

Abstract

Biological membranes are tricky to investigate. They are complex in terms of molecular composition and structure, functional over a wide range of time scales, and characterized by nonequilibrium conditions. Because of all of these features, simulations are a great technique to study biomembrane behavior. A significant part of the functional processes in biological membranes takes place at the molecular level; thus computer simulations are the method of choice to explore how their properties emerge from specific molecular features and how the interplay among the numerous molecules gives rise to function over spatial and time scales larger than the molecular ones. In this review, we focus on this broad theme. We discuss the current state-of-the-art of biomembrane simulations that, until now, have largely focused on a rather narrow picture of the complexity of the membranes. Given this, we also discuss the challenges that we should unravel in the foreseeable future. Numerous features such as the actin-cytoskeleton network, the glycocalyx network, and nonequilibrium transport under ATP-driven conditions have so far received very little attention; however, the potential of simulations to solve them would be exceptionally high. A major milestone for this research would be that one day we could say that computer simulations genuinely research biological membranes, not just lipid bilayers.
1. Introduction
Biological membranes are everywhere. All our cells are surrounded by a biological membrane. So also are the tiny organelles such as the nucleus that contains our genetic code and the endoplasmic reticulum that synthesizes most of our proteins. Biological membranes keep us alive when they transfer oxygen from our lungs to our bloodstream. Biomembranes also control our mood, because they host the receptors of signaling molecules such as dopamine in our brain.
It is quite intriguing that membranes can play such crucial roles in maintaining life, yet these membranes are basically just soft, few nanometers thick lipid interfaces. However, the more closely one looks at them, the more complex they turn out to be. It is quite justified to note that despite about 100 years of research, we still do not understand exactly what biological membranes really look like.
We know that they are made up of thousands of different lipids. We know that they host numerous proteins that carry out many of the cellular functions. And we know that all the communication between the outside and the inside of cells is controlled by biomembranes. However, we also know that biological membranes are constantly being modified as their content and heterogeneous structure change constantly during our life. In essence, biomembranes are characterized by a series of transient structures that evolve under nonequilibrium conditions. To fully understand what is going on in biomembranes, one should be able to unravel all the possible processes, starting from reactions on a scale of angstroms to large-scale events taking place on a scale of micrometers.
One of the methods of choice to accomplish this aim is computer simulation. By carrying out simulations on well-defined model systems and using experimental data as input, one can generate new information by predicting novel phenomena and by helping to interpret experimental observations. By bridging different simulation techniques to each other, one can also investigate multiscale phenomena, such as how a single chemical reaction in a protein leads to macroscopic motion of a cell.
At present, the field of biomolecular simulations is undergoing a paradigm shift. The quality of atomistic simulation models has reached a level where computer simulations are a major complement to experimental research. At the same time, increased computing resources have made millisecond atomistic simulations possible; this is a crucial point given that the activation of many membrane receptors takes place on a millisecond time scale. Furthermore, both quantum mechanics/molecular mechanics (QM/MM; see Table 1 for a complete list of abbreviations) and coarse-grained molecular simulation models have developed so dramatically that there are now a number of reliable ways to explore multiscale phenomena by means of multiscale simulations.
Table 1. List of Abbreviations and Their Full Forms Used in This Article Given in Alphabetical Order.
| abbreviation | full form |
|---|---|
| ABC | ATP binding cassette |
| AH | amphipathic helix |
| ANTH | AP180 N-terminal homology |
| APP | amyloid precursor protein |
| AQP0 | Aquaporin-0 |
| BAR | bin/amphiphysin/RVS domain |
| BK | big potassium channels |
| BMP | bis(monoacylglycero)phosphate |
| CEs | cholesteryl esters |
| CL | cardiolipin |
| CETP | cholesteryl ester transfer protein |
| Ci-VSP PD | voltage sensitive phosphatase PTEN domain |
| CO | cholesteryl oleate |
| COMT | catechol O-methyl transferase |
| CP | actin-capping protein |
| cPLA2 | PLA2 cytosolic |
| Cpx | complexin |
| CRAC | cholesterol recognition amino acid consensus motif |
| cryoEM | cryogenic electron microscopy |
| CTB | cholera toxin |
| CYP | cytochrome P450 |
| dDAT | Drosophila dopamine transporter |
| DHA | docosahexaenoic acid |
| EGFR | epidermal growth factor receptor |
| EM | electron microscopy |
| ENTH | epsin N-terminal homology domain |
| EPR | electron paramagnetic resonance |
| Exo70 | exocyst complex component 7 |
| FRET | Förster resonance energy transfer |
| GABA(A) | γ-aminobutyric acid receptor type A |
| GABAAR | pentameric ligand gated ion-channel |
| GIVA | PLA2 family in human group IVA |
| GltPH | orthologous bacterial aspartate transporter |
| GPCR | G protein-coupled receptors |
| GPMVs | Giant plasma membrane-derived vesicles |
| GRP1 | general receptor of phosphoinositides 1 |
| GVIA | PLA2 calcium-independent group VIA |
| hDAT | human dopamine transporter |
| HDL | high-density lipoprotein |
| Tm | temperature of the main phase transition |
| HMMM | highly mobile membrane mimetic |
| HOPS | protein sorting complex |
| Hsc70 | 70 kDa heat shock protein |
| hSERT | human serotonin transporter |
| HVR | hypervariable region |
| IF | inward-facing |
| KV | voltage-gated potassium channels |
| LAT | linker of activation of T cells |
| LC3 | microtubule-associated protein light chain 3 |
| Ld | liquid-disordered |
| LDL | low-density lipoprotein |
| LDs | lipid droplets |
| LeuT | bacterial leucine transporter |
| Lo | liquid-ordered |
| MATs | monoamine transporters |
| MB-COMT | membrane-bound COMT |
| MD | molecular dynamics |
| MD/MC | molecular dynamics/Monte Carlo |
| MOA | monoamine oxidase |
| MscL | conductance mechanosensitive ion channel |
| Munc18a | mammalian uncoordinated-18 protein |
| nAChR | nicotinic acetylcholine receptor |
| NhaA | Na+/H+ antiporter |
| nHDL | nascent HDL |
| NMR | nuclear magnetic resonance |
| NPC1 | Niemann–Pick C protein 1 |
| NPC2 | Niemann–Pick C protein 2 |
| NSF | N-ethylmaleimide sensitive factor |
| NSS | neurotransmitter-sodium-symporter |
| OF | outward-facing |
| Osh | oxysterol binding protein |
| PH | pleckstrin homology |
| PKCα | protein kinase C-α |
| PLA2 | phospholipase A2 |
| PMF | potential of mean force |
| PTEN-like | phosphatase tensin-type domain |
| QM/MM | quantum-mechanics/molecular mechanics |
| S1P1 | sphingosine 1-phosphate receptor 1 |
| SANS | small angle neutron scattering |
| SAXS | small angle x-ray scattering |
| SC | stratum corneum |
| S-COMT | water-soluble COMT |
| Sec3 | exocyst complex component 1 |
| SNARE | soluble N-ethylmaleimide-sensitive factor attachment protein receptor |
| STED | stimulated emission depletion microscopy |
| STxB | Shiga toxin |
| Syt1 | synaptotagmin-1 |
| TAGs | triacylglycerols |
| TAP | antigen translocation complex |
| TGs | triglycerides |
| TO | triolein |
| TRPV1 | transient receptor potential vanilloid subtype 1 |
| t-SNARE | membrane-attached SNARE |
| v-SNARE | vesicle-attached SNARE |
| WALP | tryptophan (W)-alanine (A)-leucine (L) peptide |
| α-TTP | α-tocopherol transfer protein |
| β2AR | β2-adrenergic receptor |
In this review, we describe the challenge that we must face. The question is, when we want to understand how biological membranes function, then what exactly should we be able to simulate?
In this spirit, we here discuss the recent progress in computer simulations of biomembranes. It is worth clarifying that when we talk about biomembranes, then we mean the real biological membrane in our living cells. So, no one has simulated it yet, although many would like to.
We discuss how the quite extreme diversity of different lipids govern membrane behavior, cellular signaling, mass transport, formation of functional membrane domains, storage of energy, and metabolism. We discuss several themes that have so far received little attention but that are of exceptionally central importance for understanding the functioning of biomembranes, such as glycosylation and other post-translational modifications and the machinery associated with, for example, the actin cytoskeleton and the glycocalyx. We close the review by introducing some of the future challenges and goals. The aim is that, someday, we will be able to simulate the functioning of biomembranes and cells in a multiscale manner, using simulation models that match reality.
2. Historical Overview
It is always fascinating to see retrospectively how science has uncovered the secrets of nature. Let us here briefly discuss two themes that are central to this review: the understanding of biological membranes in terms of theoretical models proposed over the years and the history of computer simulations as the basis for simulating lipid systems together with other biomolecules.
2.1. Brief History of Cell Membrane Models
Several models have been suggested during the previous 95 years to explain the structural and dynamical aspects of biological membranes. Already in 1925, Gorter and Grendel1 suggested that lipids form biomolecular layers. Then in the 1950s, Robertson2 proposed a three-layer structure, where two layers of proteins were attached to a lipid layer in the middle. A few years later Lenard and Singer suggested a revised model,3 where the proteins were now allowed to span a lipid bilayer structure. This picture was yet considered incomplete, and in 1972 Singer and Nicolson proposed the famous “fluid mosaic” model4 that is nowadays generally known also as the Singer–Nicolson model.
The fluid mosaic model depicts reality, as we understand it today, already in a quite convincing manner. It suggests that lipids form a fluid two-dimensional matrix that hosts proteins to carry out their functions. In the fluid mosaic model, lipids as well as proteins are assumed to be more or less randomly distributed in the plane of a lipid bilayer structure, thereby providing a considerable degree of freedom for lateral and rotational diffusion. Meanwhile, this assumption also implies that the model does not account for lateral heterogeneity despite the fact that the idea of lateral heterogeneities and membrane domains in model membranes was suggested already in 1970.5−7 After the fluid mosaic model was proposed, more and more data were published in favor of membrane heterogeneity and lateral lipid composition fluctuations, leading to the ideas of lateral segregation, domain formation, and lipid–protein interactions driving the formation of functional membrane regions (see discussion in refs (8 and 9), for example). Further, while a number of additional models to account for aspects such as hydrophobic matching, and the role of the cytoskeleton and the glycocalyx network were further proposed in the 1980s and 1990s (for a comprehensive discussion on this theme, see the reviews by Ole Mouritsen and Luis Bagatolli10,11), it is fair to say that the next major paradigm was suggested by Simons and Ikonen in 1997 in terms of the raft hypothesis.12 In the raft model, based on observations reported some years earlier,13 one assumes membranes to be comprised of cholesterol- and sphingolipid-rich domains functionally associated with specific proteins involved in cellular functions, in particular cell signaling. The idea of domains rich in cholesterol was not new, and it had been explored theoretically prior to the suggestion of the raft hypothesis. In particular, the theoretical work by Hjort Ipsen et al. had predicted that under certain thermodynamic conditions cholesterol generates the coexistence of cholesterol-rich liquid-ordered (Lo) and cholesterol-poor liquid-disordered (Ld) phases.14 This picture has largely been the basis of the idea that lipid rafts are highly ordered cholesterol-rich membrane domains or even membrane phases. Yet, this picture is not complete.
Is it justified to consider ordered protein-free membranes or membrane domains as lipid rafts? If one can show that protein-free membrane domains have a biological function and if ordered protein-free membrane phases (compare with nanoscale membrane regions) exist in real cell membranes, then perhaps yes. Otherwise, quite likely not. The core of the matter lies in that lipid rafts are considered, after two decades of research, as fluctuating and functional nanoscale assemblies of proteins and lipids.15 This picture would also intuitively make sense, since a lipid raft would then correspond to a concept of a protein that together with specified lipids would form a protein–lipid complex, that is, a functional protein–lipid unit. Given that individual (integral) membrane proteins are about 3–6 nm in size, and the dynamical lipid pool bound to the protein increases the lateral size of this complex by ∼5 nm,16 then the minimal size of a lipid raft would be on the order of 10 nm. Experimental data are not incompatible with this assessment: there was a quite long period when the resolution of super-resolution microscopy increased steadily, and every time the spatial resolution was improved, the size estimates for rafts also decreased. Currently, when the spatial resolution of, for example, stimulated emission depletion microscopy (STED) is in the range of 10–20 nm,17 the raft size is also predicted to be in the same range.
There will be more refined models for cell membranes, but the lipid raft model serves currently as a quite reasonable paradigm to understand the conditions where cell functions take place in biological membranes. In the following sections, we discuss many of these processes quite in detail.
2.2. History of Computer Simulations with Applications to Lipids
The era of computer simulations can be traced back to the dates when the first computers were deployed. The development of the first electronic computer (ENIAC; Electronic Numerical Integrator and Computer) that operated from 1945 to 1952 changed the world quite completely. The second computer known as MANIAC (Mathematical Analyzer, Numerical Integrator, and Computer) operated between 1952 and 1958 and was followed by, surprisingly, MANIAC 2 and MANIAC 3.
The people who pioneered this process can also be considered as the pioneers of the computer simulation field. For instance, Nicholas Metropolis, who was one of the figures developing and using ENIAC,18 originally suggested19 an obvious name for the Monte Carlo simulation method.20,21 A very essential detail at this point is that the first molecular dynamics (MD) simulations were reported not earlier than in 1957. Therefore, the computational science between 1945 and 1957 was based on Monte Carlo simulations that rely on stochastic sampling, and therefore on the use of random numbers to solve problems in a stochastic manner. Unfortunately, the quality of random numbers is typically considered to be self-evident, and is often not appreciated, so let us make an exception here and discuss this important topic briefly before we move on to focus on lipid simulations.
Together with John von Neumann, Nicholas Metropolis also studied randomness of the decimals of π and e,22 and developed the first algorithm for generating pseudorandom numbers that are needed in all stochastic simulation techniques we use these days: the so-called midsquare method.23,24 In this method, an arbitrary n-digit integer is squared, creating a 2n-digit product. A new integer is formed by extracting the middle n digits from the product. We do not encourage using the midsquare technique, however, since the properties of the pseudorandom number sequences generated with the method are bad24 (consider a seed of 50 with n = 2, for example), but it was the first step in the right direction to find efficient and practical means to generate huge numbers of pseudorandom numbers for stochastic computer simulation applications. This pioneering work has evolved to a research field of its own,25 with an objective to develop deterministic algorithms that generate pseudorandom number sequences that mimic truly random behavior as closely as possible, but with extreme efficiency. Importantly, these sequences are used not only in Monte Carlo sampling methods but also in MD simulations that often are based on using stochastic noise to model the effect of a thermodynamic heat bath, that is, temperature. Given this, the quality of the pseudorandom number generators used in simulation packages is critical to the reliability of the simulation results. The essence of the matter is that if the quality of pseudorandom number sequences used in any stochastic simulation method, including MD, is low, then the validity of the results may be compromised. As a matter of fact, when articles of simulation studies using stochastic noise are reported, it is truly peculiar how seldom the name of the pseudorandom number generator used in the stochastic simulations is mentioned at all.
The footsteps of the above work by Metropolis and von Neumann also generated the first MD simulations, which were conducted on hard-sphere models and reported in 1957.26 The first MD simulation of argon as a representative of a real liquid was published in 1964.27 It took a few years more until the first simulations on biological molecules were reported. In 1977, MacCammon et al. studied a small protein (bovine pancreatic trypsin inhibitor) for 8.8 ps.28 The simulation time was quite limited but so was also the environment, since the simulation was conducted in vacuum. Due to progress in the design of faster computers, the simulation time scales have increased rapidly, and the first 1 μs simulation on protein folding was performed already in 1998.29
Meanwhile, the era of lipid simulations started in 1980 as has been reviewed by Rich Pastor.30 The first MD simulations were based on simplified models of monolayers,31 lipid bilayers without solvent (water),32 and small micelles immersed in the water phase.33 A few years later, Larry Scott and his co-workers started combining Monte Carlo and MD for simulations of lipid membranes.34−36 Using Monte Carlo can sometimes be a clever and efficient trick, since it allows the use of unphysical moves that may foster the sampling of the phase space.
Yet, it is a fact that the most common method used today for simulations of complex membranes is MD. Further, while this is a matter of taste, it may be fair to say that the field of lipid simulations as we understand it today started in early 1990s, when a number of exciting articles of hydrated lipid bilayers were published in a row.37−40 At the same time, the experts started to pay significant attention to the accuracy of the simulation models. Let us consider one of these trends in more detail here.
In 1994, Egberts et al.41 designed a model for DPPC (for the naming convention of lipids, see Figure 1) based on the GROMOS-87 force field. However, since their initial choice of the force field parameters resulted in the gel phase, they reduced the partial charges of the lipids by a factor of 2, adjusted the van der Waals parameters for the CH2/CH3 groups, and modified the dihedral potential of the acyl chains into the Ryckaert–Belleman representation to reproduce the structure of the acyl chains. The changes corrected the problem and resulted in a model that generated a lipid bilayer in the liquid-disordered phase. Then, three years later Berger et al. published a lipid simulation model that is still in common use and known as the “minimal standard” in lipid simulations.42 As is well-known, the tough part in the development of classical simulation models is the derivation of Lennard-Jones interaction parameters. To this end, Berger et al. systematically reparametrized the nonbonded interactions used in the Egberts paper by applying OPLS (optimized potential for liquid simulations) parameters that were earlier used in a model for DMPC.43 They systematically adjusted the Lennard-Jones parameters for CH2/CH3 groups by simulating bulk pentadecane and then fitted the Lennard-Jones parameters to find a match with experimental data for volume and heat of vaporization.
Figure 1.

Naming convention for common lipids (upper panel) and names with abbreviations of the lipids discussed in the manuscript (lower panel).
In a way, the “Berger lipids” have become a classic. This model is not bad, rather it is surprisingly good given that it was developed with minor resources about 20 years ago. It was essentially the most accurate force field from 1997 until about 2010, when CHARMM36 lipids were published. Many senior scientists working today on lipid simulations recall nostalgically the times when they used the Berger model in 1 ns (or may be even 100 ns) simulations and then used the data of a single simulation to publish a solid paper.
Starting from about 2000, the progress in lipid membrane simulations has been rapid. For recent reviews, see, for instance, the one in ref (44) and the review describing the progress in lipid force field development by Lyubartsev and Rabinovich.45 In other reviews, some methodological topics are discussed including MD simulations and modeling tools,46 free energy calculations,47 artifacts frequently encountered in simulations,48 and problems about validating MD results.49−51
2.3. What is Currently Feasible by Computer Simulations?
The answer to this question depends on the quality and the nature of the simulation models, the amount of computer resources available for simulations, and also how efficiently the simulation software is integrated to the hardware.
As to classical atomistic simulations, protein folding has been explored over millisecond time scales.52 Nanoscale domain formation in lipid membranes has been investigated in systems of ∼1000 lipids over ∼1 μs per system, the total simulation time being ∼10 μs.53 The dynamics of membrane proteins and their allosteric interactions with cholesterol have been investigated over simulation times of >100 μs.54 Since the activation process of membrane proteins usually takes place in time scales of milliseconds, it is expected that in a few years the consideration of these events in full atomic detail becomes feasible.
In coarse-grained simulations, the scales depend very much on the extent of coarse graining. In the MARTINI model, using an explicit solvent, large highly protein-crowded membranes have been explored over multi-millisecond time scales.55 In related simulations of lipid membranes with integral membrane proteins, several papers have reported data for large membranes over times from ∼10 μs56 to milliseconds.57 In implicit solvent simulations, the scales would obviously be much longer.
3. Biomembranes as the Target of Simulations: Native Membranes Whose Behavior We Would Like To Understand
Biological membranes are highly complex systems composed of various types of macromolecules, in particular lipids, proteins, and carbohydrates. Even the smallest membrane-bound intracellular organelle with a diameter of ∼100 nm is huge compared to atomic size scales. This makes them almost impossible to model and simulate as a whole. Considering that a real cell is approximately 10 μm in size, the challenge faced by computer simulations is indeed enormous.
Due to these unfortunate facts, biological membranes are explored via simulations by focusing on smaller fragments instead of being examined as a whole. This divide-and-conquer approach allows capturing of the entire complexity of real cell membranes and the processes that take place in their vicinity.
Here we provide the background for sections 4–10. We present the most important molecular components of which the biomembranes are formed, lipids, proteins, and glycoconjugates. Of these components, glucoconjugates have received the least attention in membrane simulations. We further discuss post-translational modifications such as glycosylation and their roles in membrane function. Again, it turns out that these structural modifications have received very little attention in membrane simulations until now. Finally, we draw the reader’s attention to the themes where the biomembrane structure is really challenging, such as the glycocalyx and the cell wall. Understanding how these complex structures function would be extremely important, but currently the simulation research done to explore these systems has been very modest.
The reader is requested to keep in mind that we focus on providing a concise overview of the complexity of biomembranes in this section. Simulations to explore these themes are discussed in later sections.
3.1. Lipids
3.1.1. Diversity of Lipids
Lipids, which are the main building blocks of, for example, membranes, lipid droplets, and lipoproteins, constitute a large and diverse group of biological compounds. The physical and chemical properties characterized by lipids are very diverse; thus there are also very distinct molecules all known as lipids. For instance, various lipids are intermediate metabolites on synthetic pathways, such as 17 different sterols acting as precursors of cholesterol; signaling molecules, such as steroid hormones; and cofactors of enzymatic proteins such as retinol; as well as products of pathological oxidation processes. In practice, lipids are essentially surface-active (amphiphilic) fatty molecules that have a biological function. Here for the definition of lipids we emphasize the importance of biological function. While soaps and detergents are also surface-active molecules, they lack the biological function and thus are not lipids.
The Lipid Maps Structure Database (LMSD) (http://lipidmaps.org/data/structure/) of biological lipids contains structures of nearly 21 000 known and 22 000 predicted lipids. Not all of these lipids are abundant in biological membranes. Nonetheless, they are found in trace amounts in biological membrane samples. The number of typical lipid types found in cells is ∼1000, though there are significant differences between different organs or clades. For a recent review of lipid diversity, please read refs (58−61). In spite of this variety, artificial lipids are actively developed for pharmaceutical applications62−69 or as research tools.70−72 For reviews, see refs (73−75).
Importantly, lipids play diverse roles not only in membrane structures but also in physiological processes in whole organisms. For example, cholesterol-derived bile acids participate in the digestion of fats76 and steroid-glycosides such as saponins act as protective elements in plants.
In this section, we discuss the lipids and the lipid groups that are the main building blocks of biological membranes. We introduce the key building blocks needed to construct simulation models for lipid membrane structures. To demonstrate the diversity of lipids in a real setting, we also discuss recent lipidomics studies that highlight the variety of lipids that the simulation models should include, if the aim would be to simulate membranes whose content would match reality.
3.1.2. Main Lipid Classes
Glycerol-Based Lipids
Backbone
Glycerol-based lipids are the most common lipids in both prokaryotes and eukaryotes. Glycerol-based lipids are predominantly esters of the glycerol group, which is the backbone of the lipid. The glycerol group is linked to fatty acid chains and a polar headgroup (Figure 2). Typical membrane lipids in this class have two chains attached to the sn-1 and sn-2 positions, while the headgroup is connected to the sn-3 position. There are also lipids known as lyso-lipids with a single chain, such as lyso-phosphatidylcholine (lyso-PC), which acts as a modulator of membrane curvature. Lipids with three hydrophobic chains (triglycerides) are the storage form of lipids found, for example, in lipid droplets and lipoproteins. If the ester linkage to the glycerol group is replaced with an ether link (Figure 2), one ends up with ether lipids such as plasminogens, where the ether bond is a part of the vinyl group (Figure 2).77
Figure 2.

Glycerol-based lipids. Upper panels show the glycerol backbone with possible modifications, the middle panel shows the most common headgroups, and the bottom panel shows phosphatidylinositol headgroups.
Headgroups
The simplest headgroup of glycerol-based lipids is the unesterified hydroxyl group. Lipids with this group are called diacylglycerols (DAGs) (Figure 2). DAGs are a minor component of cell membranes, but they play a central role in lipid synthesis. Further, they also play an essential role in signaling as second messengers.78,79 As the headgroup of DAGs is smaller than their hydrophobic part, they induce spontaneous negative curvature. Due to this quite unique property, DAGs are involved in membrane fusion and fission.79
Another simple headgroup is the phosphate group present in phosphatidic acid (PA), which is perhaps the simplest phospholipid (Figure 2) but also the one with the most complex biophysical properties.80,81 PA, similarly to DAG, is only a minor component in cell membranes, their concentration in intracellular membranes being ∼1 mol %. Nevertheless, it is a crucial metabolite in the synthesis of other phospholipids and a second messenger in cellular signaling.82−85 PA is also vital for mitochondrial function due to its role in mitochondrial fusion and fission.86 Due to the relatively small headgroup of PA, diacyl-PA has a conical shape promoting spontaneous negative membrane curvature, but its monoacyl form, the lyso-PA, has a cylindrical shape, which promotes a flat membrane topology.87 Specific lipases interconvert these two forms during membrane fusion and fission. PA is also characterized by complex protonation behavior, since at physiological pH there are two possible protonation states with a charge of −1 and −2. The equilibrium between these states is likely influenced by membrane lipid composition, proteins, divalent cations, and modifications to the chemical structure of the molecule (e.g., the lyso form).88,89
Phosphatidylcholines (PCs) are the main structural lipids in animals and fungi, while in plants and bacteria, they are less common. Most biophysical studies of lipid bilayers have been based on PC membranes that are commonly used as a control. The headgroup of PC is relatively large and zwitterionic, containing a positively charged tetramethylammonium moiety along with a negatively charged phosphate group. Like in all naturally occurring phospholipids including PA and phospholipid groups discussed below, the headgroup is linked to a glycerol backbone via an ester bond with a phosphate group. Nevertheless, lipids with reverse order of charge, where the tetramethylammonium group attaches the headgroup to the glycerol backbone, were synthesized providing a better understanding of PC’s electrostatic properties.90
Another common zwitterionic lipid is phosphatidylethanolamine (PE), which is not only the second most common lipid in animals but also commonly found in bacteria. The headgroup of PE is composed of a positively charged amine group and a negatively charged phosphate group. PE is a precursor of PC, which is synthesized by substitution of hydrogen atoms of the amine group with methyl groups. Interestingly, in some bacteria intermediates between PE and PC with one or two methyl groups act as structural lipids.91
Phosphatidylserine (PS) is the main negatively charged lipid in animals, accounting for 8–15 mol % of all phospholipids in cells.92,93 The headgroup consists of a negatively charged phosphate group esterified with serine, thus having another negatively charged group, a carboxylic group, and a positively charged amine group. PS is located mostly in the inner leaflet of the cell membrane, and its translocation to the outer leaflet signals apoptosis.94,95 PS directly interacts with and regulates the functions of numerous proteins, such as protein kinase C, Raf-1, a serine-threonine kinase, AMPA, a glutamate receptor, and proteins related to exocytosis.92
Another important negatively charged lipid is phosphatidylglycerol (PG). The PG headgroup consists of a negatively charged phosphate group esterified with a neutral glycerol molecule. While PG is a minor component of animal membranes,96 its concentration is high in plants and bacteria.91,97
Phosphatidylinositols (PIPs) constitute a group of lipids found in small quantities in cells but having significant functions related to cellular signaling, membrane trafficking, cell division and migration, and endo- and exocytosis.98−100 The headgroup of PIPs is based on inositol, a hexane ring substituted with hydroxyl groups attached to each carbon, which can be esterified by phosphatidic acid. In biological PIPs, there are up to three phosphates. Phosphate groups in water solution may be singly or doubly deprotonated; thus, PIP molecules may adopt various protonation states.
Glycerol-based glycolipids are common components of plant membranes. They are especially enriched in photosynthetic thylakoid membranes (Figure 3).101 They may comprise up to 50 mol % of lipids in higher plants. In Prochlorococcus, the main phytoplankton organism, they may comprise even 94–99 mol % of all lipids. Further, they are also components of cyanobacterial membranes. The headgroup of this class of glycolipids is a carbohydrate moiety directly linked to the glycerol backbone. Most common glycerol-based glycolipid species are monogalactosyldiacylglycerol (MGDG) and digalactosyldiacylglycerol (DGDG).101 Other sugars, such as glucose, mannose, glucuronosyl, and sulfoquinovosyldiacylglycerol are also present in bacterial glycolipids. MGDGs that are not membrane-forming lipids are believed to play a role similar to that of cardiolipin in mitochondria.102
Figure 3.

Most common headgroups of plants and bacteria in glycerol-based glycolipid headgroups (see ref (101)). The dotted line indicates the attachment point for DAG.
Carboxylic Acids: Lipid Chains
Biological samples contain diverse types of not only lipid headgroups but also hydrocarbon chains. For instance, analysis of free fatty acids in human plasma has identified 31 different species (Figure 4).103 The most common ones are oleic acid (18:1, O, 37 mol %), palmitic acid (16:0, P, 30 mol %), stearic acid (18:0, S, 10 mol %), linoleic acid (18:2, Li, 7 mol %), palmitoleic acid (16:1, 7 mol %), myristic acid (14:0, M, 3 mol %), and arachidonic acid (20:4, A, 1 mol %).103 The shortest fatty acid that has been observed is lauric acid (12:0, L), and the longest one is cerotic acid (26:0). All of the identified fatty acids have an even number of carbons with four exceptions: pentadecanoic acid (15:0), margaric acid (17:0), heptadecenoic acid (17:1 ω-7), and tricosanoic acid (23:0). The number of unsaturated bonds has been found to range from zero (10 cases) to six (docosahexaenoic acid (22:6 ω-3, D or DHA). The polyunsaturated fatty acid content covers about 3.5 mol % of all fatty acids.
Figure 4.

Most common free fatty acids from human serum (see ref (103)): (A) saturated fatty acids, (B) unsaturated 22 carbon long fatty acids, (C) unsaturated 20 carbon long fatty acids, (D) unsaturated 18 carbon long fatty acids, and (E) remaining unsaturated fatty acids. Arrows in panel A show the last carbon atoms; those in panels B, C, and D show the positions of unsaturated bonds. In panel E, the left arrow shows the position of a double bond, while the right arrow shows the last carbon atom.
Combinations of 31 different free fatty acids in lipids with two chains leads to 496 possible pairs of chains, thus 961 lipid species for each possible headgroup. In reality, though, the relevant number of different chain pairs is much smaller, since some combinations are much more frequent than the others. For instance, usually lipids contain 16–18 carbon-long saturated or moderately unsaturated chains. These “standard” plasma fatty acid compositions are, however, not universal, as they depend on many factors such as diet. Moreover, in different tissues and cells, there are different fatty acid profiles. For instance, in the brain tissue, the chain composition in PCs mirrors the standard fatty acid profile, while PE and PS are highly enriched in polyunsaturated fatty acids.104
Fatty acid profiles in fungus also have similar features: the predominant chain types are C16 and moderately unsaturated (1–3 double bonds) C18 chains.105 A comparison of the fatty acid profiles of 43 fungus species has shown that nearly 90% of all chains belong to the above-mentioned dominant cases, but their proportions vary significantly.106 For example, palmitic, oleic, and linoleic acid contents range from 15% to 28%, 16% to 41%, and 9% to 27%, respectively.106 In different groups of fungi up to 45% of fatty acids are polyunsaturated fatty acids (PUFAs).106 Differences in the main fatty acid profiles were also observed in different spore types of the same fungal species.107 A more detailed analysis of fatty acid profiles from the edible fungus Laetiporus sulphurous revealed the presence of 30 different fatty acids with the chain length ranging from 10 to 24; it also showed that the fatty acid with the highest degree of unsaturation has six double bonds (22:6).108 Further, fungi have a high concentration of unusual fatty acids. For example, the fraction of branched fatty acid chains may reach 50% in the Conidiobolus species,105 and a relatively large fraction of branched chains has been found in samples of soil microbes.109
An analysis of the fatty acid composition in diverse organisms (including animals, plants, algae, insects, and fungus) has suggested a general feature that 16 and 18 carbon chains (palmitic, palmitoleic, stearic, oleic, and linoleic acid) with some amount of myristic acid are overall the most abundant ones.110
In nature, the double bond in the unsaturated fatty acids mainly adopts a cis conformation. On the other hand, trans unsaturated fatty acids are also found in bacterial membranes in small amounts, as they are produced in response to environmental stress. Trans unsaturated fatty acids also occur in human food as a result of industrial oil treatment (see refs (111 and 112) and references therein).
Unsaturated fatty acids are targets for oxidation by free radicals, resulting in numerous oxidized moieties including hyperoxide, hydroxyl, carboxylic, aldehyde, or ketone groups. Oxidized lipids have significantly different properties than the native (unmodified) ones,113−118 and are involved in pathological mechanisms of numerous illnesses.119−121
Cardiolipin
Cardiolipin has to be discussed as a special case since it is truly unique. It is highly branched and charged. Its glycerol moiety connects two phosphatidic acid moieties together (Figure 2). Due to the two phosphate groups, there are two protonation states at the physiological pH, due to which the molecule assumes a charge of either −1 or −2. The older literature suggests that cardiolipin is mostly singly charged.122−124 However, the latest studies have shown definitively that both phosphate groups are deprotonated; therefore cardiolipin is doubly charged in a lipid bilayer environment.125−127
Cardiolipin has typically four acyl chains. Thus, considering the 32 different fatty acid types, one expects theoretically up to ∼106 possible molecular species.128 When oxidation products are also taken into account, this number grows to ∼107. Yet the actual number of observed species is much lower. The most common cardiolipin in mammals is tetra-linoleoyl-cardiolipin, which accounts for 80% of all cardiolipins in heart and skeletal muscle tissues. Meanwhile, neural tissues exhibit a larger diversity of cardiolipin species. For instance, about 100 different cardiolipin species have been identified in the rat brain;129 however this number ranges between 20 and 40 in other organs.130
Sphingolipids
Sphingolipids constitute a class of lipids with a sphingosine backbone (Figure 5). Typically, sphingosine acyl chains are 16–18 carbons long and have a trans unsaturated bond between carbons 4 and 5. A common modified form, sphinganine, lacks the double bond and may have additional hydroxyl groups attached to carbons 4 and 5.131 An acyl chain attached to the amide group in the sphingosine backbone has usually a length of 16 to 24 carbons.132 The acyl chains are typically saturated; the only unsaturated species is nervonic acid 24:1.132
Figure 5.

Sphingolipid structures.
Sphingolipids differ from other lipids also in terms of their headgroup diversity. While sphingomyelin has phosphatidylcholine as a headgroup, ceramide’s headgroup is only a hydroxyl group (for review of sphingolipid structure and biophysical properties, see refs (132−134)). This makes ceramide an analogue of DA, a glycerol-based lipid discussed above. Ceramide actually acts as a precursor for a large group of glycolipids.
Further, sphingolipids known as glycosphingolipids have a headgroup that is comprised of carbohydrates. They are present in the extracellular leaflet in small quantities amounting to a few mol %, but they are quite enriched in specific tissues and cell types such as neural tissues, where they can constitute up to 30 mol % of all lipids. Gangliosides, which in turn are derived from glycosphingolipids, have a headgroup of complex carbohydrates with at least one sialic acid residue. These lipids act as receptors for various extracellular proteins including bacterial toxins, and they also regulate transmembrane protein function by interacting with their extracellular domains.
Sphingolipids are present in all eukaryotes but rarely in prokaryotes.135 Although the cellular concentration of sphingolipids is small (on the order of a few mol %), they accumulate in the plasma membrane, predominately in the extracellular leaflet. Sphingolipids together with cholesterol form nanoscale dynamical ordered domains (rafts), which are thought to play vital functions in cellular processes including signaling, cell adhesion, intercellular communication, and apoptosis.15,133,136,137 Unsurprisingly, specific species of sphingolipids, for example, sphingosine 1-phosphate, are involved in numerous illnesses, particularly in the central nervous system.138
Sterols
Sterols constitute a group of lipids with a steroid ring, which is a group of biological compounds with a core made up of 17 carbon atoms arranged in a specific four fused ring configuration. Some common sterols are shown in Figure 6 with the steroid core and atom numbering indicated on the chemical structure of cholesterol. Typical sterols have a hydroxyl group attached to carbon 3 (Figure 6); however other hydrophilic groups such as ketone are also found in certain sterols. The remaining part of the molecule is typically hydrophobic and characterized by considerable structural diversity in terms of groups attached to the steroid ring, number and positions of double bonds, and the structure of the short chain at the end of the molecule. The Lipid Maps database currently lists 2828 compounds belonging to the steroid family, but only a small fraction of these compounds function as structural elements in biological membranes. The majority of these compounds are signaling molecules, surfactants (bile acids and saponins), or metabolites or are of unknown function.
Figure 6.

Structures of the most common sterols in animals, plants, and fungi. The structure of the steroid core is comprised of four rings (labeled ABCD in the cholesterol molecule) that are fused to one another. The carbon atoms of cholesterol are numbered.
Cholesterol is perhaps the most widely known sterol due to its ubiquity and vital functions in vertebrate membranes. Membranes in the eye lens, where the concentration of cholesterol can be as large as 70 mol %,139,140 represent the extreme cases of cholesterol enrichment in biomembranes. Despite this, crystals of cholesterol-rich membranes have not been captured. Unusually high levels of cholesterol have also been observed in neural tissues and in the skin.141−143
In cells, the highest level of cholesterol is observed in the plasma membrane, where it constitutes 10–50 mol % of the lipid content.144 However, its distribution between the cytosolic and extracellular leaflets has remained controversial.145 In intracellular membranes, the cholesterol content is much lower. For instance, in mitochondrial membranes, the cholesterol concentration is essentially negligible.
Cholesterol affects numerous physicochemical properties of lipid bilayers by, for example, increasing their mechanical strength and decreasing their permeability.146−150 In artificial membranes, cholesterol has been observed to modulate membrane phase behavior by leading to the formation of the Lo phase, which is liquid-like due to its fluidity (lack of translational order) but also highly ordered due to conformational order in the lipid acyl chain region strengthened by cholesterol. Membranes in the liquid-disordered (Ld) phase lack the conformational order that is therefore the fingerprint of cholesterol. Depending on cholesterol concentration and temperature, Lo domains can separate from the Ld ones in model membranes. By analogy, separation of the phases is also assumed to occur in biological membranes, but the cases where this has been shown to take place have been rare.151,152
These cholesterol-rich ordered domains, assuming that they exist in biomembranes, are often considered as the basis for a concept of rafts.12,15,153−155 In this context, it is important to stress that there is considerable evidence in favor of the idea that cell membranes are signaling platforms for numerous cellular processes17,156,157 and that there are nanoscale membrane regions where proteins and lipids function together to realize signaling processes.158−161 However, what exactly is a raft is still unclear. The current understanding of rafts highlights the importance of their functionality,15 so in this spirit we consider that lipid membranes or lipid domains without proteins should not be called rafts, unless one can show that these lipid regions have a biological function.
Cholesterol can get oxidized due to both normal metabolism and pathological states (for a review, see ref (162)). In both cases, behavior of the oxidized sterol and its effect on the physicochemical properties of the membrane are significantly different from those of cholesterol.163,164
Moving on, the majority of invertebrates lost the ability to synthesize sterols during the evolution.165−167 Therefore, they depend on dietary uptake of sterols, which are then enzymatically modified to suit their needs.168
In plants, more than 200 sterols (phytosterols) have been identified until now. While no single sterol species seems to dominate the plant sterol profile, sitosterol, campesterol, and stigmasterol are the most common ones followed by brassicasterol, gramisterol, obtusifoliol, cycloartenol, amyrin, stigmastanol, and sitostanol.169 Sitosterol and campesterol are also noteworthy, as they constitute 90 mass % of phytosterols (plant sterols) in the human diet.170 In Arabidopsis thaliana, of all sterols, 64 mol % are sitosterols, 11 mol % are campesterols, 6 mol % are stigmasterols, 3 mol % are isofucosterols, and 2 mol % are brassicasterols.171 Despite the commonly held belief that plants do not contain cholesterol, the remaining 14% of sterols in Arabidopsis thaliana are, actually, cholesterols.171 Phytosterols are of medical interest because even though they are as abundant as cholesterol in the human diet, their plasma concentration does not contribute to cardiovascular diseases.172 On the contrary, they are likely cholesterol-lowering agents.173 Nevertheless, the inability to expel sitosterol leads to its accumulation in a rare genetic syndrome called sitosterolemia.174 Finally, phytosterols are also of interest to the food industry and actively studied due to their potential in improving food quality.175
Ergosterol is the principal fungal sterol, typically constituting 80% of fungal membrane sterols.176 However, it does not dominate the fungal sterol profiles in the manner of cholesterol for the vertebrates. In fact, other sterols like cholesterol, 4-methyl cholesterol, 24-ethyl cholesterol, brassicasterol, and some precursors of these sterols are frequently found in fungi.177 The current knowledge of fungal sterols is limited, as only 175 species have been examined.177 This is surprising given that many antifungal drugs target ergosterol synthesis.178 The synthesis of ergosterol is intriguing also from the evolutionary perspective. Fungi developed an ergosterol biosynthetic pathway, although a cholesterol biosynthesis pathway already had existed. This happened even though synthesis of ergosterol is energetically more expensive and at the same time ergosterol is a less potent modifier of lipid bilayer properties, such as ordering and condensing effects.
Prokaryotes, leaving aside a few exceptions, are traditionally thought not to utilize sterols. An exception to this is mycoplasma, a parasitic bacterium with a minimalistic genome that takes all its lipids from the host organism.179 Recent studies showed that at least 34 bacterial species can synthesize and use sterols,180 but their functions remain unknown. Bacteria also often feature another group of lipids with sterol-like structures called hopanoids.91,181 In bacteria, hopanoids seem to serve the same function that sterols serve in other organisms by inducing the formation of ordered phases and rafts.182,183
While most sterols are natural and serve biological functions, there exist few synthetic sterols predominately synthesized to be used as research tools. Since only a few natural sterols are intrinsically fluorescent (e.g., dehydroergosterol and cholestenone),184,185 sterols with attached fluorescent labels, for example, BODIPY,186 or photoreactive groups (so-called click groups)187 were developed. A second group of synthetic cholesterol analogues is derived by modifying the functional groups of cholesterol, with the purpose of investigating the important functions of cholesterol structure. These cholesterol derivatives include enantiomeric cholesterol,188 epicholesterol,189 sterols without methyl groups on the rings,190−193 and sterols with a modified side chain.194
Other Lipid Classes
Lipopolysaccharides are bacterial lipids present in the outer membrane of Gram-negative bacteria, protecting them from harmful environmental factors. Lipopolysaccharides have three functional units: lipid A, core polysaccharide, and O-antigen polysaccharide (see Figure 7).195 The lipid A backbone is a β(1–6) linked glucosamine disaccharide phosphorylated at positions 1 and 4. A typical lipid A moiety has six hydrocarbon chains, mostly 14 carbons long, although some lipid A moieties have four chains with different lengths. The core polysaccharide is composed of up to 15 sugar units and is connected to the sugar moiety of lipid A via a bacteria-specific sugar, 3-deoxy-d-manno-octulosonic acid (Kdo).
Figure 7.

Structures of lipid A, carotenoid (zeaxanthin), quinone, and bolaamphiphile (caldarchaeol).
Isoprenoids are lipids based on isoprene units with every second bond in the chain unsaturated, which makes them rigid (Figure 7). Examples of isoprenoids include carotenoids, pigments of plants, and photosynthetic bacteria. Polyprenyls, on the other hand, have unsaturated bonds spaced with two saturated bonds, which makes them very flexible (Figure 7). Polyprenyls act as ubiquinone substrates for redox reactions in respiration and photosynthesis. Polyprenyl chains are also present in glycosylphosphatidylinositol (GPI) lipids used to anchor peripheral membrane proteins.
Archaea is a phyla not belonging to either bacteria or eukaryotes, but having many unusual molecular mechanisms (Figure 7).195−197 They have glycerol-based lipids with ether links. Typically, archaeal lipid chains are branched or polyprenyls. A common archaea-specific lipid type that can be considered as an example is bola-amphiphile. These lipids contain chains connected to two headgroups located in the opposite bilayer leaflets, implying that these lipids span the lipid bilayer. Sometimes, bola-amphiphiles contain five carbon rings in their chains.
3.1.3. Lipids in Cells
Distribution of Lipids between Cellular Compartments
Eukaryotic cells are complex structures with membrane-bound subcellular compartments and organelles. Each cell is surrounded by the cell membrane. Two close and connected membranes surround the nucleus. Each mitochondria is surrounded by a double membrane with the inner membrane forming stack-like folded structures, called the cristae. Other organelles like the Golgi apparatus, the endoplasmic reticulum, endosomes, and exosomes are stack-like structures or vesicles wrapped by membranes. To serve the different functions that each of these membranes have, their lipid composition differs significantly (Figure 8). Extensive discussion of subcellular distribution of lipids can be found in refs (144 and 198) and in the special issue of Biochimica et Biophysica Acta - Molecular and Cell Biology of Lipids (2016).199 Here, we discuss only the most important features of the subcellular distribution of lipids.
Figure 8.

Cellular distribution of phospholipids. Composition is given in mol %. Reproduced with permission from ref (198). Copyright 2018 American Society for Biochemistry and Molecular Biology.
Cholesterol is the main component of the cell membrane. Here, the molar ratio of cholesterol to phospholipids may be as large as one. On the other hand, in the intracellular membranes, like those that enclose mitochondria, endoplasmic reticulum, or endosomes, this molar ratio is only 0.1. The only organelles with a higher amount of cholesterol are the Golgi apparatus and exosomes, which are formed in the Golgi apparatus. Sphingomyelins, known as the partner of cholesterol in the formation of raft domains, are present in high concentrations in the cell membrane, and in lower concentrations in exosomes and the Golgi apparatus. That is, the spatial distribution of sphingomyelin overlaps with that of cholesterol. PCs are the main phospholipids in all types of animal cell membranes, amounting to 30–60 mol % of all phospholipids. The second most common lipid in the intracellular membranes is PE, amounting to 15–30 mol % of all phospholipids. The second most common phospholipid in the cell membrane is sphingomyelin. PIPs and PSs are present in most cellular membranes with a concentration of 5–15 mol %, except in the mitochondrial membrane, where PS concentration is very small. In contrast, cardiolipins and PGs are predominately found in mitochondria.
Asymmetry of the Cell Membrane
Cell membranes have an asymmetric distribution of lipids between the intracellular and extracellular leaflets.200,201 Sphingomyelins, PCs, and gangliosides are present mainly in the extracellular leaflet, while PE, PS, PG, PIP, and PA lipids are almost exclusively located in the intracellular leaflet. Certain transmembrane enzymes actively maintain this asymmetry by translocating lipids from the extracellular to the intracellular leaflet (flippases) and vice versa (floppases).200,202−204 Scramblases, on the other hand, facilitate the passive mixing of the leaflets without directionality and substrate specificity.204
While cholesterol is present in both leaflets of the plasma membrane, its transmembrane distribution is still a controversial subject, since experiments have not been able to measure it directly. To understand the background of this issue, one has to consider how the asymmetric distribution of lipids is typically measured. To avoid causing artifacts, an approach that has been found useful and reliable is to employ enzymes that break lipids apart. For instance, to measure the distribution of PCs, one can expose the extracellular leaflet of a plasma membrane to phosholipases that break PCs to lyso-PCs and fatty acids. Right after the exposure, the plasma membrane content can be resolved through mass spectrometry. By repeating the same experiment separately on the cytosolic leaflet, one can resolve how PCs are distributed between the two leaflets. In a similar way, one can measure the distribution of other lipids by using, for instance, sphingomyelinases for SM.
The problem is that for cholesterol there is no enzyme of this kind. What is used instead is, for example, techniques that depend on indirect analyses that largely use membrane-perturbing probes, non-natural cholesterol derivatives, or detergents at low temperatures to extract cholesterol. Since these treatments alter the membrane structure quite severely, it is not clear how well the resulting data for cholesterol’s asymmetric distribution really matches reality. The results found for cholesterol distribution are therefore quite perplexing. Mondal et al. found in studies of the CHO cell line that 60–70% of the plasma membrane sterol is in the cytoplasmic leaflet.205 Meanwhile, in a recent work Liu et al. used a new imaging method using tunable orthogonal cholesterol sensors and found that the concentration of plasma membrane cholesterol in the inner plasma membrane leaflet was ∼12-fold lower than that in the outer leaflet.206 For comparison, for ergosterol Solanko et al. used a technique where they quantitatively replaced endogenous ergosterol by (fluorescent) dehydroergosterol and found that the majority (∼80%) of ergosterols in the yeast plasma membrane are located in the cytoplasmic leaflet.207 If the reader considers these results to be confusing, he/she is not the only one. At the moment, it would be too daring to make strong and generic conclusions about the distribution of cholesterol in the plasma membrane.
It is quite certain that the cholesterol distribution depends on cell type, membrane curvature, the proteins in the membrane (as many of them act as sensors of cholesterol) and the activity of flippases and floppases translocating cholesterol. The cholesterol transporter ABC1 transfers cholesterol from the cell membrane to the high density lipoprotein particle.208,209 The lack of known enzymes that actively transport cholesterol between the leaflets suggests that the cholesterol distribution solely depends on the distribution of other membrane components. Besides, due to the fast flip–flop rate of cholesterol, metastable distributions resulting from kinetic barriers are unlikely. Cholesterol has a high affinity for sphingomyelin, which is mainly located in the extracellular leaflet. Recent studies showed that the length of the acyl chain modulates the affinity of cholesterol for sphingomyelin.210,210 Long chains interdigitating into the opposite leaflet promote localization of cholesterol into the opposite leaflet. However, to make the situation more complicated, cholesterol has high affinity also for PS,211−213 which is mainly located in the intracellular leaflet. The interplay of these factors can in part explain the conflicting results about the transmembrane cholesterol distribution.
The asymmetry of the plasma membrane is well established. However, there are also other biomembranes that are characterized by an asymmetric lipid distribution, too, such as the membranes of the synaptic vesicles,214 and cellular organelles including the endoplasmic reticulum,215 and the Golgi apparatus.216,217
Example of Lipidomics 1: Cells and Exosomes Derived from Them
Although the combinations of different headgroups and acyl chains yield a vast number of possible lipid species, cells incorporate only a small subset of them. This is demonstrated by a lipidomics study of PC3-prostate cancer cells and exosomes. A total of 277 different lipid species were found, of which 60 were found only in cells, 190 in both cells and exosomes, and 27 only in exosomes.96 In cells, 72% were phospholipids, 19% were cholesterol, 7% were sphingomyelins, and the remaining 2% were DAG, ceramide, and glycolipids. For comparison, exosomes were enriched in cholesterol (44%) and sphingomyelin (16%), while they were depleted in phospholipids (38%). Exosomes contained also more PS and more lipids with longer acyl chains (20-carbon-long instead of the most frequent 18-carbon-long) and sphingomyelin with 22- and 24-carbon-long chains. Subsequent studies showed that long sphingomyelin chains penetrate deeply into the opposite leaflet, modifying its structure.218,219
Example of Lipidomics 2: Remodeling of the Postsynaptic Cell Membrane During Neural Development
There are known examples of membranes whose compositions change during their lifetime; this is particularly well understood for the membranes of eye lenses.220 These changes are observed over a period of many years and result from aging. What is even more exciting, lipid composition can be modulated during the cellular development. For the first 60 postnatal days, postsynaptic membranes isolated from rat brains were shown to change in their lipid composition.221 During this period, the amount of cholesterol, sphingolipids, and PE plasmalogens increased and the amount of PC decreased. Moreover, unsaturation and acyl chain length were observed to increase for glycerol phospholipids. In particular, the amount of polyunsaturated ω-3 chains increased, while the amount of saturated and monounsaturated chains decreased over a period of 60 days. The changes in the lipid composition may lead to ordered domain formation and stabilization as shown in biophysical experiments.
Example of Lipidomics 3: Effect of Obesity on Lipid Profile
Obesity is characterized by the accumulation of an excessive amount of fat in the adipose tissue. Obesity frequently leads to cardiovascular illnesses and metabolic complications, such as insulin resistance. In order to understand the effects of excessive fat accumulation in the adipose tissue, lipidomics analysis of adipose tissue samples was performed.222 Samples originated from identical twins chosen such that one of the twins was morbidly obese, while the second one had normal weight. The results of lipidomics studies showed substantial remodeling of the lipid profile in adipose tissues in response to the excessive fat accumulation. In particular, concentration of palmitoleic and arachidonic fatty acids was elevated. Other lipid types with increased concentration were PEs and plasmalogens. In particular, PE plasmalogen species with arachidonic acid chains were accumulated. Surprisingly, the physicochemical properties of lipid bilayers designed to mimic the observed lipid profiles of the normal and obese individuals were found to be very similar.
3.2. Membrane Proteins
3.2.1. Membrane Proteins in Numbers
Proteins are the second main component of biological membranes. They contribute up to 25–75% of the membrane mass223 with high variability among the membrane types. For example, in synaptic vesicles, it has been estimated that proteins constitute 25 mol % of the inner and 18 mol % of the outer leaflet224 and that there are 20 lipids per transmembrane helix. Membrane proteins are traditionally divided into 8 classes: single-pass membrane proteins, which have a single transmembrane helix (4 classes), multipass transmembrane proteins, GPI-anchored proteins, lipid-anchored proteins, and peripheral membrane proteins.223 Most transmembrane proteins have α-helical transmembrane segments, but some are also of the β-barrel type.
In the human genome, about 26% of genes encode membrane proteins.225 In HeLa cells (the oldest stable human cell line, derived from epithelial cancer),226 30% of the expressed proteins are membrane proteins, contributing to only 7–8% of the total protein mass.227 Genomic analysis of 29 prokaryotic and eukaryotic species showed that 15–30% of genes encode α-helical membrane proteins;228 for humans, this number is 25–30%.229 Interestingly, no correlation between the number of transmembrane proteins and the complexity of the organism has been found except for 7-helix transmembrane proteins, which are more frequent in eukaryotes, and 6- and 12-helix proteins, which are more frequent in prokaryotes. β-barrel transmembrane proteins are less frequent than the helical ones with less than 3% of the prokaryotic genome coding them.230 (For a review of the function and evolution of β-barrel transmembrane proteins, see Chaturvedi and Mahalakshmi.231)
Further analysis of the human genome has indicated that there are at least 3399 helical membrane protein species classified into 230 families.232 Among these proteins, the largest group is comprised of membrane receptors coded by 1352 genes. The largest group of receptors is the G protein-coupled receptor (GPCR) family encoded by 901 genes, followed by receptors of the immunoglobulin superfamily encoded by 149 genes, and tyrosine kinase type receptors encoded by 72 genes. More than 60% of existing drugs target membrane proteins, mainly GPCRs, highlighting their pharmacological importance.233,234 The second most abundant group of membrane proteins are transporters and channels, which are coded by 817 genes.232 These two major groups are followed by membrane enzymes with 533 members, adhesion proteins with 187 members, and 181 members with unknown functions.232
The number of transmembrane helices in membrane proteins ranges from 1 to 14.235 Constituting 43–45% of helical membrane proteins, the single pass or bitopic membrane proteins, which have a single transmembrane helix, are the most numerous. The recently created Membrane Database lists about 6000 members of bitopic membrane proteins from various organisms.236 Interestingly, the number of bitopic proteins and their functional diversity have increased during evolution with increasing organismal complexity.237 Human cell membranes encompass more bitopic proteins than those of lower organisms, particularly those that function as receptors. Indeed, the majority of human bitopic membrane proteins are receptors, enzymes, and adhesion proteins. Interestingly, bitopic membrane proteins are more often associated with the cell membrane than the subcellular membranes. Bitopic membrane proteins frequently form homodimers (for an extensive review, see ref (238)) Recently, a number of bioinformatics tools for dimer structure prediction and databases for storing experimentally and theoretically predicted structures were created.239−241 Analysis of interfaces between two transmembrane helices revealed the existence of a few conserved dimerization motifs, with the most frequent being the GXXXG motif.242,243
The second most frequent transmembrane proteins are those with seven transmembrane helices constituting 10–15% of all membrane proteins.235 The most prominent members of this group are the GPCRs. Finally, proteins with 2, 4, and 12 transmembrane helices each represent 5–10% of membrane proteins.235
3.2.2. Insight into Structure of Integral Membrane Proteins
Despite the abundance and medical importance of membrane proteins, information on their three-dimensional structures is somewhat limited. In the PDB database only about 2% of the entries include membrane proteins. We note that this number also includes extramembrane domains of bitopic proteins or structures of single transmembrane helices. As of 18 April 2018, the database of three-dimensional membrane protein structures maintained by Stephan White (UC Irvin) included 2506 entries of PDB records and 772 unique proteins (http://blanco.biomol.uci.edu/mpstruc/). Information concerning membrane protein structures is collected in a few independent databases. An extensive comparison of these can be found in Shimizu et al.244
The primary method for resolving the protein structure is X-ray crystallography.245,246 Determining the structure of proteins is already difficult in itself, but determining the structure of membrane proteins with crystallography is all the more difficult. Since the first protein structure was solved in 1955 for myoglobin,247 it took 27 years to solve the first membrane protein structure–the photosynthetic reaction center.248 Prior to crystallization, membrane proteins have to be extracted from the membrane, purified, expressed, and embedded in lipid structures such as micelles. Finally, the challenge is crystallization. Alternatively, proteins may be embedded into cubic phases, which have repetitive crystal-like structures. Membrane protein crystallography typically employs about ten different detergents249 to embed proteins into micelles, and monoolein to form cubic phases.250 Recently, lipids such as cholesterol or its more soluble derivative, cholesteryl hemisuccinate, have frequently been used as cosolvents.163,251,252 To make proteins to form crystals, the proteins are usually heavily modified, including removal of glycosylation, introduction of stabilizing point mutations, inclusion of scaffolding proteins such as antibodies, and removal of flexible loops (Figure 9).253 These modifications can alter the native structure, causing, for example, the arrangement of protein domains to change or the protein structure to partially unfold. Insulin receptor structures strikingly exemplify such crystallographic artifacts. While the heavily modified crystal structure captured a U-shaped arrangement of the dimer,254 electron microscopy of the unmodified protein contested that by revealing a T-shaped arrangement.254 Recent simulations of Manna et al. provide another illustrative example of this issue.255 They used the crystal structure of β2AR (one member of the GPCR protein family) as the basis of their atomistic simulations and studied what happens when the structural modifications made to the protein structure for crystallization were reversed one at a time. It turned out that the final protein structure without structural modifications was clearly different from the underlying protein structure that included the modifications.
Figure 9.

An example of membrane protein (β2AR) engineering toward successful crystallization. Removal of stabilizing point mutations (PDB 3D4S):256 (A) mutation E1223.41W stabilizing the structure, (B) mutation N187E introduced to avoid glycosylation. The altered proteins are shown on the left panels and the native protein on the right. The spheres colored according to the atom type represent the native and mutated residues. The neighboring residues are shown in stick representation. A water molecule is shown as a ball-and-stick representation. Reproduced with permission from ref (255). Copyright 2015 American Chemical Society.
The second most common method to solve protein structure is nuclear magnetic resonance (NMR) spectroscopy.257 Currently, there are about 100 structures of membrane proteins or their fragments in the PDB database that have been determined by NMR. The majority of these are small proteins with one or two transmembrane helices. A few larger proteins, such as the GPCR chemokine receptor,258 could also be structurally characterized with NMR. Electron paramagnetic resonance (EPR) spectroscopy combined with site-directed spin labeling is capable of providing information about intramolecular distances allowing the determination of protein structure. However, it is rarely used, since it requires many site-directed mutations to resolve the whole structure. (For a review, see Bordignon and Bleicken.259)
In recent years, cryo-electron microscopy (CryoEM) has emerged as an important method in structural biology.260−263 As CryoEM does not require crystals, the proteins do not need to be modified. Besides, sample preparation is less complicated in comparison to X-ray crystallography. Recent developments that allow the use of nanodiscs have given CryoEM yet another advantage, as nanodiscs are better membrane mimetics than micelles.264−267 The main drawback of CryoEM is its low resolution. Nevertheless, at least 124 membrane protein structures have been solved with this method (based on the database “Membrane proteins of known 3D structure”268). Computational methods such as MD or machine learning have also been used to predict the structures of membrane proteins; however, this task is still challenging. (For a review, see Almeida et al.269)
3.2.3. Lipids as a Structural Element of Transmembrane Proteins
Lipids are often tightly bound to membrane proteins. Therefore, they are extracted from membranes together with proteins and appear in crystal structures. In early studies of bacteriorhodopsin, it was estimated that the protein might be cocrystallized with at least ten lipids, and electron density was observed for six of these.270 More recent mass spectroscopy studies showed that the stabilizing effect of protein–lipid binding on the protein structure depended on both the number and the type of bound lipids.271 For instance, the binding of a single cardiolipin to Aquaporin Z appears to stabilize the protein to a similar degree as the binding of three PC molecules.271 Lipids not only stabilize membrane proteins but also regulate their function in various ways (for reviews, see refs (272 and 273)).
Table 2 lists the PDB data bank entries of membrane proteins that contain lipids resolved as a part of the protein structure. While more than 245 reports of such cases exist in the PDB data bank, the number of distinct proteins is slightly over 100 (May 2018). A previous report from 2014 listed 100 entries with noncovalently bound lipids in protein structures,274 highlighting the accumulating evidence for tight lipid–protein association. Such structures predominantly belong to helical proteins with only two single helices and five β-barrel proteins.
Table 2. Membrane Proteins the Structures of Which Have Been Solved Together with Lipids.
| protein | lipids | PDB ID | year | ref |
|---|---|---|---|---|
| (1) Light-Harvesting Complex 1 | ||||
| Blastochloris viridis | cardiolipin | 5Y5S | 2018 | (1378) |
| PG | ||||
| PE | ||||
| spirilloxanthin | ||||
| ubiquinone-8 | ||||
| menaquinone-8 | ||||
| Thermochromatium tepidum | dihydroneurosporene | 6ET5 | 2018 | (1379) |
| menaquinone-9 | ||||
| ubiquinone-9 | ||||
| Thermochromatium tepidum | PE | 4V8K, 3WMM | 2014 | (1380) |
| PG | ||||
| spirilloxanthin | ||||
| ubiquinone-8 | ||||
| menaquinone-8 | ||||
| (2) Photosynthetic Reaction Center | ||||
| Heliobacterium modesticaldum | PG | 5V8K | 2017 | (1381) |
| diaponeurosporene | ||||
| Thermochromatium tepidum | PE | 1EYS | 2000 | (1382) |
| spirilloxanthin | ||||
| menaquinone-8 | ||||
| Rhodobacter sphaeroides | cardiolipin | 4N7K | 2014 | (1383) |
| PC | ||||
| GLU-GAL-DG | ||||
| spheroidene | ||||
| Rhodobacter sphaeroides | cardiolipin | 2J8C, 2UXM, 2UXL, 2UXK, 2UXJ, 2UX5, 2UX4, 2UX3, 2UWW, 2UWV, 2UWU, 2UWT, 2UWS, 2J8D | 2007 | (1384) |
| PC | ||||
| GLU-GAL-DG | ||||
| spheroidene | ||||
| ubiquinone-10 | ||||
| Rhodobacter sphaeroides | cardiolipin | 1OGV | 2003 | (1385) |
| ubiquinone-10 | ||||
| Rhodobacter sphaeroides | ubiquinone-10 | 1AIJ, 1AIG | 1997 | (1386) |
| Rhodobacter sphaeroides | spirilloxanthin | 1PSS, 1PST | 1994 | (1387) |
| ubiquinone-10 | ||||
| Rhodobacter sphaeroides | ubiquinone-10 | 2RCR | 1991 | (1388) |
| Blastochloris viridis | DAG | 4CAS | 2013 | (1389) |
| octaprenyl-pyrophosphate | ||||
| dihydroneurosporene | ||||
| menaquinone-7 | ||||
| Blastochloris viridis | dihydroneurosporene | 2WJN, 2WJM | 2009 | (1390) |
| menaquinone-7 | ||||
| Rhodopseudomonas viridis | diaponeurosporene | 1PRC, 2JBL, 1DXR | 1995 | (1391) |
| menaquinone-7 | ||||
| ubiquinone-1 | ||||
| (3) Light-Harvesting Complex of Photosystem II | ||||
| Pisum sativum (Pea) | PG | 2BHW | 2005 | (1392) |
| DGDG | ||||
| violaxanthin | ||||
| lutein | ||||
| neoxanthin | ||||
| Spinacia oleracea (spinach) complex CP29 | violaxanthin | 3PL9 | 2011 | (1393) |
| lutein | ||||
| neoxanthin | ||||
| Spinacia oleracea (spinach) | PG | 1RWT | 2004 | (1394) |
| DGDG | ||||
| violaxanthin | ||||
| lutein | ||||
| neoxanthin | ||||
| Rhodospirillum molischianum | lycopene | 1LGH | 1996 | (1395) |
| Rhodopseudomonas acidophila | rhodopin b-d-glucoside | 1NKZ, 1KZU | 2003 | (1396,1397) |
| 1997 | ||||
| (4) Supercomplex of Light-Harvesting Complex of Photosystem II of C2S2M2-type | ||||
| Pisum sativum (Pea) | PG | 5XNL, 5XNO, 5XNN, 5XNM | 2017 | (279) |
| MGDG | ||||
| DGDG | ||||
| sulfoquinovosyl-DG | ||||
| violaxanthin | ||||
| lutein | ||||
| neoxanthin | ||||
| β-carotene | ||||
| plastoquinone-9 | ||||
| (5) Photosystem II | ||||
| Cyanidium caldariu (red alga) | PG | 4YUU | 2016 | (1398) |
| MGDG | ||||
| DGDG | ||||
| sulfoquinovosyl-DG | ||||
| β-carotene | ||||
| plastoquinone-9 | ||||
| Thermosynechococcus vulcanus | PG | 5WS5, 5WS6, 5GTI, 5GTH | 2017 | (1399) |
| MGDG | ||||
| DGDG | ||||
| sulfoquinovosyl-DG | ||||
| β-carotene | ||||
| plastoquinone-9 | ||||
| Thermosynechococcus vulcanus | PG | 4UB6, 4UB8 | 2015 | (1400) |
| MGDG | ||||
| DGDG | ||||
| sulfoquinovosyl-DG | ||||
| β-carotene | ||||
| plastoquinone-9 | ||||
| Thermosynechococcus vulcanus, Thermosynechococcus elongatus (strain BP-1) | PG | 4IL6 | 2013 | (1401) |
| MGDG | ||||
| DGDG | ||||
| sulfoquinovosyl-DG | ||||
| β-carotene | ||||
| plastoquinone-9 | ||||
| Thermosynechococcus vulcanus | PG | 3WU2, 3ARC | 2011 | (1402) |
| MGDG | 5V2C | 2017 | (1403) | |
| DGDG | ||||
| sulfoquinovosyl-DG | ||||
| β-carotene | ||||
| plastoquinone-9 | ||||
| Thermosynechococcus elongatus (strain BP-1) | PG | 4RVY, 4PBU | 2014 | (1404) |
| MGDG | ||||
| DGDG | ||||
| sulfoquinovosyl-DG | ||||
| β-carotene | ||||
| plastoquinone-9 | ||||
| Thermosynechococcus vulcanus | PG | 3A0B, 3A0H | 2009 | (1405) |
| MGDG | ||||
| DGDG | ||||
| β-carotene | ||||
| plastoquinone-6 | ||||
| Thermosynechococcus vulcanus, Thermosynechococcus elongatus (strain BP-1) | β-carotene | 1IZL | 2003 | (1406) |
| Thermosynechococcus elongatus (strain BP-1) | PG | 5KAF, 5TIS, 5KAI | 2016 | (1407) |
| MGDG | ||||
| DGDG | ||||
| sulfoquinovosyl-DG | ||||
| β-carotene | ||||
| plastoquinone-9 | ||||
| Thermosynechococcus elongatus (strain BP-1) | PG | 5E7C, 5E79 | 2016 | (1408) |
| MGDG | ||||
| DGDG | ||||
| sulfoquinovosyl-DG | ||||
| β-carotene | ||||
| plastoquinone-9 | ||||
| Thermosynechococcus elongatus (strain BP-1) | PG | 4V62, 3BZ2, 3BZ1 | 2009 | (1409) |
| MGDG | ||||
| DGDG | ||||
| sulfoquinovosyl-DG | ||||
| β-carotene | ||||
| plastoquinone-9 | ||||
| Thermosynechococcus elongatus (strain BP-1) | PG | 2AXT | 2005 | (1410) |
| MGDG | ||||
| DGDG | ||||
| sulfoquinovosyl-DG | ||||
| β-carotene | ||||
| plastoquinone-9 | ||||
| Thermosynechococcus elongatus (strain BP-1) | β-carotene | 1S5L | 2004 | (1411) |
| plastoquinone-9 | ||||
| (6) Supercomplex of Photosystem I with Light-Harvesting Complex I | ||||
| Pisum sativum (pea) | PG | 4XK8 | 2015 | (1412) |
| MGDG | ||||
| DGDG | ||||
| β-carotene | ||||
| violaxanthin | ||||
| lutein | ||||
| phylloquinone | ||||
| (7) Photosystem I | ||||
| Pisum sativum (pea) | MGDG | 2WSC, LW5, 2WSF, 2WSE | 2010 | (1413) |
| β-carotene | ||||
| phylloquinone | ||||
| Thermosynechococcus elongatus (strain BP-1) | MGDG | 1JB0 | 2001 | (1414) |
| β-carotene | 2012 | |||
| phylloquinone | 2011 | |||
| MGDG | 4FE1 | 2012 | (1415) | |
| β-carotene | ||||
| phylloquinone | ||||
| PG | ||||
| MGDG | 3PCQ | 2011 | (1416) | |
| β-carotene | ||||
| phylloquinone | ||||
| diaponeurosporene | ||||
| (8) Photosystem I Supercomplex with Light-Harvesting Complexes I and II | ||||
| Zea mays | PG | 5ZJI | 2018 | (1417) |
| MGDG | ||||
| DGDG | ||||
| violaxanthin | ||||
| β-carotene | ||||
| phylloquinone | ||||
| Cyanidioschyzon merolae | PG | 5ZGB | 2018 | (1418) |
| DGDG | 5ZGH | |||
| β-carotene | ||||
| phylloquinone | ||||
| (9) Mammalian Respiratory Supercomplex | ||||
| Sus scrofa | PC | 5GUP | 2016 | (278) |
| PE | ||||
| cardiolipin | ||||
| (10) Cytochrome Oxidase | ||||
| Thermus thermophilus | DAG | 2YEV | 2012 | (1419) |
| native Thermus lipid (5PL) | ||||
| (10) Cytochrome c Oxidase | ||||
| Paracoccus denitrificans | PC | 1QLE | 1997 | (1420) |
| Bos taurus | PC | 5W97, 5WAU | 2017 | (1421) |
| PE | ||||
| PG | ||||
| cardiolipin | ||||
| triacylglycerol | ||||
| cholic acid | ||||
| Bos taurus | PC | 5XDQ | 2018 | (1422) |
| PE | ||||
| PG | ||||
| cardiolipin | ||||
| triacylglycerol | ||||
| cholic acid | ||||
| Bos taurus | PC | 5Z84, 5ZCQ, 5ZCP, 5ZCO, 5Z86, 5Z85 | 2018 | (1423) |
| PE | ||||
| PG | ||||
| cardiolipin | ||||
| tristearoylglycerol | ||||
| cholic acid | ||||
| Bos taurus | PC | 1V54, 1V55 | 2003 | (1424) |
| PE | ||||
| PG | ||||
| cardiolipin | ||||
| triacylglycerol | ||||
| cholic acid | ||||
| (11) Cytochrome b6f | ||||
| Nostoc sp. | PC | 4H44, 4H13, 4H0L | 2013 | (1425) |
| PA | ||||
| sulfoquinovosyl-DG | ||||
| β-carotene | ||||
| Chlamydomonas reinhardtii | MGDG | 1Q90 | 2003 | (1426) |
| sulfoquinovosyl-DG | ||||
| β-carotene | ||||
| Mastigocladus laminosus | MGDG | 2E74, 2E76, 2E75 | 2007 | (1427) |
| sulfoquinovosyl-DG | ||||
| β-carotene | ||||
| Mastigocladus laminosus | PC | 2D2C | 2006 | (1428) |
| β-carotene | ||||
| Mastigocladus laminosus | PC | 1VF5 | 2003 | (1429) |
| β-carotene | ||||
| plastoquinone-9 | ||||
| (12) Cytochrome bc1 Complex | ||||
| Rhodobacter sphaeroides | PE | 2FYN, 2FYU | 2006 | (1430) |
| Saccharomyces cerevisiae | PG | 3CX5, 3CXH | 2008 | (1431) |
| PA | ||||
| cardiolipin | ||||
| Saccharomyces cerevisiae | PC | 1KB9 | 2001 | (1286) |
| PE | ||||
| PG | ||||
| PIP | ||||
| cardiolipin | ||||
| ubiquinone-6 | ||||
| Gallus gallus | PE | 1BCC, 3BCC, 3H1J, 3H1I, 3H1H, 2BCC | 1998 | (1432) |
| ubiquinone-10 | ||||
| PE | 3CWB | 2008 | (1433) | |
| ubiquinone-10 | ||||
| cardiolipin | ||||
| Bos taurus | PE | 1PPJ, 2A06, 1PP9 | 2005 | (1434) |
| cardiolipin | ||||
| (13) Complex II | ||||
| Gallus gallus | PE | 2FBW, 1YQ4, 1YQ3 | 2006 | (1435) |
| 2H89, 2H88 | (1436) | |||
| Sus scrofa | PE | 1ZOY, 1ZP0 | 2005 | (1437) |
| Escherichia coli (strain K12) | cardiolipin | 2ACZ, 2AD0 | 2006 | (1438) |
| Escherichia coli | PE | 1NEK, 1NEN | 2003 | (1439) |
| cardiolipin | ||||
| (14) Formate Dehydrogenase | ||||
| Escherichia coli | cardiolipin | 1KQF, 1KQG | 2002 | (1440) |
| (15) Complex I | ||||
| Ovis aries | PC | 5LNK | 2016 | (277) |
| PE | ||||
| cardiolipin | ||||
| Mus musculus | PC | 6G2J | 2018 | (1441) |
| PG | ||||
| cardiolipin | ||||
| lipid fragment | ||||
| Yarrowia lipolytica | PE | 6GCS | 2018 | (1442) |
| cardiolipin | ||||
| lipid fragment | ||||
| (17) Nitrate Reductase | ||||
| Escherichia coli | PG | 1Y4Z, 1Y5N, 1Y5L, 1Y5I | 2005 | (1438) |
| Escherichia coli | PA | 1SIW | 2004 | (1443) |
| Escherichia coli | PG | 1Q16 | 2003 | (1444) |
| PA | ||||
| (18) Gastric Proton Pump | ||||
| Sus scrofa | PC | 5YLU, 5YLV | 2018 | (1445) |
| (19) Copper-Transporting PIB-ATPase | ||||
| Legionella pneumophila | PC (headgroup only) | 4BBJ | 2014 | (1446) |
| (20) Na+,K+-ATPase | ||||
| Squalus acanthias | cholesterol | 5AVQ, 5AW9, 5AW8, 5AW7, 5AW6, 5AW5, 5AW4, 5AW3, 5AW2, 5AW1, 5AW0, 5AVZ, 5AVY, 5AVX, 5AVW, 5AVV, 5AVU, 5AVT, 5AVS, 5AVR | 2015 | (1447) |
| Squalus acanthias | cholesterol | 3A3Y | 2009 | (1448) |
| Squalus acanthias | cholesterol | 2ZXE | 2009 | (1449) |
| Sus scrofa | cholesterol | 4RES | 2015 | (1450) |
| Sus scrofa | PC | 3WGU | 2013 | (1451) |
| cholesterol | ||||
| Sus scrofa | cholesterol | 4HQJ | 2013 | (1452) |
| Sus scrofa | PS | 4HYT | 2013 | (1453) |
| cholesterol | ||||
| Sus scrofa | PC | 3B8E, 3KDP | 2007 | (1454) |
| (21) Ca2+-ATPase | ||||
| Oryctolagus cuniculus | PC | 5XA7, 5XAB, 5XAA, 5XA9, 5XA8 | 2017 | (1455) |
| Oryctolagus cuniculus | PC | 4NAB | 2013 | (1456) |
| Oryctolagus cuniculus | PE | 3W5A, 3W5D, 3W5C, 3W5B | 2013 | (1457) |
| Oryctolagus cuniculus | PE | 2AGV, 2ZBG, 2ZBF, 2ZBE, 2ZBD, 2Z9R, 2EAU, 2EAT, 2EAS, 2EAR, 2DQS | 2005 | (1458) |
| (22) Chloroplast ATP Synthase c-Ring | ||||
| Pisum sativum | DGDG | 3V3C | 2012 | (1459) |
| (22) Rotor of the V-type ATPase | ||||
| Enterococcus hirae | PG | 2BL2 | 2005 | (1460) |
| (23) Potassium-Importing KdpFABC Membrane Complex | ||||
| Escherichia coli | PC | 5MRW | 2017 | (1461) |
| (24) Niemann−Pick C1 Protein | ||||
| Homo sapiens | cholesterol | 3JD8 | 2018 | (857) |
| (25) Multidrug Resistance Protein 1 | ||||
| Bos taurus | cholesterol | 6BHU | 2018 | (1462) |
| (26) ABC Transporter | ||||
| Homo sapiens | cardiolipin | 4AYT, 4AYX, 4AYW, 3ZDQ | 2013 | (1463) |
| (27) ABC Maltose Transporter | ||||
| Escherichia coli | PG | 4KHZ, 4KI0 | 2013 | (1464) |
| Escherichia coli | PG | 4JBW | 2013 | (1465) |
| Escherichia coli | PG | 3RLF, 3PUX, 3PUW, 3PUV | 2011 | (1466) |
| (28) ABC Core-Lipopolysaccharide Transporter MsbA | ||||
| Escherichia coli | PE | 6BPL, 6BPP | 2018 | (1467) |
| lauric acid | ||||
| myristic acid | ||||
| (29) Bile Acid Transporter Homolog | ||||
| Neisseria meningitidis | PE | 3ZUY, 3ZUX | 2011 | (1468) |
| (30) Mitochondrial ADP/ATP Carrier | ||||
| Saccharomyces cerevisiae | cardiolipin | 4C9G, 4C9Q, 4C9J, 4C9H | 2014 | (1469) |
| Bos taurus | cardiolipin | 2C3E | 2005 | (1470) |
| Bos taurus | cardiolipin | 1OKC, 1YMJ, 1YM6 | 2003 | (1471) |
| (31) Sodium/Proton Antiporters | ||||
| Pyrococcus abyssi | PE | 4CZ8, 4CZA, 4CZ9 | 2014 | (1472) |
| (32) Mitochondrial Calcium Uniporter MCU | ||||
| Cyphellophora europaea | PG | 6DNF | 2018 | (1473) |
| (33) Betaine Transporter BetP | ||||
| Escherichia coli | PG | 4C7R | 2013 | (1474) |
| Corynebacterium glutamicum | PG | 4DOJ, 4AIN | 2012 | (1475) |
| (34) Translocator Protein (TSPO) | ||||
| Rhodobacter sphaeroides | PA | 4UC1, 4UC3, 4UC2 | 2015 | (1476) |
| (35) Sodium-Dependent Citrate Symporter | ||||
| Salmonella enterica | PE | 5A1S | 2015 | (1477) |
| (36) Serotonin Transporter | ||||
| Homo sapiens | cholesterol | 6AWN, 6AWQ, 6AWP, 6AWO | 2018 | (1478) |
| Homo sapiens | cholesterol | 5I6X, 5I75, 5I74, 5I73, 5I71, 5I6Z, 5I66 | 2016 | (820) |
| (37) Cationic Amino Acid Transporter | ||||
| Geobacillus kaustophilus | cholesterol | 5OQT, 6F34 | 2018 | (1479) |
| (38) Dopamine Transporter | ||||
| Drosophila melanogaster | cholesterol | 4XNU, 4XNX | 2015 | (1480) |
| Drosophila melanogaster | cholesterol | 4XP1, 4XPT, 4XPH, 4XPG, 4XPF, 4XPB, 4XPA, 4XP9, 4XP6, 4XP5, 4XP4 | 2015 | (819) |
| Drosophila melanogaster | cholesterol | 4M48 | 2013 | (818) |
| (39) Multidrug Transporter MdfA | ||||
| Escherichia coli | deoxycholic acid | 4ZP0, 4ZP2, 4ZOW | 2015 | (1481) |
| (40) Multidrug Efflux Transporter AcrB | ||||
| Escherichia coli | PE | 5JMN | 2016 | (1482) |
| (41) γ-Secretase | ||||
| Homo sapiens | PC | 5A63 | 2015 | (1483) |
| (42) Zinc Metalloprotease | ||||
| Homo sapiens | PC | 4AW6, 2YPT | 2013 | (1484) |
| (43) Rhomboid Protease GlpG | ||||
| Escherichia coli | PC | 2XTV, 2XTU | 2011 | (1485) |
| Escherichia coli | PG | 2IRV | 2007 | (1486) |
| (44) Bacterial Vitamin C Transporter UlaA/SgaT | ||||
| Escherichia coli | triglyceride | 4RP9, 4RP8 | 2015 | (1487) |
| (45) Methyltransferase Icmt | ||||
| Escherichia coli | cardiolipin | 4A2N | 2011 | (1488) |
| palmitic acid | ||||
| Escherichia coli | PG | 5AZC, 5AZB | 2016 | (1489) |
| Tribolium castaneum | undecane | 5V7P | 2018 | (1490) |
| lipid fragment | ||||
| (46) 4-Amino-4-deoxy-l-arabinose Transferase | ||||
| Cupriavidus metallidurans | PC (headgroup only) | 5EZM, 5F15 | 2016 | (1491) |
| decaprenyl-phosphate | ||||
| (47) Oligosaccharyltransferase | ||||
| Saccharomyces cerevisiae | PC | 6C26 | 2018 | (1492) |
| Saccharomyces cerevisiae | PC | 6EZN | 2018 | (1493) |
| PE | ||||
| (48) Serotonin Receptor | ||||
| Mus musculus | PC | 6BE1 | 2018 | (1494) |
| (49) Synaptic GABAA Receptor | ||||
| Homo sapiens | cholesteryl hemisuccinate | 6D6U, 6D6T | 2018 | (1495) |
| (50) GluN1/GluN2B δ-ATD NMDA Receptor | ||||
| Xenopus laevis | cholesteryl hemisuccinate | 5UN1 | 2018 | (1496) |
| (51) α4β2 Nicotinic Receptor | ||||
| Homo sapiens | cholesteryl hemisuccinate | 6CNJ, 6CNK | 2018 | (1497) |
| (52) Ligand-Gated Ion Channel GLIC | ||||
| Gloeobacter violaceus | PC | 4F8H | 2012 | (1498) |
| Gloeobacter violaceus | PC | 3P50, 3P4W | 2011 | (1128) |
| Gloeobacter violaceus | PC | 4HFI, 4ILC, 4ILB, 4ILA4IL9, 4IL4 | 2013 | (1499) |
| Gloeobacter violaceus | PC | 3EAM | 2009 | (1041) |
| (53) Lipid-Gated Cation Channel TRPC3 | ||||
| Homo sapiens | DAG | 6CUD | 2018 | (1500) |
| PE | ||||
| (54) Ammonia Channel (AmtB) | ||||
| Escherichia coli | PG | 4NH2 | 2014 | (271) |
| (55) Proton-Gated Urea Channel | ||||
| Helicobacter pylori | PA | 3UX4 | 2012 | (1501) |
| (56) Urea Transporter B | ||||
| Bos taurus | ceramide | 4EZC, 4EZD | 2012 | (1502) |
| (57) Aquaporin Z | ||||
| Escherichia coli | PE | 2ABM | 2008 | (1503) |
| PG | ||||
| (58) Aquaporin 5 | ||||
| Homo sapiens | PS | 3D9S | 2008 | (1003) |
| (59) Aquaporin 4 | ||||
| Rattus norvegicus | PE | 2ZZ9 | 2009 | (1504) |
| (60) Aquaporin 0 | ||||
| Ovis aries | PE | 3M9I | 2010 | (1505) |
| Ovis aries | PC | 2B6O, 2B6P | 2005 | (1506) |
| (61) Mechanotransduction Ion Channel | ||||
| Drosophila melanogaster | PC | 5VKQ | 2017 | (1507) |
| (62) Calcium-Activated Cation TRP Channel | ||||
| Homo sapiens | PA | 5MKE, 5MKF | 2017 | (1508) |
| palmitic acid | ||||
| (63) Lysosomal Calcium-Permeable Channel TRPML3 | ||||
| Callithrix jacchus | PE | 5W3S | 2017 | (1509) |
| cholesteryl succinate | ||||
| (64) Transient Receptor Potential Melastatin 4 (TRPM4) | ||||
| Homo sapiens | cholesteryl succinate | 6BQR, 6BQV | 2018 | (267) |
| (65) nvTRPM2 Channel in Complex with Ca2+ | ||||
| Nematostella vectensis | POPC | 6CO7 | 2018 | (1510) |
| cholesterol | ||||
| (66) TRPC4 Ion Channel | ||||
| Mus musculus | PA | 5Z96 | 2018 | (1511) |
| cholesteryl hemisuccinate | ||||
| Danio rerio | PA | 6G1K | 2018 | (1512) |
| cholesteryl hemisuccinate | ||||
| (67) Voltage-Gated Calcium Channel CavAb | ||||
| Arcobacter butzleri | PC | 5KLB, 5KMH, 5KMF, 5KMD, 5KLS, 5KLG | 2016 | (1513) |
| (68) Voltage-Gated Calcium Channel CavAb | ||||
| Arcobacter butzleri | PC | 4MS2, 4MW8, 4MW3, 4MVZ, 4MVU, 4MVS, 4MVR, 4MVQ, 4MVO, 4MVM, 4MTO, 4MTG, 4MTF | 2013 | (1514) |
| (69) Trimeric Intracellular Cation Channel TRIC-B1 and TRIC-B2 | ||||
| Caenorhabditis elegans | PIP2 | 5EGI | 2016 | (1515) |
| (70) Voltage-Gated Sodium Channel | ||||
| Homo sapiens | PC | 5EK0 | 2015 | (1516) |
| Homo sapiens | PG | 6AGF | 2018 | (1517) |
| alpha proteobacterium HIMB114 | PC | 4DXW | 2012 | (1518) |
| Arcobacter butzleri | PC | 5VB2, 5VB8 | 2017 | (1519) |
| Arcobacter butzleri | PC | 4EKW | 2012 | (1520) |
| Arcobacter butzleri | PC | 3RVY, 3RW0, 3RVZ | 2011 | (1521) |
| Arcobacter butzleri | PC | 6C1E, 6C1P, 6C1M, 6C1K | 2018 | (1522) |
| (71) Magnesium Channel TRPM7 | ||||
| Mus musculus | cholesteryl hemisuccinate | 5ZX5, 6BWF, 6BWD | 2018 | (1523) |
| (72) G Protein-Gated Inward Rectifier K1 (GIRK) Channel 2 | ||||
| Mus musculus | PIP2 | 4KFM | 2013 | (1524) |
| (73) Inward Rectifier Kir2.2 Channel | ||||
| Gallus gallus | PIP2 | 3SPI, 3SPJ, 3SPH, 3SPG, 3SPC | 2011 | (1525) |
| (74) High-Conductance Ca2+-Activated K+ Channel | ||||
| Aplysia californica | PG | 5TJI | 2017 | (1526) |
| Aplysia californica | PG | 5TJ6 | 2017 | (1527) |
| (75) Voltage-Gated K+ Channel Eag1 | ||||
| Homo sapiens | cholesteryl succinate | 5K7L | 2016 | (1528) |
| (76) Voltage-Gated K+ Channel Kv1.2-2.1 Chimera | ||||
| Rattus norvegicus | PG | 5WIE | 2017 | (1529) |
| (77) Voltage-Gated K+ Channel | ||||
| Rattus norvegicus | PG | 3LNM | 2010 | (1530) |
| (78) Voltage Dependent Potassium Channel (Kv1.2−Kv2.1 Chimera) | ||||
| Rattus norvegicus | PG | 2R9R | 2007 | (1531) |
| (79) SthK Cyclic Nucleotide-Gated Potassium Channel | ||||
| Spirochaeta thermophila | PG | 6CJQ | 2018 | (1532) |
| (80) Organellar Two-Pore Channel | ||||
| Mus musculus | PIP2 | 6C9A, 6C96 | 2018 | (1533) |
| (81) Two Pore Domain Potassium Ion Channel TREK2 | ||||
| Homo sapiens | PC | 4BW5, 4XDL, 4XDK, 4XDJ | 2015 | (1534) |
| (82) Two-Pore Channel TPC1 | ||||
| Arabidopsis thaliana | PA | 6E1M, 6E1P, 6E1N, 6E1K, 6CX0 | 2018 | (1535) |
| oleic acid | ||||
| (83) Potassium Channel KcsA | ||||
| Streptomyces lividans | DAG | 5VK6, 5VKH, 5VKE | 2017 | (1536) |
| Streptomyces lividans | DAG | 5EC1, 5EC2, 5EBW, 5EBM, 5EBL | 2016 | (1537) |
| Streptomyces lividans | DAG | 2JK5, 4UUJ, 2W0F | 2014 | (1538) |
| Streptomyces lividans | DAG | 4MSW | 2013 | (1539) |
| Streptomyces lividans | DAG | 3IGA, 3GB7 | 2009 | (1540) |
| Streptomyces lividans | DAG | 1K4C, 1K4D | 2001 | (1541) |
| (84) KcsA Potassium Channel | ||||
| Streptomyces coelicolor | DAG | 6BY2, 6BY3 | 2018 | (1542) |
| (85) Patched 1 Receptor | ||||
| Homo sapiens | cholesteryl hemisuccinate | 6DMB, 6DMY, 6DMO | 2018 | (1543) |
| (86) Metabotropic Glutamate Receptor 1 (GPCR) | ||||
| Homo sapiens | cholesterol | 4OR2 | 2014 | (1544) |
| oleic acid | ||||
| (87) Corticotropin-Releasing Factor Receptor (GPCR) | ||||
| Homo sapiens | PG | 4K5Y | 2013 | (1545) |
| (88) P2Y1 Receptor 1 (GPCR) | ||||
| Homo sapiens | cholesterol | 4XNV, 4XNW | 2015 | (1546) |
| cholesteryl succinate | ||||
| (89) P2Y1 Receptor 1 (GPCR) | ||||
| Homo sapiens | cholesterol | 4PXZ, 4PY0 | 2014 | (1547) |
| Homo sapiens | cholesterol | 4NTJ | 2014 | (1548) |
| (90) Serotonin Receptor 5-HT2B (GPCR) | ||||
| Homo sapiens | cholesterol | 5TVN | 2017 | (1549) |
| Homo sapiens | DAG | 4NC3 | 2013 | (1550) |
| cholesterol | ||||
| palmitic acid | ||||
| oleic acid | ||||
| (91) Chimeric Protein of Serotonin Receptor 5-HT2B−BRIL (GPCR) | ||||
| Homo sapiens, Escherichia coli | cholesterol | 4IB4 | 2013 | (1551) |
| palmitic acid | ||||
| oleic acid | ||||
| (92) Cannabinoid Receptor CB1 (GPCR) | ||||
| Homo sapiens | cholesterol | 5XRA, 5XR8 | 2017 | (1552) |
| oleic acid | ||||
| (93) μ-Opioid Receptor (GPCR) | ||||
| Mus musculus | cholesterol | 5C1M | 2015 | (1553) |
| Mus musculus | cholesterol | 4DKL | 2012 | (1554) |
| (94) κ-Opioid Receptor (GPCR) | ||||
| Homo sapiens | cholesterol | 6B73 | 2018 | (1555) |
| (95) M1 and M4 Muscarinic Acetylcholine Receptor (GPCR) | ||||
| Homo sapiens | cholesteryl hemi-succinate | 5CXV, 5DSG | 2016 | (1556) |
| (96) Endothelin Receptor Type-B (GPCR) | ||||
| Homo sapiens | cholesterol | 5X93, 5XPR | 2017 | (1557) |
| (97) Viral Chemokine Receptor (GPCR) | ||||
| human cytomegalovirus | cholesterol | 4XT1, 4XT3 | 2015 | (1558) |
| (98) CC Chemokine Receptor 9 (CCR9) | ||||
| Homo sapiens | cholesterol | 5LWE | 2016 | (1559) |
| oleic acid | ||||
| (99) A1 and A2A Adenosine Receptors | ||||
| Homo sapiens | cholesterol | 5MZJ, 5N2S, 5N2R, 5MZP | 2017 | (1560) |
| Homo sapiens | cholesterol | 4EIY | 2012 | (1034) |
| oleic acid | ||||
| (100) β2 Adrenoceptor | ||||
| Homo sapiens | cholesterol | 5X7D | 2017 | (1561) |
| Homo sapiens | cholesterol | 3PDS | 2011 | (1562) |
| Homo sapiens | cholesterol | 3NY8, 3NYA, 3NY9 | 2010 | (1563) |
| Homo sapiens | cholesterol | 3D4S | 2008 | (256) |
| Homo sapiens | cholesterol | 2RH1 | 2007 | (1004) |
| Meleagris gallopavo | cholesterol | 2Y00, 2Y03, 2Y02, 2Y01 | 2011 | (1564) |
| (101) Isorhodopsin | ||||
| Todarodes pacificus | PC | 3AYN, 3AYM | 2011 | (1565) |
| palmitic acid | ||||
| retinal | ||||
| Todarodes pacificus | PC | 2Z73 | 2008 | (1566) |
| palmitic acid | ||||
| retinal | ||||
| (102) Rhodopsin | ||||
| Bos taurus | oleic acid | 6FK6, 6FKD, 6FKC, 6FKB, 6FKA, 6FK9, 6FK8, 6FK7 | 2018 | (1567) |
| Bos taurus | PE | 2X72 | 2011 | (1568) |
| PA | ||||
| palmitic acid | ||||
| retinal | ||||
| Bos taurus | PE | 3C9L, 3C9M | 2008 | (1569) |
| palmitic acid | ||||
| retinal | ||||
| Bos taurus | PE | 1GZM, 3C9L | 2004 | (1002) |
| palmitic acid | ||||
| retinal | ||||
| (103) Tetraspanin CD81 | ||||
| Homo sapiens | cholesterol | 5TCX | 2016 | (1570) |
| (104) STRA6 Receptor for Retinol Uptake in Complex with Calmodulin | ||||
| Danio rerio | cholesterol | 5SY1, 5K8Q | 2016 | (1571) |
| (105) Cruxrhodopsin-3 | ||||
| Haloarcula vallismortis | bacterioruberin | 4L35, 4JR8 | 2014 | (1572) |
| (106) Blue Light-Absorbing Proteorhodopsin | ||||
| uncultured bacterium | DAG | 4JQ6, 4KNF, 4KLY | 2013 | (1573) |
| retinol | ||||
| (107) Proton Pumping Rhodopsin AR2 | ||||
| Acetabularia acetabulum | cholesterol | 3AM6 | 2011 | (1574) |
| retinal | ||||
| (108) Xanthorhodopsin | ||||
| Salinibacter ruber | PC | 3DDL | 2008 | (1575) |
| salinixanthin | ||||
| (109) Archaerhodopsin-2 | ||||
| Halobacterium sp. | PC | 3WQJ | 2014 | (1576) |
| PG | ||||
| DAG | ||||
| bacterioruberin | ||||
| squalene | ||||
| (110) Sensory Rhodopsin II | ||||
| Natronomonas pharaonis | DAG | 3QDC, 3QAP | 2011 | (1577) |
| retinal | ||||
| eicosane (lipid fragment) | ||||
| (111) Sensory Rhodopsin | ||||
| Nostoc sp. | PE | 1XIO | 2004 | (1578) |
| retinal | ||||
| (112) Light-Driven Chloride Pump Halorhodopsin | ||||
| Natronomonas pharaonis | PC | 3A7K | 2010 | (1579) |
| PA | ||||
| DAG | ||||
| bacterioruberin | ||||
| retinal | ||||
| (113) Bacteriorodopsin | ||||
| Halobacterium salinarum | DAG | 6G7H, 6G7L, 6G7K, 6G7J, 6G7I | 2018 | (1580) |
| retinal | ||||
| Halobacterium salinarum | DAG | 5B6V, 5H2P, 5H2O, 5H2N, 5H2M, 5H2L, 5H2K, 5H2J, 5H2I, 5H2H, 5B6Z, 5B6Y, 5B6W, 5B6X | 2016 | (1581) |
| retinal | ||||
| Halobacterium salinarum | oxysterol | 5B35, 5B34 | 2016 | (1582) |
| retinal | ||||
| Halobacterium salinarum | DAG | 4X31, 4X325J7A | 2015 | (1583) |
| retinal | ||||
| Halobacterium salinarum | DAG | 4FPD | 2013 | (1584) |
| retinal | ||||
| Halobacterium salinarum | DAG | 3T45 | 2012 | (1585) |
| retinal | ||||
| Halobacterium salinarum | DAG | 3NS0, 3NSB | 2011 | (1586) |
| retinal | ||||
| Halobacterium salinarum | DAG | 1C8R, 1C8S | 1999 | (1587) |
| retinal | ||||
| trimethyl-tetracosane (lipid fragment) | ||||
| Halobacterium salinarum | DAG | 1C3W | 1999 | (1588) |
| retinal | ||||
| trimethyl-tetracosane (lipid fragment) | ||||
| Halobacterium salinarum | retinal | 1QHJ, 1QKP, 1QKO | 1999 | (1589) |
| trimethyl-tetracosane (lipid fragment) | ||||
| Halobacterium salinarum | DAG | 5B6V, 5H2P, 5H2O, 5H2N, 5H2M, 5H2L, 5H2K, 5H2J, 5H2I, 5H2H, 5B6Z, 5B6Y, 5B6W, 5B6X | 2016 | (1581) |
| retinal | ||||
| Halobacterium salinarum | oxysterol | 5B35, 5B34 | 2016 | (1582) |
| retinal | ||||
| Halobacterium salinarum | DAG | 4X31, 4X325J7A | 2015 | (1583) |
| retinal | ||||
| Halobacterium salinarum | DAG | 4FPD | 2013 | (1584) |
| retinal | ||||
| Halobacterium salinarum | DAG | 3T45 | 2012 | (1585) |
| retinal | ||||
| Halobacterium salinarum | DAG | 3NS0, 3NSB | 2011 | (1586) |
| retinal | ||||
| Halobacterium salinarum | DAG | 1C8R, 1C8S | 1999 | (1587) |
| retinal | ||||
| trimethyl-tetracosane (lipid fragment) | ||||
| Halobacterium salinarum | DAG | 1C3W | 1999 | (1588) |
| retinal | ||||
| trimethyl-tetracosane (lipid fragment) | ||||
| Halobacterium salinarum | retinal | 1QHJ, 1QKP, 1QKO | 1999 | (1589) |
| trimethyl-tetracosane (lipid fragment) | ||||
| Halobacterium salinarum | DAG | 1KG8, 1KGB, 1KG9 | 2001 | (1590) |
| retinal | ||||
| Halobacterium salinarum | DAG | 1M0K, 1M0L | 2002 | (1591) |
| retinal | ||||
| trimethyl-tetracosane (lipid fragment) | ||||
| Halobacterium salinarum | PA-phosphate | 2BRD | 1996 | (270) |
| retinal | ||||
| (114) Xanthorhodopsin | ||||
| Salinibacter ruber | PC | 3DDL | 2008 | (1575) |
| Salinixanthin | ||||
| (115) Archaerhodopsin-2 | ||||
| Halobacterium sp. | PC | 3WQJ | 2014 | (1576) |
| PG | ||||
| DAG | ||||
| bacterioruberin | ||||
| squalene | ||||
| (116) Sensory Rhodopsin II | ||||
| Natronomonas pharaonis | DAG | 3QDC, 3QAP | 2011 | (1577) |
| retinal | ||||
| eicosane (lipid fragment) | ||||
| (117) Sensory Rhodopsin | ||||
| Nostoc sp. | PE | 1XIO | 2004 | (1578) |
| retinal | ||||
| (118) Light-Driven Chloride Pump Halorhodopsin | ||||
| Natronomonas pharaonis | PC | 3A7K | 2010 | (1579) |
| PA | ||||
| DAG | ||||
| bacterioruberin | ||||
| retinal | ||||
| (119) Fragaceatoxin C | ||||
| Actinia fragacea | PC | 4TSY, 4TSQ, 4TSP, 4TSO, 4TSN, 4TSL, 3W9P, 3VWI | 2015 | (1592) |
| (120) Murine Voltage-Dependent Anion Channel 1 (mVDAC1) | ||||
| Mus musculus | PC | 4C69 | 2014 | (1593) |
| Mus musculus | PC | 3EMN | 2008 | (1594) |
| (121) Omp85 | ||||
| Escherichia coli | PC | 4C00, 4BZA | 2013 | (1595) |
| (122) CmeC Outer Membrane Channel | ||||
| Campylobacter jejuni | lipid fragment | 4MT4 | 2014 | (1596) |
| (123) Outer Membrane Carboxylate Channels OccK2 | ||||
| Pseudomonas aeruginosa | PE | 3SZD, 3T24, 3T20, 3T0S, 3SZV, 3SYS, 3SYB, 3SY9, 3SY7 | 2012 | (1597) |
| (124) Lanosterol 14-α-Demethylase | ||||
| Saccharomyces cerevisiae | lanosterol | 4LXJ, 5EQB, 4K0F5V5Z, 5JLC, 5HS1, 5ESN, 5ESM, 5ESL, 5ESK, 5ESJ, 5ESI, 5ESH, 5ESG, 5ESF, 5ESE, 4ZE3, 4ZE2, 4ZE1, 4ZE0, 4ZDZ, 4ZDY, 4WMZ | 2014 | (1598) |
| (125) N-Acylphosphatidyl-ethanolamine-Hydrolyzing Phospholipase D | ||||
| Homo sapiens | PE | 4QN9 | 2015 | (1599) |
| (126) Phosphoglycosyl Transferase | ||||
| Campylobacter concisus | PE | 5W7L | 2018 | (1600) |
| (127) Gasdermin | ||||
| Mus musculus | cardiolipin | 6CB8 | 2018 | (1601) |
Most frequent lipids among the membrane protein structures are PCs appearing in 59 entries for 36 distinct proteins (Table 2), followed by zwitterionic PE found in 42 entries for 28 distinct proteins. As to charged lipids, PG is the most frequent with 41 entries for 28 distinct proteins. Inositol and PIP2 are present in six structures and PS in only two. DAG appears in 35 structures, PA in 12, and oleic or palmitic acids in 15. DAG, PA, and oleic and palmitic acids occur in small quantities in biological membranes; thus, in most cases they are fragments of larger lipids, where the remaining part of a molecule is not resolved.
Cardiolipin is a unique lipid due to its role in the formation of respiratory supercomplexes in mitochondria.275 Planas-Iglesias et al. built up a database of proteins known to bind cardiolipin. Out of the total 62 proteins, 21 were cocrystallized with cardiolipin and seven were not membrane proteins.276 The database revealed distinct contributions from the cardiolipin headgroup and acyl chains to its protein-binding mechanism. Acyl chains appear to bind the helical regions of the proteins, while the headgroups show a preference for unstructured flexible regions. Positively charged amino acids are essential for binding the negatively charged cardiolipin headgroup. After the report by Planas-Iglesias et al. was published, cardiolipin has also been found in respiratory complex 1277 and in mammalian respiratory supercomplex structures.278
Cholesterol is the most common sterol observed in the crystal structures of membrane proteins. In this context, Table 2 lists 53 entries belonging to 33 distinct proteins. However, the reader is advised to realize that actually in some of the cases it is not cholesterol that is in the structure but cholesteryl hemisuccinate. Cellular experiments with reactive cholesterol analogues (so-called clickable cholesterols) reveal 250 cholesterol binding proteins, including various enzymes, channels, and receptors.187
Some less common lipids have also been cocrystallized with membrane proteins. Mono- and digalactosyl-diacyl-glycerol (MGDG and DGDG) are common lipids in plants and the predominant lipids in chloroplasts. These lipids are therefore present in structures of photosynthetic proteins.279 Carotenoids constitute a group of isoprenoid lipids also involved in photosynthesis. They have also been cocrystallized with photosynthetic complexes, reaction centers, and antennas. The cocrystallized carotenoids are not limited to the most common one, β-carotene, but also include others, such as violaxanthin, neoxanthin, and lutein. Retinol is a short carotenoid found in complex with rhodopsins and bacteriorhodopsins. Moreover, polyprenyl lipids such as ubiquinone are present in the structures of photosynthetic and respiratory chain proteins.
3.2.4. Peripheral Membrane Proteins
Peripheral membrane proteins do not have transmembrane segments but are attached to the membrane surface via polar interactions with lipids or transmembrane proteins or via an anchor that links the protein to the lipid membrane.
Peripheral membrane proteins are hard to identify, since membrane protein binding and recognition employ several mechanisms. Usually, two such mechanisms are necessary to establish stable docking of the protein to the membrane surface. The most common membrane docking mechanisms involve specific interactions with certain lipids, hydrophobic patches on the protein surface, recognition of physical properties of the bilayer, and protein lipidation. Protein–membrane binding may also require conformational changes in the structure of the protein as it approaches the membrane surface. Additionally, numerous intracellular proteins with a net positive charge form some interaction with the intracellular membranes rich in anionic lipids such as PS, PG, cardiolipin, and PIPs. Given these reasons, it is not very surprising that the perception of many proteins has changed over time. For instance, L-DOPA decarboxylase, which for a long time was considered to be water-soluble, was later discovered to be membrane-associated.280 For the same reasons, the number of peripheral membrane proteins is hard to estimate.
A few protein domains involved in membrane recognition and binding have been characterized.281 The C1 domain was first recognized in protein kinase C as a diacyl-glycerol binding protein.282 This small domain composed of 50 residues appears in about 66 human proteins. It interacts with the membrane via nonspecific electrostatic interactions with anionic lipids, and by means of several hydrophobic residues that insert themselves into the membrane core.
There are about 14 known protein domains that bind PIPs (For a recent review, see refs (281 and 283)). Among them, the 100–150 residue-long PH domain is the most common one, existing in ∼250 proteins. The PH domain can likely bind different PIP species; however binding is inhibited by Ca2+ and Mg2+ due to changes induced on the PIP headgroup.284 FYVE domains are only 70–80 amino acids long, recognizing PIP3. Membrane docking of these two domains is stabilized by hydrophobic residues that insert themselves into the membrane core located around lysine or arginine residues, which interact electrostatically with PIP phosphate groups. Additionally, dimerization can stabilize docking of the protein. The PX domain has a docking mechanism similar to that of FYVE, but it binds various PIPs. The ENTH (epsin N-terminal homology domain) and ANTH domains are also PIP-binding domains. During the docking process, a conformational rearrangement leads to the formation of an amphipathic helix, which stabilizes interactions with lipids. On the other hand, the C2 domain requires Ca2+ cations to anchor to the membrane. While it has no specific lipid-binding pocket, it interacts with various PIP species. The BAR domain is another compelling case in that it requires both PIP and bilayer curvature in order to be able to bind to a membrane with high affinity. Other domains known to dock to lipid bilayers are FERM, GOLPH3, PDZ, PROPPINS, PTB, and Tubby.
Glycolipids, in particular gangliosides, are a group of lipids that participate in signaling and recognition. Although the majority of their binding partners are transmembrane proteins, including protein kinase receptors, some water-soluble proteins also recognize and bind them. Galectins are such small proteins that have a carbohydrate recognition domain with affinity for β-galactoside-containing oligosaccharides.285 Dimers or oligomers of galectins connected via flexible linkers allow them to bind several targets simultaneously. Gangliosides also act as receptors for bacterial toxins such as cholera toxin.286 Also, HIV entry into the host cell is mediated by interactions of gangliosides with the virus protein gp120.287
PA is another intriguing signaling lipid involved in many cellular processes. Despite the large number of proteins interacting with PA, no common binding motif or domain has been found.288 Several 3–4 residue long sequences rich in arginines and lysines were found in certain proteins. Direct interaction of PA with arginine and lysine lowers the pKa of the headgroup further leading to its deprotonation. The small headgroup with a charge of −2 further strengthens PA electrostatic interactions.88,289 This mechanism, called the electrostatic-hydrogen bonding switch, has been documented for several proteins.290 Moreover, sequences composed of hydrophobic residues were also found to participate in PA binding. Interestingly, membrane binding of PA-binding proteins also depends on membrane curvature.291
Cardiolipin, as an anionic lipid, has important functions in mitochondria.275,276 Due to its double negative charge, cardiolipin is also expected to attract peripheral proteins. Examples of such proteins are mitochondrial creatine kinase, nucleoside diphosphate kinase,292 and cytochrome c.293 Further, microtubule-associated protein is involved in recognition of cardiolipin on damaged mitochondria.294 Phosphatidylglycerols might play a role akin to that of cardiolipin in mitochondria. Similarly, some peripheral proteins recognize phosphatidylglycerols selectively.295
PE has a zwitterionic headgroup, which can be recognized by proteins in the phosphatidylethanolamine-binding protein family,296 which has 400 known members. Nevertheless, anchoring of these proteins to the membrane also requires negatively charged lipids.297
Membrane curvature is a property of lipid bilayers, which may be recognized or induced by peripheral membrane proteins. (For a review, see ref (298)). In addition to the above-mentioned BAR domain proteins, various amphipathic helices recognize curved bilayer regions.
A frequent mechanism for protein anchoring to the membrane surface involves post-transcriptional modifications, for example, lipidation, which we discuss in more detail below in the section on post-translational protein modifications.
As with transmembrane proteins, lipids are also present in crystal structures of peripheral membrane proteins. For example, the glycan of ganglioside GM1 is present in the crystal structures of cholera toxin,299 capsid protein VP1 of Simian virus 40300 and polyomaviruses,301,302 AB5 toxin,303 complement factor H,304 galectin 3 CRD domain,305Agrocybe aegerita lectin AAL,306 and endoglycoceramidases.307 Interestingly, distinct conformations of glycans are observed in complement factor H and cholera toxin. PIP2 is another good example. PIP2 is present in crystal structures of focal adhesion protein vinculin,308 clathrin membrane adapter protein epsin, which has an ENTH domain, and Sla2, which has an ANTH domain.309 Furthermore, PIP2 is present also in rabphilin, which has a C2 domain and is involved in vesicular transport,310 multifunctional scaffold protein Kibra, which also has a C2 domain,311 Arf GTPase-activating protein ASAP1, which has a PH domain, HIV-1 Gag protein responsible for virus assembly,312 alpha-tocopherol transfer protein,313 cytoskeleton regulating protein neurofibromin 2,314 and phosphatidylinositol 3-kinase α.315 GM1 and PIP2 exemplify the importance of lipids for the functions of peripheral proteins and reveal the amazing range of cellular processes, where protein–lipid interactions are crucial.
3.2.5. Protein Oligomerization and Large Membrane-Associated Protein Complexes
Membrane proteins are known to form dimers, oligomers, or larger complexes incorporating multiple different proteins. The simplest case is the above-discussed dimerization of bitopic receptors, where multiple possible arrangements of transmembrane helices are possible. Other well-documented cases are known from the available crystal structures. For example, cytochrome bc1 (respiratory complex 3) is a functional dimer of three proteins, namely, cytochrome b, cytochrome c, and Riske protein.
Here we discuss examples of protein complex formation ranging from small-scale dimerization or oligomerization to formation of large supercomplexes composed of hundreds of proteins and lipids (see Figure 10).
Figure 10.

Dimer interactions in the chemokine receptor 4 (CXCR4) crystal structure. The structures include a small molecule antagonist (A, B) or a cyclic peptide CVX15 (C, D). (A, C) The surface representation of the CXCR4 dimer and (B, D) the dimer interface. The surface involved in dimerization is highlighted in dark blue and yellow, respectively. Reproduced with permission from ref (316). Copyright 2010 American Association for the Advancement of Science.
GPCRs, the largest class of transmembrane receptors, form functional oligomers (for recent reviews, see refs (317−319)). In some cases, oligomerization has been established to be obligatory for their function, such as among class-C GPCRs. For instance, metabotropic glutamate (mGlu) receptors form covalently bound dimers. When extracted and reconstituted in nanodiscs as monomers, they fail to activate G protein.320 Another receptor of this family, γ-aminobutyric acid receptor (GABAB), exists as a heterodimer of GABAB-R1 and GABAB-R2. Contrary to family C, family-A GPCR receptors can activate G protein when reconstituted as monomers.321 However, there is some evidence that they form homo- and hetero-oligomers in the cellular environment.319 Homodimers of the dopamine D2 receptor were shown to behave as a single signaling unit and simultaneously bind a single G protein.322 Other studies showed that one-third of the muscarinic M1 receptors organize into dimers in the cell membrane.323 While β1-adrenergic receptors form transient dimers, β2-adrenergic receptors form stable dimers.324 Dimerization of β2-adrenergic receptors seems to be necessary for their export from the reticulum to the plasma membrane.325 Moreover, β1-adrenergic326 and CXCR4316 receptors were crystallized as dimers revealing their dimerization interfaces. Another layer of complexity arises from the formation of hetero-oligomers, which leads to cross-talk between different signaling pathways. An example of such hetero-oligomers is the dopamine D2 receptor oligomerizing with the adenosine A2A receptor.327
Mechanisms underlying GPCR oligomerization are not well understood. However, the involvement of lipids has been discussed in the literature. These lipid-mediated mechanisms include nonspecific ones driven by a tendency to minimize the hydrophobic mismatch between the proteins and the membrane, as well as those that involve specific lipid interactions mainly with cholesterol and polyunsaturated lipids.328 Dimerization or oligomerization is not limited to GPCRs and has been shown also for other classes of transmembrane proteins, for example, bile acid transporter ASBT329 and receptor tyrosine kinase.330,331
Mitochondrial proteins are organized into complexes of various sizes. The mitochondrial respiratory chain consists of five complexes located in mitochondrial cristae, which are elongated tubes or sheets on the inner mitochondrial membrane protruding into the mitochondrial matrix (see Figure 11). Complexes 1, 3, and 4 have been shown to be organized into a supercomplex, which keeps the orientation and the position of each protein unit optimal for an efficient flow of substrates.332,333 The supercomplex also includes 200–400 lipids, including cardiolipin.35 ATP synthase (complex 5) is organized into dimers forming rows at the tip of the cristae, where the membrane curvature is maximal. This supraorganization ensures that ATP synthase uses the proton gradient generated by the supercomplex with utmost efficiency. The elongation of the cristae is controlled by other macromolecular structures connecting the inner and the outer mitochondrial membranes at the contact sites.334,335
Figure 11.

Membrane-associated protein complexes. (A1) The V-shaped dimer of the mitochondrial ATP synthase. Protomer fitted with atomistic details is taken from PDB 2WSS(338) (α-subunits, cyan; β-subunits, blue; γδε subunits, gray; OSCP, green) and PDB 3U2Y(339) (yellow, c10-ring). Reproduced with permission from ref (333). Copyright 2015 Kühlbrandt. (A2) Organization of mitochondrial cristae. Reproduced with permission from ref (340). Copyright 2015 Davies et al. (A3, left) Cryo-EM structure of the respiratory chain supercomplex composed of NADH dehydrogenase (complex 1, blue), a cytochrome bc1 dimer (complex 3, pink), and cytochrome c oxidase (complex 4, green). (A3, right) The ubiquinol binding sites of complexes 1 and 3. Arrows indicate the electron path through the supercomplex.341 Reproduced with permission from ref (332). Copyright 2011 John Wiley and Sons. (B) Translocon complex. (B1) The pretranslocation state of the ribosome with an A-site tRNA (red) and a P-site tRNA (green). An arrow shows the direction of tRNA’s traversal motion.342 (B2) Insertion of a nascent protein by the ribosome into a nanodisc membrane working with the SecYE translocon. The protein and P-site tRNA are shown in green. An arrow shows the direction of the protein’s movement.343 (B3) Bacterial ribosome with erythromycin (in red circle) at its binding site.344 Reproduced with permission from ref (345). Copyright 2015 Elsevier. (C) Formation of COPII carrier formation. Sar1 is GTPase, Sec23 and Sec24 proteins are the inner-layer coat components, and Sec13 and Sec31 are the outer-layer coat components. Reproduced with permission from ref (346). Copyright 2013 Elsevier. (D) Nuclear pore complex. The tomographic structure (EMD-3105)347 embedded into double membrane of nucleus envelope; cytoplasmic filaments and nuclear basket, which are not part of the experimental structure, are shown in cartoon representation. Reproduced with permission from ref (348). Copyright 2016 Elsevier.
Transmembrane protein synthesis is coupled to the insertion into the lipid bilayer. To accomplish this coupling, ribosomes associate with the reticular membrane–protein complex, the translocon. Translocon is composed of several units, the most crucial of which is the protein-conducting channel, Sec61.336 This highly conserved channel facilitates the translocation of the protein through the lipid bilayer as ribosome synthesizes the protein. Other important units of translocon are the signal peptidase complex, which cleaves the signal peptides, oligosaccharyltransferase complex, which catalyzes the cotranslational N-glycosylation, and two other complexes with unknown functions: the translocating chain-associated membrane protein and the translocon-associated protein complex. The overall structure of the ribosome-translocon complex is known (see Figure 11); however, not all parts of the complex are resolved in atomistic detail.337
Coat protein complex II (COPII) vesicle is responsible for the delivery of the secretory proteins from the endoplasmic reticulum to the cell membrane via the Golgi apparatus and for the transport of membrane proteins synthesized in the endoplasmic reticulum to other cellular compartments.346,349 Three different proteins responsible for inducing the membrane curvature and closing and coating the vesicle organize the formation and the disassembly of COPII vesicles, which have a size of ∼60–90 nm (Figure 11). Cargo proteins are thought to participate in vesicle formation, and additional proteins are involved in the initiation of the vesicle formation.
Another interesting lipid–protein ensemble is the nuclear pore complex, which controls the transport of proteins and RNA between the cytosol and the nucleus (Figure 11). The mass of the complex formed by ∼30 different protein species and 50–100 individual proteins is about 50–112 MDa.348,350 The complex has an 8-fold symmetry and is assembled of many preformed supercomplexes. The nucleus is surrounded by two membranes, which are connected at the nuclear pore via a highly curved bilayer. The central part of the pore is filled by flexible, unstructured proteins, which form a hydrogel responsible for the pore selectivity. The pore is permeable to small molecules. Proteins larger than 40 kDa need to associate with the transport protein responsible for the pore selectivity before they enter or exit the nucleus. This translocation mechanism is not well understood.351
3.2.6. Post-translational Modifications of Membrane Proteins
Membrane proteins are subject to post-translational modifications. The number of possible modifications is immense. Over 200 different types of post-translational amino acid modifications are known, and new ones are still systematically discovered.352 Analysis of data in Swiss-Port (2011_7) revealed 87,308 post-translational modifications detected experimentally, and 234,938 predicted on 530,264 proteins.353 The most frequent modifications are phosphorylation, acetylation, N-linked glycosylation, amidation, hydroxylation, methylation, O-linked glycosylation, and ubiquitylation.353 Two out of three proteins in the cell are estimated to be phosphorylated.354 Post-translational modifications are challenging for analytical chemistry methods, as only a small fraction of the given protein might be modified.355 Moreover, many modifications are reversible (e.g., phosphorylation, acetylation, methylation, palmitoylation, and GlcNAcylation). Experimental identifications of these modifications are made even more challenging and error prone by complicated modifications such as lysine methylation, which can lead to three different products.356 The main experimental method used for identification of protein modifications is mass spectroscopy,355 but bioinformatics-based prediction tools are also quite successful and commonly used.357 Most of the post-translational modifications are observed in all types of proteins in all cellular compartments; however, tyrosine sulfation occurs only in the transmembrane and secretory proteins.358 Multiple types of post-translational modifications may take place on a given protein during various stages of its activity (a review summarizing the post-translational modifications in class-C GPCRs is given in ref (359), see Figure 12). Also, multiple post-translational modifications regulate biological function at the organismal level (e.g., biological rhythm, see ref (360)).
Figure 12.

