

Abstract
Senescence is a state of growth arrest induced not only in normal cells but also in cancer cells by aging or stress, which triggers DNA damage. Despite growth suppression, senescent cancer cells promote tumor formation and recurrence by producing cytokines and growth factors; this state is designated as the senescence‐associated secretory phenotype. In this study, we examined the susceptibility of senescent human breast cancer cells to immune cell‐mediated cytotoxicity. Doxorubicin (DXR) treatment induced senescence in 2 human breast cancer cell lines, MDA‐MB‐231 and BT‐549, with the induction of γH2AX expression and increased expression of p21 or p16. Treatment with DXR also induced the expression of senescence‐associated β‐galactosidase and promoted the production of pro‐inflammatory cytokines. Importantly, DXR‐treated senescent MDA‐MB‐231 cells showed increased sensitivity to 2 types of immune cell‐mediated cytotoxicity: cytotoxicity of activated CD4+ T cells and Ab‐dependent cellular cytotoxicity by natural killer cells. This increased sensitivity to cytotoxicity was partially dependent on tumor necrosis factor‐related apoptosis‐inducing ligand and perforin, respectively. This increased sensitivity was not observed following treatment with the senescence‐inducing cyclin‐dependent kinase‐4/6 inhibitor, abemaciclib. In addition, treatment with DXR, but not abemaciclib, decreased the expression of antiapoptotic proteins in cancer cells. These results indicated that DXR and abemaciclib induced senescence in breast cancer cells, but that they differed in their sensitivity to immune cell‐mediated cytotoxicity. These findings could provide an indication for combining anticancer immunotherapy with chemotherapeutic drugs or molecular targeting drugs.
Keywords: abemaciclib, breast cancer, cytotoxicity, doxorubicin, senescence
1. INTRODUCTION
Breast cancer is the most common type of cancer in women worldwide.1 At present, the main treatment for breast cancer involves chemotherapy consisting of anthracycline and cyclophosphamide in combination with taxanes.2 Furthermore, patients with intractable triple‐negative breast cancer (TNBC) can benefit from chemotherapy, such as doxorubicin (DXR).3 Doxorubicin is an anthracycline drug that induces DNA damage in cancer cells, leading to apoptosis.4
Some cancer cells survive as senescent cells through the DNA damage response after chemotherapy.5 Senescence is a state in which cells undergo irreversible cell cycle arrest in response to various stresses.6 Senescence is induced not only in normal cells but also in cancer cells when anticancer agents trigger DNA damage.7, 8 Recent studies have indicated that senescent cells show growth arrest and increased secretion of secretory proteins, including inflammatory cytokines, chemokines, growth factors, and MMPs, which is designated as the senescence‐associated secretory phenotype (SASP).9 Although growth arrest in senescent cancer cells has a tumor suppressive effect, their SASP features are reported to contribute to tumor promotion and recurrence.10
Immune checkpoint blockade (ICB) Ab therapy has attracted attention as a promising anticancer therapy against some types of cancer.11 To increase its therapeutic efficacy, ICB Ab therapy must be combined with other types of treatment, including chemotherapy and/or radiotherapy.12, 13 Alternatively, chemotherapy has been shown to modulate immune responses both positively and negatively. Chemotherapeutic drugs exert several immune‐enhancing effects, including relief of immune suppressor cells14, 15 and the induction of immunogenic cancer cell death.16 Conversely, they induce immunosuppressive conditions, such as immune suppression when administered at higher doses, subsequent recruitment of immune suppressor cells,17 and the induction of immune checkpoint molecules on cancer cells.18 Clinically, the combination of ICB Ab therapy with chemotherapy has been reported to show therapeutic efficacy.12, 13 However, the mutual effects of anticancer immunotherapy and chemotherapy and how chemotherapeutic drugs influence the sensitivity of cancer cells to cytotoxicity by immune cells have not been fully elucidated.
Although senescent cancer cells with SASP play a crucial role in tumor recurrence and promotion after chemotherapy,4, 10 these cells reduce their growth and vigor. These lines of evidence suggest that senescent cancer cells after chemotherapy could be ideal targets for anticancer immunotherapy and that preferential killing of senescent cancer cells by immune cells might account for suppression of tumor recurrence by immunotherapy after chemotherapy. Therefore, after confirming that the DNA damage‐inducing drug DXR can induce senescence in human breast cancer cells, we examined their susceptibility to 2 different types of immune cell‐mediated cytotoxicity. We also examined the sensitivity of cancer cells treated with either DXR or the cyclin‐dependent kinase (CDK)4/6 inhibitor abemaciclib, which induces senescence.
2. MATERIALS AND METHODS
2.1. Cell lines and reagents
Three human breast cancer cell lines, MDA‐MB‐231, BT‐549, and MCF‐7 (kindly provided by Dr. K. Takenaga, Faculty of Medicine, Shimane University, Izumo, Japan), were maintained in DMEM (Sigma‐Aldrich) supplemented with 10% FCS (Invitrogen) and 20 μg/mL gentamicin (Sigma‐Aldrich) at 37°C in a humidified atmosphere of 5% CO2 in air. Doxorubicin (Sigma‐Aldrich) was diluted first in distilled water and subsequently in PBS. Anti‐tumor necrosis factor‐related apoptosis‐inducing ligand (TRAIL) Ab was purchased from BioLegend. Concanamycin A (CMA) was purchased from Santa Cruz Biotechnology. Recombinant TRAIL was purchased from PeproTech. Anti‐CD3 Ab (clone UCHT1) was purchased from Ancell.
2.2. Preparation of effector immune cells
Anti‐epidermal growth factor receptor (EGFR) chimeric antigen receptor (CAR)‐T cells were purchased from ProMab. After initial thawing, these cells were cultured in RPMI‐1640 supplemented with 2% human serum and 300 U/mL interleukin (IL)‐2 for 10 days. The cultured cells were aliquoted in vials and kept at −70°C. For each experiment, these T cells were expanded in anti‐CD3 Ab‐coated plates with 300 U/mL IL‐2 for 7‐10 days.
2.3. Cell viability assay
Cell viability was measured using the 2‐(2‐methoxy‐4‐nitrophenyl)‐3‐(4‐nitrophenyl)‐5‐(2, 4‐disulfophenyl)‐2H‐tetrazolium monosodium salt (WST‐8) assay (Nacalai Tesque). Briefly, cells were seeded in flat‐bottomed 96‐well plates and DXR was added at the indicated doses. Two days later, 10 μL WST‐8 solution was added to each well after removal of half of the medium, and the plates were incubated for an additional 3 hours. The plates were then read at a wavelength of 450 nm using a microplate reader (Beckman Coulter).
