerebroventricular delivery, intraperitoneal administration to rats also reduced ischemic damage to the cortex 24 h after transient MCAO (Fig. 4A) (volume of cortical infarct expressed as percentage of total ischemic hemisphere: vehicle, 14.76 ± 3.83; tBHQ, 7.50 ± 3.04; *p < 0.05; two-tailed t test), whereas no significant protection was observed in the sub-cortex (Fig. 4B). In this case, ischemia was induced 24 h after the first of three tBHQ injections (Fig. 4E). We did not observe differences in core temperature between vehicle and tBHQ-treated groups before, during, or 30 min after the ischemic period (Table 1). Again, a similar level of sensorimotor impairment was detected between vehicle and tBHQ groups during ischemia, but a significant improvement in the tBHQ-treated animals was observed 24 h after ischemia (Fig. 4C). Importantly, tBHQ injection did not significantly reduce body weight compared with controls, suggesting that the protective effect was not caused by reduced caloric intake (Fig. 4D, Table 1) (Yu and Mattson, 1999).
Figure 4.

Intraperitoneal injection of tBHQ reduced sensorimotor deficit and stroke damage 24 h after ischemia-reperfusion. Sprague Dawley rats were injected with tBHQ intraperitoneally (16.7 mg/kg; 3 times) or vehicle, as described in Figure 3, followed by focal ischemia-reperfusion. The animals were killed 24 h after initiation of transient MCAO (1.5 h), and brain slices were stained with TTC. A, tBHQ treatment significantly reduced stroke damage in anterior cortical regions of the affected hemisphere. B, Similar to intracerebroventricular delivery of tBHQ, no sparing of subcortical tissue was observed. C, Acute sensorimotor deficits assessed during the stroke (1.5 h) were similar, suggesting that animals from both groups received the same degree of ischemia. However, sensorimotor deficit was significantly lower in the tBHQ-treated group when evaluated 24 h after stroke. Avg. Neurol. Score, Average neurological score. D, Average animal weight in the two treatment groups was not different when examined both 24 h after intraperitoneal injection of tBHQ and 24 h after stroke. E, A schematic diagram summarizing the injection and stroke timeline. The short arrows represent time points of tBHQ injection. Data represent mean ± SEM collected from n = 11 animals per treatment group. *p < 0.05 and **p < 0.01, two-tailed t test or one-way ANOVA with Bonferroni's post hoc test.
To assess the long-term benefit of acute tBHQ injection, we examined a second group of rats for 1 month after ischemia. These animals were injected with tBHQ as with 24 h survival groups, and no additional tBHQ supplementation was given after stroke. Arterial blood-gas levels were examined before, during, and 15 min after ischemia-reperfusion. We found no differences in blood pH, pCO2, pO2, Cl-, or hematocrit between vehicle and tBHQ-treated groups (Table 1). Neurological scoring was performed during ischemia to verify successful occlusion, and on a weekly basis thereafter. tBHQ-treated animals had, on average, lower neurological scores throughout the entire survival period (**p = 0.002; two-way ANOVA) (Fig. 5B) but recovered sensorimotor function more quickly within the first 4 d of reperfusion, compared with vehicle controls. Histological examination showed that ischemic damage in anterior cortex was significantly reduced with tBHQ treatment even 1 month after the ischemic event (volume of cortical infarct expressed as percentage of total ischemic hemisphere: vehicle, 24.9 ± 3.9; tBHQ, 10.1 ± 4.5; *p < 0.05; two-tailed t test) (Fig. 5A,C). No attenuation of damage was observed in the subcortex (Fig. 5D). Together, these data suggest that the effects of systemic tBHQ administration extend beyond the blood-brain barrier (possibly even more so in an ischemia-compromised state), providing neuroprotection and preservation of sensorimotor function for up to 1 month after stroke.
Figure 5.

Intraperitoneal injection of tBHQ reduces sensorimotor deficit and stroke damage 1 month after stroke onset. As with 24 h survival animals, long-term survival experiments were initiated with the same intraperitoneal tBHQ injection regimen (16.7 mg/kg; 3 times) or vehicle before ischemia-reperfusion. No additional injections were given after stroke. Sensorimotor function was evaluated on a weekly basis. After 1 month, the animals were perfused, and brains were cryosectioned for cresyl violet staining. A, Representative examples are shown from each treatment group (infarct border is demarcated by black line). B, tBHQ treatment significantly improved the average neurological score (Avg. Neurol. Score) throughout the entire survival period (**p = 0.002; two-way ANOVA). C, Infarction of the anterior cortical regions was attenuated in the tBHQ-treated animals. D, No significant subcortical protection was observed. Data represent mean ± SEM collected from n = 6 for tBHQ and n = 5 animals for vehicle control groups. *p < 0.05, two-tailed t test or one-way ANOVA with Bonferroni's post hoc test.
Basal and inducible phase 2 enzyme activities are suppressed in the Nrf2-/- mouse brain
Induction of Nrf2-dependent gene expression normally occurs in the ischemic penumbra after stroke, raising the possibility that Nrf2 activation may be an endogenous response to limit stroke damage (van Lookeren Campagne et al., 1999; Campagne et al., 2000; Laxton et al., 2001; Liverman et al., 2004). Notably, data from gel-shift assays indicated that binding to the ARE sequence found in Nrf2 target genes such as metallothionein-1 and metallothionein-2 was upregulated after transient forebrain ischemia in rat (Campagne et al., 2000).
Before testing, the hypothesis that Nrf2 modifies sensitivity to ischemia in vivo, we first verified the Nrf2-/- phenotype and the effect of tBHQ administration in our Nrf2-/- mouse colony. Basal and inducible phase 2 enzyme levels were examined in the liver and multiple brain regions, again using GSH, GST, and NQO1 as markers. As observed in previous studies, all three markers were suppressed in the livers of Nrf2-/- mice when compared with Nrf2+/+ controls (Fig. 6B,D,F) (Chan and Kwong, 2000; Hayes et al., 2000; Kwak et al., 2001a; Chanas et al., 2002). In brain, basal GST and NQO1 activities were modestly but significantly reduced in multiple brain regions of Nrf2-/- mice, compared with Nrf2+/+ controls (Fig. 6A,C). Interestingly, basal brain GSH levels were not altered, suggesting that the constitutive synthesis of brain GSH does not rely on Nrf2 function, at least in the young adult mice examined in this study (Fig. 6E). LDH is not regulated by Nrf2 and, accordingly, no difference in activity was observed between genotypes (Fig. 6G,H).
Figure 6.
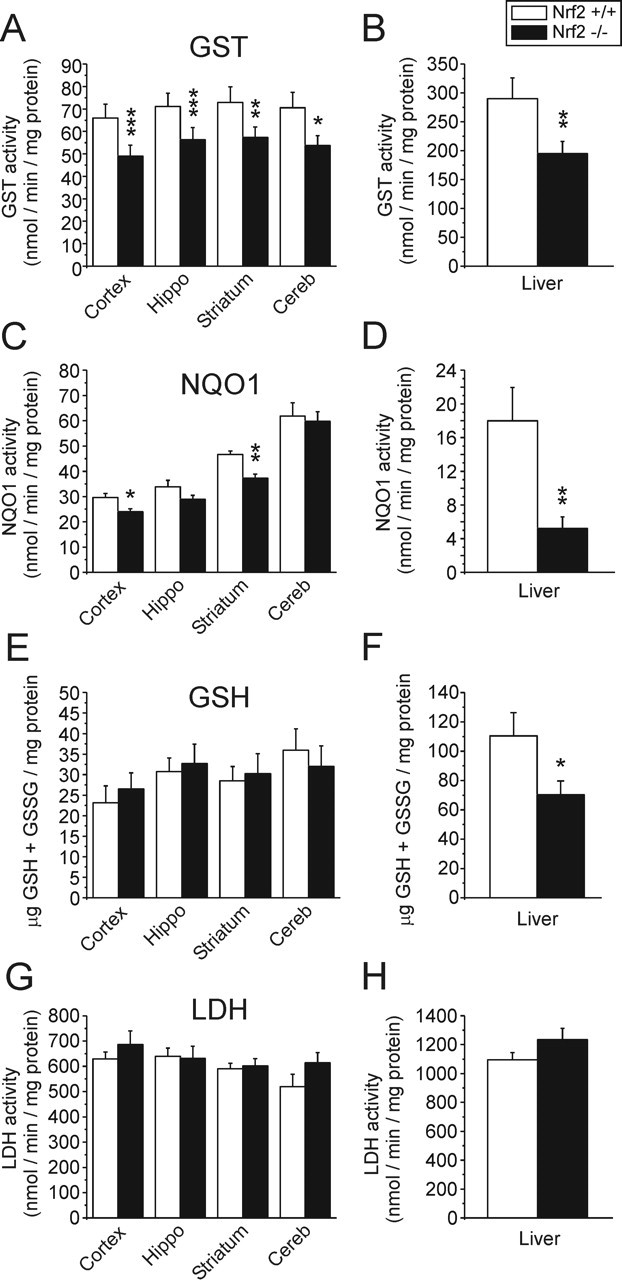
Reduced basal GST and NQO1 activity, but not GSH content, in the Nrf2-/- brain. Nrf2+/+ and Nrf2-/- littermate pairs were examined for markers of Nrf2 activity. Brains were dissected in cold PBS, homogenized, and immediately assayed for GSH, GST, NQO1, and LDH. A, B, GST activity in Nrf2-/- mice was lower in liver and all brain regions examined, compared with Nrf2+/+ mice. C, D, NQO1 activity was significantly reduced in the Nrf2-/- liver, cortex, and striatum. E, F, Total GSH content, although reduced in the Nrf2-/- liver, was not reduced in any brain regions examined. G, H, Activity levels of LDH (not an Nrf2 target) were not reduced in liver or brains of Nrf2-/- mice. Data represent the mean ± SEM of six mice per genotype. *p < 0.05, **p < 0.01, and ***p < 0.001; paired two-tailed t test. Hippo, Hippocampus; Cereb, cerebellum.