(A) Post-translational modifications of GPCRs. The figure shows a cellular compartment where modifications are performed. Reproduced with permission from ref (359). Copyright 2015 Elsevier. (B) Structure and process of formation of a GPI anchor. Abbreviations: GlucN, glucosylamine; GalNac, N-acetyl galactose; EtNP, phosphatidylethanolamine; ± indicates nonmandatory fragments. Reproduced with permission from ref (361). Copyright 2012 Elsevier.
Glycosylation occurs in half of the eukaryotic proteins.362 It affects, for example, the intracellular transport of membrane proteins, receptor interactions with their ligands and galectins as well as gangliosides, and dimerization and signaling of the tyrosine kinase receptor.363 Mass of the attached carbohydrates is often substantial; for example, the mass of glycans decorating the epidermal growth factor receptor, a protein composed of 1180 amino acids, is about 40 kDa for ∼220 hexose units in 15 N-glycan arms.364 Abnormal glycosylation is a regular feature of cell membrane proteins in numerous cancers. Particularly, increased sialylation, core fucosylation, branching, O-glycan truncation, and high mannose N-glycosylation are common in cancers.365 Disturbed glycosylation of Notch proteins is known to induce developmental disorders as well as cancer and other diseases.366 Similarly, glycosylation of voltage-gated calcium channels was implicated in numerous pathologies.367
O-GlcNAcylation is an O-type glycosylation, where acetylglucosamine (GlcNAc) is attached to a serine or a threonine. As observed in most of the cellular compartments and also on transmembrane proteins, O-GlcNAcylation has numerous functions (For a review, see refs (368 and 369)). However, pathological O-GlcNAcylation occurs in diabetes,370 cancer,371 and neurodegeneration.372 Nuclear pore complex is extensively O-GlcNAcylated with nearly half of the proteins modified.373 In particular, intrinsically disordered nucleoporins are heavily O-GlcNAcylated, which is considered to be a mechanism for preventing aggregation and degradation of these proteins. The roles and effects of O-GlcNAcylation are not well understood. For example, it may induce both protective and pathological effects during cardiac hypertrophy development.374
Ubiquitination is a post-translational modification, in which a small protein, ubiquitin (8.5 kDa), is attached to the protein. The main function of ubiquitination is the control of protein degradation. Examples of membrane proteins for which ubiquitination has recently been shown to control degradation include the epithelial Na+ channel and arginine transporter, Can1.375,376 Ubiquitination also controls protein trafficking and DNA repair.377,378 While the most frequent sites of modification are the lysine side chains, alternative attachment points exist: the N-terminal amine group, the hydroxyl groups of serine and threonine residues, and the thiol groups of cysteine residues.378
Lipidation is particularly important for peripheral membrane proteins as it is involved in membrane binding, but it also occurs in transmembrane proteins. There are many possible lipids used in lipidation, including acyl chains, isoprenoids, sterols, phospholipids, and glycosylphosphatidylinositol (GPI). The most common acyl chains attached to proteins are the myristoyl and the palmitoyl chains (For extensive reviews, see refs (379 and 380)). N-myristoylation is an attachment of the myristoyl chain to the N-terminus, which contains the consensus sequence Met-Gly-X-X-X-Ser/Thr.379 First, the methionine is removed, and then myristoyl is attached via the amide group to the glycine. Myristoylation has been identified in about 150 mammalian proteins.379 Myristoylation is not likely to drive protein docking to the membrane surface on its own,379 as it contributes only 8 kcal/mol to the membrane binding free energy.381 For this reason, a second contribution is also needed, which might be the presence of hydrophobic or basic amino acids, favorable interactions with other membrane proteins, or a second lipidation.379
S-Palmitoylation is the attachment of a palmitoyl chain to the cysteine residues. While there seems to be no consensus sequence for S-palmitoylation, bioinformatics tools can predict S-palmitoylation sites.382 It is estimated that 10% of human proteins are palmitoylated.383 Palmitoylation, unlike myristoylation, can drive protein docking to the membrane on its own.379 N-Palmitoylation occurring at the N-terminus of the protein is a rare type of palmitoylation. Cholesterolylation, also uncommon, is known as a second lipidation after N-palmitoylation of the hedgehog signaling protein.384,385 Other fatty chains used to lipidate proteins are octanoyl, palmitoleic, stearoyl, and arachidonic acids.379
Prenylation is an attachment of the isoprenoid chain to the C-terminus of proteins favoring their membrane association. The two types of prenylation are farnesylation and geranylgeranylyzation. Farnesyl is a 15-carbon and geranyl–geranyl a 20-carbon isoprenoid. In prenylation, isoprenoids are attached to a cysteine side chain at the C-terminus, which has the consensus sequence of C-a-a-X, where C is cysteine and “a” is an aliphatic amino acid.386−388 Before prenylation, the last three residues are removed, and the C-terminal carboxylic group of cysteine is methylated making the C-terminus hydrophobic. In mammals, there are about 50 known farnesylated and about 170 known geranylgeranylated proteins, among which are Ras and Rab proteins involved in cellular signaling.389 For this reason, prenylation is targeted in cancer therapies.389,390
Approximately 150 proteins are GPI-anchored. GPI is a glycosylated phosphatidylinositol molecule with a glycosyl moiety composed of a glucosamine and three subsequent mannose units.361,391 At the first and the third mannose, ethanolamine-phosphate groups are attached (Figure 12). The ethanolamine-phosphate moiety located at the third mannose acts as the connection point for the protein C-terminus. Polar parts of GPI may differ between organisms or even between cells, having additional carbohydrates, ethanolamine-phosphate groups, or palmitoyl.391 GPI anchor is essential for protein trafficking and sorting.392 The synthesis of GPI is complex and requires substantial remodeling of the molecule, including, for instance, the replacement of the polyunsaturated chain of PI (the stem of the GPI anchor) with a saturated one.391 In humans, 26 genes are involved in the synthesis and maturation of the GPI anchor.393 Thirteen known mutations of these genes lead to inherited genetic disorders, which manifests with various syndromes. These genes are also associated with cancers and prion diseases.393
Lipidation of proteins with phospholipids has been shown only for yeast Apg8 protein located in the autophagosome and for its mammalian analog LC3. The attached lipid was phosphatidylethanolamine.394,395 Interestingly, this modification occurs only for membranes with large defects, such as in highly curved bilayers.396
Finally, oxidation is another protein modification, which can be enzymatically catalyzed as a part of a healthy cellular function. But, oxidation can also result from pathological oxidative stress due to free radicals, metal-catalyzed reactions, or ozone.397−400 Indeed, ozone or nitrogen dioxide inhalation may lead to protein oxidation in lungs.401 A possible consequence of oxidation is protein backbone cleavage or side chain modifications to many amino acids (see Table 3).399,400,402 Oxidized proteins are effectively repaired or degraded; however, they can accumulate in pathological states,403−405 such as in cardiovascular diseases,406 Alzheimer disease,407 and aging.408
Table 3. Oxidation Products of Amino Acids.
| amino acid | product |
|---|---|
| (1) Oxidative Modifications of Amino Acid Residue Side Chains399,400,409,410 | |
| arginine | γ-glutamyl semialdehyde, glyco-oxidation adducts |
| cysteine | disulfides, cysteic acid, glutathione cysteine, lipid peroxidation adducts, amino acid oxidation adducts, sulfenic acid, sulfinic acid, sulfonic acid, nitroso-cysteine |
| glutamic acid | oxalic acid, pyruvate adducts, hydro-peroxides |
| histidine | aspartate |
| asparagine | |
| 2-oxo-histidine | |
| 4-hydroxy-glutaminic acid | |
| lipid peroxidation adducts | |
| amino acid oxidation adducts | |
| hydro-peroxides | |
| isoleucine | hydro-peroxides |
| leucine | 3-hydroxy-leucine, 4-hydroxy-leucine |
| 5-hydroxy-leucine, hydro-peroxides | |
| lysine | α-amino-adipic semialdehyde, chloramines, hydro-peroxides |
| lipid peroxidation adducts, amino acid oxidation adducts | |
| glyco-oxidation adducts | |
| methionine | methionine sulfoxide, methionine sulfone |
| phenylalanine | 2-hydroxy-phenylalanine, 3-hydroxy-phenylalanine |
| 4-hydroxy-phenylalanine, 3,4-dihydroxy-phenylalanine | |
| proline | glutamic semialdehyde, 4-hydroxyproline, 5-hydroxyproline |
| pyroglutamic acid, 2-pyrrolidone, hydro-peroxides | |
| threonine | 2-amino-3-ketobutyric acid |
| tryptophan | 2-hydroxy-tryptophan, 4-hydroxy-tryptophan |
| 5-hydroxy-tryptophan, 6-hydroxy-tryptophan | |
| 7-hydroxy-tryptophan, n-formyl-kynurenine | |
| 3-hydroxy-kynurenine, kynurenine | |
| 6-nitro-tryptophan, tryptophan-tryptophan cross-linkages | |
| hydro-peroxides | |
| tyrosine | 3,4-dihydroxy-phenylalanine, 3-nitro-tyrosine, 3-chloro-tyrosine |
| 3,5-dichloro-tyrosine, tyrosine–tyrosine cross-linkages | |
| tyrosine-oxygen-tyrosine, 2,3-dihydroxy-phenylalanine | |
| hydro-peroxides | |
| (2) Metal-Catalyzed Oxidation of Amino Acid Residues in Proteins400 | |
| histidine | aspartate, asparagine, oxo-histidine |
| proline | hydroxy-proline, glutamate, γ-glutamyl-semialdehyde |
| arginine | γ-glutamyl-semialdehyde |
| lysine | amino-adipic-semialdehyde |
| threonine | amino-ketobutyrate |
| tyrosine | Tyr–Tyr (dityrosine) |
| cysteine | ESESE (disulfide cross-links) |
3.3. Cell Membranes at the Border of Cytoplasm and the Extracellular Environment
3.3.1. Integrins
Integrins are heterodimeric, bitopic membrane proteins located in the cell membrane of animal cells. Integrin heterodimers are composed of subunits α and β. There are 18 α subunits and 8 β subunits, which combine to form 24 distinct known dimers.411 Integrins mainly facilitate the cell–extracellular matrix adhesion (see Figure 13). They are also receptors that pass signals in and out of the cell. Integrin activation is a complex process regulated by numerous proteins. The key proteins involved in integrin activation are talin and kindlin, which directly interact with the β subunit. The inactivating proteins are sharpin, nisharin, and MDG1, which interact directly with the α subunit.412 Other proteins interacting with integrins are signaling and recycling proteins. Integrins are indirectly connected to the cytoskeleton, to actin filaments. The mechanisms of integrin regulation are far from understood. The complexity of the process is highlighted by a proteomics analysis, which indicated that over 300 proteins are involved in integrin adhesion.413 Integrins act also as receptors for extracellular matrix components, including collagen fibers in the interstitial matrices and laminins in pericellular matrices.414 We note that the extracellular matrix is another complex structure that is not understood, and its nonuniformity in the body aggravates this.414
Figure 13.
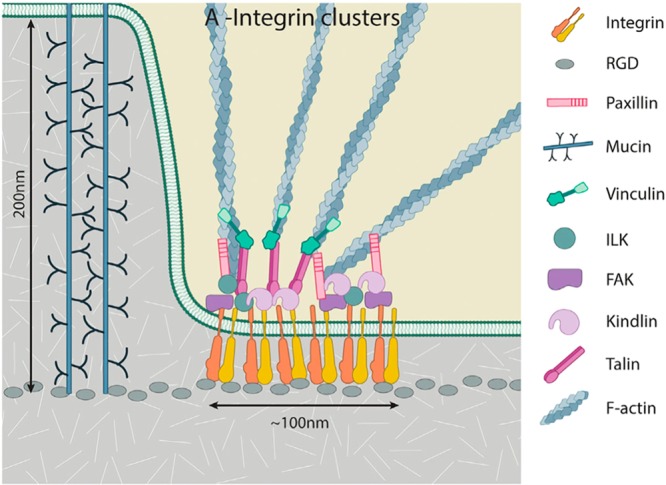
Simplified cartoon representation of components of the modular nascent cell-matrix adhesions. RGD, substrate for integrin binding. Reproduced with permission from ref (415). Copyright 2016 Wiley Periodicals, Inc.
3.3.2. Glycocalyx
Glycocalyx is an extracellular structure formed by carbohydrate polymers, glycoproteins, and glycolipids (Figure 14). Glycocalyx is most frequently discussed in the context of vessel endothelial cells, where it extends 150–400 nm away from the membrane and is organized in 100 nm-wide units along the membrane plane. Glycocalyx and its components are also found in many other organs and cells, including intestinal epithelium, mucus, smooth muscle cells, osteocytes, and cancer cells.416 The main function of glycocalyx is to act as a barrier between a cell and its environment, but it is also involved in mechanosensing.417−419 Glycocalyx is of great medical interest due to its involvement in numerous diseases, for example, atherosclerosis, stroke, sepsis, diabetes and related renal diseases, hypertension, pulmonary edema, and cancer.416,420,421
Figure 14.

Glycocalyx and its components. (A) Electron microscopy image of the endothelial glycocalyx in a coronary capillary. Reproduced with permission from ref (420). Copyright 2005 Wolters Kluwer Health, Inc. (B) A cartoon representation of proteoglycans and glycoproteins on the surface of endothelial cells. The red line represents the cytoskeleton. Reproduced with permission from ref (419). Copyright 2006 John Wiley and Sons.
The main components of glycocalyx are the proteoglycan proteins that anchor long carbohydrate polymers (Figure 14). Carbohydrate polymers forming the glycocalyx are heparin sulfate, chondroitin sulfate, hyaluronic acid, dermatan sulfate, and keratan sulfate.419,422,423 The major carbohydrate polymer, heparin sulfate contributes up to 50–90% of proteoglycans in the glycocalyx. Heparin sulfate and chondroitin sulfate are anchored to the cell membrane by a transmembrane protein called syndecan, which can accommodate five polymer chains.422 The syndecan, which has four subtypes, is also important for the above-mentioned mechanosensing function of glycocalyx, as it is connected to the cytoskeleton.422,424 Especially heparin sulfate is expected to participate in mechanosensing via interactions with the extracellular matrix.416 Currently the structure of syndecans is not known; only the structure of a 28-residue-long intracellular domain of syndecan-4 was solved in solution in the dimeric form (PDB 1EJP).425
The second group of proteins anchoring heparin sulfate and chondroitin sulfate to a membrane are GPI-anchored proteins, called glypicans, which have six subtypes.422 Only the structure of human glypican 1 is known (PDB 4ACR,4264AD7,4264BWE,427 and 4YWT(428)). However, not all proteoglycan proteins that are attached to heparin or chondroitin sulfate are membrane-associated, such as perlecan, versican, decorin, biglycan, and mimecan.422 Hyaluronic acid is anchored to the cell membrane by the CD44 receptor, but it may be also loosely associated with the glycocalyx structure without specific interactions.422,423 Currently, the structure of CD44 is not known; only the structure of the human and murine hyaluronan binding domain composed of about 180 residues was solved (e.g., PDB 1UUH,4292JPC430), while the structure of the remaining ∼560 residues is not known.
Glycoproteins and glycolipids are also considered to be a part of the glycocalyx, but their size is insignificant when compared to above-discussed carbohydrate polymers, as glycolipids can contain one sugar monomers and each N- or O-glycan linked to a glycoprotein has about ten sugar monomers.
3.3.3. Cell–Cell Junctions
Cells are frequently directly connected via different types of cell junctions: tight junctions, adherens junctions, desmosome, and gap junctions (Figure 15). Each of these connections has a different function, but they are dynamically coupled to each other and are located at specific regions in the cell membrane.431,432
Figure 15.

Cell–cell junctions. (A) Overview of cell–cell junctions and main proteins involved in their formation. Reproduced with permission from ref (439). Copyright 2017 John Wiley and Sons. (B, left) Tight junction location in the cell membrane providing a barrier between an apical membrane and a basolateral membrane. PC is shown in red, sphingolipids and cholesterol are shown in green, and tight junction proteins are shown in purple. Reproduced with permission from ref (433). Copyright 2008 Elsevier. (B, right) Molecular architecture of a tight junction formed by two claudin molecules. Reproduced with permission from ref (436). Copyright 2016 Springer Nature. (C) Maturation of an adherens junction via dimerization and aggregation. Reproduced with permission from ref (440). Copyright 2017 Cold Spring Harbor Laboratory Press. (D) Three-dimensional model of the E-cadherin–catenin cell adhesion complex associated with F-actin based on the crystal structures: E-cadherin ectodomain EC1–5 trans-dimer (purple or dark yellow) bound to calcium (yellow spheres) (PDB 3Q2 V),441 p120 catenin (blue) bound to E-cadherin (PDB: 3L6X),442 β-catenin (green) bound to E-cadherin (PDB 1I7W),443 αE-catenin (red) (PDB 1DOW,4444IGG,445 and 4K1N),446 and F-actin (light and dark gray) (PDB 3B63).447 Reproduced with permission from ref (440). Copyright 2017 Cold Spring Harbor Laboratory Press. (E) Schematic representation of connexins and gap junction channels. Reproduced with permission from ref (448). Copyright 2007 The Society for Investigative Dermatology. (F) Molecular architecture of a gap junction channel (PDB 2ZW3).449 Reproduced with permission from ref (449). Copyright 2009 Springer Nature.
Tight junctions are cell–cell connections located at the borders of the membrane compartments in polarized cells. The most generic examples of tight junctions can be found in the epithelial and endothelial cells, where the tight junctions separate apical and basolateral membranes.433−435 Tight junctions are formed by a few classes of proteins, among which the most important are the transmembrane proteins claudins and occludins (Figure 15).436 According to some tight junction models, lipids also participate in the junction structure.436 The junctions bring two cell membranes into close contact, which is sometimes referred to as a kiss point. Tight junctions control material flow into the paracellular space, which is of particular importance for the blood–brain barrier.437,438 Moreover, tight junctions act as fences preventing the flow of lipids and proteins between distinct membrane domains of polarized cells.435 Tight junctions are also connected with the cytoskeleton and participate in mechanotransduction.439
Adherens junctions are formed by the transmembrane proteins cadherins and nectins, while desmosomes are formed by specific desmosomal cadherins called desmogleins and are considered by some authors as a separate type of junction (Figure 15).438,439,450 In these junctions, the distance between two connected membranes is longest among all junction types, being ∼34 nm for desmosomes.451 In mature junctions, multiple cadherins form clusters that, together with a strong association with the cytoskeleton, provide mechanical strength to the junctions.440 Adherence junctions are of primary importance for tissue integrity and their mechanical properties.439,451 Desmosomes are the strongest cellular junctions; thus, they are especially important for tissues like skin and organs like heart, which are subject to mechanical stress.450,452 It is, therefore, not surprising that mutations of desmosomal proteins cause myocardial, skin, and hair pathologies.453−455
The main components of the gap junctions are proteins that belong to two groups: connexins and pannexins, both of which are proteins with four-helical transmembrane domains (Figure 15).456,457 These proteins form channels connecting cytoplasms of two cells, whose opening and closure are controlled by various mechanisms.458 The diameter of the channel in its narrowest part is ∼1.4 nm, thus allowing the passage of water, ions, and small solutes including second messengers.448,459 Membranes of the two cells connected via gap junctions are at a distance of 2–4 nm.448,449 This distance results in a gap when compared to tight junctions, where the kiss point is formed. Typically, in cells gap junctions form plaques gathering a large number of individual channels together.460
Connexins and pannexins are present in vertebrates, while in nonvertebrates gap junctions are formed by innexins, which are related to pannexins.456 In humans, there are 21 known connexin isoforms, and cells express many of them at the same time. To form a pore, six connexin molecules are needed in each membrane. They may be of different isoforms allowing for very many combinations.448,461,462 Gap junctions are not selective for ions, and thus they play a role in electrical coupling between cells, which is of high importance in nerve tissues and muscles.463 On the other hand, gap junctions are selective for second messengers, which is likely achieved by the huge number of combinations to form a channel due to simultaneous expression of many connexin isoforms.459,463 This also explains why mutations of only one isoform can lead to cancer or other disorders.463−469
Plant and fungal cells are surrounded by a cell wall, but direct cell–cell communication analogous to gap junctions exists. In plants, analogs of gap junctions are called plasmodesmata, which are elongated patches of cytoplasm passing through the cell wall;470,471 in fungi, they are called septal pores.471 The molecular architecture of these connections is not understood yet.
3.3.4. Cell Wall
The cell wall is a structure that surrounds plant,472−476 bacterial,477−480 and fungal cells.481−484 That is, the majority of cells on earth have cell walls. The main function of the cell wall is to provide mechanical protection from external forces. It is also involved in signaling and sensing the external environment; however the exact mechanisms are not known.
The cell wall is composed of carbohydrate polymers, which are specific for each group of the organisms. In plants, the main polymer is cellulose, while in fungi it is glucan and chitin, and in bacteria, it is peptidoglycan. Synthesis and organization of the cell wall is a complex process, which is still not clear. However, cell walls are of pharmaceutical importance as targets for antibiotics and antifungal drugs. The cell membrane plays important roles in cell wall synthesis, anchoring, and establishing its connection with the cytoskeleton.
In plants, the cell wall is composed of cellulose fibers, which are connected with matrix carbohydrates like pectin, hemicellulose, and a polyphenolic compound, lignin (Figure 16).472−475 Lignin and matrix carbohydrates are synthesized in the intracellular compartment, after which they are actively transported through the cell membrane to the cell wall. Cellulose fibers, in turn, are synthesized by cellulose synthase, which is a large enzymatic complex located on the cell membrane. Cellulose synthase has a diameter of ∼30 nm in the region spanning the cell membrane and is connected to the cytoskeleton. The cell wall of plants is anchored to the cell membrane by numerous GPI-anchored proteins, which bind directly to either cellulose or other carbohydrates. Other groups of proteins that anchor the cell wall to the cell membrane are arabinogalactan proteins, wall-associated kinase receptors, and analogs of integrin. The cell wall is connected to the cytoskeleton via transmembrane proteins belonging to the class-1 formin family.
Figure 16.

Cartoon representation of the complexity of the plant cell wall. The cell membrane is shown in brown; matrix polysaccharides are shown as thin brown, blue, green, red, and black chains. The question mark represents an unknown bonding partner of integrin-like receptors. Adapted with permission from ref (475). Copyright 2015 Oxford University Press.
The fungal cell wall is similar in complexity to the plant cell wall, but its structure is even less understood.484−486 The main carbohydrate polymers of fungal cell walls are α-1,3-, β-1,3-, and β-1,6-glucans and chitin, which form a mesh-like structure. Their synthesis takes place at the cell membrane.484 Additional components of the mesh structure are melanin and phenolic pigments, whose structures are not well characterized.484 Fungal cell wall is attached to the cell membrane predominantly via GPI-anchored proteins.482
A bacterial cell wall structure in comparison to fungal and plant cell walls is less complicated and better understood. However, the entire envelope surrounding bacterial cells (including their cell wall, the outer membrane in Gram-negative bacteria, and glycocalyx) makes bacterial cell–environment barrier the most complex among all groups of organisms (Figure 17a,b). Due to the importance of bacteria in human health, the bacterial cell envelope structure, the synthesis of its structural units, and the assembly process are of primary interest, as they are potential targets for antibiotics, too.487,488
Figure 17.

Cartoon representation of the bacterial cell wall structure. (A) Gram-negative bacteria and (B) Gram-positive bacteria. For the legend, see panels C and D. (C) Chemical structure of the peptidoglycan unit. (D) Chemical structure of teichoic acid. (E) Synthesis of peptidoglycan. Proteins are shown as gray ellipses.
The main component of the bacterial cell wall is peptidoglycan, a polymer whose basic unit is disaccharide–pentapeptide N-acetyl-glucosaminyl-(β,1→4)-N-acetyl-muramyl-l-alanyl-d-glutaminyl(γ)-l-(meso)diaminopimelyl-d-alanyl-d-alanine (Figure 17c). In peptidoglycans, carbohydrates form linear structures, while pentapeptides are crossed-connected via short peptides called bridges. Due to this, peptidoglycan has mesh-like structure that is not as rigid as the cell wall of plants but allows changes of cell shape necessary in active movements of bacteria. The exact structure of the mesh differs between bacterial species due to different lengths of the bridge peptide and number of formed connections (See ref (489)). The second component of the cell wall of Gram-positive bacteria is teichoic acids, polymers of 3-phosphate glycerol or 5-phosphate ribitol or both, attached to a peptidoglycan MurNac residue via two sugar units (sequence) (Figure 17e).490 Alternatively, teichoic acids remain connected with lipids. Teichoic acids may comprise 50% of the cell wall mass. Elements of the bacterial cell wall are recognized by innate immune systems;491 thus, bacteria have developed numerous modifications to the basic structure described here.492−494
Synthesis of the bacterial cell wall elements is an almost entirely membrane-associated process, where Lipid I and II act as platforms for the peptidoglycan unit formation (Figure 17e).495 The precursor of Lipid I is a polyprenol chain with a single phosphate group and UDP-N-acetylmuramic acid pentapeptide synthesized in the cytoplasm. The next step of the synthesis produces Lipid II by attachment of GlucNac to Lipid I and the peptide forming a bridge between them (Figure 17c). Synthesis of Lipid II takes place at the inner leaflet of the membrane; thus, it has to translocate to the outer leaflet of the cell membrane. However, it is not clear which proteins are involved in this process. The leading candidate as a Lipid II floppase is the MurJ protein, although it is possible that more proteins are involved in the process.496 At the outer leaflet, a peptidoglycan fragment is used to extend the cell wall, and a phosphorylated polyprenol moiety translocates back to the inner leaflet of the cell membrane. Synthesis of teichoic acids is substantially similar to the synthesis of Lipid II, as it also takes place at the inner leaflet of the cell membrane and uses phosphorylated polyprenol as the base to which elements of teichoic acids are gradually attached.490 The ABC transporters, TagG or TarG, translocate the mature construct to the outer leaflet of the cell membrane, where teichoic acid is attached to the MurNAc residues of peptidoglycan. Interestingly, an enzyme responsible for this final step of teichoic acid integration into the cell wall has not been identified yet.
Current state of knowledge of the molecular architecture of the cell wall and its connection with the cell membrane is not impressive. This is particularly highlighted by the lack of knowledge of the structures of the main proteins involved in organization of the cell wall. This is surprising considering the pharmacological importance of the cell wall as an antimicrobial target. Current drugs typically target the synthesis of the cell wall elements, which takes place inside cells. This allows the bacteria to develop resistance using ABC transporters. Here, one can imagine drugs that prevent the assembly of the cell wall by acting outside the cell membrane. In this case, development of resistance would be much more complicated.
3.4. Challenges
As discussed in this section, biological membranes are complex structures made up of lipids, proteins, and carbohydrates. All of these components have specific functions, but they also depend on each other. The primary role of lipids is to form the membrane matrix, which provides a 2-dimensional environment for various proteins to carry out their functions. Lipids also specifically regulate protein activation through allosteric interactions, and they act as primary receptors for peripheral membrane proteins. Proteins, in turn, affect lipids. Enzymes modulate lipids to, for example, induce membrane curvature. Local lipid composition is modulated by flippases, floppases, and scramblases, which establish and maintain asymmetric lipid compositions across the bilayer leaflets. Carbohydrates are also a part of these interactions as modifications in the glycolipid headgroups or post-transcriptional modifications of proteins.
The properties of lipid bilayers depend on where they are located in a cell. For example, the cell membrane is thick and rigid, while reticular membranes are thinner and more fluid. All of the cellular membranes are connected together via a complex interaction network, which is far from understood.
To construct realistic computational models of biological membranes, we need to know their exact lipid, protein, and carbohydrate composition as well as the structures of the proteins. Recent studies have given us a growing number of transmembrane protein structures thanks to the improvements in protein crystallography and cryo-EM methods. Cryo-EM is particularly promising, since its resolution is now close to the atomistic level. Despite this progress, structures remain to be characterized for most transmembrane proteins, and too little is known of the structures of peripheral proteins, which are likely different in water-soluble and membrane-bound forms.
Omics-type studies provide valuable information concerning molecular composition. However, when performed on crude tissue samples, these studies are not sufficient to build realistic models of the biological membranes. Therefore, we need data specifically collected for individual organelles, or membrane types. The number of published studies is still quite modest due to experimental difficulties to get sufficiently good samples from specific organelles, which are not contaminated by other cellular components. Post-transcriptional modifications of proteins pose yet another difficulty for the construction of realistic simulation models. For example, protein glycosylation is a specific process, and for most proteins, the exact glycosylation sequence is unknown. Besides, the glycosylation sequence for each protein depends also on the cell type.
Finally, large-scale structures of systems where membranes are involved, such as glycocalyx, cell wall, and the connection of the membranes with the cytoskeleton and the extracellular matrix are weakly understood. These structures are particularly challenging to simulate due to their dynamic nature, multiple partners interacting via weak and redundant interactions, and the presence of unstructured carbohydrates. To characterize these complex structures, support given by new experimental methods is likely needed.
4. Are Biomembranes Comprised of Transient Functional Domains?
In this section, we discuss how computer simulations have tackled a number of open questions regarding the laterally heterogeneous structure of the plasma membrane, and therefore how scientific computing has helped to bridge together studies on model membranes and living cells. First, however, we complement our previous discussion in sections 2 and 3 by briefly reviewing the current paradigm of plasma membrane heterogeneity provided by the lipid raft model. We briefly discuss the state-of-the-art experiments on living cells, model membranes, and plasma membrane-derived vesicles, all of which approach this topic from different perspectives. Notably, we consider the convincing evidence suggesting that the nanoscale heterogeneities in the plasma membrane are associated with the phase behavior of model lipid membranes. We close the outlook on experimental results by discussing some open questions that might benefit from computer simulation studies.
Next, we take a bottom-up approach and review recent simulation studies by focusing on membrane heterogeneity. Here, we first concentrate on studies on lipid phase behavior. We start from single-component membranes and then move on to recent studies on binary mixtures containing either two kinds of phospholipids or a phospholipid with cholesterol. Moving on to ternary mixtures, we consider studies of uniform raft-like membranes, studies with preformed raft-like domains, and studies that capture spontaneous phase separation into ordered raft-like and disordered non-raft like phases. Next, we consider heterogeneities in complex membranes whose composition mimics the content found in plasma membranes. Finally, we discuss simulation studies that suggest protein–lipid interactions to give rise to the formation of distinct functional domains. We mainly focus on the development that has taken place during the past decade, unless earlier studies are of significant interest.
We close the section by discussing the limitations of the current simulation studies and provide our opinion on how these limitations could be overcome in the future by tightly integrating experimental and simulation efforts.
4.1. Heterogeneity in the Plasma Membrane and Model Membranes
As discussed in section 2, a reasonably accurate picture of the structure of cellular membranes was provided by the fluid mosaic model introduced by Singer and Nicholson back in 1972.4 This model remains mostly valid even today, although the discoveries made during the last four decades have also called for some updates.497 Namely, according to the current paradigm, cellular membranes display both transmembrane and lateral heterogeneity. While the former is discussed in detail in our description of the target membrane in section 3, the latter is captured in the raft concept,12 which states that specific lipids and proteins cluster to form nanoscopic ordered domains. More precisely, the Keystone Symposium on Lipid Rafts and Cell Function held in 2006 defined rafts to be heterogeneous and dynamic entities containing sphingomyelin and cholesterol.498 More recent interpretations15 have stressed that rafts are functional, which emphasizes the importance of accounting for proteins, too. The diameter of rafts has often been estimated to be 10–200 nm, yet rafts have been considered to be able to coalesce via protein–lipid and protein–protein interactions. A schematic picture of a lipid raft based on current understanding is shown in Figure 18.
Figure 18.

Schematic drawing of the organization of the plasma membrane. Saturated lipids, shown in reddish colors, segregate with cholesterol to form a domain that structurally resembles the liquid-ordered phase. Glycolipids, GPI-anchored proteins, and lipidated proteins partition preferably into this domain, while many other membrane proteins reside in the regions outside the rafts, resembling a liquid-disordered phase. Reproduced with permission from ref (17). Copyright 2017 Springer Nature.
Multiple roles have been assigned to rafts during the last two decades since their introduction in 1997. Most importantly, they provide membrane proteins with specific cholesterol-rich environments, where they can perform their functions associated with, for example, signaling and trafficking.15 During the last two decades, our picture of rafts has evolved due to a significant amount of experimental and theoretical work. The modern view considers rafts as transient and fairly ordered functional domains, whose formation is driven mainly by lipid–lipid interactions.17 These favorable lipid–lipid interactions include, in addition to cholesterol and sphingomyelin, also interactions of cholesterol with other saturated lipids and gangliosides.17 Additionally, the roles of hydrophobic mismatch, cortical actin, and the possibility of protein-induced domain formation have been highlighted.17
Curiously, even though the resolution of experimental techniques has steadily improved since the introduction of the raft concept, there are still no direct observations of their existence in the membranes of living cells. This controversy has also sparked alternative explanations for the numerous indirect findings.499 Perhaps, the most convincing pieces of evidence come from super-resolution fluorescence correlation spectroscopy (FCS) measurements. Independent studies by two groups employing different experimental setups suggest that sphingomyelin and GPI-anchored proteins are transiently trapped in small (∼10 nm) domains in the plasma membranes of living cells and that this phenomenon is cholesterol-dependent.500,501
There is a plethora of evidence showing that cholesterol is a crucial player in promoting lateral heterogeneity in the plasma membrane. It can induce the Lo phase14 when mixed with lipids whose chains are adequately saturated, such as sphingomyelin (SM). Since cholesterol and sphingomyelin are also the main components of rafts, their association with the Lo phase is imminent, and membranes are assumed to resemble the coexistence of Lo-like raft domains with Ld-like regions, although with the two phases being less distinct than in model membranes. However, the plasma membrane is an incredibly complex entity502 with the combinations of the various types of acyl chains and headgroups adding up to thousands of distinct lipid moieties. This picture is further complicated by the presence of numerous kinds of membrane proteins, the interactions between the membrane and the actin cytoskeleton, membrane asymmetry, and the interplay of the two membrane leaflets with their distinct solvent environments. It is clear that understanding the phase behavior and nanodomain-forming properties of biomembranes requires the use of simplified model membranes with a much more limited number of lipid types.
The phase behavior of a lipid membrane is captured in a phase diagram, which usually maps lipid composition and temperature to either a single phase or coexistence of two or more phases. Constructing such a diagram requires that numerous points in the phase space are sampled, that is, that a model system is set up and experimentally characterized at each point. Therefore, realistic mixtures of thousands of lipids are way beyond the reach of this approach. Instead, phase diagrams have been mainly measured for synthetic liposomes formed by two or three lipid components, one of them usually being cholesterol (see the thorough reviews of the available phase diagrams by Marsh,503,504 Veatch and Keller,505 and Feigenson.506 Importantly, some cholesterol-containing lipid mixtures show Lo/Ld phase coexistence under specific compositions and temperatures. The domain structure can be readily visualized using fluorescence microscopy by tagging two molecules with distinct partitioning preference (Lo or Ld) with different probes and locating these probes by illumination. This leads to beautiful pictures that show micrometer-sized domains in mixtures of either (1) two phospholipids with different chain lengths mixed with cholesterol,507,508 (2) two phospholipids with differing degrees of chain unsaturation combined with cholesterol,509,510 or (3) a phospholipid with unsaturated chains and sphingomyelin with a saturated chain mixed with cholesterol.511,512 The common factor in these mixtures is that one of the two types of noncholesterol lipids has a high temperature of the main phase transition (high-Tm), while the other has a low temperature of the main phase transition (low-Tm). Interestingly, the demixing to two distinct phases occurs usually at up to the ambient temperature, which is somewhat lower than the body temperature and falls between the values of high-Tm and low-Tm. This behavior strongly indicates that in Lo/Ld coexistence, the role of cholesterol is to melt the gel phase into the Lo phase, as in its absence the mixture would exhibit gel/Ld coexistence.
However, it is still unclear how the picture of nanoscopic Lo-like rafts in an otherwise Ld-like membrane is related to the microscopic phase separation observed in model membranes. Curiously, in addition to large-scale phase separation seen in ternary lipid mixtures, some binary mixtures of cholesterol and a lipid with saturated chains reveal heterogeneous behavior with a resemblance to both Lo and Ld phases.513,514 However, no micrometer-sized phase separation is observed in such systems using fluorescence microscopy. These observations have recently been explained by the presence of ordered cholesterol-rich nanodomains, whose size is below the diffraction limit.514 These nanodomains with a size similar to that postulated for lipid rafts could arise through multiple physical mechanisms.515 However, the coupling of macroscopic phase separation and nanodomain formation is still somewhat controversial, and this picture is further complicated by two very recent independent reports of nanoscopic substructures within the Lo phase of phase-separated model membranes.516,517
One promising approach to study the phase behavior of the plasma membrane is to induce the budding of vesicles out from the plasma membrane of a living cell. These giant plasma membrane-derived vesicles (GPMVs) provide a quasi-model system: they are well fit for biophysical characterization techniques, and their composition is assumed to be a faithful representation of the real plasma membrane. However, they are free of the many complexities present in vivo, such as the actin cytoskeleton or the involvement of active processes. Interestingly, GPMVs undergo phase separation at below ambient temperature, while they are microscopically homogeneous at body temperature.518 This behavior is consistent with that of ternary model membranes, suggesting that the latter indeed capture the essential features of the plasma membrane.
It is unclear how well the GPMVs capture the heterogeneity present in a plasma membrane of a living cell, notably the absence of active processes and the actin cytoskeleton. Very recently, a landmark study demonstrated reversible phase separation in the vacuole of live yeast152 suggesting that these factors cannot suppress the formation of domains. However, contrasting results were reported on live rat basophilic leukemia cells, which did not exhibit the miscibility phase transition, that is, the demixing of lipids into two liquid phases when cooled down.152
The observation that both GPMVs and synthetic liposomes undergo phase separation only slightly below the body temperature suggests that transient heterogeneities in the size range postulated for lipid rafts might result from critical fluctuations, that is, compositional fluctuations near a critical point.519 Indeed, such phenomena have been observed for both GPMVs520 and synthetic liposomes,521 further supporting the use of model systems to understand the heterogeneity in the plasma membrane.
Unfortunately, the used model systems are often compositionally symmetric, which is not the case in the plasma membrane, as discussed in section 3. Notably, while the extracellular leaflet, rich in cholesterol, sphingomyelin, and phosphatidylcholines with somewhat unsaturated chains, is assumed to display heterogeneities, the intracellular leaflet, rich in phosphatidylethanolamine, phosphatidylserine, and other anionic lipids, all with similarly unsaturated chains, is not expected to do so. Currently, relatively little is known about the coupling between membrane leaflets, and it is unclear whether domain formation in the extracellular leaflet can induce structural heterogeneity in the intracellular one.522
Before moving on, the extensive theoretical work should also be highlighted. Phase diagrams calculated using different approaches reveal the physical parameters necessary for determining whether a system undergoes phase separation, forms nanoscopic domains, and ends up as a microemulsion or as a modulated phase.515,523 However, these studies are not discussed further in this review.
Concluding, recent evidence strongly supports the presence of cholesterol- and sphingomyelin-rich nanoscopic domains in the plasma membrane. These domains likely arise due to critical composition fluctuations near a phase coexistence region, which can lead to transient domain formation in a system that is homogeneous in equilibrium. Due to this analogy, we shamelessly refer to ordered nanodomains as being “Lo” in the following sections, even though such structures do not correspond to an equilibrium phase.
The limited ability of experiments to directly probe nanoscale phenomena in soft matter has left a plethora of questions open: What is the mechanism behind phase separation or raft formation? How are the structures of the two distinct plasma membrane leaflets correlated? How do the details in lipid structures regulate the tendency to phase separate or to form nanodomains? How do additional molecules, such as drugs, affect the lateral heterogeneities of the membrane? Can membrane proteins induce heterogeneity in their environment, hence providing themselves with a functional domain? As discussed below in detail, all these questions have recently been subject to examination by biomolecular computer simulations.
4.2. Gel Phases and Transition Temperatures
To understand the complete phase behavior of mixed lipid membranes, one has to know how single-component membranes act under different conditions. Such membranes already display surprisingly complex phase behavior as a function of temperature. Our understanding of the phase transitions between the various gel and ripple phases is largely based on differential scanning calorimetry, while structural information on the phases comes mainly from scattering experiments. Contrastingly, above Tm, the liquid-disordered phase is characterized by a plethora of experimental techniques including NMR. Scattering techniques require a model that estimates the lipid densities based on the measured electron density profiles. For heterogeneous systems, such as the ripple phase, details of the structure remain unknown. Computer simulations are therefore highly desired to provide an atomistic picture of membrane structure and the phase transitions in such systems. While the various gel phases have little importance in living cells, it is crucial that a simulation model provides a reasonably accurate estimate for the main transition temperature. Otherwise the model is unlikely to correctly reproduce the phase behavior of more complex lipid mixtures, where—as discussed above—the Lo phase forms when cholesterol melts the gel phase. Unfortunately, many simulation force fields rest on a weak foundation, since they are parametrized only to reproduce liquid phase properties.
4.2.1. Phase Behavior of Single-Component Bilayers
Perhaps due to the difficulties in sampling the essentially immobile gel phase, very few MD studies have probed phase transitions involving the gel phase. Yet, these studies have revealed how the current models struggle in capturing the phase behavior accurately.
United-atom simulations of the melting of DPPC and DPPE bilayers were unable to capture the formation of the ripple phase,524 and even the gel phase structure provided by the Berger model was partially incorrect.525 However, in another study on DPPC bilayers using a similar model yet a larger system size, the formation of the ripple phase was observed.526 Moreover, its structure agreed well with experimental data.
Marrink et al. studied the formation of the gel phase in DPPC bilayers using the coarse-grained MARTINI model.527 They observed the establishment of an untilted gel phase at temperatures below 295 K, some 20 degrees lower than the experimentally resolved Tm of DPPC. Moreover, the lipid chains were expected to adopt a tilted conformation, which could be induced by adjusting the sizes of the beads therein. However, considering the level of detail in this model, this agreement is already quite impressive.
Rodgers et al. systematically studied the phase behavior of phosphatidylcholine lipids with different acyl chain lengths.528 They performed temperature scans similar to those in experimental calorimetric studies. The MARTINI model only captured a single transition between gel and fluid phases, while a dissipative particle dynamics model was able to reproduce all expected phases. Notably, the melting points of lipids were again found to be somewhat lower than in reality.
4.2.2. Phase Transitions in the Presence of Cholesterol
Arnarez et al. performed temperature scans of DPPC membranes with increasing concentrations of cholesterol using the MARTINI model.529 They found that the hysteresis in the temperature-dependence of the enthalpy vanished with increasing content of cholesterol. This behavior is in excellent agreement with the experimental observation that at sufficient concentration cholesterol induces the Lo phase both below and above Tm.513
In contrast, a study by Waheed et al., also using the coarse-grained MARTINI model, did not report disappearance of hysteresis during the phase transition of DPPC even at high cholesterol concentrations.530 While the reason for this discrepancy is unclear, it might result from the different cholesterol model employed in these two studies.
We note that in addition to coarse-grained models, also modern atomistic lipid models derived to reproduce the properties of the liquid phases struggle in describing the main transition temperatures of lipid bilayers.531,532
Concluding, the conventional force fields have been derived to describe the liquid phases of lipid membranes. However, they severely struggle to capture the behavior below Tm, and also the value of Tm itself. This limitation is of concern since cholesterol can melt the gel phase into the Lo phase, which is crucial for the formation of Lo/Ld phase coexistence in ternary mixtures. This issue, including other model-related ones, is discussed in more detail below.
4.3. Binary Mixtures of Non-cholesterol Lipids
Mixtures of two lipid moieties with different transition temperatures due to varying levels of chain unsaturation, different chain lengths, or different headgroups all have relatively similar phase diagrams. At temperatures approximately between the two transition temperatures, the systems undergo a separation into gel and liquid phases with compositions defined by the tie line. This region is of interest since therein the inclusion of cholesterol can lead to the separation into the Lo and Ld phases. To our best knowledge, all bilayer-forming binary mixtures of phospholipids with a different Tm display this somewhat ideal behavior, which leaves very few open questions for computer simulations. However, for the realistic Lo/Ld phase separation in ternary mixtures, it is crucial that the properties of the gel/Ld coexistence region are correctly captured in the same system in the absence of cholesterol. Unfortunately, this coexistence in binary mixtures has been studied relatively little using simulations.
Faller and Marrink studied mixtures of DLPC and DSPC in the coarse-grained scheme.533 In a narrow temperature and composition window, they observed the formation of Ld-like domains in an otherwise gel phase membrane.
Pyrkova et al. studied atomistic membranes containing different ratios of DOPC and DPPC in the Ld phase.534 They observed nonideal mixing in the nanoscale and postulated that the heterogeneities might serve as seeds for the formation of larger domains by external stimuli. Although this study did not consider temperatures between the Tm values of DPPC and DOPC and hence the gel/Ld coexistence, the observed heterogeneity is likely the two-component equivalent of critical fluctuations, where small transient domains arise near phase coexistence.
Similar nonidealities were also observed in two coarse-grained simulations of Ld phase membranes consisting of DPPC and a lipid with two unsaturated chains of different lengths.535,536
Baoukina et al. studied membranes formed by POPG, DOPC, and DPPC, mimicking the composition of the cholesterol-free pulmonary surfactant.537 At temperatures below Tm of DPPC, the system displayed coexistence of DPPC-rich gel domains in an Ld-like membrane rich in DOPC and POPG. The former formed by a nucleation mechanism, and the gel domains coalesced into large ones. We see no reason not to expect similar behavior also in a binary DPPC/DOPC mixture below the Tm of DPPC.
Concluding, only a few studies exist to our knowledge of binary systems with a low-Tm and a high-Tm lipid. Moreover, these mixtures were studied at a temperature above the Tm values of both lipid components, and therefore only transient heterogeneities, with likely resemblance to critical fluctuations, were observed. This observation calls for a systematic study of the gel/Ld coexistence in a binary mixture, and the effects of introducing cholesterol in this mixture as the third component.
4.4. Binary Mixtures with Cholesterol
The presence of cholesterol substantially complicates the phase behavior of lipid membranes. It induces the ordering of lipid chains with most potent effects on saturated chains.538 At large enough concentrations in such systems, it induces the Lo phase, both above and below the Tm of the other lipid moiety, as long as the chains of this other lipid are saturated enough.513 Since the literature on the ordering effects of cholesterol up to 2008 has been reviewed carefully by Róg et al.,149 we focus here on more recent studies. We also omit reports of cholesterol effects on the bulk properties of the membrane. Still, due to their systematic and extensive nature, there is reason to mention the recent studies probing the effects of cholesterol in binary mixtures with phospholipids with different headgroups and acyl chains,539 and in mixtures of sphingomyelin and cholesterol.540
4.4.1. Phospholipid–Cholesterol Interactions Arising from Details in Cholesterol Structure
Using atomistic (united atom) simulations, Martinez-Seara et al. discovered that the unique ordering effects of cholesterol result from a very distinct spatial and orientational structure that it forms with phospholipids.541 The presence of the two out-of-plane methyl groups in the ring structure of cholesterol was found to be crucial for these effects. Even more importantly, a smoother cholesterol analog was unable to induce similar ordering patterns as cholesterol. This highlights the importance to understand the effects arising from chemical details in sterol structure, since based on this study it is clear that the order induced by a sterol around it depends on the details of the sterol structure. After all, there are ∼20 sterols, and the phase diagrams they give rise to are sterol dependent, and also the allosteric binding of sterols to membrane receptor proteins depends on the details of the sterol structure.54
Garg et al. studied the solubility limits of cholesterol in POPC and POPS bilayers in the coarse-grained MARTINI scheme and found a good match between simulations and scattering experiments.542
Using a set of free energy calculations combined with theoretical predictions, Díaz-Tejada et al. evaluated the preference of cholesterol to interact with phospholipids of different levels of chain unsaturation.543 To this end, they simulated binary lipid mixtures using the coarse-grained MARTINI model. Their analyses revealed that, despite cholesterol preferring saturated environments, the interactions among cholesterol molecules are repulsive despite the level of saturation of the chains of the surrounding phospholipids. This results in a homogeneous distribution of cholesterol molecules in the membrane plane, independent of the degree of saturation of the phospholipid chains. Furthermore, the spontaneous demixing resulting in a cholesterol-depleted domain is very unlikely, as the free energy of creating such domain was found to be high and surprisingly similar for DPPC and DOPC lipids, yet larger for DLiPC. At low cholesterol concentrations, entropy is behind this unfavorable demixing, while at higher cholesterol concentrations enthalpic effects become dominant.
4.4.2. Phase Behavior of Phospholipid–Cholesterol Mixtures
Before moving to macroscopically phase-separating mixtures in the next subsection, we note that simulations have thoroughly probed the suggested heterogeneous behavior with a resemblance to Lo and Ld phases513 in the binary mixture of DPPC and cholesterol.
Waheed et al. performed united atom and coarse-grained temperature scans in a DPPC bilayer with various cholesterol concentrations and observed no signs of domain formation resembling neither the Lo/Ld coexistence nor the gel/Lo coexistence.530
More recently, Zhang et al. performed a very similar study on DPPC/cholesterol mixtures, but they sampled the phase diagram at fixed temperatures.544 Again, no spontaneous demixing of lipids occurred. However, in simulations starting from cholesterol-poor and cholesterol-rich regions, no mixing was observed at low temperatures indicating a coexistence of gel and Lo regions, which might also result from the slow dynamics of the gel phase. At higher temperatures, no Lo/Ld-like heterogeneity was observed.
The third and most recent coarse-grained study of the phase behavior of the DPPC/cholesterol mixtures was performed by Wang et al.545 They focused more carefully on low cholesterol concentrations and witnessed nonideal behavior in the condensation effect in the region where experiments reported Lo/Ld-like heterogeneity. However, no visible domains were observed in the system, except in the gel/Lo coexistence region.
To our best knowledge, the only atomistic simulation that has probed the phase space of the DPPC/cholesterol system was performed by Javanainen et al.53 Within the single-phase regions of the phase diagram, the system showed expected behavior in line with experiments. However, within the phase diagram region initially associated with Lo/Ld coexistence,513 the spontaneous formation of a nanodomain was observed. The nanodomain contained a cholesterol-free hexagonally packed core. The formation of such small domains could explain why some experiments report Lo/Ld phase coexistence for this system under some conditions, while fluorescence microscopy does not detect macroscopic phases.53 Some recent experimental and simulation studies are also in favor of this view.514
Concluding, many simulations, mostly using coarse-grained models, have been used to probe the phase behavior of phospholipid–cholesterol systems extensively. However, only one atomistic study reported heterogeneities in the form of visible nanodomains. As such domains have been reported in many experiments, it is worth questioning whether the interactions between phospholipids and cholesterol are described in sufficient detail in the coarse-grained scale. Given that the details in sterol structure can be critically decisive for membrane structure541 and these tiny details cannot be easily included in coarse-grained sterol models, the importance to validate predictions based on coarse-grained models with atomistic simulation data is more than justified. Yet, it is positive that the solubility of cholesterol can be reproduced in the coarse-grained level, and the recent cholesterol parametrization546 seems to capture the specific asymmetry-induced effects on lateral membrane packing observed at the atomistic level. These details in cholesterol structure are likely important for correctly capturing the phase behavior of both binary and ternary mixtures.
4.5. Ternary Lipid Mixtures
As discussed above, the picture where ordered rafts float in an otherwise disordered membrane is often associated with the macroscopic Lo/Ld phase separation. This separation is observed in model membranes, plasma-membrane derived vesicles, and the vacuole of living yeast152 at temperatures lower than the body temperature, yet not in live rat basophilic leukemia cells.547 While higher temperatures suppress macroscopic separation, critical fluctuations can give rise to transient nanoscopic domains. Moreover, the interactions with the cytoskeleton or the nonequilibrium conditions induced by active processes might further affect the phase and favor the formation of nanoscopic domains instead of microscopic ones, as demonstrated by the differences between live cells and GPMVs.518,547 Molecular dynamics simulations have been extensively employed to probe the factors that affect the phase behavior of ternary mixtures. Here, we first describe studies of homogeneous membranes with a raft-like composition. Next, we consider studies on preformed raft-like domains, and finally move on to simulations where spontaneous phase separation into Lo/Ld coexistence is observed.
4.5.1. Simulations of Raft-Like Mixtures
Based on experimental studies on both the plasma membrane and model membranes, raft models in simulation studies usually contain sphingomyelin, cholesterol, and a phosphatidylcholine lipid such as POPC.548 Alternatively, sphingomyelin is often replaced by DPPC that is more commonly employed in model membranes, yet is scarce in the plasma membrane. The structural and dynamic properties of raft-like ternary mixtures have been characterized computationally in numerous studies.549−551 Overall, these studies report tight yet heterogeneous at nanoscale lateral structure resulting from the favorable packing of cholesterol and sphingomyelin.
Yang et al. carefully analyzed the interactions among DPPC, POPC, and cholesterol using atomistic simulations.552 They considered compositions corresponding to the two phases in a phase-separated system. In this mixture, the saturated chains interacted most favorably with each other, whereas the interactions between saturated and unsaturated chains were least favored. Cholesterol was found to interact with saturated chains favorably. These observations are in line with the experimentally observed push–pull mechanism of lipid raft formation.553
Using a bright approach of mixed MD/MC (Molecular Dynamics/Monte Carlo) simulations, de Joannis et al. resolved the relative affinities of DOPC and DPPC to interact with cholesterol.554 They simulated the ternary mixture at different cholesterol concentrations with a fixed chemical potential difference between DOPC and DPPC, which demonstrated the expected preference of DPPC toward cholesterol. Moreover, probing the DPPC/DOPC ratios at different cholesterol concentrations and comparing the observed compositions with experimental tie lines allowed the evaluation of the difference in the chemical potentials of DPPC and DOPC. Here, the tie lines were not well reproduced, which might result from the limitations of the united atom simulation model. Moreover, no separation into fluid and gel phases was observed in the absence of cholesterol, even though simulations were performed below the Tm of DPPC. This behavior likely results from the limitations in the system size and the inability of the used model to accurately capture the transition to the gel phase.525
Sodt et al. simulated in atomistic detail raft-like Lo phase membranes with compositions corresponding to the Lo phase in a phase-separated DOPC/DPPC/cholesterol mixture.555 They observed a hexagonal substructure of saturated chains with interstitial cholesterols and unsaturated chains. Curiously, this structure resembles the one found for nanodomains in binary DPPC/cholesterol mixtures.53 The structure of the Lo phase is shown in Figure 19.
Figure 19.
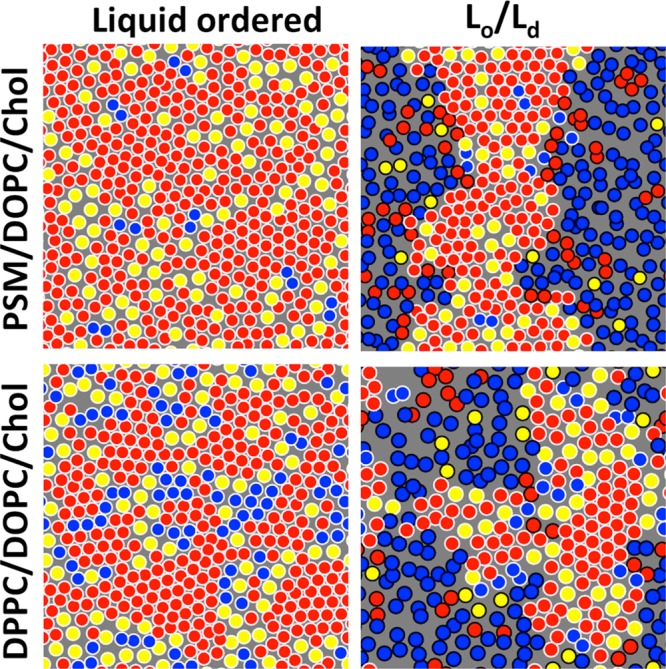
Lipid organization in the liquid-ordered phase (left column) and in the liquid-ordered/liquid-disordered coexistence (right column). The figure highlights the structural similarity of lipid organization in PSM/DOPC/cholesterol (top) and DPPC/DOPC/cholesterol (bottom) mixtures. Here, saturated lipid chains are shown in red, unsaturated ones in blue, and cholesterol in yellow. The hexagonal packing of the ordered regions and the partitioning of cholesterol to domain boundaries are evident. Reprinted with permission from ref (556). Copyright 2015 Elsevier.
In a follow-up study, Sodt et al. considered single-phase systems corresponding to the Lo phase composition in phase-separated DOPC/PSM/cholesterol and POPC/PSM/cholesterol mixtures.556 As highlighted in Figure 19, the behavior of DOPC/PSM/cholesterol resembled that of DOPC/DPPC/cholesterol, yet there were a few notable differences. While the Lo phase had a hexagonal substructure with cholesterol perturbing hexagonal ordering and the formation of a gel phase, the interactions between PSM and cholesterol differed from those between DPPC and cholesterol. PSM favored the rough face of cholesterol, and their mutual association due to hydrogen bonding broke the hexagonal ordering of lipid chains. DPPC, however, preferred the smooth cholesterol face instead, and the hexagonal order was perturbed by cholesterols located at a favorable position between a saturated and an unsaturated chain. Curiously, cholesterol favored interactions with DOPC over PSM and DPPC in the Lo phase in DOPC/PSM/cholesterol and DOPC/DPPC/cholesterol mixtures, yet in POPC/PSM/cholesterol mixture cholesterol preferred to interact with PSM.
Concluding, simulations of ternary lipid mixtures provide us information on the structure of rafts and of the specific lipid–lipid interactions underlying their formation. Such studies, often performed at atomistic resolution where the essential structural details are present, provide reasonable agreement with experimental data. This finding also suggests that data obtained from lower level models should be compared against atomistic simulations in addition to experimental data.
4.5.2. Simulations Considering Preformed Domains
Studying phase separation using MD simulations is currently almost exclusive to coarse-grained simulations due to the prolonged time and large size scales involved. However, the behavior of preformed ordered domains in a disordered membrane has also been studied in atomistic resolution.
Pandit et al. simulated a small Lo-like domain consisting of SM and cholesterol and evaluated the effects of the domain boundary on the properties of the surrounding Ld phase formed by DOPC.557 They observed long-range perturbations in membrane structure, yet the short simulation time due to atomistic resolution did not allow for a proper relaxation of the compositions of the domain and its surroundings, which leads to an overestimation of boundary effects. In another study, Pandit et al. studied the phase separation process in the same ternary mixture.558 While the atomistic simulation was too short to track the whole separation process, the role of cholesterol in promoting domain formation was found to be its specific positioning and orientation between DOPC and SM molecules.
More recently, in addition to the single-phase simulations discussed in the previous section, Sodt et al. employed atomistic models to study Ld phase membranes with an embedded ordered domain.555 Their structures are shown in Figure 19. Here, the compositions of the domain and its surroundings were realistic, as they were adjusted based on the tie lines measured for this DOPC/DPPC/cholesterol mixture.521 During the simulations. spanning multiple microseconds, lipids readily exchanged between the domain and its surroundings, yet the domain remained distinguishable through the simulation. The hexagonal structure, also observed in the pure Lo phase, was also present in the phase-separated system. In agreement with the simulations by Pandit et al. mentioned above,558 they observed cholesterol positioning at the boundary of the domain with the smooth face of the cholesterol ring facing the ordered domain.
As discussed above, Sodt et al. also extended their study to mixtures containing sphingomyelin instead of DPPC and POPC instead of DOPC.556 In the system with a preformed ordered domain, cholesterol favored interactions with sphingomyelin, as expected. The hexagonal structure, observed in the pure Lo phase system was also readily visible in the phase-separated system, even though there were also differences as discussed earlier. A snapshot of this system, shown in Figure 19, allows for a direct comparison of the molecular level organization in PSM/DOPC/cholesterol and DPPC/DOPC/cholesterol membranes.
Apajalahti et al. studied the dynamics of lipids in phase-separated DLiPC/DPPC/cholesterol and DAPC/DPPC/cholesterol membranes in the coarse-grained scheme.559 As expected, they observed the lateral diffusion to be distinctly different in the Lo and Ld phases. However, the lifetimes of the correlated lipid motion as lipids migrated in a concerted manner as lipid clusters (see section 5) were quite alike in both phases. In general, this correlation had a spatial range of about 10 nm and a lifetime of the order of a microsecond, indicating that prolonged and extended simulations are required to study phase-separated membranes in proper equilibrium conditions.
Concluding, atomistic studies on preformed domains can provide information on the structure of the domain and its boundaries as well as the lipid exchange between the domain and its surroundings. Notably, when the composition of the domain and its environment are in proper equilibrium, a feat only recently achieved in atomistic resolution, the simulation can provide insight into these questions and hence help explain experimental findings. Indeed, we consider that the two studies by Sodt et al.,555,556 also discussed in the caption of Figure 19, currently provide the most detailed description of phase coexistence available from molecular dynamics simulations.
4.5.3. Simulations That Have Captured Spontaneous Phase Separation
The coarse-grained MARTINI model has been commonly employed to study the phase behavior of ternary lipid mixtures, as reviewed recently.560,561 While the model has its limitations in describing such mixtures, discussed in this section, it has also provided substantial insight on how external conditions, proteins, or other molecules affect the phase behavior and related kinetics and energetics. The discussion below mainly considers coarse-grained studies, yet when atomistic data exist for comparison, it is also reviewed.
Requirements for Phase Separation and Related Issues
The seminal study by Risselada et al. a decade ago demonstrated the spontaneous Lo/Ld phase separation of a mixture containing DLiPC, a low-Tm lipid with two diunsaturated chains and hence containing two kinks; DPPC, a high-Tm lipid with two saturated chains; and cholesterol.562 The phenomenon was reproduced in both planar bilayers and vesicles. The two phases and their boundary region were carefully characterized, and phase registration across the leaflets was observed.
While this work has sparked numerous central follow-up studies discussed extensively in this section, too, some related methodological issues are also worth noting right away. First of all, the use of DLiPC/DPPC/cholesterol instead of DOPC/DPPC/cholesterol complicates comparison to experiments since, to the best of our knowledge, the former has not been characterized. Without the experimental phase diagram and the tie lines, it is hard to predict the compositions of the Lo and Ld phases in the coexistence phase. However, the phases are likely not as exclusive as MARTINI predicts: essentially all DPPC was observed to be in the Lo phase, whereas the Ld phase consisted almost solely of DLiPC. As an example, experiments in turn suggest that in DOPC/DPPC/cholesterol mixtures the ratio of DPPC in the Lo and Ld phases is approximately two.563 The seemingly odd choice of using DLiPC/DPPC/cholesterol mixtures instead of DOPC/DPPC/cholesterol systems in MARTINI simulations is discussed further in the next subsection.
It is also worth noting that the Lo phase in many of the early MARTINI studies on phase coexistence was extremely ordered, almost to the level of the gel phase.546 While the new cholesterol parameters improve the situation significantly,546 plenty of the studies reviewed below were performed using the original model, which hardly allows any molecules to partition to the Lo phase.
Experiments only report Lo/Ld phase coexistence at temperatures where the cholesterol-free mixture would exhibit gel/Ld coexistence, that is, below the Tm of the high-Tm lipid. In the MARTINI model, however, the coexistence in the DLiPC/DPPC/cholesterol mixture also takes place at simulation temperatures above the Tm of DPPC in the MARTINI model,528 as mentioned earlier. This discrepancy, together with the almost exclusive partitioning of cholesterol to the Lo phase, suggests that the phospholipid–cholesterol interactions are somewhat imbalanced compared to reality. As discussed earlier in this section, this error might be rooted already in the incorrect phase behavior of single-component membranes.
Recently, Pantelopulos et al. studied the minimum size of a membrane required for the phase separation process.564 They simulated DLiPC/DPPC/cholesterol membranes of different sizes and observed that only systems with over ∼1500 lipids showed phase separation with well-defined phase boundaries, while many structural properties (possibly) likely converge only in systems composed of over 10000 lipids. Interestingly, in large enough systems, nanoscopic domains were found to exist in a mostly phase-separated membrane.
Baoukina et al. simulated a mixture of DOPC/DPPC/cholesterol at different temperatures.537 Notably, the DOPC model was modified and the used model actually resembled that of DLiPC, hence resulting in the spontaneous separation into Lo and Ld phases. Moreover, at higher temperatures, nanoscale heterogeneities were observed. As the temperature was lowered, larger yet transient clusters started to form, likely similar in nature to critical fluctuations. These clusters were coupled between the leaflets.
Despite the limitations, however, the MARTINI model can provide qualitative information on phase coexistence. Moreover, this information quite often seems to have predictive value and to agree well with experiment. Therefore, even with its shortcomings, it is currently the best model to describe spontaneous phase separation and factors affecting it in complex lipid mixtures at sufficiently large time and size scales.
Effect of Lipid Chain Length, Unsaturation, and Cholesterol Content
The coarse-grained MARTINI approach has been used to study the effects of composition on phase behavior thoroughly. Here, we describe studies on symmetric lipids with different chain lengths and saturation levels, whereas the next subsection focuses on studies on the effects of lipid chain asymmetry.
As a first striking observation on the effects of lipid saturation on phase separation, many teams have reported that the DOPC/DPPC/cholesterol mixture, a standard model system in laboratory experiments, does not undergo spontaneous phase separation in simulations using the MARTINI model.565−569 This finding raises the question of whether phase separation in MARTINI occurs for the right physical reason.
Davis et al. studied the effect of lipid chain bead types on phase behavior and noticed that phase separation was driven by the different Lennard-Jones interactions between saturated and unsaturated segments of the lipid chains.565 Therefore, phase separation in the DOPC/DPPC/cholesterol system could be induced by changing the bead types corresponding to the double bond region in the DOPC chains to the same types as in DLiPC chains. Notably, another kink in the chains, due to the second double bond in linoleic acid of DLiPC, was not required. These findings suggest that phase separation in MARTINI is not driven by entropy, as expected, but instead by enthalpy.
This study was extended by Hakobyan and Heuer, who studied the effect of chain kinks and Lennard-Jones parameters on the phase behavior of the DLiPC/DPPC/cholesterol mixture further.569 Interestingly, while the kinks in the DLiPC chains were found to be less relevant for phase separation, introducing them in the DPPC chains resulted in random lipid mixing. Removing the kinks from the DLiPC chains, however, resulted in its gelation and hence in the system showing gel/Lo phase coexistence. Curiously, unlike in the Lo/Ld coexistence of DLiPC/DPPC/cholesterol mixture, the leaflet registration vanished in the gel/Lo coexistence.
Very recently, Carpenter et al. refined the MARTINI model to capture the phase separation in the DOPC/DPPC/cholesterol mixture.570 By adjusting the bonded parameters of the CG models to match those observed in all-atom simulations, they were able to fine-tune the phase behavior of the pure lipid membranes, which resulted in an agreement between the simulations and the experiments on the ternary system. Unfortunately, this comes with a cost: the bond lengths and the related force constants in the refined model vary from bond to bond, hence suggesting that each lipid should be carefully parametrized based on atomistic data, instead of relying on universal parameters for such bonds, one of the key strengths of the MARTINI model.
Domański et al. studied the effect of lipid chain length on phase behavior using the MARTINI model.567 They hypothesized that the shortening of the acyl chains of the saturated DPPC lipid would lead to weaker phase separation due to lower hydrophobic mismatch. Indeed, replacing 4-bead chains of DPPC by 3-bead chains of DMPC, they observed the reduction in line tension of the phase boundary. Further shortening of the chains of the saturated lipid to two beads resulted in the disappearance of phase coexistence and the emergence of smaller-scale heterogeneities with a characteristic size scale of a couple of nanometers. The effect of cholesterol concentration on phase behavior was also studied, and lowering of cholesterol concentration was found to result in a systematic decrease in line tension. However, despite getting less stable upon a reduction of the amount of cholesterol present, phase separation was still observed even at a cholesterol concentration of 6 mol %.
Lin et al. also studied the effect of chain unsaturation on phase separation using lipids with symmetric chains.568 In agreement with other studies listed above, they also observed that DOPC/DPPC/cholesterol mixtures do not phase separate. Replacing DOPC by either DLiPC, DAPC, or DDPC stabilizes phase separation, and the effect is stronger the higher the degree of chain unsaturation is. Moreover, lipids with a higher unsaturation level interacted less with cholesterol. This difference in behavior led to a larger order parameter difference and height mismatch between the phases and therefore also to an increase in line tension. The effect of height mismatch was further studied in the DPPC/DLiPC/cholesterol system. Replacing DPPC by DMPC with two saturated chains, comparable in length to those of DLiPC, had essentially no effect on phase separation indicating that it is driven mainly by differences in lipid order instead of membrane thickness. This finding was supported by the absence of phase separation in a mixture of DPPC/DMPC/cholesterol, where both phospholipids have similar affinities for cholesterol but different chain lengths. These observations are in disagreement with the findings of Fowler et al., who reported stricter phase boundaries in systems with higher thickness mismatch of the two phases,571 as well as the findings of Domański et al., who reported the decrease of line tension upon the shortening of the DPPC chains.567 Finally, Lin et al. also considered DDPC/DLiPC/DPPC/cholesterol mixtures at increasing DPPC-to-DLiPC ratios.568 The Ld phase formed by DDPC and DLiPC had a substructure where DLiPC resided at the Lo/Ld interface, while DDPC formed the core of the Ld phase. This peculiar organization was likely driven by the less favorable interaction of cholesterol with DDPC than with DLiPC. The key observations were complemented by experiments, which showed higher mixing temperatures for a DDPC/DPPC/cholesterol system as compared to the DOPC/DPPC/cholesterol mixture, favoring the view that a higher level of unsaturation of the low-Tm lipid stabilizes phase separation.
Concluding, the effects of lipid structure and model parameters on the stability of phase separation have been thoroughly investigated using the MARTINI model. While the model has its limitations, we see no reason to question the observed trends resolved from the systematic probing of structural features. These trends provide us with at least qualitative information on how the plasma membrane lipidome and changes therein could modulate domain formation. Notably, the studies that have resolved factors dictating whether a membrane forms nanodomains or undergoes phase separation are of interest in the light of the raft concept. More such studies are discussed in the next subsection.
4.5.4. Macroscopic Separation or Nanodomain Formation?
Different Behavior of Lipids with Symmetric and Asymmetric Chains
Experimental work suggests that when mixed with a phospholipid with saturated chains, such as DPPC, and cholesterol, DOPC with two unsaturated chains induces macroscopic phase separation, whereas POPC with one unsaturated and one saturated chain induces nanodomain formation.506,572 Moreover, between these microscopically separating “Type 2” and nanodomain forming “Type 1” mixtures, quaternary mixtures with varying amounts of both DOPC and POPC can also induce a modulated phase. This peculiar behavior and the role of lipid chain saturation symmetry have been extensively studied using MARTINI simulations.
The group of Gerald Feigenson employed coarse-grained simulations to provide a detailed view of experimental findings regarding Type 1 and Type 2 mixtures. They studied the effect of replacing DLiPC by PLiPC on the phase behavior of the standard DPPC/DLiPC/cholesterol mixture.573 In line with experiments, membranes with the lipid with two unsaturated chains (DLiPC in simulations and DOPC in experiments) phase-separated into Ld and Lo regions, while membranes with the lipid with one saturated and one unsaturated lipid (PLiPC in simulations and POPC in experiments) formed transient nanodomains. However, no modulated phases were detected in simulations at intermediate compositions, likely due to the limitations imposed by the flat geometry and the size of the simulated membrane. The united-atom simulations performed on fine-grained structures revealed fairly long-ranged perturbations of membrane properties at the edge of a domain or phase. They also carefully probed quaternary mixtures with an increasing ratio of DLiPC to PLiPC and provided insight into the transition from the Type 1 to the Type 2 mixture. Interestingly, this transition was fairly sharp, and membrane registration also rapidly increased during the transition.573 The membrane morphologies at selected PLiPC/DLiPC ratios are shown Figure 20A.
Figure 20.

Factors affecting the nature of phase separation in coarse-grained simulations. (A) Morphologies of domains in DLiPC/PLiPC/DPPC/cholesterol mixtures as a function of DLiPC/(DLiPC + PLiPC) ratio (ρ). The mixtures shown here are those with ρ = (i) 0, (ii) 0.5, and (iii) 1. The membrane is tessellated with areas assigned to cholesterol shown in yellow, DPPC in blue, PLiPC in green, and DLiPC in red. The domain boundaries are shown as black lines. The phase separation becomes stronger with increasing ρ. Adapted with permission from ref (573). Copyright 2015 American Chemical Society. (B) The partitioning of POPC (orange) at the Lo/Ld phase boundary. The liquid-ordered phase is formed mainly by DPPC (cyan) and cholesterol (gray), whereas the liquid-disordered phase consists mainly of DLiPC (red). Adapted with permission from ref (574). Copyright 2010 Elsevier. (C) The effect of transmembrane peptides on the alignment of Lo and Ld phases across leaflets in the DPPC/PLiPC/DLiPC/cholesterol mixture. Aligned Lo phases are shown in white, aligned Ld phases in black, and nonaligned regions in gray. Here, ρ = 0.6 (see panel A). Data are shown for (i) the peptide-free system and (ii) a system with 4 mol % WALP-23. Adapted with permission from ref (575). Copyright 2016 American Chemical Society. (D) Alignment of Lo and Ld phases across leaflets is modulated by lipid chain length. The lipid with two saturated chains is shown in blue, cholesterol in yellow, and the lipid with two unsaturated chains in red. Here, the saturated chains were either (i) 4 beads (corresponding to 16–18 carbons) or (ii) 5 beads (corresponding to 20–22 carbons) long. Adapted from with permission from ref (576). Copyright 2011 American Chemical Society. (E) The alignment of Lo and Ld phases is modulated by chain interdigitation. The Lo phase is mainly formed by DPPC (orange) and cholesterol (white), whereas the Ld phase consists of mainly DLiPC (green). The GM1 lipids (blue) partition to the Lo phase. With a long saturated chain, GM1 perturbs the phase alignment (panel i), whereas with a shorter chains this effect vanishes (panel ii). Adapted with permission from ref (219). Copyright 2017 Elsevier. (F) Mechanism of phase separation depends on hydrophobic mismatch. Dark blue and red highlight regions with alignment of the lipids with two unsaturated and two saturated chains, respectively, whereas in regions colored in light blue and red, this alignment is not present. The time evolution of the alignment is demonstrated by data measured at 0, 2, 4, and 10 μs of simulation time. Here, DLiPC serves as the lipid with two unsaturated chains, whereas the lipid with saturated chains is either (i) DLPC (3 beads per chain, corresponding to 12–14 carbons) or (ii) DAPC (5 beads per chain, corresponding to 20–22 carbons). Adapted with permission from ref (571). Copyright 2016 American Chemical Society.
In a follow-up paper, combining experiments on various quaternary mixtures with coarse-grained simulations, this transition was concluded to take place at a fixed line tension value.577 Here, the simulations performed at intermediate mixtures with both DLiPC and PLiPC present revealed complex and dynamic morphologies for the domains that did not coalesce.
Rosetti and Pastorino evaluated the roles of lipid chain symmetry and saturation level on phase separation using the MARTINI model.566 They again observed that a DOPC/DPPC/cholesterol membrane showed uniform lipid mixing, while DLiPC/DPPC/cholesterol and DAPC/DPPC/cholesterol mixtures spontaneously phase-separated. The phase boundary in the latter was stricter, as was also reflected by a 2-fold increase in line tension. Mixtures with DPPC, cholesterol, and a lipid with one unsaturated and one saturated chain were also studied. In both PLiPC/DPPC/cholesterol and PAPC/DPPC/cholesterol membranes, only short-scale heterogeneity was observed. This behavior, associated with nanodomains506,572 (see also below), was stronger in the PAPC/DPPC/cholesterol with a higher level of unsaturation, as expected, and it could be further strengthened by lowering the simulation temperature.566 Curiously, in line with other simulation studies discussed below,573 membrane registration was only observed in phase-separated membranes.566
Domanski et al. studied the effect of lipid composition on phase behavior in the coarse-grained MARTINI scheme.567 They observed that mixtures of PLiPC, DPPC, and cholesterol only form transient heterogeneities with a characteristic size of 2.5 nm.
Curiously, the study by Pantelopulos et al. discussed earlier demonstrated the possibility for the presence of small nanodomains in a membrane under large-scale phase separation.564 This behavior seems to somewhat disagree with the concept of exclusively Type 1 and Type 2 mixtures.
Concluding, in line with experiments, replacing the low-Tm lipid with two unsaturated chains by one with only one unsaturated chain seems to favor nanodomain formation instead of phase separation, which might have importance for the formation of rafts with similar structural features. The mechanism behind this behavior is discussed in more detail in the next section.
Linactant-Induced Nanodomains
One possible mechanism through which “hybrid” lipids with one unsaturated and one saturated chain might promote nanodomain formation is that they can partition to phase boundaries and act as a linactant. Linactant is a two-dimensional analogy to surfactant and is hence a molecule that reduces the line tension of an interface, thus disfavoring the coalescence of small domains into larger ones and all the way to microscopic phases. Hybrid lipids reduce the line tension of phase boundaries by partitioning efficiently into both phases and reducing their structural dissimilarity.578 However, this picture has also been challenged by experiments.579
This concept was studied by Schäfer and Marrink, who included POPC, PLiPC, and a single-tailed lyso-PC into the ternary phase-separating mixture of DLiPC, DPPC, and cholesterol.574 They observed that POPC indeed preferred the phase boundary, where it decreased the line tension, whereas PLiPC partitioned to the Ld phase and had no effect on the phase stability. Lyso-PC was found at the interface, yet with its one tail, it had no significant impact on line tension. A snapshot of the membrane with POPC present and partitioning at the phase boundary is shown in Figure 20B.
The concept of hybrid lipids acting as linactants was studied further by Rosetti et al.580 They simulated a mixture of DAPC, DPPC, and cholesterol with increasing amounts of DAPC replaced by PAPC. The DAPC/DPPC/cholesterol system phase separated spontaneously, and PAPC was observed to partition to the boundary, where it systematically decreased line tension. With the thickness of PAPC being intermediate between DPPC and DAPC, it relieved the stress resulting from the hydrophobic mismatch.
Concluding, lipids with asymmetric chains can destabilize phase coexistence and hence promote the formation of transient nanoscopic ordered domains. While the plasma membrane is exceptionally complex in composition, it is evident that lipid chains with different saturation levels can regulate the lifetimes and sizes of raft-like ordered domains.
Phase Behavior Affected by Proteins
The plasma membrane is crowded with proteins,581 which has implications on both membrane dynamics and structure. Moreover, recent observations from single-molecule tracking experiments suggest that cellular membranes are partitioned by actin-linked pickets and fences, which prevents the free diffusion of lipids and proteins.582 In this model, pickets are proteins immobilized by their interaction with the actin cytoskeleton.
Fischer et al. simulated the DLiPC/DPPC/cholesterol mixture in the coarse-grained scheme and examined the role of immobile obstacles on phase separation.583 To this end, they restrained individual DPPC lipids either in the plane of the membrane in both leaflets or in all dimensions but only in one leaflet. Hence, the first setup allowed undulations perpendicular to the membrane, while they were suppressed in the second one. Immobilized lipids prevented full phase separation, which was otherwise observed. When fluctuations were present, multiple domains formed in the membrane, and they did not coalesce. Suppressing the fluctuations and inducing immobilized lipids only in one leaflet resulted in intermediate behavior between macroscopic phase separation and nanodomain formation.
Domański et al. studied the effect of multiple tryptophan (W)–alanine (A)–leucine (L) peptides (WALP) or one GPCR bacteriorhodopsin on the phase behavior of ternary lipid mixtures.567 The phase separation of the DLiPC/DPPC/cholesterol system was not substantially affected by the presence of a small concentration of WALP peptides or a single bacteriorhodopsin. However, WALP peptides somewhat stabilized the separation by increasing the line tension of the phase boundaries, whereas a single bacteriorhodopsin did not have such an effect. Interestingly, the presence of a large number of WALP peptides in the PLiPC/DPPC/cholesterol increased the size of heterogeneities from a couple of nanometers to 6 nm, indicating critical behavior between nanodomain formation and phase separation. This effect was strongly concentration-dependent, as a smaller amount of WALP peptides did not result in an impact of similar magnitude, even though the size scale of heterogeneities was somewhat increased.
Ackerman and Feigenson studied the effect of the presence of WALP peptides of different lengths on the tendency of quaternary lipid mixtures to phase-separate or to form nanodomains.575 Here, they used the DLiPC/PLiPC/DPPC/cholesterol mixtures from their earlier studies. They found that shorter peptides stabilized phase separation at all DLiPC/PLiPC ratios by partitioning to the Ld phase. However, all peptides, regardless of their length, promoted membrane registration. Both effects were found to be stronger for shorter peptides and larger peptide concentrations. The differences in phase alignment with different PLiPC/DLiPC ratios in the presence and absence of WALP peptides are demonstrated in Figure 20C.
Concluding, immobile objects, such as pickets, seem to strongly destabilize phase separation, while freely diffusing membrane-spanning objects such as peptides can promote phase separation via the hydrophobic matching mechanism. These two phenomena highlight the importance of considering the effects of proteins on membrane phase behavior.
4.5.5. Interleaflet Coupling and Phase Separation Mechanism
It is still debated whether the putative domain formation in the extracellular leaflet of the plasma membrane induces heterogeneity also in the cytosolic leaflet, which by itself does not form domains or phase-separate.522 Another critical question is whether the positioning of such domains is coupled across the membrane leaflets, that is, whether the membrane shows registration.522 Model membranes and theoretical work584 have been employed to resolve these questions, yet MD simulations can again prove to be helpful in providing a more detailed picture of these effects.
Coupling of Membrane Properties across Leaflets
Perlmutter and Sachs simulated an extensive set of coarse-grained membranes based on the DLiPC/DPPC/cholesterol mixture.576 In one of their setups, one leaflet had this phase-separating composition, whereas the other leaflet consisted solely of DLiPC. Curiously, phase separation in one leaflet induced heterogeneity in the opposing leaflet, as the regions facing the Lo phase showed increasing lipid order. When DPPC was replaced by lipids with longer chains in the phase-separating leaflet, this effect was strengthened due to the further disordering of the DLiPC lipids facing the Ld regions in the non-phase-separating leaflet. Symmetric phase-separating membranes were also studied either with DPPC (4 coarse-grained beads per fatty acid, corresponding to 16–18 carbons) or with saturated lipids with longer chains (5 or 6 coarse-grained beads per fatty acid, corresponding to 20–22 or 24–26 carbons, respectively). Surprisingly, while the most commonly employed DLiPC/DPPC/cholesterol mixture showed strong registration between the leaflets, strong antiregistration was observed for other tested lipids with longer saturated chains. This effect was also dependent on the ratios of the lipids present in the mixtures. Snapshots of the registered and antiregistered membranes are shown in Figure 20D. This study highlights the role of curvature stress in membrane registration and demonstrates that hydrophobic mismatch might have a more significant role in interleaflet coupling instead of in intraleaflet separation.
Tian et al. simulated asymmetric bilayers using an all-atom model.585 The leaflets reflected the typical compositions of the Lo and Ld phases in a phase-separating POPC/DSPC/cholesterol mixture and were first equilibrated as parts of symmetric bilayers. Interestingly, the leaflets retained their properties in the asymmetric bilayer, suggesting that their structural parameters are not mutually coupled. However, the removal of lipids from one leaflet induced stress in the membrane, which also affected the results in the opposing leaflet.
Manna et al. studied the effect of long-chained GM1 lipids, located in one of the leaflets, on the phase behavior of the DLiPC/SM/cholesterol mixture.219 Based on lipidomics data, such long-chained GM1 is present in the plasma membrane,586 whereas the acyl chains of other lipid types are much shorter. The coarse-grained simulations revealed that a relatively modest concentration of GM1 with a 30-carbon long chain was able to perturb both the degree of phase separation and its rate in the opposing leaflet, as well as to decrease membrane registration. Snapshots demonstrating the effect of long-chained GM1 on domain registration are shown in Figure 20E. These observations were suggested to have possible implications for signaling across the plasma membrane.
Two very recent studies employing the MARTINI model have revealed the crucial role of cholesterol for domain registration. Weiner and Feigenson observed that the amount of midplane cholesterol in the membrane correlates with the tendency for domain antiregistration.587 Increasing the length of the chains of the lipid with saturated chains resulted in the antiregistration, in line with the results of Perlmutter and Sachs,576 and this was coupled with an increased cholesterol concentration in the membrane core. Moreover, similar effects were recovered when the concentration of cholesterol was increased. A possible explanation for the observed effect is that the midplane cholesterol compromises the coupling between lipid chains and hence favors antiregistration. Curiously, midplane cholesterol most prefers to locate itself in domain boundary regions.
Along the same lines, Thallmair et al. observed that restricting cholesterol flip–flops in quaternary lipid mixtures and more complex plasma membrane models resulted in a decrease in the level of nanodomain registration.588 However, no such effects were observed for mixtures undergoing larger scale phase separation. The suppression of cholesterol flip–flop had no effects on average membrane properties. Moreover, the level of registration only decreased when cholesterol was not allowed to partition to the membrane midplane, whereas preventing complete flip–flops had no such effect. This finding is in line with those by Weiner and Feigenson,587 suggesting that the cholesterol molecules residing in the membrane midplane are crucial for the decrease in membrane registration.
Mechanism of Phase Separation
The effect of hydrophobic mismatch between the lipid phases on the phase separation mechanism and domain registration was studied by Fowler et al.571 They observed that in the case of a high mismatch, phase separation and registration proceeded via an antiregistered intermediate, which was absent in the case of a low mismatch. These two mechanisms are highlighted in Figure 20F. However, large membranes in all studied cases eventually relaxed to a registered conformation, yet smaller membranes were found to favor antiregistration. Interestingly, the threshold for this effect lies at the putative size range of lipid rafts.
As discussed above, phase separation in the coarse-grained MARTINI model is driven by enthalpy.565,569 Before this observation, Hakobyan and Heuer compared the phase separation process of the DLiPC/DPPC/cholesterol mixture using coarse-grained and united-atom models.589 The coarse-grained membranes readily phase-separated during the 12 μs simulation time, whereas the united-atom membrane displayed only partial demixing during the 9 μs simulation. To fill this gap, a united-atom simulation of a preformed DPPC/cholesterol domain in a DLiPC membrane served as a model for complete phase separation. Interestingly, when the number of DPPC–DPPC neighbors, characterizing the degree of phase separation, was plotted against simulation time that was scaled based on the differences in lateral diffusion coefficients, the phase separation kinetics seemed strikingly similar. This finding suggests that the underlying phase separation mechanisms are alike in both models. Indeed, the structural parameters extracted from the united-atom system with a preassembled domain agree reasonably well with those of the phase-separated coarse-grained system. Even though the mechanism seems similar, it is unclear whether the right factors drive it. In the united-atom model, cholesterol has special arrangements around saturated and unsaturated lipid chains, which provides an entropic contribution to the separation process. However, no such tendencies were observed in the coarse-grained model supporting the idea that phase separation is driven by enthalpy therein.
Concluding, coarse-grained simulations have demonstrated that structural details can affect membrane registration, which can result from at least the mechanical properties of the leaflets or direct interactions by interdigitating lipid chains. Moreover, the antiregistered membrane might serve as an intermediate step on the way to registration. Most importantly, simulations have demonstrated that the formation of a domain in one leaflet can induce heterogeneity in the other leaflet that does not phase separate by itself. Since the model used to demonstrate this feature captures the essential elements of the plasma membrane structure, this finding has significant implications for our understanding of the structure of lipid rafts.
4.5.6. Membrane-Partitioning Solutes Affecting Phase Separation
The ability of specific molecules to strengthen or weaken the phase separation has been examined using simulations. These simulations have revealed many possible mechanisms that are summarized in Figure 21.
Figure 21.
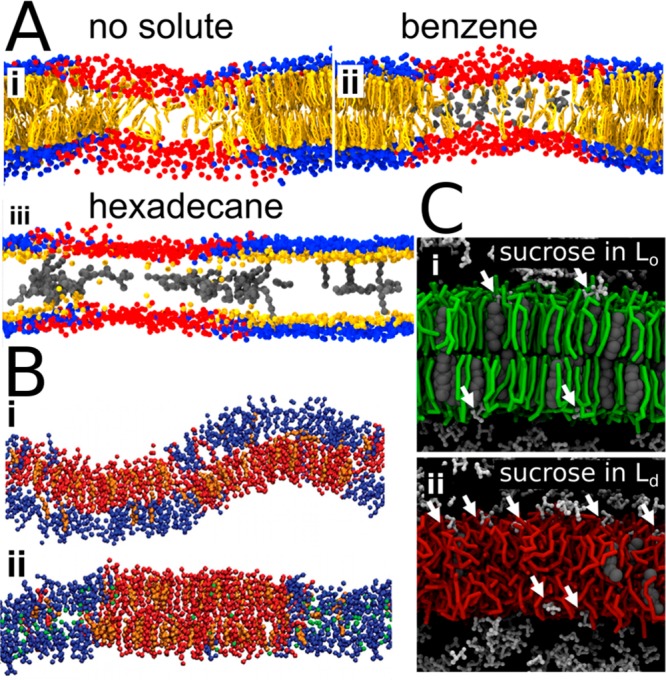
Mechanisms that stabilize or destabilize phase separation in ternary bilayers. (A) Aromatic and aliphatic compounds have different effects on phase separated membranes. Cholesterol is shown in yellow, DPPC headgroups in blue, DLiPC headgroups in red, and hydrophobic compounds in gray. Aromatic benzene partitions to the Ld phase and stabilizes separation (panel ii), whereas aliphatic hexadecane partitions at the phase boundary and destabilizes separation (panel iii). Note that in panel iii, cholesterol is not fully shown to reveal the presence of hexadecane in the Lo phase. Adapted with permission from ref (590). Copyright 2014 Barnoud et al. (https://creativecommons.org/licenses/by/4.0/legalcode). (B) Effect of chloroform on domain registration. DSPC is shown in red, DLiPC in blue, cholesterol in orange, and chloroform in green. In the absence of chloroform (panel i), the phases are unaligned, whereas in the presence of chloroform (panel ii), the alignment is present. Adapted with permission from ref (591). Copyright 2015 Reigada et al. published by the Royal Society. (C) Mechanism of disruption of phase separation induced by surface-bound molecules. Sucrose has fewer sufficiently large defects as biding sites in the Lo phase (panel i) than in the Ld phase (panel ii). Therefore, the presence of sucrose favors lipid mixing. A similar effect is not observed for smaller glucose, which binds the Lo and Ld phases with equal magnitudes. Adapted from ref (592). Copyright 2014 American Chemical Society.
Rossi et al. studied the effect of polystyrene on lipid membranes and observed that it partitions to the Ld phase and stabilizes phase separation.593 This study was extended to other plastics with very different effects, either stabilizing or destabilizing phase coexistence, based on their chemical nature.594 The authors later continued to study some hydrocarbon compounds and observed different behaviors for aliphatic and aromatic molecules.590 While the former partitioned at the phase boundary and destabilized the separation, the latter partitioned exclusively to the Ld phase and stabilized it. These mechanisms are highlighted in Figure 21A.
Reigada et al. studied the effects of chloroform on mixtures of DSPC, DLiPC, and cholesterol.591 They observed that it promotes phase registration across the leaflets via an entropic effect, while it does not affect the phase separation kinetics or the degree of separation within individual leaflets. This mechanism is highlighted in Figure 21B.
In addition to partitioning into the bilayer, certain molecules can affect membrane phase behavior by phase-dependent attachment to the membrane surface. Moiset et al. studied the effect of mono- and disaccharides on the phase behavior of DPPC/DLiPC/cholesterol mixture in the coarse-grained scheme.592 They observed that nonreducing disaccharides, but not reducing disaccharides or monosaccharides, destabilized phase separation. Interestingly, this behavior, confirmed by experiments, was explained by the differences in the surface topology of the membrane: The membrane has more surface defects able to accommodate nonreducing disaccharides when no phase separation is present. Hence, in such conditions, demixing of the phases is favored. This mechanism is highlighted in Figure 21C.
Muddana et al. studied the phase behavior of the DLiPC/DPPC system when cholesterol was replaced by a non-lipid amphiphile.595 In line with experimental observations, vitamin E destabilized phase separation, while benzyl alcohol and Triton-X 100 stabilized it. Moreover, the MD simulations provided an atomistic picture of the underlying mechanisms: Vitamin E acted as a linactant, benzyl alcohol partitioned to the DLiPC-rich phase and further decreased order therein, whereas Triton-X showed nonspecific partitioning yet promoted hydrophobic mismatch by thickening the DPPC-rich phase.
Concluding, the MARTINI model reveals numerous mechanisms through which different molecules can disturb phase separation and regulate membrane heterogeneity. The information provided by these studies can be harnessed to understand how the complex mixture of molecules present in the cell membrane and its environment can affect raft formation. Besides, these findings are essential for understanding how both undesired particles, such as plastic nanoparticles, as well as desired ones, such as pharmaceuticals, can affect membrane organization and thereby have also implications on health.
4.5.7. More Complex Lipid Mixtures
Membranes with a Realistic Lipid Composition
Some recent simulations have considered even more complex membrane compositions, mimicking the composition of real membranes. These initiatives are partly driven by more and more lipidomics data becoming available in the literature.
Ingólfsson et al. simulated a model for the plasma membrane consisting of a total of 63 lipid moieties distributed asymmetrically between the leaflets.596 They applied a soft harmonic potential, modeling the interactions with the cytoskeleton, to a fraction of the lipid headgroups. This trick helped them keep the thickness of the water slab reasonably thin and therefore the computational requirements reasonable. The simulation reproduced many central phenomena suggested by experiments. Transient nanoscopic domains, coupled across the leaflets, were observed. While gangliosides formed clusters in the extracellular leaflet, phosphatidylinositol displayed similar behavior on the cytosolic side. However, the former is likely enhanced by the artificially strong clustering tendency of the ganglioside model.597 Finally, cholesterol equilibrated by spontaneous flip–flops, which resulted in its enrichment on the extracellular leaflet. Snapshots of this complex membrane are shown in Figure 22A.
Figure 22.
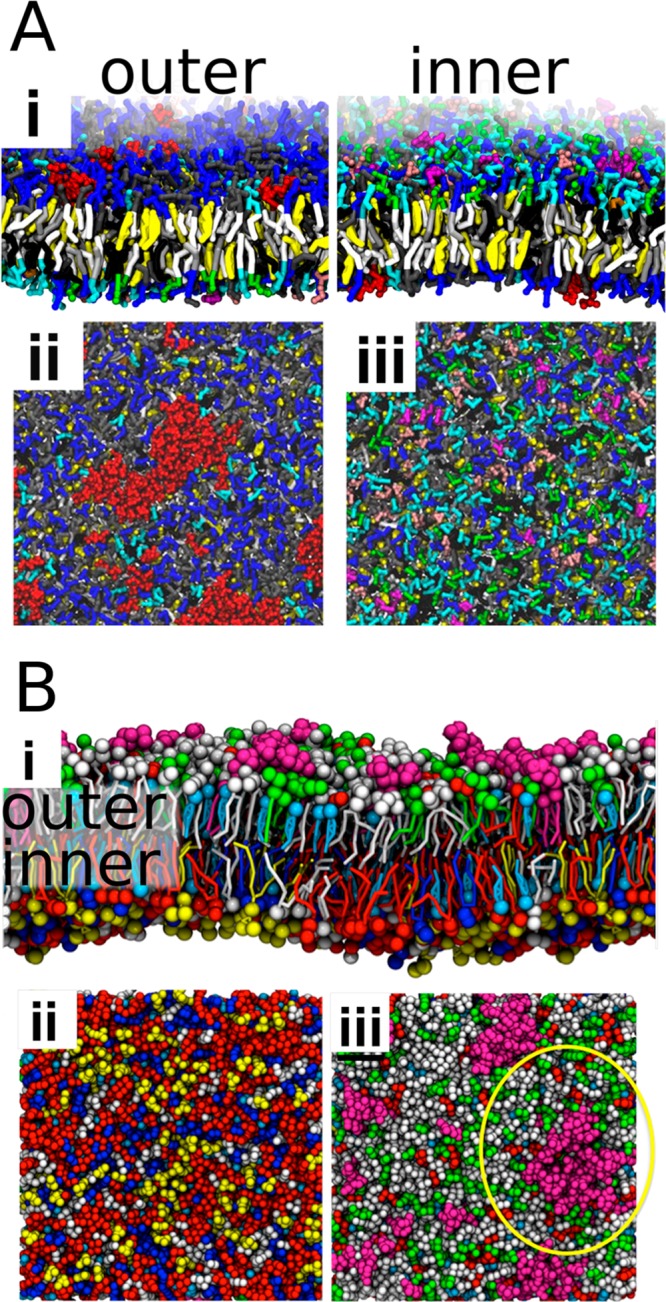
Small and transient domains in complex biomembranes. (A) In a membrane with a total of 63 lipid types, GM1 (shown in red) clusters at the outer leaflet (ii), while phosphoinositides, shown in magenta, form small clusters in the inner leaflet (iii). Adapted with permission from ref (596). Copyright 2014 American Chemical Society. (B) Similar clustering of GM3 (shown in purple) in the outer leaflet (iii) and to a lesser degree that of phosphoinositides (yellow) in the inner leaflet (ii) in a 7-component membrane. Adapted with permission from ref (598). Copyright 2014 Koldsø et al. (https://creativecommons.org/licenses/by/4.0/legalcode).
In a follow-up study, Ingólfsson et al. adjusted the composition of their plasma membrane model to match that of brain plasma membranes,599 meaning that the levels of polyunsaturated fatty acids (PUFAs) and cholesterol were increased. An increase in cholesterol content orders the membrane, while PUFAs have an opposing effect. These effects partially cancel each other, and the behavior of the brain plasma membrane model does not differ significantly from that of the average plasma membrane model. However, the domain sizes and their lifetimes were more significant in the averaged plasma membrane. Similarly, domain registration was more prominent therein. Interestingly, the domain size was found to heavily depend on the undulations of the bilayer: while suppressing undulations by harmonic restraints modeling the interactions with a cytoskeleton resulted in a decrease in domain size in the brain plasma membrane model, an opposite effect was observed for the average plasma membrane model. This finding highlights the importance of often overlooked curvature on membrane heterogeneity. Indeed, local curvature seems to be a crucial parameter in defining domain registration.587,591 Moreover, the lipid sorting of the tethers pulled from this membrane model and their subsequent softening was observed by Baoukina et al.600 All in all, the picture provided by these studies by Ingólfsson and co-workers is currently the most comprehensive model of a protein-free plasma membrane.
Other Complex Lipid Mixtures
The role of curvature on lipid sorting was further evaluated by Koldsø et al., who simulated asymmetric 7-component membranes in the coarse-grained scheme.598 GM3 gangliosides and to a lesser degree also phosphoinositides were found to assemble into nanodomains, yet the former effect is again likely overestimated by the exaggerated interactions between gangliosides in the used model.597 The mechanism behind this clustering was found to be dynamic curvature fluctuations, which sorted GM3 to concave regions in the extracellular leaflet, whereas phosphoinositides localized in the concave areas in the intracellular leaflet together with cholesterol. In both leaflets, PE lipids also partitioned to these concave regions. A snapshot of this membrane, highlighting the clustered lipids, is shown in Figure 22B. The model of Koldsø and co-workers contained lipids with one saturated and one monounsaturated chain, yet the effect of the presence of lipids with two monounsaturated chains was also evaluated. In line with the inability of such lipids to induce phase separation within the MARTINI model,565 no signs of large-scale lateral heterogeneity were observed.
Guixà-González et al. studied brain cortex tissue membranes with compositions characteristic for healthy individuals and individuals with neurodegenerative or mental disorders.57 These membranes contained a total of seven lipid components, including phosphatidylcholines with saturated and monounsaturated chains, sphingomyelin, and cholesterol. Additionally, SDPC with a PUFA chain, whose membrane levels are affected by the diseases mentioned above, was present in the mixture. In the absence of any proteins, only transient heterogeneities were observed, as the polyunsaturated chains formed clusters of irregular shape but were unable to undergo separation on a larger scale.
Flinner and Schleiff considered membranes mimicking the composition of the red blood cell.601 These membranes contained an asymmetric distribution made up of a total of 9 different lipids of four different families, PE, PC, PS, and SM, and with different chains. This mixture was found to show transient domain formation and dissociation, which was found to be driven by fatty acids. Indeed, changing all headgroups to PC had no significant effect on lateral heterogeneity. However, in the absence of cholesterol, no domain formation was observed, as expected.
Concluding, coarse-grained simulations have been used to probe very complex lipid mixtures that aim to mimic the lipid composition of the plasma membrane. These studies have revealed the formation of small and transient lipid clusters, which might bear a resemblance to rafts in the plasma membrane. However, the current paradigm views rafts not as lipid entities but instead as lipid–protein platforms. This role of proteins in the formation of raft-like domains is discussed in detail in section 4.5.8.
4.5.8. Membrane Proteins Induce the Formation of Local Domains
Instead of being purely lipid assemblies, heterogeneity also arises from specific protein–lipid interactions that shape the composition in the immediate vicinity of a membrane protein. Such interactions were recently reviewed by Hedger and Sansom.602 In the following, we focus on protein-induced domains in multicomponent plasma membrane models. These can form in many ways: a single protein can alter its neighborhood by favorable interactions with certain lipid types. Additionally, charged lipids can glue proteins together into aggregates. Moreover, these two mechanisms together can result in the formation of large aggregates with specific lipid and protein compositions.
Koldsø and Sansom simulated the transmembrane domain of the cytokine receptor gp130 in POPC membranes where a fraction of the lipids in the intracellular leaflet was replaced by anionic POPS.603 They observed the heterogeneous distribution of the PC and PS lipids around the gp130 transmembrane domain, which was further affected by the presence of extra amino acids in the gp130 structure beyond the transmembrane domain.
In a follow-up study, Koldsø et al. included a total of 16 gp130 transmembrane domains embedded into the asymmetric 7-component system earlier described.598 Here, the basic amino acids, located within the intracellular leaflet, again clustered anionic lipids, this time the PIP2 present in the model. On the extracellular leaflet, GM3 assembled around the gp130 transmembrane domain. Interestingly, this localization of the GM3 and PIP2 near the transmembrane domain did not affect the size of the lipid clusters compared to the protein-free membranes.
In yet another study, Koldsø and Sansom simulated larger versions of the protein-free and the gp130 transmembrane domain-containing plasma membrane models and complemented the study with a large plasma membrane model containing multiple copies of the GPCR sphingosine 1-phosphate receptor 1 (S1P1).56 All these membranes consisted of more than two million MARTINI beads, making them currently the most extensive simulations of the plasma membrane outside of studies focusing on long-time lipid dynamics.604 Snapshots of the membranes containing gp130 or S1P1 are shown in Figure 23A. The earlier results for gp130 transmembrane domain were reproduced, and favorable interactions with S1P1 were observed for cholesterol and PIP2. These changes in local lipid composition induced by the presence of the proteins could result in more extensive heterogeneities through protein aggregation.
Figure 23.

Protein-induced lateral heterogeneity. (A) Lateral heterogeneity induced by membrane proteins. The gp130 TMD (i), shown in red, gets solvated by GM3 (light blue) and PIP2 (orange). The S1P1 (ii), shown in purple, interacts favorably with PIP2 and cholesterol (green). Adapted with permission from ref (56). Copyright 2015 American Chemical Society. (B) Preferential solvation of dopamine D2 (blue) and adenosine A2A (red) receptors by SDPC (yellow) with a polyunsaturated chain. The other four lipid types are shown in gray. Reproduced with permission from ref (57). Copyright 2016 Guixà-González et al. (https://creativecommons.org/licenses/by/4.0/legalcode). (C) The formation of a Lo domain surrounded by the influenza hemagglutinin proteins due to sterical hindrance. DLiPC is shown in dark blue, DPPC in cyan. Adapted with permission from ref (605). Copyright 2013 Parton et al. (https://creativecommons.org/licenses/by/4.0/legalcode).
The same group also studied the effect of membrane composition on inwardly rectifying potassium (Kir) channel clustering.606 These channels were found to interact favorably with PIP2, PS, and GM3 lipids, inducing heterogeneity in their environment. However, a more detailed exploration of these environments was omitted.
As discussed earlier in this section, in the studies of Ackerman and Feigenson575 and Domanski et al.,567 WALP peptides were found to promote phase separation and membrane registration via the hydrophobic mismatch mechanism. Especially the increase in the size of heterogeneities in the PLiPC/DPPC/cholesterol membranes is a reliable indicator of protein-induced domain formation, even though the driving force is not based on specific lipid–protein interactions as in most of the other studies discussed in this section.
Sharma et al. simulated the self-assembly of membranes mimicking the composition of synaptic vesicles.607 They biased the self-assembly protocol to induce membrane asymmetry corresponding to experimental data. Notably, PC and SM were located in the intravesicular leaflet, whereas the cytoplasmic leaflet consisted mainly of PE, PS, and PIP2. Cholesterol was present in both leaflets at equal amounts due to rapid equilibration via flip–flops. This self-assembly was demonstrated in the presence of a 48-residue long segment of syntaxin-1A, which was readily embedded into the forming bilayer. Favorable interactions between syntaxin-1A and PIP2 were observed in the self-assembled structures leading to local heterogeneity.
Several factors (lipid composition, protein structure, mutations, and cations) affecting syntaxin-1A clustering were resolved in a multidisciplinary study by van den Bogaart et al.608 In addition to pure DOPC, they evaluated syntaxin-1A clustering in membranes where one leaflet was enriched with either PI(4,5)P2 or PI(3)P. Moreover, PIP2 was also included in a mixture of DOPC and DOPS. The simulations showed, in line with experiments, that the presence of PIP2 results in the formation of syntaxin-1A clusters, and PIP2 acts as a bridge between the monomers.
In the study by Guixà-González et al., described in the previous subsection,57 the inclusion of adenosine A2A and dopamine D2 receptors in the brain cortex tissue membranes induced the formation of SDPC- and cholesterol-rich coronas in their immediate vicinity. These coronas were observed to promote receptor oligomerization with implications for their signaling. Moreover, this oligomerization led to the formation of protein–lipid assemblies rich in SDPC and cholesterol. These assemblies are evident in Figure 23B, which shows a snapshot of the simulation of an SDPC-rich membrane.
Similar enrichment by polyunsaturated fatty acids was also observed in the environment of glycophorin A, when it was embedded in the complex mixture corresponding to the composition of the red blood cell plasma membrane.601 While glycophorin A had persistent interactions with sphingomyelin and cholesterol, it still preferred to partition into the Ld phase together with polyunsaturated fatty acids.
In all of the examples discussed, a single protein can induce heterogeneity in its vicinity due to specific protein–lipid interactions. However, proteins can also affect membrane heterogeneity in other ways. Parton et al. studied clusters of influenza hemagglutinin proteins in a phase-separating DLiPC/DPPC/cholesterol membrane.605 They observed that the proteins destabilized phase separation compared to the protein-free case. Interestingly, protein clusters were able to sterically cage slowly diffusing DPPC lipids as compared to the faster moving DLiPC ones. This hindrance resulted in the formation of a Lo domain surrounded by the transmembrane segments of the protein. A snapshot of this domain is shown in Figure 23C.
Finally, we must highlight the major effort of Corradi et al. to build on the multicomponent systems by Ingólfsson et al.596 by including 10 distinct membrane proteins in the membrane and observing their interactions with the over 60 lipid components.609 They observed specific interactions determined by lipid charge and chain unsaturation. Moreover, interactions with cholesterol varied substantially between the proteins. All these effects led to both specific direct lipid–protein contacts as well as longer-range changes in membrane structure characterized by thickness and curvature.
Concluding, membrane proteins can induce heterogeneity via different mechanisms. Direct protein–lipid interactions reshape the neighborhoods of proteins, while the coalescence of many such lipid shells leads to the formation of larger functional domains, especially if protein aggregation has functional implications. Proteins can also affect heterogeneity via physical mechanisms, such as promoting hydrophobic mismatch between the phases thereby stabilizing the separation. This field will undoubtedly grow as new protein structures become available and will likely move on to employ even more complex lipid mixtures to resolve favorable lipid–protein interactions. We believe that the coarse-grained framework, introduced by Corradi et al.,609 will provide a useful approach for such attempts.
4.6. Challenges
Our somewhat limited understanding of heterogeneity in the plasma membrane is primarily based on experiments carried out in live cells, plasma membrane extracts, or model membranes. Despite these efforts, numerous details in the plasma membrane organization remain a mystery. Recently, both theoretical efforts and computer simulations have been extensively employed to understand how lipids arrange in membranes under different conditions. The progress in the simulation field has especially benefited from the rapid development of simulation models and computer infrastructure. Currently, atomistic models can probe the structure of preformed domains and their boundaries in detail. The coarse-grained MARTINI model captures the spontaneous separation of membrane lipids into two liquid phases. On the basis of these possibilities, different factors affecting phase separation have been probed by numerous groups. The studies have revealed different mechanisms that might regulate the size scale of heterogeneities and favor small domains in the plasma membrane environment. Moreover, they have provided insight on the interleaflet coupling between membrane leaflets, a feature that is extremely hard to tackle experimentally. Coarse-grained simulations have also probed complex lipid membranes, where, in line with experiments, small and transient nanodomains are observed. Moreover, simulations have demonstrated how lipid–protein interactions, driven by different mechanisms, can result in the formation of membrane heterogeneities in the protein’s immediate neighborhood and lead to longer-range structural perturbations.
While all these efforts have advanced the field substantially, many challenges still remain. So far, the spontaneous phase separation or nanodomain formation in ternary or more complex systems has not been demonstrated using atomistic models. While the limitations in the force fields cannot be ruled out, the biggest reason for this is the limited simulation time scale, which is likely in the 10–100 μs regime. Moreover, the system sizes need to be fairly big, too.
For coarse-grained models, these scales do not usually pose a problem, yet simulations large enough to capture multiple coexisting nanodomains that do not coalesce or long enough to probe the formation of transient domains in complex mixtures are still scarce. Unfortunately, for coarse-grained models, the issues with methodology are more severe. The MARTINI model predicts a very sharp separation that is readily seen when a simulation trajectory is visualized. However, tie lines measured experimentally suggest more subtle differences in the compositions of the coexisting phases, down to a level where it cannot be distinguished by the naked eye. This calls for algorithms to detect the phase coexistence, and the models could be improved based on experimental tie lines. Moreover, the canonical mixture of DOPC, DPPC, and cholesterol used in numerous experiments does not phase-separate in the standard MARTINI model. Fortunately, attempts to improve the situation have been proposed recently.
It is also worth highlighting that to provide correct phase behavior of ternary and more complex mixtures, the phase behavior of the underlying single-component and binary mixtures should be correct. This means that the melting points should be reproduced with reasonable accuracy. Moreover, to provide liquid–liquid coexistence upon the addition of cholesterol, the gel–fluid coexistence should be captured in its absence. Unfortunately, many models do not capture the main transition points of lipids, and for the MARTINI model, the values of Tm of high-Tm phospholipids are drastically underestimated. It seems that phase separation and domain formation are driven by enthalpy rather than entropy in MARTINI. Due to the decrease in degrees of freedom, this behavior is likely common for all coarse-grained models. Hence, the temperature-dependence is not captured well with the models suggesting that they are used at the temperature in which they were parametrized. Unfortunately, below the Tm of a high-Tm phospholipid, this might no longer be the case.
To conclude, there are many methodological issues that need to be solved before simulations can be considered as an equal counterpart to experimental work. The time scales of atomistic simulations need to be extended significantly to probe spontaneous phase separation. As long as this task remains too challenging, coarse-grained models are a tempting option, given that their parametrization is critically evaluated in the light of their shortcomings. Still, the coarse-grained models are an indispensable tool for providing qualitative information on phase separation and to explain experimental observations.
5. Lateral Diffusion in the Membrane Plane: Is It Biologically Relevant?
Systems without internal motion are frozen, that is, crystalline. If a system is frozen, it cannot be alive, can it? And if a system is not alive, it is likely not relevant in a biological sense.
The process maintaining a biological system in motion is diffusion. In biological membranes, diffusion takes place as lipids, proteins, and the other molecules hosted by the lipid membrane move in two dimensions in the membrane plane. This motion, as surprising as it may be, is largely based on a very simple phenomenon: a random walk.
The first observations of random walk were made in 1828 by the botanist Robert Brown,610 who explored the pollen of plants and observed that, when placed in water, the pollen particles conducted irregular “swarming” motion that we today call Brownian motion. Theoretically, Brownian motion describes the motion of a colloidal particle in a liquid, resulting from numerous random molecular collisions with tiny liquid molecules. Each of these collisions has only a marginal effect on the colloid. However, when the collision rate is large and since each of the collisions leads to an exchange of momentum between the liquid molecules and the colloid particle, the overall effect on the colloidal particle is perceptible as a random movement.
In biological membranes, the size of membrane-associated proteins is much greater than the size of the surrounding solvent and lipid particles. One can then assume that the idea of random walks is valid for membrane proteins, too. For lipids in a membrane, the separation of mass scales is not so obvious, meaning that lipids move in an environment of largely identical particles–other lipids. Given this, there is reason to assume that the motion of lipids at time scales where they collide with one another is not a pure random walk, but the motions of the lipids next to one another are somehow correlated. At long times, when the “memory” of these time-dependent correlations has died out, the random walk picture is expected to emerge again. Altogether, there is an analogy from colloidal particles to biological molecules, thus providing us with some grounds to use the random walk concept to describe diffusion in biomembrane systems.
Lipids and proteins use thermal energy as a driving force to move about until they find their partners in functional complexes. As we discuss in section 7, lipids regulate the function of many membrane proteins. Some of the modulation mechanisms are based on specific lipid–protein interactions, such as allosteric binding. The allosteric binding in turn is possible if and only if the specific lipid in question finds the binding pocket of its protein host, and this requires diffusion to take place. In the same spirit, for membrane protein complexes comprised of several proteins, the formation of the functional complex requires the proteins to diffuse in the membrane plane until they find their partners.
It is noteworthy that the typical length scales of the domain structures in membranes are in the 10–1000 nm range, that is, on scales where diffusional processes can be effective on the time scales that are relevant in biology. This can be rationalized by considering the time needed to diffuse over a distance L in a time scale tD. Based on dimensional analysis and the definition of diffusion, this time scale for diffusion in two dimensions is on average tD = L2/(4D). Given that a typical diffusion coefficient D for a lipid in a cell membrane is about 10–7 cm2/s,611 the time needed to diffuse over a domain of size 100 nm is ∼250 μs, or a distance of 1 μm in 25 ms. These times are amazingly short given that the diffusion is not driven by any external source of energy but is fully based on thermal fluctuations. Meanwhile, the diffusion time scales as tD ≈ L2, which is bad news for long-range transport.
For membrane proteins, one can perform the same analysis but with certain reservations. The diffusion coefficient describing membrane protein diffusion depends on the actin cytoskeleton network, the cell type, and also the cell studied (within the same cell type) due to heterogeneities between different cells and then obviously also on thermodynamic conditions.612 Anyhow, using a value of ∼10–9 cm2/s that is quite often in the right ballpark,613 one finds the diffusion time tD to be around 25 ms for L = 100 nm and 2.5 s for L = 1 μm.
In brief, diffusion based on thermal fluctuations without an external energy source is an exceptionally efficient means to transport molecules and molecular complexes along the membrane surface, if the transport takes place over a relatively short distance. For long-range transport, this conclusion is not as evident.
Based on this picture, it is tempting to ask whether Nature has over evolutionary times developed strategies to compartmentalize and structure biomembranes on scales from nanometers to micrometers in order to take advantage of thermally driven diffusion. Regardless of the answer, what is clear is that diffusion is an exceptionally important dynamic process that renders most biological processes possible.
Here, we do not discuss topics covered elsewhere, such as how the diffusion coefficient depends on membrane lipid content611,614 or how anomalous diffusion contributes to membrane functions.615 Instead, we focus on selected questions that aim to unravel how diffusion of lipids and proteins takes place in biomembranes as their complexity is increased systematically.
What is the mechanism of lipid diffusion at the molecular level? How do lipids and proteins move together in the membrane plane? How do cellular conditions such as protein crowding influence the diffusion process? How do the protein networks that are bound to membrane structures affect the diffusion rates? Are there valid theories able to grasp the essential physics of lateral diffusion and its dependence on the relevant thermodynamic variables?
In this section, our aim is to discuss recent computational research that has unraveled these questions. In part, they represent examples of the groundbreaking knowledge needed to understand the core of the diffusion process. Further, they highlight the profound challenge to understand the underlying physical bases of diffusion in more and more complex membrane environments. We close this section by identifying a few major questions where molecular simulations would be very helpful to understand the physical laws underlying a number of biologically relevant dynamical processes.
5.1. Mechanisms of Lipid and Protein Diffusion in Lipid Membranes
5.1.1. Concerted Lipid Motion
As discussed in section 5.2.1, the picture that has been an established way to think of how lipids diffuse in a membrane has been based on the concept of free volume pockets. For a number of years, one has assumed based on theories developed originally for hard-sphere colloids that lipids move in the membrane plane by “jumping” from one position to another. First, a lipid is assumed to reside in its cage, surrounded by other lipids in a quite tightly packed membrane. Then, if a thermally driven density fluctuation causes the membrane to locally expand, thus creating more space for the lipid in question in its vicinity, or if another lipid moves to a new position next to the lipid in question, then the lipid can “jump” to a new position over a distance of about its own size, that is ∼1 nm. While this picture is appealing, it is hard to find experimental evidence that would prove its validity.
Atomistic simulations suggested a different picture. Falck et al.616 carried out atomistic simulations on single-component phospholipid bilayers and inspected the lateral trajectories of all individual lipids. Examples of center of mass trajectories covering the last 30 ns of one of the simulations are shown in Figure 24. The relation L2 ≈ 4Dt for the distance L traveled by the lipid during time t, with the observed diffusion coefficient of D = 1.5 × 10–7 cm2/s and L = 0.7 nm for the linear size of a lipid, suggests that each lipid should diffuse over L in about 8 ns. Therefore, if jump diffusion were the predominant diffusion mechanism, Figure 24 should show several jumps for each of the lipids. However, most trajectories do not contain such jumps. The trajectory labeled A in Figure 24 contains one of the fewer than 10 rapid jump-like events that were observed in any of the simulations analyzed in the study.
Figure 24.
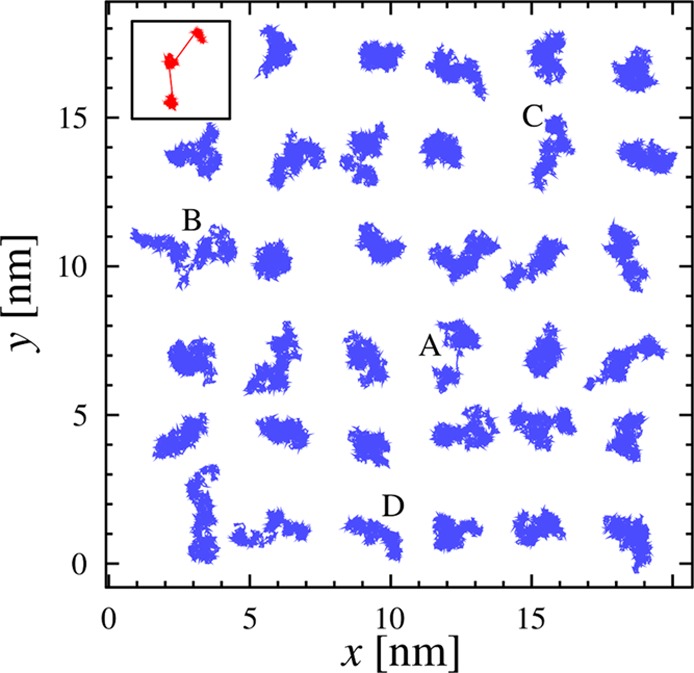
Examples of center of mass trajectories for selected lipids in the plane of a DPPC bilayer, showing the last 30 ns of the simulation. The red inset (top left) highlights an example of how a trajectory is expected to look like if the diffusion were jump-dominated. The diffusion of lipids marked with A–D are discussed further in Figure 25. Adapted with permission from ref (616). Copyright 2008 American Chemical Society.
However, a detailed analysis by Falck et al.616 demonstrated that none of these jump-like events were a real “jump”, where the lipid in question would have migrated over its own size when the neighboring lipids would have stayed more or less still. Therefore, the diffusion mechanism of lipids is not based on jumps. It turned out to be more complicated and based on concerted motion of various lipids. Figure 25 depicts this motion for a representative case during a period of 1 ns. The data show that the movement of the lipids undergoing jump-like motion is actually highly correlated with the motion of their neighbors’ movement; one can observe several lipids to move as loosely defined local clusters.
Figure 25.

