

Abstract
Cellular organelles are not static but show dynamism—a property that is likely relevant for their function. In addition, they interact with other organelles in a highly dynamic manner. In this review, we analyze the proteins involved in the interaction between mitochondria and other cellular organelles, especially the endoplasmic reticulum, lipid droplets, and lysosomes. Recent results indicate that, on one hand, metabolic alterations perturb the interaction between mitochondria and other organelles, and, on the other hand, that deficiency in proteins involved in the tethering between mitochondria and the ER or in specific functions of the interaction leads to metabolic alterations in a variety of tissues. The interaction between organelles is an emerging field that will permit to identify key proteins, to delineate novel modulation pathways, and to elucidate their implications in human disease.
Keywords: contact sites, diabetes, endoplasmic reticulum, insulin resistance, lipid droplets
Subject Categories: Metabolism
Glossary
- ABHD5
1‐acylglycerol‐3‐phosphate O‐acyltransferase
- ACSL1
Acyl‐CoA synthase long chain family member 1
- Agrp
Agouti‐related protein
- AKT
Protein kinase B
- ATAD3A
ATPase family, AAA domain containing 3A
- ATF4
Activating transcription factor 4
- ATF6
Activating transcription factor 6
- ATG12
Autophagy related 12
- ATG14
Autophagy related 14
- ATG16L1
Autophagy related 16 like 1
- ATG5
Autophagy related 5
- ATGL
Adipose triglyceride lipase
- BAK
Bcl‐2 homologous antagonist/killer
- BAP31
B‐cell receptor‐associated protein 31
- BAT
Brown adipose tissue
- BAX
BCL2‐associated X
- BCL2
Apoptosis regulator B‐cell lymphoma 2
- BECN1
Beclin 1
- BioID
Proximity dependent biotin identification
- BiP
Binding immunoglobulin protein
- Ca2+
Calcium
- Ccl2
C‐C motif chemokine ligand 2
- CDIP
Cell death‐inducing p53‐target protein 1
- CEBPA
CCAAT/enhancer‐binding protein alpha
- CHOP
CCAAT/enhancer‐binding protein homologous protein
- CIDEA
Cell death‐inducing DFFA‐like effector a
- CYP11A1
Cytochrome P450 family 11 subfamily A member 1
- CypD
Cyclophilin D
- DRP1
Dynamin‐related protein 1
- ECI2
Enoyl‐CoA delta isomerase 2
- eIF2α
Eukaryotic initiation factor 2 alpha
- ER
Endoplasmic reticulum
- ERK
Extracellular signal‐regulated kinase
- ERLIN2
ER lipid raft associated 2
- ERMES
ER‐mitochondria encounter structure
- FACL4
Fatty acid‐CoA ligase 4
- FDB
Flexor digitorum brevis
- FGF21
Fibroblast growth factor 21
- FIS1
Mitochondrial fission 1
- FUNDC1
FUN14 domain‐containing protein 1
- GLUT4
Glucose transporter type 4
- GM1
Gangliosidosis‐1
- GRP75
Glucose‐regulated protein 75
- GRP94
Glucose‐regulated protein 94
- GTPase
GTP hydrolase
- GTP
Guanosine 5′‐triphosphate
- H2O2
Hydrogen peroxide
- HDL
High‐density lipoprotein
- HFD
High‐fat diet
- Il6
Interleukin 6
- IMM
Inner mitochondrial membrane
- INF2
Inverted formin 2
- IP3R1/2/3
Inositol 1,4,5‐triphosphate receptors 1, 2 and 3
- IRE1α
Inositol‐requiring enzyme 1 alpha
- JNK
c‐Jun N‐terminal kinase
- LC3
Microtubule‐associated protein 1A/1B‐light chain 3
- LD
Lipid droplet
- M1
Classically activated macrophages
- MAM
Mitochondria‐associated membranes
- MAPK
Mitogen‐activated protein kinase
- MCU
Mitochondrial calcium uniporter
- MDV
Mitochondria‐derived vesicle
- MEF
Mouse embryonic fibroblasts
- MEK
Mitogen‐activated protein kinase kinase
- MERC
Mitochondria–ER contact sites
- MFF
Mitochondria fission factor
- MFN1
Mitofusin 1
- MFN2
Mitofusin 2
- MiD49/51
Mitochondrial dynamics proteins 49 and 51
- MOSPD2
Motile sperm domain containing 2
- MPTP
Mitochondrial permeability transition pore
- mRNA
Messenger ribonucleic acid
- mtDNA
Mitochondrial deoxyribonucleic acid
- mTORC1
Mammalian target of rapamycin complex 1
- mTORC2
Mammalian target of rapamycin complex 2
- mTOR
Mammalian target of rapamycin
- OMM
Outer mitochondrial membrane
- OPA1
Optic atrophy 1
- ORP5
Oxysterol‐binding protein–related protein 5
- ORP8
Oxysterol‐binding protein–related protein 8
- OSBP
Oxysterol‐binding protein
- OXPHOS
Oxidative phosphorylation
- PACS2
Phosphofurin acidic cluster sorting protein 2
- PC
Phosphatidylcholine
- PDK4
Pyruvate dehydrogenase kinase 4
- PDZD8
PDZ domain‐containing protein 8
- PEMT
Phosphatidylethanolamine N‐methyltransferase
- PE
Phosphatidylethanolamine
- PERK
Protein kinase RNA‐like endoplasmic reticulum kinase
- PI3K
Phosphatidylinositol‐3‐kinase
- PI4P
Phosphatidylinositol 4‐phosphate
- PISD
Phosphatidylserine decarboxylase proenzyme
- PLIN1
Perilipin 1
- PLIN5
Perilipin 5
- POMC
Pro‐opiomelanocortin
- PPARγ
Peroxisome proliferator‐activated receptor gamma
- PS
Phosphatidylserine
- PSS1
Phosphatidylserine synthase‐1
- PSS2
Phosphatidylserine synthase‐2
- PTPIP51
Protein tyrosine phosphatase interacting protein 51
- RAB5
Ras‐related protein Rab‐5
- RAB7
Ras‐related protein Rab‐7
- ROS
Reactive oxygen species
- RYR1/2/3
Ryanodine receptors 1, 2, and 3
- SEC61
Protein transport protein Sec61
- SERCA
Sarco/endoplasmic reticulum Ca2+‐ATPase
- SLC
Solute carrier protein
- SMP
Synaptotagmin‐like mitochondrial‐lipid‐binding domain
- SNAP23
Synaptosomal‐associated protein 23
- SREBP1C
Sterol regulatory element‐binding protein 1
- STARD1/3/4
Steroidogenic acute regulatory lipid transfer domain proteins 1, 3, and 4
- StAR
Steroidogenic acute regulatory protein
- START
StAR‐related lipid transfer
- STX17
Syntaxin 17
- TAG
Triacylglycerides
- TBC1D15
TBC1 domain family member 15
- TCHP
Trichoplein
- TG2
Transglutaminase 2
- Tnfa
Tumor necrosis factor alpha
- TOM20
Translocase of outer mitochondrial membrane 20
- TOM22
Translocase of outer mitochondrial membrane 22
- TOM40
Translocase of outer mitochondrial membrane 40
- TOM70
Translocase of outer mitochondrial membrane 70
- TSPO
Translocator protein
- UCP1
Uncoupling protein 1
- UPR
Unfolded protein response
- VAMP4
Vesicle‐associated membrane protein 4
- VAPB
VAMP‐associated Protein B
- VDAC1
Voltage‐dependent anion channel 1
- VDAC2
Voltage‐dependent anion channel 2
- VDAC
Voltage‐dependent anion channels
- VPS13A
Vacuolar protein sorting‐associated protein 13 A
- VPS15
Vacuolar protein sorting‐associated protein 15
- VPS34
Vacuolar protein sorting‐associated protein 34
- Vps39
Vacuolar protein sorting‐associated protein 39
- WASF3
Wiskott–Aldrich syndrome protein family member 3
- WAT
White adipose tissue
- XBP1
X‐box binding protein 1
- Ypt7
GTP‐binding protein YPT7
Contacts between mitochondria and other organelles
Mitochondria are highly dynamic and social organelles. They undergo continuous morphological changes to maintain cellular homeostasis; i.e., they fuse in response to specific physiological conditions, they divide to facilitate their removal by autophagy, and they maintain dynamic contacts with other membranous compartments of the cell. Mitochondria communicate with the endoplasmic reticulum (ER), lipid droplets (LDs), Golgi apparatus, lysosomes, melanosomes, and peroxisomes by establishing physical contacts (Fig 1). Fluorescent labeling of all these organelles in vivo has recently revealed cellular regions where three or more of these organelles physically interact 1. Mitochondria and ER actively communicate, and their contact sites are important hubs for lipid trafficking, mitochondrial dynamics, Ca2+ signaling, ER stress, apoptosis, and macroautophagy. Mitochondria–ER contact sites are also referred to as MERCs or when studied at a biochemical level MAMs (mitochondria‐associated membranes). The contacts between mitochondria and LD functionally support triacylglyceride synthesis 2, and they are sustained by the interaction between MFN2 and PLIN1 3. Apposition of the Golgi apparatus and mitochondria has been demonstrated by microscopy techniques; however, the molecular features of this interaction remain poorly understood 4, 5. Mitochondria incorporate Ca2+ excess from the Golgi apparatus and have been proposed as a source of ATP for this organelle 4. The contacts between mitochondria and lysosomes have been described to regulate mitochondrial fission, as well as lysosomal dynamics, by RAB7 GTP hydrolysis 6. Melanosomes, which are lysosome‐related organelles that store pigments, also interact with mitochondria 7. These sites are associated with the process of melanogenesis 7. MFN2 has been found at these areas of juxtaposition, and its knockdown reduces these interorganelle connections 7. Peroxisomes and mitochondria interact through TOM20 in the mitochondria and ECI2 in peroxisomes. This interaction has functional implications in steroid biosynthesis in mouse Leydig cells 8. Of all the mitochondrial contacts, those with ER are the best characterized to date, and some metabolic implications of those contacts have been also documented. The contacts between mitochondria and LDs or lysosomes are currently gaining insight and relevance and their potential metabolic implications are in the spotlight. In this review, we focus mainly on the molecular biology of mitochondria–ER, mitochondria–LD, and mitochondria–lysosome contacts and their involvement in metabolism.
Figure 1. Contacts between mitochondria and other organelles.

Mitochondria interact with other membranous compartments in the cell. Mitochondria interact with the Golgi apparatus; however, the identities of the proteins involved in this interaction have not been discovered yet. Mitochondria are also in contact with lysosomes, but the mediators of these contacts remain unknown. MFN2 in mitochondria interacts with melanosomes. ECI2 and TOM20 bridge the peroxisome to the mitochondria. Mitochondria are anchored to lipid droplets by the MFN2–PLIN1 interaction. Mitochondria–ER contacts harbor a singular architecture and are hubs for several cellular processes such as Ca2+ signaling and lipid trafficking (see further details in Figs 2 and 3).
Architecture of mitochondria–ER tethers
The structural scaffold of ER‐mitochondria contact sites consists of proteins inserted in the outer mitochondrial membrane (OMM) that interact with those in the ER membrane. Available data indicate that ER bridging to mitochondria is governed by the following protein complexes (Fig 2): VAPB in the ER and PTPIP51 in mitochondria 9; inositol 1,4,5‐triphosphate receptors (IP3R1/2/3) in the ER and GRP75, together with VDAC1 in mitochondria 10; BAP31 in the ER and FIS1 in mitochondria 11; and MFN2 both in the ER and mitochondria 12. These proteins not only shape mitochondria–ER contacts but also participate in the functions associated with these domains. PTPIP51 and VAPB are necessary to maintain Ca2+ transport between the ER and mitochondria 13. Recently, MOSPD2 has been proposed as a new tethering protein in the ER that interacts with PTPIP51 14. MOSPD2 is located at the contact sites of the ER with other organelles 14. IP3Rs, GRP75, and VDAC1 form the gate through which Ca2+ leaves the ER and enters mitochondria 10. These proteins are core components of a bigger complex specialized in calcium channeling. Tethering capacity and calcium flux are sustained by the mitochondrial proteins PDK4 and TG2 15. The BAP31‐FIS1 interaction is established at the MAM, upstream of apoptosis induction 11. Moreover, this complex also participates in mitochondrial fission: FIS1 is a receptor for DRP1 in mitochondria, the major player in mitochondrial fission 16, and BAP31, once cleaved, is able to induce mitochondrial fission 17. MFN2 is localized both in the mitochondria and in the ER, and in both cases, it is able to homooligomerize and to heteroligomerize with MFN1 to tether both organelles or to promote mitochondrial fusion 18, 19. Furthermore, the lack of MFN2 results in reduced mitochondrial Ca2+ uptake and in autophagosome formation arrest 12, 20. Although several studies argue that MFN2 is an organelle spacer 21, 22, rather than a tether, we consider that the available data strongly support the role of MFN2 as a tether 12, 23, 24. A recent study has suggested that another mitochondria–ER tethering complex could exist containing BiP in the ER membrane toward the ER lumen, WASF3 at the cytoplasm, and ATAD3A in the inner mitochondrial membrane (IMM) 25 (Fig 2). ATAD3A, WASF3, and BiP co‐immunoprecipitate and silencing of ATAD3A downregulates BiP and WASF3 25. Given the location of these proteins in the cell, it is likely that they are part of a larger complex whose components remain undescribed.
Figure 2. The architecture of mitochondria–ER contact sites: tethering complexes.

Mitochondria are bridged to the ER by several protein complexes. In the ER, VAPB or MOSPD2 bind to PTPIP51 in mitochondria. IP3R in the ER is anchored to VDAC in the OMM by the cytosolic protein GRP75. MFN2 is present both at the ER and in the OMM. From the ER, MFN2 interacts with either MFN1 or MFN2 in the mitochondria. BAP31 in the ER partners up with FIS1 in the mitochondria. BiP in the ER, WASF3 in the cytosol, and ATAD3A in the IMM have been suggested to form a complex that tethers both organelles.
Partial or total ablation of tethering proteins influences the architecture of mitochondria–ER contact sites. GRP75 silencing decreases the interaction between IP3R1 and VDAC1 both in HT22 mouse hippocampal neurons cells and in HuH7 human hepatocarcinoma cells 26, 27. This effect is also observed upon MFN2 knockdown in HuH7 cells and H9c2 rat cardiomyoblasts 27, 28. VDAC1 partial ablation diminishes the number of interaction spots between GRP75 and IP3R1 in HuH7 cells 27. IP3R1 silencing in hepatocytes does not alter the protein levels of the other IP3Rs 29. Pdk4 ablation in mice results in decreased MAM formation in skeletal muscle 15. Tg2 ablation in mouse embryonic fibroblasts (MEF) decreases the quantity of mitochondria–ER contact sites 30. Vapb knockdown does not affect PTPIP51 expression or vice versa in NSC34 mouse motor neuron‐like cells; however, it does reduce mitochondria–ER association 13. In line with this, Ptpip51 silencing in rat neonatal cardiomyocytes reduces mitochondria–ER contacts 31. Moreover, the downregulation of Vapb or Ptpip51 in NSC34 cells does not affect total MFN2 expression 13. MFN2 knockdown in human lung cancer H838 cells leads to an increase in ATAD3A localization to the MAM 32. Both total and partial ablation of Mfn2 in MEF cells increases the distance between the ER and mitochondria 23. In agreement with this finding, in flexor digitorum brevis (FDB) muscles, mitochondria and ER apposition is reduced upon temporal Mfn2 depletion 33. It is likely that additional tethers or spacers will be identified in the future and that they will allow a more global view of the proteins involved in the maintenance of mitochondria–ER contacts. In addition, further studies are needed to determine whether the ablation of a single protein modifies the expression of proteins or of genes involved in a different tether and to know whether these structures cooperate.
Functions linked to the mitochondria–ER contacts
The interface between ER and mitochondria harbors processes that are essential for the cell (Fig 3; Table 1). In this review, we have classified these processes in six groups: traffic of lipids, mitochondrial dynamics, Ca2+ signaling, ER stress, apoptosis initiation, and autophagosome formation.
Figure 3. Cellular functions at mitochondria–ER contact sites.

The main processes that take place at the MAM are as follows: phospholipid trafficking, mitochondrial dynamics, Ca2+ signaling, unfolded protein response (UPR), apoptosis initiation, and autophagosome formation. MAMs are hubs for phospholipid exchange between the ER and mitochondria. Mitochondria take phosphatidylserine from the ER, which is supplied with phosphatidylethanolamine by mitochondria. Mitochondrial dynamics processes are regulated at the interface between the ER and mitochondria. The mitochondrial fission effector DRP1 is recruited by MFF and MiD49/51 to the mitochondrial surface, and it interacts with STX17 in the ER membrane. The ER wraps around the mitochondrion, which is finally excised into two daughter mitochondria after mtDNA replication. Mitochondrial fusion is promoted by TCHP binding to MFN2. This interaction separates MFN2 tethers and promotes the fusogenic function of mitochondrion‐bound MFN2. The ER is the cellular Ca2+ reservoir. IP3R, GRP75, and VDAC form a Ca2+ channeling complex that allows Ca2+ flux from the ER to mitochondria. MCU transports the intermembrane Ca2+ to the mitochondrial matrix. The UPR is regulated at the interface between mitochondria and the ER. One of the key regulators is MFN2, which inhibits the UPR by interacting with PERK. It is not known whether PERK interacts with MFN2 in mitochondria, MFN2 in the ER or with both. MAMs are also involved in the initiation of apoptosis. Sustained pro‐apoptotic stimuli lead to BCL2 sequestration by the reticulum proteins BAP31 and CDIP in order to initiate apoptotic signaling cascades. Autophagosomes arise from mitochondria–ER contact sites. Proteins involved in autophagosome formation are recruited to these locations. STX17 in the ER attracts ATG14L and the PI3K complex. The mitochondrial component involved in this process is still not known.
Table 1.
Mitochondria–ER contact sites proteins
| Protein | Location | Function in the MAM |
|---|---|---|
| ATAD3A | IMM | ER‐mt tethering 25, lipid trafficking 52 |
| ATF6 | ER | UPR 106 |
| ATG14 | ER | Autophagosome formation 20 |
| BAK | OMM | Apoptosis 126 |
| BAP31 | ER | ER‐mt tethering 11, Apoptosis 11 |
| BAX | Cyt, OMM | Apoptosis 126 |
| BCL2 | OMM | Apoptosis 126 |
| BiP | ER | ER‐mt tethering 25 |
| CDIP1 | ER | Apoptosis 126 |
| CypD | IMM | Apoptosis 125 |
| DRP1 | OMM | Mitochondrial dynamics 252 |
| FIS1 | OMM | ER‐mt tethering 11, Mitochondrial dynamics 16, Apoptosis 11 |
| FUNDC1 | OMM | Ca2+ signaling 76, Mitochondrial dynamics 68 |
| GRP75 | Mt | ER‐mt tethering 10, Ca2+ signaling 10 |
| INF2 | ER | Mitochondrial dynamics 59 |
| IP3R | ER | ER‐mt tethering 10, Ca2+ signaling 10 |
| IRE1α | ER | UPR 106 |
| MCU | IMM | Ca2+ signaling 253 |
| MFF | OMM | Mitochondrial dynamics 62 |
| MFN1 | OMM | ER‐mt tethering 12, Mitochondrial dynamics 69 |
| MFN2 | ER, OMM | ER‐mt tethering 12, Mitochondrial dynamics 69, UPR 97 |
| MiD49 | OMM | Mitochondrial dynamics 62 |
| MiD51 | OMM | Mitochondrial dynamics 62 |
| ORP5 | ER | Lipid trafficking 43 |
| ORP8 | ER | Lipid trafficking 43 |
| PACS2 | Cyt | ER‐mt tethering maintenance 55 |
| PDZD8 | ER | Lipid trafficking(?) 46 |
| PERK | ER | UPR 106 |
| PTPIP51 | OMM | ER‐mt tethering 9 |
| RyR | ER | Ca2+ signaling 8 |
| SEC61 | ER | Ca2+ signaling 42 |
| SERCA1 | ER | UPR 114 |
| STARD1 | Cyt | Lipid trafficking 48 |
| STX17 | ER | Autophagosome formation 20, Mitochondrial dynamics 65 |
| TSPO | OMM | Lipid trafficking 52 |
| VAPB | ER | ER‐mt tethering 9 |
| VDAC1 | OMM | ER‐mt tethering 10, Ca2+ signaling 10, Lipid trafficking 53 |
| WASF3 | Cyt | ER‐mt tethering 25 |
Lipid trafficking
Intracellular lipid transport can occur by flip‐flop from one side of a bilayer to the other, by vesicular trafficking, by lipid transfer proteins, or by diffusion within a bilayer 34. MAMs are hubs for non‐vesicular phospholipid and cholesterol transport, and this process is linked to the synthesis of phospholipid and cholesterol intermediates. In this chapter, we analyze the progress in the traffic of lipids between ER and mitochondria and we highlight the remaining unexplored questions. Most of the advances so far have been obtained in cellular models, and in vivo assays would provide these findings with more robustness.
