

ABBREVIATIONS
- AUC
area under the curve
- BIA
budget impact analysis
- CBER
Center for Biologics Evaluation and Research
- CE
cost‐effectiveness
- CEA
cost‐effectiveness analysis
- DOD
Department of Defense
- EID(s)
emerging infectious disease(s)
- ID
individual donation
- OBRR
Office of Blood Research and Review
- PI
pathogen inactivation
- PR
pathogen reduced
- ROS
reactive oxygen species
- QALY
quality‐adjusted life‐year
- PRT(s)
pathogen reduction technology(‐ies)
- TA‐GVHD
transfusion‐associated graft‐versus‐host disease
- TT
transfusion transmission
- TTI(s)
transfusion‐transmitted infection(s)
- WB
whole blood
- WNV
West Nile virus
- ZIKV
Zika virus
On November 29, 2018, experts in the field of infectious diseases, pathogen reduction technologies (PRTs) and other participants from blood centers, academia, and industry gathered at the Food and Drug Administration (FDA) White Oak Campus in Silver Spring, Maryland, for a 2‐day public workshop entitled “Pathogen Reduction Technologies for Blood Safety.” The workshop opened with welcome remarks from Dr. Nicole Verdun, Director, Office of Blood Research and Review (OBRR), Center for Biologics Evaluation and Research (CBER), FDA, followed by introductory remarks from Dr. Peter Marks, Director, CBER, FDA. The first day of the workshop focused on blood‐borne infectious agents and their impact on blood safety, experiences of the American Red Cross, and other blood establishments in implementing FDA‐approved pathogen inactivation (PI) technology for plasma and platelets (PLTs) in the United States and novel PRTs under consideration for whole blood (WB) and red blood cells (RBCs).
The second day opened with welcome remarks from Dr. Chintamani Atreya, Associate Director for Research, OBRR, CBER, FDA. The focus was on emerging innovations relevant to PRTs and potential alternatives to PRTs. The workshop concluded with remarks on insights for future research and development in this area for blood and blood product safety from infectious agents.
A brief introduction of each session by the session moderator followed by a summary of the speaker presentation as submitted by the moderator and speaker are reported here.
SESSION 1: BLOOD‐BORNE INFECTIOUS AGENTS AND THEIR IMPACT ON BLOOD SAFETY
Introduction. Moderator—Simone Glynn, MD, MPH
The first session titled “Blood‐Borne Infectious Agents and Their Impact on Blood Safety” provides a state‐of‐the‐science overview of the risks to blood safety posed by infectious agents. Additionally, this session addresses the strategies used to mitigate these risks in the United States including the introduction of increasingly sensitive laboratory screening testing platforms and PRTs for PLTs and plasma products. In a first part, the session includes a general overview of the evolution of responses to established, emerging, and reemerging transfusion‐transmitted infectious diseases in the past 50 years. Further, it addresses the need for ongoing surveillance for and systematic responses to emerging infectious diseases (EIDs), optimally with sensitive metagenomics, multiplexed nucleic acid amplification technology (NAT) and serologic testing strategies in sentinel global donor populations. This is followed by a review of the major policy issues pertaining to the development and implementation of PRTs, which if successfully adopted will provide insurance against known and unknown pathogens that may enter the blood supply. It will be noted that these technologies, if applied to all blood components or WB, may allow for the relaxation of redundant donor laboratory screening, modified donor questioning and/or deferral, and simplified handling of postdonation information while preserving or enhancing the safety of the blood supply. The session ends with an overview of the current status of approved pathogen‐reduced (PR) PLT and plasma products in the United States with attention provided to their current effectiveness and safety profile. Major reasons for the slow adoption of the currently approved PR products in the United States are discussed including cost‐effectiveness (CE) considerations.
Risk to the blood safety from infectious agents—Michael Busch, MD
Speaker's summary: Blood donor screening began in the 1940s with testing for syphilis, followed in the early 1970s by testing for hepatitis B surface antigen. The discovery of human immunodeficiency virus (HIV), human T‐lymphotropic viruses, and hepatitis C virus (HCV) and introduction of progressively more sensitive serological assays targeting virus‐specific antibodies and antigens for these “classic” transfusion‐transmitted infections (TTIs) in the 1980s and 1990s was effective in interdicting the majority of infectious blood donations.1 Implementation of NAT screening for HIV, HCV, and hepatitis B virus (HBV) further reduced the residual risk of infectious window period donations, such that per‐unit risks are less than one in 1,000,000 in the United States (Fig. 1).2, 3, 4
Figure 1.
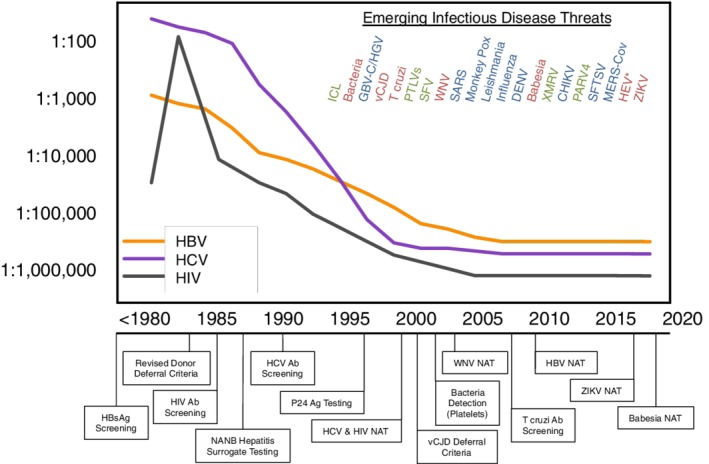
Per‐unit infectious risk for HBV, HCV, and HIV from 1980 to 2018 and emerging infectious agents that were investigated for potential TT over the past 25 years. Agents proven to be blood safety threats for which interventions were implemented are in red, agents that were established to be legitimate infectious agents but not TT or associated with diseases are in blue, and alleged threats that were determined to not cause human infections or due to artifacts are shown in green. No interventions for HEV have yet been implemented in the United States, but interventions have been implemented in some countries where HEV incidence is higher. ICL = idiopathic CD4+ T‐lymphocytopenia; HGV = hepatitis G virus; SFV = simian foamy virus; PTLV = primate T‐cell lymphotrophic viruses; SARS = severe acute respiratory syndrome; PARV‐4 = parvovirus type 4; SFTSV = severe fever with thrombocytopenia virus; MERS‐CoV = Middle Eastern respiratory syndrome coronavirus; Ab = antibody; Ag = antigen; HBsAg = hepatitis B surface antigen; vCJD = variant Creutzfeldt‐Jakob disease. This figure was updated from Perkins and Busch.1 [Color figure can be viewed at http://wileyonlinelibrary.com]
We now recognize that in addition to classic TTIs that establish chronic infection, agents that cause acute transient infections may also be TTI at significant rates if there are large epidemics or recurrent seasonal transmission.5, 6, 7 Salient examples of EIDs where interventions were implemented in the United States include nationwide screening of donors for Trypanosoma cruzi using a one‐time antibody testing strategy,8 NAT testing for West Nile virus (WNV)9, 10 and Zika virus (ZIKV),11, 12 and testing for Babesia microti in endemic regions.13, 14 Testing for bacterial contamination of PLT components was instituted in the 2000s to prevent septic transfusion reactions.15 Donor deferrals were also implemented to reduce other risks including variant Creutzfeldt‐Jakob disease and several other agents.16 Fig. 1 lists the EIDs for which interventions were implemented over the past two decades.
The emergence of EIDs has proven to be unpredictable, as is their risk to blood safety.5, 6, 7 EIDs of concern span all pathogen classes, with 60% being from zoonotic sources. The AABB has developed “Fact Sheets” (available at http://www.aabb.org/tm/eid/Pages/default.aspx) that provide information on agent classification, background on pathogenesis and clinical syndromes, modes of transmission [including vectors/reservoirs], likelihood of transfusion transmission [TT] and information on known transmission cases, feasibility and predicted success of interventions that could be used for donor qualification [questioning], tests available for donor screening, and efficacy of PRTs).
Proactive surveillance and research to evaluate responses to potential EID threats to blood safety have been adopted through collaborative initiatives of NIH, FDA, CDC, AABB, and blood research organizations.5, 6, 7 An unintended consequence has been the identification of agents found through viral discovery programs using metagenomics technologies that can theoretically be transmitted by transfusion but which, upon subsequent investigation, prove not to be (Fig. 1). The most striking example of this was xenotrophic murine leukemia–related virus, which was reported to be associated with prostate cancer and later chronic fatigue syndrome and to be present in the blood of asymptomatic blood donors.17 Intensive research consuming much time and money subsequently determined that xenotrophic murine leukemia–related virus did not affect humans and was a laboratory contaminant from cell lines that contained this murine virus.18 These experiences led to the US National Heart, Lung and Blood Institute (NHLBI) and FDA to convene workshops focused on proactive but rational and systematic responses to EIDs.6, 7 The Alliance of Blood Operators developed a “risk‐based decision‐making” process that includes formalized methods for quantifying risk and evaluating interventions.19
The 2015 to 2017 ZIKV pandemic is illustrative of the ongoing challenge of balancing timely and precautionary responses to emerging TTI threats with logistic and economic considerations. As the epidemic expanded in the Americas and the association of ZIKV with severe fetal outcomes emerged along with several cases of probable TT, the FDA mandated implementation of individual‐donation (ID)‐NAT or PRT of PLTs throughout the United States in 2016.11, 12 After 2 years of ID‐NAT with very low yield at an annual cost of $137 million, the FDA ZIKV policy was revised to allow for minipool NAT with ID‐NAT triggered during future epidemics, similar to successful screening for WNV. Nonetheless, ZIKV testing now represents a theoretical benefit at a high cost, precipitating consideration of regional screening policies and an urgent need to further define “tolerable risk” in the blood safety arena.20
Pathogen reduction: an overview of policy issues—Steve Kleinman, MD
Speaker's summary: Pathogen inactivation and/or reduction should be viewed in the context of shifting the blood safety paradigm from reactive to proactive thereby providing insurance against known and unknown pathogens that may enter the blood supply or are currently un(der)recognized. Based on the positive experience of PI for plasma derivatives (e.g., no HIV, HCV, or HBV transmission since 1987), it seems reasonable to apply this safety paradigm to blood components. Of note, a consensus conference held in Canada in 2007 issued recommendations in favor of rapid adoption of a PI technology even if it could not be applied to the full range of blood components. Despite these recommendations, PI technology for PLTs has been slow to be adopted in the United States. As described in Table 1, the reasons for this are many but it appears that the predominant impediment has been cost.
Table 1.
Reasons for slow adoption of PR PLTs in the United States
| Current safety of the volunteer blood supply |
| Success of surveillance and screening in dealing with emerging pathogens |
| Inability of current technologies to inactivate all agents (small nonencapsulated viruses, spores, high titers of virus, and prions) |
| Efficacy concerns |
| No single method to treat all components |
| Regulatory requirements |
| Cost |
When evaluating current blood safety risks and the need for additional interventions, it is important to understand that most often these risks are expressed on a per‐unit basis and represent the likelihood of the agent surviving during storage in a particular type of blood component (e.g., plasma, PLTs, or RBCs). The risk of TT of an agent and/or the occurrence of symptomatic or serious disease is likely to be less than this per‐unit exposure risk. On the other hand, these risk estimates are for single‐unit transfusions; risk will be higher (i.e., multiplied by the number of units) for the majority of patients who receive multiple units, either in single exposures or over a treatment course.
Assuming that therapeutic product efficacy is maintained and cost issues can be addressed, the goal is to have all blood components (RBCs, PLTs, plasma) or WB (before component separation) treated by PI—this could then allow for the relaxation of redundant donor laboratory screening, modification of donor questioning and/or deferral, simplified handling of postdonation information, and elimination of the need for irradiation of cellular components to prevent transfusion‐associated graft‐versus‐host disease (TA‐GVHD). Potential blood screening changes include eliminating syphilis, T. cruzi, cytomegalovirus (CMV), and Babesia testing; modifying the menu of HBV tests; eliminating off‐season WNV and ZIKV testing; and eliminating ID‐NAT. A fully PI‐treated blood supply would shape the response to threats from new enzyme immunoassays in that there would be less pressure to develop laboratory screening assays.
Additional important considerations in evaluating the role of PI in blood safety policy are that not all infectious agents are inactivated by PI technology (nonenveloped viruses and prions show variable resistance) and each manufacturer's process must be independently evaluated for quantitative levels of inactivation of numerous known pathogens as well as for therapeutic efficacy of the treated component and potential adverse effects in the recipient. The health care reimbursement system must also be able to accommodate the cost.
Current status of pathogen‐reduced platelets in the United States—Edward Snyder, MD, FACP, and Sara Rutter, MD
Speaker's summary: Pathogen‐reduced PLTs manufactured using a synthetic psoralen compound (amotosalen) are approved by the FDA for use by all patient demographics.21, 22, 23, 24 Currently this is the only PLT PR manufacturing system approved by the FDA in the United States. It requires ultraviolet (UV)‐A light activation of the psoralen photochemical to enable it to function as the inactivation agent.21, 22, 23, 24, 25 Approval is limited to single‐donor PLTs collected using either of two apheresis devices and stored in a PLT additive solution, PAS‐C, or in autologous donor plasma, depending on the apheresis device used for manufacture. Both PR products have a 5‐day shelf life at 20 to 24°C.23
The psoralen product currently is being evaluated in PIPER, a Phase IV postmarketing study. Other manufacturing systems are under varying degrees of development.26, 27 One of these systems uses a different light‐activated photochemical, riboflavin, and is currently being evaluated in the United States in a Phase III randomized clinical trial, MIPLATE.28 A third PR technology uses a shorter wavelength of UV light (UVC), as the sole mechanism of inactivation.27, 29, 30 It, too, is being evaluated in CAPTURE, a Phase III clinical trial in Europe.
Major benefits of PR PLTs include:
Despite FDA approval and the acknowledged benefits of the technology, however, the medical field has been slow to adopt and integrate PR technology into day‐to‐day hospital operations.
Reasons for this slow adoption include concerns over the possibility of:
Lower posttransfusion corrected count increment in PR PLTs versus conventional PLTs;
Lower hemostatic efficacy of PR PLTs versus conventional PLTs;
Higher risk of TA‐GVHD since gamma or x‐radiation of the PR PLTs is not recommended;
Toxicity from repeated administration of psoralen—in adults and especially in neonates and children;27, 30
An increase in the incidence of transfusion reactions due to PR PLTs;
Occurrence of skin rashes in neonates exposed to blue light therapy for hyperbilirubinemia;30
Increased cost associated with use of PR PLTs versus conventional PLTs.27
Many of these concerns have been addressed in published studies. Importantly, clinical reports have shown PR PLTs to be clinically acceptable.31, 32, 33, 34, 35, 36 FDA has provided draft guidance, but to date the Agency has stopped short of encouraging use of PR technology.21 Thus, it is left up to individual hospitals as to whether they adopt, or refrain from, use of PR PLT technology.
Currently the biggest ongoing credible blood‐borne pathogen threat to the nation's blood supply comes from bacterial contamination.37 While PR technology can address this, there are other options for mitigating risk of bacterial contamination of PLTs, including performing secondary bacterial cultures and point‐of‐release immunologic testing. However, should a new viral or other nonbacterial agent threaten the national blood supply, the time to ramp up adequate PR manufacturing infrastructure to meet such a nonbacterial threat would likely be substantial.38 More widespread adoption of PR technology now would do much to ameliorate the concern over this scenario.