We next examined tBHQ-dependent induction of these markers in mice with different Nrf2 genotypes. We initially found that all genotypes responded poorly to intraperitoneal injections of tBHQ administered at the same dose used for rats (three injections of 16.7 mg/kg), thus exhibiting hyperactivity, increased sensitivity to tactile stimulus, and vertical jumping. As an alternative, we delivered tBHQ by dietary supplementation, as described previously for other Nrf2 inducers such as sulforaphane and oltipraz (McMahon et al., 2001; Ramos-Gomez et al., 2001; Fahey et al., 2002; Wu et al., 2004). Studies conducted by the World Health Organization found that C14-labeled tBHQ could be detected in the brain after dietary administration to rodents, suggesting that tBHQ is able to pass the blood-brain barrier through this route (National Toxicology Program, 1997; Food and Agriculture Organization of the United Nations/World Health Organization, 1999). In previous studies, we found that 5% tBHQ in diet was not consumed by Nrf2-expressing mice, whereas 1% tBHQ was consumed and highly protective (in Nrf2-expressing mice but not in knock-outs) during 3-nitropropionic acid toxicity in vivo, a model involving striatal-specific oxidative stress (Shih et al., 2005). A diet of 0.5% tBHQ was only partially neuroprotective against 3-nitropropionic acid (Shih et al., 2005). For these reasons, a concentration of 1% tBHQ was used in all additional feeding experiments for the current study.
Nrf2-/- mice were less tolerant of dietary tBHQ administration, losing ∼20% body weight during 1 week of tBHQ feeding and consuming approximately one-half as much food as tBHQ-fed Nrf2+/+ and Nrf2+/- mice (Fig. 7A,B). In total, Nrf2+/+, Nrf2+/-, and Nrf2-/- mice consumed 12.5 ± 1.4, 12.7 ± 1.3, and 7.3 ± 0.5 mg of tBHQ per gram of body weight, respectively, over 1 week. The 1% tBHQ diet increased cortical GSH levels in Nrf2+/+ and Nrf2+/- mice, consistent with intraperitoneal delivery of tBHQ in rats (Fig. 7C). Importantly, the induction of cortical GSH was not observed in Nrf2-/- mice, providing evidence that tBHQ-induced changes in cortical GSH are Nrf2 dependent (Fig. 7C). In addition, a tBHQ-induced increase in liver GST and NQO1 activities was observed in Nrf2-expressing mice, but not Nrf2-/- mice, consistent with the effects of other Nrf2 inducers used in vivo (Fig. 7D,E) (Kwak et al., 2001a; McWalter et al., 2004). As with rat intraperitoneal injection, tBHQ feeding did not induce a detectable increase in GST or NQO1 activity measured by homogenate assay from brain tissue. No changes were observed with the negative control enzyme LDH in any of the tissues examined (Fig. 7F). Interestingly, we observed an increase in liver GSH levels in tBHQ-fed Nrf2-/- mice. We believe that this Nrf2-independent change may reflect an inherent hepatic toxicity associated with tBHQ feeding, perhaps because of inefficient detoxification from the body by the same pathways regulated by Nrf2 (phase 2 genes). Importantly, tBHQ feeding in the absence of stroke did not cause any overt toxicity to brain tissue of Nrf2-/- mice, as assessed by the lack of FluoroJade staining (data not shown) and normal levels of LDH (Fig. 7F). Furthermore, because GSH content and antioxidant enzyme activities were unchanged in the brains of tBHQ-fed Nrf2-/- mice, it is unlikely that other Nrf2-independent stress responses are evoked by tBHQ toxicity in brain. Together, these results indicate that phase 2 induction in response to tBHQ is abrogated in Nrf2-/- mice, and these mice exhibit some tBHQ-induced systemic toxicity but not necessarily brain toxicity.
Figure 7.

Effect of dietary tBHQ administration on brain and liver GSH content and phase 2 enzyme activity. Nrf2-/- mice and Nrf2-expressing controls were fed 1% tBHQ for 1 week and were subsequently examined for markers of Nrf2 activity. A, B, Summary of animal weight and food consumption from tBHQ feeding experiments. On average, Nrf2-/- mice lost ∼20% of their starting body weight and consumed approximately one-half as much food as Nrf2-/- mice fed control diet. tBHQ feeding did not cause loss of body weight in Nrf2-expressing mice. Brains from tBHQ-fed mice were dissected in cold PBS, homogenized, and immediately assayed for GSH, GST, NQO1, and LDH. C, Consistent with intraperitoneal injection of tBHQ in rats, dietary tBHQ also produced a significant increase in GSH levels within the cortex and striatum of Nrf2-expressing mice. No increase in GSH was observed in the brains of Nrf2-/- mice, consistent with the Nrf2-specific action of tBHQ. However, an increase of GSH was detected in the livers of Nrf2-/- mice, which may be associated with tBHQ toxicity observed in this genotype. D, E, Increases in both GST and NQO1 activity were only detected in the livers of Nrf2-expressing mice but not Nrf2-/- mice. F, LDH levels were not altered by tBHQ feeding in any genotype. Cx, Cortex; Str, striatum; Liv, liver. Data represent the mean ± SEM from the number of mice indicated in figure legends (in parentheses). *p < 0.05, two-tailed t test.
Loss of Nrf2 function exacerbates cortical infarction 7 d, but not 24 h, after permanent ischemia
Given the apparent changes in basal markers of ARE-mediated gene expression, we next examined whether Nrf2-/- mice were more susceptible to ischemic injury. In our hands, the intraluminal suture model of MCAO (as used with rats) did not produce consistent strokes with Nrf2 colony mice because of increased patency of the posterior communicating arteries leading to strong collateral blood flow from the vertebral arteries (Kitagawa et al., 1998; Maeda et al., 1998). Furthermore, bilateral occlusion of the common carotid arteries was also unable to achieve global ischemia in these mice for the same reason (data not shown) (Fujii et al., 1997; Kitagawa et al., 1998). Thus, we found it necessary to use a focal ischemia model involving permanent occlusion of the distal middle cerebral artery by cauterization, as described previously (Cechetto et al., 1989). Using this model, we could consistently reduce regional cerebral blood flow by ∼80% in mice expressing Nrf2 as well as in knock-outs (measured by laser Doppler) (Fig. 8B), resulting in a cortical infarct that affected ∼10-15% of the ipsilateral cortical volume (Fig. 8A). Twenty-four hours after permanent focal ischemia, we observed no significant difference in infarct size between Nrf2-/- and Nrf2+/+ mice by TTC vital staining (Fig. 8C,E). However, it is conceivable that 24 h (in contrast to 7 d) is too early to observe cell death specific to the Nrf2-/- genotype within the permanently occluded infarct core because the penumbra is still evolving as collateral blood flow develops, and late reperfusion may actually promote secondary generation of oxidative stress (Chan, 2001). In addition, we may not observe a difference between genotypes at 24 h after stroke because this time is likely insufficient for Nrf2-dependent mRNA and protein expression in Nrf2+/+ mice. Therefore, we also evaluated a different group of mice at 7 d after permanent occlusion. At this time point, we then observed a large increase (approximately twofold) in infarct volume with Nrf2-/- mice but not with Nrf2+/+ controls (Fig. 8A,D,E). No significant difference in body core temperature (Table 1) or reduction of regional cerebral blood flow was observed between genotypes during the induction of ischemia (Fig. 8B). We propose that this continued expansion of infarct size, between the 24 h and 7 d time points, reflects cell death that is sensitive to the Nrf2-/- genotype. These results suggest a model in which, at short time points (24 h), Nrf2-mediated gene induction is not complete or impaired in permanent ischemia models, whereas over time (7 d), Nrf2 activity in perhaps surviving glial cells restricts infarct expansion in Nrf2-expressing mice but not in Nrf2-/- mice.