Trajectories of selected lipids and their immediate neighbors during 1 ns time intervals. The red trajectory in each panel is part of the trajectory labeled A, B, C, or D in Figure 24. For each lipid, the 1 ns interval was chosen to be the one during which the lipid migrated the longest distance during the simulation. The blue trajectories are those of the nearest neighbors, which in practice are the lipids whose centers of mass are within 1 nm of that of lipid A, B, C, or D at any time during the 1 ns period. The black arrows indicate the total displacements during the 1 ns period. Adapted with permission from ref (616). Copyright 2008 American Chemical Society.
The above observations are consistent with related earlier findings by Ayton and Voth who used a coarse-grained model to study lateral diffusion on a mesoscopic level.617 More recent coarse-grained simulation studies by Apajalahti et al.559 are also in agreement with the above picture and suggest the sizes of the dynamical lipid clusters that undergo concerted motion to be larger than ∼10 nm. Experimentally, the predictions by Falck et al.616 have been later tested by the Unruh group using quasielastic neutron scattering, which is the method of choice to explore membrane dynamics over time scales on the order of 1 ns. Their data618,619 are consistent with the concerted lipid diffusion mechanism predicted by Falck et al.616 Similar observations have also been made in quasielastic neutron scattering experiments by Armstrong et al.620 and in simulation studies by Chavent et al.621 and Starr et al.622
The simulation results therefore provide a confirmed and validated picture for the mechanism of lateral diffusion of lipids in membranes. In essence, lateral diffusion is not dominated by jumps. Instead, the mechanism is based on predominately continuous, correlated, and concerted motion of neighboring lipids. The correlations persist over tens of nanometers, manifesting themselves as two-dimensional flow patterns originating from thermal density fluctuations complemented by local momentum conservation despite the dissipative nature of the membrane embedded in a water bath. These flow patterns are expected to play a role in the molecular mechanisms of several key processes in membranes, such as in the formation of lipid domain structures, allosteric binding of lipids with their host proteins, and exposing membrane proteins to lateral pressure.
5.1.2. Concerted Motion of Protein–Lipid Complexes
It is now clear that lipids move in a concerted fashion as loosely defined correlated transient clusters. Then, what happens if an integral membrane protein is embedded in the lipid membrane? How does the protein move, and how does the presence of the protein interfere with the lateral dynamics of the lipids in a membrane?
These questions were explored by Niemelä et al.16 using atomistic as well as coarse-grained simulation models. In atomistic simulations, they focused on resolving the lateral diffusion of a voltage-gated channel Kv1.2 in a one-component POPC membrane. As Figure 26 depicts, they found that the protein and the neighboring lipids form a transient complex: about 100 lipids move laterally together with the protein. On average, these lipids close to the protein move much more slowly than the other lipids farther away from the protein.
Figure 26.
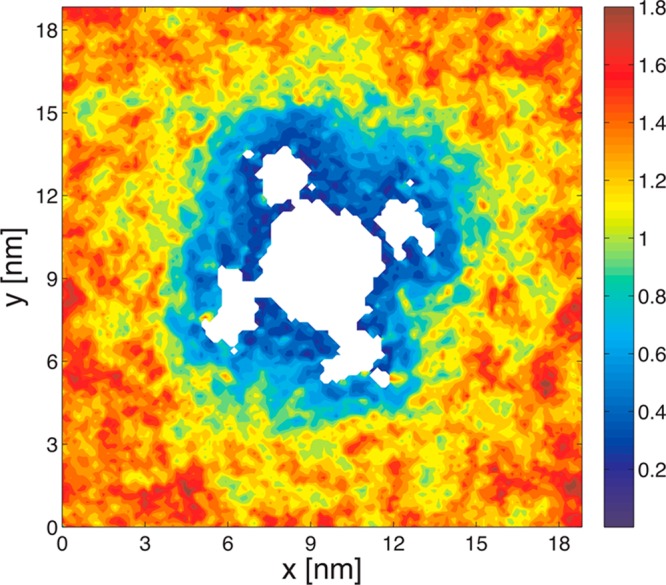
Mean lateral displacements of lipids in one of the two membrane leaflets over a time scale Δt = 50 ns. The protein is shown in white, and its center of mass is centered. The scale bar on the right indicates dark blue to be characterized by very slow diffusion, and dark red to correspond to fast lateral diffusion. The blue region around the protein highlights the slowed-down lipids that move together with the protein in a concerted fashion. Adapted with permission from ref (16). Copyright 2010 American Chemical Society.
The study revealed that the effects on the layer of lipids in direct contact with Kv1.2 (annular lipids) were strongest, yet a significant slowing-down effect was observed also for several other layers of lipids up to a distance of ∼7 nm from the protein center of mass (about 4 nm from the lipid–protein interface). The annular lipids as the closest neighbors of the protein were found to be largely immobile. As the lateral distance from the protein increased, so did also the lipid diffusion coefficient. However, only lipids that were as far as ∼7 nm from the protein center of mass were observed to have a diffusion coefficient that was close to the value found in a protein-free membrane (or very far from the protein surface).16
The implications of these observations are quite exciting. For Kv1.2 studied in the above case,16 the number of lipids whose lateral diffusion was slowed down significantly by the protein was about 50–100. These lipids essentially diffuse together with the protein as a dynamic protein–lipid complex. Given that biological membranes are highly crowded with proteins, a typical lipid–protein ratio being roughly 50:1–100:1,581,623 the results by Niemelä et al.16 highlight an important conclusion that in native biological membranes there are no free lipids but they are all subject to a significant interplay with proteins. Moving on, since in biological membranes the average distance from the surface of one protein to the surface of its protein neighbor is just a few nanometers, one can conclude that in protein-crowded membranes the membrane-mediated protein–protein interactions are significant and very likely the lateral diffusion of membrane proteins is also correlated. Further, since for colloids such as membrane proteins the diffusion coefficient D and the system’s viscosity η are scaled through the relation D ≈ 1/η, it is apparent that a moving protein senses a viscosity that differs quite substantially from the one in a protein-free membrane.
The results of Niemelä et al.16 have been confirmed in a number of other simulation studies for other protein systems, such as aquaporin-0624 and hemagglutinin.605 Experimental verification of this phenomenon remains to be done, since it is not clear which technique would be appropriate to do so. Meanwhile, theoretically the physical basis is clear. Based on fluid dynamics, particles close to a surface do not move along with a flow when adhesion forces are stronger than cohesion forces. Therefore, as long as the lipid–protein attraction at the protein surface is greater than the lipid–lipid interaction, the velocity of the lipids next to the protein surface goes down to zero. The size of the dynamical lipid cluster bound to the protein therefore also depends on the relative strengths of lipid–protein and lipid–lipid interactions, and on the shape of the protein as it may form regions that confine lipid motion.
The take-home message of these simulation predictions is that in protein-crowded biological membranes, the dynamics of lipids and proteins are not two separate issues but they have to be considered together.
5.2. Theoretical Descriptions for Lateral Diffusion in Model Membranes
5.2.1. Free Volume Theory for Diffusion of Lipids
Theoretical understanding of lateral diffusion in protein-free lipid membranes is not exceptionally strong. The only theoretical description that has received considerable attention is the free volume theory, which was originally presented in 1959 by Cohen and Turnbull625 to describe diffusion of hard spheres (colloids) in a liquid environment. Later, it has been extended to describe diffusion also in lipid membranes.626−630 The free volume theory is based on several assumptions. The main assumption is that diffusion takes place as jumps, where the diffusing particle moves rapidly over a distance close to its own size. This obviously requires that there is a free volume pocket available next to the diffusing particle. Since consideration of this process is theoretically quite tough in any interacting many-body system, the derivation in the free volume theory is done in a mean-field fashion in terms of the average free volume. The diffusion coefficient D is then described by
where vf is the free volume available and v* is called the critical volume, which represents the minimum size of the free volume pocket required for a jump. The parameter A is a constant, and γ is a numerical factor accounting for possible overlap of free volume (0.5 < γ < 1).625 This description was later extended by Macedo and Litovitz631 to account for energetic barriers that need to be overcome in a diffusion process, thereby complementing the above equation by an activation term.
The same idea has been applied to describe diffusion in lipid membranes626−630 in two dimensions, for which reason the free volume theory in this context is often called the free area theory. The most common description of the free area theory applied to describe diffusion of lipids in membranes is the one discussed by Almeida et al.,626 who considered lateral diffusion as a two-dimensional process. Then the critical volume is replaced with a critical area, a*, above which lateral diffusion can take place. Following the results of MacCarthy and Kozak,628 Almeida et al.626 considered that γa* = a0, where a0 is the cross-sectional close-packed area of the lipid. The expression for the lipid diffusion coefficient then turns out to be
Here δ is the distance between the neighboring lipids (“cages” surrounded by other lipids), kB is the Boltzmann constant, a(T) is the area per lipid at temperature T, and Ea is the activation energy of a diffusion jump. In practice, this equation is fitted to diffusion data to yield values for the close-packed cross-sectional area of a lipid a0 and the activation energy Ea.
The validity of the free area theory was tested by Javanainen et al.632 in a lung surfactant mixture of DPPC, POPC, POPG, and cholesterol. They studied lateral diffusion of lipids in Langmuir monolayers, where the area per lipid can be varied systematically. Since Javanainen et al.632 also computed the membrane-spanning close-packed cross-sectional area profiles of every lipid type in the membrane, they were able to resolve the value that a0 should have to render diffusion possible in the first place.
When the free area theory was fitted to diffusion data, one found for all lipid types an area parameter of a0 = 40 Å2. Meanwhile, when a0 was determined from the close-packed cross-sectional area profiles, one found values that were smaller and also lipid-type dependent. For cholesterol, a0 was ∼25 Å2, and for other lipids, the values ranged from 25 to 35 Å2, with values increasing with increasing average area per lipid. Altogether, one can conclude that there is no quantitative agreement; therefore, the values found by fitting the free area theory to actual diffusion data should not be taken literally. However, the ballpark seems to be correct.632
Javanainen et al.632 also considered the activation energy by repeating the diffusion simulations at several temperatures. The activation energy was then determined from the Arrhenius (activated) form,633 resulting in the Arrhenius diffusion barrier, EA, that is assumed to describe the rate-limiting barrier in a diffusion process. Alternatively, pragmatically speaking, EA describes how significantly diffusion speeds up for increasing temperature. The Arrhenius barriers that were found were dependent on the lipid type and the average area per lipid. At small areas, the Arrhenius diffusion barrier ranged between 24 (POPG) and 43 kJ/mol (cholesterol). At large areas, it varied between 18 (POPC) and 31 kJ/mol (cholesterol). These values are close to those measured in experiments.634 Meanwhile, the diffusion activation barrier, Ea, found by fitting the diffusion data to the free area theory was about 14 kJ/mol. The quantitative disagreement between EA and Ea is so obvious that clearly the activation barrier Ea in the free area theory does not correspond to the Arrhenius barrier EA.
We are not aware of other analyses where the validity of the free area theory would have been tested. However, one can discuss the validity of the assumptions that are its basis. The main assumption is that diffusion of lipids takes place as jumps. As discussed in section 5.1.1, this is not the case. There are no jumps. Instead, lipids undergo concerted motions where many lipids move as loosely defined lipid clusters, and this diffusion mechanism has been confirmed by quasielastic neutron scattering experiments.618,619 Moving on, in the free area theory, one assumes that a diffusing lipid is a hard rod with a well-defined close-packed area a0, which is independent of temperature and composition of the membrane. The results by Javanainen et al.632 and Falck et al.635 have shown that this assumption is too simplified. Further, in the free area theory, the diffusing lipid needs to break loose from the interactions with its nearest neighbors by overcoming the activation energy Ea. The above discussion shows, however, that the interpretation of Ea is not a simple feat. It is not clear how Ea should be interpreted in terms of the interactions between the lipids, ions, and water.
Concluding, the free volume theory is an appropriate theoretical framework to consider diffusion in hard-sphere systems such as colloids; however there is quite a bit of convincing evidence suggesting that it is not an appropriate approach to describe the diffusion of lipids in membranes.
The take-home message is that there is clearly room for developing a new theory to describe lipid diffusion, not an easy task but certainly an appealing and rewarding challenge.
5.2.2. Saffman–Delbruck Theory for Diffusion of Membrane Proteins
In section 5.1.2, we mentioned that for colloids the diffusion coefficient is inversely proportional to the viscosity of the solvent hosting the colloid, that is, D ∝ 1/η. Would it be possible to use the same relation to describe the lateral diffusion of integral membrane proteins in a lipid membrane environment? To some extent yes, since intuitively this inverse coupling has to be valid. However, based on the seminal work by Saffman and Delbrück,636 there are also other factors influencing the diffusion of these proteins.
Saffman and Delbrück636 considered the lateral diffusion of an individual membrane protein in a protein-poor membrane. Instead of describing lipids in any molecular detail, they used a continuum-level approach, where the lipid membrane was described as a thin layer of viscous fluid, surrounded by another, less viscous liquid. As to the protein, the key variable is its size R measured as its lateral radius.
The Saffman–Delbrück (SD) relation for the protein diffusion coefficient then arises as636
where μm and μf are the viscosities of the membrane and the surrounding fluid (typically water), respectively, h is the hydrophobic thickness of the membrane, and γ ≈ 0.5772 is the Euler–Mascheroni constant.
The SD relation is currently the widely accepted paradigm for describing protein diffusion under dilute (protein-poor) conditions, since a number of experiments on membrane protein diffusion have shown agreement with the SD prediction D ∝ ln(1/R).637−639 Results of computer simulations on membrane protein diffusion have also been reported to be in line with the SD relation.640−642 Despite this, fits to experimental data measured in living cells by Oswald et al. have revealed that the values found for several of the physical observables in the SD relation are nonphysical.639 In particular, Oswald et al.639 concluded that the membrane viscosity they found by fitting the diffusion data to the SD relation was 1–2 orders of magnitude larger than the membrane viscosity found in vitro. The interpretation of the parameters in the SD relation can therefore be more challenging than one might assume.
However, this is surprising given that in section 5.1.2 we found membrane proteins to diffuse as dynamical complexes with ∼100 lipids. Would it be possible that the protein size R in the SD relation is not the hard-core size of the protein but rather the size of the dynamical protein–lipid complex? Further, since the diffusion of lipids next to a membrane protein is slowed down to a significant degree, the membrane viscosity sensed by the protein (and the lipids in its vicinity) is much higher compared to the viscosity sensed by lipids far from the protein. Intuitively, this could be consistent with the experimental observations by Oswald et al.
Javanainen et al.55 and Jeon et al.643 considered this possibility in molecular-scale simulations (see Figure 27 (top)). By varying the proteins, and therefore their size, they found the simulation data for protein diffusion to match the SD relation D ∝ ln(1/R). For the protein size R, they observed that the fit of the simulation data to the SD equation was better when it was made with the concerted dynamical protein–lipid cluster, compared to the bare protein without lipids. The radius R was then ∼1–2 nm larger than the size of the bare protein.
Figure 27.
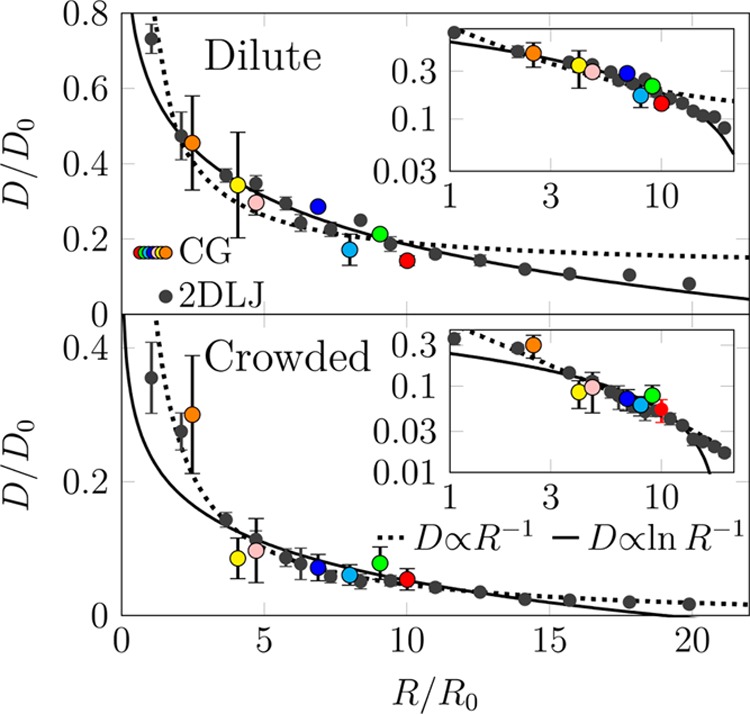
Diffusion coefficients of integral membrane proteins (D) versus protein/inclusion size (R). Results are shown for a polydisperse mixture of proteins based on simulations of a coarse-grained (MARTINI) model (colored points) and a system comprised of two-dimensional disks (gray). The results are shown in dimensionless units. The lines are fits to the Saffman–Delbrück relation (full line) and the Stokes law (dotted line). (top) In the dilute case, the data fully agree with the SD model. (bottom) In the crowded case, the Stokes-like 1/R dependence of the diffusivity is evident. Different colors of the markers stand for different proteins. Adapted with permission from ref (55). Copyright 2017 American Chemical Society.
Simulation results therefore suggest that the protein size in the SD equation is not the hard-core cross-sectional lateral size of the protein. Rather, it describes the average radius of a protein–lipid cluster that is dynamically constrained to move in a concerted fashion (cf. section 5.1.2). In practice, in addition to the protein, this is expected to include a lipid corona that is at least a few shells of lipids thick.
The appealing view that arises here is that protein motion taking place in soft membrane systems is not just about protein motion. Rather what happens is that proteins move as quite massive protein–lipid clusters whose size and inertia are larger than those of the protein alone. Biologically, this makes sense since exploiting this strategy fosters the stability of the protein such that its membrane microenvironment comprised of lipids around it changes very slowly, and once the protein has reached circumstances where it has the lipids needed for its correct membrane partitioning and activation, these conditions will be maintained for sufficiently long times when the protein is doing its job.
5.3. Beyond Saffman–Delbruck: Influence of Protein Crowding
However, cases where cell membranes would be poor in proteins are rare. Usually the situation is quite the opposite, as cell membranes are typically very rich in proteins.581,623,644 Obviously the diffusion of proteins and lipids slows down for increasing protein crowding due to blocking effects that become more pronounced with increasing protein coverage.56,640,644−649 However, the exciting question is, how does the size dependence D(R) for the membrane protein diffusion coefficient change for increasing crowding with proteins? Is the SD relation still valid?
Figure 27 (bottom) shows the results of extensive coarse-grained simulations of Javanainen et al.55 Intriguingly, when protein crowding increases, there is a crossover from the SD relation, D ∝ ln(1/R), to behavior characterized by the Stokes law D ∝ 1/R. The change in diffusion is profound in spite of the fact that the distribution of membrane protein sizes is limited, ranging from the size of individual proteins (∼1 nm) to the size of the largest protein complexes (of the order of 10 nm). In essence, while in the dilute limit the protein size does not matter much, in crowded settings the protein size plays a decisive role in protein diffusion. The crossover between these highly different behaviors takes place when the lipid–protein ratio is around 200–300, that is, for significantly smaller protein concentrations than in native cell membranes.
The simulation results provide compelling evidence that protein diffusion under protein-poor and protein-rich conditions is distinctly different. In the dilute limit, hydrodynamics is important, and consequently proteins of all sizes diffuse largely at the same speed. However, in membranes crowded with proteins, there is no space or time for the formation of hydrodynamic modes, thus diffusion is instead dominated by protein–protein collisions, which cause large proteins or protein complexes to be largely immobile, while small proteins are able to move rapidly.
5.4. Confinement by the Actin Cytoskeleton Network
The actin cytoskeleton comprises a highly dynamic protein network regulated by a myriad of binding proteins. It is involved in numerous cellular functions, including motility, programmed cell death, and cell division. Given that the cytoskeleton is bridged to the plasma membrane, the cytoskeleton underlying the membrane is likely to influence the organization of membrane proteins and lipids and therefore also influence their lateral diffusion. This is particularly likely for membrane proteins and their complexes that can be quite significant in size, extending deep into the cytosolic side of a membrane and therefore interacting with the cytoskeleton protein network.
Due to the complexity of the cytoskeleton network and its size, it is not a simple feat to simulate its effects on lateral diffusion. An excellent step in this direction was carried out by Koldsø et al.,650 who used coarse-grained (MARTINI) simulations to study the influence of compartmentalization caused by cytoskeletal immobilization on lateral diffusion. It was found that it leads to reduced and anomalous diffusion of both proteins and lipids, and this in turn was observed to result in a reduced rate of protein dimerization within the membrane and of migration of membrane proteins between different compartments. The study represents a promising molecular-scale approach for future work to explore diffusion under complex and realistic conditions that would include the effects of the cytoskeleton.
Partly in the same context, it would be fascinating to understand how peripheral proteins diffuse along the membrane surface. The physical context is here very different from the one with transmembrane proteins, since peripheral proteins are bound to the lipid membrane or membrane receptors via physical, often electrostatic, forces or via linkers that anchor them to the membrane, and the viscosity of the solvent surrounding them is much lower compared to that in a membrane. Recently, not much has been done, however. A nice exception is the work by Yamamoto et al.,651,652 who elucidated the localization and dynamics of membrane-binding lipid recognition proteins on the (cytosolic) surface of PIP-containing lipid bilayers. They observed that the anomalous diffusion and long-term correlated interaction of the protein with the membrane may contribute to an enhanced probability of encounter with target complexes on cell membrane surfaces and that the dynamic and transient interactions between the protein and membrane lipids can give rise to fluctuating diffusivity.
The scales associated with cytoskeleton-induced compartmentalization are large; therefore also lateral diffusion should be considered over time and spatial scales that are far beyond the molecular scales. A question arises whether highly coarse-grained simulation approaches would be more meaningful to consider lateral diffusion under these conditions compared to molecular simulation models. Recent work by Kalay et al. supports this view.653 They modeled the membrane as a two-dimensional fluid composed of hard particles simulated through MD. They showed that the diffusion sharply slows down with increasing fraction of immobile particles. Similar physics-based and highly simplified two-dimensional simulation models have been used recently in other contexts, too,55 and there is much literature published in the 1980s and 1990s, where models of the same type have been exploited to study surface diffusion problems, including the work by Saxton with lattice models for membranes.654−656 It would be highly justified to focus for a moment on the knowledge that was then created and use it now to develop new and simple simulation models for considerations of membrane dynamics over large scales. If combined with state-of-the-art techniques for the derivation of intermolecular free energy functionals for these coarse-grained models (using atomistic/molecular simulation models and experimental data with, for example, force matching and inverse Monte Carlo techniques), one could generate simple but realistic large-scale models with reasonable effort.
5.5. Challenges
Without lateral diffusion, cells would not function. Given this, it may be fair to say that diffusion is a biologically relevant process.
Computer simulation studies on lateral diffusion started from solving the easy problems, such as, how lipid unsaturation or the amount of membrane cholesterol influences the diffusion rate inside a membrane domain whose lipid content is clearly specified. These are examples of clarifying nanoscale conditions, where diffusion can play a significant role. Further examples of exploring diffusion in the nanoscale include elucidation of the effects of membrane-binding peripheral proteins on the diffusion and reorganization of lipids in a membrane, studies of diffusion of second messengers inside a membrane in processes such as complex I activation that requires ubiquinone binding to the protein inside the membrane hydrophobic core, and computational research needed to understand the links between short-time anomalous diffusion and the biological functions. Unraveling these questions will need some brains, but the resources and simulation models to do the job we mainly have already.
Meanwhile, there are numerous extremely interesting and important research topics that also call for clarification but are so resource-intensive that the simulation models to be used to unravel the underlying biological processes have to be considered very carefully. These cases are challenging because they take place on remarkably large, often macroscopic, scales. For instance, interpretation of super-resolution microscopy diffusion experiments sounds like an easy task, since the scales studied even in experiments are small. Wrong. If the experiments are done with living cells, the membrane environment is such a mess that bridging molecular simulations to super-resolution microscopy experiments requires the simulations to be done under conditions that mimic living matter as closely as possible. To sample the process adequately, the simulation system overall has to describe reality to a sufficient degree. This is the reason why in this section we have discussed diffusion in membranes of increasing complexity. Starting from diffusion in well-defined membrane domains at short scales, moving on to multicomponent membranes with diffusion between domains of varying sizes, moving further on to include the effects of proteins under crowded conditions, seeing the formation of functional membrane protein oligomers as a result of their diffusion, accounting for the effects of the actin cytoskeleton network, and then understanding all this complexity under nonequilibrium conditions with mass transport into and out of the cell membrane, that is a challenging task. The good news is that it will be done. The bad news is that maybe not today.
At the same time, certain practical and methodological problems remain to be solved. One encountered problem is sampling that is often too modest. Here, one has to keep in mind that diffusion is well-defined only in the long-time hydrodynamic limit. This can be a major problem especially in the determination of the diffusion coefficient of membrane proteins, because their diffusion is so slow that achieving the linear behavior of the mean-squared displacement is a challenge in atomistic as well as coarse-grained simulations. With coarse-grained models, another persistent problem is the adhesive nature of membrane proteins, meaning that in membranes, proteins tend to stick to each other too tightly.657 If the interaction is too strong, it can lead to binding that looks irreversible at simulation times. This would alter lateral membrane dynamics. If at the same time the dimerization of proteins takes place at a wrong interface, then the chances for coarse-grained simulation models to describe the dynamics of complex formation would be compromised. More work is needed to overcome these issues. Moving on, a number of recent studies have investigated the significance of finite-size effects on lateral diffusion.604,658−660 This is an issue that should be resolved in detail especially for diffusion problems where the dynamics takes place over large scales in a compartmentalized system. The work by Vögele et al.604 is an exceptionally inspirational study that demonstrates the role of hydrodynamic interactions in finite-size effects and consequently stresses the need to establish and use simulation thermostats such as dissipative particle dynamics that conserve momentum. Finally, comparison of simulation and experimental diffusion data is based on the assumption that simulations are able to mimic the conditions used in experimental measurements, and that one understands what actually is measured in experiments. In this context, one of the major issues is the use of fluorescent dyes or other markers, which, without doubt, always interfere with the membrane dynamics. Recent simulation studies have provided new information on how experimental conditions can be optimized to minimize the adverse effects of probes.70,661 This information also helps to build simulation models that bridge simulations to experiments in the most fruitful fashion.
What is needed are game changers: simple simulation models that correctly account for the principles of physics that dictate system behavior under nonequilibrium conditions on large scales in space and time and are thus able to describe biologically relevant conditions and processes in the meso- and macroscale.
6. Mass Transport
An essential feature of cellular membranes is their “selective permeability” in mass transport. This term refers to a vital property of biological membranes that lets some solutes pass though the membrane without much resistance but prevents others from entering or leaving the cell. The selective permeability realized through the synergy between all components of the membranes, ranging from lipids to proteins, is the main determinant of mass transport, which we discuss in this section.
Two essential features determine the transport of a certain solute through the membrane. The first one is the electrochemical gradient of the solute, which depends on two components: the difference in (i) solute concentration and (ii) charge between the inside and the outside of a cell. The solutes move effectively under the influence of the thermodynamic force imposed by their electrochemical gradients. Therefore, the electrochemical gradient of the solute determines the directionality of its diffusion and does not depend on the properties of the membrane. On the other hand, the permeability of the solute depends also on the membrane, which poses a free energy barrier against the diffusion of the solute depending on the physicochemical properties of the solute and the membrane. These two features, namely, the electrochemical gradient of the solute across the membrane and the free energy barrier imposed by the membrane, are the determinants of passive permeability through a membrane. We note here that membranes not only are composed of lipids but also contain various protein and carbohydrate compounds that are collectively involved in determining the free energy barrier against solutes. Some proteins, for instance, act as channels that facilitate the permeation of specific solutes, for which the lipid phase of the membrane constitutes a large free energy barrier.662
While the aforementioned mode of mass transport may sometimes be facilitated by proteins, it is a passive process, as its direction is along the electrochemical gradient and does not need external energy. However, living cells also need to internalize certain molecules, such as nutrients, and get rid of others, such as waste and toxic compounds produced in the cell. These cellular processes essentially move the solutes against their electrochemical gradients and, thus, require external energy. Biological membranes are equipped with machinery that couples various sources of energy stored in the electrochemical gradients of some molecules and ions, or chemical bonds of molecules such as ATP to facilitate and derive these active mass transport processes.662
Overall, biological membranes regulate and facilitate mass transport by an intricate interplay between their various components that may or may not require harnessing energy from external sources. In this section, we review recent computational studies on the role of biomembranes in mass transport in three major modes depending on the machinery involved in it:
Passive nonfacilitated permeation: This route refers to the diffusion of molecules down their electrochemical gradient across the membrane without the help of membrane proteins. Particularly, small hydrophobic molecules permeate the membrane primarily via this route.
-
Facilitated transport: Most molecules cannot efficiently pass through the membrane due to their size or electrochemical properties. Besides, some need to be transported against their electrochemical gradients. Specialized membrane-associated proteins facilitate the permeation of such molecules.
Channel proteins: Specialized channel proteins facilitate selective permeation of various molecules across the membrane, including ions and water. The cells regulate many important cellular processes, such as signaling, via the intricate gating mechanisms of channel proteins. Gating requires protein conformational changes, which can be triggered by membrane potential, ligand binding, light, and temperature depending on the type of the channel. Regardless of the gating mechanism, an open membrane channel forms a selectively permeable but continuous pathway between the two sides of the membrane.
-
Carrier proteins: Larger and more complex molecules and those that have to be transported against their electrochemical gradient (active transport) require another special class of membrane proteins, called carriers. In contrast to channels, carrier proteins are only open to one side of the membrane at a given time. The carrier facilitated transport, therefore, requires a series of conformational changes, in which the protein visits many conformational states. This universal transport mechanism of carriers is called the alternating-access model. Carriers are classified based on the energy-coupling mechanism required by the transport process.
Passive transport: Not all carriers facilitate active transport. Some are, indeed, responsible for downhill permeation of large polar molecules. Most such carriers are uniporters, as they bind and carry one substrate at a time.
Secondary active transport: Some carriers use the energy stored in the electrochemical gradients of one or more of their substrates to transport the other(s) against their gradient. These carriers are called secondary-active transporters, and are referred to as symporters or antiporters, depending on whether the substrates move in the same or opposite directions during the transport, respectively.
Primary active transport: Still some other carriers catalyze ATP hydrolysis and couple the released energy to transport of their substrates against their electrochemical gradients. Due to this direct coupling with the primary energy source, ATP, they are called primary active transporters.
Lipid transport facilitated by membrane transport proteins: Both channels and carriers are integral membrane proteins. Here, we add to these traditional categories a group of soluble and peripheral membrane proteins that facilitate lipid transport between discontinuous membranes. These proteins bind to a donor membrane superficially to capture hydrophobic molecules dissolved in the membrane, usually lipids. They dissociate and carry the hydrophobic molecule through the aqueous phase to an acceptor membrane.
Vesicular (bulk) transport: This mode of mass transport involves wrapping of molecules to be transported in vesicles. The processes are called endocytosis or exocytosis, depending on whether the cargo is taken into the cell or expelled from the cell, respectively. Two common molecular processes are essential for vesicular transport: vesicle fusion and budding. Both processes require recruitment of various proteins, and localization of specific lipids.
In this section, we first review the computational studies that aimed to describe passive diffusion through the membranes, focusing in particular on how biological membrane components change membrane permeability. We, then, move on to discuss facilitated transport, which has been the target of extensive computational investigations. Although most simulations have been performed in the membrane environment, only a subset of them focused on lipids and membranes as an active part of the transport process. We therefore limit our discussion to this subset, aiming to categorize common lipid interaction patterns in channels, carriers, and peripheral lipid transport proteins. Finally, we review the recent computational studies that have explored the bulk transport machinery involved in vesicle fusion and budding, again concentrating on the role of lipids and their interactions with proteins. Mass transport through pores formed due to external factors like electric field, ion imbalance, antimicrobial peptides, and surfactants is not discussed in this section; extensive reviews of these topics are given elsewhere.663,664
6.1. Passive Non-Facilitated Mass Transport through Membranes Depends on Membrane Lipid Composition and Phase
6.1.1. Water Permeation
Water permeation through membranes is still not fully understood at the molecular level due to the rarity of water permeation events observed in unbiased MD simulations and due to inaccurate water models. Awoonor-Williams and Rowley665 and Sinoda666 summarized the relatively up-to-date understanding of the general features of water permeability through the membranes. The free energy for water permeation through a generic (DPPC) bilayer slowly increases in the head and ester group region (0–5 kJ/mol), followed by a steep increase from the ester group region to the center of the bilayer (5–27 kJ/mol), slightly decreasing again at the very center (27–25 kJ/mol). The diffusivity, on the other hand, is reduced around the headgroup region and increases again in the hydrophobic core.665 Qiao et al. proposed thermal fluctuations of the potential energy as the driving force for water permeation through the membrane.667
An accurate description of water permeation through the membrane requires that one accounts for polarizability. Despite its higher computational cost, the use of polarizable force fields can provide advantages in modeling water permeation, although this approach does not necessarily provide estimates that are more accurate. On the other hand, the coarse-grained MARTINI model, which provides a huge gain in computational efficiency, appears to describe water permeability of membranes reasonably well, although the model lacks chemical details not only for the lipids but also for water, as it groups four water molecules into a single bead.665
Lipid peroxidation has been shown to affect not only the membrane physical properties but also its water permeability. A number of computational studies have shown that water penetrates deeper into the membranes that contain oxidized lipids,115,117,668,669 sometimes leading to membrane damage.115,117,669−671 Lis et al.671 investigated the effect of increasing oxidation on water permeability of DOPC membranes using MD simulations performed with a united-atom force field. High oxidation resulted in pore formation within the simulation time scale, leading to massive water flow through the membrane. Nevertheless, even a low degree of oxidation leads to increased water permeability, despite not causing pore formation within the simulation time scales. Predictably, the pore formation rate depends on the degree of oxidation. Once the pore forms, though, water permeation appears to be independent of the oxidation level.671 Lee et al.672 simulated coarse-grained models of a membrane composed of unsaturated lipids, cholesterol, and relatively low concentrations of oxidized lipids. Their simulations also captured increased water permeation without pore formation. The increased water permeability is likely a consequence of oxidation-induced changes in the membrane properties, such as increasing curvature, disorder, and lateral lipid movements.672 Overall, these results highlight an important mechanism for oxidation caused damage; that is, via interfering with the selective permeability of biomembranes.
A common aspect of the MD studies of membranes has been the use of flat bilayers with periodic boundary conditions. While this is a reasonable approach and sometimes the only affordable way to simulate the system, passive water permeation through curved bilayers, which are biologically relevant, remains rather unexplored. In a recent study, Su et al.673 explored the osmotic permeability of closed vesicular systems using coarse-grained simulations. Their results suggest that the water flux depends on where the salt is placed. A higher flux is observed when the salt is placed outside the vesicle than when it is placed inside the vesicle. This is likely due to the looser lipid structure in the former case. Moreover, water flux increased linearly with increasing salt concentration and vesicle surface area regardless of the position of the salt and showed an Arrhenius dependence on temperature.673 These results suggest that curved membranes and closed structures might exhibit different behavior than the commonly studied flat bilayers.
Composition of the membrane determines its permeability to water and other particles. There is a consensus that cholesterol decreases water permeability of membranes.665,674−676 Saito et al.674 attributed this effect to the reduction of cavity density in the membrane center by cholesterol. Their simulations of DPPC and PSM membranes showed that higher cholesterol concentrations make these membranes resemble each other in many physical attributes and water permeability.674 Issack and Peslherbe676 suggested that at higher cholesterol concentrations, not only is water partitioning to the center of the membrane lower, but also the free energy barriers shift outward. On the other hand, cholesterol does not appear to influence diffusivity of water substantially, except in the very center of the bilayer.676
Only a few computational studies has investigated water permeation through the gel-phase lipid bilayer. Qiao and Olvera de la Cruz667 performed atomistic simulations of DPPC and DLPS bilayers in the liquid-crystalline and ordered gel phases. The phase of the lipids emerged as a significant determinant of water permeability.667 Using MD-based permeability calculations, Hartkamp et al.677 showed that the lipid headgroups and the acyl chain packing density substantially affect the permeation through gel-phase bilayers. They also investigated the effects of alcohol and emollients, which cause nontrivial effects on the permeability.677
Research on permeation through the gel phase lipids is physiologically very relevant, since, as a model for the skin barrier, it carries implications on drug delivery through the transdermal route. Ceramides are an important component of the stratum corneum (SC) as they support its function as a barrier. The other major components of SC are cholesterol and free fatty acids. Das et al.680 performed one of the earliest simulation studies of SC model bilayers, composed of ceramides and the other main SC lipid components. Their results showed a substantially higher barrier for water in these fully hydrated SC models when compared to phospholipid bilayers.680 Later, Gupta et al.681 explored the effect of ceramide chain length on water permeability of pure ceramide bilayers, showing that increasing chain length decreases the water permeability.681 In another study,682 the same authors used a more realistic SC model containing ceramides, cholesterol, and free fatty acids. This study showed that the main barriers against permeation of water and many other small molecules are at the membrane center and headgroup region.682 Recently, Regno et al.679 explored the permeation pathways modeling the “bricks and mortar” arrangement of the stratum corneum (Figure 28A). They used atomistic simulations of two types of systems: high hydration (Figure 28B), which is a regular single bilayer with a slab of water on both sides, and low hydration (Figure 28C), which is composed of two bilayers stacked on top of each other with little water between layers. The membrane models contained ceramides, cholesterol, and free fatty acids in different compositions. While water permeates through the cholesterol-rich regions in fully hydrated membranes, no continuous water pathway exists in low hydration membranes, which are closer models for the SC skin barrier.679 This finding may appear counterintuitive considering cholesterol’s role in increasing packing and reducing membrane permeability.665 However, in contrast to fluid membranes, cholesterol introduces free volume and disorder in gel-phase membranes, enhancing their water permeability.679
Figure 28.

Permeation pathways in the “bricks and mortar” model for the stratum corneum. (A) Schematic representation of the skin layers (bottom). Adapted with permission from ref (678). Copyright 2014 Elsevier. The “bricks and mortar” model of the SC (top left) and its main components (top right) are also shown. (B) Full- and (C) low-hydration bilayer models used in MD simulations. Adapted with permission from ref (679). Copyright 2018 PCCP Owner Societies.
6.1.2. Amino Acid and Peptide Permeation
The titration state of the amino acid along with the lipid headgroup and chain saturation appear to determine the mechanism and the free energy cost of amino acid insertion into phospholipid membranes.683−690 Protein conformational changes and flexibility were suggested to determine the membrane permeation of pharmaceutically important cyclic peptides.691−693 Moreover, MD simulations were also used to characterize the insertion of antimicrobial peptides into the bacterial membranes.694−698
6.1.3. Gas Permeation
Membrane permeability of gas molecules has been studied using both atomistic699,700 and coarse-grained simulations701 and recently reviewed by Mayne et al.702 Overall, the free energy profile for bilayer permeation of nonpolar gas molecules, such as O2, CO, and NO, features a well of about −(3–4) kJ/mol in the membrane center.699 The profile for polar CO2, on the other hand, contains two wells of about the same depth at the headgroup regions.699 This suggests that nonpolar gas molecules accumulate in the center of the membrane, while polar ones like CO2 accumulate in the headgroup regions. Dotson et al.703 recently showed that while cholesterol enhances oxygen diffusion in the membrane center, it decreases the membrane’s permeability for oxygen by reducing its solubility within the membrane.703 Although various experimental704−706 and computational studies707−711 have implicated membrane channel proteins as potential routes for gas permeation, permeation through the lipid phase is likely the dominant pathway for gas permeation, since it does not feature high barriers.
Despite their simplicity, the accuracy of models used for gas simulations is a matter of concern. Recently, Javanainen et al.712 evaluated 14 different models of O2 based on their ability to reproduce available experimental data on density, heat of vaporization, free energy of hydration, and free energy of solvation in hexadecane. All models reproduce the trends of these properties qualitatively, but not quantitatively. The authors also proposed two new models that improve agreement with experiments on the density, heat of vaporization, and free energy of hydration but still fail to capture the water–oil partitioning. These results suggest that partitioning of oxygen and other nonpolar gas molecules into lipid bilayers may require including electronic polarizability explicitly.712
Cholesterol appears to modulate the permeability of membranes to gas molecules as well. The eye lens, in particular, is exceptionally rich in cholesterol. The cholesterol content of the lens has been suggested to prevent cataract development by maintaining a low oxygen partial pressure in the lens. Plesnar et al.713 performed atomistic MD simulations to estimate the permeability of model membranes with three different compositions to O2. Placing 200 O2 molecules in the bulk aqueous phase, they simulated a pure cholesterol bilayer and a 1:1 POPC/cholesterol mixture, both of which model cholesterol bilayer domains detected experimentally in the eye lens, as well as a pure POPC bilayer as a control. They estimated the permeability coefficient from the position-dependent concentration and diffusion constants as previously proposed by Subczynski et al.714 (We refer the reader to section 6.1.5 for an extended discussion on the methods to estimate the permeability coefficient from MD simulations.) The simulation study by Plesnar et al.713 confirm that cholesterol enhances the barrier against O2 diffusion. In the pure cholesterol bilayer, which was used as a model of the experimentally detected pure cholesterol bilayer domains in the eye lens, the permeability was estimated to be almost 10 times smaller.
6.1.4. Small Molecule Permeation
Understanding the molecular mechanisms underlying small molecule permeation through biological membranes is important for pharmaceutical and health purposes. Particularly, accurate and high-throughput methods for estimating thermodynamic and dynamic quantities related to small molecule permeation may aid in drug design. On the other hand, universal rules are hard to establish in this context due to the vast diversity of drugs and lipids.
Many studies have been performed to derive general features and interaction types that determine the membrane permeability for various groups of small molecules, such as β-blocker drugs,715 lipophilic drugs,716 and antibacterial hydrophobic molecules.717 We recommend a recent review by Lopes et al.718 on drug–membrane interactions for an extensive survey of experiments and simulations.
Cholesterol has been shown to increase the barrier for small molecule or drug permeation in many computational studies including those on the anticancer drugs, such as doxorubicin,719 5-FU,720 and ibuprofen,721 likely due to increased lipid packing as in the case of water permeability.674 However, this phenomenon is not universal. For example, while cholesterol impairs the membrane permeability for the hydrophilic cancer drug pirarubicin, its effect for the hydrophobic ellipticine, another cancer drug, is negligible.722 This is likely because hydrophilic pirorubicin brings water to the membrane core, for which the free energy cost increases with cholesterol concentration.722
6.1.5. Estimation of Passive Permeability of Solutes from Simulations
The solubility–diffusion model relates the passive permeability to free energy and diffusivity along the membrane normal making it possible to estimate the permeability coefficients from simulations.665 On the other hand, passive permeability calculations have always been a challenge, as they require calculating both the free energy and diffusivity. Improving efficiency without losing accuracy has been the goal in many computational studies. Below, we briefly summarize recent advances in this area, pointing out the essential physical and chemical contributions to permeability and the bottlenecks in its estimation.
As mentioned earlier, passive permeability calculations require estimating two position dependent quantities, the diffusivity and the free energy (or the potential of mean force (PMF)), each of which are challenging in themselves. Recently, statistical methods based on maximum likelihood and Bayesian inference have been developed to estimate diffusivity.723−725 Lee et al. systematically assessed various methods for estimating PMF profiles and the position-dependent diffusivities from MD simulations to calculate the rate of permeation using the inhomogeneous solubility-diffusion model. They concluded that all tested enhanced sampling methods (umbrella sampling with and without replica exchange, adaptive biasing force with single and multiple-walkers) for PMF estimation are comparable in accuracy. The diffusivity estimates appear to be sensitive to the choice of parameters in both tested methods, one based on generalized-Langevin and the other on Bayesian inference. Interestingly, the permeability only requires calculating the PMF and the diffusivities at the water–membrane interface and at the membrane center.723 This technical finding reveals the location of essential interactions that determine the permeability. Nitschke et al. further suggested that membrane permeation PMFs are robust, and calculations can be speeded up using smaller systems, shorter cutoffs, and multiple solute molecules simultaneously.726
Votapka et al.727 derived a variant of the inhomogeneous solubility-diffusion model as a function of the mean first passage time. This variant allows the use of milestoning methods to estimate the mean first passage time of membrane crossing and the permeability.727 Dickson et al.728 suggested that the permeation rates depend on the kinetic rate constants of three steps: membrane entry, flip–flop at the center, and membrane exit. Kinetic rate constants of each step estimated from MD simulations were used to build a structure-kinetic relationship. This model suggested that the changes in desolvation and hydrogen bonding for leaving the membrane determine the rate of flip–flop, while membrane partitioning determines the rate of membrane exit.728
Common sampling errors in permeation of small molecules through the bilayers were suggested to originate from “hidden sampling barriers” at the headgroup region and from the bending modulus of the membranes. Besides, the estimates exhibit strong dependence on the initial configuration of the system.47,729 Neale et al.730 suggested coupling umbrella sampling windows via Hamiltonian exchange to eliminate systematic sampling errors and achieve fast statistical convergence. Comer et al.725 put forward slow orientational relaxation as a potential source of error and, to circumvent it, included explicit sampling of the orientation of small molecules.
Filipe et al.731 assessed various formalisms for obtaining rate constants from MD simulations concentrating on a physiologically interesting application: translocation of cholesterol across bilayers. They compared the translocation rates obtained from explicit events in long unrestrained simulations with the estimates obtained with these formalisms. They also derived a new procedure that uses time intervals between transitions based on the explicit relaxation frequencies method. The authors conclude that while most formalisms result in fair estimates, the method based on explicit relaxation frequencies outperforms them and is the most reliable. Although the method requires complementing biased simulations with many unbiased simulations initiated at the transition state and is not computationally cheap, it eliminates a degree of uncertainty by not requiring depth-dependent transverse diffusion coefficients.731
Overall, the new developments along with various simplification schemes have made it easier and more reliable to calculate permeability of small molecules through lipid bilayers. Important contributions appear to arise at the water–membrane interface and at the center of the bilayer. The slow degrees of freedom, such as orientation relaxation, appear as a major source of sampling error.
6.2. Lipids Modulating Membrane Protein Structure, Function, and Dynamics in Facilitated Transport
Evidence has been accumulating on the regulation of membrane transport by membrane lipids directly through the site-specific protein–lipid interactions and indirectly through the physical properties of membranes. These effects were termed “ligand-like allosteric modulation” and “hydrophobic mismatch-driven modulation” in a recent review by LeVine et al.732 We group the latter within the allosteric effects of membrane’s physical properties, which also includes lateral tension, surface pressure, curvature, etc. Indeed, many channels and transporters cocrystallized with detergents or lipids (Table 2) support the idea of an integral role of site-specific protein–lipid interactions. Mechanosensitive channels, on the other hand, directly exemplify how channels respond to physical properties of membranes.733 Protein–lipid binding appears to manifest commonalities across various membrane transport protein families in characterized structures and support function by linking functional units, improving overall stability as well as the stability of different functional conformational states.734 MD simulations have provided extensive insights into protein–lipid interactions in various types of membrane proteins including membrane channels and transporters.602,735 Especially, cholesterol has been established as a key molecule for modulation of various types of channels and transporters.736
6.2.1. Channels: The Gatekeepers in Facilitated Diffusion Are Actively Modulated by Membranes
One of the topics that has received considerable attention in simulation studies of transport across lipid membranes is the diffusion facilitated by numerous channels. Given this, we here discuss the progress in this field quite extensively.
Aquaporins: The Water Channels
Aquaporins are transmembrane channels that facilitate the conduction of water and other small molecules across cell membranes. Among the membrane proteins characterized structurally to date, aquaporins form the group that is the most extensively studied through MD-based methods. Water permeation via aquaporins has been studied and reviewed over the last two decades shedding light to the selectivity and gating mechanisms.737−749 Despite accumulated experimental evidence on the potential role of lipids and membrane properties modulating aquaporin activity,750 there are only a few computational studies on this aspect.
Using multiscale simulations, Stansfeld et al. investigated lipid interactions with all aquaporins whose structures were known at the time. Although the simulations did not identify highly specific lipid binding sites, they showed common lipid interaction patterns across the studied proteins. Moreover, the annular lipids surrounding the protein appear to be fluid, exchanging frequently with the bulk lipids rather than being tightly associated with the protein surface.751
Electron crystallography of aquaporin-0 (AQP0) characterized annular lipids that surround AQP0. Aponte-Santamaría et al.624 evaluated the structure and the dynamics of the annular lipids using atomistic MD simulations on a system consisting of AQP0 embedded in a DMPC bilayer. They further assessed the consistency between MD-generated lipid conformations with the electron crystallography data. The results suggested that the annular lipids are similar in MD simulations and electron crystallography.624 In agreement with the results of Stansfeld et al.,751 there seems to be no specific lipid binding. The differences in protein mobility on the extracellular and intracellular sides appear to lead to asymmetry in the degree of localization of lipids in each leaflet, with the more flexible residues on the intracellular side hindering lipid localization in the intracellular leaflet. Furthermore, lipid positions are determined by packing of the acyl chains against the rough surface of the protein and the effect of the protein on lipid behavior extends beyond the first lipid shell.624
Later, Briones et al.752 studied the dependence of the localization and ordering of annular lipids around the AQP0 on the temperature, lipid phase, and protein flexibility using atomistic MD. They simulated DMPC-embedded AQP0 in various conditions to control protein and lipid flexibility. The authors demonstrated that the restricted protein flexibility due to restraining its heavy-atom positions enhances lipid localization and ordering around the AQP0. On the other hand, decreased lipid motion, probed in gel-phase DMPC simulations, did not affect the lipid localization. The authors suggested that the annular lipids provides a compensatory mechanism against the hydrophobic mismatch (Figure 29). Aquaporin was also observed to induce local fluid and gel-like areas, which was proposed as a potential mechanism driving its interactions with other membrane components.752
Figure 29.
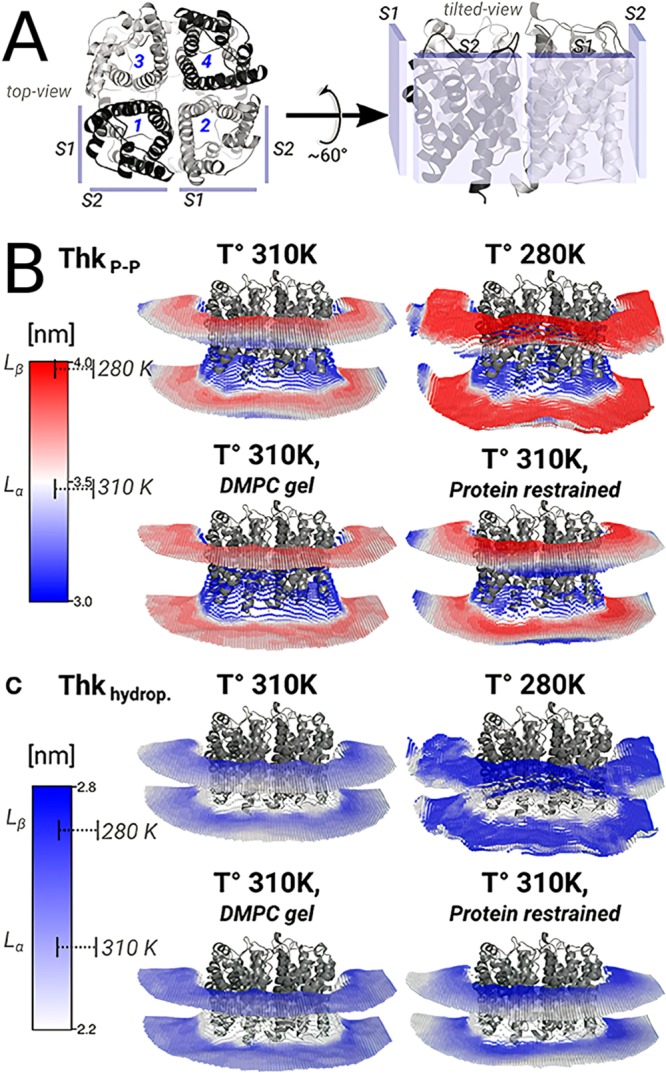
Membrane thickening near the AQP0 surface. (A) AQP0 monomers from the top and side views. (B) Average interphosphate thickness (ThkP–P) and (C) average hydrophobic distance (Thkhydrop) around AQP0 tetramer. The average ± standard deviation of the thickness values for a pure DMPC bilayer at 310 K (fluid-phase, Lα) and at 280 K (gel-phase, Lβ) are indicated on the color bars. Reprinted from ref (752). Copyright Briones et al. (https://creativecommons.org/licenses/by/4.0/legalcode).
Mechanosensitive Channels: Prime Examples of Protein Modulation by Physical Properties of the Membrane and Site-Specific Interactions with Lipids
The interaction between the large conductance mechanosensitive ion channel (MscL) family and the bacterial membranes is a prime example of the crosstalk between lipids and proteins as they modulate each other. MscLs are mechanosensitive channels activated by membrane tension. That is, they open in response to osmotic shock, releasing the built-up osmotic pressure by discharging ions, water, and even small proteins.753 Thus, MscLs constitute an essential survival kit for bacteria. These channels are particularly interesting in terms of interactions between biomembranes and proteins as they not only manifest site-specific interactions with the membrane lipids but also react to the physical properties of the membrane.
Many computational studies on MscL have concentrated on studying how the physical properties of the membranes change MscL activity as a part of its function.754−758 Samuli Ollila et al.759 estimated the mechanical energy change during channel gating using coarse-grained MD simulations of the open and closed states of MscL in a DOPC bilayer. They decomposed this free energy into contributions arising from the area dilation of the membrane and the protein shape change. Their results suggest that the protein adopts different conformations by expanding at the water–lipid interfaces based on the pressure profile of the membrane.759 Coarse-grained simulations of MscL embedded in pressurized liposomes by Louhivuori et al.760 captured the spontaneous activation and deactivation of MscL. Channel opening appeared to proceed asymmetrically in contrast to the symmetric “iris-like” opening model. The study captured the release of liposomal stress in sub-millisecond time scales upon MscL activation.760 Interestingly, Mukherjee et al.761 showed that l-α-lysophosphatidylcholine (LPC), an inverted cone-shaped amphipathic lipid with a single acyl chain, can activate MscL in the absence of lateral bilayer tension. Found in small amounts in the cell membrane, LPC does this likely by affecting the interfacial line tension between the protein and the bilayer.761
The site-specific interactions with lipids and how they may be involved in the activation mechanism of MscL have also been characterized in various studies.762 Vanegas and Arroyo763 studied the mechanism of force transduction mediated by protein–lipid interactions using MD simulations. According to this study, the asymmetric distribution of stress on the protein surface concentrating on the cytoplasmic side exerts outward forces on the protein. This force pattern appears to arise from interactions between the lipid headgroups and a protein motif that contains lysines. Interestingly, similar motifs exist in the human mechanosensitive channel K2P1 and bovine rhodopsin.763 Atomistic simulations of Sawada et al.764 pointed out a particular phenylalanine (F78) as the tension sensor based on its strong interaction with membrane lipids. Coupling between membrane tension and gating involves the first two transmembrane helices.764 An experimental study supported by atomistic MD implicated the amphipathic N-terminal helix of MscL in stabilizing the closed state and coupling the channel to the membrane through a tension-induced gating mechanism.765
The related mechanosensitive channel of small conductance (MscS) has also been investigated computationally.766−769 Lipid–protein interactions in the mechanosensing mechanism of MscS were suggested to play a crucial role. Mutagenesis combined with MD simulations identified the membrane tension sensor region of MscS as two transmembrane helices, where several residues specifically interact with the lipids in a conformational-state-dependent manner.770 Overall, the coarse-grained and atomistic simulations implicate specific lipid–protein interactions to be involved in mechanosensitive channel function.
Voltage-Gated Potassium Channel Function is Modulated by PIP2 and PUFAs
The voltage-gated potassium (KV) channels control electrical signal propagation in nerves, muscle contraction, and hormone secretion.771 Apart from being important membrane channels, they constitute the third largest signaling protein family.772 They switch between open and closed conformations tuning K+ flux in response to changes in the voltage across the cell membrane. Recently, lipid motion between different binding sites on the protein surface has been implicated in the modulation of channel function. Chen et al.773 combined MD simulations with mutagenesis and electrophysiology experiments to identify the PIP2 interaction sites on the KV channel KCNQ2. The simulations captured PIP2 binding to the S4–S5 linker and infrequent migration to the S2–S3 linker from the S4–S5 linker. This migration was linked to deactivation kinetics and appeared to depend on the functionally important basic residues on the linker.773 Yazdi et al.774 employed atomistic MD simulations to explore interactions of the Shaker KV channel in the open and closed states with polyunsaturated fatty acids (PUFAs). The simulations showed that the open state of the channel is stabilized by PUFA interactions with helices S3 and S4 of the voltage-sensing domain and the linker connecting them. The larger conformational flexibility of PUFAs likely allows them to adapt to the channel surface. In contrast, both PUFAs and saturated lipids interacted less with the closed state of the channel.774 Moreover, a small-molecule binding site determined at the interface between the bilayer and helices S3 and S4 by combined MD and experimental approaches775 supports the role of this region for lipid–protein interactions.
Other Channels
Since the structural characterization of the K+ channel by the team of MacKinnon,776−778 channels have been extensively studied using computational methods for various mechanistic features including their gating and selectivity.779−782 Henin et al.783 identified cholesterol binding sites in intersubunit cavities of the pentameric ligand gated ion-channel GABAAR transmembrane domain using homology models of a human GABAAR and suggested a cholesterol-induced pore opening mechanism.783
6.2.2. Carriers: Facilitators of Primary and Secondary Active Transport
Carrier proteins are involved in both passive and active membrane transport. Due to their central role in these processes, they have been the targets of extensive structural characterization efforts. Regardless of their energy coupling mechanism, carrier proteins go through a transport cycle (Figure 30, the dark green region). The transport cycle has long been suggested to involve a common mode, called the alternating-access model.784−786 In essence, this model asserts that the binding site of the carrier is only accessible from one side of the membrane at a given time. This is achieved by the carrier protein going through several conformational states usually referred to as inward-facing (IF) or outward-facing (OF) and further qualified as open or occluded (Figure 30) based on the accessibility of the binding site.
Figure 30.
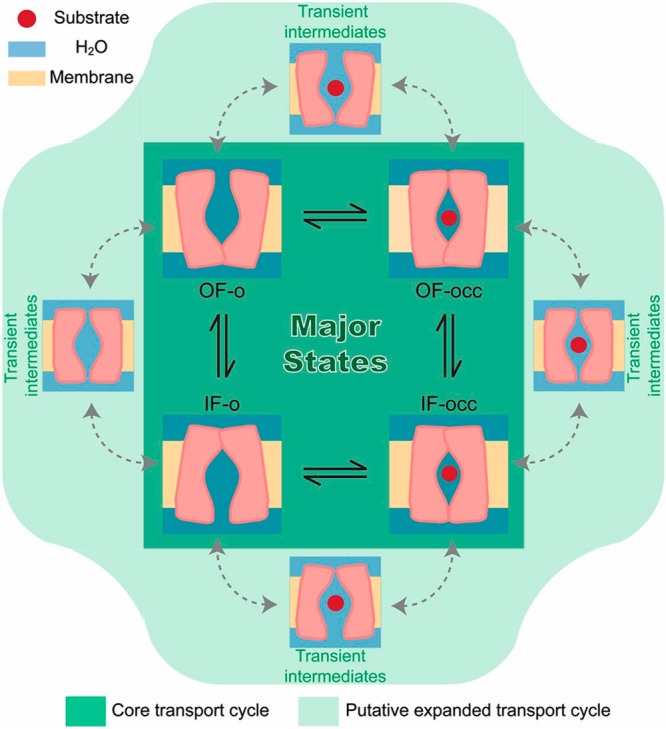
A schematic representation of the transport cycle of a carrier (dark green). The alternating-access mechanism requires interconversion between functional conformational states: outward-facing open (OF-o), outward-facing occluded (OF-occ), inward-facing open (IF-o), and inward-facing occluded (IF-occ). Uncoupled water or ion transport can occur due to channel-like transient intermediates (light green). Reprinted with permission from ref (787). Copyright 2013 Li et al.
Indeed, the absence of alternating-access would essentially turn the carrier into a large channel or a nondiscriminating pore. This would result in the leakage of the cellular contents, such as nutrients and salts, as well as in the entry of harmful substances. Moreover, it would waste energy in active transport by allowing species that are transported against their gradient to leak back. On the other hand, various computational studies have identified uncoupled water transport through channel-like intermediates or water wires in carriers from diverse families (Figure 30, the light green region). These results explain at least some of the experimental findings that showed coupled and uncoupled water and ion transport.787−790 While the simulations point out imperfect sealing during the alternating-access mechanism, size and other physical properties of the channels as well as their transiency are unlikely to render them deleterious for the cell.787−790 Taken together with the water permeation through other membrane channels (including but not limited to aquaporins) and the sheer amount of mass transport proteins in cellular membranes, the passive water permeability of biomembranes is unlikely to be only due to permeation through the lipid phase.
Nowadays it is unexceptional to find structures of membrane transporters in various conformational states representing a different stage of their transport cycle. Moreover, low-resolution experiments have been used to tap into the dynamics of the transporters. The intrinsically dynamic conformational landscape of the membrane carriers, which is absent in most membrane channels, makes MD a valuable tool to investigate their transport cycle. Indeed, MD-based methods, such as Molecular Dynamics flexible fitting,791,792 have been instrumental in generating atomistic models for membrane transporters793−796 and channels797,798 based on low resolution cryoelectron microscopy or X-ray scattering densities, exploiting homology models or available structures in alternative conformational states. The long time scales of the transport cycle of carriers bring about challenges for simulation studies. These challenges, on the other hand, have been partly circumvented by the improving computational resources and the developments in MD-based methods. Various enhanced sampling methods have successfully been used to sample the transport cycle along carefully chosen collective variables.799−803 Recently, Latorraca et al.804 captured the spontaneous OF-o to IF-o conformational transition in unbiased MD simulations of the sugar transporter SemiSWEET, one of the smallest carrier proteins characterized to date. The simulations depict the alternating access mechanism at the atomistic scale, revealing a coupling between the intracellular and the extracellular gates of the substrate-binding pocket, which prevents their simultaneous opening.804
The use of MD simulations in exploring the conformational changes during the transport cycle as well as their dependence on substrate/ion binding in secondary active transporters and ATP hydrolysis in primary active transporters has been extensively reviewed.805,806 Here, we review the recent computational studies on carriers from various families focusing on protein–lipid interactions and the modulation of their transport activity by membranes.
Cholesterol Modulates Carrier Proteins, Particularly Those Involved in Nerve Signaling
Cholesterol is of particular interest for modulation of membrane proteins. Groulef et al.736 reviewed the MD studies aiming to delineate how cholesterol affects the structure, dynamics, and function of membrane proteins.736 We focus here on the effects of cholesterol on membrane transport proteins, leaving aside other membrane proteins not directly involved in mass transport.
An overarching theme of computational studies of both primary and secondary active membrane carriers has been the characterization of the conformational transition pathways. On the other hand, only a few studies directly explored how the functional conformational changes depend on membrane cholesterol. Before we go on for an in-depth discussion, we note that the reason for the scarcity of computational studies directly exploring this phenomenon is likely 2-fold. First, the time scales required for the conformational transitions are too long for MD. Unfortunately, the presence of cholesterol is unlikely to raise the stakes for capturing these slowly progressing dynamic events; on the contrary, it is likely to hinder it. Second, often the structures of membrane transporters are from prokaryotes. A quick query for the term “transporter” in the database “Membrane proteins of known three dimensional structure”268 results in over 300 matches, and only a few of them are of eukaryotic origin, let alone being of animal origin.
One of the few computational studies that investigated the modulatory effect of cholesterol on membrane transporters, was an atomistic and coarse-grained simulation study of sarco- and endoplasmic reticulum Ca2+-ATPase.807 This study suggested a nonspecific effect of cholesterol, as cholesterol did not appear to interact specifically with the known inhibitor binding sites.
Cholesterol is an essential component of the neural membranes and is enriched in the synapses, which are active sites of neurotransmitter release and clearance. A family of secondary active transporters, referred to as the solute carrier 6 or the neurotransmitter-sodium-symporter (NSS) family, couples the Na+ and Cl– gradients to uphill transport of neurotransmitters to clear them from the synaptic cleft. A subset of NSS transporters is the monoamine transporters (MATs) responsible for serotonin, dopamine, and epinephrine clearance. These transporters are of major clinical interest as potential drug targets against disorders related to monoamine dysregulation, including drug addiction, hyperactivity, schizophrenia, and depression.808 Interestingly, the site-specific interactions of cholesterol with conserved sites on the monoamine transporters influence their conformational state.
Until recently, computational studies of this important family have focused on the archetypal NSS orthologue, bacterial leucine transporter (LeuT).809 LeuT has been the target of extensive structural and computational investigations, as it features a conserved structural fold (the LeuT-fold) shared among diverse transporter families with little sequence and functional homology.810 The importance of lipid environment in transporter shape and conformation has been highlighted by structural differences observed in MD simulations of LeuT.811 Hydrophobic mismatch quantified by a hybrid Continuum-Molecular Dynamics approach was suggested to allosterically regulate the functional conformational transitions of LeuT812,813 exemplifying the hydrophobic-mismatch driven allosteric modulation.732 Several computational studies also captured intermediate conformational states.814−816 On the other hand, due to its bacterial origin, LeuT is not suitable for studying key site-specific lipid interactions in MAT members.
Structural characterization of two MATs bound to cholesterol in different regions of the protein led to the subsequent MD studies of cholesterol binding. These structures belong to the human818 and drosophila819 dopamine transporter (hDAT and dDAT, respectively), and to the human serotonin transporter (hSERT).820 In each of these structures, one cholesterol was bound to a distinct site. Based on CG simulations of a LeuT-based dimeric hSERT homology model embedded in a raft-like membrane, Ferraro et al.821 identified six cholesterol-binding spots on the serotonin transporter (hSERT), four of which appear to be important for ion binding and transport. For this purpose, they developed a protocol to determine the hot spots based on residence times and spatial distribution of cholesterol.821 Laursen et al.822 performed coarse-grained simulations of the recently obtained crystal structure of SERT820 and characterized cholesterol binding to a site that was previously identified in dDAT. Their mutagenesis experiments showed that the mutations that inhibit cholesterol binding to this site promote the IF conformation, whereas those that promote cholesterol binding lead to the OF conformation.822 A study by Zeppelin et al.817 concentrated on the cholesterol interaction with hDAT (Figure 31A). Atomistic simulations of hDAT captured the transition to an IF conformation in the absence of cholesterol, as well as immobilization of two helices on the intracellular side in the presence of cholesterol. They also extended the study to three other MAT members: dDAT, hSERT, and human norepinephrine transporter. The coarse-grained simulations revealed a common set of cholesterol binding spots in all MAT members (Figure 31 B).817 Overall, these studies strongly suggest that the binding of cholesterol to conserved surface spots on MATs regulates the conformational transitions important for the transport cycle of these proteins.
Figure 31.
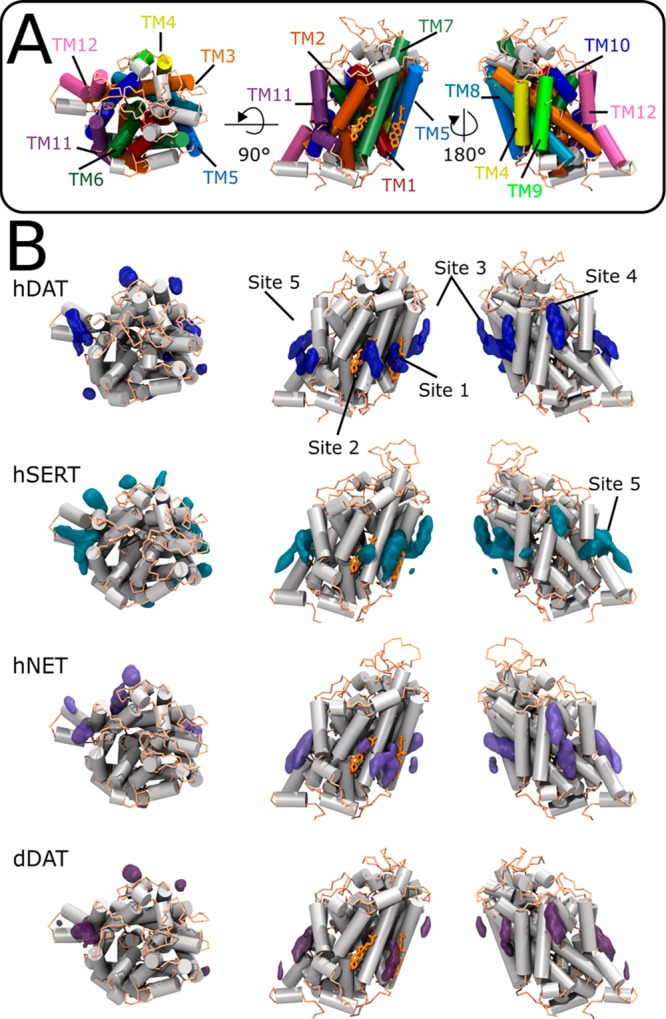
Annular cholesterol-binding sites on various monoamine transporters (MATs). (A) The cartoon representation of hDAT from the top (left) and the side views (center and right). The two cocrystallized cholesterol molecules are shown in orange licorice representation. (B) Cholesterol occupancy isosurfaces are shown for various MATs shown from the top (left) and side views (center and right). Cholesterol in 5 conserved sites is depicted in orange licorice. Reprinted from with permission from ref (817). Copyright 2018 Zeppelin et al. (https://creativecommons.org/licenses/by/4.0/legalcode)
PIP2 Modulates the Conformational State and Oligomerization of Monoamine Transporters
The N-terminus of DAT regulates the reverse transport of the neurotransmitter, namely, its release to the synaptic cleft. However, the structure of this important segment has not been solved. Khelashvili et al.823 performed an ab initio structure prediction of the 57-residue-long N-terminal segment. They carried out atomistic simulations of the selected model in water with and without temperature replica exchange in the membrane environment and attached to the transmembrane region. This segment appears to interact with the anionic PIP2 or PS lipids in the bilayers, which favor various binding modes of the N-terminal segment on the membrane surface.823 In a later study on the full-length hDAT embedded in PIP2-containing and PIP2-free membranes, Khelashvili et al.824 captured a spontaneous transition toward the IF state, as well as release of Na+ and destabilization of dopamine driven by PIP2 interactions with the N-terminal segment and an intracellular loop.824
PIP2 has also been implicated by computational studies in stabilizing the oligomeric forms of dopamine and serotonin transporters. However, these studies are largely inconclusive or indirect in identifying the lipid–protein interactions in oligomerization. Recent evidence has suggested the functional forms of these transporters as dimers, despite the crystal structures in the monomeric state. Cheng et al.825 modeled the dimeric state of hDAT. Although not present in the simulations, authors hypothesized that PIP2 may be involved in stabilizing the dimeric interface.825 The energetics of the identified dimeric interfaces was quantified using Hamiltonian replica exchange umbrella sampling simulations. The simulations hinted at the potential roles of PIP2 and cholesterol in oligomerization, but without any certainty.826 A largely experimental study by Anderluh et al.827 asserted that direct PIP2 binding mediates SERT oligomerization. In this study, MD simulations were used to model PIP2 interactions with the experimentally characterized region.827 It is worth noting that coarse-grained MD of LeuT dimers suggested phospholipid and anionic cardiolipin binding to the dimer interface.828 The collective motions of LeuT and hDAT were suggested to change upon dimerization.829 Taken together with their likely role in oligomerization, anionic lipids, especially PIP2, at least play indirect roles in the conformational changes required by the transport cycle of MATs. Yet, the direct effect of these lipids on oligomerization and the transport cycle remains to be explored.
The Effects of Detergent Micelles on Structure and Function of Carrier Proteins
The extensive treatments performed during crystallization make it harder to relate the obtained structure of the protein to its function in its natural environment. MD simulations have been instrumental in relaxing the structures, originally obtained using detergents, in their natural environment. However, computational studies on the direct effects of the unnatural crystallization environments are rare. Recently, Cheng et al.830 developed strategies to construct micelle models for computer simulations and tested them on two systems, one of which is the voltage-sensing domain of KV, to facilitate the computational study of protein–micelle complexes.830
The bacterial secondary active transporter LeuT, discussed above in the context of site-specific lipid protein interactions especially with MATs, has also been examined to characterize the effects of unnatural lipid environment on the structure of protein and the annular lipids. Atomistic simulations by Khelashvili et al.831 showed the formation of a detergent shell that coats the transmembrane region of LeuT. At high detergent-to-protein ratios, the simulations captured the penetration of the detergent molecules into the LeuT structure, occupying the second substrate-binding site.831 LeVine et al.832 later investigated whether the detergent in the micellar environment strips away or preserves the annular lipids around the protein. The simulations suggest that at low detergent concentrations, the annular lipids remain undisrupted, and the detergent molecules cannot penetrate into the protein. They further showed that larger detergents do not penetrate into the protein even at high concentrations.832
The micellar environment also influences the functional conformational states of LeuT. Sohail et al.811 simulated IF-open LeuT in micellar and bilayer environments. In the detergent micelle, which reproduces the crystallization environment, LeuT structure remains stable and open.811 In the bilayer, however, one of the transmembrane helices moves substantially, resulting in a different IF-open conformation. These results confirmed by lanthanide-based resonance energy transfer experiments indicate that the environment used during crystallization might affect the captured conformational state.811 The consequences of the unnatural detergent environment were acknowledged in a recent study by Adhikary et al.833 To study the protein in a more natural setting without labeling, they reconstituted LeuT into nanodiscs and studied the protein conformational dynamics using hydrogen–deuterium exchange coupled with mass spectrometry. This study, which exploited MD for the interpretation of the experimental data, confirms that the IF to OF conformational transitions described in detergent micelles take place also in the lipid environment. Besides, the study also characterized the differences between IF and OF states in the accessibility and the conformation of several transmembrane helices and extracellular loops.833
Lipids are Active Facilitators of the Elevator Mechanism of the Glutamate Transporter
Another group of neurotransmitter transporters is comprised of the glutamate transporters, which facilitate the clearance of excitatory amino acids, glutamate and aspartate, from the neural synapse. As there are no experimental structures available for the human members, their structures are inferred from the orthologous bacterial aspartate transporter, GltPH, which features another conserved fold, called the NhaA-fold.810 Remarkably, GltPH is a homotrimer assembled symmetrically around a central axis normal to the membrane. Each monomer contains an N-terminal scaffold domain that forms the trimeric interface and a C-terminal transport domain, which moves up and down against the scaffold during the transport cycle in an elevator-like mechanism.786 Lezon and Bahar834 proposed that the elevator-like mechanism can be realized based on the collective motions estimated by the elastic network models only when the surrounding lipids are incorporated into the model. This observation suggests that the membrane plays an active role in the transport cycle of GltPH.834 An experimental study by Akyuz et al.835 combined with atomistic MD simulations supported the elevator-like mechanism by studying two mutations that make the bacterial protein similar to its human orthologue. These mutations were characterized by a decreased substrate binding affinity and an enhanced transport rate. MD simulations showed that the mutant proteins exhibited increased insertion of lipid chains into the interface between the scaffold and the transport domains, strongly suggesting that lipids facilitate the elevator-like mechanism.835
ATP Binding-Cassette (ABC) Transporters Maintain Membrane Asymmetry
Lipid distribution in cellular membranes is inhomogeneous laterally and asymmetric between leaflets. While the electrochemical potential of a specific lipid and the physical properties of the membranes partially dictate its location, active transport of lipids is necessary to maintain the asymmetry.836,837 Here, the active transport is mainly primary; that is, it is directly coupled to ATP hydrolysis. Some ATP binding cassette (ABC) transporters are directly involved in the transport of sphingolipids, phospholipids, sterols, bile salts, and fatty acids. Moreover, lipid environment acts as a reservoir for other types of amphipathic and hydrophobic substrates. Lipid molecules also influence ABC transporter function, and some may even be tightly bound, thereby acting as an essential unit of the functional protein.838 Montigny et al.836 recently reviewed the proposed mechanisms of flippases and scramblases involved in active and passive phospholipid transport, and the regulatory role of lipids in their function.
MsbA is a bacterial ABC exporter, a lipid flippase, characterized structurally in both IF and OF conformations. The conformational changes and the coupling between ATP hydrolysis in the nucleotide binding domains and the transmembrane domains have been investigated using MD simulations.839−841 Ward et al.842 explored the distribution of lipids around MsbA using a multiscale coarse-grained approach, which optimized the coarse-grained parameters based on atomistic simulations. Three conformational states of MsbA were embedded in mixed DOPC/DOPE bilayers separately. The simulations captured weakly bound annular lipids around the transmembrane region of MsbA penetrating into the substrate-binding chamber.842 Bechara et al.843 studied the endogenous composition of annular lipids around TmrAB, a potential glycolipid flippase related to MsbA, using both experiments and MD simulations of a homology model of TmrAB embedded in a POPE bilayer. They showed that annular lipids have a constant composition and that negatively charged lipids tightly associate with the protein.843
P-glycoprotein is another member of the ABC transporter superfamily existing in animals, fungi, and bacteria. P-glycoprotein not only moves various compounds across the plasma membrane but also acts as a flippase.844 Domicevica et al.845 built a complex coarse-grained lipid bilayer model composed of POPC, POPE, POPS, sphingomyelin, cholesterol, and POPIP asymmetrically distributed in each leaflet in keeping with the composition of brain epithelial cells. Coarse-grained simulations of the membrane embedded P-glycoprotein complemented by shorter atomistic simulations captured the formation of an annular lipid belt composed of anionic PS in the inner leaflet. Cholesterol also appeared to interact specifically with three sites on the protein.845 Barreto-Ojeda et al.846 explored possible lipid access pathways to the binding cavity. They employed coarse-grained simulations of the inward-facing conformation of P-glycoprotein embedded in bilayers with different POPC/POPE compositions. The simulations showed that only lipids of the lower leaflet can access the cavity without any PC or PE specificity. They also found that there are key lipid binding residues that are mostly basic at the portals and mostly hydrophobic and aromatic in the cavity.846
G Protein-Coupled Receptors Act as Lipid Scramblases
Although their main function is signaling, and therefore is discussed in detail in section 7, several G protein-coupled receptors have been implicated for their scramblase function.837 While the scramblase activity of G proteins can be categorized as neither carrier-like nor channel-like, we note here that it is a mode of passive mass transport, distinct from the above-described flippase activity in that it is passive and thus not directional.
A few computational studies characterized this function of GPCRs focusing on rhodopsin. Nieminen suggested that rhodopsin lowers the free energy for POPC flip–flop.848,849 Verchère et al.850 showed that several polar residues on bacteriorhodopsin exposed to the membrane interact with the polar lipid headgroups facilitating the phospholipid flip–flop.850 Recently, Morra et al.847 suggested a similar “credit-card” mechanism for retinal-free oxin (Figure 32A). They employed atomistic simulations and generated kinetic models based on the Markov State Model analysis for the translocation process. Their results revealed a hydrophilic groove between two helices, which opens up while the lipid headgroups translocate through it with the chainsexposed to the membrane environment (Figure 32B–D).847 Along these lines, Sapay et al.851 suggested that model transmembrane helices can facilitate lipid flip–flops by stabilizing the membrane against deformation during the flip–flop.851 Overall, these results suggest that the protein surface can act as a rail against which the phospholipids can slide, leading to faster flip–flop and thus membrane scrambling.
Figure 32.
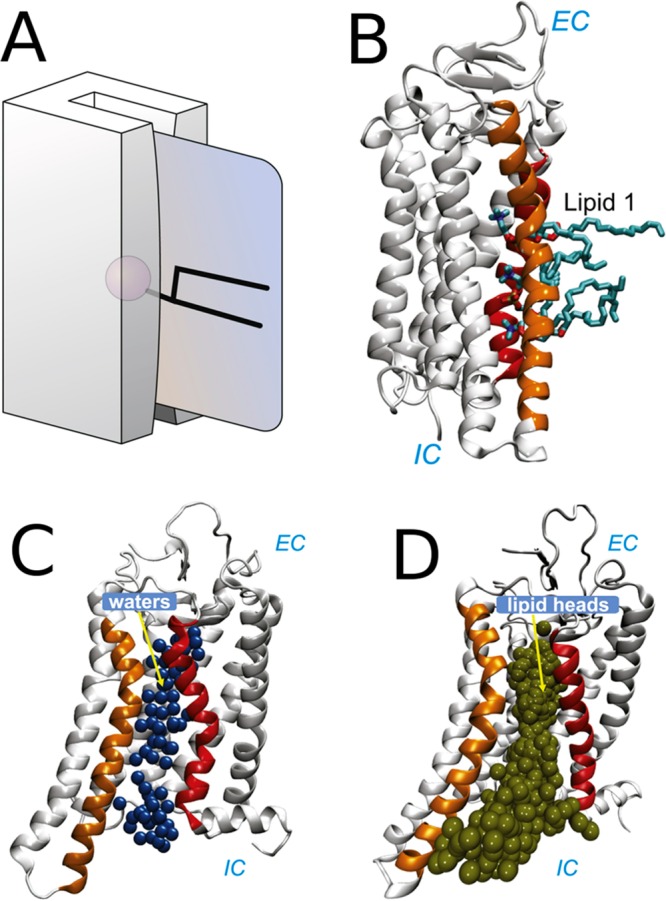
Lipid scramblase function of GPCRs and its mechanism. (A) Schematic representation of the “credit-card mechanism” for lipid flip–flop. Adapted with permission from ref (837). Copyright 2006 Springer Nature. (B) The polar groove formed between the transmembrane helices is filled with water molecules. (C) MD snapshot showing lipids with their headgroups inserted into the groove between two transmembrane helices. (D) Continuous lipid translocation pathway characterized in simulations indicated by overlaid lipid phosphorus atoms. Adapted with permission from ref (847). Copyright 2018 Elsevier.
6.2.3. Peripheral Lipid Transport Proteins
A group of soluble proteins classified under the lipid transport proteins facilitates the lipid transfer through the aqueous phase in a cell. They achieve this by extracting lipids from the membrane by associating superficially with membranes and carrying them through the aqueous phase in their buried binding pockets.852
Membrane association and lipid binding of lipid transport proteins have been investigated using computer simulations. Phosphatidylinositol-transfer proteins exchange phosphatidylinositol molecules between membranes regulating PIPs signaling. Grabon et al.853 characterized the lipid-binding mode and a lipid uptake mechanism based on atomistic MD simulations. The simulations identified a set of conserved residues aiding in lipid uptake, reducing the free energy barrier and functional conformational changes.853 Moreover, two computational studies complementing experiments investigated the conformational effect of lipid binding in two different members of oxysterol-binding protein (Osh), sterol/PI(4)P exchangers.775,817
Cholesterol efflux in the late endosomes and lysosomes is facilitated by a “tag-team duo” formed by Niemann-Pick C (NPC) proteins 1 and 2, which get their name from the genetic Niemann-Pick C disease. NPC1 is a transmembrane protein on the limiting membrane of the lysosomes/late endosomes. NPC2, on the other hand, is a small soluble protein. NPC2 shuttles cholesterol between the internal membranes of the organelles rich in a unique lysosomal/late endosomal anionic lipid, called bis(monoacylglycero)phosphate (BMP). Moreover, NPC2 carries cholesterol and hands it to the NPC1 N-terminal domain, which through an unknown mechanism expels the cholesterol through the glycocalyx-coated limiting membrane.855 Estiu et al.856 estimated an optimized pathway for cholesterol hand-off from NPC2 to NPC1’s N-terminal domain.856
Enkavi et al.854 investigated the membrane-binding properties of NPC2 using all-atom simulations and free energy calculations. The simulations captured NPC2-membrane binding in two competitively favorable modes (Figure 33). The first binding mode (Prone) is associated with cholesterol uptake and release as it places the cholesterol-binding pocket in direct contact with the membrane. The second binding mode (Supine), on the other hand, places the cholesterol-binding pocket away from the membrane surface. The Supine mode appears to be more favorable overall, and the productive binding mode (Prone) forms only in BMP-containing membranes. Interestingly, sphingomyelin hinders the formation of this mode, counteracting BMP.854 The effect of BMP is shown to be specific in that another anionic lipid, PG, cannot reproduce the effects. These simulations suggest a mechanism by which BMP and sphingomyelin regulate the NPC2-mediated cholesterol transport.
Figure 33.

Two membrane-binding modes of NPC2 and their interactions with BMP characterized in extensive biased simulations. (A) Normalized contact frequency (NCF) is used as a measure of specific interactions with BMP. The Prone mode, which places the opening of the cholesterol-binding pocket in direct contact with the membrane surface, exhibits extensive specific interactions with BMP, while the Supine mode, which places cholesterol away from the membrane, does not form such extensive specific interactions. (B) The MD snapshots showing the two binding modes. The protein is shown in cartoon representation colored based on the NCF values; the membrane is represented as a yellow transparent surface. BMP residues near the protein and the protein residues with high NCF are shown in licorice representation. Reproduced with permission from ref (854). Copyright 2017 Enkavi et al. (https://creativecommons.org/licenses/by/4.0/legalcode).
With the recent discovery of the cryoEM structure857 and several insightful X-ray structures858,859 of NPC1, the whole process of cholesterol transport by the full-length NPC1 and NPC2 can be investigated using MD.
6.3. Membrane–Protein Interactions in Vesicular (Bulk) Transport
Vesicular transport has a central role in trafficking molecules both within a cell involving various organelles, such as the endoplasmic reticulum, Golgi apparatus, endosomes, and lysosomes and in the uptake (endocytosis) and release (exocytosis) of vesicle-enclosed material by the cell, as in the case of neurotransmitter release. Various types of molecules are required for different vesicular transport pathways. Clathrin facilitates membrane invagination in the vesicle budding process, enabling endocytosis of extracellular material on the plasma membrane. The soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complex, on the other hand, is responsible for vesicle fusion at the neuronal synapse allowing docking and subsequent fusion of vesicles.860 Both budding and fusion events in vesicular transport require extensive membrane remodeling. Here, we review the computational studies performed on the vesicular transport machinery involved in vesicle fusion and budding.
6.3.1. Vesicle Fusion
Vesicle fusion requires complex machinery. Some of the proteins in the machinery are crystallized and extensive simulations have been performed to investigate their interactions with each other and the membrane to shed light on the vesicle fusion mechanism.
The SNARE Complex
One of the most important steps in neural signaling is the release of neurotransmitters at the synaptic cleft. This is an exocytotic process, in which the synaptic vesicles filled with neurotransmitters fuse with the membrane, releasing the contents to the synaptic cleft.
The SNARE protein complex is the main engine that facilitates the fusion of synaptic vesicles with the synaptic membrane. A schematic representation of the complexation process and the elements involved in it are depicted in Figure 34. The complex forms when a membrane-attached (t-SNARE) and a vesicle-attached (v-SNARE) domain associate during the membrane fusion process. The product of the complexation is referred to as the trans-SNARE complex. This complex is composed of four helices contributed by three proteins: one helix from the v-SNARE protein, synaptobrevin, and three helices from the t-SNARE proteins syntaxin and SNAP-25. The energy released during the formation of the trans-SNARE complex compensates for the repulsive forces between the membranes and brings them together.
Figure 34.
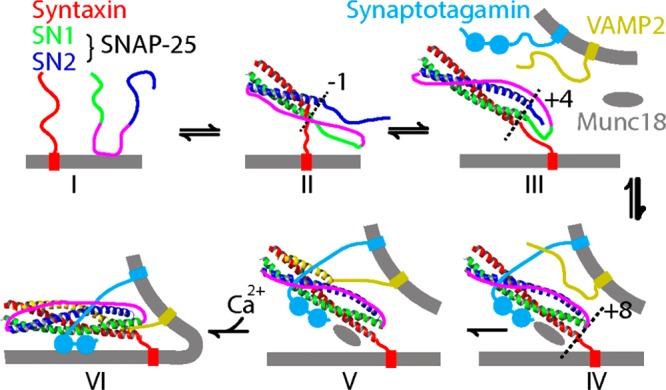
Schematic model for SNARE-mediated membrane fusion. The monomers of t-SNARE (I) assemble into the t-SNARE complex (II) and folds (III). The t-SNARE forms a complex with the v-SNARE (VAMP2), also binding synaptotagmin and Munc18. (IV) The complex formed by t- and v-SNARE zips up (V) and initiates fusion (VI). Reproduced with permission from ref (861). Copyright 2016 Zhang et al.
The mechanism of complexation of t- and v-SNAREs is likened to zipping up, and the resulting trans-SNARE has a coiled-coil structure. The zipping up of the SNAREs not only brings the membranes together but also mechanically distorts them to produce a fusion pore. Figure 35 shows schematic representations of structures that precede the formation of the fusion pore. The review by Risselada and Grubmüller863 depicts a mechanism for this complex process based on molecular simulations and experiments. In essence, the SNARE complex takes active part in all stages of fusion. (i) SNARE complex zipping brings the membranes together. (ii) A stalk intermediate forms, in which one or more splayed lipids connect adjacent monolayers. Here, the zipping releases energy to bring the lipid headgroups sufficiently close, while the transmembrane domain of SNARE perturbs the lipid packing facilitating the stalk formation. The splayed lipid was suggested to form the main fusion barrier. (iii) The stalk expands as driven by the assembly of SNARE complexes. (iv) Finally, the SNARE complexes facilitate the formation of the fusion pore by inhibiting a metastable hemifusion diaphragm (Figure 35). The penetration of the negatively charged C-termini of the transmembrane domains into the membrane further helps the opening of a fusion pore.863
Figure 35.

Schematic representation of intermediate structures in membrane fusion. Adapted with permission from ref (862). Copyright 2006 Rockefeller University Press.
The hemifusion diaphragm (Figure 35) is an important intermediate in membrane fusion, in which the two bilayers are connected by another bilayer. The hemifusion diaphragm contains a highly negative membrane curvature at the junction, where a trilayer structure forms. The simulation studies by Gardner and Abrams864 using a solvent-free coarse-grained model originally developed by Cooke et al.865 showed that the lipid composition modulates the dynamics of the hemifusion diaphragm. The lipids with negative intrinsic curvature help stabilize the trilayer structure at the junction. In the presence of such lipids, the hemifusion diaphragm can relax via lipid flip–flops, while lateral diffusion dominates the relaxation mechanism in the presence of lipids with neutral intrinsic curvature.864
The fusion pores have been generally thought to be toroidal.866 Various computational studies aimed to characterize the shape and the structure of the fusion pore in detail. Yoo et al.867 compared the coarse-grained and continuum models for membrane bending in fusion pores. Their results characterize the fusion pore as a metastable structure with neither toroidal nor catenoidal shape. The fusion pore formation likely follows a rim-pore structure near the rim of the extended hemifusion diaphragm (Figure 35).866 Risselada et al.866 investigated the shape of the rim pore and the free energy associated with the rim pore expansion using coarse-grained simulations. Their analytical free energy model and the simulations support a nontoroidal shaped rim-pore formation. Sharma and Lindau869 commented on a nanodisc based study by Bao et al.868 that showed fusion pores to be formed of both lipids and proteins. They included coarse-grained simulation models in the commentary proposing a molecular picture.869
The number of SNARE molecules for completion of membrane fusion is not clearly known. Recently, Fortoul et al.870 combined a coarse-grained model of the SNARE complex calibrated based on all-atom MD simulations and laser tweezer force measurements with a continuum model for a membrane that incorporated membrane deformation and hydration or electrostatic repulsion. The changes in the equilibrium SNARE membrane-docked configuration were investigated with a varying number of SNARE complexes. The model suggested that the distance between the membrane and the vesicle can be reduced with addition of more SNARE complexes up to 4–6 complexes in total.870
Zheng871 used nonequilibrium MD simulations to investigate the unzipping of t- and v-SNAREs. All-atom steered MD simulations in implicit solvent captured mostly syntaxin unfolding and thus failed to reproduce the AFM-based unzipping mechanism, showing sequential unzipping of the linker domain, the C-terminal domain, and the N-terminal domain. While coarse-graining based on the Go-model872 also failed to capture the correct mechanism, coarse-graining based on a modified elastic network model was successful.871 On the other hand, a new set of all-atom steered MD in explicit solvent simulations published shortly after the first set captured a three-stage unzipping mechanism in agreement with experiments.873 The short time scales of the simulations and an insufficient number of repeats do not allow generalization to an equilibrium regime, however, which can also explain some of the artifacts such as unfolding of helices.
As mentioned above, synaptobrevin II is an important component of v-SNARE, which by binding to t-SNARE proteins syntaxin and SNAP-25 results in the formation of a stable α-helical four-helix bundle in the coiled-coil SNARE complex (Figure 34). The synaptobrevin II contains an α-helical transmembrane domain and an unstructured cytoplasmic domain, which includes a conserved juxtamembrane region connected to the helical transmembrane domain with a linker region.874
Zipping of the SNARE coiled-coil generates a force that brings membranes together overcoming the repulsive barrier. This is accompanied by a change in the synaptobrevin helical transmembrane domain position.875 Biochemical studies have earlier shown that extending the C-terminus of synaptobrevin with charged residues inhibits membrane fusion,875 likely by preventing the motion of the helical transmembrane domain. Similarly, attaching protein domains to the C-termini of SNARE proteins was suggested to arrest membrane fusion in a hemifused state.876
Lindau et al.877 performed coarse-grained MD simulations to investigate how the force produced by zipping leads to fusion pore formation. They showed that piconewton forces applied on the extravesicular end of the helical transmembrane domain of synaptobrevin suffice to dislodge the C-terminus from the inner leaflet of the vesicle membrane. Based on the coarse-grained simulations and free energy calculations, Lindau et al.877 proposed a model for fusion pore formation, where the zipping of two SNARE complexes pulls the two C-terminal domains into the membrane and leads to consecutive tilting of the transmembrane domain. The tilting results in a state where two helical transmembrane domains become parallel to the plane of the membrane and a stalk is formed, after which the fusion pore formation completes.877
Blanchard et al.878 used atomistic MD simulations to characterize the prefusion state of membrane-embedded synaptobrevin truncated to include only the linker and the transmembrane domain and reported the insertion depth and the protein orientation. In this study,878 the authors employed a membrane model that aims to bypass the time scale limitations due to slow lipid diffusion, called the highly mobile membrane mimetic (HMMM) model.879 In HMMM, the membrane core is represented as an organic solvent, and headgroups as short-tailed lipids. The simulations initiated at various tilt angles in the HMMM converged to a tilt angle and the same insertion depth of W89 and W90, with the transmembrane and linker regions forming a continuous helix kinked at G100.878 To verify their results, the authors later converted the HMMM model to a pure POPC bilayer, in which the highly tilted state and the conformation were maintained. Although biochemical studies had shown that extending the C-terminus of synaptobrevin with charged residues hinders fusion,875 the extension does not change the membrane-embedded configuration, suggesting a different mechanism of action for the extension.878 Another set of microsecond-scale all-atom MD simulations of truncated synaptobrevin (transmembrane domain, linker, and juxtamembrane domain) and several mutants also confirmed these results about the role of G100 and the tilt angle of the protein.874 These simulations by Han et al.874 showed also that the transmembrane domain drives folding of juxtamembrane domain along with the connecting linker. This allows synaptin to adopt to different membrane thickness. In addition, the linker and juxtamembrane domain perturb and partially dehydrate the membrane.874 The lipid-regulated conformational dynamics of synaptobrevin homologue Ykt6 was also studied recently using computer simulations.880
Han et al.881 used the coarse-grained-based docking assay for the transmembrane (DAFT) components method to investigate the oligomerization of wild-type synaptobrevin II. They showed that synaptobrevin II forms stable right-handed dimers in agreement with mutagenesis experiments. The docked structures, later verified using all-atom simulations, revealed that poly-Val and poly-Leu mutants result in increased and decreased dimerization propensity, respectively.881
Syntaxin-1A is a part of the t-SNARE complex implicated in synaptic vesicle docking at presynaptic active exocytosis sites (Figure 34). PIP2 forms microdomains of about 73 nm size that sequester syntaxin 1A. In an experimental study combined with coarse-grained MD, the basic residues at the SNAP-25 C-terminus were found to be essential for tightly zipped SNARE complex. Furthermore, this conformational preference of SNARE is correlated with reduced release rates in point mutations of these basic residues.882
Munc18a (mammalian uncoordinated-18 protein) (Figure 34) is a member of the Sec1/munc18 protein family, which is involved in vesicle fusion by binding SNARE proteins. Phosphorylation of Munc18a by protein kinase C reduces its affinity for syntaxin-1A.883 To unravel the mechanism for this affinity reduction due to phosphorylation, Bar-On et al.883 investigated the conformational dynamics of munc18a and its phosphomimetic mutants computationally. The simulations showed that phosphomimetic mutants of Munc18a arrest it in a closed-cavity conformation, which makes syntaxin 1A binding unfavorable.883
Tethering proteins have been thought to play a role in initial recognition and attachment of membranes by SNAREs. Moreover, the transition from the hemifusion intermediate to a fusion pore manifests a huge free energy barrier (Figure 35), and a recent study by D’Agostino et al.884 combining in vivo and in vitro fusion of yeast vacuoles with MD simulation showed that the tethering proteins are required to overcome this barrier. The study884 suggests that tethering proteins are necessary for fusion pore formation; and SNAREs and tethering proteins form “a single, non-dissociable device”. The coarse-grained MD simulations in this study884 were instrumental in showing how binding of the tethering complex for vacuole and lysosome fusion (the homotypic fusion and protein sorting complex (HOPS)) to the headgroup region of the SNARE complex affects the hemifusion stalk geometry (Figure 35). It was shown that the presence of HOPS, approximated by a single bead of 14 nm diameter, decreases the free energy cost by half in the progression from a hemifusion intermediate to the fusion pore.884
Binding of the cytosolic protein complexin (Cpx) to the SNARE complex inhibits spontaneous fusion, and thus, also premature vesicle fusion in the absence of an action potential. The equilibrium and nonequilibrium MD simulations of the SNARE/Cpx complex showed that the accessory helix of Cpx acts as a “fusion clamp” by binding to synaptobrevin C-terminus and preventing full SNARE zipping.885 This model was further used to predict mutations that can enhance Cpx function in preventing full assembly of the SNARE complex. These simulations and mutations further support the role of Cpx in stabilizing the partially unzipped state of the SNARE complex and preventing its full zipping.886
SNARE molecules are recycled in an ATP-dependent manner by the 20S supercomplex formed by the ATPase NSF (N-ethylmaleimide sensitive factor) and SNAPs (soluble NSF attachment protein). The collective motions of the individual molecules, estimated using normal-mode analysis, were used to put forward a SNARE disassembly mechanism.887
Synaptotagmin
Synaptotagmins are a family of Ca2+ sensing proteins in the presynaptic axon terminal. The synaptotagmin family has 17 isoforms in humans, which have differing functions and varying Ca2+ binding affinity. They contain two membrane-targeting domains (C2A and C2B (Figure 34 A)) with varying lipid selectivity and affinities and are generally involved in Ca2+ binding. Synaptotagmin 1 (Syt1) C2B interacts with SNAP25 of the SNARE complex (Figure 34), inhibiting vesicle fusion before Ca influx. On the other hand, Ca2+ influx results in insertion of the C2 domains into the membrane, which removes the fusion clamp leading to fusion.888
Syt1 was shown to induce the bending of the presynaptic membrane upon binding of Ca2+, ultimately resulting in fusion pore formation with the help of the SNARE complex. Among the two C2 domains, only the isolated C2B domain can bend the membrane despite their similarities in structure, sequence, and membrane binding affinity. On the other hand, C2B contains a C-terminal helix rich in lysine and hydrophobic residues.889 Wu et al.889 hypothesized that the C-terminal helix plays a role in membrane bending and this requires a transition from the conformation captured by electron paramagnetic resonance (EPR) and NMR (helix-up) to another one, in which the C-terminal helix is in contact with the membrane (helix down) (Figure 36A). The authors tested this idea by rotating the helix in pulling simulations (through 16 repeats) to induce the necessary conformational transition from the helix-up configuration to the helix-down configuration, followed by a 200 ns long equilibration to test the stability of the final configuration. The all-atom MD results indicated that C2A and the helix-up conformation of C2B cannot bend the membrane, whereas the helix-down conformation of C2B can, when placed on the membrane. The authors further showed, by calculating the PMF using umbrella sampling simulations, that the helix-down conformation is energetically favorable when bound to an anionic membrane (Figure 36B). The simulations captured that the negatively charged lipids accumulate around the C2B Ca-loop and the C-terminal helix. The membrane bending, however, appeared to be due to insertion of some hydrophobic residues into the membrane. While the simulations for membrane binding, lipid distribution, and PMF calculations were based on the HMMM model, membrane bending and pressure profile calculations were performed using a full membrane model.889
Figure 36.

Conformational transition between helix-up and helix-down configurations in Syt1. (A) Ca2+-bound C2A domain (left) and C2B in helix-up (center) and helix-down conformations (right). (B) Potential of mean force for the transition from the helix-up to the helix-down conformation on an anionic membrane. The helix-down conformation (iii) is more stable than the helix-up conformation (i). A shallow barrier (ii) separates the two conformations. The free energy further reduces as the helix binds to the membrane surface (iv). Adapted with permission from ref (889). Copyright 2014 Biophysical Society. Published by Elsevier Inc.
A combined Förster resonance energy transfer, circular dichroism, and implicit solvent MD simulation study by Fealey et al.890 recently showed that the structure of Syt1’s intrinsically disordered region depends on phosphorylation and the dielectric constant. The study showed that lowering the dielectric constant of the environment, which mimics conditions near the membrane surface, stabilizes helical structures, while phosphorylation of T112, which is known to modulate exocytosis, destabilizes them. The mechanism involves formation of a distinct set of salt bridges in a reduced dielectric and upon phosphorylations that either stabilize or destabilize the transient secondary structures.890
While Syt1 works in the neurons, Syt7 functions in the regulation of insulin secretion and manifests stronger sensitivity to Ca2+. Chon et al.891 performed atomistic MD simulations to characterize the membrane-bound form of the Syt7 isoform. This study891 is consistent with an experimental study by Osterberg et al.888 published in the same issue. Performed without input from the experimental study,888 the simulation results helped to explain the underlying differences between membrane binding of the two isoforms (Syt7 and Syt1). EPR measurements by Osterberg et al.888 showed that Syt7 C2A inserts deeper into the membrane when compared to Syt1 C2A. The atomistic MD study by Chon et al.891 further showed that Syt7 C2A binds three Ca2+ ions and binds the membrane with two Ca2+ binding loops.
The Exocyst Complex
The exocyst complex is an evolutionarily conserved octameric protein that plays a crucial role in exocytosis. It targets post-Golgi secretory vesicles to the plasma membrane and facilitates their localization and tethering before vesicle fusion, which is mediated by the SNARE complex. As such, it plays crucial roles in many physiological processes, including morphogenesis, cell cycle progression, and tumor invasion.892
In a combined biochemical and MD simulation study, Zhao et al.893 showed that Exo70, a subunit of the exocyst complex, induces tubular membrane invagination on synthetic vesicles and protrusions on the cell surface. Coarse-grained MD simulations performed using two different dimer models of Exocyst complex component 7 (Exo70) (parallel and antiparallel) showed that wild-type dimers induce sufficient negative membrane curvature to induce tubulation, whereas monomers and a mutant that is deficient in lipid binding do not. The study established Exo70 as a membrane bending protein.893
The exocyst interacts with the membrane in a highly controlled manner. Two of its eight subunits, Exocyst complex component 1 (Sec3) and Exo70, are thought to interact with the target plasma membrane region before the rest of the complex is recruited. Sec3 and Exo70p have been suggested to bind PIP2 in this process.894 Pleskot et al.894 studied the molecular details of membrane binding of the individual subunits using coarse-grained MD. The simulations showed that both Exo70p and Sec3p-N spontaneously associate with the membrane. In both cases, PIP2 molecules clustered around positively charged residues, almost all of which are conserved in eukaryotes. Another set of simulations also included a small GTP-binding protein, Rho1p, along with Sec3p-N, which were crystallized together in the active state. Simultaneous binding of proteins and involvement of PIP2 suggest a mechanism for how small GTPases together with PIP2 can localize the exocyst.894
Although crystal structures of a few subunits of the exocyst complex were known previously, the structure of the octamer has only recently been characterized using cryoEM in combination with the cross-linking mass spectroscopy at 4.4 Å resolution.895 With the recently characterized cryoEM structure of the full-exocyst complex, we expect that more simulation studies on the mechanism and membrane interaction of the complex will follow.
6.3.2. Vesicle Budding
Various proteins are involved in endocytosis and exocytosis processes each playing a role in different parts of the cell or at different stages of the vesiculation. Here we review the computational studies performed on various proteins involved in vesicle formation, focusing on the crosstalk between membranes and proteins, specifically the lipid–protein interactions.
Clathrin-mediated endocytosis is a complex process that recruits a variety of proteins on the membrane surface. Recently, Milosevic896 reviewed this process in the recycling of synaptic vesicles. First, PIP2 and the cytosolic domains of synaptic vesicle proteins recruit the clathrin adaptors to the membrane. The adaptor proteins, then, recruit the clathrin light and heavy chains. The light and heavy chains polymerize to form a triskelion shape evolving into a coated pit that invaginates the membrane. The GTPase dynamin assembles into a collar-like coat in the neck of the pit and cleaves the coated pit using the energy from GTP hydrolysis. Then, synaptojanin-1 hydrolyzes PIP2, promoting the dissociation of the adaptors. The disassembly of clathrin is facilitated by 70 kDa heat shock protein (Hsc70), an ATPase, and auxilin.896
Bin/Amphiphysin/RVS (BAR) domain-containing proteins detect membrane curvature in the above-described process and work together with dynamin in cleavage of the coated pit.897 Proteins containing the Bin/Amphiphysin/Rvs (BAR) domain are responsible for curving the membrane in various biological functions, including endocytosis and trafficking. BAR domains manifest a crescent shape with diverse curvatures, lengths, and affinity for different membranes. Membrane sculpting properties of BAR domains have been investigated using simulations at various resolutions. Membrane curvature due to the binding of N-BAR domain (Bar domains that contain a N-terminal amphipathic helix)898 as well as the roles of N-terminal amphipathic helices899,900 and PIP2 lipids in the membrane-association900 have been captured in atomistic simulations. Takemura et al.901 suggested that salt-bridge formation between the lipids and the inverse BAR (I-BAR) domain increases the local lipid density and leads to curvature generation.
Arkhipov et al.902 investigated the collective effects of BAR domain lattices using coarse-grained models. The shape-based and residue-based coarse-graining schemes successfully captured membrane bending902 and tubulation by lattices made of N-BAR domains.903 Yu and Schulten904 used atomistic simulations and a shape-based coarse graining scheme to model F-BAR domains. Their simulations captured tubulation induced by F-BAR domain lattices.904 Lyman et al.905 suggested that water between the membrane and the positively charged concave surface of BAR screens electrostatic interactions. Electrostatics, therefore, is unlikely to be the main driving force in curvature sensing and membrane binding of BAR domains.905 Recently, microscopy, mathematical modeling, and coarse-grained MD simulations suggested that the BAR domains are not densely packed on the tube, covering only 30–40% of the tube surface, and that the amphipathic helices are not needed for forming the BAR scaffold.906 Moreover, Ayton et al.907 used mesoscale continuum models to investigate N-BAR domain induced vesiculation and tubulation. As additional reading, protein mediated membrane remodeling studied by multiscale simulations was reviewed recently elsewhere.908
BIN1 (M-Amphiphysin2) coordinates dynamin-mediated vesicle fission and is involved in the biogenesis of muscle cell T-tubules. While the N-BAR domain of amphiphysin dimerizes and binds to the negatively charged regions of the membrane, C-terminal SRC homology 3 (SH3) domain binds to the proline-rich domain of dynamin, a eukaryotic GTPase involved in cleavage of the clathrin-coated vesicles.909 Picas et al.909 showed that BIN1 clusters PIs (phosphoinositides, referring mostly to phosphatidylinositol 4,5-bisphosphate here) and the PI clustering results in dynamin recruitment. In this process, while the N-BAR domain controls the dynamin recruitment kinetics, the SH3 domain drives the dynamin accumulation on the membranes. The PI clustering appears to be a general property of proteins that interact with PI (tested also on PI-binding ANTH/ENTH domains). While curvature generation is not necessary for PI clustering, it amplifies the effect. The study also included coarse-grained MD simulations, which showed spontaneous association of the N-BAR (and ANTH/ENTH domains) with the membrane (Figure 37D), which results in increased PIP2 local concentration and density (Figure 37A,C,E). All domains induced clustering of up to 9 PIs with a diffusion-driven mechanism (Figure 37B), in which PIs remain free to dissociate, and thus interact with the recruited proteins, such as dynamin.909
Figure 37.