Overall, the use of PR technology is slowly increasing, and data addressing many of the above‐listed concerns are being reported, at least in abstract form.31, 32, 33, 34, 35, 36 However, the lack of an extensive degree of published US data, especially for pediatric and transplant recipients, coupled with the absence of a strong FDA endorsement of the technology and the high cost of this technology, has hampered widespread acceptance of PR PLTs.30 The possibility of another blood‐borne threat to the safety of the national blood supply seems inevitable. How well we mitigate that threat may well depend on how these issues regarding PR blood products are resolved. It is critical that early adopters of PR technology in the United States publish their experience with utilization of PR PLTs for patient care, especially their pediatric experience.
Pathogen reduction technology for plasma in the United States—James P. AuBuchon, MD, FACP, FRCP (Edin)
Speaker's summary: Two methods of PR plasma are currently licensed and available in the United States, solvent/detergent (S/D)‐treated plasma (SD plasma; Octaplas, Octapharma) and amotosalen/UV‐treated plasma (Intercept plasma, Cerus Corporation). Plasma treated with riboflavin and UV (Mirasol plasma; Terumo BCT) is part of a similar system being developed for other components and is also included in this summary.
Each of the techniques results in a reduction of the content and/or activity of the pro‐ and anticoagulant proteins in plasma. In general, these reductions do not exceed 20% to 30%, and for many proteins the reduction is less than this. The most notable reductions are in fibrinogen and Factors (F)VIII and FV across all platforms; F IX and FXI, protein C, and large von Willebrand factor multimers with Mirasol; and protein S and antiplasmin with Octaplas.39, 40, 41, 42, 43, 44, 45 There have been few investigations of these treatments on complement components; in an analysis of Intercept plasma, C3a was found to be reduced.45 It has been noted that the reductions, while significant, usually resulted in contents within the reference range.39 The pooled nature of SD plasma greatly reduces the variability in content that can easily be demonstrated between different donors' plasma samples.46 Cryoprecipitate may be prepared from Intercept and Mirasol plasma units to meet the minimum content requirements, although the effect of the treatment is still evident.47, 48
In vitro analyses of the clotting system have generally demonstrated substantial retention of clinically relevant function.42, 44 As might be expected from the reduced contents noted, the resulting fibrin strands are thinner (with resulting increased clot density and reduced permeability) with longer lag time for formation or prolonged time to lysis.49
Multiple clinical trials have demonstrated expected outcomes with the use of these PR plasma samples. Prophylactic transfusion of Intercept plasma into congenitally deficient patients yielded expected increases in the deficient factor in circulation with anticipated half‐lives.39 Use of large volumes of Intercept plasma in plasma exchanges for thrombotic thrombocytopenic purpura or idiopathic thrombocytopenic purpura resulted in expected outcomes without generating adverse events or (new) coagulopathy.50, 51, 52 Use of large volumes (approx. 2 L) of Intercept plasma in liver transplantation yielded the same outcomes as with quarantine plasma.50, 51, 52, 53 The current formulation of Octaplas has not been reported to be associated with thrombotic events when used in large volumes as had been seen with the original version of SD plasma.54
A theoretical question has been raised whether the reduced content these components might place massive transfusion recipients at increased risk of inadequate hemostasis and death.55 One in vitro mixing study suggested that a 50% plasma replacement would be necessary before altering coagulation kinetics.56 Several large, historically controlled experiences with Intercept plasma in trauma situations, however, have failed to show any impact on the need for other blood components, time to discharge or mortality.32
These PR plasma samples have not been associated with increased adverse events after transfusion.57 Because of the pooled basis of SD plasma, it is believed to carry a reduced risk of transfusion‐related acute lung injury (TRALI) because of dilution of potentially offending antibodies. There have been no TRALI cases reported in more than 10 million Octaplas units transfused.58, 59 If the existing risk of TRALI is greater than one in 5000, removal of this risk in itself makes use of SD plasma cost‐effective.60 This pooling, however, does increase the risk of early and wide dissemination of a nascent nonenveloped virus.61
To date, there has been little uptake of PR plasma in the United States outside of Octaplas for patients with severe allergic reactions to single‐donor plasma. This is probably due to the perception of viral safety (and lack of bacterial contamination risk) in standard plasma and the increased cost of these newer plasmas. Widespread introduction likely will follow only after implementation of similar systems for PLTs and RBCs despite admonishments from a consensus conference and demonstration of the importance of plasma transmission of new pathogens.
SESSION 2: IMPLEMENTATION OF PRT FOR BLOOD PRODUCTS IN THE UNITED STATES
Introduction. Moderator—Willy Albert Flegel, MD
Four years have passed since approval of a PRT devices for PLTs and plasma products by the FDA. The five presentations of this session addressed the implementation of these devices in the United States, their impact on PLT quality, the availability of PRT plasma in the United States, and the health economic considerations.
Experiences from the nation's largest blood product supplier62 and the blood bank at the NIH Clinical Center63 were documented. Both reports stressed the relevance of strict volume and cell limits, not required without PRT, and their effect on collection procedures and failures. The approaches were almost diametrically opposed, reflecting the different donor settings: while the American Red Cross preferred small‐volume over large‐volume kits (2/3 vs. 1/3), the NIH Blood Bank exclusively used dual‐storage kits. The American Red Cross is boosting PRT PLTs to meet the steadily increasing demand. The NIH Blood Bank has transitioned to 100% PRT PLT production, which was well received by the attending physicians and nurses.
Quality variables are expected to change, as PRT affects all treated cells. The risks must be monitored and balanced while the technologies for PRT and PLT additive solution (PAS) continue to evolve.64 PRT plasma from individual donors, although FDA‐licensed devices are available, had not been introduced in patient care by the end of 2018. Similar to lyophilized plasma, which is not available from single‐donor sources,65 a PRT plasma alternative pooled from many donors does exist: S/D‐treated plasma66 has a history of worldwide use since 1992. Five randomized controlled trials showed no difference in efficacy, but trial sizes ranged from 49 to 293 patients for a total of only 552 patients. No TRALI has been reported from passively collected data, which may not reflect all incidence.
Cost‐effectiveness estimates for PRT PLTs and plasma were reported.67 These products are considered no less cost‐effective than other widely adopted interventions in the context of blood safety technologies. A budget gap is likely to remain until PRTs become available for WB or RBCs. Low‐ and middle‐income countries may require a mixed approach of needs assessment and targeted interventions.68 While PRT PLTs have been implemented nationwide in some countries69 in an effort to improve patient safety, reimbursement was noted as a key factor in the United States stalling the quicker implementation of PRT PLT and plasma transfusions.
Experience implementing pathogen reduction technology—David Angus Reeve, MBA, MHA
Speaker's summary: Red Cross implemented PI technology with the treatment of apheresis PLTs in March 2015. The program was Initiated in Puerto Rico under a clinical study. The organization initiated routine production of PR PLTs in July 2016; it will have 15 manufacturing sites producing pathogen inactivated products by early 2019.
Implementation challenges
The most significant challenge with during the initial experience was that a limited percentage of PLT collections were eligible for PI based on approved guard bands. There was also a goal to do no harm to the PLT supply due to the growing demand for single‐donor PLTs by converting triples to doubles or doubles to singles to qualify for PI. Finally, a licensed INTERCEPT kit for PAS triples does not yet exist.
The initial performance against guard bands was unsatisfactory; more than 30% apheresis PLTs produced in Red Cross are from triples leaving only doubles or singles to qualify. Most of the single‐ and double‐plateletpheresis units failed to natively meet PI input requirements because of targeted programing set points to maximize yield on collections devices and the default values for first‐time donors.
Overcoming implementation challenges
Red Cross initiated evaluation of mitigation strategies designed to increase percentage of units that qualify for PI. Volume reduction was deployed by removing product volume from a homogenous mixture of Amicus‐PAS apheresis PLTs meet Intercept guard bands. Red Cross developed and validated a software tool that aids staff in selecting options for volume reduction.
Additional mitigations were developed in advance of an INTERCEPT triple kit. Staff split triple collections into three individual storage containers before PRT and use single (small‐volume or large‐volume) INTERCEPT kits to treat products individually. Large‐volume doubles may be split into two small‐volume or large‐volume kits; the collections team adjusted Amicus settings to optimize storage volumes to 625 mL for doubles and 780 mL for triples, which included a minimum 10‐mL volume buffer and a minimum PLT yield of 3.4.
Results of mitigations
Approximately 65% of PLT products met the guard bands during the operational trial. Presplitting largely obviated the need for conducting volume reduction. The number of PR units currently labeled is less than 50% due to combination of demand, staffing, aggregates, and units exceeding 24 hours. Additional observations included a radical shift in type of PI kit used from predominantly dual‐storage kits to small‐volume kits; the use of large‐volume kits remained the same. Before implementation of mitigations, the split rate of PI products fell to 1.30. After implementation of mitigations the split rate of PI products increased to 2.10. Programming of Amicus devices was standardized, and collection volume increased; triples became eligible and products were not downgraded.
The labor required for PRT is greater than using the BacT/ALERT system with a single bottle. Time studies compared both processes. Demonstrated an 11.1% increase in the time required to complete tasks for unmitigated PI products as compared to the traditional process. There was a 22% increase in time required to complete tasks for mitigated PI products compared to traditional process. Productivity for the unmitigated PI process was poor because the volume of products eligible for treatment was low with batch sizes of two to four products. Productivity improved 52% after implementation of mitigation steps due to significant increase in products eligible for treatment with Increased batch sizes of eight to 12 products.
Conclusion
Pathogen reduction of 100% products is challenging but not impossible based on current guard bands. Mitigations required to meet guard bands are feasible but labor‐intensive and time‐consuming. Implementation of PRT will require adjustment of set points and collection variables on apheresis devices.
Implementation in a hospital‐based blood center and acceptance by hospital staff—Willy Albert Flegel, MD
Speaker's summary: The NIH Clinical Center at the National Institutes of Health is the nation's largest hospital devoted entirely to clinical research.63 Approximately 1600 studies are in progress, focusing on Phase I and II clinical trials. The Department of Transfusion Medicine is a full‐service blood bank providing blood donation, clinical apheresis, transfusion service, human leukocyte antigen (HLA) and infectious disease testing for the hospital, and cellular engineering70 in support of cell therapies.71
In 2016, we transfused 3930 PLT, 4561 RBC, 614 plasma, and 59 granulocyte products to 668 patients. All blood components are 25 Gy irradiated; RBC products are also leukoreduced72, 73 since 2009 and none is transfused older than 35 days74, 75 since 2014. We transitioned to PR PLTs (100% collected by apheresis) with PAS in January 2016.
Within 1 month of the device's approval by the FDA, the NIH Clinical Center decided in January 2015 to implement PRT76 and concurrently a PAS for all PLT products, finally bringing this long‐anticipated technology77, 78 to the bedside in the United States. A retrospective evaluation of 1007 successful collections during 6 months in early 2015 showed 99.7% of the collections that met any guard bands specified by PRT fell within the guard band of the PRT dual‐storage kit. We decided to exclusively use this kit and prepared to adjust the variables for approximately 5% of our collections to meet the guard band specifications. If successful, we anticipated a loss of less than 1% of collections due to failures to meet the guard band.
Once agreements with the device providers were signed (Intercept, Cerus Corporation; and Intersol, Fresenius Kabi), the initial tasks of the implementation team involved writing of validation plans and standard operating procedures; ordering, installing, and validating required equipment; and reconfiguring space to house equipment and fit the new work flow. Computer upgrades to accommodate changes were made by July. Training for PRT began in August and for PAS in September. Adjusting collection variables and validation of PRT and PAS processes continued for the next 3 months.
The first apheresis PLT product with PRT and PAS was released on January 11, 2016. A dual inventory of PLTs produced by the new or the previous processes lasted for less than 1 week, because we promptly transitioned all our PLT collections to PRT and PAS. The fine tuning of collection variables was critical and needs to be monitored and maintained continuously. This remains an ongoing task for our donor staff during each PLT collection. We are closing in on our original goal of less than 1% guard band failures (Table 2) while producing 100% PRT PLTs from all plateletpheresis collections at the NIH Clinical Center.
Table 2.
Loss of PLT collections due to guard band failure
| Collections (n) | 2016 | 2017 | 2018 | Total | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Jan* ‐Mar | Apr ‐ Jun | Jul ‐Sep | Oct ‐ Dec | Jan ‐ Mar | Apr ‐ Jun | Jul ‐Sep | Oct ‐ Dec | Jan ‐ Mar | Apr ‐ Jun | Jul ‐Sep | Oct ‐ Dec | ||
| Overall | 463 | 564 | 579 | 544 | 542 | 565 | 610 | 565 | 484 | 536 | 501 | 516 | 6468 |
| Outside guard bands | 34 | 26 | 28 | 16 | 19 | 20 | 16 | 14 | 14 | 10 | 5 | 7 | 209 |
| Failure rate (%)† | 7.3 | 4.6 | 4.8 | 2.9 | 3.5 | 3.5 | 2.6 | 2.5 | 2.9 | 1.9 | 1.0 | 1.4 | 3.2 |
| PLT retention‡ | 93% ± 1.2% (n = 828) | 94% ± 1.5% (n = 794) | 91% ± 1.4% (n = 966) | NA | |||||||||
beginning January 11, 2016. A collection with high platelet yield can be split and result in 2 transfusions, often in different patients.
Failure to meet the guard band of the Dual Storage kits: loss of successful platelet collections (%).
Retention rate of platelets after the PRT process (mean ± standard deviation; number of PRT platelet products tested for quality assurance to document the loss of platelets) n/a − not applicable.
To bridge shortages of supply or serve patients with rare HLA antibodies, we must import PLTs that remain almost invariably produced without PRT. Their irradiation with 25 Gy is required before they can enter the inventory for release to patients. We do not provide bacterial testing upon release and retired our previous precaution to prevent bacterial contamination: 4 to 5 mL sampling within 24 hours of collection and release into inventory after 12 hours of negative culture in single standard aerobic bottles (BACTEC, Becton, Dickinson and Co.) while monitoring the culture for 7 days. With PRT products, the elimination of irradiation (possible without variance notification since March 17, 2016)79 and bacterial culture resulted in substantial savings of consumables and handling, which were however exceeded by the costs of the new technologies and the increased hands‐on time for the production staff. Reports of transfusion reactions ranged from nine to 19 annually without discernible trend, certainly no increase from 2015 to 2018; no severe transfusion reactions occurred.
Education and notification are important for the acceptance by hospital staff. Within 2 weeks before the first new product was released for transfusion, we notified several external customers and provided the revised circular of information; there were no calls received. The prescribers of the NIH Clinical Center were informed through the office of the deputy director for clinical care with a focus on improved patient safety; there were a few calls. The nursing staff was informed through the nursing education leadership. We provided photos of current versus new bags, highlighted the lack of irradiation labels for new bags only, and explained the new electronic transfusion documentation. Some institutions are reluctant to introduce PRT PLTs for neonates, children, and pregnant women because of the theoretical risks associated with the toxicity of psoralen and its photo products, for which there has been no evidence to date in the doses applied. The few concerns raised at NIH, for example, why the cost increase was justified, abated once the ZIKV occurred in the continental United States and PRT PLTs were recognized as safe without any action needed.