2.4. Flow cytometric analysis
Apoptosis was measured using an annexin V‐FITC Apoptosis Detection Kit (BioVision). To examine the expression of human EGFR on cancer cells, cells were stained with either FITC‐conjugated anti‐EGFR Ab (NeoMarkers) or isotype‐matched control Ab (Beckman Coulter). To examine the expression of human epidermal growth factor receptor 2 (HER2) on cancer cells, cells were stained with either FITC‐conjugated anti‐HER2 Ab (BD Bioscience) or isotype‐matched FITC‐conjugated mouse IgG (Beckman Coulter). To examine the expression of TRAIL on natural killer (NK) cells, cells were stained with anti‐CD56‐allophycocyanin (APC) Ab (EXBIO) and anti‐TRAIL‐phycoerythrin (PE) Abs. To examine the expression of TRAIL on in vitro expanded T cells, these cells were stained with PE‐conjugated anti‐TRAIL Ab. To determine the subset of T cells, expanded T cells were stained with anti‐CD4‐PE and anti‐CD8‐APC Abs. All analyses were undertaken by flow cytometry (FACSCalibur; Becton Dickinson).
2.5. Preparation of human NK cells and Ab‐dependent cell‐mediated cytotoxicity assay
Natural killer cells were prepared from peripheral blood of healthy donors. Peripheral blood mononuclear cells were prepared from blood using Lymphocyte Separation Solution (Nacalai Tesque). Natural killer cells were enriched using an NK Cell Purification Kit (Invitrogen). For the Ab‐dependent cell‐mediated cytotoxicity assay (ADCC) assay, prepared NK cells were cultured with the following Abs: anti‐HER2 Ab (trastuzumab), anti‐EGFR Ab (cetuximab), and anti‐CD20 Ab (rituximab). In some experiments, anti‐TRAIL Ab or CMA, as a perforin inhibitor, was added 1 hour before the assay. The study protocol was approved by the Ethics Review Board of Shimane University Faculty of Medicine (approval no. 20140918‐2).
2.6. Detection of senescence‐associated β‐Gal
Senescence‐associated β‐galactosidase (SA β‐Gal) was detected by flow cytometry and confocal imaging. For flow cytometry, untreated or DXR‐treated cancer cells were cultured with SPiDER β‐Gal (Dojindo Molecular Technologies) for 30 minutes and analyzed by flow cytometry. For confocal imaging, cancer cells were cultured on round coverslips in 24‐well plates with or without DXR for 48 hours. After incubation with SPiDER β‐Gal and Hoechst 33342 (5 μg/mL) for 30 minutes, cells were fixed with 4% paraformaldehyde and placed on slide glasses with 4 μL mounting medium for fluorescence analysis (VECTASHIELD; Vector Laboratories). Confocal imaging was carried out using a laser scanning microscope (FV1000‐D; Olympus).
2.7. Assay of cytotoxicity with flow cytometry
To examine apoptotic cancer cells, they were cocultured with activated T cells or purified NK cells with the indicated Abs in 96‐well plates for 6‐7 hours. After harvesting, whole cells were stained with anti‐CD45‐APC, followed by annexin V‐FITC. To evaluate the percentages of apoptotic cancer cells, annexin V‐FITC+ cells among CD45‐APC− cells were calculated by flow cytometry.
2.8. Immunoblotting assay
Cancer cells were lysed with RIPA Buffer (Fujifilm Wako Pure Chemical) containing a protease inhibitor cocktail (Nacalai Tesque) and a phosphatase inhibitor cocktail (Nacalai Tesque). Equal amounts of protein were resolved on 4%‐12% gradient or 12% SDS‐PAGE gels, followed by transfer onto PVDF membranes. After blocking the membranes, the blots were incubated with the indicated primary Abs: anti‐p21Cip1/Waf1 (#2947; Cell Signaling Technology), anti‐p16Ink4a (SPC‐1280; StressMarq Biosciences), anti‐γH2AX(Ser139) (#9718; Cell Signaling Technology), anti‐cFLIP (ALX‐804‐428; Enzo Life Sciences), anti‐survivin (#2803; Cell Signaling Technology), anti‐XIAP (#2042; Cell Signaling Technology), anti‐Bcl‐2 (#658701; BioLegend), anti‐Bcl‐xL (#2764; Cell Signaling Technology), anti‐Mcl‐1 (sc‐819; Santa Cruz Biotechnology), and anti‐β‐actin (#622102; BioLegend). After washing, membranes were incubated at room temperature for 60 minutes with either goat anti‐rabbit or horse anti‐mouse HRP‐conjugated secondary Ab (#7074 and #7076; Cell Signaling Technology) to detect the primary Abs. Protein bands were visualized using an ImageQuant LAS‐4000 system (Fujifilm).
2.9. Enzyme‐linked immunosorbent assay
The levels of human IL‐6 and IL‐8 in the supernatants were determined using an ELISA kit (PeproTech). The levels of soluble TRAIL in the supernatants of activated T cells were determined using an ELISA kit (PeproTech).
2.10. Statistical analysis
Data were statistically evaluated using the unpaired 2‐tailed Student's t test. In all analyses, P < .05 was taken to indicate statistical significance.
3. RESULTS
3.1. Doxorubicin induces senescence in MDA‐MB‐231 and BT‐549 cells
We first examined the effects of DXR on 3 human breast cancer cell lines. Doxorubicin decreased the cell viability of all cell lines in a dose‐dependent manner (Figure 1A). BT‐549 cells were the most sensitive to DXR, and MCF‐7 cells were the most resistant to DXR. In addition, DXR induced the expression of γH2AX, a DNA damage marker, in MDA‐MB‐231 and BT‐549 cells, but not in MCF‐7 cells (Figure 1B). Furthermore, DXR increased the expression levels of 21Waf1 in MDA‐MB‐231 and MCF‐7 cells and that of p16ink4A in BT‐549 cells (Figure 1C). We next examined whether senescence could be induced in DXR‐treated breast cancer cell lines. In confocal imaging, untreated MDA‐MB‐231 cells were weakly positive for SA β‐Gal, and DXR treatment increased the levels of expression (Figure 1D). Treatment with DXR induced the expression of SA β‐Gal in BT‐549 and MCF‐7 cells. In addition, DXR‐treated MDA‐MB‐231 and BT‐549 cells produced higher levels of IL‐6 and IL‐8 compared to untreated cells, whereas MCF‐7 cells failed to produce these cytokines (Figure 1E). Taken together, these results indicate that DXR induces typical senescence in both MDA‐MB‐231 and BT‐549 cells, but that senescence in DXR‐treated MCF‐7 cells is not apparent.
