

Abstract
Huntington's disease (HD) is an autosomal dominant neurodegenerative condition characterized by movement disorders, psychiatric disturbance, and cognitive decline. There are no treatments to halt or reverse the disease. Mammalian neurogenesis persists into adulthood in the subventricular zone (SVZ) and dentate gyrus (DG) of the hippocampus. In 2001, our laboratory published the hypothesis that neurogenesis is impaired in neurodegenerative diseases and that this may contribute to disease progression. Since then, it has been shown that neurogenesis is reduced in the DG of transgenic HD mice but increased in the SVZ of HD patients. We sought to characterize neurogenesis further. We found that, in the DG of the transgenic R6/2 mouse model of HD, newborn cell proliferation and morphology, but not differentiation or survival, was compromised. In R6/2 mice, neurogenesis failed to upregulate in the DG in response to seizures. Basal SVZ neurogenesis was similar between R6/2 mice and their wild-type littermates. There was no difference in the in vitro growth of adult neural precursor cells (NPCs) between genotypes. These results suggest that abnormal neurogenesis in the R6/2 mouse is not attributable to an intrinsic impairment of the NPC itself but is attributable to the environment in which the cell is located.
Keywords: Huntington's disease, R6/2, neurogenesis, seizures, quinolinic acid, doublecortin
Introduction
Huntington's disease (HD) is an inherited neurodegenerative condition characterized by movement disorders, psychiatric disturbance, and cognitive decline. The striatum is particularly vulnerable to degeneration, although other brain areas also dysfunction and degenerate early in the disease (Vonsattel et al., 1985; Lawrence et al., 1998; Rosas et al., 2003).
Mammalian neurogenesis persists into adulthood in the subventricular zone (SVZ) and the subgranular zone (SGZ) of the dentate gyrus (DG) (Altman and Das, 1965; Kaplan and Hinds, 1977) in many species, including humans (Eriksson et al., 1998; Roy et al., 2000; Palmer et al., 2001) (for review, see Gage, 2000). Adult neurogenesis is relevant because newborn neurons integrate into existing circuitry, are functional, and contribute to olfaction (Gheusi et al., 2000; Carleton et al., 2003) and hippocampal-dependent learning and memory (Shors et al., 2001, 2002; van Praag et al., 2002).
Neurogenesis can be upregulated in response to a variety of stimuli such as ischemia, seizures, and acute lesions, even in non-neurogenic areas (Kempermann et al., 1997; Fallon et al., 2000; Magavi et al., 2000; Arvidsson et al., 2002; Nakatomi et al., 2002; Parent et al., 2002). We wanted to know whether neurogenesis could be upregulated in response to a chronic lesion (as might be found in the environment of a brain with a neurodegenerative disease such as HD) or, conversely, whether this environment might be nonpermissive for neurogenesis (Armstrong and Barker, 2001). Neurogenesis is reduced in the DG of transgenic HD mice (Gil et al., 2004, 2005; Lazic et al., 2004) but increased in the SVZ of HD patients (Curtis et al., 2003, 2005). In light of this, we sought to characterize neurogenesis further using the R6/2 mouse model of HD (Mangiarini et al., 1996). In particular, we wanted to address whether the abnormalities in HD neurogenesis were attributable to a deficit of the neural precursor cells (NPCs) or a nonpermissive NPC environment. Because neurogenesis is a complex process involving proliferation, migration, differentiation, and survival, we measured all of these parameters in both the DG and SVZ/striatum. It is important to know whether neurogenesis can be upregulated in response to cell dysfunction and death in R6/2 mice, because upregulation of neurogenesis would be required in the disease state and in therapies aimed to enhance neurogenesis. To examine stimulated (upregulated) neurogenesis, we induced seizures by injecting kainic acid (KA) systemically into mice and measured neurogenesis in the DG. We chose to study seizures to stimulate neurogenesis, because this is a well characterized paradigm known to upregulate neurogenesis through a mechanism dependent on altered environmental signals. We also produced a neurotoxic lesion by injecting quinolinic acid (QA) directly into the striatum and measured neurogenesis in the SVZ/striatum. Basal neurogenesis was measured using vehicle-treated animals. We compared proliferation, longevity, differentiation fate, and survival of NPCs in vitro to establish whether changes in neurogenesis were attributable to the intrinsic properties of the NPC or the microenvironment in which it is located.
Materials and Methods
Animals. A colony of R6/2 transgenic mice was established in the Cambridge Centre for Brain Repair (University of Cambridge, Cambridge, UK). The line was maintained by backcrossing to CBA × C57BL/6 F1 animals. Wild-type (WT) littermates were used as control animals. The mice were housed in mixed genotype single-sex cages in a temperature- and humidity-controlled room on a 12 h light/dark cycle with availability to food and water ad libitum. Genotyping was confirmed by PCR, using a method described previously (Hickey and Morton, 2000). Experiments were conducted according to the United Kingdom Animals (Scientific Procedures) Act 1986, under appropriate Home Office personal and project licenses.
KA treatments. A total of 67 mice were used: 34 WT and 33 R6/2; 17 male and 50 female; 35 8-week-old and 32 13-week-old mice. Three mice died during these experiments. KA (20 mg/kg dissolved in sterile water, pH 7.4; Sigma, Gillingham, UK) or vehicle was administered intraperitoneally on the morning of the experiment start date. Thirty-three mice received KA. Seizures were rated according to a modified Racine scale, which assesses the severity of seizure (Racine et al., 1972; Morton and Leavens, 2000). Briefly, seizures were rated as follows; grade 1, mouth movements; grade 2, head nodding; grade 3, forelimb clonus; grade 4, rearing with clonus; grade 5, tonic-clonic seizures.
Bromodeoxyuridine labeling and tissue processing. Because the peak of cell proliferation occurs ∼7 d after seizures (Mohapel et al., 2004), mice were given an injection of the thymidine analog bromodeoxyuridine (BrdU) (50 mg/kg) on days 7 and 8 after KA treatment, to label dividing cells. BrdU injections were administered intraperitoneally 12 h apart, for 2 d (i.e., a total of four injections). Mice were killed on days 9, 16, and 32 after KA treatment. Mice were anesthetized using intraperitoneal sodium pentobarbital (Merial, Oxford, UK) and then perfused through the heart with a prewash solution (18 g/liter disodium hydrogen orthophosphate and 9 g/liter sodium chloride, in distilled water, pH 7.3) and then 4% paraformaldehyde (in distilled water, pH 7.4). Brains were postfixed with 4% paraformaldehyde for 24 h and then cryoprotected in 30% sucrose/0.1 m PBS. Brains were sectioned coronally at 40 μm and stored in PBS/azide at 4°C.
Immunohistochemistry. One in every 12 sections was processed immunohistochemically either free-floating or, for fluorojade-B and acetylcholinesterase, mounted on Superfrost-plus glass slides (VWR Scientific, Poole, UK). For antigens labeled with biotinylated antibodies, sections were quenched (10% methanol, 10% hydrogen peroxide, and distilled water) for 5 min, washed, and incubated in a blocking buffer [3% serum in 0.1 m Tris-buffered saline (TBS) with 0.2% Triton X-100] for 60 min at room temperature. Sections were incubated in primary antibody with 1% serum in TBS with 0.2% Triton X-100 overnight at 4°C. Biotinylated secondary antibodies (1:200 in 1% serum in TBS with 0.2% Triton X-100) were applied for 2 h, washed, and visualized using an avidin–biotin–peroxidase system (Vectastain ABC kit; Vector Laboratories, Peterborough, UK) and diaminobenzidine (Sigma). For BrdU immunolabeling, sections were incubated in 2 m HCl at 37°C for 30 min and then washed for 30 min in 0.1 m borate buffer, pH 8.5, before block. For sections in which primary antibodies were visualized using fluorescence-conjugated secondary antibodies, sections were not quenched, and secondary antibody (1:1000, Alexa-488 or Alexa-568; Invitrogen, Paisley, UK) was added for 2 h at room temperature. The sections were washed and then mounted using Vectashield (Vector Laboratories) or Fluorosave (Calbiochem, Beeston, UK) mounting media. The following primary antibodies were used: mouse anti-neuronal nuclear antigen (NeuN) (1:50; Chemicon, Southampton, UK), rat anti-BrdU (1:50; Accurate Chemicals, Westbury, NY), goat anti-doublecortin (DCX) (1:400; Santa Cruz Biotechnology, Heidelberg, Germany), and rat anti-CD11b (5C6 clone) (1:1000; Serotec, Oxford, UK). The following secondary antibodies were used: horse anti-mouse (1:200; Vector Laboratories), goat anti-rat (1:200; Serotec), donkey anti-goat (1:200; Santa Cruz Biotechnology), and the appropriate Alexa-488 or Alexa-568. Fluorojade-B staining (Chemicon) was performed according to Schmued and Hopkins (2000).
Quantification of numbers of DCX-positive cells. All slides were blind coded for assessment. DCX is a microtubule-associated phosphoprotein (Gleeson et al., 1999) expressed in NPCs and immature hippocampal granule cells for ∼3 weeks after birth (Brown et al., 2003b; Kempermann et al., 2004; Rao and Shetty, 2004) and has been validated as a marker for neurogenesis in the DG (Rao and Shetty, 2004; Couillard-Despres et al., 2005). DCX-positive cells were counted using the optical fractionator method (for review, see Gundersen et al., 1999) and CAST-grid package (version 1.09; Olympus Optical, Glostrup, Denmark). The DG was visualized using the 4× objective lens of a light microscope (BX50; Olympus Optical), and the area of interest was highlighted manually. Cells were counted using the 20× objective lens, within a specified field/counting grid (“dissector”) of area 6044 μm2, which was moved across the section systematically on a motorized stage. Cells that overlapped two of the sides of the grid were counted, and cells on the other two sides were not. One in 12 sections were analyzed, and, if any sections were damaged or missing, a correction factor was applied, i.e., the cell count was multiplied by the ratio of sections that should have been counted to the number of sections present. Total cell counts were multiplied by the ratio of reference volume to sampling volume to obtain an estimate of the total number of DCX-positive cells in the DG of each brain.
Quantification of morphological features of DCX-positive cells in the DG. Morphological measurements of DCX-positive cells were performed using a light microscope under a 20× objective lens (Leica/Leitz DMRB; Leica, Nussloch, Germany) and the LUCIA package. Measurements were taken from five areas of each DG, and three DG fields from each animal were examined. The longest measurement from each area was chosen and averaged. The degree of dendritic branching of DCX-positive cells was estimated using an arbitrary visual scale from 0 to 7. Immature DCX-positive cells, with no dendritic branching, were not included in the grading assessment. Sections were assigned a grade according to the maximum grade of branching exhibited. In other words, if some cells exhibited branching grade 5 but some cells exhibited branching grade 2, the section would be assigned an overall grade of 5. The grades of branching were assessed as follows: grade 0, no dendritic branching; grade 1, one or two first-order dendritic branches (in which “first order” refers to a dendritic branch emanating from the apical dendrite, “second order” refers to a branch emanating from a first-order branch, and “third order” refers to a branch emanating from a second-order branch); grade 2, more than two first-order dendritic branches; grade 3, more than two first-order dendritic branches and one or two second-order dendritic branches; grade 4, more than two first-order and second-order dendritic branches; grade 5, more than two first-order and second-order dendritic branches and one or two third-order dendritic branches; grade 6, more than two third-order dendritic branches; grade 7, many third-order dendritic branches.