PI clustering resulting from membrane binding of N-BAR and ANTH/ENTH domains. (A) The spontaneous membrane binding of N-BAR (left), ENTH (middle), and ANTH (right) domains induce PIP2 clustering in CG MD simulations. A snapshot of protein and the trajectory of individual PIP2 molecules (top), and PIP2 density (bottom) are shown from top view. (B) Number of PIP2 molecules bound to each domain averaged (±standard deviation) over independent runs. (C) PIP2 local concentration vs membrane-bound protein density for the N-BAR homodimer. (D) The distance between the ENTH domain and the membrane along the membrane normal (black) and the minimum distance between ENTH and PIP2 molecules. Spontaneous lipid binding and dissociation events are indicated with arrows. (E) Time evolution of PIP2 local concentration. Adapted with permission from ref (909). Copyright 2014 Springer Nature.
Auxilin I is a protein tyrosine phosphatase involved in clathrin-mediated endocytosis in neurons. By binding to clathrin-coated vesicles, auxilin I recruits HSPA8/Hsc70, which starts uncoating clathrin-coated vesicles. Multiscale simulations of the wild-type and mutant auxilin 1 showed that negatively charged lipids (especially PIP2) enhance auxilin-membrane binding and the role of phosphatase tensin-type (PTEN-like) domain in the process.910
Dynamin mediates vesicle scission in a GTP-hydrolysis-dependent manner by oligomerizing around the neck of clathrin-coated pits. Crystal structures of the nucleotide-free dynamin dimer911 and tetramer912 have been characterized. These structural studies911,912 were complemented with MD simulations for interpretation. Docking in combination with MD and linear interaction energy calculations was used to explore the binding sites of a clathrin inhibitor (bolinaquinone).913
A combined experimental and MD study by Pinot et al.914 showed that polyunsaturated phospholipids enhance the activity of dynamin and endophilin, another BAR domain protein, in deforming synthetic membranes and vesiculation and, thus, accelerate endocytosis. The study914 showed that polyunsaturated phospholipids make the membrane easy to deform under a pulling force. Pinot et al.914 assessed the conformation of the membrane lipids using coarse-grained MD, in which they applied a pulling force to a phosphatidylcholine bilayer. Under the pulling force, a tube extended, from which a vesicle separated at some point. During this process, the polyunsaturated chain conformationally adapted to membrane curvature and filled the voids.914
Epsin is involved in clathrin-coated vesicle-mediated transport by both promoting membrane curvature and recruiting accessory proteins.915 The epsin N-terminal homology (ENTH) domain was indicated in membrane remodeling by specifically interacting with PIP2 and inserting the N-terminal amphipathic helix into the membrane.916 Tourdot et al.916 developed multiscale computational models that combined coarse-grained MD simulations with continuum Monte Carlo field-based approaches to investigate the protein–membrane interactions and the resulting membrane curvature. The coarse-grained MD simulations by Tourdot et al.916 showed that PIP2 is necessary for membrane-binding of the ENTH domain of epsin, which induced positive curvature on the membrane.916 Combination of EPR and multiscale MD simulations suggested that ENTH domains aggregate and form dimers or higher order oligomers, with arrangements depending on the properties of the local curvature.915
Overall, the simulation studies presented here concentrated on individual elements involved in vesiculation. MD has made important contributions in understanding the membrane-sculpting role by BAR domains. Moreover, lipid–protein interactions have been characterized for many proteins involved in different stages of vesicle budding, especially the role of PIP2 lipids.
6.4. Challenges
In this section, we covered the role of biomembranes in mass transport. Cells have evolved various mechanisms for mass transport, regulated in particular by membrane-associated proteins. Biomembranes are not only barriers against mass transport but also actively involved in controlling what gets in to and what gets out of the cells.
Here, we first described how various small molecules, including water, can get through pure lipid bilayers in a nonfacilitated manner. These nonfacilitated transport mechanisms have many implications for drug design and delivery. A particular theme that comes up here is the role of cholesterol in decreasing membrane permeability by modulating the structural properties of the membranes. On the other hand, this property appears to depend on the membrane phase. Despite advances in MD methodology, estimation of passive permeability of small molecules is a challenging task and requires large resources. This issue is further aggravated by the complexity of the real membranes in terms of lipids and other components. With increasing resources in the future, it may be possible to use MD as a high-throughput method for estimation of passive permeability of biomedically important molecules.
We, then, moved on to describe the contributions of MD simulations in discovering how protein–lipid interactions are involved in the regulation of channel and carrier proteins that facilitate mass transport. Lipids appear to modulate these proteins both by site-specific interactions and by altering the physical properties of the membranes. Specific lipid–protein interactions have been characterized in various secondary transporters, regulating the localization of the protein and the transport dynamics. The regulatory roles of membrane physical properties are well characterized through experimental and computational studies, especially for mechanosensitive channels.
Considering the alternating access mechanism for primary and secondary active transporters and gating mechanisms of various channels, the physical properties of the membrane likely constitute a general mechanism for functional modulation of mass transport. For example, lateral pressure changes may hypothetically induce opening of channels and may even change their specificity by rearranging residues in the selectivity filters. Similarly, differential lipid packing in opposing leaflets would lead to different conformational states stabilized in membrane transporters, modulating their efficiency in transport. On the other hand, the effect of site-specific protein–lipid interactions and the physical properties of the membrane on the mechanism and the energetics of carriers and channels have not been extensively studied computationally. Here, the particular challenge is that the required conformational changes fall beyond the time scales of simulations, so their investigation is usually performed using biased and enhanced sampling methods, which usually require identification of various relevant slow degrees of freedom as collective variables. Obtaining quantitative results requires huge computational resources. Lipids constitute another level of complexity: their slow dynamics may result in convergence problems and dependence on the initial configuration used in the simulations.
Finally, we covered recent simulations of vesicle budding and fusion machinery. These processes have been studied mostly using coarse-grained models due to the vastness of the systems. Most atomistic simulations performed until now have not investigated the whole process but instead considered how individual members of the complexes might function in isolation. Overall, models studied by MD simulations have established themselves for investigating mass transport, due to their resolution and ability to capture dynamics. Both vesicle fusion and budding are membrane-remodeling processes. Studying membrane remodeling using simulations requires large systems even for the coarse-grained simulations. Besides, each step requires various protein–protein and protein–lipid interactions and protein conformational changes, each of which are challenging to tackle with both coarse-grained and atomistic simulations. Therefore, limitations of time and length scales are the major barrier against progress.
7. Lipids Contribute to Signaling, but How?
Cells are separated from their native environment by biological membranes. The plasma membrane of eukaryotic and prokaryotic cells is a bilayer composed of lipids, into which a plethora of different proteins is embedded. Lipids are involved in the formation of basic structures in cells and tissues, regulating the intertissue communication, immunological functions, energy metabolism, and ultimately, the cell survival. Plasma membrane contains thousands of different lipid types. It is safe to assume that they all have an important role to play in cells, yet their functions are not well understood. The understanding of lipid properties and the interplay between lipids and other components of biological membranes is currently the biggest challenge in cellular biology and biophysics. A substantial amount of research has been carried out over the years to clarify the importance of lipid classes. This knowledge is of great importance; however, it is just the tip of the iceberg. Each lipid class has its own subclasses that differ in the length of hydrocarbon chains, the degree of saturation, charge, structural modifications, and many others. About 42000 lipid species have been identified917,918 as biologically relevant to date (statistics from April 2018). However, studies on the molecular mechanisms by which lipids regulate cellular processes and signaling have been restricted to a rather small number of systems. Meanwhile, only recently the role of lipids in modulating the activation of integral proteins has been appreciated.54,734,919−926 This idea of lipids as modulators of protein activation has often been discussed to arise from specific interactions, where lipids are bound to the protein and act as its integral partners, or from membrane-mediated interactions, where the physical properties of the membrane control, in part, protein conformations and their activation.
In this section, we discuss the knowledge generated by computer simulations regarding the role of lipids in different aspects of cellular signaling. First, we focus on lipid signaling in the absence of proteins. We discuss the interleaflet translocation (so-called flip–flop) of lipids and briefly review the MD studies where spontaneous lipid flip–flops have been reported. We further discuss data based on free energy methods commonly used to calculate the free energy barrier for interleaflet lipid translocation. Next, we discuss the role of lipid–protein interactions in the regulation of receptor signaling. In this part, we focus our attention on computational studies where specific lipid-binding sites have been revealed. G protein-coupled receptors and transmembrane channels are here used as the main context of our discussion. We close this section by discussing how computer simulations can help us in understanding the role of protein post-translational modifications in signaling, since it turns out that here, too, the role of lipids in the signaling processes can be significant.
7.1. Lipid Signaling: Translocation of Lipids across Biological Membranes
Since the seminal work by Bretscher in the early 1970s,927 it has been known that the erythrocyte membrane is enriched in phosphatidylcholine and sphingomyelin in the outer (exoplasmic) leaflet and in phosphatidylserine, phosphatidylethanolamine, and phosphatidylinositol in the inner (cytoplasmic) leaflet. Currently, it is established that biological membranes are not only asymmetric with respect to the composition of leaflets144 but also laterally heterogeneous.497,928 Recent advances in structural biology929 and lipidomics61 have revealed that the composition of cell membranes varies substantially according to cell age, origin, metabolic state, and spatial location resulting in a rather complex matrix of lipid–protein interactions. Several functional roles for asymmetric lipid distribution in plasma membranes have been suggested. For instance, regulatory proteins such as spectrin,930,931 protein kinase C,932 and annexin933 appear to localize to the cytoplasmic face of the membrane due to their interactions with phosphatidylserine. The disruption of lipid asymmetry leads to exposure of phosphatidylserine on the outer surface of the plasma membrane, which in turn serves as a trigger for macrophage recognition of apoptotic cells.934,935 Several severe diseases and fatal conditions, such as Alzheimer’s disease,936 cancer,937,938 and atherosclerosis939 have been associated with the disruption of lipid asymmetry.
Passive transbilayer diffusion of lipids (also known as flip–flop, transbilayer translocation, or transbilayer movement) is one of the multiple mechanisms contributing to the maintenance of lipid homeostasis in biological membranes. The term flip–flop was first used by Kornberg and McConnell940 to describe “the transverse motion of phospholipids” in lipid membranes. The spontaneous translocation of phospholipids across a bilayer has been postulated to be extremely slow (with a rate of about 10–15 s–1), corresponding on average to a single lipid flip–flop event every 24 h.940,941 However, the data reported in the literature lacks consensus. For instance, the published cholesterol flip–flop times range broadly from 3 s to tens of minutes.942,943 More recent studies have suggested that the flip–flop halftime for cholesterol is shorter than 1 s.944 Bruckner et al.,945 using an NMR-based technique, showed that cholesterol residence time in a single leaflet of a bilayer is below 10 ms. However, in 2011, Garg and co-workers946 reported based on neutron scattering that the cholesterol flip–flop halftime is in the range of hundreds of minutes. These striking discrepancies can be attributed to the sensitivity of flip–flop processes to chemical and physical membrane properties, lack of standardized experimental procedures, and the intrinsic difficulty to detect flip–flops in the first place.
Theoretical and computational studies can alleviate the above-mentioned difficulties by providing detailed insight in the nanoscale. During the last 15 years, this approach has been used quite successfully to provide molecular-scale understanding for the energetics and kinetics of lipid flip–flops in biological membranes. Here, we summarize these efforts and the lessons we can learn from them.
7.1.1. Spontaneous Lipid Flip–Flop
Classical MD simulations (combined with Monte Carlo approaches) have been used to describe lipid flip–flop processes ever since the pioneering work by Imparato et al.947 published in 2003. In this study, lipids were modeled as flexible chains of beads, and water was represented as a single bead. The combination of Lennard-Jones and soft repulsive potentials was used to model hydrophilic and hydrophobic interactions. This simplistic model was able to reproduce the experimentally observed fact that the flip–flop rate decreases with increasing lipid chain length.948,949
In the early atomistic simulations of phospholipids, only a few isolated lipid flip–flop events were observed under specific conditions such as in the vicinity of membrane defects (transient water pores). A spontaneous lipid flip–flop was first noted during the self-assembly of a DPPC vesicle950 and later confirmed by direct observations of lipid translocation events under a variety of conditions: in the presence of membrane perturbing agents such as dimethyl sulfoxide (DPPC, DMPC, or POPC)951 or butanol (DPPC)952 or antimicrobial peptides (DPPC)694 and in physiologically relevant conditions, such as under an ion concentration imbalance across a lipid membrane (POPC).953 The study by Gurtovenko and Vattulainen954 is particularly appealing since it proposed a water pore-facilitated mechanism for phospholipid flip–flop. Based on 50 successful flip–flop events, the data demonstrated that lipid flip–flops occur spontaneously in protein-free phospholipid membranes under physiological conditions through transient water pores on a time scale of tens of nanoseconds (see Figure 38). Gurtovenko and Vattulainen suggested that the appearance of a transient defect in a lipid bilayer inevitably leads to diffusive translocation of lipids through the pore, and the diffusive motion through the defect is driven by thermal fluctuations. While the flip–flop process takes place very rapidly, in a time scale of 10–50 ns, the rate is dictated by the time needed to create a pore.
Figure 38.
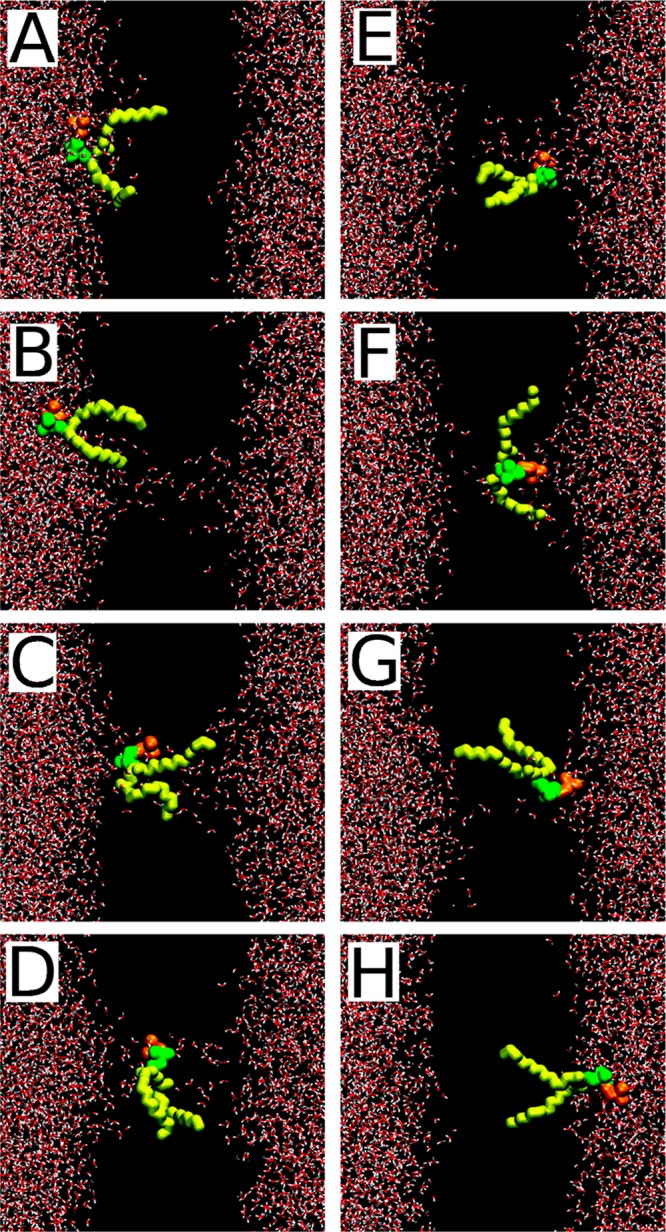
Snapshots from the molecular dynamics simulations showing the pore-mediated lipid flip–flop. Only the flip–flopping lipid is shown; water is shown in red and white, acyl chains of the flip–flopped lipid are shown in yellow, and its choline and phosphate groups are shown in orange and green, respectively. Reproduced with permission from ref (954). Copyright 2007 American Chemical Society.
For sterols, similarly only a few isolated flip–flop events have been observed in atomistic simulations. Probably the first spontaneous sterol flip–flop found through atomistic MD simulation was reported by Róg et al.955 in 2008. In the 200 ns long simulation, the authors observed two flip–flop events of cholestenone in a DPPC bilayer, while no flip–flop events were observed for cholesterol. Interestingly, the cholestenone flip–flop occurred without bilayer perturbations in terms of a push-in mechanism:956 a sterol molecule first moves toward the bilayer center, then it rotates at the center of the lipid bilayer, and finally diffuses to the opposite leaflet. In another study,957 the authors examined the behavior of cholesterol in a bilayer composed mainly of monounsaturated phospholipids with varying chain length (diC14:1-PC and diC22:1-PC). The simulations uncovered that in the bilayers composed of short-chain lipids, cholesterol exhibited broad orientational distributions. During 50 ns of atomistic simulations, a single cholesterol flip–flop was observed. The authors concluded that cholesterol flip–flop is an intrinsic feature of disordered bilayers. More recently, Choubey et al.958 studied cholesterol translocation in DPPC bilayers using large-scale atomistic MD simulations. Over a period of 15 μs of simulation, the authors observed 24 translocation events with a rate constant of 3 × 104 s–1. Once a flip–flop event had triggered, it took a cholesterol molecule an average of 73 ns to migrate from one bilayer leaflet to the other. Extensive 24 μs all-atom simulations have also been performed by Baker and Abrams,959 who simulated three model membrane systems composed of ∼50 mol % cholesterol, ∼50 mol % DPPC, and a single α-helical transmembrane protein. The authors observed one spontaneous cholesterol flip–flop. They concluded that the discrepancy with the results reported by Choubey et al.,958 where cholesterol flip–flops were more frequent, stemmed from the higher cholesterol content (50 mol %) in the study by Baker and Abrams; it is known that cholesterol typically increases the free energy barrier of sterol flip–flop.960 Meanwhile, seven cholesterol flip–flop events were reported in the atomistic simulations of a DPPC bilayer containing 10 mol % cholesterol.53 Interestingly, cholesterol molecules positioned themselves at the boundary regions of fluid, cholesterol-rich nanodomains. This type of sorting is expected to promote cholesterol availability for cholesterol-binding membrane proteins, translocation, and thereby transmembrane transport of cholesterol. The authors highlighted that this behavior was unique for cholesterol and was not observed for cholesterol analogs. It is worth mentioning that all spontaneous flip–flop events that were observed occurred at the boundary of cholesterol-rich nanodomains.
Very recently, Kulig et al.164 proposed a new mechanism for the translocation of selected sterols. In this study, the authors explored the behavior of tail- and ring-oxidized sterols in POPC bilayers using all-atom MD simulations (Figure 39). Unlike in other flip–flop mechanisms,945,961−964 where a sterol molecule changes its orientation by rotation to accommodate its hydroxyl group at the membrane–water interface, in the bobbing mechanism, this molecular rotation was not needed (Figure 39). The mechanism was observed for tail-oxidized sterols (such as 27-hydroxycholesterol), where the chemical structure contains an additional hydroxyl group at the end of the sterol tail, rendering the molecule hydrophilic at both of its ends. In unbiased MD simulations of 27-hydroxycholesterol, Kulig et al. observed about 20 translocation events during simulations of 800 ns, while no flip–flop events were detected in analogous systems with cholesterol or ring-oxidized sterols (7β-hydroxycholesterol).
Figure 39.
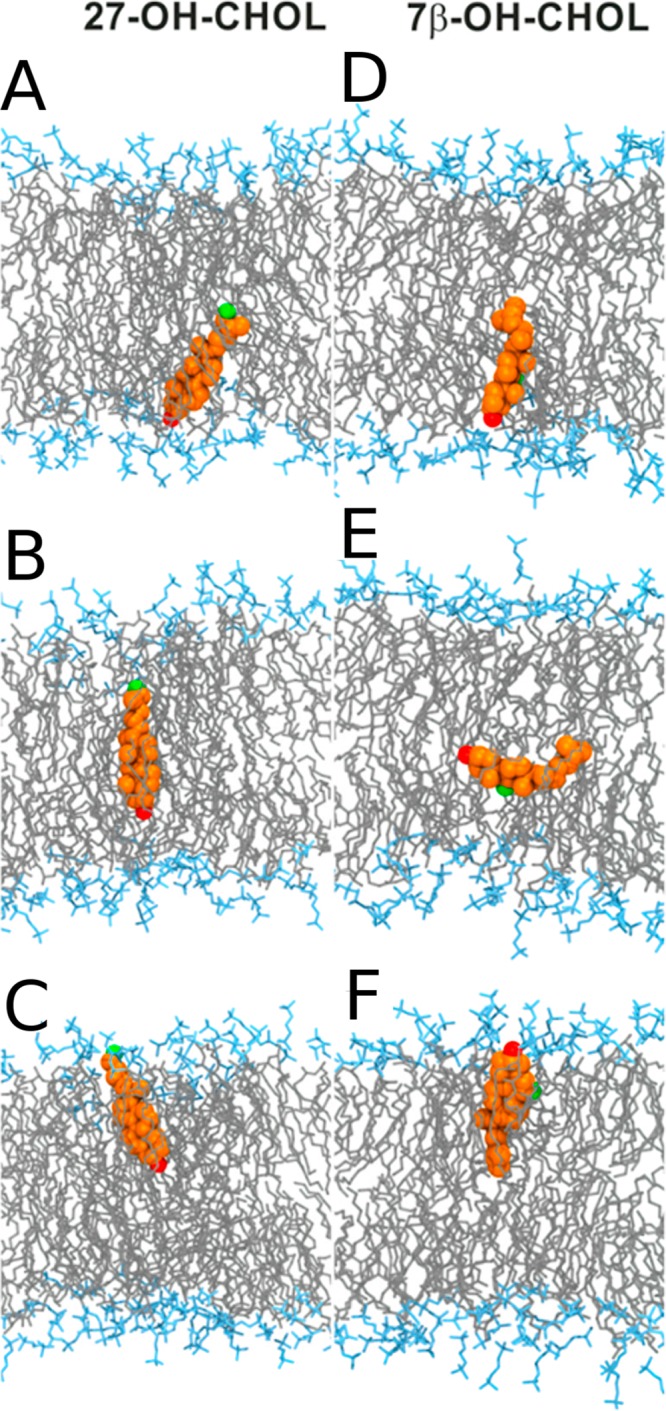
Snapshots from the MD simulation showing the translocation process of (A–C) 27-hydroxycholesterol and (D–F) 7β-hydroxycholesterol across a membrane. Shown here are the 7β- and 27-hydroxyl groups (green), the 3β-hydroxyl group (red), the oxysterol ring/tail structures (orange), the POPC headgroup (cyan), and lipid chains (gray). Reproduced with permission from ref (164). Copyright 2018 American Chemical Society.
The development of the MARTINI force field965,966 has enabled MD simulations to readily reach time scales beyond microseconds even for system sizes that are considerably larger than those used in typical atomistic simulations. Consequently, the long time scales sampled in MARTINI simulations have improved chances to observe sufficiently many flip–flop events and therefore to more accurately estimate the lipid flip–flop rates. In this spirit, recent work by Ingólfsson et al.596 reported MARTINI simulations of an idealized mammalian plasma membrane. Mixtures of about 20 000 lipids, containing more than 60 different lipid types, were simulated over a time scale of 40 μs. Thousands of flip–flop events of cholesterol, ceramide, and diacylglycerol were observed. The rate of cholesterol flip–flop was reported to be intermediate between the rates found for saturated and unsaturated lipids. The rate of diacylglycerol flip–flop was comparable to that of cholesterol, and it was 2 orders of magnitude faster than for ceramide.
Ogushi et al.967 investigated the flip–flop dynamics of cholesterol, PODAG, and SCER in DAPC, SAPC, and POPC bilayers. Surprisingly, they found that the flip–flop rate followed a slightly different order (cholesterol ≫ PODAG ≫ SCER) compared to the trend reported by Ingólfsson et al.596 The difference may stem from the tiny samples used in the Ogushi et al.967 study, where bilayers contained only 42 lipids.
The large-scale coarse-grained MD simulations have also been used to explore the effect of lipid headgroup charge, acyl chain saturation, spontaneous membrane curvature, and surface tension on cholesterol distribution and dynamics in model membranes.212,968,969 Systems containing 1000 to 4000 lipids were simulated for 12–15 μs. The authors concluded that the distribution and the flip–flop dynamics of cholesterol were strongly dependent on the lipid composition, with the mean flip–flop times ranging between 80 and 250 ns. These studies also confirmed that the flip–flop rate decreases with lipid chain saturation and that the headgroup charge has similar, though weaker, effects.
7.1.2. Free Energy Methods
Despite significant efforts, the statistically valid sampling of lipid transbilayer movement events by brute force MD simulations is still out of reach, especially in the case of phospholipids. To improve sampling, the strategy commonly known as umbrella sampling is often used. This technique allows determination of the free energy profile (or potential of mean force) of lipid motion across a membrane. In this method, the lipid bilayer is usually divided into 40–50 windows along the membrane normal direction. In each of them, one carries out an MD simulation where the molecule is constrained to stay in a given window, and the full set of simulations allows one to compute the free energy profile across the membrane. There are excellent reviews970,971 discussing the umbrella sampling method in detail.
Based on atomistic and coarse-grained umbrella sampling simulations961,972,973 for a series of PC lipids, the time of lipid flip–flop of short-chained saturated lipids (such as DLPC) was orders of magnitude faster than for longer lipids (such as DPPC). The lower cost of pore formation and the larger stability of pores in thinner bilayers can explain this behavior. In unsaturated lipid bilayers, the flip–flop rate was much slower than in saturated ones.961 The authors attributed this behavior, which is contradictory to the observation that the presence of double bonds increases bilayer fluidity, hence facilitating pore formation, to the force field used for the double bond description. Meanwhile, experimental data have shown that pore formation is reduced with increasing cholesterol content.974 This suggests that for increasing membrane fluidity, pore formation and therefore also flip–flop rates would actually increase.
Fluorescence measurements and united-atom MD simulations using the Berger force field975,976 showed that the oxidation of PC lipids in mixed PC/PS membranes, characterized by the truncated sn-2 acyl chain, enhanced the flip–flop of POPS and stabilized the formation of water pores (Figure 40). The authors suggested that “oxidative attack on membrane phospholipids could represent a causative factor inducing as such the loss of phospholipid asymmetry in apoptosis and cancer, resulting in the exposure of PS on the outer surface of the affected cells.” Similarly, Razzokov et al.977 found that an increase in POPC peroxidation (without acyl chain truncation) could also lead to a decrease of the free energy barrier for PS translocation through mixed POPC/POPS bilayers.
Figure 40.

Flip–flop of the POPS molecule in the oxidized membrane. Color-coding: PoxnoPC lipids (yellow), O atoms (pink), and H atoms of water (gray), POPS acyl chains (blue), and O atoms of the carboxylic group of POPS headgroup (red). Reproduced with permission from ref (975). Copyright 2011 Biophysical Society. Published by Elsevier Inc.
Very recently, Lin et al.978 calculated the PMF profiles for a DPPC bilayer in the presence of ion imbalance. Based on the coarse-grained simulations, the authors reported that, under these conditions, the free energy barrier of lipid flip–flop on the extracellular leaflet of the bilayer is much lower than that on the intracellular leaflet. The PMF calculations suggested that the asymmetric free energy profile was due to the ionic imbalance across the membrane. The asymmetry of lipid flip–flop and the possible subsequent lipid accumulation on the intracellular leaflet under physiological conditions has substantial implications for fusion of vesicles, protein translocation, signaling, and transport of molecular cargo in cell membranes.
The PMF calculations were also used for cholesterol, palmitoyl-ceramide, and palmitoyl-oleoyl-diacylglycerol in pure POPC bilayers in a 1:1:1 mixture of PSM, cholesterol, and POPC.979 For all three lipid types, the translocation process happened without the assistance of water defects (see Figure 41). Interestingly, the flip–flop barriers were significantly higher in raft-like bilayers (composed of PSM, POPC, and cholesterol in a 1:1:1 molar ratio), which translated into orders of magnitude slower flip–flop times. This result is of possible biological importance and was explained by higher stiffness and lower fluidity of the raft-like bilayers. In a pure POPC bilayer, the flip–flop rate followed the order: cholesterol > diacylglycerol > ceramide. Similarly, atomistic MD simulations in raft-like bilayers (POPC/PSM/cholesterol in the 2:1:1 ratio; POPC/PSM/cholesterol/cholestenone with the 2:1:0.8:0.2 ratio) were performed to compare the behavior of cholesterol and its analog cholestenone.184 It was concluded that cholestenone reduced membrane order, shortened the flip–flop duration, and desorbed more readily from membranes than cholesterol.
Figure 41.
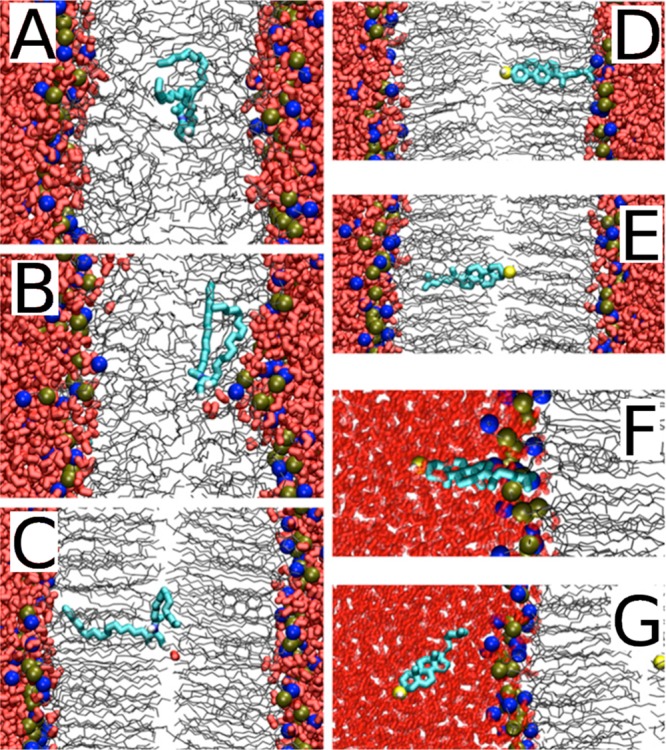
Snapshots from the MD simulation showing the translocation of ceramide and cholesterol in model membranes. In the snapshots, water is shown in red thick licorice, lipid chains in gray lines, and phosphorus and nitrogen atoms in tan and blue balls. The restrained ceramide or cholesterol are thick cyan lines, and the oxygen of the restrained cholesterol is a yellow ball. Reproduced with permission from ref (979). Copyright 2012 the American Society for Biochemistry and Molecular Biology, Inc.
More recently, Kulig et al.164 calculated the PMF profiles for the translation of oxysterol molecules (7β-hydroxycholesterol and 27-hydroxycholesterol) in POPC bilayers. The study showed that the free energy barriers of oxysterol flip–flop were strongly dependent on the chemical structure of the translocating molecule. The tail-oxidized sterols tended to translocate faster (with the flip–flop rate of 1.56 × 10–3 ns–1 as estimated from the unbiased MD simulation) compared to the ring-oxidized sterols (where no spontaneous translocation event was observed). The behavior of ring-oxidized sterols was very similar to that of cholesterol. Given that the tail-oxidized sterols often act as signaling molecules in living cells, the paper hypothesized that the higher flip–flop rate of tail-oxidized sterols has biological implications. Indeed, 25-hydroxycholesterol, for instance, transfers rapidly from cell membranes to the endoplasmic reticulum with a rate that is a hundred times larger compared to that of cholesterol.980,981
The one-dimensional PMF often used to describe the energetics and kinetics of the lipid flip–flop assumes that the reaction coordinate of the process is one-dimensional. In reality, other degrees of freedom can also play a role: large-scale membrane deformations and undulations and molecular-scale features such as local density fluctuations that can drastically change the translocation path and the immediate environment where the flip–flop of the molecule in question takes place. Nevertheless, there have been only a few attempts to go beyond the one-dimensional description of lipid flip–flops. One of these attempts was successfully implemented by Parisio et al.,964 who calculated the rates and free energy barriers associated with the flip–flop of various steroids in DPPC bilayers. In this work, the flip–flop process was described as diffusion on the free energy surface defined by lipid orientation and rotation. As variables to describe the translocation process, the authors used the position of the center of mass of a steroid molecule along the bilayer normal and the tilt angle of the steroid’s long molecular axis with respect to the membrane normal. The free energy surface was calculated using the atomistic description of steroid molecules and an implicit representation for the water-membrane environment. The flip–flop rate was estimated by solving the Master Equation that governs the transitions between the free energy minima, with transition rates evaluated from the Kramers theory.982−984 In this model, the anisotropy and the nonuniformity of the environment were taken into account in terms of gradients of density, dielectric permittivity, acyl chain order parameters, and lateral pressure across the lipid bilayer.985 The study concluded that the steroid flip–flop occurs, in general, in a nonconcerted way, with the rotation and translation taking place as two separate steps. Two possible translocation pathways were uncovered. In the first pathway, the steroid rotated and then crossed the bilayer midplane in an upside-down orientation (also known as the rotating flip–flop mechanism).956 In the second pathway, the steroid first diffused to the opposite leaflet without substantial changes in its orientation, and then rotated. A common feature in both pathways was their initial phase that started by the embedding of the polar steroid headgroup to the hydrocarbon interior of the bilayer, by either rotation or translation of the molecule. This embedding turned out to be the rate-limiting step in both pathways due to its high free energy barrier. The values of the flip–flop rates calculated by Parisio et al.964 seem to be generally faster than the measured ones.
A very similar approach was applied by Wei and Pohorille986 to the case of protonated oleic acid in a POPC bilayer. About 50 flip–flop events were observed over a period of several microseconds of atomistic simulations. Good agreement with other studies was found confirming that the flip–flop of fatty acids can be described, in a similar way to lipid flip–flops, as a two-dimensional diffusive process. Atomistic two-dimensional umbrella sampling simulations were also reported by Jo et al.,963 where two-dimensional PMFs were calculated for cholesterol as a function of sterol position inside the bilayer and its tilt angle with respect to the bilayer normal. Flip-flop occurred through a sliding mechanism956 (Figure 42), and the free energy barrier was shown to be significantly lower in a polyunsaturated membrane (DAPC) than in saturated (DPPC, POPC) bilayers.
Figure 42.
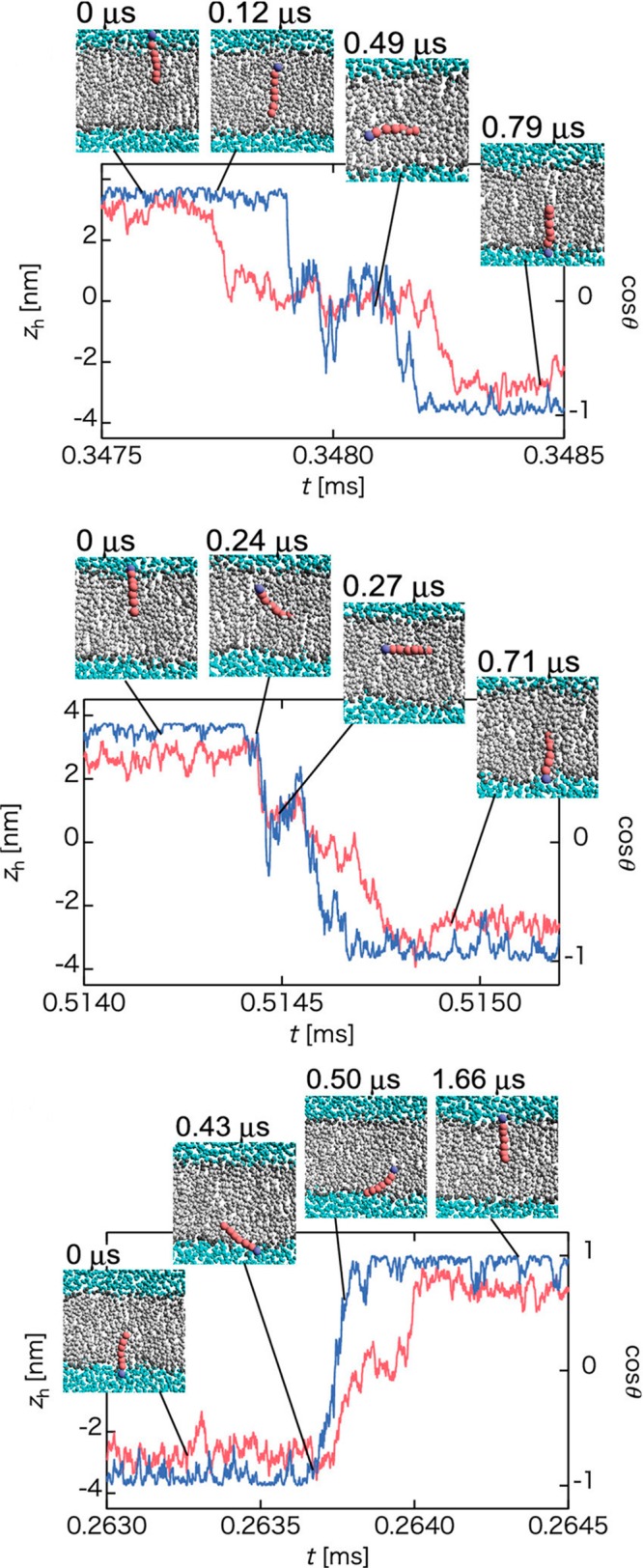
Different mechanisms of lipid flip–flop. (top) Push-in flip–flop. (middle) Sliding flip–flop. (bottom) Rotation flip–flop. Water particles are shown as blue spheres and lipid molecules performing a flip–flop are colored, whereas other lipid molecules are gray. Adapted with permission from Arai et al.956 Copyright 2014 AIP Publishing.
As described above, the calculations of one-dimensional and, to a smaller extent, of two-dimensional PMF profiles have become standard practice for an approximate estimation of flip–flop rates and lipid kinetics. Thus, caution is needed with regard to the degrees of freedom taken into consideration in the PMF calculations. The common assumption that the movement of the lipid directly along the bilayer normal is the only slow degree of freedom (and therefore also the rate limiting step) is not always the case. Inadequate sampling of the other slow degrees of freedom such as rotation and the diffusion of other molecules to organize the membrane structure in the immediate vicinity of the translocating molecule to be optimal for its translocation, may lead to inaccurate estimation of the free energy barrier. These issues are discussed in refs (987 and 988). To alleviate some of the limitations of the umbrella sampling method, other free energy based methods and enhanced sampling techniques can be used.802,989−991 Finally, alternative ways of rate constant evaluation from molecular dynamics simulations have been proposed.731
Concluding, lipid membrane composition and its lateral and interleaflet asymmetry is an essential and handy tool used by cells to modulate and control biological processes that take place in biological membranes and in their vicinity. Mechanical properties of biological membranes and the transport across them are dependent on membranes’ molecular composition. The lipid flip–flop is one of the passive mechanisms used in living cells to maintain the homeostasis between bilayer leaflets. However, despite the fact that lipid flip–flop has been the subject of intensive experimental and computational investigations since the beginning of the 1970s, a clear and comprehensive picture, as well as detailed understanding of the factors regulating translocation is still missing. This is, in part, due to experimental difficulties to monitor the time evolution of lipid asymmetry. An important deficiency is also the lack of appropriate protocols to measure translocation in a standardized fashion. The molecule that suffers from this situation most is cholesterol, since unlike many other lipids, for cholesterol there are no enzymes (such as sphingomyelinases for sphingomyelin or phospholipases for phospholipids) that would break it down, such that its asymmetric transmembrane distribution could be explored under native conditions. Currently, despite many attempts, there is still considerable confusion as to the distribution of cholesterol across cell membrane structures. Interested readers are referred to refs (145 and 201). Meanwhile, the tremendous progress in the development of computer simulation software and hardware has brought us to a situation where detailed studies of spontaneous lipid flip–flop events in atomistic resolution are possible. Free energy based methods combined with molecular dynamics simulations provide a method of choice to calculate the free energy barriers of lipid translocation, and nonbiased MD simulations over extended time scale will provide adequate sampling of average flip–flop rates. While MD simulations also have their limitations, it is clear that in the coming years, new knowledge on molecular aspects of flip–flop processes will be provided. When combined with experiments, the future work not only will clarify the details of passive flip–flops but also will bring out ideas of how these processes are actually involved in signaling.
7.2. Specific Lipid Binding Regulates Protein Activity
Lipid–protein interactions are involved in many biological processes and are of prime interest in, for example, drug discovery, as about 60% of pharmacological targets are integral or peripheral proteins.992,993 Advances in lipidomics61 and the structural biology of membrane proteins929,994 have revealed a vast complexity of biological membranes. The Protein Data Bank995 contains three-dimensional structures of ∼4654 membrane proteins (or their subdomains), and about 42000 lipid species have been identified917,918 as biologically relevant (statistics from April 2018). However, studies on the molecular mechanisms by which lipids regulate protein structure and function, and ultimately cellular physiology, have been restricted to a rather small number of proteins. Here, we summarize recent theoretical studies that have identified specific lipid interaction sites on the surface of membrane proteins. For practical reasons, we focus our attention on selected proteins in the context of G protein-coupled receptors and transmembrane channels.
7.2.1. G Protein-Coupled Receptors
G protein-coupled receptor (GPCR) proteins form a prominent family of cell surface receptors that respond to an array of chemically diverse ligands and transduce extracellular signals into intracellular responses.996 Due to the significant involvement of GPCRs in the regulation of physiological processes, these receptors are of great medical importance as they are targeted by 30–40% of the currently marketed drugs.993 GPCRs comprise the largest protein superfamily in mammalian genomes. They share a common seven-transmembrane topology (see Figure 43) and mediate cellular response to a variety of extracellular signals ranging from photons and small molecules to peptides and proteins. The mechanism by which these receptors translate extracellular signals into cellular changes includes the activation of the receptor by an agonist, the dissociation of the G protein into the α and βγ subunits, and activation or inhibition of various downstream effector molecules by the G protein subunits. Signal transduction by GPCRs is fundamental for most physiological processes, spanning from vision, smell, and taste to neurological, cardiovascular, endocrine, and reproductive functions, thus making the GPCR superfamily a major target for therapeutic intervention and an essential subject of experimental and computational studies.
Figure 43.

Schematic structure and topology of GPCRs. Reproduced with permission from ref (997). Copyright 2016 American Chemical Society.
The early experimental studies, nicely reviewed by Oates and Watts,998 have shown that cholesterol is a major player in providing a preferred environment for the functions of GPCRs. This was originally attributed to cholesterol’s ability to order lipid bilayers149 and to the receptor’s requirement for cholesterol-rich domains in order to function properly.999,1000 The presence of specific cholesterol binding sites was also postulated1001 and later confirmed by cocrystallized cholesterol molecules in many GPCR crystal structures1002−1004 (see also Table 2). It was postulated that cholesterol can modulate various aspects of GPCR signaling including ligand binding,1005−1007 stability,924,925 and oligomerization.1008,1009 Yet cholesterol is not the only lipid modulating GPCR function. Experiments have shown that polyunsaturated fatty acids (PUFAs), in particular those containing docosahexaenoic acid, enhance the function of rhodopsin.1010−1014 Similarly, phosphatidylethanolamine headgroups have been demonstrated to influence the photochemical activity of rhodopsin directly and therefore participate in the molecular mechanism of vision.1013 MD simulations have been successfully applied to identify lipids modulating the receptor function in a range of GPCRs including rhodopsin,1015−1020 the serotonin 1A and serotonin 2A receptor,1021−1025 the cannabinoid 2 receptor,1026 the sphingosine 1-phosphate receptor 1,56 the A2A adenosine receptor,57,1027−1029 the dopamine D2 receptor,57 the β2-adrenergic receptor54,1030−1032 and the β1-adrenergic receptor.1033 Below, we discuss some of them in detail.
In the study by Lee and Lyman,1029 the ligand-bound A2A receptor was simulated by means of atomistic MD simulations. Two 800 ns simulations of A2A in a POPC/cholesterol (with a 4:1 molar ratio) bilayer were performed. The study reported three distinct cholesterol-binding sites on the surface of A2A (see Figure 44). Once bound, cholesterol molecules occupied each of the binding sites for the rest of the simulation. One of the theoretically predicted cholesterol binding sites has been experimentally corroborated in the high-resolution crystal structure of the A2A receptor published by Liu et al.1034 Two other binding sites have not been experimentally confirmed yet, which may be due to the relatively weak (on the order of RT, as estimated using the Boltzmann equation) and dynamic nature of lipid–protein interactions in these sites.
Figure 44.
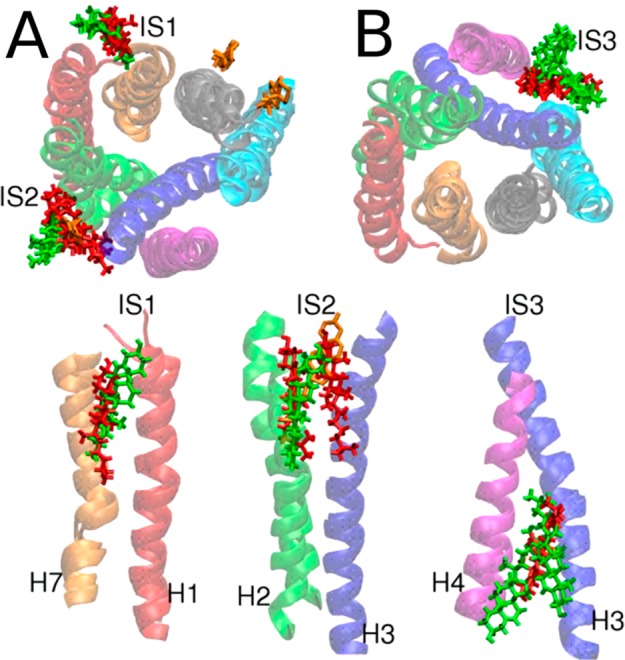
Predicted cholesterol-binding sites. (A, top panel) Cholesterol in the extracellular leaflet shown from the top. (B, top panel) Cholesterol the intracellular leaflet shown from the bottom. (bottom panels) Side views showing the positions of the cholesterols for each interaction site. The cholesterols resolved in the high-resolution structure of Liu et al.1034 are shown as orange sticks. Reproduced with permission from ref (1029). Copyright 2012 American Chemical Society.
Coarse-grained MD simulations were used to analyze the molecular nature of GPCR–cholesterol interactions for the serotonin 1A receptor.1025 In this study, the receptor was embedded in a POPC bilayer with different cholesterol contents (10, 30, or 50 mol %). Multiple interaction sites were identified based on the calculated cholesterol occupancy times. More detailed analysis revealed relatively low propensity to bind cholesterol in these sites. Interestingly, the highly conserved cholesterol recognition amino acid consensus (CRAC) motif, present on the transmembrane helix 5, was reported to have high cholesterol occupancy, which may confirm its putative role as a cholesterol-binding motif. However, studies on a closely related serotonin 2A receptor1023 identified different cholesterol binding sites, raising the question of how to evaluate the relevance of simulation-based predictions unless biochemical (mutagenesis) data are available.
Very recently, the study by Morra et al.847 employed large-scale all-atom simulations (over 50 μs of simulation time) of an opsin embedded in a lipid membrane composed of POPC and POPG in a molar ratio of 9:1. The MD simulations revealed a lipid translocation pathway on the surface of opsin, enabling passive flip–flop from one leaflet to another. The lipid pathway was localized in the region between transmembrane helices 6 and 7. The relative movement of these helices and specific repositioning of amino acid chains created the appropriate environment for the penetrating lipids. The pathway started on the IC side by random change in conformation in Y3067.53 that repositioned the tyrosine side chain away from transmembrane helix 6 followed by the disengagement of the E2496.32/K3117.58 pair of residues. These conformational changes resulted in ∼10 Å separation between the IC ends of transmembrane helices 6 and 7, increasing hydration and enabling lipid headgroup penetration. The opening of the narrowest region between TM6 and TM7 (on the level of C2646.47) widened by ∼5 Å resulting in a continuous, water-containing hydrophilic pathway. Finally, lipids moved in a manner consistent with the credit card reader mechanism suggested previously in another study.837 Overall the protein-aided lipid translocation event took place on a time scale of about 33 μs, which is consistent with the experimentally estimated rate of lipid scrambling,1035,1036 highlighting that specific lipid–protein interactions can modulate the translocation of lipids in biological membranes.
Manna et al.54 used extensive all-atom simulations (over a period larger than 100 μs) to study how the allosteric binding of cholesterol to the β2-adrenergic receptor (β2AR) modulates its conformation and dynamics. The study identified three cholesterol-binding sites on the surface of β2AR, two on the intracellular side (IC1 and IC2) and one on the extracellular side (EC1) (see Figure 45). The reported IC1 binding site matched the location of cholesterol observed in the crystal structure of β2AR.256,1004 The occupancy of two cholesterol molecules reported at EC1 was in turn in good agreement with the crystal structure of the adenosine A2A receptor,1034 which is closely related to β2AR, while IC2 is a prediction that remains to be tested through experiments. Altogether, the study provided pioneering evidence that cholesterol binds to β2AR allosterically and the binding alters the conformational state of the receptor.
Figure 45.
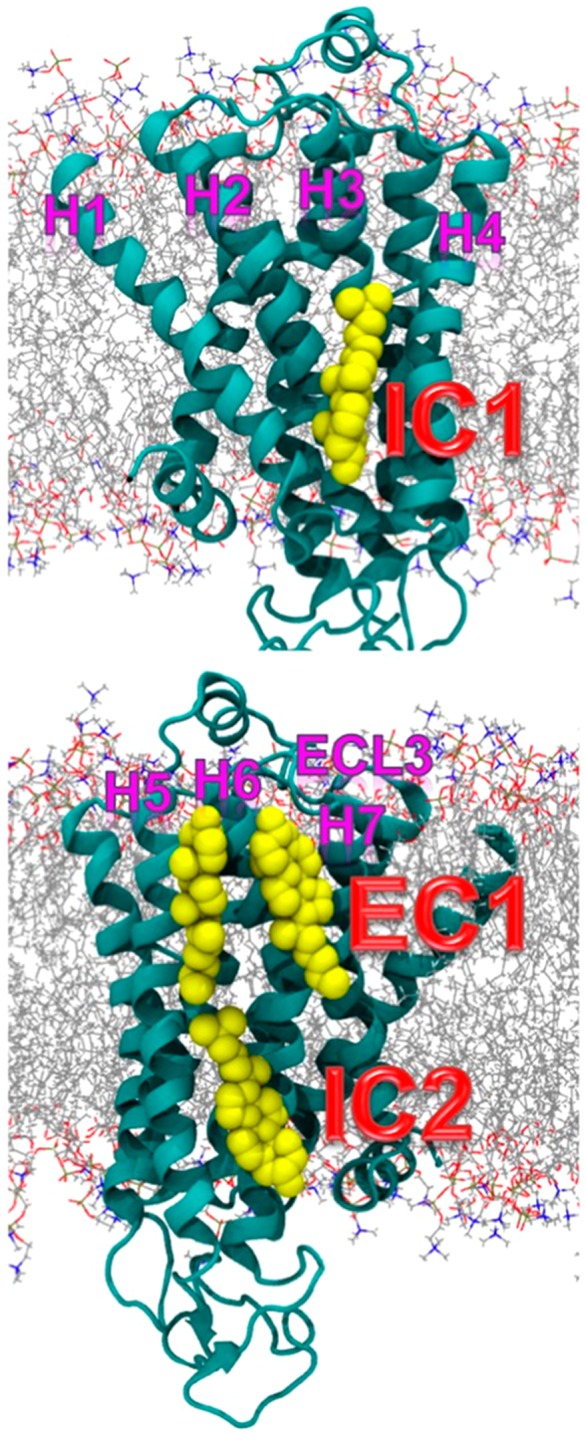
Schematic representation of three main cholesterol interaction sites in β2AR. Adapted with permission from ref (54). Copyright 2016 Manna et al. (https://creativecommons.org/licenses/by/4.0/legalcode).
Genheden et al.1037 investigated the deep cholesterol binding sites on the surface of β2AR and A2A receptors. The coarse-grained MD simulations detected a number of deep cholesterol binding sites on both receptors. The authors showed that the requirements for cholesterol binding are modest: just a potential hydrogen bond partner close to a cleft or hole on the surface.
Yuan et al.1038 studied the activation of the sphingosine 1-phosphate receptor 1 (S1P1) using all-atom MD simulations. The study reported five 700 ns long MD simulations of S1P1 either in apo form or bound to antagonist ML056 or natural agonist sphingosine 1-phosphate (S1P). Detailed analysis of the simulations showed that, upon S1P binding, lysophospholipid triggered movements of the intracellular ends of some of the transmembrane helices. The activation mechanism then proceeded in four stages (see Figure 46). First, it involved the binding of S1P to its binding site, which led to the flipping of W2696.48 (step 1). This was followed by alterations in the conformation of the side chain of F2656.44 located next to W2696.48 (step 2), and rearrangement of the centrally situated residues including N631.50, D912.50, S3047.46, and N3077.49, which facilitated the redirected flow of water molecules inside the receptor (step 3). Finally, one observed the influx of water molecules at the intracellular part of the receptor, leading to limited motions of the cytoplasmic ends of the transmembrane helices (step 4). These movements led to the opening of the protein structure, subsequently allowing G protein binding.
Figure 46.
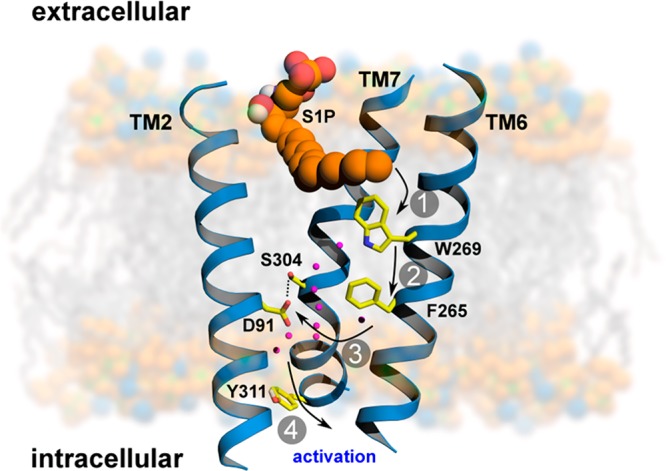
Activation mechanism of S1P1. Steps 1 and 2, binding of agonist (S1P). Step 3, rearrangement of centrally located residues facilitate the redirected flow of water molecules inside a receptor. Step 4, motion of cytoplasmic parts of transmembrane helices. Reproduced with permission from ref (1038). Copyright 2013 Yuan et al. (https://creativecommons.org/licenses/by/4.0/legalcode).
Hurst et al.1026 studied the interactions of the cannabinoid CB2 receptor with sn-2-arachidonoylglycerol (2-AG) using atomistic MD simulations. The results of this study suggest that 2-AG (which contains the polyunsaturated arachidonic acid chain (20:4, n-6)) can enter CB2 at the interface of the transmembrane helices 6 and 7, causing the formation of an opening between the helices. This association with the interface of the transmembrane helices 6 and 7 appeared to be very specific: even when 38 2-AGs were placed all around CB2, partitioning occurred only at the interface of the transmembrane helices 6 and 7.
7.2.2. Channels
Pentameric ligand-gated ion channels constitute a family of integral membrane proteins whose permeability to specific ions is increased upon agonist binding. As their name suggests, pentameric LGICs consist of five subunits that are arranged in the (pseudo) C5 symmetry around a central pore. The extracellular domain has a β-sandwich immunoglobulin-like structure, while the transmembrane domain is composed of four helices (M1 to M4) that span the membrane (see Figure 47). Relatively few computational studies have investigated interactions between pentameric ligand-gated ion channels and cholesterol. Baier et al.1039 used docking and short MD simulations to identify cholesterol-binding sites on the surface of the nicotinic acetylcholine receptor (nAChR). Three cholesterol molecules could be docked on the transmembrane segments of each nAChR subunit, rendering a total of 15 cholesterol molecules per nAChR. Cheng et al.1040 investigated interactions of nAChRs with membrane cholesterol and anionic lipids (POPA) in atomistic MD simulations that lasted over 10 ns. The penetration of the intersubunit cavities by phosphatidic acid acyl chains was observed.
Figure 47.
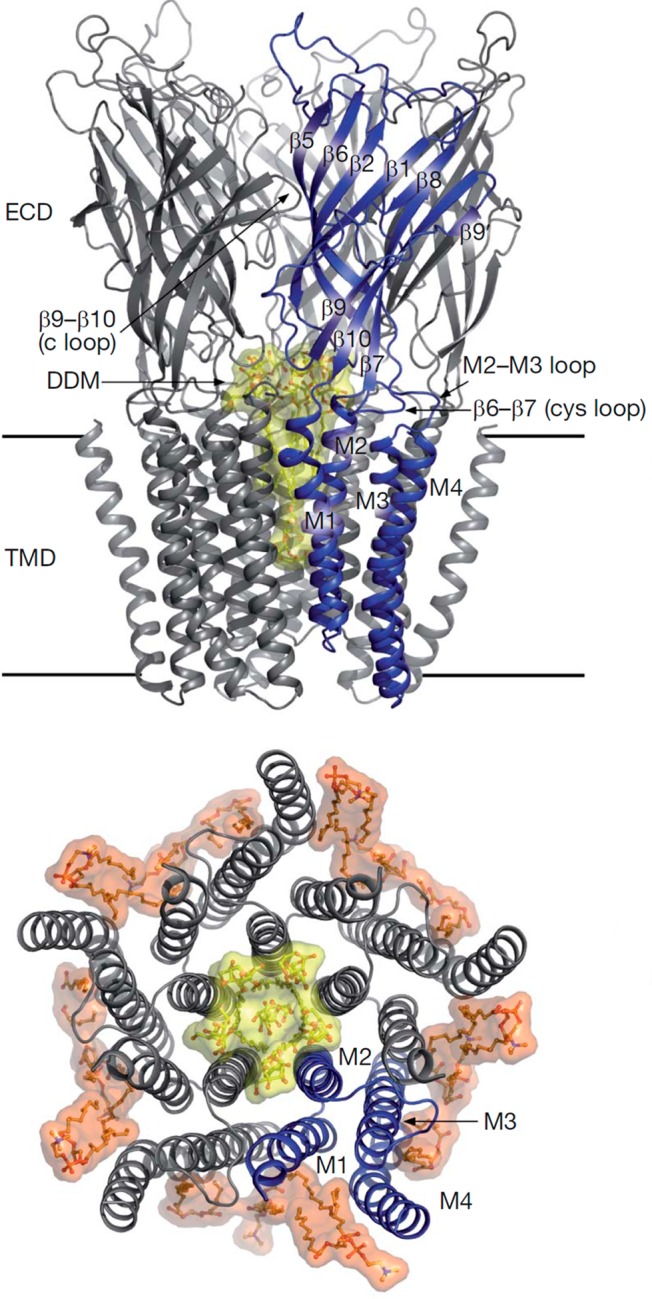
Schematic representation of GLIC. (top) GLIC viewed from the plane of the membrane. (bottom) Transmembrane part of GLIC viewed from the extracellular side. Lipids are depicted in orange. Adapted with permission from ref (1041). Copyright 2008 Springer Nature.
More recently, Hénin et al.783 focused on γ-aminobutyric acid receptor type A (GABA(A)) and used docking to identify five cholesterol-binding sites. Subsequent atomistic MD simulations were performed starting from the configuration where sterols were bound in all of these sites. Over the course of 200 ns long simulations, three cholesterol-binding sites remained occupied, though cholesterol molecules underwent some reorientation. Cholesterol molecules, bound in the remaining two binding sites predicted by docking, were observed to exchange, unbind, and spontaneously rebind.
In a study by Singh et al.,1042 the structural basis for cholesterol inhibition of large conductance Ca2+ and voltage-gated K+ channels, also known as “big potassium” (BK) channels, was investigated in the BK channel-forming protein Cbv1 by a combination of MD simulations, site-directed mutagenesis, and single-channel electrophysiology. The study showed cholesterol action to be mediated by the cytosolic C tail domain, in which seven CRAC motifs were present. MD simulations identified the interactions involved in the ion channel–cholesterol recognition. Simulations of four versions of the cytosolic C tail domain with cholesterol placed in the vicinity of the CRAC motif were further conducted. The study concluded that the sensitivity of the BK channel for cholesterol arises from the membrane-adjacent CRAC4 motif, in which a specific sequence of amino acid residues senses cholesterol. However, cumulative truncations or amino acid substitutions in other CRAC motifs progressively blunted cholesterol sensitivity, documenting the involvement of multiple CRACs in the cholesterol–BK channel interaction.
Rosenhouse-Dantsker et al.1043 used a combination of molecular docking, MD simulations, and mutagenesis studies to determine putative cholesterol binding sites for the inwardly rectifying potassium (Kir2.1) channel (see Figure 48). Based on clustering of the results from cholesterol docking, six possible binding sites were suggested, and 50 ns MD simulations were performed for each of the binding modes. During the simulations, five of the initially proposed sites merged into two distinct binding regions located between the adjacent subunits of the channel, while a sixth position led to the dissociation of cholesterol from the protein surface (see Figure 48). The location of cholesterol-binding regions suggested that cholesterol modulates the channel function by affecting the pore-lining transmembrane helix that is responsible for the channel gating either directly or through the interface between the N- and C-termini of the channel. The study highlights the importance of combining docking with MD simulations, rather than relying on the docking results alone.
Figure 48.

Cholesterol binding to the Kir2.1 channel. (left) A ribbon representation of two adjacent subunits of Kir2.1 (pink and gray). (right) A schematic model showing the location of the two cholesterol-binding sites. Reproduced with permission from ref (1043). Copyright 2013 the American Society for Biochemistry and Molecular Biology.
Very recently, Duncan et al.606 reported large-scale coarse-grained MD simulations of Kir2.2 channels embedded in membranes of varying degree of lipid complexity and protein crowding. In this study, 144 copies of Kir2.2 were embedded in lipid membranes whose lipid composition was quite complex. The authors reported that in the simulations containing PIP2, headgroup interactions with Kir2.2 channels extended further from the transmembrane domain onto the cytoplasmic domain of the channel as compared to a PIP2-deficient system. The radial distribution functions indicated that in membranes with PIP2, the principal lipids surrounding the Kir2.2 channels were PIP2 in the cytoplasmic leaflet and GM3 in the outer leaflet. In the inner leaflet, phosphatidylserine was the second most popular lipid in the annular shell around Kir2.2. In the absence of PIP2, the abundance of phosphatidylserine at the protein surface was increased, but not to the extent of PIP2 in the PIP2-containing simulation. The study highlighted the preference of Kir2.2 to interact with PIP2 in the inner leaflet and with GM3 in the outer leaflet, as has also been shown for the EGF receptor.1044 Further, the study suggested that the oligomerization of Kir2.2 channels is affected by the lipid environment: in a pure PC bilayer, Kir2.2 oligomers appeared to be bigger and less branched as compared to those in a membrane whose lipid composition was considerably more complex.
Schmidt et al.1045 also studied PIP2 binding to Kir2.2 channels using multiscale MD simulations. The method was based on coarse-grained simulations to predict the initial PIP2 binding events that were then refined by subsequent atomistic simulations. The simulations were then compared to the crystal structures of Kir2.2 with either PIP2 (PDB entry 3SPI) or phosphatidic acid (PDB entry 3SPC) to validate the computational predictions. The multiscale simulations that used the protein coordinates from 3SPI as the starting structure predicted the PIP2 headgroup to attach to the same cluster of basic residues that was observed also in the crystal structure. Similarly, when the ligand-free 3SPC structure was used as the starting point for the multiscale simulations of PIP2 binding, there was good agreement between the multiscale simulations and the crystal structure.
O’Connor and Klauda carried out MD simulations of an aquaporin-0 tetramer in a pure DMPC bilayer and a mixed DMPC/cholesterol bilayer (1:1 ratio)1046 to investigate how aquaporin-0 has evolved to prefer membranes with high levels of cholesterol (aquaporin-0 has been found in ocular lens fiber membranes in high concentrations). During 100 ns simulations, aquaporin-0 remained stable both in the presence and in the absence of cholesterol. However, in the presence of cholesterol, larger hydrogen bond occupancies and longer hydrogen bond lifetimes were observed between aquaporin-0 and the surrounding lipids as compared to a cholesterol-free membrane, indicating that the presence of cholesterol modulates the dynamical properties of the channel. Interestingly, several aromatic residues on the surface of the protein were identified to form stable interactions with cholesterol. These observations suggest that the membrane-exposed parts of aquaporin-0 have evolved to favor cholesterol-rich membranes.
Finally, there is reason to mention the relevance of mechanosensitive channels as, for example, sensors for osmotic homeostasis. Recent simulation work in this context has been quite limited, though, so it is not discussed here in considerable length. Interested readers are referred to refs (764, 1047, and 1048).
7.2.3. Other Membrane Proteins
Specific lipid binding sites on the surface of membrane proteins and the role of lipids in modulating protein activation have been addressed in a number of MD studies not described in detail in this review. These include lipid binding to sarco- and endoplasmic reticulum Ca2+-ATPases,807 receptor tyrosine kinases,919,1044,1049−1053 capsaicin receptor TRPV1,1054 nAChR,1055−1057 voltage-sensitive potassium channel Kv7.1,1058 UraA H+-uracil symporter,1059 voltage-gated potassium channel,773 neurotensin receptor 1,1060 and integrin.1061,1062
Concluding, extensive multiscale MD simulations enable us to characterize the interactions of membrane proteins with lipids, thereby extending and complementing the information obtained from experimental studies. It is now possible to identify the lipid binding sites on integral proteins and, in part, to identify specific lipid compositions needed to activate membrane proteins or maintain their function. MD simulations can also provide atomistic details of protein selectivity, and they can be used to explore how multiple ligands compete. In crowded and complex environments similar to those studied in vivo, simulations of large ensembles of proteins can offer valuable insights into the actual effects of lipid–protein interactions in multicomponent membranes.
7.3. Structural Modifications of Proteins Modulate Their Interactions with Biomembranes
In living cells, proteins generate diverse functions related to immune defense, catalysis, and transport. The human gene set has been estimated to include ∼25 000 genes.1063 At the same time, the human proteome is considered to be much larger, including more than 1 million proteins.1064 Protein diversity stems from not only the alternative splicing of the mRNA but also post-translational and other structural modifications of proteins. These modifications increase the variety of possible protein structures and functions through chemical moieties covalently attached to the amino acid residues. Over 200 different types of, for example, post-translational modifications have been identified as functionally active. These modifications include in particular phosphorylation, glycosylation, methylation, acetylation, and amidation; see UniProt1065 for a more detailed list of post-translational modifications. They modulate many aspects of cellular physiology, such as metabolism, immunity, signal transduction, and protein stability.1066−1070
A relatively small number of computational studies has investigated the role of post-translational modifications on the structure and function of the proteins, and only a few studies have tried to elucidate the role of post-translational modifications in lipid–protein interactions.
Fonseca-Maldonado et al.1071 used mutagenesis and MD simulations (GROMOS96 force field) to study the effect of glycan content and glycosylation position on the stability of xylanase A. The majority of glycosylated mutants presented there increased thermostability as compared to the deglycosylated counterparts. The study showed that the position of glycosylation rather than the number of glycosylation events determined the increase in thermostability. MD simulations also indicated that the clustered glycan chains tended to favor less stabilizing glycan–glycan interactions, whereas the more dispersed glycosylation patterns preferred stabilizing protein–glycan interactions.
Lu et al.1072 used Langevin dynamics simulations on a coarse-grained off-lattice 46-β barrel model protein that was glycosylated by glycans with different hydrophobicities and glycosylation sites to examine the effect of glycans on protein folding and stability. The glycan portion of the glycosylated protein contained 14 beads that had tunable hydrophobicity. For relatively hydrophilic glycans introduced at flexible peptide residues, conformational stability and folding was observed to increase. Meanwhile, when glycans were introduced at conformationally restricted peptide residues, only those glycans that were hydrophilic or very weakly hydrophobic contributed to increasing protein stability; however they also hindered protein folding due to increasing free energy barriers between the folded and unfolded states.
Subedi et al.1073 studied the extracellular domain of human CD16A (Fc γ receptor IIIa) that contains five N-glycosylation sites. They employed NMR experiments and all-atom MD simulations to identify the glycosylation site that contributed to antibody binding. A preliminary 850 ns simulation of the extracellular domain of CD16A revealed that based on the analysis of root-mean-square fluctuations (Figure 49) the N45 N-glycan experienced less motion than other N-glycans at the N38, N74, N162, and N169 positions. The GlcNAc residue in the N45 position established two critical interactions with E68 and D64 that were maintained for the duration of the 1 μs production simulation. The importance of these contacts was verified in a separate 1 μs all-atom simulation of deglycosylated CD16A, where the authors observed that the N45 N-glycan enhanced the expression of CD16A. The CD16A mutants without the N45 N-glycans were shown to express with a meager yield.
Figure 49.
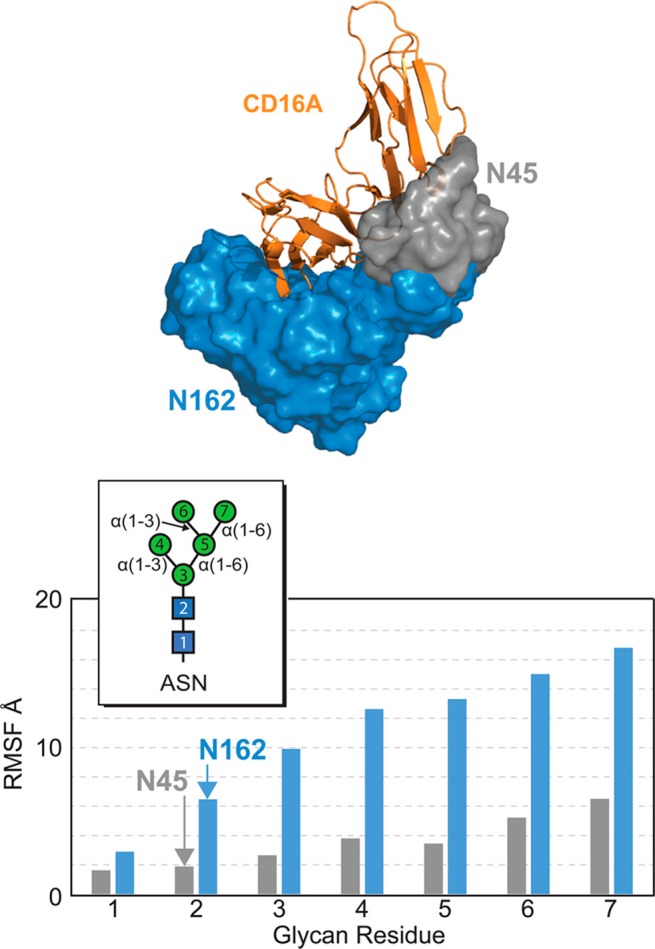
All-atom molecular dynamics simulation of N-glycosylated CD16A. The N162 N-glycan samples a large space (blue cloud) unlike the N45 N-glycan with restricted mobility (gray cloud). Reproduced with permission from ref (1073). Copyright 2017 American Chemical Society.
Recently, Kaszuba et al.919 performed extensive atomistic MD simulations of the monomeric N-glycosylated human epidermal growth factor receptor (EGFR) in biomimetic lipid bilayers. The receptor was embedded in a membrane composed of a ternary lipid mixture of DOPC, SSM, and cholesterol. This plasma membrane composition was designed to mimic, as tightly as possible, the experimental conditions used for biochemical reconstitution of EGFR in proteoliposomes. MD simulations showed that the presence of glycan residues (Man3GlcNAc2) significantly altered the relative arrangement of the individual receptor subdomains and their alignment on the lipid membrane. The glycan moieties were found to function as molecular buffers, lifting several domains of EGFR away from the plane of the lipid bilayer and rearranging them with respect to each other (Figure 50). The elevation of the EGFR structure above the membrane surface was particularly substantial in the case of subdomains DI and DIII (located in the ectodomain of the receptor), which together contribute to the ligand binding. The study reported that only the N-glycosylated receptor adopted a conformation that was in good agreement with previous experimental Förster resonance energy transfer (FRET) studies regarding the distance between the EGFR ectodomain and the lipid membrane.1074,1075 In essence, the study by Kaszuba et al. provided compelling evidence that glycosylation can play a critical role in ligand binding and thus membrane receptor activation.
Figure 50.

Snapshots from the MD simulation (1000 ns) of the glycosylated (left) and nonglycosylated (right) EGFR in DOPC/SM/cholesterol membranes. The receptor structure is color-coded throughout the panels as follows: TM domain (orange); subdomains DI (blue), DII (green), DIII (yellow), and DIV (red); intracellular TKD (salmon); glycans (purple). Adapted with permission from ref (919). Copyright 2015 Kaszuba et al.
The effect of glycosylation and lipid–protein interactions was also studied by Polley et al.1076 using atomistic MD simulations. The authors focused on the membrane-bound CD2 protein, which is a small cell adhesion receptor expressed by human T-cells and natural killer cells. The receptor was embedded in big patches of a multicomponent liquid-ordered membrane composed of sphingomyelin (d16:1/16:0), DOPC, and cholesterol. As a control system, one used a single-component DOPC bilayer in the liquid-disordered phase. The study reported that the orientation of the ectodomain of CD2 depended critically on CD2 glycosylation as well as local lipid composition (see Figure 51). In the liquid-disordered bilayer without glycosylation, the ectodomain lay down on the membrane surface. For the same liquid-disordered bilayer with glycosylation, the ectodomain of CD2 was positioned more upright, indicating that the presence of glycans plays a role in CD2 orientation. Without glycosylation in the liquid-ordered system, the ectodomain of CD2 fluctuated around an upright position; however, the most significant change was observed when the liquid-ordered bilayer hosted glycosylated CD2, where the protein assumed a constitutively upright position. Detailed analysis of MD results showed that the tilting of the membrane protein ectodomain was driven by electrostatic interactions between CD2 and lipids around it. The tilt of the CD2 ectodomain largely disappeared when the positively charged amino acids, which interacted with the DOPC headgroups, were deprotonated. The effect of glycosylation on the CD2 orientation appeared to be due to steric effects; glycans in the ectodomain of CD2 acted as a steric barrier, preventing association with the membrane surface. The influence of these glycans was enhanced in the liquid-ordered membrane environment, presumably due to mechanical effects such as the cholesterol-induced increase in order and reduction of membrane elasticity. Altogether, the study by Polley et al. demonstrated how glycosylation and lipid–protein interactions can influence receptor conformation and visibility for the receptor’s ligands in a concerted fashion.
Figure 51.

Simulation snapshots of nonglycosylated CD2 in (top, left) Ld and (top, right) Lo bilayers and of glycosylated CD2 in (bottom, left) Ld and (bottom, right) Lo bilayers. Color code: DOPC (orange), cholesterol (yellow), SM (cyan), CD2 domain 1 (dark blue), CD2 domain 2 (red), CD2 transmembrane helix (black), and lipid carbohydrate chains (light blue). The glycosylated protein structure was prepared with the DoGlycans tool.1077 Reproduced with permission from ref (1076). Copyright 2017 American Chemical Society (https://creativecommons.org/licenses/by/4.0/legalcode).
Lakkaraju et al.1078 employed fluorescence microscopy, radiolabeling, and atomistic MD simulations to study the role of palmitoylation in calnexin localization. Calnexin is a major endoplasmic reticulum chaperone involved in glycoprotein folding. Its palmitoylation leads to preferential localization of calnexin to the perinuclear rough endoplasmic reticulum, at the expense of endoplasmic reticulum tubules. Using MD simulations, the authors analyzed whether the presence of palmitoylation modifies the structure of the transmembrane domain or the cytosolic tail of calnexin. It appeared that palmitoylation did not affect the proline-induced kink and only marginally affected the calnexin tilt. The MD simulations, however, predicted a palmitoylation-dependent orientation of the cytosolic tail with respect to the helix axis. These simulations, combined with the asymmetric nature of calnexin transmembrane domain, revealed the exciting possibility that the presence of palmitoyl chain may affect the conformation of calnexin, which, in turn, could alter its affinity for membrane domains or its ability to interact with proteins in the membrane or the cytoplasm.
Thukral et al.1079 used coarse-grained MD simulations to unravel the molecular mechanism underlying recruitment and insertion of lipid-anchored microtubule-associated protein light chain 3 (LC3) into lipid membranes. LC3 is a crucial protein required during autophagy and the authors studied one of its unique covalent modifications. The cytosolic form of LC3 was reversibly linked to phosphatidylethanolamine, resulting in lipid-modified LC3 that associates with the autophagosome membrane. Fifteen independent simulations were conducted starting from a structure of LC3-PE placed about 8 nm from the center of a bilayer composed of POPC. Spontaneous insertion of the hydrophobic anchor of the protein into the membrane was observed in 14 of 15 trajectories. In these 14 cases, once the PE was inserted into the hydrophobic core, the lipid–protein interactions became stable, and no dissociation events were observed. In most of the cases, the insertion occurred within 4 μs of the simulation. The initial step of the protein insertion into the lipid bilayer involved protein docking to the surface of the membrane. After the protein was bound entirely, a subsequent stepwise insertion of the acyl chains progressed in a concerted manner, with the two acyl chains inserting themselves to the membrane one after the other. Finally, the lipid anchor of the protein was completely buried in the membrane along with the acyl chain of POPC lipids (Figure 52). Based on several spontaneous insertion events and in vitro experimental studies, the authors were able to identify the critical residues facilitating the LC3 membrane-targeting process, namely, the basic residue patch consisting of K65, R68, and R69 in the α-helix III region.
Figure 52.

Schematic snapshots showing the mutated residues at the pre- and postinsertion states of the LC3 protein. Adapted with permission from ref (1079). Copyright 2015 Biophysical Society. Published by Elsevier Inc.
The atomic details of the association of K-Ras with POPC/POPG (4:1 molar ratio) bilayers1080 have also been studied extensively using all-atom MD simulations. K-Ras is farnesylated at the C-terminal cysteine and has a stretch of eight lysine residues immediately upstream of the farnesylated cysteine. Using MD simulations, Janosi and Gorfe showed that K-Ras was tethered to the lipid bilayer surface by specific lysine–POPG salt bridges and by nonspecific farnesyl–phospholipid van der Waals interactions. Unexpectedly, however, only five of the eight K-Ras lysine residues were found to interact with the lipid bilayer directly. Interestingly, the positively charged K-Ras anchors pulled the negatively charged POPG lipids together, leading to the clustering of the POPG lipids around the proteins. A subsequent study by Prakash et al.1081 showed that GTP-bound K-Ras also interacted with a negatively charged POPC/POPS membrane and that this interaction involved multiple distinct orientations of the protein. Among these, two highly populated states accounted for ∼54% of the conformers whose catalytic domain interacted directly with the bilayer. In one of these states, membrane binding involved helices 3 and 4 of the catalytic domain in addition to the farnesyl tag and poly(lysine) motif. In the other orientation, β-strands 1–3 and helix 2 on the opposite face of the catalytic domain contributed to the membrane binding.
Edler et al.1082 employed atomistic and coarse-grained MD simulations of the truncated C-terminal hypervariable region (HVR206−215) of Rab5 in three model membranes of increasing complexity (a pure POPC bilayer, a ternary membrane with cholesterol and palmitoyl-sphingomyelin, and a six-component early endosome model membrane composed of POPC, cholesterol, palmitoyl-sphingomyelin, POPE, POPS, and phosphatidylinositol 3-phosphate (PI(3)P)). Rab5 is a crucial regulator of endosomal trafficking processes and a marker for the early endosomes. HVR206−215 of Rab5 was post-translationally modified at two cysteine residues to accommodate two geranylgeranyl anchors (C20 carbon chain length), thus associating Rab5 with the membrane. The study reported that the two adjacent geranylgeranyl chains in one HVR206−215 moiety showed anticorrelated motions. That is, if one chain was deeply inserted into the bilayer, the other was allowed to bend toward the membrane surface. The average insertion depth of the geranylgeranyl chains depended on the thickness of the surrounding membrane and was always 38–39% of the bilayer thickness. This indicates that the lipid anchor did not penetrate the central region of the lipid bilayer, which is in agreement with 2H solid-state NMR experiments showing that the anchors adapt their insertion depths to the surrounding lipids.1083
Zhang et al.1084 used all-atom MD simulations to study the behavior of palmitoyl and farnesyl anchors in raft-like and non-raft-like lipid membranes. A peptide derived from helix VIII (residues 316–328) of rhodopsin was selected as a model peptide to bear farnesyl or palmitoyl anchors. The lipid model membranes were composed of DSPC, POPE, and cholesterol in a 2:1:1 molar ratio to mimic the inner leaflet of functional membrane domains in living cells. Detailed analysis of the simulations showed that the palmitoyl anchors behaved in a more ordered fashion, were packed tighter with the lipids in the raft-like membrane, and diffused at a slower rate than farnesyl anchors. Authors reported that the palmitoyl chains preferred to associate with the saturated chains of lipids and cholesterol molecules, while farnesyl chains were more prone to association with the saturated and unsaturated chains. For non-raft-like membranes, these two lipid anchors had roughly the same preference for all lipids. Additionally, the presence of the palmitoyl anchors caused cholesterol to orient itself in more perpendicular manner to the membrane surface, ordered the lipids next to the anchor, and reduced lateral lipid motion significantly, compared to farnesyl anchors. On the other hand, POPE and DSPC became much less ordered, cholesterol became more tilted, and the bilayer became more fluid when the two types of lipid anchors were inserted in non-raft-like membranes.
The above examples clearly demonstrate the emerging crucial role of MD simulations in unraveling the role of post-translational protein modifications in lipid–protein interactions and protein function. MD simulations enable us to identify the atomistic mechanisms underlying protein recruitment into specific lipid bilayers, which, in turn, allow us to understand the function of integral proteins in critical cellular processes.
7.4. Challenges
It is now established that cell membranes are not merely elastic sheets hosting membrane and peripheral proteins to carry out their function, but the lipid membranes themselves actively participate in cellular physiology, including, for example, metabolism, immunity, and signal transduction. Given this, cells use lipid membrane composition and its lateral and interleaflet asymmetry as tools to modulate and control biological processes where biological membranes are involved. Understanding how membrane lipids control protein conformation and activation is therefore crucial to comprehend how plasma membranes work, and to clarify how, for example, changes in membrane lipid content modulated by disease and diet influence membrane protein activation.
As to the topics discussed in this section, it is obvious that there are challenges, which need to be addressed in the foreseeable future. While there are thousands of simulation articles published on membrane proteins, only a few of them have elucidated the importance of post-translational and other structural modifications, such as glycosylation. Yet experiments show very clearly that they can play a decisive role for ligand binding and therefore activation. It is hard to understand why glycosylation would not be included in a simulation model if the protein in question is known to be glycosylated in native conditions. Moving on, it is generally considered that lipid translocation is one of the means used by lipids in cellular signaling. In translocation of phosphatidylserine, the biological context is understood (apoptosis), but what is the signaling pathway associated with the translocation of other lipid types? These and the many other challenging questions remain to be addressed.
As this section has demonstrated, MD simulations can be extremely valuable to better understand the molecular-scale interplay between membrane proteins and their lipid environment. Improving quality of the simulation models, increasing computing capacity together with specialized simulation engines, and the continuously increasing extent and accuracy of membrane protein structures will strengthen the added value given by simulations even further.
8. How Lipids Influence Membrane Protein Activation and Function?
The structure and function of membrane proteins are firmly connected to their lipid environment.926 Due to the lateral and interleaflet heterogeneity of cell membranes, proteins are sorted between different lipid domains and recruited to specific locations as they form functional complexes.144,1085,1086 This compartmentalization is extremely important for many cellular processes, such as signaling,1087 membrane fusion,1088 and protein trafficking.1089 The post-translational pathway of membrane-protein trafficking to the plasma membrane involves the transport of proteins between different membrane compartments. Proteins are modified in these membrane compartments (such as, endoplasmic reticulum or Golgi apparatus) usually through post-translational modifications by the linkage of complex oligosaccharide chains. Intracellular transport is often mediated by vesicular intermediates that bud from one cellular compartment and fuse with the next one. Lipids are present at almost every step of protein trafficking and have been shown to modulate the protein activation and function. For example, phosphatidylinositols have been demonstrated to control many cellular events, such as the organization and dynamics of critical signaling pathways, the actin cytoskeleton rearrangement, and the intracellular vesicle trafficking.1090−1092
In this section, we discuss how lipids influence the function and activation of membrane proteins. First, we highlight some recent computational research that has allowed one to better understand the effects of annular lipids in protein aggregation and signaling. We review recent advances in computational studies on lipid-mediated protein sorting and compartmentalization. We show how computer simulations can aid in the elucidation of the role of lipids in recruiting proteins into specific lipid nanodomains or membrane phases.
Next, we focus on the allosteric binding of lipids and its effect on the structure and dynamics of membrane receptors. We show how cholesterol and charged lipids can inhibit ion channel function and alter the dynamics of G protein-coupled receptors.
In the second part of this section, we move on to lipid-mediated interactions between biological membranes and drug molecules. Here, we focus our attention on recent MD simulation studies that have explored the interactions of anesthetics and neurotransmitters with biological membranes.
We close this section by reviewing how phosphatidylinositols and other charged lipids modulate the binding of peripheral proteins. We show that many of these proteins interact electrostatically with negatively charged lipids. It turns out that this interaction can play different roles given that anionic lipids not only attract peripheral proteins toward the membrane surface but also can dictate the reorientation of proteins on the bilayer surface.
8.1. Lipids Form a Lipid Corona around Membrane Proteins
The number of lipid molecules surrounding the membrane-spanning domain has been deduced for several proteins from EPR spin labeling studies, which quantify the association of immobilized lipids with membrane proteins.1093 Interestingly, an almost complete annular lipid shell was resolved for the trimeric bacteriorhodopsin. Based on this structure, 24 lipids were calculated to cover the membrane-spanning region of the trimer, and six additional lipid molecules were needed to fill the central pore.270,1094 However, most of the membrane protein structures that have been resolved contain only a few lipid molecules (see Table 2), which belong to the first annular shell. This may be the result of exhaustive purification procedures in the presence of detergents that are necessary to obtain purified material for crystallization. The annular lipids mediate interactions between the membrane protein and the bulk lipid bilayer and are essential for the vertical positioning of the protein in a bilayer, to flatten the protein surface that is often rough, and to integrate the protein tightly into the membrane. More importantly, an increasing number of studies have reported the dominant role of annular lipids in the stabilization of protein structures and the modulation of protein functions. Here, we therefore discuss how recent computational studies have unraveled questions dealing with the functional role of annular lipids.
Schmidt et al.1095 employed biophysical, biochemical, and computational techniques to quantify the influence of anionic versus zwitterionic lipids on the stability of the integrin αIIbβ3 transmembrane complex. The study showed that the headgroup of anionic lipids competed for electrostatic interactions within the integrin αIIbβ3 transmembrane complex, but the net stability of its critical internal electrostatic interaction was not compromised, and an overall transmembrane complex stabilization was induced. The observed stabilization in the presence of anionic lipids, the magnitude being up to 0.5 kcal/mol, is a significant contribution to the threshold of the integrin αIIbβ3 receptor activation, which is 1.5 ± 0.2 kcal/mol in a POPC bilayer. It is worth mentioning that the integrin αIIbβ3 does not possess a high-affinity anionic lipid-binding site, which means that the annular lipids were fully responsible for the observed effects. The results suggest that anionic lipids enhanced the membrane protein stability by merely stabilizing the transmembrane helix–helix association.
The role of interfacial lipids in stabilizing membrane protein oligomers was explored by Gupta et al.828 using mass spectroscopy combined with biochemical and computational techniques. Several transmembrane proteins with low oligomeric stability, as determined experimentally, were embedded into complex lipid bilayers containing cardiolipin, POPG, POPE, and POPC. The coarse-grained simulations revealed that cardiolipin acted as a “bidentate” ligand, bridging the subunits of LeuT protein together and shifting the equilibrium from a monomer to a functional dimer for the Vibrio splendidus sugar transporter SemiSWEET. They also showed that lipid binding was obligatory for the dimerization of NhaA (Na+/H+ antiporter). The results suggest that interfacial lipids modulate the functions of these proteins since, for both SemiSWEET and NhaA, the existence of a stable dimeric state is considered to be critical for their mechanistic pathways.785
Eggensperger et al.1096 used all-atom MD simulations to study the role of the annular lipid belt in allosteric modulation and inhibition of the antigen translocation complex (TAP). Computer simulations started from the 96-lipid POPE/POPG (in 3:1 molar ratio) or POPC/POPG (in 3:1 molar ratio) nanodisc that hosted TAP. Subsequently, 74 lipids were removed in seven consecutive steps leading to a 22-lipid nanodisc with TAP. Simulations showed that 22 lipids were sufficient to form the annular belt around the TAP complex. Interestingly, this lipid belt was shown to be essential for high-affinity interactions and inhibition of TAP by viral inhibitors.
An ABC transporter, TmrAB, was studied using mass spectroscopy and all-atom MD simulations by Bechara et al.843 who identified and monitored the composition of a part of the annular lipid belt bound to a heterodimeric membrane protein complex. Twenty-four different phospholipid species were detected together with various tetra- and hepta-acylated lipid A species in four independent protein preparations. Interestingly, most of the phospholipids were found to be unsaturated. MD simulations of model structures of TmrAB explored in pure POPE and POPC bilayers revealed that about 20 phospholipid headgroups surrounded the protein and formed an annular belt around the protein (Figure 53). Mass spectroscopy showed that a subset of annular lipids was invariant in composition, with negatively charged lipids binding tightly to TmrAB. The authors hypothesized that the lipid belt is vital for the ATPase activity of TmrAB, which is in line with previous experimental observations that phospholipids stimulate and stabilize the ATPase activity of several different ABC transporters.
Figure 53.

MD simulations of inward and outward models of TmrAB in a lipid bilayer. Models of TmrAB inward (left) and outward (right), based on structures of Sav1866 (PDB ID 2HYD) and TM287/288 (PDB ID 3QF4). Reproduced with permission from ref (843). Copyright 2015 Springer Nature.
All-atom and coarse-grained simulations were employed by Guixà-González et al.57 to study the mechanism by which polyunsaturated lipids modulate the aggregation of GPCRs. They performed extensive MD simulations of the self-assembly process of A2A adenosine and D2 dopamine receptors simultaneously embedded in multicomponent lipid bilayers, reaching an exceptionally long simulation time of nearly 4 ms. The simulations uncovered that the kinetics of GPCR aggregation is modulated by the membrane docosahexaenoic acid (DHA, 22:6ω3) level: low DHA (as observed in many pathological conditions) substantially decreased the receptors’ ability to oligomerize (see Figure 54). The authors observed the formation of a lipid shell around the receptors, and this shell was found to be mostly populated by phospholipids with DHA chains. Interestingly, the assembly of the DHA shell around the receptors is in agreement with both experiments1097 and simulations,1020 showing aggregation of DHA around rhodopsin. The paper therefore suggested that the DHA solvation shell could play two critical roles in modulation of GPCR oligomerization: (a) receptors covered by DHA will partition into DHA-rich domains, which reduces the effective sampling area of receptors in the bilayer and increases the number of receptor–receptor encounters, therefore speeding up the oligomerization process; (b) the presence of the DHA shell enhances the ability of receptors to engage in protein–protein contacts, which, in turn, increases the receptor’s effective oligomerization radius.
Figure 54.

GPCR aggregation in high and low DHA membranes. Snapshots of (left) healthy (high DHA) and (right) disease-like (low DHA) systems after 60 μs of CG-MD simulation. Reproduced with permission from ref (57). Copyright 2016 Guixà-González et al. (https://creativecommons.org/licenses/by/4.0/legalcode).
Coarse-grained simulations of the sphingosine 1-phosphate receptor 1 (S1P1)56 in a model lipid bilayer provided further insight into the possible lipid modulation of GPCR oligomerization. Analysis of the trajectories showed that the main protein–lipid interactions present in the system were those with PIP2, cholesterol, and GM3. The intracellular side of the receptor contained basic residues, which formed frequent interactions with PIP2 headgroups. Given that the interactions between PIP2 and S1P1 were almost evenly distributed around the receptor surface, the negatively charged lipids seemed to form an annulus around the protein (see Figure 55). These strong annular interactions were found in all 144 S1P1 receptors for over 75% of the simulation time. Similarly, lipid-exposed hydrophobic residues of the receptor were observed to form interactions with cholesterol over the entire simulation time, and the annulus of cholesterol was also developed around the receptor (see Figure 55). Interestingly, the interaction between PIP2 and S1P1 was stable and specific, with the lipids remaining bound to the receptor throughout the whole simulation. In contrast, cholesterol interactions were more transient involving dissociation and association events (in a sub-microsecond time scale) throughout the simulation. The formation of dimers, trimers, and occasionally higher oligomers was observed. The detailed analysis suggested that cholesterol may mediate the protein–protein interactions.
Figure 55.
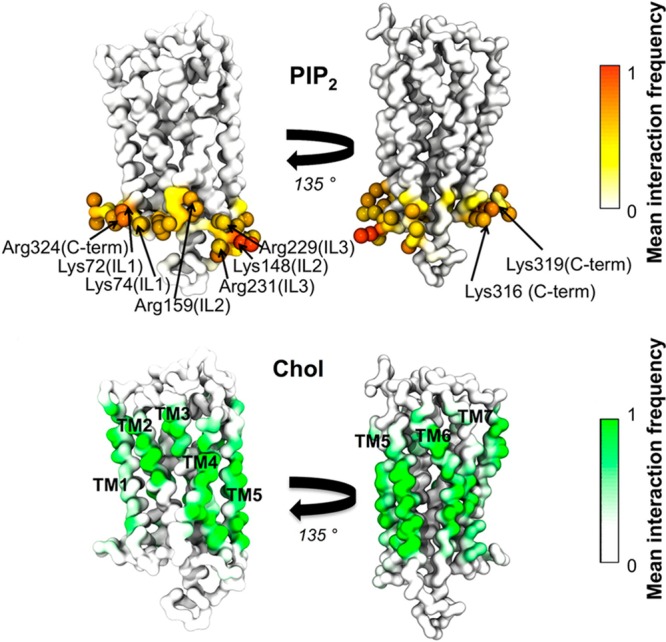
Protein–lipid interactions in the GPCR system. The color scale shows the mean fraction of time there is an interaction with all 144 repeats of the S1P1 receptor. (top) Interactions between the phosphoryl headgroup of PIP2 and the receptor. (bottom) Interactions between the cholesterol molecule and the S1P1 receptor. Reproduced with permission from ref (56). Copyright 2015 American Chemical Society.
The above studies demonstrate the functional roles of annular lipids in many cellular processes spanning from protein stability alternation, through receptor activation modulation, to membrane protein dimerization effectors. Due to the constant progress in computational techniques development, all these cellular functions of annular lipids can now be studied at the molecular level using MD simulations. In the future, MD-based studies may help in answering questions regarding the role of annular lipids in the plasma membrane, the formation of annular lipid belts, and their cellular function.
8.2. Lipid-Mediated Protein Sorting
Although both lipids and integral membrane proteins have been studied quite extensively, the molecular properties that dictate the specific interactions between them and the cellular functions that emerge from their interplay remain poorly understood. These include particular protein–protein and protein–lipid interactions, and indirect lipid-mediated interactions. Here, we discuss recent computational studies highlighting the importance of lipid-mediated protein sorting.
Coarse-grained MD simulations and confocal fluorescence microscopy were employed by Schäfer et al.1098 to investigate peptide sorting under different hydrophobic mismatch conditions. The simulations uncovered that model transmembrane helices, such as WALP peptides, do not like to reside in the liquid-ordered phase. Instead, they were observed to partition into the liquid-disordered phase spontaneously. The authors suggested that the lateral sorting process was governed by the collective motion of both lipids and peptides, which facilitated the trafficking of transmembrane helices by the transient formation of islands of unsaturated lipids disrupting the ordered domain. The driving force for the observed protein sorting was the enthalpic cost associated with the presence of a cylindrical object (the transmembrane helix) inside the ordered lipid phase. De Jong et al.1099 extended this study to investigate the role of lipid anchors in steering the sorting of membrane proteins. They considered the peripheral proteins N-Ras and H-Ras, which have a farnesyl anchor together with one or two palmitoyl anchors, respectively. The simulations showed that peripheral Ras proteins partition to the Lo or Ld phase depending on the lipid anchor type. Double-palmitoylated H-Ras preferred the Lo phase, whereas singly palmitoylated N-Ras resided at the phase boundaries. Interestingly, depalmitoylated H-Ras, with only a farnesyl anchor remaining, partitioned into the Ld phase. It is worth mentioning that the addition of GM1 decreased the preference of the transmembrane peptides toward the Ld phase even further. It was hypothesized that this indicates the essential role of GM1 in the recruitment of transmembrane proteins to membrane rafts.
Similarly, Li and Gorfe1100 used coarse-grained MD simulations to study the molecular mechanism by which H-Ras proteins form nanoclusters in model lipid membranes. Two different conformations of H-Ras, representing active and inactive states of the protein, were embedded into the lower leaflet of a mixed lipid bilayer composed of 3480 DPPC, 2304 DLiPC, and 1536 cholesterol molecules (corresponding to the ratio of 5:3:2). The authors reported that irrespective of the initial conformation, a single, large aggregate was formed at the end of the simulations and that the aggregation did not seem to require lipid domain formation. However, the two binding modes (see Figure 56), which were characterized by a different orientation of the G domain with respect to the membrane, differed in dynamics and organization during and after aggregation.
Figure 56.

Two binding modes of H-Ras. (left) Perpendicular orientation of the catalytic domain with respect to the membrane plane. (right) Semiparallel orientation of H-Ras with respect to the membrane plane. Reproduced with permission from ref (1100). Copyright 2013 Li and Gorfe. (https://creativecommons.org/licenses/by/4.0/legalcode)
Yoo and Cui studied gramicidin A (gA) dimer association with biological membranes1101 using coarse-grained MD simulations in combination with the potential of mean force and stress field calculations. Three lipid bilayers were studied to probe the effects of hydrophobic mismatch, with the degree of negative mismatch increasing along the sequence of DMPC, DPPC, and DSPC. It was observed that the association of gA dimers strongly depended on the degree of hydrophobic mismatch, the estimated binding free energy being >10 kcal/mol in a DSPC bilayer. Analysis of configuration entropy, lipid orientation, and stress distribution revealed that the annular lipids made a substantial contribution to the association of two gA dimers. The study highlights the importance of annular lipids and their energetic contributions in processes, where a significant reorganization of lipids near proteins is involved.
Parton et al.605 conducted coarse-grained MD simulations of hemagglutinin clusters in domain-forming bilayers to investigate the association of hemagglutinin with lipid rafts under a high local protein concentration. The simulations revealed an increase in the proportion of raft-type lipids (saturated phospholipids and cholesterol) in a membrane patch spanned by the protein cluster. Interestingly, the lateral diffusion of unsaturated lipids was significantly reduced within the cluster, while saturated lipids were unaffected. They hypothesized that steric crowding, resulting from the presence of slowly diffusing proteins, increased the chemical potential of unsaturated lipids within the cluster region. This led to the conclusion that the local aggregation of hemagglutinin was sufficient to drive the association of the protein with raft-type lipids.
More recently, coarse-grained MD simulations were used to investigate the interactions of the binding units of cholera toxin (CTB) with mixed GM1/DPPC membranes.1102 The simulations showed alteration of the lipid structure induced by CTB binding. Detailed analysis of the trajectories revealed that CTB bound explicitly to GM1 mostly via multivalent electrostatic interactions between the charged residues of CTB and the oligosaccharide headgroups of GM1. Interestingly, an entirely bound CTB could cross-link up to 15 GM1 molecules into the protein-binding pocket. The cross-linking gave rise to the formation of CTB-sized and less ordered nanodomains that were enriched in GM1. Such a protein-bound nanodomain resembled a lipid raft with an order that was distinct from the bulk lipids.
Computer simulations were utilized by Pezeshkian et al.1103,1104 to explore the clustering of the B subunit of the parental Shiga toxin from Shigella dysenteriae (STxB). Using a wide range of computational techniques, the authors found that the lipid-chain length mismatch and the compositional mismatch between the local membrane environment near the toxin particles and the bulk membrane were not required for STxB clustering. What is more, the simulations and experiments showed that reducing the rigidity of the bound STxB nanoparticles or displacing them from the membrane surface eliminated the clustering process. Strikingly, this was accompanied by a loss of STxB nanoparticle-mediated suppression of membrane fluctuations, strongly suggesting that the fluctuation-induced forces generated the attraction between the STxB nanoparticles that sufficed to facilitate the clustering. The paper hypothesized that this generic sorting mechanism should be operational between any sufficiently rigid nanoparticles that can bind tightly to a fluid phase lipid bilayer.
López et al.1105 used coarse-grained MD simulations to study the influence of different lipid bilayer compositions on the membrane association of the CD3ϵ cytoplasmic tails of the cluster of differentiation 3 T-cell receptor. The simulations suggested that the CD3ϵ tail preferentially bound to the negatively charged lipids (PG and PI) and that this binding was strengthened by the canonical immunoreceptor tyrosine-based activation motif. In contrast, zwitterionic lipids provided a less favorable environment for binding. The study revealed that the external shell of local positive charges, present in the zwitterionic lipids (choline groups), and the tight lipid packing in more ordered lipids (such as DPPC) was responsible for the decreased binding of the CD3ϵ tail. The study also demonstrated that the entire CD3ϵ chain preferentially localized in the boundary region between the Lo and Ld domains when incorporated as a monomer (see Figure 57). However, the chemical potential of the CD3ϵ chain was affected by the addition of GM1, which shuttled the CD3ϵ chain into the Lo domain (Figure 57) through specific interactions with the extracellular domain.
Figure 57.

Preferential sorting of the entire CD3ϵ chain in mixed ordered and disordered membranes. The CD3ϵ chain (magenta) was inserted into a preequilibrated Lo–Ld membrane patch composed of DPPC (green), cholesterol (white), DUPC (red), and POPG (orange) in a 4:3:2:1 molar ratio. Reproduced with permission from ref (1105). Copyright 2015 Biophysical Society. Published by Elsevier Inc.
Very recently, coarse-grained simulations were employed to explore the behavior of the transmembrane domain of amyloid precursor protein (APP) that was affected by the lipid raft microenvironment. Sun et al.1106 showed that the transmembrane domain of APP was attached at the boundary of the lipid raft and that this anchoring relied on the conserved hydrophobic motif of V710xxA713xxxV717xxxV721. However, monomers and homodimers of the transmembrane domain of APP were not observed to enter the lipid raft. Moreover, the dimerization of the transmembrane domain of APP was disrupted by the lipid raft. The study suggested that the driving forces of this disruption were derived from the combined regulation of the saturated lipids and cholesterol molecules rather than from the binding competition of cholesterol to the transmembrane domain of APP.
Lorent et al.1107 quantified the raft affinity for dozens of protein transmembrane domains using both experiments and coarse-grained simulations. They suggested that transmembrane domain area and length and the presence of palmitoylation affected protein raft partitioning. They found that plasma membrane proteins had a higher raft affinity than those of intracellular membranes. These findings are consistent with the hypothesis that trafficking to the plasma membrane is partially mediated by raft domains and explains the observation that plasma membrane is enriched in proteins containing long and thin transmembrane domains.1108
Coarse-grained and atomistic MD simulations complemented by free energy calculations were employed by Lin et al.1109 to study the sorting of the linker of activation of T cells (LAT) between coexisting membrane domains. MD simulations revealed that palmitoylation and hydrophobic length were essential for preferential interactions between the transmembrane domain of LAT and the lipids in the ordered phase: The simulations showed that long palmitoylated peptides interacted more preferentially with liquid-ordered domain lipids as compared to shorter and depalmitoylated peptides. Interestingly, an enrichment of peptides at the Lo/Ld interface was observed. It was suggested that this effect stemmed from “an inherent polarity of the peptide with respect to its interaction with the coexisting phases.”
Kaiser et al.1110 used atomistic MD simulations and biochemical experiments to study the lateral sorting of transmembrane peptides by cholesterol-mediated hydrophobic mismatch. The authors demonstrated that the presence of cholesterol forces the membrane, containing high amounts of transmembrane peptides, to undergo collective rearrangement. This research suggests that steric confinement of lipid acyl chains by cholesterol may be one of the physiological roles of cholesterol.
Grau et al.1111 explored the role of hydrophobic mismatch on transmembrane helix packing in a cellular environment using atomistic MD and biochemical experiments. Using the dimerization of glycophorin A as a test case, they showed that biological membranes can accommodate transmembrane homodimers with a wide range of hydrophobic lengths. This study indicates that biological membranes can adapt to structural deformations through compensatory mechanisms that emerge from their complex structure and composition to alleviate membrane stress.
Summarizing, lipid-mediated protein sorting and compartmentalization are crucial in many cellular processes, and their disruption leads to severe pathological conditions. However, lipid-mediated protein sorting is still poorly understood, and more studies bridging computational and experimental approaches are needed to understand the molecular mechanisms in sufficient detail.
8.3. Allosteric Modulation of Membrane Proteins by Lipids
For decades, the development of ligands in traditional drug discovery has focused on targeting membrane receptors’ orthosteric binding sites defined as sites where the endogenous ligands bind to elicit signal transduction. This approach has guided the development of classical orthosteric ligands that directly activate the target receptor (agonists) or block the actions of the endogenous ligand (antagonists/inverse agonists). However, in recent years there has been increasing therapeutic interest for effector molecules that bind to other than the receptor’s (orthosteric) active site. These sites are known as allosteric. Many allosteric modulators have little or no agonist or inverse agonist activity themselves, but they affect the receptor’s response to endogenous agonists and other ligands. Allosteric modulators usually elicit a conformational change in the receptor structure while still allowing, in many cases, simultaneous binding of orthosteric ligands–thus modulating the pharmacological characteristics of the orthosteric agent.
There is a growing number of experimental studies demonstrating the putative role of membrane lipids as allosteric modulators of membrane protein function. For example, the lipid sensitivity of nAChR has been known since the earliest attempts to isolate and reconstitute nAChR function in model membranes. nAChR must be purified in the presence of lipids and then placed in a bilayer with an appropriate lipid composition1112 to recover agonist-induced channel flux. Both anionic lipids, such as phosphatidylserine or phosphatidic acid, and neutral lipids, such as cholesterol or diacylglycerol, appear to be necessary for the receptor’s activity.1113−1115 Lipids also play an essential role in regulating receptor kinase EnvZ function both by the interaction with the transmembrane segments and by the interplay with nonembedded peripheral domains. Specifically, the dynamics and the activity of a glycine-rich motif that is critical for phosphotransfer from ATP1116 is allosterically modulated by the lipid membrane. Furthermore, it has been shown that the lipid composition has a profound regulatory effect on kinase domain activation of the human epidermal growth factor receptor,1117 where the ganglioside GM3 inhibited the autophosphorylation of the EGFR kinase domain. These results demonstrated that GM3 exhibits the potential to regulate the allosteric structural transition from an inactive to a signaling EGFR dimer by preventing the autophosphorylation of the intracellular kinase domain in response to ligand binding.
Here we summarize the computational studies where lipids have been shown to act as allosteric modulators, thereby modulating the structure and the dynamics of membrane proteins. In this context, lipids bound to specific lipid binding sites reviewed in section 7 can also be seen as potential allosteric modulators, though it is not always clear whether these interactions modulate receptor activity.
Coarse-grained molecular dynamics simulations were used to study the interaction of the β2-adrenergic receptor with membrane lipids and to explore the direct and indirect membrane effects that could be important in receptor dimerization.1030 The analysis of the local membrane thickness around the receptor, embedded in a bilayer with 0 and 50 mol % cholesterol, showed hydrophobic mismatch at helices 1/7 and 4/5, which was partially alleviated in the presence of cholesterol. In the absence of cholesterol, the receptor dimerization was observed via the interface involving transmembrane helices 4 and 5, while in the presence of cholesterol dimers with a different interface (involving transmembrane helices 1 and 2) were formed. Detailed analysis of the trajectories also revealed several cholesterol binding sites;1118 one of them was found on the transmembrane helix 4. The authors hypothesized that in the cholesterol-containing bilayer this site was occupied by sterol and the dimerization through transmembrane helices 4 and 5 was disrupted. The result seems to suggest the alleged role of cholesterol in GPCR dimerization involving direct competition of protein–lipid and protein–protein interactions.
Manna et al.54 used extensive all-atom simulations to clarify the mechanisms responsible for the modulatory role of cholesterol on the β2-adrenergic receptor. In a cholesterol-free DOPC bilayer, the receptor was found to adopt a wide range of conformations both on the ligand binding side and on the G protein side. In the presence of cholesterol, the situation changed dramatically, and the conformational flexibility of the receptor was significantly reduced. The receptor stayed predominantly in one conformation, and no further opening/closing of the ligand-binding site or G protein-binding sites was observed. The restriction of receptor flexibility occurred in bilayers with cholesterol concentration above 5 mol %. At lower levels (2 and 5 mol %), the distribution of the receptor’s conformation was broader and comparable to that of a cholesterol-free membrane. The authors ruled out the possibility that changes in physical membrane properties (such as bilayer thickness and lipid acyl chain order) would be responsible for the restriction of the conformational dynamics of the receptor and concluded that the cause of the observed changes in β2-adrenergic receptor conformation and dynamics is the specific binding of cholesterol to the receptor.
In their atomistic study on the effect of membrane properties on the A2A adenosine receptor, Ng et al.1028 demonstrated that the bulk properties of lipid membranes could also modulate the dynamics of the receptor. In this study, the distinct chemical nature of POPC and POPE lipids resulted in different dynamic behavior of the receptor. The principal component analysis revealed that different receptor dynamics stemmed from a combination of the different loop movements (overall more mobile in POPC as compared to POPE bilayer) and the divergent interhelical motions between the two lipid systems. The authors hypothesized that this effect could be a consequence of protein adaptation to the changing hydrophobic thickness in POPC and POPE bilayers.
Zhang et al.1119 employed all-atom molecular dynamics simulations, mutagenesis, and electrophysiological measurements to study the effect of PIP2 on the voltage sensitivity of a voltage-gated potassium channel. As a case study, they used KCNQ2, which is a member of the KQT family of slowly activated outwardly rectifying potassium channels. The analysis of both experimental and computational data suggested that PIP2 upregulated both the current amplitude and the voltage sensitivity of KCNQ2 and that PIP2 preferentially interacted with the S4–S5 linker of the open-state KCNQ2 channel. On the other hand, in the closed state, PIP2 only interacted with the S2–S3 loop (see Figure 58). Interestingly, the disruption of the interaction of PIP2 with the S4–S5 linker might decrease both the voltage sensitivity and the current amplitude, whereas disruption of the interaction with the S2–S3 loop only altered the current amplitude of the channel. The results strongly indicated the involvement of PIP2 allosteric modulation in the mechanism of action of KCNQ2.
Figure 58.
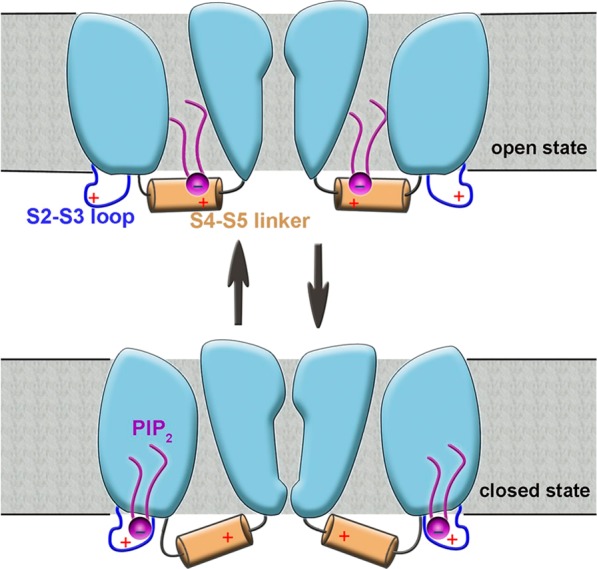
Mechanism of PIP2 action on the KCNQ2 channel. PIP2 is anchored at the S2–S3 loop in the closed state (bottom). Upon channel activation, PIP2 interacts with the S4–S5 linker (top). PIP2 is shown in magenta. Reproduced with permission from ref (1119). Copyright 2013 Zhang et al.
Delemotte et al.1120 used atomistic MD simulations to study the membrane-bound Kv1.2 channel subjected to a hyperpolarized potential. They used biased and unbiased MD simulations to reveal the initial steps of the voltage sensor domain participation in the channel deactivation mechanism. The simulations showed that the conformational changes taking place within the voltage sensor domain involved a zipper-like motion of the basic residues, resulting in sequential ion pairing with nearby counter charges from the voltage sensor domain and lipid headgroups from both the upper and lower membrane leaflets. The presence of lipid headgroups was essential, and it was shown to have a dramatic effect on the voltage-gated channel function in general. For example, the activation of K+ voltage-gated channels may be disrupted when they are embedded in bilayers formed by cationic lipids.1121 What is more, the removal of the lipid headgroups by enzymes also resulted in immobilization of the voltage sensor domain motion, thereby inhibiting channel function.1122,1123
Lingwood et al.1124 employed a set of experimental and computational methods to study the modulation of glycosphingolipids by cholesterol. Glycosphingolipids are essential communication devices used by cells for signaling, microbial and cellular adhesion processes, and immunological recognition. The paper showed that membrane cholesterol is a critical molecule in regulating glycolipid conformation and therefore receptor function. Cholesterol changed receptor availability by inducing a membrane-parallel glycolipid headgroup configuration. This feature seemed to have a crucial role in the presentation of erythrocyte blood groups and in the exposure of sperm sugar residues during conversion to the fertile state.
The interactions between serotonin 1A receptor and ganglioside GM1 were studied by Prasanna et al.1125 using coarse-grained MD simulations. Ten independent simulations, each 10 μs long, were used to examine the molecular basis of the interaction of GM1 with the serotonin 1A receptor. Simulations showed that GM1 interacted with the serotonin 1A receptor predominantly at the extracellular loop 1 and precisely at the sphingolipid-binding domain. This motif consisted of a specific combination of aromatic, basic, and turn-inducing residues, and was evolutionarily conserved. The interaction of the sphingolipid-binding domain with GM1 appeared to stabilize the specific conformation of the receptor. The population of this conformation also increased in the presence of cholesterol. The results suggest a direct role of lipid–GPCR interaction in modulating ligand binding and receptor function.
Here, we reviewed the recent progress in elucidating allosteric modulation utilizing MD simulations. The above-discussed studies demonstrate the significance of computational approaches in unraveling the molecular mechanisms of protein allosteric regulation by lipids. Generally speaking, regulation of protein structure and function is crucial to all organisms. All cellular processes are carefully controlled. If the control machinery fails, then the outcome is often a disease. While allosteric regulation is one of many means of cellular control, it is also a particularly interesting and not well understood one, stressing the importance of exploiting computer simulations to further our understanding of lipid-based allosteric modulation mechanisms.
8.4. Lipids Mediate Interactions of Drugs and Other Small Molecules with Proteins in Biomembranes
Interaction of drugs and small molecules with biological membranes is a critical and somewhat complicated process in living cells. For instance, 60% of currently marketed pharmaceuticals target peripheral and membrane proteins,993 making the binding of small molecules with membranes one of the critical steps in pharmacodynamics. Further, many membrane-bound transporters and membrane proteins have their binding sites located deep in a bilayer, thus the understanding of the dynamics of drugs bound to membranes is crucial both for drug discovery and drug delivery. Below, we review recent MD simulation studies in this context, paying particular attention to the interplay of anesthetics and neurotransmitters with biological membranes.
8.4.1. Anesthetics
Arcario et al. performed atomistic MD simulations of desflurane1126,1127 to study its binding to the Gloeobacter violaceus ligand-gated ion channel. Simulations revealed two anesthetic binding sites: the TM1 site, which was previously observed in the experimental studies,1128 and the TM2 site, which has not yet been confirmed experimentally. Interestingly, to reach both binding sites, the desflurane must first partition to the lipid membrane. Moreover, the study observed asymmetric binding of desflurane to the channel, and this binding promoted a specific conformational change of the protein, resulting in dehydration of the central ion pore and creation of a 13.5 kcal/mol barrier to ion translocation.
The role of the lipid bilayer in capsaicin binding to the transient receptor potential vanilloid subtype 1 (TRPV1) was studied by Hanson et al.1129 TRPV1 is a heat-sensitive ion channel also involved in pain sensation, with an intracellular capsaicin binding site. Atomistic MD simulations of capsaicin in a POPC bilayer showed capsaicin to undergo translocation from the extracellular to the intracellular leaflet, thereby crossing a barrier of 6 kBT as estimated by the potential of mean force calculations. The spontaneous capsaicin translocation was also observed in unbiased simulations. The results suggest a possible mechanism for capsaicin binding to TRPV1: capsaicin penetrates the lipid bilayer, and then flips from the extracellular to the intracellular leaflet to access the TRPV1 binding site.
An impressive set of over 500 independent coarse-grained MD simulations were carried out by Melo et al.1130 to study the effect of alcohol on the opening of the mechanosensitive MscL channel. The simulations demonstrated the MscL opening time to increase with the addition of unbranched alcohols. This effect saturated at longer alcohol chain lengths and higher alcohol concentrations. Fluorescence assays verified the theoretical predictions. Detailed analysis indicated that the alcohol-induced stabilization of the closed state of the MscL channel originated not from specific alcohol–MscL interactions but from the combination of the alcohol-induced changes to a number of bilayer properties (area per lipid, bilayer thickness, area compressibility, lipid diffusion, and average lipid chain order parameter) and modulation of the MscL–bilayer interface.
Jerabek et al.1131 studied the membrane-mediated effect of ketamine on ion channels. MD simulations showed that the membrane thickness and lateral area per lipid were not affected by the addition of ketamine (up to 8 mol %). However, significant changes in the lateral pressure were observed within the membrane–water interface due to the insertion of ketamine. It was hypothesized that these changes would be substantial enough to affect the probability of membrane ion channel opening.
Mojumdar and Lyubartsev1132 examined the structural and dynamical properties of a local anesthetic (articaine) in DMPC bilayers. MD simulations for the neutral and protonated forms of articaine inserted in fully hydrated lipid bilayers revealed that while some properties of these two anesthetics were very similar (for example, the location in a lipid bilayer and the effect on the lipid headgroup angle), some other properties differed substantially (for example, the orientation of the charged form in the membrane and the hydrogen bonding network). Interestingly, the presence of the neutral articaine molecules in model bilayers increased the electrostatic dipole potential inside the bilayer due to the dipole moments of the articaine’s carbonyl groups and their preferential orientation in the bilayer. Given that the neutral form of articaine is predominant at neutral pH and the electrostatic potential of the lipid bilayer governs the functionality of ion channels, it is possible that the anesthetic effect of articaine stems from its perturbation of the membrane dipole moment, which, in turn, disrupts the normal operation of voltage-gated ion channels.
Articaine was also a subject of research in another MD study conducted by Skjevik et al.1133 The simulations showed that the increased ability of articaine to reach the bone tissue in comparison with other local anesthetics is due to its ability to form intramolecular hydrogen bonds. The authors hypothesized that this H-bonding network may be responsible for the higher lipophilicity of articaine, which, in turn, would improve its therapeutic efficiency.
Cascales et al.1134 performed a series of MD simulations of benzocaine insertion into symmetric lipid bilayers composed of different ratios of DPPC and DPPS. Biased MD simulations at different temperatures indicated that a maximum in the free energy profile was located at the membrane–water interface region. The free energy barrier appeared to be exponentially dependent on the DPPS content in the lipid bilayer. However, the minimum free energy within the lipid bilayer remained almost independent of the lipid composition. By repeating the simulations at different temperatures, the authors demonstrated that the spontaneity of benzocaine insertion into the lipid bilayer was due to an increase in entropy associated with the insertion process.
Propofol is an anesthetic widely used for inducing general anesthesia, and it binds to pentameric ligand-gated ion channels with high affinity.1128 Hansen et al.1135 used MD simulations and experiments to study propofol behavior in DPPC bilayers. The simulations revealed that the depth of propofol molecules inside the lipid bilayers coincided with the depth of the protein’s binding sites. Additionally, the drug molecules in the membrane ordered the lipid acyl chains, but contrary to that with cholesterol, the ordering effect was observable only for carbon atoms near the membrane–water interface.
The interactions between enfuvirtide and lipid bilayers were the subject of studies conducted by do Canto et al.1136 Enfuvirtide is a peptide, which reduces HIV progress by inhibiting the fusion of the HIV envelope with the cell membrane. The simulations indicated that enfuvirtide interacted less with model lipid bilayers as compared to other more efficient peptides. It was hypothesized that the ability to communicate with biological membranes is crucial in the development of potent anti-HIV drugs; low affinity to lipid bilayers implies low local concentration of the peptide, which, in turn, affects the bilayer surface’s ability to act efficiently as a reservoir for the antifusion peptide. This suggestion was later confirmed in a study by Leonis et al.,1137 where a favorable conformation of darunavir (human immunodeficiency virus type 1 protease inhibitor) was maintained by the hydrogen bonds formed between lipids and water.
The above examples show that anesthetics can fulfill their function in the cells either by directly modulating protein structures or by altering the physical properties of membranes in which they reside. The latter mode of action can change the fluidity, order, or lateral pressure profile of lipid bilayers, which, in turn, affects the bioavailability and efficiency of a given drug. Further, for completeness, there is reason to mention that the anesthetic–membrane interactions have recently been addressed also in many other MD studies not described in detail in this review. These include the effects of bromoform,1138 sevoflurane,1139 tetracaine and caffeine,1140 lidocaine and chloroform,1108,1127 articaine,1141 xenon,1142 bupivacaine,1143 benzocaine,1144,1145 diethyl ether and enflurane,1146−1148 halothane,1146,1149,1150 and alkylphenols.1151
8.4.2. Neurotransmitters
Neurotransmitters are endogenous small molecules unevenly distributed in the neuronal tissues with concentrations reaching rather high levels inside the synaptic vesicles1152,1153 and in the synaptic clefts.1154 Such high concentrations of neurotransmitters should allow neurotransmitter partitioning into synaptic membranes, if the partition coefficients and rates of diffusion were sufficiently high. Indeed, Seeger et al.1155 showed experimentally that the interaction between serotonin and neutral membranes results in a decrease of the membrane main phase transition temperature. Neurotransmitters may also affect neuronal signaling through direct ligand–neuroreceptor interactions as well as through an indirect lipid-mediated mechanism.1153,1156,1157 Here, we review recent computational studies that have shed light into the molecular mechanisms of neurotransmitter modes of action.
Peters et al.1158 used MD simulations to study the interactions between a series of neurotransmitters (γ-aminobutyrate, glycine, acetylcholine, and glutamate) and a DPPC bilayer. The simulations uncovered that the charged neurotransmitters demonstrated the lowest lipid membrane affinity, while zwitterionic and polar neurotransmitters showed moderate affinity. The membrane affinity of neurotransmitters was ranked as follows: acetylcholine ≈ glutamate ≪ γ-aminobutyrate < glycine. Glycine penetrated the bilayer the most, with the deepest location being close to the glycerol backbone of lipids. However, even at this position, it was strongly hydrated. Free energy profiles obtained from umbrella sampling simulations showed a minimum of about 0.48–0.72 kcal/mol close to the membrane–water interface for acetylcholine and glutamate, while for γ-aminobutyrate and glycine, a minimum of, respectively, 0.48 and 1.2 kcal/mol was observed in the vicinity of the lipid glycerol backbone.
MD simulations in combination with thermodynamic measurements were applied to study the interaction of serotonin with DPPC and DOPC bilayers.1159 Serotonin is a monoamine neurotransmitter, which is commonly considered to be a significant contributor to the feeling of happiness.1160,1161 Peters et al.1159 demonstrated through MD simulations and thermodynamic measurements that serotonin interacted strongly with DPPC and DOPC bilayers, which is highly unusual for a hydrophilic solute like serotonin (the oil–water partitioning coefficient for serotonin is well below one). The authors hypothesized that this behavior stems from specific neurotransmitter–lipid interactions. Indeed, as the most prominent interaction the MD simulations identified the salt-bridge between the amine group of serotonin and the lipid phosphate group. This salt-bridge anchored the positively charged serotonin at the membrane–water interface, with the aromatic ring system pointing inward. The most probable serotonin location was identified to be between the phosphate and the carbonyl groups of the lipid bilayer. Interestingly, the deprotonated form of serotonin showed the opposite orientation, with the amine group pointing toward the membrane core.
The interactions of dopamine and its precursor L-dopa with membrane lipids were investigated by Orlowski et al.1162 using atomistic MD simulations. They demonstrated that both dopamine and L-dopa interact with lipid headgroups via hydrogen bonds and electrostatic interactions. These interactions anchored the neurotransmitter molecules to the membrane–water interface. Interestingly, the strength of neurotransmitter–lipid interactions strongly depended on the lipid type. In the mixed-lipid bilayer containing anionic phosphatidylserine, one observed complete adsorption of both dopamine and L-dopa at the bilayer surface. However, this adsorption was incomplete at the surface of a pure zwitterionic PC bilayer. The study hypothesized that the excessive association of dopamine and L-dopa with the lipid bilayers containing phosphatidylserine may limit the free use of dopamine as a synaptic transmitter, which, in turn, could be a possible molecular mechanism responsible for some of the neurodegenerative disorders. This hypothesis agrees with a post-mortem study on the brain lipid composition, where it was shown that, in schizophrenic patients, the levels of phosphatidylcholine, sphingomyelin, and galactocerebrosides were decreased, while the phosphatidylserine level was increased.1163,1164
Wang et al.1165 drew similar conclusions regarding the role of anionic lipids. In this study, based on dialysis equilibrium studies, calorimetric measurements, and MD simulations, the authors studied an anionic (glutamate), a cationic (acetylcholine), and two zwitterionic (γ-aminobutyric acid and glycine) neurotransmitters in lipid bilayers of different compositions. The simulations revealed that the zwitterionic neurotransmitters were attracted to lipid membranes containing anionic lipids, with their local concentration at the membrane–water interface being 5–10 times larger than in the aqueous bulk. This attraction was mainly driven by electrostatic interactions of the neurotransmitter amine group and the lipid phosphate group.
Shen et al.1166 used atomistic MD simulations to study the insertion dynamics of several neurotransmitters (methionine enkephalin, leucine enkephalin, dopamine, acetylcholine, and aspartic acid) at the atomic scale. The simulation results showed that the methionine enkephalin, leucine enkephalin, and dopamine were able to insert freely into both POPC and POPE membranes and that the aromatic residues guided this insertion. However, only a limited number of neurotransmitter molecules were able to diffuse into the lipid membrane, suggesting the existence of an intrinsic mechanism by which the lipid membrane is protected from being destroyed by extreme insertion of neurotransmitters. In contrast, acetylcholine and aspartic acid were observed to diffuse freely in the solution and never be incorporated into the lipid bilayers.
Experiments and MD simulations were also employed to study the effect of melatonin on the DPPC and DOPC bilayers.1141,1168 The simulations revealed that melatonin increased the lipid headgroup area and decreased the thickness of model bilayers by disordering the lipid hydrocarbon chains, thus increasing membrane fluidity. When both melatonin and cholesterol were present in the systems, the compression isotherms were close to those of the pure lipid monolayer systems, indicating that melatonin was able to alleviate cholesterol’s effects.
An extensive set of atomistic simulations were conducted by Postila et al.1167 to probe the neurotransmitter interactions with postsynaptic lipid membranes. In this study, 13 neurotransmitters were simulated in multicomponent model bilayers mimicking postsynaptic membranes. The simulations revealed that neurotransmitters explored in this study fall into two categories: membrane-binding (dopamine, serotonin, epinephrine, norepinephrine, melatonin, and adenosine) and non-membrane-binding (γ-aminobutyric acid, acetylcholine, serine, aspartate, and glutamate). Interestingly, this classification agrees with the location of the neurotransmitter-binding sites in the synaptic receptors. G protein-coupled receptors were found to be receptors for membrane-binding neurotransmitters with a membrane-buried ligand-binding site (see Figure 59). In contrast, the non-membrane-binding neurotransmitters were demonstrated to have extracellular ligand-binding sites positioned above the cell membrane surface (see Figure 59). Atomistic MD simulations provided substantial evidence that the neurotransmission process follows either the membrane-independent or the membrane-dependent mechanism, and that the receptor’s ligand-binding site position has a decisive role in the selection between these two mechanisms.
Figure 59.