Ongoing tasks are to improve the prompt availability of predonation PLT counts to better control the collections process and reduce guard band failures. We are optimizing reports and documentation in pursuit of a biologics license application and FDA‐CBER permission to eventually ship the biologics product across state lines. Many patients depend on PLT transfusions for their quality of life, and PLTs with PRT and PAS enabled us to enhance patient safety.80
Impact of pathogen reduction technology on platelet quality, count, and clinical implications—Dana V. Devine, PhD
Speaker's summary: Laboratory investigations of PI‐treated PLT components readily show that the approaches used generate changes to in vitro quality variables. This is an expected result of PI as the treatments themselves are not specific to pathogens and human cells are also targets. Processing alone causes a loss of PLTs as the steps involve transfer of the PLT concentrate to a treatment bag and transfer back to storage bag in some cases. PLT count reduction of 5% to 10% are typical, and production planning must accommodate this loss.
Treated PLTs show evidence of activation,81, 82, 83, 84 loss of various RNA species,85, 86 and an accelerated decrease in the pH with storage.84 PI treatment may also cause the release of PLT cytokines, including EGF, PDGF, and RANTES.87
Overall, the use of PI and/or reduction technologies results in PLT responses that can be interpreted as a reduction in PLT product quality in laboratory tests. It is unclear whether we are using the best tests to perform quality monitoring of PR PLTs as they are the same as those used for untreated PLTs.
Results of in vitro laboratory markers should not be presumed to parallel the clinical efficacy of the PI‐treated transfusion product. PR PLTs show a 15% to 25% decrease in survival and recovery in normal volunteers and, not surprisingly, this translates into a shorter intertransfusion intervals in patients receiving prophylactic transfusions with this product.88, 89 Unlike animal models, patients treated with PI PLTs have an increased risk of alloimmunization for reasons yet to be determined. Importantly, studies in hematologic oncology patients do not reveal an increased risk of bleeding with the use of PI‐treated PLTs, although the use of AS may be a confounding factor.90, 91, 92 These differences compared to transfusion with untreated PLTs are the tradeoff for increased safety.
There are still unknowns with respect to the use of PI‐treated PLTs in actively bleeding patients, particularly those with massive hemorrhage.55 Whether use of large volumes of PI‐treated products, including PI‐treated PLTs will amplify the negative effect of PI on PLT efficacy and overall hemostasis remains to be determined. Limited retrospective studies in surgical patients have not given cause for alarm with respect to adverse reactions or efficacy.32, 93
Important research questions remain to be answered: 1) Can we develop strategies to minimize damage to PLTs and RBCs including the use of better ASs and storage conditions after treatment? 2) What are the best quality control measures for PI PLTs? 3) Will the use of multiple types of PI‐treated products in trauma really create a problem? This needs to be assessed clinically; however, such studies are expensive and take a long time to complete. 4) Do we need to adapt transfusion practice to accommodate PI‐treated products? This may be particularly important for pediatric patients. 5) How do we best calculate the risk and benefit of PI as it is an expensive technology that may result in increased PLT use?
We must remind ourselves that PI for cellular products is a major paradigm shift for transfusion safety. The systems currently available are the first generation and we will certainly see improvements over time that minimize the negative side of the balance sheet.
Using solvent/detergent‐treated pooled plasma (Octaplas): implementation at University of Minnesota—Claudia Cohn, MD, PhD
Speaker's summary: Octaplas, an FDA‐approved blood component, is S/D‐treated plasma that is manufactured by combining multiple ABO‐matched plasma units from healthy US donors. The plasma units, which are frozen within 8 hours of collection, are pooled (approx. 380 L from 630 to 1520 donors in the United States) and then filtered to remove residual cells and treated with a combination of 1% tri(n‐butyl)‐phosphate and 1% octoxynol to inactivate enveloped viruses. The residual S/D is removed by oil extraction and chromatography. The final product is sterile‐filtered, aliquoted into 200‐mL bags, and stored frozen.94
Octaplas is indicated for replacement of coagulation factors in patients with acquired deficiencies due to liver disease or undergoing cardiac surgery or liver transplant and for plasma exchange in patients with thrombotic thrombocytopenic purpura. Octaplas is contraindicated in patients with immunoglobulin A deficiency and severe deficiency of protein S. All other contraindications, including a history of hypersensitivity to fresh‐frozen plasma and/or plasma‐derived products, are common to conventional plasma components. Octaplas may be stored for up to three years at not more than − 18°C. After thawing, Octaplas may be stored for 24 hours at +1 to 6°C or for 8 hours at room temperature.
All plasma used to make Octaplas is tested for viral markers in compliance with US regulation; however, additional steps further mitigate the risk of infectious disease transmission. The S/D treatment inactivates enveloped viruses with a log‐kill reduction factor of at least 5 to 6 for common viruses such as HIV, HBV and HCVC, WNV, ZIKV, and Dengue virus. Additional testing is performed for nonenveloped viruses, so that the pooled plasma may not contain more than 10.0 IU/μL parvovirus B19 DNA and must be negative for hepatitis A virus (HAV) by NAT polymerase chain reaction (PCR) and hepatitis E virus (HEV) RNA by NAT PCR (sensitivity of ≤2.5 log IU/mL). Furthermore, Octaplas contains prespecified levels of neutralizing antibodies for HAV and parvovirus B19. The final sterile filtration step (0.2 μm) could remove additional infectious agents.
Pooling has the added benefit of diluting allergens by approximately 1000‐fold, which reduces the risk of allergic reactions. Multiple studies have shown a comparative reduction in the rate of allergic reactions (Table 3).95, 96, 97, 98, 99, 100, 101 Pooling likely mitigates the risk of TRALI by diluting and neutralizing antibodies to human neutrophil antigen and HLA. The S/D process also removes bioactive lipids, which have been implicated as a causative factor in TRALI. While proving a negative is impossible, there have been more than 10 million units of Octaplas transfused with zero incidence of TRALI reported.59
Table 3.
A comparison of allergic reaction rates for S/D versus conventional plasma
| S/D plasma | Conventional plasma | Study author |
|---|---|---|
| Allergic reactions/patients (%) | ||
| 0/36 (0%) | 0/31 (0%) | Haubelt3 |
| 1/81 (1.2%) | NA | Solheim5 |
| 16/509 (3.1%) | 16/172 (9.3%) | Scully4 |
| 7/81 (8.6%) | 8/27 (29.6%) | Toussaint‐Hacquard6 |
| 20/981 (2.0%) | NA | Vendramin7 |
| 1/35 (0.3%) | NA | Witt* 8 |
| Allergic reactions/10,000 transfusions | ||
| 4.86 | 7.14 | Bost2 |
Pediatric patients.
The levels of coagulation factors, protease inhibitors, and cofactors are tested for each lot before release. Older versions of S/D plasma were associated with hyperfibrinolysis and bleeding;102, 103, 104 however, changes to the manufacturing process have mitigated the risk of these adverse events.105 In several studies conducted with the current version of Octaplas, no additional risk of these or other adverse events was identified.96, 105, 106, 107, 108, 109, 110 No clinically relevant differences in efficacy were identified; however, most of these studies were small and may not have been powered to show significant differences.96, 105, 106, 107, 108, 109, 110, 111
The University of Minnesota has chosen to maintain an inventory of Octaplas for patients who have a history of moderate to severe allergic transfusion reactions. It is especially helpful for patients who are undergoing plasma exchange, as they must contend with multiple units of conventional plasma. The extra time and effort saved by the reduction of reactions plus the increased comfort and safety experienced by the patient make the added cost of Octaplas worthwhile. In conclusion, the efficacy and safety profiles of Octaplas make it a good alternative to conventional plasma, and it is beneficial for patients with a history of allergic transfusion reactions.
Health economic considerations for pathogen reduction technology—Brian Custer, MPH, PhD
Speaker's summary: Pathogen reduction technologies must be evaluated with two different health economic considerations in mind: effect on available finances or budgets (budget impact analysis [BIA]) and an assessment of value for money (CE analysis [CEA]).112 BIA is a type of cost accounting study focused on implementation or maintenance of a health care intervention over a relatively short period of time, typically 1 to 5 years. CEA is an assessment of the ratio of costs to benefits, comparing at least two different interventions.113 Results are often reported as cost per quality‐adjusted life‐year (QALY). The CE plane can be used to graphically display the results for CEA separately for costs and effects in a way that provides several insights into the relative efficiency of each of the interventions included in the analysis and thus is highly informative to the broader decision making process for PRT.114
The health economics of PRT in the United States continues to be one of the barriers to broader adoption. For PRT there are several technologies, each with different mechanisms of action and inactivation capacity, which have been reviewed in detail in several previous publications.26, 27, 58, 115, 116, 117, 118 These differences are important for the health economics of each technology. Although RBC inactivation and WB inactivation technologies are in development119, 120, 121, 122 and clinical studies are under way,121, 123 current health economic evidence has not been published for these methods. Only two PRTs, S/D‐treated plasma, and amotosalen plus UV light for PLTs and plasma are approved for use in the United States.
Budget impact
An exemplary combined BIA and CEA conducted by the Canadian Agency for Drugs and Technologies in Health has been reported.124 However, the health economics of PLT PRT are arguably more important to define because BIA and CEA for PLTs are highly dependent on the level of clinically apparent bacterial contamination and PLT preparation method in use in a given setting. A recently published BIA focused on an analysis for a mid‐sized US hospital that was assumed to acquire 5500 apheresis PLT components per year from an external blood supplier. Several assumptions of this analysis sought to mimic a realistic situation: it was assumed that approximately 60% of acquired PLTs are irradiated and 20% are tested for CMV by the blood supplier. The model predicted minimal cost increase for PRT compared to conventional PLTs after including cost offsets such as elimination of bacterial detection and irradiation and differential reimbursement for treated and untreated PLTs.38 Another BIA conducted for Italy concluded further studies based on actual numbers of PLT transfusion complications and their societal cost at a local level are needed to establish the full cost to benefit ratio of PLT PRT.125
CE
Several studies have examined the cost‐effectiveness of plasma PRT. Compared to a benchmark of $50,000 to $100,000/QALY in most of health care, CE results for plasma PRT are expected to fall in the range of $800,000 to $1,200,000/QALY regardless of the technology. CE of PLT PRT is more variable. Without removal of bacterial culture and using hemovigilance data, estimates of $750,000 to $1,000,000/QALY have been reported. If all bacterial contamination, whether leading to clinically apparent sepsis is considered and culture is discontinued, results might approach $250,000/QALY. CE of PRT for both PLTs and plasma if in addition to current interventions is estimated to be approximately $1,250,000/QALY. The cost offsets that might be feasible by discontinuing other interventions in use alongside the cost of implementation need to be considered jointly as part of the value proposition of PRT.
Emerging agents will potentially shift ratios to being more cost‐effective but may not approach accepted thresholds in health care. Analyses focused on the cost offsets that can be achieved are being published and are expected to increase in the near future.37, 126 Reimbursement is also a barrier to adoption. Assessment of budget impact, CE, and pathways to reimbursement PRT are necessary considerations for successful widespread use of PRT.
SESSION 3. PATHOGEN REDUCTION TECHNOLOGIES FOR WHOLE BLOOD AND RED BLOOD CELLS
Introduction. Moderator—Raymond P. Goodrich, PhD
Technologies for the treatment of blood products to prevent TT of diseases have been in development now for more than 30 years.127 A review of the issues associated with these technologies can now be considered from more than a hypothetical framework and rather in the context of the experiences and observations from their use in routine production of PLT and plasma products.128 Such knowledge may also be extrapolated to the ongoing development efforts on methods for the treatment of WB or RBC products, which was the main topic for presentations in this section of the workshop.
Recent developments have focused on the use of PRT methods or modified methods using non–light‐activated chemistry, to treat either WB or the RBC component separated from WB. Several of these products are in clinical phases of evaluation. The challenges faced are expected to be comparable to those experienced for the now broadly approved and implemented methods for treating PLTs and plasma with added complications and challenges due to the number of units required to be treated and the nature of the patients who receive RBC products compared to PLT and plasma components. This session was intended to discuss some of the historical experience with PRT methods and to provide an overview on the latest information available from the development of methods to produce WB or RBC products treated with PRT methods.
Optimal pathogen reduction system for blood safety: is it a dream?—Raymond P. Goodrich, PhD., Colorado State University, CO
Speaker's summary: Experience from the implementation of PRT methods for PLTs and plasma has lessons to teach us as we extend the application of these processes into the production of WB and RBC products.