Quantification of numbers of BrdU-positive cells in the DG. BrdU-positive cells at day 9 were counted using the 40× objective lens of a florescence microscope (Leitz, Wetzlar, Germany) and the Open Lab package (Improvision, Lexington, MA). Every cell was counted throughout the thickness of the section, and total counts were multiplied by the frequency of sections, i.e., 12, to obtain an estimate of the total number of BrdU-positive cells in the DG of each brain.
Quantification of density of CD11b immunoreactivity (microglia). CD11b was semiquantified using the intensity of immunoreactivity because individual cells were not easily identified and not amenable to counting. This was performed by selecting an area of DG using the 20× objective lens of a light microscope (Leica/Leitz DMRB) and measuring the mean intensity of immunoreactivity using the LUCIA package. Sections were kept in chromogen for 3 min per mouse, and illumination, contrast, and brightness were kept consistent for each image in parallel.
Quantification of numbers of BrdU/NeuN-positive cells. It was necessary to count cells that colocalized for BrdU and NeuN using a confocal microscope to avoid spurious counting of “satellite cells” (Kuhn et al., 1997; Kornack and Rakic, 2001). BrdU and NeuN were considered to be colocalized if nuclear colocalization was seen over the extent of the nucleus in consecutive 1 μm z-stacks, profiles of green and red fluorescence coincided, and when colocalization was confirmed in x–y, x–z, and y–z cross-sections produced by orthogonal reconstructions from z-series using Leica software (version 2.61). Confocal images were taken using a 40× oil objective lens from a Leica microscope. Fluorophores were excited at 488 nm (argon laser) and 568 nm (helium/neon laser). Counting was performed using 1 μm z-stacks, three DG fields were taken for analysis, and five areas were selected (while visualizing the NeuN immunoreactivity), one from the genu of the DG, two from the upper blade and two from the lower blade, ensuring that the areas did not overlap so that cells were not counted twice. In effect, most of the BrdU-positive cells in the DG were counted using this method. This was done for all brains from days 16 and 32 after KA treatment.
SVZ neurogenesis in response to a QA lesion: surgical procedures. Twenty-eight mice, 11 weeks old, were used in this experiment; 16 WT and 12 R6/2. Half of each genotype received a QA injection, and half received vehicle (0.1 m PBS). Weights ranged from 16 to 25 g. Each animal received a subcutaneous injection of analgesic (Rimadyl, 5 mg/kg; Pfizer, Kent, UK) and antibiotic (Terramycin, 60 mg/kg; Pfizer). QA (Sigma) was dissolved in 0.1 m PBS. Under halothane anesthesia, QA (0.66 μl, 40 nmol) or PBS (0.66 μl) was injected using a 1 μl Hamilton syringe, into the striatum using the following coordinates: 0.7 mm rostral to bregma, 1.9 mm lateral to the midline, and 2.5 mm ventral to the dura, with the tooth bar set at zero (Hansson et al., 1999). The injection was given over 2 min, and the syringe was left in place for an additional 3 min and then slowly removed. Body temperature was controlled using a heat pad maintained at 37°C.
BrdU labeling and tissue processing. The BrdU labeling protocol was similar to that described above for the KA experiment. The time points for BrdU administration were chosen on the basis of previous studies showing maximal proliferation at days 6 and 7 (Jin et al., 2001; Arvidsson et al., 2002; Parent et al., 2002). Tissue processing and immunohistochemistry was performed as in the KA experiment, but animals were killed on day 8 and day 31. Acetylcholinesterase staining was performed according to the modified Koelle method (Koelle, 1955).
Quantification of lesion area. Lesion areas, identified by fluorojade-B, were measured using a fluorescence microscope (Leitz); the lesion area on each section was delineated manually using the Open Lab package (Improvision). The lesion area in each section was multiplied by 12 (frequency of section) and 40 μm (section thickness) to calculate lesion volume (in cubic millimeters).
Quantification of numbers of BrdU-positive cells in the SVZ and striatum. Using a light microscope (Leica) under a 25× objective lens, all BrdU-positive cells in the lateral and medial wall of the SVZ from anterior to the septodiencephalic level were counted, because they tended to cluster. Cells at the dorsolateral corner of the SVZ, extending laterally, were not counted. Sections were counterstained with fluorojade-B to identify the lesion. BrdU-positive cells in the striatum were counted using the optical fractionator method described above for the KA experiment.
Quantification of numbers and migration of DCX-positive cells in the striatum. Neuroblast migration was measured as the distance of the furthest lateral DCX-positive neuroblast from the dorsolateral corner of the SVZ. Measurements from each section were taken and averaged using a light microscope (10× objective lens; Leica/Leitz DMRB) and the LUCIA package. The total number of DCX-positive cells in the striatum, including the peri-SVZ area, were counted using a 20× objective lens of a light microscope (BX50; Olympus Optical) and multiplied by 12 (the frequency of sections) to obtain an estimate of the total.
Quantification of numbers of BrdU/NeuN-positive cells in the striatum. For BrdU/NeuN quantification, sections were viewed for NeuN, and six representative samples were taken for counting using confocal images. Three sections were randomly chosen from within the core of the lesion and three sections around the edge of the lesion. Numbers of BrdU/NeuN-positive cells are thus arbitrary and not intended as an estimate of the total number but sufficient for comparison between genotypes and QA-treated versus vehicle-treated groups. z-Series were performed at 1 μm intervals, and images were viewed using a 40× oil objective lens. Cells were considered double labeled according to the criteria described above for the KA experiment. All of the genotype comparisons are made using cell counts from the ipsilateral side of the brain.
Neurosphere assay. Female R6/2 mice and their WT littermates were killed by CO2 asphyxiation, and pups (postnatal day 3) were killed by decapitation. Dissections were conducted in PBS with 0.6% glucose, pH 7.4, at 4°C under sterile conditions. Meninges and blood vessels were removed. For the SVZ and DG dissections, each brain was sliced into 2 mm coronal sections. Tissue was washed in PBS/glucose, digested in papain–dispase–DNase solution [0.01% papain, Lorne Laboratories (Twyford, UK); 0.1% dispase, Roche Diagnostics (Hertfordshire, UK); 0.01% DNase, Lorne Laboratories] for 4 min, filtered through 40 μm nylon mesh cell filters (Falcon, Suffolk, UK), and then washed three times. Cell viability was calculated using the trypan blue dye (Sigma) exclusion method. Cells were seeded at a density of 1 × 105 cells/ml in 25 and 75 cm2 flasks in Neurobasal medium (Invitrogen) with 2% B27 (Invitrogen), 1% penicillin/streptomycin/fungizone (PSF) (0.1 g/liter; Invitrogen), 1% l-glutamine (Sigma), 20 ng/ml basic fibroblast growth factor-heparin (R & D Systems, Oxford, UK), and 20 ng/ml epidermal growth factor (Sigma) (Wachs et al., 2003).
Flasks were incubated at 37°C, 95% air/5% CO2, in a humidified atmosphere. Media was replaced on day 4 and added on day 7, and neurospheres were dissociated (“passaged”) on day 10. To dissociate the neurospheres, calcium-free HBSS (Invitrogen) at pH 7.5 was added to centrifuged neurospheres, and the mixture was gently aspirated. A total of 200 μl of media, pH 11, was then added for 7 min, and finally 200 μlof media, pH 1.8, was added (Sen et al., 2004). Cells were washed, counted using the trypan blue dye exclusion method, and reseeded.
For differentiation, dissociated cells were plated on glass coverslips in 1 ml wells precoated with 0.01 μl/ml poly-d-lysine (Sigma) and, for adult cultures, 1 μg/ml laminin (Sigma). Cells were seeded at 5 × 104 cells per 500 μl in Neurobasal medium with 2% B27, 1% PSF, 1% l-glutamine, and 1% fetal calf serum (FCS) (Invitrogen). An additional 500 μl of media was added on day 4, and cells were fixed in 4% paraformaldehyde after 7 d. Cells were incubated in blocking solution (5% serum in 0.1 m PBS with 0.2% Triton X-100) for 30 min at room temperature and then incubated in primary antibody in 1% serum (in PBS with 0.2% Triton X-100) overnight at 4°C. After washing in PBS, secondary fluorescence-conjugated antibodies were diluted in the fluorescent nuclear stain, bisbenzimine/Hoechst-33258 (1:5000 in PBS; Sigma), and applied for 2 h at room temperature. After washing, coverslips were mounted on glass slides with 1:1 PBS/glycerol. Galactocerebroside (Gal C) staining was performed on live cells: cells were incubated with primary antibody and 10% FCS for 60 min, washed, and fixed, and the secondary antibody was then applied (1:500, Alexa-488- and Alexa-568-conjugated secondary antibodies; Invitrogen). For BrdU staining, ice-cold methanol was applied to fixed cells for 20 min, and the cells were washed, exposed to 2 m HCl at 37°C, washed in 0.1 m borate buffer at pH 8.5, and then stained in the usual way. The following primary antibodies were used: anti-β-tubulin (1:500; Sigma), anti-glial fibrillary acid protein (GFAP) (1:500; DakoCytomation, Ely, UK), anti-Gal C (gift from P. Gaughwin, Cambridge Centre for Brain Repair), anti-nestin (1:500; Chemicon), anti-BrdU (1:50; Accurate Chemicals), and anti-DCX (1:400; Santa Cruz Biotechnology). Mutant huntingtin (htt) protein was detected using an antibody raised against a human GST-exon-1 fusion protein carrying 53 glutamines (gift from Dr. G. Bates, Guy's Hospital, London, UK) and used at a dilution of 1:1000. Primary antibodies were visualized using Alexa-488- and Alexa-568-conjugated secondary antibodies (1:1000; Invitrogen). Two to four coverslips were stained with each antibody. Slides were examined using a counting grid (0.0625 mm2) under the 40× objective lens of a fluorescence microscope (Leitz), five random fields were counted from each coverslip, cell counts were expressed as a percentage of Hoechst-positive cells, and images were taken and stored using the OpenLab package (Improvision).
Statistical analysis. Some data were normalized by log transformation or square root (NPC proliferation in vitro) to allow parametric statistical analysis, and any transformation is indicated on the appropriate figure. Normalization was confirmed by appearance on a quantile–quantile plot and using the Kolmogorov–Smirnov Z test. ANOVA was used to test for differences in BrdU, DCX, and BrdU/NeuN (using age, day, genotypes, treatment, and gender as independent variables), using a full factorial analysis (although it was not possible to model all four- and five-way interactions). Post hoc tests were done using an ANOVA with a Bonferroni's correction for multiple comparisons. When data could not be normalized, or ordinal scales were used, nonparametric tests were used (two-tailed Mann–Whitney U test), as indicated in the text. Correlations were made using Pearson's correlation coefficient (parametric data) or Spearman's rank correlation coefficient (nonparametric data). α was set at 0.05, and analysis was performed using SPSS for Windows (version 11.5; SPSS, Chicago, IL).