Mechanisms of neurotransmitter release. (A) Classical membrane-independent mechanism and (B) membrane-dependent mechanism. Reproduced with permission from ref (1167). Copyright 2016 Postila et al. (https://creativecommons.org/licenses/by/4.0/legalcode).
Very recently, Mokkila et al.1169 studied the effect of physiologically relevant ions (Na+, K+, Ca2+) on dopamine–membrane interactions using atomistic MD simulations. Cations are essential components of neurosignaling; hence the influx of Ca2+ ions into the presynaptic neuron is a trigger for neurotransmitter release.1170 This work showed that high calcium levels could efficiently decrease dopamine binding to phosphatidylcholine lipid bilayers. Based on these results and the fact that the intravesicular space of presynaptic vesicle is kept at a pH of 5.6, assuring that the phosphatidylserine present in the inner leaflet is efficiently rendered neutral, the authors argued that the lipid content and the low pH inside the vesicles, together with an active inward pumping of divalent cations prevents neurotransmitter accumulation at the inner leaflet surfaces of the presynaptic vesicles. The dopamine–lipid bilayer simulations described in this study allow one to draw the conclusion that both the inner leaflet phospholipid composition and the divalent cation content of the presynaptic vesicles are somehow optimized for neurotransmitter release into the synaptic cleft.
In summary, the above examples show that MD simulations can be successfully applied to study not only the lipid bilayer–protein interactions but also the membrane–small molecule interplay in cellular membranes. The simulations of model membranes can elucidate how neurotransmitters and anesthetics interact with lipid bilayers, thus expanding our understanding of their therapeutic actions. Finally, the atomistic insight into membrane–small molecule interactions enables us to understand the membrane-independent as well as the membrane-dependent mechanisms of anesthesia and neurosignaling.
8.5. Lipids Guide the Binding of Peripheral Proteins to Biological Membranes
Peripheral membrane proteins constitute a family of a structurally diverse subset of membrane proteins involved in cellular signaling.1171 They play a pivotal role in the initiation of key biological processes such as the blood coagulation cascade1172 or viral fusion.1173 The essentiality of lipid membranes in the binding and activity modulation of peripheral membrane proteins is well established, but more work is needed to clarify the coupling between specific protein–lipid interactions and observable protein action.
Experimental studies on peripheral proteins, especially the determination of three-dimensional membrane-bound structures, are challenging. Even when the crystal structure has been solved, the dynamic properties such as the activation mechanism upon ligand binding or large-scale conformational changes due to allosteric protein–lipid interactions cannot be addressed, since after all the crystal structure is static. The difficulty to crystallize membrane-bound peripheral proteins and to solve their three-dimensional structure using, for example, X-ray diffraction stems from the transient character of interactions between peripheral proteins and biomembranes, and the fact that peripheral proteins function at the membrane–water interface where the electrostatic environment changes rapidly. There are also other experimental methods able to shed light on protein structure, such as NMR, EPR, FRET, and small-angle X-ray scattering (SAXS), but the information they provide about protein–lipid interactions is usually insufficient to determine the protein–membrane interface, which is crucial to understand the mechanism of action of peripheral proteins.
The binding of peripheral membrane proteins with lipid membranes is classically described as an electrostatically driven process followed by the insertion of hydrophobic groups into the lipid bilayer. Electrostatic forces between the negatively charged bilayers and clusters of basic amino acids on the protein surface bring the protein into a specific, binding-prone orientation relative to the lipid bilayer. In essence, electrostatic forces play a significant role in the function of peripheral membrane proteins.1174,1175 Experimental and computational studies have revealed that nonspecific electrostatic interactions can contribute up to a few kilocalories per mole to the overall protein–membrane affinity and that each basic amino acid contributes up to 1 kcal/mol to the total binding free energy.1175−1177
Here, we discuss recent computational studies trying to address the question–how do biological membranes guide and modulate the binding of peripheral proteins? We focus our attention on anionic lipids, with the strongest interest in phosphatidylinositols.
8.5.1. Phosphatidylinositols
Several MD studies have recently investigated the interactions of various peripheral membrane proteins with phosphatidylinositols (PIs). The importance of the PI lipid interactions with peripheral proteins is highlighted by the fact that, for example, they appear to be specifically involved in the control of many cellular events, such as the organization and dynamics of critical signaling pathways, the actin cytoskeleton rearrangement, and the intracellular vesicle trafficking.1090−1092 What is more, the unique feature of PIs, which can be synthesized rapidly and degraded in discrete membrane domains, makes them ideal regulators of fast and dynamic mechanisms of cell regulation.
A neuron-specific membrane-binding protein, called auxilin-1, was studied by Kalli et al.910 This protein is involved in the late stage of clathrin-mediated endocytosis, which enables extracellular material, including membrane receptors and ligands, to be imported into cells through the formation of clathrin-coated vesicles.1178 Auxilin-1 binds to the newly budded clathrin-coated vesicles and facilitates the recruitment of the uncoating enzyme that removes the clathrin coat. To fulfill its function, auxilin-1 has to attach correctly to the lipid bilayer surface. Kalli et al.910 suggested a model for the protein–membrane encounter of the auxilin-1 phosphatase and tensin homologue (PTEN)-like domain using multiscale MD simulations. They showed that the negatively charged lipids (especially PIP2) enhance the binding of the auxilin PTEN-like domain to lipid bilayers and facilitate its correct orientation with respect to the membrane. Subsequent in silico mutations of three basic residues in the C2 subdomain of auxilin were observed to perturb its interaction with the lipid bilayer and result in changing the protein orientation. Interestingly, the interaction of membrane-bound auxilin-1 with negatively charged lipid headgroups resulted in clustering of PIP2 in the adjacent bilayer leaflet. Similarly, Kalli et al.1179 reported the clustering of PI lipids around a voltage sensitive phosphatase PTEN domain (Ci-VSP PD). Simulations suggested that PIP3 lipids bind to Ci-VSP PD in the vicinity of the protein active sites and that this binding is structurally similar to that of auxilin PTEN. The authors proposed a novel mechanism of association of PTEN with a lipid bilayer, including protein reorientation to optimize its interactions with PIP molecules. In contrast to auxilin PTEN, Ci-VSP binds directly to PIP-containing membranes without a subsequent reorientation step.
The pleckstrin homology (PH) domain of the general receptor of phosphoinositides 1 (GRP1) selectively binds to PIP3 lipids. The GRP1 PH domain has been the focus of a number of computational studies1180−1183 using a wide range of simulation methodologies. MD simulations revealed both translational and orientational electrostatic steering of the PH domain toward the PIP3-containing bilayer surface. Computational studies also suggested that the background anionic phosphatidylserine lipids play a critical role in the initial stages of protein recruitment to the membrane surface through nonspecific electrostatic interactions. The experimentally observed preference of GRP1-PH for PIP3 over PIP2 and the presence of a noncanonical PIP-interaction site observed previously in other PH domains but not in GRP1-PH were also revealed. These studies demonstrate how combining Brownian MD, coarse-grained MD simulations, and umbrella sampling can elucidate the molecular mechanisms and energetics of interactions between peripheral membrane proteins and complex cellular membranes.
Similarly, the BAR-PH domain of ACAP1 protein was a subject of computational studies by Chan et al.1184 The ACAP1 protein belongs to the GTPase-activating proteins that act on the ADP-ribosylation factor (ARF) family that also serves as a vital component of a clathrin complex for endocytic recycling. The authors investigated the molecular details of the protein–lipid interactions of ACAP1BAR-PH protein using both restrained and unbiased atomistic MD simulations as well as free-energy umbrella sampling calculations. The simulations revealed that the PH domains of an ACAP1BAR-PH dimer underwent orientation changes during the membrane binding process. The study showed two PIP2-binding pockets on the PH domain. Preference of the PH domain binding to PIP2 lipids over POPS lipids was explained by the comparison of the PMF profiles generated by umbrella sampling simulations (see Figure 60). Exploring the free energy surface of the protein–lipid systems uncovered a much deeper free energy well when PIP2 was in either binding pocket of the protein, compared to the case of POPS.
Figure 60.

PH domain binding to PIP2 lipids. (left) Simulation snapshot of a PH domain in the bound state with PIP2. The PH domain is shown in gray; DOPC phosphorus atoms are depicted as pink spheres. (right) Potential of mean force profiles for the PH domain of the ACAP1BAR-PH protein dimer bound to PIP2 at pocket 1 and pocket 2. Reproduced with permission from ref (1184). Copyright 2017 American Chemical Society.
Recently, Yamamoto et al.1185 reported MD simulations of the membrane localization and interactions of 13 different PH domains (see Figure 61). The study showed that the β1/β2 loop region of PH domains is the primary PIP-binding site. The simulations were also able to identify secondary (noncanonical) lipid-binding sites on PH domains, suggesting that multiple lipid interactions were crucial for the binding of PH domains to membranes. In addition to the PIP binding sites, the clustering of PIP lipids around the PH domains was observed. To this end, the authors developed a computational protocol employing both coarse-grained and atomistic MD simulations that could yield structural data for PH/PIP complexes. These structures are directly comparable to the structures obtained from NMR and X-ray crystallography. The authors showed that a high-throughput coarse-grained simulation approach, generating ensembles of simulations, can successfully be used to study the structural and dynamic features of the association of peripheral membrane proteins with model membranes.
Figure 61.

Pleckstrin homology domain–PIP complexes. Alignments of the PH/PIP complexes were derived from the MD simulations. Reproduced with permission from ref (1185). Copyright 2016 Yamamoto et al. (https://creativecommons.org/licenses/by/4.0/legalcode).
Atomistic MD simulations of a homologue oxysterol binding protein (Osh4) were used to characterize the structure and molecular mechanism of protein attachment to various model lipid membranes.1186 A single binding conformation was found regardless of the nature of the negatively charged lipid used. Membrane binding of Osh4 was relatively fast (about 100 ns) and remained stable for at least 1 μs. The study showed that Osh4 interaction with the lipid membrane primarily involves electrostatic interactions, but a hydrophobic anchor (phenylalanine loop) and specific hydrogen bonding also promote stable Osh4–membrane interactions.
Pleskot et al.1187 studied the interactions of the actin-capping protein (CP) with phosphatidic acid and PIP2. CP binds to the barbed end of actin filaments and inhibits both addition and loss of actin monomers at this end. The ability of CP to bind filaments is highly regulated by signaling phospholipids, which inhibit CP activity. Using homology modeling, molecular docking, and coarse-grained MD simulations, the authors addressed the high-affinity interactions between plant CP and membranes containing PA. A similar study has been done for systems containing animal CP and PIP2.1187 Computational studies identified some structural differences in the C-terminal part of the protein, leading to different binding of membrane lipids by animal and plant CP. The study was also able to locate the PA-binding domain of plant CP and experimentally showed that it was sufficient to drive membrane binding in vitro.
Actin-capping protein is not the only protein taking part in the organization and dynamics of the actin cytoskeleton modulated by membrane phosphatidylinositols.1189 Combination of mutagenesis and biochemical experiments together with atomistic MD simulations has been utilized in a very recent study to elucidate how mouse twinfilin-1 interacts with phosphatidylinositols.1188 The study revealed that the high-affinity lipid-binding site is located at the C terminal region of twinfilin and that the actin depolymerizing factor homology domains bind to phosphatidylinositol-rich domains only with low-affinity. MD simulations showed that twinfilin is initially tethered to the membrane through its C-terminal tail and that this interaction leads to subsequent association of actin depolymerizing factor homology domains with the bilayer, resulting in inhibition of their actin-binding properties. This study shows that atomistic MD simulations combined with biochemical experiments can provide a detailed molecular mechanism of the twinfilin inhibition by phosphatidylinositols (see Figure 62). Since twinfilin promotes actin filament disassembly and inhibits their growth, the authors hypothesized that phosphatidylinositols could control twinfilin localization and regulate its activity during lamellipodial protrusions and retractions.
Figure 62.
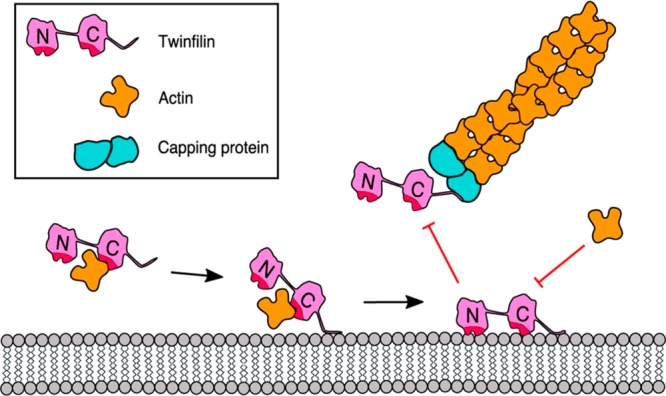
Model of the inhibition of twinfilin-1 by a phosphoinositide-rich membrane. Reproduced with permission from ref (1188). Copyright 2018 the American Society for Biochemistry and Molecular Biology.
Multiscale MD simulations using both atomistic and coarse-grained resolutions were used to investigate the interaction and the binding of α-tocopherol transfer protein (α-TTP) to PIP2 lipids,1190 both when they were bound in the protein’s cavity and while they were embedded in the plasma membrane. The simulations revealed that a single PIP2 molecule is able to stabilize the anchoring of α-TTP to the membrane surface. Protein binding occurred by direct interaction of the negatively charged lipid headgroup with the positively charged patch of amino acids at the surface of the protein near the opening of the ligand-binding cavity. This electrostatic attraction turned out to be very stable. When PIP2 was incorporated into the α-TTP binding cavity, the fast detachment from the membrane into the bulk water was observed, consistent with the loss of the anchoring group and the putative role of α-TTP lipid exchange between the endosome and the plasma membrane.313
PROPPIN (β-propellers that bind polyphosphoinositides)-membrane binding was studied by Busse et al.1191 using isothermal titration calorimetry, stopped-flow measurements, mutagenesis studies, and MD simulations. The simulations revealed that the 6CD loop is an anchor for PROPPIN-membrane binding based on the observation that this was the region of the protein that inserted most deeply into the lipid bilayer. Subsequent mutagenesis studies showed that both hydrophobic and electrostatic interactions play a role in membrane insertion of the 6CD loop. The authors proposed a model for the PROPPIN–membrane binding in which PROPPINs are initially attracted to the membrane surface through nonspecific electrostatic interactions and then retained at the membrane through specific PIP binding.
Ghosh et al.1192 used MD simulations to identify the AtSfh1 pattern of Sec16-nodulin as a PIP2-binding region. The study showed that the nodulin peptide binds firmly to the lipid membrane. The protein interacted with the lipid membrane mostly via H-bond interactions with PIP2 molecules. The simulations predicted that a single peptide could bind to one, two, or three PIP2 molecules with similar probabilities (about 30%). Interestingly, the number of H-bonds established between the peptide and PI(3,5)P2 differed significantly as compared to PI(4,5)P2. Based on this observation, the authors speculated that the binding of PI(3,5)P2 is weaker than that of PI(4,5)P2.
The lipid membrane anchoring of HIV-1 myristoylated matrix domain protein was simulated using coarse-grained MD simulations.1193 Preferential binding of PIP2 headgroups to the highly basic region of the matrix domain was reported. Interestingly, the matrix domain was able to confine PIP2 lipids all around its surface after having found a stable orientation at the membrane surface.
In summary, many peripheral proteins contain clusters of basic residues on their surface that interact electrostatically with negatively charged lipids such as PI. The computational studies presented above suggest different roles for PI in the interactions of peripheral membrane proteins with lipid bilayers. Anionic lipids seem to attract peripheral proteins toward the membrane surface and then also bind specifically to the proteins. Further, PI-facilitated reorientation of the protein and clustering of PI lipids near protein surfaces have been observed.
8.5.2. Other Charged Lipids
The effect of ganglioside GM1 binding to cholera toxin was studied by Basu and Mukhopadhyay1194 using atomistic MD simulations. Starting from the initial structure where the five units of the cholera toxin B subunit bind with five GM1 lipids (Figure 63), the simulations showed that only three of the five units remained bound and the whole cholera toxin tilted such that the three binding subunits reached deeper into the membrane. Interestingly, after binding of cholera toxin to the GM1-containing lipid layer, the lipid curvature and the protein orientation changed. Lipids that were lying just under the cholera toxin pentameric hole moved to the corner of the lipid monolayer and thus made an empty void so that cholera toxin could pass through it. Therefore, these results suggest a putative mechanism for the early stage alteration of lipid structure, allowing penetration of the lipid bilayer by cholera toxin. No such behavior of the LT1 mutant (E. coli type I heat labile toxin; closely related to cholera toxin regarding structure and function1195) was observed.
Figure 63.

Snapshots from the MD simulations depicting (left) the CT-monolayer and (right) the mutated-LT1-monolayer systems. POPC and GM1 molecules are shown as silver lines and cyan bonds, respectively. Individual subunits such as B1, B2, B3, B4, and B5 are presented separately as red, blue, green, mauve, and yellow spheres, respectively. Reproduced with permission from ref (1194). Copyright 2014 American Chemical Society.
Very recently, Rissanen et al.71 revisited the cholera toxin binding to GM1 and acyl-chain labeled bodipy-GM1. The paper explored the partitioning of these gangliosides in the Lo and Ld phases using atomistic MD simulations and cholera toxin binding experiments. MD simulation data showed that GM1 headgroup localization and geometry were sensitive to the membrane environment and that the presence of the bodipy label caused a deeper penetration of bodipy-GM1 into the membrane in the Lo domains. It was hypothesized that these changes could render the bodipy-GM1 headgroup partially inaccessible to cholera toxin. Indeed, the experimental binding data showed that the overall binding of cholera toxin to bodipy-GM1 was reduced in comparison to GM1, especially in the Lo liposomes. This study confirmed at the molecular level that the presence of a fluorophore on the acyl chain, rather than changing the headgroup geometry, largely excluded the bodipy-GM1 molecules from the Lo membrane environments, where native GM1 lipids seemed to be enriched.
All-atom MD simulations were used to address how membrane binding modulates the structural and dynamical properties of talin to activate integrin optimally.1196 The talin head domain consists of four subdomains, namely, the F0, F1, F2, and F3 subdomains. The F2 and F3 subdomains are crucial for the membrane binding process and the activation of integrin at the membrane surface.1197 It was shown that the F3 subdomain is sufficient for integrin activation;1198 nevertheless, the interactions between talin’s positively charged membrane orientation patch in the F2 subdomain (see Figure 64) and anionic lipids enhance the rate of integrin binding and activation. Arcario and Tajkhorshid1196 performed five independent simulations of the talin F2F3 subdomain binding to the PS bilayers. In all simulations, the talin F2F3 subdomain spontaneously bound to the membrane, and the basic membrane orientation patch residues played a crucial role in attracting talin to the anionic bilayer surface. The authors reported that the membrane binding of talin (see Figure 64) proceeded through three distinct steps: initial electrostatic recruitment of the F2 subdomain to anionic lipids, insertion of an initially buried (and conserved) hydrophobic anchor into the membrane, and an association of the F3 subdomain with the membrane surface through a massive conformational change.
Figure 64.

Membrane binding modes of talin F2F3. (A) Initial configuration of the talin F2F3 (green cartoon). (B–D) The three distinct stages in talin interactions with lipid membrane. (E) The C-α center of mass trajectory of each membrane binding moiety (i.e., MOP, FAP, and membrane anchor) projected onto the membrane normal. The dashed line indicates the average position of the upper leaflet phosphate moieties. Reproduced with permission from ref (1196). Copyright 2014 Biophysical Society. Published by Elsevier Inc.
Interactions of protein kinase C-α (PKCα) C1A and C1B domains were the subject of the experimental and computational studies reported by Li et al.1199 and Ziemba et al.1200 Atomistic MD simulations coupled to experiments uncovered the mechanism of PKCα activation, where both C1A and C1B domains bound to the bilayer during activation but after PKCα association with the lipid membrane. In this mechanism, the C1A domain was recruited first with strong interactions to PS (coactivator) and DAG (activator) lipids, followed by the C1B domain, which was preferentially bound to phorbol-12-myristate-13-acetate (activator).
An extensive set of atomistic MD simulations and free energy calculations were exploited by Enkavi et al. to clarify the molecular mechanism of the Niemann-Pick Protein C2 (NPC2) binding to the lipid bilayers and to characterize the role of lipids associated with the binding process.854 NPC2 is the critical protein involved in cholesterol efflux from late endosomes/lysosomes. Two competitively favorable membrane-binding orientations of NPC2 with a low interconversion barrier were observed in atomistic simulations. The detailed descriptions of these binding modes are given in section 6.2.3. Atomistic simulations revealed that sphingomyelin neutralized the effect of bis(monoacylglycero)phosphate by hindering the Prone binding mode (associated with cholesterol uptake and release) without affecting the Supine orientation (not taking part in cholesterol transport). This study clearly shows that lipids modulate NPC2-mediated cholesterol transport either by favoring or disfavoring the Prone binding mode.
Zhang et al.1201 studied the binding of the RecA protein at the surface of model membranes. RecA is a DNA repair protein that plays an essential role in homologous recombination and mediates the bacterial SOS response. The highly mobile membrane mimetic model and all-atom simulations were used to study how different anionic lipids (PG/CL versus PS) perturb RecA binding to the membrane. Contrary to PG- and CL-containing membranes, ionic H-bonds between the carboxylate group of phosphatidylserine and several lysine residues in the C-terminal region of RecA were shown to stabilize the parallel binding orientation.
Summarizing, our understanding of how peripheral membrane proteins interact with lipid membranes has significantly improved during recent years, as more accurate experimental and computational techniques have been developed. However, resolving the molecular mechanisms and the details of the binding process remains challenging. The simulation data presented in this section show convincingly that specific structural features are often significant in determining the properties of charged lipids. While MD simulations can predict the molecular mechanisms of the association of peripheral membrane proteins with model lipid bilayers, the challenge is to combine the multiscale MD approach and experimental techniques and to use them in a concerted manner to obtain a comprehensive view of what happens at both small and large scales in time and space in a cellular context.
8.6. Challenges
The main challenge in cellular biology is to understand the interplay between proteins and the other molecular components that modulate protein activation in cell membranes. Cell membranes are complex mixtures of lipids, proteins, and carbohydrates arranged into two asymmetric leaflets. Lipids have been shown to play a significant role in maintaining a healthy body, for example, by regulating neurotransmission, ligand binding, cell signaling, and protein sorting. However, a molecular-scale picture of how lipids are involved in these essential biochemical processes is still often lacking. Yet, it is known that an altered lipid content may lead to diseases and pathological conditions, such as cancer, diabetes, or respiratory syndromes.1202−1205 This section, therefore, discussed the role of lipids in membrane protein activation and function as it has been revealed by recent computational studies.
The discussed examples show clearly that the MD simulation method is an exceptionally useful tool to unravel the complexity and consequences of protein–lipid interactions. Computer simulations have been able to clarify the role of lipids in, for example, allosteric regulation of membrane receptors, preferential binding of peripheral proteins to the cellular membranes, protein sorting mechanisms, and neurotransmission. In all of these processes, MD simulations have been able to reveal both direct and indirect effects of lipids, as well as to differentiate between these two types of lipid modulation mechanisms.
Meanwhile, there are numerous interesting research topics that also call for clarification, but they are so resource-intensive that they are almost impossible to address by the current supercomputers. For instance, what is the molecular picture of membrane protein oligomerization, and how exactly do the lipid coronas facilitate the formation of protein supercomplexes? What is the role of protein–lipid interactions in membrane fusion and fission? Given that there are thousands of different lipid species in cells, which spend the minimum amount of energy to function, it is safe to assume that this plethora of different lipids is, indeed, needed for cell survival. However, what is the functional role of the numerous different lipid species in cellular metabolism?
During the past decade, the applicability of MD simulations has accelerated tremendously due to increasing computational power, advances in the development of massively parallel algorithms, and the development of accurate simulation models. This progress continues at a rapid pace, and it is likely that atomistic simulations of lipid-assisted membrane protein activation over multi-millisecond time scales are feasible sooner than one might even hope.
9. How Do Biomembranes Use Lipids to Store Energy?
Cells store energy (i) in the covalent chemical bonds of energy storage molecules and (ii) in the electrochemical ion gradients. Lipids and biomembranes are directly involved in both modes of energy storage. In the first mode, lipids carry the energy directly in their chemical bonds. These storage lipids such as triacylglycerols and cholesteryl esters (CEs) are transported within carrier particles, such as lipid droplets and lipoproteins. In the second mode, biomembranes act as an interface between two compartments across which cells build up electrochemical gradients akin to a reservoir created by a dam.
In this section, we first focus on the lipid storage organelles known as lipid droplets. We then move on to lipoproteins and cover their different structural stages. In the second part of this section, we concentrate on the second mode of energy storage. We review how lipids, especially cardiolipin, modulate the machinery involved in the storage of energy in terms of electrochemical gradients. The discussion below concentrates on recent progress in simulating these phenomena.
9.1. Energy Storage in Chemical Bonds
9.1.1. Lipid Droplets
Lipid droplets (LDs) are the cytoplasmic storage organelles for neutral lipids, such as triglycerides, CEs, and retinyl esters. They play crucial roles in energy and lipid metabolisms. LDs are linked to many diseases, such as obesity, atherosclerosis, and fatty liver disease. Indeed, LD accumulation likely serves protective functions in lipid metabolism malfunction. Recent advances have highlighted that LDs have many biological roles other than being passive cytosolic storage units. LDs are highly dynamic and tightly regulated, serving as hubs not only for fatty acid trafficking but also for protein trafficking and maturation. Moreover, LDs emerge as essential modulators of nuclear function and lipid signaling.1206 The modulatory roles of LDs mainly depend on regulated lipid and protein exchange between LDs and other organelles, mostly nuclei and mitochondria.1206 LDs might, also, act as general protein sequestration sites that take part in protein delivery and storage and foster protein–protein interactions and protein complex assembly.1206 Furthermore, LDs are involved in immune response, and they store antiviral and antibacterial proteins. On the downside, hepatitis C virus (HCV) maturation requires LD accumulation.1206
Energy storage and lipid metabolism make LDs indispensable components of adipose tissue, the liver, and the intestine. Similarly, LDs are also abundant in the nervous system where they support signaling among other membrane functions. LD overaccumulation in neurons and glia is associated with neurodegeneration. We refer the reader to three excellent reviews on general LD biology,1207 LD biogenesis,1208 and emerging roles of LDs.1206 In this section, we cover the computational studies of LDs performed in recent years.
Lipid Droplet Models: Simulations of Atomistic Lamellar Lipid Trilayer Models and Coarse-Grained Spherical Bodies
Due to the complexity of the lipid droplets and scarcity of atomic-scale information on the structure of LDs, most studies of LDs require either coarse-grained or simplified atomistic models. An approach commonly used in atomistic LD models is the lamellar trilayer model, in which two monolayers of phospholipids sandwich a cholesteryl ester/triglyceride phase. Complex models loyal to the natural spherical shape of LDs require coarse-graining. Using coarse-grained simulation approaches, LDs can be modeled as spherical bodies with cholesteryl CEs, triglycerides (TGs), or triacylglycerols (TAGs) forming a region in the core, sheeted with a monolayer of phospholipids.
Lamellar lipid trilayers have been used as models of LDs and similar emulsion droplets. Henneré et al.1209 performed simulations of POPC at the water–triglyceride (trilinoleoyl-glycerol) interface. In this lamellar trilayer model, the headgroup conformations and interactions were similar to those in a POPC bilayer, but the acyl chain properties showed small differences.1209 Henneré et al.1210 later increased the complexity of the system by introducing more lipid types to the monolayer and the triglyceride phase to mimic the composition of a commercial lipid emulsion. This more complex lipid composition in the monolayer resulted in the increase of acyl chain disorder, head group hydration, and area per lipid, when compared to bilayers of the same composition.1210 The trilayer system of Koivuniemi et al.1211 consisting of POPC monolayers sandwiching a cholesteryl oleate (CO) phase showed that CO slightly interdigitates into the POPC monolayers but does not substantially affect the POPC structure.1211 Lamellar trilayer models have also been employed in several studies that aimed to characterize the effect of curvature on protein–LD interactions,1212 interfacial tension of high-density lipoprotein (HDL) and low-density lipoprotein (LDL) models,1213 the surface properties of LDs, protein targeting,1214,1215 and the tear-fluid lipid layer.1216,1217
Coarse-Grained Simulations on Lipid Droplet Formation
To investigate LD formation, Khandelia et al.1218 explored the distribution of TAGs inside a POPC bilayer with coarse-grained simulations using triolein (TO) as model TAG. They initiated simulations where they replaced some POPC in the bilayer with TO amounting to two different concentrations: one above (5.2% w/w) and one below (2.3% w/w) the TAG solubility in POPC (3% w/w). To verify the observations further, they also simulated the self-assembly of a random dispersion of POPC, TAG, and water molecules. The simulations showed that the number of TOs that remain intercalated within POPC is limited by its solubility, and the rest of the TOs partition into disk-shaped aggregates in the center of the membrane. The blisters (Figure 65) formed due to aggregation were stable and exchanged TOs with the interface. These results suggest a possible mechanism for the growth of LDs inside the endoplasmic reticulum membrane and lipid signaling in cancer cells, which have high amounts of TAGs.1218
Figure 65.
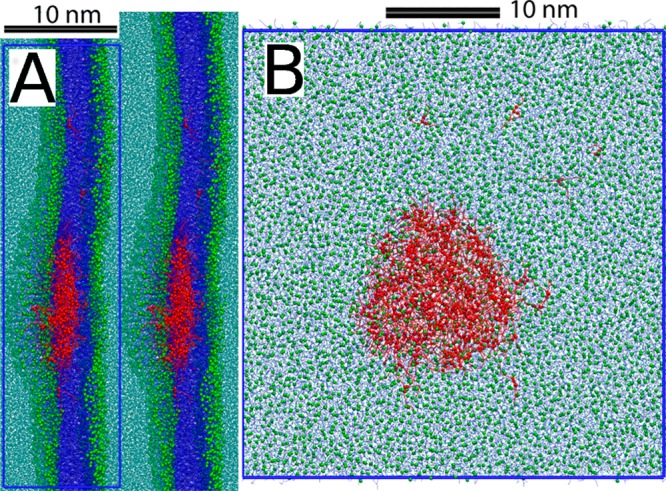
Formation of a blister of about 17 nm diameter due to TO aggregation captured in the simulations (A) from the side view and (B) from the top view. Reproduced with permission from ref (1218). Copyright 2010 Khandelia et al. (https://creativecommons.org/licenses/by/4.0/legalcode).
Coarse-Grained Simulations on the Distribution of Lipids in Lipid Droplets
Chaban and Khandelia1219 studied the distribution of lipids inside the triolein LDs. Coarse-grained simulations of spherical LD models composed of different mixtures of POPC, POPE, TO, and cholesterol showed that cholesterol is mostly located inside the lipid core within the TO phase. Cholesterol did not form crystals and existed primarily alone with only a small proportion forming dimers. POPC and POPE formed a monolayer on the surface of the LD, minimizing the surface tension at the LD–water interface. Further, POPE did not affect the distribution of lipids, and the thickness of the monolayer was almost equal to the thickness of one leaflet in a bilayer with the same composition.1219
The storage form of cholesterol, CEs, is a significant component of LDs and has a lower solubility in the bilayer than cholesterol. Investigating CE distribution in the LD is essential, as the surface exposure of CEs possibly determines the efficiency of acid lipases, which operate on the LD surface. Thus, Chaban and Khandelia followed up on the above work1219 by incorporating CO, a CE family member, instead of cholesterol in the spherical LD models.1220 They reported that CO forms a single phase with the TO in the LD core and is little soluble in the monolayer. Due to the hydrophilic groups, the LD core manifests long-range order that decays toward the center of the LD. The authors also observed that the excess of POPC and POPE forms inverted micelles in the LD core. The intercalation of TO into the monolayer and some degree of water–TO interactions in both works by Chaban and Khandelia1219,1220 suggest a potential mechanism for LD fusion.
Binding of Proteins on the Lipid Droplet Surface
Recently, LDs emerged as protein sequestration and exchange sites.1208 The proteins that bind to the endoplasmic reticulum and the Golgi apparatus also bind the LDs specifically. Many cytosolic LD-binding proteins contain an amphipathic helix (AH), which recognizes the endoplasmic reticulum and the Golgi apparatus by the lipid-packing defects. The unique surface properties of LDs have recently been suggested to underlie their specific interactions with proteins. Bacle et al.1215 simulated united-atom and coarse-grained trilayer LD models composed of POPC and TO. While TO was not found to affect the structural and dynamic properties of the POPC monolayer, it interdigitated substantially into the POPC monolayer. The interdigitation led to differences in lateral pressure profiles, as well as to a decrease in deep lipid-packing defects and an increase in shallow ones. Moreover, the lipid-packing defects depended on the surface tension and the LD size. For most other properties, the LD surface appeared to be similar to that of the bilayer with the same composition, which explains how the same protein could bind both ER or Golgi apparatus and the LDs. On the other hand, LDs can accommodate a broader range of surface tensions (0–20 mN/m) when compared to the bilayers (10–3–10–1 mN/m). It is likely that LD-specific proteins sense the defects modulated by surface tension. In addition, these properties might play a role in LD biogenesis, whereby a decrease in the lipid defect content due to the growing size of budding LDs might recruit LD-specific proteins that replace the ER-specific ones.1215
The results of Bacle et al., which suggest a role of lipid packing defects on LD-binding of amphipathic helices,1215 were confirmed more directly by Prévost et al.1214 in a combined MD and experimental study. They employed an all-atom trilayer model of LDs, containing TO and CO in the center and POPC, DOPE, and SAPIP in the monolayers. The simulations suggested that the LD surface is, indeed, distinct from the bilayer surface of the same composition. Furthermore, the simulations showed that the LD surface exhibits more substantial and longer-lasting packing defects. Besides, they probed AH binding to the trilayer models directly by placing the peptide (the AH of the LD-binding metabolic enzyme CCTα) in a random coil conformation away from the membrane surface. Three simulations out of four resulted in membrane association, with large hydrophobic residues inserting first into a packing defect. In one simulation, they also observed the initial stages of AH folding. They further tested other peptides with different numbers of hydrophobic residues. The simulations collectively suggested that the large hydrophobic residues recognize the packing defects on the LD surface and drive the LD binding. The predictions of the simulations were tested by in vitro experiments, which confirmed the role of lipid packing defects and the hydrophobic residues in LD surface recognition and binding. In essence, the simulations suggest that AHs containing large hydrophobic residues bind LDs, recognizing them by the large lipid packing defects.1214
9.1.2. Lipoproteins
High levels of low-density lipoprotein (“bad” cholesterol) have been implicated in cardiovascular diseases, leading to arterial plaques. On the other hand, high-density lipoprotein particles (known as “good” cholesterol) are critical in reverse cholesterol transport, and their low levels have been linked to an increased risk of atherosclerosis. Here, we review the use of all-atom and coarse-grained MD simulations that have been used to model and explore the structure–function relationship in LDL and HDL particles. A review from 2016 by Pan and Segrest covers the computational studies of plasma lipoprotein lipids.1221 We also refer the reader to the book chapter titled “Modeling of Lipid Membranes and Lipoproteins”1222 and to the review cited in ref (1223), in both of which Koivuniemi and Vattulainen discuss HDL in detail.
Low-Density Lipoprotein
LDL particles are among the most massive cholesterol-carrying particles in the bloodstream. Apolipoprotein B (apoB-100) is the primary apolipoprotein found in LDLs as well as in the other three lower density lipoproteins (chylomicrons, very low-density lipoprotein, and intermediate-density lipoprotein).1224 Due to their large size (21–25 nm) and the absence of known three-dimensional structure for apoB-100, only a single computational study of LDLs exists to date.
Murtola et al.1225 performed coarse-grained simulations on the first molecular-scale model for LDL consisting of COs, trioleates (TOs), unesterified cholesterols, phospholipids (PCs), and lysolipids (lysoPCs) with and without apoB-100, modeled in two different conformational states (Figure 66). Lipid–ApoB-100 interactions were shown to bring (hydrophobic) core lipids to the particle surface. The distribution of the lipids within LDL was observed to be complex. The study characterized an isotropic-like distribution of neutral (hydrophobic) lipids in the core of the particle, and a quite clear surface region comprised of polar lipids and the protein, but the interface between these regions was quite wide and not well-defined. This model should be a good starting point for further simulation studies of LDL particles.1225
Figure 66.
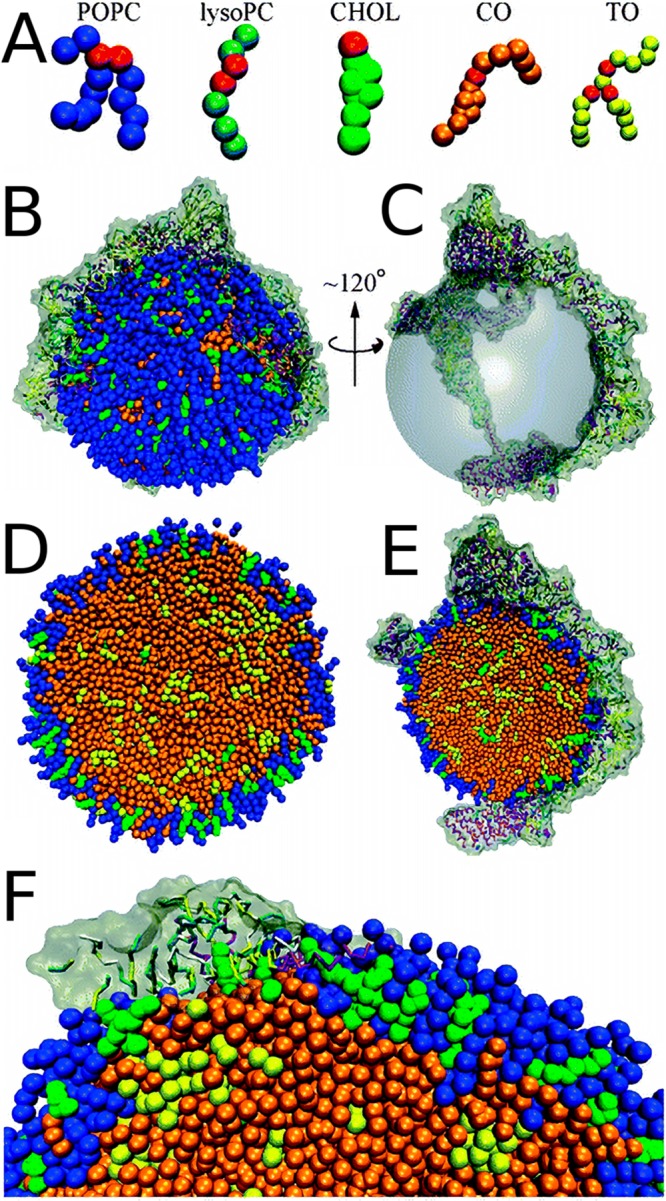
Spherical LDL model by Murtola et al.1225 (A) CG lipid models used in the simulations of LDL with glycerol moieties shown in red. (B) The full LDL model with ApoB-100 in tan. (C) The lipid core in the configuration in panel B is replaced with a transparent sphere to highlight the position of apoB-100. (D) Cross section of protein-free LDL. (E) Cross section of configuration shown in panel B. (F) The zoomed in surface of the configuration shown in panel B. Adapted with permission from ref (1225). Copyright 2011 the Royal Society of Chemistry.
High-Density Lipoprotein
High-density lipoproteins extract excess cholesterol from macrophages via pathways involving two ATP-binding cassette transporters in the reverse cholesterol transport. HDL protects against atherosclerosis through various mechanisms ranging from inhibiting lipid oxidation to anti-inflammatory and antiapoptotic functions.1226 A maturation scheme suggested for HDLs involves the following steps: (i) Lipid-free apoA-I associates with cholesterol and lipids in the bloodstream. (ii) This process leads to the formation of the nascent HDL (likely a discoidal particle). (iii) Lecithin cholesterol acyltransferase facilitates more cholesterol and lipid uptake, forming a mature spherical HDL particle.1227 Despite extensive studies on the structure and function of HDL particles, the specific protein and lipid configurations involved in each step during the maturation remain unknown, likely due to their natural diversity and flexibility. Here, we review the recent findings based on simulation studies on HDL models, covering lipid-free apo A-I, nascent, and spherical HDL models, separately.
Lipid-Free ApoA-I
Apolipoprotein A1 (apoA-I) is the major protein constituent of HDL. It stabilizes HDL particles during their maturation by forming a scaffold around the lipids (phospholipids, free cholesterol, CEs, TGs).1228 Moreover, the lipid-free monomeric apoA-I is vital for HDL biogenesis. Therefore, it is essential to characterize it structurally, which however has been quite unsuccessful despite all the efforts with X-ray crystallography and NMR.1229
Oda recently provided a historical overview of structural characterization of the lipid-free apoA-I. The study also covers potential reasons for the model diversity.1230 Various molecular modeling efforts have been undertaken to model lipid-free apoA-I in solution. These approaches were covered comprehensively in a recent review by Gogonea.1228 In short, the structural models by Silva et al.,1231 Pollard et al.,1232 Segrest et al.,1233 and Zhang et al.1234 feature several bundled α-helices that differ in their locations. These models conform to various sets of experimental data. On the other hand, the model by Legerstedt et al.1235 is the only one that also features β-strands, suggesting conformational switching from β-strands to α-helix upon lipid binding. While all four models can explain low-resolution experimental data to a reasonable extent, their validity is yet to be tested against high-resolution experimental data. Recently, a consensus time-averaged model of human apolipoprotein A-I in its lipid-free monomeric state was proposed by Melchior et al.1229 based on cross-link-derived constraints, SAXS, hydrogen–deuterium exchange, and crystallography data. Although the model was suggested by the authors to be consistent with much of the historically accumulated experimental data and carries implications about the lipid binding and HDL biogenesis, it still lacks the atomic scale resolution.1229
Among these lipid-free apoA-I models, those by Segrest et al.1233 and Zhang et al.1234 are particularly interesting as they capitalize on MD simulations to explore the configuration space of the protein as a tool for molecular modeling and refinement. Segrest et al.1233 and Zhang et al.1234 not only assessed the stability of the previous models1231,1232 using MD, but also employ high-temperature MD and iterative MD, respectively, to propose presumably improved models. The Zhang model contains a four-helix N-terminal domain. Similarly, the Segrest model is formed of α-helices bundled together with a loosely packed interior, representing a molten globule. Segrest et al.1233 further studied the lipid binding of this model by initiating a simulation with the coarse-grained version of their model in the presence of dispersed POPC. These simulations revealed the formation of a monolayer of POPC on the surface of the monomeric apoA-I. We note that the simulations were used as a tool for exploration of the protein conformational space to generate test conformations against low-resolution experimental data, that is, for molecular modeling purposes.
MD simulations have been used for other variants of apolipoproteins, for example, in modeling human apoA-IV,1236 in characterizing the effect of lipid concentration on apoC-II,1237 and identifying a lipid-binding motif in apolipoprotein B.1238
Nascent HDL Models
Nascent HDL (nHDL) particles manifest shape and size diversity since each particle can incorporate different numbers of apoA-I and lipids of various types. Therefore, any working model of nHDL has to account for and accommodate such intrinsic diversity. The majority of the models fit into a broad category termed the discoidal models of nHDL, which are mainly composed of a patch of a circular bilayer wrapped around by two apoA-I chains. The distinguishing feature of each discoidal model is the different conformation of the apoA-I chains. A second, more recent nHDL model was proposed based on small-angle neutron scattering (SANS) with contrast variation: the Double Superhelix model, in which the protein forms an open-ended spiral around an oblate lipid phase, which, unlike that in the discoidal models, is not exclusively a bilayer, but a lamellar–micellar mixture. Here, we concentrate on the simulation studies of nHDL since 2010 and refer the reader to the review by Gogonea1228 for the historical development of the models and a detailed account of the experimental protocols.
Discoidal HDL
Various crude models have been proposed for the structure of nHDL molecules: the picket-fence model, the double-belt model, the hairpin model, the looped belt model, and the solar flare model.1228 Among these, the double-belt discoidal model has been most extensively studied using MD simulations. Many modeling approaches for discoidal HDLs include an MD simulated annealing protocol, where the system is heated up and cooled down in successive stages. Simulated annealing allows crossing kinetic barriers resulting in better structural optimization. Gu et al.1239 combined experiments and the simulated annealing protocol to generate PL-rich HDL models with unesterified cholesterol wrapped by truncated apoA-I. In their model, apoA-I forms a flexible saddle-shaped double-belt structure around a central membrane. This saddle-shaped structure, by incrementally twisting, can accommodate a variable number of lipids in the central membrane.1239 Li et al.1240 explained the size heterogeneity of discoidal HDL by the presence of a hinged domain formed by a helix. Biased MD simulations were used to probe the independent rotation of two α-helical apoA-I monomers around the central lipid phase to assess the helix–helix registration between the monomers. These simulations, while supporting the existing double-helix models, also propose a mechanism for how two well-known naturally occurring apoA-I mutants (Paris, R151C, and Milano, R173C) can manifest different registrations.1241 In a later study, the conformational effects of these mutants, which contain extra disulfide bridges between the monomers, was also tested using unbiased MD.1242 The conformational states of the N- and C- termini of apoA-I were also modeled and studied.1243,1244 The N-terminal domain was suggested to drive particle expansion and the C-terminal domain, to be involved in the lipid exchange.1244 Segrest et al.1245 compared their previous discoidal models to the lipid-free apoA-I crystal structure and concluded their models to be valid. The most recent models of discoidal HDLs were generated by Pourmousa et al.1246 To characterize the tertiary structure of apoA-I in high and low cholesterol nHDL models, Pourmousa et al.1246 combined multi-microsecond atomistic MD, molecular modeling, and cross-linking experiments. The simulations captured how apoA-I adapts to growing particle size due to the N-terminal and C-terminal domains.1246
Segrest et al.1247 also studied the mechanism of discoidal HDL generation by the lipid-poor apoA-I. nHDL formation involves the ATP-binding cassette transporter A1 (ABCA1), which uses ATP to transport phospholipids from the inner leaflet to the outer leaflet. Surface density increase on the outer leaflet simulated using coarse-grained MD resulted in lipid monolayer pleating. The membrane-attached discoidal structures formed due to pleating appeared to be stabilized by apoA-I and influenced by membrane cholesterol (see Figure 67 for the details of the mechanism).1247,1248
Figure 67.

CG MD simulations capture how apoA-I stabilizes pleats formed on the outer leaflet due to different lipid densities between the leaflets. The left and right panels show the same configuration from two different angles. (A) First, a wedge-like structure forms, (B) which grows in time, (C) forming into a hemidisk. (D) ApoA-I wrapped disk connected to the bilayer with a stalk. Adapted with permission from ref (1247). Copyright 2015 Elsevier.
Several studies also concentrated on apoA-I mimetics and their conformations. A picket-fence arrangement was suggested for synthetic amphipathic peptides, which are indicated as potential therapeutics for cardiovascular diseases based on the MD simulations.1249,1250 For another apo A-I mimetic, reverse-4F, similar discoidal structures were captured, which tend to aggregate.1251
The Double Superhelix Model of nHDL
The double superhelix model of nHDL has been controversial as it substantially differs from the more widely accepted discoidal models listed previously. Here, we discuss the two separate studies by Gogonea et al.1252 and Jones et al.1253 that have employed MD simulations to assess the stability and the validity of the models. These two studies arrive at two different conclusions, the first being in favor and the latter being against the model.
To test the validity of the double superhelix model, Gogonea et al.1252 performed 80 ns long MD simulations using the united-atom force field GROMOS87 with POPC, cholesterol, and SPC water molecules in the NVT ensemble at 300 K. Based on root-mean-square deviation and solvent accessible surface area calculations, the protein conformation did not change substantially after 10 ns, while the lipid core evolved from a prolate ellipsoid to a spheroid. This shape change was accompanied by compression of the helix along its axis and the formation of a void in the lipid core. The authors rationalized this change in particle dimensions, which contradicts SANS and electron microscopy (EM), by referring to previous MD simulations of micelles, where such voids and shape changes were also observed and attributed the effect to the artifacts in the lipid force fields.1252
On the other hand, Jones et al.1253 contested the Double Superhelix model. In contrast to the simulations of Gogonea et al.,1252 Jones et al. performed simulations in the NPT ensemble using the all-atom Charmm22 force field for the protein and the lipids and the TIP3P water model. Moreover, they used a simulated annealing protocol, in which the system was heated up to 500 K and gradually cooled ddown to 310 K twice in a total of 60 ns. They also performed coarse-grained simulations of the complex using the MARTINI force field for ∼20 μs. In addition, they obtained the simulated model from Gogonea et al. and extended the simulations for 10 ns using the force field and simulation parameters used for the simulated annealing simulations. Both the coarse-grained and the simulated annealing simulations resulted in the formation of discoidal bilayers with the termini of the belt still separated, likely due to the short time scales of the simulations and kinetic trapping. Subjecting the Gogonea et al.1252 system to the same conditions, which involve pressurizing the system in the NPT ensemble, results in loss of voids seen in the model and expulsion of waters from the interior within 5 ns. These studies suggest that the original Double Superhelix model is unstable and that the discoidal structures are more likely to represent nHDL configurations. These simulation studies were also supported by the negative stain and cryo-electron microscopy tomography experiments, which showed that the reconstituted apoA-I/HDL particles are discoidal, and by FRET experiments, which showed the distances between the terminal domain residues to be much shorter than those predicted by the Double Superhelix model.1253
Spherical HDL Models
The reverse cholesterol transport proceeds by the maturation of nHDL to spherical HDL (sHDL). Lecithin cholesteryl acyltransferase, a plasma enzyme, esterifies the free cholesterol on the nHDL surface producing CEs, which translocates to the core of the particle. As the particle assumes a spherical shape with the accumulation of cholesteryl ester molecules in the lipid particle core, more apoA-I associates with the surface of the particle, amounting to as many as 7 apoA-I per particle.1228
In 2010, Yetukuri et al.1254 performed a lipidomics profiling of HDL fractions from subjects with high and low HDL using ultraperformance liquid chromatography–mass spectrometry (UPLC/MS).1254 They determined that subjects with low HDL have elevated triacylglycerols and diminished lysophosphatidylcholines and sphingomyelins. They combined these lipidomics data with molecular modeling and coarse-grained MD simulations to study the spatial distribution of lipids within the HDL particles with three different compositions corresponding to those of the subjects with low, high, and normal HDL (referred to as low, high and normal HDL systems, respectively, from here on) (Figure 68). The spherical HDL models were prepared by placing the almost entirely α-helical full-length model of apoA-I around a pre-equilibrated lipid droplet, whose composition was later adjusted to the desired state. In 8 μs of coarse-grained simulations, they observed a smaller particle size for the low HDL system (Figure 68A). In the high HDL system, cholesterols appeared to preferentially interact with apoA-I (Figure 68A, center). While the number of contacts between apoA-I and sphingomyelin (SM), cholesterol, or lysophosphatidylcholine (lysoPC) increases from low to high HDL, those of CEs, TAG, and PC decreases (Figure 68B). TAGs accumulated near the surface relatively more in the low HDL systems as indicated by the tail of the radial distribution function and higher number of contacts with water (Figure 68C). Although the simulations did not account for the potential particle-size dependent variation of the number of apoA-I adsorbed onto the surface of the particles, this effect is only likely to result in even more significant differences in the spatial distribution of lipids.1254 A later study explored the impact of drug treatment on the lipidomic profiles. Drug treatment increased the amount of apoA-II and sphingomyelins and decreased the amount of lysophosphatidylcholines. The coarse-grained MD studies based on the experimental findings revealed that a growing number of apoA-IIs remodels the HDL by reducing the neutral lipids, like CEs, on the HDL surface.1255
Figure 68.

SHDL models based on the lipid compositions of subjects with different levels of HDL and the CG simulations. (A) The CG simulation snapshots after 8 μs equilibration. ApoA-I proteins are shown in red and green, cholesterol in yellow, and other lipids in gray. Waters are not shown for clarity. The localization of cholesterols near apoA-I in the high HDL-C case is visible in the second panel from the left. (B) Unnormalized number of contacts between apoA-Is and lipids. The errors bars denote the standard deviation. (C) Radial distribution function of TG molecules around the center of mass of the HDL particle. The inset shows the average number of water contacts of TG. Reproduced with permission from ref (1254). Copyright 2010 the American Society for Biochemistry and Molecular Biology.
Another study that investigated the structure and dynamics of sHDL particles, emphasizing the importance of lipid composition and the role of lipids, came in the same year. Vuorela et al.1256 performed multi-microsecond coarse-grained MD simulations of two lipid droplet models: a lipid droplet without apoA-I and the full HDL particle including two apoA-I molecules surrounding the lipid phase. The main finding of this study is that the widely accepted two-layer model cannot explain the observed conformation and dynamics of the lipids. Instead, the study proposes a three-layer model. The models were designed to mimic the size and lipid composition of human serum HDL, composed of free cholesterol, CEs, TGs, phospholipids, and lysolipids. The lipid phase appears to be divided into three regions; the core, the surface, and the intermediate region characterized by different dynamics, compositions, and conformational entropy of the components. The center is mainly composed of TGs and CEs, while POPC and lysophosphatidylcholine headgroups form the surface layer. Cholesterol, some CEs, and the acyl chains of phospholipids constitute the intermediate region. While the presence of apoA-I did not alter the shape of the particle substantially, it affected the surface lipids, especially decreasing the conformational order of CEs and cholesterol. The study puts forward the conformational entropy of the lipids as a factor in the formation of sHDL.1256
Simulations of reconstituted and circulating HDL particles with different lipid compositions and numbers of apoA-I suggested that apoA-I covers most of the HDL surface. Moreover, the simulations capture that circulating and reconstituted sHDL contain an equal amount of core lipids, but circulating sHDL has fewer surface lipids. The differences in lipid compositions appear to result in substantial differences in the particle surface, and the authors concluded that the reconstituted sHDL may not be suitable models for the circulating sHDL.1257
Not only the lipid composition but also pH has been suggested to affect properties of bilayers such as their thickness and negative curvature with implications on surface properties of LDs and lipoproteins.1258
The surface pressure and interfacial tension are essential determinants of the shape and the protein/enzyme adsorption onto the surface of HDL and LDL particles. Ollila et al.1213 used coarse-grained MD simulations to estimate the quantities mentioned above for protein-free lipid droplets (using both trilayer and spherical models) and in HDL and LDL particles mimicking physiological conditions. While the MARTINI model appears to underestimate the surface pressure, the dependence of interfacial tension on the curvature of the particle could be assessed. Presence of apoA-I on the surface was not found to affect the interfacial tension in the HDL and LDL models. The high interfacial tension indicates that HDL and LDL represent kinetically stabilized metastable states.1213
Koivuniemi et al.1259 investigated the effect of core lipid composition and apoA-I on the interfacial tension and the concentration of core lipids in the surface monolayer, building upon the study by Ollila et al.1213 The simulations performed with different lipid compositions revealed that TGs intercalate into the monolayer and reside close to the water molecules much more often when compared to CEs. TGs also exhibited slower exchange between the monolayer and the core when compared to CEs. The higher water accessibility of TGs makes them better targets for enzymatic activity, such as transportation by cholesteryl ester transfer protein (CETP). Furthermore, CEs lowered the interfacial tension stabilizing the droplets. In contrast, TGs helped maintain the interfacial tension despite changes in the composition of the surface monolayer.1259
Cholesteryl ester transfer protein facilitates the transfer of CEs from HDL to LDL and VDL, as well as TG transfer in the opposite direction. Although CETP deficiency is linked to high HDL cholesterol levels,1260 it is also implicated in increased coronary heart disease incidence.1261
Koivuniemi et al.1212 investigated the lipid exchange mechanism of CETP using atomistic and coarse-grained simulations. The simulations captured CETP binding to the HDL surface with its charged and tryptophan residues. The inherent flexibility of CETP appeared to enable its binding to lipoproteins with varying curvatures. Moreover, a closed to open conformational transition was proposed to be modulated by a helix (helix X) acting as the lid for the CETP tunnel. CETP appeared to have a flexible structure, which was suggested to enable its binding to the curved lipoproteins. The tryptophan residues inserted into the headgroup region and salt bridges between the POPC and the charged residues stabilized the binding. Coarse-grained simulations also captured the formation of a hydrophobic patch connecting the core to the surface. Reduction of the surface to core lipid ratio resulted in the interactions between core CEs and the CETP. Helix X appeared to form a mobile lid, controlling the accessibility of the CETP tunnel.1212 In a follow-up study, Äijänen et al.1262 investigated the mechanism of CETP inhibition by the drug candidate anacetrapib. The authors showed that the anacetrapib binding site is located in a tunnel near the N-terminal opening inside CETP and that anacetrapib locks CEs inside the protein preventing their translocation.1262
Cilpa-Karhu et al.1263 performed atomistic simulations to explore the interactions of CETP with the double superhelix model of nascent HDL as well as the spherical HDL. We note here that the authors employed a noncanonical combination for force fields, transferring parameters between GROMOS and Berger. Their results suggest a different mechanism than the curvature-matching model suggested by Koivuniemi et al.,1212 in which CETP does not penetrate below the headgroup region. Their model involves CETP in an upright orientation. MD simulations captured CETP penetrating the HDL particle surface and the opening of the N barrel domain end of CETP. Moreover, instead of helix X proposed as a lid by Koivuniemi et al., Cilpa-Karhu et al. suggested the N barrel domain of CETP as a lid for lipid transfer. Based on these results, the authors proposed a tunnel mechanism for the transfer of CEs from HDL.1263
Combining electron microscopy with MD, Zhang et al.1264 showed that CETP binding to HDL particles involves hydrophobic interactions and does not depend on the individual lipid or protein components of HDL and that CETP is only bound to one HDL at a time. The experiments showed that CETP interaction with HDL/liposome surface does not involve apoA-I or apoA-II and is stronger for smaller liposomes. Coarse-grained MD simulations in this study were used to study the dependence of surface hydrophobicity on the liposome radius. The results confirmed that smaller liposomes have higher hydrophobicity than the larger ones. Altogether, instead of a shuttle mechanism, the MD and experimental studies favor the tunnel mechanism for the transfer of CEs from HDL to LDL particles, where CETP dissociates from the HDL surface after getting loaded and associates with LDL. Their results are also consistent with the results of Cilpa-Karhu et al.1257 about the role of the N barrel domain of CETP.1264
Karilainen et al.1265 studied the effect of attaching the fluorescent probe BODIPY to CEs in HDL particles. They compared the structural and dynamical properties of BODIPY-CEs to unlabeled CEs in spherical HDL models using unbiased all-atom simulations. To characterize the partitioning of the labeled and unlabeled CEs, they also performed replica exchange umbrella sampling simulations (REUS) in which they chose the distance between the center of mass of HDL and the center of mass of the CEs or BODIPY as the reaction coordinate. The simulations did not capture any significant effect of BODIPY on CE partitioning between the water phase and HDL. On the other hand, labeled CEs oriented so that the BODIPY moiety interacted with the headgroup region. The labeled CEs were localized closer to the surface of the particle due to the competing effects of the hydrophilic BODIPY and the hydrophobic ester. Moreover, the BODIPY moiety appeared to slow down the diffusion of CEs. These results suggest that the BODIPY probe widely used in experiments may not be inert and may lead to qualitatively different observations in studies of spherical HDLs.1265
Oxysterols are oxidized cholesterol derivatives. Since oxidation impairs cholesterol uptake from the cells by HDL and is likely to play a signaling role in cholesterol transport, Karilainen et al.1266 investigated the effect of oxidation on spontaneous cholesterol partitioning into the spherical HDL using REUS all-atom simulations. Reconstructing the free energy profiles for transfer of cholesterol and an oxysterol (7-ketocholesterol) from the water phase into the HDL particle, they showed that both cholesterol and the oxysterol exhibit comparable minima and localize to the surface of the particle. The authors proposed the surface localization of the oxysterols as a potential mechanism for how oxysterols may be involved in signaling.1266
9.2. Energy Storage in Electrochemical Gradients
Physiologically important ions are not in electrochemical equilibrium across the cellular membranes. Active membrane transport proteins embedded in cellular membranes create and maintain this nonequilibrium condition. Without such active maintenance, the ionic gradients would dissipate over time due to downhill diffusion of ions across the membrane through ion channels and, despite being slower, also through the lipid bilayer. Dissipation of ionic gradients is, indeed, equivalent to cell death.1267
Two types of active membrane transporters are involved in the creation, maintenance, and modulation of ionic gradients. Primary active membrane transport proteins, such as Na+/K+ and Ca2+/H+ ATPase pumps catalyze ATP hydrolysis and couple the released energy to pumping ions against their electrochemical gradients. Secondary active membrane transport proteins, such as Na+/Ca+, Cl–/HCO3–, or Na+/H+ exchangers, on the other hand, couple the energy stored in the established electrochemical gradient of one ion to transporting another ion up its electrochemical gradient.1267 Secondary active transport proteins also exemplify a major mechanism of how cells use the energy stored in electrochemical gradients; that is, for uptake of important molecular species like nutrients or expulsion of deadly chemicals such as antibiotics against their concentration gradients. We refer the reader to the comprehensive review of the computational studies on active membrane transporters that we presented in section 6.
The mechanism mentioned above for generating ion gradients as intermediate energy storage relies on energy stored in ATP or other electrochemical gradients. All living cells, on the other hand, have to create ATP from an external source. Chemiosmotic theory, proposed originally for ATP synthesis in mitochondria by the Nobel Laureate Peter D. Mitchell in 1961, states that the proton gradients act as the energy reservoir in the last stage of aerobic cellular respiration. The chemiosmotic coupling is now established as a standard mechanism for ATP production by mitochondria in eukaryotes, by chloroplasts in plants and algae, and by bacteria and archaea.662
Here, we review the computational studies performed on the characteristic components of the mitochondrial membranes. We first describe the simulations performed on cardiolipin and cardiolipin-containing membranes, as cardiolipin is an essential and unique component of mitochondrial membranes. Then, we move on to examine lipids that modulate the electron transfer chain’s components, membrane enzymes that intricately couple redox-reactions to generation of proton gradient across the inner mitochondrial membrane. For in-depth understanding of cellular respiration, we refer the reader to reviews on the electron transport chain members.1268,1269
9.2.1. Cardiolipin and Cardiolipin-Containing Membranes
Cardiolipin (CL) is a unique component of the inner mitochondrial membrane serving various structural and functional roles. Here, we cover the computational studies performed on CL and CL-containing membranes. These include quantum mechanical studies aimed at generating force field parameters and exploring the titration states. Various coarse-grained and atomistic studies have aimed to characterize the mechanical properties of CL-containing membranes. The structural and mechanical properties of the mitochondrial inner membrane are of particular interest, as the invaginations and buckles of the inner membrane, called cristae, serve as the metabolic platforms for ATP production. The effects of counterions and lipid oxidation have also been discussed briefly in the literature. The unique titration properties and their association with oxidative phosphorylation proteins have suggested that CL increases the efficiency of these machines by acting as a “proton sink” on the membrane surface.1270 Last, we discuss how simulation studies provides insight into the role of CL as a “proton sink”.
Many groups have undertaken efforts to parametrize CLs for simulations. Lemmin et al.1271 parametrized CL at different physiological protonation states, that is, with unprotonated or singly protonated phosphate groups. The parametrization scheme was designed to be consistent with the AMBER and CHARMM force fields. The models reproduced the various biophysical properties of the membranes, such as the lateral diffusion coefficient and the area per lipid, and captured the titration state-dependent (therefore, pH-dependent) changes in lipid packing. This effect was discussed in the context of membrane shape and formation of a proton sink, whose implications about on-surface proton diffusion we have briefly touched on above. The simulations also showed differential CL interactions with monovalent and divalent cations.1271
Dahlberg et al.1272 explored the CL headgroup structure and the proton exchange mechanism between the phosphate groups using density functional theory calculations of the CL headgroup, and its 2′-deoxy derivative, also considering the effect of solvation. They calculated a large value gap between the two pKa values. The proton exchange between the two phosphate groups appeared to have a barrier of about 4–5 kcal/mol.1272 These calculations are in agreement with the long established model of CL titration state and supports its role as proton sink.123 We note, however, that a recent biophysical study contested this, showing that both CL headgroup phosphates are strong acids and remain unprotonated in the physiological pH range.126
Aguayo et al.1273 developed a new parameter set for two versions of CL (tetramyristoyl and tetraoleoyl) for simulations of CL-containing mixed-lipid membranes combining all-atom headgroups with united atom acyl chains within the CHARMM force field framework. The models were reported to agree well with other simulations. The presence of CLs in mixed bilayers results in membrane thickening and acyl chain ordering. The new models reduced the number of atoms by half, while still capturing important hydrogen-bonding interactions. This is especially helpful for building sufficiently large models to study the unique mechanical properties of mitochondrial membranes.1273
Structure of CL-containing bilayers has been investigated in only a few studies up to date. Earlier MD investigations of CL-containing membranes have shown that CL increases membrane order and reduces lipid diffusivity.1274−1276 Dahlberg and Maliniak1277 investigated the mechanical properties of CL bilayers with varying lipid compositions including zwitterionic DOPC or DOPE, and different CL headgroup charges. Their coarse-grained simulations showed that low CL charge reduces the bending moduli and results in curved microdomains.1277 Boyd et al.1278 reported that CL increases bilayer deformability and gets enriched in highly negatively curved regions of the membrane. These findings were based on coarse-grained simulations, where they probed membrane buckling by applying lateral pressure on the mitochondrial bilayer models. These effects appeared to be unique for CL, as they could not be reproduced by the other inverted conical lipid in mitochondria, namely, phosphatidylethanolamine (PE).1278
Pan et al.1279 performed atomistic MD simulations of CL membranes constrained at different surface areas and selected the model that best reproduces the experimental scattering data. The chosen MD-based model also revealed important Na+–membrane and lipid–lipid interactions.1279 An earlier computational investigation by Pöyry et al.1274 suggested that Na+ and Ca2+ ions do not significantly influence the structural properties of CL-containing membranes but alter their dynamical properties, such as rotational and lateral diffusion of lipids.1274
Boscia et al.1280 showed that CL increases the bending modulus of the membranes and stiffens them. They obtained X-ray structures of pure gel and fluid tetramyristoylcardiolipin (TMCL) and simulated pure DMPC and DMPC and TMCL mixed membranes to help in interpretation of the X-ray results. Due to their bacterial ancestry, mitochondrial membranes contain little cholesterol. The findings of Boscia et al.1280 revealed that CL indeed takes on various functional roles of cholesterol. While TMLC appeared not to exhibit domain formation and aggregation, it increased the thickness of the membrane and reduced water leakage, which are established roles of cholesterol. Besides, the phosphocholines formed an umbrella covering the TMCL headgroup, as they do for cholesterol.1280
CL oxidation can potentially cause various malfunctions due to altered protein interactions and membrane properties, and diminished capacity as a proton sink. On the other hand, CL has to function in an environment conducive to oxidation. Vähäheikkilä et al.113 explored the effect of cardiolipin oxidation on the bilayer properties. They parametrized oxidized linoleic acids and performed atomistic simulations of mixtures of DLPC, DLPE, and CL. Oxidation resulted in thinning of the membrane and increase in its area. The oxidized chains moved closer to the membrane–water interface. Moreover, the oxidized chains appeared to influence the chain order of unoxidized lipids.113 These results suggest that CL oxidation may effect activation of mitochondrial proteins that bind CL, for example, in an allosteric fashion.
Kaurola et al.1281 investigated the dynamics and distribution of quinones and quinols and their chain length variants (collectively referred to as Qs from here on) in a three-component mitochondrial membrane model composed of PC, PE, and CL with atomistic MD simulations (Figure 69A). The simulations showed that both quinones and quinols partition below the lipid headgroups near the acyl groups (Figure 69B). Quinones, however, exhibit a strong peak also in the middle of the bilayer (Figure 69B). Free energy calculations also support these conclusions (Figure 69C). In addition, Qs mostly interact with PC molecules instead of PE or CL likely due to the large headgroup of PC shielding Qs from unfavorable water interactions (the “umbrella effect”). Furthermore, translocation of quinones through the bilayer is very fast (10–100 ns), while that of quinols is an order of magnitude slower. This is relevant for the efficiency of the electron transport chain, as quinones formed on the P-side of the membrane, which is named after the periplasmic side for bacteria corresponding to the intermembrane space in mitochondria, need to translocate rapidly to the N-side to be reduced.1281
Figure 69.

Dynamics and distribution of quinones and quinols in the membrane. (A) The chemical structures of (left) ubiquinone (oxidized Q, Qox) and ubiquinol (reduced and protonated Q, QH2), where n indicates the variable isoprenoid chain lengths; (right, top to bottom) cardiolipin, DLPC, and DLPE. (B) Density profiles of (top) QH2 and (bottom) Qox for different isoprenoid chain lengths. The density of phosphorus atoms of DLPC is shown in gray. (C) Free energy profiles for (top) QH2 and (bottom) Qox along the membrane normal for quinone with 1 and 10 isoprene monomers. The origin is set to the center of mass of the membrane. Adapted with permission from ref (1281). Copyright 2016 Elsevier.
CL has long been suggested to form “a proton trap for oxidative phosphorylation”.123 Bulk-to-bulk proton motive force was shown to be inadequate for the experimentally measured ATP yields. Thus, protons were suggested to accumulate on the membrane surface, separated from the bulk by a free energy barrier. Wolf et al.1282 applied an MD protocol that accounts for excess proton in the Grotthuss proton shuttling mechanism. Their findings revealed that excess protons have a high affinity for the membrane surface, where they interact strongly with the oxygen atoms in the headgroup phosphate and carbonyl groups. The simulations characterized three proton diffusion modes resulting in anomalous diffusion: (i) binding to a lipid and diffusing with it; (ii) hopping between water clusters associated with the lipid headgroups; and (iii) escaping to the bulk and diffusing through the bulk. The findings imply that on-surface diffusion of protons can be enhanced with respect to the counterproductive bulk diffusion in the presence of phosphatidylethanolamines and CL. These lipids have smaller headgroups than DMPC used in the study; thus, they can mediate proton hopping between headgroup-associated water clusters more efficiently.1282 The effect of lipid headgroup on the behavior of excess protons was also studied using the multistate empirical valence bond method.1283
9.2.2. Cardiolipin Interactions with Oxidative Phosphorylation Proteins
The role of CLs in stabilizing integral membrane proteins including all respiratory chain complexes responsible for generating the proton motive force is discussed in detail in a recent review by Musatov and Sedlák1270 The review concludes that CL acts both as a “glue” stabilizing the functional assembly of subunits and as a linker allowing communication between distant sites on the proteins.
CL not only stabilizes cytochrome bc1 complex and cytochrome c oxidase,1285 but it is also involved in proton transport.123,1286 The interactions of CL with these two complexes characterized by MD put forward how CLs might be involved in the proton transfer. Pöyry et al.1284 performed atomistic simulations of a bacterial cytochrome bc1 dimeric complex embedded in a three-component membrane model composed of POPC, POPE, and CL to explore the CL interactions with the protein. The lipid environment appeared to influence the structure of the complex, resulting in opening up on the P side. This conformational change is likely driven by the positively charged residues on the P side adapting to the negatively charged lipids. Remarkably, the simulations revealed that the CLs move to the dimer interface and localize close to catalytic Qi-sites, occupying similar positions captured in the crystal structures (Figure 70A). The proximity to the active site suggests a mechanism for CL involvement in proton transfer via hydrogen-bonded networks (Figure 70B).1284 Coarse-grained simulations of cytochrome bc1 from bovine and yeast mitochondria by Arnarez et al.1287 also identified six reproducible buried and surface CL binding sites. The surface binding sites were located on the M-site region, supporting CLs role as a proton sink. Arnarez et al.1287 used coarse-grained simulations to characterize the CL binding site, also, on the cytochrome c oxidase. They identified seven CL binding sites, two of which were shown through PMF calculation to have strong affinity. These two sites appeared to be in proximity to two of the three known proton channels (D- and H-), implicating CL in proton uptake.1287 The role of lipids in proton uptake was also supported by Sharma et al.,1288 who performed atomistic simulations of cytochrome c oxidase to investigate the effect of conserved bound phospholipids on subunit III. They captured water wire formation between the phospholipid lipid headgroups and the proton uptake site, enhancing its proton affinity.1288 Overall, the simulations have not only helped characterize important CL binding sites but also strongly support its role as a “proton sink”.
Figure 70.