Figure 8.

Loss of Nrf2 function in vivo increased cortical damage after permanent focal ischemia. Male mice (10-16 weeks of age) of various Nrf2 genotypes were subjected to permanent focal ischemia by cauterization of the distal middle cerebral artery, producing an infarct restricted to the cortex. Twenty-four hours or 7 d after stroke onset, mice were killed for evaluation of infarct size using cresyl violet staining. A, Representative examples for each genotype are shown from the 7 d survival group (infarct borders are demarcated by black line). B, To ensure that all genotypes received complete occlusion of regional cerebral blood flow, surgeries were guided by laser Doppler continuous measurements of blood flow through the skull above the temporal ridge. No difference in blood flow reduction was observed between the genotypes. C, E, A separate group of animals, killed 24 h after stroke, showed no significant differences in cortical damage between genotypes (visualized with TTC; data not shown). D, E, However, 7 d after stroke, Nrf2-/- mice sustained considerably more damage, which usually extended to medial and dorsal cortical regions, whereas Nrf2+/+ and Nrf2+/- controls exhibited no increase in infarct size beyond the 24 h time point. Data represent the mean ± SEM collected from n = 9, 3, and 8 mice for Nrf2+/+, Nrf2+/-, and Nrf2-/- groups, respectively (7 d survival). n = 8 and 5 mice for Nrf2+/+ and Nrf2-/- groups, respectively (24 h survival). *p < 0.05, **p < 0.01, and ***p < 0.001; paired two-tailed t test or one-way ANOVA with Bonferroni's post hoc test.
Given that the infarct size of Nrf2+/+ mice did not increase in size between 24 h and 7 d, we speculated that the potentially salvageable penumbral region associated with this permanent ischemia model was small or predominantly survived even 7 d after ischemia, attributable, in part, to the action of Nrf2-dependent factors (Hata et al., 2000a,b). To examine this possibility, we fed mice with 1% tBHQ before permanent focal ischemia and evaluated infarct size 7 d after ischemia. Consistent with Nrf2-dependent events already limiting infarct size, 1 week of tBHQ feeding did not offer additional reduction of infarct size in Nrf2+/+ and Nrf2+/- mice (supplemental Fig. 2 and supplemental Table 1, available at www.jneurosci.org as supplemental material). In contrast, the continuing growth of infarct size in Nrf2-/- mice suggests that loss of Nrf2 function promotes periinfarct (penumbral) death of neurons well after ischemic onset. However, in Nrf2-/- mice, this delayed neuronal death should be insensitive to the Nrf2-dependent effects of tBHQ. Consistent with this idea, tBHQ feeding also offered no protection to Nrf2-/- mice (supplemental Fig. 2 and supplemental Table 1, available at www.jneurosci.org as supplemental material). Nevertheless, in this second cohort of animals, Nrf2-/- mice still exhibited larger infarcts with either control or tBHQ diet, compared with Nrf2-expressing mice, as observed before. Our data, showing an inability of tBHQ to protect against the core of the ischemic infarct in Nrf2+/+ and Nrf2+/- mice, is consistent with tBHQ effects being restricted to a salvageable penumbra (that is perhaps not well developed in a permanent occlusion model) (Memezawa et al., 1992a,b; Hata et al., 2000a). Nevertheless, this negative result also fails to directly link Nrf2 and tBHQ in protection against stroke damage in mice.
Loss of Nrf2 function abrogates tBHQ-mediated neuroprotection during an endothelin-1 model of ischemia-reperfusion
To better test the effect of tBHQ in penumbral brain regions (with partial reduction in blood flow), we asked whether the neuroprotective effect of tBHQ was abrogated in Nrf2-/- mice used in an ET-1-induced ischemia-reperfusion model, which produces a mild insult with a larger penumbra. ET-1 is a potent vasoconstrictive peptide that can be either applied directly to cerebral vessels to produce large regions of ischemia, or injected into the parenchyma to produce a localized and quantifiable infarct. In this way, variability in cerebral vasculature can be circumvented, and ischemia can be reliably achieved (Yanagisawa et al., 1988; Fuxe et al., 1992; Sharkey et al., 1993; Hughes et al., 2003; Gilmour et al., 2004). It is well known that ET-1 application produces a long-lasting but moderate reduction in blood flow (to ∼30-50% of baseline for 2-16 h, dependent on dose), followed by complete reperfusion, effectively modeling the ischemic penumbra (Fuxe et al., 1992; Biernaskie et al., 2001; Zhang et al., 2005). Although the intraluminal suture model allows more abrupt and controlled reperfusion, the gradual reperfusion observed in ET-1-induced stroke may be a more adequate reflection of human stroke (Biernaskie et al., 2001). Infarction caused by intraparenchymal ET-1 injection can be reversed by vasodilatory drugs, suggesting that damage is directly caused by loss of blood flow (Fuxe et al., 1992). Tissue damage resulting from ET-1-induced ischemia has injury mechanisms similar to intraluminal suture MCAO. For example, brain glucose levels decrease concurrently with an increase in lactate, suggesting metabolic inhibition (Gramsbergen et al., 2004). Furthermore, extracellular glutamate and dopamine levels accumulate (Bogaert et al., 2000), and peri-infarct depolarizations occur (Kleeberg et al., 2004). ET-1-induced ischemic injury can also be attenuated by strategies known to be protective during intraluminal suture MCAO, such as blockade of NMDA receptors (MK801 [(+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine maleate]) (Sharkey et al., 1994), voltage-gated sodium channels (AM-36) (Callaway et al., 2004), and voltage-gated calcium channels (nimodipine) (Bogaert et al., 2001), as well as by free radical scavengers (ebselen and coenzyme Q10) (Dawson et al., 1995; Ostrowski et al., 1998), and hypothermia (Van Hemelrijck et al., 2003). In this study, ET-1 was directly injected into the cortical parenchyma to create a localized and reproducible region of ischemia. Infarct sizes were similar to those observed in previous studies, as assessed by FluoroJade-positive staining of degenerating neurons. (Fig. 9A) (Zhang et al., 2005).
Figure 9.

tBHQ-mediated protection of cortical tissue from transient ischemia is lost in Nrf2-/- mice. Male Nrf2+/+ and Nrf2-/- mice were fed 1% tBHQ or control diet for 1 week before intracortical ET-1 microinjection. Mice were maintained for 1 additional week after ET-1 injection (during which respective diets were continued) and were then killed for evaluation of infarct size. A-D, Representative FluoroJade-stained images of ischemia-induced neurodegeneration after ET-1 injection (bregma = -0.9 mm). Scale bar, 500 μm. E, Summary of animal weight during tBHQ prefeeding (1-7 d) and after ET-1 injection (8-15 d). F, Average infarct size, as assessed by the FluoroJade-positive area, was significantly reduced with tBHQ-fed Nrf2+/+ mice, compared with its control diet group. In contrast, the average infarct size of tBHQ-fed Nrf2-/- mice showed a trend toward exacerbation when compared with its control diet group. Data represent the mean ± SEM from the number of mice indicated in figure legends (in parentheses). *p < 0.05, unpaired two-tailed t test.