At the ER, phosphatidylcholine (PC) is converted into phosphatidylserine (PS) by PSS1 35. Mitochondria are not able to synthesize PS and therefore receive it from the ER 36, 37. The decarboxylation of PS in the mitochondria by PISD produces phosphatidylethanolamine (PE) 36, 38. This newly synthesized PE can then be translocated to the ER 37, where it is converted to PC by PEMT 39 or, less likely, to PS by PSS2 35. The discovery that newly synthesized PS and PE are transported between ER and mitochondria in a non‐vesicular manner 37, 40 was a key finding in the lipid trafficking field and an outstanding contribution to the understanding of the molecular biology of mitochondria and ER apposition. It is not fully understood how PS is transported from the ER to mitochondria in mammalian cells; however, members of the oxysterol‐binding protein (OSBP) family have been proposed to participate in this process. OSBP family proteins contain an OSBP‐related ligand‐binding domain that has the structure of a beta‐barrel and binds PI4P and, in some cases, sterols 34. Two members of this family, ORP5 and ORP8, localize at the ER membrane facing the cytosol (Fig 3). ORP5 was first discovered to catalyze the exit of cholesterol from endosomes 41. Later, it was found that ORP5 and ORP8 counter‐exchange PS and phosphatidylinositol 4‐phosphate (PI4P) between the ER and plasma membrane 42. ORP5 and ORP8 are present at mitochondria–ER contact sites, where they interact with the tethering protein PTPIP51 43 and could be responsible for PS transport to mitochondria. In addition to the OSBP family, in yeast, the ERMES complex, which has no homology with any mammalian complex, tethers mitochondria to the ER and transfers PS from the ER to mitochondria 44. Mmm1 and Mdm12, components of the ERMES complex, have been found to harbor an SMP domain that is responsible for phospholipid transfer 44, 45. This domain shows functional orthology with the SMP domain of mammalian proteins that localize at the ER, such as extended synaptotagmins and PDZD8 45, 46. Expression of a chimeric form of Mmm1 containing the PDZD8 SMP domain in deficient MMM1 yeast rescues mitochondrial defects. PDZD8 localizes at the ER fraction of MAMs 46. The fact that PDZD8 harbors an SMP domain makes it a potential candidate for phospholipid exchange between the ER and mitochondria.
Sterols, oxysterol, and bile acids are synthesized in mitochondria. These molecules originate from cholesterol that is imported into mitochondria from several sources: ER, LD, and endosomes (reviewed by Elustondo et al 47). Here, we focus on cholesterol transport from the ER, which is performed by STARD1 48. STARD1 contains a lipid binding domain known as START, which particularly in this protein has specificity for sterols 49, 50. In mitochondria–ER contact sites, STARD1 is recruited to the OMM where it forms a complex with the OMM proteins TOM22 and VDAC2 and the ER proteins BiP, ERLIN2, and SLC 48. STARD1 incorporates ER‐cholesterol into the OMM so that it can be transported to the IMM, where CYP11A1 further processes it 51. The complex that moves cholesterol from the OMM to the IMM is formed by VDAC1 and TSPO in the OMM and by ATAD3A and CYP11A1 in the IMM 52. STARD1 interacts with members of this complex, namely VDAC1 and TSPO 53, thus linking cholesterol incorporation from the ER to its processing in the mitochondrial matrix.
The modification of proteins involved in lipid trafficking in the MAM influences other protein complexes and functions that take place at the MAM, and it also affects the interaction between mitochondria and the ER. Moreover, the alteration of tethering proteins has an impact on lipid transport between the two organelles. ORP5 and ORP8 depletion from HeLa cells alters mitochondrial morphology and respiration; however, the number of mitochondria–ER contact sites is not affected 43. Overexpression of ORP5 and ORP8 increases mitochondrial Ca2+ concentration in HeLa cells after histamine treatment, although depletion of these proteins does not alter Ca2+ signaling 54. Also, when the tethering protein PTPIP51 is overexpressed in HeLa cells, the presence of ORP5/8 at mitochondria–ER contact sites is increased 43. However, it has not been assessed whether PS transport is increased upon PTPIP51 overexpression. Depletion of PDZD8 leads to reduced contact surface between the ER and mitochondria and to reduced Ca2+ flux into mitochondria in HeLa cells 46. The expression of a loss‐of‐function mutant of PACS2, a cytosolic protein involved in tether maintenance, in A7 human skin melanoma cells diminishes the levels of PSS1 and FACL4, a fatty acid metabolism enzyme 55. Regarding cholesterol trafficking proteins, TSPO downregulation is observed upon the deletion of the tethering protein VDAC1 in U87 MG human glioma cells 56. Furthermore, TSPO was demonstrated to inhibit mitochondrial Ca2+ uptake by promoting VDAC1 phosphorylation 57. The available evidence for the role of different proteins in lipid trafficking is limited to cultured cells, and further studies should be done to validate their function in the context of the whole animal.
Mitochondrial dynamics
Mitochondria change their morphology in order to efficiently adapt to the energetic demands of the cell, to respond to stress conditions (such as nutrient deprivation), or to react to apoptotic stimuli. Mitochondria–ER contacts are crucial for mitochondrial fission, since ER tubules surround and constrict mitochondria at the sites of division 58. This constriction is mediated by actin filaments that accumulate between mitochondria and the ER and that are polymerized at the ER membrane by INF2 59. INF2‐mediated actin polymerization leads to an accumulation of myosin type II 60, an increase in mitochondria–ER contacts, and the subsequent stimulation of mitochondrial Ca2+ uptake before constriction of the IMM and mitochondrial division 61. The main driver of mitochondrial fission is the dynamin‐related GTPase DRP1, which moves from the cytosol to the OMM, where it interacts with membrane proteins such as FIS1, MFF, MiD49, and MiD51 62. MFF, MiD49, and MiD51 recruit DRP1 at the mitochondrial surface to form trimeric complexes in which MiD49/51 compete with MFF for DRP1 interaction 63. DRP1 activity can be regulated by redox signals. PDIA1 modifies DRP1 to negatively regulate its activity and to maintain mitochondrial reactive oxygen species (ROS) at low levels 64. Conversely, an increase in mitochondrial ROS results in oxidation of DRP1 and in increased mitochondrial fission which favors mitochondrial ROS accumulation 64. However, further insight is necessary for a deep characterization of how redox states of cells influence mitochondrial dynamics. At mitochondria–ER contact sites, the ER protein STX17 interacts with mitochondria‐bound DRP1 to support fission 65 (Fig 3). Under starvation conditions, STX17 releases DRP1 to initiate autophagosome formation and to promote the elongation of mitochondria to be protected from autophagy 65, 66. Recent findings have revealed that mitochondria–ER contacts are sites for mitochondrial DNA (mtDNA) synthesis and that nascent mtDNA stays in the daughter mitochondria after fission 67. How the mitochondrial fission machinery is coupled to mtDNA replication has not been explained to date. Mitochondrial fission is a process coupled to mitochondrial removal by mitophagy. The link between these two processes is the OMM protein FUNDC1, which localizes at mitochondria–ER contact sites and interacts with calnexin in the ER and DRP1 on the mitochondrial surface 68. FUNDC1 promotes both autophagosome recruitment and mitochondrial fission 68.
Furthermore, the interaction between the ER and mitochondria is a key player in the regulation of mitochondrial fusion. This process occurs first by fusion of the OMMs, followed by fusion of the IMMs 69. The key players in these two processes are the dynamin‐like GTPases MFN1 and MFN2 in the OMM and OPA1 in the IMM 69. Mitofusins present in mitochondria interact with each other to fuse the OMMs. After OMM fusion, OPA1 oligomerizes and IMMs fuse. A cleaved isoform of OPA1 is associated with mitochondrial fission rather than fusion 70. It is still not clear how many MFN1, MFN2, and OPA1 molecules oligomerize to carry out this function. Dimeric and tetrameric interaction models have been proposed 71, 72, 73. As mentioned before, MFN2 is present at both sides of the MAM, and it participates in mitochondria–ER tethering complexes 74. These two roles of MFN2 are mutually exclusive and determined by its interaction with TCHP (Fig 3). TCHP, a MAM protein localized at the ER, prevents mitochondria–ER tethering and favors mitochondrial elongation when it is bound to MFN2 74.
The alteration of mitochondrial dynamics proteins has an impact on MAM functions. Moreover, changes in certain tethering proteins alter mitochondrial morphology. Regarding the mitochondrial fission machinery, the induction of INF2‐mediated actin polymerization with ionomycin in U2OS human osteosarcoma cells leads to an increase in mitochondrial Ca2+ and mitochondria–ER contact sites 61. Moreover, the chemical inhibition of DRP1 in PC12 rat pheochromocytoma cells leads to a reduction in ER Ca2+ release compared to untreated cells and concomitantly decreases mitochondrial Ca2+ intake 75. Fundc1 overexpression in mouse cardiomyocytes increases Ca2+ release from the ER to mitochondria 76. The role of FUNDC1 in Ca2+ release will be further assessed in this review. Concerning mitochondrial fusogenic proteins, the lack of MFN1 and MFN2 produces an aberrant distribution of pro‐apoptotic proteins in the OMM, thus reducing apoptotic signaling 77. Mfn2 ablation in mouse hearts increases the levels of the anti‐apoptotic protein BCL2 78. Loss of function of the tethering protein VAPB in Caenorhabditis elegans impairs mitochondrial dynamics 79, although this has not been reported in mammals to date. The depletion of the tether maintainer PACS2 in A7 cells results in mitochondrial fragmentation and uncoupling from the ER 55.
Ca2+ signaling
Before MAMs were studied at the molecular level, there was evidence that regions of mitochondria in close proximity to the ER participated in Ca2+ signaling 80. The ER lumen is the cell Ca2+ storage area and the sites of proximity of the ER to mitochondria harbor high Ca2+ microdomains. The ER incorporates Ca2+ from the cytoplasm by SERCA1/2/3 ATPases 81, 82. ATP is hydrolyzed by SERCA transporters in order to allow Ca2+ entry to the ER lumen 81. Ca2+ is released from the ER by the IP3R1/2/3 and the ryanodine receptors (RyR1/2/3), transferred to the mitochondrial intermembrane space by VDAC porins, and finally introduced into the mitochondrial matrix by the mitochondrial Ca2+ uniporter (MCU) complex (reviewed by Giorgi et al 83). Ca2+ transport at the MAMs is depicted in Fig 3. In the OMM, TOM70 recruits IP3R to favor Ca2+ transference to mitochondria 84 and, in the cytosolic part of the MAM, GRP75 couples IP3Rs to VDAC 10, thereby allowing rapid Ca2+ flux into mitochondria. CypD, a protein involved in apoptosis initiation and in mitochondrial ATP synthase modulation 85, interacts with and maintains the VDAC1‐GRP75‐IP3R1 complex 28. Another Ca2+ channel in the ER is the SEC61 complex, a translocon at the ER from which Ca2+ leaks passively to the cytosol 86, 87, and once in the cytosol, it can be sequestered into mitochondria by VDAC. Ca2+ import to mitochondria stimulates the translocation of cristae accumulated H2O2 to MAMs, which results in the appearance of redox nanodomains at the mitochondria–ER interface which enhance Ca2+ efflux from the ER 88. The accumulations of ROS at the MAMs arise as a consequence of active Ca2+ exit from the ER and do not occur with passive Ca2+ leakage. Mitochondrial ROS can regulate as well Ca2+ flux to the mitochondrial matrix by MCU oxidation, which increases the MCU oligomerization and thus its activity 89. Insulin signaling modulates IP3R Ca2+ flux to mitochondria via mTORC2. After insulin stimuli, mTORC2 in the MAM phosphorylates AKT 90, which in turn phosphorylates IP3Rs to reduce Ca2+ release from the ER 91, 92. BiP limits ER Ca2+ leakage through the Sec61 complex by binding to the ER lumenal region of Sec61α 93.
Alterations in Ca2+ trafficking between the ER and mitochondria affect mitochondrial morphology. Mitochondria of brown adipose tissue (BAT) of mice fed on Ca2+ excess for 3 days are larger and fewer than in control mice 94. MFN1 and MFN2 are increased in the BAT of these mice, whereas DRP1 is decreased 94. Mitochondria–ER contacts are also increased in BAT after Ca2+ treatment 94. Moreover, MCU ablation in U2OS cells prevents mitochondrial division 61. In addition to its role in mitochondrial dynamics, FUNDC1 interacts with the ER Ca2+ channel IP3R2 and promotes Ca2+ flux to mitochondria 76. The depletion of Fundc1 in mouse cardiomyocytes and H9c2 myoblasts leads to a decrease in IP3R2 and colocalization between mitochondria and the ER 76. The authors of this study proposed that FUNDC1 and IP3R2 act together as a tethering complex of mitochondria and the ER. FUNDC1 ablation also decreases the levels of the MAM maintenance protein PACS2 76. Inhibition of ER Ca2+ uptake by SERCA initiates UPR and eventually provokes apoptosis 95. PDK4 inhibition decreases Ca2+ flux in C2C12 myoblasts 15. Tg2 ablation impairs Ca2+ flux in MEF 30. The alteration of mitochondria–ER tethers also has an impact on Ca2+ trafficking 76. Disruption of PTPIP51 and VAPB tethering complex impairs Ca2+ homeostasis in HEK293 cells 9. The knockdown of either VAPB or PTPIP51 decreases Ca2+ uptake into mitochondria 9, and overexpression of these proteins increases Ca2+ flux to mitochondria 96. MFN2 depletion has been reported to cause both increase and decrease in Ca2+ uptake 33, 97. In FDB muscles, mitochondrial Ca2+ uptake decreased upon temporal Mfn2 depletion 33. However, the protein levels of Ca2+ transport proteins (MCU, SERCA1, RyR1) remained unchanged 33. In contrast to this result, Mfn2 depletion in MEF cells caused Ca2+ overload in mitochondria 97.
ER stress
When protein folding efficiency is disturbed at the ER, misfolded proteins accumulate in the lumen and cause ER stress. This can happen as a result of certain conditions, such as nutrient deprivation, hypoxia, loss of Ca2+ homeostasis, free fatty acids, and GM1 ganglioside accumulation 98, 99, 100, 101, 102, 103. Why stress conditions lead to protein misfolding remains unknown. The accumulation of large amounts of these misfolded proteins activates the unfolded protein response (UPR) in order to restore protein homeostasis or to induce apoptosis 104, 105. The UPR has three main branches, which are interconnected after the signal transducers in the ER, namely PERK, ATF6, and IRE1α, have been stimulated 106. What is known about UPR branches has been discovered by treating cells and animals with exogenous compounds or unphysiological harvesting conditions to provoke protein misfolding; nevertheless, the natural cause of protein folding defects and their accumulation is not known. The activation of PERK induces eIF2α phosphorylation. Phosphorylated eIF2α inhibits global protein translation and activates ATF4, which translocates to the nucleus to induce the expression of survival genes 107. Prolonged UPR activation induces apoptosis through the activation of CHOP by ATF4 108. The ATF6 branch of the UPR starts with the translocation of ATF6 from the ER to the Golgi apparatus for cleavage 109. Cleaved ATF6 is a transcription factor that induces ER‐associated degradation genes 110 and XBP1 111. IRE1α activation induces splicing of XBP1 mRNA to enhance cell survival 111, activation of MAPK 112 to modulate autophagy and apoptosis, and IRE1α‐dependent mRNA decay 113. ER stress signaling can be amplified at MAMs by SERCA1 truncated isoform (ER), which acts upstream of the PERK–eIF2α–ATF4–CHOP pathway 114. Moreover, the location of PERK at MAMs contributes to the maintenance of mitochondria–ER contact sites and to the enhancement of ROS‐mediated mitochondrial apoptosis signaling 115. UPR mission is to restore cellular homeostasis by correcting protein folding and recovering damaged ER environment 116. As long as protein folding efficiency is not resolved, UPR is activated 116. ER stress results in increased mitochondria coupling to ER, which increases ATP production, oxygen consumption, and mitochondrial Ca2+ uptake 117. Chronic UPR signaling initiates a signaling cascade in the MAM that eventually leads to apoptosis 116. This signaling pathway is discussed in the following section.
The alteration of ER stress proteins, especially the IRE1α branch, impairs lipid handling. The hepatic depletion of Xbp1 in mice decreases circulating levels of fatty acids, triglycerides, and sterols, compared to control mice 118. Lack of XBP1 in the liver impairs cholesterol processing for the generation of bile acids in these mice 119. This phenotype is recovered by overfeeding the mice with cholesterol 119. A separate study using liver Xbp1 knockout mice also reports decreased plasma levels of cholesterol and triglycerides compared to control mice 120. This effect is prevented by knocking down IRE1α 120. The modification of mitochondria–ER tethering proteins leads to ER stress. MFN2 interacts with PERK and represses its activity 97. MFN2 loss of function in MEF cells dysregulates the three branches of the UPR by enhancing the PERK–eiF2α–ATF4–CHOP pathway 97. Ablation of Mfn2 results in the continuous activation of PERK, and PERK silencing in these cells causes ROS production, the restoration of mitochondrial Ca2+ levels, and an improvement of mitochondrial morphology 97. However, it is not known whether PERK interacts with mitochondrial MFN2, ER MFN2, or both (Fig 3). The increase in mitochondria coupling to ER upon ER stress 117 is coherent with the upregulation of MFN2 expression observed in MEFs upon ER‐stress induction with thapsigargin and tunicamycin 121. In the same study, the authors show that ablation of Mfn2 in MEFs upregulates ER stress markers (BiP, GRP94, and ATF4) 121. Specific ablation of Mfn2 in mouse cardiac myocytes also causes an increase in the expression of BiP, GRP94, and ATF4 121. Another tethering protein, VAPB, represses the UPR by binding to ATF6 122. Overexpression of VAPB both in HEK293 and NSC34 cells decreases ATF6/XBP1‐induced luciferase activity upon tunicamycin stimulation, even in combination with ATF6 overexpression 122.
Apoptosis initiation and ER stress‐mediated apoptosis
When cells cannot adequately handle certain stress stimuli, they activate pathways that lead to cell death. A complex formed by BAP31 and FIS1 at mitochondria–ER contact sites is able to transfer apoptotic signals back and forth from the mitochondria to the ER 11. In response to apoptotic stimulus, the FIS1‐BAP31 complex recruits procaspase‐8 to be activated 11. Active caspase 8 cleaves BAP31 into a pro‐apoptotic form that, together with FIS1, promotes Ca2+ release from the ER 11, 123 and mitochondrial fission 17. Mitochondrial Ca2+ increase leads to CYPD activation in the IMM to open the permeability transition pore from which molecules that drive apoptosis are released 124, 125. Under sustained ER stress conditions, CDIP and BAP31 interact at the ER side of the MAM to sequester the anti‐apoptotic factor BCL2 located at the OMM, in order to promote apoptosis (Fig 3) 126. The protein PACS2, which localizes in MAMs, has been reported to promote the translocation of the pro‐apoptotic protein BID to mitochondria 55. The CDIP1‐BAP31‐BCL2 complex, together with the truncated form of Bid and caspase‐8 activation, promotes BAX and BAK oligomerization 126. BAX translocates from the cytosol to the OMM, where BAK locates constitutively. The activation of these two molecules occurs after their oligomerization 127. Although not yet demonstrated, it is believed that cytochrome c exits mitochondria from the pores formed by these oligomers 128. After cytochrome c release, an apoptotic protease cascade is initiated 129.
Since MAMs are hubs for apoptosis initiation and this process involves Ca2+ signaling at the MAM and mitochondrial fission, the alteration of some apoptosis initiators has an impact on these MAM functions. BCL2 loss‐of‐function mutations in Jurkat T cells decreases mitochondrial Ca2+ uptake 130. When Bak is knocked out in MEF cells, mitochondria are not fragmented in response to apoptotic stimulus 131. Overexpression of Bax promotes MFN2‐mediated mitochondrial fusion in MEFs 132. It has been proposed that BAX plays a dual role: The soluble cytoplasmic form promotes mitochondrial fusion and, when activated and recruited to the OMM, it participates in apoptosis 132.