Mechanism of CL involvement in proton transfer in the cytochrome bc1 dimeric complex. (A, left) CL molecule (cyan) and cytochrome bc1 (CIII) dimer (cartoon) interface captured in MD. (A, right) The small figures show the divergent interactions of CL with nearby residues captured in different simulations. (B) Putative proton conduction pathways (magenta) mediated through waters and a lysine residue between the CL (cyan) and the water in the quinone-binding site. The protein is not shown, and the heme is shown in black. The right-most figure shows the same configuration as that on its left except colored based on the electrostatic potential (red, negative; blue, positive). Reprinted with permission from ref (1284). Copyright 2013 Elsevier.
There is a growing consensus on the formation of higher order supercomplexes by the respiratory chain elements, also called a respirasome. This organization is likely linked to CL in mitochondria (or PE in bacteria).1289 Coarse-grained simulations of cytochrome bc1 complex by Arnarez et al.1290 had earlier hinted at CL driven supercomplex formation.1290 In a later study, Arnarez et al.1291 investigated how membrane CL glues the respiratory supercomplexes using self-assembly coarse-grained simulations. They embedded 9 cytochrome bc1 dimers and 27 cytochrome c oxidase monomers in a CL-containing bilayer to mimic the crowded environment. The simulations captured oligomerization events that reproduced the known interfaces as well as an alternative one. The oligomerization interfaces featured bound CLs, and free energy calculations showed that CLs bind stronger to the identified binding sites than any other mitochondrial lipid.1291
CL has also been implicated in mitochondria-mediated apoptosis functioning together with cytochrome c. A recent study combining NMR, fluorescence spectroscopy, and coarse-grained MD showed that CL is required for membrane binding of cytochrome c, which leads to CL clustering initiating apoptosis.1292
9.3. Challenges
Simulation studies have made significant advances in the last two decades due to the increasing computational power and better models. In this section, we reviewed how recent simulations have contributed to our understanding of energy storage mechanisms. Energy storage along with its production is one of the hallmarks of living cells, and biomembranes are involved in almost all its aspect. An organelle called a lipid droplet is the major site of energy storage in mammalian cells and is linked to metabolic disorders. MD simulations have made great contributions to modeling the LD surface and characterizing the physical properties of its membranes. However, the mechanisms leading to lipid droplet formation are weakly understood, if at all. Experimental evidence suggests that there are proteins such as seipin playing key roles in lipid droplet formation. With very recent characterization of the structure of seipin,1293,1294 simulation studies can now investigate the mechanism of seipin-mediated LD formation. Similarly, simulations have been of utmost importance in understanding lipoproteins, particularly in generating models that conform to low resolution experimental data. Plasma lipoproteins are integral to energy and cholesterol metabolism, and computational research to understand lipoprotein structure and dynamics has been quite active in this context. On the other hand, lipoproteins remain challenging to investigate computationally, due to their large size, their molecular complexity, and the lack of sufficient structural information on how their components interact. The overall structure of LDL and that of apoB-100, the primary protein component of the LDL, remain virtually unknown, despite some low resolution models.1295−1297 Moreover, the interactions of LDL and HDL with their receptors constitute an essential step in their endocytosis and, thus, their metabolism. However, these processes are still challenging for simulation studies. We expect that with improving MD methodologies, models capturing more complexity are likely to come. The machinery interacting with these lipid storage particles have various health related implications. With better models and improving time scales, MD simulations can be used to investigate the interactions of enzymes and other biomolecules with these particles.
In the second part of this section, we covered how simulations contributed to elucidating the mechanism of ATP-generation by cells. For this, we concentrated on the mitochondrial energy production mechanism. Due to the availability of immense biochemical information distilled through years, this machinery is very well characterized structurally and functionally but does not fail to surprise us with how much we do not know. Various levels of computational theory have been applied to characterize the dynamical nature of the electron transport chain components. Here, we limited our discussion to lipids and lipid–protein interactions. Cardiolipin appears as a fundamental functional part of these complexes in the mitochondria, being involved in electron transport and protein organization. However, how CLs are directly involved in proton uptake has not been studied directly. This requires quantum chemical studies, and it would definitely benefit from developing MD methodologies like the constant-pH simulations. Needless to say, the role of the lipid environment is also established now as an active part of energy storage in the gradients of protons and other ions, on top of its passive role as a barrier in creating and maintaining the electrochemical gradient reservoirs.
10. Are Biomembranes Metabolic Platforms?
A group of transmembrane proteins, transmembrane enzymes, are encoded by 533 genes in the human genome.232 Among others, these include 194 transferases, 178 hydrolases, and 123 oxidoreductases. Yet, enzymatic activity is not limited to these 533 enzymes, but it is also part of the function of some membrane receptors, such as tyrosine kinase type receptors and active transporters. Additionally, there are many peripheral membrane enzymes; however, their exact number is unknown.
Quite often enzymes that are well-known and established, later turn out to be membrane associated. For example, L-Dopa decarboxylase, the enzyme responsible for dopamine synthesis, was shown to be associated with membranes in the cellular environment, and this observation was made long after its discovery.280 There are also enzymes that are not present in humans but still abundant in nature, such as those involved in photosynthesis, which is an entirely membrane-associated process similar to oxidation. Further, the complexity and the size of these enzymes can be quite large, such as in the case of the respiratory supercomplex and photosynthetic reaction centers and antennas.
Here, we discuss computational studies of membrane enzymes with an emphasis on the role of membrane and specific lipid–protein interactions on the functions of this group of proteins. The main objective of this section is to show that membranes should not be neglected, as has been done in many previous studies of membrane enzymes.
10.1. Do Lipids Always Play a Role in Enzyme Activity?
Computational studies of transmembrane or peripheral enzymes often consider those aspects of the enzyme activity in which lipids seemingly do not play any significant role. For example, mainly due to practical limitations, enzymatic catalysis mechanisms studied with quantum-mechanical calculations typically ignore the environment beyond the catalytic pocket. The lipid environment is usually also disregarded in the design of inhibitors that would block the catalytic pocket of enzymes. In this respect, MD simulations are often used in combination with protein crystallography to provide insight into the binding modes of substrates and inhibitors, as well as to reveal possible effects of point mutations (see, for example, ref (1298)). However, because membranes likely participate in substrate and inhibitor entrance into the binding cavities of membrane enzymes, omitting the membrane environment can be misleading. In fact, computational studies of various substrates in a membrane environment, such as quinones and quinols in the respiratory and photosynthetic membrane complexes, have captured the locations used to enter the catalytic cavities and the optimal positions of these molecules.1281 Further examples are discussed below.
Moving on, channels present in membrane enzymes for gas molecules such as oxygen, hydrogen, nitric oxide, and water, are frequently studied in MD simulations. However, most of these studies, although performed in lipid environment, have not considered the involvement of lipids.1269,1299−1310 Similarly, proton and electron channels studied by classical simulations have typically disregarded lipids,1269,1311−1313 though lipids such as cardiolipin have been shown to be potential proton donors in cytochrome bc1,1314 and lipids associated with cytochrome c oxidase likely play a structural role in maintaining the oxygen patches inside the protein.1288
Considering the examples discussed in this section related to membrane-associated enzymes where the specific role of lipids has been recognized, it is clear that lipids may play a role in all of the above-mentioned processes. Moreover, lipids can also act as allosteric regulators. For example, cholesterol has been shown to significantly reduce the conformational space explored by β2AR.54 For this reason, one can imagine that the channels for gas molecules observed inside the proteins located in single-component membranes may not exist at all in membranes whose lipid composition is realistic. Thus, the take-home message is that future studies should account for the lipid involvement in the function of enzymes.
10.2. Membrane Enzymes Involved in Xenobiotic and Small Molecule Catabolism
Cytochrome P450 (CYP) proteins constitute a family of single-helical membrane enzymes with multiple functions in both catabolism and anabolism.1315 In humans, there are 41 CYPs, which are involved in the synthesis of cholesterol, steroid hormones, bile acids, vitamin D3, and second messengers derived from polyunsaturated lipids as well as from the degradation of naturally occurring small molecules such as neurotransmitters, various xenobiotics, and drugs.1316−1318 In fact, enzymes of the CYP family form a major group responsible for drug metabolism catabolizing approximately 50% of all approved drugs.1317−1320 Several crystallographic structures of CYPs have been solved, but the structures that have been determined are mostly for the water-soluble domains of the proteins. Unfortunately, only one of the crystallographic structures includes also the structure of the transmembrane helix.1321 For this reason, a large number of computational studies of CYPs have been performed without the transmembrane domain in the absence of the membrane (see, for example, ref (1322)). Here, we concentrate on simulation studies, in which the membrane was included.
Lonsdale et al. performed coarse-grained simulations to identify the optimal arrangement of the transmembrane and water-soluble domains of CYP around the membrane and then initiated atomistic simulations from the configurations most frequently found in the coarse-grained simulations.1323 Atomistic simulations showed that the flexibility of the membrane-bound protein does not differ significantly from the protein simulated in water. However, the tunnels used by the substrates and the reaction products to enter and exit the catalytic pocket were different between the membrane-bound and the aqueous structures. In other studies, MD simulations were used to compare the tunnels in the human and human parasite Trypanosoma brucei CYP51s. The subtle differences captured in the simulations may aid in optimization of drugs to specifically and exclusively target the parasite CYP51.1324
In another study, association of CYP 12A with lipids induced the opening of the tunnel 2A, which allows the substrates to enter the protein from the membrane.1325 A similar observation was made for CYP 17A1.1326 Extensive unbiased MD simulations, randomly accelerated MD, and free energy calculations characterized the entrance pathway and the binding pockets of testosterone in CYP 3A4 (see Figure 71), as well as the free energy landscape along the entry pathway.1327
Figure 71.
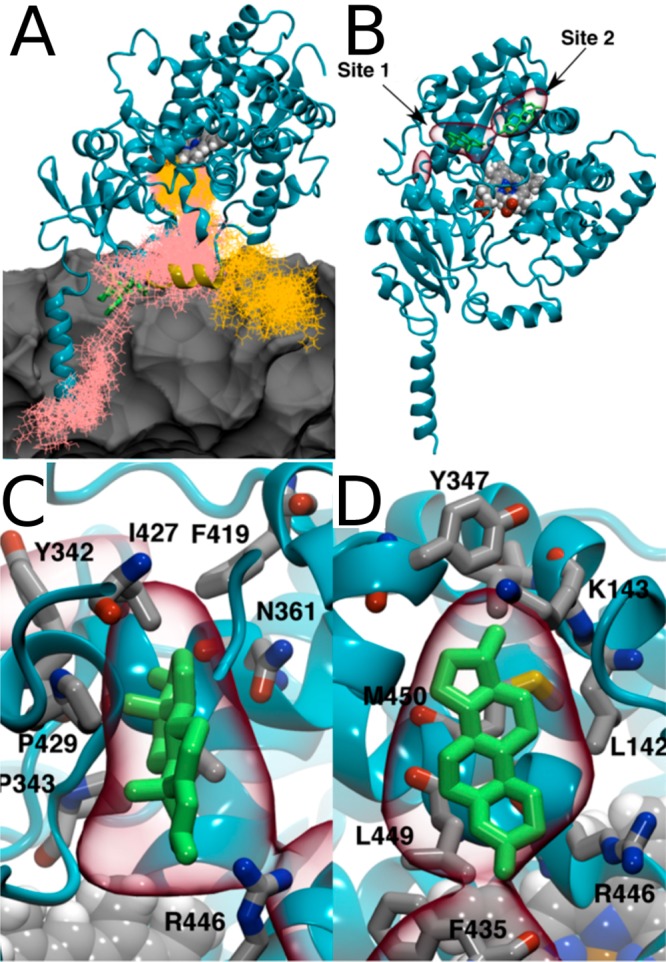
Snapshots of two testosterone molecules (pink and orange) in the windows prior to their docking into the catalytic pockets. Lipids are shown as a gray surface (A). Location of testosterone binding sites (B) and their close views (C, D) with volumetric maps (red lines) highlighting the highest testosterone mass-weighted density over 25 μs of a MD simulation. Representative testosterone configurations are shown in green stick representation. Reproduced with permission from ref (1327). Copyright 2018 the American Society for Biochemistry and Molecular Biology.
A few other studies have also pointed out the importance of the membrane composition in protein stability and CYP positioning in the membrane (Figure 72). Negatively charged PS and PG were shown to promote the penetration of CYP deeper into the membrane. PS and PG also promoted a more tilted CYP orientation, when compared to simulations of CYP in bilayers composed only of PC or PE.1328 In the presence of cholesterol, the cholesterol molecules were observed to accumulate in the vicinity of the protein, causing reorientation of the water-soluble domain. This, in turn, led to tighter interactions with the membrane and to a decrease in the protein flexibility.1329
Figure 72.
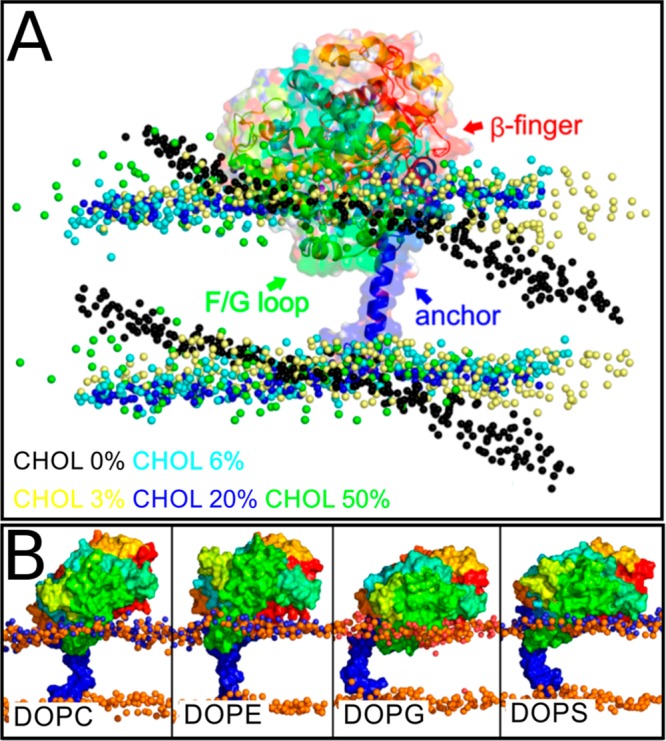
Changes in the CYP catalytic domain orientation toward a lipid bilayer due to an altered lipid composition: (A) Effect of cholesterol concentration. Reproduced with permission from ref (1329). Copyright 2015 American Chemical Society. (B) Effect of lipid headgroup charge. Reproduced with permission from ref (1328). Copyright 2016 American Chemical Society.
Due to the importance of CYPs in xenobiotic metabolism, studies of point mutations may be of practical importance. As various point mutations of CYP 2C19 were explored through simulations, it was observed how they reduced the enzyme’s affinity toward the substrate.1330 Possible mechanisms of the reduction of the enzyme activity by the point mutations include local deformation of the protein secondary structure, affecting the shape and the dynamics of the catalytic pocket, and the formation of new stable networks of hydrogen bonds, affecting the shape of the catalytic pocket. Other studies of CYP 1A1 showed that point mutations facilitate the interactions of organic pollutants with the protein.1331
Like many other proteins, CYPs can form not only homodimers but also heterodimers with other membrane proteins. MD simulations were recently used to explore the formation of homodimers by cytochrome P450 belonging to the aromatase class (P450arom or CYP19) and in particular to characterize the dimerization interface.1332 In another study on a heterodimeric complex, Jeřábek et al. elucidated the dimerization of CYP 1A2 with cytochrome b5, predicting the relative orientation of the proteins in the complex.1333
Monoamine oxidase (MAO) is a single transmembrane helix enzyme playing important roles in the nervous system in degradation of monoamine type neurotransmitters and in xenobiotic catabolism. Therefore, inhibition of MAO has important medical applications in the treatment of Parkinson’s disease and major depressive disorder.1334,1335 There are two forms of MAO: A and B. Comparison of MAO-A simulated in water and at the membrane–water interface showed that the membrane is necessary for opening a channel for the substrate and the reaction products.1336 Similar observations were made for MAO-B, for which the bilayer was shown to control the behavior of two loops at the entrance of the active site.1337 In the same study, interactions between the protein and various reversible inhibitors were also considered providing drug candidates with the optimal binding mode.1337 In recent studies, where MAO-A and MAO-B inhibition by flavonoids originating from medicinal plants were investigated, MD simulations provided information about the flavonoid binding modes.1338
Catechol O-methyl transferase (COMT) is an interesting example of enzymes that have both water-soluble (S-COMT) and membrane-bound (MB-COMT) forms. COMT and MAO are primary enzymes that initialize the degradation of key neurotransmitters. They, therefore, prevent the accumulation of neurotransmitters in the neurons, which is potentially toxic. MAO and COMT are particularly important, as they prevent dopamine oxidation and thus, the formation of oxidized dopamine, which is a toxic compound with destructive effects in, for example, mitochondria function and vesicular transport.1339 While both forms of COMT have the same catalytic domain, MB-COMT also contains a transmembrane helix and a short loop linking it to the catalytic domain. Extensive MD simulations of the transmembrane helix connected to the linker showed that the linker is highly mobile and flexible. The simulations also identified the formation of a specific salt bridge, which appeared to be the only structural motif in the linker.1340 These studies allowed reconstruction of the whole MB-COMT structure, thus rendering full MB-COMT simulations possible.1341 In these follow-up simulations, after binding to its cofactor S-adenosyl methionine, MB-COMT associated with the lipid bilayer in a stable fashion such that the entrance of the catalytic pocket faced toward the lipids.1341 Moreover, inhibitors having a high affinity for MB-COMT were found to adopt an orientation, in which the catechol group is exposed toward water and thus toward the entrance of the catalytic site.1341
Membrane association of CYP, MAO, and COMT seems to be related to their involvement in xenobiotic catabolism. The majority of drugs that target them are amphipathic or hydrophobic molecules that are known to position either at the membrane–water interface or in the hydrophobic core of the membrane, often affecting the membrane structure. Examples of such drugs that have been studied recently through simulations include itraconazole,1342 piroxicam,1343,1344 indocyanine,1345 fluoroquinolones,1346 porphyrins,1347,1348 and some others.1135,1349,1350 (For reviews, see refs (1351 and 1352)) These enzymes are also involved in the catabolism of natural compounds such as amphipathic neurotransmitters and steroids, which also reside preferentially at the membrane–water interface.1159,1162,1166,1167,1169,1353−1359
For the enzymes discussed in this section, the importance of membrane–protein cohabitation is clear. Existing studies have shown that the membrane lipid composition affects protein conformation by, for example, opening the entrance to the catalytic pocket and modifying the orientation of proteins toward the membrane surface. On the other hand, these enzymes are responsible for the catabolism of xenobiotic and naturally occurring small molecules, which frequently aggregate at the membrane interface, affecting membrane structure. For this reason, preventing such aggregation is crucial, since membrane properties may affect the functioning of many transmembrane proteins. Here, CYP, MAO, and COMT may be seen as membrane vacuum cleaners serving to maintain the native state of a membrane.
10.3. Enzymes Involved in Lipid Metabolism
Phospholipase A2 (PLA2) is a key enzyme responsible for phospholipid hydrolysis. This and other lipases are water-soluble peripheral membrane proteins. An MD study of two members of the human PLA2 family, the cytosolic group IVA (GIVA) PLA2 (cPLA2) and calcium-independent group VIA (GVIA) PLA2 (iPLA2), showed that the binding of these enzymes to the membrane surface induces protein conformational changes, leading to the opening and expansion of the binding site.1360 Thus, the membrane acts as an allosteric regulator of the protein. Interestingly, the PLA2 enzymes seem to have more than one allosteric site for membrane interactions. The analysis of PAPC binding modes in the binding cavities of cPLA2 and iPLA2 indicated that both proteins employ the same catalytic mechanism despite the large structural differences between them.1360 Subsequent studies showed the same effect of the membrane on a third enzyme in the same family: group V secreted PLA2.1361 Additionally, it was shown that the part of the binding cavity that hosts the acyl chain in the sn-2 position, but not the part that hosts the headgroup, determines the phospholipid specificity of the enzyme.1361
Monoacylglycerol lipase, in turn, is a peripheral membrane enzyme that cleaves monoacylglycerols involved in the production of pro-inflammatory molecules. Extensive MD simulations and free energy calculations showed that the higher affinity of the enzyme for substrates containing arachidonic acyl chains compared to those containing the palmitoyl chain is related to the lid domain plasticity and its interplay with membrane fluidity.1362 This specificity for the arachidonic acid is important for the enzyme function, as arachidonic acid is a precursor for pro-inflammatory molecules. These studies allowed identifying key residues (F159 and I179) involved in specific interactions with unsaturated chains. Interestingly, F159 is conserved across orthologues of the enzyme across numerous species.1362 Finally, the simulation studies provided also a model for the key steps in the catalytic cycle of this enzyme.1362
Another peripheral lipase, the M37 protein from Photobacterium lipolyticum, is an interesting interfacial enzyme as it preserves its activity at low temperatures and is stable in nonaqueous solvents. The simulations by Willems et al. showed that although M37 preferentially binds to anionic bilayers, it is not activated upon binding to the bilayers.1363 On the other hand, M37 also binds triglyceride surfaces, which results in structural changes characterized by the opening of the entrance to the catalytic site.
Glutathione peroxidase 4 is a water-soluble peripheral membrane enzyme responsible for reduction of hydroperoxides in lipids. MD simulations showed that lipid chains oxidized with hydroperoxide groups change their conformation to expose the polar OOH-group toward the water phase.113,117,669,1364 MD simulations also showed that the interaction of glutathione peroxidase 4 with lipids is driven by electrostatics, particularly between the positively charged residues of the protein and the negatively charged phosphate moieties of the lipids.1364 For this reason, interaction with cardiolipin, which has two phosphate groups, was stronger than that with PC.1364 Consideration of peroxides at the most common positions along the acyl chains (9 and 13) indicated that glutathione peroxidase 4 predominately reduces the peroxide group at the chain of the bound lipid molecule upon binding to the lipid headgroups; however, reduction of peroxide located at other positions or neighboring lipids is still possible, but due to steric obstacles less likely.
Vitamin K is a fat-soluble compound broadly categorized with lipids. In a recent study, the structure of the vitamin K epoxide reductase was predicted via homology modeling and shown to be stable in a microsecond simulation.1365 The structure was then used to explore vitamin K and its possible inhibitor binding modes.
10.4. Steps of Membrane Protein Biosynthesis
Protein biosynthesis and folding are processes characterized by large time and size scales. These are typically not accessible with the current computational resources and methodologies. Nevertheless, simulations can be used to explore some steps of these processes, such as the entering of the unfolded proteins into the translocon (for a review, see ref (1366)). Early simulation studies have considered, for example, the size of the translocon pore,1367 the free energy of peptide insertion,1368 and translocon’s arrangement in a bilayer.1369−1371
The folding of proteins begins at the exit channel of the ribosome. Coarse-grained simulations verified that the volume of the ribosome exit tunnel is large enough to accommodate the initial steps of folding.1372 Extensive atomistic simulations showed that the folding of helices in the translocon channel is a fast process occurring in a time scale of 100–1000 ns (Figure 73). This stems from the significant reduction of conformational space due to the shape of the channel, which promotes helix formation.1373 Meanwhile, these studies also showed that translocation of the helix from the translocon pore to the lipid bilayer requires time scales much longer than the microseconds often probed in the simulations. As a matter of fact, free energy calculations suggested that it occurs in the time scale of seconds.1374 The mechanism and the energetics of the translocation process have been elucidated by coarse-grained simulations of translocon models.1375 These studies showed, among others, that the opening of translocon due to ribosome attachment reduces the kinetic barrier of the insertion. Thus, the membrane insertion energy of residues does not correspond to the free energy difference between the translocon channel and the lipid environment.
Figure 73.
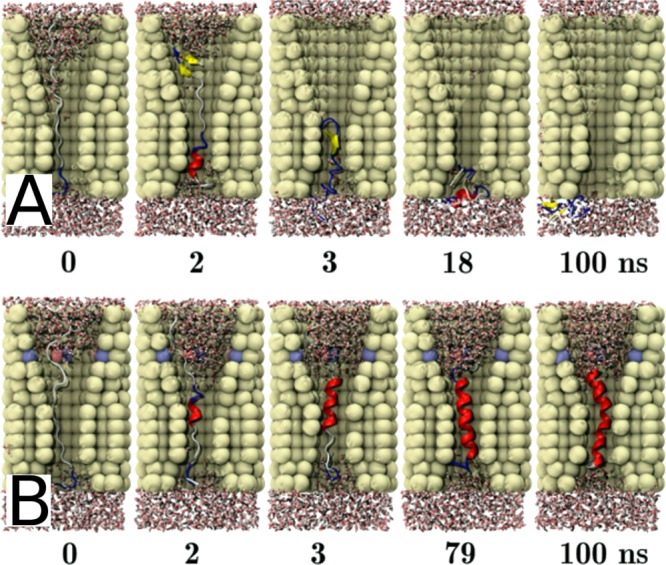
Example of peptide folding in the environment mimicking a translocon channel. Channel thickness was 1.2 nm. (A) In the absence of water in the channel, the peptide did not fold into a helix. (B) In the presence of water, on the other hand, the helix formed within 100 ns. Reproduced with permission from ref (1373). Copyright 2015, Springer Science Business Media New York.
Considering the size of the ribosome–translocon complex, it is clear that in the near future, atomistic simulations cannot reach the time scales taken by the processes related to protein synthesis. For this reason, it is imperative to develop specific coarse-grained simulation strategies. Nielsen et al. recently used this approach, as they developed a coarse-grained model based on experimental and extensive simulation data to study protein translocation and integration into a lipid bilayer.1376 There is reason to encourage more research to be done in the same spirit.
10.5. Challenges
Although membrane enzymes constitute a large group of proteins with numerous vital functions, the current understanding of the role of lipids in their activity seems to be very limited. The number of MD simulation studies in this field is not impressive. This is particularly obvious in the light of the literature discussed in sections 6 and 7, which highlight the quite extensive simulation work done to understand how lipids modulate membrane proteins. Nevertheless, a limited number of studies have shown that membrane enzymes are not very different from the other classes of membrane proteins and that lipids regulate their activity, too.
Enzymatic functions usually involve specific chemical reactions, which can only be studied using quantum-mechanical (QM) calculations. Although classical MD cannot provide insight into enzymatic reactions, hybrid methods (QM/MM) aim to bring the best of the two worlds together. QM/MM achieves this by limiting the QM calculations to a small region of interest, while handling the rest of the system using classical molecular mechanics. Therefore, developing QM/MM methodologies is crucial for future studies (for the current state of QM/MM development, see the review by Crespo-Otero and Barbatti1377).
The fact that only a limited number of studies have explored membrane associated enzymatic complexes is partly due to the lack of structural data to construct atomistic models. Further, even if the structures of individual proteins are known, it is difficult to predict their quaternary association. Protein–protein docking methods are not reliable for this task, and the spontaneous assembly of the complexes occurs in time scales far beyond those that are currently accessible to MD.
11. Concluding Remarks
In this review, we have provided a comprehensive overview of the role of biomembranes in numerous cellular processes. We have briefly introduced the key experimental findings related to each of these processes, followed by an exhaustive listing and discussion of the most relevant computational studies.
After the introduction, in section 2, we reviewed the history of biomembrane models, ending up in the current paradigm that sets its foundations around the raft concept. We also briefly described the development of computer simulation methods used to study biomembranes and concluded that the molecular dynamics method is currently by far the most widely used technique in the field. We also discussed the length and time scales achievable using classical molecular dynamics simulations.
Next, in section 3, we thoroughly described the complexity of the biomembrane structure. Our aim here was to provide a description of a target membrane, that is, the membrane including all the known features that should eventually be included in simulation models. These features include lipid diversity, heterogeneity, and asymmetry; protein diversity, post-translational modifications, and multiprotein assemblies; the membrane-lining actin cytoskeleton and the glycocalyx; and their mutual interactions, especially between lipids and proteins. While the list seems long and even discouraging, it is also delighting to see that simulation approaches have numerous uncharted territories left to explore in the future.
The following six sections each focused on a different function associated with biomembranes. In section 4, we reviewed the studies on membrane phase behavior, discussing both phase-separating model systems and more complex mixtures in which the formation of nanoscopic domains is either driven by lipid–lipid interactions or induced by membrane proteins. We concluded that the comparison between simulations and experiments is not straightforward due to methodological issues, the limitations in the reached time scales, and somewhat controversial experimental data. Next, in section 5, we reviewed simulation studies of membrane dynamics. We concluded that the dynamics in protein-free systems is well described by a concept of concerted flows, whereas in systems with dilute concentrations of proteins the Saffman–Delbrück description succeeds, as long as the effects of the lipid shell are accounted for. However, in more crowded systems these models often fail, and the little studied effects of the actin cytoskeleton and membrane domains are likely not simply described either. In section 6, we reviewed simulation studies on mass transport in biomembranes, covering passive permeation and transport by channels and carriers. Moreover, we discussed protein-mediated lipid flip–flops and lipid transport by peripheral proteins. All these topics are readily studied using all-atom simulations and provide vital information to support experimental work. In the second part, we reviewed vesicular transport including the budding and fusion of vesicles, topics currently within the reach of coarse-grained models. Our examples highlight how biomembranes, being usually a barrier for permeation, can facilitate transport through themselves through vesiculation or by regulating the function of proteins responsible for transbilayer diffusion. In section 7, we reviewed the literature on how simulations have helped us understand the role of biomembranes in signaling. We discussed spontaneous lipid flip–flops and the regulation of protein function by lipid–protein interactions and post-translational modifications. Current computing resources are usually adequate to tackle these problems efficiently using all-atom models, especially with the aid of biased simulations. In section 8, we discussed how membrane lipids can regulate protein function. We considered their effects on protein aggregation, signaling, and sorting. Moreover, we reviewed drug–membrane interactions, binding of peripheral proteins on membrane lipids, and the allosteric modulation of membrane proteins by their neighboring lipids. While all-atom simulations readily probe the favorable interactions, studies on conformational changes of proteins are still not entirely feasible. Moreover, studies on protein aggregation and partition rely on coarse-grained models. In section 9, we discussed energy storage in terms of lipid droplets and lipoproteins, as well as ion gradients across membranes. For the large droplets, as for all nonplanar membrane systems, coarse-grained models are currently required. Finally, in section 10, we reviewed the rather limited number of molecular dynamics studies that have characterized the role of membranes in enzymatic activity related to lipid metabolism and catabolism of small molecules and xenobiotics. Here, the role of lipids in regulating the access of the substrate and inhibitor molecules to the catalytic pockets is of crucial pharmacological interest.
The vast number of topics and studies we have covered in this review proves that computer simulations have rapidly evolved into an indispensable tool to support experiments and theoretical work, and to lead the development of selected topics. It is fairly impressive considering that the first membrane simulations were performed only 25 years ago and covered time scales with little relevance for biological functions. How did we get so far so fast? First, the development of computing power has been rapid over the last decades. Apart from the rapid increase in single-chip performance, improved parallelization schemes, the use of graphics processing units, and the integration to hardware development via the use of advanced instruction sets have contributed greatly to the push from the simulations spanning a few picoseconds to the microsecond or even millisecond regime. That is 8 orders of magnitude, arguably the most rapid development in any field in the history of modern science.
Not all computing capacity has been exploited to push up the time and size scales. The simulation engines have also implemented new and improved algorithms that increase the accuracy of the simulations. Improved models for proteins and lipids constantly appear in the literature, and parametrizations of new molecule types such as sugars are also popping up regularly. These developments are driven by improved experimental input as well as more accurate quantum-mechanical calculations used in parametrization. In addition to pushing the size and length scales further, enhanced sampling methods are also constantly under development, providing faster convergence and hence more trustworthy data with the same amount of resources. It is also worth mentioning that the multiscale methods also bridge the more detailed models with more efficient ones to utilize the computing capacity optimally. Such multiscale methods include combinations of atomistic and coarse-grained models, as well as combining quantum-mechanical calculations with classical molecular mechanics in QM/MM. Finally, we are starting to see the impact of machine learning methods on the field. Tools for parametrizing molecules, finding reaction coordinates for free energy calculations, evaluating protein–protein interactions, and predicting the effect of mutations have recently surfaced, and we expect to see many more applications in the near future. These developments will hopefully aid us in improving the extremely vast and complex force fields that are soon becoming impossible to maintain by manual efforts only, and to determine proper reaction coordinates to study protein activation–a task that currently resembles looking for a needle in a haystack. Still, despite the rapid developments of the classical molecular dynamics method, other techniques such as physics-based phase field models and techniques that incorporate hydrodynamics such as the lattice-Boltzmann methods are likely required on the way to a cell-level description.
Another factor that has driven the simulation field forward is the rapid development of experimental methods. Improved crystallization techniques and cryo-EM supply protein structures at record rates, and these structures are often resolved with some bound lipid molecules. Moreover, new lipidomics techniques provide information on the lipid composition of different tissues, and especially on how these compositions are affected by disease. Together, they allow for straightforward computational studies of lipid–protein interactions. What is more, current super-resolution microscopy methods can probe the plasma membranes of living cells at a resolution that already overlaps with the size scales of current simulations, providing means for seamless comparison of simulation and experiment. While benefiting from the experimental input, simulations can also pay back by characterizing the effects of probes and other required nonidealities and therefore help improve the experimental methodology. Fortunately, many laboratory experiments are becoming less intrusive, further benefiting the interplay with simulations.
We would also like to specially thank the raft concept. Despite its historical shortcomings, it has reached an odd status in the scientific community. While the discussion on the existence of rafts seems to continue, rafts still dominate the way we consider biomembranes today, and the concept has sparked an enormous amount of studies. Importantly, the research built around rafts has brought up many interesting findings on biomembranes, fostered the rapid development of super-resolution applications, and, most notably, elevated the lipid membrane from merely an embedding matrix into the limelight as a functionally relevant counterpart of proteins. This development has also given a massive boost to the biomembrane simulation field, rendering it a central tool in many related fields.
Perhaps everyone thinking of rafts should instead consider what has been observed through biomembrane experiments and simulations. It is established that biomembranes are heterogeneous and rich in proteins. For many membrane proteins, there is compelling evidence that they have allosteric binding pockets for specific lipids. The proteins also favor certain lipids and fatty acids in their immediate vicinity, and together, these proteins and lipids form dynamic complexes that partition to membrane environments that foster the protein’s activation and function. Further, quite often it is not just a single protein doing the job, but the proteins form oligomers that together with selected lipids form the functional unit. The size of the dynamical protein–lipid complex depends on the protein oligomer and the lipid–protein interactions, but roughly speaking it ranges from about 5 to 20 nm, being possibly larger if the number of proteins in the oligomers is very large. It is a matter of taste what these dynamical, transient, and functional protein–lipid nanocomplexes are called, but there is quite a bit of evidence supporting the view that they exist.
Unfortunately, bridging simulation and experiment is not always a walk in a park. While we struggle to understand even the behavior of simple model membranes, we are still heading full speed toward more and more complex membrane systems, trying to capture the in vivo behavior probed by many experiments nowadays. This is understandable since complexity seems to sell papers better than reliability and the most interesting phenomena take place in extremely complicated membranes. Sadly, the model is always only as good as its weakest part, and the current hastiness can become costly in case the models are not constantly improved. In case we lose the hard-earned status as a reliable tool to back up experiments, it will be incredibly difficult to reclaim it.
So let us review for what the current models and the available scales are good. Considering the topics covered in this review and listed in the beginning of this section, it is safe to say that all-atom simulations can nowadays be used to study the dynamics of fairly simple membranes, the passive interaction of drugs and proteins with membranes, and the interaction of membrane proteins and lipids. With the use of enhanced sampling techniques, all-atom models can be extended to studies of protein conformational changes, permeation of molecules through membranes either in the absence or in the presence of proteins, and lipid flip–flops. Moving on to coarse-grained models, dynamics and phase behavior of more complex membranes can be studied along with membrane fusion, membrane protein oligomerization, and sorting.
The listing above suggests that further developments in computing power and model quality need to be made before certain topics can be reliably studied using simulations. Such problematic cases include monolayers at water–air interphases where surface tension is not well reproduced by the used models. Similarly, the interactions between membranes and peripheral proteins are likely affected by the poor description of the headgroup structure and especially their response to ionic environments. The coarse-grained MARTINI model used to study the aggregation of membrane proteins and water-soluble proteins suffers from excessive protein–protein interactions undermining such studies. Similarly, studies on phase behavior with the same model seem to be haunted by occasionally poor agreement with experimentally observed behavior. While biased simulation techniques can be used to study protein conformational changes, finding the right reaction coordinates is often hard. Hence, it would be desirable to see spontaneous conformational changes in unbiased simulations. Unfortunately, this might not be only a time scale issue, since the membrane proteins seem to be overstabilized in many force fields. The proper sampling of time scales is also important in studies of membrane dynamics, where only averages over a substantial amount of long trajectories are useful. The importance of sampling becomes crucial in crowded environments, where membrane components can be in an extremely immobile state. Similar issues also arise when considering crystalline structures such as the waxes in tear film lipid layer or the ceramide-based models of stratum corneum. Large scales are also required to see the proper mixing and hence domain formation in complex biomembranes, as well as in studies involving large vesicles.
Special attention must also be paid to the fact that the equilibrium state modeled by most simulation studies is fairly irrelevant for most processes. Instead and except for diffusion, most mass transport takes place via nonequilibrium processes, such as endo- and exocytosis and the function of carrier proteins such as lipoproteins. Moreover, the components in lipid bilayers are constantly recycled meaning that the membranes are in a steady state instead of being in equilibrium. The largest challenge in modeling such nonequilibrium processes is likely sampling as statistically significant results can only be extracted from a large number of samples, which requires simulating the process numerous times. Sometimes the nonequilibrium conditions at the nanoscale are apparent even in the macroscale: Due to our breathing and blinking, the lipid structures at the interfaces in the eye and the lungs undergo repeated cycles of spreading to and buckling away from the interface. Fortunately, the current simulation scales are reaching a point in which these phenomena can be modeled at realistic speeds.
How about the features that our models currently lack? Most biomembrane simulations are still performed with simple and symmetric lipid compositions, whereas cellular membranes have long been known to be compositionally complex and asymmetric. Implementing the former means that simulations have to be extended to allow for a proper mixing of lipid molecules, while the latter feature can be readily incorporated as soon as experiments can provide us with trustworthy data on asymmetry, especially with regard to the distribution of cholesterol. Moreover, the roles of the actin cytoskeleton and the glycocalyx, lining the membrane at both sides, are very rarely considered in simulations, partly due to their large sizes and in part due to the lack of structures of proteins building these networks. Perhaps the easiest things to implement are post-translational modifications in proteins. While these modifications do not call for a large increase in the required computing resources, their importance for protein function can be significant. Fortunately, databases for these modifications are readily available, and the tools for incorporating them in simulation models are also rapidly improving.
Finally, we want to point out that exceptional scientists are required to overcome all these limitations. Fortunately, research groups engaging in software, tool, and model development are filled with bright minds that work to benefit everyone in the field. Technical aspects aside, we also need the people that are able to ask the right scientific questions. Here, the persistent efforts of computational scientists have raised molecular dynamics simulations to a level where they can be considered as equal counterparts to experimental work. This interplay leads to an exchange of interesting ideas and hence to better science. Furthermore, the ongoing attempts to make simulation software and tools more accessible to people without programming skills has lured numerous talented scientists to the field, leading to a rapid increase in the size of the community. All in all, we believe that the near future will turn out to be an extremely prosperous era for the field as more and more complex systems are explored at ever larger and longer scales by more accurate models and by an ever growing number of bright scientists.
Acknowledgments
We thank every one of our collaborators and colleagues with whom we have discussed this work during the many years. We also acknowledge the European Research Council (Advanced Grant Project CROWDED-PRO-LIPIDS (Grant No. 290974)), the Academy of Finland (Centre of Excellence program (Grant No. 307415)), and the Sigrid Juselius Foundation for financial support. CSC–IT Center for Science (Espoo, Finland) is thanked for ample computing resources that have rendered our research possible. M.J. also thanks Emil Aaltonen Foundation for financial support.
Biographies
Giray Enkavi completed his Ph.D. studies in “Biophysics and Computational Biology” in December 2013 at the University Illinois Urbana–Champaign, USA, under the supervision of Prof. Emad Tajkhorshid (Computational Structural Biology and Molecular Biophysics group). Since August 2014, he has been working as a postdoctoral researcher in the Biological Physics and Soft Matter Group directed by Prof. Ilpo Vattulainen. The central aspect of Giray Enkavi’s research during his Ph.D. and postdoctoral work has been biomodeling and simulation studies of membrane proteins. In his Ph.D. studies, he focused on membrane channels and transporters, and in his postdoctoral studies, he has been investigating cholesterol transport.
Matti Javanainen received his Ph.D. from Tampere University of Technology in 2018 under the supervision of Prof. Ilpo Vattulainen. His Thesis work considered diffusion in protein-crowded membranes, yet his research has also covered other topics related to lipid membranes and membrane proteins. At present, he works as a postdoctoral fellow in the group of Prof. Pavel Jungwirth at the Institute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences in Prague. His current work considers the interactions of charged objects with biomembranes.
Waldemar Kulig is a senior researcher in the Department of Physics, University of Helsinki. He received his Ph.D. from the Jagiellonian University of Cracow in 2011 under the supervision of Prof. Piotr Petelenz. Next, as a postdoctoral researcher, he worked at the Fritz Haber Center for Molecular Dynamics, the Hebrew University of Jerusalem, under the supervision of Prof. Noam Agmon. In 2013, he moved to Prof. Ilpo Vattulainen’s group at the Tampere University of Technology. Waldemar’s research interests revolve around lipid–lipid and protein–lipid interactions, properties of phospholipid bilayers, effect of oxidative stress, cellular signaling, and development of simulation methods.
Tomasz Róg is a senior researcher at the University of Helsinki, Department of Physics, and a member of the Center of Excellence in Biomembrane Research. He completed M.Sc. studies in 1996 in the Faculty of Biological and Earth Sciences of the Jagiellonian University. Next, he continued studies in the same faculty, and in 2000, he received Ph.D. in Biological Sciences specialized in Biophysics. He was awarded a fellowship by the Foundation for Polish Science (http://www.fnp.org.pl/) in 2002 and 2003. In 2005–2007, he worked as a Marie Curie Fellow at Helsinki University of Technology (currently Aalto University). From 2008 to 2013, he was awarded the 5-year Academy Research Fellow project by the Academy of Finland, which he carried out at Tampere University of Technology. His research interests include cholesterol biophysics, lipid bilayer structure and dynamics, lipid–lipid and lipid–protein interactions, membrane protein function, biophysical pharmacology, and methodological aspects of molecular dynamics simulations.
Ilpo Vattulainen (born 1968) defended his Ph.D. thesis in Helsinki University of Technology in 1998 and then moved to the MEMPHYS group in Denmark for a Marie Curie Fellowship. He founded the Biological Physics group in 2001 and was awarded a 5-year fellowship by the Academy of Finland for 2002–2007. More recently he has been appointed to a professorship at Tampere University of Technology (2006−) and University of Helsinki (2016−), and he is a Mercator Fellow at TU Dresden/Heidelberg (2018–2021). His group of about 25 people is affiliated with a centre of excellence (Academy of Finland). The group focuses on computational and theoretical studies of phenomena dealing with biomembranes, proteins, drugs, sterols, carbohydrates, and nanomaterials. He loves nature, that is, wilderness.
Author Contributions
G.E., M.J., W.K., T.R., and I.V. contributed equally to this work.
The authors declare no competing financial interest.
References
- Gorter E.; Grendel F. On Bimolecular Layers of Lipoids on the Chromocytes of the Blood. J. Exp. Med. 1925, 41, 439–443. 10.1084/jem.41.4.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson J. The Ultrastructure of Cell Membranes and Their Derivatives. Biochem. Soc. Symp. 1959, 16, 3–43. [PubMed] [Google Scholar]
- Lenard J.; Singer S. J. Protein Conformation in Cell Membrane Preparations as Studied by Optical Rotatory Dispersion and Circular Dichroism. Proc. Natl. Acad. Sci. U. S. A. 1966, 56, 1828–1835. 10.1073/pnas.56.6.1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer S. J.; Nicolson G. L. The Fluid Mosaic Model of the Structure of Cell Membranes. Science 1972, 175, 720–731. 10.1126/science.175.4023.720. [DOI] [PubMed] [Google Scholar]
- Phillips M. C.; Ladbrooke B. D.; Chapman D. Molecular Interactions in Mixed Lecithin Systems. Biochim. Biophys. Acta, Biomembr. 1970, 196, 35–44. 10.1016/0005-2736(70)90163-X. [DOI] [PubMed] [Google Scholar]
- Oldfield E.; Chapman D. Dynamics of Lipids in Membranes: Heterogeneity and the Role of Cholesterol. FEBS Lett. 1972, 23, 285–297. 10.1016/0014-5793(72)80300-4. [DOI] [PubMed] [Google Scholar]
- Shimshick E. J.; McConnell H. M. Lateral Phase Separation in Phospholipid Membranes. Biochemistry 1973, 12, 2351–2360. 10.1021/bi00736a026. [DOI] [PubMed] [Google Scholar]
- Mouritsen O. G.; Bloom M. Mattress Model of Lipid-Protein Interactions in Membranes. Biophys. J. 1984, 46, 141–153. 10.1016/S0006-3495(84)84007-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sackmann E.Physical Basis for Trigger Processes and Membrane Structures. In Biological Membranes; Chapman D., Ed.; Academic Press: London, 1984. [Google Scholar]
- Bagatolli L.; Mouritsen O. Is the Fluid Mosaic (and the Accompanying Raft Hypothesis) a Suitable Model to Describe Fundamental Features of Biological Membranes? What May Be Missing?. Front. Plant Sci. 2013, 4, 457. 10.3389/fpls.2013.00457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouritsen O. G.; Bagatolli L. A. Lipid Domains in Model Membranes: A Brief Historical Perspective. Essays Biochem. 2015, 57, 1–19. 10.1042/bse0570001. [DOI] [PubMed] [Google Scholar]
- Simons K.; Ikonen E. Functional Rafts in Cell Membranes. Nature 1997, 387, 569–572. 10.1038/42408. [DOI] [PubMed] [Google Scholar]
- Simons K.; Van Meer G. Lipid Sorting in Epithelial Cells. Biochemistry 1988, 27, 6197–6202. 10.1021/bi00417a001. [DOI] [PubMed] [Google Scholar]
- Hjort Ipsen J.; Karlström G.; Mourtisen O. G.; Wennerström H.; Zuckermann M. J. Phase Equilibria in the Phosphatidylcholine-Cholesterol System. Biochim. Biophys. Acta, Biomembr. 1987, 905, 162–172. 10.1016/0005-2736(87)90020-4. [DOI] [PubMed] [Google Scholar]
- Lingwood D.; Simons K. Lipid Rafts As a Membrane-Organizing Principle. Science 2010, 327, 46–50. 10.1126/science.1174621. [DOI] [PubMed] [Google Scholar]
- Niemelä P. S.; Miettinen M. S.; Monticelli L.; Hammaren H.; Bjelkmar P.; Murtola T.; Lindahl E.; Vattulainen I. Membrane Proteins Diffuse as Dynamic Complexes with Lipids. J. Am. Chem. Soc. 2010, 132, 7574–7575. 10.1021/ja101481b. [DOI] [PubMed] [Google Scholar]
- Sezgin E.; Levental I.; Mayor S.; Eggeling C. The Mystery of Membrane Organization: Composition, Regulation and Roles of Lipid Rafts. Nat. Rev. Mol. Cell Biol. 2017, 18, 361–374. 10.1038/nrm.2017.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstine H. H.; Goldstine A. The Electronic Numerical Integrator and Computer (ENIAC). Math. Tables Aids Comput. 1946, 2, 97–110. 10.2307/2002620. [DOI] [Google Scholar]
- Metropolis N.Los Alamos Science. Spec. Issue 1987, 125. [Google Scholar]
- Metropolis N.; Ulam S. The Monte Carlo Method. J. Am. Stat. Assoc. 1949, 44, 335–341. 10.1080/01621459.1949.10483310. [DOI] [PubMed] [Google Scholar]
- Metropolis N.; Rosenbluth A. W.; Rosenbluth M. N.; Teller A. H.; Teller E. Equation of State Calculations by Fast Computing Machines. J. Chem. Phys. 1953, 21, 1087–1092. 10.1063/1.1699114. [DOI] [Google Scholar]
- Metropolis N. C.; Reitwiesner G.; von Neumann J. Statistical Treatment of Values of First 2,000 Decimal Digits of e and of π Calculated on the ENIAC. Math. Comput. 1950, 4, 109–109. 10.1090/S0025-5718-1950-0037598-8. [DOI] [Google Scholar]
- Hull T.; Dobell A. Random Number Generators. SIAM Rev. 1962, 4, 230–254. 10.1137/1004061. [DOI] [Google Scholar]
- Knuth D. E.; Knuth D. E.. Seminumerical Algorithms, 2nd ed., 25th print.; The art of computer programming; Addison-Wesley: Reading, Mass., 1996. [Google Scholar]
- L’Ecuyer P.Random Number Generation. In Handbook of Computational Statistics: Concepts and Methods; Gentle J. E., Härdle W. K., Mori Y., Eds.; Springer Handbooks of Computational Statistics; Springer-Verlag: Berlin Heidelberg, 2012. [Google Scholar]
- Alder B. J.; Wainwright T. E. Phase Transition for a Hard Sphere System. J. Chem. Phys. 1957, 27, 1208–1209. 10.1063/1.1743957. [DOI] [Google Scholar]
- Rahman A. Correlations in the Motion of Atoms in Liquid Argon. Phys. Rev. 1964, 136, A405–A411. 10.1103/PhysRev.136.A405. [DOI] [Google Scholar]
- McCammon J. A.; Gelin B. R.; Karplus M. Dynamics of Folded Proteins. Nature 1977, 267, 585–590. 10.1038/267585a0. [DOI] [PubMed] [Google Scholar]
- Duan Y.; Kollman P. A. Pathways to a Protein Folding Intermediate Observed in a 1-Microsecond Simulation in Aqueous Solution. Science 1998, 282, 740–744. 10.1126/science.282.5389.740. [DOI] [PubMed] [Google Scholar]
- Pastor R. W. Molecular Dynamics and Monte Carlo Simulations of Lipid Bilayers. Curr. Opin. Struct. Biol. 1994, 4, 486–492. 10.1016/S0959-440X(94)90209-7. [DOI] [Google Scholar]
- Kox A. J.; Michels J. P. J.; Wiegel F. W. Simulation of a Lipid Monolayer Using Molecular Dynamics. Nature 1980, 287, 317–319. 10.1038/287317a0. [DOI] [Google Scholar]
- van der Ploeg P.; Berendsen H. J. C. Molecular Dynamics Simulation of a Bilayer Membrane. J. Chem. Phys. 1982, 76, 3271–3276. 10.1063/1.443321. [DOI] [Google Scholar]
- Jönsson B.; Edholm O.; Teleman O. Molecular Dynamics Simulations of a Sodium Octanoate Micelle in Aqueous Solution. J. Chem. Phys. 1986, 85, 2259–2271. 10.1063/1.451122. [DOI] [Google Scholar]
- Scott H. L. Lipid-Cholesterol Interactions. Monte Carlo Simulations and Theory. Biophys. J. 1991, 59, 445–455. 10.1016/S0006-3495(91)82238-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu S. W.; Jakobsson E.; Scott H. L. Combined Monte Carlo and Molecular Dynamics Simulation of Hydrated Lipid-Cholesterol Lipid Bilayers at Low Cholesterol Concentration. Biophys. J. 2001, 80, 1104–1114. 10.1016/S0006-3495(01)76088-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu S. W.; Jakobsson E.; Subramaniam S.; Scott H. L. Combined Monte Carlo and Molecular Dynamics Simulation of Fully Hydrated Dioleyl and Palmitoyl-Oleyl Phosphatidylcholine Lipid Bilayers. Biophys. J. 1999, 77, 2462–2469. 10.1016/S0006-3495(99)77082-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venable R. M.; Zhang Y.; Hardy B. J.; Pastor R. W. Molecular Dynamics Simulations of a Lipid Bilayer and of Hexadecane: An Investigation of Membrane Fluidity. Science 1993, 262, 223–226. 10.1126/science.8211140. [DOI] [PubMed] [Google Scholar]
- Heller H.; Schaefer M.; Schulten K. Molecular Dynamics Simulation of a Bilayer of 200 Lipids in the Gel and in the Liquid Crystal Phase. J. Phys. Chem. 1993, 97, 8343–8360. 10.1021/j100133a034. [DOI] [Google Scholar]
- Marrink S. J.; Berkowitz M.; Berendsen H. J. C. Molecular Dynamics Simulation of a Membrane/Water Interface: The Ordering of Water and Its Relation to the Hydration Force. Langmuir 1993, 9, 3122–3131. 10.1021/la00035a062. [DOI] [Google Scholar]
- Berendsen H. J. C.; Marrink S.-J. Molecular Dynamics of Water Transport through Membranes: Water from Solvent to Solute. Pure Appl. Chem. 1993, 65, 2513–2520. 10.1351/pac199365122513. [DOI] [Google Scholar]
- Egberts E.; Marrink S.-J.; Berendsen H. J. C. Molecular Dynamics Simulation of a Phospholipid Membrane. Eur. Biophys. J. 1994, 22, 423–436. 10.1007/BF00180163. [DOI] [PubMed] [Google Scholar]
- Berger O.; Edholm O.; Jähnig F. Molecular Dynamics Simulations of a Fluid Bilayer of Dipalmitoylphosphatidylcholine at Full Hydration, Constant Pressure, and Constant Temperature. Biophys. J. 1997, 72, 2002–2013. 10.1016/S0006-3495(97)78845-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essex J. W.; Hann M. M.; Richards W. G. Molecular Dynamics Simulation of a Hydrated Phospholipid Bilayer. Philos. Trans. R. Soc. London B. Biol. Sci. 1994, 344, 239–260. 10.1098/rstb.1994.0064. [DOI] [PubMed] [Google Scholar]
- Vattulainen I.; Róg T. Lipid Membranes: Theory and Simulations Bridged to Experiments. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 2251–2253. 10.1016/j.bbamem.2016.06.007. [DOI] [PubMed] [Google Scholar]
- Lyubartsev A. P.; Rabinovich A. L. Force Field Development for Lipid Membrane Simulations. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 2483–2497. 10.1016/j.bbamem.2015.12.033. [DOI] [PubMed] [Google Scholar]
- Javanainen M.; Martinez-Seara H. Efficient Preparation and Analysis of Membrane and Membrane Protein Systems. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 2468–2482. 10.1016/j.bbamem.2016.02.036. [DOI] [PubMed] [Google Scholar]
- Neale C.; Pomès R. Sampling Errors in Free Energy Simulations of Small Molecules in Lipid Bilayers. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 2539–2548. 10.1016/j.bbamem.2016.03.006. [DOI] [PubMed] [Google Scholar]
- Wong-ekkabut J.; Karttunen M. The Good, the Bad and the User in Soft Matter Simulations. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 2529–2538. 10.1016/j.bbamem.2016.02.004. [DOI] [PubMed] [Google Scholar]
- Ollila O. H. S.; Pabst G. Atomistic Resolution Structure and Dynamics of Lipid Bilayers in Simulations and Experiments. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 2512–2528. 10.1016/j.bbamem.2016.01.019. [DOI] [PubMed] [Google Scholar]
- Botan A.; Favela-Rosales F.; Fuchs P. F. J.; Javanainen M.; Kanduč M.; Kulig W.; Lamberg A.; Loison C.; Lyubartsev A.; Miettinen M. S.; et al. Toward Atomistic Resolution Structure of Phosphatidylcholine Headgroup and Glycerol Backbone at Different Ambient Conditions. J. Phys. Chem. B 2015, 119, 15075–15088. 10.1021/acs.jpcb.5b04878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catte A.; Girych M.; Javanainen M.; Loison C.; Melcr J.; Miettinen M. S.; Monticelli L.; Määttä J.; Oganesyan V. S.; Ollila O. H. S.; et al. Molecular Electrometer and Binding of Cations to Phospholipid Bilayers. Phys. Chem. Chem. Phys. 2016, 18, 32560–32569. 10.1039/C6CP04883H. [DOI] [PubMed] [Google Scholar]
- Piana S.; Klepeis J. L.; Shaw D. E. Assessing the Accuracy of Physical Models Used in Protein-Folding Simulations: Quantitative Evidence from Long Molecular Dynamics Simulations. Curr. Opin. Struct. Biol. 2014, 24, 98–105. 10.1016/j.sbi.2013.12.006. [DOI] [PubMed] [Google Scholar]
- Javanainen M.; Martinez-Seara H.; Vattulainen I. Nanoscale Membrane Domain Formation Driven by Cholesterol. Sci. Rep. 2017, 7, 1143. 10.1038/s41598-017-01247-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manna M.; Niemelä M.; Tynkkynen J.; Javanainen M.; Kulig W.; Müller D. J.; Rog T.; Vattulainen I. Mechanism of Allosteric Regulation of β2-Adrenergic Receptor by Cholesterol. eLife 2016, 5, e18432. 10.7554/eLife.18432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javanainen M.; Martinez-Seara H.; Metzler R.; Vattulainen I. Diffusion of Integral Membrane Proteins in Protein-Rich Membranes. J. Phys. Chem. Lett. 2017, 8, 4308–4313. 10.1021/acs.jpclett.7b01758. [DOI] [PubMed] [Google Scholar]
- Koldsø H.; Sansom M. S. P. Organization and Dynamics of Receptor Proteins in a Plasma Membrane. J. Am. Chem. Soc. 2015, 137, 14694–14704. 10.1021/jacs.5b08048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guixà-González R.; Javanainen M.; Gómez-Soler M.; Cordobilla B.; Domingo J. C.; Sanz F.; Pastor M.; Ciruela F.; Martinez-Seara H.; Selent J. Membrane Omega-3 Fatty Acids Modulate the Oligomerisation Kinetics of Adenosine A2A and Dopamine D2 Receptors. Sci. Rep. 2016, 6, 19839. 10.1038/srep19839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harayama T.; Riezman H. Understanding the Diversity of Membrane Lipid Composition. Nat. Rev. Mol. Cell Biol. 2018, 19, 281–296. 10.1038/nrm.2017.138. [DOI] [PubMed] [Google Scholar]
- Shahidi F.; Zhong Y. Lipid Oxidation and Improving the Oxidative Stability. Chem. Soc. Rev. 2010, 39, 4067–4079. 10.1039/b922183m. [DOI] [PubMed] [Google Scholar]
- Yin H.; Xu L.; Porter N. A. Free Radical Lipid Peroxidation: Mechanisms and Analysis. Chem. Rev. 2011, 111, 5944–5972. 10.1021/cr200084z. [DOI] [PubMed] [Google Scholar]
- Shevchenko A.; Simons K. Lipidomics: Coming to Grips with Lipid Diversity. Nat. Rev. Mol. Cell Biol. 2010, 11, 593–598. 10.1038/nrm2934. [DOI] [PubMed] [Google Scholar]
- Dhawan V.; Magarkar A.; Joshi G.; Makhija D.; Jain A.; Shah J.; Reddy B. V. V.; Krishnapriya M.; Róg T.; Bunker A.; et al. Stearylated Cycloarginine Nanosystems for Intracellular Delivery – Simulations, Formulation and Proof of Concept. RSC Adv. 2016, 6, 113538–113550. 10.1039/C6RA16432C. [DOI] [Google Scholar]
- Magarkar A.; Róg T.; Bunker A. A Computational Study Suggests That Replacing PEG with PMOZ. May Increase Exposure of Hydrophobic Targeting Moiety. Eur. J. Pharm. Sci. 2017, 103, 128–135. 10.1016/j.ejps.2017.03.008. [DOI] [PubMed] [Google Scholar]
- Lehtinen J.; Magarkar A.; Stepniewski M.; Hakola S.; Bergman M.; Róg T.; Yliperttula M.; Urtti A.; Bunker A. Analysis of Cause of Failure of New Targeting Peptide in PEGylated Liposome: Molecular Modeling as Rational Design Tool for Nanomedicine. Eur. J. Pharm. Sci. 2012, 46, 121–130. 10.1016/j.ejps.2012.02.009. [DOI] [PubMed] [Google Scholar]
- Pathak P.; Dhawan V.; Magarkar A.; Danne R.; Govindarajan S.; Ghosh S.; Steiniger F.; Chaudhari P.; Gopal V.; Bunker A.; et al. Design of Cholesterol Arabinogalactan Anchored Liposomes for Asialoglycoprotein Receptor Mediated Targeting to Hepatocellular Carcinoma: In Silico Modeling, in Vitro and in Vivo Evaluation. Int. J. Pharm. 2016, 509, 149–158. 10.1016/j.ijpharm.2016.05.041. [DOI] [PubMed] [Google Scholar]
- Magarkar A.; Róg T.; Bunker A. Molecular Dynamics Simulation of PEGylated Membranes with Cholesterol: Building Toward the DOXIL Formulation. J. Phys. Chem. C 2014, 118, 15541–15549. 10.1021/jp504962m. [DOI] [Google Scholar]
- Magarkar A.; Karakas E.; Stepniewski M.; Róg T.; Bunker A. Molecular Dynamics Simulation of PEGylated Bilayer Interacting with Salt Ions: A Model of the Liposome Surface in the Bloodstream. J. Phys. Chem. B 2012, 116, 4212–4219. 10.1021/jp300184z. [DOI] [PubMed] [Google Scholar]
- Stepniewski M.; Pasenkiewicz-Gierula M.; Róg T.; Danne R.; Orlowski A.; Karttunen M.; Urtti A.; Yliperttula M.; Vuorimaa E.; Bunker A. Study of PEGylated Lipid Layers as a Model for PEGylated Liposome Surfaces: Molecular Dynamics Simulation and Langmuir Monolayer Studies. Langmuir 2011, 27, 7788–7798. 10.1021/la200003n. [DOI] [PubMed] [Google Scholar]
- Ríos-Marco P.; Marco C.; Gálvez X.; Jiménez-López J. M.; Carrasco M. P. Alkylphospholipids: An Update on Molecular Mechanisms and Clinical Relevance. Biochim. Biophys. Acta, Biomembr. 2017, 1859, 1657–1667. 10.1016/j.bbamem.2017.02.016. [DOI] [PubMed] [Google Scholar]
- Mobarak E.; Javanainen M.; Kulig W.; Honigmann A.; Sezgin E.; Aho N.; Eggeling C.; Rog T.; Vattulainen I. How to Minimize Dye-Induced Perturbations While Studying Biomembrane Structure and Dynamics: PEG Linkers as a Rational Alternative. Biochim. Biophys. Acta, Biomembr. 2018, 1860, 2436–2445. 10.1016/j.bbamem.2018.07.003. [DOI] [PubMed] [Google Scholar]
- Rissanen S.; Grzybek M.; Orłowski A.; Róg T.; Cramariuc O.; Levental I.; Eggeling C.; Sezgin E.; Vattulainen I. Phase Partitioning of GM1 and Its Bodipy-Labeled Analog Determine Their Different Binding to Cholera Toxin. Front. Physiol. 2017, 8, 252. 10.3389/fphys.2017.00252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Björkbom A.; Róg T.; Kaszuba K.; Kurita M.; Yamaguchi S.; Lönnfors M.; Nyholm T. K. M.; Vattulainen I.; Katsumura S.; Slotte J. P. Effect of Sphingomyelin Headgroup Size on Molecular Properties and Interactions with Cholesterol. Biophys. J. 2010, 99, 3300–3308. 10.1016/j.bpj.2010.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kepczynski M.; Róg T. Functionalized Lipids and Surfactants for Specific Applications. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 2362–2379. 10.1016/j.bbamem.2016.02.038. [DOI] [PubMed] [Google Scholar]
- Bunker A.; Magarkar A.; Viitala T. Rational Design of Liposomal Drug Delivery Systems, a Review: Combined Experimental and Computational Studies of Lipid Membranes, Liposomes and Their PEGylation. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 2334–2352. 10.1016/j.bbamem.2016.02.025. [DOI] [PubMed] [Google Scholar]
- Faller R. Molecular Modeling of Lipid Probes and Their Influence on the Membrane. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 2353–2361. 10.1016/j.bbamem.2016.02.014. [DOI] [PubMed] [Google Scholar]
- Shiffka S. J.; Kane M. A.; Swaan P. W. Planar Bile Acids in Health and Disease. Biochim. Biophys. Acta, Biomembr. 2017, 1859, 2269–2276. 10.1016/j.bbamem.2017.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braverman N. E.; Moser A. B. Functions of Plasmalogen Lipids in Health and Disease. Biochim. Biophys. Acta, Mol. Basis Dis. 2012, 1822, 1442–1452. 10.1016/j.bbadis.2012.05.008. [DOI] [PubMed] [Google Scholar]
- Toker A. The Biology and Biochemistry of Diacylglycerol Signalling. EMBO Rep. 2005, 6, 310–314. 10.1038/sj.embor.7400378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrasco S.; Mérida I. Diacylglycerol, When Simplicity Becomes Complex. Trends Biochem. Sci. 2007, 32, 27–36. 10.1016/j.tibs.2006.11.004. [DOI] [PubMed] [Google Scholar]
- Zegarlińska J.; Piaścik M.; Sikorski A. F.; Czogalla A. Phosphatidic Acid - a Simple Phospholipid with Multiple Faces. Acta Biochim. Polym. 2018, 65, 163–171. 10.18388/abp.2018_2592. [DOI] [PubMed] [Google Scholar]
- Kwolek U.; Kulig W.; Wydro P.; Nowakowska M.; Róg T.; Kepczynski M. Effect of Phosphatidic Acid on Biomembrane: Experimental and Molecular Dynamics Simulations Study. J. Phys. Chem. B 2015, 119, 10042–10051. 10.1021/acs.jpcb.5b03604. [DOI] [PubMed] [Google Scholar]
- Athenstaedt K.; Daum G. Phosphatidic Acid, a Key Intermediate in Lipid Metabolism. Eur. J. Biochem. 1999, 266, 1–16. 10.1046/j.1432-1327.1999.00822.x. [DOI] [PubMed] [Google Scholar]
- Wang X.; Devaiah S. P.; Zhang W.; Welti R. Signaling Functions of Phosphatidic Acid. Prog. Lipid Res. 2006, 45, 250–278. 10.1016/j.plipres.2006.01.005. [DOI] [PubMed] [Google Scholar]
- Testerink C.; Munnik T. Phosphatidic Acid: A Multifunctional Stress Signaling Lipid in Plants. Trends Plant Sci. 2005, 10, 368–375. 10.1016/j.tplants.2005.06.002. [DOI] [PubMed] [Google Scholar]
- Phospholipases in Plant Signaling. In Signaling and Communication in Plants; Wang X., Ed.; Springer Berlin Heidelberg: Berlin, Heidelberg, 2014; Vol. 20, 10.1007/978-3-642-42011-5. [DOI] [Google Scholar]
- Mejia E. M.; Hatch G. M. Mitochondrial Phospholipids: Role in Mitochondrial Function. J. Bioenerg. Biomembr. 2016, 48, 99–112. 10.1007/s10863-015-9601-4. [DOI] [PubMed] [Google Scholar]
- Kooijman E. E.; Chupin V.; de Kruijff B.; Burger K. N. J. Modulation of Membrane Curvature by Phosphatidic Acid and Lysophosphatidic Acid: Biophysical Properties of Phosphatidic Acid and Lysophosphatidic Acid. Traffic 2003, 4, 162–174. 10.1034/j.1600-0854.2003.00086.x. [DOI] [PubMed] [Google Scholar]
- Kooijman E. E.; Tieleman D. P.; Testerink C.; Munnik T.; Rijkers D. T. S.; Burger K. N. J.; de Kruijff B. An Electrostatic/Hydrogen Bond Switch as the Basis for the Specific Interaction of Phosphatidic Acid with Proteins. J. Biol. Chem. 2007, 282, 11356–11364. 10.1074/jbc.M609737200. [DOI] [PubMed] [Google Scholar]
- Kooijman E. E.; Burger K. N. J. Biophysics and Function of Phosphatidic Acid: A Molecular Perspective. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2009, 1791, 881–888. 10.1016/j.bbalip.2009.04.001. [DOI] [PubMed] [Google Scholar]
- Magarkar A.; Róg T.; Bunker A. Molecular Dynamics Simulation of Inverse-Phosphocholine Lipids. J. Phys. Chem. C 2014, 118, 19444–19449. 10.1021/jp505633y. [DOI] [Google Scholar]
- Sohlenkamp C.; Geiger O. Bacterial Membrane Lipids: Diversity in Structures and Pathways. FEMS Microbiol. Rev. 2016, 40, 133–159. 10.1093/femsre/fuv008. [DOI] [PubMed] [Google Scholar]
- Steimle A.; Autenrieth I. B.; Frick J.-S. Structure and Function: Lipid A Modifications in Commensals and Pathogens. Int. J. Med. Microbiol. 2016, 306, 290–301. 10.1016/j.ijmm.2016.03.001. [DOI] [PubMed] [Google Scholar]
- Leventis P. A.; Grinstein S. The Distribution and Function of Phosphatidylserine in Cellular Membranes. Annu. Rev. Biophys. 2010, 39, 407–427. 10.1146/annurev.biophys.093008.131234. [DOI] [PubMed] [Google Scholar]
- Bevers E. M.; Williamson P. L. Getting to the Outer Leaflet: Physiology of Phosphatidylserine Exposure at the Plasma Membrane. Physiol. Rev. 2016, 96, 605–645. 10.1152/physrev.00020.2015. [DOI] [PubMed] [Google Scholar]
- Rysavy N. M.; Shimoda L. M. N.; Dixon A. M.; Speck M.; Stokes A. J.; Turner H.; Umemoto E. Y. Beyond Apoptosis: The Mechanism and Function of Phosphatidylserine Asymmetry in the Membrane of Activating Mast Cells. Bioarchitecture 2014, 4, 127–137. 10.1080/19490992.2014.995516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llorente A.; Skotland T.; Sylvänne T.; Kauhanen D.; Róg T.; Orłowski A.; Vattulainen I.; Ekroos K.; Sandvig K. Molecular Lipidomics of Exosomes Released by PC-3 Prostate Cancer Cells. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2013, 1831, 1302–1309. 10.1016/j.bbalip.2013.04.011. [DOI] [PubMed] [Google Scholar]
- Nakamura Y. Plant Phospholipid Diversity: Emerging Functions in Metabolism and Protein–Lipid Interactions. Trends Plant Sci. 2017, 22, 1027–1040. 10.1016/j.tplants.2017.09.002. [DOI] [PubMed] [Google Scholar]
- Schink K. O.; Tan K.-W.; Stenmark H. Phosphoinositides in Control of Membrane Dynamics. Annu. Rev. Cell Dev. Biol. 2016, 32, 143–171. 10.1146/annurev-cellbio-111315-125349. [DOI] [PubMed] [Google Scholar]
- Marat A. L.; Haucke V. Phosphatidylinositol 3-phosphates—at the Interface between Cell Signalling and Membrane Traffic. EMBO J. 2016, 35, 561–579. 10.15252/embj.201593564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choy C. H.; Han B.-K.; Botelho R. J. Phosphoinositide Diversity, Distribution, and Effector Function: Stepping Out of the Box. BioEssays 2017, 39, 1700121. 10.1002/bies.201700121. [DOI] [PubMed] [Google Scholar]
- Hölzl G.; Dörmann P. Structure and Function of Glycoglycerolipids in Plants and Bacteria. Prog. Lipid Res. 2007, 46, 225–243. 10.1016/j.plipres.2007.05.001. [DOI] [PubMed] [Google Scholar]
- Domonkos I.; Laczkó-Dobos H.; Gombos Z. Lipid-Assisted Protein–Protein Interactions That Support Photosynthetic and Other Cellular Activities. Prog. Lipid Res. 2008, 47, 422–435. 10.1016/j.plipres.2008.05.003. [DOI] [PubMed] [Google Scholar]
- Quehenberger O.; Armando A. M.; Dennis E. A. High Sensitivity Quantitative Lipidomics Analysis of Fatty Acids in Biological Samples by Gas Chromatography–Mass Spectrometry. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2011, 1811, 648–656. 10.1016/j.bbalip.2011.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H.-Y.; Huang B. X.; Spector A. A. Phosphatidylserine in the Brain: Metabolism and Function. Prog. Lipid Res. 2014, 56, 1–18. 10.1016/j.plipres.2014.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan P. J.; Griffin P. F. S.; Lösel D. M.; Tyrrell D. The Lipids of Fungi. Prog. Chem. Fats Other Lipids 1975, 14, 49–89. 10.1016/0079-6832(75)90002-6. [DOI] [PubMed] [Google Scholar]
- Kosa G.; Zimmermann B.; Kohler A.; Ekeberg D.; Afseth N. K.; Mounier J.; Shapaval V. High-Throughput Screening of Mucoromycota Fungi for Production of Low- and High-Value Lipids. Biotechnol. Biofuels 2018, 11, 66. 10.1186/s13068-018-1070-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mysyakina I. S.; Sergeeva Y. E.; Bokareva D. A. Lipid Composition of the Spores of Zygomycetous and Ascomycetous Fungi during Cessation of the Exogenous Dormancy State. Microbiology 2018, 87, 51–59. 10.1134/S0026261718010125. [DOI] [Google Scholar]
- Sinanoglou V. J.; Zoumpoulakis P.; Heropoulos G.; Proestos C.; Ćirić A.; Petrovic J.; Glamoclija J.; Sokovic M. Lipid and Fatty Acid Profile of the Edible Fungus Laetiporus Sulphurous. Antifungal and Antibacterial Properties. J. Food Sci. Technol. 2014, 52, 3264–3272. 10.1007/s13197-014-1377-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelles L. Fatty Acid Patterns of Phospholipids and Lipopolysaccharides in the Characterisation of Microbial Communities in Soil: A Review. Biol. Fertil. Soils 1999, 29, 111–129. 10.1007/s003740050533. [DOI] [Google Scholar]
- Sawangkeaw R.; Ngamprasertsith S. A Review of Lipid-Based Biomasses as Feedstocks for Biofuels Production. Renewable Sustainable Energy Rev. 2013, 25, 97–108. 10.1016/j.rser.2013.04.007. [DOI] [Google Scholar]
- Kulig W.; Pasenkiewicz-Gierula M.; Róg T. Cis and Trans Unsaturated Phosphatidylcholine Bilayers: A Molecular Dynamics Simulation Study. Chem. Phys. Lipids 2016, 195, 12–20. 10.1016/j.chemphyslip.2015.07.002. [DOI] [PubMed] [Google Scholar]
- Róg T.; Murzyn K.; Gurbiel R.; Takaoka Y.; Kusumi A.; Pasenkiewicz-Gierula M. Effects of Phospholipid Unsaturation on the Bilayer Nonpolar Region: A Molecular Simulation Study. J. Lipid Res. 2004, 45, 326–336. 10.1194/jlr.M300187-JLR200. [DOI] [PubMed] [Google Scholar]
- Vähäheikkilä M.; Peltomaa T.; Róg T.; Vazdar M.; Pöyry S.; Vattulainen I. How Cardiolipin Peroxidation Alters the Properties of the Inner Mitochondrial Membrane?. Chem. Phys. Lipids 2018, 214, 15–23. 10.1016/j.chemphyslip.2018.04.005. [DOI] [PubMed] [Google Scholar]
- Cwiklik L.; Jungwirth P. Massive Oxidation of Phospholipid Membranes Leads to Pore Creation and Bilayer Disintegration. Chem. Phys. Lett. 2010, 486, 99–103. 10.1016/j.cplett.2010.01.010. [DOI] [Google Scholar]
- Beranova L.; Cwiklik L.; Jurkiewicz P.; Hof M.; Jungwirth P. Oxidation Changes Physical Properties of Phospholipid Bilayers: Fluorescence Spectroscopy and Molecular Simulations. Langmuir 2010, 26, 6140–6144. 10.1021/la100657a. [DOI] [PubMed] [Google Scholar]
- Boonnoy P.; Jarerattanachat V.; Karttunen M.; Wong-ekkabut J. Bilayer Deformation, Pores, and Micellation Induced by Oxidized Lipids. J. Phys. Chem. Lett. 2015, 6, 4884–4888. 10.1021/acs.jpclett.5b02405. [DOI] [PubMed] [Google Scholar]
- Jurkiewicz P.; Olżyńska A.; Cwiklik L.; Conte E.; Jungwirth P.; Megli F. M.; Hof M. Biophysics of Lipid Bilayers Containing Oxidatively Modified Phospholipids: Insights from Fluorescence and EPR Experiments and from MD Simulations. Biochim. Biophys. Acta, Biomembr. 2012, 1818, 2388–2402. 10.1016/j.bbamem.2012.05.020. [DOI] [PubMed] [Google Scholar]
- Van der Paal J.; Neyts E. C.; Verlackt C. C. W.; Bogaerts A. Effect of Lipid Peroxidation on Membrane Permeability of Cancer and Normal Cells Subjected to Oxidative Stress. Chem. Sci. 2016, 7, 489–498. 10.1039/C5SC02311D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deigner H.-P.; Hermetter A. Oxidized Phospholipids: Emerging Lipid Mediators in Pathophysiology. Curr. Opin. Lipidol. 2008, 19, 289–294. 10.1097/MOL.0b013e3282fe1d0e. [DOI] [PubMed] [Google Scholar]
- Fruhwirth G. O.; Loidl A.; Hermetter A. Oxidized Phospholipids: From Molecular Properties to Disease. Biochim. Biophys. Acta, Mol. Basis Dis. 2007, 1772, 718–736. 10.1016/j.bbadis.2007.04.009. [DOI] [PubMed] [Google Scholar]
- Pham-Huy L. A.; He H.; Pham-Huy C. Free Radicals, Antioxidants in Disease and Health. Int. J. Biomed. Sci. IJBS 2008, 4, 89–96. [PMC free article] [PubMed] [Google Scholar]
- Kates M.; Syz J.-Y.; Gosser D.; Haines T. H. pH-Dissociation Characteristics of Cardiolipin and Its 2′-Deoxy Analogue. Lipids 1993, 28, 877–882. 10.1007/BF02537494. [DOI] [PubMed] [Google Scholar]
- Haines T. H.; Dencher N. A. Cardiolipin: A Proton Trap for Oxidative Phosphorylation. FEBS Lett. 2002, 528, 35–39. 10.1016/S0014-5793(02)03292-1. [DOI] [PubMed] [Google Scholar]
- Nichols-Smith S.; Kuhl T. Electrostatic Interactions between Model Mitochondrial Membranes. Colloids Surf., B 2005, 41, 121–127. 10.1016/j.colsurfb.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Kooijman E. E.; Swim L. A.; Graber Z. T.; Tyurina Y. Y.; Bayır H.; Kagan V. E. Magic Angle Spinning 31P NMR Spectroscopy Reveals Two Essentially Identical Ionization States for the Cardiolipin Phosphates in Phospholipid Liposomes. Biochim. Biophys. Acta, Biomembr. 2017, 1859, 61–68. 10.1016/j.bbamem.2016.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sathappa M.; Alder N. N. The Ionization Properties of Cardiolipin and Its Variants in Model Bilayers. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 1362–1372. 10.1016/j.bbamem.2016.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olofsson G.; Sparr E. Ionization Constants pKa of Cardiolipin. PLoS One 2013, 8, e73040. 10.1371/journal.pone.0073040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maguire J. J.; Tyurina Y. Y.; Mohammadyani D.; Kapralov A. A.; Anthonymuthu T. S.; Qu F.; Amoscato A. A.; Sparvero L. J.; Tyurin V. A.; Planas-Iglesias J.; et al. Known Unknowns of Cardiolipin Signaling: The Best Is yet to Come. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2017, 1862, 8–24. 10.1016/j.bbalip.2016.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H.; Mancuso D. J.; Jiang X.; Guan S.; Yang J.; Yang K.; Sun G.; Gross R. W.; Han X. Shotgun Lipidomics Reveals the Temporally Dependent, Highly Diversified Cardiolipin Profile in the Mammalian Brain: Temporally Coordinated Postnatal Diversification of Cardiolipin Molecular Species with Neuronal Remodeling †. Biochemistry 2008, 47, 5869–5880. 10.1021/bi7023282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X.; Yang K.; Yang J.; Cheng H.; Gross R. W. Shotgun Lipidomics of Cardiolipin Molecular Species in Lipid Extracts of Biological Samples. J. Lipid Res. 2006, 47, 864–879. 10.1194/jlr.D500044-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson K.-A. Sphingolipid Long Chain Bases. Lipids 1970, 5, 878–891. 10.1007/BF02531119. [DOI] [PubMed] [Google Scholar]
- Ramstedt B.; Slotte J. P. Membrane Properties of Sphingomyelins. FEBS Lett. 2002, 531, 33–37. 10.1016/S0014-5793(02)03406-3. [DOI] [PubMed] [Google Scholar]
- Slotte J. P. Biological Functions of Sphingomyelins. Prog. Lipid Res. 2013, 52, 424–437. 10.1016/j.plipres.2013.05.001. [DOI] [PubMed] [Google Scholar]
- Manna M.; Róg T.; Vattulainen I. The Challenges of Understanding Glycolipid Functions: An Open Outlook Based on Molecular Simulations. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2014, 1841, 1130–1145. 10.1016/j.bbalip.2013.12.016. [DOI] [PubMed] [Google Scholar]
- Slotte J. P. Molecular Properties of Various Structurally Defined Sphingomyelins – Correlation of Structure with Function. Prog. Lipid Res. 2013, 52, 206–219. 10.1016/j.plipres.2012.12.001. [DOI] [PubMed] [Google Scholar]
- Gulshan K.; Smith J. D. Sphingomyelin Regulation of Plasma Membrane Asymmetry, Efflux and Reverse Cholesterol Transport. Clin. Lipidol. 2014, 9, 383–393. 10.2217/clp.14.28. [DOI] [Google Scholar]
- Müller C. P.; Reichel M.; Mühle C.; Rhein C.; Gulbins E.; Kornhuber J. Brain Membrane Lipids in Major Depression and Anxiety Disorders. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2015, 1851, 1052–1065. 10.1016/j.bbalip.2014.12.014. [DOI] [PubMed] [Google Scholar]
- Karunakaran I.; van Echten-Deckert G. Sphingosine 1-Phosphate – A Double Edged Sword in the Brain. Biochim. Biophys. Acta, Biomembr. 2017, 1859, 1573–1582. 10.1016/j.bbamem.2017.03.008. [DOI] [PubMed] [Google Scholar]
- Li L. K.; So L.; Spector A. Membrane Cholesterol and Phospholipid in Consecutive Concentric Sections of Human Lenses. J. Lipid Res. 1985, 26, 600–609. [PubMed] [Google Scholar]
- Subczynski W. K.; Raguz M.; Widomska J.; Mainali L.; Konovalov A. Functions of Cholesterol and the Cholesterol Bilayer Domain Specific to the Fiber-Cell Plasma Membrane of the Eye Lens. J. Membr. Biol. 2012, 245, 51–68. 10.1007/s00232-011-9412-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain M.; Ngoy S.; Sheth S. A.; Swanson R. A.; Rhee E. P.; Liao R.; Clish C. B.; Mootha V. K.; Nilsson R. A Systematic Survey of Lipids across Mouse Tissues. Am. J. Physiol.-Endocrinol. Metab. 2014, 306, E854–E868. 10.1152/ajpendo.00371.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwai I.; Han H.; Hollander L. d.; Svensson S.; Öfverstedt L.-G.; Anwar J.; Brewer J.; Bloksgaard M.; Laloeuf A.; Nosek D.; et al. The Human Skin Barrier Is Organized as Stacked Bilayers of Fully Extended Ceramides with Cholesterol Molecules Associated with the Ceramide Sphingoid Moiety. J. Invest. Dermatol. 2012, 132, 2215–2225. 10.1038/jid.2012.43. [DOI] [PubMed] [Google Scholar]
- Boncheva M. The Physical Chemistry of the Stratum Corneum Lipids. Int. J. Cosmet. Sci. 2014, 36, 505–515. 10.1111/ics.12162. [DOI] [PubMed] [Google Scholar]
- van Meer G.; Voelker D. R.; Feigenson G. W. Membrane Lipids: Where They Are and How They Behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giang H.; Schick M. On the Puzzling Distribution of Cholesterol in the Plasma Membrane. Chem. Phys. Lipids 2016, 199, 35–38. 10.1016/j.chemphyslip.2015.12.002. [DOI] [PubMed] [Google Scholar]
- Ohvo-Rekilä H.; Ramstedt B.; Leppimäki P.; Peter Slotte J. Cholesterol Interactions with Phospholipids in Membranes. Prog. Lipid Res. 2002, 41, 66–97. 10.1016/S0163-7827(01)00020-0. [DOI] [PubMed] [Google Scholar]
- Róg T.; Vattulainen I. Cholesterol, Sphingolipids, and Glycolipids: What Do We Know about Their Role in Raft-like Membranes?. Chem. Phys. Lipids 2014, 184, 82–104. 10.1016/j.chemphyslip.2014.10.004. [DOI] [PubMed] [Google Scholar]
- de Meyer F.; Smit B. Effect of Cholesterol on the Structure of a Phospholipid Bilayer. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 3654–3658. 10.1073/pnas.0809959106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Róg T.; Pasenkiewicz-Gierula M.; Vattulainen I.; Karttunen M. Ordering Effects of Cholesterol and Its Analogues. Biochim. Biophys. Acta, Biomembr. 2009, 1788, 97–121. 10.1016/j.bbamem.2008.08.022. [DOI] [PubMed] [Google Scholar]
- Magarkar A.; Dhawan V.; Kallinteri P.; Viitala T.; Elmowafy M.; Róg T.; Bunker A. Cholesterol Level Affects Surface Charge of Lipid Membranes in Saline Solution. Sci. Rep. 2015, 4, 5005. 10.1038/srep05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushik S.; Massey A. C.; Cuervo A. M. Lysosome Membrane Lipid Microdomains: Novel Regulators of Chaperone-Mediated Autophagy. EMBO J. 2006, 25, 3921–3933. 10.1038/sj.emboj.7601283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayermann S. P.; Rayermann G. E.; Cornell C. E.; Merz A. J.; Keller S. L. Hallmarks of Reversible Separation of Living, Unperturbed Cell Membranes into Two Liquid Phases. Biophys. J. 2017, 113, 2425–2432. 10.1016/j.bpj.2017.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingwood D.; Kaiser H.-J.; Levental I.; Simons K. Lipid Rafts as Functional Heterogeneity in Cell Membranes. Biochem. Soc. Trans. 2009, 37, 955–960. 10.1042/BST0370955. [DOI] [PubMed] [Google Scholar]
- Simons K.; Gerl M. J. Revitalizing Membrane Rafts: New Tools and Insights. Nat. Rev. Mol. Cell Biol. 2010, 11, 688–699. 10.1038/nrm2977. [DOI] [PubMed] [Google Scholar]
- Owen M. C.; Kulig W.; Rog T.; Vattulainen I.; Strodel B. Cholesterol Protects the Oxidized Lipid Bilayer from Water Injury: An All-Atom Molecular Dynamics Study. J. Membr. Biol. 2018, 251, 521–534. 10.1007/s00232-018-0028-9. [DOI] [PubMed] [Google Scholar]
- Schuck S. Polarized Sorting in Epithelial Cells: Raft Clustering and the Biogenesis of the Apical Membrane. J. Cell Sci. 2004, 117, 5955–5964. 10.1242/jcs.01596. [DOI] [PubMed] [Google Scholar]
- Diaz-Rohrer B. B.; Levental K. R.; Simons K.; Levental I. Membrane Raft Association Is a Determinant of Plasma Membrane Localization. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 8500–8505. 10.1073/pnas.1404582111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons K.; Toomre D. Lipid Rafts and Signal Transduction. Nat. Rev. Mol. Cell Biol. 2000, 1, 31–39. 10.1038/35036052. [DOI] [PubMed] [Google Scholar]
- Allen J. A.; Halverson-Tamboli R. A.; Rasenick M. M. Lipid Raft Microdomains and Neurotransmitter Signalling. Nat. Rev. Neurosci. 2007, 8, 128–140. 10.1038/nrn2059. [DOI] [PubMed] [Google Scholar]
- Agarwal S. R.; Gratwohl J.; Cozad M.; Yang P.-C.; Clancy C. E.; Harvey R. D. Compartmentalized CAMP Signaling Associated With Lipid Raft and Non-Raft Membrane Domains in Adult Ventricular Myocytes. Front. Pharmacol. 2018, 9, 332. 10.3389/fphar.2018.00332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshney P.; Yadav V.; Saini N. Lipid Rafts in Immune Signalling: Current Progress and Future Perspective. Immunology 2016, 149, 13–24. 10.1111/imm.12617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulig W.; Cwiklik L.; Jurkiewicz P.; Rog T.; Vattulainen I. Cholesterol Oxidation Products and Their Biological Importance. Chem. Phys. Lipids 2016, 199, 144–160. 10.1016/j.chemphyslip.2016.03.001. [DOI] [PubMed] [Google Scholar]
- Kulig W.; Jurkiewicz P.; Olżyńska A.; Tynkkynen J.; Javanainen M.; Manna M.; Rog T.; Hof M.; Vattulainen I.; Jungwirth P. Experimental Determination and Computational Interpretation of Biophysical Properties of Lipid Bilayers Enriched by Cholesteryl Hemisuccinate. Biochim. Biophys. Acta, Biomembr. 2015, 1848, 422–432. 10.1016/j.bbamem.2014.10.032. [DOI] [PubMed] [Google Scholar]
- Kulig W.; Mikkolainen H.; Olżyńska A.; Jurkiewicz P.; Cwiklik L.; Hof M.; Vattulainen I.; Jungwirth P.; Rog T. Bobbing of Oxysterols: Molecular Mechanism for Translocation of Tail-Oxidized Sterols through Biological Membranes. J. Phys. Chem. Lett. 2018, 9, 1118–1123. 10.1021/acs.jpclett.8b00211. [DOI] [PubMed] [Google Scholar]
- Kurzchalia T. V.; Ward S. Why Do Worms Need Cholesterol?. Nat. Cell Biol. 2003, 5, 684–688. 10.1038/ncb0803-684. [DOI] [PubMed] [Google Scholar]
- Clayton R. B. The Utilization of Sterols by Insects. J. Lipid Res. 1964, 5, 3–19. [PubMed] [Google Scholar]
- Hannich J. T.; Umebayashi K.; Riezman H. Distribution and Functions of Sterols and Sphingolipids. Cold Spring Harbor Perspect. Biol. 2011, 3, a004762–a004762. 10.1101/cshperspect.a004762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janson E. M.; Grebenok R. J.; Behmer S. T.; Abbot P. Same Host-Plant, Different Sterols: Variation in Sterol Metabolism in an Insect Herbivore Community. J. Chem. Ecol. 2009, 35, 1309–1319. 10.1007/s10886-009-9713-6. [DOI] [PubMed] [Google Scholar]
- Gachumi G.; El-Aneed A. Mass Spectrometric Approaches for the Analysis of Phytosterols in Biological Samples. J. Agric. Food Chem. 2017, 65, 10141–10156. 10.1021/acs.jafc.7b03785. [DOI] [PubMed] [Google Scholar]
- Ostlund R. E. Phytosterols in Human Nutrition. Annu. Rev. Nutr. 2002, 22, 533–549. 10.1146/annurev.nutr.22.020702.075220. [DOI] [PubMed] [Google Scholar]
- Valitova J. N.; Sulkarnayeva A. G.; Minibayeva F. V. Plant Sterols: Diversity, Biosynthesis, and Physiological Functions. Biochemistry 2016, 81, 819–834. 10.1134/S0006297916080046. [DOI] [PubMed] [Google Scholar]
- Genser B.; Silbernagel G.; De Backer G.; Bruckert E.; Carmena R.; Chapman M. J.; Deanfield J.; Descamps O. S.; Rietzschel E. R.; Dias K. C.; et al. Plant Sterols and Cardiovascular Disease: A Systematic Review and Meta-Analysis. Eur. Heart J. 2012, 33, 444–451. 10.1093/eurheartj/ehr441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silbernagel G.; Genser B.; Nestel P.; März W. Plant Sterols and Atherosclerosis:. Curr. Opin. Lipidol. 2013, 24, 12–17. 10.1097/MOL.0b013e32835b6271. [DOI] [PubMed] [Google Scholar]
- Ajagbe B. O.; Othman R. A.; Myrie S. B. Plant Sterols, Stanols, and Sitosterolemia. J. AOAC Int. 2015, 98, 716–723. 10.5740/jaoacint.SGEAjagbe. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He W.-S.; Zhu H.; Chen Z.-Y. Plant Sterols: Chemical and Enzymatic Structural Modifications and Effects on Their Cholesterol-Lowering Activity. J. Agric. Food Chem. 2018, 66, 3047–3062. 10.1021/acs.jafc.8b00059. [DOI] [PubMed] [Google Scholar]
- Corrêa R. C. G.; Peralta R. M.; Bracht A.; Ferreira I. C. F. R. The Emerging Use of Mycosterols in Food Industry along with the Current Trend of Extended Use of Bioactive Phytosterols. Trends Food Sci. Technol. 2017, 67, 19–35. 10.1016/j.tifs.2017.06.012. [DOI] [Google Scholar]
- Weete J. D.; Abril M.; Blackwell M. Phylogenetic Distribution of Fungal Sterols. PLoS One 2010, 5, e10899. 10.1371/journal.pone.0010899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhingra S.; Cramer R. A. Regulation of Sterol Biosynthesis in the Human Fungal Pathogen Aspergillus Fumigatus: Opportunities for Therapeutic Development. Front. Microbiol. 2017, 8, 92. 10.3389/fmicb.2017.00092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornspan J. D.; Rottem S. The Phospholipid Profile of Mycoplasmas. J. Lipids 2012, 2012, 1–8. 10.1155/2012/640762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J. H.; Yin X.; Welander P. V. Sterol Synthesis in Diverse Bacteria. Front. Microbiol. 2016, 7, 990. 10.3389/fmicb.2016.00990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belin B. J.; Busset N.; Giraud E.; Molinaro A.; Silipo A.; Newman D. K. Hopanoid Lipids: From Membranes to Plant–Bacteria Interactions. Nat. Rev. Microbiol. 2018, 16, 304–315. 10.1038/nrmicro.2017.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sáenz J. P.; Sezgin E.; Schwille P.; Simons K. Functional Convergence of Hopanoids and Sterols in Membrane Ordering. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 14236–14240. 10.1073/pnas.1212141109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sáenz J. P.; Grosser D.; Bradley A. S.; Lagny T. J.; Lavrynenko O.; Broda M.; Simons K. Hopanoids as Functional Analogues of Cholesterol in Bacterial Membranes. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 11971–11976. 10.1073/pnas.1515607112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuvonen M.; Manna M.; Mokkila S.; Javanainen M.; Rog T.; Liu Z.; Bittman R.; Vattulainen I.; Ikonen E. Enzymatic Oxidation of Cholesterol: Properties and Functional Effects of Cholestenone in Cell Membranes. PLoS One 2014, 9, e103743. 10.1371/journal.pone.0103743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pourmousa M.; Róg T.; Mikkeli R.; Vattulainen l.; Solanko L. M.; Wüstner D.; List N. H.; Kongsted J.; Karttunen M. Dehydroergosterol as an Analogue for Cholesterol: Why It Mimics Cholesterol So Well—or Does It?. J. Phys. Chem. B 2014, 118, 7345–7357. 10.1021/jp406883k. [DOI] [PubMed] [Google Scholar]
- Hölttä-Vuori M.; Uronen R.-L.; Repakova J.; Salonen E.; Vattulainen I.; Panula P.; Li Z.; Bittman R.; Ikonen E. BODIPY-Cholesterol: A New Tool to Visualize Sterol Trafficking in Living Cells and Organisms. Traffic 2008, 9, 1839–1849. 10.1111/j.1600-0854.2008.00801.x. [DOI] [PubMed] [Google Scholar]
- Hulce J. J.; Cognetta A. B.; Niphakis M. J.; Tully S. E.; Cravatt B. F. Proteome-Wide Mapping of Cholesterol-Interacting Proteins in Mammalian Cells. Nat. Methods 2013, 10, 259–264. 10.1038/nmeth.2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X.; Covey D. F. Total Synthesis of Ent-Cholesterol via a Steroid C,D-Ring Side-Chain Synthon. J. Org. Chem. 2002, 67, 4893–4900. 10.1021/jo025535k. [DOI] [PubMed] [Google Scholar]
- Róg T.; Pasenkiewicz-Gierula M. Effects of Epicholesterol on the Phosphatidylcholine Bilayer: A Molecular Simulation Study. Biophys. J. 2003, 84, 1818–1826. 10.1016/S0006-3495(03)74989-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Róg T.; Pöyry S.; Vattulainen I. Building Synthetic Sterols Computationally – Unlocking the Secrets of Evolution?. Front. Bioeng. Biotechnol. 2015, 3, 121. 10.3389/fbioe.2015.00121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pöyry S.; Róg T.; Karttunen M.; Vattulainen I. Significance of Cholesterol Methyl Groups. J. Phys. Chem. B 2008, 112, 2922–2929. 10.1021/jp7100495. [DOI] [PubMed] [Google Scholar]
- Róg T.; Pasenkiewicz-Gierula M.; Vattulainen I.; Karttunen M. What Happens If Cholesterol Is Made Smoother. Biophys. J. 2007, 92, 3346–3357. 10.1529/biophysj.106.095497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mydock-McGrane L.; Rath N. P.; Covey D. F. Synthesis of a Smoothened Cholesterol: 18,19-Di-nor-Cholesterol. J. Org. Chem. 2014, 79, 5636–5643. 10.1021/jo500813n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer T.; Baek D. J.; Bittman R.; Haralampiev I.; Müller P.; Herrmann A.; Huster D.; Scheidt H. A. Membrane Properties of Cholesterol Analogs with an Unbranched Aliphatic Side Chain. Chem. Phys. Lipids 2014, 184, 1–6. 10.1016/j.chemphyslip.2014.08.002. [DOI] [PubMed] [Google Scholar]
- Benvegnu T.; Brard M.; Plusquellec D. Archaeabacteria Bipolar Lipid Analogues: Structure, Synthesis and Lyotropic Properties. Curr. Opin. Colloid Interface Sci. 2004, 8, 469–479. 10.1016/j.cocis.2004.01.005. [DOI] [Google Scholar]
- Koga Y. Thermal Adaptation of the Archaeal and Bacterial Lipid Membranes. Archaea 2012, 2012, 1–6. 10.1155/2012/789652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprott G. D.Archaeal Membrane Lipids and Applications. eLS; John Wiley & Sons, Ltd, Ed.; John Wiley & Sons, Ltd: Chichester, UK, 2011; Vol. 15, 10.1002/9780470015902.a0000385.pub3. [DOI] [Google Scholar]
- Yang Y.; Lee M.; Fairn G. D. Phospholipid Subcellular Localization and Dynamics. J. Biol. Chem. 2018, 293, 6230–6240. 10.1074/jbc.R117.000582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon A. K.; Levine T. P. The Cellular Lipid Landscape. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2016, 1861, 755–756. 10.1016/j.bbalip.2016.05.004. [DOI] [PubMed] [Google Scholar]
- Daleke D. L. Regulation of Transbilayer Plasma Membrane Phospholipid Asymmetry. J. Lipid Res. 2003, 44, 233–242. 10.1194/jlr.R200019-JLR200. [DOI] [PubMed] [Google Scholar]
- Kobayashi T.; Menon A. K. Transbilayer Lipid Asymmetry. Curr. Biol. 2018, 28, R386–R391. 10.1016/j.cub.2018.01.007. [DOI] [PubMed] [Google Scholar]
- Panatala R.; Hennrich H.; Holthuis J. C. M. Inner Workings and Biological Impact of Phospholipid Flippases. J. Cell Sci. 2015, 128, 2021–2032. 10.1242/jcs.102715. [DOI] [PubMed] [Google Scholar]
- Andersen J. P.; Vestergaard A. L.; Mikkelsen S. A.; Mogensen L. S.; et al. P4-ATPases as Phospholipid Flippases—Structure, Function, and Enigmas. Front. Physiol. 2016, 7, 275. 10.3389/fphys.2016.00275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hankins H. M.; Baldridge R. D.; Xu P.; Graham T. R. Role of Flippases, Scramblases and Transfer Proteins in Phosphatidylserine Subcellular Distribution. Traffic 2015, 16, 35–47. 10.1111/tra.12233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondal M.; Mesmin B.; Mukherjee S.; Maxfield F. R. Sterols Are Mainly in the Cytoplasmic Leaflet of the Plasma Membrane and the Endocytic Recycling Compartment in CHO Cells. Mol. Biol. Cell 2009, 20, 581–588. 10.1091/mbc.e08-07-0785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S.-L.; Sheng R.; Jung J. H.; Wang L.; Stec E.; O’Connor M. J.; Song S.; Bikkavilli R. K.; Winn R. A.; Lee D.; et al. Orthogonal Lipid Sensors Identify Transbilayer Asymmetry of Plasma Membrane Cholesterol. Nat. Chem. Biol. 2017, 13, 268–274. 10.1038/nchembio.2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solanko L. M.; Sullivan D. P.; Sere Y. Y.; Szomek M.; Lunding A.; Solanko K. A.; Pizovic A.; Stanchev L. D.; Pomorski T. G.; Menon A. K.; et al. Ergosterol Is Mainly Located in the Cytoplasmic Leaflet of the Yeast Plasma Membrane. Traffic 2018, 19, 198–214. 10.1111/tra.12545. [DOI] [PubMed] [Google Scholar]
- Brooks-Wilson A.; Marcil M.; Clee S. M.; Zhang L.-H.; Roomp K.; van Dam M.; Yu L.; Brewer C.; Collins J. A.; Molhuizen H. O. F.; et al. Mutations in ABC1 in Tangier Disease and Familial High-Density Lipoprotein Deficiency. Nat. Genet. 1999, 22, 336–345. 10.1038/11905. [DOI] [PubMed] [Google Scholar]
- Phillips M. C. Is ABCA1 a Lipid Transfer Protein?. J. Lipid Res. 2018, 59, 749–763. 10.1194/jlr.R082313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtney K. C.; Pezeshkian W.; Raghupathy R.; Zhang C.; Darbyson A.; Ipsen J. H.; Ford D. A.; Khandelia H.; Presley J. F.; Zha X. C24 Sphingolipids Govern the Transbilayer Asymmetry of Cholesterol and Lateral Organization of Model and Live-Cell Plasma Membranes. Cell Rep. 2018, 24, 1037–1049. 10.1016/j.celrep.2018.06.104. [DOI] [PubMed] [Google Scholar]
- Maekawa M.; Fairn G. D. Complementary Probes Reveal That Phosphatidylserine Is Required for the Proper Transbilayer Distribution of Cholesterol. J. Cell Sci. 2015, 128, 1422–1433. 10.1242/jcs.164715. [DOI] [PubMed] [Google Scholar]
- Yesylevskyy S. O.; Demchenko A. P. How Cholesterol Is Distributed between Monolayers in Asymmetric Lipid Membranes. Eur. Biophys. J. 2012, 41, 1043–1054. 10.1007/s00249-012-0863-z. [DOI] [PubMed] [Google Scholar]
- Falkovich S. G.; Martinez-Seara H.; Nesterenko A. M.; Vattulainen I.; Gurtovenko A. A. What Can We Learn about Cholesterol’s Transmembrane Distribution Based on Cholesterol-Induced Changes in Membrane Dipole Potential?. J. Phys. Chem. Lett. 2016, 7, 4585–4590. 10.1021/acs.jpclett.6b02123. [DOI] [PubMed] [Google Scholar]
- Michaelson D. M.; Barkai G.; Barenholz Y. Asymmetry of Lipid Organization in Cholinergic Synaptic Vesicle Membranes. Biochem. J. 1983, 211, 155–162. 10.1042/bj2110155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins J. A.; Dawson R. M. C. Asymmetry of the Phospholipid Bilayer of Rat Liver Endoplasmic Reticulum. Biochim. Biophys. Acta, Biomembr. 1977, 470, 342–356. 10.1016/0005-2736(77)90126-2. [DOI] [PubMed] [Google Scholar]
- Ma Z.; Liu Z.; Huang X. Membrane Phospholipid Asymmetry Counters the Adverse Effects of Sterol Overloading in the Golgi Membrane of Drosophila. Genetics 2012, 190, 1299–1308. 10.1534/genetics.111.137687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bick R. J.; Van Winkle B. W.; Taffet G. E. Cardiac Sarcoplasmic Reticulum Membrane Lipid Asymmetries. Ann. N. Y. Acad. Sci. 1998, 853, 365–367. 10.1111/j.1749-6632.1998.tb08301.x. [DOI] [PubMed] [Google Scholar]
- Róg T.; Orłowski A.; Llorente A.; Skotland T.; Sylvänne T.; Kauhanen D.; Ekroos K.; Sandvig K.; Vattulainen I. Interdigitation of Long-Chain Sphingomyelin Induces Coupling of Membrane Leaflets in a Cholesterol Dependent Manner. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 281–288. 10.1016/j.bbamem.2015.12.003. [DOI] [PubMed] [Google Scholar]
- Manna M.; Javanainen M.; Monne H. M.-S.; Gabius H.-J.; Rog T.; Vattulainen I. Long-Chain GM1 Gangliosides Alter Transmembrane Domain Registration through Interdigitation. Biochim. Biophys. Acta, Biomembr. 2017, 1859, 870–878. 10.1016/j.bbamem.2017.01.033. [DOI] [PubMed] [Google Scholar]
- Li L. K.; So L.; Spector A. Age-Dependent Changes in the Distribution and Concentration of Human Lens Cholesterol and Phospholipids. Biochim. Biophys. Acta, Lipids Lipid Metab. 1987, 917, 112–120. 10.1016/0005-2760(87)90291-8. [DOI] [PubMed] [Google Scholar]
- Tulodziecka K.; Diaz-Rohrer B. B.; Farley M. M.; Chan R. B.; Di Paolo G.; Levental K. R.; Waxham M. N.; Levental I. Remodeling of the Postsynaptic Plasma Membrane during Neural Development. Mol. Biol. Cell 2016, 27, 3480–3489. 10.1091/mbc.e16-06-0420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietiläinen K. H.; Róg T.; Seppänen-Laakso T.; Virtue S.; Gopalacharyulu P.; Tang J.; Rodriguez-Cuenca S.; Maciejewski A.; Naukkarinen J.; Ruskeepää A.-L.; et al. Association of Lipidome Remodeling in the Adipocyte Membrane with Acquired Obesity in Humans. PLoS Biol. 2011, 9, e1000623. 10.1371/journal.pbio.1000623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butt A. H.; Rasool N.; Khan Y. D. A Treatise to Computational Approaches Towards Prediction of Membrane Protein and Its Subtypes. J. Membr. Biol. 2017, 250, 55–76. 10.1007/s00232-016-9937-7. [DOI] [PubMed] [Google Scholar]
- Takamori S.; Holt M.; Stenius K.; Lemke E. A.; Grønborg M.; Riedel D.; Urlaub H.; Schenck S.; Brügger B.; Ringler P.; et al. Molecular Anatomy of a Trafficking Organelle. Cell 2006, 127, 831–846. 10.1016/j.cell.2006.10.030. [DOI] [PubMed] [Google Scholar]
- Fagerberg L.; Jonasson K.; von Heijne G.; Uhlén M.; Berglund L. Prediction of the Human Membrane Proteome. Proteomics 2010, 10, 1141–1149. 10.1002/pmic.200900258. [DOI] [PubMed] [Google Scholar]
- Scherer W. F.; Syverton J. T.; Gey G. O. Studies on the Propagation in Vitro of Poliomyelitis Viruses. IV. Viral Multiplication in a Stable Strain of Human Malignant Epithelial Cells (Strain HeLa) Derived from an Epidermoid Carcinoma of the Cervix. J. Exp. Med. 1953, 97, 695–710. 10.1084/jem.97.5.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaraj N.; Wisniewski J. R.; Geiger T.; Cox J.; Kircher M.; Kelso J.; Paabo S.; Mann M. Deep Proteome and Transcriptome Mapping of a Human Cancer Cell Line. Mol. Syst. Biol. 2011, 7, 548–548. 10.1038/msb.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Rost B. Comparing Function and Structure between Entire Proteomes. Protein Sci. 2001, 10, 1970–1979. 10.1110/ps.10101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vit O.; Petrak J. Integral Membrane Proteins in Proteomics. How to Break Open the Black Box?. J. Proteomics 2017, 153, 8–20. 10.1016/j.jprot.2016.08.006. [DOI] [PubMed] [Google Scholar]
- Garrow A. G.; Agnew A.; Westhead D. R. TMB-Hunt: An Amino Acid Composition Based Method to Screen Proteomes for Beta-Barrel Transmembrane Proteins. BMC Bioinf. 2005, 6, 56. 10.1186/1471-2105-6-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaturvedi D.; Mahalakshmi R. Transmembrane β-Barrels: Evolution, Folding and Energetics. Biochim. Biophys. Acta, Biomembr. 2017, 1859, 2467–2482. 10.1016/j.bbamem.2017.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almén M.; Nordström K. J.; Fredriksson R.; Schiöth H. B. Mapping the Human Membrane Proteome: A Majority of the Human Membrane Proteins Can Be Classified According to Function and Evolutionary Origin. BMC Biol. 2009, 7, 50. 10.1186/1741-7007-7-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H.; Flynn A. D. Drugging Membrane Protein Interactions. Annu. Rev. Biomed. Eng. 2016, 18, 51–76. 10.1146/annurev-bioeng-092115-025322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yıldırım M. A.; Goh K.-I.; Cusick M. E.; Barabási A.-L.; Vidal M. Drug—Target Network. Nat. Biotechnol. 2007, 25, 1119–1126. 10.1038/nbt1338. [DOI] [PubMed] [Google Scholar]
- Dobson L.; Reményi I.; Tusnády G. E. The Human Transmembrane Proteome. Biol. Direct 2015, 10, 31. 10.1186/s13062-015-0061-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomize A. L.; Lomize M. A.; Krolicki S. R.; Pogozheva I. D. Membranome: A Database for Proteome-Wide Analysis of Single-Pass Membrane Proteins. Nucleic Acids Res. 2017, 45, D250–D255. 10.1093/nar/gkw712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pogozheva I. D.; Lomize A. L. Evolution and Adaptation of Single-Pass Transmembrane Proteins. Biochim. Biophys. Acta, Biomembr. 2018, 1860, 364–377. 10.1016/j.bbamem.2017.11.002. [DOI] [PubMed] [Google Scholar]
- Bocharov E. V.; Mineev K. S.; Pavlov K. V.; Akimov S. A.; Kuznetsov A. S.; Efremov R. G.; Arseniev A. S. Helix-Helix Interactions in Membrane Domains of Bitopic Proteins: Specificity and Role of Lipid Environment. Biochim. Biophys. Acta, Biomembr. 2017, 1859, 561–576. 10.1016/j.bbamem.2016.10.024. [DOI] [PubMed] [Google Scholar]
- Polyansky A. A.; Chugunov A. O.; Volynsky P. E.; Krylov N. A.; Nolde D. E.; Efremov R. G. PREDDIMER: A Web Server for Prediction of Transmembrane Helical Dimers. Bioinformatics 2014, 30, 889–890. 10.1093/bioinformatics/btt645. [DOI] [PubMed] [Google Scholar]
- Lomize A. L.; Pogozheva I. D. TMDOCK: An Energy-Based Method for Modeling α-Helical Dimers in Membranes. J. Mol. Biol. 2017, 429, 390–398. 10.1016/j.jmb.2016.09.005. [DOI] [PubMed] [Google Scholar]
- Lomize A. L.; Hage J. M.; Pogozheva I. D. Membranome 2.0: Database for Proteome-Wide Profiling of Bitopic Proteins and Their Dimers. Bioinformatics 2018, 34, 1061–1062. 10.1093/bioinformatics/btx720. [DOI] [PubMed] [Google Scholar]
- Mueller B. K.; Subramaniam S.; Senes A. A Frequent, GxxxG-Mediated, Transmembrane Association Motif Is Optimized for the Formation of Interhelical Cα-H Hydrogen Bonds. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, E888–E895. 10.1073/pnas.1319944111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cymer F.; Veerappan A.; Schneider D. Transmembrane Helix–Helix Interactions Are Modulated by the Sequence Context and by Lipid Bilayer Properties. Biochim. Biophys. Acta, Biomembr. 2012, 1818, 963–973. 10.1016/j.bbamem.2011.07.035. [DOI] [PubMed] [Google Scholar]
- Shimizu K.; Cao W.; Saad G.; Shoji M.; Terada T. Comparative Analysis of Membrane Protein Structure Databases. Biochim. Biophys. Acta, Biomembr. 2018, 1860, 1077–1091. 10.1016/j.bbamem.2018.01.005. [DOI] [PubMed] [Google Scholar]
- Cross T. A.; Murray D. T.; Watts A. Helical Membrane Protein Conformations and Their Environment. Eur. Biophys. J. 2013, 42, 731–755. 10.1007/s00249-013-0925-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moraes I.; Evans G.; Sanchez-Weatherby J.; Newstead S.; Stewart P. D. S. Membrane Protein Structure Determination — The next Generation. Biochim. Biophys. Acta, Biomembr. 2014, 1838, 78–87. 10.1016/j.bbamem.2013.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendrew J. C.; Bodo G.; Dintzis H. M.; Parrish R. G.; Wyckoff H.; Phillips D. C. A Three-Dimensional Model of the Myoglobin Molecule Obtained by X-Ray Analysis. Nature 1958, 181, 662–666. 10.1038/181662a0. [DOI] [PubMed] [Google Scholar]
- Deisenhofer J.; Epp O.; Miki K.; Huber R.; Michel H. Structure of the Protein Subunits in the Photosynthetic Reaction Centre of Rhodopseudomonas Viridis at 3Å Resolution. Nature 1985, 318, 618–624. 10.1038/318618a0. [DOI] [PubMed] [Google Scholar]
- Stetsenko A.; Guskov A. An Overview of the Top Ten Detergents Used for Membrane Protein Crystallization. Crystals 2017, 7, 197. 10.3390/cryst7070197. [DOI] [Google Scholar]
- Caffrey M. A Comprehensive Review of the Lipid Cubic Phase or in Meso Method for Crystallizing Membrane and Soluble Proteins and Complexes. Acta Crystallogr., Sect. F: Struct. Biol. Commun. 2015, 71, 3–18. 10.1107/S2053230X14026843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y.; Kenworthy A. K.; Sanders C. R. Cholesterol as a Co-Solvent and a Ligand for Membrane Proteins: Cholesterol-Protein Interactions. Protein Sci. 2014, 23, 1–22. 10.1002/pro.2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulig W.; Tynkkynen J.; Javanainen M.; Manna M.; Rog T.; Vattulainen I.; Jungwirth P. How Well Does Cholesteryl Hemisuccinate Mimic Cholesterol in Saturated Phospholipid Bilayers?. J. Mol. Model. 2014, 20, 2121. 10.1007/s00894-014-2121-z. [DOI] [PubMed] [Google Scholar]
- Loll P. J. Membrane Proteins, Detergents and Crystals: What Is the State of the Art?. Acta Crystallogr., Sect. F: Struct. Biol. Commun. 2014, 70, 1576–1583. 10.1107/S2053230X14025035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutmann T.; Kim K. H.; Grzybek M.; Walz T.; Coskun Ü. Visualization of Ligand-Induced Transmembrane Signaling in the Full-Length Human Insulin Receptor. J. Cell Biol. 2018, 217, 1643–1649. 10.1083/jcb.201711047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manna M.; Kulig W.; Javanainen M.; Tynkkynen J.; Hensen U.; Müller D. J.; Rog T.; Vattulainen I. How To Minimize Artifacts in Atomistic Simulations of Membrane Proteins, Whose Crystal Structure Is Heavily Engineered: β2-Adrenergic Receptor in the Spotlight. J. Chem. Theory Comput. 2015, 11, 3432–3445. 10.1021/acs.jctc.5b00070. [DOI] [PubMed] [Google Scholar]
- Hanson M. A.; Cherezov V.; Griffith M. T.; Roth C. B.; Jaakola V.-P.; Chien E. Y. T.; Velasquez J.; Kuhn P.; Stevens R. C. A Specific Cholesterol Binding Site Is Established by the 2.8 Å Structure of the Human β2-Adrenergic Receptor. Structure 2008, 16, 897–905. 10.1016/j.str.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radoicic J.; Lu G. J.; Opella S. J. NMR Structures of Membrane Proteins in Phospholipid Bilayers. Q. Rev. Biophys. 2014, 47, 249–283. 10.1017/S0033583514000080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S. H.; Das B. B.; Casagrande F.; Tian Y.; Nothnagel H. J.; Chu M.; Kiefer H.; Maier K.; De Angelis A. A.; Marassi F. M.; et al. Structure of the Chemokine Receptor CXCR1 in Phospholipid Bilayers. Nature 2012, 491, 779–783. 10.1038/nature11580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordignon E.; Bleicken S. New Limits of Sensitivity of Site-Directed Spin Labeling Electron Paramagnetic Resonance for Membrane Proteins. Biochim. Biophys. Acta, Biomembr. 2018, 1860, 841–853. 10.1016/j.bbamem.2017.12.009. [DOI] [PubMed] [Google Scholar]
- Bai X.; McMullan G.; Scheres S. H. How Cryo-EM Is Revolutionizing Structural Biology. Trends Biochem. Sci. 2015, 40, 49–57. 10.1016/j.tibs.2014.10.005. [DOI] [PubMed] [Google Scholar]
- Murata K.; Wolf M. Cryo-Electron Microscopy for Structural Analysis of Dynamic Biological Macromolecules. Biochim. Biophys. Acta, Gen. Subj. 2018, 1862, 324–334. 10.1016/j.bbagen.2017.07.020. [DOI] [PubMed] [Google Scholar]
- Mio K.; Sato C. Lipid Environment of Membrane Proteins in Cryo-EM Based Structural Analysis. Biophys. Rev. 2018, 10, 307–316. 10.1007/s12551-017-0371-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinothkumar K. R. Membrane Protein Structures without Crystals, by Single Particle Electron Cryomicroscopy. Curr. Opin. Struct. Biol. 2015, 33, 103–114. 10.1016/j.sbi.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. C.; Khalid S.; Pollock N. L.; Knowles T. J.; Edler K.; Rothnie A. J.; Thomas O. R. T.; Dafforn T. R. Encapsulated Membrane Proteins: A Simplified System for Molecular Simulation. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 2549–2557. 10.1016/j.bbamem.2016.02.039. [DOI] [PubMed] [Google Scholar]
- Lee S. C.; Knowles T. J.; Postis V. L. G.; Jamshad M.; Parslow R. A.; Lin Y.; Goldman A.; Sridhar P.; Overduin M.; Muench S. P.; et al. A Method for Detergent-Free Isolation of Membrane Proteins in Their Local Lipid Environment. Nat. Protoc. 2016, 11, 1149–1162. 10.1038/nprot.2016.070. [DOI] [PubMed] [Google Scholar]
- Nasr M. L.; Baptista D.; Strauss M.; Sun Z.-Y. J.; Grigoriu S.; Huser S.; Plückthun A.; Hagn F.; Walz T.; Hogle J. M.; et al. Covalently Circularized Nanodiscs for Studying Membrane Proteins and Viral Entry. Nat. Methods 2017, 14, 49–52. 10.1038/nmeth.4079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Autzen H. E.; Myasnikov A. G.; Campbell M. G.; Asarnow D.; Julius D.; Cheng Y. Structure of the Human TRPM4 Ion Channel in a Lipid Nanodisc. Science 2018, 359, 228–232. 10.1126/science.aar4510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White S.Membrane Proteins of Known 3D Structure. http://blanco.biomol.uci.edu/mpstruc/ (accessed Aug 5, 2018).
- Almeida J. G.; Preto A. J.; Koukos P. I.; Bonvin A. M. J. J.; Moreira I. S. Membrane Proteins Structures: A Review on Computational Modeling Tools. Biochim. Biophys. Acta, Biomembr. 2017, 1859, 2021–2039. 10.1016/j.bbamem.2017.07.008. [DOI] [PubMed] [Google Scholar]
- Grigorieff N.; Ceska T. A.; Downing K. H.; Baldwin J. M.; Henderson R. Electron-Crystallographic Refinement of the Structure of Bacteriorhodopsin. J. Mol. Biol. 1996, 259, 393–421. 10.1006/jmbi.1996.0328. [DOI] [PubMed] [Google Scholar]
- Laganowsky A.; Reading E.; Allison T. M.; Ulmschneider M. B.; Degiacomi M. T.; Baldwin A. J.; Robinson C. V. Membrane Proteins Bind Lipids Selectively to Modulate Their Structure and Function. Nature 2014, 510, 172–175. 10.1038/nature13419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poveda J. A.; Marcela Giudici A.; Lourdes Renart M.; Morales A.; González-Ros J. M. Towards Understanding the Molecular Basis of Ion Channel Modulation by Lipids: Mechanistic Models and Current Paradigms. Biochim. Biophys. Acta, Biomembr. 2017, 1859, 1507–1516. 10.1016/j.bbamem.2017.04.003. [DOI] [PubMed] [Google Scholar]
- Ciardo M. G.; Ferrer-Montiel A. Lipids as Central Modulators of Sensory TRP Channels. Biochim. Biophys. Acta, Biomembr. 2017, 1859, 1615–1628. 10.1016/j.bbamem.2017.04.012. [DOI] [PubMed] [Google Scholar]
- Yeagle P. L. Non-Covalent Binding of Membrane Lipids to Membrane Proteins. Biochim. Biophys. Acta, Biomembr. 2014, 1838, 1548–1559. 10.1016/j.bbamem.2013.11.009. [DOI] [PubMed] [Google Scholar]
- Paradies G.; Paradies V.; De Benedictis V.; Ruggiero F. M.; Petrosillo G. Functional Role of Cardiolipin in Mitochondrial Bioenergetics. Biochim. Biophys. Acta, Bioenerg. 2014, 1837, 408–417. 10.1016/j.bbabio.2013.10.006. [DOI] [PubMed] [Google Scholar]
- Planas-Iglesias J.; Dwarakanath H.; Mohammadyani D.; Yanamala N.; Kagan V. E.; Klein-Seetharaman J. Cardiolipin Interactions with Proteins. Biophys. J. 2015, 109, 1282–1294. 10.1016/j.bpj.2015.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiedorczuk K.; Letts J. A.; Degliesposti G.; Kaszuba K.; Skehel M.; Sazanov L. A. Atomic Structure of the Entire Mammalian Mitochondrial Complex I. Nature 2016, 538, 406–410. 10.1038/nature19794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M.; Gu J.; Guo R.; Huang Y.; Yang M. Structure of Mammalian Respiratory Supercomplex I1III2IV1. Cell 2016, 167, 1598–1609.e10. 10.1016/j.cell.2016.11.012. [DOI] [PubMed] [Google Scholar]
- Su X.; Ma J.; Wei X.; Cao P.; Zhu D.; Chang W.; Liu Z.; Zhang X.; Li M. Structure and Assembly Mechanism of Plant C2S2M2 Type PSII-LHCII Supercomplex. Science 2017, 357, 815–820. 10.1126/science.aan0327. [DOI] [PubMed] [Google Scholar]