Figure 1.
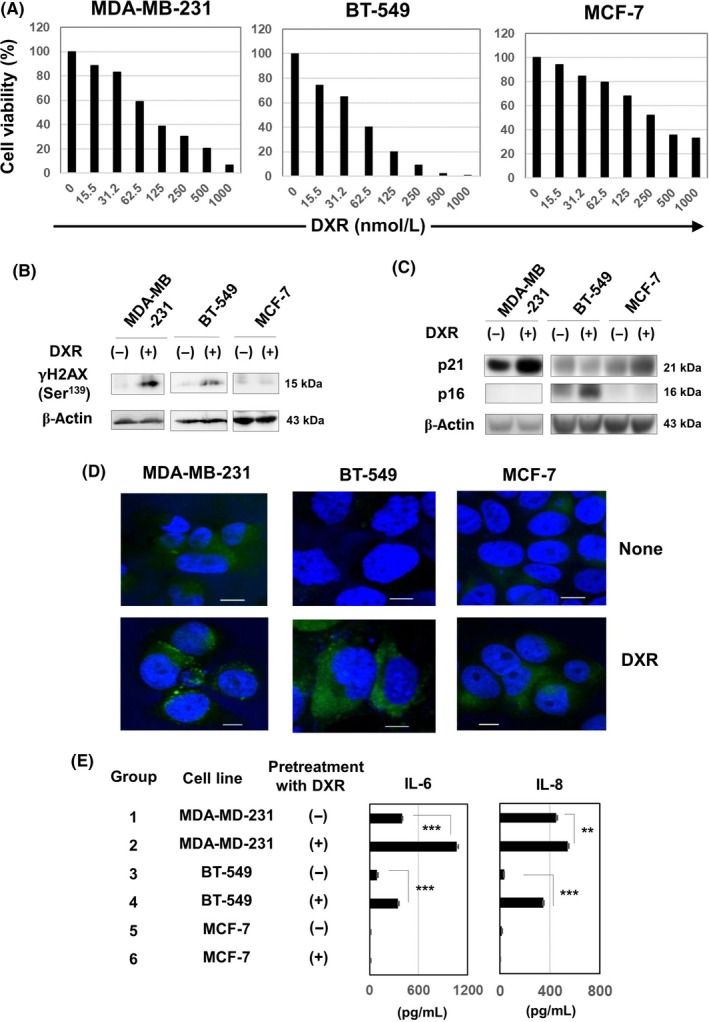
Doxorubicin (DXR) induces senescence in human breast cancer cells. A, Three breast cancer cell lines were cultured with the indicated doses of DXR (nmol/L) for 72 h. Medium alone (background) was subtracted. In these experiments, cell viability (%) was determined using the WST‐8 assay. The results are shown as the means of 3 wells. B, Three breast cancer cell lines were cultured with DXR (250 nmol/L for MDA‐MB‐231, 100 nmol/L for BT‐549, and 200 nmol/L for MCF‐7) for 48 h. Using the tumor lysates, immunoblotting analysis was carried out using anti‐γH2AX Ab. β‐Actin was used as a control. C, Similarly, 3 breast cancer cell lines were cultured with DXR for 48 h. Immunoblotting analysis was undertaken using anti‐p21 and anti‐p16 Abs. β‐Actin was used as a control. D, To examine the expression of senescence‐associated β‐Gal, cancer cells were treated with DXR (250 nmol/L for MDA‐MB‐231, 100 nmol/L for BT‐549, and 200 nmol/L for MCF‐7) for 2 d and stained with SPiDER β‐Gal. Confocal imaging was carried out on untreated or DXR‐treated cancer cells. Scale bar = 10 μm. E, Similarly, 3 cancer cell lines were treated with or without DXR for 2 d. After harvesting, cancer cells were cultured without DXR for 2 d. Thereafter, the levels of interleukin (IL)‐6 and IL‐8 in the supernatants were examined by ELISA. **P < .01, ***P < .005
3.2. Increased sensitivity of DXR‐treated MDA‐MB‐231 and BT‐549 cells to T cells
We next examined whether DXR‐induced senescence could influence the sensitivity of breast cancer cells to immune cell‐mediated cytotoxicity. We attempted to utilize anti‐EGFR CAR‐T cells as effector immune cells because the 3 breast cancer cell lines examined here expressed EGFR on their cell surface (Figure S1). These T cells were used for assays after 2‐day culture in anti‐CD3 Ab‐coated wells with 300 U/mL IL‐2, and subsequently with IL‐2 alone for 7‐10 days. However, expanded T cells were unexpectedly positive for CD4 (Figure 2A). Nevertheless, we undertook experiments using these T cells. Either untreated or DXR‐treated MDA‐MB‐231 cells were cocultured with activated T cells, and the percentages of apoptotic cancer cells were examined by flow cytometry by gating on CD45− cancer cells. Treatment with DXR was shown to significantly increase the susceptibility of MDA‐MB‐231 cells to T cells (Figure 2B). We further examined whether similar results could be obtained with BT‐549 and MCF‐7 cells. Treatment with DXR significantly increased the susceptibility of BT‐549 cells, but no such increase was observed with MCF‐7 cells (Figure 2C). The data for the 3 cell lines are summarized in Figure 2D. These results indicate that DXR‐induced senescent MDA‐MB‐231 and BT‐549 cells have increased susceptibility toward activated T cells, but that no such increase occurs in MCF‐7 cells.
Figure 2.