Autophagosome formation
In order to preserve cellular homeostasis, damaged or unnecessary components of the cell must be degraded or recycled. This process is achieved by autophagy. Autophagosomes engulf damaged or needless components and can originate from ER–mitochondria contact sites 20. The ER side of the MAM region, where proteins related to autophagosome formation start to accumulate, is known as the isolation membrane. This structure protrudes from the ER and finally closes around the cellular components to be eliminated, forming a vesicle named the autophagosome 133. After starvation stimulus, ATG14 is recruited to the MAM by STX17 20. This results in accumulation of the components of the class III PI3K complex (ATG14, BECN1, VPS34, and VPS15) at the MAM 20 (Fig 3) and contributes to the initiation and nucleation of the isolation membrane. Next, the ATG16L1 complex (ATG5‐ATG12‐ATG16L) is recruited to the isolation membrane, where it binds PE to LC3 134. Lipidated LC3 molecules associate with the isolation membrane and remain attached once the autophagosome is closed 133. How the isolation membrane closes is still poorly understood.
Alterations in tethering proteins have an impact on autophagosome formation. The absence of MFN2 or PACS2 at the MAM impedes STX17‐mediated ATG14 recruitment to this area in HeLa cells 20. MFN2 and PACS2 seem to be crucial for autophagosome biogenesis; nevertheless, their role in this process has not been described yet. Moreover, the tightening of mitochondria–ER contact sites by overexpression of VAPB and PTPIP51 results in decreased autophagosome formation after torin‐1 or rapamycin stimulus in HeLa and HEK293T cells 135. Conversely, the opposite effect is observed when PTPIP51 and VAPB are ablated in HeLa and HEK293T cells 135. Future work will unravel what is the precise role of the MAM in autophagosome formation and which MAM proteins recruit the autophagosome biogenesis machinery. Moreover, since membrane contact sites from other organelles have also been proposed as autophagosome factories 20, 136, 137, 138, a remaining open question in the field is whether autophagosomal content to be degraded is influenced by the origin of the autophagosome.
Consequences of metabolic challenge in mitochondria–ER contacts
Human, animal, and cellular studies have revealed that metabolic alterations can perturb mitochondria–ER contact sites. In this section, we review the observations that document the impact of altered metabolic homeostasis on the architecture and functioning of MAMs, and also the impact of nutrient availability and lysosomal storage disorders.
The impact of metabolic disorders on the architecture and functioning of mitochondria–ER contacts
Some studies have reported alterations in mitochondria–ER contacts in liver and in muscle cells upon metabolic dysregulation. Surprisingly, there is considerable discrepancy in the data available. Thus, liver analysis of ob/ob obese mice and mice subjected to a high‐fat diet (HFD) showed an increased abundance of MFN2, IP3R, and PACS2 in MAM fractions, in parallel to increased mitochondria–ER contacts, and an excess of mitochondrial Ca2+ accumulation 29. Moreover, a forced increase in the mitochondria–ER contacts, induced by expression of an artificial linker in livers from control mice, caused an increased mitochondrial Ca2+ uptake and impaired glucose homeostasis 29. Conversely, Ip3r1 knockdown in obese mice caused a reduced Ca2+ flux, and similarly, Pacs2 knockdown led to a decreased physical interaction between the ER and mitochondria, and to improved glucose homeostasis 29.
In contrast to the results obtained by Arruda et al, Tubbs et al 27 detected that mitochondria–ER contacts (measured by quantifying VDAC1–IP3R1 interaction) were decreased in hepatocytes isolated from mice subjected to diet‐induced diabetes and from ob/ob mice. Moreover, the overexpression of CypD in these hepatocytes increased mitochondria–ER contacts and improved the effects of insulin. A third study revealed that HepG2 hepatoma cells treated with palmitate showed reduced mitochondrial Ca2+ flux, lower mitochondria–ER contacts, and impaired insulin sensitivity 139. Under these conditions, Mfn2 overexpression ameliorated the mitochondria–ER contact area and insulin sensitivity. We do not yet know whether the opposite observations are a consequence of differences in the methodology used (proximity ligation assays or transmission electron microscopy, or tissue sections versus isolated hepatocytes), or whether they are based on subtle differences in the nutritional state of the animals studied. Regarding the impact of metabolic alterations in liver ER stress and ER Ca2+ homeostasis, the main studies of the field show more consensus. Livers of obese mice show increased ER stress 140, 141 and reduced cellular SERCA2b levels 140 or impaired SERCA activity in the ER 141. Glucose tolerance was increased and ER stress was alleviated in obese and diabetic mice by liver exogenous expression of SERCA2b 140, 141.
In the skeletal muscle of ob/ob mice or in mice subjected to a high‐fat, high‐sucrose diet, Tubbs et al found impaired insulin signaling and decreased levels of MAM proteins, accompanied by a decrease in mitochondria–ER contacts (measured by quantifying VDAC1–IP3R1 interaction). In human myotubes from healthy patients, mitochondria–ER contacts were also diminished after palmitate treatment 142. Mitochondrial Ca2+ concentration was somewhat decreased compared to untreated cells. Mfn2 or Grp75 overexpression reversed the effects of palmitate on MAM proteins and on insulin signaling 142. Moreover, myotubes from obese patients and from obese patients with type 2 diabetes showed decreased mitochondria–ER contacts compared to those of healthy patients 142. In contrast to these results, Arruda et al 29 found an increase in the MAM proteins MCU and RyR in soleus muscles from ob/ob and HFD‐fed mice, suggesting an enrichment in MAMs under these conditions. In line with these results, Thoudam et al 15 found increased levels of IP3R1, VDAC1, and GRP75 in the MAM fraction of skeletal muscle of HFD‐fed mice and ob/ob mice. Furthermore, they observed increased levels of MFN2 in HFD‐fed mice. Moreover, using proximity ligation assays, they detected increased IP3R1–GRP75–VDAC1 interactions in HFD‐fed mice and ob/ob mice 15. Transmission electron microscopy of muscle MAM surface revealed increased MAM area in HFD‐fed mice compared to animals on a chow diet 15. Interestingly, the authors quantified the distance between juxtaposed ER and mitochondria and found that in HFD‐fed mice the proportion of loose contacts between the ER and mitochondria was increased. As mentioned in the liver studies, we do not know whether the differing observations detected in muscles of obese mice are due to differences in the methodology used or to differences in the nutritional state of the animals studied. In any case, further studies are required to clarify the nature and kinetics of the changes that occur in mitochondria–ER contacts during metabolic dysregulation in muscle and in liver.
Insulin‐resistant conditions such as obesity and type 2 diabetes are characterized by altered muscle expression of MFN2, which may participate in the alterations in mitochondria–ER contacts. Thus, Mfn2 is repressed in the muscles of obese Zucker rats 143. Obesity induced by a HFD during 40 weeks also reduces MFN2 and MFN1 expression in muscle 144. In addition, the muscles of obese subjects also show a reduced expression of MFN2 compared with lean subjects 143, 145. In contrast, bariatric surgery‐induced body weight loss was reported to increase MFN2 gene expression in the skeletal muscle of morbidly obese subjects 146, 147, 148 in parallel with increased insulin sensitivity 146, 147, 148. Type 2 diabetic patients also show reduced MFN2 expression in the skeletal muscle compared with control subjects 145, 149, and this occurs both in obese and non‐obese type 2 diabetic patients 145. The dysregulation of MFN2 is unlikely to be a consequence of reduced insulin action because the expression of this gene in lean, obese, or type 2 diabetic subjects is not altered in response to 3 h of hyperinsulinemia during clamp studies. Neither is the expression of this protein affected when cultured muscle cells are chronically incubated in the presence of insulin 145, 150. Induced insulin resistance in rats by high sucrose diet provokes slower contraction and elongation of cardiomyocytes 151. ER Ca2+ uptake by SERCA is impaired in these cardiomyocytes although the levels of this protein remain unchanged 151.
Studies in beta‐cells also indicate the existence of alterations in response to metabolic stress. Thivolet et al 152 reported an increased IP3R2 and decreased VDAC1 expression, accompanied by reduced mitochondria–ER contact sites in beta‐cells from type 2 diabetic patients. In addition, Min6‐B1 beta‐cells exposed to palmitate show ER stress, reduced mitochondria–ER contacts, and impaired insulin secretion 152. Zhang et al 153 reported increased VDAC1 abundance accompanied by mislocalization of part of VDAC1 to the plasma membrane. INS1E beta‐cells also respond to elevated glucose concentrations in the culture medium by upregulating VDAC1 154. High glucose environment increases Bax mRNA levels and stimulates BAX‐dependent apoptosis in mouse islets 155.
POMC neurons respond to a HFD by reducing the number of mitochondria–ER contacts compared to mice on a regular diet 156. Under these conditions, MFN2 levels are reduced in HFD mice 156. In HFD‐fed mice, stimulation of DRP1‐dependent mitochondrial fission in the dorsal vagal complex induces ER stress and insulin resistance 157, and inhibition of DRP1 restores ER stress, insulin resistance, and hepatic glucose metabolism.
An increase in mitochondria–ER contacts, increased expression of IP3R1, IP3R2, and PACS2 protein levels, and greater mitochondrial Ca2+ uptake and apoptosis has been documented in oocytes from HFD‐treated mice 158. These changes compromise oocyte maturation.
In conclusion, available data suggest that metabolic stress linked to insulin resistance, obesity, or type 2 diabetes affects mitochondria–ER contacts, and may occur in various cells or tissues. Liver and muscle have been deeply analyzed in this context, and the observations annotated by the authors differ across the studies that we have discussed. Whether the consequence of this metabolic stress in liver and muscle is to increase or to reduce the surface of contact between the ER and the mitochondria is still under debate. Moreover, current data regarding the expression levels of the tethering proteins MFN2, IP3R, VDAC, and GRP75 under conditions of insulin resistance, obesity, or type 2 diabetes annotated across several studies are not uniform. As a consequence, the functional impact of the modification of these contacts is not known. An example that illustrates this lack of consensus is the persistent discrepancy on the observations regarding Ca2+ influx into mitochondria under metabolic stress conditions. In order to solve this puzzle, there is need of a precise characterization of the molecular mechanisms involved in the response to metabolic stress as well as the establishment of standard procedures to perform this characterization.
The adaptation of mitochondria–ER contact sites to nutrient availability
Mitochondria–ER contact sites respond to changes in nutrient availability by modifying Ca2+ signaling, initiating autophagy or activating the UPR. The first evidence for this response was observed by Sood et al 159 in postprandial mouse livers. Five hours postprandial livers showed larger mitochondria–ER contact sites than 2 h postprandial livers. In agreement with this study, livers of fed mice show reduced mitochondria–ER contacts compared to those of overnight fasted mice, and this occurs without a significant modification of the ER or mitochondrial content 160. The interactions between VDAC1 and IP3R1 are reduced in fed compared to fasted mice. VDAC1 and GRP75 are also decreased upon feeding in mouse liver, whereas IP3R1 protein expression remains unchanged 160. In a different study, mice refed after 22 h of fasting showed a 50% decrease in hepatic IP3R1 levels compared to fasted animals 161. Interestingly, primary hepatocytes cultured in the presence of high glucose (17 mM) also show a reduced VDAC1–IP3R1 interaction compared to cells cultured in the presence of 5 mM glucose. In conclusion, these data suggest that glucose availability is a key signal in the regulation of mitochondria–ER contacts in liver cells.
Genetic obesity appears to alter the dynamics of mitochondria–ER contacts. Thus, the livers of obese ob/ob mice do not reproduce the mitochondria–ER uncoupling during fed to fasted transition. MAMs are reduced in fasting conditions in ob/ob compared to lean mice, and no differences are detectable compared to obese mice analyzed during fed conditions 160. Moreover, the interaction between IP3R1 and VDAC1 is reduced in fasted ob/ob mice compared to lean mice and no significant differences are observed in ob/ob mice when comparing fed and fasted states 160. In keeping with these data, ob/ob hepatocytes cultured in the presence of high glucose do not show a reduced VDAC1–IP3R1 interaction compared to cells cultured in the presence of 5 mM glucose, again indicating that obesity disrupts glucose‐induced control over mitochondria–ER contacts.
Nutrient deprivation causes the inhibition of mTOR activity, which activates autophagy 162 and promotes the formation of autophagosomes. In this regard, autophagosomes originate at mitochondria–ER contact sites 20, and the disruption of MAMs by depletion of Mfn2 or Pacs2 results in impaired autophagosome formation 20, 163. Moreover, glucose and amino acid deprivation causes mitochondrial elongation 66, which occurs through inhibition of DRP1 and activation of MFN1, thus protecting mitochondria against autophagic degradation. In all, available data allow us to propose that the modulation of mitochondria–ER contact sites is linked to the modulation of autophagosome formation and mitochondrial morphology through mechanisms that require extensive research efforts. It is likely that signals such as mTOR, and ER stress response, which are modulated by nutrient availability 164, 165, participate in mitochondria–ER contacts during fed to fasted transitions.
In summary, ER and mitochondria contact surface increases in response to lack of nutrients. This adaptive increase in contact sites is impaired under obesogenic conditions. However, it is still uncertain whether these changes in the MAM are accompanied by a modification in the expression levels of tethering proteins. Enhanced autophagosome formation and ER‐stress response are probably the functional consequences of MAM enlargement.
Metabolic impact of alterations in proteins participating in mitochondria–ER contacts
The concept that metabolic homeostasis is determined by modulation of mitochondria–ER contact sites arose after several reports documenting that Mfn2 deficiency disrupts metabolism in cells and in animal models 97, 156, 166, 167. In addition, the metabolic effects caused by Mfn2 deficiency differ to those detected upon Mfn1 ablation, thereby indicating that they are probably due to compromised mitochondria–ER contacts rather than to impaired mitochondrial fusion. Subsequent mouse studies have revealed that ablation of proteins participating in mitochondria–ER contacts causes three distinct phenotypes, namely (i) reduced glucose tolerance and insulin signaling; (ii) improved glucose tolerance; and (iii) disrupted lipid metabolism. In this chapter, we analyze the data currently available with respect to these three categories (Fig 4).
Figure 4. Metabolic impact of alterations in proteins participating in mitochondria–ER contacts.

(A) GRP75, MFN2, BAP31, or CypD depletion leads to deficient insulin signaling and glucose intolerance. The lack of GRP75, MFN2, BAP31, or CypD at mitochondria–ER contact sites causes deficient insulin signaling and glucose intolerance, probably through a mechanism that involves ERUPR or mtUPR. (B) IP3R1, VDAC1, FUNDC1, or PACS2 ablation results in enhanced insulin signaling and improved glucose tolerance. The lack of IP3R1, VDAC1, FUNDC1, or PACS2 at mitochondria–ER contact sites potentiates insulin signaling and improves glucose tolerance. The mechanism by which FUNCD1 or PACS2 causes these effects is mediated by the release of FGF21. As a result of decreased mitochondrial Ca2+ accumulation, IP3R1 or VDAC1 ablation may result in enhanced insulin signaling and improved glucose tolerance. (C) Mitochondria–ER contacts response to protein ablation. The deletion of proteins that participate in the MAM results in the activation of signaling pathways that either enhance or impair insulin sensitivity and glucose tolerance. It has been proposed that these signaling pathways are related to UPR, FGF21, and an adaptive mitochondrial response that may lead to an improved or to a worsened response to insulin and glucose handling. (D) ORP8 or ATAD3A deficiency causes lipid metabolism alterations. Various alterations in lipid metabolism are observed upon ORP8 and ATAD3A ablation. ORP8 deficiency causes an increase in circulating HDL, cholesterol, triglycerides, and phospholipids. On the other hand, a lack of ATAD3A results in impaired cholesterol and lipid metabolism, reduced cholesterol esters, and decreased steroidogenesis.
Proteins whose depletion causes deficient insulin signaling and glucose intolerance
There is extensive evidence of a major metabolic role of MFN2 in mouse tissues. Thus, muscle‐specific ablation of Mfn2 causes age‐dependent glucose intolerance and deficient insulin signaling 166, 167 (Fig 4A). Mfn2‐deficient muscles also show reduced muscle autophagy, muscle atrophy, and loss of muscular function 167. Mfn2 repression in cultured muscle cells also reduces insulin signaling 142.
Mfn2 deficiency in liver is also associated with disrupted insulin signaling, enhanced hepatic glucose production, enhanced expression of gluconeogenic genes, and glucose intolerance 166. Mfn2 repression also reduces insulin signaling in human liver cells 27. Notably, the phenotype linked to Mfn2 deficiency is opposite to what occurs in mice upon ablation of Mfn1, which show protection against HFD‐induced insulin resistance, and enhanced mitochondrial respiration 168. These observations suggest that the mechanisms linked to Mfn2 deficiency are not a consequence of alterations in mitochondrial fusion but are rather linked to its tethering function. Mfn2 ablation in adipose tissues obtained by crossing Mfn2 loxP/loxP mice with mice expressing the Cre recombinase under the adiponectin promoter leads to enhanced body weight and fat mass, which was linked to a reduction in energy expenditure and in BAT thermogenesis 3. In keeping with these data, BAT‐specific Mfn2 deletion through Ucp1‐Cre causes BAT lipohypertrophy and cold intolerance 169. The effects linked to Mfn2 ablation in adipose depots are not detected upon ablation of Mfn1 3. These findings thus support the notion that the alterations detected in BAT are not dependent on mitochondrial fusion, but on a different function of MFN1 and MFN2.
Specific ablation of Mfn2 in pro‐opiomelanocortin (POMC) neurons of the hypothalamus results in defective POMC processing, leptin resistance, hyperphagia, reduced energy expenditure, and obesity 156. These data establish MFN2 in POMC neurons as an essential regulator of systemic energy balance. Along the same lines, interfering with mitochondrial fusion mechanisms in Agrp neurons by selectively knocking down Mfn2 results in altered mitochondrial size and density in these cells. Agrp‐specific Mfn2 knockout mice gain less weight when fed a HFD due to decreased fat mass 170. Available data in POMC neurons also indicate that the effects caused by Mfn2 ablation differ greatly from what is detected for Mfn1 ablation. Thus, mice lacking MFN1 in POMC neurons exhibit attenuated hypothalamic gene expression programs during the fast‐to‐fed transition 171. This loss of mitochondrial flexibility in POMC neurons alters glucose sensing, causing abnormal glucose homeostasis as a result of defective insulin secretion by pancreatic β cells 171. In conclusion, available data in conditional knockout mouse models indicate that Mfn2 ablation causes defects in metabolism that are very different to those effects that result from ablation of Mfn1. These observations are compatible with MFN2 exerting metabolic effects in tissues via modulation of the mitochondria–ER contact sites.
In connection with the effects of the ER triggered by Mfn2 deficiency, a potent UPR has been documented both in cells and in tissues 97, 156, 166. Thus, Mfn2 ablation in liver or in skeletal muscle causes chronic activation of the UPR, which involves the three UPR arms, i.e., the PERK, IRE‐1a, and the ATF6 pathways 166. Furthermore, treatment of liver‐specific Mfn2 knockout mice with an ER stress blocker restores insulin sensitivity and glucose homeostasis 166, thereby suggesting that the functional link between MFN2 and the UPR has metabolic relevance.
Ablation of GRP75, the bridge between IP3R and VDAC, has been reported to cause a reduction in VDAC1/IP3R1 interactions in human liver cells and in myotubes 27, 142. Moreover, GRP75 deficiency reduces insulin signaling and insulin action in HuH7 liver cells 27, 142 (Fig 4A). The depletion of GRP75 in medullary thyroid carcinoma cells induces the MEK/ERK pathway and increases oxidative stress 172. Both in GRP75 knockdown SH‐SY5Y bone marrow neuroblasts and in fibroblasts derived from a Parkinson's disease patient with GRP75 loss of function, increased mitochondrial UPR (UPRMT) was reported 173.
Hepatocyte‐specific deletion of the tethering protein BAP31 is linked to metabolic defects in mice. Bap31‐deficient mice show enhanced body weight, increased food intake, and greater liver steatosis after exposure to a HFD 174. Another study with these mice reported the same effects upon tunicamycin treatment, as well as increased levels of ER‐stress markers 175. In Bap31‐deficient mice, although not statistically significant, a trend toward increased p‐eIF2α, ATF4, and CHOP signaling was observed 175. Moreover, hepatocytes of these mice show increased lipogenic gene expression and SREBP1C expression, and activation of SREBP signaling. Bap31‐deficient mice show impaired glucose tolerance and reduced insulin responsiveness under normal diet or a HFD challenge (Fig 4A).
Depletion of the modulator of the permeability transition pore, CypD, in mice has been reported to enhance hepatic gluconeogenesis, as assessed by the pyruvate tolerance test 27. In addition, CypD deficiency is associated with deficient insulin signaling and a reduced number of VDAC1/IP3R1 interactions in human liver cells 27. The metabolic effects detected under conditions of CypD deficiency in mice and in human liver cells were characterized by UPRER 176. In conclusion, these data are coherent with a model in which CYPD plays an important role in the maintenance of mitochondria–ER contact sites, which, upon dysregulation, trigger ER stress and metabolic alterations, namely deficient insulin signaling and insulin resistance (Fig 4A).