Feeding Nrf2+/+ mice with tBHQ reduced ET-1-induced infarction by 66.1 ± 15.5% when compared with Nrf2+/+ mice on control diet (Fig. 9A,B,F) (*p < 0.05; unpaired t test; n = 6 for both groups). Consistent with the protective effects of tBHQ being Nrf2 dependent, in vivo tBHQ feeding did not reduce infarct size in Nrf2-/- mice (Fig. 9C,D,F). In fact, tBHQ appeared to exacerbate injury with infarcts, on average, 280.8 ± 132.2% larger than in Nrf2-/- mice receiving control diet (n = 5 for control diet; n = 6 for tBHQ diet); however, the apparent increase in sensitivity was not significantly different from controls because of high infarct variability between Nrf2-/- mice on tBHQ diets. Again, tBHQ feeding reduced body weight of Nrf2-/- mice by ∼20%, highlighting the increased sensitivity of the Nrf2-/- genotype (Fig. 9E) and as observed previously with 3-nitropropionic acid toxicity in vivo (Shih et al., 2005). Nrf2+/+ mice did not lose significant body weight during tBHQ feeding, suggesting that caloric restriction did not account for neuroprotection (Fig. 9E). Interestingly, when using ET-1-induced ischemia, the infarct size of Nrf2-/- mice fed the control diet was un-expectedly smaller than that of Nrf2+/+ mice on control diet (Fig. 9A,C and data not shown) (*p < 0.05; unpaired t test). This result could be explained by differential sensitivity of the Nrf2-/- genotype to ET-1-induced vasoconstriction. Supporting this proposal, we re-examined our microarray data obtained using adenovirus to overexpress Nrf2 in astrocytes and found that the endothelin B receptor was upregulated ∼30-fold by Nrf2 (Shih et al., 2003) (http://www.pharmacy.wisc.edu/facstaff/sciences/JohnsonGroup/microdata.cfm), possibly altering ET-1-induced signaling (see Discussion). In summary, using the ET-1 model, we show that dietary administration of tBHQ ameliorates damage caused by ET-1-induced ischemia-reperfusion in Nrf2+/+ mice. Importantly, tBHQ offers no protection to Nrf2-/- mice from ET-1 injection, confirming the Nrf2-specific action of tBHQ in vivo.
Discussion
Our findings show that Nrf2 plays an important role in limiting ischemic injury. We observed the neuroprotective effects of tBHQ, a well characterized Nrf2 inducer, with two different models of ischemia-reperfusion (MCAO and ET-1 vasoconstriction) in rats and mice, using three different routes of administration (intracerebroventricular, intraperitoneal, and dietary). An important finding was that prophylactic tBHQ treatment improves functional recovery up to 1 month after transient MCAO in rats, suggesting that previous Nrf2 activation may reduce neuronal death during delayed apoptosis and inflammation long after stroke onset. Conversely, loss of Nrf2 function in vivo exacerbates ischemic damage and abrogates the protective effects of tBHQ (in the ET-1 model), consistent with a specific role for Nrf2. Together, these results highlight the potential of Nrf2 activation by small molecule inducers such as tBHQ as a new prophylactic strategy for stroke. Similarly, natural Nrf2 inducers such as sulforaphane are also relatively nontoxic and may also be of value (Fahey and Talalay, 1999; Kraft et al., 2004).
Nrf2 dependency of tBHQ-mediated neuroprotection
There is abundant in vitro evidence demonstrating that tBHQ is dependent on Nrf2 for its ability to induce phase 2 enzymes and confer protection (Venugopal and Jaiswal, 1996; Alam et al., 1999). Brain astrocytes derived from Nrf2-/- mice do not induce phase 2 genes and fail to protect neurons from in vitro toxicity paradigms when treated with small molecule inducers of Nrf2 activity (Fig. 1) (Lee et al., 2003a,b; Kraft et al., 2004). Our recent studies on mitochondrial stress (systemic 3-nitropropionic acid) show that tBHQ action is Nrf2 dependent in vivo because its protective effect was lost in Nrf2-/- mice (Shih et al., 2005). However, in these studies, tBHQ-mediated enhancement of peripheral 3-nitropropionic acid detoxification (through liver and intestines) may have contributed to reduction of striatal toxicity. In the current study, Nrf2-/- mice exhibited lower phase 2 enzyme levels in brain and liver, and the neuroprotective effects of tBHQ were lost in an ET-1-induced ischemia model (Figs. 6, 7, 9), providing additional evidence that tBHQ function is dependent on Nrf2 in vivo. Furthermore, the effect of tBHQ is physiologically relevant to brain because it results in attenuated local ischemic brain damage, as opposed to potentially metabolizing toxins such as 3-nitropropionic acid peripherally. Our findings also dispute the possibility that tBHQ provides neuroprotection through its well known function as an antioxidant. If this were the case, neuroprotection through the intrinsic antioxidant properties of tBHQ would have been Nrf2-independent and thus observed in Nrf2-/- mice.
There remain some important caveats that must be considered. First, Nrf2-/- mice were found to be less tolerant to tBHQ feeding, consuming less food and losing more weight than Nrf2 expressing controls (Fig. 7A,B) (Shih et al., 2005). We speculate that the reduced tolerance to tBHQ is attributable to systemic toxicity (i.e., liver) associated with poor tBHQ metabolism and detoxification, which may sensitize Nrf2-/- mice to toxic tBHQ side effects. However, it is worth noting that we found no evidence of tBHQ toxicity in the brains of Nrf2-/- mice (based on lack of FluoroJade staining and normal levels of LDH and anti-oxidant enzyme activities in brain tissue).
Previous studies have also used Nrf2-/- mice to demonstrate the Nrf2 dependency of small molecule inducers in vivo (nonbrain related). For example, inducers such as sulforaphane and oltipraz (an anti-shistosomal drug) have been administered through diet (at similar concentrations as we use tBHQ) and were found to require Nrf2 for protection from toxicity paradigms such as liver/gastrointestinal tract carcinogenesis and acute lung injury (Kwak et al., 2001b; Ramos-Gomez et al., 2001; Fahey et al., 2002). Furthermore, McMahon et al. (2001) found that butylated hydroxyanisole, a direct precursor of tBHQ, also exhibited some toxicity in Nrf2-/- mice. Butylated hydroxyanisole was a potent inducer of phase 2 enzymes in Nrf2+/+ mice, but not Nrf2-/- mice, confirming its Nrf2-dependent action (McMahon et al., 2001). To clearly dissociate potential systemic toxicity of tBHQ from its effects on CNS glia, future experiments would benefit from inducible and tissue-specific Nrf2 knock-out strategies.
A second caveat is that Nrf2-/- mice may have reduced sensitivity to the ET-1-induced ischemia model. Indeed, the ET-B subtype of endothelin receptor (GenBank accession number S65355) was found to be highly upregulated (∼30-fold) by Nrf2 overexpression in astrocyte cultures (Shih et al., 2003) (also see http://www.pharmacy.wisc.edu/facstaff/sciences/Johnson-Group/microdata.cfm). ET-B has been shown to modulate cerebral blood flow by inducing both vasodilation or vasoconstriction in different circumstances (Clozel et al., 1992; Moreland et al., 1992; Touzani et al., 1997). This suggests that loss of Nrf2 function may decrease basal expression of ET-B, leading to the reduced response we observed during exogenous ET-1 injection (Stenman et al., 2002). However, it is important to note that exogenous ET-1 injection still produced an ischemic infarct in Nrf2-/- mice that was not attenuated by tBHQ, despite reduced sensitivity to ET-1.
Nrf2 activation protects the ischemic penumbra but not the stroke core
Nrf2 activity attenuates damage caused by transient MCAO in rats. Within this injury paradigm, tBHQ administration was able to salvage the cortical penumbra but not the striatal stroke core. It is well known that, during proximal MCAO, death of striatal tissue results from more severe ischemia than within the surrounding cortex (Tyson et al., 1984; Nedergaard et al., 1986; Fisher and Garcia, 1996). Similarly, we also found that tBHQ was unable to protect mice against permanent focal ischemia (supplemental Fig. 2 and supplemental Table 1, available at www.jneurosci.org as supplemental material). Permanent ischemia also tends to result in a more severe infarction on a faster timescale (Memezawa et al., 1992a,b; Lipton, 1999; Mao et al., 1999). Permanent ischemia may also produce a smaller and more short-lived penumbra than ischemia-reperfusion (Hata et al., 2000a,b). This idea is supported by our findings using ET-1-induced stroke, in which a partial reduction of blood flow produces a larger penumbra and the protective effect of tBHQ can be clearly observed. Interestingly, although tBHQ did not reduce damage to the striatal stroke core during transient MCAO, it significantly attenuated striatal neurodegeneration during 3-nitropropionic acid toxicity (Shih et al., 2005). Because 3-nitropropionic acid produces gradual and partial metabolic inhibition (Beal et al., 1993; Sato et al., 1997; Brouillet et al., 1998; Kim et al., 2000), it is in some ways similar to the salvageable cortical stroke penumbra.