- Poulikakos P.; Vassilacopoulou D.; Fragoulis E. G. L-DOPA Decarboxylase Association with Membranes in Mouse Brain. Neurochem. Res. 2001, 26, 479–485. 10.1023/A:1010952610387. [DOI] [PubMed] [Google Scholar]
- Moravcevic K.; Oxley C. L.; Lemmon M. A. Conditional Peripheral Membrane Proteins: Facing up to Limited Specificity. Structure 2012, 20, 15–27. 10.1016/j.str.2011.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colón-González F.; Kazanietz M. G. C1 Domains Exposed: From Diacylglycerol Binding to Protein–Protein Interactions. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2006, 1761, 827–837. 10.1016/j.bbalip.2006.05.001. [DOI] [PubMed] [Google Scholar]
- Stahelin R. V.; Scott J. L.; Frick C. T. Cellular and Molecular Interactions of Phosphoinositides and Peripheral Proteins. Chem. Phys. Lipids 2014, 182, 3–18. 10.1016/j.chemphyslip.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilkova E.; Pleskot R.; Rissanen S.; Sun S.; Czogalla A.; Cwiklik L.; Róg T.; Vattulainen I.; Cremer P. S.; Jungwirth P.; et al. Calcium Directly Regulates Phosphatidylinositol 4,5-Bisphosphate Headgroup Conformation and Recognition. J. Am. Chem. Soc. 2017, 139, 4019–4024. 10.1021/jacs.6b11760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johannes L.; Wunder C.; Shafaq-Zadah M. Glycolipids and Lectins in Endocytic Uptake Processes. J. Mol. Biol. 2016, 428, 4792–4818. 10.1016/j.jmb.2016.10.027. [DOI] [PubMed] [Google Scholar]
- Wernick N. L. B.; Chinnapen D. J.-F.; Cho J. A.; Lencer W. I. Cholera Toxin: An Intracellular Journey into the Cytosol by Way of the Endoplasmic Reticulum. Toxins 2010, 2, 310–325. 10.3390/toxins2030310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammache D.; Yahi N.; Maresca M.; Piéroni G.; Fantini J. Human Erythrocyte Glycosphingolipids as Alternative Cofactors for Human Immunodeficiency Virus Type 1 (HIV-1) Entry: Evidence for CD4-Induced Interactions between HIV-1 Gp120 and Reconstituted Membrane Microdomains of Glycosphingolipids (Gb3 and GM3). J. Virol. 1999, 73, 5244–5248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stace C.; Manifava M.; Delon C.; Coadwell J.; Cockcroft S.; Ktistakis N. T. PA Binding of Phosphatidylinositol 4-Phosphate 5-Kinase. Adv. Enzyme Regul. 2008, 48, 55–72. 10.1016/j.advenzreg.2007.11.008. [DOI] [PubMed] [Google Scholar]
- Capelluto D. G. S.; Zhao X.; Lucas A.; Lemkul J. A.; Xiao S.; Fu X.; Sun F.; Bevan D. R.; Finkielstein C. V. Biophysical and Molecular-Dynamics Studies of Phosphatidic Acid Binding by the Dvl-2 DEP Domain. Biophys. J. 2014, 106, 1101–1111. 10.1016/j.bpj.2014.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton J. M.; Takkellapati S.; Lawrence R. T.; McQueeney K. E.; Boroda S.; Mullins G. R.; Sherwood S. G.; Finck B. N.; Villén J.; Harris T. E. Lipin 2 Binds Phosphatidic Acid by the Electrostatic Hydrogen Bond Switch Mechanism Independent of Phosphorylation. J. Biol. Chem. 2014, 289, 18055–18066. 10.1074/jbc.M114.547604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putta P.; Rankenberg J.; Korver R. A.; van Wijk R.; Munnik T.; Testerink C.; Kooijman E. E. Phosphatidic Acid Binding Proteins Display Differential Binding as a Function of Membrane Curvature Stress and Chemical Properties. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 2709–2716. 10.1016/j.bbamem.2016.07.014. [DOI] [PubMed] [Google Scholar]
- Schlattner U.; Tokarska-Schlattner M.; Ramirez S.; Brückner A.; Kay L.; Polge C.; Epand R. F.; Lee R. M.; Lacombe M.-L.; Epand R. M. Mitochondrial Kinases and Their Molecular Interaction with Cardiolipin. Biochim. Biophys. Acta, Biomembr. 2009, 1788, 2032–2047. 10.1016/j.bbamem.2009.04.018. [DOI] [PubMed] [Google Scholar]
- Sinibaldi F.; Howes B. D.; Piro M. C.; Polticelli F.; Bombelli C.; Ferri T.; Coletta M.; Smulevich G.; Santucci R. Extended Cardiolipin Anchorage to Cytochrome c: A Model for Protein–Mitochondrial Membrane Binding. JBIC, J. Biol. Inorg. Chem. 2010, 15, 689–700. 10.1007/s00775-010-0636-z. [DOI] [PubMed] [Google Scholar]
- Chu C. T.; Ji J.; Dagda R. K.; Jiang J. F.; Tyurina Y. Y.; Kapralov A. A.; Tyurin V. A.; Yanamala N.; Shrivastava I. H.; Mohammadyani D.; et al. Cardiolipin Externalization to the Outer Mitochondrial Membrane Acts as an Elimination Signal for Mitophagy in Neuronal Cells. Nat. Cell Biol. 2013, 15, 1197–1205. 10.1038/ncb2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X.; Shi Y.; Liu G.; Guo Y.; Yang Y. Activation of ROP6 GTPase by Phosphatidylglycerol in Arabidopsis. Front. Plant Sci. 2018, 9, 347. 10.3389/fpls.2018.00347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Mulla F.; Bitar M. S.; Taqi Z.; Yeung K. C. RKIP: Much More than Raf Kinase Inhibitory Protein. J. Cell. Physiol. 2013, 228, 1688–1702. 10.1002/jcp.24335. [DOI] [PubMed] [Google Scholar]
- Vallée B. S.; Tauc P.; Brochon J.-C.; Maget-Dana R.; Lelièvre D.; Metz-Boutigue M.-H.; Bureaud N.; Schoentgen F. Behaviour of Bovine Phosphatidylethanolamine-Binding Protein with Model Membranes.: Evidence of Affinity for Negatively Charged Membranes. Eur. J. Biochem. 2001, 268, 5831–5841. 10.1046/j.0014-2956.2001.02528.x. [DOI] [PubMed] [Google Scholar]
- Nguyen N.; Shteyn V.; Melia T. J. Sensing Membrane Curvature in Macroautophagy. J. Mol. Biol. 2017, 429, 457–472. 10.1016/j.jmb.2017.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merritt E. A.; Sarfaty S.; Hol W. G. J.; Jobling M. G.; Holmes R. K.; Chang T.; Hirst T. R. Structural Studies of Receptor Binding by Cholera Toxin Mutants. Protein Sci. 1997, 6, 1516–1528. 10.1002/pro.5560060716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neu U.; Woellner K.; Gauglitz G.; Stehle T. Structural Basis of GM1 Ganglioside Recognition by Simian Virus 40. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 5219–5224. 10.1073/pnas.0710301105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ströh L. J.; Gee G. V.; Blaum B. S.; Dugan A. S.; Feltkamp M. C. W.; Atwood W. J.; Stehle T. Trichodysplasia Spinulosa-Associated Polyomavirus Uses a Displaced Binding Site on VP1 to Engage Sialylated Glycolipids. PLoS Pathog. 2015, 11, e1005112. 10.1371/journal.ppat.1005112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ströh L. J.; Maginnis M. S.; Blaum B. S.; Nelson C. D. S.; Neu U.; Gee G. V.; O’Hara B. A.; Motamedi N.; DiMaio D.; Atwood W. J.; et al. The Greater Affinity of JC Polyomavirus Capsid for α2,6-Linked Lactoseries Tetrasaccharide c than for Other Sialylated Glycans Is a Major Determinant of Infectivity. J. Virol. 2015, 89, 6364–6375. 10.1128/JVI.00489-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng N. M.; Littler D. R.; Paton A. W.; Le Nours J.; Rossjohn J.; Paton J. C.; Beddoe T. EcxAB Is a Founding Member of a New Family of Metalloprotease AB5 Toxins with a Hybrid Cholera-like B Subunit. Structure 2013, 21, 2003–2013. 10.1016/j.str.2013.08.024. [DOI] [PubMed] [Google Scholar]
- Blaum B. S.; Frank M.; Walker R. C.; Neu U.; Stehle T. Complement Factor H and Simian Virus 40 Bind the GM1 Ganglioside in Distinct Conformations. Glycobiology 2016, 26, 532–539. 10.1093/glycob/cwv170. [DOI] [PubMed] [Google Scholar]
- Bian C.-F.; Zhang Y.; Sun H.; Li D.-F.; Wang D.-C. Structural Basis for Distinct Binding Properties of the Human Galectins to Thomsen-Friedenreich Antigen. PLoS One 2011, 6, e25007. 10.1371/journal.pone.0025007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L.; Sun H.; Zhang Y.; Li D.-F.; Wang D.-C. Structural Insights into the Recognition Mechanism between an Antitumor Galectin AAL and the Thomsen-Friedenreich Antigen. FASEB J. 2010, 24, 3861–3868. 10.1096/fj.10-159111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y.-B.; Chen L.-Q.; Li Z.; Tan Y.-M.; Feng Y.; Yang G.-Y. Structural Insights into the Broad Substrate Specificity of a Novel Endoglycoceramidase I Belonging to a New Subfamily of GH5 Glycosidases. J. Biol. Chem. 2017, 292, 4789–4800. 10.1074/jbc.M116.763821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinthalapudi K.; Rangarajan E. S.; Brown D. T.; Izard T. Differential Lipid Binding of Vinculin Isoforms Promotes Quasi-Equivalent Dimerization. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 9539–9544. 10.1073/pnas.1600702113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Alai M. M.; Heidemann J.; Skruzny M.; Gieras A.; Mertens H. D. T.; Svergun D. I.; Kaksonen M.; Uetrecht C.; Meijers R. Epsin and Sla2 Form Assemblies through Phospholipid Interfaces. Nat. Commun. 2018, 9, 328. 10.1038/s41467-017-02443-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coudevylle N.; Montaville P.; Leonov A.; Zweckstetter M.; Becker S. Structural Determinants for Ca2+ and Phosphatidylinositol 4,5-Bisphosphate Binding by the C2A Domain of Rabphilin-3A. J. Biol. Chem. 2008, 283, 35918–35928. 10.1074/jbc.M804094200. [DOI] [PubMed] [Google Scholar]
- Posner M. G.; Upadhyay A.; Ishima R.; Kalli A. C.; Harris G.; Kremerskothen J.; Sansom M. S. P.; Crennell S. J.; Bagby S. Distinctive Phosphoinositide- and Ca2+-Binding Properties of Normal and Cognitive Performance–Linked Variant Forms of KIBRA C2 Domain. J. Biol. Chem. 2018, 293, 9335–9344. 10.1074/jbc.RA118.002279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saad J. S.; Miller J.; Tai J.; Kim A.; Ghanam R. H.; Summers M. F. Structural Basis for Targeting HIV-1 Gag Proteins to the Plasma Membrane for Virus Assembly. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 11364–11369. 10.1073/pnas.0602818103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kono N.; Ohto U.; Hiramatsu T.; Urabe M.; Uchida Y.; Satow Y.; Arai H. Impaired α-TTP-PIPs Interaction Underlies Familial Vitamin E Deficiency. Science 2013, 340, 1106–1110. 10.1126/science.1233508. [DOI] [PubMed] [Google Scholar]
- Chinthalapudi K.; Mandati V.; Zheng J.; Sharff A. J.; Bricogne G.; Griffin P. R.; Kissil J.; Izard T. Lipid Binding Promotes the Open Conformation and Tumor-Suppressive Activity of Neurofibromin 2. Nat. Commun. 2018, 9, 1338. 10.1038/s41467-018-03648-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller M. S.; Schmidt-Kittler O.; Bolduc D. M.; Brower E. T.; Chaves-Moreira D.; Allaire M.; Kinzler K. W.; Jennings I. G.; Thompson P. E.; Cole P. A.; et al. Structural Basis of nSH2 Regulation and Lipid Binding in PI3Kα. Oncotarget 2014, 5, 5198–5208. 10.18632/oncotarget.2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B.; Chien E. Y. T.; Mol C. D.; Fenalti G.; Liu W.; Katritch V.; Abagyan R.; Brooun A.; Wells P.; Bi F. C.; et al. Structures of the CXCR4 Chemokine GPCR with Small-Molecule and Cyclic Peptide Antagonists. Science 2010, 330, 1066–1071. 10.1126/science.1194396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferré S. The GPCR Heterotetramer: Challenging Classical Pharmacology. Trends Pharmacol. Sci. 2015, 36, 145–152. 10.1016/j.tips.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordomí A.; Navarro G.; Aymerich M. S.; Franco R. Structures for G-Protein-Coupled Receptor Tetramers in Complex with G Proteins. Trends Biochem. Sci. 2015, 40, 548–551. 10.1016/j.tibs.2015.07.007. [DOI] [PubMed] [Google Scholar]
- Gaitonde S. A.; González-Maeso J. Contribution of Heteromerization to G Protein-Coupled Receptor Function. Curr. Opin. Pharmacol. 2017, 32, 23–31. 10.1016/j.coph.2016.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Moustaine D.; Granier S.; Doumazane E.; Scholler P.; Rahmeh R.; Bron P.; Mouillac B.; Baneres J.-L.; Rondard P.; Pin J.-P. Distinct Roles of Metabotropic Glutamate Receptor Dimerization in Agonist Activation and G-Protein Coupling. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 16342–16347. 10.1073/pnas.1205838109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whorton M. R.; Bokoch M. P.; Rasmussen S. G. F.; Huang B.; Zare R. N.; Kobilka B.; Sunahara R. K. A Monomeric G Protein-Coupled Receptor Isolated in a High-Density Lipoprotein Particle Efficiently Activates Its G Protein. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 7682–7687. 10.1073/pnas.0611448104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y.; Moreira I. S.; Urizar E.; Weinstein H.; Javitch J. A. Allosteric Communication between Protomers of Dopamine Class A GPCR Dimers Modulates Activation. Nat. Chem. Biol. 2009, 5, 688–695. 10.1038/nchembio.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hern J. A.; Baig A. H.; Mashanov G. I.; Birdsall B.; Corrie J. E. T.; Lazareno S.; Molloy J. E.; Birdsall N. J. M. Formation and Dissociation of M1 Muscarinic Receptor Dimers Seen by Total Internal Reflection Fluorescence Imaging of Single Molecules. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 2693–2698. 10.1073/pnas.0907915107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorsch S.; Klotz K.-N.; Engelhardt S.; Lohse M. J.; Bünemann M. Analysis of Receptor Oligomerization by FRAP Microscopy. Nat. Methods 2009, 6, 225–230. 10.1038/nmeth.1304. [DOI] [PubMed] [Google Scholar]
- Parmar V. K.; Grinde E.; Mazurkiewicz J. E.; Herrick-Davis K. Beta2-Adrenergic Receptor Homodimers: Role of Transmembrane Domain 1 and Helix 8 in Dimerization and Cell Surface Expression. Biochim. Biophys. Acta, Biomembr. 2017, 1859, 1445–1455. 10.1016/j.bbamem.2016.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J.; Chen S.; Zhang J. J.; Huang X.-Y. Crystal Structure of Oligomeric β1-Adrenergic G Protein–Coupled Receptors in Ligand-Free Basal State. Nat. Struct. Mol. Biol. 2013, 20, 419–425. 10.1038/nsmb.2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonaventura J.; Navarro G.; Casadó-Anguera V.; Azdad K.; Rea W.; Moreno E.; Brugarolas M.; Mallol J.; Canela E. I.; Lluís C.; et al. Allosteric Interactions between Agonists and Antagonists within the Adenosine A2A Receptor-Dopamine D2 Receptor Heterotetramer. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, E3609–E3618. 10.1073/pnas.1507704112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gahbauer S.; Böckmann R. A. Membrane-Mediated Oligomerization of G Protein Coupled Receptors and Its Implications for GPCR Function. Front. Physiol. 2016, 7, 494. 10.3389/fphys.2016.00494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chothe P. P.; Czuba L. C.; Moore R. H.; Swaan P. W. Human Bile Acid Transporter ASBT (SLC10A2) Forms Functional Non-Covalent Homodimers and Higher Order Oligomers. Biochim. Biophys. Acta, Biomembr. 2018, 1860, 645–653. 10.1016/j.bbamem.2017.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valley C. C.; Lewis A. K.; Sachs J. N. Piecing It Together: Unraveling the Elusive Structure-Function Relationship in Single-Pass Membrane Receptors. Biochim. Biophys. Acta, Biomembr. 2017, 1859, 1398–1416. 10.1016/j.bbamem.2017.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung I. Optical Measurement of Receptor Tyrosine Kinase Oligomerization on Live Cells. Biochim. Biophys. Acta, Biomembr. 2017, 1859, 1436–1444. 10.1016/j.bbamem.2017.03.026. [DOI] [PubMed] [Google Scholar]
- Althoff T.; Mills D. J.; Popot J.-L.; Kühlbrandt W. Arrangement of Electron Transport Chain Components in Bovine Mitochondrial Supercomplex I1III2IV1: Cryo-EM of Respiratory Chain Supercomplex. EMBO J. 2011, 30, 4652–4664. 10.1038/emboj.2011.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kühlbrandt W. Structure and Function of Mitochondrial Membrane Protein Complexes. BMC Biol. 2015, 13, 89. 10.1186/s12915-015-0201-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schorr S.; van der Laan M. Integrative Functions of the Mitochondrial Contact Site and Cristae Organizing System. Semin. Cell Dev. Biol. 2018, 76, 191–200. 10.1016/j.semcdb.2017.09.021. [DOI] [PubMed] [Google Scholar]
- Ikon N.; Ryan R. O. Cardiolipin and Mitochondrial Cristae Organization. Biochim. Biophys. Acta, Biomembr. 2017, 1859, 1156–1163. 10.1016/j.bbamem.2017.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer S.; Dudek J.; Zimmermann R.; Förster F. Organization of the Native Ribosome–Translocon Complex at the Mammalian Endoplasmic Reticulum Membrane. Biochim. Biophys. Acta, Gen. Subj. 2016, 1860, 2122–2129. 10.1016/j.bbagen.2016.06.024. [DOI] [PubMed] [Google Scholar]
- Pfeffer S.; Brandt F.; Hrabe T.; Lang S.; Eibauer M.; Zimmermann R.; Förster F. Structure and 3D Arrangement of Endoplasmic Reticulum Membrane-Associated Ribosomes. Structure 2012, 20, 1508–1518. 10.1016/j.str.2012.06.010. [DOI] [PubMed] [Google Scholar]
- Rees D. M.; Leslie A. G. W.; Walker J. E. The Structure of the Membrane Extrinsic Region of Bovine ATP Synthase. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 21597–21601. 10.1073/pnas.0910365106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symersky J.; Pagadala V.; Osowski D.; Krah A.; Meier T.; Faraldo-Gómez J. D.; Mueller D. M. Structure of the c10 Ring of the Yeast Mitochondrial ATP Synthase in the Open Conformation. Nat. Struct. Mol. Biol. 2012, 19, 485–491. 10.1038/nsmb.2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies K. M.; Strauss M.; Daum B.; Kief J. H.; Osiewacz H. D.; Rycovska A.; Zickermann V.; Kuhlbrandt W. Macromolecular Organization of ATP Synthase and Complex I in Whole Mitochondria. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 14121–14126. 10.1073/pnas.1103621108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapuente-Brun E.; Moreno-Loshuertos R.; Acin-Perez R.; Latorre-Pellicer A.; Colas C.; Balsa E.; Perales-Clemente E.; Quiros P. M.; Calvo E.; Rodriguez-Hernandez M. A.; et al. Supercomplex Assembly Determines Electron Flux in the Mitochondrial Electron Transport Chain. Science 2013, 340, 1567–1570. 10.1126/science.1230381. [DOI] [PubMed] [Google Scholar]
- Bock L. V.; Blau C.; Schröder G. F.; Davydov I. I.; Fischer N.; Stark H.; Rodnina M. V.; Vaiana A. C.; Grubmüller H. Energy Barriers and Driving Forces in TRNA Translocation through the Ribosome. Nat. Struct. Mol. Biol. 2013, 20, 1390–1396. 10.1038/nsmb.2690. [DOI] [PubMed] [Google Scholar]
- Frauenfeld J.; Gumbart J.; van derSluis E. O.; Funes S.; Gartmann M.; Beatrix B.; Mielke T.; Berninghausen O.; Becker T.; Schulten K.; et al. Cryo-EM Structure of the Ribosome–SecYE Complex in the Membrane Environment. Nat. Struct. Mol. Biol. 2011, 18, 614–621. 10.1038/nsmb.2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sothiselvam S.; Liu B.; Han W.; Ramu H.; Klepacki D.; Atkinson G. C.; Brauer A.; Remm M.; Tenson T.; Schulten K.; et al. Macrolide Antibiotics Allosterically Predispose the Ribosome for Translation Arrest. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 9804–9809. 10.1073/pnas.1403586111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perilla J. R.; Goh B. C.; Cassidy C. K.; Liu B.; Bernardi R. C.; Rudack T.; Yu H.; Wu Z.; Schulten K. Molecular Dynamics Simulations of Large Macromolecular Complexes. Curr. Opin. Struct. Biol. 2015, 31, 64–74. 10.1016/j.sbi.2015.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venditti R.; Wilson C.; De Matteis M. A. Exiting the ER: What We Know and What We Don’t. Trends Cell Biol. 2014, 24, 9–18. 10.1016/j.tcb.2013.08.005. [DOI] [PubMed] [Google Scholar]
- von Appen A.; Kosinski J.; Sparks L.; Ori A.; DiGuilio A. L.; Vollmer B.; Mackmull M.-T.; Banterle N.; Parca L.; Kastritis P.; et al. In Situ Structural Analysis of the Human Nuclear Pore Complex. Nature 2015, 526, 140–143. 10.1038/nature15381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz T. U. The Structure Inventory of the Nuclear Pore Complex. J. Mol. Biol. 2016, 428, 1986–2000. 10.1016/j.jmb.2016.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgese N. Getting Membrane Proteins on and off the Shuttle Bus between the Endoplasmic Reticulum and the Golgi Complex. J. Cell Sci. 2016, 129, 1537–1545. 10.1242/jcs.183335. [DOI] [PubMed] [Google Scholar]
- Jovanovic-Talisman T.; Zilman A. Protein Transport by the Nuclear Pore Complex: Simple Biophysics of a Complex Biomachine. Biophys. J. 2017, 113, 6–14. 10.1016/j.bpj.2017.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakiyama Y.; Panatala R.; Lim R. Y. H. Structural Dynamics of the Nuclear Pore Complex. Semin. Cell Dev. Biol. 2017, 68, 27–33. 10.1016/j.semcdb.2017.05.021. [DOI] [PubMed] [Google Scholar]
- Jensen O. N. Interpreting the Protein Language Using Proteomics. Nat. Rev. Mol. Cell Biol. 2006, 7, 391–403. 10.1038/nrm1939. [DOI] [PubMed] [Google Scholar]
- Khoury G. A.; Baliban R. C.; Floudas C. A. Proteome-Wide Post-Translational Modification Statistics: Frequency Analysis and Curation of the Swiss-Prot Database. Sci. Rep. 2011, 1, 90. 10.1038/srep00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey S. J.; James D. E.; Mann M. Protein Phosphorylation: A Major Switch Mechanism for Metabolic Regulation. Trends Endocrinol. Metab. 2015, 26, 676–687. 10.1016/j.tem.2015.09.013. [DOI] [PubMed] [Google Scholar]
- Huang J.; Wang F.; Ye M.; Zou H. Enrichment and Separation Techniques for Large-Scale Proteomics Analysis of the Protein Post-Translational Modifications. J. Chromatogr. A 2014, 1372, 1–17. 10.1016/j.chroma.2014.10.107. [DOI] [PubMed] [Google Scholar]
- Prabakaran S.; Lippens G.; Steen H.; Gunawardena J. Post-Translational Modification: Nature’s Escape from Genetic Imprisonment and the Basis for Dynamic Information Encoding: Information Encoding by Post-Translational Modification. Wiley Interdiscip. Rev. Syst. Biol. Med. 2012, 4, 565–583. 10.1002/wsbm.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gianazza E.; Parravicini C.; Primi R.; Miller I.; Eberini I. In Silico Prediction and Characterization of Protein Post-Translational Modifications. J. Proteomics 2016, 134, 65–75. 10.1016/j.jprot.2015.09.026. [DOI] [PubMed] [Google Scholar]
- Yang Y.-S.; Wang C.-C.; Chen B.-H.; Hou Y.-H.; Hung K.-S.; Mao Y.-C. Tyrosine Sulfation as a Protein Post-Translational Modification. Molecules 2015, 20, 2138–2164. 10.3390/molecules20022138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nørskov-Lauritsen L.; Bräuner-Osborne H. Role of Post-Translational Modifications on Structure, Function and Pharmacology of Class C G Protein-Coupled Receptors. Eur. J. Pharmacol. 2015, 763, 233–240. 10.1016/j.ejphar.2015.05.015. [DOI] [PubMed] [Google Scholar]
- Hirano A.; Fu Y.-H.; Ptáček L. J. The Intricate Dance of Post-Translational Modifications in the Rhythm of Life. Nat. Struct. Mol. Biol. 2016, 23, 1053–1060. 10.1038/nsmb.3326. [DOI] [PubMed] [Google Scholar]
- Fujita M.; Kinoshita T. GPI-Anchor Remodeling: Potential Functions of GPI-Anchors in Intracellular Trafficking and Membrane Dynamics. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2012, 1821, 1050–1058. 10.1016/j.bbalip.2012.01.004. [DOI] [PubMed] [Google Scholar]
- Apweiler R. On the Frequency of Protein Glycosylation, as Deduced from Analysis of the SWISS-PROT Database. Biochim. Biophys. Acta, Gen. Subj. 1999, 1473, 4–8. 10.1016/S0304-4165(99)00165-8. [DOI] [PubMed] [Google Scholar]
- Ferreira I. G.; Pucci M.; Venturi G.; Malagolini N.; Chiricolo M.; Dall’Olio F. Glycosylation as a Main Regulator of Growth and Death Factor Receptors Signaling. Int. J. Mol. Sci. 2018, 19, 580. 10.3390/ijms19020580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M.; Hasegawa Y.; Gao C.; Kuroki Y.; Taniguchi N. N-Glycans of Growth Factor Receptors: Their Role in Receptor Function and Disease Implications. Clin. Sci. 2016, 130, 1781–1792. 10.1042/CS20160273. [DOI] [PubMed] [Google Scholar]
- Christiansen M. N.; Chik J.; Lee L.; Anugraham M.; Abrahams J. L.; Packer N. H. Cell Surface Protein Glycosylation in Cancer. Proteomics 2014, 14, 525–546. 10.1002/pmic.201300387. [DOI] [PubMed] [Google Scholar]
- Pakkiriswami S.; Couto A.; Nagarajan U.; Georgiou M. Glycosylated Notch and Cancer. Front. Oncol. 2016, 6, 37. 10.3389/fonc.2016.00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazniewska J.; Weiss N. Glycosylation of Voltage-Gated Calcium Channels in Health and Disease. Biochim. Biophys. Acta, Biomembr. 2017, 1859, 662–668. 10.1016/j.bbamem.2017.01.018. [DOI] [PubMed] [Google Scholar]
- Yang X.; Qian K. Protein O-GlcNAcylation: Emerging Mechanisms and Functions. Nat. Rev. Mol. Cell Biol. 2017, 18, 452–465. 10.1038/nrm.2017.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worth M.; Li H.; Jiang J. Deciphering the Functions of Protein O-GlcNAcylation with Chemistry. ACS Chem. Biol. 2017, 12, 326–335. 10.1021/acschembio.6b01065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson S. B.; Hart G. W. New Insights: A Role for O-GlcNAcylation in Diabetic Complications. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 150–161. 10.3109/10409238.2015.1135102. [DOI] [PubMed] [Google Scholar]
- Copeland R. J.; Han G.; Hart G. W. O-GlcNAcomics-Revealing Roles of O-GlcNAcylation in Disease Mechanisms and Development of Potential Diagnostics. Proteomics: Clin. Appl. 2013, 7, 597–606. 10.1002/prca.201300001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wani W. Y.; Chatham J. C.; Darley-Usmar V.; McMahon L. L.; Zhang J. O-GlcNAcylation and Neurodegeneration. Brain Res. Bull. 2017, 133, 80–87. 10.1016/j.brainresbull.2016.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruba A.; Yang W. O-GlcNAc-ylation in the Nuclear Pore Complex. Cell. Mol. Bioeng. 2016, 9, 227–233. 10.1007/s12195-016-0440-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailleux F.; Gélinas R.; Beauloye C.; Horman S.; Bertrand L. O-GlcNAcylation, Enemy or Ally during Cardiac Hypertrophy Development?. Biochim. Biophys. Acta, Mol. Basis Dis. 2016, 1862, 2232–2243. 10.1016/j.bbadis.2016.08.012. [DOI] [PubMed] [Google Scholar]
- Gournas C.; Saliba E.; Krammer E.-M.; Barthelemy C.; Prévost M.; André B. Transition of Yeast Can1 Transporter to the Inward-Facing State Unveils an α-Arrestin Target Sequence Promoting Its Ubiquitylation and Endocytosis. Mol. Biol. Cell 2017, 28, 2819–2832. 10.1091/mbc.e17-02-0104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You H.; Ge Y.; Zhang J.; Cao Y.; Xing J.; Su D.; Huang Y.; Li M.; Qu S.; Sun F.; et al. Derlin-1 Promotes Ubiquitylation and Degradation of the Epithelial Na+ Channel, ENaC. J. Cell Sci. 2017, 130, 1027–1036. 10.1242/jcs.198242. [DOI] [PubMed] [Google Scholar]
- Lorenz S.; Cantor A. J.; Rape M.; Kuriyan J. Macromolecular Juggling by Ubiquitylation Enzymes. BMC Biol. 2013, 11, 65. 10.1186/1741-7007-11-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDowell G. S.; Philpott A. Non-Canonical Ubiquitylation: Mechanisms and Consequences. Int. J. Biochem. Cell Biol. 2013, 45, 1833–1842. 10.1016/j.biocel.2013.05.026. [DOI] [PubMed] [Google Scholar]
- Resh M. D. Fatty Acylation of Proteins: The Long and the Short of It. Prog. Lipid Res. 2016, 63, 120–131. 10.1016/j.plipres.2016.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng T.; Thinon E.; Hang H. C. Proteomic Analysis of Fatty-Acylated Proteins. Curr. Opin. Chem. Biol. 2016, 30, 77–86. 10.1016/j.cbpa.2015.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peitzsch R. M.; McLaughlin S. Binding of Acylated Peptides and Fatty Acids to Phospholipid Vesicles: Pertinence to Myristoylated Proteins. Biochemistry 1993, 32, 10436–10443. 10.1021/bi00090a020. [DOI] [PubMed] [Google Scholar]
- Kumari B.; Kumar R.; Kumar M. PalmPred: An SVM Based Palmitoylation Prediction Method Using Sequence Profile Information. PLoS One 2014, 9, e89246. 10.1371/journal.pone.0089246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc M.; David F.; Abrami L.; Migliozzi D.; Armand F.; Bürgi J.; van der Goot F. G. SwissPalm: Protein Palmitoylation Database. F1000Research 2015, 4, 261. 10.12688/f1000research.6464.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter J. A.; Young K. E.; Beachy P. A. Cholesterol Modification of Hedgehog Signaling Proteins in Animal Development. Science 1996, 274, 255–259. 10.1126/science.274.5285.255. [DOI] [PubMed] [Google Scholar]
- Mann R. K.; Beachy P. A. Novel Lipid Modifications of Secreted Protein Signals. Annu. Rev. Biochem. 2004, 73, 891–923. 10.1146/annurev.biochem.73.011303.073933. [DOI] [PubMed] [Google Scholar]
- Wang M.; Casey P. J. Protein Prenylation: Unique Fats Make Their Mark on Biology. Nat. Rev. Mol. Cell Biol. 2016, 17, 110–122. 10.1038/nrm.2015.11. [DOI] [PubMed] [Google Scholar]
- Palsuledesai C. C.; Distefano M. D. Protein Prenylation: Enzymes, Therapeutics, and Biotechnology Applications. ACS Chem. Biol. 2015, 10, 51–62. 10.1021/cb500791f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benetka W.; Koranda M.; Eisenhaber F. Protein Prenylation: An (Almost) Comprehensive Overview on Discovery History, Enzymology, and Significance in Physiology and Disease. Monatsh. Chem. 2006, 137, 1241. 10.1007/s00706-006-0534-9. [DOI] [Google Scholar]
- Berndt N.; Hamilton A. D.; Sebti S. M. Targeting Protein Prenylation for Cancer Therapy. Nat. Rev. Cancer 2011, 11, 775–791. 10.1038/nrc3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu N.; Shen N.; Wang X.; Jiang S.; Xue B.; Li C. Protein Prenylation and Human Diseases: A Balance of Protein Farnesylation and Geranylgeranylation. Sci. China: Life Sci. 2015, 58, 328–335. 10.1007/s11427-015-4836-1. [DOI] [PubMed] [Google Scholar]
- Fujita M.; Kinoshita T. Structural Remodeling of GPI Anchors during Biosynthesis and after Attachment to Proteins. FEBS Lett. 2010, 584, 1670–1677. 10.1016/j.febslet.2009.10.079. [DOI] [PubMed] [Google Scholar]
- Zurzolo C.; Simons K. Glycosylphosphatidylinositol-Anchored Proteins: Membrane Organization and Transport. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 632–639. 10.1016/j.bbamem.2015.12.018. [DOI] [PubMed] [Google Scholar]
- Pawar K.; Singh B. Understanding Clinical Significance of GPI Anchored Proteins in Mammalian System: Special Emphasis on Genetic Disorders of GPI Anchor Biosynthesis Pathway. Int. J. Med. Res. Health Sci. 2017, 6, 53–58. [Google Scholar]
- Ichimura Y.; Kirisako T.; Takao T.; Satomi Y.; Shimonishi Y.; Ishihara N.; Mizushima N.; Tanida I.; Kominami E.; Ohsumi M.; et al. A Ubiquitin-like System Mediates Protein Lipidation. Nature 2000, 408, 488–492. 10.1038/35044114. [DOI] [PubMed] [Google Scholar]
- Kabeya Y. LC3, GABARAP and GATE16 Localize to Autophagosomal Membrane Depending on Form-II Formation. J. Cell Sci. 2004, 117, 2805–2812. 10.1242/jcs.01131. [DOI] [PubMed] [Google Scholar]
- Nath S.; Dancourt J.; Shteyn V.; Puente G.; Fong W. M.; Nag S.; Bewersdorf J.; Yamamoto A.; Antonny B.; Melia T. J. Lipidation of the LC3/GABARAP Family of Autophagy Proteins Relies on a Membrane-Curvature-Sensing Domain in Atg3. Nat. Cell Biol. 2014, 16, 415–424. 10.1038/ncb2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma V. K.; Graham N. J. D. Oxidation of Amino Acids, Peptides and Proteins by Ozone: A Review. Ozone: Sci. Eng. 2010, 32, 81–90. 10.1080/01919510903510507. [DOI] [Google Scholar]
- Lushchak V. I. Free Radical Oxidation of Proteins and Its Relationship with Functional State of Organisms. Biochemistry 2007, 72, 809–827. 10.1134/S0006297907080020. [DOI] [PubMed] [Google Scholar]
- Stadtman E. R.; Levine R. L. Free Radical-Mediated Oxidation of Free Amino Acids and Amino Acid Residues in Proteins. Amino Acids 2003, 25, 207–218. 10.1007/s00726-003-0011-2. [DOI] [PubMed] [Google Scholar]
- Shacter E. Quantification and Significance of Protein Oxidation in Biological Samples. Drug Metab. Rev. 2000, 32, 307–326. 10.1081/DMR-100102336. [DOI] [PubMed] [Google Scholar]
- Kelly F. J.; Mudway I. S. Protein Oxidation at the Air-Lung Interface. Amino Acids 2003, 25, 375–396. 10.1007/s00726-003-0024-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barelli S.; Canellini G.; Thadikkaran L.; Crettaz D.; Quadroni M.; Rossier J. S.; Tissot J.-D.; Lion N. Oxidation of Proteins: Basic Principles and Perspectives for Blood Proteomics. Proteomics: Clin. Appl. 2008, 2, 142–157. 10.1002/prca.200780009. [DOI] [PubMed] [Google Scholar]
- Dunlop R. A.; Brunk U. T.; Rodgers K. J. Oxidized Proteins: Mechanisms of Removal and Consequences of Accumulation. IUBMB Life 2009, 61, 522–527. 10.1002/iub.189. [DOI] [PubMed] [Google Scholar]
- Bader N.; Grune T. Protein Oxidation and Proteolysis. Biol. Chem. 2006, 387, 1351. 10.1515/BC.2006.169. [DOI] [PubMed] [Google Scholar]
- Costa V.; Quintanilha A.; Moradas-Ferreira P. Protein Oxidation, Repair Mechanisms and Proteolysis in Saccharomyces Cerevisiae. IUBMB Life 2007, 59, 293–298. 10.1080/15216540701225958. [DOI] [PubMed] [Google Scholar]
- Brennan M.-L.; Hazen S. L. Amino Acid and Protein Oxidation in Cardiovascular Disease. Amino Acids 2003, 25, 365–374. 10.1007/s00726-003-0023-y. [DOI] [PubMed] [Google Scholar]
- Cheignon C.; Tomas M.; Bonnefont-Rousselot D.; Faller P.; Hureau C.; Collin F. Oxidative Stress and the Amyloid Beta Peptide in Alzheimer’s Disease. Redox Biol. 2018, 14, 450–464. 10.1016/j.redox.2017.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Vlies D.; Woudenberg J.; Post J. A. Protein Oxidation in Aging: Endoplasmic Reticulum as a Target. Amino Acids 2003, 25, 397–407. 10.1007/s00726-003-0025-9. [DOI] [PubMed] [Google Scholar]
- Trnková L.; Dršata J.; Boušová I. Oxidation as an Important Factor of Protein Damage: Implications for Maillard Reaction. J. Biosci. 2015, 40, 419–439. 10.1007/s12038-015-9523-7. [DOI] [PubMed] [Google Scholar]
- Zhang W.; Xiao S.; Ahn D. U. Protein Oxidation: Basic Principles and Implications for Meat Quality. Crit. Rev. Food Sci. Nutr. 2013, 53, 1191–1201. 10.1080/10408398.2011.577540. [DOI] [PubMed] [Google Scholar]
- Ginsberg M. H. Integrin Activation. BMB Rep 2014, 47, 655–659. 10.5483/BMBRep.2014.47.12.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morse E. M.; Brahme N. N.; Calderwood D. A. Integrin Cytoplasmic Tail Interactions. Biochemistry 2014, 53, 810–820. 10.1021/bi401596q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manninen A.; Varjosalo M. A Proteomics View on Integrin-Mediated Adhesions. Proteomics 2017, 17, 1600022. 10.1002/pmic.201600022. [DOI] [PubMed] [Google Scholar]
- Theocharis A. D.; Skandalis S. S.; Gialeli C.; Karamanos N. K. Extracellular Matrix Structure. Adv. Drug Delivery Rev. 2016, 97, 4–27. 10.1016/j.addr.2015.11.001. [DOI] [PubMed] [Google Scholar]
- Changede R.; Sheetz M. Integrin and Cadherin Clusters: A Robust Way to Organize Adhesions for Cell Mechanics. BioEssays 2017, 39, e201600123. 10.1002/bies.201600123. [DOI] [PubMed] [Google Scholar]
- Tarbell J. M.; Cancel L. M. The Glycocalyx and Its Significance in Human Medicine. J. Intern. Med. 2016, 280, 97–113. 10.1111/joim.12465. [DOI] [PubMed] [Google Scholar]
- Curry F. E.; Adamson R. H. Endothelial Glycocalyx: Permeability Barrier and Mechanosensor. Ann. Biomed. Eng. 2012, 40, 828–839. 10.1007/s10439-011-0429-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alphonsus C. S.; Rodseth R. N. The Endothelial Glycocalyx: A Review of the Vascular Barrier. Anaesthesia 2014, 69, 777–784. 10.1111/anae.12661. [DOI] [PubMed] [Google Scholar]
- Tarbell J. M.; Pahakis M. Y. Mechanotransduction and the Glycocalyx. J. Intern. Med. 2006, 259, 339–350. 10.1111/j.1365-2796.2006.01620.x. [DOI] [PubMed] [Google Scholar]
- Nieuwdorp M.; Meuwese M. C.; Vink H.; Hoekstra J. B.; Kastelein J. J.; Stroes E. S. The Endothelial Glycocalyx: A Potential Barrier between Health and Vascular Disease. Curr. Opin. Lipidol. 2005, 16, 507–511. 10.1097/01.mol.0000181325.08926.9c. [DOI] [PubMed] [Google Scholar]
- Salmon A. H.; Satchell S. C. Endothelial Glycocalyx Dysfunction in Disease: Albuminuria and Increased Microvascular Permeability. J. Pathol. 2012, 226, 562–574. 10.1002/path.3964. [DOI] [PubMed] [Google Scholar]
- Reitsma S.; Slaaf D. W.; Vink H.; van Zandvoort M. A. M. J.; oude Egbrink M. G. A. The Endothelial Glycocalyx: Composition, Functions, and Visualization. Pfluegers Arch. 2007, 454, 345–359. 10.1007/s00424-007-0212-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinbaum S.; Tarbell J. M.; Damiano E. R. The Structure and Function of the Endothelial Glycocalyx Layer. Annu. Rev. Biomed. Eng. 2007, 9, 121–167. 10.1146/annurev.bioeng.9.060906.151959. [DOI] [PubMed] [Google Scholar]
- Leonova E. I.; Galzitskaya O. V. Structure and Functions of Syndecans in Vertebrates. Biochemistry 2013, 78, 1071–1085. 10.1134/S0006297913100015. [DOI] [PubMed] [Google Scholar]
- Shin J.; Lee W.; Lee D.; Koo B. K.; Han I.; Lim Y.; Woods A.; Couchman J. R.; Oh E. S. Solution Structure of the Dimeric Cytoplasmic Domain of Syndecan-4. Biochemistry 2001, 40, 8471–8478. 10.1021/bi002750r. [DOI] [PubMed] [Google Scholar]
- Svensson G.; Awad W.; Håkansson M.; Mani K.; Logan D. T. Crystal Structure of N-Glycosylated Human Glypican-1 Core Protein: Structure of Two Loops Evolutionarily Conserved in Vertebrate Glypican-1. J. Biol. Chem. 2012, 287, 14040–14051. 10.1074/jbc.M111.322487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awad W.; Svensson Birkedal G.; Thunnissen M. M. G. M.; Mani K.; Logan D. T. Improvements in the Order, Isotropy and Electron Density of Glypican-1 Crystals by Controlled Dehydration. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2013, 69, 2524–2533. 10.1107/S0907444913025250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awad W.; Adamczyk B.; Örnros J.; Karlsson N. G.; Mani K.; Logan D. T. Structural Aspects of N-Glycosylations and the C-Terminal Region in Human Glypican-1. J. Biol. Chem. 2015, 290, 22991–23008. 10.1074/jbc.M115.660878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teriete P.; Banerji S.; Noble M.; Blundell C. D.; Wright A. J.; Pickford A. R.; Lowe E.; Mahoney D. J.; Tammi M. I.; Kahmann J. D.; et al. Structure of the Regulatory Hyaluronan Binding Domain in the Inflammatory Leukocyte Homing Receptor CD44. Mol. Cell 2004, 13, 483–496. 10.1016/S1097-2765(04)00080-2. [DOI] [PubMed] [Google Scholar]
- Banerji S.; Wright A. J.; Noble M.; Mahoney D. J.; Campbell I. D.; Day A. J.; Jackson D. G. Structures of the Cd44–Hyaluronan Complex Provide Insight into a Fundamental Carbohydrate-Protein Interaction. Nat. Struct. Mol. Biol. 2007, 14, 234–239. 10.1038/nsmb1201. [DOI] [PubMed] [Google Scholar]
- Campbell H. K.; Maiers J. L.; DeMali K. A. Interplay between Tight Junctions & Adherens Junctions. Exp. Cell Res. 2017, 358, 39–44. 10.1016/j.yexcr.2017.03.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejana E. Endothelial Cell–Cell Junctions: Happy Together. Nat. Rev. Mol. Cell Biol. 2004, 5, 261–270. 10.1038/nrm1357. [DOI] [PubMed] [Google Scholar]
- Giepmans B. N. G.; van IJzendoorn S. C. D. Epithelial Cell–Cell Junctions and Plasma Membrane Domains. Biochim. Biophys. Acta, Biomembr. 2009, 1788, 820–831. 10.1016/j.bbamem.2008.07.015. [DOI] [PubMed] [Google Scholar]
- Balda M. S.; Matter K. Tight Junctions as Regulators of Tissue Remodelling. Curr. Opin. Cell Biol. 2016, 42, 94–101. 10.1016/j.ceb.2016.05.006. [DOI] [PubMed] [Google Scholar]
- Shin K.; Fogg V. C.; Margolis B. Tight Junctions and Cell Polarity. Annu. Rev. Cell Dev. Biol. 2006, 22, 207–235. 10.1146/annurev.cellbio.22.010305.104219. [DOI] [PubMed] [Google Scholar]
- Zihni C.; Mills C.; Matter K.; Balda M. S. Tight Junctions: From Simple Barriers to Multifunctional Molecular Gates. Nat. Rev. Mol. Cell Biol. 2016, 17, 564–580. 10.1038/nrm.2016.80. [DOI] [PubMed] [Google Scholar]
- Bauer H.-C.; Krizbai I. A.; Bauer H.; Traweger A. You Shall Not Pass”—Tight Junctions of the Blood Brain Barrier. Front. Neurosci. 2014, 8, 392. 10.3389/fnins.2014.00392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tietz S.; Engelhardt B. Brain Barriers: Crosstalk between Complex Tight Junctions and Adherens Junctions. J. Cell Biol. 2015, 209, 493–506. 10.1083/jcb.201412147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sluysmans S.; Vasileva E.; Spadaro D.; Shah J.; Rouaud F.; Citi S. The Role of Apical Cell-Cell Junctions and Associated Cytoskeleton in Mechanotransduction: Junctions, Cytoskeleton and Mechanotransduction. Biol. Cell 2017, 109, 139–161. 10.1111/boc.201600075. [DOI] [PubMed] [Google Scholar]
- Mège R. M.; Ishiyama N. Integration of Cadherin Adhesion and Cytoskeleton at Adherens Junctions. Cold Spring Harbor Perspect. Biol. 2017, 9, a028738. 10.1101/cshperspect.a028738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison O. J.; Jin X.; Hong S.; Bahna F.; Ahlsen G.; Brasch J.; Wu Y.; Vendome J.; Felsovalyi K.; Hampton C. M.; et al. The Extracellular Architecture of Adherens Junctions Revealed by Crystal Structures of Type I Cadherins. Structure 2011, 19, 244–256. 10.1016/j.str.2010.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiyama N.; Lee S.-H.; Liu S.; Li G.-Y.; Smith M. J.; Reichardt L. F.; Ikura M. Dynamic and Static Interactions between p120 Catenin and E-Cadherin Regulate the Stability of Cell-Cell Adhesion. Cell 2010, 141, 117–128. 10.1016/j.cell.2010.01.017. [DOI] [PubMed] [Google Scholar]
- Huber A. H.; Weis W. I. The Structure of the Beta-Catenin/E-Cadherin Complex and the Molecular Basis of Diverse Ligand Recognition by Beta-Catenin. Cell 2001, 105, 391–402. 10.1016/S0092-8674(01)00330-0. [DOI] [PubMed] [Google Scholar]
- Pokutta S.; Weis W. I. Structure of the Dimerization and Beta-Catenin-Binding Region of Alpha-Catenin. Mol. Cell 2000, 5, 533–543. 10.1016/S1097-2765(00)80447-5. [DOI] [PubMed] [Google Scholar]
- Rangarajan E. S.; Izard T. Dimer Asymmetry Defines α-Catenin Interactions. Nat. Struct. Mol. Biol. 2013, 20, 188–193. 10.1038/nsmb.2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiyama N.; Tanaka N.; Abe K.; Yang Y. J.; Abbas Y. M.; Umitsu M.; Nagar B.; Bueler S. A.; Rubinstein J. L.; Takeichi M.; et al. An Autoinhibited Structure of α-Catenin and Its Implications for Vinculin Recruitment to Adherens Junctions. J. Biol. Chem. 2013, 288, 15913–15925. 10.1074/jbc.M113.453928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong Y.; Topf M.; Sali A.; Matsudaira P.; Dougherty M.; Chiu W.; Schmid M. F. Crystallographic Conformers of Actin in a Biologically Active Bundle of Filaments. J. Mol. Biol. 2008, 375, 331–336. 10.1016/j.jmb.2007.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meşe G.; Richard G.; White T. W. Gap Junctions: Basic Structure and Function. J. Invest. Dermatol. 2007, 127, 2516–2524. 10.1038/sj.jid.5700770. [DOI] [PubMed] [Google Scholar]
- Maeda S.; Nakagawa S.; Suga M.; Yamashita E.; Oshima A.; Fujiyoshi Y.; Tsukihara T. Structure of the Connexin 26 Gap Junction Channel at 3.5 Å Resolution. Nature 2009, 458, 597–602. 10.1038/nature07869. [DOI] [PubMed] [Google Scholar]
- Garrod D.; Chidgey M. Desmosome Structure, Composition and Function. Biochim. Biophys. Acta, Biomembr. 2008, 1778, 572–587. 10.1016/j.bbamem.2007.07.014. [DOI] [PubMed] [Google Scholar]
- Delva E.; Tucker D. K.; Kowalczyk A. P. The Desmosome. Cold Spring Harbor Perspect. Biol. 2009, 1, a002543–a002543. 10.1101/cshperspect.a002543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Jassar C.; Bikker H.; Overduin M.; Chidgey M. Mechanistic Basis of Desmosome-Targeted Diseases. J. Mol. Biol. 2013, 425, 4006–4022. 10.1016/j.jmb.2013.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahoney M. G.; Sadowski S.; Brennan D.; Pikander P.; Saukko P.; Wahl J.; Aho H.; Heikinheimo K.; Bruckner-Tuderman L.; Fertala A.; et al. Compound Heterozygous Desmoplakin Mutations Result in a Phenotype with a Combination of Myocardial, Skin, Hair, and Enamel Abnormalities. J. Invest. Dermatol. 2010, 130, 968–978. 10.1038/jid.2009.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broussard J. A.; Getsios S.; Green K. J. Desmosome Regulation and Signaling in Disease. Cell Tissue Res. 2015, 360, 501–512. 10.1007/s00441-015-2136-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitoiu D.; Etheridge S. L.; Kelsell D. P. Insights into Desmosome Biology from Inherited Human Skin Disease and Cardiocutaneous Syndromes. Cell Commun. Adhes. 2014, 21, 129–140. 10.3109/15419061.2014.908854. [DOI] [PubMed] [Google Scholar]
- Panchin Y. V. Evolution of Gap Junction Proteins - the Pannexin Alternative. J. Exp. Biol. 2005, 208, 1415–1419. 10.1242/jeb.01547. [DOI] [PubMed] [Google Scholar]
- Beyer E. C.; Berthoud V. M. Gap Junction Gene and Protein Families: Connexins, Innexins, and Pannexins. Biochim. Biophys. Acta, Biomembr. 2018, 1860, 5–8. 10.1016/j.bbamem.2017.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshima A. Structure and Closure of Connexin Gap Junction Channels. FEBS Lett. 2014, 588, 1230–1237. 10.1016/j.febslet.2014.01.042. [DOI] [PubMed] [Google Scholar]
- Valiunas V.; Cohen I. S.; Brink P. R. Defining the Factors That Affect Solute Permeation of Gap Junction Channels. Biochim. Biophys. Acta, Biomembr. 2018, 1860, 96–101. 10.1016/j.bbamem.2017.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk M. M.; Bell C. L.; Kells Andrews R. M.; Murray S. A. Molecular Mechanisms Regulating Formation, Trafficking and Processing of Annular Gap Junctions. BMC Cell Biol. 2016, 17, S22. 10.1186/s12860-016-0087-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall D. H. Gap Junctions in C. Elegans : Their Roles in Behavior and Development: Gap Junctions in C. Elegans. Dev. Neurobiol. 2017, 77, 587–596. 10.1002/dneu.22408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu K.; Stopfer M. Gap Junctions. Curr. Biol. 2013, 23, R1026–R1031. 10.1016/j.cub.2013.10.067. [DOI] [PubMed] [Google Scholar]
- Cronier L.; Crespin S.; Strale P.-O.; Defamie N.; Mesnil M. Gap Junctions and Cancer: New Functions for an Old Story. Antioxid. Redox Signaling 2009, 11, 323–338. 10.1089/ars.2008.2153. [DOI] [PubMed] [Google Scholar]
- Meda P. Gap Junction Proteins Are Key Drivers of Endocrine Function. Biochim. Biophys. Acta, Biomembr. 2018, 1860, 124–140. 10.1016/j.bbamem.2017.03.005. [DOI] [PubMed] [Google Scholar]
- Srinivas M.; Verselis V. K.; White T. W. Human Diseases Associated with Connexin Mutations. Biochim. Biophys. Acta, Biomembr. 2018, 1860, 192–201. 10.1016/j.bbamem.2017.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rovegno M.; Sáez J. C. Role of Astrocyte Connexin Hemichannels in Cortical Spreading Depression. Biochim. Biophys. Acta, Biomembr. 2018, 1860, 216–223. 10.1016/j.bbamem.2017.08.014. [DOI] [PubMed] [Google Scholar]
- Mesnil M.; Aasen T.; Boucher J.; Chépied A.; Cronier L.; Defamie N.; Kameritsch P.; Laird D. W.; Lampe P. D.; Lathia J. D.; et al. An Update on Minding the Gap in Cancer. Biochim. Biophys. Acta, Biomembr. 2018, 1860, 237–243. 10.1016/j.bbamem.2017.06.015. [DOI] [PubMed] [Google Scholar]
- Boucher J.; Monvoisin A.; Vix J.; Mesnil M.; Thuringer D.; Debiais F.; Cronier L. Connexins, Important Players in the Dissemination of Prostate Cancer Cells. Biochim. Biophys. Acta, Biomembr. 2018, 1860, 202–215. 10.1016/j.bbamem.2017.06.020. [DOI] [PubMed] [Google Scholar]
- Kim Y.; Davidson J. O.; Green C. R.; Nicholson L. F. B.; O’Carroll S. J.; Zhang J. Connexins and Pannexins in Cerebral Ischemia. Biochim. Biophys. Acta, Biomembr. 2018, 1860, 224–236. 10.1016/j.bbamem.2017.03.018. [DOI] [PubMed] [Google Scholar]
- Burch-Smith T. M.; Zambryski P. C. Plasmodesmata Paradigm Shift: Regulation from Without Versus Within. Annu. Rev. Plant Biol. 2012, 63, 239–260. 10.1146/annurev-arplant-042811-105453. [DOI] [PubMed] [Google Scholar]
- Bloemendal S.; Kück U. Cell-to-Cell Communication in Plants, Animals, and Fungi: A Comparative Review. Naturwissenschaften 2013, 100, 3–19. 10.1007/s00114-012-0988-z. [DOI] [PubMed] [Google Scholar]
- Wolf S.; Hématy K.; Höfte H. Growth Control and Cell Wall Signaling in Plants. Annu. Rev. Plant Biol. 2012, 63, 381–407. 10.1146/annurev-arplant-042811-105449. [DOI] [PubMed] [Google Scholar]
- Hamann T. The Plant Cell Wall Integrity Maintenance Mechanism – A Case Study of a Cell Wall Plasma Membrane Signaling Network. Phytochemistry 2015, 112, 100–109. 10.1016/j.phytochem.2014.09.019. [DOI] [PubMed] [Google Scholar]
- Kim S.-J.; Brandizzi F. The Plant Secretory Pathway for the Trafficking of Cell Wall Polysaccharides and Glycoproteins. Glycobiology 2016, 26, 940–949. 10.1093/glycob/cww044. [DOI] [PubMed] [Google Scholar]
- Liu Z.; Persson S.; Sanchez-Rodriguez C. At the Border: The Plasma Membrane-Cell Wall Continuum. J. Exp. Bot. 2015, 66, 1553–1563. 10.1093/jxb/erv019. [DOI] [PubMed] [Google Scholar]
- Lehman T. A.; Smertenko A.; Sanguinet K. A. Auxin, Microtubules, and Vesicle Trafficking: Conspirators behind the Cell Wall. J. Exp. Bot. 2017, 68, 3321–3329. 10.1093/jxb/erx205. [DOI] [PubMed] [Google Scholar]
- Siegel S. D.; Liu J.; Ton-That H. Biogenesis of the Gram-Positive Bacterial Cell Envelope. Curr. Opin. Microbiol. 2016, 34, 31–37. 10.1016/j.mib.2016.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhar S.; Kumari H.; Balasubramanian D.; Mathee K. Cell-Wall Recycling and Synthesis in Escherichia Coli and Pseudomonas Aeruginosa – Their Role in the Development of Resistance. J. Med. Microbiol. 2018, 67, 1–21. 10.1099/jmm.0.000636. [DOI] [PubMed] [Google Scholar]
- Riedlová K.; Nekardová M.; Kačer P.; Syslová K.; Vazdar M.; Jungwirth P.; Kudová E.; Cwiklik L. Distributions of Therapeutically Promising Neurosteroids in Cellular Membranes. Chem. Phys. Lipids 2017, 203, 78–86. 10.1016/j.chemphyslip.2016.12.004. [DOI] [PubMed] [Google Scholar]
- Mistou M.-Y.; Sutcliffe I. C.; van Sorge N. M. Bacterial Glycobiology: Rhamnose-Containing Cell Wall Polysaccharides in Gram-Positive Bacteria. FEMS Microbiol. Rev. 2016, 40, 464–479. 10.1093/femsre/fuw006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopke A.; Brown A. J. P.; Hall R. A.; Wheeler R. T. Dynamic Fungal Cell Wall Architecture in Stress Adaptation and Immune Evasion. Trends Microbiol. 2018, 26, 284–295. 10.1016/j.tim.2018.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Free S. J.Fungal Cell Wall Organization and Biosynthesis. In Advances in Genetics; Friedmann T., Dunlap J. C., Goodwin S. F., Eds.; Elsevier: 2013; Vol. 81, pp 33–82. 10.1016/B978-0-12-407677-8.00002-6. [DOI] [PubMed] [Google Scholar]
- Kock C.; Dufrêne Y. F.; Heinisch J. J. Up against the Wall: Is Yeast Cell Wall Integrity Ensured by Mechanosensing in Plasma Membrane Microdomains?. Appl. Environ. Microbiol. 2015, 81, 806–811. 10.1128/AEM.03273-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimi A.; Miyazawa K.; Abe K. Cell Wall Structure and Biogenesis in Aspergillus Species. Biosci., Biotechnol., Biochem. 2016, 80, 1700–1711. 10.1080/09168451.2016.1177446. [DOI] [PubMed] [Google Scholar]
- Gow N. A. R.; Latge J.-P.; Munro C. A.. The Fungal Cell Wall: Structure, Biosynthesis, and Function. In The Fungal Kingdom; Heitman J., Howlett B. J., Crous P. W., Stukenbrock E. H., James T. Y., Gow N. A. R., Eds.; American Society of Microbiology Press: Washington, DC, 2017; pp 267–292. [Google Scholar]
- Orlean P. Architecture and Biosynthesis of the Saccharomyces Cerevisiae Cell Wall. Genetics 2012, 192, 775–818. 10.1534/genetics.112.144485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugg T. D. H.; Braddick D.; Dowson C. G.; Roper D. I. Bacterial Cell Wall Assembly: Still an Attractive Antibacterial Target. Trends Biotechnol. 2011, 29, 167–173. 10.1016/j.tibtech.2010.12.006. [DOI] [PubMed] [Google Scholar]
- Oppedijk S. F.; Martin N. I.; Breukink E. Hit ’em Where It Hurts: The Growing and Structurally Diverse Family of Peptides That Target Lipid-II. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 947–957. 10.1016/j.bbamem.2015.10.024. [DOI] [PubMed] [Google Scholar]
- Kim S. J.; Chang J.; Singh M. Peptidoglycan Architecture of Gram-Positive Bacteria by Solid-State NMR. Biochim. Biophys. Acta, Biomembr. 2015, 1848, 350–362. 10.1016/j.bbamem.2014.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swoboda J. G.; Campbell J.; Meredith T. C.; Walker S. Wall Teichoic Acid Function, Biosynthesis, and Inhibition. ChemBioChem 2010, 11, 35–45. 10.1002/cbic.200900557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau M. A.; Fisher J. F.; Mobashery S. Messenger Functions of the Bacterial Cell Wall-Derived Muropeptides. Biochemistry 2012, 51, 2974–2990. 10.1021/bi300174x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Münch D.; Sahl H.-G. Structural Variations of the Cell Wall Precursor Lipid II in Gram-Positive Bacteria — Impact on Binding and Efficacy of Antimicrobial Peptides. Biochim. Biophys. Acta, Biomembr. 2015, 1848, 3062–3071. 10.1016/j.bbamem.2015.04.014. [DOI] [PubMed] [Google Scholar]
- Vollmer W.Peptidoglycan. In Molecular Medical Microbiology; Tang Y.-W., Sails A., Eds.; Elsevier: 2015; pp 105–124, 10.1016/B978-0-12-397169-2.00006-8. [DOI] [Google Scholar]
- Yadav A. K.; Espaillat A. Bacterial Strategies to Preserve Cell Wall Integrity Against Environmental Threats. Front. Microbiol. 2018, 9, 2064 10.3389/fmicb.2018.02064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radkov A. D.; Hsu Y.-P.; Booher G.; VanNieuwenhze M. S. Imaging Bacterial Cell Wall Biosynthesis. Annu. Rev. Biochem. 2018, 87, 991–1014. 10.1146/annurev-biochem-062917-012921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz N. Filling Holes in Peptidoglycan Biogenesis of Escherichia Coli. Curr. Opin. Microbiol. 2016, 34, 1–6. 10.1016/j.mib.2016.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolson G. L. The Fluid—Mosaic Model of Membrane Structure: Still Relevant to Understanding the Structure, Function and Dynamics of Biological Membranes after More than 40 Years. Biochim. Biophys. Acta, Biomembr. 2014, 1838, 1451–1466. 10.1016/j.bbamem.2013.10.019. [DOI] [PubMed] [Google Scholar]
- Pike L. J. Rafts Defined: A Report on the Keystone Symposium on Lipid Rafts and Cell Function. J. Lipid Res. 2006, 47, 1597–1598. 10.1194/jlr.E600002-JLR200. [DOI] [PubMed] [Google Scholar]
- Sevcsik E.; Schütz G. J. With or without Rafts? Alternative Views on Cell Membranes. BioEssays 2016, 38, 129–139. 10.1002/bies.201500150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggeling C.; Ringemann C.; Medda R.; Schwarzmann G.; Sandhoff K.; Polyakova S.; Belov V. N.; Hein B.; von Middendorff C.; Schönle A.; et al. Direct Observation of the Nanoscale Dynamics of Membrane Lipids in a Living Cell. Nature 2009, 457, 1159–1162. 10.1038/nature07596. [DOI] [PubMed] [Google Scholar]
- Regmi R.; Winkler P. M.; Flauraud V.; Borgman K. J. E.; Manzo C.; Brugger J.; Rigneault H.; Wenger J.; García-Parajo M. F. Planar Optical Nanoantennas Resolve Cholesterol-Dependent Nanoscale Heterogeneities in the Plasma Membrane of Living Cells. Nano Lett. 2017, 17, 6295–6302. 10.1021/acs.nanolett.7b02973. [DOI] [PubMed] [Google Scholar]
- Bernardino de la Serna J.; Schütz G. J.; Eggeling C.; Cebecauer M. There Is No Simple Model of the Plasma Membrane Organization. Front. Cell Dev. Biol. 2016, 4, 106. 10.3389/fcell.2016.00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh D. Liquid-Ordered Phases Induced by Cholesterol: A Compendium of Binary Phase Diagrams. Biochim. Biophys. Acta, Biomembr. 2010, 1798, 688–699. 10.1016/j.bbamem.2009.12.027. [DOI] [PubMed] [Google Scholar]
- Marsh D. Cholesterol-Induced Fluid Membrane Domains: A Compendium of Lipid-Raft Ternary Phase Diagrams. Biochim. Biophys. Acta, Biomembr. 2009, 1788, 2114–2123. 10.1016/j.bbamem.2009.08.004. [DOI] [PubMed] [Google Scholar]
- Veatch S. L.; Keller S. L. Seeing Spots: Complex Phase Behavior in Simple Membranes. Biochim. Biophys. Acta, Mol. Cell Res. 2005, 1746, 172–185. 10.1016/j.bbamcr.2005.06.010. [DOI] [PubMed] [Google Scholar]
- Feigenson G. W. Phase Diagrams and Lipid Domains in Multicomponent Lipid Bilayer Mixtures. Biochim. Biophys. Acta, Biomembr. 2009, 1788, 47–52. 10.1016/j.bbamem.2008.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feigenson G. W.; Buboltz J. T. Ternary Phase Diagram of Dipalmitoyl-PC/Dilauroyl-PC/Cholesterol: Nanoscopic Domain Formation Driven by Cholesterol. Biophys. J. 2001, 80, 2775–2788. 10.1016/S0006-3495(01)76245-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korlach J.; Schwille P.; Webb W. W.; Feigenson G. W. Characterization of Lipid Bilayer Phases by Confocal Microscopy and Fluorescence Correlation Spectroscopy. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 8461–8466. 10.1073/pnas.96.15.8461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veatch S. L.; Keller S. L. Separation of Liquid Phases in Giant Vesicles of Ternary Mixtures of Phospholipids and Cholesterol. Biophys. J. 2003, 85, 3074–3083. 10.1016/S0006-3495(03)74726-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veatch S. L.; Keller S. L. Organization in Lipid Membranes Containing Cholesterol. Phys. Rev. Lett. 2002, 89, 268101. 10.1103/PhysRevLett.89.268101. [DOI] [PubMed] [Google Scholar]
- Dietrich C.; Bagatolli L. A.; Volovyk Z. N.; Thompson N. L.; Levi M.; Jacobson K.; Gratton E. Lipid Rafts Reconstituted in Model Membranes. Biophys. J. 2001, 80, 1417–1428. 10.1016/S0006-3495(01)76114-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veatch S. L.; Keller S. L. Miscibility Phase Diagrams of Giant Vesicles Containing Sphingomyelin. Phys. Rev. Lett. 2005, 94, 148101. 10.1103/PhysRevLett.94.148101. [DOI] [PubMed] [Google Scholar]
- Vist M. R.; Davis J. H. Phase Equilibria of Cholesterol/Dipalmitoylphosphatidylcholine Mixtures: Deuterium Nuclear Magnetic Resonance and Differential Scanning Calorimetry. Biochemistry 1990, 29, 451–464. 10.1021/bi00454a021. [DOI] [PubMed] [Google Scholar]
- Rheinstädter M. C.; Mouritsen O. G. Small-Scale Structure in Fluid Cholesterol–Lipid Bilayers. Curr. Opin. Colloid Interface Sci. 2013, 18, 440–447. 10.1016/j.cocis.2013.07.001. [DOI] [Google Scholar]
- Schmid F. Physical Mechanisms of Micro- and Nanodomain Formation in Multicomponent Lipid Membranes. Biochim. Biophys. Acta, Biomembr. 2017, 1859, 509–528. 10.1016/j.bbamem.2016.10.021. [DOI] [PubMed] [Google Scholar]
- Winkler P. M.; Regmi R.; Flauraud V.; Brugger J.; Rigneault H.; Wenger J.; García-Parajo M. F. Transient Nanoscopic Phase Separation in Biological Lipid Membranes Resolved by Planar Plasmonic Antennas. ACS Nano 2017, 11, 7241–7250. 10.1021/acsnano.7b03177. [DOI] [PubMed] [Google Scholar]
- Wu H.-M.; Lin Y.-H.; Yen T.-C.; Hsieh C.-L. Nanoscopic Substructures of Raft-Mimetic Liquid-Ordered Membrane Domains Revealed by High-Speed Single-Particle Tracking. Sci. Rep. 2016, 6, 20542. 10.1038/srep20542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgart T.; Hammond A. T.; Sengupta P.; Hess S. T.; Holowka D. A.; Baird B. A.; Webb W. W. Large-Scale Fluid/Fluid Phase Separation of Proteins and Lipids in Giant Plasma Membrane Vesicles. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 3165–3170. 10.1073/pnas.0611357104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honerkamp-Smith A. R.; Veatch S. L.; Keller S. L. An Introduction to Critical Points for Biophysicists; Observations of Compositional Heterogeneity in Lipid Membranes. Biochim. Biophys. Acta, Biomembr. 2009, 1788, 53–63. 10.1016/j.bbamem.2008.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veatch S. L.; Cicuta P.; Sengupta P.; Honerkamp-Smith A.; Holowka D.; Baird B. Critical Fluctuations in Plasma Membrane Vesicles. ACS Chem. Biol. 2008, 3, 287–293. 10.1021/cb800012x. [DOI] [PubMed] [Google Scholar]
- Veatch S. L.; Soubias O.; Keller S. L.; Gawrisch K. Critical Fluctuations in Domain-Forming Lipid Mixtures. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 17650–17655. 10.1073/pnas.0703513104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May S. Trans-Monolayer Coupling of Fluid Domains in Lipid Bilayers. Soft Matter 2009, 5, 3148–3156. 10.1039/b901647c. [DOI] [Google Scholar]
- Mitra E. D.; Whitehead S. C.; Holowka D.; Baird B.; Sethna J. P. Computation of a Theoretical Membrane Phase Diagram and the Role of Phase in Lipid-Raft-Mediated Protein Organization. J. Phys. Chem. B 2018, 122, 3500–3513. 10.1021/acs.jpcb.7b10695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leekumjorn S.; Sum A. K. Molecular Studies of the Gel to Liquid-Crystalline Phase Transition for Fully Hydrated DPPC and DPPE Bilayers. Biochim. Biophys. Acta, Biomembr. 2007, 1768, 354–365. 10.1016/j.bbamem.2006.11.003. [DOI] [PubMed] [Google Scholar]
- Tjörnhammar R.; Edholm O. Reparameterized United Atom Model for Molecular Dynamics Simulations of Gel and Fluid Phosphatidylcholine Bilayers. J. Chem. Theory Comput. 2014, 10, 5706–5715. 10.1021/ct500589z. [DOI] [PubMed] [Google Scholar]
- de Vries A. H.; Yefimov S.; Mark A. E.; Marrink S. J. Molecular Structure of the Lecithin Ripple Phase. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 5392–5396. 10.1073/pnas.0408249102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrink S. J.; Risselada J.; Mark A. E. Simulation of Gel Phase Formation and Melting in Lipid Bilayers Using a Coarse Grained Model. Chem. Phys. Lipids 2005, 135, 223–244. 10.1016/j.chemphyslip.2005.03.001. [DOI] [PubMed] [Google Scholar]
- Rodgers J. M.; Sørensen J.; de Meyer F. J.-M.; Schiøtt B.; Smit B. Understanding the Phase Behavior of Coarse-Grained Model Lipid Bilayers through Computational Calorimetry. J. Phys. Chem. B 2012, 116, 1551–1569. 10.1021/jp207837v. [DOI] [PubMed] [Google Scholar]
- Arnarez C.; Webb A.; Rouvière E.; Lyman E. Hysteresis and the Cholesterol Dependent Phase Transition in Binary Lipid Mixtures with the Martini Model. J. Phys. Chem. B 2016, 120, 13086–13093. 10.1021/acs.jpcb.6b09728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waheed Q.; Tjörnhammar R.; Edholm O. Phase Transitions in Coarse-Grained Lipid Bilayers Containing Cholesterol by Molecular Dynamics Simulations. Biophys. J. 2012, 103, 2125–2133. 10.1016/j.bpj.2012.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogata K.; Nakamura S. Improvement of Parameters of the AMBER Potential Force Field for Phospholipids for Description of Thermal Phase Transitions. J. Phys. Chem. B 2015, 119, 9726–9739. 10.1021/acs.jpcb.5b01656. [DOI] [PubMed] [Google Scholar]
- Pluhackova K.; Kirsch S. A.; Han J.; Sun L.; Jiang Z.; Unruh T.; Böckmann R. A. A Critical Comparison of Biomembrane Force Fields: Structure and Dynamics of Model DMPC, POPC, and POPE Bilayers. J. Phys. Chem. B 2016, 120, 3888–3903. 10.1021/acs.jpcb.6b01870. [DOI] [PubMed] [Google Scholar]
- Faller R.; Marrink S.-J. Simulation of Domain Formation in DLPC–DSPC Mixed Bilayers. Langmuir 2004, 20, 7686–7693. 10.1021/la0492759. [DOI] [PubMed] [Google Scholar]
- Pyrkova D. V.; Tarasova N. K.; Pyrkov T. V.; Krylov N. A.; Efremov R. G. Atomic-Scale Lateral Heterogeneity and Dynamics of Two-Component Lipid Bilayers Composed of Saturated and Unsaturated Phosphatidylcholines. Soft Matter 2011, 7, 2569–2579. 10.1039/c0sm00701c. [DOI] [Google Scholar]
- Wong B. Y.; Faller R. Phase Behavior and Dynamic Heterogeneities in Lipids: A Coarse-Grained Simulation Study of DPPC–DPPE Mixtures. Biochim. Biophys. Acta, Biomembr. 2007, 1768, 620–627. 10.1016/j.bbamem.2006.12.009. [DOI] [PubMed] [Google Scholar]
- Rosetti C.; Pastorino C. Polyunsaturated and Saturated Phospholipids in Mixed Bilayers: A Study from the Molecular Scale to the Lateral Lipid Organization. J. Phys. Chem. B 2011, 115, 1002–1013. 10.1021/jp1082888. [DOI] [PubMed] [Google Scholar]
- Baoukina S.; Mendez-Villuendas E.; Bennett W. F. D.; Tieleman D. P. Computer Simulations of the Phase Separation in Model Membranes. Faraday Discuss. 2013, 161, 63–75. 10.1039/C2FD20117H. [DOI] [PubMed] [Google Scholar]
- Ohvo-Rekilä H. Cholesterol Interactions with Phospholipids in Membranes. Prog. Lipid Res. 2002, 41, 66–97. 10.1016/S0163-7827(01)00020-0. [DOI] [PubMed] [Google Scholar]
- Boughter C. T.; Monje-Galvan V.; Im W.; Klauda J. B. Influence of Cholesterol on Phospholipid Bilayer Structure and Dynamics. J. Phys. Chem. B 2016, 120, 11761–11772. 10.1021/acs.jpcb.6b08574. [DOI] [PubMed] [Google Scholar]
- Wang E.; Klauda J. B. Examination of Mixtures Containing Sphingomyelin and Cholesterol by Molecular Dynamics Simulations. J. Phys. Chem. B 2017, 121, 4833–4844. 10.1021/acs.jpcb.7b01832. [DOI] [PubMed] [Google Scholar]
- Martinez-Seara H.; Róg T.; Karttunen M.; Vattulainen I.; Reigada R. Cholesterol Induces Specific Spatial and Orientational Order in Cholesterol/Phospholipid Membranes. PLoS One 2010, 5, e11162. 10.1371/journal.pone.0011162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg S.; Castro-Roman F.; Porcar L.; Butler P.; Bautista P. J.; Krzyzanowski N.; Perez-Salas U. Cholesterol Solubility Limit in Lipid Membranes Probed by Small Angle Neutron Scattering and MD Simulations. Soft Matter 2014, 10, 9313–9317. 10.1039/C4SM01219D. [DOI] [PubMed] [Google Scholar]
- Díaz-Tejada C.; Ariz-Extreme I.; Awasthi N.; Hub J. S. Quantifying Lateral Inhomogeneity of Cholesterol-Containing Membranes. J. Phys. Chem. Lett. 2015, 6, 4799–4803. 10.1021/acs.jpclett.5b02414. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Lervik A.; Seddon J.; Bresme F. A Coarse-Grained Molecular Dynamics Investigation of the Phase Behavior of DPPC/Cholesterol Mixtures. Chem. Phys. Lipids 2015, 185, 88–98. 10.1016/j.chemphyslip.2014.07.011. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Gkeka P.; Fuchs J. E.; Liedl K. R.; Cournia Z. DPPC-Cholesterol Phase Diagram Using Coarse-Grained Molecular Dynamics Simulations. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 2846–2857. 10.1016/j.bbamem.2016.08.005. [DOI] [PubMed] [Google Scholar]
- Melo M. N.; Ingólfsson H. I.; Marrink S. J. Parameters for Martini Sterols and Hopanoids Based on a Virtual-Site Description. J. Chem. Phys. 2015, 143, 243152. 10.1063/1.4937783. [DOI] [PubMed] [Google Scholar]
- Lee I.-H.; Saha S.; Polley A.; Huang H.; Mayor S.; Rao M.; Groves J. T. Live Cell Plasma Membranes Do Not Exhibit a Miscibility Phase Transition over a Wide Range of Temperatures. J. Phys. Chem. B 2015, 119, 4450–4459. 10.1021/jp512839q. [DOI] [PubMed] [Google Scholar]
- de Almeida R. F. M.; Fedorov A.; Prieto M. Sphingomyelin/Phosphatidylcholine/Cholesterol Phase Diagram: Boundaries and Composition of Lipid Rafts. Biophys. J. 2003, 85, 2406–2416. 10.1016/S0006-3495(03)74664-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemelä P. S.; Ollila S.; Hyvönen M. T.; Karttunen M.; Vattulainen I. Assessing the Nature of Lipid Raft Membranes. PLoS Comput. Biol. 2007, 3, e34. 10.1371/journal.pcbi.0030034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bera I.; Klauda J. B. Molecular Simulations of Mixed Lipid Bilayers with Sphingomyelin, Glycerophospholipids, and Cholesterol. J. Phys. Chem. B 2017, 121, 5197–5208. 10.1021/acs.jpcb.7b00359. [DOI] [PubMed] [Google Scholar]
- Aittoniemi J.; Niemelä P. S.; Hyvönen M. T.; Karttunen M.; Vattulainen I. Insight into the Putative Specific Interactions between Cholesterol, Sphingomyelin, and Palmitoyl-Oleoyl Phosphatidylcholine. Biophys. J. 2007, 92, 1125–1137. 10.1529/biophysj.106.088427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.; Martí J.; Calero C. Pair Interactions among Ternary DPPC/POPC/Cholesterol Mixtures in Liquid-Ordered and Liquid-Disordered Phases. Soft Matter 2016, 12, 4557–4561. 10.1039/C6SM00345A. [DOI] [PubMed] [Google Scholar]
- Wang C.; Krause M. R.; Regen S. L. Push and Pull Forces in Lipid Raft Formation: The Push Can Be as Important as the Pull. J. Am. Chem. Soc. 2015, 137, 664–666. 10.1021/ja5115437. [DOI] [PubMed] [Google Scholar]
- de Joannis J.; Coppock P. S.; Yin F.; Mori M.; Zamorano A.; Kindt J. T. Atomistic Simulation of Cholesterol Effects on Miscibility of Saturated and Unsaturated Phospholipids: Implications for Liquid-Ordered/Liquid-Disordered Phase Coexistence. J. Am. Chem. Soc. 2011, 133, 3625–3634. 10.1021/ja110425s. [DOI] [PubMed] [Google Scholar]
- Sodt A. J.; Sandar M. L.; Gawrisch K.; Pastor R. W.; Lyman E. The Molecular Structure of the Liquid-Ordered Phase of Lipid Bilayers. J. Am. Chem. Soc. 2014, 136, 725–732. 10.1021/ja4105667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sodt A. J.; Pastor R. W.; Lyman E. Hexagonal Substructure and Hydrogen Bonding in Liquid-Ordered Phases Containing Palmitoyl Sphingomyelin. Biophys. J. 2015, 109, 948–955. 10.1016/j.bpj.2015.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandit S. A.; Vasudevan S.; Chiu S. W.; Jay Mashl R.; Jakobsson E.; Scott H. L. Sphingomyelin-Cholesterol Domains in Phospholipid Membranes: Atomistic Simulation. Biophys. J. 2004, 87, 1092–1100. 10.1529/biophysj.104.041939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandit S. A.; Jakobsson E.; Scott H. L. Simulation of the Early Stages of Nano-Domain Formation in Mixed Bilayers of Sphingomyelin, Cholesterol, and Dioleylphosphatidylcholine. Biophys. J. 2004, 87, 3312–3322. 10.1529/biophysj.104.046078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apajalahti T.; Niemelä P.; Govindan P. N.; Miettinen M. S.; Salonen E.; Marrink S.-J.; Vattulainen I. Concerted Diffusion of Lipids in Raft-like Membranes. Faraday Discuss. 2010, 144, 411–430. 10.1039/B901487J. [DOI] [PubMed] [Google Scholar]
- Bennett W. F. D.; Tieleman D. P. Computer Simulations of Lipid Membrane Domains. Biochim. Biophys. Acta, Biomembr. 2013, 1828, 1765–1776. 10.1016/j.bbamem.2013.03.004. [DOI] [PubMed] [Google Scholar]
- Baoukina S.; Tieleman D. P.. Computer Simulations of Phase Separation in Lipid Bilayers and Monolayers. In Methods in Membrane Lipids; Owen D. M., Ed.; Springer New York: New York, NY, 2015; Vol. 1232, pp 307–322, 10.1007/978-1-4939-1752-5_21. [DOI] [PubMed] [Google Scholar]
- Risselada H. J.; Marrink S. J. The Molecular Face of Lipid Rafts in Model Membranes. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 17367–17372. 10.1073/pnas.0807527105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uppamoochikkal P.; Tristram-Nagle S.; Nagle J. F. Orientation of Tie-Lines in the Phase Diagram of DOPC/DPPC/Cholesterol Model Biomembranes. Langmuir 2010, 26, 17363–17368. 10.1021/la103024f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantelopulos G. A.; Nagai T.; Bandara A.; Panahi A.; Straub J. E. Critical Size Dependence of Domain Formation Observed in Coarse-Grained Simulations of Bilayers Composed of Ternary Lipid Mixtures. J. Chem. Phys. 2017, 147, 095101. 10.1063/1.4999709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis R. S.; Sunil Kumar P. B.; Sperotto M. M.; Laradji M. Predictions of Phase Separation in Three-Component Lipid Membranes by the MARTINI Force Field. J. Phys. Chem. B 2013, 117, 4072–4080. 10.1021/jp4000686. [DOI] [PubMed] [Google Scholar]
- Rosetti C.; Pastorino C. Comparison of Ternary Bilayer Mixtures with Asymmetric or Symmetric Unsaturated Phosphatidylcholine Lipids by Coarse Grained Molecular Dynamics Simulations. J. Phys. Chem. B 2012, 116, 3525–3537. 10.1021/jp212406u. [DOI] [PubMed] [Google Scholar]
- Domański J.; Marrink S. J.; Schäfer L. V. Transmembrane Helices Can Induce Domain Formation in Crowded Model Membranes. Biochim. Biophys. Acta, Biomembr. 2012, 1818, 984–994. 10.1016/j.bbamem.2011.08.021. [DOI] [PubMed] [Google Scholar]
- Lin X.; Lorent J. H.; Skinkle A. D.; Levental K. R.; Waxham M. N.; Gorfe A. A.; Levental I. Domain Stability in Biomimetic Membranes Driven by Lipid Polyunsaturation. J. Phys. Chem. B 2016, 120, 11930–11941. 10.1021/acs.jpcb.6b06815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakobyan D.; Heuer A. Key Molecular Requirements for Raft Formation in Lipid/Cholesterol Membranes. PLoS One 2014, 9, e87369. 10.1371/journal.pone.0087369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter T. S.; López C. A.; Neale C.; Montour C.; Ingólfsson H. I.; Di Natale F.; Lightstone F. C.; Gnanakaran S. Capturing Phase Behavior of Ternary Lipid Mixtures with a Refined Martini Coarse-Grained Force Field. J. Chem. Theory Comput. 2018, 14, 6050–6062. 10.1021/acs.jctc.8b00496. [DOI] [PubMed] [Google Scholar]
- Fowler P. W.; Williamson J. J.; Sansom M. S. P.; Olmsted P. D. Roles of Interleaflet Coupling and Hydrophobic Mismatch in Lipid Membrane Phase-Separation Kinetics. J. Am. Chem. Soc. 2016, 138, 11633–11642. 10.1021/jacs.6b04880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh S. L.; Amazon J. J.; Feigenson G. W. Toward a Better Raft Model: Modulated Phases in the Four-Component Bilayer, DSPC/DOPC/POPC/CHOL. Biophys. J. 2013, 104, 853–862. 10.1016/j.bpj.2013.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ackerman D. G.; Feigenson G. W. Multiscale Modeling of Four-Component Lipid Mixtures: Domain Composition, Size, Alignment, and Properties of the Phase Interface. J. Phys. Chem. B 2015, 119, 4240–4250. 10.1021/jp511083z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schäfer L. V.; Marrink S. J. Partitioning of Lipids at Domain Boundaries in Model Membranes. Biophys. J. 2010, 99, L91–L93. 10.1016/j.bpj.2010.08.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ackerman D. G.; Feigenson G. W. Effects of Transmembrane α-Helix Length and Concentration on Phase Behavior in Four-Component Lipid Mixtures: A Molecular Dynamics Study. J. Phys. Chem. B 2016, 120, 4064–4077. 10.1021/acs.jpcb.6b00611. [DOI] [PubMed] [Google Scholar]
- Perlmutter J. D.; Sachs J. N. Interleaflet Interaction and Asymmetry in Phase Separated Lipid Bilayers: Molecular Dynamics Simulations. J. Am. Chem. Soc. 2011, 133, 6563–6577. 10.1021/ja106626r. [DOI] [PubMed] [Google Scholar]
- Usery R. D.; Enoki T. A.; Wickramasinghe S. P.; Weiner M. D.; Tsai W.-C.; Kim M. B.; Wang S.; Torng T. L.; Ackerman D. G.; Heberle F. A.; et al. Line Tension Controls Liquid-Disordered + Liquid-Ordered Domain Size Transition in Lipid Bilayers. Biophys. J. 2017, 112, 1431–1443. 10.1016/j.bpj.2017.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmieri B.; Grant M.; Safran S. A. Prediction of the Dependence of the Line Tension on the Composition of Linactants and the Temperature in Phase Separated Membranes. Langmuir 2014, 30, 11734–11745. 10.1021/la502347a. [DOI] [PubMed] [Google Scholar]
- Heberle F. A.; Doktorova M.; Goh S. L.; Standaert R. F.; Katsaras J.; Feigenson G. W. Hybrid and Nonhybrid Lipids Exert Common Effects on Membrane Raft Size and Morphology. J. Am. Chem. Soc. 2013, 135, 14932–14935. 10.1021/ja407624c. [DOI] [PubMed] [Google Scholar]
- Rosetti C. M.; Montich G. G.; Pastorino C. Molecular Insight into the Line Tension of Bilayer Membranes Containing Hybrid Polyunsaturated Lipids. J. Phys. Chem. B 2017, 121, 1587–1600. 10.1021/acs.jpcb.6b10836. [DOI] [PubMed] [Google Scholar]
- Dupuy A. D.; Engelman D. M. Protein Area Occupancy at the Center of the Red Blood Cell Membrane. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 2848–2852. 10.1073/pnas.0712379105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusumi A.; Nakada C.; Ritchie K.; Murase K.; Suzuki K.; Murakoshi H.; Kasai R. S.; Kondo J.; Fujiwara T. Paradigm Shift of the Plasma Membrane Concept from the Two-Dimensional Continuum Fluid to the Partitioned Fluid: High-Speed Single-Molecule Tracking of Membrane Molecules. Annu. Rev. Biophys. Biomol. Struct. 2005, 34, 351–378. 10.1146/annurev.biophys.34.040204.144637. [DOI] [PubMed] [Google Scholar]
- Fischer T.; Jelger Risselada H.; Vink R. L. C. Membrane Lateral Structure: The Influence of Immobilized Particles on Domain Size. Phys. Chem. Chem. Phys. 2012, 14, 14500. 10.1039/c2cp41417a. [DOI] [PubMed] [Google Scholar]
- Schick M. Strongly Correlated Rafts in Both Leaves of an Asymmetric Bilayer. J. Phys. Chem. B 2018, 122, 3251–3258. 10.1021/acs.jpcb.7b08890. [DOI] [PubMed] [Google Scholar]
- Tian J.; Nickels J.; Katsaras J.; Cheng X. Behavior of Bilayer Leaflets in Asymmetric Model Membranes: Atomistic Simulation Studies. J. Phys. Chem. B 2016, 120, 8438–8448. 10.1021/acs.jpcb.6b02148. [DOI] [PubMed] [Google Scholar]
- Iwabuchi K.; Nakayama H.; Iwahara C.; Takamori K. Significance of Glycosphingolipid Fatty Acid Chain Length on Membrane Microdomain-Mediated Signal Transduction. FEBS Lett. 2010, 584, 1642–1652. 10.1016/j.febslet.2009.10.043. [DOI] [PubMed] [Google Scholar]
- Weiner M. D.; Feigenson G. W. Presence and Role of Midplane Cholesterol in Lipid Bilayers Containing Registered or Antiregistered Phase Domains. J. Phys. Chem. B 2018, 122, 8193–8200. 10.1021/acs.jpcb.8b03949. [DOI] [PubMed] [Google Scholar]
- Thallmair S.; Ingólfsson H. I.; Marrink S. J. Cholesterol Flip-Flop Impacts Domain Registration in Plasma Membrane Models. J. Phys. Chem. Lett. 2018, 9, 5527–5533. 10.1021/acs.jpclett.8b01877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakobyan D.; Heuer A. Phase Separation in a Lipid/Cholesterol System: Comparison of Coarse-Grained and United-Atom Simulations. J. Phys. Chem. B 2013, 117, 3841–3851. 10.1021/jp312245y. [DOI] [PubMed] [Google Scholar]
- Barnoud J.; Rossi G.; Marrink S. J.; Monticelli L. Hydrophobic Compounds Reshape Membrane Domains. PLoS Comput. Biol. 2014, 10, e1003873. 10.1371/journal.pcbi.1003873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reigada R.; Sagues F. Chloroform Alters Interleaflet Coupling in Lipid Bilayers: An Entropic Mechanism. J. R. Soc., Interface 2015, 12, 20150197–20150197. 10.1098/rsif.2015.0197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moiset G.; López C. A.; Bartelds R.; Syga L.; Rijpkema E.; Cukkemane A.; Baldus M.; Poolman B.; Marrink S. J. Disaccharides Impact the Lateral Organization of Lipid Membranes. J. Am. Chem. Soc. 2014, 136, 16167–16175. 10.1021/ja505476c. [DOI] [PubMed] [Google Scholar]
- Rossi G.; Barnoud J.; Monticelli L. Polystyrene Nanoparticles Perturb Lipid Membranes. J. Phys. Chem. Lett. 2014, 5, 241–246. 10.1021/jz402234c. [DOI] [PubMed] [Google Scholar]
- Bochicchio D.; Panizon E.; Monticelli L.; Rossi G. Interaction of Hydrophobic Polymers with Model Lipid Bilayers. Sci. Rep. 2017, 7, 6357. 10.1038/s41598-017-06668-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muddana H. S.; Chiang H. H.; Butler P. J. Tuning Membrane Phase Separation Using Nonlipid Amphiphiles. Biophys. J. 2012, 102, 489–497. 10.1016/j.bpj.2011.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingólfsson H. I.; Melo M. N.; van Eerden F. J.; Arnarez C.; Lopez C. A.; Wassenaar T. A.; Periole X.; de Vries A. H.; Tieleman D. P.; Marrink S. J. Lipid Organization of the Plasma Membrane. J. Am. Chem. Soc. 2014, 136, 14554–14559. 10.1021/ja507832e. [DOI] [PubMed] [Google Scholar]
- Gu R.-X.; Ingólfsson H. I.; de Vries A. H.; Marrink S. J.; Tieleman D. P. Ganglioside-Lipid and Ganglioside-Protein Interactions Revealed by Coarse-Grained and Atomistic Molecular Dynamics Simulations. J. Phys. Chem. B 2017, 121, 3262–3275. 10.1021/acs.jpcb.6b07142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koldsø H.; Shorthouse D.; Hélie J.; Sansom M. S. P. Lipid Clustering Correlates with Membrane Curvature as Revealed by Molecular Simulations of Complex Lipid Bilayers. PLoS Comput. Biol. 2014, 10, e1003911. 10.1371/journal.pcbi.1003911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingólfsson H. I.; Carpenter T. S.; Bhatia H.; Bremer P.-T.; Marrink S. J.; Lightstone F. C. Computational Lipidomics of the Neuronal Plasma Membrane. Biophys. J. 2017, 113, 2271–2280. 10.1016/j.bpj.2017.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baoukina S.; Ingólfsson H. I.; Marrink S. J.; Tieleman D. P. Curvature-Induced Sorting of Lipids in Plasma Membrane Tethers. Adv. Theory Simul. 2018, 1, 1800034. 10.1002/adts.201800034. [DOI] [Google Scholar]
- Flinner N.; Schleiff E. Dynamics of the Glycophorin A Dimer in Membranes of Native-Like Composition Uncovered by Coarse-Grained Molecular Dynamics Simulations. PLoS One 2015, 10, e0133999. 10.1371/journal.pone.0133999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedger G.; Sansom M. S. P. Lipid Interaction Sites on Channels, Transporters and Receptors: Recent Insights from Molecular Dynamics Simulations. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 2390–2400. 10.1016/j.bbamem.2016.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koldsø H.; Sansom M. S. P. Local Lipid Reorganization by a Transmembrane Protein Domain. J. Phys. Chem. Lett. 2012, 3, 3498–3502. 10.1021/jz301570w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vögele M.; Köfinger J.; Hummer G. Hydrodynamics of Diffusion in Lipid Membrane Simulations. Phys. Rev. Lett. 2018, 120, 268104. 10.1103/PhysRevLett.120.268104. [DOI] [PubMed] [Google Scholar]
- Parton D. L.; Tek A.; Baaden M.; Sansom M. S. P. Formation of Raft-Like Assemblies within Clusters of Influenza Hemagglutinin Observed by MD Simulations. PLoS Comput. Biol. 2013, 9, e1003034. 10.1371/journal.pcbi.1003034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan A. L.; Reddy T.; Koldsø H.; Hélie J.; Fowler P. W.; Chavent M.; Sansom M. S. P. Protein Crowding and Lipid Complexity Influence the Nanoscale Dynamic Organization of Ion Channels in Cell Membranes. Sci. Rep. 2017, 7, 16647. 10.1038/s41598-017-16865-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S.; Kim B. N.; Stansfeld P. J.; Sansom M. S. P.; Lindau M. A Coarse Grained Model for a Lipid Membrane with Physiological Composition and Leaflet Asymmetry. PLoS One 2015, 10, e0144814. 10.1371/journal.pone.0144814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Bogaart G.; Meyenberg K.; Risselada H. J.; Amin H.; Willig K. I.; Hubrich B. E.; Dier M.; Hell S. W.; Grubmüller H.; Diederichsen U.; et al. Membrane Protein Sequestering by Ionic Protein–Lipid Interactions. Nature 2011, 479, 552–555. 10.1038/nature10545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corradi V.; Mendez-Villuendas E.; Ingólfsson H. I.; Gu R.-X.; Siuda I.; Melo M. N.; Moussatova A.; DeGagné L. J.; Sejdiu B. I.; Singh G.; et al. Lipid–Protein Interactions Are Unique Fingerprints for Membrane Proteins. ACS Cent. Sci. 2018, 4, 709–717. 10.1021/acscentsci.8b00143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Einstein A. In Investigations on the Theory of the Brownian Movement; Fürth R., Ed.; Cowper, A. D., Translator; Dover Publications, 1956. [Google Scholar]
- Vattulainen I.; Mouritsen O. G.. Diffusion in Membranes. In Diffusion in Condensed Matter; Heitjans P., Kärger J., Eds.; Springer: Berlin, Heidelberg, 2005; pp 471–509, 10.1007/3-540-30970-5_12. [DOI] [Google Scholar]
- Wieser S.; Weghuber J.; Sams M.; Stockinger H.; Schütz G. J. Cell-to-Cell Variability in the Diffusion Constants of the Plasma Membrane Proteins CD59 and CD147. Soft Matter 2009, 5, 3287–3294. 10.1039/b902266j. [DOI] [Google Scholar]
- Milo R.; Phillips R.. Cell Biology by the Numbers; Garland Science, Taylor & Francis Group: New York, NY, 2016. [Google Scholar]
- Bag N.; Yap D. H. X.; Wohland T. Temperature Dependence of Diffusion in Model and Live Cell Membranes Characterized by Imaging Fluorescence Correlation Spectroscopy. Biochim. Biophys. Acta, Biomembr. 2014, 1838, 802–813. 10.1016/j.bbamem.2013.10.009. [DOI] [PubMed] [Google Scholar]
- Metzler R.; Jeon J.-H.; Cherstvy A. G. Non-Brownian Diffusion in Lipid Membranes: Experiments and Simulations. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 2451–2467. 10.1016/j.bbamem.2016.01.022. [DOI] [PubMed] [Google Scholar]
- Falck E.; Róg T.; Karttunen M.; Vattulainen I. Lateral Diffusion in Lipid Membranes through Collective Flows. J. Am. Chem. Soc. 2008, 130, 44–45. 10.1021/ja7103558. [DOI] [PubMed] [Google Scholar]
- Ayton G. S.; Voth G. A. Mesoscopic Lateral Diffusion in Lipid Bilayers. Biophys. J. 2004, 87, 3299–3311. 10.1529/biophysj.104.047811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch S.; Smuda C.; Pardo L. C.; Unruh T. Molecular Mechanism of Long-Range Diffusion in Phospholipid Membranes Studied by Quasielastic Neutron Scattering. J. Am. Chem. Soc. 2010, 132, 3232–3233. 10.1021/ja907581s. [DOI] [PubMed] [Google Scholar]
- Lautner L.; Pluhackova K.; Barth N. K. H.; Seydel T.; Lohstroh W.; Böckmann R. A.; Unruh T. Dynamic Processes in Biological Membrane Mimics Revealed by Quasielastic Neutron Scattering. Chem. Phys. Lipids 2017, 206, 28–42. 10.1016/j.chemphyslip.2017.05.009. [DOI] [PubMed] [Google Scholar]
- Armstrong C. L.; Trapp M.; Peters J.; Seydel T.; Rheinstädter M. C. Short Range Ballistic Motion in Fluid Lipid Bilayers Studied by Quasi-Elastic Neutron Scattering. Soft Matter 2011, 7, 8358–8362. 10.1039/c1sm05691c. [DOI] [Google Scholar]
- Chavent M.; Reddy T.; Goose J.; Dahl A. C. E.; Stone J. E.; Jobard B.; Sansom M. S. P. Methodologies for the Analysis of Instantaneous Lipid Diffusion in MD Simulations of Large Membrane Systems. Faraday Discuss. 2014, 169, 455–475. 10.1039/C3FD00145H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr F. W.; Hartmann B.; Douglas J. F. Dynamical Clustering and a Mechanism for Raft-like Structures in a Model Lipid Membrane. Soft Matter 2014, 10, 3036–3047. 10.1039/c3sm53187b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson K.; Mouritsen O. G.; Anderson R. G. W. Lipid Rafts: At a Crossroad between Cell Biology and Physics. Nat. Cell Biol. 2007, 9, 7–14. 10.1038/ncb0107-7. [DOI] [PubMed] [Google Scholar]
- Aponte-Santamaria C.; Briones R.; Schenk A. D.; Walz T.; de Groot B. L. Molecular Driving Forces Defining Lipid Positions around Aquaporin-0. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 9887–9892. 10.1073/pnas.1121054109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen M. H.; Turnbull D. Molecular Transport in Liquids and Glasses. J. Chem. Phys. 1959, 31, 1164–1169. 10.1063/1.1730566. [DOI] [Google Scholar]
- Almeida P. F. F.; Vaz W. L. C.; Thompson T. E. Lateral Diffusion in the Liquid Phases of Dimyristoylphosphatidylcholine/Cholesterol Lipid Bilayers: A Free Volume Analysis. Biochemistry 1992, 31, 6739–6747. 10.1021/bi00144a013. [DOI] [PubMed] [Google Scholar]
- Galla H. J.; Hartmann W.; Theilen U.; Sackmann E. On Two-Dimensional Passive Random Walk in Lipid Bilayers and Fluid Pathways in Biomembranes. J. Membr. Biol. 1979, 48, 215–236. 10.1007/BF01872892. [DOI] [PubMed] [Google Scholar]
- MacCarthy J. E.; Kozak J. J. Lateral Diffusion in Fluid Systems. J. Chem. Phys. 1982, 77, 2214–2216. 10.1063/1.444032. [DOI] [Google Scholar]
- O’Leary T. J. Lateral Diffusion of Lipids in Complex Biological Membranes. Proc. Natl. Acad. Sci. U. S. A. 1987, 84, 429–433. 10.1073/pnas.84.2.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaz W. L. C.; Clegg R. M.; Hallmann D. Translational Diffusion of Lipids in Liquid Crystalline Phase Phosphatidylcholine Multibilayers. A Comparison of Experiment with Theory. Biochemistry 1985, 24, 781–786. 10.1021/bi00324a037. [DOI] [PubMed] [Google Scholar]
- Macedo P. B.; Litovitz T. A. On the Relative Roles of Free Volume and Activation Energy in the Viscosity of Liquids. J. Chem. Phys. 1965, 42, 245–256. 10.1063/1.1695683. [DOI] [Google Scholar]
- Javanainen M.; Monticelli L.; de la Serna J. B.; Vattulainen I. Free Volume Theory Applied to Lateral Diffusion in Langmuir Monolayers: Atomistic Simulations for a Protein-Free Model of Lung Surfactant. Langmuir 2010, 26, 15436–15444. 10.1021/la102454m. [DOI] [PubMed] [Google Scholar]
- Arrhenius S. Über Die Reaktionsgeschwindigkeit Bei Der Inversion von Rohrzucker Durch Säuren. Z. Phys. Chem. 1889, 4U, 226–248. 10.1515/zpch-1889-0416. [DOI] [Google Scholar]
- Filippov A.; Orädd G.; Lindblom G. The Effect of Cholesterol on the Lateral Diffusion of Phospholipids in Oriented Bilayers. Biophys. J. 2003, 84, 3079–3086. 10.1016/S0006-3495(03)70033-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falck E.; Patra M.; Karttunen M.; Hyvönen M. T.; Vattulainen I. Lessons of Slicing Membranes: Interplay of Packing, Free Area, and Lateral Diffusion in Phospholipid/Cholesterol Bilayers. Biophys. J. 2004, 87, 1076–1091. 10.1529/biophysj.104.041368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saffman P. G.; Delbrück M. Brownian Motion in Biological Membranes. Proc. Natl. Acad. Sci. U. S. A. 1975, 72, 3111–3113. 10.1073/pnas.72.8.3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramadurai S.; Holt A.; Krasnikov V.; van den Bogaart G.; Killian J. A.; Poolman B. Lateral Diffusion of Membrane Proteins. J. Am. Chem. Soc. 2009, 131, 12650–12656. 10.1021/ja902853g. [DOI] [PubMed] [Google Scholar]
- Weiß K.; Neef A.; Van Q.; Kramer S.; Gregor I.; Enderlein J. Quantifying the Diffusion of Membrane Proteins and Peptides in Black Lipid Membranes with 2-Focus Fluorescence Correlation Spectroscopy. Biophys. J. 2013, 105, 455–462. 10.1016/j.bpj.2013.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oswald F.; Varadarajan A.; Lill H.; Peterman E. J. G.; Bollen Y. J. M. MreB-Dependent Organization of the E. Coli Cytoplasmic Membrane Controls Membrane Protein Diffusion. Biophys. J. 2016, 110, 1139–1149. 10.1016/j.bpj.2016.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goose J. E.; Sansom M. S. P. Reduced Lateral Mobility of Lipids and Proteins in Crowded Membranes. PLoS Comput. Biol. 2013, 9, e1003033. 10.1371/journal.pcbi.1003033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guigas G.; Weiss M. Size-Dependent Diffusion of Membrane Inclusions. Biophys. J. 2006, 91, 2393–2398. 10.1529/biophysj.106.087031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guigas G.; Weiss M. Influence of Hydrophobic Mismatching on Membrane Protein Diffusion. Biophys. J. 2008, 95, L25–L27. 10.1529/biophysj.108.136069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon J.-H.; Monne H. M.-S.; Javanainen M.; Metzler R. Anomalous Diffusion of Phospholipids and Cholesterols in a Lipid Bilayer and Its Origins. Phys. Rev. Lett. 2012, 109, 188103. 10.1103/PhysRevLett.109.188103. [DOI] [PubMed] [Google Scholar]
- Guigas G.; Weiss M. Effects of Protein Crowding on Membrane Systems. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 2441–2450. 10.1016/j.bbamem.2015.12.021. [DOI] [PubMed] [Google Scholar]
- Weiss M.Crowding, Diffusion, and Biochemical Reactions. In New Models of the Cell Nucleus: Crowding, Entropic Forces, Phase Separation, and Fractals; Hancock R., Jeon K. W., Eds.; International Review of Cell and Molecular Biology; Academic Press, 2014; Vol. 307, Chapter 11, pp 383–417, 10.1016/B978-0-12-800046-5.00011-4. [DOI] [PubMed] [Google Scholar]
- Jeon J.-H.; Javanainen M.; Martinez-Seara H.; Metzler R.; Vattulainen I. Protein Crowding in Lipid Bilayers Gives Rise to Non-Gaussian Anomalous Lateral Diffusion of Phospholipids and Proteins. Phys. Rev. X 2016, 6, 021006. 10.1103/PhysRevX.6.021006. [DOI] [Google Scholar]
- Peters R.; Cherry R. J. Lateral and Rotational Diffusion of Bacteriorhodopsin in Lipid Bilayers: Experimental Test of the Saffman-Delbrück Equations. Proc. Natl. Acad. Sci. U. S. A. 1982, 79, 4317–4321. 10.1073/pnas.79.14.4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casuso I.; Khao J.; Chami M.; Paul-Gilloteaux P.; Husain M.; Duneau J.-P.; Stahlberg H.; Sturgis J. N.; Scheuring S. Characterization of the Motion of Membrane Proteins Using High-Speed Atomic Force Microscopy. Nat. Nanotechnol. 2012, 7, 525–529. 10.1038/nnano.2012.109. [DOI] [PubMed] [Google Scholar]
- Munguira I.; Casuso I.; Takahashi H.; Rico F.; Miyagi A.; Chami M.; Scheuring S. Glasslike Membrane Protein Diffusion in a Crowded Membrane. ACS Nano 2016, 10, 2584–2590. 10.1021/acsnano.5b07595. [DOI] [PubMed] [Google Scholar]
- Koldsø H.; Reddy T.; Fowler P. W.; Duncan A. L.; Sansom M. S. P. Membrane Compartmentalization Reducing the Mobility of Lipids and Proteins within a Model Plasma Membrane. J. Phys. Chem. B 2016, 120, 8873–8881. 10.1021/acs.jpcb.6b05846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto E.; Akimoto T.; Kalli A. C.; Yasuoka K.; Sansom M. S. P. Dynamic Interactions between a Membrane Binding Protein and Lipids Induce Fluctuating Diffusivity. Sci. Adv. 2017, 3, e1601871. 10.1126/sciadv.1601871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto E.; Kalli A. C.; Akimoto T.; Yasuoka K.; Sansom M. S. P. Anomalous Dynamics of a Lipid Recognition Protein on a Membrane Surface. Sci. Rep. 2016, 5, 18245. 10.1038/srep18245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalay Z.; Fujiwara T. K.; Otaka A.; Kusumi A. Lateral Diffusion in a Discrete Fluid Membrane with Immobile Particles. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2014, 89, 022724. 10.1103/PhysRevE.89.022724. [DOI] [PubMed] [Google Scholar]
- Saxton M. J. Lateral Diffusion in an Archipelago. Single-Particle Diffusion. Biophys. J. 1993, 64, 1766–1780. 10.1016/S0006-3495(93)81548-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton M. J. The Spectrin Network as a Barrier to Lateral Diffusion in Erythrocytes. A Percolation Analysis. Biophys. J. 1989, 55, 21–28. 10.1016/S0006-3495(89)82776-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton M. J. Lateral Diffusion in an Archipelago. Distance Dependence of the Diffusion Coefficient. Biophys. J. 1989, 56, 615–622. 10.1016/S0006-3495(89)82708-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javanainen M.; Martinez-Seara H.; Vattulainen I. Excessive Aggregation of Membrane Proteins in the Martini Model. PLoS One 2017, 12, e0187936. 10.1371/journal.pone.0187936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camley B. A.; Lerner M. G.; Pastor R. W.; Brown F. L. H. Strong Influence of Periodic Boundary Conditions on Lateral Diffusion in Lipid Bilayer Membranes. J. Chem. Phys. 2015, 143, 243113. 10.1063/1.4932980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venable R. M.; Ingólfsson H. I.; Lerner M. G.; Perrin B. S.; Camley B. A.; Marrink S. J.; Brown F. L. H.; Pastor R. W. Lipid and Peptide Diffusion in Bilayers: The Saffman–Delbrück Model and Periodic Boundary Conditions. J. Phys. Chem. B 2017, 121, 3443–3457. 10.1021/acs.jpcb.6b09111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vögele M.; Hummer G. Divergent Diffusion Coefficients in Simulations of Fluids and Lipid Membranes. J. Phys. Chem. B 2016, 120, 8722–8732. 10.1021/acs.jpcb.6b05102. [DOI] [PubMed] [Google Scholar]
- Gurtovenko A. A.; Javanainen M.; Lolicato F.; Vattulainen I.. The Devil Is in the Details: What Do We Really Track in Single-Particle Tracking iSCAT Experiments? 2018, Manuscript in preparation. [DOI] [PubMed] [Google Scholar]
- Lehninger A. L.; Nelson D. L.; Cox M. M.. Lehninger Principles of Biochemistry, 6th ed.; W.H. Freeman: New York, 2013. [Google Scholar]
- Casciola M.; Tarek M. A Molecular Insight into the Electro-Transfer of Small Molecules through Electropores Driven by Electric Fields. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 2278–2289. 10.1016/j.bbamem.2016.03.022. [DOI] [PubMed] [Google Scholar]
- Kirsch S. A.; Böckmann R. A. Membrane Pore Formation in Atomistic and Coarse-Grained Simulations. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 2266–2277. 10.1016/j.bbamem.2015.12.031. [DOI] [PubMed] [Google Scholar]
- Awoonor-Williams E.; Rowley C. N. Molecular Simulation of Nonfacilitated Membrane Permeation. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 1672–1687. 10.1016/j.bbamem.2015.12.014. [DOI] [PubMed] [Google Scholar]
- Shinoda W. Permeability across Lipid Membranes. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 2254–2265. 10.1016/j.bbamem.2016.03.032. [DOI] [PubMed] [Google Scholar]
- Qiao B.; Olvera de la Cruz M. Driving Force for Water Permeation Across Lipid Membranes. J. Phys. Chem. Lett. 2013, 4, 3233–3237. 10.1021/jz401730s. [DOI] [Google Scholar]
- Conte E.; Megli F. M.; Khandelia H.; Jeschke G.; Bordignon E. Lipid Peroxidation and Water Penetration in Lipid Bilayers: A W-Band EPR Study. Biochim. Biophys. Acta, Biomembr. 2013, 1828, 510–517. 10.1016/j.bbamem.2012.09.026. [DOI] [PubMed] [Google Scholar]
- Wong-Ekkabut J.; Xu Z.; Triampo W.; Tang I. M.; Tieleman D. P.; Monticelli L. Effect of Lipid Peroxidation on the Properties of Lipid Bilayers: A Molecular Dynamics Study. Biophys. J. 2007, 93, 4225–4236. 10.1529/biophysj.107.112565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khandelia H.; Mouritsen O. G. Lipid Gymnastics: Evidence of Complete Acyl Chain Reversal in Oxidized Phospholipids from Molecular Simulations. Biophys. J. 2009, 96, 2734–2743. 10.1016/j.bpj.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lis M.; Wizert A.; Przybylo M.; Langner M.; Swiatek J.; Jungwirth P.; Cwiklik L. The Effect of Lipid Oxidation on the Water Permeability of Phospholipids Bilayers. Phys. Chem. Chem. Phys. 2011, 13, 17555. 10.1039/c1cp21009b. [DOI] [PubMed] [Google Scholar]
- Lee H.; Malmstadt N. Effect of Low Levels of Lipid Oxidation on the Curvature, Dynamics, and Permeability of Lipid Bilayers and Their Interactions with Cationic Nanoparticles. J. Phys. D: Appl. Phys. 2018, 51, 164002. 10.1088/1361-6463/aab4b8. [DOI] [Google Scholar]
- Su J.; Zhao Y.; Fang C.; Shi Y. Asymmetric Osmotic Water Permeation through a Vesicle Membrane. J. Chem. Phys. 2017, 146, 204902. 10.1063/1.4983749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito H.; Shinoda W. Cholesterol Effect on Water Permeability through DPPC and PSM Lipid Bilayers: A Molecular Dynamics Study. J. Phys. Chem. B 2011, 115, 15241–15250. 10.1021/jp201611p. [DOI] [PubMed] [Google Scholar]
- Mathai J. C.; Tristram-Nagle S.; Nagle J. F.; Zeidel M. L. Structural Determinants of Water Permeability through the Lipid Membrane. J. Gen. Physiol. 2008, 131, 69–76. 10.1085/jgp.200709848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issack B. B.; Peslherbe G. H. Effects of Cholesterol on the Thermodynamics and Kinetics of Passive Transport of Water through Lipid Membranes. J. Phys. Chem. B 2015, 119, 9391–9400. 10.1021/jp510497r. [DOI] [PubMed] [Google Scholar]
- Hartkamp R.; Moore T. C.; Iacovella C. R.; Thompson M. A.; Bulsara P. A.; Moore D. J.; McCabe C. Composition Dependence of Water Permeation Across Multicomponent Gel-Phase Bilayers. J. Phys. Chem. B 2018, 122, 3113–3123. 10.1021/acs.jpcb.8b00747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieg P.; Fürstenberger G. The Role of Lipoxygenases in Epidermis. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2014, 1841, 390–400. 10.1016/j.bbalip.2013.08.005. [DOI] [PubMed] [Google Scholar]
- Del Regno A.; Notman R. Permeation Pathways through Lateral Domains in Model Membranes of Skin Lipids. Phys. Chem. Chem. Phys. 2018, 20, 2162–2174. 10.1039/C7CP03258G. [DOI] [PubMed] [Google Scholar]
- Das C.; Noro M. G.; Olmsted P. D. Simulation Studies of Stratum Corneum Lipid Mixtures. Biophys. J. 2009, 97, 1941–1951. 10.1016/j.bpj.2009.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta R.; Dwadasi B. S.; Rai B. Molecular Dynamics Simulation of Skin Lipids: Effect of Ceramide Chain Lengths on Bilayer Properties. J. Phys. Chem. B 2016, 120, 12536–12546. 10.1021/acs.jpcb.6b08059. [DOI] [PubMed] [Google Scholar]
- Gupta R.; Sridhar D. B.; Rai B. Molecular Dynamics Simulation Study of Permeation of Molecules through Skin Lipid Bilayer. J. Phys. Chem. B 2016, 120, 8987–8996. 10.1021/acs.jpcb.6b05451. [DOI] [PubMed] [Google Scholar]
- Li L.; Vorobyov I.; Allen T. W. Potential of Mean Force and pKa Profile Calculation for a Lipid Membrane-Exposed Arginine Side Chain. J. Phys. Chem. B 2008, 112, 9574–9587. 10.1021/jp7114912. [DOI] [PubMed] [Google Scholar]
- MacCallum J. L.; Bennett W. F. D.; Tieleman D. P. Distribution of Amino Acids in a Lipid Bilayer from Computer Simulations. Biophys. J. 2008, 94, 3393–3404. 10.1529/biophysj.107.112805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L.; Vorobyov I.; Allen T. W. The Different Interactions of Lysine and Arginine Side Chains with Lipid Membranes. J. Phys. Chem. B 2013, 117, 11906–11920. 10.1021/jp405418y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panahi A.; Brooks C. L. Membrane Environment Modulates the pKa Values of Transmembrane Helices. J. Phys. Chem. B 2015, 119, 4601–4607. 10.1021/acs.jpcb.5b00289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson A. C. V.; Lindahl E. Titratable Amino Acid Solvation in Lipid Membranes as a Function of Protonation State. J. Phys. Chem. B 2009, 113, 245–253. 10.1021/jp8048873. [DOI] [PubMed] [Google Scholar]
- Bonhenry D.; Tarek M.; Dehez F. Effects of Phospholipid Composition on the Transfer of a Small Cationic Peptide Across a Model Biological Membrane. J. Chem. Theory Comput. 2013, 9, 5675–5684. 10.1021/ct400576e. [DOI] [PubMed] [Google Scholar]
- Gleason N. J.; Vostrikov V. V.; Greathouse D. V.; Koeppe R. E. Buried Lysine, but Not Arginine, Titrates and Alters Transmembrane Helix Tilt. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 1692–1695. 10.1073/pnas.1215400110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonhenry D.; Dehez F.; Tarek M. Effects of Hydration on the Protonation State of a Lysine Analog Crossing a Phospholipid Bilayer – Insights from Molecular Dynamics and Free-Energy Calculations. Phys. Chem. Chem. Phys. 2018, 20, 9101–9107. 10.1039/C8CP00312B. [DOI] [PubMed] [Google Scholar]
- Witek J.; Keller B. G.; Blatter M.; Meissner A.; Wagner T.; Riniker S. Kinetic Models of Cyclosporin A in Polar and Apolar Environments Reveal Multiple Congruent Conformational States. J. Chem. Inf. Model. 2016, 56, 1547–1562. 10.1021/acs.jcim.6b00251. [DOI] [PubMed] [Google Scholar]
- Witek J.; Mühlbauer M.; Keller B. G.; Blatter M.; Meissner A.; Wagner T.; Riniker S. Interconversion Rates between Conformational States as Rationale for the Membrane Permeability of Cyclosporines. ChemPhysChem 2017, 18, 3309–3314. 10.1002/cphc.201700995. [DOI] [PubMed] [Google Scholar]
- Wang C. K.; Swedberg J. E.; Harvey P. J.; Kaas Q.; Craik D. J. Conformational Flexibility Is a Determinant of Permeability for Cyclosporin. J. Phys. Chem. B 2018, 122, 2261–2276. 10.1021/acs.jpcb.7b12419. [DOI] [PubMed] [Google Scholar]
- Leontiadou H.; Mark A. E.; Marrink S. J. Antimicrobial Peptides in Action. J. Am. Chem. Soc. 2006, 128, 12156–12161. 10.1021/ja062927q. [DOI] [PubMed] [Google Scholar]
- Li J.; Liu S.; Lakshminarayanan R.; Bai Y.; Pervushin K.; Verma C.; Beuerman R. W. Molecular Simulations Suggest How a Branched Antimicrobial Peptide Perturbs a Bacterial Membrane and Enhances Permeability. Biochim. Biophys. Acta, Biomembr. 2013, 1828, 1112–1121. 10.1016/j.bbamem.2012.12.015. [DOI] [PubMed] [Google Scholar]
- Sengupta D.; Leontiadou H.; Mark A. E.; Marrink S.-J. Toroidal Pores Formed by Antimicrobial Peptides Show Significant Disorder. Biochim. Biophys. Acta, Biomembr. 2008, 1778, 2308–2317. 10.1016/j.bbamem.2008.06.007. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Chen C. H.; Hu D.; Ulmschneider M. B.; Ulmschneider J. P. Spontaneous Formation of Structurally Diverse Membrane Channel Architectures from a Single Antimicrobial Peptide. Nat. Commun. 2016, 7, 13535. 10.1038/ncomms13535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S.; Kar R. K.; Nanga R. P. R.; Mroue K. H.; Ramamoorthy A.; Bhunia A. Accelerated Molecular Dynamics Simulation Analysis of MSI-594 in a Lipid Bilayer. Phys. Chem. Chem. Phys. 2017, 19, 19289–19299. 10.1039/C7CP01941F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jedlovszky P.; Mezei M. Calculation of the Free Energy Profile of H2O, O2, CO, CO2, NO, and CHCl3 in a Lipid Bilayer with a Cavity Insertion Variant of the Widom Method. J. Am. Chem. Soc. 2000, 122, 5125–5131. 10.1021/ja000156z. [DOI] [Google Scholar]
- Pártay L. B.; Jedlovszky P.; Hoang P. N. M.; Picaud S.; Mezei M. Free-Energy Profile of Small Solute Molecules at the Free Surfaces of Water and Ice, as Determined by Cavity Insertion Widom Calculations. J. Phys. Chem. C 2007, 111, 9407–9416. 10.1021/jp0719816. [DOI] [Google Scholar]
- Yuan H.; Jameson C. J.; Murad S. Exploring Gas Permeability of Lipid Membranes Using Coarse-Grained Molecular Dynamics. Mol. Simul. 2009, 35, 953–961. 10.1080/08927020902763839. [DOI] [Google Scholar]
- Mayne C. G.; Arcario M. J.; Mahinthichaichan P.; Baylon J. L.; Vermaas J. V.; Navidpour L.; Wen P.-C.; Thangapandian S.; Tajkhorshid E. The Cellular Membrane as a Mediator for Small Molecule Interaction with Membrane Proteins. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 2290–2304. 10.1016/j.bbamem.2016.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dotson R. J.; Smith C. R.; Bueche K.; Angles G.; Pias S. C. Influence of Cholesterol on the Oxygen Permeability of Membranes: Insight from Atomistic Simulations. Biophys. J. 2017, 112, 2336–2347. 10.1016/j.bpj.2017.04.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boron W. F.; Endeward V.; Gros G.; Musa-Aziz R.; Pohl P. Intrinsic CO2 Permeability of Cell Membranes and Potential Biological Relevance of CO2 Channels. ChemPhysChem 2011, 12, 1017–1019. 10.1002/cphc.201100034. [DOI] [PubMed] [Google Scholar]
- Herrera M.; Garvin J. L. Aquaporins as Gas Channels. Pfluegers Arch. 2011, 462, 623–630. 10.1007/s00424-011-1002-x. [DOI] [PubMed] [Google Scholar]
- Endeward V.; Arias-Hidalgo M.; Al-Samir S.; Gros G. CO2 Permeability of Biological Membranes and Role of CO2 Channels. Membranes 2017, 7, 61. 10.3390/membranes7040061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H.; Jameson C. J.; Murad S. Diffusion of Gases across Lipid Membranes with OmpA Channel: A Molecular Dynamics Study. Mol. Phys. 2010, 108, 1569–1581. 10.1080/00268976.2010.484396. [DOI] [Google Scholar]
- Geyer R. R.; Musa-Aziz R.; Enkavi G.; Mahinthichaichan P.; Tajkhorshid E.; Boron W. F. Movement of NH3 through the Human Urea Transporter B: A New Gas Channel. Am. J. Physiol.-Ren. Physiol. 2013, 304, F1447–F1457. 10.1152/ajprenal.00609.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Tajkhorshid E. Nitric Oxide Conduction by the Brain Aquaporin AQP4. Proteins: Struct., Funct., Genet. 2009, 78, 661–670. 10.1002/prot.22595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Cohen J.; Boron W. F.; Schulten K.; Tajkhorshid E. Exploring Gas Permeability of Cellular Membranes and Membrane Channels with Molecular Dynamics. J. Struct. Biol. 2007, 157, 534–544. 10.1016/j.jsb.2006.11.008. [DOI] [PubMed] [Google Scholar]
- Hub J. S.; Winkler F. K.; Merrick M.; de Groot B. L. Potentials of Mean Force and Permeabilities for Carbon Dioxide, Ammonia, and Water Flux across a Rhesus Protein Channel and Lipid Membranes. J. Am. Chem. Soc. 2010, 132, 13251–13263. 10.1021/ja102133x. [DOI] [PubMed] [Google Scholar]
- Javanainen M.; Vattulainen I.; Monticelli L. On Atomistic Models for Molecular Oxygen. J. Phys. Chem. B 2017, 121, 518–528. 10.1021/acs.jpcb.6b11183. [DOI] [PubMed] [Google Scholar]
- Plesnar E.; Szczelina R.; Subczynski W. K.; Pasenkiewicz-Gierula M. Is the Cholesterol Bilayer Domain a Barrier to Oxygen Transport into the Eye Lens?. Biochim. Biophys. Acta, Biomembr. 2018, 1860, 434–441. 10.1016/j.bbamem.2017.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subczynski W. K.; Hyde J. S.; Kusumi A. Oxygen Permeability of Phosphatidylcholine—Cholesterol Membranes. Proc. Natl. Acad. Sci. U. S. A. 1989, 86, 4474–4478. 10.1073/pnas.86.12.4474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.; Ren X.; Meng F. Molecular Dynamics Simulation of Six β-Blocker Drugs Passing across POPC Bilayer. Mol. Simul. 2016, 42, 56–63. 10.1080/08927022.2015.1008470. [DOI] [Google Scholar]
- Ghaemi Z.; Alberga D.; Carloni P.; Laio A.; Lattanzi G. Permeability Coefficients of Lipophilic Compounds Estimated by Computer Simulations. J. Chem. Theory Comput. 2016, 12, 4093–4099. 10.1021/acs.jctc.5b01126. [DOI] [PubMed] [Google Scholar]
- Li J.; Beuerman R. W.; Verma C. S. Molecular Insights into the Membrane Affinities of Model Hydrophobes. ACS Omega 2018, 3, 2498–2507. 10.1021/acsomega.7b01759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes D.; Jakobtorweihen S.; Nunes C.; Sarmento B.; Reis S. Shedding Light on the Puzzle of Drug-Membrane Interactions: Experimental Techniques and Molecular Dynamics Simulations. Prog. Lipid Res. 2017, 65, 24–44. 10.1016/j.plipres.2016.12.001. [DOI] [PubMed] [Google Scholar]
- Yacoub T. J.; Reddy A. S.; Szleifer I. Structural Effects and Translocation of Doxorubicin in a DPPC/Chol Bilayer: The Role of Cholesterol. Biophys. J. 2011, 101, 378–385. 10.1016/j.bpj.2011.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khajeh A.; Modarress H. Effect of Cholesterol on Behavior of 5-Fluorouracil (5-FU) in a DMPC Lipid Bilayer, a Molecular Dynamics Study. Biophys. Chem. 2014, 187–188, 43–50. 10.1016/j.bpc.2014.01.004. [DOI] [PubMed] [Google Scholar]
- Khajeh A.; Modarress H. The Influence of Cholesterol on Interactions and Dynamics of Ibuprofen in a Lipid Bilayer. Biochim. Biophys. Acta, Biomembr. 2014, 1838, 2431–2438. 10.1016/j.bbamem.2014.05.029. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Bennett W. F. D.; Zheng T.; Ouyang P.-K.; Ouyang X.; Qiu X.; Luo A.; Karttunen M.; Chen P. Effect of Cholesterol on Cellular Uptake of Cancer Drugs Pirarubicin and Ellipticine. J. Phys. Chem. B 2016, 120, 3148–3156. 10.1021/acs.jpcb.5b12337. [DOI] [PubMed] [Google Scholar]
- Lee C. T.; Comer J.; Herndon C.; Leung N.; Pavlova A.; Swift R. V.; Tung C.; Rowley C. N.; Amaro R. E.; Chipot C.; et al. Simulation-Based Approaches for Determining Membrane Permeability of Small Compounds. J. Chem. Inf. Model. 2016, 56, 721–733. 10.1021/acs.jcim.6b00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaemi Z.; Minozzi M.; Carloni P.; Laio A. A Novel Approach to the Investigation of Passive Molecular Permeation through Lipid Bilayers from Atomistic Simulations. J. Phys. Chem. B 2012, 116, 8714–8721. 10.1021/jp301083h. [DOI] [PubMed] [Google Scholar]
- Comer J.; Schulten K.; Chipot C. Calculation of Lipid-Bilayer Permeabilities Using an Average Force. J. Chem. Theory Comput. 2014, 10, 554–564. 10.1021/ct400925s. [DOI] [PubMed] [Google Scholar]
- Nitschke N.; Atkovska K.; Hub J. S. Accelerating Potential of Mean Force Calculations for Lipid Membrane Permeation: System Size, Reaction Coordinate, Solute-Solute Distance, and Cutoffs. J. Chem. Phys. 2016, 145, 125101. 10.1063/1.4963192. [DOI] [PubMed] [Google Scholar]
- Votapka L. W.; Lee C. T.; Amaro R. E. Two Relations to Estimate Membrane Permeability Using Milestoning. J. Phys. Chem. B 2016, 120, 8606–8616. 10.1021/acs.jpcb.6b02814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson C. J.; Hornak V.; Pearlstein R. A.; Duca J. S. Structure–Kinetic Relationships of Passive Membrane Permeation from Multiscale Modeling. J. Am. Chem. Soc. 2017, 139, 442–452. 10.1021/jacs.6b11215. [DOI] [PubMed] [Google Scholar]
- Kopelevich D. I. One-Dimensional Potential of Mean Force Underestimates Activation Barrier for Transport across Flexible Lipid Membranes. J. Chem. Phys. 2013, 139, 134906. 10.1063/1.4823500. [DOI] [PubMed] [Google Scholar]
- Neale C.; Madill C.; Rauscher S.; Pomès R. Accelerating Convergence in Molecular Dynamics Simulations of Solutes in Lipid Membranes by Conducting a Random Walk along the Bilayer Normal. J. Chem. Theory Comput. 2013, 9, 3686–3703. 10.1021/ct301005b. [DOI] [PubMed] [Google Scholar]
- Filipe H. A. L.; Javanainen M.; Salvador A.; Galvão A. M.; Vattulainen I.; Loura L. M. S.; Moreno M. J. Quantitative Assessment of Methods Used To Obtain Rate Constants from Molecular Dynamics Simulations—Translocation of Cholesterol across Lipid Bilayers. J. Chem. Theory Comput. 2018, 14, 3840–3848. 10.1021/acs.jctc.8b00150. [DOI] [PubMed] [Google Scholar]
- LeVine M. V.; Cuendet M. A.; Khelashvili G.; Weinstein H. Allosteric Mechanisms of Molecular Machines at the Membrane: Transport by Sodium-Coupled Symporters. Chem. Rev. 2016, 116, 6552–6587. 10.1021/acs.chemrev.5b00627. [DOI] [PubMed] [Google Scholar]
- Denning E. J.; Beckstein O. Influence of Lipids on Protein-Mediated Transmembrane Transport. Chem. Phys. Lipids 2013, 169, 57–71. 10.1016/j.chemphyslip.2013.02.007. [DOI] [PubMed] [Google Scholar]
- Koshy C.; Ziegler C. Structural Insights into Functional Lipid–Protein Interactions in Secondary Transporters. Biochim. Biophys. Acta, Gen. Subj. 2015, 1850, 476–487. 10.1016/j.bbagen.2014.05.010. [DOI] [PubMed] [Google Scholar]
- Chavent M.; Duncan A. L.; Sansom M. S. Molecular Dynamics Simulations of Membrane Proteins and Their Interactions: From Nanoscale to Mesoscale. Curr. Opin. Struct. Biol. 2016, 40, 8–16. 10.1016/j.sbi.2016.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grouleff J.; Irudayam S. J.; Skeby K. K.; Schiøtt B. The Influence of Cholesterol on Membrane Protein Structure, Function, and Dynamics Studied by Molecular Dynamics Simulations. Biochim. Biophys. Acta, Biomembr. 2015, 1848, 1783–1795. 10.1016/j.bbamem.2015.03.029. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Shaikh S. A.; Tajkhorshid E. Exploring Transmembrane Diffusion Pathways With Molecular Dynamics. Physiology 2010, 25, 142–154. 10.1152/physiol.00046.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozu M.; Alvarez H. A.; McCarthy A. N.; Grigera J. R.; Chara O. Molecular Dynamics of Water in the Neighborhood of Aquaporins. Eur. Biophys. J. 2013, 42, 223–239. 10.1007/s00249-012-0880-y. [DOI] [PubMed] [Google Scholar]
- Hub J. S.; Grubmüller H.; de Groot B. L.. Dynamics and Energetics of Permeation Through Aquaporins. What Do We Learn from Molecular Dynamics Simulations? In Aquaporins; Beitz E., Hofmann F., Eds.; Springer Berlin Heidelberg: Berlin, Heidelberg, 2009; Vol. 190, pp 57–76, 10.1007/978-3-540-79885-9_3. [DOI] [PubMed] [Google Scholar]
- de Groot B. L.; Grubmüller H. The Dynamics and Energetics of Water Permeation and Proton Exclusion in Aquaporins. Curr. Opin. Struct. Biol. 2005, 15, 176–183. 10.1016/j.sbi.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Sachdeva R.; Singh B. Insights into Structural Mechanisms of Gating Induced Regulation of Aquaporins. Prog. Biophys. Mol. Biol. 2014, 114, 69–79. 10.1016/j.pbiomolbio.2014.01.002. [DOI] [PubMed] [Google Scholar]
- Mangiatordi G.; Alberga D.; Trisciuzzi D.; Lattanzi G.; Nicolotti O. Human Aquaporin-4 and Molecular Modeling: Historical Perspective and View to the Future. Int. J. Mol. Sci. 2016, 17, 1119. 10.3390/ijms17071119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosinska Eriksson U.; Fischer G.; Friemann R.; Enkavi G.; Tajkhorshid E.; Neutze R. Subangstrom Resolution X-Ray Structure Details Aquaporin-Water Interactions. Science 2013, 340, 1346–1349. 10.1126/science.1234306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajkhorshid E. Control of the Selectivity of the Aquaporin Water Channel Family by Global Orientational Tuning. Science 2002, 296, 525–530. 10.1126/science.1067778. [DOI] [PubMed] [Google Scholar]
- Jensen M. Ø.; Tajkhorshid E.; Schulten K. The Mechanism of Glycerol Conduction in Aquaglyceroporins. Structure 2001, 9, 1083–1093. 10.1016/S0969-2126(01)00668-2. [DOI] [PubMed] [Google Scholar]
- Savage D. F.; O’Connell J. D.; Miercke L. J. W.; Finer-Moore J.; Stroud R. M. Structural Context Shapes the Aquaporin Selectivity Filter. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 17164–17169. 10.1073/pnas.1009864107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliva R.; Calamita G.; Thornton J. M.; Pellegrini-Calace M. Electrostatics of Aquaporin and Aquaglyceroporin Channels Correlates with Their Transport Selectivity. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 4135–4140. 10.1073/pnas.0910632107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padhi S.; Priyakumar U. D. Microsecond Simulation of Human Aquaporin 2 Reveals Structural Determinants of Water Permeability and Selectivity. Biochim. Biophys. Acta, Biomembr. 2017, 1859, 10–16. 10.1016/j.bbamem.2016.10.011. [DOI] [PubMed] [Google Scholar]
- de Groot B. L.; Grubmüller H. Water Permeation Across Biological Membranes: Mechanism and Dynamics of Aquaporin-1 and GlpF. Science 2001, 294, 2353–2357. 10.1126/science.1062459. [DOI] [PubMed] [Google Scholar]