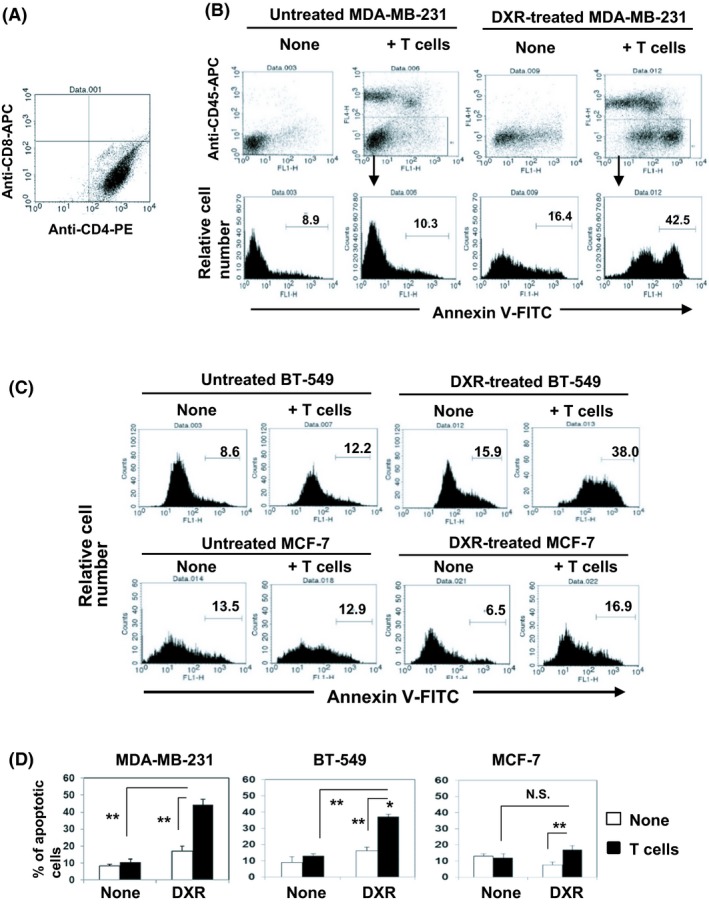
Treatment with doxorubicin (DXR) increases the sensitivity of MDA‐MB‐231 and BT‐549 cells to activated T cells. A, In vitro expanded T cells were examined by flow cytometry after staining with anti‐CD4‐phycoerythrin (PE) and anti‐CD8‐allophycocyanin (APC). B, MDA‐MB‐231 cells were cultured with DXR at a dose of 250 nmol/L for 2 d. Untreated or DXR‐treated MDA‐MB‐231 cells were cultured with in vitro expanded activated T cells in 96‐well plates for 6 h. After harvesting, whole cells were first stained with anti‐CD45‐APC, followed by annexin V‐FITC. A representative result of flow cytometry is shown. C, BT‐549 or MCF‐7 cells were cultured with DXR at doses of 100 and 200 nmol/L, respectively, for 2 d. Thereafter, the percentages of annexin V+ cancer cells were evaluated. A representative result of flow cytometry is shown. D, Results of 3 wells are shown. **P < .01. N.S., not significant
3.3. Involvement of TRAIL in enhanced sensitivity of DXR‐treated cancer cells to T cells
T cells exert cytotoxicity against cancer cells through several mechanisms, including perforin/granzyme and TRAIL/death receptors (DRs).19 We next examined the mechanisms involved in increased sensitivity of DXR‐treated breast cancer cells to T cells. The addition of anti‐TRAIL Ab decreased the percentage of apoptotic cells in cultures of MDA‐MB‐231 and BT‐549 cells (Figures 3A and S2). In contrast, the addition of CMA, a perforin inhibitor,20 showed no effect on the percentage of apoptotic cancer cells. Flow cytometric analysis indicated that in vitro expanded T cells expressed TRAIL at a considerable level (Figure 3B). In addition, soluble TRAIL was detected in the culture supernatant of in vitro expanded T cells (186.6 ng/mL at a dose of 2 × 105 cells/mL for 2 days). We also examined the expression of DR4, DR5, decoy receptor (DcR)1, and DcR2 on untreated and DXR‐treated cancer cell lines (Figure S3). Untreated MDA‐MB‐231 cells were positive for DR4 and DR5, but DXR treatment decreased their expression. No clear expression of DR4 or DR5 was observed on BT‐549 cells. MCF‐7 cells were positive for DR4 and DR5, but DXR treatment decreased their expression. No DcR1 or DcR2 expression was detected on the 3 cell lines. We next compared the sensitivity of untreated and DXR‐treated breast cancer cells to recombinant TRAIL. Pretreatment with DXR increased the sensitivity of both MDA‐MB‐231 and BT‐549 cells toward recombinant TRAIL, whereas no such increase in sensitivity was observed in MCF‐7 cells (Figure 3C). We next examined the expression of a panel of antiapoptotic proteins involved in extrinsic and/or intrinsic apoptosis pathways (Figure 3D). Treatment with DXR decreased the expression of cFLIPs in MDA‐MB‐231 cells and of cFLIPL in BT‐549 cells. Doxorubicin treatment also decreased the expression of Mcl‐1 in MDA‐MB‐231 cells.
Figure 3.
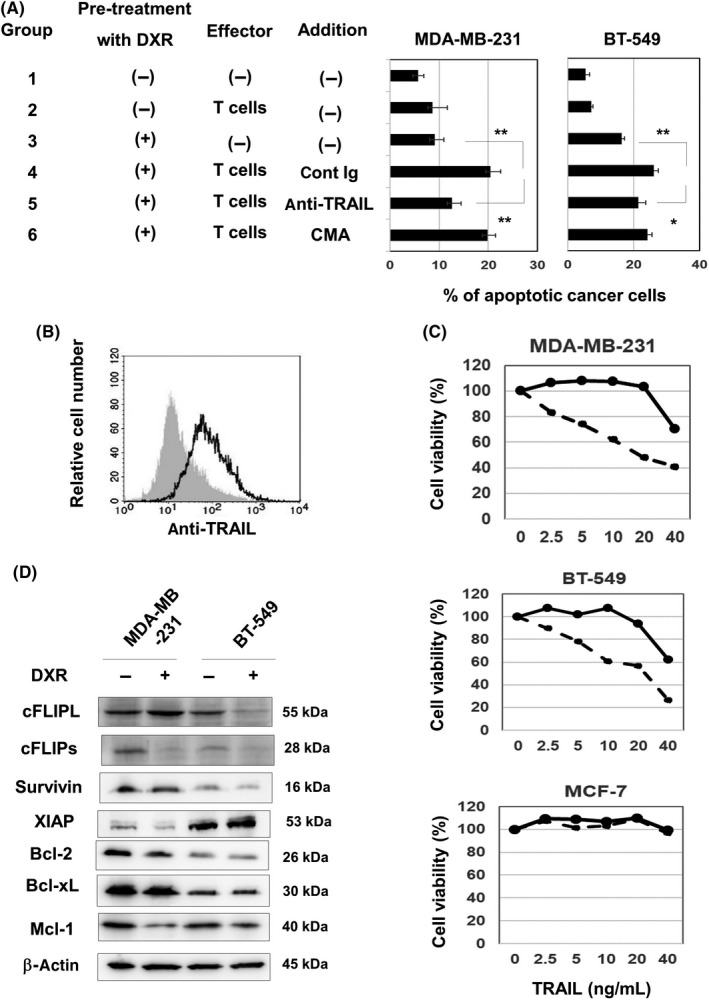
Tumor necrosis factor‐related apoptosis‐inducing ligand (TRAIL) is involved in increased sensitivity of doxorubicin (DXR)‐treated MDA‐MB‐231 cells to activated T cells. A, Cancer cells were treated with or without DXR (250 nmol/L for MDA‐MB‐231 and 100 nmol/L for BT‐549) for 2 d. After harvesting, cancer cells were cultured with expanded activated T cells for 6 h. In some groups, anti‐TRAIL Ab, control mouse IgG, or concanamycin A (CMA) was added 1 h before the addition of cancer cells. After harvesting these cells, whole cells were first stained with anti‐CD45‐APC, followed by annexin V‐FITC and analyzed by flow cytometry, as Figure 2B. Results of 3 wells are shown. *P < .05, **P < .01. B, In vitro expanded T cells were examined by flow cytometry after staining with phycoerythrin (PE)‐conjugated anti‐TRAIL Ab or control mouse IgG, followed by FITC‐conjugated anti‐mouse IgG. Gray background is the control. C, Cancer cells were treated with or without DXR (250 nmol/L for MDA‐MB‐231, 100 nmol/L for BT‐549, and 200 nmol/L for MCF‐7) for 2 d. After harvesting, cancer cells were cultured with recombinant TRAIL for 2 d. Cell viability (%) was determined using the WST‐8 assay. Bold and dotted lines are untreated and death receptor‐treated cancer cells, respectively. Results of 3 wells are shown. D, Two breast cancer cell lines were cultured with DXR (250 nmol/L for MDA‐MB‐231 and 100 nmol/L for BT‐549) for 48 h. Immunoblotting analysis was undertaken using the lysates from untreated and DXR‐treated cancer cells with the indicated Abs. β‐Actin was used as a control