A shared feature of the absence of MFN2, GRP75, BAP31, or CYPD is the activation of UPR 97, 156, 166, 173, 175, 176. ER stress was initially proposed as a mechanism that drives insulin resistance‐related diseases 177, and altered reticulum proteostasis alteration has also been in the spotlight as a possible driver of metabolic diseases 178. Insulin resistance observed upon ablation of MFN2, GRP75, BAP31, or CYPD may arise as a result of altered mitochondria and/or ER protein homeostasis. The signaling pathways activated upon loss of protein homeostasis in both organelles converge in ATF4 and CHOP upregulation 179, 180. JNK activation is as well a consequence of ER stress that impairs insulin signaling, eventually leading to insulin resistance 177. ER stress provokes hyperactivation of JNK, which phosphorylates and inhibits the insulin receptor IRS1 177. MFN2, BAP31, and CYPD ablation provoke an increase in activated JNK 156, 175, 176; however, there are no data available on the levels of activated JNK in the absence of GRP75. Another common pathway that could be involved in insulin resistance upon depletion of Mfn2, Grp75, Bap31, or CypD is the MEK/ERK signaling cascade. MFN2 is a repressor of the MEK/ERK signaling pathway by its interaction with RAS 181. ERK is hyperactivated upon Mfn2 ablation in MEFs 182. Ablation of GRP75 stimulates the MEK/ERK signaling pathway in medullary thyroid carcinoma cells 172. Moreover, the ERK signaling pathway is more active in CypD knockout mice hearts than in hearts of control animals 183. Moreover, BAP31 depletion in human embryonic stem cells leads to a mild increase in ERK phosphorylation 184. Active ERK phosphorylates PPARγ, which in turn stimulates the expression of genes related to diabetes 185, 186.
Proteins whose depletion enhances insulin signaling and improves glucose tolerance
In addition to participating in mitophagy, FUNDC1 mediates the formation of mitochondria–ER contact sites and promotes Ca2+ flux from the ER to mitochondria through binding to IP3R2 in cardiac cells 76. Cardiomyocyte‐specific ablation of Fundc1 results in decreased mitochondrial and cytosolic Ca2+ levels 76. Fundc1 ablation in mouse muscle causes mitochondrial dysfunction characterized by lower ATP and enhanced ROS 187. In addition, Fundc1‐ablated muscles show a reduced capacity to exercise, probably as a consequence of reduced fat oxidation 187. Nevertheless, Fundc1 knockout mice show improved glucose tolerance, insulin responsiveness, and less adiposity upon treatment with a HFD (Fig 4B). The process responsible for this reduced susceptibility to obesity is the activation of adaptive thermogenesis of adipose tissue driven by ROS‐dependent muscle expression of FGF21 187.
The downregulation of hepatic Pacs2 in ob/ob mice increases maximal mitochondrial respiration and reduces JNK 29. Under these conditions, Pacs2 deficiency improves glucose tolerance and increases hepatic insulin signaling 29. In oocytes from obese mice, genetic repression of Pacs2 decreases Ca2+ influx into mitochondria and ROS production 158. In Pacs2 knockout mice, liver expression of FGF21 is increased and mice are resistant to diet‐induced obesity 188 (Fig 4B).
FGF21 is a systemic regulator of energy homeostasis and insulin sensitivity 189. Its expression is activated upon detection of low protein and high carbohydrate levels. In mouse models of diabetes, this protein improves insulin sensitivity and reduces circulating glucose and triglyceride levels 190. FGF21 upregulation in Fundc1‐ and Pacs2‐deficient mice explains the improvement in glucose handling and insulin signaling observed in these mice.
The inhibition of the anti‐apoptotic protein BCL2 mimics glucose stimulation by increasing mitochondrial activity and ATP production in pancreatic b‐cells 191. Pancreatic islets isolated from Bcl2 −/− mice show enhanced insulin secretion in response to glucose stimulation 191. The knockout of Bak and Bax in β‐cells does not involve metabolic changes, indicating a role for BCL2 in metabolism besides its anti‐apoptotic function 191. In keeping with these results, induced insulin resistance in HepG2 cells upregulates BCL2 192. How Bcl2 suppression leads to improved response to glucose has not been elucidated.
In contrast to the above data on the ablation of the Ca2+ channeling protein GRP75, adenoviral‐induced hepatic deficiency of Ip3r1 enhances mitochondrial respiration, lowers JNK activity, enhances insulin signaling, and improves glucose tolerance in mice 29 (Fig 4B). Analysis of the Ip3r1 heterozygous mutant (opt/C) mouse indicates defects in beta‐pancreatic cells, with reduced beta‐cell mass, and impaired glucose tolerance 193. In oocytes from obese mice, genetic repression of Ip3r1 reduces Ca2+ influx into mitochondria and also leads to a decrease in ROS production 158.
In line with these results, the downregulation of VDAC1 in pancreatic beta‐cells has a protective effect against high glucose concentrations and maintains cellular reductive capacity 153. VDAC1 depletion in cancer cells has been shown to reprogram metabolism toward a decrease in energy production, accompanied by growth arrest 194, 195. Moreover, insulin release and ATP production in response to high glucose concentrations are improved in VDAC1‐depleted cells 153. In pancreatic islets from db/db mice, VDAC1 inhibition leads to enhanced ATP generation and glucose‐stimulated insulin secretion in response to high glucose levels 196 (Fig 4B).
To explain the hepatic increase in IP3R1‐observed obese mice, in 2014 Arruda et al hypothesized that excessive Ca2+ accumulation in mitochondria was the cause of impaired glucose metabolism and insulin sensitivity. In the same study, they showed improved glucose tolerance when they knocked down IP3R1 29. Since the ablation of VDAC1 in pancreatic cells also has a protective effect against high glucose concentrations 153, it is possible that decreased mitochondrial Ca2+ accumulation drives an improvement in insulin signaling.
Another important player in insulin signaling at the MAMs is mTORC2. mTORC2 signaling is essential for an adequate response to insulin 197. Moreover, mTORC2 is implicated in the regulation of MAM integrity and its ablation results in decreased MAM formation and insulin resistance 90, 197. Insulin enhances mTORC2 localization at the MAMs 90. Here, mTORC2 activation induces inhibitory phosphorylation of IP3R and PACS2 by AKT 90. PACS2 ablation and IP3R ablation could have a synergistic effect with mTOCR2 signaling in response to insulin stimuli.
Surprisingly, current data indicate that the repression of some proteins involved in mitochondria–ER contacts enhances glucose tolerance and insulin sensitivity by inducing the expression of FGF21, and perhaps by an independent mechanism related to an adaptive response of mitochondria which implies reduced mitochondrial Ca2+ or mTORC2 signaling. Further studies are required to determine whether those processes are indeed independent or whether they share common mechanisms.
Why the deletion of certain proteins that participate in mitochondria–ER contacts results in enhanced or in impaired insulin sensitivity and glucose tolerance is not known yet. We believe that the absence of these proteins stimulates several signaling pathways that are related to UPR, FGF21, and an adaptive mitochondrial response (Fig 4C). The final output, i.e., the observed phenotype, will depend in each case on the sum of all the pathways that have been activated. We propose that a sum of stimuli that results in UPR and therefore JNK activation will provoke impaired response to insulin and glucose handling. On the other hand, if the combination of all the stimuli leads to increased circulating FGF21 and/or an adaptive mitochondrial response (which would include decreased mitochondrial Ca2+ accumulation), the phenotype observed will be an improved response to insulin and glucose tolerance.
Proteins whose depletion alters lipid metabolism
Skeletal muscle‐specific Atad3 knockout mice show muscle atrophy in combination with mitochondrial abnormalities that include lack of cristae, reduced OXPHOS complexes and OPA1 expression, and progressive mtDNA depletion 198. Fibroblasts derived from patients suffering from ATAD3 gene cluster deletions show impaired cholesterol metabolism and mtDNA damage, as well as impaired lipid metabolism 199 (Fig 4D). In agreement with these data, muscle ATAD3 deficiency reduces the levels of cholesterol esters in muscle, probably due to reduced Acetyl‐CoA acetyltransferase 199. The effects of ATAD3 ablation on substrate handling have not been analyzed. Moreover, ATAD3 ablation decreases steroidogenesis in a mouse cell line derived from Leydig cell tumor 200.
ORP5 and ORP8 depletion leads to altered mitochondria morphology and function in HeLa cells 43. The global ablation of Orp8 in mice causes a marked elevation of high‐density lipoprotein (HDL) cholesterol and phospholipids, which occurs in the absence of changes in apolipoprotein A‐I 201. Moreover, the secretion of nascent HDL particles is enhanced in primary ORP8‐deficient hepatocytes, thereby suggesting altered biosynthesis of HDL 201 (Fig 4D). No information on the impact of ORP8 depletion on glucose homeostasis is available.
In conclusion, available data suggest that some proteins of mitochondria–ER contacts play a pivotal role in the modulation of lipid metabolism. Future studies should address the mechanisms by which given proteins specifically modulate lipid metabolism in the absence of changes in energy metabolism.
Architectural, functional, and metabolic aspects of mitochondria–LD contact sites
Mitochondria and LDs are in active communication in highly metabolic tissues such as BAT, skeletal muscle, and heart 3, 202, 203, 204. It has been reported that the properties of the mitochondria surrounding LDs differ to those of mitochondria in the cytoplasm 2, 205, which suggests a context‐specific metabolic behavior of mitochondria. A study conducted by Benador et al 2 in BAT revealed that mitochondria surrounding LDs oxidize pyruvate, generate ATP, and use fatty acids for triacylglycerol (TAG) synthesis, whereas cytosolic mitochondria oxidize fatty acids. In addition, it has been reported in cultured cells that, under nutrient deprivation conditions, mitochondria and LDs interact in order to favor fatty acid oxidation 206, 207. This observation is in keeping with prior findings indicating that lack of nutrients enhances fatty acid oxidation 208. The variable impact of the interaction of LD with mitochondria on beta‐oxidation will require a precise molecular explanation.
Proteins involved in mitochondria–LD contacts
The protein components of mitochondria–LD contact sites have been only partially characterized (Table 2). A tethering complex identified to operate in BAT is MFN2‐PLIN1 3 (MFN2 is located in the mitochondria and PLIN1 in LDs; Fig 5). Another mitochondrial protein that interacts with LD proteins is ACSL1 (acyl‐CoA synthase long chain family member 1). BioID technology has revealed the interaction between ASCL1 and SNAP23 and VAMP4 in LDs 209 (Fig 5). The LD protein SNAP23 was first suggested to mediate the interaction between mitochondria and LDs since it was observed that its ablation reduced the contacts between these two organelles 210. Moreover, SNAP23, together with VAMP4, is involved in LD fusion 211. The LD protein PLIN5 has been described to localize at mitochondria–LD contacts; however, the mitochondrial partner of this protein is unknown 212, 213. PLIN5 interacts with ATGL and ABDH5 214 on the LD surface (Fig 5).
Table 2.
Mitochondria–LD contact sites proteins
| Protein | Location | Function in the mitochondria–LD contacts |
|---|---|---|
| ABDH5 | LD | Lipolysis 214 (coupled to fatty acid transport into mitochondria?) |
| ACSL1 | OMM | Mitochondria–LD tether (?) |
| ATGL | LD | Lipolysis 214 (coupled to fatty acid transport into mitochondria?) |
| MFN2 | OMM | Mitochondria–LD tether 3 (?), Fatty acid transport into mitochondria 3 (?) |
| PLIN1 | LD | Mitochondria–LD tether 3 (?), Fatty acid transport into mitochondria 3 (?) |
| PLIN5 | LD, OMM | Fatty acid transport into mitochondria 214 (?) |
| SNAP23 | LD | Mitochondria–LD tether 210 (?), LD fusion 211 |
| VAMP4 | LD | Mitochondria–LD tether (?), LD fusion 211 |
Figure 5. The architecture of mitochondria–LD contact sites.

Mitochondria establish contacts with lipid droplets (LDs). Although these contacts are poorly studied, several proteins have been found to participate in them. MFN2 in mitochondria interacts with the LD protein PLIN1. Mitochondrial ASCL1 has been found to form a complex with SNAP23 and VAMP4, both present on the LD surface. Moreover, PLIN5 has been found both on the surface of LDs and in the OMM. It is known that PLIN5 interacts with ATGL; however, the protein complex through which PLIN5 anchors LDs to mitochondria is still uncharacterized.
Functional implications of the interaction between mitochondria and LDs
The associations between mitochondria and LDs influence LD size and mitochondrial dynamics. Thus, the interaction of mitochondria with LDs promotes the expansion of the latter 2. PLIN5 overexpression increases mitochondrial recruitment to LDs and LD total area relative to control cells 2. Mitochondrial morphology is also somehow regulated by interaction with LDs 2: Mitochondria associated with LDs are more elongated than free mitochondria and contain reduced levels of DRP1 and cleaved OPA1 2. In turn, mitochondrial dynamics also influence LD size and distribution. Thus, impaired mitochondrial fusion causes an heterogeneous distribution of lipids throughout the mitochondrial network, greater accumulation of fatty acids in LDs, and increased fatty acid release from the cell 206. This increase in fatty acids accumulation and release probably occurs due to impaired fatty acid oxidation in fragmented mitochondria.
PLIN5 is likely to promote the mitochondrial uptake of fatty acids since it interacts with adipose triglyceride lipase (ATGL) and its activator ABHD5 on the LD surface 214, leading to enhanced lipolysis. During glucose deprivation, ACSL1 interacts with SNAP23 and VAMP4 and thus increases mitochondria–LD contact sites 209. Under these conditions, ACSL1 promotes the synthesis of acyl‐CoA from fatty acids released by LDs, which are then channeled through mitochondrial beta‐oxidation. Such a process may sustain the thermogenic capacity of BAT during its activation 215.
The MFN2–PLIN1 complex may be key for the maintenance of fatty acid oxidation in BAT and thus for thermogenesis 3. Moreover, proteomic analysis of the LD‐enriched fraction of BAT has identified the mitochondrial thermogenic protein UCP1 upon cold treatment 205, thereby suggesting increased cooperation of mitochondria and LDs for heat production.
Mitochondria–LD contacts respond to metabolic alterations
Mitochondria–LD contacts are modulated by nutrient availability in various tissues. Thus, glucose deficiency promotes LD interaction with mitochondria in monkey kidney Vero cells and in mouse primary hepatocytes 209, 216. The interaction of ACSL1 with SNAP23 and VAMP4 is enhanced upon glucose deprivation in mouse primary hepatocytes 209. In agreement with these observations, SNAP23 expression increases in rat livers during fasting 217. Proteomic analysis of purified LDs from mouse livers revealed that PLIN5 and ACSL1 proteins are more abundant in fasted mice 218. It is likely that the enhanced mitochondria–LD contacts favor fatty acid oxidation in mitochondria under conditions of nutrient deprivation or during physiological fasting.
Mitochondria–LD contacts are also modified in BAT under conditions of thermogenic activity. In this regard, Benador et al 2 reported a 50% decrease in mitochondria contacts with LDs in primary cultures of BAT from mice maintained at 6°C compared to BAT of mice under thermoneutral conditions. In contrast, Boutant et al 3 observed that PLIN1–MFN2 interaction is enhanced upon treatment with an adrenergic agonist in brown adipocytes. In agreement with the latter findings, Yu et al 205 detected increased expression of PLIN1 and ATGL in BAT of mice subjected to cold. No explanation for the reduced mitochondria–LD interaction upon cold exposure, under conditions which are linked to greater beta‐oxidation, has been put forward to date.
The effects of a HFD, obesity, diabetes, and exercise on mitochondria–LD apposition have not been studied in depth. However, it has been reported that these conditions have an impact on proteins that operate at the interface of these two organelles. PLIN1 protein levels are decreased in the WAT of mice on a HFD in comparison with that of animals on a normal chow diet 219. In skeletal muscle, two independent studies found that PLIN5 protein expression is increased in mice upon exposure to a HFD 220, 221. These findings are in agreement with a study performed in human muscle biopsies, in which PLIN5 protein levels were higher in obese and diabetic patients compared to those of healthy subjects 222.
Exercise influences the expression of proteins located at the mitochondria–LD interface. Both control and HFD‐fed mice subjected to chronic exercise show increased levels of muscle PLIN1 221. Furthermore, the levels of this protein are higher in trained HFD‐fed mice compared to trained mice under a normal diet. Interestingly, triglycerides tended to accumulate more in the muscle of trained animals and of those on HFD.
In turn, PLIN5 is upregulated in the primary myotubes of physically active subjects upon lipolytic stimulation 223. In agreement with this, PLIN5‐positive LDs are higher in muscle sections from trained individuals and total PLIN5 is higher in these subjects 222. Upregulation of PLIN1 and PLIN5 in skeletal muscle of trained subjects may participate in the increased contacts that occur between mitochondria and LDs.
Metabolic impact of alterations in proteins participating in mitochondria–LD contacts
Disruption of the contacts between mitochondria and LDs may affect metabolism. However, information in this regard is scarce. Some animal and cellular models lacking proteins involved in these contacts have been generated, but the effects observed may not be fully attributable to the disruption of mitochondria–LD contact sites.
The specific ablation of Mfn2 in BAT in mice impairs lipolysis, fatty acid oxidation, and respiration, and thus decreases the thermogenic capacity of this tissue 3. Moreover, that study showed that the lack of MFN2 disrupts fatty acid flux into mitochondria, probably as a result of impairment of mitochondria and LD contacts. Mfn2‐deficient BAT shows an enhanced expression of FGF21 when mice are subjected to a HFD, leading to increased circulating levels of FGF21. Whether FGF21 induction is due to cellular stress that is specifically dependent on alterations in mitochondria–LD contacts or to other biological effects of MFN2 remains unknown. However, under these conditions, the enhanced levels of FGF21 protect Mfn2 knockout mice against lipid accumulation in the liver and lead to improved hepatic fatty acid oxidation.
Liver‐specific Plin5 knockout mice show reduced mitochondria–LDs contacts in hepatocytes, decreased fatty acid oxidation, and reduced triglyceride secretion 224. Treatment of these mice with a HFD induces greater accumulation of TAG, the activation of JNK, and insulin resistance. Partial ablation of Plin5 in muscle leads to insulin resistance with improved insulin responsiveness under HFD feeding 220. Complete ablation of Plin5 in skeletal muscle results in glucose intolerance, insulin resistance in adipose tissue, and reduced circulating insulin levels 225. After HFD feeding, Plin5 knockout animals show decreased ER stress markers (p‐IRE1α, spliced XBP1, Atf4), reduced activation of JNK, and diminished levels of proinflammatory markers (Tnfa, Il6, Ccl2), as well as decreased levels of muscle, circulating, and hepatic FGF21 compared to wild‐type mice. These metabolic alterations are reversed upon injection of recombinant FGF21. Moreover, in agreement with these findings, Plin5 overexpression in skeletal muscle causes increased energy metabolism and accumulation of more TAG in skeletal muscle under conditions of normal glucose and insulin tolerance 226. Upon HFD, the expression of inflammatory markers in the liver is lower and muscle and plasma levels of FGF21 are increased. Moreover, when mice overexpressing Plin5 are subjected to a HFD, they show upregulation of browning markers (Ucp1, Cidea, Adiponectin, Cebpa) in WAT, in comparison with wild‐type mice. In line with these results, another study found that overexpression of PLIN5 reduces lipolysis and fatty acid oxidation and increases glycogen synthesis and glucose oxidation in human primary myotubes 220. Nevertheless, augmented energy demand by forskolin application increases lipid oxidation in conditions of PLIN5 overexpression to a higher rate than in control myotubes. Laurens et al proposed that higher energy demands increase the contacts between mitochondria and LDs in order to optimize fatty acid oxidation and that HFD uncouples LD from mitochondria.
Total Plin1 knockout mice are resistant to insulin 219. Livers of Plin1 knockout mice show spontaneous hepatosteatosis, impaired glucose metabolism, increased synthesis of TAG, and decreased fatty acid oxidation 227. Furthermore, these mice suffer from hypertrophic cardiomyopathy 228 and increased atherosclerosis 229. The depletion of Plin1 causes increased circulating levels of proinflammatory cytokines, TAGs, and free fatty acids accompanied by white adipose tissue inflammation with higher M1 macrophage infiltration 219. This proinflammatory phenotype is driven by enhanced lipolysis in adipocytes, which leads to increased production and the release of prostaglandins.