Identifying the signals that activate endogenous Nrf2 in response to ischemia
In this study, we found that Nrf2-/- mice were more sensitive than controls during permanent focal ischemia, suggesting that Nrf2-mediated phase 2 gene induction may be a physiological response to mitigate additional damage. Previous studies using gel-shift assays found that ischemia in rats selectively upregulates ARE-mediated gene expression in vivo (mediated by Nrf2), whereas binding activities of other stress response elements were unchanged [including metal response element, interleukin-6, and STAT (signal transducer and activator of transcription) response elements] (Campagne et al., 2000). Ischemia-induced ARE binding was specific because it could be blocked by mutation of the consensus ARE sequence. In addition, oligemia (blood flow reduction without acute tissue damage that occurs in shock, migraine, and stroke penumbra) also activates Nrf2 (Liverman et al., 2004). Robust induction of phase 2 enzymes such as NQO1 and metallothionein-1 can be observed in astrocytes, vascular endothelial cells, and pia-mater cells of the infarct border within hours/days after stroke onset (van Lookeren Campagne et al., 1999; Campagne et al., 2000; Laxton et al., 2001). Thus, the timing and location of phase 2 gene induction suggests that activation of endogenous Nrf2 may be necessary for limiting oxidative stress within the ischemic penumbra. Many components of ischemic injury may trigger this activation of endogenous Nrf2. For example, mitochondrial inhibition, endoplasmic reticulum stress induced by glucose deprivation, release of inflammatory mediators, and increased oxidative stress are events relevant to ischemic injury and have all been shown to promote Nrf2 activity (Ishii et al., 2000; Kang et al., 2002; Cullinan and Diehl, 2004; Itoh et al., 2004; Zhang et al., 2004; Calkins et al., 2005).
Our preliminary in vitro studies suggest that increased extracellular glutamate may be a novel mediator of Nrf2 activation (A. Y. Shih and T. H. Murphy, unpublished observation). This finding is highly relevant to ischemia because glutamate released during anoxic depolarization is thought to cause neuronal excitotoxicity (Dirnagl et al., 1999; Lee et al., 1999). Peri-infarct depolarizations could further propagate anoxic glutamate release into penumbral zones (Hossmann, 1996) and activate Nrf2 within surviving astrocytes. Clearly, additional studies are required to determine the physiological relevance of glutamate-mediated Nrf2 activation because in vitro glial culture systems are not necessarily comparable with the temporal and spatial aspects of glutamate release during in vivo stroke. It is conceivable that activation of a “glutamate-Nrf2 signal” within astrocytes could exist to limit oxidative stress created during excitotoxicity (Reynolds and Hastings, 1995). Nrf2-targeted genes may then strengthen neuronglia metabolic coupling, enabling reestablishment of neuronal function after ischemia (Shih et al., 2003; Kraft et al., 2004).
Conclusion
Nrf2 inducers represent a new generation of drugs that can single-handedly activate multiple cellular defense effectors. As an improvement on previous approaches, future therapies could target these “molecular switches” that control the coordinated expression of protective gene families.
Footnotes
This work was supported by a Canadian Institutes of Health Research and by a Michael Smith Foundation for Health Research (MSFHR) Studentship Award to A.Y.S. and by operating grants from the Heart and Stroke Foundation of British Columbia and Yukon and the Canadian Stroke Network (T.H.M.). T.H.M. is an MSFHR senior scholar. We are grateful to Sophie Imbeault and Dr. George S. Robertson for helpful comments, Heidi Erb and Lei Jiang for excellent technical assistance, Dr. Yuet Wai Kan for Nrf2-/- mouse production, and Dr. Jeff Johnson for distributing the mice. We also thank the staff at the UBC Animal Research Unit and Steve Callaghan at the Adenovirus Core Facility of the University of Ottawa (Ottawa, Ontario, Canada) for adenovirus production.
Correspondence should be addressed to Dr. Timothy H. Murphy, 4N1-2255 Wesbrook Mall, University of British Columbia, Vancouver, British Columbia, Canada V6T 1Z3. E-mail: thmurphy@interchange.ubc.ca.
Copyright © 2005 Society for Neuroscience 0270-6474/05/2510321-15$15.00/0
References
- Ahlgren-Beckendorf JA, Reising AM, Schander MA, Herdler JW, Johnson JA (1999) Coordinate regulation of NAD(P)H:quinone oxidoreductase and glutathione-S-transferases in primary cultures of rat neurons and glia: role of the antioxidant/electrophile responsive element. Glia 25: 131-142. [PubMed] [Google Scholar]
- Alam J, Stewart D, Touchard C, Boinapally S, Choi AM, Cook JL (1999) Nrf2, a cap'n'collar transcription factor, regulates induction of the heme oxygenase-1 gene. J Biol Chem 274: 26071-26078. [DOI] [PubMed] [Google Scholar]
- Beal MF, Brouillet E, Jenkins BG, Ferrante RJ, Kowall NW, Miller JM, Storey E, Srivastava R, Rosen BR, Hyman BT (1993) Neurochemical and histologic characterization of striatal excitotoxic lesions produced by the mitochondrial toxin 3-nitropropionic acid. J Neurosci 13: 4181-4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bederson JB, Pitts LH, Germano SM, Nishimura MC, Davis RL, Bartkowski HM (1986) Evaluation of 2,3,5-triphenyltetrazolium chloride as a stain for detection and quantification of experimental cerebral infarction in rats. Stroke 17: 1304-1308. [DOI] [PubMed] [Google Scholar]
- Benson AM, Hunkeler MJ, Talalay P (1980) Increase of NAD(P)H:quinone reductase by dietary antioxidants: possible role in protection against carcinogenesis and toxicity. Proc Natl Acad Sci USA 77: 5216-5220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biernaskie J, Corbett D, Peeling J, Wells J, Lei H (2001) A serial MR study of cerebral blood flow changes and lesion development following endothelin-1-induced ischemia in rats. Magn Reson Med 46: 827-830. [DOI] [PubMed] [Google Scholar]
- Bogaert L, Scheller D, Moonen J, Sarre S, Smolders I, Ebinger G, Michotte Y (2000) Neurochemical changes and laser Doppler flowmetry in the endothelin-1 rat model for focal cerebral ischemia. Brain Res 887: 266-275. [DOI] [PubMed] [Google Scholar]
- Bogaert L, O'Neill MJ, Moonen J, Sarre S, Smolders I, Ebinger G, Michotte Y (2001) The effects of LY393613, nimodipine and verapamil, in focal cerebral ischaemia. Eur J Pharmacol 411: 71-83. [DOI] [PubMed] [Google Scholar]
- Brouillet E, Guyot MC, Mittoux V, Altairac S, Conde F, Palfi S, Hantraye P (1998) Partial inhibition of brain succinate dehydrogenase by 3-nitropropionic acid is sufficient to initiate striatal degeneration in rat. J Neurochem 70: 794-805. [DOI] [PubMed] [Google Scholar]
- Calkins MJ, Jakel RJ, Johnson DA, Chan K, Kan YW, Johnson JA (2005) Protection from mitochondrial complex II inhibition in vitro and in vivo by Nrf2-mediated transcription. Proc Natl Acad Sci USA 102: 244-249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callaway JK, Castillo-Melendez M, Giardina SF, Krstew EK, Beart PM, Jarrott B (2004) Sodium channel blocking activity of AM-36 and sipatrigine (BW619C89): in vitro and in vivo evidence. Neuropharmacology 47: 146-155. [DOI] [PubMed] [Google Scholar]
- Campagne MV, Thibodeaux H, van Bruggen N, Cairns B, Lowe DG (2000) Increased binding activity at an antioxidant-responsive element in the metallothionein-1 promoter and rapid induction of metallothionein-1 and -2 in response to cerebral ischemia and reperfusion. J Neurosci 20: 5200-5207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cechetto DF, Wilson JX, Smith KE, Wolski D, Silver MD, Hachinski VC (1989) Autonomic and myocardial changes in middle cerebral artery occlusion: stroke models in the rat. Brain Res 502: 296-305. [DOI] [PubMed] [Google Scholar]