3.4. Perforin is involved in enhanced sensitivity of DXR‐treated cancer cells to ADCC by NK cells and anti‐HER2 Ab
We next examined the sensitivity of DXR‐treated MDA‐MB‐231 cells to ADCC induced by NK cells and anti‐HER2 Ab. The percentage of CD56+ cells in prepared NK cells was approximately 90%, and CD56+ NK cells were weakly positive for TRAIL (Figure 4A). Although MDA‐MB‐231 is known as a TNBC cell line,21 these cells expressed HER2 at a low level and DXR treatment decreased this level of HER2 expression (Figure 4B). Nevertheless, DXR treatment slightly increased the sensitivity of NK cells (Group 2 vs Group 5) and markedly augmented the sensitivity to ADCC with anti‐HER2 Ab (Group 3 vs Group 6) (Figures 4C and S4A). In addition, the increased sensitivity was partially but significantly inhibited by CMA (Group 4 vs Group 5) but not by anti‐TRAIL Ab (Group 2 vs Group 3) (Figures 4D and S4B).
Figure 4.
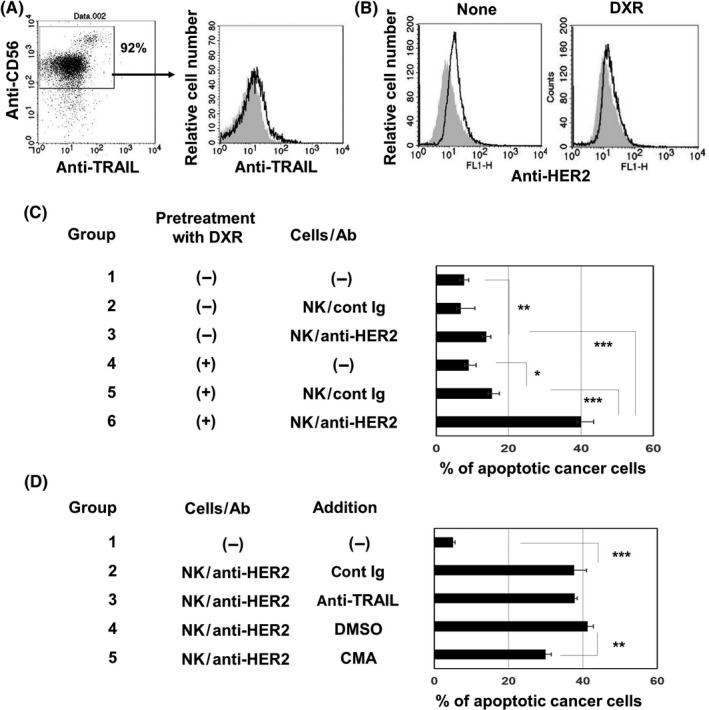
Perforin is involved in increased sensitivity of doxorubicin (DXR)‐treated MDA‐MB‐231 cells to cytotoxicity by natural killer (NK) cells and anti‐ human epidermal growth factor receptor 2 (HER2) Ab. A, Left panel, NK cells prepared from PBMCs of a healthy donor were stained with allophycocyanin‐conjugated anti‐CD56 Ab and phycoerythrin (PE)‐conjugated anti‐tumor necrosis factor‐related apoptosis‐inducing ligand (TRAIL) Ab. Right panel, histogram of TRAIL expression after gating on CD56+ cells is shown. PE‐conjugated mouse IgG was used as a control (gray background). B, MDA‐MB‐231 cells were stained with anti‐HER2‐FITC. FITC‐conjugated mouse IgG was used as an isotype‐matched control (gray background). C, MDA‐MB‐231 cells were treated with or without DXR (250 nmol/L) for 2 d. After harvesting, cancer cells were cultured with prepared NK cells with the indicated Ab for 6 h. After harvesting these cells, whole cells were first stained with anti‐CD45‐APC, followed by annexin V‐FITC and analyzed by flow cytometry, as Figure 2B. Results of 3 wells are shown. *P < .05, **P < .01, ***P < .005. D, Similarly, DXR‐treated MDA‐MB‐231 cells were cultured with prepared NK cells with the indicated Ab for 6 h. In some groups, either anti‐TRAIL Ab, control mouse IgG, concanamycin A (CMA), or DMSO as a vehicle control was added 1 h before the addition of cancer cells. After harvesting these cells, whole cells were first stained with anti‐CD45‐APC, followed by annexin V‐FITC and analyzed by flow cytometry, as Figure 2B. Results of 3 wells are shown. **P < .01, ***P < .005
3.5. Different sensitivities of senescent cancer cells induced by DXR or abemaciclib to cytotoxicity
We finally examined whether the senescent phenotype is needed for the increased sensitivity to cell‐mediated cytotoxicity. We utilized the CDK4/6 inhibitor abemaciclib as a senescence inducer.22, 23 Abemaciclib treatment increased cell size as well as the expression of SA β‐Gal in MDA‐MB‐231 cells (Figure 5A), but decreased their expression of EGFR (Figure 5B). In addition, abemaciclib treatment increased the sensitivity of cancer cells to T cells (Group 2 vs Group 6), whereas the enhancement was apparently lower than that of DXR treatment (Group 2 vs Group 4) (Figures 5C and S5). Similar results were observed in terms of sensitivity to ADCC by NK cells and anti‐EGFR Ab (Group 8 vs Group 9). We also examined the expression of antiapoptotic proteins and found that, in contrast to the decreased expression of cFLIP and Mcl‐1 in DXR‐treated MDA‐MB‐231 cells (Figure 3D), abemaciclib treatment showed no definite effect on the expression of these proteins (Figure 5D). In addition, the expression of γH2AX was not detected in abemaciclib‐treated MDA‐MB‐231 cells (Figure 5E).