Snap23 knockdown in HL‐1 cardiomyocytes causes insulin resistance in vitro 211. In the same study, wild‐type HL‐1 cells exposed to oleic acid showed increased recruitment of SNAP23 to LDs and insulin resistance. Insulin resistance was rescued by Snap23 overexpression. Since SNAP23 also localizes to the plasma membrane where it translocates GLUT4, insulin resistance might be caused by the absence of this protein in the plasma membrane.
Functional and metabolic aspects of mitochondria–lysosome contact sites
The interaction between mitochondria and lysosomes has been widely studied upon stress or mitochondrial damage conditions. Damaged mitochondria are degraded by selective autophagy (mitophagy), and this process implies the fusion of the mitophagosome with a lysosome (reviewed by Nguyen et al 230). Moreover, mitochondria generate vesicles that trigger lysosomes, mitochondria‐derived vesicles (MDVs), and this process is considered as a way for mitochondria to degrade proteins or oxidized components (reviewed by Sugiura et al 231). Besides these studies performed under stress conditions and revealing indirect interaction between mitochondria and lysosomes, little is known about physical apposition of mitochondria and lysosomes in basal conditions. In this section, we review the recent advances on this field and we analyze the functional aspects of the apposition between lysosomes and mitochondria and its crosstalk with metabolism.
Composition and functions of contacts between mitochondria and lysosomes
Over the past decade, although physical interaction between mitochondria and lysosomes had not yet been described, there was evidence for mitochondria and lysosome crosstalk since disruption of mitochondria affects lysosomal function and dynamics and lysosomal damage triggers mitochondrial homeostasis impairment (reviewed by Raimundo et al 232). Recently, cutting‐edge microscopy studies have detected the establishment of physical contacts between lysosomes and mitochondria in healthy cells in the absence of damage stimulus 1, 233. Besides, the latest discoveries in the field point to RAB7 as a master coordinator of these contacts 6, 7, 234, 235, 236 (Fig 6).
Figure 6. Mitochondria–lysosome interface.
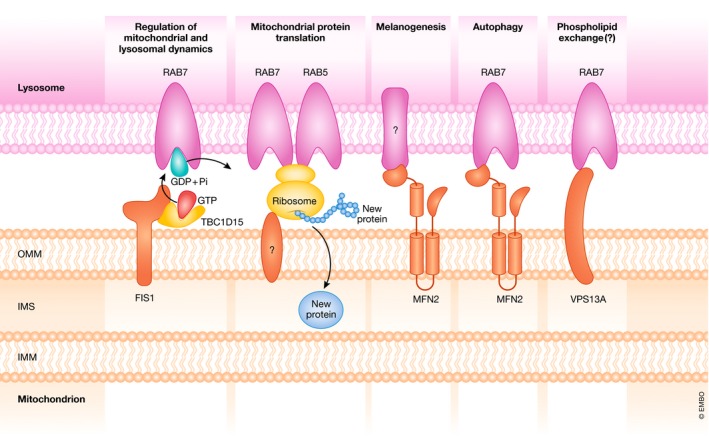
Several processes take place at the contacts between mitochondria and lysosomes: (A) Regulation of mitochondrial and lysosomal dynamics by RAB7, TBC1D15, and FIS1 coordination; (B) mitochondrial protein translation in ribosomes anchored to the endosomal surface by interaction with RAB7 and RAB5 in close proximity to mitochondria; (C) melanogenesis in melanosomes that interact with mitochondria through MFN2; (D) autophagosome to lysosome fusion, which is supported by MFN2–RAB7 interaction; and (E) possible phospholipid exchange through VPS13A interaction with RAB7.
Lysosome–mitochondria contacts are promoted by GTP‐bound lysosomal RAB7 6. When a constitutively active GTP‐bound RAB7 mutant is expressed, contacts between lysosomes and mitochondria increase and these are more stable over time. TBC1D15 is a GTPase activating protein that governs RAB7 activity 237. Recruitment of TBC1D15 to mitochondria by FIS1 favors GTP hydrolysis and separates lysosomes from mitochondria 6 (Fig 6). TBC1D15 inactive mutants can still be recruited to mitochondria; however, they do not uncouple mitochondria from lysosomes and lysosomes become larger 6. Interaction between RAB7 and TBC1D15 has not been described yet. Confocal microscopy has revealed colocalization between lysosomes and sites for mitochondrial division 6 in which, as we described before, ER tubules participate 58. These findings suggest that the interface between these two organelles plays a major role in the regulation of mitochondrial and lysosomal dynamics.
It has been discovered in retinal ganglion cells of Xenopus laevis embryos that late endosomes can associate with ribosomes in axons through RAB7 and RAB5 and protein translation can occur at the endosomal membrane 235. Some mitochondrial proteins such as VDAC2 can be translated at the surface of these late endosomes and then be transferred to mitochondria 235 (Fig 6). Mutations in rab7 associated with Charcot–Marie–Tooth disease cause downregulation of mitochondrial protein translation, mitochondrial elongation, and diminished mitochondrial membrane potential in the axons 235. Currently, the proteins in the interorganellar surface that contribute to mitochondrial incorporation of the newly synthesized peptides remain unknown.
Melanogenesis is a process influenced by the apposition between melanosomes and mitochondria 7. Melanosomes, which are lysosomal related organelles that accumulate melanin in pigmented cells, establish contacts with mitochondria through MFN2 in melanocytes 7 (Fig 6). However, the melanosomal component of these junctions remains unknown. MFN2 ablation decreases the interaction between mitochondria and melanosomes and arrests melanogenesis activation 7.
The interface between mitochondria and lysosomes could play a role in autophagy. It is known that MFN2 interacts with RAB7 in mouse hearts 234 (Fig 6). This interaction increases upon autophagy induction by starvation and has been proposed to be key for autophagosome–lysosome fusion 234.
A recent study indicates that lysosomal RAB7 immunoprecipitates and partially colocalizes with mitochondrial VPS13A in HeLa cells and that the absence of VPS13A causes impaired lysosomal degradation 236. Moreover, a different study has identified a hydrophobic cavity in the structure of this protein and has proved its capacity to bind and transfer phospholipids between two membranes 238. However, it has not been elucidated whether mitochondria and lysosomes exchange phospholipids in a non‐vesicular manner (Fig 6).
A long DRP1 isoform present in mitochondria, lysosomes, and plasma membrane is highly enriched in mitochondrial contacts with lysosomes 239. This isoform is mainly expressed in the brain and conserves mitochondrial division capacity 239. When overexpressed in MEF, DRP1 long isoform colocalizes with RAB7; nevertheless, it is not known whether RAB7 and DRP1 interact 239. GTPase activity and oligomerization capacity of DRP1 long isoform are necessary for its localization to lysosomes 239. The role of this isoform of DRP1 in membrane junctions remains uncharacterized, although a role in lysosomal dynamics has already been discarded 239.
Besides the participation of mitochondria–lysosome contacts in organelle dynamics regulation, mitochondrial protein translation and melanogenesis, and their possible contribution to autophagy and phospholipid exchange, communication between mitochondria and lysosomes has been associated with iron translocation to mitochondria 240 and lipofuscin deposit formation in lysosomes 241. Nevertheless, mechanisms for these processes are poorly described and it is not known whether they take place in a vesicular or non‐vesicular manner.
The effects of mitochondria–lysosome contact modification on metabolism
Little is known about how the modification of the contacts between mitochondria and lysosomes affects metabolism. RAB7 role in fat storage has been reported in C. elegans, in which neuronal silencing of rab7 results in reduced fat storage 242. Rab7 knockdown in mouse bone marrow cells decreases glucose consumption and ROS excess 243. The knockout of TBC1D15 decreases glucose uptake in L02 human fetal hepatocytes 244. Moreover, TBC1D15 ablation reduces GLUT4 mRNA and protein levels and this effect is more dramatic in the presence of insulin 244. In the absence of TBC1D15, GLUT4 colocalizes with lysosomes, where it could be degraded 244. The authors suggest that TBC1D15 is necessary for GLUT4 translocation to the plasma membrane. There are no studies linking these observations on RAB7 and TBC1D15 with mitochondria–lysosome interaction.
Impact of metabolism on mitochondria–lysosome contacts
The influence of metabolic homeostasis on mitochondria–lysosome contacts in mammalian cells is an unexplored field. Related findings have been discovered in yeast 245. Yeast vacuoles interact with mitochondria through Ypt7, the yeast homologue of RAB7, and Vps39 in the vacuole 245 and Tom40 in the mitochondria 246. In the absence of glucose, vacuole–mitochondria contacts are lost, but upon addition of glucose to the growth medium, these contacts slowly reappear 245. This study is the first evidence that glucose levels modulate mitochondria–lysosome contacts in yeast cells; nevertheless, more insight is necessary into how glucose deprivation affects these contacts in mammalian cells.
Under insulin or amino acid stimuli in mammalian cells, lysosomal mTOCRC1 is activated and stimulates mitochondrial oxidative pathways and mtDNA synthesis 247. RAB7 interacts with mTOR in mouse bone marrow cells 243. It is not known whether this interaction occurs in other cell types. Given the importance of mTOR in response to nutrient availability and RAB7 in the interaction between mitochondria and lysosomes, this complex is a potential candidate to modulate mitochondria–lysosome contacts in response to metabolism.
Contacts of mitochondria with peroxisomes and the Golgi apparatus
Peroxisomes and mitochondria establish physical contacts, and this interaction influences cell metabolism 1, 8, 248. ECI2, a peroxisomal enoyl‐CoA isomerase, is so far the only protein established as part of a mitochondria–peroxisome tethering complex 8. A proximity ligation assay has revealed that peroxisomal ECI2 and mitochondrial TOM20 interact in MA‐10 Leydig‐like cells 8, although more precise analyses of this interaction such as co‐immunoprecipitation or two‐hybrid screening have not been performed. Overexpression of ECI2 results in increased steroid biosynthesis 8, a metabolic process that takes place in mitochondria. Moreover, MAMs associate with peroxisomes during antiviral immune signaling 249, 250. Beta‐oxidation needs the interplay between both compartments: Very long chain fatty acids (> 16 carbons) are shortened in peroxisomes (to 6–8 carbons fatty acids) and then preferentially translocated to mitochondria for complete oxidation 251. How shortened fatty acids are shuttled from peroxisomes to mitochondria remains unknown.
Golgi apparatus and mitochondria also communicate with each other by physical interaction 1, 4. Nevertheless, the key players of this apposition remain unknown. The existence of Ca2+ gradients from the Golgi apparatus to mitochondria has been discovered in pancreatic acinar cells 4, although the existence of calcium channels for active or passive calcium transport has not been clarified yet.
Future perspectives
The interaction between organelles is an emerging field that is gaining importance due to the physiological implications of organelle contact sites. In this regard, mitochondria contacts with the ER are the better characterized ones to date and their functional and metabolic aspects have already been highlighted. Nevertheless, the full composition of tethering complexes still needs to be discovered. Mitochondria interacting with lipid droplets show different metabolic behavior compared with those mitochondria that are free in the cytosol. Studies on the interaction of LD and mitochondria have generated two hypotheses: The first hypothesis is that mitochondria–LD interaction favors beta‐oxidation, and the second hypothesis is that mitochondria–LD interaction results in increased TAG synthesis. More insight is needed to determine whether metabolic behavior of mitochondria associated with LDs depends on the energetic demands of the tissue or the whole body. Lysosome–mitochondria contacts in mammalian cells are still poorly described but currently in the spotlight of research. Whether reciprocal dynamics regulation exerted between mitochondria and lysosomes has any metabolic implications is yet to be discovered. The key players in the communication between mitochondria and peroxisomes have not been deciphered. Future characterization of this interaction will be metabolically relevant since peroxisomes and mitochondria interact during beta‐oxidation of fatty acids. Although several studies have revealed an interaction between mitochondria and the Golgi apparatus, the functional and structural features of this interaction remain unknown. How Ca2+ is transferred from the Golgi apparatus to mitochondria and what Ca2+ flux between these organelles signals for has not been discovered yet. It is possible that metabolic features of mitochondria could depend on their interaction with other organelles, which we hypothesize would be determined by the organ or tissue energetic requirements. A tissue‐/cell‐specific characterization of contact sites is lacking, but it will be fundamental for a better understanding of contact sites influence on metabolism. Future discoveries on organelle contact sites will fill these gaps and transform our understanding of cellular physiology and metabolism regulation and likely will provide us with new tools for targeting metabolic disorders.
Conflict of interest
The authors declare that they have no conflict of interest.
Box: In need of answers.
What is the precise composition of tethering (and spacer) complexes responsible for the contacts of mitochondria with other organelles (ER, lipid droplets, lysosomes, Golgi, peroxisomes, etc.)?
How do the tethering (and spacer) complexes undergo modulation in a tissue‐specific manner in response to hormonal or nutritional alterations? What are the mechanisms involved?
Do the tethering complexes functionally interact with other tethering/spacer complexes or with functional proteins present in contacts? If so, what is the nature of the mechanisms involved?
Identification of the precise functions operating in the different contact sites, and characterization of the complexes involved in the catalysis present in those contacts. Analysis of the modes of regulation of the functional complexes in response to hormonal and/or nutritional cues.
What are the mechanisms by which mitochondria within a specific cell type show a heterogeneous interaction with specific organelles such as lipid droplets?
Because some of the proteins involved in tethering or in function in contact sites are present in multiple locations in cells, there is a need to design strategies to identify the specific function of the protein located in the contact site rather in other locations.
How do alterations in proteins present in contact sites impact on metabolic homeostasis? Why do some proteins mediate enhanced anabolism whereas others mediate inhibited anabolism, and other alterations in lipid metabolism? What is the physiological meaning, and what the mechanisms in place?
Acknowledgements
I.G. is a recipient of a PhD fellowship from the Ministerio de Economía y Competitividad (MINECO). This study was supported by research grants from the MINECO (SAF2016‐75246R), the Generalitat de Catalunya (Grant 2017SGR1015), INFLAMES (PIE‐14/00045, Instituto de Salud Carlos III, CIBERDEM (“Instituto de Salud Carlos III”), the Marató de TV3, the Fundación BBVA, and the Fundación Ramon Areces. We gratefully acknowledge institutional funding from MINECO through the Centres of Excellence Severo Ochoa Award, and from the CERCA Programme of the Generalitat de Catalunya.
EMBO Reports (2019) 20: e47928
See the Glossary for abbreviations used in this article.
References
- 1. Valm AM, Cohen S, Legant WR, Melunis J, Hershberg U, Wait E, Cohen AR, Davidson MW, Betzig E, Lippincott‐Schwartz J (2017) Applying systems‐level spectral imaging and analysis to reveal the organelle interactome. Nature 546: 162–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Benador IY, Veliova M, Mahdaviani K, Petcherski A, Wikstrom JD, Assali EA, Acín‐Pérez R, Shum M, Oliveira MF, Cinti S et al (2018) Mitochondria bound to lipid droplets have unique bioenergetics, composition, and dynamics that support lipid droplet expansion. Cell Metab 27: 869–885.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Boutant M, Kulkarni SS, Joffraud M, Ratajczak J, Valera‐Alberni M, Combe R, Zorzano A, Cantó C (2017) Mfn2 is critical for brown adipose tissue thermogenic function. EMBO J 36: 1543–1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dolman NJ, Gerasimenko JV, Gerasimenko OV, Voronina SG, Petersen OH, Tepikin AV (2005) Stable Golgi‐mitochondria complexes and formation of Golgi Ca2+ gradients in pancreatic acinar cells. J Biol Chem 280: 15794–15799 [DOI] [PubMed] [Google Scholar]
- 5. Marsh BJ, Mastronarde DN, Buttle KF, Howell KE, McIntosh JR (2001) Organellar relationships in the Golgi region of the pancreatic beta cell line, HIT‐T15, visualized by high resolution electron tomography. Proc Natl Acad Sci USA 98: 2399–2406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wong YC, Ysselstein D, Krainc D (2018) Mitochondria‐lysosome contacts regulate mitochondrial fission via RAB7 GTP hydrolysis. Nature 554: 382–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Daniele T, Hurbain I, Vago R, Casari G, Raposo G, Tacchetti C, Schiaffino MV (2014) Mitochondria and melanosomes establish physical contacts modulated by Mfn2 and involved in organelle biogenesis. Curr Biol 24: 393–403 [DOI] [PubMed] [Google Scholar]
- 8. Fan J, Li X, Issop L, Culty M, Papadopoulos V (2016) ACBD2/ECI2‐mediated peroxisome‐mitochondria interactions in leydig cell steroid biosynthesis. Mol Endocrinol 30: 763–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. De Vos KJ, Mórotz GM, Stoica R, Tudor EL, Lau KF, Ackerley S, Warley A, Shaw CE, Miller CCJ (2012) VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum Mol Genet 21: 1299–1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Szabadkai G, Bianchi K, Várnai P, De Stefani D, Wieckowski MR, Cavagna D, Nagy AI, Balla T, Rizzuto R (2006) Chaperone‐mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J Cell Biol 175: 901–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Iwasawa R, Mahul‐Mellier AL, Datler C, Pazarentzos E, Grimm S (2011) Fis1 and Bap31 bridge the mitochondria–ER interface to establish a platform for apoptosis induction. EMBO J 30: 556–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. de Brito OM, Scorrano L (2008) Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456: 605–610 [DOI] [PubMed] [Google Scholar]
- 13. Stoica R, De Vos KJ, Paillusson S, Mueller S, Sancho RM, Lau KF, Vizcay‐Barrena G, Lin WL, Xu YF, Lewis J et al (2014) ER‐mitochondria associations are regulated by the VAPB‐PTPIP51 interaction and are disrupted by ALS/FTD‐associated TDP‐43. Nat Commun 5: 3996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Di Mattia T, Wilhelm LP, Ikhlef S, Wendling C, Spehner D, Nominé Y, Giordano F, Mathelin C, Drin G, Tomasetto C et al (2018) Identification of MOSPD2, a novel scaffold for endoplasmic reticulum membrane contact sites. EMBO Rep 19: e45453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Thoudam T, Ha C, Leem J, Chanda D, Park J, Kim H, Jeon J‐H, Choi Y‐K, Liangpunsakul S, Huh YH et al (2019) PDK4 augments ER – mitochondria contact to dampen skeletal muscle insulin signaling during obesity. Diabetes 68: 571–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yoon Y, Krueger EW, Oswald BJ, McNiven MA (2003) The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin‐like protein DLP1. Mol Cell Biol 23: 5409–5420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Breckenridge DG, Stojanovic M, Marcellus RC, Shore GC (2003) Caspase cleavage product of BAP31 induces mitochondrial fission through endoplasmic reticulum calcium signals, enhancing cytochrome c release to the cytosol. J Cell Biol 160: 1115–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC (2003) Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol 160: 189–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Santel A, Fuller MT (2001) Control of mitochondrial morphology by a human mitofusin. J Cell Sci 114: 867–874 [DOI] [PubMed] [Google Scholar]