Results
Seizure severity is similar between genotypes, but younger R6/2 mice have less severe seizures than younger WT mice
KA-induced seizures lasted for up to 3 h, and their severity was graded according to a modified Racine scale (Racine et al., 1972; Morton and Leavens, 2000). All but two mice had seizures that could be graded. Two mice (both 13 weeks old, one WT and one R6/2) did not display gradable seizures but became immobile and hunched, although rousable. Such behavior has been associated with EEG-confirmed seizure activity (Mohapel et al., 2004). One 8-week-old WT mouse and two R6/2 mice (aged 8 and 13 weeks) died during these experiments. The WT mouse died after a series of tonic-clonic seizures; the R6/2 mice died after one prolonged tonic-clonic seizure.
There was no difference in scoring on the modified Racine scale between genotypes (p = 0.11) (Fig. 1). However, when each age group was considered separately, younger R6/2 mice had less severe seizures than younger WT mice (p < 0.05), but older R6/2 mice had similar scoring on the modified Racine scale to older WT mice (p = 0.829). This is consistent with the findings of Morton and Leavens (2000). There was no correlation between the scoring on the modified Racine scale and neurogenesis, in both genotypes, as measured by the number of DCX-positive cells in the DG (p = 0.388; r = 0.155).
Figure 1.

Comparison of seizure severity between genotypes and age. R6/2 and WT mice were given an injection of KA, and seizures were rated on the modified Racine scale. Mice that died (18-week-old WT mouse and 2 R6/2 mice aged 8 weeks and 13 weeks) are included in the figure. There was no difference in scoring on the modified Racine scale by genotype (p = 0.11), although when each age group is considered separately, younger R6/2 mice had less severe seizures (p < 0.05) than older mice (p = 0.829). NS, Not statistically significant (p > 0.05); the asterisk denotes statistical significance (p < 0.05).
A previous study showed that intraperitoneal KA at a dose of 20 mg/kg causes seizures but no cell loss (Morton and Leavens, 2000). This was confirmed in the present study using fluorojade-B. This dose of KA was chosen to allow a comparison of neurogenesis in response to seizures without the complicating factor of differences in cell loss between genotypes (Morton and Leavens, 2000).
Neurogenesis is lower in R6/2 mice and in older mice
The use of DCX to study neurogenesis has many advantages (Rao and Shetty, 2004; Couillard-Despres et al., 2005), although transcription or handling of DCX in R6/2 mice may be altered and be erroneously interpreted as changes in neurogenesis. Intensity of immunolabeling in individual cells, however, was the same between genotypes, and DCX and BrdU cell counts in the DG correlated closely with one another (p < 0.001; r = 0.776). Importantly, the duration of DCX expression remains the same in aging, environmental enrichment and seizure-induced hippocampal neurogenesis (Rao and Shetty, 2004; Couillard-Despres et al., 2005).
Mice were killed on days 9, 16, and 32 after KA treatment, and there was no difference in the number of DCX-positive cells in the DG between these time points (F(2,33) = 0.601; p = 0.554). There were fewer DCX-positive cells in the DG of R6/2 mice compared with WT mice (F(1,33) = 14.422; p = 0.001) (Fig. 2) (supplemental Table 1, available at www.jneurosci.org as supplemental material). There was a genotype × treatment interaction effect (F(1,33) = 5.408; p < 0.05), suggesting that neurogenesis was greater in the KA-treated group compared with the vehicle-treated group in WT mice, consistent with previous studies (Parent et al., 1997; Scott et al., 1998) but that neurogenesis was the same in the KA-treated group compared with the vehicle-treated group in R6/2 mice. Thus, neurogenesis can be upregulated in WT mice but not R6/2 mice.
Figure 2.

Comparison of neurogenesis as measured by DCX-positive cells in the DG between genotypes in vehicle-treated and KA-treated groups. R6/2 mice had fewer DCX-positive cells in the DG than WT mice (F(1,33) = 14.422; p = 0.001). There was a genotype × treatment interaction effect (F(1,33) = 5.408;p < 0.05), suggesting that neurogenesis can be upregulated in WT mice but not in R6/2 mice. Data have been log transformed, bars show means, and error bars show means ± 1.0 SEM. NS, Not statistically significant (p > 0.05); the asterisk denotes statistical significance (p < 0.05).
There was no significant difference in DCX-positive cells between genotypes in vehicle-treated mice (p = 1.000). That is, basal neurogenesis was similar between WT and R6/2 mice.
There were also fewer DCX-positive cells in the DG of older mice compared with younger mice (F(1,33) = 15.407; p < 0.001). The decline in neurogenesis with age is well recognized (Kuhn et al., 1996) and has been demonstrated for transgenic HD mice (Lazic et al., 2004; Gil et al., 2005).
It is important to consider sex differences in studies of neurogenesis because, for example, NPC proliferation is higher in female rodents compared with males (Tanapat et al., 1999) (for review, see Abrous et al., 2005). We included mice of both sexes for logistical reasons. Seventeen male mice were included in the study, but the numbers of male and female mice were spread evenly across all groups, sex of the mice did not affect neurogenesis (F(1,33) = 2.872; p = 0.100), and there was no interaction between sex and the other variables.
The morphology of DCX-positive cells is altered in R6/2 mice
DCX-positive cells in the DG possessed the morphology of stage 5 immature postmitotic granule cells and had a globular cell body with many fine dendritic processes extending into the molecular layer (Kempermann et al., 2004) (Fig. 3A). Dendritic growth into the molecular layer was greater for the upper blade of the DG than for the lower blade (observation only, not quantified). There were also DCX-positive cells with short bipolar processes arranged tangentially in the SGZ. These cells may correspond to type 2b cells (Kempermann et al., 2004). These short tangential cells could not be discerned individually and thus were not counted.
Figure 3.

Morphological characteristics of DCX-positive cells in the DG. A, Photomicrograph of the DG. B, Line drawing of photomicrograph in A, illustrating morphological measurements. C, Photomicrographs illustrating the morphology of DCX-positive cells in R6/2 and WT mice. Scale bars, 100 μm.
R6/2 mice had shorter newborn granule neurons than WT mice (as measured from mid cell body to the tip of the apical dendrite), in both vehicle-treated and KA-treated groups (F(1,33) = 31.225; p < 0.001) (Fig. 4A). Older mice had shorter cells than younger mice (F(1,33) = 4.391; p < 0.05). Mice from the KA-treated group compared with the vehicle-treated group had cells of the same length (F(1,33) = 3.837; p = 0.059).
Figure 4.

Comparison of the morphological characteristics of DCX-positive cells in the DG. A, R6/2 mice have shorter newborn cells, from mid cell body to the tip of the apical dendrites, than those from WT mice (F(1,33) = 31.225; p < 0.001).B, The degree of branching of dendritic arbor from three DG sections was assessed on an arbitrary visual eight-point scale (for details, see Material and Methods). R6/2 mice had a lower degree of branching than WT mice (p < 0.001). C, Newborn cells from R6/2 mice migrated shorter distances into the GCL from the SGZ than those from WT mice (F(1,33) = 15.128; p < 0.001). D, Newborn cells in R6/2 mice had a shorter distance to the first branch of the apical dendrite than those from WT mice (F(1,33) = 13.397; p < 0.001). Bars show means, and error bars show means ± 1.0 SEM.
Newborn granule cells in R6/2 mice had a lesser degree of dendritic branching than those from WT mice, as assessed on an eight-point visual scale (p < 0.001) (Fig. 4B). There was a trend for branching to be greater in the KA-treated group in WT mice, but this did not reach statistical significance (WT vehicle treated vs KA treated, p = 0.069; R6/2 vehicle treated vs KA treated, p = 0.760).
DCX-positive cells from R6/2 mice migrated shorter distances from the SGZ into the granule cell layer (GCL) than those in WT mice in both vehicle-treated and KA-treated groups (F(1,33) = 15.128; p < 0.001) (Fig. 4C). Migration of newborn neurons was not greater in WT mice of the KA-treated group compared with the vehicle-treated group (p = 0.174).
In DCX-positive cells from both genotypes, the first branch emanated from a relatively thick apical dendrite, and branching occurred in the GCL, as described previously (Rao and Shetty, 2004). However, newborn cells in R6/2 mice had a shorter apical dendrite from the cell body to the first dendritic branch in both vehicle-treated and KA-treated groups when compared with WT mice (F(1,33) = 13.397; p < 0.001) (Fig. 4D).
Regarding vehicle-treated mice only compared with WT mice, R6/2 mice had shorter DCX-positive cells (p = 0.003), less dendritic branching (p < 0.01), and a trend for shorter initial apical dendrites (p = 0.060).
In summary, newborn granule cells of R6/2 mice had shorter apical dendrites and more sparse dendritic arbor, which emanated close from the cell body, and cells migrated shorter distances when compared with newborn granule cells of WT mice (Fig. 3C).
Cell proliferation 1 d after BrdU injection is lower in R6/2 mice compared with WT mice
BrdU-positive cells in the DG at day 9 after KA treatment (i.e., 1 d after BrdU injection) were fewer in number in R6/2 mice compared with WT mice (F(1,10) = 8.886; p < 0.02; n = 19) (Fig. 5) (supplemental Table 1, available at www.jneurosci.org as supplemental material), consistent with previous findings (Gil et al., 2004, 2005; Lazic et al., 2004). Proliferation was higher in mice aged 8 weeks than those aged 13 weeks (F(1,10) = 38.076; p < 0.001). DCX and BrdU cell counts in the DG correlated closely with one another (p < 0.001; r = 0.776). There was no correlation between BrdU-positive cell number in the DG and scores on the modified Racine scale (p = 0.476; r = 0.274).
Figure 5.

Comparison of cell proliferation in the DG between genotypes. BrdU-positive cells in the DG at day 9 after KA-treatment (1 d after the last BrdU injection) were fewer in number in R6/2 mice compared with WT mice (F(1,10) = 8.886; p < 0.02). There was a genotype × treatment interaction effect (F(1,10) = 7.573; p = 0.02), suggesting that proliferation was higher in KA-treated WT versus vehicle-treated WT mice but the same in KA-treated R6/2 versus vehicle-treated R6/2 mice. Data have been log transformed, bars show means, and error bars show means ± 1.0 SEM. The asterisk denotes statistical significance (p < 0.05). NS, Not statistically significant.
There was no significant difference in BrdU-positive cells between genotypes in vehicle-treated mice (p = 1.000). That is, basal NPC proliferation was similar between WT and R6/2 mice.
When both vehicle-treated and KA-treated mice were analyzed, there was a genotype × treatment interaction effect (F(1,10) = 7.573; p = 0.02), suggesting that proliferation was greater in KA-treated WT mice compared with vehicle-treated WT mice and that proliferation was the same in KA-treated R6/2 mice compared with vehicle-treated R6/2 mice. This suggests that NPC proliferation is upregulated only in WT mice, not in R6/2 mice, consistent with the DCX data. R6/2 mice are reported to have handling-induced seizures later in life (Mangiarini et al., 1996), so the fact that neurogenesis was the same in R6/2 mice after seizure is important to know when interpreting results from neurogenesis experiments in R6/2 mice. However, handling-induced seizures in our colony are extremely low (fewer than 1 in 100 mice exhibit such seizures in their lifetime).