Figure 5.
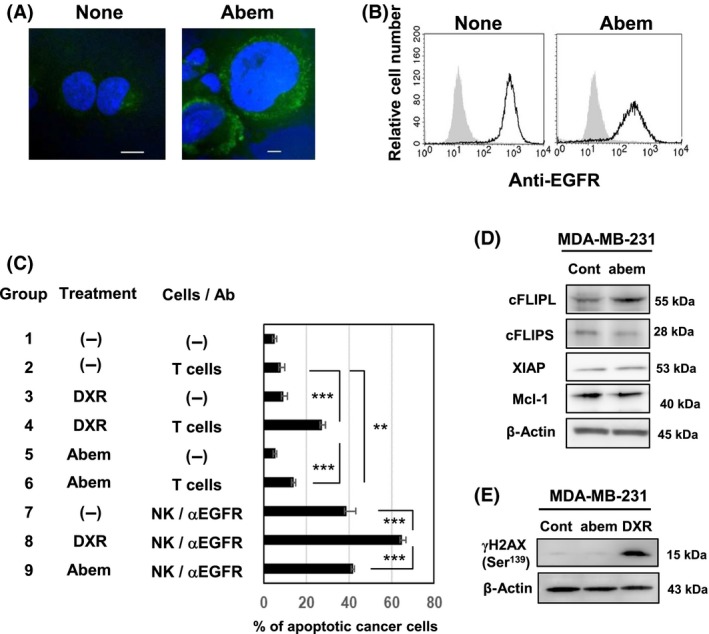
Abemaciclib treatment does not increase the sensitivity of MDA‐MB‐231 cells to immune cell‐mediated cytotoxicity. A, Confocal imaging was carried out on untreated or abemaciclib‐treated MDA‐MB‐231 cells. Scale bar = 10 μm. B, MDA‐MB‐231 cells were stained with anti‐epidermal growth factor receptor (EGFR) Ab‐FITC. Gray background shows staining with FITC‐conjugated mouse IgG as a control. C, MDA‐MB‐231 cells were treated without or with doxorubicin (DXR; 250 nmol/L) or abemaciclib (Abem; 1 μmol/L) for 48 h. After harvesting, cancer cells were cultured with activated T cells and prepared natural killer (NK) cells and with anti‐EGFR Ab for 6 h. After harvesting these cells, whole cells were first stained with anti‐CD45‐APC, followed by annexin V‐FITC and analyzed by flow cytometry, as Figure 2B. Results of 3 wells are shown. **P < .01, ***P < .005. D, MDA‐MB‐231 cells were cultured with or without abemaciclib (1 μmol/L) for 48 h. Immunoblotting analysis was carried out using the lysates with the indicated Abs. β‐Actin was used as a control. E, MDA‐MB‐231 cells were cultured with or without abemaciclib (1 μmol/L) or DXR (250 nmol/L) for 48 h. Immunoblotting analysis was undertaken using anti‐γH2AX Ab. β‐Actin was used as a control
4. DISCUSSION
A major mechanism underlying the anticancer effects of chemotherapeutic drugs is the induction of DNA damage. In this study, we showed that DXR induced the expression of γH2AX and typical senescence in MDA‐MB‐231 and BT‐549 cells, but not in MCF‐7 cells. One possible explanation why γH2AX was not detected and SASP was not induced in DXR‐treated MCF‐7 cells is that MDA‐MB‐231 and BT‐549 cells carry mutant p53, but MCF‐7 cells have WT p53.24 The tumor suppressor p53 acts to restrict proliferation in response to DNA damage or deregulation of mitogenic oncogenes, leading to apoptosis or cellular senescence.25 In general, growth arrest is achieved and maintained in either the G1 or G2/M phase of the cell cycle, in part by the increased expression of specific CDK inhibitors, including p16ink4A and p21Waf1.26 Senescence growth arrest is largely induced by either or both of the p53/p21Waf1 and p16ink4A/pRb tumor suppressive pathways; p21 Waf1 is a downstream effector of p53, whereas p16ink4A is a positive upstream regulator of pRb.6 Doxorubicin treatment increased p21 expression in MCF‐7 cells, probably through p53 activation, whereas no change was observed in γH2AX expression. We assumed that cell death of DXR‐treated MCF‐7 cells occurred too rapidly to detect γH2AX expression. In addition, it has been reported that SASP is limited by WT p53.27 Therefore, MCF‐7 cells were limited in their acquisition of the SASP features due to WT p53, whereas p53‐mutated MDA‐MB‐231 and BT‐549 cells showed a strong SASP response.
As for the reason why the level of SA β‐Gal expression in DXR‐treated MCF‐7 cells was low, we propose that this observation was due to their inability to produce IL‐6 and IL‐8, as reported previously.28 Indeed, we observed no production of these cytokines by untreated and DXR‐treated MCF‐7 cells. Doxorubicin induced senescence‐like changes in MCF‐7 cells, while they lacked SASP. Interestingly, treatment of MCF‐7 cells with IL‐6 and/or IL‐8 can induce senescence, leading to tumor aggressiveness.29 Interleukin‐6 and IL‐8 are known to be responsible for the maintenance and propagation of the SASP response in the tumor microenvironment9, 27 and regulation of the inflammatory phenotype.30, 31 Clinically, elevated serum levels of IL‐6 and IL‐8 have been used independently as prognostic markers for breast cancer,32 and both cytokines are essential for cell growth in TNBC cells.33 These cytokines are likely to play crucial roles in the pathogenesis of breast cancer.
