

Abstract
Long‐term memory formation is attributed to experience‐dependent gene expression. Dynamic changes in histone methylation are essential for the epigenetic regulation of memory consolidation‐related genes. Here, we demonstrate that the plant homeodomain finger protein 2 (PHF2) histone demethylase is upregulated in the mouse hippocampus during the experience phase and plays an essential role in memory formation. PHF2 promotes the expression of memory‐related genes by epigenetically reinforcing the TrkB–CREB signaling pathway. In behavioral tests, memory formation is enhanced by transgenic overexpression of PHF2 in mice, but is impaired by silencing PHF2 in the hippocampus. Electrophysiological studies reveal that PHF2 elevates field excitatory postsynaptic potential (fEPSP) and NMDA receptor‐mediated evoked excitatory postsynaptic current (EPSC) in CA1 pyramidal neurons, suggesting that PHF2 promotes long‐term potentiation. This study provides insight into the epigenetic regulation of learning and memory formation, which advances our knowledge to improve memory in patients with degenerative brain diseases.
Keywords: CREB, hippocampus, learning and memory, lysine methylation, PHF2
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics; Neuroscience; Signal Transduction
Introduction
Learning and remembering are neural processes by which humans and other animals modify their behavior based on past experience, reacting to changes in environment, or after social interactions. The formation of long‐term memory is inevitably associated with dynamic gene expression 1, 2, 3. In this process, post‐translational modifications (PTMs) of receptors, signaling molecules and transcription factors, contribute significantly to the regulatory processes involved in memory formation 4.
Histone PTMs induce remodeling of the chromatin structure, thereby epigenetically regulating gene expression and eliciting profound effects on synaptic plasticity, memory formation, and other long‐lasting complex behavior. Histone acetylation, which usually facilitates gene transcription, has been implicated in synaptic plasticity and learning 5, 6, 7, 8. This role of histone acetylation has also been supported by studies showing that histone deacetylase inhibitors enhance the consolidation of associative memory in rodents 9, 10, 11. On the other hand, histone methylation plays variable roles in gene transcription, depending on specific methylated residues of histones. For example, di/tri‐methylation at histone 3 lysine 9 (H3K9) and H3K4 elicits gene repression and activation, respectively. A growing body of evidence supports the involvement of histone methylation in learning and memory. The haploinsufficiency of euchromatin histone methyltransferase1 (EHMT1) is clinically associated with intellectual disability in Kleefstra syndrome 12. The loss of the gene Kmt2b in forebrain excitatory neurons induces impaired hippocampal memory formation in mice 13. Compared to histone methyltransferases, histone demethylases have been investigated less extensively as modulators in memory formation. For example, LSD1+8a, which is a neuron‐specific variant of LSD1/KDM1A, was reported to participate in the balance of memory formation and emotional behavior as a co‐regulator of the activity‐evoked transcription of immediate early genes 14. Based on these reports, it appears that histone demethylases might participate in learning and memory processes as epigenetic regulators, but their precise roles have not been clearly defined.
Plant homeodomain finger protein 2 (PHF2) is a Jumonji‐C (JmjC) domain‐containing demethylase that specifically targets H3K9me2. Of its domains, the plant homeodomain (PHD) recognizes methylated lysine residues and JmjC demethylates the residues using ferrous iron, oxygen, and 2‐oxoglutarate 15. Functionally, PHF2 is believed to act as an epigenetic activator by erasing the repressive mark H3K9me2 16. PHF2 has been reported to act on many transcription factors, such as HNF4 17, CEBPα 18, NF‐κB 19, p53 20, and Runx2 21. Usually, PHF2 functions to promote cell differentiation or to strengthen the cellular response to external stress. Yet, little is known about the role of PHF2 in learning and memory.
Long‐term memory formation requires de novo protein synthesis at the transcriptional levels, and the post‐training transcription of memory‐related genes may be epigenetically programed. Therefore, investigation of the roles of histone demethylases in hippocampal neurons should provide important information that leads to a better understanding of learning and memory formation. In this study, we found that the PHF2 histone demethylase is upregulated in mouse hippocampus after training and facilitates memory formation by epigenetically activating CREB‐driven transcription of memory‐related genes.
Results
PHF2 is upregulated in the mouse hippocampus after contextual fear conditioning
As shown in Appendix Fig S1A, mice were subjected to hippocampus‐dependent contextual fear conditioning (CFC) training. To identify the histone methyltransferases (HMTs) and histone demethylases (HDMs) related to learning and memory, we performed LC/MASS‐based proteomic analyses in the hippocampal tissues obtained from untrained and CFC‐trained mice. The heat map of the proteomics data is presented in Fig 1A. Hierarchical clustering was performed using only the genes that showed a significant difference (P < 0.056) in protein expression between the trained and untrained groups. A total of 233 upregulated and 291 downregulated hippocampal proteins were identified in the trained group relative to controls, which is summarized in Dataset EV1. Among these proteins, 2 HDMs (PHF2 and KDM1A) and 1 HMT (ASH2L) were found to be upregulated after training, with no HDMs or HMTs significantly downregulated. We also confirmed significant induction of Phf2 and Ash2l mRNA levels from the same hippocampal tissues used for proteomics, whereas this was not found for Kdm1a (Fig 1B). Further analysis was performed by dividing the mice into untrained (NT), contextual‐only trained (CTO), tone and shock trained (T+S), and CFC‐trained (T) groups. We found that ASH2L showed no significant change in any of the groups, while KDM1A showed significant increased expressions only in the CTO and T+S groups (Fig 1C, left). However, PHF2 showed the most significant increase in the T group when compared to the NT group. Furthermore, we examined the histone methylation levels and confirmed that H3K9me2 showed a significant decrease in the T group (Fig 1C, right). Since PHF2 is known to be a histone demethylase, and considering the significant increase in the T group, we focused on the role of PHF2 in learning and memory. The increased expression of immediate early genes (IEG) plays an important role in the long‐term synaptic plasticity necessary for associative memory consolidation in many behavioral paradigms. Thus, we analyzed the correlation between c‐FOS and PHF2 levels in the hippocampal CA1 region of each group by immunofluorescence staining, revealing evidence that PHF2 may regulate IEG expression in the process of memory consolidation (Fig 1D). Additionally, we examined gene expression in the hippocampus of WT and PHF2 t/g mice by QuantSeq analysis (Appendix Fig S2). The heat map depicts the changes in gene expression between WT and PHF2 t/g hippocampal tissue (Appendix Fig S2A), and the Venn diagrams show the number of genes that were upregulated or downregulated between WT and PHF2 t/g, revealing 119 upregulated and 56 downregulated genes in the PHF2 t/g group relative to controls (Appendix Fig S2B). Gene ontology analysis for RNA‐seq was followed from KEGG pathway analyses (Appendix Fig S2C and D). These results prompted us to test the possibility that PHF2‐mediated H3K9 demethylation plays a role in hippocampal learning and related memory processes.
Figure 1. PHF2 is upregulated in mouse hippocampus after training.
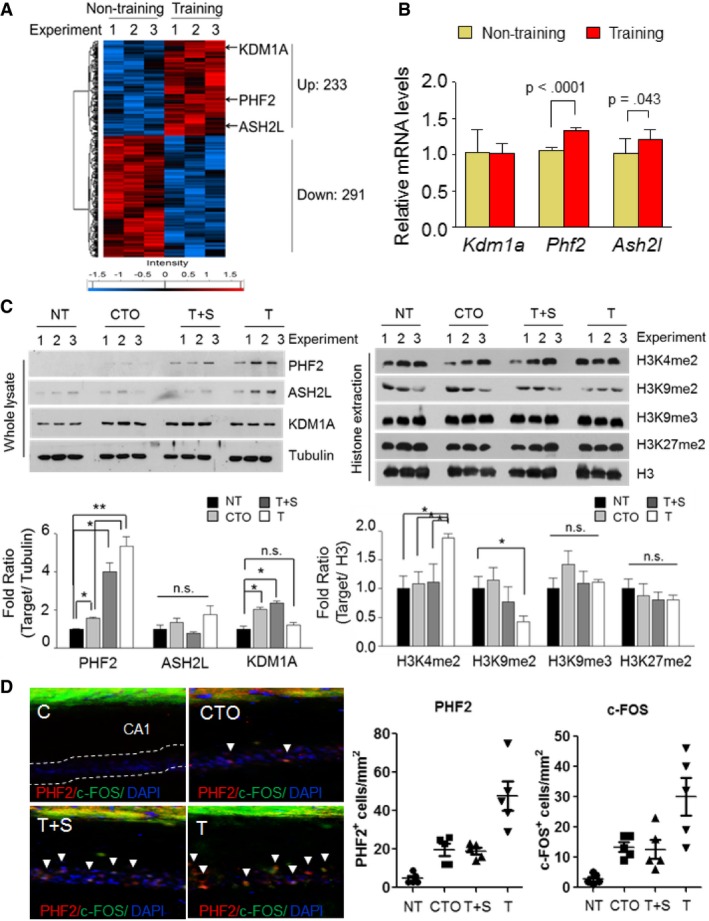
- Hierarchical clustering of 524 DEPs (P < 0.056, Pearson's correlation) was performed as indicated, delineating samples into two groups (not training and training). Log2‐transformed protein expression values were normalized by Z‐score transformation to a scale of −2 to 2 prior to clustering. Color bars show a heat map of Z‐scores and log2‐transformed ion intensities.
- RT–qPCR analysis revealed a significant upregulation of Phf2 and Ash2l expression in the same hippocampal lysates of the untrained and trained mice used for proteomic analysis. Relative mRNA levels were normalized to 18S rRNA levels.
- Immunoblot assays of hippocampal lysates from four groups: untrained (NT), contextual‐only control (CTO), tone + shock (T+S), and CFC trained (T), were performed. The T group showed increased levels of PHF2, but there were no statistical changes in ASH2L expression (left). Decreased methylation of H3K9me2 with no changes in H3K4me2, H3K9me3, or H3K27me2 was observed in the T group (right). Quantification of the immunoblot analysis (Bottom). The protein levels were normalized against tubulin and quantified as fold change relative to the NT group. H3Kme protein levels were normalized against H3 and quantified as fold change relative to the NT group.
- Immunofluorescence of PHF2 and c‐Fos levels in the hippocampus of WT mice divided into untrained (NT), contextual‐only control (CTO), tone + shock (T+S), and CFC trained (T) (left panel). Scale bar, 200 μm. Quantification of PHF2 and c‐Fos double positive stained cells (arrowheads) by immunofluorescence analysis (right panel).
PHF2 is essential for hippocampus‐dependent learning and memory
To further examine the role of PHF2 in learning and memory, we injected GFP‐expressing sh‐control (sh‐Con) or sh‐PHF2 lentiviruses into the bilateral hippocampal CA1 regions of 4‐month‐old mice. Time‐dependent knocked‐down expression of PHF2 by sh‐PHF2 lentivirus was verified by immunoblotting after transfection into the primary neurons (Appendix Fig S3A). The appropriate injection of lentivirus was confirmed by measuring the GFP fluorescence (Fig EV1A). Knockdown efficiency of PHF2 was analyzed by immunoblotting or immunofluorescence staining using anti‐PHF2 antibody in the hippocampal tissues at the indicated times (Appendix Fig S3B), or on the third day after stereotactic injection of sh‐PHF2 lentiviruses (I and II) or si‐PHF2 into the CA1 region (Fig EV1B, Appendix Fig S3C, Appendix Fig S4, and Appendix Fig S5). As expected, PHF2 knockdown significantly increased the hippocampal level of H3K9me2, but not of H3K4me2, H3K9me3, or H3K27me2 (Figs 2A and EV1C). Interestingly, Phf2 and 13 memory‐related genes (Nr2b, Glur1, Glur2, Trkb, CamkII, Creb, Cdk5, Shank3, Psd95, Bdnf, Egr1, c‐Fos, and Npas4) were all downregulated in the PHF2‐depleted hippocampi, but two of them (Glur1 and Npas4) did not statistically differ (Fig 2B). We observed the general behavior of mice after hippocampal injection with shRNAs. The anxiety and locomotor activity of mice in the sh‐Con and sh‐PHF2 groups did not differ in either the open field test (Fig EV1D), or the rotarod test (Fig EV1E). We next examined the role of PHF2 in CFC memory. The day after CFC training, freezing behavior, which is indicative of associative memory consolidation, was observed in the mice re‐exposed to the conditioning context. Mice lacking PHF2 in the hippocampus showed a shorter freezing time than control mice (Fig 2C). This result suggests that PHF2 is essential for the formation of contextual fear memory. Next, we analyzed spatial learning and memory in mice using the Morris water maze test, as shown in Appendix Fig S1B. The probe test was actuated by designating the 2 days that constituted statistical significance as the end point. Mice were placed in the area opposite to the platform in the water tank. In the sh‐Con group, the latency before escape from the original placement site was shorter on training days 3 and 4 than it was on training day 1. The control mice typically learned to swim out of their original location to a platform by day 3. On the other hand, the escape latency time was significantly prolonged in the sh‐PHF2 group (Fig 2D, left), suggesting that the mice lacking PHF2 in the hippocampus were less competent in terms of their learning and memory abilities. Additionally, we evaluated spatial memory using the probe trial test and found that PHF2‐knocked‐down mice stayed longer at their original location, but shorter at the target area (Fig 2E, left; Movie EV1). To exclude the possibility of PHF2 affecting motor ability during the water maze test, swimming ability was tested between sh‐Con and sh‐PHF2 groups, but no differences were observed (Fig 2F, left). Compared with control mice, mice in the sh‐PHF2 group approached or crossed over the vestige of the pre‐existing platform less frequently (Fig 2G, left). To further confirm the effect of PHF2 knockdown on spatial memory formation, we injected PHF2‐silencing siRNA, instead of the shRNA lentivirus, bilaterally into mouse hippocampi. Subsequently, the Morris water maze test revealed that mice in the si‐PHF2 group were less able to learn and to remember compared to mice in the si‐Con group (Fig 2D, E, and G, right). As expected, hippocampal injection with si‐PHF2 did not affect swimming ability (Fig 2F, right). We additionally confirmed whether reduction in learning and memory due to PHF2 knockdown could be rescued by ectopic expression of PHF2. Equal injection of GFP‐Con siRNA or GFP‐PHF2 siRNA vector was verified by measuring both GFP and PHF2 fluorescence (Appendix Fig S5). In the Morris water maze test, the escape latency time was shorter in the co‐transfected siPHF2+F/S‐PHF2 group than in the si‐PHF2 + F/S vector group. In addition, starting on training day 2, co‐transfected siPHF2 + F/S‐PHF2 group of mice stayed longer in the target quadrant (Fig 2H). This increase in the number of platform crossings observed in the si‐PHF2 + F/S‐PHF2 mice group with no difference in swimming ability (Fig 2I) confirmed the role of PHF2 in memory formation recovery (Fig 2J). Overall, PHF2 is probably required for hippocampus‐dependent memory formation.
Figure EV1. Mice lacking PHF2 show no difference in basal behavior tests.
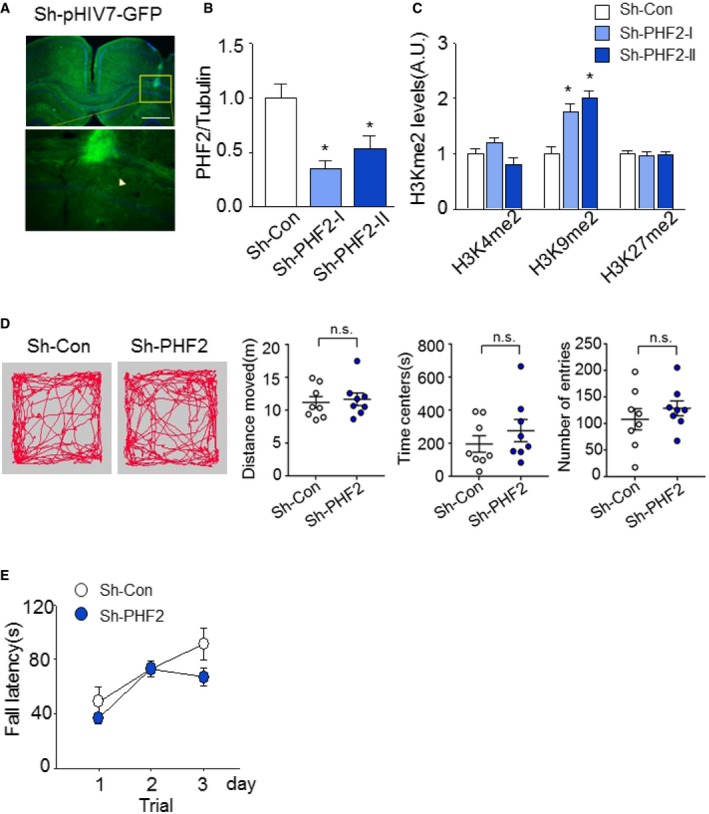
- Stereotaxic injection of GFP‐expressing lentiviral particles into bilateral hippocampus CA1 in WT mice. Scale bar, 200 μm.
- Quantification of the immunoblot analysis. PHF2 protein levels of hippocampal tissues in sh‐PHF2 (I, II) mice were normalized against tubulin and quantified as fold change relative to that seen in sh‐Con mice.
- The H3Kme2 protein levels in sh‐PHF2 (I, II) mice were normalized against H3 and quantified as fold change relative to that seen in sh‐Con mice.
- Open field test shows that basal anxiety and locomotor activity in sh‐PHF2 mice were not altered in comparison with sh‐Con mice. Path traces of single‐trial open field tests were recorded for all sh‐Con and sh‐PHF2 mice (left). Distance moved, time in center, and number of entries were scored (right).
- Rotarod test showed no differences in motor activity for sh‐Con and sh‐PHF2 mice.
Figure 2. PHF2 is essential for hippocampus‐dependent learning and memory.
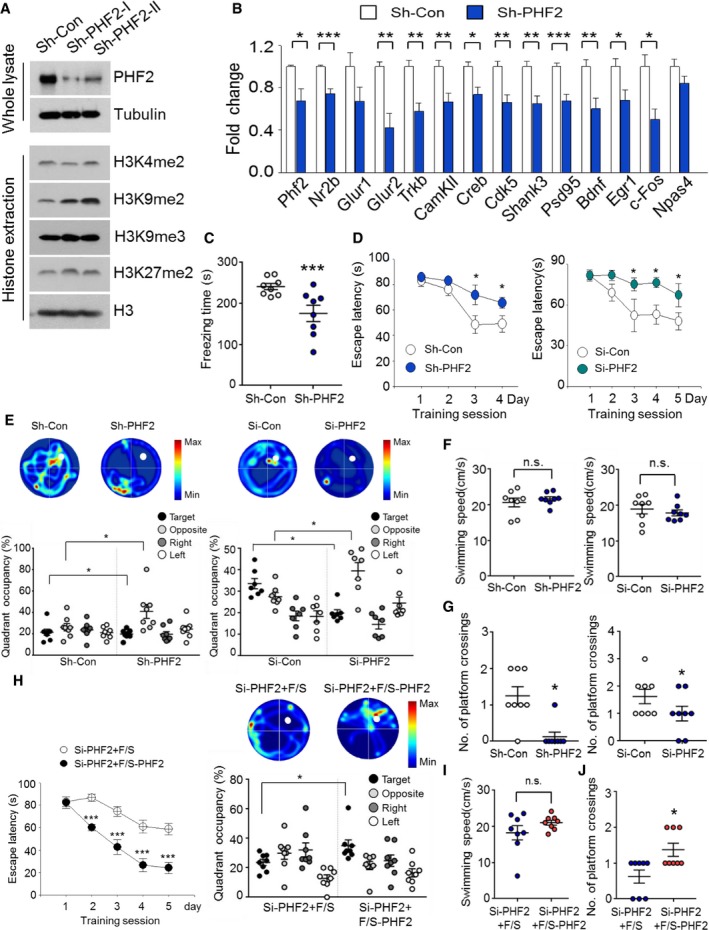
- Immunoblot analysis of hippocampal lysates collected from sh‐Con and sh‐PHF2 mice revealed decreased PHF2 expression in sh‐PHF2 mice. Increased methylation of H3K9me2 was observed in sh‐PHF2 mice, with no changes in either H3K4me2 or H3K9me3.
- RT–qPCR analysis revealed strong downregulation of memory formation‐related genes in the hippocampus of sh‐PHF2 mice, relative to sh‐Con mice. Relative mRNA levels were normalized to 18S rRNA levels.
- Re‐exposure of mice kept at the home cage for 24 h after T+S to the context elicited less freezing in sh‐PHF2 mice relative to that in sh‐Con mice.
- Morris water maze training revealed increase in escape latency for both sh‐ and si‐PHF2 mice compared to that of control mice when trained for 4 and 5 days, respectively.
- Heat plots of search intensity during probe trial were conducted for both sh‐ and si‐PHF2 mice on days 5 and 6, respectively. Both sh‐ and si‐PHF2 mice spent less time in the target quadrant during the probe trial. See also Movie EV1.
- Swimming ability in sh‐ and si‐PHF2 mice did not differ from that of the control group.
- A decrease in the number of platform crossings was observed for both sh‐ and si‐PHF2 mice compared to that of the sh‐Con mice.
- Morris water maze training showed decreased escape latency for si‐PHF2 + F/S‐PHF2 mice compared to that of si‐PHF2 + F/S mice when trained for 5 days (left). Heat plots of search intensity during probe trial were conducted on day 6. Si‐PHF2 + F/S‐PHF2 mice spent more time in the target quadrant than si‐Con + F/S mice during the probe trial (right).
- Swimming ability did not differ between si‐PHF2 + F/S and si‐PHF2 + F/S‐PHF2 mice.
- The number of platform crossings increased in si‐PHF2 + F/S‐PHF2 mice, relative to si‐Con + F/S mice.
PHF2 transgenic mice show high hippocampus‐dependent learning and memory
To confirm the role of PHF2 in learning and memory, we generated homozygous transgenic mice expressing human PHF2 (PHF2 t/g) 21. Expression of the transgene in mouse hippocampi was verified by Western blotting (Fig 3A, Top) and ImageJ‐based densitometry (Fig EV2A). Immunofluorescence images showed that PHF2 was widely overexpressed at the hippocampal CA1 region, and PHF2 expression in hippocampal CA1 of PHF2 t/g mouse was stronger than that of wild‐type mice (Fig EV2B). Given that the hippocampal level of H3K9me2 alone was reduced in the transgenic mice (Fig 3A, Bottom and Fig EV2C), transgenic PHF2 might be functionally active in the hippocampus. Basically, PHF2 overexpression did not substantially affect the anxiety, general locomotor activity, or swimming ability of mice (Fig EV2D–F). We initially analyzed the mRNA levels of the genes related to memory formation. In PHF2 t/g mice, Phf2 and 9 of 13 memory‐related genes (Nr2b, Glur1, CamkII, Creb, Cdk5, Shank3, Psd95, Bdnf, and Erg1) were significantly upregulated, whereas four genes (Glur2, Trkb, c‐Fos, and Npas4) were not (Fig 3B). The day after CFC training, PHF2 t/g mice showed a longer freezing time than did their wild‐type littermates (Fig 3C), suggesting that PHF2 facilitated the formation of contextual fear memory. However, PHF2 did not affect cued‐dependent memory formation (Appendix Fig S6). In the Morris water maze test, the escape latency time was shorter in the PHF2 t/g group than in the wild‐type littermate group, starting on training day 5 (Fig 3D). Compared to wild‐type littermate, PHF2 t/g mice stayed longer in the target quadrant (Fig 3E; Movie EV2). Additionally, the closer proximity (Fig 3F) and increasing number of platform crossings (Fig 3G) of the PHF2 t/g group indicate higher efficiency in searching for platform area. In summary, these results suggest that PHF2 potentiates hippocampus‐dependent memory formation.
Figure 3. PHF2 transgenic mice show high hippocampus‐dependent learning and memory.
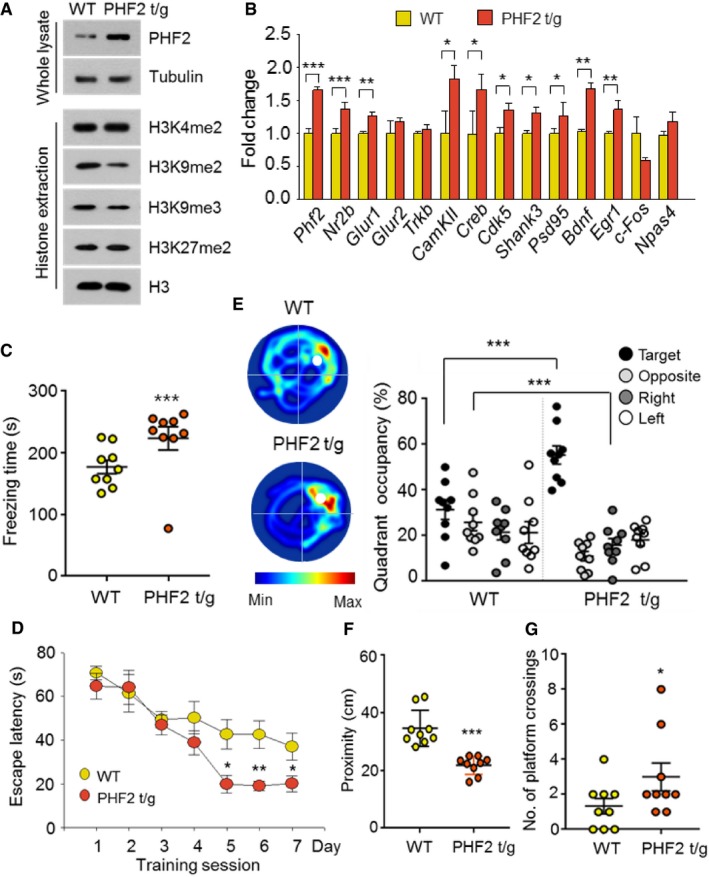
- Immunoblot analysis revealed overexpression of PHF2 and decreased methylation of H3K9me2 in PHF2 t/g mice, with no changes in H3K4me2, H3K9me3, or H3K27me2 relative to WT mice.
- RT–qPCR data analysis revealed upregulation of memory formation‐related genes in PHF2 t/g mice.
- The context elicited increased freezing time in PHF2 t/g mice relative to that in WT littermate mice.
- Results from the Morris water maze showed that the escape latency of PHF2 t/g mice was shorter than that of WT littermate mice.
- Heat plots of search intensity during probe trial were conducted on day 8 (left). PHF2 t/g mice spent more time in the target quadrant than WT littermate mice (right). See also Movie EV2.
- Decrease in proximity to the platform for PHF2 t/g mice relative to WT littermate mice was observed.
- An increase in the number of platform crossings was observed for PHF2 t/g mice relative to WT littermate mice.
Figure EV2. PHF2 t/g mice show no difference in basal behavior tests.
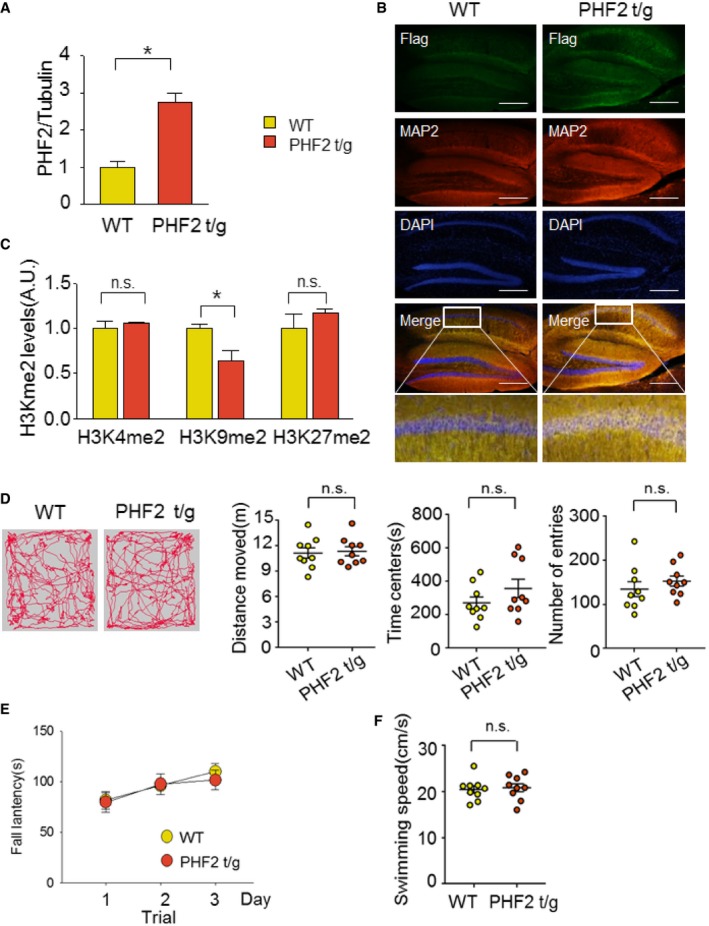
- Quantification of the immunoblot analysis. PHF2 protein levels of PHF2 t/g mice were normalized against tubulin and quantified as fold change relative to that seen in WT littermate mice.
- Immunofluorescence of hippocampal coronal sections of WT littermate and Flag‐PHF2 t/g mice stained with Flag (green), MAP2 (red), and DAPI (blue). Ectopic expression of Flag‐PHF2 in the CA1 region of the hippocampus of PHF2 t/g compared to WT littermate mice. Scale bar, 200 μm.
- H3Kme2 protein levels in PHF2 t/g mice were normalized against H3 and quantified as fold change relative to levels in WT mice.
- Path traces of single‐trial open field tests for WT littermate and PHF2 t/g mice (left). No differences in distance moved, time spent in the center, or number of entries to the center area were observed between the two groups (right).
- No difference in rotarod motor activity was observed between WT littermate and PHF2 t/g mice.
- No difference in swimming ability was observed between WT littermate and PHF2 t/g mice.
PHF2 facilitates long‐term potentiation in CA1 pyramidal neurons
Long‐lasting synaptic plasticity underlies learning and memory formation. To understand the mechanism underlying the PHF2 enhancement of hippocampus‐dependent memory, long‐term potentiation (LTP) was induced in the Schaffer collateral‐CA1 synapses of hippocampus by high‐frequency stimulation (100 Hz; HFS). Field excitatory postsynaptic potential (fEPSP) was significantly impaired in hippocampi transduced with sh‐PHF2 (mean ± SEM, 118.0% ± 7.8), compared to those transduced with sh‐Con (165.3% ± 9.2) (Fig 4A). This result suggests that PHF2 is required for hippocampal LTP that is attributable to synaptic plasticity. The baseline synaptic properties, including the fiber volley–fEPSP relationship and the paired‐pulse ratio, were not different between the two groups (Fig EV3A and B). Next, we examined whether hippocampal LTP is enhanced in PHF2 t/g mice showing improved learning and memory. The basal synaptic properties revealed that the basal synaptic transmission was normal in PHF2 t/g mice (Fig EV3C and D). Interestingly, we observed a weak but significant increase of LTP when PHF2 was transiently overexpressed in the hippocampi (F/S, 111% ± 4.11%; Flag‐PHF2, 120.5% ± 2.75%) (Fig EV4A and B). The hippocampi of PHF2 t/g mice also showed a robust enhancement of early LTP compared with those of their wild‐type littermates (Fig 4B). After a brief post‐tetanic potentiation was observed, the LTP in PHF2 t/g mice was reinforced in a time‐dependent way and reached over twofold level of the LTP in the wild‐type group (WT, 135.9% ± 6.0; PHF2 t/g, 308.7% ± 34.8). Such a kinetic profile of early LTP has not been reported previously, although it is evident in the late LTP generated through brain‐derived neurotrophic factor (BDNF)–TrkB signaling 22. Indeed, BDNF induced the gradual enhancement of LTP in wild‐type hippocampal neurons (Fig EV3E), as shown in PHF2 t/g neurons. A protein synthesis inhibitor cycloheximide abolished the time‐dependent LTP enhancement by BDNF, but did not affect the initial LTP (Fig EV3E). This supports the previous notion that TrkB signaling enhances LTP through the de novo syntheses of memory‐related proteins. Therefore, we tested the possibility of whether PHF2 t/g neurons induce LTP due to pre‐activation of the TrkB‐target genes. When the PHF2 t/g hippocampal neurons were pretreated with cycloheximide or a Trk kinase inhibitor K252a, the increment in early LTP was almost completely abolished (Fig 4C). This result suggests that the LTP enhancement in PHF2 t/g hippocampal neurons is attributable to TrkB‐induced de novo protein synthesis. All fEPSP data were statistically analyzed and are presented as bar graphs in Fig 4D.
Figure 4. PHF2 facilitates long‐term potentiation via the TrkB pathway.
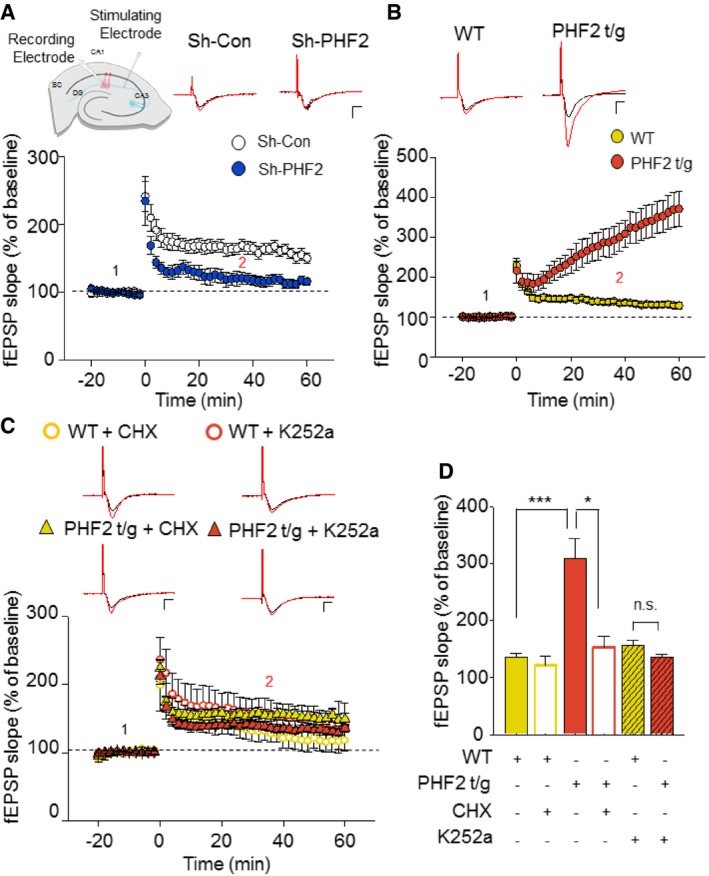
- LTP fEPSP recordings of sh‐Con (white) and sh‐PHF2 (blue) hippocampal slices induced by 100 Hz HFS showed impaired LTP in sh‐PHF2 mice relative to sh‐Con mice. Representative baseline (1: black) and 40 min post‐HFS (2: red) raw fEPSP traces of sh‐Con and sh‐PHF2 (right). Scale bar: 1 mV/5 ms.
- LTP fEPSP recordings from WT (yellow) and PHF2 t/g (orange) hippocampal slices. PHF2 t/g mice produced an enhanced potentiation after HFS. Representative raw traces (right) of WT and PHF2 t/g during baseline (1: black) and 40 min post‐HFS (2: red). Scale bar, 1 mV/5 ms.
- Bath application (10 min pre‐incubation prior to baseline recording) of a protein synthesis blocker, CHX (60 μM) on PHF2 t/g slices (yellow triangles, n = 5), significantly blocked the enhancement of LTP but had no effect on WT slices (open yellow, n = 5). Pre‐incubation with K252a (200 nM), a TrkB inhibitor, 10 min prior to baseline recordings, also inhibited the enhancement of LTP in PHF2 t/g hippocampal slices (orange triangle, n = 15). K252a showed no effect on WT slices (open orange circle, n = 10). Representative raw traces of WT and PHF2 t/g during baseline (1: black) and 40 min post‐HFS (2: red) under the influence of CHX or K252a. Scale bars, 1 mV/5 ms.
- Comparison of LTP at 40 min post‐HFS induction.
Figure EV3. BDNF‐HFS mimics the protein synthesis‐dependent enhancement of LTP observed in PHF2 t/g mice.
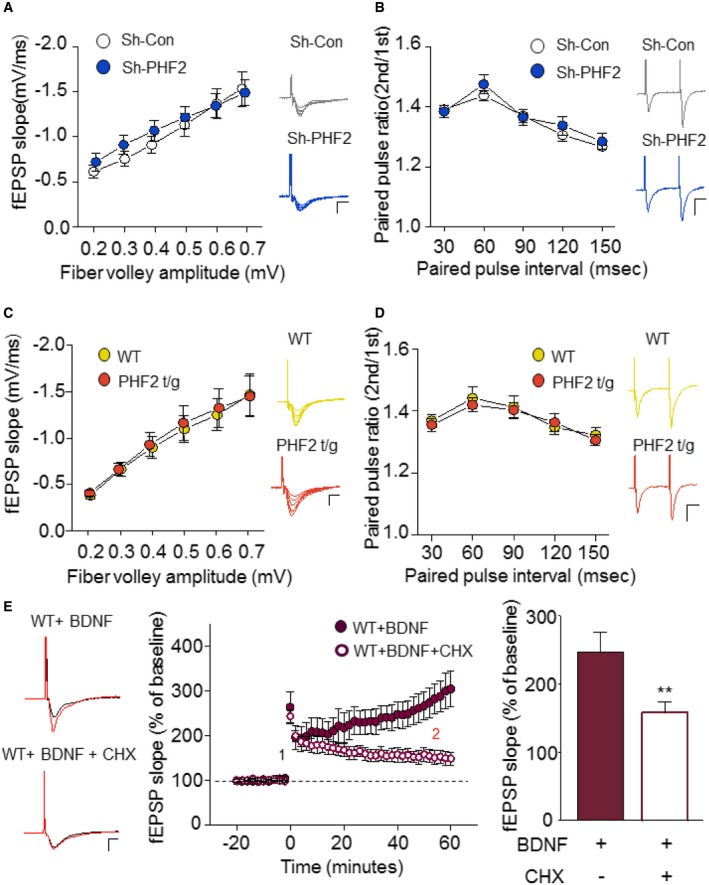
- Input–output of the SC‐CA1 synapse of sh‐Con or sh‐PHF2 expressing mice is represented by fiber volley and fEPSP slope, respectively. Each point represents a mean for a narrow range of fiber volley amplitudes. Representative raw traces are shown for each fiber volley amplitude. Scale bar, 1 mV/5 ms.
- Relationship of PPR of the SC‐CA1 synapses in the hippocampus of sh‐Con (white) and sh‐PHF2 (blue) mice. Example raw traces for PPR are shown for 60‐ms intervals. Scale bar: 1 mV/20 ms.
- SC‐CA1 synapses of WT and PHF2 t/g mice show no differences in input–output relationship. Representative raw traces are shown for each fiber volley amplitude. Scale bar, 1 mV/5 ms.
- PPR is shown for PHF2 t/g mice in comparison with WT mice. Example raw traces for PPR are shown for 60‐ms intervals. Scale bar: 1 mV/20 ms.
- WT hippocampal slices were pre‐incubated with BDNF (20 ng/ml) for > 1 h before HFS induction. BDNF‐HFS‐induced LTP enhancement was blocked by simultaneous application of CHX (60 μM). Example traces of baseline (1: black) and 40 min after LTP induction (2: red) are shown for BDNF‐treated and BDNF + CHX‐treated WT hippocampal slices (Middle). Scale bar, 1 mV/5 ms. Comparison of LTP at 40 min post‐HFS induction (right).
Figure EV4. Transient ectopic expression of PHF2 facilitates long‐term potentiation (LTP) in the hippocampal Schaffer collateral‐CA1 synapses.
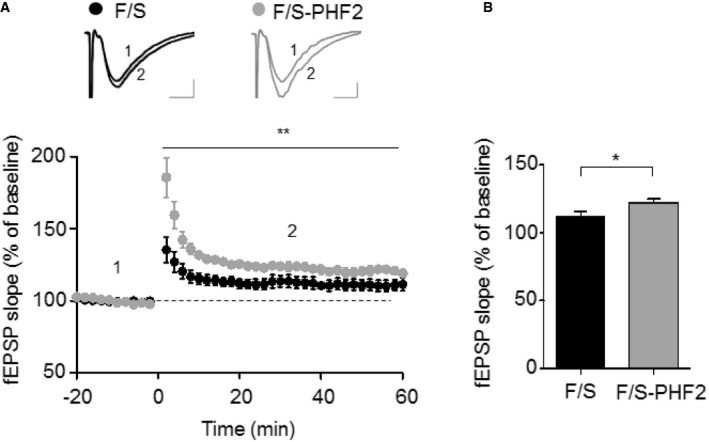
- Field excitatory postsynaptic potentials (fEPSPs) induced by a single 1‐s training at 100 Hz HFS. LTP was significantly greater in slices derived from transiently Flag‐PHF2 expressing mice compared to those from control mice (P < 0.05). Measurements correspond to the time points indicated on the time course graph in this figure. Error bars show the standard error of the mean (**P = 0.0055, 2‐way ANOVA). Scale bar, 0.25 mV/5 ms.
- LTP of control mice was 111 ± 4.11% of baseline during the 10 min before and after time point 2 (n = 5 slices from 3 mice). In Flag‐PHF2 overexpressing mice, fEPSPs were 120.5 ± 2.75% of baseline during the 10 min before and after time point 2 (n = 9 slices from 4 mice). Error bars show the standard error of the mean (*P = 0.0465, 2‐way ANOVA).
PHF2 acts as an epigenetic co‐activator of CREB in the hippocampus
PHF2 is an epigenetic regulator that interacts with and regulates the activity transcription factors. To identify the transcription factors interacting with PHF2, we reanalyzed our proteomics data (Dataset EV1) obtained from the hippocampi of CFC‐trained mice and listed the results in Appendix Fig S7A. Of these proteins, we focused on CREB, TCF4, and GTF2l because they are known to express memory‐related genes. Of the three candidates, we determined that PHF2 was co‐precipitated only with CREB, and vice versa (Fig 5A). The CREB–PHF2 interaction was also confirmed by co‐immunoprecipitating ectopically expressed proteins (Fig 5B). When four domains of PHF2 were co‐expressed with CREB, the C‐terminus (the 4th fragment) of PHF2 (Appendix Fig S8) was identified as the CREB‐interacting domain (Fig 5C). The post‐training induction of CREB was evaluated by Western blotting. The hippocampal level of phospho‐CREB was enhanced to a greater extent than total CREB (Appendix Fig S7B), suggesting that hippocampal CREB was activated during learning and memory formation. To examine whether PHF2 epigenetically regulates the CREB‐driven transcription of target genes Bdnf and Nr2b, chromatin immunoprecipitation (ChIP)‐qPCR analyses were performed. The Gapdh gene was also examined as the representative non‐target gene of CREB. We found that both PHF2 and CREB were more abundantly recruited to CREB target genes in the hippocampal tissues of CFC‐trained mice than in those of untrained mice (Fig 5D and E). Additionally, CREB recruitment to target genes depended on PHF2 expression (Fig 5G and H). To examine the role of PHF2 as an epigenetic regulator of CREB‐driven transcription, we analyzed the binding of H3K9me2 to the promoters of CREB target genes. The binding levels of H3K9me2 to the promoters of CREB target genes were reduced after CFC training (Fig 5F), and it was inversely correlated with PHF2 expression (Fig 5I). These results indicate that PHF2 works actively as a H3K9 demethylase at the promoter region of CREB target genes. To understand how PHF2 regulates the CREB binding to DNA, we measured the CREB and phospho‐CREB proteins in hippocampal tissues. Both CREB and phospho‐CREB were up‐ and downregulated by PHF2 overexpression and knockdown, respectively (Fig 5J, Top). The induction of total CREB protein may be significant; however, the phosphorylation of CREB seems to be more actively regulated by PHF2 (Fig 5J, Bottom). Next, we used a domain study to assess the interaction between PHF2 and CREB, confirming the involvement of the C‐terminal region in this interaction (Fig 5K). Additionally, we confirmed that deletion of the C‐terminal region of PHF2 disrupts CREB‐dependent transcription due to reduced interaction between CREB and PHF2‐CREB‐dependent promoter recruitment (Fig 5L and M). Given that hippocampal memory consolidation is mediated by a positive autoregulatory CREB–BDNF feedback loop 23, PHF2 may activate this autoregulatory loop, i.e., PHF2 activation of CREB promotes the self‐induction of CREB, and induce BDNF and NR2B expression, which stimulates the phosphorylation of CREB.
Figure 5. PHF2 acts as an epigenetic co‐activator of CREB in the hippocampus.
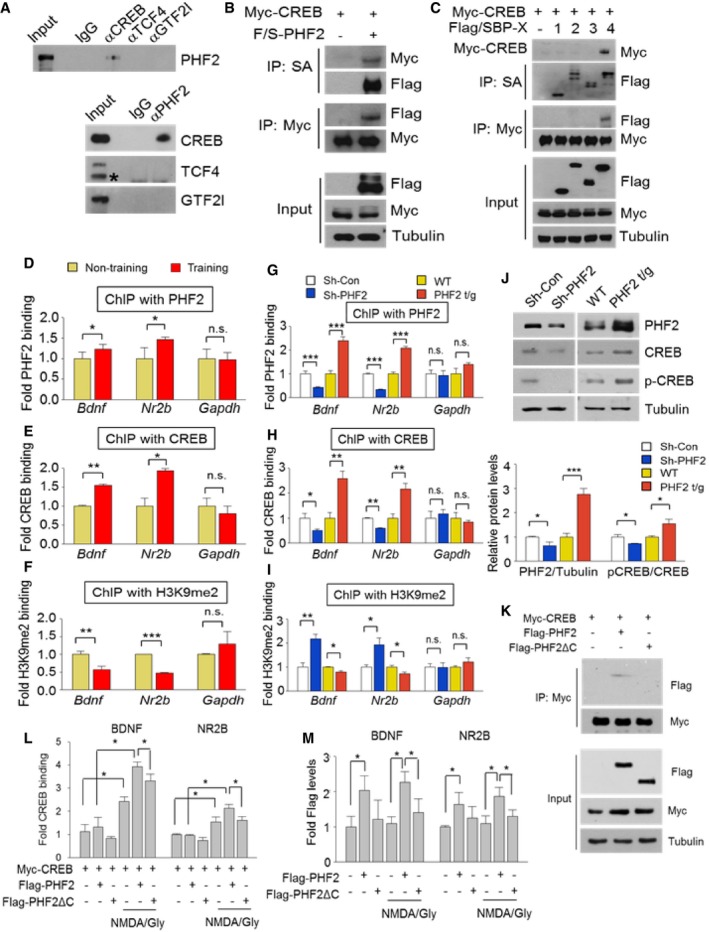
-
AHippocampal lysates prepared from WT mice were immunoprecipitated to evaluate the association of PHF2 with memory‐related transcription factors. PHF2 bound to CREB. The asterisk indicates a specific band.
-
BCo‐immunoprecipitation using Myc‐CREB and Flag‐PHF2 confirmed the interaction between CREB and PHF2 in HEK 293T cells.
-
CExamination of the binding domains of PHF2 and CREB. HEK 293T cells that had been co‐transfected with Myc‐CREB and one of four F/S PHF2 fragments were subjected to streptavidin pulldown, after which the precipitated proteins were immunoblotted.
-
D–FChIP analysis showed that the binding of PHF2 (D), CREB (E) to Bdnf and Nr2b promoters increased in CFC‐trained mice. The binding of H3K9me2 (F) showed the opposite pattern.
-
G–IChIP analysis showed that the binding of PHF2 (G), CREB (H) to Bdnf and Nr2b promoters decreased in sh‐PHF2 mice and did not increase in PHF2 t/g mice. The binding of H3K9me2 (I) showed the opposite pattern.
-
JImmunoblot analysis revealed decreased expression of PHF2, p‐CREB, and CREB in Sh‐PHF2 mice relative to Sh‐Con, whereas expression levels were increased in PHF2 t/g mice relative to WT littermate mice (Top). Quantification of the immunoblot analysis (Bottom).
-
KCo‐immunoprecipitation using Flag‐PHF2, Flag‐PHF2 c‐terminal deletion mutant (Flag‐PHF2ΔC), and Myc‐CREB confirmed the interaction between CREB and PHF2 in HEK 293T cells. Flag‐PHF2ΔC could not bind to CREB.
-
LChIP analysis showed that the binding of CREB to Bdnf and Nr2b promoters decreased in the c‐terminal deleted PHF2 mutant expressing cells.
-
MChIP analysis showed that the binding of PHF2 to Bdnf and Nr2b promoters decreased in the c‐terminal deleted PHF2 mutant expressing cells.
PHF2 activates CREB by NMDA receptor‐dependent signaling
BDNF–TrkB signaling and N‐methyl‐D‐aspartate (NMDA) receptors induce synaptic plasticity in hippocampal neurons to promote memory consolidation 22, 24, 25, 26. CREB is phosphorylated through this signaling cascade and then transactivates many genes essential for synaptic plasticity 27, 28. In the present study, we found that PHF2 enhanced memory formation through the BDNF–TrkB‐CREB signaling pathway (Figs 4C and D, and Fig 5D–I, and EV3E). When the NMDA receptor was stimulated by NMDA and glycine, the CREB in hippocampal neurons was noticeably phosphorylated, but this was attenuated by a CaMKII inhibitor KN‐62. However, PHF2 knockdown attenuated the NMDA‐induced phosphorylation of CREB and total CREB levels (Fig 6A), as well as NMDA‐mediated inductions of Bdnf, Nr2b, Glur2, and Trkb genes (Fig 6B). Additionally, PHF2 and CREB were recruited together to the CREB target genes under the NMDAR signaling activation, which was blocked by PHF2 knockdown (Fig 6C and D). Concomitantly, the H3K9me2 levels at the CREB target genes were inversely related to PHF2 recruitment (Fig 6E). ChIP‐ChIP sequential analysis using two antibodies against PHF2 and CREB showed that PHF2 and CREB are recruited together to the same regions of the Bdnf and Nr2b promoters (Fig 6F). These results suggest that PHF2 acted as a positive regulator for synaptic plasticity‐related gene expression through the activation of CREB. Next, we performed whole‐cell patch‐clamp recordings to analyze the evoked NMDAR‐ and AMPAR‐mediated excitatory postsynaptic current (EPSC). As an index of release probability at presynaptic terminals onto CA1 pyramidal neurons, we measured paired‐pulse ratios of NMDAR‐ and AMPAR‐mediated EPSCs. Given that the paired‐pulse ratio was consistent regardless of PHF2 expression, PHF2 did not affect the vesicle release from presynaptic terminals (Fig EV5A–D). In contrast, NMDAR‐mediated eEPSC was reduced in PHF2 knocked‐down pyramidal neurons but enhanced in PHF2 t/g neurons (Figs 7A and C, and 7B and D, left). AMPAR‐mediated eEPSC was also reduced by PHF2 knockdown, whereas it was not significantly increased in PHF2 t/g neurons (Fig 7A and C, and 7B and D, right). On the other hand, the ratio of NMDAR‐/AMPAR‐mediated postsynaptic responses was determined in a PHF2‐dependent manner (Fig 7E), suggesting that PHF2 enhanced hippocampal LTP by specifically promoting NMDAR‐mediated synaptic plasticity.
Figure 6. PHF2 epigenetically regulates CREB‐driven transcription of target genes in hippocampal neurons.
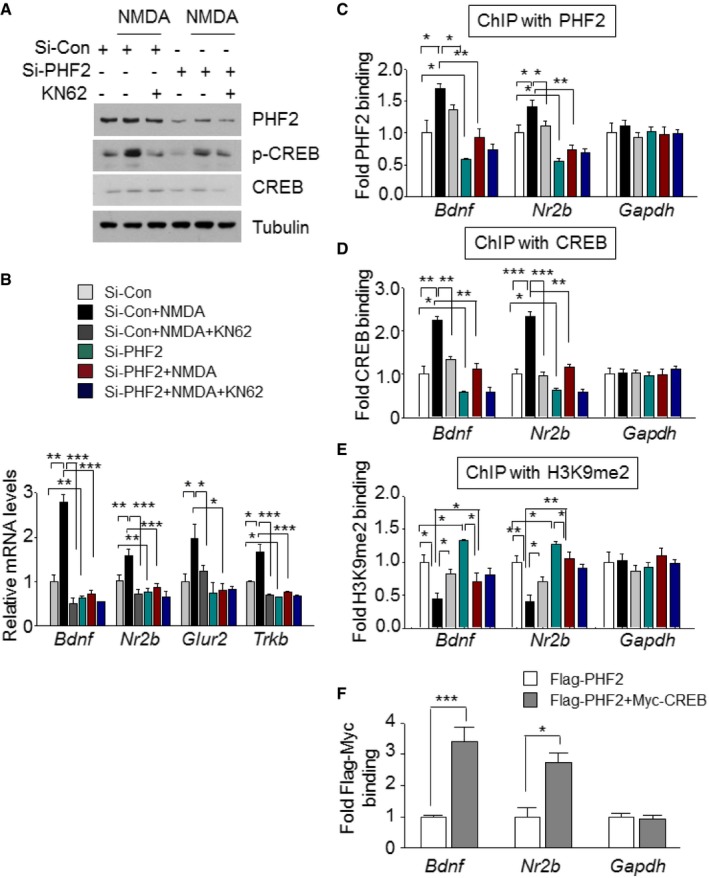
-
ASamples were collected 3 min after stimulation with NMDA and glycine, and the expressions of p‐CREB, CREB, and PHF2 were examined by immunoblotting.
-
BThe mRNA levels of memory formation‐related genes were analyzed using RT–qPCR normalized to 18S rRNA levels.
-
C–EChIP analysis represented the binding of PHF2 (C), CREB (D), and H3K9me2 (E) to Bdnf and Nr2b promoters in neurons.
-
FChIP‐ChIP analysis showed that PHF2 and CREB are recruited together to the same region of Bdnf and Nr2b promoters.
Figure EV5. Basal synaptic transmission of NMDA‐ and AMPA‐mediated EPSCs do not differ by PHF2 expression levels.
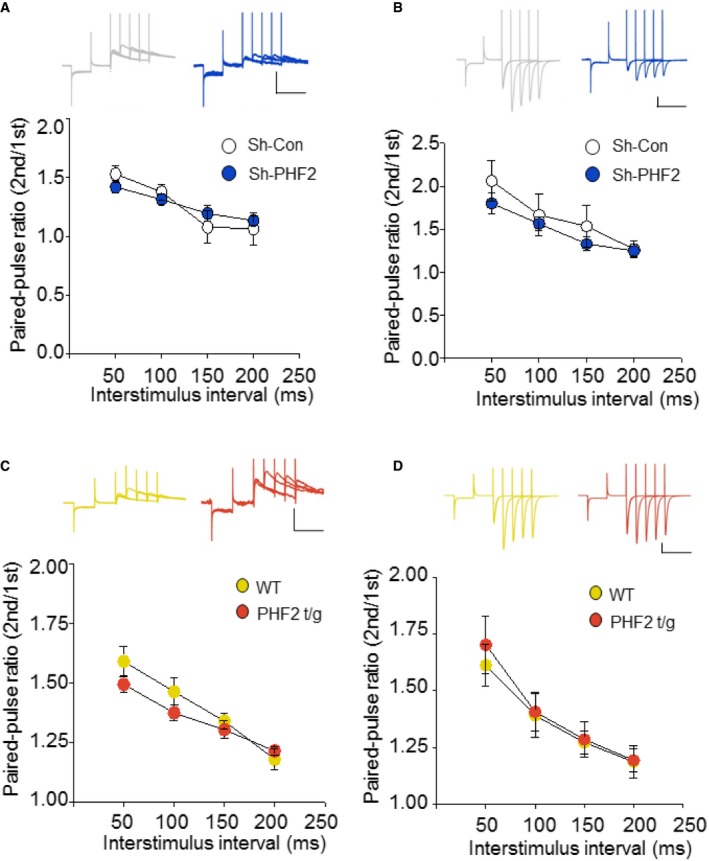
-
A, BRecording of NMDAR‐ (A) and AMPAR‐mediated (B) PPR at 50, 100, 150, and 200 ms intervals. Insets indicate raw trace of PPR (Top). Scale bars, 100 pA/150 ms.
-
C, DRecording of NMDAR‐ (C) and AMPAR‐mediated (D) PPR at 50, 100, 150, and 200 ms intervals. Insets indicate raw trace of PPR (Top). Scale bars, 100 pA/150 ms.
Figure 7. PHF2 promotes NMDAR function by activating CREB .
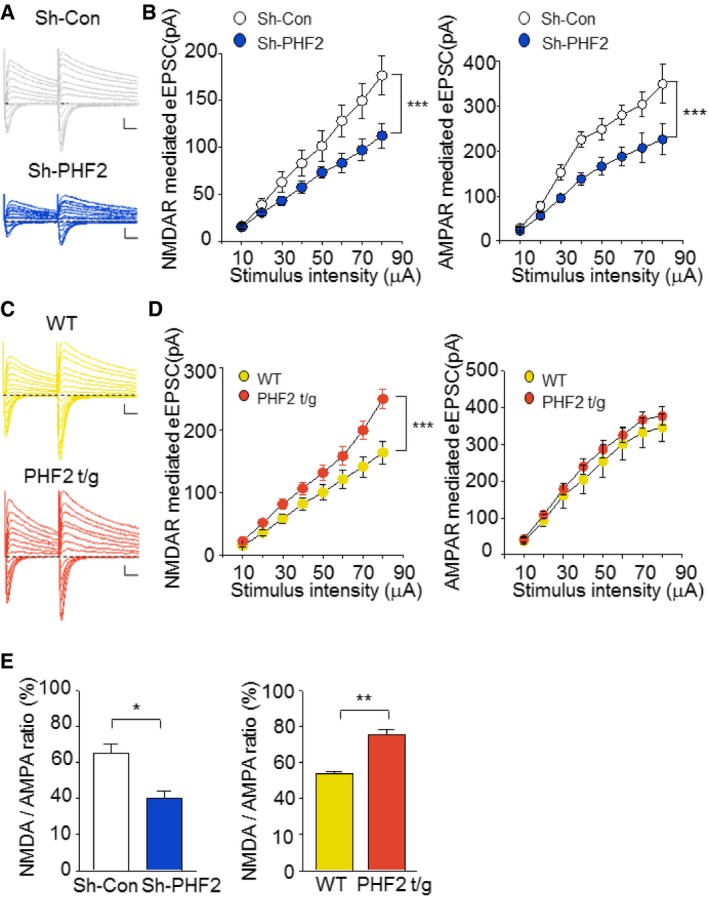
- Raw traces of NMDA‐ and AMPA‐mediated EPSCs of sh‐Con (gray) and sh‐PHF2 (blue) in response to different stimulus inputs. Scale bars, 50 ms/100 pA.
- Recording of evoked NMDAR‐ (left) and AMPAR‐mediated (right) EPSCs in CA1 pyramidal neurons. Input–output curve of NMPAR‐ and AMPAR‐mediated EPSCs at different stimulus intensities in sh‐Con and sh‐PHF2 expressing neurons.
- Raw traces of NMDA‐ and AMPA‐mediated EPSCs from WT (yellow) and PHF2 t/g (orange) mice subjected to different stimulus inputs. Scale bars, 50 ms/100 pA.
- Recording of evoked NMDAR‐ (left) and AMPAR‐mediated (right) EPSCs in CA1 pyramidal neurons. Input–output curve of NMPAR‐ and AMPAR‐mediated EPSCs at different stimulus intensities in WT and PHF2 t/g mice.
- Bar graph shows decreased NMDA to AMPA ratio in sh‐PHF2 mice relative to that in sh‐Con mice, whereas the ratio in PHF2 t/g mice increased.
Discussion
In this study, we found that PHF2 positively regulates memory formation by reinforcing CREB‐mediated synaptic plasticity. Among the histone‐modifying enzymes, PHF2 was shown to be induced in the hippocampi of trained mice and was essential for CREB‐dependent memory‐related gene transcription. PHF2 guides CREB to the promoters of target genes and also provides a transcription‐favorable chromatin structure by erasing repressive H3K9me2‐mediated histone methylation. PHF2 epigenetically activates CREB‐driven transcription, thereby inducing transcription of BDNF, NR2A/B, and other genes related to synaptic plasticity following spatial learning and CFC. Notably, NMDA receptor activation in synaptic plasticity was coupled with PHF2‐mediated activation of CREB.
A subset of histone methyltransferases has been shown to regulate gene expression programs necessary for memory formation. KMT2A haploinsufficient mice exhibited impaired memory formation 29, whereas mice lacking KMT2B in the dorsal dentate gyrus of the hippocampus exhibited memory defects stemming from the deregulation of memory‐related genes 13. EHMT‐mutated Drosophila exhibited defects in both short‐ and long‐term courtship memory 30. Likewise, EHMT1 heterozygous knockout mice showed deficits in learning and memory with abnormal hippocampal dendrite branching 31. Although many studies have investigated the role of histone methyltransferases as epigenetic regulators of memory formation, the role of histone demethylases in learning and memory remains poorly understood. Given that the histone methylation status is counterbalanced by methyltransferases and demethylases, we sought to identify histone demethylases responsible for memory formation, identifying PHF2 as a critical regulator of memory consolidation via its modulation activity of memory‐related gene expression.
Here, we showed that PHF2 was essential for CREB‐mediated transcription of memory‐related genes. A series of publications have suggested that PHF2 promotes gene expression in response to external stress. As memory is regarded as a neuronal response to environmental or social changes, the potentiation of memory formation via the activation of CREB appears to be consistent with the reported functions of PHF2.
CREB has many important roles in the nervous system, affecting neurogenesis and neuronal differentiation, neuroprotection, axonal outgrowth, circadian rhythms, and addiction 32, 33, 34, 35, 36, 37. In particular, numerous studies across multiple species have built a strong consensus that CREB is critical for synaptic plasticity and memory formation. Deletion of CREB in mice resulted in significant deficits in spatial and contextual memory formation with impaired LTP 38, 39. In contrast, when CREB was overexpressed in flies, mice, and rats, they displayed enhanced learning and memory ability 40, 41, 42. In terms of epigenetic regulation, the transcriptional activity of CREB is enhanced in response to reductions in histone methylation. BDNF and nerve‐growth factor (NGF) have been shown to enhance CREB‐driven gene expression via Siah‐mediated degradation of the H3K9 methyltransferase SUV39H1 43. Accordingly, SUV39H1 is regarded as a co‐repressor of CREB, providing indirect regulation of memory‐related gene expression. In the present study, PHF2 was found to be induced in hippocampal neurons of trained mice and was identified as a co‐activator of CREB function. Therefore, CREB‐mediated memory formation may be dynamically regulated by the reciprocal expression of SUV39H1 and PHF2. Further research will be necessary to identify the mechanisms underlying post‐training induction of PHF2.
Phosphorylation‐mediated activation of CREB is mediated by a number of protein kinases, such as PKA, PKC, MAPK, CamKII, CamKIV, and RSK2, and is reversed by protein phosphatases PP1 and calcineurin 44, 45. The role of CREB phosphorylation in memory formation is coordinated by a variety of signaling processes, including an increase in intracellular Ca2+ level through voltage‐ or ligand‐gated channels such as NMDAR, an increase in cAMP levels through G protein‐coupled receptors, and the activation of receptor tyrosine kinase by neurotrophins such as BDNF. In this study, we demonstrated that PHF2 plays an essential role in the NMDA‐induced phosphorylation of CREB. Because it is unknown whether PHF2 directly regulates protein phosphorylation, it is possible that CREB phosphorylation occurs as a result of PHF2 activation of CREB‐driven gene expression. It is also thought that PHF2 can promote binding of CREB to its target genes by increasing the level of phospho‐CREB capable of interacting with DNA. However, we cannot exclude the possibility that PHF2 per se helps CREB access its target genes. In addition to its demethylase activity, PHF2 functions in enhancing the recruitment of transcription factors. Several reports have shown that PHF2 facilitates the recruitment of TP53, C/EBP‐α, and C/EBP‐δ to their target genes 18, 20, 46. Likewise, other JmjC demethylases JMJD2A and JMJD3 have also been reported to increase the DNA binding of ETV1, TBX21, and SMAD3 47, 48. Despite this evidence, the precise mechanism by which PHF2 enhances CREB recruitment to target genes remains as an open question.
Brain‐derived neurotrophic factor is a neurotrophin that exerts a wide range of functions, including promotion of neuronal survival, differentiation, proliferation, and regeneration 49, 50, 51, 52, as well as inducing synaptic plasticity associated with long‐term potentiation and depression 53. Indeed, memory deficits in many neuropsychiatric diseases are associated with deregulation of the BDNF‐TrkB signaling pathway 54. Our results suggest that PHF2 accelerates the positive feedback cycling between CREB and BDNF by epigenetically activating CREB‐driven transcription of BDNF. This study may provide a better understanding of how synaptic plasticity is potentiated upon neurotrophin response.
Taken together, the most remarkable outcome of this study is that overexpression of PHF2 improves memory formation in mice. This implies that PHF2 may be a potential target for therapeutic intervention in human diseases associated with memory impairment.
Materials and Methods
Animals
Male C57BL6/J mice (16 weeks of age) were purchased from the Orient (Sungnam, Korea). Transgenic mice were generated as described previously 21. Transgenic mice were created by injecting the CMV‐Flag‐PHF2 vector into fertilized eggs from C57BL6 mice. Transgenic lines were established from 9 founders that were identified via PCR‐based genotyping. Among the transgenic lines, mice with high PHF2 expressed brains were singled out for breeding. First, selected heterozygote males were bred with heterozygote females which produced homozygote, heterozygote, and wild littermates. Since it is difficult to differentiate between hetero and homo mice with genotyping, we separated homo mice from hetero mice by breeding each of hetero and homo mice with wild‐type mice and confirming the genotypes of their offspring. In other words, hetero mice bred with wild‐type mice would have both wild type and hetero genotype of offspring, while homo mice bred with wild‐type mice would only have hetero genotype of offspring. Through this process, we particularly selected the homo mice and this transgenic line was used and maintained for this experiment. All experiments were done in compliance with the guide included in the Seoul National University Laboratory Animal Maintenance Manual (approve No. SNU‐150602‐5‐1, SNU‐150116‐4‐1, and SNU‐130501‐2).
Proteomics
Cell lysis and protein digestion
Hippocampus tissues were lysed in SDS‐lysis buffer (4% SDS and 1 mM TCEP in 0.1 M Tris pH 7.5) with a sonicator for 5 min. After measurement of protein concentration using BCA reducing agent‐compatible kit, 100 μg of proteins was precipitated overnight at −20°C using acetone. Proteins were digested by FASP procedure with some modifications in steps 1 and 2. Protein pellets were resolved in SDT buffer (4% SDS and 0.1 M DTT in 0.1 M TEAB pH 8.0) and loaded onto a 30 K amicon filter (Millipore). Buffer was exchanged with UA solution (8 M urea in 0.1 M TEAB pH 8.5) by centrifugation at 14,000 g. Reduced cysteines were alkylated with 50 mM IAA solution for 30 min at room temperature (RT) in the dark. After exchanging buffer with 50 mM TEAB, protein digestion was performed at 37°C overnight with trypsin/LysC mixture at a 100:1 protein‐to‐protease ratio.
Tandem mass tag labeling
Tandem mass tag (TMT) 6‐plex labeling was performed according to the manufacturer's instruction with some modification. Briefly, peptide concentration was measured by tryptophan assay 3. MT reagents (0.8 mg) were dissolved in anhydrous acetonitrile (ACN) of which 10 μl was added to the peptides (50 μg) along with acetonitrile to achieve a final acetonitrile concentration of approximately 30% (v/v). Control samples were labeled with 126, 128, and 130, while training samples were labeled with 127, 129, and 131. For normalization and labeling quality check, 500 ng of peptides derived from ovalbumin standard protein was spiked into all channels. Following the incubation at room temperature for 1 h, the reaction was quenched with hydroxylamine to a final concentration of 0.3% (v/v). The TMT‐labeled samples were pooled at a 1:1:1:1:1:1 ratio. The sample was dried using speed‐Vac and subjected to C18 solid‐phase extraction for desalting.
Offline High‐pH reverse‐phased fractionation
The TMT‐labeled peptide pooled mixtures were fractionated using an Agilent 1260 bioinert HPLC (Agilent, Santa Clara, CA) equipped with an analytical column (4.6 × 250 mm, 5 μm). High‐pH reverse‐phase liquid chromatography was performed at a flow rate of 0.8 ml/min during 60‐min gradient using Solvent A (15 mM Ammonium hydroxide in water) and solvent B (15 mM Ammonium Hydroxide in 90% ACN). Peptides were separated with a gradient of 5–35% acetonitrile at 0.2 ml/min. Total 96 fractions were collected every minute within the time period of 40 min and were non‐contiguously concatenated into 12 fractions.
LC‐MS/MS analysis
MS analysis was performed using Quadrupole Orbitrap mass spectrometers, Q‐exactive plus (Thermo) coupled to an Ultimate 300 RSLC systems (Dionex) via a nano electrospray source. Fractionated samples were separated on a 2‐column system with a trap column and an analytical column (75 μm inner diameter, 50 cm length) with 240‐min gradients from 7% to 32% acetonitrile at 300 nl/min and analyzed on the mass spectrometer. The column temperature was constantly set to 60°C by using column heater. The survey scans (350–1,650 m/z) were acquired with a resolution of 70,000 at m/z 200. Top 20 methods were used to select up to the 20 most abundant precursor ions with an isolation windows of 1.2 m/z. The selected precursor ions were subjected to high‐energy collisional dissociation (HCD) fragmentation at a normalized collision energy of 32 with a resolution of 35,000 at m/z 200. The maximum ion injection time for the full scan and MS/MS scan was 20 and 100 ms, respectively.
Data analysis
MS raw files were processed using Proteome Discoverer 2.1 software interfaced SEQUEST‐HT search engine, based on Mouse Uniprot database, including forward and reverse protein sequences and common contaminants. Searches were performed using a 10 ppm precursor ion tolerance for total protein level analysis. The MS/MS ion tolerance was set to 20 ppm. TMT tags on lysine residues and peptide N termini (+229.163 Da) and carbamido‐methylation of cysteine residues (+57.021 Da) were set as fixed modifications, while oxidation of methionine residues (+15.995 Da) was set as a variable modification. The false discovery rate (FDR) for all PSM, peptides, and protein assignments was performed by the included Percolator software package. Reporter ion quantification was performed in the MS2 channel with a 20 ppm mass tolerance. Only the PSMs that contained all six reporter ions were considered. Each reporter ion channel was summed across all quantified proteins and normalized assuming equal protein loading of all six samples.
Bioinformatics analysis
Bioinformatics analysis was performed in the Perseus software5. Statistical analysis of proteome was performed on logarithmized intensities of TMT‐reporter ions. To identify significantly expressed proteins, we have performed the two‐sample t‐test analysis with Benjamini–Hochberg FDR cutoff of 0.05. The gene ontology of analyzed proteins was explicated using the DAVID bioinformatics tool (https://david.ncifcrf.gov/). Pathways were analyzed using the KEGG database.
Quantseq
RNA isolation
Total RNA was isolated using Trizol reagent (Invitrogen). RNA quality was assessed by Agilent 2100 bioanalyzer using the RNA 6000 Nano Chip (Agilent Technologies, Amstelveen, The Netherlands), and RNA quantification was performed using ND‐2000 Spectrophotometer (Thermo Inc., DE, USA).
Library preparation and sequencing
For control and test RNAs, the construction of library was performed using QuantSeq 3′ mRNA‐Seq Library Prep Kit (Lexogen, Inc., Austria) according to the manufacturer's instructions. In brief, each 500 ng total RNA was prepared, an oligo‐dT primer containing an Illumina‐compatible sequence at its 5′ end was hybridized to the RNA, and reverse transcription was performed. After degradation of the RNA template, second strand synthesis was initiated by a random primer containing an Illumina‐compatible linker sequence at its 5′ end. The double‐stranded library was purified by using magnetic beads to remove all reaction components. The library was amplified to add the complete adapter sequences required for cluster generation. The finished library is purified from PCR components. High‐throughput sequencing was performed as single‐end 75 sequencing using NextSeq 500 (Illumina, Inc., USA).
Data analysis
QuantSeq 3′ mRNA‐Seq reads were aligned using Bowtie2 55. Bowtie2 indices were either generated from genome assembly sequence or the representative transcript sequences for aligning to the genome and transcriptome. The alignment file was used for assembling transcripts, estimating their abundances and detecting differential expression of genes. Differentially expressed genes were determined based on counts from unique and multiple alignments using coverage in Bedtools 56. The RT (Read Count) data were processed based on Quantile normalization method using EdgeR within R 57 using Bioconductor 58. Gene classification was based on searches done by DAVID (https://david.ncifcrf.gov/) and Medline databases (http://www.ncbi.nlm.nih.gov/).
Stereotaxic microinjection
For gene silencing, control (sh‐Con) or PHF2‐targeting shRNA (sh‐PHF2) was inserted into the pHIV7‐GFP vector using AgeI and EcoRI enzymes. The viral vector was co‐transfected with pMD2‐VSVG, pRSV‐Rev, and pMDLg/pRRE helper DNA into HEK293T cells. Then, the viral supernatant was collected after 48–72 h of culture. Virus particles were concentrated using high speed ultracentrifugation at 75,600 g for 90 min at 4°C. Male C57BL/6J mice at the age of 4 months (Orient, Gyeonggi‐do, Korea) were anesthetized with isoflurane and placed in stereotaxic frame. A hole was drilled above the CA1 region (AP = −2.0 mm, ML = ± 2.0 mm, DV = 1.7 mm from bregma). Bilateral microinjections of lentiviral particle (5 × 109/ml for CA1) were delivered over a period of 10 min through glass micropipettes. Mice were microinjected with lentiviral particles, siRNAs (1.0 μg), or DNA vector (4 μg) with in vivo‐jetPEI transfection reagent (Polyplus, France), and 3 days later, trained for behavior tests and LTP recordings. The sequences of shRNAs and siRNAs are summarized in Supplementary information, Appendix Table S1.
The open field test
The open field box consists of a square black box (40 cm × 40 cm × 30 cm), which is made out of black plexiglass with an outlined center area. Each of the mice was placed in the center of an open field apparatus and was allowed to move freely for 30 min. Total distance (locomotor activity) and center distance (the distance traveled in the center of the area) were video recorded and analyzed by a computerized mouse tracking system (Ethovision XT 8.5, Noldus, Netherlands).
Rotarod
Mice were placed on a rotating rod (Rotamex‐5; Columbus Instruments, Columbus, OH). The rotarod was set at a rotating speed of 1 rpm (revolution of per minute) with an incremental acceleration of 1 rpm every 6 s up to a maximum speed of 40 rpm reached in 195 s. A day before each test, the motor response in the mice was observed for a period of 2 min after placing them on the rotating bar. For three consecutive days, one trial was performed per day. The time of the mice falling off the rod was recorded.
Morris water maze test
The water maze model was performed in a circular tank (diameter 1.5 m/depth 60 cm) filled with opaque water (40–50 cm, 21–23°C). A platform (10 cm) was submerged below the water's surface in the center of the target quadrant. The swimming path of the mice was recorded by a video camera and analyzed by Ethovision XT 8.5 software (Noldus, Netherlands). The mice start three different points of the tank. For each training session, the mice were allowed to search for the platform for 90 s. If the mice did not find the platform within 90 s, they were gently guided back to the platform. Mice were allowed to remain on the platform for 30 s. Three training trials were given every day; the latency for each trial was recorded for analysis. During the memory test (probe test), the platform was removed from the tank, and the mice were allowed to swim in the maze for 1 min.
Fear conditioning test
Mice were transported to the laboratory at least 60 min prior to the fear conditioning test. The training consisted of a 280 s exposure of mice to the conditioning box (context) followed by a tone (80 db, 2 kHz, 20 s). A foot shock (0.7 mA/s) was administered during the last 2 s of tone presentation and co‐terminate with the tone. Then, context‐dependent freezing test was performed 24 h later by re‐exposing the mice to the conditioning context for 5 min. Cued‐dependent freezing test was also performed by re‐exposing the mice to a novel context for 3 min followed by an additional 3‐min exposure to a tone. The freezing of the mice was recorded by a video camera and analyzed by Ethovision XT 8.5 software (Noldus, Netherlands). The number of observations indicating freezing obtained as a mean from both observers was expressed as a percentage of the total number of observations.
Slice preparation
Hippocampal slices, 300 μm thick for whole‐cell recordings and 400 μm thick for field‐potential recordings, were prepared from 12‐ to 16‐week‐old mice. In brief, the brain was rapidly removed and placed in an ice‐cold high‐sucrose solution which containing (in mM): 212.7 sucrose, 5 KCl, 1.23 NaH2PO4, 0.5 CaCl2, 10 MgSO4, 26 NaHCO3, and 10 d‐glucose bubbled with 95% O2/5% CO2. The isolated brain was glued onto the stage, and transverse sections were cut using a vibrating blade microtome (Leica VT 1200). The slices were transferred to a reservoir chamber and incubated at 34°C for 30 min in artificial cerebrospinal fluid (aCSF) containing (in mM): 124 NaCl, 2.5 KCl, 1.3 MgCl2, 2.5 CaCl2, 2.5 NaH2PO4, 26.2 NaHCO3, and 20 d‐glucose saturated with carbogen (95% O2/5% CO2). Slices were allowed to recover at room temperature (22–24°C).
Field‐potential recording
To obtain extracellular field recordings, we transferred transverse hippocampal slices to an interface‐type chamber, perfused continuously with aCSF at a rate of 3–4 ml/min, and maintained at 32°C for 1 h. Low‐resistance recording electrodes (1–2 MΩ) were pulled using a horizontal puller (Sutter Instruments, Novato, CA) and filled with aCSF and placed into the striatum radiatum of the hippocampal CA1 region to record field excitatory postsynaptic potentials (fEPSPs). Presynaptic stimulations were introduced to the Schaffer collateral fibers via a concentric bipolar electrode (FHC, Bowdoin, ME). The test stimuli were given every 30 s with a 100 μsec pulse duration and were adjusted to elicit 30% of the maximal fEPSP. Basal synaptic transmission was measured by producing an input (fiber volley amplitude)/output (fEPSP slope) curve. Paired‐pulse facilitation (PPF) was measured by delivering pairs of presynaptic fiber stimulation pulses with varying inter‐pulse intervals (30, 60, 90, 120, 150 ms) and at different input intensities. A high‐frequency stimulation (HFS; one 1 s, 100 Hz frequency) protocol was used for LTP induction. Baseline recordings lasted for 20 mins prior to HFS LTP induction and further monitored and recorded for 60 min after HFS. Recordings were made using an A‐M systems microelectrode AC amplifier (Model 1800, A‐M Systems, Sequim, WA) controlled by Clampex 10.2 via Digidata 1440A data acquisition system (Molecular Devices, Sunnyvale, CA), and analysis was performed using Clampfit 10.2 software.
Whole‐cell recording
Whole‐cell recordings of hippocampal CA1 pyramidal neurons made using patch pipettes pulled from borosilicate glass on a Flaming–Brown micropipette puller model P‐1000 (Sutter Instruments, USA). Patch pipettes had a resistance of 4–6 MΩ. Pipettes were filled with an internal solution containing 135 CsMeS, 10 CsCl, 10 HEPES, 0.2 EGTA, 4 Mg‐ATP, 0.4 Na2‐GTP, pH 7.3, and 295 mOsm. Signals were recorded using a patch‐clamp amplifier (Multiclamp 700B, Axon Instruments) and were digitized via Digidata 1550A (Axon Instruments) controlled by Clampex 10.2 software. Signals were amplified, sampled at 10 kHz, and filtered to 2 or 5 kHz. Evoked excitatory postsynaptic currents (eEPSCs) were recorded on pyramidal neurons at CA1 of hippocampus, identified by large apical dendrites. Stimulating electrodes filled with ACSF were placed along the Schaffer collateral in hippocampus. To block inhibitory synaptic responses, SR95531 (20 μM, Tocris) was both applied. To isolate NMDA receptor‐mediated eEPSC, AMPA receptors were blocked with NBQX (100 μM, Tocris). For AMPA receptor‐ and NMDA receptor‐mediated eEPSC, patch recording of CA1 pyramidal neuron was held in voltage clamp at −70 mV and +40 mV respectively, with stimulation in increments of 10 μA between 10 and 80 μA. NMDA to AMPA ratio was calculated by current ratio of eEPSCs between +40 mV and −70 mV at 80 μA stimulation strength. Paired‐pulse ratio of eEPSC was recorded with two pulses in increments of interval 50 ms between 50 and 200 ms.
Cell culture and transfection
Neurons were cultured from embryonic day 16–17 C57BL/6 mouse embryos. Briefly, the cortical and the hippocampus were dissected from the rest of the brain, minced, and incubated for 20 min at 37°C with trypsin and 100 μ/ml DNase in Neurobasal medium (Invitrogen, Carlsbad, CA) supplemented with 1% B‐27 additives (Invitrogen, Carlsbad, CA), 2% N2 (Invitrogen, Carlsbad, CA), 0.5 mM l‐glutamine (Sigma‐Aldrich, St. Louis, MO), and an antibiotic mixture. Tissue was resuspended in medium, filtered twice through 70‐μm pore nylon mesh strainer, and then plated in Neurobasal medium. For gene silencing, control (sh‐Con) or PHF2‐targeting shRNA (sh‐PHF2) was inserted into the pHIV7‐GFP vector using AgeI and EcoRI enzymes. The viral vector was co‐transfected with pMD2‐VSVG, pRSV‐Rev, and pMDLg/pRRE helper DNA into HEK293T cells. Then, the viral supernatant was collected after 48–72 h of culture. Cells were transfected with siRNA against PHF2 and control siRNA using LipofectamineMAX (Invitrogen, Carlsbad, CA) or infected with lentiviral particles (10 MOI).
Immunoblotting and immunoprecipitation
Total proteins were electrophoresed on 8, 10, or 15% SDS/polyacrylamide gel and transferred to Immobilon‐P membranes. The membranes were pre‐incubated in 5% nonfat milk for 60 min and then sequentially incubated with a primary antibody (diluted 1:250–1:5,000 in 5% nonfat milk) overnight at 4°C. Then, the membranes were incubated with a secondary antibody conjugated with horseradish peroxidase (diluted 1:5,000 in 5% nonfat milk) at room temperature for 1 h. Primary antibodies against PHF2, GTF2I, KDM1A, H3K4me2, H3K9me2, H3K9me3, H3K27me2, H3, ASH2L, and p‐CREB were purchased from Cell Signaling (Danvers, MA); anti‐PHF2, anti‐CREB, anti‐TCF4, and anti‐β‐tubulin antibodies were from Santa Cruz biotechnology (Santa Cruz, CA). For immunoprecipitation, cell lysates (1 mg protein) were incubated with 1–1.5 mg of antibody for 12–16 h and further incubated with 20 μml of Protein A/G‐sepharose beads (GE Healthcare Bio‐Sciences, Piscataway, NJ) for 2 h. Precipitated immune complexes were eluted in a denaturing SDS sample buffer and then subjected to immunoblotting.
RT–qPCR
Total RNA was isolated using TRIZOL reagent (Invitrogen, Carlsbad, CA). cDNAs were synthesized from 1 μg of total RNAs using the cDNA synthesis kit provided by Applied Biological Materials Inc (Richmond, BC) containing OneScript® RTase, RNase inhibitor, dNTP, and random primers at 42°C for 50 min. cDNAs were amplified using the ABI StepOne™ Real‐Time PCR (Applied Biosystems, Foster City, CA). The thermal cycling conditions were composed of 50°C for 2 min followed by an initial denaturation step at 95°C for 10 min, 40 cycles at 95°C for 30 s, 60°C for 30 s, and 72°C for 30 s. The experiments were carried out in triplicate for each data point. Relative quantification of mRNA levels was analyzed using the delta‐delta Ct method. The nucleotide sequences of PCR primers are summarized in Appendix Table S2.
Immunofluorescence
Brains were fixed in 4% paraformaldehyde for 24 h, cryoprotected in 30% sucrose for 3–4 days, embedded in OCT, and then serially sectioned as 50 μm. Sections were pre‐incubated in PBS containing 0.3% Triton X‐100 for 30 min and then blocked for 2 h in PBS containing 0.3% Triton X‐100 buffer constituted of 20% horse serum. With primary antibodies of anti‐PHF2 (1:250, Santa Cruz), anti‐c‐Fos (1:250, Cell signaling), anti‐p‐CREB (1:150, Cell signaling), and anti‐GFP (1:500, Invitrogen), sections were incubated for 48 h at 4°C. After incubation, sections were washed and sequentially incubated 1 h in biotinylated anti rabbit (1:500, Vector Laboratories) and 1 h in both Streptavidin 488 and Alexa 594‐conjugated anti‐goat (Invitrogen, Carlsbad, CA). Nuclei were counterstained with DAPI (1:1,000). Stained tissues were mounted on glass sides for evaluation under a fluorescence microscope (Olympus).
Chromatin immunoprecipitation
Chromatins were cross‐linked with 1% formaldehyde for 15 min, followed by washing with ice‐cold PBS. Hippocampus tissues or neuron cells were collected by scraping and centrifugation, and pellets were lysed in the FA lysis buffer (50 mM HEPES, pH 7.5, 140 mM NaCl, 1 mM EDTA, 1% Triton X‐100, 0.1% sodium deoxycholate, 0.1% SDS, and a protease inhibitor cocktail). The lysates were sonicated to chop chromosomal DNAs into 400–800 bp pieces. The lysates were spun down and diluted fivefold in a chromatin RIPA buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 2 mM EDTA, 1% NP‐40, 0.5% sodium deoxycholate, 0.1% SDS, and a protease inhibitor cocktail). The samples were precleaned with protein A/G beads, and 1% of each sample was used for the input control. The samples were immunoprecipitated with anti‐PHF2, anti‐CREB, anti‐H3K9me2, or control IgG. The complexes were washed with a buffer (20 mM Tris, pH 8.0, 0.1% SDS, 1% Triton X‐100, 2 mM EDTA, and 150 mM NaCl) and eluted with an elution buffer (1% SDS, 100 mM NaHCO3). Immunoprecipitated DNAs were subjected to PCR amplification using specific primers for CREB binding elements in the BDNF, NR2A, NR2B, or GAPDH genes. The sequences of PCR primers are summarized in Table EV2. For ChIP‐ChIP assay, Flag‐PHF2 expressed in primary neuron cells was bound to Flag‐affinity beads for 4 h at 4°C and eluted with Flag peptide. The eluted Flag‐PHF2 was secondarily bound to Myc beads for overnight at 4°C. The complexes were washed with a buffer (20 mM Tris, pH 8.0, 0.1% SDS, 1% Triton X‐100, 2 mM EDTA, and 150 mM NaCl) and eluted with an elution buffer (1% SDS, 100 mM NaHCO3). Immunoprecipitated DNAs were subjected to PCR amplification using specific primers for Flag‐CREB binding elements in the BDNF, NR2B, or GAPDH genes (Appendix Fig S9).
Statistical analysis
RT–qPCR and ChIP data were expressed as means and standard deviations and behavioral data as means and standard errors. They were statistically analyzed by unpaired, two‐sided Student's t‐test using the Microsoft Excel 2010 software. Field‐potential recording data and whole‐cell recording data were expressed as means and standard errors, and analyzed by unpaired, two‐sided Student's t‐test using the Clampfit 10.2 (Molecular Devices, CA) and OriginPro (OriginLab, MA) programs. The significance of differences between groups was analyzed by ANOVA with a Bonferroni post hoc test. Significance was reported at P < 0.05.
Author contributions
H‐JK conceived, designed, and performed behavior experiments, molecular experiments, and analyzed experiments; SWH and JJS performed electrophysiology experiments and analyzed experiments; JS, JBP, and S‐YK performed molecular experiment and behavior experiments; M‐HK analyzed behavior experiments; DHH performed and analyzed proteomics experiment; J‐WP wrote the manuscript and edited; JMP and SJK provide expertise, wrote manuscript and edited. Y‐SC conceived, analyzed, wrote the manuscript, edited, and supervised.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Movie EV1
Movie EV2
Source Data for Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
This work was supported by a National Research Foundation grants from the Korean government (2016R1AB4013377, 2018R1A2B6007241, 2018R1A5A2025964). H‐J Kim, J Seo, JB Park, and S‐Y Kim received a scholarship from the BK21‐plus education program provided by the National Research Foundation of Korea.
EMBO Reports (2019) 20: e45907
Contributor Information
Joo Min Park, Email: joominp@ibs.re.kr.
Sang Jeong Kim, Email: sangjkim@snu.ac.kr.
Yang‐Sook Chun, Email: chunys@snu.ac.kr.
Data availability
The Quantseq data have been deposited in the NCBI GEO repository (http://www.ncbi.nlm.nih.gov/geo) with the accession number GSE129148.
References
- 1. Davis HP, Squire LR (1984) Protein synthesis and memory: a review. Psychol Bull 96: 518–559 [PubMed] [Google Scholar]
- 2. Hernandez PJ, Abel T (2008) The role of protein synthesis in memory consolidation: progress amid decades of debate. Neurobiol Learn Mem 89: 293–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. McGaugh JL (2000) Memory‐a century of consolidation. Science 287: 248–251 [DOI] [PubMed] [Google Scholar]
- 4. Routtenberg A (2005) Long‐lasting memory from evanescent networks. Trends Neurosci 28: 12–19 [DOI] [PubMed] [Google Scholar]
- 5. Korzus E, Rosenfeld MG, Mayford M (2004) CBP histone acetyltransferase activity is a critical component of memory consolidation. Neuron 42: 961–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kumar A, Choi KH, Renthal W, Tsankova NM, Theobald DE, Truong HT, Russo SJ, Laplant Q, Sasaki TS, Whistler KN et al (2005) Chromatin remodeling is a key mechanism underlying cocaine‐induced plasticity in striatum. Neuron 48: 303–314 [DOI] [PubMed] [Google Scholar]
- 7. Levenson JM, O'Riordan KJ, Brown KD, Trinh MA, Molfese DL, Sweatt JD (2004) Regulation of histone acetylation during memory formation in the hippocampus. J Biol Chem 279: 40545–40559 [DOI] [PubMed] [Google Scholar]
- 8. Vecsey CG, Hawk JD, Lattal KM, Stein JM, Fabian SA, Attner MA, Cabrera SM, McDonough CB, Brindle PK, Abel T et al (2007) Histone deacetylase inhibitors enhance memory and synaptic plasticity via CREB:CBP‐dependent transcriptional activation. J Neurosci 27: 6128–6140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Federman N, Fustiñana MS, Romano A (2009) Histone acetylation is recruited in consolidation as a molecular feature of stronger memories. Learn Mem 16: 600–606 [DOI] [PubMed] [Google Scholar]
- 10. Fischer A, Sananbenesi F, Wang X, Dobbin M, Tsai LH (2007) Recovery of learning and memory is associated with chromatin remodelling. Nature 447: 178–182 [DOI] [PubMed] [Google Scholar]
- 11. Stefanko DP, Barrett RM, Ly AR, Reolon GK, Wood MA (2009) Modulation of long‐term memory for object recognition via HDAC inhibition. Proc Natl Acad Sci USA 106: 9447–9452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kleefstra T, van Zelst‐Stams WA, Nillesen WM, Cormier‐Daire V, Houge G, Foulds N, van Dooren M, Willemsen MH, Pfundt R, Turner A et al (2009) Further clinical and molecular delineation of the 9q subtelomeric deletion syndrome supports a major contribution of EHMT1 haploinsufficiency to the core phenotype. J Med Genet 46: 598–606 [DOI] [PubMed] [Google Scholar]
- 13. Kerimoglu C, Agis‐Balboa RC, Kranz A, Stilling R, Bahari‐Javan S, Benito‐Garagorri E, Halder R, Burkhardt S, Stewart AF, Fischer A (2013) Histone‐methyltransferase MLL2 (KMT2B) is required for memory formation in mice. J Neurosci 33: 3452–3464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rusconi F, Grillo B, Toffolo E, Mattevi A, Battaglioli E (2017) NeuroLSD1: splicing‐generated epigenetic enhancer of neuroplasticity. Trends Neurosc 40: 28–38 [DOI] [PubMed] [Google Scholar]
- 15. Fortschegger K, Shiekhattar R (2011) Plant homeodomain fingers form a helping hand for transcription. Epigenetics 6: 4–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nakayama J, Rice JC, Strahl BD, Allis CD, Grewal SI (2001) Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science 292: 110–113 [DOI] [PubMed] [Google Scholar]
- 17. Baba A, Ohtake F, Okuno Y, Yokota K, Okada M, Imai Y, Ni M, Meyer CA, Igarashi K, Kanno J et al (2011) PKA‐dependent regulation of the histone lysine demethylase complex PHF2‐ARID5B. Nat Cell Biol 13: 668–675 [DOI] [PubMed] [Google Scholar]
- 18. Okuno Y, Ohtake F, Igarashi K, Kanno J, Matsumoto T, Takada I, Kato S, Imai Y (2013) Epigenetic regulation of adipogenesis by PHF2 histone demethylase. Diabetes 62: 1426–1434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Stender JD, Pascual G, Liu W, Kaikkonen MU, Do K, Spann NJ, Boutros M, Perrimon N, Rosenfeld MG, Glass CK (2012) Control of proinflammatory gene programs by regulated trimethylation and demethylation of histone H4K20. Mol Cell 48: 28–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lee KH, Ju UI, Song JY, Chun YS (2014) The histone demethylase PHF2 promotes fat cell differentiation as an epigenetic activator of both C/EBPα and C/EBPδ. Mol Cells 37: 734–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kim HJ, Park JW, Lee KH, Yoon H, Shin DH, Ju UI, Seok SH, Lim SH, Lee ZH, Kim HH et al (2014) Plant homeodomain finger protein 2 promotes bone formation by demethylating and activating Runx2 for osteoblast differentiation. Cell Res 4: 1231–1249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kang H, Schuman EM (1995) Long‐lasting neurotrophin‐induced enhancement of synaptic transmission in the adult hippocampus. Science 267: 1658–1662 [DOI] [PubMed] [Google Scholar]
- 23. Bambah‐Mukku D, Travaglia A, Chen DY, Pollonini G, Alberini CM (2014) A positive autoregulatory BDNF feedback loop via C/EBPβ mediates hippocampal memory consolidation. J Neurosci 34: 12547–12559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Carroll RC, Zukin RS (2002) NMDA‐receptor trafficking and targeting: implications for synaptic transmission and plasticity. Trends Neurosci 25: 571–577 [DOI] [PubMed] [Google Scholar]
- 25. Tsien JZ, Huerta PT, Tonegawa S (1996) The essential role of hippocampal CA1 NMDA receptor‐dependent synaptic plasticity in spatial memory. Cell 87: 1327–1338 [DOI] [PubMed] [Google Scholar]
- 26. Tyler WJ, Alonso M, Bramham CR, Pozzo‐Miller LD (2002) From acquisition to consolidation: on the role of brain‐derived neurotrophic factor signaling in hippocampal‐dependent learning. Learn Mem 9: 224–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Barco A, Alarcon JM, Kandel ER (2002) Expression of constitutively active CREB protein facilitates the late phase of long‐term potentiation by enhancing synaptic capture. Cell 108: 689–703 [DOI] [PubMed] [Google Scholar]
- 28. Finkbeiner S, Tavazoie SF, Maloratsky A, Jacobs KM, Harris KM, Greenberg ME (1997) CREB: a major mediator of neuronal neurotrophin responses. Neuron 19: 1031–1047 [DOI] [PubMed] [Google Scholar]
- 29. Gupta S, Kim SY, Artis S, Molfese DL, Schumacher A, Sweatt JD, Paylor RE, Lubin FD (2010) Histone methylation regulates memory formation. J Neurosci 30: 3589–3599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kramer JM, Kochinke K, Oortveld MA, Marks H, Kramer D, de Jong EK, Asztalos Z, Westwood JT, Stunnenberg HG, Sokolowski MB et al (2011) Epigenetic regulation of learning and memory by Drosophila EHMT/G9a. PLoS Biol 9: e100569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Balemans MC, Kasri NN, Kopanitsa MV, Afinowi NO, Ramakers G, Peters TA, Beynon AJ, Janssen SM, van Summeren RC, Eeftens JM et al (2013) Hippocampal dysfunction in the Euchromatin histone methyltransferase 1 heterozygous knockout mouse model for Kleefstra syndrome. Hum Mol Genet 22: 852–866 [DOI] [PubMed] [Google Scholar]
- 32. Barco A, Kandel ER (2006) The role of CREB and CBP in brain function In Transcription factors in the nervous system: development, brain function and disease. Thiel G. (ed.), pp 206–241. Weinheim, Germany: WILEY‐VCH Press; [Google Scholar]
- 33. Carlezon WA Jr, Duman RS, Nestler EJ (2005) The many faces of CREB. Trends Neurosci 28: 436–445 [DOI] [PubMed] [Google Scholar]
- 34. Mioduszewska B, Jaworski J, Kaczmarek L (2003) Inducible cAMP early repressor (ICER) in the nervous system–a transcriptional regulator of neuronal plasticity and programmed cell death. J Neurochem 87: 1313–1320 [DOI] [PubMed] [Google Scholar]
- 35. Montminy M, Koo SH, Zhang X (2004) The CREB family: key regulators of hepatic metabolism. Ann Endocrinol (Paris) 65: 73–75 [DOI] [PubMed] [Google Scholar]
- 36. Persengiev SP, Green MR (2003) The role of ATF/CREB family members in cell growth, survival and apoptosis. Apoptosis 8: 225–228 [DOI] [PubMed] [Google Scholar]
- 37. Silva AJ, Kogan JH, Frankland PW, Kida S (1998) CREB and memory. Ann Rev Neurosci 21: 127–148 [DOI] [PubMed] [Google Scholar]
- 38. Bourtchuladze R, Frenguelli B, Blendy J, Cioffi D, Schutz G, Silva AJ (1994) Deficient long‐term memory in mice with a targeted mutation of the cAMP‐responsive element‐binding protein. Cell 79: 59–68 [DOI] [PubMed] [Google Scholar]
- 39. Kida S, Josselyn SA, Peña de Ortiz S, Kogan JH, Chevere I, Masushige S, Silva AJ (2002) CREB required for the stability of new and reactivated fear memories. Nature Neurosci 5: 348–355 [DOI] [PubMed] [Google Scholar]
- 40. Bozon B, Kelly A, Josselyn SA, Silva AJ, Davis S, Laroche S (2003) MAPK, CREB and zif268 are all required for the consolidation of recognition memory. Philos Trans R Soc Lond B Biol Sci 358: 805–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Josselyn SA, Kida S, Silva AJ (2004) Inducible repression of CREB function disrupts amygdala‐dependent memory. Neurobiol Learn Mem 82: 159–163 [DOI] [PubMed] [Google Scholar]
- 42. Matynia A, Kushner SA, Silva AJ (2002) Genetic approaches to molecular and cellular cognition: a focus on LTP and learning and memory. Annu Rev Genet 36: 687–720 [DOI] [PubMed] [Google Scholar]
- 43. Sen N, Snyder SH (2011) Neurotrophin‐mediated degradation of histone methyltransferase by S‐nitrosylation cascade regulates neuronal differentiation. Proc Natl Acad Sci USA 108: 20178–20183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bito H, Deisseroth K, Tsien RW (1996) CREB phosphorylation and dephosphorylation: a Ca(2+)‐ and stimulus duration‐dependent switch for hippocampal gene expression. Cell 87: 1203–1214 [DOI] [PubMed] [Google Scholar]
- 45. Wu GY, Deisseroth K, Tsien RW (2001) Activity‐dependent CREB phosphorylation: convergence of a fast, sensitive calmodulin kinase pathway and a slow, less sensitive mitogen‐activated protein kinase pathway. Proc Natl Acad Sci USA 98: 2808–2813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lee KH, Park JW, Sung HS, Choi YJ, Kim WH, Lee HS, Chung HJ, Shin HW, Cho CH, Kim TY et al (2015) Histone demethylase acts as a tumor suppressor in association with p53 in cancer. Oncogene 34: 2897–2909 [DOI] [PubMed] [Google Scholar]
- 47. Kim TD, Jin F, Shin S, Lightfoot SA, Grande JP, Johnson AJ, van Deursen JM, Wren JD, Janknecht R (2016) Histone demethylase JMJD2A drives prostate tumorigenesis through transcription factor ETV1. J Clin Invest 126: 706–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Li Q, Zou J, Wang M, Ding X, Chepelev I, Zhou X, Zhao W, Wei G, Cui J , Zhao K et al (2014) Critical role of histone demethylase Jmjd3 in the regulation of CD4+ T‐cell differentiation. Nat Commun 5: 5780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Alderson RE, Altman AL, Barde YA, Lindsay RM (1990) Brain‐derived neurotrophic factor increases survival and differentiated functions of rat septal cholinergic neurons in culture. Neuron 5: 297–306 [DOI] [PubMed] [Google Scholar]
- 50. Islam O, Loo TX, Heese K (2009) Brain‐derived neurotrophic factor (BDNF) has proliferative effects on neural stem cells through the truncated TRK‐B receptor, MAP kinase, AKT, and STAT‐3 signaling pathways. Curr Neurovasc Res 6: 42–53 [DOI] [PubMed] [Google Scholar]
- 51. Knusel B, Winslow JW, Rosenthal A, Burton LE, Seid DP, Nikolics K, Hefti F (1991) Promotion of central cholinergic and dopaminergic neuron differentiation by brain‐derived neurotrophic factor but not neurotrophin‐3. Proc Natl Acad Sci USA 88: 961–965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhang JY, Luo XG, Xian CJ, Liu ZH, Zhou XF (2000) Endogenous BDNF is required for myelination and regeneration of injured sciatic nerve in rodents. Eur J Neurosci 12: 4171–4180 [PubMed] [Google Scholar]
- 53. Aarse J, Herlitze S, Manahan‐Vaughan D (2016) The requirement of BDNF for hippocampal synaptic plasticity is experience‐dependent. Hippocampus 26: 739–751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Autry AE, Monteggia LM (2012) Brain‐derived neurotrophic factor and neuropsychiatric disorders. Pharmacol Rev 64: 238–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Langmead B, Salzberg SL (2012) Fast gapped‐read alignment with Bowtie 2. Nature 9: 357–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Quinlan AR, Hall IM (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. R Development Core Team (2016) R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; http://www.R-project.org [Google Scholar]
- 58. Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J et al (2014) Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 5: R80 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Dataset EV1
Movie EV1
Movie EV2
Source Data for Appendix
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 5
Source Data for Figure 6
Data Availability Statement
The Quantseq data have been deposited in the NCBI GEO repository (http://www.ncbi.nlm.nih.gov/geo) with the accession number GSE129148.