There will be a measurable reduction in cell or protein quality after treatment
Preclinical studies and clinical trials with PRTs conducted in the preceding 18 years have repeatedly demonstrated changes in both in vitro and in vivo variables.129 These include changes in metabolic variables in treated PLTs, changes in aggregation, and adhesion function and changes in PLT proteomic and metabolomics measurements.130 Similar results with plasma products have shown reduced coagulation factor levels after treatment.131 Clinical studies evaluating the performance of these products have demonstrated noninferiority with regard to prevention of bleeding in patients with hematologic malignancies. Estcourt and colleagues, summarizing meta‐analyses for 12 clinical trials with these products, indicated that “We found moderate‐quality evidence that pathogen‐reduced platelet transfusions do not affect all‐cause mortality, the risk of clinically significant or severe bleeding, or the risk of a serious adverse event.”132, 133 The authors also noted that, “We found high‐quality evidence that pathogen‐reduced platelet transfusions increase the risk of platelet refractoriness and the platelet transfusion requirement.”132, 133
Additives will be added to the blood supply that are not common blood additives or routinely present in the human body
Most PRT methods that have been implemented and are approved for use in the United States and outside the United States are based on the use of chemical additives to the blood products, which can be activated in specific ways to prevent nucleic acid replication of targeted pathogens and white blood cells (WBCs) in these products.25 The nature of the compounds used in these approaches varies considerably based on method and include the use of riboflavin134 and vitamin B2 as well as synthetically derived compounds such as amotosalen and amustaline, which are based on psoralen, acridine, and mustard hydrochloride derivatives, respectively.135 In many cases, these agents and their breakdown products exhibit considerable toxicity and genotoxicity that must be considered in terms of likely patient population and effectiveness of both handling and removal methods intended to reduce exposure to patients and health care workers.136 Processing requirements necessitated by the nature of the agents used have complicated the practical logistics of delivering blood products to patients.118
Not all pathogens will be eliminated by the application of these processes
Variable levels of PR are observed for bacteria and enveloped viruses depending on the method that is being applied.137 Knowledge about what levels of inactivation are necessary to prevent disease transmission are not clear or uniform for all agents.138, 139 Examples are now available from completed and ongoing routine use studies that suggest that while disease transmission may be significantly curtailed, some breakthrough events may still be anticipated including cases where pathogen loads in donated products may exceed inactivation potential123 or where the PRT method may prove ineffective against specific types of resistant agents such as nonenveloped viruses.140 Not all methods will deliver the same outcomes in pathogen load reduction capabilities; hence continued field evaluation of what is needed to significantly reduce the probability of disease transmission in areas where such diseases are endemic will be necessary to fully determine the extent of effectiveness.131
Process control will be essential to assure reproducibility and reliability of these methods
Significant efforts have been made by manufacturers to develop techniques that can be practically applied to treatment of PLT and plasma products in the routine blood bank setting. Such devices and processes need to account for practical factors including throughput, product specifications, media, product losses, timing of process steps, record keeping, and costs to manufacture disposables and equipment. Validation of these methods has required significant investment on the part of manufacturers yet concerns in each of these categories related to implementation remain and continue to be identified as these methods enter routine clinical use.142
These processes will add cost
Routine implementation in high‐income countries where endemic disease and exposure are rare is likely to continue to be debated on the basis of cost and benefit. Advances in adoption are likely to result when these processes can serve low‐income nations with significant blood safety concerns with affordable products made with high quality and low cost. Questions about implementation with patients who are most vulnerable and thus may benefit the most from these methods need to be asked. Such groups may include those receiving chronic transfusion support or pediatric patients where cost–benefit and risk–benefit calculations are likely to have their most favorable outcomes.143 Providing products to regions with high endemic disease rates can greatly improve cost–benefit analyses but will require partnerships with nongovernmental organizations and longer‐term considerations of sustainability in these environments when supplementation of costs is no longer feasible.144
Clinical experience with pathogen reduction for red blood cells: completing the triad—Richard J. Benjamin, MD, PhD, FRCPath
Speaker's summary: Unique technologies are needed to adequately balance the need for robust, broad‐spectrum PI and the disparate characteristics of RBCs, PLTs, and plasma proteins. INTERCEPT RBCs incorporate treatment of RBCs in AS with 0.2 mmol/L amustaline (S‐303) in the presence of 20 mmol/L glutathione (GSH), in a closed system.135 Amustaline rapidly crosslinks and/or forms adducts with nucleic acids to prevent pathogen replication without generating reactive oxygen species (ROS), while the active compound degrades to undetectable concentrations (<0.75 nmol/L) during processing. An exchange step into a licensed storage solution removes the majority of the breakdown products, resulting in treated RBCs with a 35‐day shelf life.145 Extensive testing confirmed a lack of neonatal and reproductive toxicity, carcinogenicity, genotoxicity, and acute and chronic toxicity, while confirming potent inactivation of a broad range of enveloped and nonenveloped viruses, bacteria, protozoa, and WBCs.146 Animal models confirmed a lack of immunogenicity. Treated RBCs demonstrate reduced hemolysis and increased ATP levels compared to conventional irradiated RBCs. A similar closed‐system process is in development for WB collections in collaboration with the Swiss Red Cross for use in austere environments.147
INTERCEPT RBCs were successfully evaluated in a series of clinical trials demonstrating the safety, efficacy, and the performance of the system (Table 4). Radiolabeled recovery and survival studies in healthy volunteers confirm acceptable RBC recovery and life span, exceeding FDA requirements.120, 148 A Phase III randomized controlled study (STARS) involving 51 complex cardiac surgery patients in Germany demonstrated that treated RBCs met the predetermined noninferiority margin for hemoglobin (Hb) content (mean treatment difference [test–control] of 2.27 g/unit [95% CI, −2.61 to −1.92 g/unit), within the prespecified equivalence margins (±5 g/unit) and with reduced hemolysis at the end of storage.149 Subjects received a mean of 2.9 (range, 1‐8) test or control RBC components during surgery or within 7 days of surgery. Exploratory clinical endpoints, including renal and hepatic insufficiency and the 6‐minute walk test, as well as adverse events, were not different, and no patients had antibodies specific for INTERCEPT RBCs.
Table 4.
Summary of completed clinical studies with INTERCEPT RBCs
| Study161, 162, 163 | Population | Intervention | Study design | Comparison | Outcome |
|---|---|---|---|---|---|
| CLI 00062 | 28 enrolled, 26 evaluable, healthy subjects | Compare survival and recovery of autologous INTERCEPT RBCs to conventional RBCs (prototype set) | Single‐blinded, randomized crossover, controlled radiolabeled autologous RBC | Single transfusion ~10 mL of 51Cr‐labeled test or control RBCs (35‐day storage) | RBCs prepared using the INTERCEPT PI process were physiologically and metabolically suitable for transfusion after 35 days of storage, met the FDA guidance criteria for 24‐hour recovery, and did not induce antibody formation. |
| CLI 00073 | 42 enrolled, 26 evaluable, healthy subjects | Compare survival and recovery of autologous INTERCEPT RBCs to conventional RBCs (final set) | Single‐blinded, randomized crossover, controlled, radiolabeled autologous RBC | Single transfusion 10‐30 mL of autologous 51Cr‐labeled test or control RBCs (35‐day storage) | RBCs met the FDA criteria for posttransfusion RBC recovery 24 hr after transfusion. The mean life span and median life span (T50) of autologous RBCs after storage for 35 days were shorter for test RBCs compared to control RBCs, but within the published reference range, and the AUCs of test and control were not different. |
|
CLI 00070 (STARS) |
87 randomized, 51 evaluable, cardiac surgery patients | Assess the in vitro characteristics of INTERCEPT RBCs; assess the clinical safety and efficacy of INTERCEPT RBCs in transfusion support for acute anemia | Randomized, controlled, double‐blinded, parallel design, noninferiority | INTERCEPT or conventional RBCs for up to 7 days during and after surgery | The mean postproduction Hb content per component was 53.6 ± 5.6 g/component in the test and 56.3 ± 6.0 g/component in the control groups. Equivalence was declared since the 95% CI for the mean treatment difference was within the a priori defined margins (±5 g/component). INTERCEPT RBC components met EDQM guidelines for Hb content, hematocrit, and hemolysis. The safety profile of INTERCEPT RBCs was comparable to conventional RBCs. |
| CLI 00076 (SPARC) | 86 randomized, 81 evaluable, transfusion‐dependent thalassemia patients | Evaluate the efficacy and safety of INTERCEPT RBCs in subjects who require chronic transfusion support due to thalassemia major | Randomized, controlled, double blinded, two‐period, crossover, noninferiority | Six transfusion episodes of INTERCEPT or conventional RBC; two wash‐in and four evaluable episodes | Mean Hb consumption (g/kg/day) with INTERCEPT RBCs was not inferior (p < 0.001) to conventional RBCs (0.113 ± 0.04 vs. 0.111 ± 0.04, p = 0.373) by intent‐to‐treat or per‐protocol (0.112 ± 0.04 vs. 0.110 ± 0.03, p = 0.162) analysis. No antibodies specific to INTERCEPT RBCs were detected; there were no substantial differences in transfusion reactions, adverse events, or serious adverse events recorded. |
A second randomized, double‐blind, controlled crossover study (SPARC) performed in Italy and Turkey was completed in 81 transfusion‐dependent thalassemia patients who receive regular RBC transfusions to treat anemia and suppress ineffective hematopoiesis (Table 4). Each patient (≥10 years old) received six cycles (two “wash‐in” and four “efficacy evaluation” cycles) of test and control RBCs with the RBC dose determined by a physician blinded to treatment. The primary endpoint was Hb consumption (g Hb/kg/day), a measure of iron burden. Subjects met the predetermined noninferiority margin of 15% of control for Hb consumption in both the intention to treat and per‐protocol populations, receiving a mean of 12.6 test and control RBCs in the two treatment periods. Only 11 of 2006 (0.5%) RBC components were transfused off protocol to six patients. Adverse events were similarly distributed between periods, and no patients made treatment‐emergent antibodies to INTERCEPT RBCs or to RBC alloantigens.
The completed studies demonstrated the safety and efficacy of INTERCEPT RBCs while in vitro analyses demonstrated robust PI. Further US clinical studies in cardiac surgery (ReCePI) and in the general hospital population (RedeS) are under way (see http://clinicaltrials.gov). The availability of PRTs for RBCs, PLTs, and plasma would increase patient safety and revolutionize the current reactive, incremental testing approach to infectious disease threats. It may also allow a reassessment of the need for current donor deferral and testing requirements.
State of PRT for whole blood—Anna Razatos, PhD
Speaker's summary: The Mirasol PRT system is CE marked for the treatment of PLTs, plasma, and WB for transfusion and is in routine use in many countries outside of the United States. The Mirasol PRT system uses one device to treat all blood products. Mirasol consists of the photosensitizer riboflavin (vitamin B2) in combination with UV light to irreversibly damage DNA and RNA resulting in inactivation of viruses, bacteria, parasites, and WBCs. Because riboflavin and its photoproducts are nontoxic and naturally occurring, they do not require removal from the blood product after treatment.
The WB Mirasol program has been developed in partnership with the US Department of Defense (DOD). The African Investigation of Mirasol System (AIMS) clinical trial at the Komfo Anokye Teaching Hospital in Kumasi, Ghana, is the first and only clinical trial to demonstrate that PRT can effectively reduce the incidence of TTI of a blood‐borne pathogen. The test arm consisted of Mirasol‐treated nonleukoreduced WB and the control arm consisted of untreated nonleukoreduced WB.123 Sixty‐five nonparasitemic patients were exposed to parasitemic blood; 28 received Mirasol‐treated WB and 37 received untreated WB.123 The incidence of transfusion‐transmitted malaria was significantly lower for the patients receiving Mirasol‐treated WB (one [4%] of 28 patients) compared to patients receiving untreated WB (eight [22%] of 37 patients; p = 0·039).123 Moreover there were no significant differences in 1) total Hb for up to 28 days posttransfusion and 2) adverse events between Mirasol‐treated WB and untreated WB.123 The success of the AIMS clinical trial supported CE mark of the Mirasol PRT system for the treatment of WB for transfusion.
After completion of the AIMS clinical trial, the Japan International Cooperation Agency partnered with the Terumo Corporation of Japan and the AABB Consulting Services to support sustainable blood safety in Ghana with routine use of the Mirasol WB system in conjunction with the creation of the infrastructure for a hemovigilance system.150 The partnership expanded the use of Mirasol PRT for WB within Ghana; established an active hemovigilance system at two teaching hospitals, Komfo Anokye Teaching Hospital in Kumasi and Korle‐bu Teaching Hospital in Accra; and provided the necessary training and support. In September of 2018, Japan International Cooperation Agency successfully handed over the Ghana Blood Safety Program to the Ministry of Health (MOH).150
Dr. Aaron Tobian with the Makerere University–Johns Hopkins University (MU‐JHU) Collaboration will expand use of the Mirasol PRT system for WB in Africa to Uganda with the Mirasol Evaluation of Reduction in Infections Trial (MERIT).141 The primary objective was to evaluate the Mirasol WB system to reduce malaria and other TTIs in Uganda.150 Secondary objectives include an evaluation of the impact of TTI and potential benefit of PRT as well as the feasibility and sustainability of implementing Mirasol in austere environments.141
In addition to Mirasol PRT treatment of WB for transfusion, active studies are under way to evaluate componentization of Mirasol‐treated WB. Terumo BCT has initiated the PRAISE clinical trial to evaluate efficacy and safety of RBCs derived from Mirasol‐treated WB compared to conventional RBCs in patients requiring chronic transfusion support.151 Moreover, Terumo BCT has supported investigator‐initiated studies of components derived from Mirasol‐treated WB. One such study conducted by Dr. Pavel Trakhtman from the Russian Federal Center for Pediatric Hematology, Oncology and Immunology evaluated the safety and efficacy of RBCs derived from Mirasol‐treated WB in pediatric patients with malignancies.152 Finally, as part of Terumo BCT's commitment to advancing blood safety, the company is investing in innovation to further refine and improve PRT.
Pathogen reduction technologies for red blood cell products: impact on biochemical and viability variables in humans—Jose A. Cancelas, MD, PhD
Speaker's summary: Despite the implementation of improved diagnostic screening and donor selection, the residual risk of transmission of new emerging pathogens and bacterial contamination persists. PR of blood components may be capable of proactively reducing and/or eliminating the chance of disease transmission by transfusion. PR could replace the current paradigm of serial introduction of diagnostic tests as new microorganisms with potential TT are recognized. PR for RBC and WB products remains a major challenge in the development of safe blood products for transfusion and it is of major interest in the context of chronic transfusion protocols.
The first technology evaluated was the use of riboflavin/UV (80 J/mL RBC) light (Mirasol) system for PR of WB. The riboflavin/UV light PR technology (Mirasol PRT system, or Mirasol) method is based on irreversible nucleic acid damage mediated by electron transfer processes at sites where riboflavin‐guanine base chemistry occurs.153, 154 Our group and others have already published significant information on the RBC viability of PR WB‐derived RBCs in vitro in the context of high‐dose illumination resulting in significant PR.84, 154, 155, 156, 157, 158, 159, 160, 161, 162, 163, 164 These sets of data indicate that PR of WB is possible albeit at a loss of RBC viability in vitro as assessed by hemolysis, ATP levels, and potassium leakage and in vivo as assessed by 24‐hour recovery and survival analyses.155, 156 Through a two‐center clinical trial, our combined data consistently support that riboflavin/UV light–treated RBCs stored for 21 days after being generated from Mirasol‐treated WB maintain adequate levels of 24‐hour recovery as assessed by 51Cr‐tagged circulatory RBC survival analysis. As anticipated, there was decreased in vivo viability of poststorage riboflavin/UV light RBCs compared to data obtained from the same donors similarly stored untreated RBCs. The overall 24‐hour recovery, survival, T50, and area under the curve (AUC) of Mirasol RBCs were reduced by 9.9, 25.9, 36.9, and 16.5%, respectively, compared to the subjects untreated control RBCs.165 As expected, the percentage of RBC hemolysis significantly correlated with the 24‐hour recoveries of the control RBCs but not with their T50 survivals, suggesting that both hemolysis and 24‐hour recoveries measured the destruction of a population of cells that did not survive storage. In contrast, there was no similar correlation between percent hemolysis and recoveries of Mirasol RBCs nor between their T50 and percent hemolysis. However, for the Mirasol RBCs, there was a significant correlation between ATP levels and 24‐hour recoveries, but a similar association was not found for control RBCs.
The second technology tested was PRT using amustaline (S‐303). This approach to target nucleic acid–containing pathogens does not require photochemical activation through the use of a small molecule (amustaline, S‐303) that reacts rapidly with nucleic acid and then decomposes into unreactive byproducts. Published PI results show that amustaline treatment of RBCs effectively inactivates multiple blood‐borne pathogens (≥4 log) and residual WBCs.147, 166, 167, 168, 169 To prevent nonspecific reactions of amustaline with RBC membrane proteins, GSH is used in conjunction with the amustaline treatment. In two different two‐center studies we analyzed the viability of RBCs prepared with a second‐generation process and stored for 35 days was evaluated in two different blood centers in a Phase I and II clinical trial design, respectively. Both were single‐blind randomized, controlled, two‐period crossover studies where amustaline or control RBCs were prepared in random sequence and stored for 35 days followed by 51Cr tagging and 24‐hour RBC posttransfusion recovery, mean life span, median lifespan (T50), and life span AUC analyzed. The mean 24‐hour posttransfusion recovery of test and control RBCs was comparable (83.2% ± 5.2% and 84.9% ± 5.9%, respectively; p = 0.06), and consistent with the FDA criteria for acceptable RBC viability. There were differences in the T50 between test and control RBCs (33.5 and 39.7 days, 17% difference; p < 0.001) but it was within published reference ranges of 28 to 35 days. The AUC (percent surviving × days) for test and control RBCs was similar (22.6 and 23.1% surviving cells × days, respectively; p > 0.05). After infusion of test RBCs, there were no clinically relevant abnormal laboratory values or adverse events.