Neuronal fate and survival of newborn cells are the same in WT and R6/2 mice
To measure cell fate and survival of newborn cells, we counted the number of BrdU/NeuN-positive cells and the percentage of BrdU-positive cells that expressed NeuN (supplemental Table 1, available at www.jneurosci.org as supplemental material). The number of BrdU-positive cells that coexpressed NeuN (presumptive newborn neurons) at day 16 and day 32 after KA treatment was similar in R6/2 and WT mice (Mann–Whitney U test, p = 0.394; n = 45). The percentage of BrdU-positive cells coexpressing NeuN was also similar (Mann–Whitney U test, p = 0.170). Survival of BrdU/NeuN-positive cells was no different in the KA-treated group compared with the vehicle-treated group (Mann–Whitney U test, p = 0.230).
Comparison of genotypes with the same seizure grade
Most mice (55%) displayed seizures scoring 2 on the modified Racine scale. These mice were analyzed separately (n = 18) along with vehicle-injected mice (n = 49 in total). There were not enough mice displaying the other grades to analyze separately and produce valid analyses. In this group, there were significant genotype effects. That is, when compared with WT mice, R6/2 mice had fewer DCX-positive cells in the DG (F(1,20) = 9.269; p < 0.01), shorter apical dendrites (F(1,20) = 21.974; p < 0.001), shorter distance of the apical dendrite from cell body to first branch (F(1,20) = 9.272; p < 0.01), shorter migration distances into the GCL (F(1,20) = 5.500; p < 0.05), less arbor ramification (Mann–Whitney U test, p < 0.01), and no difference in the number of BrdU/NeuN-positive cells (p = 0.138). There were fewer BrdU-positive cells in the DG of R6/2 mice (F(1,8) = 6.441; p < 0.05), and there was a genotype × treatment interaction effect (F(1,8) = 7.411; p < 0.05), again suggesting that NPC proliferation can be upregulated in WT but not R6/2 mice.
In summary, seizure severity does not influence the differences in neurogenesis and newborn neuron morphology between genotypes. This is consistent with Mohapel et al. (2004), who showed that seizure severity does not influence the rate of cell proliferation but reduces newborn cell survival, possibly attributable to cell death. No cell death was seen in our study, as measured using fluorojade-B.
R6/2 mice are resistant to QA-induced striatal lesions
We next examined neurogenesis in the SVZ, the other area of constitutive neurogenesis in the brain, as well as the DG. We also examined the striatum, because it the brain region most vulnerable in HD and a possible target area of newborn neurons from the SVZ (Arvidsson et al., 2002; Parent et al., 2002).
It has been shown previously that transgenic HD mice are resistant to excitotoxic lesions (Hansson et al., 1999, 2001a,b; Hickey and Morton, 2000; Petersen et al., 2001; MacGibbon et al., 2002), although this is age, strain, and excitotoxin dependent (Bogdanov et al., 1998; Hansson et al., 2001a; Petersen et al., 2002; Schauwecker, 2002; Zeron et al., 2002). QA has been used in various doses to induce lesions in mice [1 μl of 8, 20, and 30 nmol (Zeron et al., 2002; Hansson et al., 2001b; Petersen et al., 2002, respectively], so, in the present study, a larger dose (40 nmol) was given to all animals in an attempt to induce a lesion in R6/2 mice. Fluorojade-B was used to label lesioned tissue, and the area of immunofluorescence was measured (Duckworth et al., 2005). As well as fluorojade-B, brains were stained for acetylcholinesterase, which confirmed the presence of lesions. All WT mice had extensive striatal lesions that also involved the adjacent cortex. However, lesions in the R6/2 mice were smaller at both time points (Mann–Whitney U test, p < 0.02) (Fig. 6). Furthermore, whereas all WT mice had distinct lesions, one R6/2 mouse had no lesion (at day 8), two R6/2 mice (one at day 8, one at day 31) had distinct lesions but not as large as the smallest WT mouse lesion, and the other three R6/2 mice had diffuse lesions (i.e., individual fluorojade-B-positive cells were present, but a lesion area could not be measured).
Figure 6.

R6/2 mice are resistant to QA-induced striatal toxicity. R6/2 mice had a smaller lesion area than WT mice (Mann–Whitney U test, p < 0.02). Lesion size is measured using fluorojade-B and expressed as cubic millimeters. Bars show means, and error bars show means ± 1.0 SEM. The asterisks denote statistical significance (p < 0.05).
There is no difference in basal SVZ proliferation between genotypes, but proliferation can be upregulated in WT but not R6/2 mice
There was no significant difference in the number of BrdU-positive cells in the SVZ in WT mice compared with R6/2 mice when analyzing both vehicle-treated and QA-treated mice (F(1,20) = 3.232; p = 0.087) (Fig. 7) (supplemental Table 2, available at www.jneurosci.org as supplemental material). Similarly, SVZ proliferation was the same between genotypes in vehicle-treated mice (p = 1.000). The number of BrdU-positive cells in the SVZ was significantly lower in the day 31 group (F(1,20) = 63.815; p < 0.001), suggesting that the NPCs labeled on days 6 and 7 have died, migrated to the olfactory bulb or striatum, or proliferated such that the marker has become diluted. There was a genotype × treatment interaction effect (F(1,20) = 5.211; p < 0.05), suggesting that NPC proliferation was upregulated in response to QA lesions only in WT mice. That cell proliferation in the SVZ was higher after striatal lesions in WT mice is consistent with previous studies (Jin et al., 2001; Arvidsson et al., 2002; Parent et al., 2002).
Figure 7.

Comparison of SVZ proliferation in QA-treated and vehicle-treated animals. There was no significant difference in numbers of BrdU-positive cells in the SVZ in WT mice compared with R6/2 mice in either the QA-treated or vehicle-treated group (F(1,20) = 3.232; p = 0.087). The number of BrdU-positive cells was greater in the QA-treated group versus the vehicle-treated group only in WT mice (F(1,20) = 5.211; p < 0.05), suggesting that NPC proliferation was upregulated in response to QA lesions only in WT mice. Data have been log transformed, bars show means, and error bars show means ± 1.0 SEM. NS, Not statistically significant (p > 0.05); the asterisk denotes statistical significance (p < 0.05).
There is no difference in basal neuroblast numbers and migration between genotypes, but neuroblast numbers can be upregulated in WT but not R6/2 mice
There was no difference in lateral migration of neuroblasts overall between genotypes (F(1,20) = 0.111; p = 0.742) (Fig. 8A). Similarly, lateral migration was the same between genotypes in vehicle-treated mice (p = 1.000). There was no genotype × treatment interaction effect (F(1,20) = 2.249; p = 0.149), suggesting that there was no difference between genotypes in upregulation of neuroblast migration with QA treatment. There was no significant effect of QA treatment on lateral migration of neuroblasts (F(1,20) = 1.662; p = 0.212), and, as lesion size increased, lateral migration did not increase (Pearson's correlation coefficient, p = 0.06; r = 0.514), but striatal collapse must be considered when interpreting this result. Ventral and lateral migration from the wall of the SVZ was not quantified because it was difficult to ascertain whether a neuroblast had migrated or whether it was an in situ progenitor.
Figure 8.

Comparison of neuroblasts between genotypes and treatment (i.e., intrastriatal injection of vehicle QA). A, The distance (in micrometers) of the farthest lateral DCX-positive cell, with the morphology of a migrating neuroblast, from the anterior SVZ was measured from three to four sections and averaged. There was no difference in lateral migration between genotypes (F(1,20) = 0.111; p = 0.742). There was no genotype × treatment interaction effect (F(1,20) = 2.249; p = 0.149), suggesting that there was no difference between genotypes in upregulation of neuroblast migration with QA treatment. B, Numbers of DCX-positive cells in the striatum are greater in QA-treated mice compared with the vehicle-treated mice (F(1,20) = 22.898; p < 0.001) but the same between genotypes (F(1,20) = 0.003; p = 0.956). DCX-positive cells in the striatum were greater in QA-treated versus vehicle-treated animals only in WT mice (p < 0.005) but not in R6/2 mice (p = 0.441), suggesting that neurogenesis was upregulated only in WT mice. Data have been log transformed, bars show means, and error bars show means ± 1.0 SEM. NS, Not statistically significant (p > 0.05); the asterisk denotes statistical significance (p < 0.05).
Numbers of DCX-positive cells in the striatum were greater in QA-treated mice compared with vehicle-treated mice (F(1,20) = 22.898; p < 0.001) (Fig. 8B) (supplemental Table 2, available at www.jneurosci.org as supplemental material) but the same between genotypes (F(1, 20) = 0.003, p = 0.956; genotype × treatment interaction effect, F(1,20) = 2.164, p = 0.157). Similarly, numbers of striatal neuroblasts were the same between WT and R6/2 mice in vehicle-treated mice (p = 1.000). DCX-positive cells in the striatum were greater in QA-treated versus vehicle-treated animals only in WT mice (p < 0.005) but not in R6/2 mice (p = 0.441), suggesting that neurogenesis is upregulated only in WT mice. The fact that the interaction effect was not significant, whereas the post hoc comparison was, is likely to be explained by the variability of the R6/2 QA-treated group.
There were no obvious morphological differences in SVZ and striatal DCX-positive cells between WT and R6/2 mice (observation only, not quantified). In both genotypes, fusiform-shaped DCX-positive cells were seen in the SVZ, particularly in the dorsolateral corner of the SVZ. DCX-positive cells migrated laterally and ventrally from the dorsolateral SVZ and laterally from the lateral wall of the SVZ. In QA-treated mice of both genotypes, a “ridge” of neuroblasts could be seen migrating from the SVZ (Fig. 9A,A′), reminiscent of the study by Fallon et al. (2000). In some QA-treated animals, a large number of putative proliferating neuroblasts could be seen in the contralateral side of the corpus callosum and appeared to be migrating toward the lesion (Fig. 9B,B′). Some cells, in QA-treated animals, had the morphology of migrating neuroblasts, with a single leading process (Fig. 9A′) (O'Rourke et al., 1995), whereas others were clustered in “nests” near the wall of the lateral ventricle (Fig. 9C). Cells with a more mature morphology were observed adjacent to the lateral wall of the SVZ (Fig. 9D) and rostral to the tip of the third ventricle. DCX-positive cells in the striatal parenchyma (distant from the SVZ) were usually isolated, although some clusters were observed, particularly around lesions. DCX-positive cells were seen occasionally in the cortex of both genotypes, and the cells had a more mature morphology, reminiscent of the semilunarpyramidal neurons described by Nacher et al. (2001). Many isolated cells, lightly immunoreactive for DCX and with possible astrocytic morphology, were also observed throughout the brain (these were counted but not included in the main analysis, and there was no difference between genotypes; data not shown).
Figure 9.