In this study, we examined the sensitivity of DXR‐induced senescent cancer cells to 2 different types of immune cell‐mediated cytotoxicity, ie, T cells and ADCC by NK cells. In vitro expanded anti‐EGFR CAR‐T cells were CD4+ T cells and had lost their anti‐EGFR specificity. However, these CD4+ T cells showed higher levels of cytotoxicity against DXR‐treated MDA‐MB‐231 and BT‐549 cells than against untreated cell lines in a TRAIL‐dependent manner. Death receptor 4 and/or DR5 were detected on MDA‐MB‐231 cells, whereas their expression on BT‐549 cells was not clear (Figure S3). We have no definitive explanation for this observation. However, one possibility is that very low levels of DR4 or DR5, which cannot be detected by flow cytometry, might be enough to provide the death signal to BT‐549 cells because DXR‐treated BT‐549 cells showed sensitivity to recombinant TRAIL (Figure 3C). Alternatively, DXR‐treated MDA‐MB‐231 cells showed increased sensitivity toward ADCC by NK cells with either anti‐HER2 or anti‐EGFR Ab in a perforin‐dependent manner. Due to the lack of availability of antigen‐specific CD8+ T cells, we could not examine the sensitivity of DXR‐treated breast cancer cells to antigen‐specific CD8+ T cells that can recognize MDA‐MB‐231 or BT‐549 cells. However, the mechanisms of immune cell‐mediated cytotoxicity are generally shared (ie, activation by caspase‐8, followed by the caspase cascade).34 In addition, DXR‐treated MDA‐MB‐231 and BT‐549 cells showed decreased expression of the antiapoptotic protein, cFLIP, an inhibitor of caspase‐8, and/or Mcl‐1 (Figure 3D), which play protective roles in extrinsic and intrinsic apoptosis pathways, respectively.35 Taken together, these observations suggest that DXR treatment could increase the sensitivity of breast cancer cells to breast cancer‐recognizing CD8+ T cells.
Cyclin‐dependent kinase 4/6 inhibitors have attracted a great deal of attention as a promising therapy for breast cancer.22 The effectiveness of palbociclib has been reported in phase III clinical trials in estrogen receptor‐positive breast cancers.36 In addition, CDK4/6 inhibitors are known to induce senescence.22, 23 The most notable finding in this study was that senescent MDA‐MB‐231 cells showed increased sensitivity to cytotoxicity only after DXR treatment. This difference seemed to be due to their effects on the expression of the antiapoptotic proteins cFLIP and Mcl‐1. In addition, DXR induced long‐term growth arrest, whereas abemaciclib‐treated cancer cells regrew after the removal of this agent. These results could have been due to the fact that DXR induces senescence through DNA damage, whereas abemaciclib induces senescence by inhibiting CDK4/6 function without DNA damage.
Several reports have suggested that CDK4/6 inhibitors can enhance antitumor immunity in vivo. Two studies showed that CDK4/6 inhibitors, including abemaciclib, triggered antitumor immunity through T cell activation in murine tumor models.37, 38 Similarly, abemaciclib has been reported to upregulate antigen presentation by cancer cells and increase T cell activation.39 In these preclinical studies, the combination with PD‐1/PD‐L1 blockade further enhanced the antitumor effects. Although we focused on the in vitro effects of DXR and abemaciclib on human breast cancer cells in this study, CDK4/6 inhibitors have the potential to enhance the therapeutic efficacy in combination with anticancer immunotherapy.40
In conclusion, the results of the present study indicated that both DXR and the CDK4/6 inhibitor abemaciclib induce senescence in breast cancer cells, but that they show contrasting sensitivity to immune cell‐mediated cytotoxicity. Given that senescent cells with SASP induce tumor promotion and recurrence,9, 10 targeting senescent cancer cells and noncancer cells could be a promising strategy to increase the efficacy of treatment and to control tumor recurrence. In addition, our results provide a possible strategy for combining anticancer immunotherapy with DNA damage‐inducing chemotherapeutic drugs or function‐blocking molecular targeting drugs.
DISCLOSURE
The authors have no conflict of interest.
Supporting information
ACKNOWLEDGMENTS
This study was supported in part by the Japan Society for the Promotion of Science KAKENHI (grant no. 17K07217 to M. Harada) and from the Shimane University “SUIGANN” Project.
Inao T, Kotani H, Iida Y, et al. Different sensitivities of senescent breast cancer cells to immune cell‐mediated cytotoxicity. Cancer Sci. 2019;110:2690‐2699. 10.1111/cas.14116
REFERENCES
- 1. Fitzmaurice C, Allen C, Barber RM, et al. Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability‐adjusted life‐years for 32 cancer groups, 1990 to 2015: a systematic analysis for the global burden of disease study. JAMA Oncol. 2017;3:524‐548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Apuri S. Neoadjuvant and adjuvant therapies for breast cancer. South Med J. 2017;110:638‐642. [DOI] [PubMed] [Google Scholar]
- 3. Abdel‐Fatah TM, Perry C, Dickinson P, et al. Bcl2 is an independent prognostic marker of triple negative breast cancer (TNBC) and predicts response to anthracycline combination (ATC) chemotherapy (CT) in adjuvant and neoadjuvant settings. Ann Oncol. 2013;24:2801‐2807. [DOI] [PubMed] [Google Scholar]
- 4. Tacar O, Sriamornsak P, Dass CR. Doxorubicin: an update on anticancer molecular action, toxicity and novel drug delivery systems. J Pharm Pharmacol. 2013;65:157‐170. [DOI] [PubMed] [Google Scholar]
- 5. Gewirtz DA. Growth arrest and cell death in the breast tumor cell in response to ionizing radiation and chemotherapeutic agents which induce DNA damage. Breast Cancer Res Treat. 2000;62:223‐235. [DOI] [PubMed] [Google Scholar]
- 6. Campisi J. Aging, cellular senescence, and cancer. Annu Rev Physiol. 2013;75:685‐705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ewald JA, Desotelle JA, Wilding G, et al. Therapy‐induced senescence in cancer. J Natl Cancer Inst. 2010;102:1536‐1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gonzalez LC, Ghadaouia S, Martinez A, et al. Premature aging/senescence in cancer cells facing therapy: good or bad? Biogerontology. 2016;17:71‐87. [DOI] [PubMed] [Google Scholar]
- 9. Coppé JP, Desprez PY, Krtolica A, et al. The senescence‐associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010;5:99‐118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Watanabe S, Kawamoto S, Ohtani N, et al. Impact of senescence‐associated secretary phenotype and its potential as a therapeutic target for senescence‐associated diseases. Cancer Sci. 2017;108:563‐569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Postow MA, Callahan MK, Wolchok JD. Immune checkpoint blockade in cancer therapy. J Clin Oncol. 2015;33:1974‐1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Antonia SJ, Villegas A, Daniel D, et al. Durvalumab after chemoradiotherapy in stage III non‐small‐cell lung cancer. N Engl J Med. 2017;377:1919‐1929. [DOI] [PubMed] [Google Scholar]