Both technologies represent examples of first‐generation efforts toward developing a technology that can maintain the product potency while eradicating nucleic acid–containing pathogens. If the goal is to increase the safety with a tradeoff of 10% reduction in RBC, the results provided indicate that the potency of RBCs treated with both PR products, especially riboflavin/UV light, would fall short of such goal. I believe that these results may form part of the basis for the discussion on the development of improved protocols and/or approaches to PR of WB in general and, specifically, RBCs.
SESSION 4: EMERGING INNOVATIONS RELEVANT TO PATHOGEN REDUCTION TECHNOLOGY AND ALTERNATIVES
Introduction. Moderator—Stephen J. Wagner, PhD
Most discussion in the conference was focused on the practice of and issues surrounding current PR methods approved for use in the United States or Europe. Session 3 was devoted to the development of emerging PRTs and alternatives to PR. Use of different light sources for PR without the use of photochemicals, as well as use of novel photochemicals or photochemical mechanisms that target pathogens with less collateral damage to the blood component, are emerging as improvements to current methods of PR. Alternatively, different storage conditions for components may reduce the potential for bacterial outgrowth in PLT components, reducing the need for PR. Finally, PR of WB may produce sterile WB or isolated components, simplifying PR compared to current methods.
A nucleic acid–binding photosensitizer with flexible structure for pathogen inactivation in red blood cell suspensions—Stephen J. Wagner, PhD
Speaker's summary: Photochemicals localize at nucleic acid target sites as well as off‐target locations in the supernatant or bound to RBCs or PLTs. Upon illumination, photochemicals bound to target produce singlet oxygen or other ROS or are involved with electron transfer and adduct formation, preventing further replication of the pathogen. Photochemicals localized in the supernatant can produce ROS, which can diffuse to the PLT or RBC membrane to produce oxidative damage or undergo electron transfer with other colocalized photochemicals, producing photochemical dimers. Photochemicals bound to the RBC or PLT membrane produce ROS, which can oxidize membrane components or produce covalent adducts to polyunsaturated lipids via electron transfer.170 Off‐target lesions are responsible for alterations of in vitro or in vivo properties of photochemically treated RBCs and PLTs.
Most photochemicals are composed of fused rings of conjugated double bonds. When a photochemical absorbs light, it cannot dissipate the absorbed energy via bond rotation as heat because of the rigidity of the ring structure and therefore releases energy via fluorescence, phosphorescence, or photochemical reactions. On the other hand, flexible photosensitizers can dissipate absorbed light energy through bond rotation and, therefore, cannot readily act as a photochemical unless rigidly bound to substrate in a planar geometry.
One such flexible dye is thiazole orange, which binds to the minor groove of nucleic acids and can act as a photochemical when bound but does not participate in photochemical reactions when free in solution.171 Use of 80 μmol/L thiazole orange in RBCs suspended in Erythrosol AS and 7.9 J/cm2 cool white light resulted in inactivation of more than 7 log vesicular stomatitis virus, more than 5.8 log bovine virus diarrhea virus, 5.5 log pseudorabies virus, more than 6.5 log extracellular HIV, more than 6.3 log intracellular HIV, more than 5 log of Leishmania donavani infantum, and more than 5 log T. cruzi.171, 172 Bacterial inactivation ranged from 2.3 to more than 7 log, depending on the species.171 RBC storage studies with thiazole orange demonstrated a hemolysis of 0.43% on Day 42 compared to 0.1% hemolysis of untreated controls, no change in ATP levels during 42‐day storage compared to untreated controls, and a two‐ to threefold increase in potassium leakage in thiazole orange–treated RBCs compared with untreated controls. Unilluminated RBC controls containing thiazole orange had 0.3% hemolysis on Day 42, suggesting that some of the observed hemolysis may be due to the presence of thiazole orange rather than photo‐induced hemolysis. Future development of thiazole orange as a RBC photo‐inactivating agent will require toxicology, scale‐up, and in vivo studies.
Blue light inactivation of pathogens in platelet and plasma: a pilot study—Michelle Maclean, PhD
The antimicrobial properties of 405‐nm violet‐blue light is a research area gaining increasing prominence, and due to the inherent properties of these visible light wavelengths, research has been initiated to investigate its potential for development as a novel alternative PRT. The workshop presentation provided an overview of a pilot study that has begun evaluation the antimicrobial capability and compatibility of 405‐nm violet‐blue light for decontamination of blood transfusion components.
Recent studies have demonstrated the application of violet‐blue light for safe, continuous environmental decontamination173 and its potential for wound decontamination.174 These antimicrobial applications are made possible due to the safety advantages of these non–UV light wavelengths. Unlike UV light, which induces direct DNA‐based damage and/or oxidative damage in exposed microbial cells (depending on the exact UV wavelengths used), inactivation by violet‐blue light involves the excitation of endogenous photosensitive porphyrin molecules found within exposed microbial cells.175
Although less germicidally efficient, research has demonstrated that these lower‐energy violet‐blue 405‐nm photons are particularly suited to decontamination of sensitive blood components and have potential operational advantages. A recent study176 demonstrated the antimicrobial potential of 405‐nm light for decontamination of key bacterial pathogens in blood plasma, with a complete 5‐log reduction of Staphylococcus aureus in plasma using a dose of approximately 360 J/cm2. Importantly, antiviral potential has also been demonstrated in plasma, with a successful 5‐log reduction of calicivirus after a 561 J/cm2 light exposure.177 A major finding demonstrated by the study4 was that the higher penetrability of 405‐nm light photons facilitates decontamination of prebagged transfusion products. Results showed significant antimicrobial efficacy within sealed plasma transfusion bags, with approximately 99% inactivation of S. aureus contamination in plasma achieved using low irradiance light at a dose of 144 J/cm2. Similar antimicrobial results were presented demonstrating successful decontamination of prebagged PLT suspensions, at comparative dose levels to those used with plasma.
As with all PRTs, a crucial question that must be answered is whether antimicrobial efficacy can be imparted with negligible and/or minimal effect on the quality of the blood component itself. Results of sodium dodecyl sulfate‐polyacrylamide gel electrophoresis and Western blotting analysis were presented which highlighted the potential for “safe” treatment of blood plasma and the need for determining upper and lower threshold treatment levels, which would facilitate decontamination while retaining plasma and PLT integrity. With regard to PLTs, an evaluation of the in vivo recovery of light‐treated (10 mW/cm2 for 8 hr) PLTs in a murine model demonstrated no difference to that of nontreated PLTs. These results are encouraging; however, further investigation is needed on the impact of dose delivery (high irradiance light delivered over rapid exposure periods compared to low irradiance light over longer exposures), as this will influence the optimal treatment conditions for the blood components. Discussions highlighted the potential for the technology to be utilized for rapid treatment before storage (for PLTs or plasma) or incorporation into standard storage conditions for PLTs with the aim of extending the shelf life of these sensitive components.
This ability to directly treat prebagged plasma and PLT blood products using 405‐nm light exposure, without the addition of photosensitizer molecules, is highly desirable as it would eliminate the requirement for the additional processing stages needed with other PRTs for passage through equipment and removal of chemical additives, thus reducing cross‐contamination risks and also potentially reducing processing times. Thus, further research to expand the understanding and compatibility of this technology with blood transfusion products is warranted.
Pathogen reduction in blood products: refrigerate and use pathogen reduction technology—COL Andrew P. Cap, MD, PhD
The DOD has invested significant resources into the development of PRT to improve the safety of blood transfusion on the battlefield.178 In addition to the need to reduce the risk of bacterial contamination associated with room temperature storage of PLTs, the unique challenges of combat casualty care include the need to collect WB and PLTs in remote deployed environments and transfuse these products before the availability of transfusion‐transmitted disease testing results. The risks inherent in these emergency blood transfusions can be magnified by donor exposure to endemic diseases such as malaria and potentially to biowarfare agents. The possibility of myelosuppressive radiation exposure on the battlefield could require the inactivation of WBCs in blood products to reduce the risk of TA‐GVHD. PRTs for WB and PLTs are urgently needed to support military operations.
In addition to investing in technologies that use various photosensitizers and UV light to damage nucleic acids in pathogens and WBCs, the DOD has pursued alternative blood storage approaches to not only reduce infectious risks of transfusion but also increase the availability of PLTs, since current room temperature storage practices limit shelf life to 5 to 7 days. Refrigeration has been demonstrated to significantly reduce bacterial growth as well as reduce the metabolic activity of PLTs during storage, preserving their hemostatic function. Refrigerated PLTs retain significant capacity to adhere to collagen under shear, aggregate, release granules, catalyze thrombin generation, and contract clots for up to 21 days.179 Refrigeration causes partial activation of PLTs, leading to desialylation of membrane proteins and clearance from circulation over 1‐2 days in vivo.180 Nevertheless, in a randomized trial in cardiac surgery patients, refrigerated PLTs stored for up to 14 days performed comparably to standard‐of‐care PLTs stored for less than 7days (K.M. Reddoch Cardenas and A. Cap, unpublished, 2019).181 PLTs can be refrigerated in either plasma or PAS, and preliminary results suggest that PRT‐treated PLTs can also be refrigerated without loss of hemostatic function.182, 183 Refrigeration thus offers not only an extra layer of protection from bacterial pathogens in addition to PRT, but also an opportunity to extend shelf life and availability while preserving the essential contribution of PLTs to bleeding control. It should be noted that refrigerated WB contains active PLTs that have been shown to maintain considerable hemostatic function through the entire shelf life of WB (up to 35 days in CPDA‐1).184, 185
The DOD is also supporting the development of other alternative storage modalities for PLTs such as cryopreservation and lyophilization.178, 186 Both technologies offer extended storage times (years) and bacterial pathogen risk reduction and could be combined with PRT. While the clinical safety and efficacy of these products is being established, refrigeration with or without PRT offers a practical way to improve PLT product safety and availability. WB PRT remains an important capability gap for the US military, but a technology using riboflavin and UV light has been shown to yield a product with preserved hemostatic function and to inactivate Ebola virus, among other pathogens, and reduce transfusion‐transmitted malaria; this technology is in Phase III trials.123, 184, 187 Successful development of WB PRT will be a landmark achievement in the history of transfusion medicine and will benefit both civilian and military patients.
Concluding remarks. Insights for future research and development—Paul Ness, MD
The comprehensive 2‐day conference on PRTs organized by the FDA presented the current state of the art and raised several questions whose answers will drive future development and clinical implementation of PRTs. Although the following list is by no means all‐inclusive, it represents some recurrent themes and areas that were considered during the conference and that need to be addressed going forward as issues of importance.
Blood safety and pathogen kill
Do in vitro cell kill measures correlate with clinical efficacy? The reader is reminded that the AIMS trial in Ghana showed malarial prevention with infectious loads greater than the predicted capacity of the Mirasol system123 and a review of this comprehensive topic has been published in TRANSFUSION. 139
What is the appropriate balance between pathogen kill and blood component function?
PLT issues
Do PRT PLTs stop acute hemorrhage? Most of the clinical trials have assessed prophylaxis to prevent hemorrhage in patients with thrombocytopenia with hematologic malignancies.
Can the damage to PLTs that occurs with available PRT systems be mitigated to enhance PLT recovery, survival, and function?
How effective are PRT PLTs in reducing alloimmunization? Although leukoreduction has significantly reduced alloimmunization from 15% to 5% in patients with acute leukemia, the problem of alloimmune refractoriness to PLTs has not been eliminated.
Will PRT PLTs effectively reduce PLT transfusion reactions involving WBCs such as febrile reactions and TRALI?
Plasma issues
What is the effect of PRT on procoagulant and prothrombotic constituents and their balance in patients?
Can PRT be used to prepare pooled plasma products to reduce infectivity risks while taking advantage of the ability of large pools of plasma to reduce transfusion complications such as allergic reactions, ABO antibody–induced hemolysis, and hypocoagulability due to the variable content of coagulation constituents in single bags?
Can recovered plasma containing additives required for preparation of PRT treated WB be used in plasma fractionation?
RBC issues
Can damage to RBCs with current PRT systems be limited by new processes under development or additional manufacturing steps?
Do PRT processes affect immune‐hematologic testing procedures?
Does the addition of PRT reopen the age of blood storage controversy?
Other unanswered questions
Do we really need blood storage for 42 days or could the blood transfusion systems in the United States handle shorter storage limited in required for PRT‐treated RBCs?
- Can FDA adopt guidelines for industry that would allow enhancements in blood storage solutions using PRT to be licensed and implemented that would be cost sensitive?
- How much of the new knowledge about RBC storage would need to be applied to applications for licensure of PRT RBCs? Will recovery, survival, and hemolysis be the major criteria for licensure?
- Will FDA work with blood centers to increase efficiencies with modified blood processing procedures and elimination of redundant testing or donor history?
- Will these cooperative efforts results in PRT components that hospitals view as cost‐effective and worthy of the increased expenses?
- If guidelines and requirements become too burdensome, will we ever be able to take advantage the clinical and operational advantages of PRT that will improve patient care?
ACKNOWLEDGMENTS
The organizing committee thanks CBER Director Dr. Marks and OBRR Director Dr. Verdun for their constant support and providing direction to the committee. Dr. Flegel thanks Harvey Gordon Klein, MD, for review of the manuscript and comments and acknowledges the late Sherry Lynne Sheldon, MT(ASCP)SBB, whose contribution as laboratory supervisor in the Laboratory Services Section was critical for efficiently implementing the technology in a timely manner under fiscal constraints. PRT PLTs remain S.L. Sheldon's legacy to the patients and their safety at the NIH Clinical Center. Dr. Benjamin acknowledges that INTERCEPT RBCs are being developed under contract with the Biomedical Advanced Research and Development Authority, a division of the Department of Health and Human Services.
CONFLICT OF INTEREST
SK is a consultant to Cerus, manufacturer of the Intercept system and serves on Medical Advisory Board of Creative Testing Solutions (CTS). ES is PI for the Cerus PIPER Phase IV clinical study at the Yale site—he receives no personal remuneration. SR has nothing to disclose. WF states that the views expressed do not necessarily represent the view of the National Institutes of Health, the Department of Health and Human Services, or the US Federal Government and no financial relationship relevant for this publication. RB is an employee and stockholder in Cerus Corporation, a manufacturer of pathogen reduction technologies. AR is an employee of Terumo BCT.
SR was not a speaker at the workshop but contributed to the content ES presented at the workshop. MG was a speaker at the workshop but contributed to the content MM presented at the workshop.
The content of each speaker's summary reflects the views of the author and should not be construed to represent FDA's views or policies.
Workshop transcript is available at https://www.fda.gov/BiologicsBloodVaccines/NewsEvents/WorkshopsMeetingsConferences/ucm620519.htm.
Workshop Organizing Committee: C.D. Atreya, Jennifer Scharpf, Monique Gelderman, Paul Buehler, Jaro Vostal, David Leiby, Salim Haddad, Carlos Villa, and Orieji Illoh, CBER, FDA.
External Advisors: Simone Glynn, NHLBI, NIH; Harvey Klein, Clinical Center, NIH; and Mary Homer, BARDA, ASPR, DHHS.
The work was supported by a grant (project ID Z99 CL999999) from the Intramural Research Program of the NIH Clinical Center.