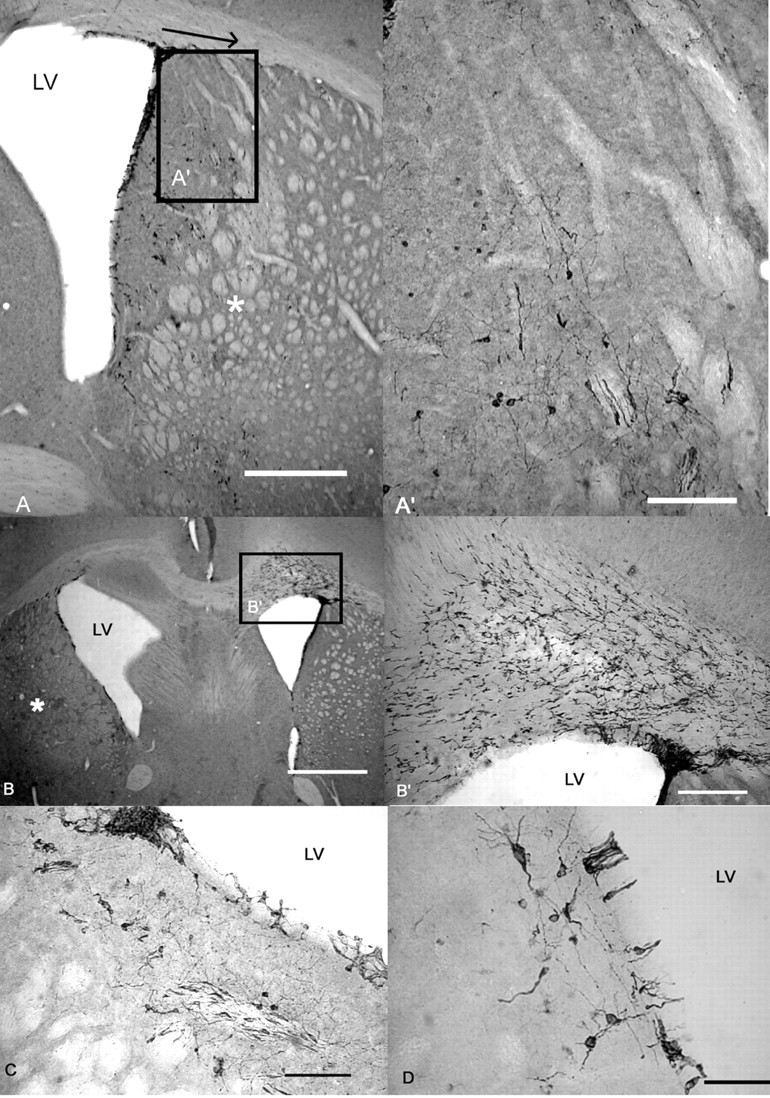
The morphology of DCX-positive cells in the SVZ and striatum. A, Photomicrograph showing the SVZ and striatum from an R6/2 mouse 31 d after lesion. DCX-positive cells migrated laterally from the SVZ toward the lesion (denoted by *). The arrow shows the extent of lateral migration, as used for quantification, from the dorsolateral corner of the SVZ to the furthest neuroblast. A′, The same area highlighted in A; A′ is shown at a higher magnification, illustrating the migratory morphology of DCX-positive cells. B, Photomicrograph showing the SVZ and striatum from an R6/2 mouse 31 d after lesion. The QA injection in this mouse produced a relatively large lesion (denoted by *). A large number of DCX-positive cells were seen in the corpus callosum, with migratory processes directed toward the contralateral lesioned striatum. B′, The same area highlighted in B; B′ is shown at a higher magnification, illustrating migration of DCX-positive cells toward the contralateral side. C, Photomicrograph showing the ipsilateral SVZ from a WT mouse 31 d after lesion, showing a nest of neuroblasts. These were found in mice of both genotypes. D, DCX-positive cells adjacent to the contralateral SVZ in a vehicle-treated WT mouse. Scale bars: A, 500μm; A′, C, 100 μm; B, 1 mm; B′, 200 μm; D,50 μm. LV, Lateral ventricle.
Striatal neurogenesis was observed in QA-treated WT but not R6/2 mice
After QA treatment, a large increase in total number of BrdU-positive cells was observed in both R6/2 and WT mice (data not shown). We did not correlate DCX and BrdU counts in the striatum, because many BrdU-positive cells were likely to be proliferating glia (Stoll et al., 1998) and because DCX has not been validated as a marker of neurogenesis in the striatum. We counted BrdU-positive cells that coexpressed NeuN to quantify neurogenesis. BrdU/NeuN colocalization was found only in QA-treated WT mice, consistent with previous studies of striatal lesions (Arvidsson et al., 2002; Parent et al., 2002). Numbers and proportions of BrdU/NeuN-positive cells in QA-treated WT mice were greater in animals with larger lesions (Spearman's rank correlation coefficient, p < 0.01, r = 0.898; p = 0.002, r = 0.935, respectively). However, no surviving newborn neurons were found in R6/2 mice. This could be a result of the smaller lesions sustained (Jin et al., 2001), although surviving BrdU/NeuN-positive cells were not seen in the R6/2 mice that sustained relatively large lesions.
In WT and R6/2 mice, NPCs proliferate at the same rate in vitro
Proliferation, morphology, migration, and overall neurogenesis in the DG were reduced in the R6/2 mouse, and SVZ/striatal neurogenesis was not upregulated in response to a striatal lesion. This could be attributable to a defect in the NPC itself, its interaction with the environment, or a nonpermissive environment in the R6/2 mouse. To address the first question, we examined NPC in vitro from R6/2 and WT mice.
Neurospheres from the SVZ of four R6/2 and five WT mice aged 8 weeks were established as free floating in culture. Neurospheres developed within 4–5 d from seeding and did not differ in gross appearance, size, or number between WT and R6/2 mice. Although the absolute number of NPCs increased from initial seeding after passage, the rate of increase was greatest at early passages and then the rate reached a plateau (Fig. 10A). Neurospheres derived from both genotypes could be maintained in culture for up to 70 d (up to nine passages), with no difference in proliferation rate between genotypes (t test, p = 0.206) (Fig. 10A, data to passage 4 shown). This result is consistent with Chu-LaGraff et al. (2001), who established neurospheres from the striatum and cortex of neonatal R6/2 mice. This finding from SVZ-derived neurospheres was confirmed using a range of methods and using different ages of mouse and different regions of the brain, including adult striatum, neonatal cerebellum, hippocampus, and cortex (data not shown). Non-SVZ tissue could not be maintained in large numbers beyond secondary neurospheres, similar to the findings by Seaberg et al. (2005).
Figure 10.

Proliferation and differentiation fate of NPC in vitro from adult SVZ. A, Comparison of the expansion rate of adult SVZ NPC from WT and R6/2 mice. The SVZ from five WT and four R6/2 mice, aged 9 weeks, were grown as free-floating neurospheres in 10 tissue culture flasks each for the first three passages and two to four each for subsequent passages. The expansion rate represents the number of dissociated cells at each passage divided by the seedingdensity. The expansion rate of WT and R6/2 mice was the same (t test, p = 0.446). Data have been transformed by square root, and each point represents means ± SEM of each culture. B, Comparison of the differentiation fate of dissociated adult SVZ NPC from WT and R6/2 mice at passage 1. Numbers of neurons or astrocytes are expressed as the percentage of Hoechst-positive cells. Each bar represents means ± SEM for each culture, and statistical comparisons are made using a two-tailed Mann–Whitney U test. There was no difference in differentiation fate between genotypes at passage 1 (for β-tubulin III-positive neurons, p = 0.960; for GFAP-positive astrocytes, p = 0.363).
Differentiation fate of NPCs is the same in WT and R6/2 mice
There was no difference in differentiation fate between genotypes at passage 1 from the SVZ cultures (Mann–Whitney U test, β-tubulin III-positive neurons, p = 0.96; GFAP-positive astrocytes, p = 0.363) (Fig. 10B). There were no obvious morphological differences in differentiated cells between genotypes. There were very few oligodendrocytes/Gal C-positive cells seen in WT cells but none in R6/2 cells. Previous quantitative analyses have shown that Gal C-positive cells are present in very low numbers in dissociated adult NPCs at passage 1 (Wachs et al., 2003). These results were confirmed using adult hippocampal tissue, i.e., there was no difference in the differentiation fate between genotypes (Mann–Whitney U test; n = 5; neurons, p = 0.23; astrocytes, p = 0.56).
There was no difference in differentiation fate between genotypes at passage 4 from SVZ cultures (Mann–Whitney U test, p = 0.241), by which time only GFAP-positive cells were seen. This is consistent with observations from previous studies (Imura et al., 2003; Jain et al., 2003), suggesting that either glial precursors are selected for passage 4 or neuronal precursors are more vulnerable to the dissociation or differentiation procedure. No culture modifications to increase neuronal yield were used in this study, such as addition of retinoic acid (Palmer et al., 1999; Takahashi et al., 1999; Monje et al., 2002). It has been suggested that the tendency to form predominantly glia with time in culture is intrinsic to the precursor cell program (Seaberg et al., 2005).
The majority of cells fixed 1 d after first passage, from both genotypes, expressed nestin, a marker for neuroepithelial stem cells (data not shown) (Frederiksen and McKay, 1988; Lendahl et al., 1990).
Cell survival, measured indirectly by counting the number of Hoechst-positive cells, after 7 d of differentiation was no different between genotypes (t test, p = 0.429).
Cells dissociated from neurosphere cultures in R6/2 mice express mutant huntingtin
R6/2 cells expressed mutant htt in a perinuclear location, whereas cells from WT neurosphere cultures did not. This is consistent with data from Chu-LaGraff et al. (2001) who showed, from Western blotting, that exon 1 is expressed in R6/2 NPCs. These results suggest that, despite expression of mutant htt by R6/2 NPCs, the intrinsic properties of proliferation, longevity in culture, differentiation, and survival is similar to those derived from WT mice.
The density of microglia in the DG is the same in WT and R6/2 mice
Neurogenesis was impaired in vivo in the R6/2 mouse but intact in vitro. This suggests that the in vivo environment of the R6/2 mouse may be nonpermissive for neurogenesis. One aspect of the local environment of R6/2 mice, which may be nonpermissive for neurogenesis, is differences in microglia. We chose to study this, following reports that activated microglia reduce neurogenesis (Monje et al., 2002; Ekdahl et al., 2003) and because numbers and morphology of microglia are decreased in R6/2 mice (Ma et al., 2003) but increased in HD patients (Sapp et al., 2001).
Microglia were identified by immunoreactivity for CD11b and mice from the 9 and 16 d time points after KA treatment, from the KA experiment, were examined. Microglia were seen in large numbers in the hilus and molecular layer but were sparse in the GCL. There was no relationship between CD11b immunoreactivity and neurogenesis in the DG, as measured by numbers of DCX-positive cells (Pearson's correlation coefficient, p = 0.229; r = 0.223). There was no significant difference in microglial density between genotypes (F(1,18) = 0.326; p = 0.575) or KA-treated mice versus vehicle-treated mice (F(1,18) = 0.446; p = 0.513). Of note, neurogenesis as measured by DCX was still greater in WT mice compared with R6/2 mice at the 9 and 16 d time points (F(1,24) = 22.094; p < 0.001). Neuronal death produces microglial activation, and it is possible that there was no significant microglial activation after seizures in our study because no cell loss was seen (Ekdahl et al., 2001).
Discussion
Cell proliferation, migration, and morphology, but not differentiation or survival, was impaired in the DG of R6/2 mice. Neurogenesis failed to upregulate after KA-induced seizures in R6/2 mice. Furthermore, although basal neurogenesis was intact in the SVZ, neurogenesis could not be upregulated in the SVZ/striatum of R6/2 mice. The differences in DG neurogenesis are unlikely to be attributable to an impairment of the NPC per se, because R6/2 NPCs were indistinguishable from WT NPCs in vitro. This suggests that the in vivo microenvironment is “hostile” to neurogenesis in the R6/2 mouse.