- 20. Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, Oomori H, Noda T, Haraguchi T, Hiraoka Y et al (2013) Autophagosomes form at ER–mitochondria contact sites. Nature 495: 389–393 [DOI] [PubMed] [Google Scholar]
- 21. Filadi R, Greotti E, Turacchio G, Luini A, Pozzan T, Pizzo P (2015) Mitofusin 2 ablation increases endoplasmic reticulum – mitochondria coupling. Proc Natl Acad Sci USA 112: E2174–E2181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Santos Leal N, Schreiner B, Moreira C, Filadi R, Wiehager B, Karlstr H, Pizzo P, Ankarcrona M (2016) Mitofusin‐2 knockdown increases ER – mitochondria contact and decreases amyloid b‐peptide production. J Cell Mol Med 20: 1686–1695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Naon D, Zaninello M, Giacomello M, Varanita T, Grespi F, Lakshminaranayan S, Serafini A, Semenzato M, Herkenne S, Hernández‐Alvarez MI et al (2016) Critical reappraisal confirms that Mitofusin 2 is an endoplasmic reticulum–mitochondria tether. Proc Natl Acad Sci USA 113: 11249–11254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mclelland G, Goiran T, Yi W, Chen CX, Lauinger ND, Krahn AI, Valimehr S, Rakovic A, Rouiller I, Durcan TM (2018) Mfn2 ubiquitination by PINK1/parkin gates the p97‐dependent release of ER from mitochondria to drive mitophagy. Elife 7: 1–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Teng Y, Ren X, Li H, Shull A, Kim J, Cowell JK (2016) Mitochondrial ATAD3A combines with GRP78 to regulate the WASF3 metastasis‐promoting protein. Oncogene 35: 333–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Honrath B, Metz I, Bendridi N, Rieusset J, Culmsee C, Dolga AM (2017) Glucose‐regulated protein 75 determines ER–mitochondrial coupling and sensitivity to oxidative stress in neuronal cells. Cell Death Discov 3: 17076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tubbs E, Theurey P, Vial G, Bendridi N, Bravard A, Chauvin MA, Ji‐Cao J, Zoulim F, Bartosch B, Ovize M et al (2014) Mitochondria‐associated endoplasmic reticulum membrane (MAM) integrity is required for insulin signaling and is implicated in hepatic insulin resistance. Diabetes 63: 3279–3294 [DOI] [PubMed] [Google Scholar]
- 28. Paillard M, Tubbs E, Thiebaut PA, Gomez L, Fauconnier J, Da Silva CC, Teixeira G, Mewton N, Belaidi E, Durand A et al (2013) Depressing mitochondria‐reticulum interactions protects cardiomyocytes from lethal hypoxia‐reoxygenation injury. Circulation 128: 1555–1565 [DOI] [PubMed] [Google Scholar]
- 29. Arruda AP, Pers BM, Parlakgül G, Güney E, Inouye K, Hotamisligil GS (2014) Chronic enrichment of hepatic endoplasmic reticulum‐mitochondria contact leads to mitochondrial dysfunction in obesity. Nat Med 20: 1427–1435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Eletto MD, Rossin F, Occhigrossi L, Pinton P, Eletto MD, Rossin F, Occhigrossi L, Farrace MG, Faccenda D, Desai R (2018) Transglutaminase type 2 regulates ER‐ mitochondria contact sites by interacting with report transglutaminase type 2 regulates ER‐mitochondria contact sites by interacting with GRP75. Cell Rep 25: 3573–3581 [DOI] [PubMed] [Google Scholar]
- 31. Qiao X, Jia S, Ye J, Fang X, Zhang C, Cao Y, Xu C, Zhao L, Zhu Y, Wang L et al (2017) PTPIP51 regulates mouse cardiac ischemia/reperfusion through mediating the mitochondria‐SR junction. Sci Rep 7: 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chiang SF, Huang CY, Lin TY, Chiou SH, Chow KC (2012) An alternative import pathway of AIF to the mitochondria. Int J Mol Med 29: 365–372 [DOI] [PubMed] [Google Scholar]
- 33. Ainbinder A, Boncompagni S, Protasi F, Dirksen RT (2015) Role of mitofusin‐2 in mitochondrial localization and calcium uptake in skeletal muscle. Cell Calcium 57: 14–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kentala H, Weber‐Boyvat M, Olkkonen VM (2016) OSBP‐related protein family: mediators of lipid transport and signaling at membrane contact sites. Int Rev Cell Mol Biol 321: 299–340 [DOI] [PubMed] [Google Scholar]
- 35. Kuge O, Nishijima M (1997) Phosphatidylserine synthase I and II of mammalian cells. Biochim Biophys Acta 1348: 151–156 [DOI] [PubMed] [Google Scholar]
- 36. McMurray W (1986) Origins of the phospholipids in animal mitochondria. Biochem Cell Biol 64: 1115–1124 [DOI] [PubMed] [Google Scholar]
- 37. Vance JE (1991) Newly made phosphatidylserine and phosphatidylethanolamine are preferentially translocated between rat liver mitochondria and endoplasmic reticulum. J Biol Chem 266: 89–97 [PubMed] [Google Scholar]
- 38. Dennis EA, Kennedy EP (1972) Intracellular sites of lipid synthesis and the biogenesis of mitochondria. J Lipid Res 13: 263–267 [PubMed] [Google Scholar]
- 39. Blusztajn JK, Zeisel SH, Wurtman RJ (1979) Synthesis of lecithin (phosphatidylcholine) from phosphatidylethanolamine in bovine brain. Brain Res 179: 319–327 [DOI] [PubMed] [Google Scholar]
- 40. Vance JE (1990) Phospholipid synthesis in a membrane fraction associated with mitochondria. J Biol Chem 265: 7248–7257 [PubMed] [Google Scholar]
- 41. Du X, Kumar J, Ferguson C, Schulz TA, Ong YS, Hong W, Prinz WA, Parton RG, Brown AJ, Yang H (2011) A role for oxysterol‐binding protein‐related protein 5 in endosomal cholesterol trafficking. J Cell Biol 192: 121–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chung J, Torta F, Masai K, Lucast L, Czapla H, Tanner LB, Narayanaswamy P, Wenk MR, Nakatsu F, De Camilli P (2015) PI4P/phosphatidylserine countertransport at ORP5‐ and ORP8‐mediated ER – plasma membrane contacts. Science 349: 428–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Galmes R, Houcine A, van Vliet AR, Agostinis P, Jackson CL, Giordano F (2016) ORP5/ORP8 localize to endoplasmic reticulum–mitochondria contacts and are involved in mitochondrial function. EMBO Rep 17: 800–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. AhYoung AP, Jiang J, Zhang J, Khoi Dang X, Loo JA, Zhou ZH, Egea PF (2015) Conserved SMP domains of the ERMES complex bind phospholipids and mediate tether assembly. Proc Natl Acad Sci USA 112: E3179–E3188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Schauder CM, Wu X, Saheki Y, Narayanaswamy P, Torta F, Wenk MR, De Camilli P, Reinisch KM (2014) Structure of a lipid‐bound extended synaptotagmin indicates a role in lipid transfer. Nature 510: 552–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hirabayashi Y, Kwon SK, Paek H, Pernice WM, Paul MA, Lee J, Erfani P, Raczkowski A, Petrey DS, Pon LA et al (2017) ER‐mitochondria tethering by PDZD8 regulates Ca2+ dynamics in mammalian neurons. Science 358: 623–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Elustondo P, Martin LA, Karten B (2017) Mitochondrial cholesterol import. Biochim Biophys Acta 1862: 90–101 [DOI] [PubMed] [Google Scholar]
- 48. Prasad M, Kaur J, Pawlak KJ, Bose M, Whittal RM, Bose HS (2015) Mitochondria‐associated endoplasmic reticulum membrane (MAM) regulates steroidogenic activity via steroidogenic acute regulatory protein (StAR)‐voltage‐dependent anion channel 2 (VDAC2) interaction. J Biol Chem 290: 2604–2616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Reitz J, Gehrig‐burger K, Iii JFS, Gimpl G (2008) Cholesterol interaction with the related steroidogenic acute regulatory lipid‐transfer (START) domains of StAR (STARD1) and MLN64 (STARD3). FEBS J 64: 1790–1802 [DOI] [PubMed] [Google Scholar]
- 50. Tsujishita Y, Hurley JH (2000) Structure and lipid transport mechanism of a StAR‐related domain. Nat Struct Biol 7: 408–414 [DOI] [PubMed] [Google Scholar]
- 51. Strushkevich N, MacKenzie F, Cherkesova T, Grabovec I, Usanov S, Park H‐W (2011) Structural basis for pregnenolone biosynthesis by the mitochondrial monooxygenase system. Proc Natl Acad Sci USA 108: 10139–10143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rone MB, Midzak AS, Issop L, Rammouz G, Jagannathan S, Fan J, Ye X, Blonder J, Veenstra T, Papadopoulos V (2012) Identification of a dynamic mitochondrial protein complex driving cholesterol import, trafficking, and metabolism to steroid hormones. Mol Endocrinol 26: 1868–1882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Liu J, Rone MB, Papadopoulos V (2006) Protein‐protein interactions mediate mitochondrial cholesterol transport and steroid biosynthesis. J Biol Chem 281: 38879–38893 [DOI] [PubMed] [Google Scholar]
- 54. Pulli I, Lassila T, Pan G, Yan D, Olkkonen VM, Törnquist K (2018) Oxysterol‐binding protein related‐proteins (ORPs) 5 and 8 regulate calcium signaling at specific cell compartments. Cell Calcium 72: 62–69 [DOI] [PubMed] [Google Scholar]
- 55. Simmen T, Aslan JE, Blagoveshchenskaya AD, Thomas L, Wan L, Xiang Y, Feliciangeli SF, Hung CH, Crump CM, Thomas G (2005) PACS‐2 controls endoplasmic reticulum‐mitochondria communication and Bid‐mediated apoptosis. EMBO J 24: 717–729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Arif T, Krelin Y, Shoshan‐Barmatz V (2016) Reducing VDAC1 expression induces a non‐apoptotic role for pro‐apoptotic proteins in cancer cell differentiation. Biochim Biophys Acta 1857: 1228–1242 [DOI] [PubMed] [Google Scholar]
- 57. Gatliff J, East DA, Singh A, Alvarez MS, Frison M, Matic I, Ferraina C, Sampson N, Turkheimer F, Campanella M (2017) A role for TSPO in mitochondrial Ca2+ homeostasis and redox stress signaling. Cell Death Dis 8: 1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK (2011) ER tubules mark sites of mitochondrial division. Science 334: 358–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Korobova F, Ramabhadran V, Higgs HN (2013) An actin‐dependent step in mitochondrial fission mediated by the ER‐associated formin INF2. Science 339: 464–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Korobova F, Gauvin TJ, Higgs HN (2014) A role for myosin II in mammalian mitochondrial fission. Curr Biol 24: 409–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Chakrabarti R, Ji WK, Stan RV, de Juan Sanz J, Ryan TA, Higgs HN (2018) INF2‐mediated actin polymerization at the ER stimulates mitochondrial calcium uptake, inner membrane constriction, and division. J Cell Biol 217: 251–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Loson OC, Song Z, Chen H, Chan DC (2013) Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol Biol Cell 24: 659–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yu R, Liu T, Jin S, Ning C, Lendahl U, Nistér M, Zhao J (2017) MIEF1/2 function as adaptors to recruit Drp1 to mitochondria and regulate the association of Drp1 with Mff. Sci Rep 7: 880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kim Y, Youn S, Sudhahar V, Das A, Chandhri R, Cuervo Grajal H, Kweon J, Leanhart S, He L, Toth PT et al (2018) Redox regulation of mitochondrial fission protein Drp1 by protein disulfide isomerase limits endothelial senescence. Cell Rep 23: 3565–3578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Arasaki K, Shimizu H, Mogari H, Nishida N, Hirota N, Furuno A, Kudo Y, Baba M, Baba N, Cheng J et al (2015) A role for the ancient SNARE syntaxin 17 in regulating mitochondrial division. Dev Cell 32: 304–317 [DOI] [PubMed] [Google Scholar]
- 66. Rambold AS, Kostelecky B, Elia N, Lippincott‐Schwartz J (2011) Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc Natl Acad Sci USA 108: 10190–10195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Lewis SC, Uchiyama LF, Nunnari J (2016) ER‐mitochondria contacts couple mtDNA synthesis with mitochondrial division in human cells. Science 353: aaf5549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wu W, Lin C, Wu K, Jiang L, Wang X, Li W, Zhuang H, Zhang X, Chen H, Li S et al (2016) FUNDC1 regulates mitochondrial dynamics at the ER–mitochondrial contact site under hypoxic conditions. EMBO J 35: 1368–1384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Chan DC (2006) Dissecting mitochondrial fusion. Dev Cell 11: 592–594 [DOI] [PubMed] [Google Scholar]
- 70. Anand R, Wai T, Baker MJ, Kladt N, Schauss AC, Rugarli E, Langer T (2014) The i‐AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J Cell Biol 204: 919–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Yan L, Qi Y, Huang X, Yu C, Lan L, Guo X, Rao Z, Hu J, Lou Z (2018) Structural basis for GTP hydrolysis and conformational change of MFN1 in mediating membrane fusion. Nat Struct Mol Biol 25: 233–243 [DOI] [PubMed] [Google Scholar]
- 72. Franco A, Kitsis RN, Fleischer JA, Gavathiotis E, Kornfeld OS, Gong G, Biris N, Benz A, Qvit N, Donnelly SK et al (2016) Correcting mitochondrial fusion by manipulating mitofusin conformations. Nature 540: 74–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Liu J, Noel JK, Low HH (2018) Structural basis for membrane tethering by a bacterial dynamin‐like pair. Nat Commun 9: 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Cerqua C, Anesti V, Pyakurel A, Liu D, Naon D, Wiche G, Baffa R, Dimmer KS, Scorrano L (2010) Trichoplein/mitostatin regulates endoplasmic reticulum‐mitochondria juxtaposition. EMBO Rep 11: 854–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Tian Y, Li B, Shi WZ, Chang MZ, Zhang GJ, Di ZL, Liu Y (2014) Dynamin‐related protein 1 inhibitors protect against ischemic toxicity through attenuating mitochondrial Ca2+ uptake from endoplasmic reticulum store in PC12 cells. Int J Mol Sci 15: 3172–3185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wu S, Lu Q, Wang Q, Ding Y, Ma Z, Mao X, Huang K, Xie Z, Zou MH (2017) Binding of FUN14 domain containing 1 with inositol 1,4,5‐trisphosphate receptor in mitochondria‐associated endoplasmic reticulum membranes maintains mitochondrial dynamics and function in hearts in vivo . Circulation 136: 2248–2266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Weaver D, Eisner V, Liu X, Várnai P, Hunyady L, Gross A, Hajnóczky G (2014) Distribution and apoptotic function of outer membrane proteins depend on mitochondrial fusion. Mol Cell 54: 870–878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Papanicolaou KN, Khairallah RJ, Ngoh GA, Chikando A, Luptak I, O'Shea KM, Riley DD, Lugus JJ, Colucci WS, Lederer WJ et al (2011) Mitofusin‐2 maintains mitochondrial structure and contributes to stress‐induced permeability transition in cardiac myocytes. Mol Cell Biol 31: 1309–1328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Han SM, Tsuda H, Yang Y, Vibbert J, Cottee P, Lee SJ, Winek J, Haueter C, Bellen HJ, Miller MA (2012) Secreted VAPB/ALS8 major sperm protein domains modulate mitochondrial localization and morphology via growth cone guidance receptors. Dev Cell 22: 348–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, Tuft RA, Pozzan T (1998) Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 280: 1763–1766 [DOI] [PubMed] [Google Scholar]
- 81. Lytton J, Westlin M, Burk SE, Shull E, Maclennan H (1992) Functional comparisons between isoforms of the sarcoplasmic or endoplasmic reticulum family of calcium pumps. J Biol Chem 267: 14483–14489 [PubMed] [Google Scholar]
- 82. Miller KK, Verma A, Snyder SH, Ross CA (1991) Localization of an endoplasmic reticulum calcium ATPase mRNA in rat brain by in situ hybridization. Neuroscience 43: 1–9 [DOI] [PubMed] [Google Scholar]
- 83. Giorgi C, Marchi S, Pinton P (2018) The machineries, regulation and cellular functions of mitochondrial calcium. Nat Rev Mol Cell Biol 19: 713–730 [DOI] [PubMed] [Google Scholar]
- 84. Filadi R, Leal NS, Schreiner B, Rossi A, Dentoni G, Pinho CM, Wiehager B, Cieri D, Calì T, Pizzo P et al (2018) TOM70 sustains cell bioenergetics by promoting IP3R3‐mediated ER to mitochondria Ca2+ transfer. Curr Biol 28: 369–382.e6 [DOI] [PubMed] [Google Scholar]
- 85. Giorgio V, Bisetto E, Soriano ME, Dabbeni‐sala F, Basso E, Petronilli V, Forte MA, Bernardi P, Lippe G (2009) Cyclophilin D modulates mitochondrial F0F1 ‐ATP synthase by interacting with the lateral stalk of the complex. J Biol Chem 284: 33982–33988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Ong HL, Liu X, Sharma A, Hegde RS, Ambudkar IS (2007) Intracellular Ca2+ release via the ER translocon activates store‐operated calcium entry. Pflugers Arch Eur J Physiol 453: 797–808 [DOI] [PubMed] [Google Scholar]
- 87. Giunti R, Gamberucci A, Fulceri R, Bánhegyi G, Benedetti A (2007) Both translocon and a cation channel are involved in the passive Ca2+ leak from the endoplasmic reticulum: a mechanistic study on rat liver microsomes. Arch Biochem Biophys 462: 115–121 [DOI] [PubMed] [Google Scholar]
- 88. Booth DM, Enyedi B, Geiszt M, Várnai P, Hajnóczky G (2016) Redox nanodomains are induced by and control calcium signaling at the ER‐mitochondrial interface. Mol Cell 63: 240–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Dong Z, Shanmughapriya S, Tomar D, Siddiqui N, Lynch S, Nemani N, Breves SL, Zhang X, Tripathi A, Palaniappan P et al (2017) Mitochondrial Ca2+ uniporter is a mitochondrial luminal redox sensor that augments MCU channel activity. Mol Cell 65: 1014–1028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Betz C, Stracka D, Prescianotto‐baschong C, Frieden M, Demaurex N, Hall MN (2013) mTOR complex 2‐Akt signaling at mitochondria associated endoplasmic reticulum membranes (MAM) regulates mitochondrial physiology. Proc Natl Acad Sci USA 110: 12526–12534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Khan MT, Wagner L, Yule DI, Bhanumathy C, Joseph SK (2006) Akt kinase phosphorylation of inositol 1,4,5‐trisphosphate receptors. J Biol Chem 281: 3731–3737 [DOI] [PubMed] [Google Scholar]
- 92. Szado T, Vanderheyden V, Parys JB, De Smedt H, Rietdorf K, Kotelevets L, Chastre E, Khan F, Landegren U, Söderberg O et al (2008) Phosphorylation of inositol 1,4,5‐trisphosphate receptors by protein kinase B/Akt inhibits Ca2+ release and apoptosis. Proc Natl Acad Sci USA 105: 2427–2432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Schäuble N, Lang S, Jung M, Cappel S, Schorr S, Ulucan Ö, Linxweiler J, Dudek J, Blum R, Helms V et al (2012) BiP‐mediated closing of the Sec61 channel limits Ca2+ leakage from the ER. EMBO J 31: 3282–3296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Golic I, Velickovic K, Markelic M, Stancic A, Jankovic A, Vucetic M, Otasevic V, Buzadzic B, Korac B, Korac A (2014) Calcium‐induced alteration of mitochondrial morphology and mitochondrial‐endoplasmic reticulum contacts in rat brown adipocytes. Eur J Histochem 58: 2377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Sehgal P, Szalai XP, Olesen XC, Praetorius XHA, Nissen XP, Christensen SB, Engedal XN, Møller JV (2017) Inhibition of the sarco/endoplasmic reticulum (ER) Ca2+ ‐ATPase by thapsigargin analogs induces cell death via ER Ca2+ depletion and the unfolded protein response. J Biol Chem 292: 19656–19673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Gomez‐Suaga P, Paillusson S, Miller CCJ (2017) ER‐mitochondria signaling regulates autophagy. Autophagy 13: 1250–1251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Muñoz JP, Ivanova S, Sánchez‐Wandelmer J, Martínez‐Cristóbal P, Noguera E, Sancho A, Díaz‐Ramos A, Hernández‐Alvarez MI, Sebastián D, Mauvezin C et al (2013) Mfn2 modulates the UPR and mitochondrial function via repression of PERK. EMBO J 32: 2348–2361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R et al (2003) An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell 11: 619–633 [DOI] [PubMed] [Google Scholar]