- Chan JY, Kwong M (2000) Impaired expression of glutathione synthetic enzyme genes in mice with targeted deletion of the Nrf2 basic-leucine zipper protein. Biochim Biophys Acta 1517: 19-26. [DOI] [PubMed] [Google Scholar]
- Chan K, Lu R, Chang JC, Kan YW (1996) NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc Natl Acad Sci USA 93: 13943-13948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan PH (2001) Reactive oxygen radicals in signaling and damage in the ischemic brain. J Cereb Blood Flow Metab 21: 2-14. [DOI] [PubMed] [Google Scholar]
- Chanas SA, Jiang Q, McMahon M, McWalter GK, McLellan LI, Elcombe CR, Henderson CJ, Wolf CR, Moffat GJ, Itoh K, Yamamoto M, Hayes JD (2002) Loss of the Nrf2 transcription factor causes a marked reduction in constitutive and inducible expression of the glutathione S-transferase Gsta1, Gsta2, Gstm1, Gstm2, Gstm3 and Gstm4 genes in the livers of male and female mice. Biochem J 365: 405-416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clozel M, Gray GA, Breu V, Loffler BM, Osterwalder R (1992) The endothelin ETB receptor mediates both vasodilation and vasoconstriction in vivo. Biochem Biophys Res Commun 186: 867-873. [DOI] [PubMed] [Google Scholar]
- Coyle JT, Puttfarcken P (1993) Oxidative stress, glutamate, and neurodegenerative disorders. Science 262: 689-695. [DOI] [PubMed] [Google Scholar]
- Cullinan SB, Diehl JA (2004) PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J Biol Chem 279: 20108-20117. [DOI] [PubMed] [Google Scholar]
- Dawson DA, Masayasu H, Graham DI, Macrae IM (1995) The neuroprotective efficacy of ebselen (a glutathione peroxidase mimic) on brain damage induced by transient focal cerebral ischaemia in the rat. Neurosci Lett 185: 65-69. [DOI] [PubMed] [Google Scholar]
- De Keyser J, Sulter G, Luiten PG (1999) Clinical trials with neuroprotective drugs in acute ischaemic stroke are we ng the right thing? Trends Neurosci 22: 535-540. [DOI] [PubMed] [Google Scholar]
- De Long MJ, Santamaria AB, Talalay P (1987) Role of cytochrome P1-450 in the induction of NAD(P)H:quinone reductase in a murine hepatoma cell line and its mutants. Carcinogenesis 8: 1549-1553. [DOI] [PubMed] [Google Scholar]
- Dinkova-Kostova AT, Holtzclaw WD, Cole RN, Itoh K, Wakabayashi N, Katoh Y, Yamamoto M, Talalay P (2002) Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc Natl Acad Sci USA 99: 11908-11913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirnagl U, Iadecola C, Moskowitz MA (1999) Pathobiology of ischaemic stroke: an integrated view. Trends Neurosci 22: 391-397. [DOI] [PubMed] [Google Scholar]
- Duffy S, So A, Murphy TH (1998) Activation of endogenous antioxidant defenses in neuronal cells prevents free radical-mediated damage. J Neurochem 71: 69-77. [DOI] [PubMed] [Google Scholar]
- Eftekharpour E, Holmgren A, Juurlink BH (2000) Thioredoxin reductase and glutathione synthesis is upregulated by t-butylhydroquinone in cortical astrocytes but not in cortical neurons. Glia 31: 241-248. [DOI] [PubMed] [Google Scholar]
- Everse J, Berger RL, Kaplan NO (1970) Physiological concentrations of lactate dehydrogenases and substrate inhibition. Science 168: 1236-1238. [DOI] [PubMed] [Google Scholar]
- Fahey JW, Talalay P (1999) Antioxidant functions of sulforaphane: a potent inducer of phase II detoxication enzymes. Food Chem Toxicol 37: 973-979. [DOI] [PubMed] [Google Scholar]
- Fahey JW, Zhang Y, Talalay P (1997) Broccoli sprouts: an exceptionally rich source of inducers of enzymes that protect against chemical carcinogens. Proc Natl Acad Sci USA 94: 10367-10372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahey JW, Zalcmann AT, Talalay P (2001) The chemical diversity and distribution of glucosinolates and isothiocyanates among plants. Phytochemistry 56: 5-51. [DOI] [PubMed] [Google Scholar]
- Fahey JW, Haristoy X, Dolan PM, Kensler TW, Scholtus I, Stephenson KK, Talalay P, Lozniewski A (2002) Sulforaphane inhibits extracellular, intracellular, and antibiotic-resistant strains of Helicobacter pylori and prevents benzo[a]pyrene-induced stomach tumors. Proc Natl Acad Sci USA 99: 7610-7615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Food and Agriculture Organization of the United Nations/World Health Organization (1975) Toxicological evaluation of some food additives, including food colours, thickening agents and others. Joint FAO/WHO Expert Committee on Food Additives. Geneva, 14-23 April 1975. FAO Nutr Meet Rep Ser 55A: 1-204. [PubMed] [Google Scholar]
- Food and Agriculture Organization of the United Nations/World Health Organization (1999) Evaluation of certain food additives and contaminants (forty-ninth report of the Joint FAO/WHO Expert Committee on Food Additives). World Health Organ Tech Rep Ser 884: i-viii, 1-96. [PubMed] [Google Scholar]
- Fisher M, Garcia JH (1996) Evolving stroke and the ischemic penumbra. Neurology 47: 884-888. [DOI] [PubMed] [Google Scholar]
- Fujii M, Hara H, Meng W, Vonsattel JP, Huang Z, Moskowitz MA (1997) Strain-related differences in susceptibility to transient forebrain ischemia in SV-129 and C57black/6 mice. Stroke 28: 1805-1811. [DOI] [PubMed] [Google Scholar]
- Fuxe K, Kurosawa N, Cintra A, Hallstrom A, Goiny M, Rosen L, Agnati LF, Ungerstedt U (1992) Involvement of local ischemia in endothelin-1 induced lesions of the neostriatum of the anaesthetized rat. Exp Brain Res 88: 131-139. [DOI] [PubMed] [Google Scholar]
- Gilmour G, Iversen SD, O'Neill MF, Bannerman DM (2004) The effects of intracortical endothelin-1 injections on skilled forelimb use: implications for modelling recovery of function after stroke. Behav Brain Res 150: 171-183. [DOI] [PubMed] [Google Scholar]
- Gramsbergen JB, Skjoth-Rasmussen J, Rasmussen C, Lambertsen KL (2004) On-line monitoring of striatum glucose and lactate in the endothelin-1 rat model of transient focal cerebral ischemia using microdialysis and flow-injection analysis with biosensors. J Neurosci Methods 140: 93-101. [DOI] [PubMed] [Google Scholar]
- Hardy S, Kitamura M, Harris-Stansil T, Dai Y, Phipps ML (1997) Construction of adenovirus vectors through Cre-lox recombination. J Virol 71: 1842-1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hata R, Maeda K, Hermann D, Mies G, Hossmann KA (2000a) Dynamics of regional brain metabolism and gene expression after middle cerebral artery occlusion in mice. J Cereb Blood Flow Metab 20: 306-315. [DOI] [PubMed] [Google Scholar]
- Hata R, Maeda K, Hermann D, Mies G, Hossmann KA (2000b) Evolution of brain infarction after transient focal cerebral ischemia in mice. J Cereb Blood Flow Metab 20: 937-946. [DOI] [PubMed] [Google Scholar]
- Hayes JD, Chanas SA, Henderson CJ, McMahon M, Sun C, Moffat GJ, Wolf CR, Yamamoto M (2000) The Nrf2 transcription factor contributes both to the basal expression of glutathione S-transferases in mouse liver and to their induction by the chemopreventive synthetic antioxidants, butylated hydroxyanisole and ethoxyquin. Biochem Soc Trans 28: 33-41. [DOI] [PubMed] [Google Scholar]
- Henthorn P, Zervos P, Raducha M, Harris H, Kadesch T (1988) Expression of a human placental alkaline phosphatase gene in transfected cells: use as a reporter for studies of gene expression. Proc Natl Acad Sci USA 85: 6342-6346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hossmann KA (1996) Periinfarct depolarizations. Cerebrovasc Brain Metab Rev 8: 195-208. [PubMed] [Google Scholar]
- Hughes PM, Anthony DC, Ruddin M, Botham MS, Rankine EL, Sablone M, Baumann D, Mir AK, Perry VH (2003) Focal lesions in the rat central nervous system induced by endothelin-1. J Neuropathol Exp Neurol 62: 1276-1286. [DOI] [PubMed] [Google Scholar]
- Ishii T, Itoh K, Takahashi S, Sato H, Yanagawa T, Katoh Y, Bannai S, Yamamoto M (2000) Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J Biol Chem 275: 16023-16029. [DOI] [PubMed] [Google Scholar]
- Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, Hatayama I, Yamamoto M, Nabeshima Y (1997) An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun 236: 313-322. [DOI] [PubMed] [Google Scholar]
- Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, Yamamoto M (1999) Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev 13: 76-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh K, Mochizuki M, Ishii Y, Ishii T, Shibata T, Kawamoto Y, Kelly V, Sekizawa K, Uchida K, Yamamoto M (2004) Transcription factor Nrf2 regulates inflammation by mediating the effect of 15-deoxy-delta(12,14)prostaglandin j(2). Mol Cell Biol 24: 36-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang KW, Choi SH, Kim SG (2002) Peroxynitrite activates NF-E2-related factor 2/antioxidant response element through the pathway of phosphatidylinositol 3-kinase: the role of nitric oxide synthase in rat glutathione S-transferase A2 induction. Nitric Oxide 7: 244-253. [DOI] [PubMed] [Google Scholar]
- Kawamura S, Yasui N, Shirasawa M, Fukasawa H (1991) Rat middle cerebral artery occlusion using an intraluminal thread technique. Acta Neurochir (Wien) 109: 126-132. [DOI] [PubMed] [Google Scholar]
- Kelly VP, Ellis EM, Manson MM, Chanas SA, Moffat GJ, McLeod R, Judah DJ, Neal GE, Hayes JD (2000) Chemoprevention of aflatoxin B1 hepatocarcinogenesis by coumarin, a natural benzopyrone that is a potent inducer of aflatoxin B1-aldehyde reductase, the glutathione S-transferase A5 and P1 subunits, and NAD(P)H:quinone oxidoreductase in rat liver. Cancer Res 60: 957-969. [PubMed] [Google Scholar]
- Kim GW, Copin JC, Kawase M, Chen SF, Sato S, Gobbel GT, Chan PH (2000) Excitotoxicity is required for induction of oxidative stress and apoptosis in mouse striatum by the mitochondrial toxin, 3-nitropropionic acid. J Cereb Blood Flow Metab 20: 119-129. [DOI] [PubMed] [Google Scholar]
- Kitagawa K, Matsumoto M, Yang G, Mabuchi T, Yagita Y, Hori M, Yanagihara T (1998) Cerebral ischemia after bilateral carotid artery occlusion and intraluminal suture occlusion in mice: evaluation of the patency of the posterior communicating artery. J Cereb Blood Flow Metab 18: 570-579. [DOI] [PubMed] [Google Scholar]
- Kleeberg J, Petzold GC, Major S, Dirnagl U, Dreier JP (2004) ET-1 induces cortical spreading depression via activation of the ETA receptor/phospholipase C pathway in vivo. Am J Physiol Heart Circ Physiol 286: H1339-H1346. [DOI] [PubMed] [Google Scholar]
- Kraft AD, Johnson DA, Johnson JA (2004) Nuclear factor E2-related factor 2-dependent antioxidant response element activation by tert-butylhydroquinone and sulforaphane occurring preferentially in astrocytes conditions neurons against oxidative insult. J Neurosci 24: 1101-1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak MK, Itoh K, Yamamoto M, Sutter TR, Kensler TW (2001a) Role of transcription factor Nrf2 in the induction of hepatic phase 2 and antioxidative enzymes in vivo by the cancer chemoprotective agent, 3H-1, 2-dimethiole-3-thione. Mol Med 7: 135-145. [PMC free article] [PubMed] [Google Scholar]
- Kwak MK, Egner PA, Dolan PM, Ramos-Gomez M, Groopman JD, Itoh K, Yamamoto M, Kensler TW (2001b) Role of phase 2 enzyme induction in chemoprotection by dithiolethiones. Mutat Res 480-481: 305-315. [DOI] [PubMed] [Google Scholar]
- Laxton AW, Sun MC, Shen H, Murphy TH, Honey CR (2001) The antioxidant enzyme quinone reductase is up-regulated in vivo following cerebral ischemia. NeuroReport 12: 1045-1048. [DOI] [PubMed] [Google Scholar]
- Lee JM, Zipfel GJ, Choi DW (1999) The changing landscape of ischaemic brain injury mechanisms. Nature 399: A7-A14. [DOI] [PubMed] [Google Scholar]
- Lee JM, Calkins MJ, Chan K, Kan YW, Johnson JA (2003a) Identification of the NF-E2-related factor-2-dependent genes conferring protection against oxidative stress in primary cortical astrocytes using oligonucleotide microarray analysis. J Biol Chem 278: 12029-12038. [DOI] [PubMed] [Google Scholar]
- Lee JM, Shih AY, Murphy TH, Johnson JA (2003b) NF-E2-related factor-2 mediates neuroprotection against mitochondrial complex I inhibitors and increased concentrations of intracellular calcium in primary cortical neurons. J Biol Chem 278: 37948-37956. [DOI] [PubMed] [Google Scholar]
- Lewen A, Matz P, Chan PH (2000) Free radical pathways in CNS injury. J Neurotrauma 17: 871-890. [DOI] [PubMed] [Google Scholar]
- Li J, Lee JM, Johnson JA (2002) Microarray analysis reveals an antioxidant responsive element-driven gene set involved in conferring protection from an oxidative stress-induced apoptosis in IMR-32 cells. J Biol Chem 277: 388-394. [DOI] [PubMed] [Google Scholar]
- Lipton P (1999) Ischemic cell death in brain neurons. Physiol Rev 79: 1431-1568. [DOI] [PubMed] [Google Scholar]
- Liverman CS, Cui L, Yong C, Choudhuri R, Klein RM, Welch KM, Berman NE (2004) Response of the brain to oligemia: gene expression, c-Fos, and Nrf2 localization. Brain Res Mol Brain Res 126: 57-66. [DOI] [PubMed] [Google Scholar]
- Lo EH, Dalkara T, Moskowitz MA (2003) Mechanisms, challenges and opportunities in stroke. Nat Rev Neurosci 4: 399-415. [DOI] [PubMed] [Google Scholar]
- Longa EZ, Weinstein PR, Carlson S, Cummins R (1989) Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 20: 84-91. [DOI] [PubMed] [Google Scholar]
- Love S (1999) Oxidative stress in brain ischemia. Brain Pathol 9: 119-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda K, Hata R, Hossmann KA (1998) Differences in the cerebrovascular anatomy of C57black/6 and SV129 mice. NeuroReport 9: 1317-1319. [DOI] [PubMed] [Google Scholar]
- Majid A, He YY, Gidday JM, Kaplan SS, Gonzales ER, Park TS, Fenstermacher JD, Wei L, Choi DW, Hsu CY (2000) Differences in vulnerability to permanent focal cerebral ischemia among 3 common mouse strains. Stroke 31: 2707-2714. [DOI] [PubMed] [Google Scholar]
- Mao Y, Yang GY, Zhou LF, Stern JD, Betz AL (1999) Focal cerebral ischemia in the mouse: description of a model and effects of permanent and temporary occlusion. Brain Res Mol Brain Res 63: 366-370. [DOI] [PubMed] [Google Scholar]
- McMahon M, Itoh K, Yamamoto M, Chanas SA, Henderson CJ, McLellan LI, Wolf CR, Cavin C, Hayes JD (2001) The cap'n'collar basic leucine zipper transcription factor Nrf2 (NF-E2 p45-related factor 2) controls both constitutive and inducible expression of intestinal detoxification and glutathione biosynthetic enzymes. Cancer Res 61: 3299-3307. [PubMed] [Google Scholar]
- McWalter GK, Higgins LG, McLellan LI, Henderson CJ, Song L, Thornalley PJ, Itoh K, Yamamoto M, Hayes JD (2004) Transcription factor Nrf2 is essential for induction of NAD(P)H:quinone oxidoreductase 1, glutathione S-transferases, and glutamate cysteine ligase by broccoli seeds and isothiocyanates. J Nutr 134: 3499S-3506S. [DOI] [PubMed] [Google Scholar]
- Memezawa H, Smith ML, Siesjo BK (1992a) Penumbral tissues salvaged by reperfusion following middle cerebral artery occlusion in rats. Stroke 23: 552-559. [DOI] [PubMed] [Google Scholar]
- Memezawa H, Minamisawa H, Smith ML, Siesjo BK (1992b) Ischemic penumbra in a model of reversible middle cerebral artery occlusion in the rat. Exp Brain Res 89: 67-78. [DOI] [PubMed] [Google Scholar]
- Moreland S, McMullen DM, Delaney CL, Lee VG, Hunt JT (1992) Venous smooth muscle contains vasoconstrictor ETB-like receptors. Biochem Biophys Res Commun 184: 100-106. [DOI] [PubMed] [Google Scholar]