- 13. Paz‐Ares L, Luft A, Vicente D, et al. Pembrolizumab plus chemotherapy for squamous non‐small‐cell lung cancer. N Engl J Med. 2018;379:2040‐2051. [DOI] [PubMed] [Google Scholar]
- 14. Ghiringhelli F, Larmonier N, Schmitt E, et al. CD4+CD25+ regulatory T cells suppress tumor immunity but are sensitive to cyclophosphamide which allows immunotherapy of established tumors to be curative. Eur J Immunol. 2004;34:336‐344. [DOI] [PubMed] [Google Scholar]
- 15. Suzuki E, Kapoor V, Jassar AS, et al. Gemcitabine selectively eliminates splenic Gr‐1+/CD11b+ myeloid suppressor cells in tumor‐bearing animals and enhances antitumor immune activity. Clin Cancer Res. 2005;11:6713‐6721. [DOI] [PubMed] [Google Scholar]
- 16. Kroemer G, Galluzzi L, Kepp O, et al. Immunogenic cell death in cancer therapy. Annu Rev Immunol. 2013;31:51‐72. [DOI] [PubMed] [Google Scholar]
- 17. Ding ZC, Munn DH, Zhou G. Chemotherapy‐induced myeloid suppressor cells and antitumor immunity: the Janus face of chemotherapy in immunomodulation. Oncoimmunology. 2014;3:e954471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Peng J, Hamanishi J, Matsumura N, et al. Chemotherapy induces programmed cell death‐ligand overexpression via the Nuclear Factor‐κB to foster an immunosuppressive tumor microenvironment in ovarian cancer. Cancer Res. 2015;75:5034‐5045. [DOI] [PubMed] [Google Scholar]
- 19. Takeda K, Hayakawa Y, Smyth MJ, et al. Involvement of tumor necrosis factor‐related apoptosis‐inducing ligand in surveillance of tumor metastasis by liver natural killer cells. Nat Med. 2001;7:94‐100. [DOI] [PubMed] [Google Scholar]
- 20. Kataoka T, Shinohara N, Takayama H, et al. Concanamycin A, a powerful tool for characterization and estimation of contribution of perforin‐ and Fas‐based lytic pathways in cell‐mediated cytotoxicity. J Immunol. 1996;156:3678‐3686. [PubMed] [Google Scholar]
- 21. Gründker C, Föst C, Fister S, et al. Gonadotropin‐releasing hormone type II antagonist induces apoptosis in MCF‐7 and triple‐negative MDA‐MB‐231 human breast cancer cells in vitro and in vivo. Breast Cancer Res. 2010;12:R49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Murphy CG, Dickler MN. The role of CDK4/6 inhibition in breast cancer. Oncologist. 2015;20:483‐490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Klein ME, Kovatcheva M, Davis LE, et al. CDK4/6 inhibitors: the mechanism of action may not be as simple as once thought. Cancer Cell. 2018;34:9‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Simon M, Mesmar F, Helguero L, et al. Genome‐wide effects of MELK‐inhibitor in triple‐negative breast cancer cells indicate context‐dependent response with p53 as a key determinant. PLoS ONE. 2017;12:e0172832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Harris SL, Levine AJ. The p53 pathway: positive and negative feedback loops. Oncogene. 2005;24:2899‐2908. [DOI] [PubMed] [Google Scholar]
- 26. Bringold F, Serrano M. Tumor suppressors and oncogenes in cellular senescence. Exp Gerontol. 2000;35:317‐329. [DOI] [PubMed] [Google Scholar]
- 27. Coppé JP, Patil CK, Rodier F, et al. Senescence‐associated secretory phenotypes reveal cell‐nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6:2853‐2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chavey C, Bibeau F, Gourgou‐Bourgade S, et al. Oestrogen receptor negative breast cancers exhibit high cytokine content. Breast Cancer Res. 2007;9:R15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ortiz‐montero P, Londoño‐vallejo A, Vernot JP. Senescence‐associated IL‐6 and IL‐8 cytokines induce a self‐ and cross‐reinforced senescence/inflammatory milieu strengthening tumorigenic capabilities in the MCF‐7 breast cancer cell line. Cell Commun Signal. 2017;15:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kuilman T, Michaloglou C, Vredeveld LC, et al. Oncogene‐induced senescence relayed by an interleukin‐dependent inflammatory network. Cell. 2008;133:1019‐1031. [DOI] [PubMed] [Google Scholar]
- 31. Acosta JC, O'Loghlen A, Banito A, et al. Chemokine signaling via the CXCR31 receptor reinforces senescence. Cell. 2008;133:1006‐1018. [DOI] [PubMed] [Google Scholar]
- 32. Bachelot T, Ray‐Coquard I, Menetrier‐Caux C, et al. Prognostic value of serum levels of interleukin 6 and of serum and plasma levels of vascular endothelial growth factor in hormone‐refractory metastatic breast cancer patients. Br J Cancer. 2003;88:1721‐1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hartman ZC, Poage GM, den Hollander P, et al. Growth of triple‐negative breast cancer cells relies upon coordinate autocrine expression of the proinflammatory cytokines IL‐6 and IL‐8. Cancer Res. 2013;73:3470‐3480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Murphy BM, Creagh EM, Martin SJ. Interchain proteolysis, in the absence of a dimerization stimulus, can initiate apoptosis‐associated caspase‐8 activation. J Biol Chem. 2004;279:36916‐36922. [DOI] [PubMed] [Google Scholar]
- 35. Kim SH, Ricci MS, El‐Deiry WS. Mcl‐1: a gateway to TRAIL sensitization. Cancer Res. 2008;68:2062‐2064. [DOI] [PubMed] [Google Scholar]
- 36. Cristofanilli M, Turner NC, Bondarenko I, et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone‐receptor‐positive, HER2‐negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA‐3): final analysis of the multicentre, double‐blind, phase 3 randomised controlled trial. Lancet Oncol. 2016;17:425‐439. [DOI] [PubMed] [Google Scholar]
- 37. Goel S, DeCristo MJ, Watt AC, et al. CDK4/6 inhibition triggers anti‐tumour immunity. Nature. 2017;548:471‐475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Deng J, Wang ES, Jenkins RW, et al. CDK4/6 inhibition augments antitumor immunity by enhancing T‐cell activation. Cancer Discov. 2018;8:216‐233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schaer DA, Beckmann RP, Dempsey JA, et al. The CDK4/6 inhibitor abemaciclib induces a T cell inflamed tumor microenvironment and enhances the efficacy of PD‐L1 checkpoint blockade. Cell Rep. 2018;22:2978‐2994. [DOI] [PubMed] [Google Scholar]
- 40. Teh JLF, Aplin AE. Arrested developments: CDK4/6 inhibitor resistance and alterations in the tumor immune microenvironment. Clin Cancer Res. 2019;25:921‐927. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials