

Abstract
Caspase‐4, the cytosolic LPS sensor, and gasdermin D, its downstream effector, constitute the non‐canonical inflammasome, which drives inflammatory responses during Gram‐negative bacterial infections. It remains unclear whether other proteins regulate cytosolic LPS sensing, particularly in human cells. Here, we conduct a genome‐wide CRISPR/Cas9 screen in a human monocyte cell line to identify genes controlling cytosolic LPS‐mediated pyroptosis. We find that the transcription factor, IRF2, is required for pyroptosis following cytosolic LPS delivery and functions by directly regulating caspase‐4 levels in human monocytes and iPSC‐derived monocytes. CASP4,GSDMD, and IRF2 are the only genes identified with high significance in this screen highlighting the simplicity of the non‐canonical inflammasome. Upon IFN‐γ priming, IRF1 induction compensates IRF2 deficiency, leading to robust caspase‐4 expression. Deficiency in IRF2 results in dampened inflammasome responses upon infection with Gram‐negative bacteria. This study emphasizes the central role of IRF family members as specific regulators of the non‐canonical inflammasome.
Keywords: caspase‐11, inflammasome, interferon regulatory factor, lipopolysaccharide, LPS
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics; Immunology; Microbiology, Virology & Host Pathogen Interaction
Introduction
Sepsis is a leading cause of mortality worldwide, accounting for over 5 million deaths annually 1. Sepsis is a deregulated host response to severe infection and is characterized by life‐threatening organ dysfunction 2. While the deregulated host immune responses include both pro‐ and anti‐inflammatory responses, the early deaths observed during sepsis are thought to be due to a systemic inflammatory response syndrome (SIRS) 3. A prominent initiator of SIRS is lipopolysaccharide (LPS) 4, a component of the envelope of Gram‐negative bacteria. This potent endotoxin engages several inflammatory receptors and is sufficient to trigger SIRS in both mice and humans 5, 6.
Immune responses to LPS are induced following either extracellular recognition by TLR4 7 or cytosolic recognition by caspase‐4/5/11 8, 9, 10. While the TLR4 pathway was long thought to be the main pathway responsible for LPS‐induced lethality 7, the discovery that caspase‐11 is activated in mice in response to cytosolic LPS has shed a new light on LPS‐induced pathologies 9, 10. Caspase‐11 and its human homologues caspase‐4 and 5 bind LPS and act both as receptor 8 and as effector of the so‐called non‐canonical inflammasomes 11. Upon LPS binding, these inflammatory caspases oligomerize and become activated, leading to the cleavage of their substrate, gasdermin D 12, 13. Cleaved gasdermin D acts as a pore forming protein triggering an inflammatory form of cell death termed pyroptosis 14. In addition, potassium efflux through gasdermin D pores activates NLRP3, leading to caspase‐1 activation and subsequent release of IL‐1β and IL‐18 11, 15, 16, 17. In contrast to the TLR4 pathway, which has been well characterized and involves multiple proteins upstream and downstream of TLR4 18, the description of the core complex involved in LPS cytosolic sensing is currently limited to two proteins: caspase‐4 (and its homologues) and gasdermin D 12. Due to the relatively recent identification of this immune complex, it remains unclear whether additional factors regulate cytosolic LPS sensing by the non‐canonical inflammasome 19. Further, most studies characterizing the non‐canonical inflammasome have employed mouse models, which do not fully recapitulate inflammasome activation in humans. In mice, cytoplasmic LPS sensing occurs by caspase‐11, which differs from its human homologue, caspase‐4, in several ways 20, 21, 22, 23, 24, 25. First, caspase‐11 is highly inducible following engagement of the TLRs/TRIF pathways and/or IFN signaling 9, 10, 26, 27. In contrast, in human monocytes and macrophages, caspase‐4 is constitutively expressed 28, 29. In addition, we recently demonstrated that caspase‐4 detects under‐acylated LPS 30 while murine macrophages did not respond to cytoplasmic delivery of under‐acylated LPS 31. The tetra‐acylated LPS from Francisella tularensis, the agent of tularemia, is thus detected in human macrophages but does not trigger any inflammasome response in murine macrophages 30.
Here, we hypothesized that factors controlling cytosolic LPS‐mediated pyroptosis remained to be identified in human monocytes. Using electroporation of Francisella novicida LPS and a CRISPR/cas9 genome‐wide screen in U937 cells, we identified interferon regulatory factor 2 (IRF2) as one of the three human genes (together with CASP4 and GSDMD) required for LPS‐mediated pyroptosis in human cells. We further showed that under steady‐state conditions, IRF2 drives sensing of cytosolic LPS by directly controlling caspase‐4 transcript levels; however, in the presence of IFN‐γ, IRF2 cooperates with IRF1 to control the inflammasome responses to cytosolic LPS and Gram‐negative bacteria. Our results thus provide insights into the regulation of caspase‐4 in human cells at both steady state and during inflammation.
Results
A genome‐wide screen identifies IRF2, gasdermin D, and caspase‐4 as the three main players in LPS sensing
Prior to this report, two genome‐wide CRISPR/cas9 screens have been performed to identify proteins involved in the cytosolic sensing of LPS. Both have been conducted in murine macrophage cell lines using Escherichia coli LPS 12, 32. To identify human‐specific factors that may contribute to cytosolic detection of tetra‐acylated LPS, we performed a genome‐wide screen in U937 cells transfected with F. novicida LPS. A library of 76,441 sgRNA, in which each gene was targeted by four independent sgRNA 33, was introduced by transduction in U937 cells expressing spCas9 30. The obtained cell library was subsequently electroporated with 10 μg of F. novicida LPS (a condition leading to approximately 40% cell death at 3 h post‐transfection 30). The surviving cells were then amplified over 4 days before going through another round of LPS electroporation. This cycle was repeated three times for a total of four electroporations (Fig 1A). SgRNA enrichment factors in the LPS‐electroporated cells compared to the mock‐electroporated cells were determined by next‐generation sequencing to identify genes which knock‐out impaired cell death/promoted cell survival following LPS electroporation. Strikingly, when focusing on the 10 most enriched sgRNAs, three genes—IRF2, CASP4, and GSDMD—were represented by several independent sgRNA (Fig 1B). Statistical analysis taking into account the score of the four independent sgRNAs strengthened the prominent enrichment of these three genes (Fig 1C). Fifteen other genes were scored as significant although the corresponding adjusted P‐values were at least 108‐fold greater than the adjusted P‐values of IRF2, CASP4, and GSDMD (Table EV1). Overall, this genome‐wide screen suggests that IRF2 is a major player of F. novicida LPS cytosolic sensing in human cells.
Figure 1. A genome‐wide CRISPR/Cas9 screen identifies IRF2, gasdermin D, and caspase‐4 as the three main players in LPS sensing.
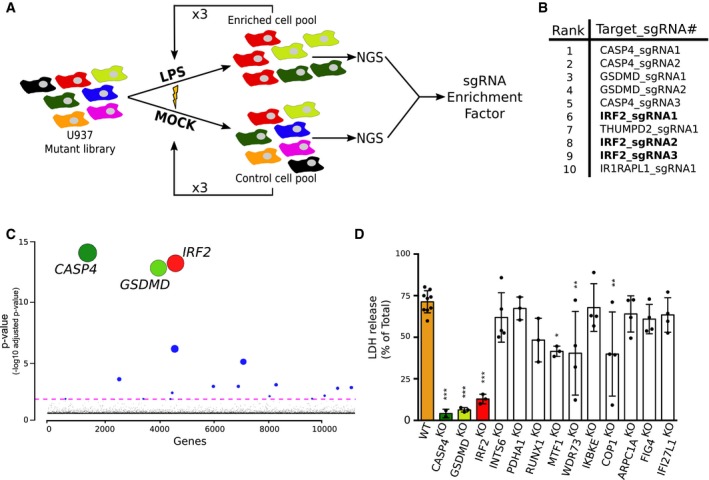
- A genome‐wide CRISPR/Cas9 screen based on LPS electroporation was performed in U937 cells. Four successive electroporation rounds were performed. DNA was extracted from surviving cells, and next‐generation sequencing (NGS) was performed to calculate the sgRNA enrichment factor.
- The 10 most enriched sgRNA in the LPS‐electroporated samples are shown.
- Graphical representation of the screen results with each gene identified in the output libraries (10,750) represented on the x‐axis and the corresponding adjusted P‐value on the y‐axis. Statistical analysis was performed using the Wald test. The colored line represents the 0.05 P‐value threshold. The size of the circle is inversely proportional to the P‐value.
- U937 cell lines were knock‐out using CRISPR/Cas9, and cell death was quantified by LDH release assay 4 h after LPS electroporation. Each dot represents the average of three technical LDH replicates; means and SD of 3–9 independent experiments are shown. One‐way ANOVA with Dunnet's multiple comparisons test was performed. Adjusted P‐value is shown (from left to right ***P < 0.001, ***P < 0.001, ***P < 0.001, P = 0.84, P = 1, P = 0.092, *P = 0.012, **P = 0.0026, P = 1, **P = 0.0022, P = 0.98, P = 0.82, P = 0.97).
To assess the role of all eighteen genes found to be significantly associated with cytosolic LPS sensing, we created stable cell lines using CRISPR/Cas9 and 2 sgRNA targeting each gene in U937 cells. Two of the identified genes (PACS1 and ZNF699) did not pass the RNA‐seq filtering step, indicating they are either not expressed, or poorly expressed in U937 cells (see Materials and Methods) (Fig EV1); therefore, they were not targeted. Following CRISPR/Cas9‐mediated editing, three other candidate genes were excluded from further analyses due to either reduced growth rate of the mutated cell line (TRIM28), inefficient mutation (DET1), or a strong bias in insertions/deletions not causing a frameshift (TFAP4) (Appendix Table S1). Out of the 13 cell lines with validated open‐reading frame disruptions (Appendix Table S1), the cell lines mutated for IRF2 (Fig EV2A), CASP4 30, and GSDMD 30 demonstrated a drastic reduction in LDH release following LPS electroporation (Fig 1D). To further corroborate these results, we generated three independent IRF2 knock‐out (KO) cell lines and again observed a strong reduction in cell death upon LPS electroporation (Fig EV2A and B). Of note, since we observed that IRF2 was required for LDH release in response to both E. coli and F. novicida LPS (Fig EV2C), we used E. coli LPS for subsequent experiments.
Figure EV1. 16 out of the 18 genes significantly enriched in the genome‐wide CRISPR/Cas9 screen are expressed above the RNA‐seq filtering threshold in U937.
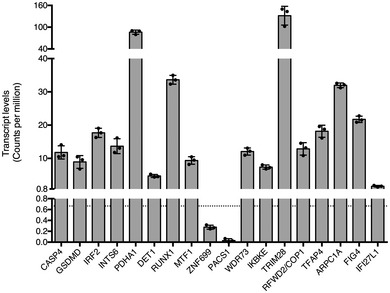
Each dot represents the transcript level of a biological U937 replicate as quantified by RNA‐seq; the bar represents the mean ± SD of a biological triplicate. The filtering threshold is shown as a horizontal dotted line and corresponds to absolute transcript counts below 30 in all the libraries.Source data are available online for this figure.
Figure EV2. IRF2 is required to promote F. novicida and E. coli LPS‐mediated cell death.
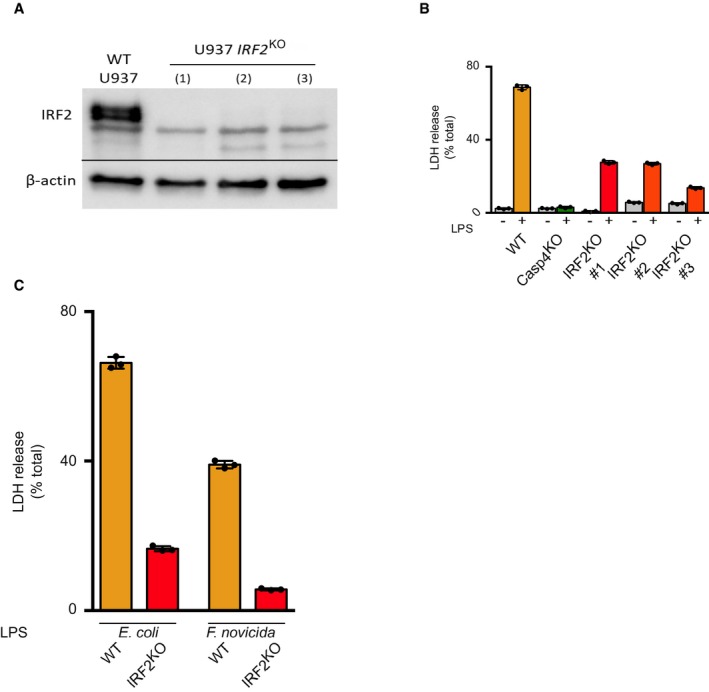
- IRF2 and β‐actin protein levels in WT and three independent IRF2 KO U937 cell lines (#1–3) were assessed by Western blotting analysis.
- LDH release was quantified from three independent IRF2 KO cell lines 4 h after E. coli LPS electroporation. Each dot represents a technical LDH replicate; the bar represents the mean ± SD of 3 LDH replicates.
- LDH release was quantified 4 h after electroporation with LPS for E. coli or F. novicida (5 μg for 4 × 105 cells). Each dot represents a technical LDH replicate; the bar represents the mean ± SD of 3 LDH replicates.
IRF2 is specifically required for caspase‐4‐mediated cell death
To confirm the role of IRF2 in mediating pyroptosis following LPS electroporation, we first reconstituted physiological IRF2 expression in IRF2 KO cells (Fig 2A). Importantly, ectopic expression of IRF2 fully restored pyroptosis after LPS electroporation in IRF2 KO cells (Fig 2B and C), demonstrating that IRF2 controls caspase‐4‐mediated pyroptosis.
Figure 2. IRF2 is specifically required for caspase‐4‐mediated cell death.
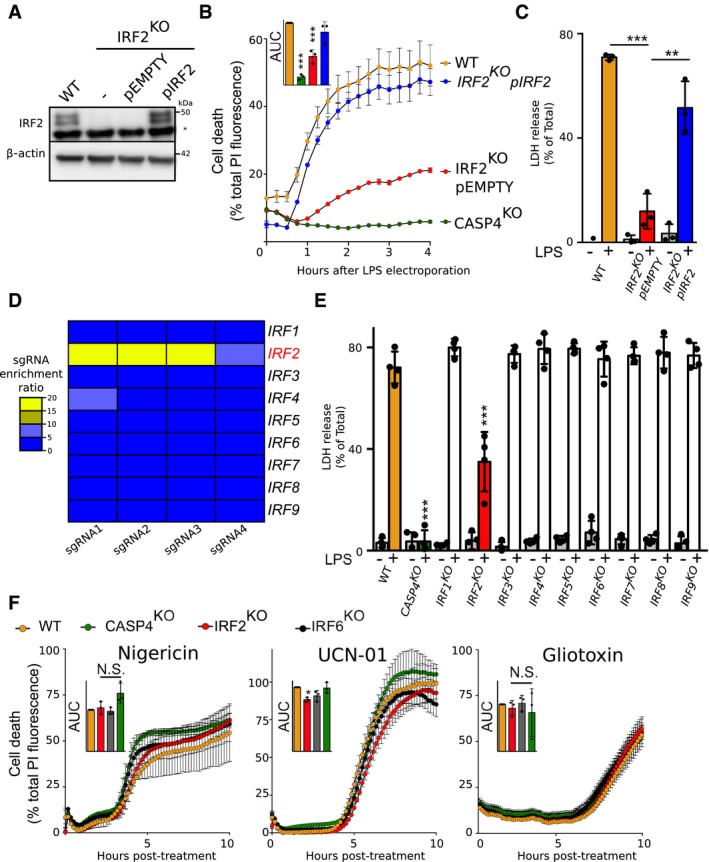
- IRF2 and β‐actin protein levels were assessed in the lysate of the indicated U937 cells by Western blotting analysis. A non‐specific band (*) is observed in the IRF2 Western blot. One experiment representative of three experiments is shown.
- Cell death induced by LPS electroporation or the indicated treatment was quantified in real time by measuring propidium iodide (PI) incorporation/fluorescence every 15 min. Cell death was normalized using untreated and TX‐100‐treated cells. The kinetics of one representative experiment and the areas under the curve (AUC) (normalized to the WT AUC) of three independent experiments are shown. Each point represents the mean of a biological triplicate of one experiment; the bar represents the mean ± SD of three independent experiments.
- Cell death was measured by LDH release assay 4 h after LPS electroporation. Each dot corresponds to the LDH triplicate of one experiment; the bar represents the mean ± SD of four independent experiments.
- Heat map representation of the enrichment factor for the 4 sgRNA targeting each IRF in the genome‐wide screen.
- Cell death was measured by LDH release assay 4 h after LPS electroporation. Each dot corresponds to the LDH triplicate of one experiment; the bar represents the mean ± SD of three independent experiments.
- Cell death was quantified by measuring PI incorporation/fluorescence every 15 min after treatment with nigericin, UCN‐01, or gliotoxin. The kinetics of one representative experiment and the areas under the curve (AUC) (normalized to the WT AUC) of three independent experiments are shown. Each point represents the mean of a biological triplicate of one experiment; the bar represents the mean ± SD of three independent experiments.
We next sought to determine whether other IRFs could be involved in caspase‐4‐mediated pyroptosis. While several IRFs are involved in the regulation of various inflammasomes in mice 34, 35, we did not observe a significant enrichment for any IRFs besides IRF2 in our CRISPR/Cas9 screen (Fig 2D). Nine IRFs are present in humans. To exclude a possible role for other IRFs in caspase‐4‐mediated pyroptosis, we generated individual IRF mutated cell lines. Importantly, only IRF2 mutations resulted in a significant reduction in LDH release upon LPS transfection (Fig 2E). This result suggests that IRF2 is specifically required among IRF family members to regulate caspase‐4‐dependent pyroptosis.
To assess whether IRF2 could be involved in other cell death pathways, we triggered necrosis using nigericin 36, 37 and apoptosis using gliotoxin 38 or a staurosporine analogue, UCN‐01 39. Of note, in the absence of phorbol 12‐myristate 13‐acetate (PMA)‐mediated differentiation and LPS priming, we found nigericin‐mediated necrosis in U937 cells to be independent of caspase‐1 and gasdermin D (Appendix Fig S1). We monitored U937 cell death kinetics in real time using propidium iodide incorporation (Fig 2F). This technique confirmed the strong pyroptosis defect of IRF2 KO cells in response to LPS transfection (Fig 2B). However, we could not observe substantial defects in apoptosis or necrosis kinetics (Fig 2F). Altogether, these experiments demonstrate that, among IRF family members, IRF2 specifically controls pyroptosis in response to cytosolic LPS in U937 monocytes.
IRF2 regulates Caspase‐4 level
IRF2 is a transcription factor described to act as transcriptional activator and repressor 40, 41. To understand the mechanisms by which IRF2 controls pyroptosis, we performed transcriptome analysis of wild‐type (WT) and IRF2 KO U937 cells, as well as IRF2 KO U937 cells reconstituted with IRF2 (Fig 3A–C). Twenty‐four genes (Table EV2) were significantly down‐regulated (fold change < 0.5) in IRF2‐deficient cells compared to WT cells and significantly up‐regulated (fold change > 2) upon reconstitution of IRF2 expression (Fig 3B). Interestingly, expression of three genes (CASP1, CASP4, and CARD16) from the inflammatory caspase locus 42 was positively regulated by IRF2 (Fig 3A and C). We did not observe any regulation of GSDMD by IRF2, either at the transcript level (Fig 3C) or at the protein level (Fig EV3A). To determine whether IRF2 directly regulated expression of genes in the inflammatory caspase locus, we analyzed IRF2 chromatin immunoprecipitation coupled to high‐throughput sequencing (ChIP‐seq) in primary human monocytes 43. We identified peaks of IRF2 binding on the promoters of both CASP4 and CASP1 genes (Fig 3D), suggesting that IRF2 directly regulates CASP1 and CASP4 expression level.
Figure 3. IRF2 regulates CASP4 transcript levels.
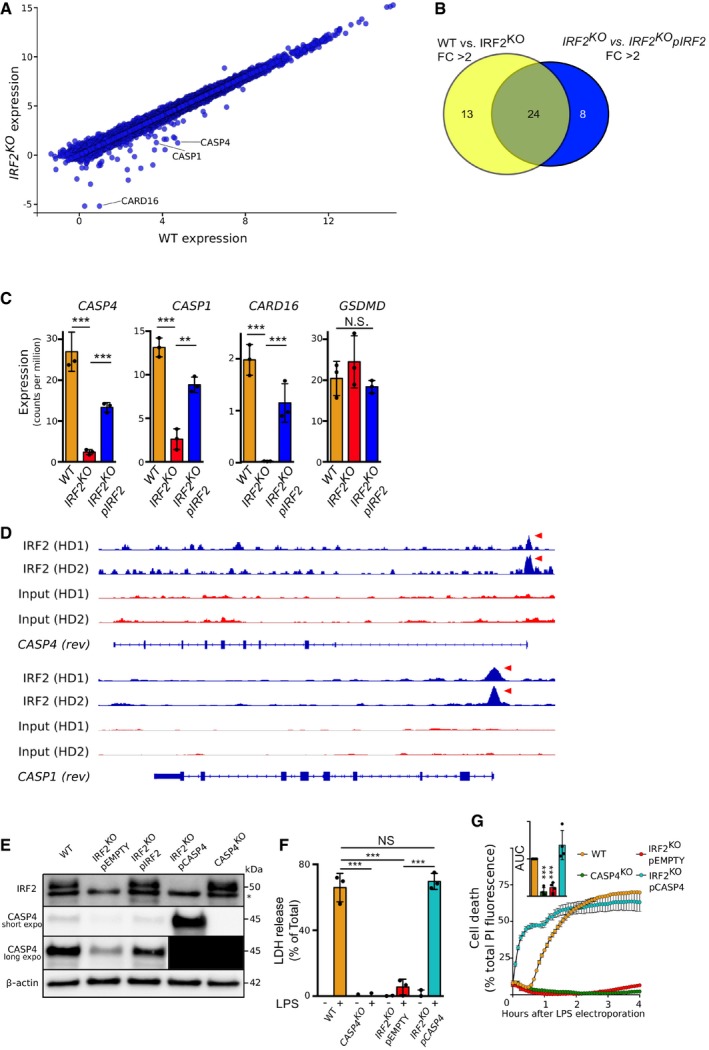
-
AmRNA transcript levels in U937 cells WT vs. IRF2KO cells were quantified by RNA‐seq.
-
BVenn diagram showing the number of genes with a transcript level fold change (FC) greater than 2 in U937 cells WT vs. IRF2KO cells (yellow) and greater than 2 in IRF2KO vs. IRF2KO pIRF2 (Blue).
-
CCASP4, CASP1, CARD16, and GSDMD transcript levels were quantified by RNA‐seq in the indicated U937 cell lines.
-
DChromatin immunoprecipitation sequencing (ChIP‐seq) in primary human monocytes from two healthy donors (HD) for IRF2 binding onto CASP1 and CASP4 promoters. Red arrows indicate promoter region with IRF2 binding.
-
EIRF2 and CASP4 protein levels were assessed by Western blotting in the indicated cell lines. The asterisk indicates a non‐specific band. For the long exposure of the CASP4 Western blot, the 2 left lanes were covered during revelation to prevent signal bleed through and were blacked out for representation purposes.
-
F, GCell death was quantified by (F) LDH release assay 4 h after LPS electroporation or (G) by measuring propidium iodide (PI) incorporation/fluorescence every 5 min. Cell death was normalized using untreated and TX‐100‐treated cells. The kinetics of one representative experiment and the areas under the curve (AUC) (normalized to the WT AUC) of four independent experiments are shown. Each point represents the mean of a biological triplicate of one experiment; the bar represents the mean ± SD of four independent experiments.
Figure EV3. IRF2 regulates GSDMD cleavage and Casp1 level in U937 cells.
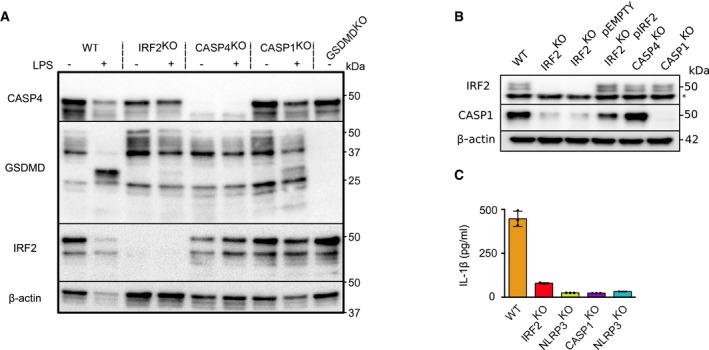
- Caspase‐4, gasdermin D, IRF2, and β‐actin protein levels in the indicated U937 cell lines were assessed by Western blotting analysis at 1 h post‐mock (−) or LPS (+) electroporation. Processing of gasdermin D (31 kDa band) is clearly observed in WT U937 cells but is barely detectable in IRF2KO cells. One experiment representative of three independent experiments is shown.
- IRF2, caspase‐1, and β‐actin protein levels in the indicated U937 cell lines were assessed by Western blotting analysis at steady state. The asterisk indicates a non‐specific band. One experiment representative of two independent experiments is shown.
- IL‐1β level in the supernatant of the indicated PMA‐differentiated U937 cell lines was quantified by ELISA at 2 h post‐nigericin treatment. Each dot represents the value of a biological replicate; the bar represents the mean ± SD of 3 biological replicates from one experiment representative of two independent experiments.
Source data are available online for this figure.
The strong and specific regulation of CASP4 mRNA levels by IRF2 (Fig 3C) suggested that IRF2 might control pyroptosis through the control of CASP4 expression. We thus assessed caspase‐4 protein levels in WT, IRF2 KO, and pIRF2‐reconstituted cell lines by Western blotting. In line with the transcriptomic data, we observed a strong decrease in caspase‐4 (Fig 3E) and caspase‐1 (Fig EV3B) protein levels in IRF2 KO U937 cells, which could be corrected upon IRF2 reconstitution. As expected, the reduction in caspase‐1 levels was associated with decreased IL‐1β release upon activation of the NLRP3 inflammasome with LPS + nigericin (Fig EV3C).
Because of the direct link between IRF2 activity and caspase‐4 expression, we next tested whether expressing CASP4 under a constitutive promoter (Fig 3E–G) could restore pyroptosis in IRF2 KO cells. Indeed, constitutive expression of CASP4 led to strong LDH release upon LPS transfection in IRF2 KO cells (Fig 3F). Interestingly, overexpression of CASP4 in IRF2 KO cells decreased the delay between LPS electroporation and membrane permeabilization as visualized by PI fluorescence, indicating that caspase‐4 levels control the cell death response kinetics (Fig 3G). Altogether, these results demonstrate that IRF2 positively regulates caspase‐4 levels in U937 monocytes, which is required for these cells to trigger pyroptosis in response to LPS electroporation.
IFN‐γ treatment induces IRF1 to cooperate with IRF2 to regulate CASP4 transcript level and LPS‐mediated pyroptosis
Inflammasome activation can be modulated by the surrounding inflammatory milieu. We therefore assessed the role of IRF2 in LPS‐induced pyroptosis in the presence of IFN‐γ, a prominent anti‐bacterial cytokine 44. Surprisingly, IFN‐γ treatment restored pyroptosis in IRF2‐deficient cells to levels observed in WT cells (Fig 4A). Since numerous IRFs are IFN‐inducible and share similar binding sites 45, we hypothesized that other IRFs could compensate for the absence of IRF2 after IFN‐γ treatment. To test this hypothesis, we generated cell lines double‐knock‐out (DKO) for IRF2 and each individual IRF (Appendix Table S1). Remarkably, the IRF1/IRF2 DKO cell line was resistant to LPS electroporation‐induced pyroptosis even in the presence of IFN‐γ, while none of the other cell lines showed any substantial difference with WT cells after IFN‐γ treatment (Fig 4B).
Figure 4. IFN‐γ treatment induces IRF1 to redundantly act with IRF2 to regulate caspase‐4 levels and LPS‐mediated pyroptosis.
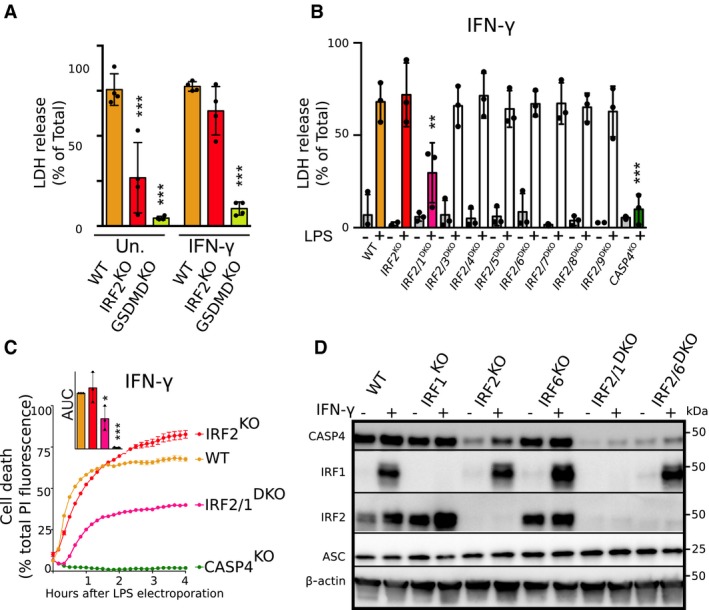
-
A–CCell death was assessed (A, B) by measuring LDH release 4 h after LPS electroporation or (C) by measuring propidium iodide (PI) incorporation/fluorescence every 10 min in the indicated U937 cell lines primed (A‐as indicated, B, C‐all samples) or not with IFN‐γ. (C) Cell death was normalized using untreated and TX‐100‐treated cells. One real‐time cell death experiment (mean and SD of triplicate) representative of three independent experiments is shown. The areas under the curve (AUC) (normalized to the WT AUC) of three independent experiments are shown. Each point represents the mean of a triplicate of one experiment; the bar represents the mean (and SD) of the three independent experiments.
-
DCaspase‐4, IRF2, and IRF1 protein levels were assessed in the lysate of the indicated U937 cell lines primed or not with IFN‐γ. Data are representative of three independent experiments.
We further validated the redundant role of IRF1 and IRF2 in the presence of IFN‐γ by monitoring LPS‐induced cell death in real time using PI incorporation. We found that while IRF2 deficiency alone was insufficient to prevent LPS‐mediated pyroptosis after IFN‐γ treatment, double‐knock‐out cells were largely resistant to cell death (Fig 4C). By blotting for caspase‐4 protein levels in IRF2 KO and IRF1/IRF2 DKO cell lines, we observed that in the presence of IFN‐γ, expression of caspase‐4 increased in IRF2 KO cells and that this increase was dependent on IRF1 (Fig 4D). In contrast, we did not observe any substantial induction of caspase‐4 protein in the presence of either Pam3CSK4, a TLR2 agonist, or extracellular LPS (Appendix Fig S2). Although we cannot exclude that other IRF‐1‐induced genes 30, 34, 46, 47 may participate in restoring the sensitivity of IRF2‐deficient cells to electroporated LPS (see GBP1 in Appendix Fig S3), our data suggest that the dual regulation of caspase‐4 levels by IRF1 and IRF2 contributes to cytosolic LPS‐mediated pyroptosis in the presence of IFN‐γ.
IRF2 controls, in a partially redundant manner with IRF1, caspase‐4‐mediated responses during infection in macrophages
To obtain robust inflammasome activation following E. coli and F. novicida infection, U937 monocytes must first be differentiated into macrophages using PMA and priming with IFN‐γ 30. Since IL‐1β secretion upon cytosolic LPS delivery is mediated by the NLRP3 inflammasome downstream of the caspase‐4‐gasdermin D cascade 16, Pam3CSK4 was used to induce proIL‐1β and prime the NLRP3 inflammasome. In IFN‐γ‐primed, PMA‐differentiated U937 macrophages, as described above, the presence of IRF1 compensated IRF2 deficiency in regulating LDH release and IL‐1β release upon cytosolic delivery of LPS (Fig 5A and B). U937 macrophages deficient in both IRF1 and IRF2 displayed a profound decrease in LPS‐mediated cytotoxicity and IL‐1β release (Fig 5A and B).
Figure 5. IRF1 cooperates with IRF2 to regulate non‐canonical inflammasome activation in PMA‐differentiated macrophages.
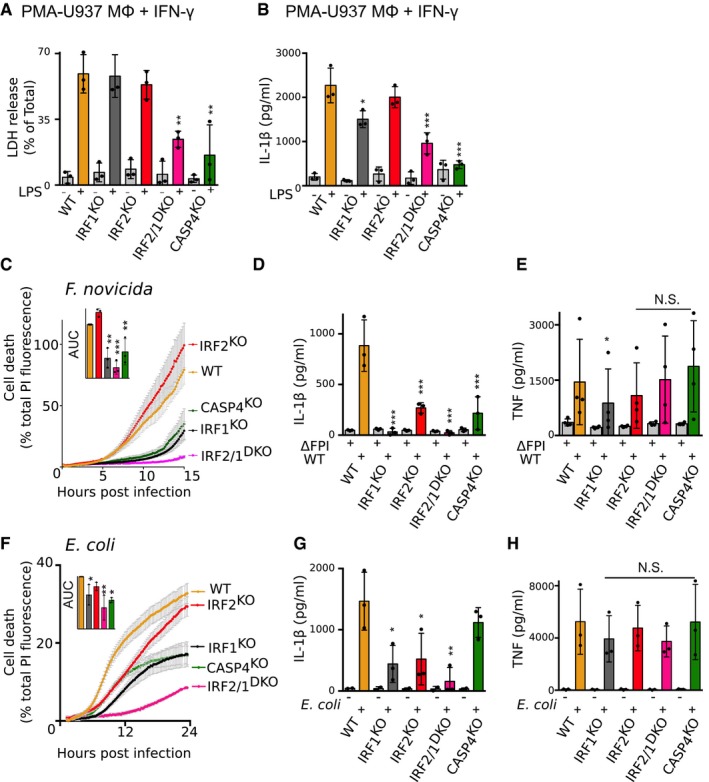
-
ACell death was quantified by LDH assay 2 h after LPS electroporation.
-
BCells were primed with Pam3CSK4. IL‐1β levels were assessed by ELISA 4 h after LPS electroporation.
-
CCell death was quantified by measuring propidium iodide (PI) incorporation/fluorescence every 5 min after infection with F. novicida or (F) E. coli. Cell death was normalized using untreated and TX‐100‐treated cells. One real‐time cell death experiment (mean and SD of triplicate) representative of three independent experiments is shown. The areas under the curve (AUC) (normalized to the WT AUC) of three independent experiments are shown. Each point represents the mean of a triplicate of one experiment; the bar represents the mean (and SD) of the three independent experiments.
-
(D–H)(D, G) IL‐1β and (E, H) TNF levels were assessed by ELISA 6 h after WT or ΔFPI mutant F. novicida (D, E) or E. coli (G, H) infection.
We then investigated the role of IRF1 and IRF2 in controlling caspase‐4‐mediated responses during infection. We used F. novicida to infect cells, as it is known to activate caspase‐4 in human macrophages 30. As previously described for murine macrophages 34, mutation of IRF1 highly delayed cell death in IFN‐γ‐primed U937 macrophages that were infected with F. novicida (Fig 5C), likely through the regulation of guanylate binding proteins (GBPs) 30, 34 (Appendix Fig S3). Mutation of IRF2 alone had no significant effect; however, in IRF1 KO cells, IRF2 deletion delayed F. novicida‐mediated cell death and fully abolished caspase‐4‐dependent cell death (Fig 5C). Of note, cell death kinetics were substantially slower in IRF1/2 DKO cells than in CASP4 KO cells suggesting that IRF1 and IRF2 control both non‐canonical and canonical inflammasome‐mediated cell death during F. novicida infection. In addition to regulating cell death, IRF1 and IRF2 deficiency also affected cytokine release after F. novicida infection. In particular, single invalidation of either IRF1 or IRF2 deeply and significantly dampened IL‐1β release (Fig 5D) while TNF levels were largely consistent across all genotypes (Fig 5E). Similar results were observed upon E. coli infection (Fig 5F–H), although the contribution of caspase‐4 to cell death and IL‐1β release was only partial in this model. Altogether, these results demonstrate that, during infection of IFN‐γ‐primed U937 macrophages, IRF2 acts in a partially redundant manner with IRF1 to control inflammasome responses. This action likely occurs through the regulation of caspase‐4 levels, as well as other innate immune proteins (Appendix Fig S2 and S3).
IRF2 controls caspase‐4 levels in human iPSC‐derived macrophages
To validate our findings in a more relevant cellular system, we deleted IRF2 in human induced pluripotent cells (iPSC). Gene deletion was confirmed at the DNA level by sequencing (Appendix Fig S4) and at the protein level in iPSC‐derived macrophages (Fig 6A). Corroborating observations made in U937 cells, deletion of IRF2 in iPSC‐derived macrophages led to a strong reduction in caspase‐4 protein levels (Fig 6A). At odds with the results obtained in U937 cells, we did not observe any impact of IRF2 deletion on caspase‐1 protein levels in iPSC‐derived macrophages, possibly reflecting interindividual variability. In line with the role of IRF2 as a transcription factor, CASP4 transcript levels were also strongly and significantly reduced in the two IRF2 knock‐out iPSC‐derived macrophage clones. NLRP3, ASC, and CASP1 did not display any reduction in their transcript levels (Fig 6B). Altogether, these results demonstrate that in human cells, IRF2 is a key regulator of caspase‐4 levels.
Figure 6. IRF2 controls caspase‐4 levels in human induced pluripotent stem cell (iPSC)‐derived macrophages.
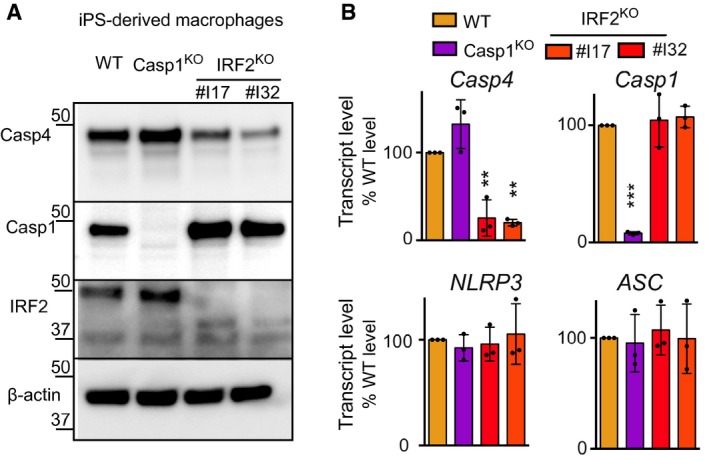
-
A, BMacrophages were derived from human iPSC clones knock‐out for the indicated genes. Two clones were selected for IRF2 KO cells. (A) Caspase‐4, caspase‐1, IRF2, and β‐actin protein levels were assessed by Western blotting analysis. One experiment representative of two independent experiments is shown. (B) The indicated mRNA levels were quantified by qRT–PCR normalized to β‐actin mRNA levels and expressed as fold change compared to WT levels. Each dot represents the average of a technical RT–PCR triplicate from one experiment; the bar represents the mean of three independent experiments. One‐way ANOVA with Dunnet's multiple comparisons test was performed (WT vs. IRF2 KO #I17 **P = 0.0020; WT vs. IRF2 KO #32 **P = 0.0013 for CASP4 transcript. WT vs. CASP1 KO ***P < 0.0001 for CASP1 transcript levels).
Source data are available online for this figure.
Discussion
Here, we describe an unbiased, genome‐wide screen used to identify genes involved in non‐canonical inflammasome activation in human cells. We found that three genes—encoding caspase‐4, gasdermin D, and IRF2—were significantly associated with non‐canonical inflammasome activation and therefore represent the core pathway controlling cytosolic LPS‐mediated cell death in human cells. This work not only supports the role of caspase‐4 as the direct LPS receptor and effector of the non‐canonical inflammasome complex 8, but also identifies IRF2 as a novel factor regulating caspase‐4 expression levels and non‐canonical inflammasome activation in human cells. Further, this screen stresses the unusual simplicity of this signaling complex, particularly when compared to the large number of proteins involved in extracellular LPS sensing by TLR4, and in its downstream signal transduction pathway 48.
The remaining 15 significant hits identified in this screen were not investigated in the present study. These genes likely include both minor contributors and false positives. We cannot rule out the existence of other regulators that may have been missed in the screen either due to redundancy or to our experimental system. For example, U937 cells may not fully model primary monocytes in terms of expression or regulation. Furthermore, electroporation of LPS was required to trigger a robust and reproducible cell death, but likely prevented the identification of host molecules required for LPS trafficking from the extracellular environment to the cytosol, or for the extraction of lipid A from its membrane environment (e.g. outer membrane vesicles—OMVs). Indeed, we and others have previously identified the GBPs as co‐factors regulating non‐canonical inflammasome activation in response to Gram‐negative infection, OMV internalization, or LPS transfection 30, 46, 47, 49. These proteins were not identified in the screen.
Expression of caspase‐11 in mouse cells is highly inducible by multiple pro‐inflammatory stimuli including TLR ligands and IFNs 27. Conversely, caspase‐4 is constitutively expressed in human monocytes and macrophages 28, 29, 50, which correlates with the high sensitivity of humans to LPS compared to mice 6, 51. Our results demonstrate that IRF2 is required for this constitutive expression and is responsible for the sensitivity of human monocytes to LPS‐mediated pyroptosis. Accordingly, IRF2 is constitutively expressed 52 in monocytes and acts as a transcriptional activator of caspase‐4 expression. In contrast, IRF‐1 expression is strongly inducible by IFN‐γ and compensates IRF‐2 deficiency in IFN‐γ‐primed cells. This result is consistent with the ability of IRF‐1 and IRF‐2 to bind to the same recognition site 53 and suggests that in the presence of IFN‐γ, IRF1 and IRF2 ensure a robust and possibly redundant control of caspase‐4 expression. Interestingly, this regulatory mechanism is not limited to the CASP4 gene; in epithelial cells, TLR3 expression is positively regulated by IRF2 at steady state, while treatment with IFN leads to IRF1‐dependent control 41. This regulation exemplifies the recurring theme of redundancy in innate immunity 54, which is thought to contribute to the robustness of the innate immune system in humans. The redundant mechanisms controlling caspase‐4 expression in the presence of IFN‐γ likely ensure the efficiency of cytosolic LPS detection during infection. Another potentially redundancy player of this system is caspase‐5, a poorly characterized human inflammatory caspase detecting LPS in the cytosol. Yet, caspase‐5 was not expressed in our U937 experimental system—even in the presence of IFN‐γ—and we were thus unable to investigate the potential role of IRF1/2 in regulating caspase‐5 levels.
IRF2−/− mice exhibit resistance to LPS‐induced lethality 55. This resistance had been associated with a greater number of apoptotic Kupffer cells in IRF2−/− mice than in WT mice upon LPS injection. Yet, while this manuscript was under review, IRF2 was demonstrated to regulate gasdermin D level in murine macrophage through direct binding of the GSDMD promoter region 56. The study also found that IRF1 could compensate for the absence of IRF2 in controlling gasdermin D levels, similar to what we found for IRF1 regulation of caspase‐4 in human cells. Interestingly, IRF2 in that study was found to control gasdermin D but not caspase‐4 levels in the human EA.hy926 cell line 57. In contrast, we did not observe any substantial regulation of GSDMD levels by IRF1/2 in U937 cells at steady state (Fig EV3A and Appendix Fig S2). However, we observed an IFN‐γ‐mediated increase in GSDMD level that was abolished in IRF1/2DKO cells (Appendix Fig S2). This result strongly suggests a role of IRF1 (and potentially IRF2) in the regulation of human GSDMD. Further studies are needed to better characterize the regulation of GSDMD by IFN‐γ and to understand its relevance in the context of cell death and cytokine secretion. At this stage, it is unclear whether the differences observed at steady state between the different human cell lines reflect interindividual variations in the promoter regions of inflammasome genes or whether IRF2 controls different subsets of genes in different cell types. Altogether, these studies strongly demonstrate that IRF2 has a conserved role in mice and humans to control inflammasome genes.
Interestingly, IRF2 expression is differentially regulated in human individuals in response to LPS 43, suggesting that single nucleotide polymorphisms (SNPs) affecting IRF2 levels might control interindividual susceptibility to Gram‐negative bacteria and septic shock. Similarly, common variants in IRF5 modulate the antimicrobial responses of human macrophages 58, 59, indicating that the polymorphisms in the various IRF genes may be an important driver of the interindividual differences in susceptibility to bacterial infections. Recently, IRF‐1 and IRF‐8 were demonstrated to regulate Gbps and Naips expression in mice 34, 35. Collectively, these data demonstrate the emerging role of IRFs as central regulators of inflammasome accessory proteins, sensors, and effectors in mice and humans.
Materials and Methods
Genome‐wide CRISPR/cas9 screen
The Brunello library obtained from Addgene (ref# 73178) is made of 76,441 sgRNAs targeting 19,114 human genes and 1,000 non‐targeting control sgRNAs, all cloned in lentiGuide‐Puro (Addgene ref# 52963). A clonal population of U937 Cas9+ was infected with a virus library titrated in order to achieve an MOI of 0.5 and a 390‐fold coverage for each sgRNA. Starting at 3 days post‐transduction, cells were treated with puromycin at 2 mg/ml for 12 days. Four replicates of 4 × 106 cells were then electroporated with 10 μg of F. novicida LPS or mock‐electroporated using the Neon electroporator (1 pulse of 1.3 KV and 30 ms width). This operation was repeated three more times with 4 days of recovery between each electroporation. DNA was extracted using a kit from Macherey‐Nagel. sgRNA containing regions were amplified by nested PCR using the following 1st set of primers: Fwd 5′‐AATGGACTATCATATGCTTACCGTAACTTGAAAGTATTTCG‐3′, Rev 5′‐CTTTAGTTTGTATGTCTGTTGCTATTATGTCTACTATTCTTTCC‐3′ and 2nd set of primers: Fwd 5′‐PRIMER FUSION+BARCODE+TCTTGTGGAAAGGACGAAACACCG‐3′, Rev 5′‐PRIMER FUSION+TCTACTATTCTTTCCCCTGCACTGT‐3′. For the mock‐transfected samples, 56 PCRs were performed using the first set of primers (1 μg of DNA per reaction in a 50 μl volume). These samples were then pooled, and 5 μl of this product was used as a template for 24 additional PCRs (using primer set 2 in a 50 μl final volume). For the LPS‐transfected samples, 36 PCRs were performed using primer set 1 (1 μg of DNA per reaction in a 50 μl volume). Again, these samples were pooled, and 5 μl of this product was used to run 16 PCRs with primer set 2. The barcodes were used to discriminate different samples in the same run of sequencing. The primer fusion was required for performing next‐generation sequencing on the Ion Proton system (Thermo Fisher). The experimental depth of sequencing was at least 15 million sequences reads per sample. Raw data were obtained as fastq files. Low‐quality sequences and sequences with < 30 reads were removed, and primers were trimmed. The sequences obtained were aligned against the reference library of sgRNA. sgRNA enrichment factor was determined by the ratio between the number of reads for a given sgRNA in LPS‐treated and control sample. Statistical analysis was performed using the R DESeq2 package. First, counts were normalized to account for differences in sequencing depth between sgRNAs; that is, each sgRNA raw count was divided by its own size factor. Then, the normalized data were tested for differential expression between the LPS‐electroporated and the mock samples using Wald significance tests. The sgRNA enrichment factor was calculated by dividing the number of normalized reads for sgRNA in LPS treatment sample by the number of normalized reads for sgRNA in mock sample.
Cell culture
The human myeloid cell line U937 (obtained from CelluloNet) was grown in RPMI 1640 media with glutaMAX‐I (#61870‐010) supplemented with 10% (vol/vol) FBS, 100 IU/ml penicillin, and 100 μg/ml streptomycin (all from Thermo Fisher Scientific). When indicated, U937 cells were treated with 100 ng/ml of phorbol 12‐myristate 13‐acetate (PMA; InvivoGen) for 48 h to trigger differentiation into macrophages.
iPSC generation and differentiation
NCRM‐1 iPSC (RRID:CVCL_1E71), available at the NIMH Repository and Genomics Resource, were matured and differentiated into macrophages (iPSC‐MC) in the absence of feeding layers and in serum‐free conditions as previously described 60. Briefly, cells were cultured in colonies in StemFlexTM medium (Thermo Fisher Scientific) and mechanically dissociated using TrypLE (Thermo Fisher Scientific) to obtain single cell suspensions. For the generation of embryoid bodies (EBs), 104 cells were spun down and cultured in the presence of 1 mM Rock inhibitor (Y27632; Calbiochem), 50 ng/ml BMP‐4 (Peprotech EC Ltd.), 20 ng/ml SCF (Miltenyi Biotec Ltd), and 50 ng/ml VEGF. After 4 days, 70 EBs were seeded in 10‐cm2 extra‐adherent CellBIND plates (Corning) and cultured in X‐VIVOTM 15 serum‐free hematopoietic cell medium (Lonza) in the presence of human 100 ng/μl IL‐3 (R&D) and 100 ng/μl M‐CSF (Thermo Fisher Scientific) to induce myeloid differentiation. Cells were harvested every 5 days and further maturated in X‐VIVOTM 15 media in the presence of M‐CSF only during 4 days for terminal macrophage differentiation.
CRISPR/Cas9
All U937 knock‐out cell lines generated by CRISPR in this study were generated in a Cas9‐expressing U937 clone obtained by transduction with the plasmid LentiCas9‐Blast (from Feng Zhang; Addgene plasmid # 52962) followed by blasticidin selection and clonal isolation using the limit dilution method. A clone strongly expressing Cas9 was selected based on Western blot analysis using anti‐Cas9 antibody (Millipore; # MAC133; 1:1,000 dilution). sgRNAs for all the genes targeted (Appendix Table S2) were cloned into the pKLV‐U6gRNA(BbsI)‐PGKpuro2ABFP vector (from Kosuke Yusa; Addgene plasmid # 50946). For each gene, two sgRNAs were used. Lentiviral particles were produced in 293T cells using pMD2.G and psPAX2 (from Didier Trono, Addgene plasmids #12259 and #12260), and pKLV‐U6gRNA(BbsI)‐PGKpuro2ABFP. U937 Cas9+ cells were transduced by spinoculation. A polyclonal population was selected using puromycin treatment. Gene invalidation was verified by Western blot analysis, visualization of DNA fragment deletion on agarose gel, or using TIDE software. All knock‐out validation details are available in Appendix Table S1.
For generation of IRF2 NCRM‐1 knock‐out cells, cells were co‐transfected with two pL‐CRISPR.SFFV.GFP (from the laboratory Benjamin Ebert; Addgene plasmid #57827) plasmids containing two different sgRNAs targeting exon 7 or 8 of IRF2. For generation of caspase‐1 NCRM‐1 knock‐out cells, cells were co‐transfected with two px330‐P2A‐GFP plasmids containing two different sgRNAs targeting exon 1 of CASP1. Transfection was performed using Amaxa™ Human Stem Cell Nucleofector™ Kit 2 (Lonza). GFP‐positive cells were sorted and grown at low density in Matrigel‐coated plates in the presence of 1 mM Rock inhibitor (Y27362; Calbiochem). Single‐clone colonies started forming after 4 days of culture, and when fully grown were amplified and validated by sequencing. Knock‐outs were then further validated at the protein level in terminally differentiated macrophages by Western blot.
Complemented cell lines
CASP4 cDNA was obtained from J.Yuan and cloned into a pAIP lentiviral vector (from A. Cimarelli) as previously described 30. An IRF2 coding sequence with mutated PAM sites was synthesized (Thermo Fisher) and cloned into pAIP using NotI and XhoI restriction enzymes. Each of these constructs was packaged into a lentivirus, and U937 IRF2 KO cells were transduced as described above. Seventy‐two hours post‐transduction, stably transduced cells were selected with 2 μg/ml Puromycin (Sigma‐Aldrich) for 10 days.
RNA‐seq
RNA was extracted from U937 cell lines, and libraries were prepared using the SENSE mRNA‐Seq Library Prep Kit V2 (Lexogen) and sequenced on an Illumina NextSeq500 (75 bp in single end) with at least 20 millions reads per sample. Obtained sequences were trimmed, filtered, and aligned using the Tophat package. Genes with an absolute transcript count inferior to 30 reads in all the libraries were filtered out. Differential expression analysis was performed after normalization using limma‐trend (package in R).
ChIP‐seq
IRF2 ChIP‐Seq raw data were kindly provided by Benjamin Fairfax 43. Adaptor sequences, low‐quality nucleotides with a Phred score below 20, and reads with lengths shorter than 25 nucleotides were removed with the Trimmomatic tool 61. Trimmed reads were aligned to the GrCH37/Hg18 human genome build with the Bowtie2 tool by using default parameters 62. Reads mapped with low mapping quality (MAPQ < 10) were discarded with samtools 63. Duplicated reads were removed with MarkDuplicates from picard tools (https://broadinstitute.github.io/picard/). Narrow peak calling was performed with MACS2 64. BigWig files were created from MACS2 bedGraph with BEDtools 65. Results were visualized with Integrative Genomic Viewer (IGV) 66.
Delivery of intracellular LPS
U937 was electroporated with LPS using the Neon® Transfection System (Thermo Fisher Scientific, # MPK10096) according to the manufacturer's protocol (1 pulse of 1.3 kV and 30 ms width). Western blot analyses were performed at 1 h post‐electroporation. Cell death assays were performed at 4 h post‐electroporation. LPS from E. coli O111:B4 (InvivoGen) was used at 5 μg for 5 × 105 cells. LPS from F. novicida was kindly provided by Wayne Conlan (National Research Council Canada).
Cytokine release measurement and cell death assay
ELISA kits were from R&D Systems (#DY210, DY201). Quantification of cell death was performed by monitoring LDH release in the supernatant using the Cytotoxicity detection kit (Roche, #11644793001). Cell death was also quantified by monitoring propidium iodide (PI) incorporation. Briefly, 5 × 104 U937 cells were seeded in black 96 flat‐bottom well plates with CO2‐independent medium (Thermo Fisher, #18045054) supplemented with 10% (vol/vol) FBS. PI was added at a final concentration of 5 μg/ml. Fluorescence was measured on a microplate fluorimeter (Tecan). Cell death was normalized by removing PI fluorescence of untreated cells and using the PI fluorescence value of 1% Triton X‐100‐treated cells as a measure of total cell death (average of the last 4 kinetics points).
Immunoblot analysis
Cells were lysed with RIPA buffer containing Mini EDTA‐free Protease Inhibitor Mixture (Roche, #11836170001). Proteins were separated by SDS/PAGE on precast 4–15% acrylamide gels (Bio‐Rad, #4561084) and transferred to TransBlot® Turbo™ Midi‐size PVDF membranes (Bio‐Rad, #1704157). Antibodies used were mouse monoclonal anti‐caspase‐4 (MBL; clone 4B9; #M029‐3; 1:1,000 dilution), rabbit monoclonal anti‐caspase‐1 (Cell Signaling; #3866; 1:1,000 dilution), rabbit monoclonal anti‐IRF1 (Cell Signaling; #8478; 1:1,000 dilution), mouse monoclonal anti‐IRF2 (Santa Cruz; #sc‐374327; dilution 1:1,000 or sc‐101069; dilution 1:1,000), anti‐gasdermin D (Cell Signaling, #96458; dilution 1:1,000). As loading control, cell lysates were reprobed with a mouse monoclonal antibody anti‐β‐actin (clone C4, Millipore; 1:5,000 dilution). Full Western blot images are presented in the raw data source files.
U937 stimulation
To induce apoptotic cell death, cells were treated with either UCN‐01, a staurosporine derivative (Sigma; #U6508) at 12.5 mM, or gliotoxin (Enzolife; #BML‐PI129) at 500 ng/ml. For necrosis induction, unprimed U937 cells were treated with Nigericin (Invivogen; #tlrl‐nig) at 50 μg/ml. When indicated, U937 cells were primed with Pam3CSK4 (Invivogen) at 1 μg/ml for 3 h, or with IFN‐γ (Immunotools) at 1000 μ/ml for 16 h before LPS electroporation. For NLRP3 activation, PMA‐differentiated macrophages were primed with Pam3CSK4 (Invivogen) at 1 μg/ml for 16 h, followed by 2 h of Nigericin at 30 μg/ml.
Bacterial strains and macrophage infection
F. novicida strain Utah (U112) was grown in tryptic soy broth (Conda) supplemented with 0.1% (w/v) cysteine. E. coli J53 strain was grown in Luria broth (Conda). U937 cells were plated at a concentration of 5 × 104 cells in 96‐well plates. Prior to infection, U937 cells were treated 48 h with PMA for differentiation into macrophages. Additionally, U937 were stimulated with 1,000 U/ml IFN‐γ (Immunotools). Bacteria were added onto macrophages at a multiplicity of infection (MOI) of 100:1 (F. novicida) or 10:1 (E. coli). The plates were centrifuged for 15 min at 1,000 × g and incubated for 1 h at 37 °C. Next, cells were washed and fresh medium with 10 μg/ml gentamicin (Thermo Fisher Scientific) was added before incubation for the indicated time.
Real‐time qRT–PCR
Total RNA was extracted with TRI Reagent® (Sigma‐Aldrich) and reverse‐transcribed with random primer combined with the ImProm‐II™ Reverse Transcription System (Promega). Quantitative real‐time PCR was performed using FastStart Universal SYBR Green Master Mix (Roche) using an Applied StepOnePlus™ Real‐Time PCR System (Thermo Fisher Scientific). Gene‐specific transcript levels were normalized against human ACTB transcript levels. Primer sequences are indicated in Appendix Table S3.
Statistical analysis
Prism 6 (GraphPad) and R (The R foundation) were used for statistical analysis of data. Statistical analysis applied to the results from the CRISPR/Cas9 screen is described above. One‐way ANOVA with Dunnet's test for multiple comparison was used to compare the values of WT cells to the values of the various knock‐out cells. When comparisons were made with different control groups (e.g. WT untreated or WT treated with IFN‐γ), Sidak's correction was applied.
Author contributions
SB and TH designed the research. SB, GG, MPG, BL, MC, KK, and AM performed the experiments. KK, OA, AC. J‐BC, CFB, SH performed the bioinformatics analyses; SH and BG generated the NGS and RNA‐seq data. MPG and RR designed and generated the iPSC data. SB and TH wrote the manuscript with input and revisions from all authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
We thank B. Fairfax (University of Oxford, U.K.) for providing the IRF2 ChIP‐seq data; W. Conlan (NRC, Ottawa, Canada) for F. novicida LPS; J. Yuan (Harvard Medical School, USA), B. Py (CIRI, Lyon, France), A. Cimarelli (CIRI, Lyon, France) for providing cDNA and plasmids; and J. Rajwani (University of Calgary) for careful reading of the manuscript. We acknowledge the contribution of SFR Biosciences (UMS3444/CNRS, US8/Inserm, ENS de Lyon, UCBL) cytometry and vectorology facilities. This work was supported by ERC starting grant 311542, an ANR grant (ANR‐16‐CE15‐0011) and an intramural CIRI grant. This work was performed within the framework of the LABEX ECOFECT (ANR‐11‐LABX‐0048) of Université de Lyon, within the program “Investissements d'Avenir” (ANR‐11‐IDEX‐0007) operated by the French National Research Agency (ANR). Experiments done in the laboratory of Dr. Ricci have been supported by the INGESTEM French National infrastructure. GG is supported by a fellowship from the Fondation pour la Recherche Médicale (SPF201809006927).
EMBO Reports (2019) 20: e48235
See also: SJ Thygesen & KJ Stacey (September 2019)
Data availability
The authors declare that the data supporting the findings of this study are available within the paper and its Appendix. The datasets produced in this study are available in the following database (RNA‐seq data: Gene Expression Omnibus GSE132178; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE132178).
References
- 1. Fleischmann C, Scherag A, Adhikari NKJ, Hartog CS, Tsaganos T, Schlattmann P, Angus DC, Reinhart K (2016) Assessment of global incidence and mortality of hospital‐treated sepsis. Current estimates and limitations. Am J Respir Crit Care Med 193: 259–272 [DOI] [PubMed] [Google Scholar]
- 2. Singer M, Deutschman CS, Seymour CW, Shankar‐Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche J‐D, Coopersmith CM et al (2016) The third international consensus definitions for sepsis and septic shock (Sepsis‐3). JAMA 315: 801–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hotchkiss RS, Monneret G, Payen D (2013) Sepsis‐induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat Rev Immunol 13: 862–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cavaillon J‐M (2017) Exotoxins and endotoxins: inducers of inflammatory cytokines. Toxicon 149: 45–53 [DOI] [PubMed] [Google Scholar]
- 5. Stinebring WR, Youngner JS (1964) Patterns of interferon appearance in mice injected with bacteria or bacterial endotoxin. Nature 204: 712 [DOI] [PubMed] [Google Scholar]
- 6. Sauter C, Wolfensberger C (1980) Interferon in human serum after injection of endotoxin. Lancet 2: 852–853 [DOI] [PubMed] [Google Scholar]
- 7. Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C et al (1998) Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 282: 2085–2088 [DOI] [PubMed] [Google Scholar]
- 8. Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, Hu L, Shao F (2014) Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514: 187–192 [DOI] [PubMed] [Google Scholar]
- 9. Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA (2013) Cytoplasmic LPS activates caspase‐11: implications in TLR4‐independent endotoxic shock. Science 341: 1250–1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi‐Takamura S, Miyake K, Zhang J, Lee WP, Muszynski A et al (2013) Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 341: 1246–1249 [DOI] [PubMed] [Google Scholar]
- 11. Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S et al (2011) Non‐canonical inflammasome activation targets caspase‐11. Nature 479: 117–121 [DOI] [PubMed] [Google Scholar]
- 12. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, Shao F (2015) Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526: 660–665 [DOI] [PubMed] [Google Scholar]
- 13. Kayagaki N, Stowe IB, Lee BL, O'Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose‐Girma M, Phung QT et al (2015) Caspase‐11 cleaves gasdermin D for non‐canonical inflammasome signalling. Nature 526: 666–671 [DOI] [PubMed] [Google Scholar]
- 14. Cookson BT, Brennan MA (2001) Pro‐inflammatory programmed cell death. Trends Microbiol 9: 113–114 [DOI] [PubMed] [Google Scholar]
- 15. Petrilli V, Papin S, Dostert C, Mayor A, Martinon F, Tschopp J (2007) Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ 14: 1583–1589 [DOI] [PubMed] [Google Scholar]
- 16. Schmid‐Burgk JL, Gaidt MM, Schmidt T, Ebert TS, Bartok E, Hornung V (2015) Caspase‐4 mediates non‐canonical activation of the NLRP3 inflammasome in human myeloid cells. Eur J Immunol 45: 2911–2917 [DOI] [PubMed] [Google Scholar]
- 17. Ruhl S, Broz P (2015) Caspase‐11 activates a canonical NLRP3 inflammasome by promoting K(+) efflux. Eur J Immunol 45: 2927–2936 [DOI] [PubMed] [Google Scholar]
- 18. Fitzgerald KA, Rowe DC, Golenbock DT (2004) Endotoxin recognition and signal transduction by the TLR4/MD2‐complex. Microbes Infect 6: 1361–1367 [DOI] [PubMed] [Google Scholar]
- 19. Broz P, Dixit VM (2016) Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol 16: 407–420 [DOI] [PubMed] [Google Scholar]
- 20. Gaidt MM, Ebert TS, Chauhan D, Schmidt T, Schmid‐Burgk JL, Rapino F, Robertson AAB, Cooper MA, Graf T, Hornung V (2016) Human monocytes engage an alternative inflammasome pathway. Immunity 44: 833–846 [DOI] [PubMed] [Google Scholar]
- 21. Stehlik C, Dorfleutner A (2007) COPs and POPs: modulators of inflammasome activity. J Immunol 179: 7993–7998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Khare S, Dorfleutner A, Bryan NB, Yun C, Radian AD, de Almeida L, Rojanasakul Y, Stehlik C (2012) An NLRP7‐containing inflammasome mediates recognition of microbial lipopeptides in human macrophages. Immunity 36: 464–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gavrilin MA, Wewers MD (2011) Francisella Recognition by Inflammasomes: differences between Mice and Men. Front Microbiol 2: 11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chavarria‐Smith J, Mitchell PS, Ho AM, Daugherty MD, Vance RE (2016) Functional and evolutionary analyses identify proteolysis as a general mechanism for NLRP1 inflammasome activation. PLoS Pathog 12: e1006052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chae JJ, Centola M, Aksentijevich I, Dutra A, Tran M, Wood G, Nagaraju K, Kingma DW, Liu PP, Kastner DL (2000) Isolation, genomic organization, and expression analysis of the mouse and rat homologs of MEFV, the gene for familial Mediterranean fever. Mamm Genome 11: 428–435 [DOI] [PubMed] [Google Scholar]
- 26. Monack DM, Hersh D, Ghori N, Bouley D, Zychlinsky A, Falkow S (2000) Salmonella exploits caspase‐1 to colonize Peyer's patches in a murine typhoid model. J Exp Med 192: 249–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rathinam VAK, Vanaja SK, Waggoner L, Sokolovska A, Becker C, Stuart LM, Leong JM, Fitzgerald KA (2012) TRIF licenses caspase‐11‐dependent NLRP3 inflammasome activation by gram‐negative bacteria. Cell 150: 606–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vigano E, Diamond CE, Spreafico R, Balachander A, Sobota RM, Mortellaro A (2015) Human caspase‐4 and caspase‐5 regulate the one‐step non‐canonical inflammasome activation in monocytes. Nat Commun 6: 8761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yuan J, Najafov A, Py BF (2016) Roles of caspases in necrotic cell death. Cell 167: 1693–1704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lagrange B, Benaoudia S, Wallet P, Magnotti F, Provost A, Michal F, Martin A, Di Lorenzo F, Py BF, Molinaro A et al (2018) Human caspase‐4 detects tetra‐acylated LPS and cytosolic Francisella and functions differently from murine caspase‐11. Nat Commun 9: 242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Aachoui Y, Leaf IA, Hagar JA, Fontana MF, Campos CG, Zak DE, Tan MH, Cotter PA, Vance RE, Aderem A et al (2013) Caspase‐11 protects against bacteria that escape the vacuole. Science 339: 975–978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Napier BA, Brubaker SW, Sweeney TE, Monette P, Rothmeier GH, Gertsvolf NA, Puschnik A, Carette JE, Khatri P, Monack DM (2016) Complement pathway amplifies caspase‐11‐dependent cell death and endotoxin‐induced sepsis severity. J Exp Med 213: 2365–2382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R et al (2016) Optimized sgRNA design to maximize activity and minimize off‐target effects of CRISPR‐Cas9. Nat Biotechnol 34: 184–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Man SM, Karki R, Malireddi RKS, Neale G, Vogel P, Yamamoto M, Lamkanfi M, Kanneganti T‐D (2015) The transcription factor IRF1 and guanylate‐binding proteins target activation of the AIM2 inflammasome by Francisella infection. Nat Immunol 16: 467–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Karki R, Lee E, Place D, Samir P, Mavuluri J, Sharma BR, Balakrishnan A, Malireddi RKS, Geiger R, Zhu Q et al (2018) IRF8 regulates transcription of naips for NLRC4 inflammasome activation. Cell 173: 920–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ojcius DM, Zychlinsky A, Zheng LM, Young JD (1991) Ionophore‐induced apoptosis: role of DNA fragmentation and calcium fluxes. Exp Cell Res 197: 43–49 [DOI] [PubMed] [Google Scholar]
- 37. Hentze H, Lin XY, Choi MSK, Porter AG (2003) Critical role for cathepsin B in mediating caspase‐1‐dependent interleukin‐18 maturation and caspase‐1‐independent necrosis triggered by the microbial toxin nigericin. Cell Death Differ 10: 956–968 [DOI] [PubMed] [Google Scholar]
- 38. Stanzani M, Orciuolo E, Lewis R, Kontoyiannis DP, Martins SL, St John LS, Komanduri KV (2005) Aspergillus fumigatus suppresses the human cellular immune response via gliotoxin‐mediated apoptosis of monocytes. Blood 105: 2258–2265 [DOI] [PubMed] [Google Scholar]
- 39. Nie C, Luo Y, Zhao X, Luo N, Tong A, Liu X, Yuan Z, Wang C, Wei Y (2014) Caspase‐9 mediates Puma activation in UCN‐01‐induced apoptosis. Cell Death Dis 5: e1495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Harada H, Fujita T, Miyamoto M, Kimura Y, Maruyama M, Furia A, Miyata T, Taniguchi T (1989) Structurally similar but functionally distinct factors, IRF‐1 and IRF‐2, bind to the same regulatory elements of IFN and IFN‐inducible genes. Cell 58: 729–739 [DOI] [PubMed] [Google Scholar]
- 41. Ren G, Cui K, Zhang Z, Zhao K (2015) Division of labor between IRF1 and IRF2 in regulating different stages of transcriptional activation in cellular antiviral activities. Cell Biosci 5: 17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Martinon F, Tschopp J (2007) Inflammatory caspases and inflammasomes: master switches of inflammation. Cell Death Differ 14: 10–22 [DOI] [PubMed] [Google Scholar]
- 43. Fairfax BP, Humburg P, Makino S, Naranbhai V, Wong D, Lau E, Jostins L, Plant K, Andrews R, McGee C et al (2014) Innate immune activity conditions the effect of regulatory variants upon monocyte gene expression. Science 343: 1246949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. MacMicking JD (2012) Interferon‐inducible effector mechanisms in cell‐autonomous immunity. Nat Rev Immunol 12: 367–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mancino A, Natoli G (2016) Specificity and function of IRF family transcription factors: insights from genomics. J Interferon Cytokine Res 36: 462–469 [DOI] [PubMed] [Google Scholar]
- 46. Pilla DM, Hagar JA, Haldar AK, Mason AK, Degrandi D, Pfeffer K, Ernst RK, Yamamoto M, Miao EA, Coers J (2014) Guanylate binding proteins promote caspase‐11‐dependent pyroptosis in response to cytoplasmic LPS. Proc Natl Acad Sci USA 111: 6046–6051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Santos JC, Dick MS, Lagrange B, Degrandi D, Pfeffer K, Yamamoto M, Meunier E, Pelczar P, Henry T, Broz P (2018) LPS targets host guanylate‐binding proteins to the bacterial outer membrane for non‐canonical inflammasome activation. EMBO J 37: e98089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rosadini CV, Kagan JC (2017) Early innate immune responses to bacterial LPS. Curr Opin Immunol 44: 14–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Finethy R, Luoma S, Orench‐Rivera N, Feeley EM, Haldar AK, Yamamoto M, Kanneganti T‐D, Kuehn MJ, Coers J (2017) Inflammasome activation by bacterial outer membrane vesicles requires guanylate binding proteins. mBio 8: e01188‐17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Casson CN, Yu J, Reyes VM, Taschuk FO, Yadav A, Copenhaver AM, Nguyen HT, Collman RG, Shin S (2015) Human caspase‐4 mediates noncanonical inflammasome activation against gram‐negative bacterial pathogens. Proc Natl Acad Sci USA 112: 6688–6693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kajiwara Y, Schiff T, Voloudakis G, Gama Sosa MA, Elder G, Bozdagi O, Buxbaum JD (2014) A critical role for human caspase‐4 in endotoxin sensitivity. J Immunol 193: 335–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Honda K, Taniguchi T (2006) IRFs: master regulators of signalling by Toll‐like receptors and cytosolic pattern‐recognition receptors. Nat Rev Immunol 6: 644–658 [DOI] [PubMed] [Google Scholar]
- 53. Tanaka N, Kawakami T, Taniguchi T (1993) Recognition DNA sequences of interferon regulatory factor 1 (IRF‐1) and IRF‐2, regulators of cell growth and the interferon system. Mol Cell Biol 13: 4531–4538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Casanova J‐L, Abel L (2018) Human genetics of infectious diseases: unique insights into immunological redundancy. Semin Immunol 36: 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cuesta N, Salkowski CA, Thomas KE, Vogel SN (2003) Regulation of lipopolysaccharide sensitivity by IFN regulatory factor‐2. J Immunol 170: 5739–5747 [DOI] [PubMed] [Google Scholar]
- 56. Kayagaki N, Lee BL, Stowe IB, Kornfeld OS, O'Rourke K, Mirrashidi KM, Haley B, Watanabe C, Roose‐Girma M, Modrusan Z et al (2019) IRF2 transcriptionally induces GSDMD expression for pyroptosis. Sci Signal 12: eaax4917 [DOI] [PubMed] [Google Scholar]
- 57. van Oost BA, Edgell CJ, Hay CW, MacGillivray RT (1986) Isolation of a human von Willebrand factor cDNA from the hybrid endothelial cell line EA.hy926. Biochem Cell Biol 64: 699–705 [DOI] [PubMed] [Google Scholar]
- 58. Hedl M, Abraham C (2012) IRF5 risk polymorphisms contribute to interindividual variance in pattern recognition receptor‐mediated cytokine secretion in human monocyte‐derived cells. J Immunol 188: 5348–5356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hedl M, Yan J, Witt H, Abraham C (2019) IRF5 is required for bacterial clearance in human M1‐polarized macrophages, and IRF5 immune‐mediated disease risk variants modulate this outcome. J Immunol 202: 920–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. van Wilgenburg B, Browne C, Vowles J, Cowley SA (2013) Efficient, long term production of monocyte‐derived macrophages from human pluripotent stem cells under partly‐defined and fully‐defined conditions. PLoS ONE 8: e71098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Langmead B, Wilks C, Antonescu V, Charles R (2018) Scaling read aligners to hundreds of threads on general‐purpose processors. Bioinformatics 35: 421–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W et al (2008) Model‐based analysis of ChIP‐Seq (MACS). Genome Biol 9: R137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Quinlan AR, Hall IM (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP (2011) Integrative genomics viewer. Nat Biotechnol 29: 24–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Data Availability Statement
The authors declare that the data supporting the findings of this study are available within the paper and its Appendix. The datasets produced in this study are available in the following database (RNA‐seq data: Gene Expression Omnibus GSE132178; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE132178).