REFERENCES
- 1. Perkins HA, Busch MP. Transfusion‐associated infections: 50 years of relentless challenges and remarkable progress. Transfusion 2010;50:2080‐99. [DOI] [PubMed] [Google Scholar]
- 2. Stramer SL, Glynn SA, Kleinman SH, et al. Detection of HIV‐1 and HCV infections among antibody‐negative blood donors by nucleic acid‐amplification testing. N Engl J Med 2004;351:760‐8. [DOI] [PubMed] [Google Scholar]
- 3. Kleinman SH, Lelie N, Busch MP. Infectivity of human immunodeficiency virus‐1, hepatitis C virus, and hepatitis B virus and risk of transmission by transfusion. Transfusion 2009;49:2454‐89. [DOI] [PubMed] [Google Scholar]
- 4. Busch MP, Bloch EM, Kleinman SH. Prevention of transfusion transmitted infections. Blood. 2019;133(17):1854‐64. [DOI] [PubMed] [Google Scholar]
- 5. Stramer SL, Hollinger FB, Katz LM, et al. Emerging infectious disease agents and their potential threat to transfusion safety. Transfusion 2009;49(Suppl 2):1S‐29S. [DOI] [PubMed] [Google Scholar]
- 6. Glynn SA, Busch MP, Dodd RY, et al. Emerging infectious agents and the nation's blood supply: responding to potential threats in the 21st century. Transfusion 2013;53:438‐54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Atreya C, Nakhasi H, Mied P, et al. FDA workshop on emerging infectious diseases: evaluating emerging infectious diseases (EIDs) for transfusion safety. Transfusion 2011;51:1855‐71. [DOI] [PubMed] [Google Scholar]
- 8. Dodd R, Groves JA, Townsend RL, et al. Impact of one‐time testing for Trypanosoma cruzi antibodies among blood donors in the United States. Transfusion 2019;59:1016‐23. [DOI] [PubMed] [Google Scholar]
- 9. Busch MP, Caglioti S, Robertson EF, et al. Screening the blood supply for West Nile virus RNA by nucleic acid amplification testing. N Engl J Med 2005;353:460‐7. [DOI] [PubMed] [Google Scholar]
- 10. Dodd RY, Foster GA, Stramer SL. Keeping blood transfusion safe from West Nile virus: American Red Cross Experience, 2003 to 2012. Transfus Med Rev 2015;29:153‐61. [DOI] [PubMed] [Google Scholar]
- 11. Williamson PC, Linnen JM, Kessler DA, et al. First cases of Zika virus‐infected US blood donors outside states with areas of active transmission. Transfusion 2017;57(3pt2):770‐8. [DOI] [PubMed] [Google Scholar]
- 12. Saa P, Proctor M, Foster G, et al. Investigational testing for Zika Virus among U.S. blood donors. N Engl J Med 2018;378:1778‐88. [DOI] [PubMed] [Google Scholar]
- 13. Moritz ED, Winton CS, Tonnetti L, et al. Screening for Babesia microti in the U.S. Blood Supply. N Engl J Med 2016;375:2236‐45. [DOI] [PubMed] [Google Scholar]
- 14. Ward SJ, Stramer SL, Szczepiorkowski ZM. Assessing the risk of Babesia to the United States blood supply using a risk‐based decision‐making approach: Report of AABB's Ad Hoc Babesia Policy Working Group (original report). Transfusion 2018;58:1916‐23. [DOI] [PubMed] [Google Scholar]
- 15. Ramirez‐Arcos S, DiFranco C, McIntyre T, et al. Residual risk of bacterial contamination of platelets: six years of experience with sterility testing. Transfusion 2017;57:2174‐81. [DOI] [PubMed] [Google Scholar]
- 16. Urwin PJ, Mackenzie JM, Llewelyn CA, et al. Creutzfeldt‐Jakob disease and blood transfusion: updated results of the UK Transfusion Medicine Epidemiology Review Study. Vox Sang 2016;110:310‐6. [DOI] [PubMed] [Google Scholar]
- 17. Lombardi VC, Ruscetti FW, Das Gupta J, et al. Detection of an infectious retrovirus, XMRV, in blood cells of patients with chronic fatigue syndrome. Science 2009;326:585‐9. [DOI] [PubMed] [Google Scholar]
- 18. Simmons G, Glynn SA, Komaroff AL, et al. Failure to confirm XMRV/MLVs in the blood of patients with chronic fatigue syndrome: a multi‐laboratory study. Science 2011;334:814‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bennett JL, Devine DV. Risk‐based decision‐making in transfusion medicine. Vox Sang 2018;113:737‐49. [DOI] [PubMed] [Google Scholar]
- 20. Bloch EM, Ness PM, Tobian AAR, et al. Revisiting blood safety practices given emerging data about Zika virus. N Engl J Med 2018;378:1837‐41. [DOI] [PubMed] [Google Scholar]
- 21. Center for Biologics Evaluation and Research . Bacterial risk control strategies for blood collection establishments and transfusion services to enhance the safety and availability of platelets for transfusion: draft guidance for industry [Internet]. Rockville (MD): Food and Drug Administration; 2018 [cited 2019 Aug 05]. Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/bacterial-risk-control-strategies-blood-collection-establishments-and-transfusion-services-enhance.
- 22. Ceros Corporation . FDA approves pathogen reduction system to treat platelets. [Press Release]. 2014. Available from: http://www.cerus.com/Investors/Press-Releases/Press-Release-Details/2014/FDA-Approves-INTERCEPT-Blood-System-for-Platelets/default.aspx.
- 23. US Food and Drug Administration . Summary of safety and effectiveness data: INTERCEPT blood system for platelets. [Press Release]. 2014. Available from: https://www.fda.gov/downloads/BiologicsBloodVaccines/BloodBloodProducts/ApprovedProducts/PremarketApprovalsPMAs/UCM431243.pdf.
- 24. Cerus Corporation . INTERCEPT Blood System for Platelets [package insert]. Concord (CA): Cerus Corporation; 2015.
- 25. Mundt J, Rouse L, Van den Bossche J, et al. Chemical and biological mechanisms of pathogen reduction technologies. Photochem Photobiol 2014;90:957‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schlenke P. Pathogen inactivation technologies for cellular blood components: an update. Transfus Med Hemother 2014;41:309‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Seltam A. Pathogen inactivation of cellular blood products‐ an additional safety layer in transfusion medicine. Front Med (Lausanne) 2017;4:219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Terumo BCT. Terumo BCT announces enrollment of the first patient in its U.S. clinical trial designed to study platelets treated with the Mirasol® pathogen reduction technology (PRT) system [Internet]. Lakewood (CO): Terumo BCT; 2017. Available from: https://www.terumobct.com/Pages/News/Press%20Releases/Terumo-BCT-Announces-Enrollment-of-the-First-Patient--in-Its-U-S--Clinical-Trial-Designed-to-Study-Platelets-Treated--With-.aspx.
- 29. Thiele T, Pohler P, Kohlmann T, et al. Tolerance of platelet concentrates treated with UVC‐light only for pathogen reduction‐ a phase I clinical trial. Vox Sang 2015;109:44‐51. [DOI] [PubMed] [Google Scholar]
- 30. Jaquot C, Delaney M. Pathogen‐inactivated blood products for pediatric patients: blood safety, patient safety, or both? Transfusion 2018;58:2095‐101. [DOI] [PubMed] [Google Scholar]
- 31. Amato M, Schennach H, Astl M, et al. Impact of platelet pathogen inactivation on blood component utilization and patient safety in a large Austrian regional medical center. Vox Sang 2017;112:47‐55. [DOI] [PubMed] [Google Scholar]
- 32. Nussbaumer W, Amato M, Schennach H, et al. Patient outcomes and amotosalen/UVA‐treated platelet utilization in massively transfused patients. Vox Sang 2017;112:249‐56. [DOI] [PubMed] [Google Scholar]
- 33. Knutson F, Osselaer J, Pierelli L, et al. A prospective, active haemovigilance study with combined cohort analysis of 19 175 transfusions of platelet components prepared with amotosalen‐UVA photochemical treatment. Swissmedic Haemovigilance Annual Report 2014. Vox Sang 2015;109:343‐52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gokhale A, Schulz W, Bahar B, et al. Transfusion reaction rates of pathogen reduced (PR) vs conventional (CONV) platelets in adults: a single academic center experience. Transfusion 2018;58(S2). https://onlinelibrary.wiley.com/doi/10.1111/trf.14903):6A‐254A. [Google Scholar]
- 35. Gehrie E, Schulz W, Young P, et al. A retrospective evaluation of the hemostatic efficacy of pathogen reduced (PR) vs conventional (CONV) platelets at an academic medical center. Transfusion 2018;58(S2). https://onlinelibrary.wiley.com/doi/10.1111/trf.14903):6A‐254A. [Google Scholar]
- 36. Schulz W, Gokhale A, McPadden J, et al. Transfusion of pathogen reduced (PR) vs conventional (CONV) platelets in pediatric patients: an assessment of platelet usage and incidence of transfusion reactions. Transfusion 2018;58(S2). https://onlinelibrary.wiley.com/doi/10.1111/trf.14903):6A‐254A. [Google Scholar]
- 37. McCullough J, Goldfinger D, Gorlin J, et al. Cost implications of implementation of pathogen‐inactivated platelets. Transfusion 2015;55:2312‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Prioli K, Katz J, Lyons N, et al. Economic implications of pathogen reduced and bacterially tested platelet components: a US hospital budget impact model. Appl Health Econ Health Policy 2018;16:889‐99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. de Alarcon P, Benjamin R, Dugdale M, et al. Fresh frozen plasma prepared with amotosalen HCl (S‐59) photochemical pathogen inactivation: transfusion of patients with congenital factor deficiencies. Transfusion 2005;45:1362‐72. [DOI] [PubMed] [Google Scholar]
- 40. Larrea L, Ortiz‐de‐Salazar M‐I, Martinez P, et al. Quantitative analysis of plasma proteins in whole‐blood derived fresh frozen plasma prepare with three pathogen reduction technologies. Transfus Apher Sci 2009;41:199‐204. [DOI] [PubMed] [Google Scholar]
- 41. Smith J, Rock G. Protein quality in Mirasol pathogen reduction technology‐treated apheresis‐derived fresh‐frozen plasma. Transfusion 2010;50:926‐31. [DOI] [PubMed] [Google Scholar]
- 42. Coene J, Devreese K, Sabot B, et al. Paired analysis of plasma proteins and coagulation capacity after treatment with three methods of pathogen reduction. Transfusion 2014;54:1321‐31. [DOI] [PubMed] [Google Scholar]
- 43. Hornsy VS, Drummond O, Morrison A, et al. Pathogen reduction of fresh plasma using riboflavin and ultraviolet light: effects on plasma coagulation proteins. Transfusion 2009;49:2167‐72. [DOI] [PubMed] [Google Scholar]
- 44. Theusinger OM, Goslings D, Studt J‐D, et al. Quarantine versus pathogen‐reduced plasma‐coagulation factor content and rotational thromboelastometry coagulation. Transfusion 2017;57:637‐45. [DOI] [PubMed] [Google Scholar]
- 45. Irsch J, Pinkosky L, Corash L, et al. Intercept plasma: comparability with conventional fresh‐frozen plasma based on coagulation function ‐ an in vitro analysis. Vox Sang 2010;98:47‐55. [DOI] [PubMed] [Google Scholar]
- 46. Solheim BG, Chetty R, Flesland O. Indications for use and cost‐effectiveness of pathogen‐reduced ABO‐universal plasma. Curr Opin Hematol 2008;15:612‐7. [DOI] [PubMed] [Google Scholar]
- 47. Cid J, Caballo C, Pino M, et al. Quantitative and qualitative analysis of coagulation factors in cryoprecipitate prepared from fresh‐frozen plasma inactivated with amotosalen and ultraviolet A light. Transfusion 2013;53:600‐5. [DOI] [PubMed] [Google Scholar]
- 48. Ettinger A, Miklauz MM, Bihm DJ, et al. Preparation of cryoprecipitate from riboflavin and UV‐light treated plasma. Transfus Apher Sci 2012;46:153‐8. [DOI] [PubMed] [Google Scholar]
- 49. Hubbard T, Backholer L, Wiltshire M, et al. Effect of riboflavin and amotosalen photoactivation systems for pathogen inactivation of fresh‐frozen plasma on fibrin clot structure. Transfusion 2016;56:41‐8. [DOI] [PubMed] [Google Scholar]
- 50. Cinqualbre J, Kientz D, Remy E, et al. Comparative effectiveness of plasma prepared with amotosalen‐UVA pathogen inactivation and conventional plasma for support of liver transplantation. Transfusion 2015;55:1710‐20. [DOI] [PubMed] [Google Scholar]
- 51. Mintz PD, Neff A, MacKenzie M, et al. A randomized, controlled Phase III trial of therapeutic plasma exchange with fresh‐frozen plasma (FFP) prepared with amotosalen and ultraviolet A light compared to untreated FFP in thrombotic thrombocytopenic purpura. Transfusion 2006;46:1693‐704. [DOI] [PubMed] [Google Scholar]
- 52. Guignier C, Benamara A, Oriol P, et al. Amotosalen‐inactivated plasma is equally well tolerated as quarantine plasma in patients undergoing large volume therapeutic plasma exchange. Transfus Clin Biol 2018;25:73‐7. [DOI] [PubMed] [Google Scholar]
- 53. Haugaa H, Taraldsrud E, Nyrerød HC, et al. Low incidence of hyperfibrinolysis and thromboembolism in 195 primary liver transplantations transfused with solvent/detergent‐treated plasma. Clin Med Res 2014;12:27‐32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yarranton H, Cohen H, Pavord SR, et al. Venous thromboembolism associated with the management of acute thrombotic thrombocytopenic purpura. Br J Haematol 2003;121:778‐85. [DOI] [PubMed] [Google Scholar]
- 55. Hess JR, Pagano MB, Barbeau JD, et al. Will pathogen reduction of blood components harm more people than it helps in developed countries. Transfusion 2016;56:1236‐41. [DOI] [PubMed] [Google Scholar]
- 56. Arbaeen AF, Schubert P, Serrano K, et al. Pathogen inactivation treatment of plasma and platelet concentrates and their predicted functionality in massive transfusion protocols. Transfusion 2017;57:1208‐17. [DOI] [PubMed] [Google Scholar]
- 57. Saadah NH, van Hout FMA, Schipperus NR, et al. Comparing transfusion reaction rates for various plasma types: a systematic review and meta‐analysis/regression. Transfusion 2017;57:2104‐14. [DOI] [PubMed] [Google Scholar]
- 58. Solheim BG, Seghatchian J. Update on pathogen reduction technology for therapeutic plasma: an overview. Transf Apher Sci 2006;35:83‐90. [DOI] [PubMed] [Google Scholar]
- 59. Hellstern P, Solheim BG. The use of solvent/detergent treatment in pathogen reduction of plamsa. Transf Med Hemother 2011;38:65‐70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Riedler GF, Haycox AR, Duggan AK, et al. Cost‐effectiveness of solvent/detergent‐treated fresh‐frozen plasma. Vox Sang 2003;85:88‐95. [DOI] [PubMed] [Google Scholar]
- 61. AuBuchon JP, Birkmeyer JD. Safety and cost‐effectiveness of solvent‐detergent treated plasma. In search of a zero‐risk blood supply. JAMA 1994;272:1210‐4. [PubMed] [Google Scholar]
- 62. Stramer SL, Dodd RY. Transfusion‐transmitted emerging infectious diseases: 30 years of challenges and progress. Transfusion 2013;53:2375‐83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gallin JI. The NIH Clinical Center and the future of clinical research. Nat Med 2011;17:1221‐3. [DOI] [PubMed] [Google Scholar]
- 64. Devine DV. Pathogen inactivation strategies to improve blood safety: let's not throw pathogen‐reduced platelets out with their bath water. JAMA Oncol 2018;4:458‐9. [DOI] [PubMed] [Google Scholar]
- 65. Booth GS, Lozier JN, Nghiem K, et al. Spray: single‐donor plasma product for room temperature storage. Transfusion 2012;52:828‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Horowitz B, Bonomo R, Prince AM, et al. Solvent/detergent‐treated plasma: a virus‐inactivated substitute for fresh frozen plasma. Blood 1992;79:826‐31. [PubMed] [Google Scholar]
- 67. Custer B, Agapova M, Martinez RH. The cost‐effectiveness of pathogen reduction technology as assessed using a multiple risk reduction model. Transfusion 2010;50:2461‐73. [DOI] [PubMed] [Google Scholar]
- 68. Custer B, Zou S, Glynn SA, et al. Addressing gaps in international blood availability and transfusion safety in low‐ and middle‐income countries: a NHLBI workshop. Transfusion 2018;58:1307‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Jutzi M, Mansouri Taleghani B, Rueesch M, et al. Nationwide implementation of pathogen inactivation for all platelet concentrates in Switzerland. Transfus Med Hemother 2018;45:151‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Klein HG. Cellular gene therapy: an overview. J Clin Apher 1994;9:139‐41. [DOI] [PubMed] [Google Scholar]
- 71. Carter CS, Leitman SF, Cullis H, et al. Technical aspects of lymphokine‐activated killer cell production. J Clin Apher 1988;4:113‐7. [DOI] [PubMed] [Google Scholar]
- 72. Klein HG. Immunologic aspects of blood transfusion. Semin Oncol 1994;21:16‐20. [PubMed] [Google Scholar]
- 73. Willy C, Reithmeier W, Kuhlmann WD, et al. Leukocyte depletion of red cell components prevents exposure of transfusion recipients to neutrophil elastase. Vox Sang 2000;78:19‐27. [DOI] [PubMed] [Google Scholar]
- 74. Klein H, Natanson C, Flegel W. Transfusion of fresh vs. older red blood cells in the context of infection. ISBT Sci Ser 2015;10:275‐85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Flegel WA, Natanson C, Klein HG. Does prolonged storage of red blood cells cause harm? Br J Haematol 2014;165:3‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Lozano M, Cid J, Prowse C, et al. Pathogen inactivation or pathogen reduction: proposal for standardization of nomenclature. Transfusion 2015;55:690. [DOI] [PubMed] [Google Scholar]
- 77. Klein HG. Will blood transfusion ever be safe enough? JAMA 2000;284:238‐40. [DOI] [PubMed] [Google Scholar]
- 78. Klein HG, Glynn SA, Ness PM, et al. Research opportunities for pathogen reduction/inactivation of blood components: summary of an NHLBI workshop. Transfusion 2009;49:1262‐8. [DOI] [PubMed] [Google Scholar]
- 79. Regan D, Markowitz MA. Changes to the 30th edition of Standards for Blood Banks and Transfusion Services. AABB Association Bulletin 2016;#16‐05:1‐4. [Google Scholar]
- 80. Snyder EL, Stramer SL, Benjamin RJ. The safety of the blood supply ‐ time to raise the bar. N Engl J Med 2015;372:1882‐5. [DOI] [PubMed] [Google Scholar]
- 81. van Rhenen DJ, Vermeij J, Mayaudon V, et al. Functional characteristics of S‐59 photochemically treated platelet concentrates derived from buffy coat. Vox Sang 2000;76:209‐14. [DOI] [PubMed] [Google Scholar]
- 82. Johnson L, Loh YS, Kwok M, et al. In vitro assessment of buffy‐coat derived platelet components suspended in SSP+ treated with the INTERCEPT Blood system. Transfus Med 2013;23:121‐9. [DOI] [PubMed] [Google Scholar]
- 83. Picker SM, Speer R, Gathof BS. Functional characteristics of buffy coat PLTs photochemically treated with amotosalen‐HCl for pathogen inactivation. Transfusion 2004;44:320‐9. [DOI] [PubMed] [Google Scholar]
- 84. Schubert P, Culibrk B, Karwal S, et al. Whole blood treated with riboflavin and ultraviolet light: quality assessment of all blood components produced by the buffy coat method. Transfusion 2015;55:815‐23. [DOI] [PubMed] [Google Scholar]
- 85. Osman A, Hitzler WE, Meyer CU, et al. Effects of pathogen reduction systems on platelet microRNAs, mRNAs, activation, and function. Platelets 2015;26:154‐63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Klein‐Bosgoed C, Schubert P, Devine DV. Riboflavin and ultraviolet illumination affects selected platelet mRNA transcript amounts differently. Transfusion 2016;56:2286‐95. [DOI] [PubMed] [Google Scholar]
- 87. Schubert P, Culibrk B, Lautens B, et al. Increased cytokine release from platelet concentrates following riboflavin/UV light treatment accelerates endothelial cell proliferation and migration. Transfusion 2013;53(suppl):S84‐040A. [Google Scholar]
- 88. van Rhenen D, Gulliksson H, Cazenave JP, et al. Transfusion of pooled buffy coat platelet components prepared with photochemical pathogen inactivation treatment: the euroSPRITE trial. Blood 2003;101:2426‐33. [DOI] [PubMed] [Google Scholar]
- 89. McCullough J, Vesole DH, Benjamin RJ, et al. Therapeutic efficacy and safety of platelets treated with a photochemical process for pathogen inactivation: the SPRINT Trial. Blood 2004;104:1534‐41. [DOI] [PubMed] [Google Scholar]
- 90. Estcourt LJ, Malouf R, Murphy MF. Pathogen reduced platelets for the prevention of bleeding in people of any age. JAMA Oncol 2018;4:571‐2. [DOI] [PubMed] [Google Scholar]
- 91. Garban F, Guyard A, Labussière H, et al. Evaluation of the Efficacy of Platelets Treated With Pathogen Reduction Process (EFFIPAP) Study Group. Comparison of the hemostatic efficacy of pathogen‐reduced platelets vs untreated platelets in patients with thrombocytopenia and malignant hematologic diseases: a randomized clinical trial. JAMA Oncol 2018;4:468‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. van der Meer PF, Ypma PF, van Geloven N, et al. Hemostatic efficacy of pathogen‐inactivated vs untreated platelets: a randomized controlled trial. Blood 2018;132:223‐31. [DOI] [PubMed] [Google Scholar]
- 93. Knutson F, Osselaer J, Pierelli L, et al. A prospective, active haemovigilance study with combined cohort analysis of 19,175 transfusions of platelet components prepared with amotosalen‐UVA photochemical treatment. Vox Sang 2015;109:343‐52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Liumbruno GM, Marano G, Grazzini G, et al. Solvent/detergent‐treated plasma: a tale of 30 years of experience. Expert Rev Hematol 2015;8:367‐74. [DOI] [PubMed] [Google Scholar]
- 95. Bost V, Odent‐Malaure H, Chavarin P, et al. A regional haemovigilance retrospective study of four types of therapeutic plasma in a ten‐year survey period in France. Vox Sang 2013;104:337‐41. [DOI] [PubMed] [Google Scholar]
- 96. Haubelt H, Blome M, Kiessling AH, et al. Effects of solvent/detergent‐treated plasma and fresh‐frozen plasma on haemostasis and fibrinolysis in complex coagulopathy following open‐heart surgery. Vox Sang 2002;82:9‐14. [DOI] [PubMed] [Google Scholar]
- 97. Scully M, Longair I, Flynn M, et al. Cryosupernatant and solvent detergent fresh‐frozen plasma (Octaplas) usage at a single centre in acute thrombotic thrombocytopenic purpura. Vox Sang 2007;93:154‐8. [DOI] [PubMed] [Google Scholar]
- 98. Solheim BG, Granov DA, Juravlev VA, et al. Universal fresh‐frozen plasma (Uniplas): an exploratory study in adult patients undergoing elective liver resection. Vox Sang 2005;89:19‐26. [DOI] [PubMed] [Google Scholar]
- 99. Toussaint‐Hacquard M, Coppo P, Soudant M, et al. Type of plasma preparation used for plasma exchange and clinical outcome of adult patients with acquired idiopathic thrombotic thrombocytopenic purpura: a French retrospective multicenter cohort study. Transfusion 2015;55:2445‐51. [DOI] [PubMed] [Google Scholar]
- 100. Vendramin C, McGuckin S, Alwan F, et al. A single‐center prospective study on the safety of plasma exchange procedures using a double‐viral‐inactivated and prion‐reduced solvent/detergent fresh‐frozen plasma as the replacement fluid in the treatment of thrombotic microangiopathy. Transfusion 2017;57:131‐6. [DOI] [PubMed] [Google Scholar]
- 101. Witt V, Pichler H, Beiglboeck E, et al. Changes in hemostasis caused by different replacement fluids and outcome in therapeutic plasma exchange in pediatric patients in a retrospective single center study. Transfus Apher Sci 2017;56:59‐65. [DOI] [PubMed] [Google Scholar]
- 102. de Jonge J, Groenland TH, Metselaar HJ, et al. Fibrinolysis during liver transplantation is enhanced by using solvent/detergent virus‐inactivated plasma (ESDEP). Anesth Analg 2002;94:1127‐31. [DOI] [PubMed] [Google Scholar]
- 103. Magner JJ, Crowley KJ, Boylan JF. Fatal fibrinolysis during orthotopic liver transplantation in patients receiving solvent/detergent‐treated plasma (Octaplas). J Cardiothorac Vasc Anesth 2007;21:410‐3. [DOI] [PubMed] [Google Scholar]
- 104. Heger A, Romisch J, Svae TE. A biochemical comparison of a pharmaceutically licensed coagulation active plasma (Octaplas) with a universally applicable development product (Uniplas) and single‐donor FFPs subjected to methylene‐blue dye and white‐light treatment. Transfus Apher Sci 2006;35:223‐33. [DOI] [PubMed] [Google Scholar]
- 105. Jilma‐Stohlawetz P, Kursten FW, Horvath M, et al. Recovery, safety, and tolerability of a solvent/detergent‐treated and prion‐safeguarded transfusion plasma in a randomized, crossover, clinical trial in healthy volunteers. Transfusion 2013;53:1906‐17. [DOI] [PubMed] [Google Scholar]
- 106. Bartelmaos T, Chabanel A, Leger J, et al. Plasma transfusion in liver transplantation: a randomized, double‐blind, multicenter clinical comparison of three virally secured plasmas. Transfusion 2013;53:1335‐45. [DOI] [PubMed] [Google Scholar]
- 107. Bindi ML, Miccoli M, Marietta M, et al. Solvent detergent vs. fresh frozen plasma in cirrhotic patients undergoing liver transplant surgery: a prospective randomized control study. Vox Sang 2013;105:137‐43. [DOI] [PubMed] [Google Scholar]
- 108. Stensballe J, Ulrich AG, Nilsson JC, et al. Resuscitation of endotheliopathy and bleeding in thoracic aortic dissections: the VIPER‐OCTA randomized clinical pilot trial. Anesth Analg 2018;127:920‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Tølløfsrud S, Noddeland H, Svennevig JL, et al. Universal fresh frozen plasma (Uniplas): a safe product in open‐heart surgery. Intensive Care Med 2003;29:1736‐43. [DOI] [PubMed] [Google Scholar]
- 110. Williamson LM, Llewelyn CA, Fisher NC, et al. A randomized trial of solvent/detergent‐treated and standard fresh‐frozen plasma in the coagulopathy of liver disease and liver transplantation. Transfusion 1999;39:1227‐34. [DOI] [PubMed] [Google Scholar]
- 111. Noddeland H, Tollofsrud S, Svennevig J, et al. Universal solvent/detergent‐treated fresh frozen plasma (Uniplas‐‐rationale and clinical properties). Thromb Res 2002;107(Suppl 1):S33‐7. [DOI] [PubMed] [Google Scholar]
- 112. Trueman P, Drummond M, Hutton J. Developing guidance for budget impact analysis. Pharmacoeconomics 2001;19:609‐21. [DOI] [PubMed] [Google Scholar]
- 113. Custer B, Hoch JS. Cost‐effectiveness analysis: what it really means for transfusion medicine decision making. Transfus Med Rev 2009;23:1‐12. [DOI] [PubMed] [Google Scholar]
- 114. Custer B. Health economics and blood safety In: Shan H. Dodd RY, editors. Blood Safety: A Guide to Monitoring and Responding to Potential New Threats. New York: Springer International Publishing; 2019. p. 53‐81. [Google Scholar]
- 115. de Sousa G, Seghatchian J. Highlights of PBTI Coimbra Conference on PRT of plasma & current opinions on pathogen reduction treatment of blood components. Transfus Apher Sci 2015;52:228‐32. [DOI] [PubMed] [Google Scholar]
- 116. Seltsam A, Muller TH. Update on the use of pathogen‐reduced human plasma and platelet concentrates. Br J Haematol 2013;162:442‐54. [DOI] [PubMed] [Google Scholar]
- 117. Prowse CV. Component pathogen inactivation: a critical review. Vox Sang 2013;104:183‐99. [DOI] [PubMed] [Google Scholar]
- 118. Di Minno G, Navarro D, Perno CF, et al. Pathogen reduction/inactivation of products for the treatment of bleeding disorders: what are the processes and what should we say to patients? Ann Hematol 2017;96:1253‐70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Yonemura S, Doane S, Keil S, et al. Improving the safety of whole blood‐derived transfusion products with a riboflavin‐based pathogen reduction technology. Blood Transfus 2017;15:357‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Cancelas JA, Gottschall JL, Rugg N, et al. Red blood cell concentrates treated with the amustaline (S‐303) pathogen reduction system and stored for 35 days retain post‐transfusion viability: results of a two‐centre study. Vox Sang 2017;112:210‐8. [DOI] [PubMed] [Google Scholar]
- 121. Drew VJ, Barro L, Seghatchian J, et al. Towards pathogen inactivation of red blood cells and whole blood targeting viral DNA/RNA: design, technologies, and future prospects for developing countries. Blood Transfus 2017;15:512‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Wiltshire M, Meli A, Schott MA, et al. Quality of red cells after combination of prion reduction and treatment with the intercept system for pathogen inactivation. Transfus Med 2016;26:208‐14. [DOI] [PubMed] [Google Scholar]
- 123. Allain JP, Owusu‐Ofori AK, Assennato SM, et al. Effect of plasmodium inactivation in whole blood on the incidence of blood transfusion‐transmitted malaria in endemic regions: the African Investigation of the Mirasol System (AIMS) randomised controlled trial. Lancet 2016;387:1753‐61. [DOI] [PubMed] [Google Scholar]
- 124. Membe SK, Coyle D, Husereau D, et al. Octaplas Compared with Fresh Frozen Plasma to Reduce the Risk of Transmitting Lipid‐Enveloped Viruses: An Economic Analysis and Budget Impact Analysis. Ottawa ON: Canadian Agency for Drugs and Technologies in Health (CADTH); 2011. [PubMed] [Google Scholar]
- 125. Cicchetti A, Coretti S, Sacco F, et al. Budget impact of implementing platelet pathogen reduction into the Italian blood transfusion system. Blood Transfus 2018;16:483‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Girona‐Llobera E, Jimenez‐Marco T, Galmes‐Trueba A, et al. Reducing the financial impact of pathogen inactivation technology for platelet components: our experience. Transfusion 2014;54:158‐68. [DOI] [PubMed] [Google Scholar]
- 127. Walsh GM, Shih AW, Solh Z, et al. Blood‐Borne Pathogens: A Canadian Blood Services Centre for Innovation Symposium. Transfus Med Rev 2016;30:53‐68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Jacquot C, Delaney M. Efforts toward elimination of infectious agents in blood products. J Intensive Care Med 2018;33:543‐50. [DOI] [PubMed] [Google Scholar]
- 129. Picker SM, Schneider V, Oustianskaia L, et al. Cell viability during platelet storage in correlation to cellular metabolism after different pathogen reduction technologies. Transfusion 2009;49:2311‐8. [DOI] [PubMed] [Google Scholar]
- 130. Schubert P, Johnson L, Marks DC, et al. Ultraviolet‐based pathogen inactivation systems: untangling the molecular targets activated in platelets. Front Med (Lausanne) 2018;5:129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Rock G. A comparison of methods of pathogen inactivation of FFP. Vox Sang 2011;100:169‐78. [DOI] [PubMed] [Google Scholar]
- 132. Butler C, Doree C, Estcourt LJ, et al. Pathogen‐reduced platelets for the prevention of bleeding. Cochrane Database Syst Rev 2013;3:CD009072. [DOI] [PubMed] [Google Scholar]
- 133. Estcourt LJ, Malouf R, Hopewell S, et al. Pathogen‐reduced platelets for the prevention of bleeding. Cochrane Database Syst Rev 2017;7:CD009072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Reddy HL, Dayan AD, Cavagnaro J, et al. Toxicity testing of a novel riboflavin‐based technology for pathogen reduction and white blood cell inactivation. Transfus Med Rev 2008;22:133‐53. [DOI] [PubMed] [Google Scholar]
- 135. Henschler R, Seifried E, Mufti N. Development of the S‐303 pathogen inactivation technology for red blood cell concentrates. Transfus Med Hemother 2011;38:33‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Nkohkwo A, Agbor G, Asongalem E, et al. Whole blood pathogen reduction technology and blood safety in sub‐Saharan Africa: a systematic review with regional discussion. Afr J Lab Med 2016;5:363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Stramer SL. Current perspectives in transfusion‐transmitted infectious diseases: emerging and re‐emerging infections. ISBT Sci Ser 2014;9:30‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Bah A, Cardoso M, Seghatchian J, et al. Reflections on the dynamics of bacterial and viral contamination of blood components and the levels of efficacy for pathogen inactivation processes. Transfus Apher Sci 2018;57:683‐8. 10.1016/j.transci.2018.09.004. [DOI] [PubMed] [Google Scholar]
- 139. McCullough J, Alter HJ, Ness PM. Interpretation of pathogen load in relationship to infectivity and pathogen reduction efficacy. Transfusion 2018;59:1132‐46. 10.1111/trf.15103. [DOI] [PubMed] [Google Scholar]
- 140. Hauser L1, Roque‐Afonso AM, Beylouné A, et al. Hepatitis E transmission by transfusion of intercept blood system‐treated plasma. Blood 2014;123:796‐7. [DOI] [PubMed] [Google Scholar]
- 141. Mirasol Evaluation of Reduction in Infections Trial (MERIT) . http://ClinicalTrials.gov. Mirasol Evaluation of Reduction in Infections Trial (MERIT) [Internet]. Identifier NCT03737669. Bethesda (MD): National Library of Medicine; 2018. Available from: https://clinicaltrials.gov/ct2/show/NCT03737669.
- 142. Lozano M, Cid J. Analysis of reasons for not implementing pathogen inactivation for platelet concentrates. Transfus Clin Biol 2013;20:158‐64. [DOI] [PubMed] [Google Scholar]
- 143. Goodrich RP, Segatchian J. Special considerations for the use of pathogen reduced blood components in pediatric patients: an overview. Transfus Apher Sci 2018;57:374‐7. [DOI] [PubMed] [Google Scholar]
- 144. JICA Hands over Blood Safety Technology Project to National Blood Service . National Blood Service Ghana. [cited 2018 Sep 17]. Available from: https://nbsghana.org/jica-hands-over-blood-safety-technology-project-to-national-blood-service/.
- 145. Winter KM, Johnson L, Kwok M, et al. Red blood cell in vitro quality and function is maintained after S‐303 pathogen inactivation treatment. Transfusion 2014;54:1798‐807. [DOI] [PubMed] [Google Scholar]
- 146. North A, Ciaravino V, Mufti N, et al. Preclinical pharmacokinetic and toxicology assessment of red blood cells prepared with S‐303 pathogen inactivation treatment. Transfusion 2011;51:2208‐18. [DOI] [PubMed] [Google Scholar]
- 147. Mufti NA, Erickson AC, North AK, et al. Treatment of whole blood (WB) and red blood cells (RBC) with S‐303 inactivates pathogens and retains in vitro quality of stored RBC. Biologicals 2010;38:14‐9. [DOI] [PubMed] [Google Scholar]
- 148. Cancelas JA, Dumont LJ, Rugg N, et al. Stored red blood cell viability is maintained after treatment with a second‐generation S‐303 pathogen inactivation process. Transfusion 2011;51:2367‐76. [DOI] [PubMed] [Google Scholar]
- 149. Brixner V, Kiessling AH, Madlener K, et al. Red blood cells treated with the amustaline (S‐303) pathogen reduction system: a transfusion study in cardiac surgery. Transfusion 2018;58:905‐16. [DOI] [PubMed] [Google Scholar]
- 150. JICA hands over blood safety technology project to MOH [Internet]. Accra, Ghana: BusinessGhana; 2018 [cited 2018 Sep 14]. Available from: https://www.businessghana.com/site/news/business/172359/JICA-hands-over-blood-safety-technology-project-to-MOH.
- 151.“Efficacy and Safety of RBCs Derived From Mirasol‐treated Whole Blood in Patients Requiring Chronic Transfusion (PRAISE).” [cited 2019 Feb 5]. Available from: https://clinicaltrials.gov/ct2/show/NCT03329404.
- 152. Trakhtman P, Kumukova I, Starostin N, et al. The pathogen‐reduced red blood cell suspension: single centre study of clinical safety and efficacy in children with oncological and haematological diseases. Vox Sang 2019;114:223‐31. [DOI] [PubMed] [Google Scholar]
- 153. Kumar V, Lockerbie O, Keil SD, et al. Riboflavin and UV‐light based pathogen reduction: extent and consequence of DNA damage at the molecular level. Photochem Photobiol 2004;80:15‐21. [DOI] [PubMed] [Google Scholar]
- 154. Marschner S, Goodrich R. Pathogen reduction technology treatment of platelets, plasma and whole blood using riboflavin and UV light. Transfus Med Hemother 2011;38:8‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Cancelas JA, Rugg N, Pratt PG, et al. Red cells from riboflavin plus UV‐light treated whole blood can be effectively radiolabeled with 51‐Cr for viabiliity studies. Transfusion 2009;49(S3):108A (S44).18954396 [Google Scholar]
- 156. Cancelas JA, Rugg N, Fletcher D, et al. In vivo viability of stored red blood cells derived from riboflavin plus ultraviolet light‐treated whole blood. Transfusion 2011;51:1460‐8. [DOI] [PubMed] [Google Scholar]
- 157. Tonnetti L, Thorp AM, Reddy HL, et al. Evaluating pathogen reduction of Trypanosoma cruzi with riboflavin and ultraviolet light for whole blood. Transfusion 2012;52:409‐16. [DOI] [PubMed] [Google Scholar]
- 158. El Chaar M, Atwal S, Freimanis GL, et al. Inactivation of Plasmodium falciparum in whole blood by riboflavin plus irradiation. Transfusion 2013;53:3174‐83. [DOI] [PubMed] [Google Scholar]
- 159. Fast LD, Nevola M, Tavares J, et al. Treatment of whole blood with riboflavin plus ultraviolet light, an alternative to gamma irradiation in the prevention of transfusion‐associated graft‐versus‐host disease? Transfusion 2013;53:373‐81. [DOI] [PubMed] [Google Scholar]
- 160. Reddy HL, Doane SK, Keil SD, et al. Development of a riboflavin and ultraviolet light‐based device to treat whole blood. Transfusion 2013;53(Suppl 1):131S‐6S. [DOI] [PubMed] [Google Scholar]
- 161. Tonnetti L, Thorp AM, Reddy HL, et al. Riboflavin and ultraviolet light reduce the infectivity of Babesia microti in whole blood. Transfusion 2013;53:860‐7. [DOI] [PubMed] [Google Scholar]
- 162. Okoye OT, Reddy H, Wong MD, et al. Large animal evaluation of riboflavin and ultraviolet light‐treated whole blood transfusion in a diffuse, nonsurgical bleeding porcine model. Transfusion 2015;55:532‐43. [DOI] [PubMed] [Google Scholar]
- 163. Owusu‐Ofori S, Kusi J, Owusu‐Ofori A, et al. Treatment of whole blood with riboflavin and UV light: impact on malaria parasite viability and whole blood storage. Shock 2015;44(Suppl 1):33‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Tonnetti L, Thorp AM, Reddy HL, et al. Reduction of Leishmania donovani infectivity in whole blood using riboflavin and ultraviolet light. Transfusion 2015;55:326‐9. [DOI] [PubMed] [Google Scholar]
- 165. Cancelas JA, Slichter SJ, Rugg N, et al. Red blood cells derived from whole blood treated with riboflavin and ultraviolet light maintain adequate survival in vivo after 21 days of storage. Transfusion 2017;57:1218‐25. [DOI] [PubMed] [Google Scholar]
- 166. Corash L. Inactivation of infectious pathogens in labile blood components: meeting the challenge. Transfus Clin Biol 2001;8:138‐45. [DOI] [PubMed] [Google Scholar]
- 167. Dupuis K, Bernard K, Jones S, et al. Helinx technology inactivates pathogens of emerging importance in red blood cell concentrates. Blood 2003;102(Suppl. 1):816a. [Google Scholar]
- 168. Hanson D, Propst M, Dupuis K. High titer leukocyte inactivation in INTERCEPT red blood cells (RBC). Vox Sang 2001;101a:81. [Google Scholar]
- 169. Mikovits JA, Hagen K, Liu W, et al. Inactivation of XMRV in platelet and RBC components prepared with the INTERCEPT blood system. Rev Antivir Ther Infect Dis 2010;8:36. [Google Scholar]
- 170. Specht KG, Kittler L, Midden WR. A new biological target of fucomarians: photochemical formation of covalent adducts with unsaturated fatty acids. Photochem Photobiol 1988;47:537‐41. [DOI] [PubMed] [Google Scholar]
- 171. Skripchenko A, Wagner SJ, Thompson‐Montgomery D, et al. Thiazole orange, a DNA binding photochemical with flexible structure, can inactivate pathogens in red cell suspensions while maintaining red cell storage properties. Transfusion 2006;46:213‐9. [DOI] [PubMed] [Google Scholar]
- 172. Wagner SJ, Skripchenko A, Salata J, et al. Inactivation of Leishmania donavani infantum and Trypanosoma cruzi in red cell suspensions by thiazole orange. Transfusion 2008;48:1363‐7. [DOI] [PubMed] [Google Scholar]
- 173. Dai T, Gupta A, Huang YY, et al. Blue light rescues mice from potentially fatal Pseudomonas aeruginosa burn infection: efficacy, safety, and mechanism of action. Antimicrob Agents Chemother 2013;57:1238‐45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Maclean M, McKenzie K, Anderson JG, et al. 405 nm light technology for the inactivation of pathogens and its potential role for environmental disinfection and infection control. J Hosp Infect 2014;88:1‐11. [DOI] [PubMed] [Google Scholar]
- 175. Maclean M, White T, Anderson JG, et al. A new proof‐of‐concept in bacterial reduction: antimicrobial action of violet‐blue light (405 nm) in ex vivo stored plasma. J Blood Transfus 2016;2016:2920514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Maclean M, MacGregor SJ, Anderson JG, et al. Inactivation of bacterial pathogens following exposure to light from a 405‐nm LED array. Appl Environ Microbiol 2009;75:1932‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Tomb RM, Maclean M, JE Coia E, et al. Investigating the influence of biologically‐relevant suspending media on 405 nm light inactivation of feline calicivirus: a surrogate of norovirus. Food Environ Virol 2017;9:159‐67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Gonzales R, Taylor AL, Atkinson AJ, et al. US Army blood program: 2025 and beyond. Transfusion 2016;56(Suppl 1):S85‐93. [DOI] [PubMed] [Google Scholar]
- 179. Reddoch‐Cardenas KM, Bynum JA, Meledeo MA, et al. Cold‐stored platelets: A product with function optimized for hemorrhage control. Transfus Apher Sci 2019;58:16‐22. [DOI] [PubMed] [Google Scholar]
- 180. Hoffmeister KM, Felbinger TW, Falet H, et al. The clearance mechanism of chilled blood platelets. Cell 2003;112:87‐97. [DOI] [PubMed] [Google Scholar]
- 181. Apelseth TO, Kristoffersen EK, Kvalheim VL, et al. Transfusion with cold stored platelets in patients undergoing complex cardiothoracic surgery with cardiopulmonary bypass circulation: effect on bleeding and thromboembolic risk. Transfusion 2017;57:3A‐4A.28097696 [Google Scholar]
- 182. Johnson L, Cameron M, Waters L, et al. The impact of refrigerated storage of UVC pathogen inactivated platelet concentrates on in vitro platelet quality parameters. Vox Sang 2019;114:47‐56. [DOI] [PubMed] [Google Scholar]
- 183. Waters L, Cameron M, Padula MP, et al. Refrigeration, cryopreservation and pathogen inactivation: an updated perspective on platelet storage conditions. Vox Sang 2018;113:317‐28. [DOI] [PubMed] [Google Scholar]
- 184. Pidcoke HF, McFaul SJ, Ramasubramanian AK, et al. Primary hemostatic capacity of whole blood: a comprehensive analysis of pathogen reduction and refrigeration effects over time. Transfusion 2013;53(Suppl 1):137S‐49S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Meledeo MA, Peltier GC, McIntosh CS, et al. Hemostatic functional preservation of refrigerated whole blood over 35 days. Transfusion 2018;58:62A. [Google Scholar]
- 186. Cap AP, Perkins JG. Lyophilized platelets: challenges and opportunities. J Trauma 2011;70(5 Suppl):S59‐60. [DOI] [PubMed] [Google Scholar]
- 187. Cap AP, Pidcoke HF, Keil SD, et al. Treatment of blood with a pathogen reduction technology using ultraviolet light and riboflavin inactivates Ebola virus in vitro. Transfusion 2016;56(Suppl 1):S6‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]