DG neurogenesis is reduced in the R6/1 (Lazic et al., 2004) and R6/2 mouse (Gil et al., 2004, 2005), and this study confirms and extends these findings, lending support to the theory that neurodegenerative diseases are associated with impaired neuroregeneration (Armstrong and Barker, 2001). Our study does not address whether impairments in neurogenesis in R6/2 mice cause (or contribute to) neurodegeneration, nor does it address the question of whether an increase in neurogenesis would be beneficial to an animal with a neurodegenerative disease. To answer such questions, other strategies will be required. For example, models in which neurogenesis can be blocked in adulthood would be invaluable for determining the functional importance of NPCs.
The finding of abnormal morphology of newborn neurons in R6/2 mice is consistent with the reduced sprouting seen in the sciatic nerve of R6/2 mice (Ribchester et al., 2004) and abnormal dendritic branching seen in medium spiny striatal neurons of R6/2 mice (Spires et al., 2004a) and HD patients (Ferrante et al., 1991). Iannicola et al. (2000) reported that hippocampal neurites in a transgenic HD mouse are shortened, and this study supports and extends these findings. We demonstrate for the first time that newborn (as opposed to mature) neurons are abnormal in the R6/2 mouse. BDNF is upregulated after seizures and causes morphological changes in granule cells (Vaidya et al., 1999; Sathanoori et al., 2004). However, BDNF levels are low in the R6/2 hippocampus (Zuccato et al., 2001; Spires et al., 2004b), and, in R6/2 mice, the increase in BDNF after seizures may not be of sufficient magnitude to increase neurogenesis and produce morphological changes in newborn granule neurons. The altered morphology of newborn granule neurons in the R6/2 mouse may also be attributable to abnormal cytoskeletal reorganization and dendritic growth (for review, see Harjes and Wanker, 2003; Jarabek et al., 2004). These morphological changes may contribute to the cognitive impairment of R6/2 mice. Because newborn neurons show enhanced long-term potentiation (LTP) (Schmidt-Hieber et al., 2004), it is tempting to speculate that fewer or abnormal newborn neurons may contribute to the impaired LTP seen in R6/2 mice (Murphy et al., 2000; Gibson et al., 2005).
The differentiation fate and survival of NPCs were intact in R6/2 mice. Lazic et al. (2004) could not demonstrate BrdU/NeuN colocalization, but Gil et al. (2005) found that, in contrast to our study, the 2 week survival of BrdU-positive cells was lower in R6/2 compared with WT mice. In agreement with our study, percentage colocalization of BrdU with NeuN was no different between genotypes. The reasons for the difference in survival rates between studies are unclear but may involve subtle differences in mouse colonies, such as the frequency of handling-induced seizures and CAG repeat length, and differences in BrdU dosing regimens and time the animals were killed (Prickaerts et al., 2004). Also, the greatest difference in survival rates between genotypes was found for 3.5-week-old mice (Gil et al., 2005), whereas the youngest mice used in our study were 9 weeks old. From our results, it is likely that the reduced neurogenesis seen in R6/2 mice was attributable to reduced cell proliferation. R6/2 mice are resistant to neurotoxic injury in an age-dependent manner (Hansson et al., 1999; Hickey and Morton, 2000), possibly attributable to differences in calcium handling (Hansson et al., 2001b). A similar mechanism may be operating to preserve survival of newborn neurons in the R6/2 mouse.
Basal SVZ proliferation was the same between genotypes, consistent with previous reports (Gil et al., 2004, 2005; Lazic et al., 2004). We show for the first time that neuroblast migration and numbers of striatal neuroblasts are also the same between genotypes.
That neurogenesis in R6/2 mice was impaired in the DG but intact in the SVZ is interesting. Environmental enrichment increases neurogenesis in the DG but not SVZ. This is partly mediated by BDNF (Brown et al., 2003a; Spires et al., 2004b), and BDNF levels are higher in the hippocampus compared with striatum (Katoh-Semba et al., 1997). Thus, the DG may be more vulnerable to deficits of BDNF, and so neurogenesis is impaired in this region rather than the SVZ. Furthermore, there are clear anatomical differences between the DG and SVZ, and there may be intrinsic differences in the potency of stem-like cells (Seaberg and van der Kooy, 2002).
Neurogenesis in the DG of HD patients has not yet been studied, but SVZ proliferation is increased in HD patients (Curtis et al., 2003, 2005). This is in contrast with data presented here. The reason for the discrepancy between humans and mouse models is unknown. One possibility is that the stimulus for SVZ proliferation in humans may be neurodegeneration in the adjacent caudate nucleus, which is minimal in the R6/2 mouse (Mangiarini et al., 1996). It may also be that intrinsic species differences, such as anatomy (Sanai et al., 2004) and neurotransmitter and growth factor milieu (Kish et al., 1987; Reynolds and Pearson, 1987; Ferrer et al., 2000; Spires et al., 2004), account for differences in neurogenesis. Finally, it is possible that SVZ proliferation may actually increase in mouse models, but their lifespan is so short that they die before this proliferation can be detected.
Neurogenesis in WT mice was upregulated in response to stimulation. It is likely that seizures (rather than a direct pharmacological action of KA itself) are responsible for increasing neurogenesis in our study, because KA does not increase NPC proliferation in the DG in vitro (C. Watts, personal communication). Neurogenesis was not upregulated in R6/2 mice, although interpretation of the QA-induced lesion data are complex given that R6/2 mice had smaller lesions. This suggests that these mice have a lower capacity to mobilize the neurogenic process in response to injury, potentially reducing their capacity for endogenous brain repair.
There was no difference in vitro between NPC from adult R6/2 and WT mice. Any differences in neurogenesis between R6/2 and WT mice are therefore likely to be attributable to intrinsic differences undetectable by the neurosphere assay, differences in the response of the NPC to the local environment, or differences in the in vivo environment. We found no difference in microglial density between genotypes, suggesting that microglial density does not account for the abnormalities in the in vivo environment in R6/2 mice that might be responsible for impaired neurogenesis.
There are many possible mechanisms for the reduction in DG neurogenesis in transgenic HD mice, including a reduction in BDNF (Zuccato et al., 2001; Spires et al., 2004b), alterations in neurotransmitter levels such as serotonin (Gould, 1999; Reynolds et al., 1999), and possibly increased glucocorticoids (Cameron and Gould, 1994). Abnormalities of glia may also account for changes in neurogenesis (Song et al., 2002), and we are currently investigating how the function of microglia and astrocytes might impact neurogenesis in R6/2 mice.
Whether or not reduced DG neurogenesis in R6/2 mice is a primary or secondary phenomenon, it is likely that changes in DG neurogenesis will impact cognition and mood (Kempermann, 2002) in HD. Furthermore, the possible lack of upregulation of striatal neurogenesis in R6/2 mice may contribute to the slow neurodegeneration in the striatum, which is characteristic of HD.
We show here that, in a model of chronic neuronal dysfunction, there are abnormalities of neurogenesis that may relate to the microenvironment rather than to the NPC itself. Furthermore, the observation that newborn neurons in R6/2 mice are morphologically abnormal raises the possibility that they are functionally abnormal, and simply increasing endogenous NPC proliferation may not be sufficient to provide therapeutic benefit. This has implications not only for pathogenesis and therapy of human neurodegenerative disease but also for all cell-based therapies used in repair of the adult brain. Thus, for example, in the case of neural transplantation, it may be necessary to alter the microenvironment into which cells are being transplanted; manipulating the microenvironmental milieu (Morton et al., 2005) may be a useful adjuvant therapy.
Footnotes
W.P. is supported by the Medical Research Council, the Raymond and Beverly Sackler Foundation, and the Ipsen Fund. A.J.M. is supported by the Hereditary Disease Foundation and HighQ. R.A.B. is supported by the Medical Research Council and Hereditary Disease Foundation. We thank P. Tyers, Y. Hooks, S. E. Lazic, and Dr. S. Ball for technical advice and assistance.
Correspondence should be addressed to Dr. Wendy Phillips, Cambridge Centre for Brain Repair, E. D. Adrian Building, Forvie Site, Cambridge CB2 2PY, UK. E-mail: wp212@cam.ac.uk.
DOI:10.1523/JNEUROSCI.3796-05.2005
Copyright © 2005 Society for Neuroscience 0270-6474/05/2511564-13$15.00/0
References
- Abrous DN, Koehl M, Le Moal M (2005) Adult neurogenesis: from precursors to network and physiology. Physiol Rev 85: 523–569. [DOI] [PubMed] [Google Scholar]
- Altman J, Das GD (1965) Autoradiographic and histological evidence of postnatal hippocampal neurogenesis in rats. J Comp Neurol 124: 319–335. [DOI] [PubMed] [Google Scholar]
- Armstrong RJ, Barker RA (2001) Neurodegeneration: a failure of neuroregeneration? Lancet 358: 1174–1176. [DOI] [PubMed] [Google Scholar]
- Arvidsson A, Collin T, Kirik D, Kokaia Z, Lindvall O (2002) Neuronal replacement from endogenous precursors in the adult brain after stroke. Nat Med 8: 963–970. [DOI] [PubMed] [Google Scholar]
- Bogdanov MB, Ferrante RJ, Kuemmerle S, Klivenyi P, Beal MF (1998) Increased vulnerability to 3-nitropropionic acid in an animal model of Huntington's disease. J Neurochem 71: 2642–2644. [DOI] [PubMed] [Google Scholar]
- Brown J, Cooper-Kuhn CM, Kempermann G, Van Praag H, Winkler J, Gage FH, Kuhn HG (2003a) Enriched environment and physical activity stimulate hippocampal but not olfactory bulb neurogenesis. Eur J Neurosci 17: 2042–2046. [DOI] [PubMed] [Google Scholar]
- Brown JP, Couillard-Despres S, Cooper-Kuhn CM, Winkler J, Aigner L, Kuhn HG (2003b) Transient expression of doublecortin during adult neurogenesis. J Comp Neurol 467: 1–10. [DOI] [PubMed] [Google Scholar]
- Cameron HA, Gould E (1994) Adult neurogenesis is regulated by adrenal steroids in the dentate gyrus. Neuroscience 61: 203–209. [DOI] [PubMed] [Google Scholar]
- Carleton A, Petreanu LT, Lansford R, Alvarez-Buylla A, Lledo PM (2003) Becoming a new neuron in the adult olfactory bulb. Nat Neurosci 6: 507–518. [DOI] [PubMed] [Google Scholar]
- Chu-LaGraff Q, Kang X, Messer A (2001) Expression of the Huntington's disease transgene in neural stem cell cultures from R6/2 transgenic mice. Brain Res Bull 56: 307–312. [DOI] [PubMed] [Google Scholar]
- Couillard-Despres S, Winner B, Schaubeck S, Aigner R, Vroemen M, Weidner N, Bogdahn U, Winkler J, Kuhn HG, Aigner L (2005) Doublecortin expression levels in adult brain reflect neurogenesis. Eur J Neurosci 21: 1–14. [DOI] [PubMed] [Google Scholar]
- Curtis MA, Penney EB, Pearson AG, Roon-Mom WM, Butterworth NJ, Dragunow M, Connor B, Faull RL (2003) Increased cell proliferation and neurogenesis in the adult human Huntington's disease brain. Proc Natl Acad Sci USA 100: 9023–9027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MA, Penney EB, Pearson J, Dragunow M, Connor B, Faull RL (2005) The distribution of progenitor cells in the subependymal layer of the lateral ventricle in the normal and Huntington's disease human brain. Neuroscience 132: 777–788. [DOI] [PubMed] [Google Scholar]
- Duckworth EA, Butler TL, De Mesquita D, Collier SN, Collier L, Pennypacker KR (2005) Temporary focal ischemia in the mouse: technical aspects and patterns of Fluoro-Jade evident neurodegeneration. Brain Res 1042: 29–36. [DOI] [PubMed] [Google Scholar]
- Ekdahl CT, Mohapel P, Elmer E, Lindvall O (2001) Caspase inhibitors increase short-term survival of progenitor-cell progeny in the adult rat dentate gyrus following status epilepticus. Eur J Neurosci 14: 937–945. [DOI] [PubMed] [Google Scholar]
- Ekdahl CT, Claasen JH, Bonde S, Kokaia Z, Lindvall O (2003) Inflammation is detrimental for neurogenesis in adult brain. Proc Natl Acad Sci USA 100: 13632–13637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson PS, Perfilieva E, Bjork-Eriksson T, Alborn AM, Nordborg C, Peterson DA, Gage FH (1998) Neurogenesis in the adult human hippocampus. Nat Med 4: 1313–1317. [DOI] [PubMed] [Google Scholar]
- Fallon J, Reid S, Kinyamu R, Opole I, Opole R, Baratta J, Korc M, Endo TL, Duong A, Nguyen G, Karkehabadhi M, Twardzik D, Patel S, Loughlin S (2000) In vivo induction of massive proliferation, directed migration, and differentiation of neural cells in the adult mammalian brain. Proc Natl Acad Sci USA 97: 14686–14691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrante RJ, Kowall NW, Richardson EP (1991) Proliferative and degenerative changes in striatal spiny neurons in Huntington's disease: a combined study using the section-Golgi method and calbindin D28 immunocytochemistry. J Neurosci 12: 3877–3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer I, Goutane E, Marin C, Rey MJ, Ribalta T (2000) Brain-derived neurotrophic factor in Huntington disease. Brain Res 866: 257–261. [DOI] [PubMed] [Google Scholar]
- Frederiksen K, McKay RD (1988) Proliferation and differentiation of rat neuroepithelial precursor cells in vivo J Neurosci 8: 1144–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gage FH (2000) Mammalian neural stem cells. Science 287: 1433–1438. [DOI] [PubMed] [Google Scholar]
- Gheusi G, Cremer H, McLean H, Chazal G, Vincent JD, Lledo PM (2000) Importance of newly generated neurons in the adult olfactory bulb for odor discrimination. Proc Natl Acad Sci USA 97: 1823–1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson H, Reim K, Brose N, Morton AJ, Jones S (2005) A similar impairment in CA3 mossy fibre LTP in the R6/2 mouse model of Huntington's disease and in the complexin II knockout mouse. Eur J Neurosci 22: 1701–1712. [DOI] [PubMed] [Google Scholar]
- Gil JM, Leist M, Popovic N, Brundin P, Petersen A (2004) Asialoerythropoetin is not effective in the R6/2 line of Huntington's disease mice. BMC Neurosci 5: 17–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil JM, Mohapel P, Araujo IM, Popovic N, Li J, Brundin P, Petersen A (2005) Reduced hippocampal neurogenesis in R6/2 transgenic Huntington's disease mice. Neurobiol Dis 20: 744–751. [DOI] [PubMed] [Google Scholar]
- Gleeson JG, Lin PT, Flanagan LA, Walsh CA (1999) Doublecortin is a microtubule-associated protein and is expressed widely by migrating neurons. Neuron 23: 257–271. [DOI] [PubMed] [Google Scholar]
- Gould E (1999) Serotonin and hippocampal neurogenesis. Neuropsychopharmacology 21: 46S–51S. [DOI] [PubMed] [Google Scholar]
- Gundersen HJ, Jensen EB, Kieu K, Nielsen J (1999) The efficiency of systematic sampling in stereology—reconsidered. J Microsc 193: 199–211. [DOI] [PubMed] [Google Scholar]
- Hansson O, Petersen A, Leist M, Nicotera P, Castilho RF, Brundin P (1999) Transgenic mice expressing a Huntington's disease mutation are resistant to quinolinic acid-induced striatal excitotoxicity. Proc Natl Acad Sci USA 96: 8727–8732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson O, Castilho RF, Korhonen L, Lindholm D, Bates GP, Brundin P (2001a) Partial resistance to malonate-induced striatal cell death in transgenic mouse models of Huntington's disease is dependent on age and CAG repeat length. J Neurochem 78: 694–703. [DOI] [PubMed] [Google Scholar]
- Hansson O, Guatteo E, Mercuri NB, Bernardi G, Li XJ, Castilho RF, Brundin P (2001b) Resistance to NMDA toxicity correlates with appearance of nuclear inclusions, behavioural deficits and changes in calcium homeostasis in mice transgenic for exon 1 of the huntington gene. Eur J Neurosci 14: 1492–1504. [DOI] [PubMed] [Google Scholar]
- Harjes P, Wanker EE (2003) The hunt for huntingtin function: interaction partners tell many different stories. Trends Biochem Sci 28: 425–433. [DOI] [PubMed] [Google Scholar]
- Hickey MA, Morton AJ (2000) Mice transgenic for the Huntington's disease mutation are resistant to chronic 3-nitropropionic acid-induced striatal toxicity. J Neurochem 75: 2163–2171. [DOI] [PubMed] [Google Scholar]
- Iannicola C, Moreno S, Olivero S, Nardacci R, Ciofi-Luzzatto A, Piacentini M (2000) Early alterations in gene expression and cell morphology in a mouse model of Huntington's disease. J Neurochem 75: 830–839. [DOI] [PubMed] [Google Scholar]
- Imura T, Kornblum HI, Sofroniew MV (2003) The predominant neural stem cell isolated from postnatal and adult forebrain but not early embryonic forebrain expresses GFAP. J Neurosci 23: 2824–2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain M, Armstrong RJ, Tyres P, Barker RA, Rosser AE (2003) GABAergic immunoreactivity is predominant in neurons derived from expanded human neural precursor cells in vitro. Exp Neurol 182: 113–123. [DOI] [PubMed] [Google Scholar]
- Jarabek BR, Yasuda RP, Wolfe BB (2004) Regulation of proteins affecting NMDA receptor-induced excitotoxicity in a Huntington's mouse model. Brain 127: 505–516. [DOI] [PubMed] [Google Scholar]
- Jin K, Minami M, Lan JQ, Mao XO, Batteur S, Simon RP, Greenberg DA (2001) Neurogenesis in dentate subgranular zone and rostral subventricular zone after focal cerebral ischemia in the rat. Proc Natl Acad Sci USA 98: 4710–4715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan MS, Hinds JW (1977) Neurogenesis in the adult rat: electron microscopic analysis of light radioautographs. Science 197: 1092–1094. [DOI] [PubMed] [Google Scholar]
- Katoh-Semba R, Takeuchi IK, Semba R, Kato K (1997) Distribution of brain-derived neurotrophic factor in rats and its changes with development in the brain. J Neurochem 69: 34–42. [DOI] [PubMed] [Google Scholar]
- Kempermann G (2002) Regulation of adult hippocampal neurogenesis— implications for novel theories of major depression. Bipolar Disord 4: 17–33. [DOI] [PubMed] [Google Scholar]
- Kempermann G, Kuhn HG, Gage FH (1997) More hippocampal neurons in adult mice living in an enriched environment. Nature 386: 493–495. [DOI] [PubMed] [Google Scholar]
- Kempermann G, Jessberger S, Steiner B, Kronenberg G (2004) Milestones of neuronal development in the adult hippocampus. Trends Neurosci 27: 447–452. [DOI] [PubMed] [Google Scholar]
- Kish SJ, Shannak K, Hornykiewicz O (1987) Elevated serotonin and reduced dopamine in subregionally divided Huntington's disease striatum. Ann Neurol 22: 386–389. [DOI] [PubMed] [Google Scholar]
- Koelle GB (1955) The histochemical identification of cholinesterase in cholinergic, adrenergic and sensory neurons. J Pharmacol Exp Ther 114: 167–184. [PubMed] [Google Scholar]
- Kornack DR, Rakic P (2001) Cell proliferation without neurogenesis in adult primate neocortex. Science 294: 2127–2130. [DOI] [PubMed] [Google Scholar]
- Kuhn HG, Dickinson-Anson H, Gage FH (1996) Neurogenesis in the dentate gyrus of the adult rat: age-related decrease of neuronal progenitor proliferation. J Neurosci 16: 2027–2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn HG, Winkler J, Kempermann G, Thal LJ, Gage FH (1997) Epidermal growth factor and fibroblast growth factor-2 have different effects on neuronal progenitors in the adult rat brain. J Neurosci 17: 5820–5829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence AD, Hodges JR, Rosser AE, Kershaw A, ffrench-Constant C, Rubinsztein DC, Robbins TW, Sahakian BJ (1998) Evidence for specific cognitive deficits in preclinical Huntington's disease. Brain 121: 1329–1341. [DOI] [PubMed] [Google Scholar]
- Lazic SE, Grote H, Armstrong RJ, Blakemore C, Hannan AJ, van Dellen A, Barker RA (2004) Decreased hippocampal cell proliferation in R6/1 Huntington's mice. NeuroReport 15: 811–813. [DOI] [PubMed] [Google Scholar]
- Lendahl U, Zimmerman LB, McKay RD (1990) CNS stem cells express a new class of intermediate filament protein. Cell 60: 585–595. [DOI] [PubMed] [Google Scholar]
- Ma L, Morton AJ, Nicholson LF (2003) Microglia density decreases with age in a mouse model of Huntington's disease. Glia 43: 274–280. [DOI] [PubMed] [Google Scholar]
- MacGibbon GA, Hamilton LC, Crocker SF, Costain WJ, Murphy KM, Robertson HA, Denovan-Wright EM (2002) Immediate-early gene response to methamphetamine, haloperidol, and quinolinic acid is not impaired in Huntington's disease transgenic mice. J Neurosci Res 67: 372–378. [DOI] [PubMed] [Google Scholar]
- Magavi SS, Leavitt BR, Macklis JD (2000) Induction of neurogenesis in the neocortex of adult mice. Nature 405: 951–955. [DOI] [PubMed] [Google Scholar]
- Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP (1996) Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 87: 493–506. [DOI] [PubMed] [Google Scholar]
- Mohapel P, Ekdahl CT, Lindvall O (2004) Status epilepticus severity influences the long-term outcome of neurogenesis in the adult dentate gyrus. Neurobiol Dis 15: 196–205. [DOI] [PubMed] [Google Scholar]