- 99. Blais JJD, Filipenko V, Bi M, Harding HHP, Ron D, Koumenis C, Wouters BG, Bell JC (2004) Activating transcription factor 4 is translationally regulated by hypoxic stress. Mol Cell Biol 24: 7469–7482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Liang S‐H, Zhang W, Mcgrath BC, Zhang P, Cavener DR (2006) PERK (eIF2α kinase) is required to activate the stress‐activated MAPKs and induce the expression of immediate‐early genes upon disruption of ER calcium homoeostasis. Biochem J 393: 201–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Tessitore A, Martin MP, Sano R, Ma Y, Mann L, Ingrassia A, Laywell ED, Steindler DA, Hendershot LM, Azzo A (2004) GM1‐ganglioside‐mediated activation of the unfolded protein response causes neuronal death in a neurodegenerative gangliosidosis. Mol Cell 15: 753–766 [DOI] [PubMed] [Google Scholar]
- 102. Kharroubi I, Ladrie L, Cardozo AK, Dogusan Z, Cnop M, Eizrik DL (2004) Free fatty acids and cytokines induce pancreatic b‐cell apoptosis by different mechanisms: role of nuclear factor‐kB and endoplasmic reticulum stress. Endocrinology 145: 5087–5096 [DOI] [PubMed] [Google Scholar]
- 103. Lai E, Bikopoulos G, Wheeler MB, Rozakis‐adcock M, Volchuk A (2008) Differential activation of ER stress and apoptosis in response to chronically elevated free fatty acids in pancreatic beta‐cells. Am J Physiol Endocrinol Metab 294: 540–550 [DOI] [PubMed] [Google Scholar]
- 104. Sidrauski C, Chapman R, Walter P (1998) The unfolded protein response: an intracellular signalling pathway with many surprising features. Trends Cell Biol 8: 245–249 [DOI] [PubMed] [Google Scholar]
- 105. Haynes CM, Titus EA, Cooper AA (2004) Degradation of misfolded proteins prevents ER‐derived oxidative stress and cell death. Mol Cell 15: 767–776 [DOI] [PubMed] [Google Scholar]
- 106. Hetz C, Saxena S (2017) ER stress and the unfolded protein response in neurodegeneration. Nat Rev Neurol 13: 477–491 [DOI] [PubMed] [Google Scholar]
- 107. Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D (2000) Regulated translation initiation controls stress‐induced gene expression in mammalian cells. Mol Cell 6: 1099–1108 [DOI] [PubMed] [Google Scholar]
- 108. Han J, Back SH, Hur J, Lin YH, Gildersleeve R, Shan J, Yuan CL, Krokowski D, Wang S, Hatzoglou M et al (2013) ER‐stress‐induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol 15: 481–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Chen X, Shen J, Prywes R (2002) The luminal domain of ATF6 senses endoplasmic reticulum (ER) stress and causes translocation of ATF6 from the er to the Golgi. J Biol Chem 277: 13045–13052 [DOI] [PubMed] [Google Scholar]
- 110. Yamamoto K, Sato T, Matsui T, Sato M, Okada T, Yoshida H, Harada A, Mori K (2007) Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6α and XBP1. Dev Cell 13: 365–376 [DOI] [PubMed] [Google Scholar]
- 111. Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K (2001) XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107: 881–891 [DOI] [PubMed] [Google Scholar]
- 112. Urano F, Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, Ron D (2008) Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 664: 664–667 [DOI] [PubMed] [Google Scholar]
- 113. Hollien J, Weissman JS (2006) Decay of endoplasmic reticulum‐localized mRNAs during the unfolded protein response. Science 313: 104–107 [DOI] [PubMed] [Google Scholar]
- 114. Chami M, Oulès B, Szabadkai G, Tacine R, Rizzuto R, Paterlini‐Bréchot P (2008) Role of SERCA1 truncated isoform in the proapoptotic calcium transfer from ER to mitochondria during ER stress. Mol Cell 32: 641–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Verfaillie T, Rubio N, Garg AD, Bultynck G, Rizzuto R, Decuypere JP, Piette J, Linehan C, Gupta S, Samali A et al (2012) PERK is required at the ER‐mitochondrial contact sites to convey apoptosis after ROS‐based ER stress. Cell Death Differ 19: 1880–1891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Hetz C, Papa FR (2018) The unfolded protein response and cell fate control. Mol Cell 69: 169–181 [DOI] [PubMed] [Google Scholar]
- 117. Bravo R, Vicencio JM, Parra V, Troncoso R, Munoz JP, Bui M, Quiroga C, Rodriguez AE, Verdejo HE, Ferreira J et al (2011) Increased ER – mitochondrial coupling promotes mitochondrial respiration and bioenergetics during early phases of ER stress Increased ER – mitochondrial coupling promotes mitochondrial respiration and bioenergetics during early phases of ER stress. J Cell Sci 124: 2143–2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Lee A, Scapa EF, Cohen DE, Glimcher LH (2008) Regulation of hepatic lipogenesis by the transcription factor XBP1. Science 1492: 1492–1497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Liu X, Henkel AS, LeCuyer BE, Hubchak SC, Schipma MJ, Zhang E, Green RM (2017) Hepatic deletion of X‐box binding protein 1 impairs bile acid metabolism in mice. J Lipid Res 58: 504–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. So JS, Hur KY, Tarrio M, Ruda V, Frank‐Kamenetsky M, Fitzgerald K, Koteliansky V, Lichtman AH, Iwawaki T, Glimcher LH et al (2012) Silencing of lipid metabolism genes through ire1α‐mediated Mrna decay lowers plasma lipids in mice. Cell Metab 16: 487–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Ngoh GA, Papanicolaou KN, Walsh K (2012) Loss of mitofusin 2 promotes endoplasmic reticulum stress. J Biol Chem 287: 20321–20332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Gkogkas C, Middleton S, Kremer AM, Wardrope C, Hannah M, Gillingwater TH, Skehel P (2008) VAPB interacts with and modulates the activity of ATF6. Hum Mol Genet 17: 1517–1526 [DOI] [PubMed] [Google Scholar]
- 123. Ng FWH, Nguyen M, Kwan T, Branton PE, Nicholson DW, Cromlish JA, Shore GC (1997) p28 Bap31, a Bcl‐2/Bcl‐XL – and procaspase‐8–associated protein in the endoplasmic reticulum. J Cell Biol 139: 327–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Hirsch T, Marzo I, Kroemer G (1997) Role of the mitochondrial permeability transition pore in apoptosis. Biosci Rep 17: 67–76 [DOI] [PubMed] [Google Scholar]
- 125. Schinzel AC, Takeuchi O, Huang Z, Fisher JK, Zhou Z, Rubens J, Hetz C, Danial NN, Moskowitz MA, Korsmeyer SJ (2005) Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc Natl Acad Sci USA 102: 12005–12010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Namba T, Tian F, Chu K, Hwang SY, Yoon KW, Byun S, Hiraki M, Mandinova A, Lee SW (2013) CDIP1‐BAP31 complex transduces apoptotic signals from endoplasmic reticulum to mitochondria under endoplasmic reticulum stress. Cell Rep 5: 331–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Peña‐Blanco A, García‐Sáez AJ (2018) Bax, Bak and beyond — mitochondrial performance in apoptosis. FEBS J 285: 416–431 [DOI] [PubMed] [Google Scholar]
- 128. Korsmeyer SJ, Wei MC, Saito M, Weiler S, Oh KJ, Schlesinger PH (2000) Pro‐apoptotic cascade activates BID, which oligomerizes BAK or BAX into pores that result in the release of cytochrome c . Cell Death Differ 7: 1166–1173 [DOI] [PubMed] [Google Scholar]
- 129. Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X (1997) Cytochrome c and dATP‐dependent formation of Apaf‐1/caspase‐9 complex initiates an apoptotic protease cascade. Cell 91: 479–489 [DOI] [PubMed] [Google Scholar]
- 130. Bassik MC, Scorrano L, Oakes SA, Pozzan T, Korsmeyer SJ (2004) Phosphorylation of BCL‐2 regulates ER Ca2+ homeostasis and apoptosis. EMBO J 23: 1207–1216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Brooks C, Wei Q, Feng L, Dong G, Tao Y, Mei L, Xie Z, Dong Z (2007) Bak regulates mitochondrial morphology and pathology during apoptosis by interacting with mitofusins. Proc Natl Acad Sci USA 104: 11649–11654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Hoppins S, Edlich F, Cleland MM, Banerjee S, Mccaffery JM, Youle RJ (2011) The soluble form of bax regulates mitochondrial fusion via MFN2 homotypic complexes. Mol Cell 41: 150–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Lamb CA, Yoshimori T, Tooze SA (2013) The autophagosome: origins unknown, biogenesis complex. Nat Rev 14: 759–774 [DOI] [PubMed] [Google Scholar]
- 134. Tanida I, Ueno T, Kominami E (2004) LC3 conjugation system in mammalian autophagy. Int J Biochem Cell Biol 36: 2503–2518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Gomez‐Suaga P, Paillusson S, Stoica R, Noble W, Hanger DP, Miller CCJ (2017) The ER‐mitochondria tethering complex VAPB‐PTPIP51 regulates autophagy. Curr Biol 27: 371–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Ge L, Schekman R (2014) The ER‐Golgi intermediate compartment feeds the phagophore membrane. Autophagy 10: 170–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Appenzeller‐Herzog C, Hauri H (2006) The ER‐Golgi intermediate compartment (ERGIC): in search of its identity and function. J Cell Sci 119: 2173–2183 [DOI] [PubMed] [Google Scholar]
- 138. Nascimbeni AC, Giordano F, Codogno P, Morel E, Dupont N, Grasso D, Maria I (2017) ER – plasma membrane contact sites contribute to autophagosome biogenesis by regulation of local PI3P synthesis. EMBO J 36: 2018–2033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Shinjo S, Jiang S, Nameta M, Suzuki T, Kanai M, Nomura Y, Goda N (2017) Disruption of the mitochondria‐associated ER membrane (MAM) plays a central role in palmitic acid–induced insulin resistance. Exp Cell Res 359: 86–93 [DOI] [PubMed] [Google Scholar]
- 140. Park SW, Zhou Y, Lee J, Lee J, Ozcan U (2010) Sarco(endo)plasmic reticulum Ca2+‐ATPase 2b is a major regulator of endoplasmic reticulum stress and glucose homeostasis in obesity. Proc Natl Acad Sci USA 107: 19320–19325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Fu S, Yang L, Li P, Hofmann O, Dicker L, Hide W, Lin X, Watkins SM, Ivanov AR (2011) Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature 473: 528–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Tubbs E, Chanon S, Robert M, Bendridi N, Bidaux G, Chauvin MA, Ji‐Cao J, Durand C, Gauvrit‐Ramette D, Vidal H et al (2018) Disruption of mitochondria‐associated endoplasmic reticulum membrane (MAM) integrity contributes to muscle insulin resistance in mice and humans. Diabetes 67: 636–650 [DOI] [PubMed] [Google Scholar]
- 143. Bach D, Pich S, Soriano FX, Vega N, Baumgartner B, Oriola J, Daugaard JR, Lloberas J, Camps M, Zierath JR et al (2003) Mitofusin‐2 determines mitochondrial network architecture and mitochondrial metabolism. A novel regulatory mechanism altered in obesity. J Biol Chem 278: 17190–17197 [DOI] [PubMed] [Google Scholar]
- 144. Liu R, Jin P, Yu L, Wang Y, Han L, Shi T, Li X (2014) Impaired mitochondrial dynamics and bioenergetics in diabetic skeletal muscle. PLoS One 9: e92810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Bach D, Naon D, Pich S, Soriano FX, Vega N, Rieusset J, Laville M, Guillet C, Boirie Y, Wallberg‐henriksson H et al (2005) Expression of Mfn2, the charcot‐marie‐tooth neuropathy type 2A gene, in human skeletal muscle: effects of type 2 diabetes, obesity, weight loss, and the regulatory role of tumor necrosis factor alpha and interleukin‐6. Diabetes 54: 2685–2693 [DOI] [PubMed] [Google Scholar]
- 146. Gastaldi G, Russell A, Golay A, Giacobino JP, Habicht F, Barthassat V, Muzzin P, Bobbioni‐Harsch E (2007) Upregulation of peroxisome proliferator‐activated receptor gamma coactivator gene (PGC1A) during weight loss is related to insulin sensitivity but not to energy expenditure. Diabetologia 50: 2348–2355 [DOI] [PubMed] [Google Scholar]
- 147. Hernández‐Alvarez MI, Chiellini C, Manco M, Naon D, Liesa M, Palacín M, Mingrone G, Zorzano A (2009) Genes involved in mitochondrial biogenesis/function are induced in response to bilio‐pancreatic diversion in morbidly obese individuals with normal glucose tolerance but not in type 2 diabetic patients. Diabetologia 52: 1618–1627 [DOI] [PubMed] [Google Scholar]
- 148. Mingrone G, Manco M, Calvani M, Castagneto M, Naon D, Zorzano A (2005) Could the low level of expression of the gene encoding skeletal muscle mitofusin‐2 account for the metabolic inflexibility of obesity? Diabetologia 48: 2108–2114 [DOI] [PubMed] [Google Scholar]
- 149. Hernández‐Alvarez MI, Thabit H, Burns N, Shah S, Brema I, Hatunic M, Finucane F, Liesa M, Chiellini C, Naon D et al (2010) Subjects with early‐onset type 2 diabetes show defective activation of the skeletal. Diabetes Care 33: 645–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Yang C, Aye CC, Li X, Diaz Ramos A, Zorzano A, Mora S (2012) Mitochondrial dysfunction in insulin resistance: differential contributions of chronic insulin and saturated fatty acid exposure in muscle cells. Biosci Rep 32: 465–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Wold LE, Dutta K, Mason MM, Ren J, Cala SE, Schwanke ML, Davidoff AJ (2005) Impaired SERCA function contributes to cardiomyocyte dysfunction in insulin resistant rats. J Mol Cell Cardiol 39: 297–307 [DOI] [PubMed] [Google Scholar]
- 152. Thivolet C, Vial G, Cassel R, Rieusset J, Madec A‐M (2017) Reduction of endoplasmic reticulum mitochondria interactions in beta cells from patients with type 2 diabetes. PLoS One 12: e0182027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Zhang E, Mohammed Al‐Amily I, Mohammed S, Luan C, Asplund O, Ahmed M, Ye Y, Ben‐Hail D, Soni A, Vishnu N et al (2018) Preserving insulin secretion in diabetes by inhibiting VDAC1 overexpression and surface translocation in β cells. Cell Metab 29: 64–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Ahmed M, Muhammed SJ, Kessler B, Salehi A (2010) Mitochondrial proteome analysis reveals altered expression of voltage dependent anion channels in pancreatic β‐cells exposed to high glucose. Islets 2: 283–292 [DOI] [PubMed] [Google Scholar]
- 155. Mckenzie MD, Jamieson E, Jansen ES, Scott CL, Huang DCS, Bouillet P, Allison J, Kay TWH, Strasser A, Thomas HE (2010) Glucose induces pancreatic islet cell apoptosis that requires the BH3‐only proteins bim and puma and multi‐bh domain protein bax. Diabetes 59: 644–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Schneeberger M, Dietrich MO, Sebastián D, Imbernón M, Castaño C, Garcia A, Esteban Y, Gonzalez‐Franquesa A, Rodríguez IC, Bortolozzi A et al (2013) Mitofusin 2 in POMC neurons connects ER stress with leptin resistance and energy imbalance. Cell 155: 172–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Filippi BM, Abraham MA, Silva PN, Rasti M, LaPierre MP, Bauer PV, Rocheleau JV, Lam TKT (2017) Dynamin‐related protein 1‐dependent mitochondrial fission changes in the dorsal vagal complex regulate insulin action. Cell Rep 18: 2301–2309 [DOI] [PubMed] [Google Scholar]
- 158. Zhao L, Lu T, Gao L, Fu X, Zhu S, Hou Y (2017) Enriched endoplasmic reticulum‐mitochondria interactions result in mitochondrial dysfunction and apoptosis in oocytes from obese mice. J Anim Sci Biotechnol 8: 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Sood A, Vijey D, Prudent J, Caron A, Lemieux P, McBride HM, Laplante M, Tóth K, Pellegrini L (2014) A mitofusin‐2 – dependent inactivating cleavage of Opa1 links changes in mitochondria cristae and ER contacts in the postprandial liver. Proc Natl Acad Sci USA 111: 16017–16022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Theurey P, Tubbs E, Vial G, Jacquemetton J, Bendridi N, Chauvin MA, Alam MR, Le Romancer M, Vidal H, Rieusset J (2016) Mitochondria‐associated endoplasmic reticulum membranes allow adaptation of mitochondrial metabolism to glucose availability in the liver. J Mol Cell Biol 8: 129–143 [DOI] [PubMed] [Google Scholar]
- 161. Báez‐Ruiz A, Cázares‐Gómez K, Vázquez‐Martínez O, Aguilar‐Roblero R, Díaz‐Muñoz M (2013) Diurnal and nutritional adjustments of intracellular Ca2+ release channels and Ca2+ ATPases associated with restricted feeding schedules in the rat liver. J Circadian Rhythms 11: 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Levine B (2005) Eating oneself and uninvited guests: autophagy‐related pathways in cellular defense. Cell 120: 159–162 [DOI] [PubMed] [Google Scholar]
- 163. Hailey DW, Rambold AS, Satpute‐Krishnan P, Mitra K, Sougrat R, Kim PK, Lippincott‐Schwartz J (2010) Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 141: 656–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Mihai AD, Schröder M (2015) Glucose starvation and hypoxia, but not the saturated fatty acid palmitic acid or cholesterol, activate the unfolded protein response in 3T3‐F442A and 3T3‐L1 adipocytes. Adipocyte 4: 188–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Shao M, Shan B, Liu Y, Deng Y, Yan C, Wu Y, Mao T, Qiu Y, Zhou Y, Jiang S et al (2014) Hepatic IRE1α regulates fasting‐induced metabolic adaptive programs through the XBP1s‐PPARα axis signalling. Nat Commun 5: 3528 [DOI] [PubMed] [Google Scholar]
- 166. Sebastian D, Hernandez‐Alvarez MI, Segales J, Sorianello E, Munoz JP, Sala D, Waget A, Liesa M, Paz JC, Gopalacharyulu P et al (2012) Mitofusin 2 (Mfn2) links mitochondrial and endoplasmic reticulum function with insulin signaling and is essential for normal glucose homeostasis. Proc Natl Acad Sci USA 109: 5523–5528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Sebastián D, Sorianello E, Segalés J, Irazoki A, Ruiz‐Bonilla V, Sala D, Planet E, Berenguer‐Llergo A, Muñoz JP, Sánchez‐Feutrie M et al (2016) Mfn2 deficiency links age‐related sarcopenia and impaired autophagy to activation of an adaptive mitophagy pathway. EMBO J 35: 1677–1693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Kulkarni SS, Joffraud M, Boutant M, Ratajczak J, Gao AW, Maclachlan C, Hernandez Alvarez MI, Raymond F, Metairon S, Descombes P et al (2016) Mfn1 deficiency in the liver protects against diet‐induced insulin resistance and enhances the hypoglycemic effect of metformin. Diabetes 65: 3552–3560 [DOI] [PubMed] [Google Scholar]
- 169. Mahdaviani K, Benador IY, Su S, Gharakhanian RA, Stiles L, Trudeau KM, Cardamone M, Enríquez‐Zarralanga V, Ritou E, Aprahamian T et al (2017) Mfn2 deletion in brown adipose tissue protects from insulin resistance and impairs thermogenesis. EMBO Rep 18: 1123–1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Dietrich MO, Liu ZW, Horvath TL (2013) Mitochondrial dynamics controlled by mitofusins regulate agrp neuronal activity and diet‐induced obesity. Cell 155: 188–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Ramírez S, Gómez‐Valadés AG, Schneeberger M, Varela L, Haddad‐Tóvolli R, Altirriba J, Noguera E, Drougard A, Flores‐Martínez Á, Imbernón M et al (2017) Mitochondrial dynamics mediated by mitofusin 1 is required for POMC neuron glucose‐sensing and insulin release control. Cell Metab 25: 1390–1399.e6 [DOI] [PubMed] [Google Scholar]
- 172. Starenki D, Hong SK, Lloyd RV, Park JI (2015) Mortalin (GRP75/HSPA9) upregulation promotes survival and proliferation of medullary thyroid carcinoma cells. Oncogene 34: 4624–4634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Burbulla LF, Fitzgerald JC, Stegen K, Westermeier J, Thost AK, Kato H, Mokranjac D, Sauerwald J, Martins LM, Woitalla D et al (2014) Mitochondrial proteolytic stress induced by loss of mortalin function is rescued by Parkin and PINK1. Cell Death Dis 5: 1–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Xu J‐L, Li L‐Y, Wang Y‐Q, Li Y‐Q, Shan M, Sun S‐Z, Yu Y, Wang B (2018) Hepatocyte‐specific deletion of BAP31 promotes SREBP1C activation, promotes hepatic lipid accumulation, and worsens IR in mice. J Lipid Res 59: 35–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Wu Z, Yang F, Jiang S, Sun X, Xu J (2018) Induction of liver steatosis in BAP31‐deficient mice burdened with tunicamycin‐induced endoplasmic reticulum stress. Int J Mol Sci 19: 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Rieusset J, Fauconnier J, Paillard M, Belaidi E, Tubbs E, Chauvin M, Durand A, Bravard A, Teixeira G, Bartosch B et al (2016) Disruption of calcium transfer from ER to mitochondria links alterations of mitochondria‐associated ER membrane integrity to hepatic insulin resistance. Diabetologia 59: 614–623 [DOI] [PubMed] [Google Scholar]