- Murphy TH, De Long MJ, Coyle JT (1991) Enhanced NAD(P)H:quinone reductase activity prevents glutamate toxicity produced by oxidative stress. J Neurochem 56: 990-995. [DOI] [PubMed] [Google Scholar]
- Murphy TH, So AP, Vincent SR (1998) Histochemical detection of quinone reductase activity in situ using LY 83583 reduction and oxidation. J Neurochem 70: 2156-2164. [DOI] [PubMed] [Google Scholar]
- Murphy TH, Yu J, Ng R, Johnson DA, Shen H, Honey CR, Johnson JA (2001) Preferential expression of antioxidant response element mediated gene expression in astrocytes. J Neurochem 76: 1670-1678. [DOI] [PubMed] [Google Scholar]
- National Toxicology Program (1997) NTP toxicology and carcinogenesis studies of t-butylhydroquinone (CAS no. 1948-33-0) in F344/N rats and B6C3F(1) mice (feed studies). Natl Toxicol Program Tech Rep Ser 459: 1-326. [PubMed] [Google Scholar]
- Nedergaard M, Gjedde A, Diemer NH (1986) Focal ischemia of the rat brain: autoradiographic determination of cerebral glucose utilization, glucose content, and blood flow. J Cereb Blood Flow Metab 6: 414-424. [DOI] [PubMed] [Google Scholar]
- Ostrowski RP, Piotrowski P, Pankowska T, Smialek M (1998) Evaluation of morphological changes after treatment with coenzyme Q10 (CoQ10) in endothelin-1 induced experimental ischemia in the rat. Folia Neuropathol 36: 185-188. [PubMed] [Google Scholar]
- Ramos-Gomez M, Kwak MK, Dolan PM, Itoh K, Yamamoto M, Talalay P, Kensler TW (2001) Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2 transcription factor-deficient mice. Proc Natl Acad Sci USA 98: 3410-3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reglodi D, Tamas A, Lengvari I (2003) Examination of sensorimotor performance following middle cerebral artery occlusion in rats. Brain Res Bull 59: 459-466. [DOI] [PubMed] [Google Scholar]
- Reynolds IJ, Hastings TG (1995) Glutamate induces the production of reactive oxygen species in cultured forebrain neurons following NMDA receptor activation. J Neurosci 15: 3318-3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rushmore TH, Kong AN (2002) Pharmacogenomics, regulation and signaling pathways of phase I and II drug metabolizing enzymes. Curr Drug Metab 3: 481-490. [DOI] [PubMed] [Google Scholar]
- Sato S, Gobbel GT, Honkaniemi J, Li Y, Kondo T, Murakami K, Sato M, Copin JC, Chan PH (1997) Apoptosis in the striatum of rats following intraperitoneal injection of 3-nitropropionic acid. Brain Res 745: 343-347. [DOI] [PubMed] [Google Scholar]
- Schmued LC, Albertson C, Slikker Jr W (1997) Fluoro-Jade: a novel fluorochrome for the sensitive and reliable histochemical localization of neuronal degeneration. Brain Res 751: 37-46. [DOI] [PubMed] [Google Scholar]
- Sharkey J, Ritchie IM, Kelly PA (1993) Perivascular microapplication of endothelin-1: a new model of focal cerebral ischaemia in the rat. J Cereb Blood Flow Metab 13: 865-871. [DOI] [PubMed] [Google Scholar]
- Sharkey J, Butcher SP, Kelly JS (1994) Endothelin-1 induced middle cerebral artery occlusion: pathological consequences and neuroprotective effects of MK801. J Auton Nerv Syst [Suppl] 49: S177-S185. [DOI] [PubMed] [Google Scholar]
- Shih AY, Johnson DA, Wong G, Kraft AD, Jiang L, Erb H, Johnson JA, Murphy TH (2003) Coordinate regulation of glutathione biosynthesis and release by Nrf2-expressing glia potently protects neurons from oxidative stress. J Neurosci 23: 3394-3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih AY, Imbeault S, Barakauskas V, Erb H, Jiang L, Li P, Murphy TH (2005) Induction of the Nrf2-driven antioxidant response confers neuroprotection during mitochondrial stress in vivo. J Biol Chem 280: 22925-22936. [DOI] [PubMed] [Google Scholar]
- Stenman E, Malmsjo M, Uddman E, Gido G, Wieloch T, Edvinsson L (2002) Cerebral ischemia upregulates vascular endothelin ET(B) receptors in rat. Stroke 33: 2311-2316. [DOI] [PubMed] [Google Scholar]
- Talalay P (1989) Mechanisms of induction of enzymes that protect against chemical carcinogenesis. Adv Enzyme Regul 28: 237-250. [DOI] [PubMed] [Google Scholar]
- Talalay P (2000) Chemoprotection against cancer by induction of phase 2 enzymes. Biofactors 12: 5-11. [DOI] [PubMed] [Google Scholar]
- Talalay P, Fahey JW, Holtzclaw WD, Prestera T, Zhang Y (1995) Chemoprotection against cancer by phase 2 enzyme induction. Toxicol Lett 82-83: 173-179. [DOI] [PubMed] [Google Scholar]
- Thimmulappa RK, Mai KH, Srisuma S, Kensler TW, Yamamoto M, Biswal S (2002) Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res 62: 5196-5203. [PubMed] [Google Scholar]
- Tietze F (1969) Enzymic method for quantitative determination of nanogram amounts of total and oxidized glutathione: applications to mammalian blood and other tissues. Anal Biochem 27: 502-522. [DOI] [PubMed] [Google Scholar]
- Touzani O, Galbraith S, Siegl P, McCulloch J (1997) Endothelin-B receptors in cerebral resistance arterioles and their functional significance after focal cerebral ischemia in cats. J Cereb Blood Flow Metab 17: 1157-1165. [DOI] [PubMed] [Google Scholar]
- Tyson GW, Teasdale GM, Graham DI, McCulloch J (1984) Focal cerebral ischemia in the rat: topography of hemodynamic and histopathological changes. Ann Neurol 15: 559-567. [DOI] [PubMed] [Google Scholar]
- Van Hemelrijck A, Vermijlen D, Hachimi-Idrissi S, Sarre S, Ebinger G, Michotte Y (2003) Effect of resuscitative mild hypothermia on glutamate and dopamine release, apoptosis and ischaemic brain damage in the endothelin-1 rat model for focal cerebral ischaemia. J Neurochem 87: 66-75. [DOI] [PubMed] [Google Scholar]
- van Lookeren Campagne M, Thibodeaux H, van Bruggen N, Cairns B, Gerlai R, Palmer JT, Williams SP, Lowe DG (1999) Evidence for a protective role of metallothionein-1 in focal cerebral ischemia. Proc Natl Acad Sci USA 96: 12870-12875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Ommen B, Koster A, Verhagen H, van Bladeren PJ (1992) The glutathione conjugates of tert-butylhydroquinone as potent redox cycling agents and possible reactive agents underlying the toxicity of butylated hydroxyanisole. Biochem Biophys Res Commun 189: 309-314. [DOI] [PubMed] [Google Scholar]
- Venugopal R, Jaiswal AK (1996) Nrf1 and Nrf2 positively and c-Fos and Fra1 negatively regulate the human antioxidant response element-mediated expression of NAD(P)H:quinone oxidoreductase1 gene. Proc Natl Acad Sci USA 93: 14960-14965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Ashraf MH, Facci M, Wang R, Paterson PG, Ferrie A, Juurlink BH (2004) Dietary approach to attenuate oxidative stress, hypertension, and inflammation in the cardiovascular system. Proc Natl Acad Sci USA 101: 7094-7099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagisawa M, Kurihara H, Kimura S, Tomobe Y, Kobayashi M, Mitsui Y, Yazaki Y, Goto K, Masaki T (1988) A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature 332: 411-415. [DOI] [PubMed] [Google Scholar]
- Yu ZF, Mattson MP (1999) Dietary restriction and 2-deoxyglucose administration reduce focal ischemic brain damage and improve behavioral outcome: evidence for a preconditioning mechanism. J Neurosci Res 57: 830-839. [PubMed] [Google Scholar]
- Zhang S, Boyd J, Delaney K, Murphy TH (2005) Rapid reversible changes in dendritic spine structure in vivo gated by the degree of ischemia. J Neurosci 25: 5333-5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Lu L, Dixon C, Wilmer W, Song H, Chen X, Rovin BH (2004) Stress protein activation by the cyclopentenone prostaglandin 15-deoxy-delta12,14-prostaglandin J2 in human mesangial cells. Kidney Int 65: 798-810. [DOI] [PubMed] [Google Scholar]