- Monje ML, Mizumatsu S, Fike JR, Palmer TD (2002) Irradiation induces neural precursor-cell dysfunction. Nat Med 8: 955–962. [DOI] [PubMed] [Google Scholar]
- Morton AJ, Leavens W (2000) Mice transgenic for the human Huntington's disease mutation have reduced sensitivity to kainic acid toxicity. Brain Res Bull 52: 51–59. [DOI] [PubMed] [Google Scholar]
- Morton AJ, Hunt M, Hodges AK, Lewis PD, Redfern AJ, Dunnett SB, Jones L (2005) A combination drug therapy improves cognition and reverses gene expression changes in a mouse model of Huntington's disease. Eur J Neurosci 21: 855–870. [DOI] [PubMed] [Google Scholar]
- Murphy KP, Carter RJ, Lione LA, Mangiarini L, Mahal A, Bates GP, Dunnett SB, Morton AJ (2000) Abnormal synaptic plasticity and impaired spatial cognition in mice transgenic for exon 1 of the human Huntington's disease mutation. J Neurosci 20: 5115–5123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nacher J, Crespo C, McEwen BS (2001) Doublecortin expression in the adult rat telencephalon. Eur J Neurosci 14: 629–644. [DOI] [PubMed] [Google Scholar]
- Nakatomi H, Kuriu T, Okabe S, Yamamoto S, Hatano O, Kawahara N, Tamura A, Kirino T, Nakafuku M (2002) Regeneration of hippocampal pyramidal neurons after ischemic brain injury by recruitment of endogenous neural progenitors. Cell 110: 429–441. [DOI] [PubMed] [Google Scholar]
- O'Rourke NA, Sullivan DP, Kaznowski CE, Jacobs AA, McConnell SK (1995) Tangential migration of neurons in the developing cerebral cortex. Development 121: 2165–2176. [DOI] [PubMed] [Google Scholar]
- Palmer TD, Markakis EA, Willhoite AR, Safar F, Gage FH (1999) Fibroblast growth factor-2 activates a latent neurogenic program in neural stem cells from diverse regions of the adult CNS. J Neurosci 19: 8487–8497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer TD, Schwartz PH, Taupin P, Kaspar B, Stein SA, Gage FH (2001) Cell culture. Progenitor cells from human brain after death. Nature 411: 42–43. [DOI] [PubMed] [Google Scholar]
- Parent JM, Yu TW, Leibowitz RT, Geschwind DH, Sloviter RS, Lowenstein DH (1997) Dentate granule cell neurogenesis is increased by seizures and contributes to aberrant network reorganization in the adult rat hippocampus. J Neurosci 17: 3727–3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent JM, Vexler ZS, Gong C, Derugin N, Ferriero DM (2002) Rat forebrain neurogenesis and striatal neuron replacement after focal stroke. Ann Neurol 52: 802–813. [DOI] [PubMed] [Google Scholar]
- Petersen A, Hansson O, Puschban Z, Sapp E, Romero N, Castilho RF, Sulzer D, Rice M, DiFiglia M, Przedborski S, Brundin P (2001) Mice transgenic for exon 1 of the Huntington's disease gene display reduced striatal sensitivity to neurotoxicity induced by dopamine and 6-hydroxydopamine. Eur J Neurosci 14: 1425–1435. [DOI] [PubMed] [Google Scholar]
- Petersen A, Chase K, Puschban Z, DiFiglia M, Brundin P, Aronin N (2002) Maintenance of susceptibility to neurodegeneration following intrastriatal injections of quinolinic acid in a new transgenic mouse model of Huntington's disease. Exp Neurol 175: 297–300. [DOI] [PubMed] [Google Scholar]
- Prickaerts J, Koopmans G, Blokland A, Scheepens A (2004) Learning and adult neurogenesis: survival with or without proliferation? Neurobiol Learn Mem 81: 1–11. [DOI] [PubMed] [Google Scholar]
- Racine RJ, Gartner JG, Burnham WM (1972) Epileptiform activity and neural plasticity in limbic structures. Brain Res 47: 262–268. [DOI] [PubMed] [Google Scholar]
- Rao MS, Shetty AK (2004) Efficacy of doublecortin as a marker to analyse the absolute number and dendritic growth of newly generated neurons in the adult dentate gyrus. Eur J Neurosci 19: 234–246. [DOI] [PubMed] [Google Scholar]
- Reynolds GP, Pearson SJ (1987) Decreased glutamic acid and increased 5-hydroxytryptamine in Huntington's disease brain. Neurosci Lett 78: 233–238. [DOI] [PubMed] [Google Scholar]
- Reynolds GP, Dalton CF, Tillery CL, Mangiarini L, Davies SW, Bates G (1999) Brain neurotransmitter deficits in mice transgenic for the Huntington's disease mutation. J Neurochem 72: 1773–1776. [DOI] [PubMed] [Google Scholar]
- Ribchester RR, Thomson D, Wood NI, Hinks T, Gillingwater TH, Wishart TW, Court FA, Morton AJ (2004) Progressive abnormalities in skeletal muscle and neuromuscular junctions of transgenic mice expressing the Huntington's disease mutation. Eur J Neurosci 20: 3092–3114. [DOI] [PubMed] [Google Scholar]
- Rosas HD, Koroshetz WJ, Chen YI, Skeuse C, Vangel M, Cudkowicz ME, Caplan K, Marek K, Seidman LJ, Makris N, Jenkins BG, Goldstein JM (2003) Evidence for more widespread cerebral pathology in early HD: an MRI-based morphometric analysis. Neurology 27: 1615–1620. [DOI] [PubMed] [Google Scholar]
- Roy NS, Wang S, Jiang L, Kang J, Benraiss A, Harrison-Restelli C, Fraser RA, Couldwell WT, Kawaguchi A, Okano H, Nedergaard M, Goldman SA (2000) In vitro neurogenesis by progenitor cells isolated from the adult human hippocampus. Nat Med 6: 271–277. [DOI] [PubMed] [Google Scholar]
- Sanai N, Tramontin AD, Quinones-Hinojosa A, Barbaro NM, Gupta N, Kunwar S, Lawton MT, McDermott MW, Parsa AT, Manuel-Garcia Verdugo J, Berger MS, Alvarez-Buylla A (2004) Unique astrocyte ribbon in adult human brain contains neural stem cells but lacks chain migration. Nature 427: 740–744. [DOI] [PubMed] [Google Scholar]
- Sapp E, Kegel KB, Aronin N, Hashikawa T, Uchiyama Y, Tohyama K, Bhide PG, Vonsattel JP, DiFiglia M (2001) Early and progressive accumulation of reactive microglia in the Huntington disease brain. J Neuropathol Exp Neurol 60: 161–172. [DOI] [PubMed] [Google Scholar]
- Sathanoori M, Dias BG, Nair AR, Banerjee SB, Tole S, Vaidya VA (2004) Differential regulation of multiple brain-derived neurotrophic factor transcripts in the postnatal and adult rat hippocampus during development, and in response to kainate administration. Brain Res Mol Brain Res 130: 170–177. [DOI] [PubMed] [Google Scholar]
- Schauwecker PE (2002) Modulation of cell death by mouse genotype: differential vulnerability to excitatory amino acid-induced lesions. Exp Neurol 178: 219–235. [DOI] [PubMed] [Google Scholar]
- Schmidt-Hieber C, Jonas P, Bischofberger J (2004) Enhanced synaptic plasticity in newly generated granule cells of the adult hippocampus. Nature 429: 184–187. [DOI] [PubMed] [Google Scholar]
- Schmued LC, Hopkins KJ (2000) Fluoro-Jade B: a high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res 874: 123–130. [DOI] [PubMed] [Google Scholar]
- Scott BW, Wang S, Burnham WM, De Boni U, Wojtowicz JM (1998) Kindling-induced neurogenesis in the dentate gyrus of the rat. Neurosci Lett 248: 73–76. [DOI] [PubMed] [Google Scholar]
- Seaberg RM, van der Kooy D (2002) Adult rodent neurogenic regions: the ventricular subependyma contains neural stem cells, but the dentate gyrus contains restricted progenitors. J Neurosci 22: 1784–1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaberg RM, Smukler SR, van der KD (2005) Intrinsic differences distinguish transiently neurogenic progenitors from neural stem cells in the early postnatal brain. Dev Biol 278: 71–85. [DOI] [PubMed] [Google Scholar]
- Sen A, Kallos MS, Behie LA (2004) New tissue dissociation protocol for scaled-up production of neural stem cells in suspension bioreactors. Tissue Eng 10: 904–913. [DOI] [PubMed] [Google Scholar]
- Shors TJ, Miesegaes G, Beylin A, Zhao M, Rydel T, Gould E (2001) Neurogenesis in the adult is involved in the formation of trace memories. Nature 410: 372–376. [DOI] [PubMed] [Google Scholar]
- Shors TJ, Townsend DA, Zhao M, Kozorovitskiy Y, Gould E (2002) Neurogenesis may relate to some but not all types of hippocampal-dependent learning. Hippocampus 12: 578–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song HJ, Stevens CF, Gage FH (2002) Astroglia induce neurogenesis from adult neural stem cells. Nature 417: 39–44. [DOI] [PubMed] [Google Scholar]
- Spires TL, Grote HE, Garry S, Cordery PM, Van Dellen A, Blakemore C, Hannan AJ (2004a) Dendritic spine pathology and deficits in experience-dependent dendritic plasticity in R6/1 Huntington's disease transgenic mice. Eur J Neurosci 10: 2799–2807. [DOI] [PubMed] [Google Scholar]
- Spires TL, Grote HE, Varshney NK, Cordery PM, Van Dellen A, Blakemore C, Hannan AJ (2004b) Environmental enrichment rescues protein deficits in a mouse model of Huntington's disease, indicating a possible disease mechanism. J Neurosci 24: 2270–2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoll G, Jander S, Schroeter M (1998) Inflammation and glial responses in ischemic brain lesions. Prog Neurobiol 56: 149–171. [DOI] [PubMed] [Google Scholar]
- Takahashi J, Palmer TD, Gage FH (1999) Retinoic acid and neurotrophins collaborate to regulate neurogenesis in adult-derived neural stem cell cultures. J Neurobiol 38: 65–81. [PubMed] [Google Scholar]
- Tanapat P, Hastings NB, Reeves AJ, Gould E (1999) Estrogen stimulates a transient increase in the number of new neurons in the dentate gyrus of the adult female rat. J Neurosci 19: 5792–5801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaidya VA, Siuciak JA, Du F, Duman RS (1999) Hippocampal mossy fiber sprouting induced by chronic electroconvulsive seizures. Neuroscience 89: 157–166. [DOI] [PubMed] [Google Scholar]
- van Praag H, Schinder AF, Christie BR, Toni N, Palmer TD, Gage FH (2002) Functional neurogenesis in the adult hippocampus. Nature 415: 1030–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP Jr (1985) Neuropathological classification of Huntington's disease. J Neuropathol Exp Neurol 44: 559–577. [DOI] [PubMed] [Google Scholar]
- Wachs FP, Couillard-Despres S, Engelhardt M, Wilhelm D, Ploetz S, Vroemen M, Kaesbauer J, Uyanik G, Klucken J, Karl C, Tebbing J, Svendsen C, Weidner N, Kuhn HG, Winkler J, Aigner L (2003) High efficacy of clonal growth and expansion of adult neural stem cells. Lab Invest 83: 949–962. [DOI] [PubMed] [Google Scholar]
- Zeron MM, Hansson O, Chen N, Wellington CL, Leavitt BR, Brundin P, Hayden MR, Raymond LA (2002) Increased sensitivity to N-methyl-d-aspartate receptor-mediated excitotoxicity in a mouse model of Huntington's disease. Neuron 33: 849–860. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Ciammola A, Rigamonti D, Leavitt BR, Goffredo D, Conti L, MacDonald ME, Friedlander RM, Silani V, Hayden MR, Timmusk T, Sipione S, Cattaneo E (2001) Loss of huntingtin-mediated BDNF gene transcription in Huntington's disease. Science 293: 493–498. [DOI] [PubMed] [Google Scholar]