- 177. Özcan U, Cao Q, Yilmaz E, Lee A‐H, Iwakoshi NN, Özdelen E, Tuncman G, Görgün C, Glimcher LH, Hotamisligil GS (2004) Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306: 457–461 [DOI] [PubMed] [Google Scholar]
- 178. Baiceanu A, Mesdom P, Lagouge M, Foufelle F (2016) Endoplasmic reticulum proteostasis in hepatic steatosis. Nat Rev Endocrinol 12: 710–722 [DOI] [PubMed] [Google Scholar]
- 179. Frakes AE, Dillin A (2017) The UPRER: sensor and coordinator of organismal homeostasis. Mol Cell 66: 761–771 [DOI] [PubMed] [Google Scholar]
- 180. Shpilka T, Haynes CM (2018) The mitochondrial UPR: mechanisms, physiological functions and implications in ageing. Nat Rev Mol Cell Biol 19: 109–120 [DOI] [PubMed] [Google Scholar]
- 181. Chen KH, Dasgupta A, Ding J, Indig FE, Ghosh P, Longo DL (2014) Role of mitofusin 2 (Mfn2) in controlling cellular proliferation. FASEB J 28: 382–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. de Brito OM, Scorrano L (2009) Mitofusin‐2 regulates mitochondrial and endoplasmic reticulum morphology and tethering: the role of Ras. Mitochondrion 9: 222–226 [DOI] [PubMed] [Google Scholar]
- 183. Klawitter J, Seres T, Pennington A, Beatty JT, Klawitter J, Christians U (2017) Ablation of cyclophilin D results in an activation of FAK, Akt, and ERK pathways in the mouse heart. J Cell Biochem 118: 2933–2940 [DOI] [PubMed] [Google Scholar]
- 184. Kim W‐T, Choi HS, Lee HM, Jang Y‐J, Ryu CJ (2014) B‐cell receptor‐associated protein 31 regulates human embryonic stem cell adhesion, stemness, and survival via control of epithelial cell adhesion molecule. Stem Cells 32: 2626–2641 [DOI] [PubMed] [Google Scholar]
- 185. Choi JH, Banks AS, Estall JL, Kajimura S, Boström P, Laznik D, Ruas JL, Chalmers MJ, Kamenecka TM, Blüher M et al (2010) Anti‐diabetic drugs inhibit obesity‐linked phosphorylation of PPARγ 3 by Cdk5. Nature 466: 451–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Banks AS, McAllister FE, Camporez JPG, Zushin PJH, Jurczak MJ, Laznik‐Bogoslavski D, Shulman GI, Gygi SP, Spiegelman BM (2015) An ERK/Cdk5 axis controls the diabetogenic actions of PPARγ. Nature 517: 391–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Fu T, Xu Z, Liu L, Guo Q, Wu H, Liang X, Zhou D, Xiao L, Liu L, Liu Y et al (2018) Mitophagy directs muscle‐adipose crosstalk to alleviate dietary obesity. Cell Rep 23: 1357–1372 [DOI] [PubMed] [Google Scholar]
- 188. Krzysiak TC, Thomas L, Choi Y, Auclair S, Qian Y, Luan S, Krasnow SM, Thomas LL, Koharudin LMI, Benos PV et al (2018) An insulin‐responsive sensor in the SIRT1 disordered region binds DBC1 and PACS‐2 to control enzyme activity. Mol Cell 72: 985–998.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Potthoff MJ (2017) FGF21 and metabolic disease in 2016: a new frontier in FGF21 biology. Nat Rev Endocrinol 13: 74–76 [DOI] [PubMed] [Google Scholar]
- 190. Kharitonenkov A, Shiyanova TL, Koester A, Ford AM, Micanovic R, Galbreath EJ, Sandusky GE, Hammond LJ, Moyers JS, Owens RA et al (2005) FGF‐21 as a novel metabolic regulator. J Clin Invest 115: 1627–1635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Luciani DS, White SA, Widenmaier SB, Saran VV, Taghizadeh F, Hu X, Allard MF, Johnson JD (2013) Bcl‐2 and Bcl‐xl suppress glucose signaling in pancreatic b‐cells. Diabetes 62: 170–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Liu X, Li L, Li J, Cheng Y, Chen J (2016) Insulin resistance contributes to multidrug resistance in HepG2 cells via activation of the PERK signaling pathway and upregulation of Bcl‐2 and P‐gp. Oncol Rep 35: 3018–3024 [DOI] [PubMed] [Google Scholar]
- 193. Ye R, Ni M, Wang M, Luo S, Zhu G, Chow RH, Lee AS (2011) Inositol 1,4,5‐trisphosphate receptor 1 mutation perturbs glucose homeostasis and enhances susceptibility to diet‐induced diabetes. J Endocrinol 210: 209–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Arif T, Paul A, Krelin Y, Shteinfer‐Kuzmine A, Shoshan‐Barmatz V (2018) Mitochondrial VDAC1 silencing leads to metabolic rewiring and the reprogramming of tumour cells into advanced differentiated states. Cancers (Basel) 10: E499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Arif T, Krelin Y, Nakdimon I, Benharroch D, Paul A, Dadon‐Klein D, Shoshan‐Barmatz V (2017) VDAC1 is a molecular target in glioblastoma, with its depletion leading to reprogrammed metabolism and reversed oncogenic properties. Neuro Oncol 19: 951–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Chen H, Gao W, Yang Y, Guo S, Wang H, Wang W, Zhang S, Zhou Q, Xu H, Yao J et al (2014) Inhibition of VDAC1 prevents Ca2+‐mediated oxidative stress and apoptosis induced by 5‐aminolevulinic acid mediated sonodynamic therapy in THP‐1 macrophages. Apoptosis 19: 1712–1726 [DOI] [PubMed] [Google Scholar]
- 197. Lamming DW, Ye L, Katajisto P, Goncalves MD, Saitoh M, Stevens DM, Davis JG, Salmon AB, Richardson A, Ahima RS et al (2012) Rapamycin‐induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science 335: 1638–1643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Peralta S, Goffart S, Williams SL, Diaz F, Garcia S, Nissanka N, Area‐Gomez E, Pohjoismäki J, Moraes CT (2018) ATAD3 controls mitochondrial cristae structure in mouse muscle, influencing mtDNA replication and cholesterol levels. J Cell Sci 131: jcs217075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Desai R, Frazier AE, Durigon R, Patel H, Jones AW, Rosa ID, Lake NJ, Compton AG, Mountford HS, Tucker EJ et al (2017) ATAD3 gene cluster deletions cause cerebellar dysfunction associated with altered mitochondrial DNA and cholesterol metabolism. Brain 140: 1595–1610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Issop L, Fan J, Lee S, Rone MB, Basu K, Mui J, Papadopoulos V (2015) Mitochondria‐associated membrane formation in hormone‐stimulated leydig cell steroidogenesis: role of ATAD3. Endocrinology 156: 334–345 [DOI] [PubMed] [Google Scholar]
- 201. Beaslas O, Metso J, Nissila E, Laurila PP, Kaiharju E, Batchu KC, Kaipiainen L, Mayranpaa MI, Yan D, Gylling H et al (2013) Osbpl8 deficiency in mouse causes an elevation of high‐density lipoproteins and gender‐specific alterations of lipid metabolism. PLoS One 8: e58856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Wang H, Sreenivasan U, Gong D‐W, O'Connell KA, Dabkowski ER, Hecker PA, Ionica N, Konig M, Mahurkar A, Sun Y et al (2013) Cardiomyocyte‐specific perilipin 5 overexpression leads to myocardial steatosis and modest cardiac dysfunction. J Lipid Res 54: 953–965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Tarnopolsky MA, Rennie CD, Robertshaw HA, Fedak‐Tarnopolsky SN, Devries MC, Hamadeh MJ (2006) Influence of endurance exercise training and sex on intramyocellular lipid and mitochondrial ultrastructure, substrate use, and mitochondrial enzyme activity. Am J Physiol Regul Integr Comp Physiol 292: R1271–R1278 [DOI] [PubMed] [Google Scholar]
- 204. Shiozaki M, Hayakawa N, Shibata M, Koike M, Uchiyama Y, Gotow T (2011) Closer association of mitochondria with lipid droplets in hepatocytes and activation of KupVer cells in resveratrol‐treated senescence‐accelerated mice. Histochem Cell Biol 136: 475–489 [DOI] [PubMed] [Google Scholar]
- 205. Yu J, Zhang S, Cui L, Wang W, Na H, Zhu X, Li L, Xu G, Yang F, Christian M et al (2015) Lipid droplet remodeling and interaction with mitochondria in mouse brown adipose tissue during cold treatment. Biochim Biophys Acta 1853: 918–928 [DOI] [PubMed] [Google Scholar]
- 206. Rambold AS, Cohen S, Lippincott‐Schwartz J (2015) Fatty acid trafficking in starved cells: regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Dev Cell 32: 678–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Nguyen TB, Louie SM, Daniele JR, Tran Q, Dillin A, Zoncu R, Nomura DK, Olzmann JA (2017) DGAT1‐dependent lipid droplet biogenesis protects mitochondrial function during starvation‐induced autophagy. Dev Cell 42: 9–21.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Cahill GF (1970) Starvation in man. N Engl J Med 282: 668–675 [DOI] [PubMed] [Google Scholar]
- 209. Young PA, Senkal CE, Suchanek AL, Grevengoed TJ, Lin DD, Zhao L, Crunk AE, Klett EL, Füllekrug J, Obeid LM et al (2018) Long‐chain acyl‐CoA synthetase 1 interacts with key proteins that activate and direct fatty acids into niche hepatic pathways. J Biol Chem 293: 16724–16740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Jägerström S, Polesie S, Wickström Y, Johansson BR, Schröder HD, Højlund K, Boström P (2009) Lipid droplets interact with mitochondria using SNAP23. Cell Biol Int 33: 934–940 [DOI] [PubMed] [Google Scholar]
- 211. Boström P, Andersson L, Rutberg M, Perman J, Lidberg U, Johansson BR, Fernandez‐Rodriguez J, Ericson J, Nilsson T, Borén J et al (2007) SNARE proteins mediate fusion between cytosolic lipid droplets and are implicated in insulin sensitivity. Nat Cell Biol 9: 1286–1293 [DOI] [PubMed] [Google Scholar]
- 212. Wang H, Sreenivasan U, Hu H, Saladino A, Polster BM, Lund LM, Gong D, Stanley WC, Sztalryd C (2011) Perilipin 5, a lipid droplet‐associated protein, provides physical and metabolic linkage to mitochondria. J Lipid Res 52: 2159–2168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Bosma M, Minnaard R, Sparks LM, Schaart G, Losen M, De Baets MH, Duimel H, Kersten S, Bickel PE, Schrauwen P et al (2012) The lipid droplet coat protein perilipin 5 also localizes to muscle mitochondria. Histochem Cell Biol 137: 205–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Granneman JG, Moore HPH, Mottillo EP, Zhu Z, Zhou L (2011) Interactions of Perilipin‐5 (Plin5) with adipose triglyceride lipase. J Biol Chem 286: 5126–5135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Calderon‐Dominguez M, Mir JF, Fucho R, Weber M, Serra D, Herrero L (2016) Fatty acid metabolism and the basis of brown adipose tissue function. Adipocyte 5: 98–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Herms A, Bosch M, Reddy BJN, Schieber NL, Fajardo A, Ruperez C, Fernandez‐Vidal A, Ferguson C, Rentero C, Tebar F et al (2015) AMPK activation promotes lipid droplet dispersion on detyrosinated microtubules to increase mitochondrial fatty acid oxidation. Nat Commun 6: 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Sadh K, Rai P, Mallik R (2017) Feeding‐fasting dependent recruitment of membrane microdomain proteins to lipid droplets purified from the liver. PLoS One 12: e0183022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Kramer DA, Quiroga AD, Lian J, Fahlman RP, Lehner R (2018) Fasting and refeeding induces changes in the mouse hepatic lipid droplet proteome. J Proteomics 181: 213–224 [DOI] [PubMed] [Google Scholar]
- 219. Sohn JH, Lee YK, Han JS, Jeon YG, Kim JI, Choe SS, Kim SJ, Yoo HJ, Kim JB (2018) Perilipin 1 (Plin1) deficiency promotes inflammatory responses in lean adipose tissue through lipid dysregulation. J Biol Chem 293: 13974–13988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Laurens C, Bourlier V, Mairal A, Louche K, Badin PM, Mouisel E, Montagner A, Marette A, Tremblay A, Weisnagel JS et al (2016) Perilipin 5 fine‐tunes lipid oxidation to metabolic demand and protects against lipotoxicity in skeletal muscle. Sci Rep 6: 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Morton TL, Galior K, McGrath C, Wu X, Uzer G, Uzer GB, Sen B, Xie Z, Tyson D, Rubin J et al (2016) Exercise increases and browns muscle lipid in high‐fat diet‐fed mice. Front Endocrinol (Lausanne) 7: 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Gemmink A, Daemen S, Brouwers B, Huntjens PR, Schaart G, Moonen‐Kornips E, Jörgensen J, Hoeks J, Schrauwen P, Hesselink MKC (2018) Dissociation of intramyocellular lipid storage and insulin resistance in trained athletes and type 2 diabetes patients; involvement of perilipin 5? J Physiol 596: 857–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Covington JD, Noland RC, Hebert RC, Masinter BS, Smith SR, Rustan AC, Ravussin E, Bajpeyi S (2015) Perilipin 3 differentially regulates skeletal muscle lipid oxidation in active, sedentary, and type 2 diabetic males. J Clin Endocrinol Metab 100: 3683–3692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Keenan SN, Meex RC, Lo JCY, Ryan A, Nie S, Magdalene K (2019) Perilipin 5 deletion in hepatocytes remodels lipid metabolism and causes hepatic insulin resistance in mice. Diabetes 68: 543–555 [DOI] [PubMed] [Google Scholar]
- 225. Montgomery MK, Mokhtar R, Bayliss J, Parkington HC, Suturin VM, Bruce CR, Watt MJ (2018) Perilipin 5 deletion unmasks an endoplasmic reticulum Stress‐Fibroblast growth factor 21 axis in skeletal muscle. Diabetes 67: 594–606 [DOI] [PubMed] [Google Scholar]
- 226. Harris LALS, Skinner JR, Shew TM, Pietka TA, Abumrad NA, Wolins NE (2015) Perilipin 5‐driven lipid droplet accumulation in skeletal muscle stimulates the expression of fibroblast growth factor 21. Diabetes 64: 2757–2768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Wei S, Liu S, Su X, Wang W, Li F, Deng J, Lyu Y, Geng B, Xu G (2018) Spontaneous development of hepatosteatosis in perilipin‐1 null mice with adipose tissue dysfunction. Biochim Biophys Acta 1863: 212–218 [DOI] [PubMed] [Google Scholar]
- 228. Liu S, Geng B, Zou L, Wei S, Wang W, Deng J, Xu C, Zhao X, Lyu Y, Su X et al (2015) Development of hypertrophic cardiomyopathy in perilipin‐1 null mice with adipose tissue dysfunction. Cardiovasc Res 105: 20–30 [DOI] [PubMed] [Google Scholar]
- 229. Langlois D, Forcheron F, Li JY, Del Carmine P, Neggazi S, Beylot M (2011) Increased atherosclerosis in mice deficient in perilipin1. Lipids Health Dis 10: 1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Nguyen TN, Padman BS, Lazarou M (2016) Deciphering the molecular signals of PINK1/parkin mitophagy. Trends Cell Biol 26: 733–744 [DOI] [PubMed] [Google Scholar]
- 231. Sugiura A, Mclelland G, Fon EA, Mcbride HM (2014) A new pathway for mitochondrial quality control: mitochondrial‐derived vesicles. EMBO J 33: 2142–2156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Raimundo N, Fernández‐mosquera L, Yambire KF, Diogo CV (2016) Mechanisms of communication between mitochondria and lysosomes. Int J Biochem Cell Biol 79: 345–349 [DOI] [PubMed] [Google Scholar]
- 233. Han Y, Li M, Qiu F, Zhang M, Zhang Y‐H (2017) Cell‐permeable organic fluorescent probes for live‐cell long‐term super‐resolution imaging reveal lysosome‐mitochondrion interactions. Nat Commun 8: 1307–1316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Zhao T, Huang X, Han L, Wang X, Cheng H, Zhao Y, Chen Q, Chen J, Cheng H, Xiao R et al (2012) Central role of mitofusin 2 in autophagosome‐lysosome fusion in cardiomyocytes. J Biol Chem 287: 23615–23625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Cioni J, Lin JQ, Holtermann AV, Franze K, Harris WA, Holt CE, Cioni J, Lin JQ, Holtermann AV, Koppers M et al (2019) Late endosomes act as mRNA translation platforms and sustain mitochondria in axons article late endosomes act as mRNA translation platforms and sustain mitochondria in axons. Cell 176: 56–72.e15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Muñoz‐Braceras S, Tornero‐Écija AR, Vincent O, Escalante R (2019) VPS13A is closely associated with mitochondria and is required for efficient lysosomal degradation. Dis Model Mech 12: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Zhang X, Walsh B, Mitchell CA, Rowe T (2005) TBC domain family, member 15 is a novel mammalian Rab GTPase‐activating protein with substrate preference for Rab7. Biochem Biophys Res Commun 335: 154–161 [DOI] [PubMed] [Google Scholar]
- 238. Kumar N, Leonzino M, Cerutti WH, Horenkamp FA, Li P, Lees JA (2018) VPS13A and VPS13C are lipid transport proteins differentially localized at ER contact sites. J Cell Biol 217: 3625–3639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Itoh K, Adachi Y, Yamada T, Suzuki TL, Otomo T, McBride HM, Yoshimori T, Iijima M, Sesaki H (2018) A brain‐enriched Drp1 isoform associates with lysosomes, late endosomes, and the plasma membrane. J Biol Chem 293: 11809–11822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Kon K, Kim J, Uchiyama A, Jaeschke H, Lemasters JJ (2010) Lysosomal iron mobilization and induction of the mitochondrial permeability transition in acetaminophen‐induced toxicity to mouse hepatocytes. Toxicol Sci 117: 101–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. König J, Ott C, Hugo M, Jung T (2017) Mitochondrial contribution to lipofuscin formation. Redox Biol 11: 673–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. Mukhopadhyay A, Pan X, Lambright DG, Tissenbaum HA (2007) An endocytic pathway as a target of tubby for regulation of fat storage. EMBO Rep 8: 931–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Ding X, Zhang W, Zhao T, Yan C, Du H (2017) Rab7 GTPase controls lipid metabolic signaling in myeloid‐ derived suppressor cells. Oncotarget 8: 30123–30137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Wu J, Cheng D, Liu L, Lv Z, Liu K (2019) TBC1D15 a ff ects glucose uptake by regulating GLUT4 translocation. Gene 683: 210–215 [DOI] [PubMed] [Google Scholar]
- 245. Hönscher C, Mari M, Auffarth K, Bohnert M, Griffith J, Geerts W, Van Der Laan M (2014) Cellular metabolism regulates contact sites between vacuoles and mitochondria. Dev Cell 30: 86–94 [DOI] [PubMed] [Google Scholar]
- 246. Gonzalez Montoro A, Kathrin A, Honscher C, Bohnert M, Becker T, Warscheid B, Reggiori F, van der Laan M, Frohlich F, Ungermann C (2018) Vps39 interacts with Tom40 to establish one of two functionally distinct vacuole‐mitochondria contact sites. Dev Cell 45: 621–636 [DOI] [PubMed] [Google Scholar]
- 247. Norambuena A, Wallrabe H, Cao R, Wang DB, Silva A, Svindrych Z, Periasamy A, Hu S, Tanzi RE, Kim DY et al (2018) A novel lysosome‐to‐mitochondria signaling pathway disrupted by amyloid‐ b oligomers. EMBO J 37: 1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Islinger M, Lüers GH, Zischka H, Ueffing M, Völkl A (2006) Insights into the membrane proteome of rat liver peroxisomes: microsomal glutathione‐S‐transferase is shared by both subcellular compartments. Proteomics 6: 804–816 [DOI] [PubMed] [Google Scholar]
- 249. Horner SM, Wilkins C, Badil S, Iskarpatyoti J (2015) Proteomic analysis of mitochondrial‐ associated ER membranes (MAM) during RNA virus infection reveals dynamic changes in protein and organelle trafficking. PLoS One 10: e0117963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250. Horner SM, Liu HM, Park HS, Briley J, Gale M (2011) Mitochondrial‐associated membranes (MAM) form innate immune synapses and are targeted by hepatitis C virus. Proc Natl Acad Sci USA 108: 14590–14595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Schrader M, Costello J, Godinho LF, Islinger M (2015) Peroxisome‐mitochondria interplay and disease. J Inherit Metab Dis 38: 681–702 [DOI] [PubMed] [Google Scholar]
- 252. Otera H, Mihara K (2011) Discovery of the membrane receptor for mitochondrial fission GTPase Drp1. Small GTPases 2: 167–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Kirichok Y, Krapivinsky G, Clapham DE (2004) The mitochondrial calcium uniporter is a highly selective ion channel. Nature 427: 360–364 [DOI] [PubMed] [Google Scholar]