- Martínez-Ballesta M. d. C.; Carvajal M. Mutual Interactions between Aquaporins and Membrane Components. Front. Plant Sci. 2016, 7, 1322. 10.3389/fpls.2016.01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stansfeld P. J.; Jefferys E. E.; Sansom M. S. P. Multiscale Simulations Reveal Conserved Patterns of Lipid Interactions with Aquaporins. Structure 2013, 21, 810–819. 10.1016/j.str.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briones R.; Aponte-Santamaría C.; de Groot B. L. Localization and Ordering of Lipids Around Aquaporin-0: Protein and Lipid Mobility Effects. Front. Physiol. 2017, 8, 124. 10.3389/fphys.2017.00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haswell E. S.; Phillips R.; Rees D. C. Mechanosensitive Channels: What Can They Do and How Do They Do It?. Structure 2011, 19, 1356–1369. 10.1016/j.str.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon J.; Voth G. A. Gating of the Mechanosensitive Channel Protein MscL: The Interplay of Membrane and Protein. Biophys. J. 2008, 94, 3497–3511. 10.1529/biophysj.107.109850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer G. R.; Gullingsrud J.; Schulten K.; Martinac B. Molecular Dynamics Study of MscL Interactions with a Curved Lipid Bilayer. Biophys. J. 2006, 91, 1630–1637. 10.1529/biophysj.106.080721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gullingsrud J.; Kosztin D.; Schulten K. Structural Determinants of MscL Gating Studied by Molecular Dynamics Simulations. Biophys. J. 2001, 80, 2074–2081. 10.1016/S0006-3495(01)76181-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalili-Araghi F.; Gumbart J.; Wen P.-C.; Sotomayor M.; Tajkhorshid E.; Schulten K. Molecular Dynamics Simulations of Membrane Channels and Transporters. Curr. Opin. Struct. Biol. 2009, 19, 128–137. 10.1016/j.sbi.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gullingsrud J.; Schulten K. Lipid Bilayer Pressure Profiles and Mechanosensitive Channel Gating. Biophys. J. 2004, 86, 3496–3509. 10.1529/biophysj.103.034322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuli Ollila O. H.; Louhivuori M.; Marrink S. J.; Vattulainen I. Protein Shape Change Has a Major Effect on the Gating Energy of a Mechanosensitive Channel. Biophys. J. 2011, 100, 1651–1659. 10.1016/j.bpj.2011.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louhivuori M.; Risselada H. J.; van der Giessen E.; Marrink S. J. Release of Content through Mechano-Sensitive Gates in Pressurized Liposomes. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 19856–19860. 10.1073/pnas.1001316107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee N.; Jose M. D.; Birkner J. P.; Walko M.; Ingólfsson H. I.; Dimitrova A.; Arnarez C.; Marrink S. J.; Koçer A. The Activation Mode of the Mechanosensitive Ion Channel, MscL, by Lysophosphatidylcholine Differs from Tension-Induced Gating. FASEB J. 2014, 28, 4292–4302. 10.1096/fj.14-251579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmore D. E.; Dougherty D. A. Investigating Lipid Composition Effects on the Mechanosensitive Channel of Large Conductance (MscL) Using Molecular Dynamics Simulations. Biophys. J. 2003, 85, 1512–1524. 10.1016/S0006-3495(03)74584-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanegas J. M.; Arroyo M. Force Transduction and Lipid Binding in MscL: A Continuum-Molecular Approach. PLoS One 2014, 9, e113947. 10.1371/journal.pone.0113947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawada Y.; Murase M.; Sokabe M. The Gating Mechanism of the Bacterial Mechanosensitive Channel MscL Revealed by Molecular Dynamics Simulations: From Tension Sensing to Channel Opening. Channels 2012, 6, 317–331. 10.4161/chan.21895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bavi N.; Cortes D. M.; Cox C. D.; Rohde P. R.; Liu W.; Deitmer J. W.; Bavi O.; Strop P.; Hill A. P.; Rees D.; et al. The Role of MscL Amphipathic N Terminus Indicates a Blueprint for Bilayer-Mediated Gating of Mechanosensitive Channels. Nat. Commun. 2016, 7, 11984. 10.1038/ncomms11984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasquez V.; Sotomayor M.; Cordero-Morales J.; Schulten K.; Perozo E. A Structural Mechanism for MscS Gating in Lipid Bilayers. Science 2008, 321, 1210–1214. 10.1126/science.1159674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotomayor M.; Vásquez V.; Perozo E.; Schulten K. Ion Conduction through MscS as Determined by Electrophysiology and Simulation. Biophys. J. 2007, 92, 886–902. 10.1529/biophysj.106.095232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotomayor M.; Schulten K. Molecular Dynamics Study of Gating in the Mechanosensitive Channel of Small Conductance MscS. Biophys. J. 2004, 87, 3050–3065. 10.1529/biophysj.104.046045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotomayor M.; van der Straaten T. A.; Ravaioli U.; Schulten K. Electrostatic Properties of the Mechanosensitive Channel of Small Conductance MscS. Biophys. J. 2006, 90, 3496–3510. 10.1529/biophysj.105.080069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malcolm H. R.; Heo Y.-Y.; Elmore D. E.; Maurer J. A. Defining the Role of the Tension Sensor in the Mechanosensitive Channel of Small Conductance. Biophys. J. 2011, 101, 345–352. 10.1016/j.bpj.2011.05.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tombola F.; Pathak M. M.; Isacoff E. Y. How Does Voltage Open an Ion Channel?. Annu. Rev. Cell Dev. Biol. 2006, 22, 23–52. 10.1146/annurev.cellbio.21.020404.145837. [DOI] [PubMed] [Google Scholar]
- Yu F. H.; Catterall W. A. The VGL-Chanome: A Protein Superfamily Specialized for Electrical Signaling and Ionic Homeostasis. Sci. Signaling 2004, 2004, re15–re15. 10.1126/stke.2532004re15. [DOI] [PubMed] [Google Scholar]
- Chen L.; Zhang Q.; Qiu Y.; Li Z.; Chen Z.; Jiang H.; Li Y.; Yang H. Migration of PIP2 Lipids on Voltage-Gated Potassium Channel Surface Influences Channel Deactivation. Sci. Rep. 2015, 5, 15079. 10.1038/srep15079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazdi S.; Stein M.; Elinder F.; Andersson M.; Lindahl E. The Molecular Basis of Polyunsaturated Fatty Acid Interactions with the Shaker Voltage-Gated Potassium Channel. PLoS Comput. Biol. 2016, 12, e1004704. 10.1371/journal.pcbi.1004704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottosson N. E.; Silverå Ejneby M.; Wu X.; Yazdi S.; Konradsson P.; Lindahl E.; Elinder F. A Drug Pocket at the Lipid Bilayer–Potassium Channel Interface. Sci. Adv. 2017, 3, e1701099. 10.1126/sciadv.1701099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKinnon R. Potassium Channels. FEBS Lett. 2003, 555, 62–65. 10.1016/S0014-5793(03)01104-9. [DOI] [PubMed] [Google Scholar]
- MacKinnon R.; Cohen S. L.; Kuo A.; Lee A.; Chait B. T. Structural Conservation in Prokaryotic and Eukaryotic Potassium Channels. Science 1998, 280, 106–109. 10.1126/science.280.5360.106. [DOI] [PubMed] [Google Scholar]
- Doyle D. A.; Cabral J. M.; Pfuetzner R. A.; Kuo A.; Gulbis J. M.; Cohen S. L.; Chait B. T.; MacKinnon R. The Structure of the Potassium Channel: Molecular Basis of K+ Conduction and Selectivity. Science 1998, 280, 69–77. 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- Bucher D.; Rothlisberger U. Molecular Simulations of Ion Channels: A Quantum Chemist’s Perspective. J. Gen. Physiol. 2010, 135, 549–554. 10.1085/jgp.201010404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musgaard M.; Paramo T.; Domicevica L.; Andersen O. J.; Biggin P. C. Insights into Channel Dysfunction from Modelling and Molecular Dynamics Simulations. Neuropharmacology 2018, 132, 20–30. 10.1016/j.neuropharm.2017.06.030. [DOI] [PubMed] [Google Scholar]
- de Groot B. L.; Koepfer D.; Song C.; Gruene T.; Sheldrick G. M.; Zachariae U. The Molecular Dynamics of Ion Channel Permeation, Selectivity and Gating. Biophys. J. 2016, 110, 9a. 10.1016/j.bpj.2015.11.101. [DOI] [Google Scholar]
- Delemotte L.; Klein M.; TAREK M. Molecular Dynamics Simulations of Voltage-Gated Cation Channels: Insights on Voltage-Sensor Domain Function and Modulation. Front. Pharmacol. 2012, 3, 97. 10.3389/fphar.2012.00097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hénin J.; Salari R.; Murlidaran S.; Brannigan G. A Predicted Binding Site for Cholesterol on the GABAA Receptor. Biophys. J. 2014, 106, 1938–1949. 10.1016/j.bpj.2014.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jardetzky O. Simple Allosteric Model for Membrane Pumps. Nature 1966, 211, 969–970. 10.1038/211969a0. [DOI] [PubMed] [Google Scholar]
- Drew D.; Boudker O. Shared Molecular Mechanisms of Membrane Transporters. Annu. Rev. Biochem. 2016, 85, 543–572. 10.1146/annurev-biochem-060815-014520. [DOI] [PubMed] [Google Scholar]
- Ryan R. M.; Vandenberg R. J. Elevating the Alternating-Access Model. Nat. Struct. Mol. Biol. 2016, 23, 187–189. 10.1038/nsmb.3179. [DOI] [PubMed] [Google Scholar]
- Li J.; Shaikh S. A.; Enkavi G.; Wen P.-C.; Huang Z.; Tajkhorshid E. Transient Formation of Water-Conducting States in Membrane Transporters. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 7696–7701. 10.1073/pnas.1218986110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eicher T.; Seeger M. A.; Anselmi C.; Zhou W.; Brandstätter L.; Verrey F.; Diederichs K.; Faraldo-Gómez J. D.; Pos K. M. Coupling of Remote Alternating-Access Transport Mechanisms for Protons and Substrates in the Multidrug Efflux Pump AcrB. eLife 2014, 3, e03145. 10.7554/eLife.03145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasseville L. J.; Cuervo J. E.; Lapointe J.-Y.; Noskov S. Y. The Structural Pathway for Water Permeation through Sodium-Glucose Cotransporters. Biophys. J. 2011, 101, 1887–1895. 10.1016/j.bpj.2011.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe S.; Rosenberg J. M.; Abramson J.; Wright E. M.; Grabe M. Water Permeation through the Sodium-Dependent Galactose Cotransporter VSGLT. Biophys. J. 2010, 99, L56–L58. 10.1016/j.bpj.2010.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGreevy R.; Singharoy A.; Li Q.; Zhang J.; Xu D.; Perozo E.; Schulten K. XMDFF: Molecular Dynamics Flexible Fitting of Low-Resolution X-Ray Structures. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2014, 70, 2344–2355. 10.1107/S1399004714013856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trabuco L. G.; Villa E.; Mitra K.; Frank J.; Schulten K. Flexible Fitting of Atomic Structures into Electron Microscopy Maps Using Molecular Dynamics. Structure 2008, 16, 673–683. 10.1016/j.str.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermaas J. V.; Trebesch N.; Mayne C. G.; Thangapandian S.; Shekhar M.; Mahinthichaichan P.; Baylon J. L.; Jiang T.; Wang Y.; Muller M. P.; et al. Microscopic Characterization of Membrane Transporter Function by In Silico Modeling and Simulation. Methods in Enzymology; Elsevier, 2016; Vol. 578, pp 373–428, 10.1016/bs.mie.2016.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coudray N.; Seyler S. L.; Lasala R.; Zhang Z.; Clark K. M.; Dumont M. E.; Rohou A.; Beckstein O.; Stokes D. L. Structure of the SLC4 Transporter Bor1p in an Inward-Facing Conformation: Structure of the SLC4 Transporter Bor1p. Protein Sci. 2017, 26, 130–145. 10.1002/pro.3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coudray N.; Valvo S.; Hu M.; Lasala R.; Kim C.; Vink M.; Zhou M.; Provasi D.; Filizola M.; Tao J.; et al. Inward-Facing Conformation of the Zinc Transporter YiiP Revealed by Cryoelectron Microscopy. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 2140–2145. 10.1073/pnas.1215455110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozzi A. T.; Bane L. B.; Weihofen W. A.; Singharoy A.; Guillen E. R.; Ploegh H. L.; Schulten K.; Gaudet R. Crystal Structure and Conformational Change Mechanism of a Bacterial Nramp-Family Divalent Metal Transporter. Structure 2016, 24, 2102–2114. 10.1016/j.str.2016.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López C. A.; Travers T.; Pos K. M.; Zgurskaya H. I.; Gnanakaran S. Dynamics of Intact MexAB-OprM Efflux Pump: Focusing on the MexA-OprM Interface. Sci. Rep. 2017, 7, 16521. 10.1038/s41598-017-16497-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diskowski M.; Mehdipour A. R.; Wunnicke D.; Mills D. J.; Mikusevic V.; Bärland N.; Hoffmann J.; Morgner N.; Steinhoff H.-J.; Hummer G.; et al. Helical Jackknives Control the Gates of the Double-Pore K+ Uptake System KtrAB. eLife 2017, 6, e24303. 10.7554/eLife.24303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C.; Noskov S. Y. The Molecular Mechanism of Ion-Dependent Gating in Secondary Transporters. PLoS Comput. Biol. 2013, 9, e1003296. 10.1371/journal.pcbi.1003296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas J. R.; Gedeon P. C.; Grant B. J.; Madura J. D. LeuT Conformational Sampling Utilizing Accelerated Molecular Dynamics and Principal Component Analysis. Biophys. J. 2012, 103, L1–L3. 10.1016/j.bpj.2012.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moradi M.; Enkavi G.; Tajkhorshid E. Atomic-Level Characterization of Transport Cycle Thermodynamics in the Glycerol-3-Phosphate:Phosphate Antiporter. Nat. Commun. 2015, 6, 8393. 10.1038/ncomms9393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori T.; Miyashita N.; Im W.; Feig M.; Sugita Y. Molecular Dynamics Simulations of Biological Membranes and Membrane Proteins Using Enhanced Conformational Sampling Algorithms. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 1635–1651. 10.1016/j.bbamem.2015.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harpole T. J.; Delemotte L. Conformational Landscapes of Membrane Proteins Delineated by Enhanced Sampling Molecular Dynamics Simulations. Biochim. Biophys. Acta, Biomembr. 2018, 1860, 909–926. 10.1016/j.bbamem.2017.10.033. [DOI] [PubMed] [Google Scholar]
- Latorraca N. R.; Fastman N. M.; Venkatakrishnan A. J.; Frommer W. B.; Dror R. O.; Feng L. Mechanism of Substrate Translocation in an Alternating Access Transporter. Cell 2017, 169, 96–107.e12. 10.1016/j.cell.2017.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stansfeld P. J.; Sansom M. S. P. Molecular Simulation Approaches to Membrane Proteins. Structure 2011, 19, 1562–1572. 10.1016/j.str.2011.10.002. [DOI] [PubMed] [Google Scholar]
- Shaikh S. A.; Li J.; Enkavi G.; Wen P.-C.; Huang Z.; Tajkhorshid E. Visualizing Functional Motions of Membrane Transporters with Molecular Dynamics Simulations. Biochemistry 2013, 52, 569–587. 10.1021/bi301086x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Autzen H. E.; Siuda I.; Sonntag Y.; Nissen P.; Møller J. V.; Thøgersen L. Regulation of the Ca2+-ATPase by Cholesterol: A Specific or Non-Specific Effect?. Mol. Membr. Biol. 2015, 32, 75–87. 10.3109/09687688.2015.1073382. [DOI] [PubMed] [Google Scholar]
- Manepalli S.; Surratt C. K.; Madura J. D.; Nolan T. L. Monoamine Transporter Structure, Function, Dynamics, and Drug Discovery: A Computational Perspective. AAPS J. 2012, 14, 820–831. 10.1208/s12248-012-9391-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loland C. J. The Use of LeuT as a Model in Elucidating Binding Sites for Substrates and Inhibitors in Neurotransmitter Transporters. Biochim. Biophys. Acta, Gen. Subj. 2015, 1850, 500–510. 10.1016/j.bbagen.2014.04.011. [DOI] [PubMed] [Google Scholar]
- Shi Y. Common Folds and Transport Mechanisms of Secondary Active Transporters. Annu. Rev. Biophys. 2013, 42, 51–72. 10.1146/annurev-biophys-083012-130429. [DOI] [PubMed] [Google Scholar]
- Sohail A.; Jayaraman K.; Venkatesan S.; Gotfryd K.; Daerr M.; Gether U.; Loland C. J.; Wanner K. T.; Freissmuth M.; Sitte H. H.; et al. The Environment Shapes the Inner Vestibule of LeuT. PLoS Comput. Biol. 2016, 12, e1005197. 10.1371/journal.pcbi.1005197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondal S.; Khelashvili G.; Shi L.; Weinstein H. The Cost of Living in the Membrane: A Case Study of Hydrophobic Mismatch for the Multi-Segment Protein LeuT. Chem. Phys. Lipids 2013, 169, 27–38. 10.1016/j.chemphyslip.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondal S.; Khelashvili G.; Weinstein H. Not Just an Oil Slick: How the Energetics of Protein-Membrane Interactions Impacts the Function and Organization of Transmembrane Proteins. Biophys. J. 2014, 106, 2305–2316. 10.1016/j.bpj.2014.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gur M.; Zomot E.; Cheng M. H.; Bahar I. Energy Landscape of LeuT from Molecular Simulations. J. Chem. Phys. 2015, 143, 243134. 10.1063/1.4936133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng M. H.; Bahar I. Complete Mapping of Substrate Translocation Highlights the Role of LeuT N-Terminal Segment in Regulating Transport Cycle. PLoS Comput. Biol. 2014, 10, e1003879. 10.1371/journal.pcbi.1003879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L.; Quick M.; Zhao Y.; Weinstein H.; Javitch J. A. The Mechanism of a Neurotransmitter:Sodium Symporter—Inward Release of Na+ and Substrate Is Triggered by Substrate in a Second Binding Site. Mol. Cell 2008, 30, 667–677. 10.1016/j.molcel.2008.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeppelin T.; Ladefoged L. K.; Sinning S.; Periole X.; Schiøtt B. A Direct Interaction of Cholesterol with the Dopamine Transporter Prevents Its Out-to-Inward Transition. PLoS Comput. Biol. 2018, 14, e1005907. 10.1371/journal.pcbi.1005907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penmatsa A.; Wang K. H.; Gouaux E. X-Ray Structure of Dopamine Transporter Elucidates Antidepressant Mechanism. Nature 2013, 503, 85–90. 10.1038/nature12533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K. H.; Penmatsa A.; Gouaux E. Neurotransmitter and Psychostimulant Recognition by the Dopamine Transporter. Nature 2015, 521, 322–327. 10.1038/nature14431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman J. A.; Green E. M.; Gouaux E. X-Ray Structures and Mechanism of the Human Serotonin Transporter. Nature 2016, 532, 334–339. 10.1038/nature17629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraro M.; Masetti M.; Recanatini M.; Cavalli A.; Bottegoni G. Mapping Cholesterol Interaction Sites on Serotonin Transporter through Coarse-Grained Molecular Dynamics. PLoS One 2016, 11, e0166196. 10.1371/journal.pone.0166196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laursen L.; Severinsen K.; Kristensen K. B.; Periole X.; Overby M.; Müller H. K.; Schiøtt B.; Sinning S. Cholesterol Binding to a Conserved Site Modulates the Conformation, Pharmacology, and Transport Kinetics of the Human Serotonin Transporter. J. Biol. Chem. 2018, 293, 3510–3523. 10.1074/jbc.M117.809046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khelashvili G.; Doktorova M.; Sahai M. A.; Johner N.; Shi L.; Weinstein H. Computational Modeling of the N-Terminus of the Human Dopamine Transporter and Its Interaction with PIP2-Containing Membranes: Modeling of DAT N-Terminus/PIP2 Interactions. Proteins: Struct., Funct., Genet. 2015, 83, 952–969. 10.1002/prot.24792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khelashvili G.; Stanley N.; Sahai M. A.; Medina J.; LeVine M. V.; Shi L.; De Fabritiis G.; Weinstein H. Spontaneous Inward Opening of the Dopamine Transporter Is Triggered by PIP2-Regulated Dynamics of the N-Terminus. ACS Chem. Neurosci. 2015, 6, 1825–1837. 10.1021/acschemneuro.5b00179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng M. H.; Garcia-Olivares J.; Wasserman S.; DiPietro J.; Bahar I. Allosteric Modulation of Human Dopamine Transporter Activity under Conditions Promoting Its Dimerization. J. Biol. Chem. 2017, 292, 12471–12482. 10.1074/jbc.M116.763565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Periole X.; Zeppelin T.; Schiøtt B. Dimer Interface of the Human Serotonin Transporter and Effect of the Membrane Composition. Sci. Rep. 2018, 8, 5080. 10.1038/s41598-018-22912-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderluh A.; Hofmaier T.; Klotzsch E.; Kudlacek O.; Stockner T.; Sitte H. H.; Schütz G. J. Direct PIP2 Binding Mediates Stable Oligomer Formation of the Serotonin Transporter. Nat. Commun. 2017, 8, 14089. 10.1038/ncomms14089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta K.; Donlan J. A. C.; Hopper J. T. S.; Uzdavinys P.; Landreh M.; Struwe W. B.; Drew D.; Baldwin A. J.; Stansfeld P. J.; Robinson C. V. The Role of Interfacial Lipids in Stabilizing Membrane Protein Oligomers. Nature 2017, 541, 421–424. 10.1038/nature20820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gur M.; Cheng M. H.; Zomot E.; Bahar I. Effect of Dimerization on the Dynamics of Neurotransmitter:Sodium Symporters. J. Phys. Chem. B 2017, 121, 3657–3666. 10.1021/acs.jpcb.6b09876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X.; Kim J.-K.; Kim Y.; Bowie J. U.; Im W. Molecular Dynamics Simulation Strategies for Protein–Micelle Complexes. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 1566–1572. 10.1016/j.bbamem.2015.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khelashvili G.; LeVine M. V.; Shi L.; Quick M.; Javitch J. A.; Weinstein H. The Membrane Protein LeuT in Micellar Systems: Aggregation Dynamics and Detergent Binding to the S2 Site. J. Am. Chem. Soc. 2013, 135, 14266–14275. 10.1021/ja405984v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeVine M. V.; Khelashvili G.; Shi L.; Quick M.; Javitch J. A.; Weinstein H. Role of Annular Lipids in the Functional Properties of Leucine Transporter LeuT Proteomicelles. Biochemistry 2016, 55, 850–859. 10.1021/acs.biochem.5b01268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adhikary S.; Deredge D. J.; Nagarajan A.; Forrest L. R.; Wintrode P. L.; Singh S. K. Conformational Dynamics of a Neurotransmitter:Sodium Symporter in a Lipid Bilayer. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, E1786–E1795. 10.1073/pnas.1613293114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lezon T. R.; Bahar I. Constraints Imposed by the Membrane Selectively Guide the Alternating Access Dynamics of the Glutamate Transporter GltPh. Biophys. J. 2012, 102, 1331–1340. 10.1016/j.bpj.2012.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akyuz N.; Georgieva E. R.; Zhou Z.; Stolzenberg S.; Cuendet M. A.; Khelashvili G.; Altman R. B.; Terry D. S.; Freed J. H.; Weinstein H.; et al. Transport Domain Unlocking Sets the Uptake Rate of an Aspartate Transporter. Nature 2015, 518, 68–73. 10.1038/nature14158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montigny C.; Lyons J.; Champeil P.; Nissen P.; Lenoir G. On the Molecular Mechanism of Flippase- and Scramblase-Mediated Phospholipid Transport. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2016, 1861, 767–783. 10.1016/j.bbalip.2015.12.020. [DOI] [PubMed] [Google Scholar]
- Pomorski T.; Menon A. K. Lipid Flippases and Their Biological Functions. Cell. Mol. Life Sci. 2006, 63, 2908–2921. 10.1007/s00018-006-6167-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann J.; Rose-Sperling D.; Hellmich U. A. Diverse Relations between ABC Transporters and Lipids: An Overview. Biochim. Biophys. Acta, Biomembr. 2017, 1859, 605–618. 10.1016/j.bbamem.2016.09.023. [DOI] [PubMed] [Google Scholar]
- Furuta T.; Yamaguchi T.; Kato H.; Sakurai M. Analysis of the Structural and Functional Roles of Coupling Helices in the ATP-Binding Cassette Transporter MsbA through Enzyme Assays and Molecular Dynamics Simulations. Biochemistry 2014, 53, 4261–4272. 10.1021/bi500255j. [DOI] [PubMed] [Google Scholar]
- Moradi M.; Tajkhorshid E. Mechanistic Picture for Conformational Transition of a Membrane Transporter at Atomic Resolution. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 18916–18921. 10.1073/pnas.1313202110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haubertin D. Y.; Madaoui H.; Sanson A.; Guérois R.; Orlowski S. Molecular Dynamics Simulations of E. Coli MsbA Transmembrane Domain: Formation of a Semipore Structure. Biophys. J. 2006, 91, 2517–2531. 10.1529/biophysj.106.084020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward A. B.; Guvench O.; Hills R. D. Coarse Grain Lipid-Protein Molecular Interactions and Diffusion with MsbA Flippase. Proteins: Struct., Funct., Genet. 2012, 80, 2178–2190. 10.1002/prot.24108. [DOI] [PubMed] [Google Scholar]
- Bechara C.; Nöll A.; Morgner N.; Degiacomi M. T.; Tampé R.; Robinson C. V. A Subset of Annular Lipids Is Linked to the Flippase Activity of an ABC Transporter. Nat. Chem. 2015, 7, 255–262. 10.1038/nchem.2172. [DOI] [PubMed] [Google Scholar]
- Sharom F. Complex Interplay between the P-Glycoprotein Multidrug Efflux Pump and the Membrane: Its Role in Modulating Protein Function. Front. Oncol. 2014, 4, 41. 10.3389/fonc.2014.00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domicevica L.; Koldsø H.; Biggin P. C. Multiscale Molecular Dynamics Simulations of Lipid Interactions with P-Glycoprotein in a Complex Membrane. J. Mol. Graphics Modell. 2018, 80, 147–156. 10.1016/j.jmgm.2017.12.022. [DOI] [PubMed] [Google Scholar]
- Barreto-Ojeda E.; Corradi V.; Gu R.-X.; Tieleman D. P. Coarse-Grained Molecular Dynamics Simulations Reveal Lipid Access Pathways in P-Glycoprotein. J. Gen. Physiol. 2018, 150, 417–429. 10.1085/jgp.201711907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morra G.; Razavi A. M.; Pandey K.; Weinstein H.; Menon A. K.; Khelashvili G. Mechanisms of Lipid Scrambling by the G Protein-Coupled Receptor Opsin. Structure 2018, 26, 356–367.e3. 10.1016/j.str.2017.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieminen T. A Study of Rhodopsin as a Potential Phospholipid Scramblase. Master’s thesis; Tampere University of Technology: Tampere, Finland, 2012. [Google Scholar]
- Manna M.; Nieminen T.; Vattulainen I.. Rhodopsin as a Potential Phospholipid Scramblase. 2019, Manuscript in preparation. [Google Scholar]
- Verchère A.; Ou W.-L.; Ploier B.; Morizumi T.; Goren M. A.; Bütikofer P.; Ernst O. P.; Khelashvili G.; Menon A. K. Light-Independent Phospholipid Scramblase Activity of Bacteriorhodopsin from Halobacterium Salinarum. Sci. Rep. 2017, 7, 9522. 10.1038/s41598-017-09835-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapay N.; Bennett W. F. D.; Tieleman D. P. Molecular Simulations of Lipid Flip-Flop in the Presence of Model Transmembrane Helices. Biochemistry 2010, 49, 7665–7673. 10.1021/bi100878q. [DOI] [PubMed] [Google Scholar]
- Wong L. H.; Čopič A.; Levine T. P. Advances on the Transfer of Lipids by Lipid Transfer Proteins. Trends Biochem. Sci. 2017, 42, 516–530. 10.1016/j.tibs.2017.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabon A.; Orłowski A.; Tripathi A.; Vuorio J.; Javanainen M.; Róg T.; Lönnfors M.; McDermott M. I.; Siebert G.; Somerharju P.; et al. Dynamics and Energetics of the Mammalian Phosphatidylinositol Transfer Protein Phospholipid Exchange Cycle. J. Biol. Chem. 2017, 292, 14438–14455. 10.1074/jbc.M117.791467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enkavi G.; Mikkolainen H.; Güngör B.; Ikonen E.; Vattulainen I. Concerted Regulation of NPC2 Binding to Endosomal/Lysosomal Membranes by Bis(Monoacylglycero)Phosphate and Sphingomyelin. PLoS Comput. Biol. 2017, 13, e1005831. 10.1371/journal.pcbi.1005831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian K.; Balch W. E. NPC1/NPC2 Function as a Tag Team Duo to Mobilize Cholesterol. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 15223–15224. 10.1073/pnas.0808256105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estiu G.; Khatri N.; Wiest O. Computational Studies of the Cholesterol Transport between NPC2 and the N-Terminal Domain of NPC1 (NPC1(NTD)). Biochemistry 2013, 52, 6879–6891. 10.1021/bi4005478. [DOI] [PubMed] [Google Scholar]
- Gong X.; Qian H.; Zhou X.; Wu J.; Wan T.; Cao P.; Huang W.; Zhao X.; Wang X.; Wang P.; et al. Structural Insights into the Niemann-Pick C1 (NPC1)-Mediated Cholesterol Transfer and Ebola Infection. Cell 2016, 165, 1467–1478. 10.1016/j.cell.2016.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Lu F.; Trinh M. N.; Schmiege P.; Seemann J.; Wang J.; Blobel G. 3.3 Å Structure of Niemann–Pick C1 Protein Reveals Insights into the Function of the C-Terminal Luminal Domain in Cholesterol Transport. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 9116–9121. 10.1073/pnas.1711716114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Saha P.; Li J.; Blobel G.; Pfeffer S. R. Clues to the Mechanism of Cholesterol Transfer from the Structure of NPC1Middle Lumenal Domain Bound to NPC2. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 10079–10084. 10.1073/pnas.1611956113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper G. M.; Hausman R. E. The Cell: A Molecular Approach. Yale J. Biol. Med. 2014, 87, 603–604. [Google Scholar]
- Zhang X.; Rebane A. A.; Ma L.; Li F.; Jiao J.; Qu H.; Pincet F.; Rothman J. E.; Zhang Y. Stability, Folding Dynamics, and Long-Range Conformational Transition of the Synaptic t-SNARE Complex. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, E8031–E8040. 10.1073/pnas.1605748113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernomordik L. V.; Zimmerberg J.; Kozlov M. M. Membranes of the World Unite!. J. Cell Biol. 2006, 175, 201–207. 10.1083/jcb.200607083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risselada H. J.; Grubmüller H. How SNARE Molecules Mediate Membrane Fusion: Recent Insights from Molecular Simulations. Curr. Opin. Struct. Biol. 2012, 22, 187–196. 10.1016/j.sbi.2012.01.007. [DOI] [PubMed] [Google Scholar]
- Gardner J. M.; Abrams C. F. Lipid Flip-Flop vs. Lateral Diffusion in the Relaxation of Hemifusion Diaphragms. Biochim. Biophys. Acta, Biomembr. 2018, 1860, 1452–1459. 10.1016/j.bbamem.2018.04.007. [DOI] [PubMed] [Google Scholar]
- Cooke I. R.; Kremer K.; Deserno M. Tunable Generic Model for Fluid Bilayer Membranes. Phys. Rev. E 2005, 72, 011506. 10.1103/PhysRevE.72.011506. [DOI] [PubMed] [Google Scholar]
- Risselada H. J.; Smirnova Y.; Grubmüller H. Free Energy Landscape of Rim-Pore Expansion in Membrane Fusion. Biophys. J. 2014, 107, 2287–2295. 10.1016/j.bpj.2014.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo J.; Jackson M. B.; Cui Q. A Comparison of Coarse-Grained and Continuum Models for Membrane Bending in Lipid Bilayer Fusion Pores. Biophys. J. 2013, 104, 841–852. 10.1016/j.bpj.2012.12.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao H.; Goldschen-Ohm M.; Jeggle P.; Chanda B.; Edwardson J. M.; Chapman E. R. Exocytotic Fusion Pores Are Composed of Both Lipids and Proteins. Nat. Struct. Mol. Biol. 2016, 23, 67–73. 10.1038/nsmb.3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S.; Lindau M. The Mystery of the Fusion Pore. Nat. Struct. Mol. Biol. 2016, 23, 5–6. 10.1038/nsmb.3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortoul N.; Singh P.; Hui C.-Y.; Bykhovskaia M.; Jagota A. Coarse-Grained Model of SNARE-Mediated Docking. Biophys. J. 2015, 108, 2258–2269. 10.1016/j.bpj.2015.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W. All-Atom and Coarse-Grained Simulations of the Forced Unfolding Pathways of the SNARE Complex: Forced Unfolding Simulations of SNARE. Proteins: Struct., Funct., Genet. 2014, 82, 1376–1386. 10.1002/prot.24505. [DOI] [PubMed] [Google Scholar]
- Go N. Theoretical Studies of Protein Folding. Annu. Rev. Biophys. Bioeng. 1983, 12, 183–210. 10.1146/annurev.bb.12.060183.001151. [DOI] [PubMed] [Google Scholar]
- Tekpinar M.; Zheng W. Unzipping of Neuronal Snare Protein with Steered Molecular Dynamics Occurs in Three Steps. J. Mol. Model. 2014, 20, 2381. 10.1007/s00894-014-2381-7. [DOI] [PubMed] [Google Scholar]
- Han J.; Pluhackova K.; Bruns D.; Böckmann R. A. Synaptobrevin Transmembrane Domain Determines the Structure and Dynamics of the SNARE Motif and the Linker Region. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 855–865. 10.1016/j.bbamem.2016.01.030. [DOI] [PubMed] [Google Scholar]
- Ngatchou A. N.; Kisler K.; Fang Q.; Walter A. M.; Zhao Y.; Bruns D.; Sørensen J. B.; Lindau M. Role of the Synaptobrevin C Terminus in Fusion Pore Formation. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 18463–18468. 10.1073/pnas.1006727107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Agostino M.; Risselada H. J.; Mayer A. Steric Hindrance of SNARE Transmembrane Domain Organization Impairs the Hemifusion-to-fusion Transition. EMBO Rep. 2016, 17, 1590–1608. 10.15252/embr.201642209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindau M.; Hall B. A.; Chetwynd A.; Beckstein O.; Sansom M. S. P. Coarse-Grain Simulations Reveal Movement of the Synaptobrevin C-Terminus in Response to Piconewton Forces. Biophys. J. 2012, 103, 959–969. 10.1016/j.bpj.2012.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchard A. E.; Arcario M. J.; Schulten K.; Tajkhorshid E. A Highly Tilted Membrane Configuration for the Prefusion State of Synaptobrevin. Biophys. J. 2014, 107, 2112–2121. 10.1016/j.bpj.2014.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohkubo Y. Z.; Pogorelov T. V.; Arcario M. J.; Christensen G. A.; Tajkhorshid E. Accelerating Membrane Insertion of Peripheral Proteins with a Novel Membrane Mimetic Model. Biophys. J. 2012, 102, 2130–2139. 10.1016/j.bpj.2012.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Y.; Seeger M.; Weng J.; Song S.; Wang W.; Tan Y.-W. Lipid Regulated Intramolecular Conformational Dynamics of SNARE-Protein Ykt6. Sci. Rep. 2016, 6, 30282. 10.1038/srep30282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J.; Pluhackova K.; Wassenaar T. A.; Böckmann R. A. Synaptobrevin Transmembrane Domain Dimerization Studied by Multiscale Molecular Dynamics Simulations. Biophys. J. 2015, 109, 760–771. 10.1016/j.bpj.2015.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Q.; Zhao Y.; Herbst A. D.; Kim B. N.; Lindau M. Positively Charged Amino Acids at the SNAP-25 C Terminus Determine Fusion Rates, Fusion Pore Properties, and Energetics of Tight SNARE Complex Zippering. J. Neurosci. 2015, 35, 3230–3239. 10.1523/JNEUROSCI.2905-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-On D.; Nachliel E.; Gutman M.; Ashery U. Dynamic Conformational Changes in MUNC18 Prevent Syntaxin Binding. PLoS Comput. Biol. 2011, 7, e1001097. 10.1371/journal.pcbi.1001097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Agostino M.; Risselada H. J.; Lürick A.; Ungermann C.; Mayer A. A Tethering Complex Drives the Terminal Stage of SNARE-Dependent Membrane Fusion. Nature 2017, 10.1038/nature24469. [DOI] [PubMed] [Google Scholar]
- Bykhovskaia M.; Jagota A.; Gonzalez A.; Vasin A.; Littleton J. T. Interaction of the Complexin Accessory Helix with the C-Terminus of the SNARE Complex: Molecular-Dynamics Model of the Fusion Clamp. Biophys. J. 2013, 105, 679–690. 10.1016/j.bpj.2013.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasin A.; Volfson D.; Littleton J. T.; Bykhovskaia M. Interaction of the Complexin Accessory Helix with Synaptobrevin Regulates Spontaneous Fusion. Biophys. J. 2016, 111, 1954–1964. 10.1016/j.bpj.2016.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W. Probing the Structural Dynamics of the SNARE Recycling Machine Based on Coarse-Grained Modeling: Coarse-Grained Modeling of SNARE Recycling Machine. Proteins: Struct., Funct., Genet. 2016, 84, 1055–1066. 10.1002/prot.25052. [DOI] [PubMed] [Google Scholar]
- Osterberg J. R.; Chon N. L.; Boo A.; Maynard F. A.; Lin H.; Knight J. D. Membrane Docking of the Synaptotagmin 7 C2A Domain: Electron Paramagnetic Resonance Measurements Show Contributions from Two Membrane Binding Loops. Biochemistry 2015, 54, 5684–5695. 10.1021/acs.biochem.5b00421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z.; Schulten K. Synaptotagmin’s Role in Neurotransmitter Release Likely Involves Ca2+-Induced Conformational Transition. Biophys. J. 2014, 107, 1156–1166. 10.1016/j.bpj.2014.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fealey M. E.; Binder B. P.; Uversky V. N.; Hinderliter A.; Thomas D. D. Structural Impact of Phosphorylation and Dielectric Constant Variation on Synaptotagmin’s IDR. Biophys. J. 2018, 114, 550–561. 10.1016/j.bpj.2017.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chon N. L.; Osterberg J. R.; Henderson J.; Khan H. M.; Reuter N.; Knight J. D.; Lin H. Membrane Docking of the Synaptotagmin 7 C2A Domain: Computation Reveals Interplay between Electrostatic and Hydrophobic Contributions. Biochemistry 2015, 54, 5696–5711. 10.1021/acs.biochem.5b00422. [DOI] [PubMed] [Google Scholar]
- Wu B.; Guo W. The Exocyst at a Glance. J. Cell Sci. 2015, 128, 2957–2964. 10.1242/jcs.156398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.; Liu J.; Yang C.; Capraro B. R.; Baumgart T.; Bradley R. P.; Ramakrishnan N.; Xu X.; Radhakrishnan R.; Svitkina T.; et al. Exo70 Generates Membrane Curvature for Morphogenesis and Cell Migration. Dev. Cell 2013, 26, 266–278. 10.1016/j.devcel.2013.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pleskot R.; Cwiklik L.; Jungwirth P.; Žárský V.; Potocký M. Membrane Targeting of the Yeast Exocyst Complex. Biochim. Biophys. Acta, Biomembr. 2015, 1848, 1481–1489. 10.1016/j.bbamem.2015.03.026. [DOI] [PubMed] [Google Scholar]
- Mei K.; Li Y.; Wang S.; Shao G.; Wang J.; Ding Y.; Luo G.; Yue P.; Liu J.-J.; Wang X.; et al. Cryo-EM Structure of the Exocyst Complex. Nat. Struct. Mol. Biol. 2018, 25, 139–146. 10.1038/s41594-017-0016-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milosevic I. Revisiting the Role of Clathrin-Mediated Endoytosis in Synaptic Vesicle Recycling. Front. Cell. Neurosci. 2018, 12, 27. 10.3389/fncel.2018.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meinecke M.; Boucrot E.; Camdere G.; Hon W.-C.; Mittal R.; McMahon H. T. Cooperative Recruitment of Dynamin and BIN/Amphiphysin/Rvs (BAR) Domain-Containing Proteins Leads to GTP-Dependent Membrane Scission. J. Biol. Chem. 2013, 288, 6651–6661. 10.1074/jbc.M112.444869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blood P. D.; Voth G. A. Direct Observation of Bin/Amphiphysin/Rvs (BAR) Domain-Induced Membrane Curvature by Means of Molecular Dynamics Simulations. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 15068–15072. 10.1073/pnas.0603917103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui H.; Ayton G. S.; Voth G. A. Membrane Binding by the Endophilin N-BAR Domain. Biophys. J. 2009, 97, 2746–2753. 10.1016/j.bpj.2009.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blood P. D.; Swenson R. D.; Voth G. A. Factors Influencing Local Membrane Curvature Induction by N-BAR Domains as Revealed by Molecular Dynamics Simulations. Biophys. J. 2008, 95, 1866–1876. 10.1529/biophysj.107.121160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemura K.; Hanawa-Suetsugu K.; Suetsugu S.; Kitao A. Salt Bridge Formation between the I-BAR Domain and Lipids Increases Lipid Density and Membrane Curvature. Sci. Rep. 2017, 7, 6808. 10.1038/s41598-017-06334-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arkhipov A.; Yin Y.; Schulten K. Four-Scale Description of Membrane Sculpting by BAR Domains. Biophys. J. 2008, 95, 2806–2821. 10.1529/biophysj.108.132563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y.; Arkhipov A.; Schulten K. Simulations of Membrane Tubulation by Lattices of Amphiphysin N-BAR Domains. Structure 2009, 17, 882–892. 10.1016/j.str.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H.; Schulten K. Membrane Sculpting by F-BAR Domains Studied by Molecular Dynamics Simulations. PLoS Comput. Biol. 2013, 9, e1002892. 10.1371/journal.pcbi.1002892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyman E.; Cui H.; Voth G. A. Water under the BAR. Biophys. J. 2010, 99, 1783–1790. 10.1016/j.bpj.2010.06.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simunovic M.; Evergren E.; Golushko I.; Prévost C.; Renard H.-F.; Johannes L.; McMahon H. T.; Lorman V.; Voth G. A.; Bassereau P. How Curvature-Generating Proteins Build Scaffolds on Membrane Nanotubes. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 11226–11231. 10.1073/pnas.1606943113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayton G. S.; Lyman E.; Krishna V.; Swenson R. D.; Mim C.; Unger V. M.; Voth G. A. New Insights into BAR Domain-Induced Membrane Remodeling. Biophys. J. 2009, 97, 1616–1625. 10.1016/j.bpj.2009.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayton G. S.; Voth G. A. Multiscale Simulation of Protein Mediated Membrane Remodeling. Semin. Cell Dev. Biol. 2010, 21, 357–362. 10.1016/j.semcdb.2009.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picas L.; Viaud J.; Schauer K.; Vanni S.; Hnia K.; Fraisier V.; Roux A.; Bassereau P.; Gaits-Iacovoni F.; Payrastre B.; et al. BIN1/M-Amphiphysin2 Induces Clustering of Phosphoinositides to Recruit Its Downstream Partner Dynamin. Nat. Commun. 2014, 5, 5647. 10.1038/ncomms6647. [DOI] [PubMed] [Google Scholar]
- Kalli A. C.; Morgan G.; Sansom M. S. P. Interactions of the Auxilin-1 PTEN-like Domain with Model Membranes Result in Nanoclustering of Phosphatidyl Inositol Phosphates. Biophys. J. 2013, 105, 137–145. 10.1016/j.bpj.2013.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faelber K.; Posor Y.; Gao S.; Held M.; Roske Y.; Schulze D.; Haucke V.; Noé F.; Daumke O. Crystal Structure of Nucleotide-Free Dynamin. Nature 2011, 477, 556–560. 10.1038/nature10369. [DOI] [PubMed] [Google Scholar]
- Reubold T. F.; Faelber K.; Plattner N.; Posor Y.; Ketel K.; Curth U.; Schlegel J.; Anand R.; Manstein D. J.; Noé F.; et al. Crystal Structure of the Dynamin Tetramer. Nature 2015, 525, 404–408. 10.1038/nature14880. [DOI] [PubMed] [Google Scholar]
- Abdel-Hamid M.; McCluskey A. Silico Docking, Molecular Dynamics and Binding Energy Insights into the Bolinaquinone-Clathrin Terminal Domain Binding Site. Molecules 2014, 19, 6609–6622. 10.3390/molecules19056609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinot M.; Vanni S.; Pagnotta S.; Lacas-Gervais S.; Payet L.-A.; Ferreira T.; Gautier R.; Goud B.; Antonny B.; Barelli H. Polyunsaturated Phospholipids Facilitate Membrane Deformation and Fission by Endocytic Proteins. Science 2014, 345, 693–697. 10.1126/science.1255288. [DOI] [PubMed] [Google Scholar]
- Lai C.-L.; Jao C. C.; Lyman E.; Gallop J. L.; Peter B. J.; McMahon H. T.; Langen R.; Voth G. A. Membrane Binding and Self-Association of the Epsin N-Terminal Homology Domain. J. Mol. Biol. 2012, 423, 800–817. 10.1016/j.jmb.2012.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tourdot R. W.; Bradley R. P.; Ramakrishnan N.; Radhakrishnan R. Multiscale Computational Models in Physical Systems Biology of Intracellular Trafficking. IET Syst. Biol. 2014, 8, 198–213. 10.1049/iet-syb.2013.0057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahy E.; Sud M.; Cotter D.; Subramaniam S. LIPID MAPS Online Tools for Lipid Research. Nucleic Acids Res. 2007, 35, W606–W612. 10.1093/nar/gkm324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sud M.; Fahy E.; Cotter D.; Brown A.; Dennis E. A.; Glass C. K.; Merrill A. H.; Murphy R. C.; Raetz C. R. H.; Russell D. W.; et al. LMSD: LIPID MAPS Structure Database. Nucleic Acids Res. 2007, 35, D527–D532. 10.1093/nar/gkl838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaszuba K.; Grzybek M.; Orłowski A.; Danne R.; Róg T.; Simons K.; Coskun Ü.; Vattulainen I. N-Glycosylation as Determinant of Epidermal Growth Factor Receptor Conformation in Membranes. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 4334–4339. 10.1073/pnas.1503262112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts A.; Volotovski I. D.; Marsh D. Rhodopsin-Lipid Associations in Bovine Rod Outer Segment Membranes. Identification of Immobilized Lipid by Spin-Labels. Biochemistry 1979, 18, 5006–5013. 10.1021/bi00589a031. [DOI] [PubMed] [Google Scholar]
- Fretten P.; Morris S. J.; Watts A.; Marsh D. Lipid-Lipid and Lipid-Protein Interactions in Chromaffin Granule Membranes. Biochim. Biophys. Acta, Biomembr. 1980, 598, 247–259. 10.1016/0005-2736(80)90003-6. [DOI] [PubMed] [Google Scholar]
- Marsh D.; Watts A.; Pates R. D.; Uhl R.; Knowles P. F.; Esmann M. ESR Spin-Label Studies of Lipid-Protein Interactions in Membranes. Biophys. J. 1982, 37, 265–274. 10.1016/S0006-3495(82)84675-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sooksawate T.; Simmonds M. A. Effects of Membrane Cholesterol on the Sensitivity of the GABAA Receptor to GABA in Acutely Dissociated Rat Hippocampal Neurones. Neuropharmacology 2001, 40, 178–184. 10.1016/S0028-3908(00)00159-3. [DOI] [PubMed] [Google Scholar]
- Saxena R.; Chattopadhyay A. Membrane Cholesterol Stabilizes the Human Serotonin1A Receptor. Biochim. Biophys. Acta, Biomembr. 2012, 1818, 2936–2942. 10.1016/j.bbamem.2012.07.032. [DOI] [PubMed] [Google Scholar]
- Zocher M.; Zhang C.; Rasmussen S. G. F.; Kobilka B. K.; Muller D. J. Cholesterol Increases Kinetic, Energetic, and Mechanical Stability of the Human β2-Adrenergic Receptor. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, E3463–E3472. 10.1073/pnas.1210373109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips R.; Ursell T.; Wiggins P.; Sens P. Emerging Roles for Lipids in Shaping Membrane-Protein Function. Nature 2009, 459, 379–385. 10.1038/nature08147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bretscher M. S. Asymmetrical Lipid Bilayer Structure for Biological Membranes. Nature. New Biol. 1972, 236, 11–12. 10.1038/newbio236011a0. [DOI] [PubMed] [Google Scholar]
- Carquin M.; D’Auria L.; Pollet H.; Bongarzone E. R.; Tyteca D. Recent Progress on Lipid Lateral Heterogeneity in Plasma Membranes: From Rafts to Submicrometric Domains. Prog. Lipid Res. 2016, 62, 1–24. 10.1016/j.plipres.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen B. P.; Nissen P. Membrane Proteins — Do We Catch up with the Breathless Pace of Soluble Protein Structural Biology?. Biochim. Biophys. Acta, Gen. Subj. 2015, 1850, 447–448. 10.1016/j.bbagen.2015.01.001. [DOI] [PubMed] [Google Scholar]
- MacDonald R. I. Temperature and Ionic Effects on the Interaction of Erythroid Spectrin with Phosphatidylserine Membranes. Biochemistry 1993, 32, 6957–6964. 10.1021/bi00078a021. [DOI] [PubMed] [Google Scholar]
- O’Toole P. J.; Wolfe C.; Ladha S.; Cherry R. J. Rapid Diffusion of Spectrin Bound to a Lipid Surface. Biochim. Biophys. Acta, Biomembr. 1999, 1419, 64–70. 10.1016/S0005-2736(99)00048-6. [DOI] [PubMed] [Google Scholar]
- de Jong K.; Rettig M. P.; Low P. S.; Kuypers F. A. Protein Kinase C Activation Induces Phosphatidylserine Exposure on Red Blood Cells. Biochemistry 2002, 41, 12562–12567. 10.1021/bi025882o. [DOI] [PubMed] [Google Scholar]
- Meers P.; Mealy T. Calcium-Dependent Annexin V Binding to Phospholipids: Stoichiometry, Specificity, and the Role of Negative Charge. Biochemistry 1993, 32, 11711–11721. 10.1021/bi00094a030. [DOI] [PubMed] [Google Scholar]
- Shiratsuchi A.; Umeda M.; Ohba Y.; Nakanishi Y. Recognition of Phosphatidylserine on the Surface of Apoptotic Spermatogenic Cells and Subsequent Phagocytosis by Sertoli Cells of the Rat. J. Biol. Chem. 1997, 272, 2354–2358. 10.1074/jbc.272.4.2354. [DOI] [PubMed] [Google Scholar]
- Bratton D. L.; Fadok V. A.; Richter D. A.; Kailey J. M.; Guthrie L. A.; Henson P. M. Appearance of Phosphatidylserine on Apoptotic Cells Requires Calcium-Mediated Nonspecific Flip-Flop and Is Enhanced by Loss of the Aminophospholipid Translocase. J. Biol. Chem. 1997, 272, 26159–26165. 10.1074/jbc.272.42.26159. [DOI] [PubMed] [Google Scholar]
- Castegna A.; Lauderback C. M.; Mohmmad-Abdul H.; Butterfield D. A. Modulation of Phospholipid Asymmetry in Synaptosomal Membranes by the Lipid Peroxidation Products, 4-Hydroxynonenal and Acrolein: Implications for Alzheimer’s Disease. Brain Res. 2004, 1004, 193–197. 10.1016/j.brainres.2004.01.036. [DOI] [PubMed] [Google Scholar]
- Liu J.; Epand R. F.; Durrant D.; Grossman D.; Chi N.; Epand R. M.; Lee R. M. Role of Phospholipid Scramblase 3 in the Regulation of Tumor Necrosis Factor-α-Induced Apoptosis. Biochemistry 2008, 47, 4518–4529. 10.1021/bi701962c. [DOI] [PubMed] [Google Scholar]
- Ndebele K.; Gona P.; Jin T.-G.; Benhaga N.; Chalah A.; Degli-Esposti M.; Khosravi-Far R. Tumor Necrosis Factor (TNF)-Related Apoptosis-Inducing Ligand (TRAIL) Induced Mitochondrial Pathway to Apoptosis and Caspase Activation Is Potentiated by Phospholipid Scramblase-3. Apoptosis 2008, 13, 845–856. 10.1007/s10495-008-0219-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamon Y.; Broccardo C.; Chambenoit O.; Luciani M.-F.; Toti F.; Chaslin S.; Freyssinet J.-M.; Devaux P. F.; McNeish J.; Marguet D.; et al. ABC1 Promotes Engulfment of Apoptotic Cells and Transbilayer Redistribution of Phosphatidylserine. Nat. Cell Biol. 2000, 2, 399–406. 10.1038/35017029. [DOI] [PubMed] [Google Scholar]
- McConnell H. M.; Kornberg R. D. Inside-Outside Transitions of Phospholipids in Vesicle Membranes. Biochemistry 1971, 10, 1111–1120. 10.1021/bi00783a003. [DOI] [PubMed] [Google Scholar]
- Nakano M.; Fukuda M.; Kudo T.; Matsuzaki N.; Azuma T.; Sekine K.; Endo H.; Handa T. Flip-Flop of Phospholipids in Vesicles: Kinetic Analysis with Time-Resolved Small-Angle Neutron Scattering. J. Phys. Chem. B 2009, 113, 6745–6748. 10.1021/jp900913w. [DOI] [PubMed] [Google Scholar]
- Lange Y.; Dolde J.; Steck T. L. The Rate of Transmembrane Movement of Cholesterol in the Human Erythrocyte. J. Biol. Chem. 1981, 256, 5321–5323. [PubMed] [Google Scholar]
- Lange Y.; Cohen C. M.; Poznansky M. J. Transmembrane Movement of Cholesterol in Human Erythrocytes. Proc. Natl. Acad. Sci. U. S. A. 1977, 74, 1538–1542. 10.1073/pnas.74.4.1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steck T. L.; Ye J.; Lange Y. Probing Red Cell Membrane Cholesterol Movement with Cyclodextrin. Biophys. J. 2002, 83, 2118–2125. 10.1016/S0006-3495(02)73972-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruckner R. J.; Mansy S. S.; Ricardo A.; Mahadevan L.; Szostak J. W. Flip-Flop-Induced Relaxation of Bending Energy: Implications for Membrane Remodeling. Biophys. J. 2009, 97, 3113–3122. 10.1016/j.bpj.2009.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg S.; Porcar L.; Woodka A. C.; Butler P. D.; Perez-Salas U. Noninvasive Neutron Scattering Measurements Reveal Slower Cholesterol Transport in Model Lipid Membranes. Biophys. J. 2011, 101, 370–377. 10.1016/j.bpj.2011.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imparato A.; Shillcock J. C.; Lipowsky R. Lateral and Transverse Diffusion in Two-Component Bilayer Membranes. Eur. Phys. J. E: Soft Matter Biol. Phys. 2003, 11, 21–28. 10.1140/epje/i2002-10125-x. [DOI] [PubMed] [Google Scholar]
- Son M.; London E. The Dependence of Lipid Asymmetry upon Phosphatidylcholine Acyl Chain Structure. J. Lipid Res. 2013, 54, 223–231. 10.1194/jlr.M032722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John K.; Schreiber S.; Kubelt J.; Herrmann A.; Müller P. Transbilayer Movement of Phospholipids at the Main Phase Transition of Lipid Membranes: Implications for Rapid Flip-Flop in Biological Membranes. Biophys. J. 2002, 83, 3315–3323. 10.1016/S0006-3495(02)75332-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries A. H.; Mark A. E.; Marrink S. J. Molecular Dynamics Simulation of the Spontaneous Formation of a Small DPPC Vesicle in Water in Atomistic Detail. J. Am. Chem. Soc. 2004, 126, 4488–4489. 10.1021/ja0398417. [DOI] [PubMed] [Google Scholar]
- Gurtovenko A. A.; Onike O. I.; Anwar J. Chemically Induced Phospholipid Translocation Across Biological Membranes. Langmuir 2008, 24, 9656–9660. 10.1021/la801431f. [DOI] [PubMed] [Google Scholar]
- Dickey A. N.; Faller R. How Alcohol Chain-Length and Concentration Modulate Hydrogen Bond Formation in a Lipid Bilayer. Biophys. J. 2007, 92, 2366–2376. 10.1529/biophysj.106.097022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandasamy S. K.; Larson R. G. Cation and Anion Transport through Hydrophilic Pores in Lipid Bilayers. J. Chem. Phys. 2006, 125, 074901. 10.1063/1.2217737. [DOI] [PubMed] [Google Scholar]
- Gurtovenko A. A.; Vattulainen I. Molecular Mechanism for Lipid Flip-Flops. J. Phys. Chem. B 2007, 111, 13554–13559. 10.1021/jp077094k. [DOI] [PubMed] [Google Scholar]
- Róg T.; Stimson L. M.; Pasenkiewicz-Gierula M.; Vattulainen I.; Karttunen M. Replacing the Cholesterol Hydroxyl Group with the Ketone Group Facilitates Sterol Flip-Flop and Promotes Membrane Fluidity. J. Phys. Chem. B 2008, 112, 1946–1952. 10.1021/jp075078h. [DOI] [PubMed] [Google Scholar]
- Arai N.; Akimoto T.; Yamamoto E.; Yasui M.; Yasuoka K. Poisson Property of the Occurrence of Flip-Flops in a Model Membrane. J. Chem. Phys. 2014, 140, 064901. 10.1063/1.4863330. [DOI] [PubMed] [Google Scholar]
- Kučerka N.; Perlmutter J. D.; Pan J.; Tristram-Nagle S.; Katsaras J.; Sachs J. N. The Effect of Cholesterol on Short- and Long-Chain Monounsaturated Lipid Bilayers as Determined by Molecular Dynamics Simulations and X-Ray Scattering. Biophys. J. 2008, 95, 2792–2805. 10.1529/biophysj.107.122465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choubey A.; Kalia R. K.; Malmstadt N.; Nakano A.; Vashishta P. Cholesterol Translocation in a Phospholipid Membrane. Biophys. J. 2013, 104, 2429–2436. 10.1016/j.bpj.2013.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker M. K.; Abrams C. F. Dynamics of Lipids, Cholesterol, and Transmembrane α-Helices from Microsecond Molecular Dynamics Simulations. J. Phys. Chem. B 2014, 118, 13590–13600. 10.1021/jp507027t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett W. F. D.; MacCallum J. L.; Hinner M. J.; Marrink S. J.; Tieleman D. P. Molecular View of Cholesterol Flip-Flop and Chemical Potential in Different Membrane Environments. J. Am. Chem. Soc. 2009, 131, 12714–12720. 10.1021/ja903529f. [DOI] [PubMed] [Google Scholar]
- Sapay N.; Bennett W. F. D.; Tieleman D. P. Thermodynamics of Flip-Flop and Desorption for a Systematic Series of Phosphatidylcholine Lipids. Soft Matter 2009, 5, 3295. 10.1039/b902376c. [DOI] [Google Scholar]
- Anglin T. C.; Conboy J. C. Kinetics and Thermodynamics of Flip-Flop in Binary Phospholipid Membranes Measured by Sum-Frequency Vibrational Spectroscopy. Biochemistry 2009, 48, 10220–10234. 10.1021/bi901096j. [DOI] [PubMed] [Google Scholar]
- Jo S.; Rui H.; Lim J. B.; Klauda J. B.; Im W. Cholesterol Flip-Flop: Insights from Free Energy Simulation Studies. J. Phys. Chem. B 2010, 114, 13342–13348. 10.1021/jp108166k. [DOI] [PubMed] [Google Scholar]
- Parisio G.; Sperotto M. M.; Ferrarini A. Flip-Flop of Steroids in Phospholipid Bilayers: Effects of the Chemical Structure on Transbilayer Diffusion. J. Am. Chem. Soc. 2012, 134, 12198–12208. 10.1021/ja304007t. [DOI] [PubMed] [Google Scholar]
- Marrink S. J.; Tieleman D. P. Perspective on the Martini Model. Chem. Soc. Rev. 2013, 42, 6801. 10.1039/c3cs60093a. [DOI] [PubMed] [Google Scholar]
- Marrink S. J.; Risselada H. J.; Yefimov S.; Tieleman D. P.; de Vries A. H. The MARTINI Force Field: Coarse Grained Model for Biomolecular Simulations. J. Phys. Chem. B 2007, 111, 7812–7824. 10.1021/jp071097f. [DOI] [PubMed] [Google Scholar]
- Ogushi F.; Ishitsuka R.; Kobayashi T.; Sugita Y. Rapid Flip-Flop Motions of Diacylglycerol and Ceramide in Phospholipid Bilayers. Chem. Phys. Lett. 2012, 522, 96–102. 10.1016/j.cplett.2011.11.057. [DOI] [Google Scholar]
- Yesylevskyy S. O.; Demchenko A. P.. Cholesterol Behavior in Asymmetric Lipid Bilayers: Insights from Molecular Dynamics Simulations. In Methods in Membrane Lipids; Owen D. M., Ed.; Springer New York: New York, NY, 2015; Vol. 1232, pp 291–306, 10.1007/978-1-4939-1752-5_20. [DOI] [PubMed] [Google Scholar]
- Yesylevskyy S. O.; Demchenko A. P.; Kraszewski S.; Ramseyer C. Cholesterol Induces Uneven Curvature of Asymmetric Lipid Bilayers. Sci. World J. 2013, 2013, 1–10. 10.1155/2013/965230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kästner J. Umbrella Sampling: Umbrella Sampling. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 932–942. 10.1002/wcms.66. [DOI] [Google Scholar]
- Maximova T.; Moffatt R.; Ma B.; Nussinov R.; Shehu A. Principles and Overview of Sampling Methods for Modeling Macromolecular Structure and Dynamics. PLoS Comput. Biol. 2016, 12, e1004619. 10.1371/journal.pcbi.1004619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett W. F. D.; Tieleman D. P. Water Defect and Pore Formation in Atomistic and Coarse-Grained Lipid Membranes: Pushing the Limits of Coarse Graining. J. Chem. Theory Comput. 2011, 7, 2981–2988. 10.1021/ct200291v. [DOI] [PubMed] [Google Scholar]
- Bennett W. F. D.; Sapay N.; Tieleman D. P. Atomistic Simulations of Pore Formation and Closure in Lipid Bilayers. Biophys. J. 2014, 106, 210–219. 10.1016/j.bpj.2013.11.4486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicol F.; Nir S.; Szoka F. C. Effect of Cholesterol and Charge on Pore Formation in Bilayer Vesicles by a pH-Sensitive Peptide. Biophys. J. 1996, 71, 3288–3301. 10.1016/S0006-3495(96)79521-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volinsky R.; Cwiklik L.; Jurkiewicz P.; Hof M.; Jungwirth P.; Kinnunen P. K. J. Oxidized Phosphatidylcholines Facilitate Phospholipid Flip-Flop in Liposomes. Biophys. J. 2011, 101, 1376–1384. 10.1016/j.bpj.2011.07.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volinsky R.; Kinnunen P. K. J. Oxidized Phosphatidylcholines in Membrane-Level Cellular Signaling: From Biophysics to Physiology and Molecular Pathology. FEBS J. 2013, 280, 2806–2816. 10.1111/febs.12247. [DOI] [PubMed] [Google Scholar]
- Razzokov J.; Yusupov M.; Vanuytsel S.; Neyts E. C.; Bogaerts A. Phosphatidylserine Flip-Flop Induced by Oxidation of the Plasma Membrane: A Better Insight by Atomic Scale Modeling. Plasma Processes Polym. 2017, 14, 1700013. 10.1002/ppap.201700013. [DOI] [Google Scholar]
- Lin J.; Dargazany R.; Alexander-Katz A. Lipid Flip-Flop and Pore Nucleation on Zwitterionic Bilayers Are Asymmetric under Ionic Imbalance. Small 2017, 13, 1603708. 10.1002/smll.201603708. [DOI] [PubMed] [Google Scholar]
- Bennett W. F. D.; Tieleman D. P. Molecular Simulation of Rapid Translocation of Cholesterol, Diacylglycerol, and Ceramide in Model Raft and Nonraft Membranes. J. Lipid Res. 2012, 53, 421–429. 10.1194/jlr.M022491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange Y.; Ye J.; Rigney M.; Steck T. L. Regulation of Endoplasmic Reticulum Cholesterol by Plasma Membrane Cholesterol. J. Lipid Res. 1999, 40, 2264–2270. [PubMed] [Google Scholar]
- Lange Y.; Ye J.; Strebel F. Movement of 25-Hydroxycholesterol from the Plasma Membrane to the Rough Endoplasmic Reticulum in Cultured Hepatoma Cells. J. Lipid Res. 1995, 36, 1092–1097. [PubMed] [Google Scholar]
- Moro G. J.; Ferrarini A.; Polimeno A.; Nordio P. L.. Models of Conformational Dynamics. In Reactive and Flexible Molecules in Liquids; Dorfmüller T., Ed.; Springer Netherlands: Dordrecht, 1989; pp 107–139, 10.1007/978-94-009-1043-0_7. [DOI] [Google Scholar]
- Langer J. S. Statistical Theory of the Decay of Metastable States. Ann. Phys. 1969, 54, 258–275. 10.1016/0003-4916(69)90153-5. [DOI] [Google Scholar]
- Kramers H. A. Brownian Motion in a Field of Force and the Diffusion Model of Chemical Reactions. Physica 1940, 7, 284–304. 10.1016/S0031-8914(40)90098-2. [DOI] [Google Scholar]
- Parisio G.; Ferrarini A. Solute Partitioning into Lipid Bilayers: An Implicit Model for Nonuniform and Ordered Environment. J. Chem. Theory Comput. 2010, 6, 2267–2280. 10.1021/ct100210u. [DOI] [PubMed] [Google Scholar]
- Wei C.; Pohorille A. Flip-Flop of Oleic Acid in a Phospholipid Membrane: Rate and Mechanism. J. Phys. Chem. B 2014, 118, 12919–12926. 10.1021/jp508163e. [DOI] [PubMed] [Google Scholar]
- Filipe H. A. L.; Moreno M. J.; Róg T.; Vattulainen I.; Loura L. M. S. How To Tackle the Issues in Free Energy Simulations of Long Amphiphiles Interacting with Lipid Membranes: Convergence and Local Membrane Deformations. J. Phys. Chem. B 2014, 118, 3572–3581. 10.1021/jp501622d. [DOI] [PubMed] [Google Scholar]
- Neale C.; Bennett W. F. D.; Tieleman D. P.; Pomès R. Statistical Convergence of Equilibrium Properties in Simulations of Molecular Solutes Embedded in Lipid Bilayers. J. Chem. Theory Comput. 2011, 7, 4175–4188. 10.1021/ct200316w. [DOI] [PubMed] [Google Scholar]
- Spiwok V.; Sucur Z.; Hosek P. Enhanced Sampling Techniques in Biomolecular Simulations. Biotechnol. Adv. 2015, 33, 1130–1140. 10.1016/j.biotechadv.2014.11.011. [DOI] [PubMed] [Google Scholar]
- Bacci M.; Vitalis A.; Caflisch A. A Molecular Simulation Protocol to Avoid Sampling Redundancy and Discover New States. Biochim. Biophys. Acta, Gen. Subj. 2015, 1850, 889–902. 10.1016/j.bbagen.2014.08.013. [DOI] [PubMed] [Google Scholar]
- Bernardi R. C.; Melo M. C. R.; Schulten K. Enhanced Sampling Techniques in Molecular Dynamics Simulations of Biological Systems. Biochim. Biophys. Acta, Gen. Subj. 2015, 1850, 872–877. 10.1016/j.bbagen.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wymann M. P.; Schneiter R. Lipid Signalling in Disease. Nat. Rev. Mol. Cell Biol. 2008, 9, 162–176. 10.1038/nrm2335. [DOI] [PubMed] [Google Scholar]
- Overington J. P.; Al-Lazikani B.; Hopkins A. L. How Many Drug Targets Are There?. Nat. Rev. Drug Discovery 2006, 5, 993–996. 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- Hunte C.; Richers S. Lipids and Membrane Protein Structures. Curr. Opin. Struct. Biol. 2008, 18, 406–411. 10.1016/j.sbi.2008.03.008. [DOI] [PubMed] [Google Scholar]
- Rose P. W.; Prlić A.; Altunkaya A.; Bi C.; Bradley A. R.; Christie C. H.; Costanzo L. D.; Duarte J. M.; Dutta S.; Feng Z.; et al. The RCSB Protein Data Bank: Integrative View of Protein, Gene and 3D Structural Information. Nucleic Acids Res. 2017, 45, D271–D281. 10.1093/nar/gkw1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce K. L.; Premont R. T.; Lefkowitz R. J. Seven-Transmembrane Receptors: Signalling. Nat. Rev. Mol. Cell Biol. 2002, 3, 639–650. 10.1038/nrm908. [DOI] [PubMed] [Google Scholar]
- Latorraca N. R.; Venkatakrishnan A. J.; Dror R. O. GPCR Dynamics: Structures in Motion. Chem. Rev. 2017, 117, 139–155. 10.1021/acs.chemrev.6b00177. [DOI] [PubMed] [Google Scholar]
- Oates J.; Watts A. Uncovering the Intimate Relationship between Lipids, Cholesterol and GPCR Activation. Curr. Opin. Struct. Biol. 2011, 21, 802–807. 10.1016/j.sbi.2011.09.007. [DOI] [PubMed] [Google Scholar]
- Staubach S.; Hanisch F.-G. Lipid Rafts: Signaling and Sorting Platforms of Cells and Their Roles in Cancer. Expert Rev. Proteomics 2011, 8, 263–277. 10.1586/epr.11.2. [DOI] [PubMed] [Google Scholar]
- Chini B. G-Protein Coupled Receptors in Lipid Rafts and Caveolae: How, When and Why Do They Go There?. J. Mol. Endocrinol. 2004, 32, 325–338. 10.1677/jme.0.0320325. [DOI] [PubMed] [Google Scholar]
- Albert A. D.; Young J. E.; Yeagle P. L. Rhodopsin-Cholesterol Interactions in Bovine Rod Outer Segment Disk Membranes. Biochim. Biophys. Acta, Biomembr. 1996, 1285, 47–55. 10.1016/S0005-2736(96)00145-9. [DOI] [PubMed] [Google Scholar]
- Li J.; Edwards P. C.; Burghammer M.; Villa C.; Schertler G. F. X. Structure of Bovine Rhodopsin in a Trigonal Crystal Form. J. Mol. Biol. 2004, 343, 1409–1438. 10.1016/j.jmb.2004.08.090. [DOI] [PubMed] [Google Scholar]
- Horsefield R.; Norden K.; Fellert M.; Backmark A.; Tornroth-Horsefield S.; Terwisscha van Scheltinga A. C.; Kvassman J.; Kjellbom P.; Johanson U.; Neutze R. High-Resolution x-Ray Structure of Human Aquaporin 5. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 13327–13332. 10.1073/pnas.0801466105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherezov V.; Rosenbaum D. M.; Hanson M. A.; Rasmussen S. G. F.; Thian F. S.; Kobilka T. S.; Choi H.-J.; Kuhn P.; Weis W. I.; Kobilka B. K.; et al. High-Resolution Crystal Structure of an Engineered Human β2-Adrenergic G Protein-Coupled Receptor. Science 2007, 318, 1258–1265. 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pucadyil T. J.; Chattopadhyay A. Cholesterol Modulates Ligand Binding and G-Protein Coupling to Serotonin1A Receptors from Bovine Hippocampus. Biochim. Biophys. Acta, Biomembr. 2004, 1663, 188–200. 10.1016/j.bbamem.2004.03.010. [DOI] [PubMed] [Google Scholar]
- Harikumar K. G.; Puri V.; Singh R. D.; Hanada K.; Pagano R. E.; Miller L. J. Differential Effects of Modification of Membrane Cholesterol and Sphingolipids on the Conformation, Function, and Trafficking of the G Protein-Coupled Cholecystokinin Receptor. J. Biol. Chem. 2005, 280, 2176–2185. 10.1074/jbc.M410385200. [DOI] [PubMed] [Google Scholar]
- Gimpl G.; Fahrenholz F. Cholesterol as Stabilizer of the Oxytocin Receptor. Biochim. Biophys. Acta, Biomembr. 2002, 1564, 384–392. 10.1016/S0005-2736(02)00475-3. [DOI] [PubMed] [Google Scholar]
- Chakraborty H.; Chattopadhyay A. Excitements and Challenges in GPCR Oligomerization: Molecular Insight from FRET. ACS Chem. Neurosci. 2015, 6, 199–206. 10.1021/cn500231d. [DOI] [PubMed] [Google Scholar]
- Oates J.; Faust B.; Attrill H.; Harding P.; Orwick M.; Watts A. The Role of Cholesterol on the Activity and Stability of Neurotensin Receptor 1. Biochim. Biophys. Acta, Biomembr. 2012, 1818, 2228–2233. 10.1016/j.bbamem.2012.04.010. [DOI] [PubMed] [Google Scholar]
- Mitchell D. C.; Straume M.; Litman B. J. Role of Sn-1-Saturated,Sn-2-Polyunsaturated Phospholipids in Control of Membrane Receptor Conformational Equilibrium: Effects of Cholesterol and Acyl Chain Unsaturation on the Metarhodopsin I.Tautm. Metarhodopsin II Equilibrium. Biochemistry 1992, 31, 662–670. 10.1021/bi00118a005. [DOI] [PubMed] [Google Scholar]
- Mitchell D. C.; Niu S.-L.; Litman B. J. Enhancement of G Protein-Coupled Signaling by DHA Phospholipids. Lipids 2003, 38, 437–443. 10.1007/s11745-003-1081-1. [DOI] [PubMed] [Google Scholar]
- Bennett M. P.; Mitchell D. C. Regulation of Membrane Proteins by Dietary Lipids: Effects of Cholesterol and Docosahexaenoic Acid Acyl Chain-Containing Phospholipids on Rhodopsin Stability and Function. Biophys. J. 2008, 95, 1206–1216. 10.1529/biophysj.107.122788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson N. J.; Brown M. F. Lipid Headgroup and Acyl Chain Composition Modulate the MI-MII Equilibrium of Rhodopsin in Recombinant Membranes. Biochemistry 1993, 32, 2438–2454. 10.1021/bi00060a040. [DOI] [PubMed] [Google Scholar]
- Brown M. F. Modulation of Rhodopsin Function by Properties of the Membrane Bilayer. Chem. Phys. Lipids 1994, 73, 159–180. 10.1016/0009-3084(94)90180-5. [DOI] [PubMed] [Google Scholar]
- Horn J. N.; Kao T.-C.; Grossfield A.. Coarse-Grained Molecular Dynamics Provides Insight into the Interactions of Lipids and Cholesterol with Rhodopsin. In G Protein-Coupled Receptors - Modeling and Simulation; Filizola M., Ed.; Springer Netherlands: Dordrecht, 2014; Vol. 796, pp 75–94, 10.1007/978-94-007-7423-0_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossfield A.; Feller S. E.; Pitman M. C. A Role for Direct Interactions in the Modulation of Rhodopsin by ω-3 Polyunsaturated Lipids. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 4888–4893. 10.1073/pnas.0508352103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitman M. C.; Grossfield A.; Suits F.; Feller S. E. Role of Cholesterol and Polyunsaturated Chains in Lipid–Protein Interactions: Molecular Dynamics Simulation of Rhodopsin in a Realistic Membrane Environment. J. Am. Chem. Soc. 2005, 127, 4576–4577. 10.1021/ja042715y. [DOI] [PubMed] [Google Scholar]
- Grossfield A.; Feller S. E.; Pitman M. C. Contribution of Omega-3 Fatty Acids to the Thermodynamics of Membrane Protein Solvation. J. Phys. Chem. B 2006, 110, 8907–8909. 10.1021/jp060405r. [DOI] [PubMed] [Google Scholar]
- Khelashvili G.; Grossfield A.; Feller S. E.; Pitman M. C.; Weinstein H. Structural and Dynamic Effects of Cholesterol at Preferred Sites of Interaction with Rhodopsin Identified from Microsecond Length Molecular Dynamics Simulations. Proteins: Struct., Funct., Genet. 2009, 76, 403–417. 10.1002/prot.22355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feller S. E.; Gawrisch K.; Woolf T. B. Rhodopsin Exhibits a Preference for Solvation by Polyunsaturated Docosohexaenoic Acid. J. Am. Chem. Soc. 2003, 125, 4434–4435. 10.1021/ja0345874. [DOI] [PubMed] [Google Scholar]
- Patra S. M.; Chakraborty S.; Shahane G.; Prasanna X.; Sengupta D.; Maiti P. K.; Chattopadhyay A. Differential Dynamics of the Serotonin1A Receptor in Membrane Bilayers of Varying Cholesterol Content Revealed by All Atom Molecular Dynamics Simulation. Mol. Membr. Biol. 2015, 32, 127–137. 10.3109/09687688.2015.1096971. [DOI] [PubMed] [Google Scholar]
- Paila Y. D.; Tiwari S.; Sengupta D.; Chattopadhyay A. Molecular Modeling of the Human Serotonin1A Receptor: Role of Membrane Cholesterol in Ligand Binding of the Receptor. Mol. BioSyst. 2011, 7, 224–234. 10.1039/C0MB00148A. [DOI] [PubMed] [Google Scholar]
- Shan J.; Khelashvili G.; Mondal S.; Mehler E. L.; Weinstein H. Ligand-Dependent Conformations and Dynamics of the Serotonin 5-HT2A Receptor Determine Its Activation and Membrane-Driven Oligomerization Properties. PLoS Comput. Biol. 2012, 8, e1002473. 10.1371/journal.pcbi.1002473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jafurulla M.; Tiwari S.; Chattopadhyay A. Identification of Cholesterol Recognition Amino Acid Consensus (CRAC) Motif in G-Protein Coupled Receptors. Biochem. Biophys. Res. Commun. 2011, 404, 569–573. 10.1016/j.bbrc.2010.12.031. [DOI] [PubMed] [Google Scholar]
- Sengupta D.; Chattopadhyay A. Identification of Cholesterol Binding Sites in the Serotonin1A Receptor. J. Phys. Chem. B 2012, 116, 12991–12996. 10.1021/jp309888u. [DOI] [PubMed] [Google Scholar]
- Hurst D. P.; Grossfield A.; Lynch D. L.; Feller S.; Romo T. D.; Gawrisch K.; Pitman M. C.; Reggio P. H. A Lipid Pathway for Ligand Binding Is Necessary for a Cannabinoid G Protein-Coupled Receptor. J. Biol. Chem. 2010, 285, 17954–17964. 10.1074/jbc.M109.041590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyman E.; Higgs C.; Kim B.; Lupyan D.; Shelley J. C.; Farid R.; Voth G. A. A Role for a Specific Cholesterol Interaction in Stabilizing the Apo Configuration of the Human A2A Adenosine Receptor. Structure 2009, 17, 1660–1668. 10.1016/j.str.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng H. W.; Laughton C. A.; Doughty S. W. Molecular Dynamics Simulations of the Adenosine A2a Receptor in POPC and POPE Lipid Bilayers: Effects of Membrane on Protein Behavior. J. Chem. Inf. Model. 2014, 54, 573–581. 10.1021/ci400463z. [DOI] [PubMed] [Google Scholar]
- Lee J. Y.; Lyman E. Predictions for Cholesterol Interaction Sites on the A2A Adenosine Receptor. J. Am. Chem. Soc. 2012, 134, 16512–16515. 10.1021/ja307532d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasanna X.; Chattopadhyay A.; Sengupta D.. Role of Lipid-Mediated Effects in β2-Adrenergic Receptor Dimerization. In Biochemical Roles of Eukaryotic Cell Surface Macromolecules; Chakrabarti A., Surolia A., Eds.; Springer International Publishing: Cham, 2015; Vol. 842, pp 247–261, 10.1007/978-3-319-11280-0_16. [DOI] [PubMed] [Google Scholar]
- Neale C.; Herce H. D.; Pomès R.; García A. E. Can Specific Protein-Lipid Interactions Stabilize an Active State of the Beta 2 Adrenergic Receptor?. Biophys. J. 2015, 109, 1652–1662. 10.1016/j.bpj.2015.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cang X.; Du Y.; Mao Y.; Wang Y.; Yang H.; Jiang H. Mapping the Functional Binding Sites of Cholesterol in β2-Adrenergic Receptor by Long-Time Molecular Dynamics Simulations. J. Phys. Chem. B 2013, 117, 1085–1094. 10.1021/jp3118192. [DOI] [PubMed] [Google Scholar]
- Cang X.; Yang L.; Yang J.; Luo C.; Zheng M.; Yu K.; Yang H.; Jiang H. Cholesterol-β1AR Interaction versus Cholesterol-β2AR Interaction. Proteins: Struct., Funct., Genet. 2014, 82, 760–770. 10.1002/prot.24456. [DOI] [PubMed] [Google Scholar]
- Liu W.; Chun E.; Thompson A. A.; Chubukov P.; Xu F.; Katritch V.; Han G. W.; Roth C. B.; Heitman L. H.; IJzerman A. P.; et al. Structural Basis for Allosteric Regulation of GPCRs by Sodium Ions. Science 2012, 337, 232–236. 10.1126/science.1219218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ploier B.; Caro L. N.; Morizumi T.; Pandey K.; Pearring J. N.; Goren M. A.; Finnemann S. C.; Graumann J.; Arshavsky V. Y.; Dittman J. S.; et al. Dimerization Deficiency of Enigmatic Retinitis Pigmentosa-Linked Rhodopsin Mutants. Nat. Commun. 2016, 7, 12832. 10.1038/ncomms12832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goren M. A.; Morizumi T.; Menon I.; Joseph J. S.; Dittman J. S.; Cherezov V.; Stevens R. C.; Ernst O. P.; Menon A. K. Constitutive Phospholipid Scramblase Activity of a G Protein-Coupled Receptor. Nat. Commun. 2014, 5, 5115. 10.1038/ncomms6115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genheden S.; Essex J. W.; Lee A. G. G. Protein Coupled Receptor Interactions with Cholesterol Deep in the Membrane. Biochim. Biophys. Acta, Biomembr. 2017, 1859, 268–281. 10.1016/j.bbamem.2016.12.001. [DOI] [PubMed] [Google Scholar]
- Yuan S.; Wu R.; Latek D.; Trzaskowski B.; Filipek S. Lipid Receptor S1P1 Activation Scheme Concluded from Microsecond All-Atom Molecular Dynamics Simulations. PLoS Comput. Biol. 2013, 9, e1003261. 10.1371/journal.pcbi.1003261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baier C. J.; Fantini J.; Barrantes F. J. Disclosure of Cholesterol Recognition Motifs in Transmembrane Domains of the Human Nicotinic Acetylcholine Receptor. Sci. Rep. 2011, 1, 69. 10.1038/srep00069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng M. H.; Xu Y.; Tang P. Anionic Lipid and Cholesterol Interactions with α4β2 nAChR: Insights from MD Simulations. J. Phys. Chem. B 2009, 113, 6964–6970. 10.1021/jp900714b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocquet N.; Nury H.; Baaden M.; Le Poupon C.; Changeux J.-P.; Delarue M.; Corringer P.-J. X-Ray Structure of a Pentameric Ligand-Gated Ion Channel in an Apparently Open Conformation. Nature 2009, 457, 111–114. 10.1038/nature07462. [DOI] [PubMed] [Google Scholar]
- Singh A. K.; McMillan J.; Bukiya A. N.; Burton B.; Parrill A. L.; Dopico A. M. Multiple Cholesterol Recognition/Interaction Amino Acid Consensus (CRAC) Motifs in Cytosolic C Tail of Slo1 Subunit Determine Cholesterol Sensitivity of Ca2+- and Voltage-Gated K+ (BK) Channels. J. Biol. Chem. 2012, 287, 20509–20521. 10.1074/jbc.M112.356261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenhouse-Dantsker A.; Noskov S.; Durdagi S.; Logothetis D. E.; Levitan I. Identification of Novel Cholesterol-Binding Regions in Kir2 Channels. J. Biol. Chem. 2013, 288, 31154–31164. 10.1074/jbc.M113.496117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedger G.; Shorthouse D.; Koldsø H.; Sansom M. S. P. Free Energy Landscape of Lipid Interactions with Regulatory Binding Sites on the Transmembrane Domain of the EGF Receptor. J. Phys. Chem. B 2016, 120, 8154–8163. 10.1021/acs.jpcb.6b01387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt M. R.; Stansfeld P. J.; Tucker S. J.; Sansom M. S. P. Simulation-Based Prediction of Phosphatidylinositol 4,5-Bisphosphate Binding to an Ion Channel. Biochemistry 2013, 52, 279–281. 10.1021/bi301350s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor J. W.; Klauda J. B. Lipid Membranes with a Majority of Cholesterol: Applications to the Ocular Lens and Aquaporin 0. J. Phys. Chem. B 2011, 115, 6455–6464. 10.1021/jp108650u. [DOI] [PubMed] [Google Scholar]
- Pliotas C.; Naismith J. H. Spectator No More, the Role of the Membrane in Regulating Ion Channel Function. Curr. Opin. Struct. Biol. 2017, 45, 59–66. 10.1016/j.sbi.2016.10.017. [DOI] [PubMed] [Google Scholar]
- Pliotas C.; Dahl A. C. E.; Rasmussen T.; Mahendran K. R.; Smith T. K.; Marius P.; Gault J.; Banda T.; Rasmussen A.; Miller S.; et al. The Role of Lipids in Mechanosensation. Nat. Struct. Mol. Biol. 2015, 22, 991–998. 10.1038/nsmb.3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arkhipov A.; Shan Y.; Das R.; Endres N. F.; Eastwood M. P.; Wemmer D. E.; Kuriyan J.; Shaw D. E. Architecture and Membrane Interactions of the EGF Receptor. Cell 2013, 152, 557–569. 10.1016/j.cell.2012.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedger G.; Sansom M. S. P.; Koldsø H. The Juxtamembrane Regions of Human Receptor Tyrosine Kinases Exhibit Conserved Interaction Sites with Anionic Lipids. Sci. Rep. 2015, 5, 9198. 10.1038/srep09198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash A.; Janosi L.; Doxastakis M. Self-Association of Models of Transmembrane Domains of ErbB Receptors in a Lipid Bilayer. Biophys. J. 2010, 99, 3657–3665. 10.1016/j.bpj.2010.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abd Halim K. B.; Koldsø H.; Sansom M. S. P. Interactions of the EGFR Juxtamembrane Domain with PIP2-Containing Lipid Bilayers: Insights from Multiscale Molecular Dynamics Simulations. Biochim. Biophys. Acta, Gen. Subj. 2015, 1850, 1017–1025. 10.1016/j.bbagen.2014.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavent M.; Seiradake E.; Jones E. Y.; Sansom M. S. P. Structures of the EphA2 Receptor at the Membrane: Role of Lipid Interactions. Structure 2016, 24, 337–347. 10.1016/j.str.2015.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poblete H.; Oyarzún I.; Olivero P.; Comer J.; Zuñiga M.; Sepulveda R. V.; Báez-Nieto D.; González Leon C.; González-Nilo F.; Latorre R. Molecular Determinants of Phosphatidylinositol 4,5-Bisphosphate (PI(4,5)P2) Binding to Transient Receptor Potential V1 (TRPV1) Channels. J. Biol. Chem. 2015, 290, 2086–2098. 10.1074/jbc.M114.613620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antollini S. S.; Barrantes F. J. Fatty Acid Regulation of Voltage- and Ligand-Gated Ion Channel Function. Front. Physiol. 2016, 7, 573. 10.3389/fphys.2016.00573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baenziger J. E.; Hénault C. M.; Therien J. P. D.; Sun J. Nicotinic Acetylcholine Receptor–Lipid Interactions: Mechanistic Insight and Biological Function. Biochim. Biophys. Acta, Biomembr. 2015, 1848, 1806–1817. 10.1016/j.bbamem.2015.03.010. [DOI] [PubMed] [Google Scholar]
- Barrantes F. J.; Bermudez V.; Borroni M. V.; Antollini S. S.; Pediconi M. F.; Baier J. C.; Bonini I.; Gallegos C.; Roccamo A. M.; Valles A. S.; et al. Boundary Lipids In The Nicotinic Acetylcholine Receptor Microenvironment. J. Mol. Neurosci. 2010, 40, 87–90. 10.1007/s12031-009-9262-z. [DOI] [PubMed] [Google Scholar]
- Taylor K. C.; Sanders C. R. Regulation of KCNQ/Kv7 Family Voltage-Gated K+ Channels by Lipids. Biochim. Biophys. Acta, Biomembr. 2017, 1859, 586–597. 10.1016/j.bbamem.2016.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalli A. C.; Sansom M. S. P.; Reithmeier R. A. F. Molecular Dynamics Simulations of the Bacterial UraA H+-Uracil Symporter in Lipid Bilayers Reveal a Closed State and a Selective Interaction with Cardiolipin. PLoS Comput. Biol. 2015, 11, e1004123. 10.1371/journal.pcbi.1004123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolivar J. H.; Muñoz-García J. C.; Castro-Dopico T.; Dijkman P. M.; Stansfeld P. J.; Watts A. Interaction of Lipids with the Neurotensin Receptor 1. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 1278–1287. 10.1016/j.bbamem.2016.02.032. [DOI] [PubMed] [Google Scholar]
- Kalli A. C.; Rog T.; Vattulainen I.; Campbell I. D.; Sansom M. S. P. The Integrin Receptor in Biologically Relevant Bilayers: Insights from Molecular Dynamics Simulations. J. Membr. Biol. 2017, 250, 337–351. 10.1007/s00232-016-9908-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orłowski A.; Kukkurainen S.; Pöyry A.; Rissanen S.; Vattulainen I.; Hytönen V. P.; Róg T. PIP2 and Talin Join Forces to Activate Integrin. J. Phys. Chem. B 2015, 119, 12381–12389. 10.1021/acs.jpcb.5b06457. [DOI] [PubMed] [Google Scholar]
- Human Genome Sequencing Consortium I. Finishing the Euchromatic Sequence of the Human Genome. Nature 2004, 431, 931–945. 10.1038/nature03001. [DOI] [PubMed] [Google Scholar]
- Nørregaard Jensen O. Modification-Specific Proteomics: Characterization of Post-Translational Modifications by Mass Spectrometry. Curr. Opin. Chem. Biol. 2004, 8, 33–41. 10.1016/j.cbpa.2003.12.009. [DOI] [PubMed] [Google Scholar]
- The Universal Protein Resource (UniProt) in 2010. Nucleic Acids Res. 2010, 38, D142–D148. 10.1093/nar/gkp846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Qian C.; Cao X. Post-Translational Modification Control of Innate Immunity. Immunity 2016, 45, 15–30. 10.1016/j.immuni.2016.06.020. [DOI] [PubMed] [Google Scholar]
- Bah A.; Forman-Kay J. D. Modulation of Intrinsically Disordered Protein Function by Post-Translational Modifications. J. Biol. Chem. 2016, 291, 6696–6705. 10.1074/jbc.R115.695056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi A.; Kishida S.; Yamamoto H. Regulation of Wnt Signaling by Protein-Protein Interaction and Post-Translational Modifications. Exp. Mol. Med. 2006, 38, 1–10. 10.1038/emm.2006.1. [DOI] [PubMed] [Google Scholar]
- Zhao S.; Xu W.; Jiang W.; Yu W.; Lin Y.; Zhang T.; Yao J.; Zhou L.; Zeng Y.; Li H.; et al. Regulation of Cellular Metabolism by Protein Lysine Acetylation. Science 2010, 327, 1000–1004. 10.1126/science.1179689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deribe Y. L.; Pawson T.; Dikic I. Post-Translational Modifications in Signal Integration. Nat. Struct. Mol. Biol. 2010, 17, 666–672. 10.1038/nsmb.1842. [DOI] [PubMed] [Google Scholar]
- Fonseca-Maldonado R.; Vieira D. S.; Alponti J. S.; Bonneil E.; Thibault P.; Ward R. J. Engineering the Pattern of Protein Glycosylation Modulates the Thermostability of a GH11 Xylanase. J. Biol. Chem. 2013, 288, 25522–25534. 10.1074/jbc.M113.485953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu D.; Yang C.; Liu Z. How Hydrophobicity and the Glycosylation Site of Glycans Affect Protein Folding and Stability: A Molecular Dynamics Simulation. J. Phys. Chem. B 2012, 116, 390–400. 10.1021/jp203926r. [DOI] [PubMed] [Google Scholar]
- Subedi G. P.; Falconer D. J.; Barb A. W. Carbohydrate–Polypeptide Contacts in the Antibody Receptor CD16A Identified through Solution NMR Spectroscopy. Biochemistry 2017, 56, 3174–3177. 10.1021/acs.biochem.7b00392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziomkiewicz I.; Loman A.; Klement R.; Fritsch C.; Klymchenko A. S.; Bunt G.; Jovin T. M.; Arndt-Jovin D. J. Dynamic Conformational Transitions of the EGF Receptor in Living Mammalian Cells Determined by FRET and Fluorescence Lifetime Imaging Microscopy: EGFR Conformations Determined in Live Cells by FLIM. Cytometry, Part A 2013, 83, 794–805. 10.1002/cyto.a.22311. [DOI] [PubMed] [Google Scholar]
- Kozer N.; Henderson C.; Jackson J. T.; Nice E. C.; Burgess A. W.; Clayton A. H. A. Evidence for Extended YFP-EGFR Dimers in the Absence of Ligand on the Surface of Living Cells. Phys. Biol. 2011, 8, 066002. 10.1088/1478-3975/8/6/066002. [DOI] [PubMed] [Google Scholar]
- Polley A.; Orłowski A.; Danne R.; Gurtovenko A. A.; Bernardino de la Serna J.; Eggeling C.; Davis S. J.; Róg T.; Vattulainen I. Glycosylation and Lipids Working in Concert Direct CD2 Ectodomain Orientation and Presentation. J. Phys. Chem. Lett. 2017, 8, 1060–1066. 10.1021/acs.jpclett.6b02824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danne R.; Poojari C.; Martinez-Seara H.; Rissanen S.; Lolicato F.; Róg T.; Vattulainen I. DoGlycans – Tools for Preparing Carbohydrate Structures for Atomistic Simulations of Glycoproteins, Glycolipids, and Carbohydrate Polymers for GROMACS. J. Chem. Inf. Model. 2017, 57, 2401–2406. 10.1021/acs.jcim.7b00237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakkaraju A. K.; Abrami L.; Lemmin T.; Blaskovic S.; Kunz B.; Kihara A.; Dal Peraro M.; van der Goot F. G. Palmitoylated Calnexin Is a Key Component of the Ribosome-Translocon Complex: Palmitoylated Calnexin Is a Key Component of the RTC. EMBO J. 2012, 31, 1823–1835. 10.1038/emboj.2012.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thukral L.; Sengupta D.; Ramkumar A.; Murthy D.; Agrawal N.; Gokhale R. S. The Molecular Mechanism Underlying Recruitment and Insertion of Lipid-Anchored LC3 Protein into Membranes. Biophys. J. 2015, 109, 2067–2078. 10.1016/j.bpj.2015.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janosi L.; Gorfe A. A. Segregation of Negatively Charged Phospholipids by the Polycationic and Farnesylated Membrane Anchor of Kras. Biophys. J. 2010, 99, 3666–3674. 10.1016/j.bpj.2010.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash P.; Zhou Y.; Liang H.; Hancock J. F.; Gorfe A. A. Oncogenic K-Ras Binds to an Anionic Membrane in Two Distinct Orientations: A Molecular Dynamics Analysis. Biophys. J. 2016, 110, 1125–1138. 10.1016/j.bpj.2016.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edler E.; Schulze E.; Stein M. Membrane Localization and Dynamics of Geranylgeranylated Rab5 Hypervariable Region. Biochim. Biophys. Acta, Biomembr. 2017, 1859, 1335–1349. 10.1016/j.bbamem.2017.04.021. [DOI] [PubMed] [Google Scholar]
- Vogel A.; Katzka C. P.; Waldmann H.; Arnold K.; Brown M. F.; Huster D. Lipid Modifications of a Ras Peptide Exhibit Altered Packing and Mobility versus Host Membrane as Detected by 2H Solid-State NMR. J. Am. Chem. Soc. 2005, 127, 12263–12272. 10.1021/ja051856c. [DOI] [PubMed] [Google Scholar]
- Zhang T.; Luo Q.; Yang L.; Jiang H.; Yang H. Characterizing the Interactions of Two Lipid Modifications with Lipid Rafts: Farnesyl Anchors vs. Palmitoyl Anchors. Eur. Biophys. J. 2018, 47, 19–30. 10.1007/s00249-017-1217-7. [DOI] [PubMed] [Google Scholar]
- Sprong H.; van der Sluijs P.; van Meer G. How Proteins Move Lipids and Lipids Move Proteins. Nat. Rev. Mol. Cell Biol. 2001, 2, 504–513. 10.1038/35080071. [DOI] [PubMed] [Google Scholar]
- Mellman I.; Nelson W. J. Coordinated Protein Sorting, Targeting and Distribution in Polarized Cells. Nat. Rev. Mol. Cell Biol. 2008, 9, 833–845. 10.1038/nrm2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen P. J. Endosomal Sorting and Signalling: An Emerging Role for Sorting Nexins. Nat. Rev. Mol. Cell Biol. 2008, 9, 574–582. 10.1038/nrm2427. [DOI] [PubMed] [Google Scholar]
- Jahn R.; Scheller R. H. SNAREs — Engines for Membrane Fusion. Nat. Rev. Mol. Cell Biol. 2006, 7, 631–643. 10.1038/nrm2002. [DOI] [PubMed] [Google Scholar]
- Santiago-Tirado F. H.; Bretscher A. Membrane-Trafficking Sorting Hubs: Cooperation between PI4P and Small GTPases at the Trans-Golgi Network. Trends Cell Biol. 2011, 21, 515–525. 10.1016/j.tcb.2011.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viaud J.; Mansour R.; Antkowiak A.; Mujalli A.; Valet C.; Chicanne G.; Xuereb J.-M.; Terrisse A.-D.; Séverin S.; Gratacap M.-P.; et al. Phosphoinositides: Important Lipids in the Coordination of Cell Dynamics. Biochimie 2016, 125, 250–258. 10.1016/j.biochi.2015.09.005. [DOI] [PubMed] [Google Scholar]
- Mayinger P. Phosphoinositides and Vesicular Membrane Traffic. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2012, 1821, 1104–1113. 10.1016/j.bbalip.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chap H. Forty Five Years with Membrane Phospholipids, Phospholipases and Lipid Mediators: A Historical Perspective. Biochimie 2016, 125, 234–249. 10.1016/j.biochi.2016.04.002. [DOI] [PubMed] [Google Scholar]
- Marsh D.; Horváth L. I. Structure, Dynamics and Composition of the Lipid-Protein Interface. Perspectives from Spin-Labelling. Biochim. Biophys. Acta, Rev. Biomembr. 1998, 1376, 267–296. 10.1016/S0304-4157(98)00009-4. [DOI] [PubMed] [Google Scholar]
- Cartailler J.-P.; Luecke H. X-Ray Crystallographic Analysis of Lipid-Protein Interactions in the Bacteriorhodopsin Purple Membrane. Annu. Rev. Biophys. Biomol. Struct. 2003, 32, 285–310. 10.1146/annurev.biophys.32.110601.142516. [DOI] [PubMed] [Google Scholar]
- Schmidt T.; Suk J.-E.; Ye F.; Situ A. J.; Mazumder P.; Ginsberg M. H.; Ulmer T. S. Annular Anionic Lipids Stabilize the Integrin αIIbβ3 Transmembrane Complex. J. Biol. Chem. 2015, 290, 8283–8293. 10.1074/jbc.M114.623504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggensperger S.; Fisette O.; Parcej D.; Schäfer L. V.; Tampé R. An Annular Lipid Belt Is Essential for Allosteric Coupling and Viral Inhibition of the Antigen Translocation Complex TAP (Transporter Associated with Antigen Processing). J. Biol. Chem. 2014, 289, 33098–33108. 10.1074/jbc.M114.592832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polozova A.; Litman B. J. Cholesterol Dependent Recruitment of Di22:6-PC by a G Protein-Coupled Receptor into Lateral Domains. Biophys. J. 2000, 79, 2632–2643. 10.1016/S0006-3495(00)76502-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer L. V.; de Jong D. H.; Holt A.; Rzepiela A. J.; de Vries A. H.; Poolman B.; Killian J. A.; Marrink S. J. Lipid Packing Drives the Segregation of Transmembrane Helices into Disordered Lipid Domains in Model Membranes. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 1343–1348. 10.1073/pnas.1009362108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jong D. H.; Lopez C. A.; Marrink S. J. Molecular View on Protein Sorting into Liquid-Ordered Membrane Domains Mediated by Gangliosides and Lipid Anchors. Faraday Discuss. 2013, 161, 347–363. 10.1039/C2FD20086D. [DOI] [PubMed] [Google Scholar]
- Li H.; Gorfe A. A. Aggregation of Lipid-Anchored Full-Length H-Ras in Lipid Bilayers: Simulations with the MARTINI Force Field. PLoS One 2013, 8, e71018. 10.1371/journal.pone.0071018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo J.; Cui Q. Membrane-Mediated Protein-Protein Interactions and Connection to Elastic Models: A Coarse-Grained Simulation Analysis of Gramicidin A Association. Biophys. J. 2013, 104, 128–138. 10.1016/j.bpj.2012.11.3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H.; Chen L.; Gao L.; Fang W. Nanodomain Formation of Ganglioside GM1 in Lipid Membrane: Effects of Cholera Toxin-Mediated Cross-Linking. Langmuir 2015, 31, 9105–9114. 10.1021/acs.langmuir.5b01866. [DOI] [PubMed] [Google Scholar]
- Pezeshkian W.; Hansen A. G.; Johannes L.; Khandelia H.; Shillcock J. C.; Kumar P. B. S.; Ipsen J. H. Membrane Invagination Induced by Shiga Toxin B-Subunit: From Molecular Structure to Tube Formation. Soft Matter 2016, 12, 5164–5171. 10.1039/C6SM00464D. [DOI] [PubMed] [Google Scholar]
- Pezeshkian W.; Gao H.; Arumugam S.; Becken U.; Bassereau P.; Florent J.-C.; Ipsen J. H.; Johannes L.; Shillcock J. C. Mechanism of Shiga Toxin Clustering on Membranes. ACS Nano 2017, 11, 314–324. 10.1021/acsnano.6b05706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López C. A.; Sethi A.; Goldstein B.; Wilson B. S.; Gnanakaran S. Membrane-Mediated Regulation of the Intrinsically Disordered CD3ϵ Cytoplasmic Tail of the TCR. Biophys. J. 2015, 108, 2481–2491. 10.1016/j.bpj.2015.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun F.; Chen L.; Wei P.; Chai M.; Ding X.; Xu L.; Luo S.-Z. Dimerization and Structural Stability of Amyloid Precursor Proteins Affected by the Membrane Microenvironments. J. Chem. Inf. Model. 2017, 57, 1375–1387. 10.1021/acs.jcim.7b00196. [DOI] [PubMed] [Google Scholar]
- Lorent J. H.; Diaz-Rohrer B.; Lin X.; Spring K.; Gorfe A. A.; Levental K. R.; Levental I. Structural Determinants and Functional Consequences of Protein Affinity for Membrane Rafts. Nat. Commun. 2017, 8, 1219. 10.1038/s41467-017-01328-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpe H. J.; Stevens T. J.; Munro S. A Comprehensive Comparison of Transmembrane Domains Reveals Organelle-Specific Properties. Cell 2010, 142, 158–169. 10.1016/j.cell.2010.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X.; Gorfe A. A.; Levental I. Protein Partitioning into Ordered Membrane Domains: Insights from Simulations. Biophys. J. 2018, 114, 1936–1944. 10.1016/j.bpj.2018.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser H.-J.; Orlowski A.; Rog T.; Nyholm T. K. M.; Chai W.; Feizi T.; Lingwood D.; Vattulainen I.; Simons K. Lateral Sorting in Model Membranes by Cholesterol-Mediated Hydrophobic Matching. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 16628–16633. 10.1073/pnas.1103742108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grau B.; Javanainen M.; García-Murria M. J.; Kulig W.; Vattulainen I.; Mingarro I.; Martínez-Gil L. The Role of Hydrophobic Matching on Transmembrane Helix Packing in Cells. Cell Stress 2017, 1, 90–106. 10.15698/cst2017.11.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidmann T.; Sobel A.; Popot J.-L.; Changeux J.-P. Reconstitution of a Functional Acetylcholine Receptor. Conservation of the Conformational and Allosteric Transitions and Recovery of the Permeability Response; Role of Lipids. Eur. J. Biochem. 1980, 110, 35–55. 10.1111/j.1432-1033.1980.tb04839.x. [DOI] [PubMed] [Google Scholar]
- daCosta C. J. B.; Medaglia S. A.; Lavigne N.; Wang S.; Carswell C. L.; Baenziger J. E. Anionic Lipids Allosterically Modulate Multiple Nicotinic Acetylcholine Receptor Conformational Equilibria. J. Biol. Chem. 2009, 284, 33841–33849. 10.1074/jbc.M109.048280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baenziger J. E.; Domville J. A.; Therien J. P. D.. The Role of Cholesterol in the Activation of Nicotinic Acetylcholine Receptors. Current Topics in Membranes; Elsevier, 2017; Vol. 80, pp 95–137, 10.1016/bs.ctm.2017.05.002. [DOI] [PubMed] [Google Scholar]
- Domville J. A.; Baenziger J. E. An Allosteric Link Connecting the Lipid-Protein Interface to the Gating of the Nicotinic Acetylcholine Receptor. Sci. Rep. 2018, 8, 3898. 10.1038/s41598-018-22150-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh M.; Wang L. C.; Ramesh R.; Morgan L. K.; Kenney L. J.; Anand G. S. Lipid-Mediated Regulation of Embedded Receptor Kinases via Parallel Allosteric Relays. Biophys. J. 2017, 112, 643–654. 10.1016/j.bpj.2016.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coskun U.; Grzybek M.; Drechsel D.; Simons K. Regulation of Human EGF Receptor by Lipids. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 9044–9048. 10.1073/pnas.1105666108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasanna X.; Chattopadhyay A.; Sengupta D. Cholesterol Modulates the Dimer Interface of the β2-Adrenergic Receptor via Cholesterol Occupancy Sites. Biophys. J. 2014, 106, 1290–1300. 10.1016/j.bpj.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q.; Zhou P.; Chen Z.; Li M.; Jiang H.; Gao Z.; Yang H. Dynamic PIP2 Interactions with Voltage Sensor Elements Contribute to KCNQ2 Channel Gating. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 20093–20098. 10.1073/pnas.1312483110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delemotte L.; Tarek M.; Klein M. L.; Amaral C.; Treptow W. Intermediate States of the Kv1.2 Voltage Sensor from Atomistic Molecular Dynamics Simulations. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 6109–6114. 10.1073/pnas.1102724108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt D.; Jiang Q.-X.; MacKinnon R. Phospholipids and the Origin of Cationic Gating Charges in Voltage Sensors. Nature 2006, 444, 775–779. 10.1038/nature05416. [DOI] [PubMed] [Google Scholar]
- Xu Y.; Ramu Y.; Lu Z. Removal of Phospho-Head Groups of Membrane Lipids Immobilizes Voltage Sensors of K+ Channels. Nature 2008, 451, 826–829. 10.1038/nature06618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramu Y.; Xu Y.; Lu Z. Enzymatic Activation of Voltage-Gated Potassium Channels. Nature 2006, 442, 696–699. 10.1038/nature04880. [DOI] [PubMed] [Google Scholar]
- Lingwood D.; Binnington B.; Róg T.; Vattulainen I.; Grzybek M.; Coskun Ü.; Lingwood C. A.; Simons K. Cholesterol Modulates Glycolipid Conformation and Receptor Activity. Nat. Chem. Biol. 2011, 7, 260–262. 10.1038/nchembio.551. [DOI] [PubMed] [Google Scholar]
- Prasanna X.; Jafurulla M.; Sengupta D.; Chattopadhyay A. The Ganglioside GM1 Interacts with the Serotonin1A Receptor via the Sphingolipid Binding Domain. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 2818–2826. 10.1016/j.bbamem.2016.08.009. [DOI] [PubMed] [Google Scholar]
- Arcario M. J.; Mayne C. G.; Tajkhorshid E. Atomistic Models of General Anesthetics for Use in in Silico Biological Studies. J. Phys. Chem. B 2014, 118, 12075–12086. 10.1021/jp502716m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arcario M. J.; Mayne C. G.; Tajkhorshid E. A Membrane-Embedded Pathway Delivers General Anesthetics to Two Interacting Binding Sites in the Gloeobacter Violaceus Ion Channel. J. Biol. Chem. 2017, 292, 9480–9492. 10.1074/jbc.M117.780197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nury H.; Van Renterghem C.; Weng Y.; Tran A.; Baaden M.; Dufresne V.; Changeux J.-P.; Sonner J. M.; Delarue M.; Corringer P.-J. X-Ray Structures of General Anaesthetics Bound to a Pentameric Ligand-Gated Ion Channel. Nature 2011, 469, 428–431. 10.1038/nature09647. [DOI] [PubMed] [Google Scholar]
- Hanson S. M.; Newstead S.; Swartz K. J.; Sansom M. S. P. Capsaicin Interaction with TRPV1 Channels in a Lipid Bilayer: Molecular Dynamics Simulation. Biophys. J. 2015, 108, 1425–1434. 10.1016/j.bpj.2015.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melo M. N.; Arnarez C.; Sikkema H.; Kumar N.; Walko M.; Berendsen H. J. C.; Kocer A.; Marrink S. J.; Ingólfsson H. I. High-Throughput Simulations Reveal Membrane-Mediated Effects of Alcohols on MscL Gating. J. Am. Chem. Soc. 2017, 139, 2664–2671. 10.1021/jacs.6b11091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerabek H.; Pabst G.; Rappolt M.; Stockner T. Membrane-Mediated Effect on Ion Channels Induced by the Anesthetic Drug Ketamine. J. Am. Chem. Soc. 2010, 132, 7990–7997. 10.1021/ja910843d. [DOI] [PubMed] [Google Scholar]
- Mojumdar E. H.; Lyubartsev A. P. Molecular Dynamics Simulations of Local Anesthetic Articaine in a Lipid Bilayer. Biophys. Chem. 2010, 153, 27–35. 10.1016/j.bpc.2010.10.001. [DOI] [PubMed] [Google Scholar]
- Skjevik Å. A.; Haug B. E.; Lygre H.; Teigen K. Intramolecular Hydrogen Bonding in Articaine Can Be Related to Superior Bone Tissue Penetration: A Molecular Dynamics Study. Biophys. Chem. 2011, 154, 18–25. 10.1016/j.bpc.2010.12.002. [DOI] [PubMed] [Google Scholar]
- Cascales J. J. L.; Costa S. D. O.; Porasso R. D. Thermodynamic Study of Benzocaine Insertion into Different Lipid Bilayers. J. Chem. Phys. 2011, 135, 135103. 10.1063/1.3643496. [DOI] [PubMed] [Google Scholar]
- Hansen A. H.; Sørensen K. T.; Mathieu R.; Serer A.; Duelund L.; Khandelia H.; Hansen P. L.; Simonsen A. C. Propofol Modulates the Lipid Phase Transition and Localizes near the Headgroup of Membranes. Chem. Phys. Lipids 2013, 175–176, 84–91. 10.1016/j.chemphyslip.2013.08.002. [DOI] [PubMed] [Google Scholar]
- do Canto A. M. T. M.; Carvalho A. J. P.; Ramalho J. P. P.; Loura L. M. S. Molecular Dynamics Simulations of T-20 HIV Fusion Inhibitor Interacting with Model Membranes. Biophys. Chem. 2011, 159, 275–286. 10.1016/j.bpc.2011.08.001. [DOI] [PubMed] [Google Scholar]
- Leonis G.; Czyżnikowska Ż.; Megariotis G.; Reis H.; Papadopoulos M. G. Computational Studies of Darunavir into HIV-1 Protease and DMPC Bilayer: Necessary Conditions for Effective Binding and the Role of the Flaps. J. Chem. Inf. Model. 2012, 52, 1542–1558. 10.1021/ci300014z. [DOI] [PubMed] [Google Scholar]
- Laurent B.; Murail S.; Shahsavar A.; Sauguet L.; Delarue M.; Baaden M. Sites of Anesthetic Inhibitory Action on a Cationic Ligand-Gated Ion Channel. Structure 2016, 24, 595–605. 10.1016/j.str.2016.02.014. [DOI] [PubMed] [Google Scholar]
- Woll K. A.; Peng W.; Liang Q.; Zhi L.; Jacobs J. A.; Maciunas L.; Bhanu N.; Garcia B. A.; Covarrubias M.; Loll P. J.; et al. Photoaffinity Ligand for the Inhalational Anesthetic Sevoflurane Allows Mechanistic Insight into Potassium Channel Modulation. ACS Chem. Biol. 2017, 12, 1353–1362. 10.1021/acschembio.7b00222. [DOI] [PubMed] [Google Scholar]
- Sierra-Valdez F. J.; Forero-Quintero L. S.; Zapata-Morin P. A.; Costas M.; Chavez-Reyes A.; Ruiz-Suárez J. C. The Influence of Non Polar and Polar Molecules in Mouse Motile Cells Membranes and Pure Lipid Bilayers. PLoS One 2013, 8, e59364. 10.1371/journal.pone.0059364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amjad-Iranagh S.; Yousefpour A.; Haghighi P.; Modarress H. Effects of Protein Binding on a Lipid Bilayer Containing Local Anesthetic Articaine, and the Potential of Mean Force Calculation: A Molecular Dynamics Simulation Approach. J. Mol. Model. 2013, 19, 3831–3842. 10.1007/s00894-013-1917-6. [DOI] [PubMed] [Google Scholar]
- Booker R. D.; Sum A. K. Biophysical Changes Induced by Xenon on Phospholipid Bilayers. Biochim. Biophys. Acta, Biomembr. 2013, 1828, 1347–1356. 10.1016/j.bbamem.2013.01.016. [DOI] [PubMed] [Google Scholar]
- Martini M. F.; Pickholz M. Molecular Dynamics Study of Uncharged Bupivacaine Enantiomers in Phospholipid Bilayers. Int. J. Quantum Chem. 2012, 112, 3341–3345. 10.1002/qua.24208. [DOI] [Google Scholar]
- Bernardi R. C.; Pascutti P. G. Hybrid QM/MM Molecular Dynamics Study of Benzocaine in a Membrane Environment: How Does a Quantum Mechanical Treatment of Both Anesthetic and Lipids Affect Their Interaction. J. Chem. Theory Comput. 2012, 8, 2197–2203. 10.1021/ct300213u. [DOI] [PubMed] [Google Scholar]
- López Cascales J. J.; Oliveira Costa S. D. Effect of the Interfacial Tension and Ionic Strength on the Thermodynamic Barrier Associated to the Benzocaine Insertion into a Cell Membrane. Biophys. Chem. 2013, 172, 1–7. 10.1016/j.bpc.2012.12.001. [DOI] [PubMed] [Google Scholar]
- Darvas M.; Hoang P. N. M.; Picaud S.; Sega M.; Jedlovszky P. Anesthetic Molecules Embedded in a Lipid Membrane: A Computer Simulation Study. Phys. Chem. Chem. Phys. 2012, 14, 12956–12969. 10.1039/c2cp41581j. [DOI] [PubMed] [Google Scholar]
- Fábián B.; Sega M.; Voloshin V. P.; Medvedev N. N.; Jedlovszky P. Lateral Pressure Profile and Free Volume Properties in Phospholipid Membranes Containing Anesthetics. J. Phys. Chem. B 2017, 121, 2814–2824. 10.1021/acs.jpcb.7b00990. [DOI] [PubMed] [Google Scholar]
- Fábián B.; Darvas M.; Picaud S.; Sega M.; Jedlovszky P. The Effect of Anaesthetics on the Properties of a Lipid Membrane in the Biologically Relevant Phase: A Computer Simulation Study. Phys. Chem. Chem. Phys. 2015, 17, 14750–14760. 10.1039/C5CP00851D. [DOI] [PubMed] [Google Scholar]
- Chau P.-L.; Tu K. M.; Liang K. K.; Todorov I. T.; Roser S. J.; Barker R.; Matubayasi N. The Effect of Pressure on Halothane Binding to Hydrated DMPC Bilayers. Mol. Phys. 2012, 110, 1461–1467. 10.1080/00268976.2012.659682. [DOI] [Google Scholar]
- Tu K. M.; Matubayasi N.; Liang K. K.; Todorov I. T.; Chan S. L.; Chau P.-L. A Possible Molecular Mechanism for the Pressure Reversal of General Anaesthetics: Aggregation of Halothane in POPC Bilayers at High Pressure. Chem. Phys. Lett. 2012, 543, 148–154. 10.1016/j.cplett.2012.06.044. [DOI] [Google Scholar]
- Woll K. A.; Murlidaran S.; Pinch B. J.; Hénin J.; Wang X.; Salari R.; Covarrubias M.; Dailey W. P.; Brannigan G.; Garcia B. A.; et al. A Novel Bifunctional Alkylphenol Anesthetic Allows Characterization of γ-Aminobutyric Acid, Type A (GABAA), Receptor Subunit Binding Selectivity in Synaptosomes. J. Biol. Chem. 2016, 291, 20473–20486. 10.1074/jbc.M116.736975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruns D.; Riedel D.; Klingauf J.; Jahn R. Quantal Release of Serotonin. Neuron 2000, 28, 205–220. 10.1016/S0896-6273(00)00097-0. [DOI] [PubMed] [Google Scholar]
- Cantor R. S. Receptor Desensitization by Neurotransmitters in Membranes: Are Neurotransmitters the Endogenous Anesthetics?. Biochemistry 2003, 42, 11891–11897. 10.1021/bi034534z. [DOI] [PubMed] [Google Scholar]
- Milutinovic P. S.; Yang L.; Cantor R. S.; Eger E. I.; Sonner J. M. Anesthetic-Like Modulation of a γ-Aminobutyric Acid Type A, Strychnine-Sensitive Glycine, and N-Methyl-d-Aspartate Receptors by Coreleased Neurotransmitters: Anesth. Anesth. Analg. 2007, 105, 386–392. 10.1213/01.ane.0000267258.17197.7d. [DOI] [PubMed] [Google Scholar]
- Seeger H. M.; Gudmundsson M. L.; Heimburg T. How Anesthetics, Neurotransmitters, and Antibiotics Influence the Relaxation Processes in Lipid Membranes. J. Phys. Chem. B 2007, 111, 13858–13866. 10.1021/jp075346b. [DOI] [PubMed] [Google Scholar]
- Sonner J. M.; Cantor R. S. Molecular Mechanisms of Drug Action: An Emerging View. Annu. Rev. Biophys. 2013, 42, 143–167. 10.1146/annurev-biophys-083012-130341. [DOI] [PubMed] [Google Scholar]
- Cantor R. S. Lateral Pressures in Cell Membranes: A Mechanism for Modulation of Protein Function. J. Phys. Chem. B 1997, 101, 1723–1725. 10.1021/jp963911x. [DOI] [Google Scholar]
- Peters G. H.; Werge M.; Elf-Lind M. N.; Madsen J. J.; Velardez G. F.; Westh P. Interaction of Neurotransmitters with a Phospholipid Bilayer: A Molecular Dynamics Study. Chem. Phys. Lipids 2014, 184, 7–17. 10.1016/j.chemphyslip.2014.08.003. [DOI] [PubMed] [Google Scholar]
- Peters G. H.; Wang C.; Cruys-Bagger N.; Velardez G. F.; Madsen J. J.; Westh P. Binding of Serotonin to Lipid Membranes. J. Am. Chem. Soc. 2013, 135, 2164–2171. 10.1021/ja306681d. [DOI] [PubMed] [Google Scholar]
- Diener E.; Biswas-Diener R.. Happiness; Blackwell Publishing Ltd.: Oxford, UK, 2008. 10.1002/9781444305159. [DOI] [Google Scholar]
- Robertson R. W. The Relationships between Leisure and Happiness. World Leis. J. 2016, 58, 242–244. 10.1080/16078055.2016.1225880. [DOI] [Google Scholar]
- Orłowski A.; Grzybek M.; Bunker A.; Pasenkiewicz-Gierula M.; Vattulainen I.; Männistö P. T.; Róg T. Strong Preferences of Dopamine and l-Dopa towards Lipid Head Group: Importance of Lipid Composition and Implication for Neurotransmitter Metabolism: Neurotransmitters and Membranes. J. Neurochem. 2012, 122, 681–690. 10.1111/j.1471-4159.2012.07813.x. [DOI] [PubMed] [Google Scholar]
- Harrison P. J.; Weinberger D. R. Schizophrenia Genes, Gene Expression and Neuropathology: On the Matter of Their Convergence. Mol. Psychiatry 2005, 10, 40–68. 10.1038/sj.mp.4001558. [DOI] [PubMed] [Google Scholar]
- Schmitt A.; Wilczek K.; Blennow K.; Maras A.; Jatzko A.; Petroianu G.; Braus D. F.; Gattaz W. F. Altered Thalamic Membrane Phospholipids in Schizophrenia: A Postmortem Study. Biol. Psychiatry 2004, 56, 41–45. 10.1016/j.biopsych.2004.03.019. [DOI] [PubMed] [Google Scholar]
- Wang C.; Ye F.; Velardez G. F.; Peters G. H.; Westh P. Affinity of Four Polar Neurotransmitters for Lipid Bilayer Membranes. J. Phys. Chem. B 2011, 115, 196–203. 10.1021/jp108368w. [DOI] [PubMed] [Google Scholar]
- Shen C.; Xue M.; Qiu H.; Guo W. Insertion of Neurotransmitters into a Lipid Bilayer Membrane and Its Implication on Membrane Stability: A Molecular Dynamics Study. ChemPhysChem 2017, 18, 626–633. 10.1002/cphc.201601184. [DOI] [PubMed] [Google Scholar]
- Postila P. A.; Vattulainen I.; Róg T. Selective Effect of Cell Membrane on Synaptic Neurotransmission. Sci. Rep. 2016, 6, 19345. 10.1038/srep19345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reigada R. Atomistic Study of Lipid Membranes Containing Chloroform: Looking for a Lipid-Mediated Mechanism of Anesthesia. PLoS One 2013, 8, e52631. 10.1371/journal.pone.0052631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mokkila S.; Postila P. A.; Rissanen S.; Juhola H.; Vattulainen I.; Róg T. Calcium Assists Dopamine Release by Preventing Aggregation on the Inner Leaflet of Presynaptic Vesicles. ACS Chem. Neurosci. 2017, 8, 1242–1250. 10.1021/acschemneuro.6b00395. [DOI] [PubMed] [Google Scholar]
- Puchkov D.; Haucke V. Greasing the Synaptic Vesicle Cycle by Membrane Lipids. Trends Cell Biol. 2013, 23, 493–503. 10.1016/j.tcb.2013.05.002. [DOI] [PubMed] [Google Scholar]
- Whited A. M.; Johs A. The Interactions of Peripheral Membrane Proteins with Biological Membranes. Chem. Phys. Lipids 2015, 192, 51–59. 10.1016/j.chemphyslip.2015.07.015. [DOI] [PubMed] [Google Scholar]
- Zwaal R. F. A.; Comfurius P.; Bevers E. M. Lipid–Protein Interactions in Blood Coagulation. Biochim. Biophys. Acta, Rev. Biomembr. 1998, 1376, 433–453. 10.1016/S0304-4157(98)00018-5. [DOI] [PubMed] [Google Scholar]
- Harrison S. C. Viral Membrane Fusion. Nat. Struct. Mol. Biol. 2008, 15, 690–698. 10.1038/nsmb.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray D.; Arbuzova A.; Hangyás-Mihályné G.; Gambhir A.; Ben-Tal N.; Honig B.; McLaughlin S. Electrostatic Properties of Membranes Containing Acidic Lipids and Adsorbed Basic Peptides: Theory and Experiment. Biophys. J. 1999, 77, 3176–3188. 10.1016/S0006-3495(99)77148-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson J. E.; Cornell R. B. Amphitropic Proteins: Regulation by Reversible Membrane Interactions (Review). Mol. Membr. Biol. 1999, 16, 217–235. 10.1080/096876899294544. [DOI] [PubMed] [Google Scholar]
- Kim J.; Mosior M.; Chung L. A.; Wu H.; McLaughlin S. Binding of Peptides with Basic Residues to Membranes Containing Acidic Phospholipids. Biophys. J. 1991, 60, 135–148. 10.1016/S0006-3495(91)82037-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Tal N.; Honig B.; Peitzsch R. M.; Denisov G.; McLaughlin S. Binding of Small Basic Peptides to Membranes Containing Acidic Lipids: Theoretical Models and Experimental Results. Biophys. J. 1996, 71, 561–575. 10.1016/S0006-3495(96)79280-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchhausen T. Imaging Endocytic Clathrin Structures in Living Cells. Trends Cell Biol. 2009, 19, 596–605. 10.1016/j.tcb.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalli A. C.; Devaney I.; Sansom M. S. P. Interactions of Phosphatase and Tensin Homologue (PTEN) Proteins with Phosphatidylinositol Phosphates: Insights from Molecular Dynamics Simulations of PTEN and Voltage Sensitive Phosphatase. Biochemistry 2014, 53, 1724–1732. 10.1021/bi5000299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumb C. N.; Sansom M. S. P. Finding a Needle in a Haystack: The Role of Electrostatics in Target Lipid Recognition by PH Domains. PLoS Comput. Biol. 2012, 8, e1002617. 10.1371/journal.pcbi.1002617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumb C. N.; He J.; Xue Y.; Stansfeld P. J.; Stahelin R. V.; Kutateladze T. G.; Sansom M. S. P. Biophysical and Computational Studies of Membrane Penetration by the GRP1 Pleckstrin Homology Domain. Structure 2011, 19, 1338–1346. 10.1016/j.str.2011.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naughton F. B.; Kalli A. C.; Sansom M. S. P. Association of Peripheral Membrane Proteins with Membranes: Free Energy of Binding of GRP1 PH Domain with Phosphatidylinositol Phosphate-Containing Model Bilayers. J. Phys. Chem. Lett. 2016, 7, 1219–1224. 10.1021/acs.jpclett.6b00153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai C.-L.; Srivastava A.; Pilling C.; Chase A. R.; Falke J. J.; Voth G. A. Molecular Mechanism of Membrane Binding of the GRP1 PH Domain. J. Mol. Biol. 2013, 425, 3073–3090. 10.1016/j.jmb.2013.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan K. C.; Lu L.; Sun F.; Fan J. Molecular Details of the PH Domain of ACAP1BAR-PH Protein Binding to PIP-Containing Membrane. J. Phys. Chem. B 2017, 121, 3586–3596. 10.1021/acs.jpcb.6b09563. [DOI] [PubMed] [Google Scholar]
- Yamamoto E.; Kalli A. C.; Yasuoka K.; Sansom M. S. P. Interactions of Pleckstrin Homology Domains with Membranes: Adding Back the Bilayer via High-Throughput Molecular Dynamics. Structure 2016, 24, 1421–1431. 10.1016/j.str.2016.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogaski B.; Klauda J. B. Membrane-Binding Mechanism of a Peripheral Membrane Protein through Microsecond Molecular Dynamics Simulations. J. Mol. Biol. 2012, 423, 847–861. 10.1016/j.jmb.2012.08.015. [DOI] [PubMed] [Google Scholar]
- Pleskot R.; Pejchar P.; Žárský V.; Staiger C. J.; Potocký M. Structural Insights into the Inhibition of Actin-Capping Protein by Interactions with Phosphatidic Acid and Phosphatidylinositol (4,5)-Bisphosphate. PLoS Comput. Biol. 2012, 8, e1002765. 10.1371/journal.pcbi.1002765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakala M.; Kalimeri M.; Enkavi G.; Vattulainen I.; Lappalainen P. Molecular Mechanism for Inhibition of Twinfilin by Phosphoinositides. J. Biol. Chem. 2018, 293, 4818–4829. 10.1074/jbc.RA117.000484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L.; Mao Y. S.; Janmey P. A.; Yin H. L.. Phosphatidylinositol 4, 5 Bisphosphate and the Actin Cytoskeleton. In Phosphoinositides II: The Diverse Biological Functions; Balla T., Wymann M., York J. D., Eds.; Springer Netherlands: Dordrecht, 2012; Vol. 59, pp 177–215, 10.1007/978-94-007-3015-1_6. [DOI] [PubMed] [Google Scholar]
- Lamprakis C.; Stocker A.; Cascella M. Mechanisms of Recognition and Binding of α-TTP to the Plasma Membrane by Multi-Scale Molecular Dynamics Simulations. Front. Mol. Biosci. 2015, 2, 36. 10.3389/fmolb.2015.00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busse R. A.; Scacioc A.; Krick R.; Pérez-Lara Á.; Thumm M.; Kühnel K. Characterization of PROPPIN-Phosphoinositide Binding and Role of Loop 6CD in PROPPIN-Membrane Binding. Biophys. J. 2015, 108, 2223–2234. 10.1016/j.bpj.2015.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh R.; de Campos M. K. F.; Huang J.; Huh S. K.; Orlowski A.; Yang Y.; Tripathi A.; Nile A.; Lee H.-C.; Dynowski M.; et al. Sec14-Nodulin Proteins and the Patterning of Phosphoinositide Landmarks for Developmental Control of Membrane Morphogenesis. Mol. Biol. Cell 2015, 26, 1764–1781. 10.1091/mbc.E14-10-1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlier L.; Louet M.; Chaloin L.; Fuchs P.; Martinez J.; Muriaux D.; Favard C.; Floquet N. Coarse-Grained Simulations of the HIV-1 Matrix Protein Anchoring: Revisiting Its Assembly on Membrane Domains. Biophys. J. 2014, 106, 577–585. 10.1016/j.bpj.2013.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu I.; Mukhopadhyay C. Insights into Binding of Cholera Toxin to GM1 Containing Membrane. Langmuir 2014, 30, 15244–15252. 10.1021/la5036618. [DOI] [PubMed] [Google Scholar]
- Rodighiero C.; Aman A. T.; Kenny M. J.; Moss J.; Lencer W. I.; Hirst T. R. Structural Basis for the Differential Toxicity of Cholera Toxin and Escherichia Coli Heat-Labile Enterotoxin: Construction of Hybrid Toxins Identifies the A2-Domain as the Determinant of Differential Toxicity. J. Biol. Chem. 1999, 274, 3962–3969. 10.1074/jbc.274.7.3962. [DOI] [PubMed] [Google Scholar]
- Arcario M. J.; Tajkhorshid E. Membrane-Induced Structural Rearrangement and Identification of a Novel Membrane Anchor in Talin F2F3. Biophys. J. 2014, 107, 2059–2069. 10.1016/j.bpj.2014.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderwood D. A.; Zent R.; Grant R.; Rees D. J. G.; Hynes R. O.; Ginsberg M. H. The Talin Head Domain Binds to Integrin β Subunit Cytoplasmic Tails and Regulates Integrin Activation. J. Biol. Chem. 1999, 274, 28071–28074. 10.1074/jbc.274.40.28071. [DOI] [PubMed] [Google Scholar]
- Calderwood D. A.; Yan B.; de Pereda J. M.; Alvarez B. G.; Fujioka Y.; Liddington R. C.; Ginsberg M. H. The Phosphotyrosine Binding-like Domain of Talin Activates Integrins. J. Biol. Chem. 2002, 277, 21749–21758. 10.1074/jbc.M111996200. [DOI] [PubMed] [Google Scholar]
- Li J.; Ziemba B. P.; Falke J. J.; Voth G. A. Interactions of Protein Kinase C-α C1A and C1B Domains with Membranes: A Combined Computational and Experimental Study. J. Am. Chem. Soc. 2014, 136, 11757–11766. 10.1021/ja505369r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziemba B. P.; Li J.; Landgraf K. E.; Knight J. D.; Voth G. A.; Falke J. J. Single-Molecule Studies Reveal a Hidden Key Step in the Activation Mechanism of Membrane-Bound Protein Kinase C-α. Biochemistry 2014, 53, 1697–1713. 10.1021/bi4016082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L.; Rajendram M.; Weibel D. B.; Yethiraj A.; Cui Q. Ionic Hydrogen Bonds and Lipid Packing Defects Determine the Binding Orientation and Insertion Depth of RecA on Multicomponent Lipid Bilayers. J. Phys. Chem. B 2016, 120, 8424–8437. 10.1021/acs.jpcb.6b02164. [DOI] [PubMed] [Google Scholar]
- Maxfield F. R.; Tabas I. Role of Cholesterol and Lipid Organization in Disease. Nature 2005, 438, 612–621. 10.1038/nature04399. [DOI] [PubMed] [Google Scholar]
- Hallman M.; Glumoff V.; Rämet M. Surfactant in Respiratory Distress Syndrome and Lung Injury. Comp. Biochem. Physiol., Part A: Mol. Integr. Physiol. 2001, 129, 287–294. 10.1016/S1095-6433(01)00324-5. [DOI] [PubMed] [Google Scholar]
- Cho W.-J.; Trikha S.; Jeremic A. M. Cholesterol Regulates Assembly of Human Islet Amyloid Polypeptide on Model Membranes. J. Mol. Biol. 2009, 393, 765–775. 10.1016/j.jmb.2009.08.055. [DOI] [PubMed] [Google Scholar]
- Allott E. H.; Howard L. E.; Cooperberg M. R.; Kane C. J.; Aronson W. J.; Terris M. K.; Amling C. L.; Freedland S. J. Serum Lipid Profile and Risk of Prostate Cancer Recurrence: Results from the SEARCH Database. Cancer Epidemiol., Biomarkers Prev. 2014, 23, 2349–2356. 10.1158/1055-9965.EPI-14-0458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welte M. A. Expanding Roles for Lipid Droplets. Curr. Biol. 2015, 25, R470–R481. 10.1016/j.cub.2015.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walther T. C.; Farese R. V. Lipid Droplets and Cellular Lipid Metabolism. Annu. Rev. Biochem. 2012, 81, 687–714. 10.1146/annurev-biochem-061009-102430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pol A.; Gross S. P.; Parton R. G. Biogenesis of the Multifunctional Lipid Droplet: Lipids, Proteins, and Sites. J. Cell Biol. 2014, 204, 635–646. 10.1083/jcb.201311051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henneré G.; Prognon P.; Brion F.; Nicolis I. Molecular Dynamics Study of a Phospholipid Monolayer at a Water/Triglyceride Interface: Towards Lipid Emulsion Modelling. Chem. Phys. Lipids 2009, 157, 86–93. 10.1016/j.chemphyslip.2008.10.002. [DOI] [PubMed] [Google Scholar]
- Henneré G.; Prognon P.; Brion F.; Rosilio V.; Nicolis I. Molecular Dynamics Simulation of a Mixed Lipid Emulsion Model: Influence of the Triglycerides on Interfacial Phospholipid Organization. J. Mol. Struct.: THEOCHEM 2009, 901, 174–185. 10.1016/j.theochem.2009.01.020. [DOI] [Google Scholar]
- Koivuniemi A.; Heikelä M.; Kovanen P. T.; Vattulainen I.; Hyvönen M. T. Atomistic Simulations of Phosphatidylcholines and Cholesteryl Esters in High-Density Lipoprotein-Sized Lipid Droplet and Trilayer: Clues to Cholesteryl Ester Transport and Storage. Biophys. J. 2009, 96, 4099–4108. 10.1016/j.bpj.2009.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koivuniemi A.; Vuorela T.; Kovanen P. T.; Vattulainen I.; Hyvönen M. T. Lipid Exchange Mechanism of the Cholesteryl Ester Transfer Protein Clarified by Atomistic and Coarse-Grained Simulations. PLoS Comput. Biol. 2012, 8, e1002299. 10.1371/journal.pcbi.1002299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ollila O. H. S.; Lamberg A.; Lehtivaara M.; Koivuniemi A.; Vattulainen I. Interfacial Tension and Surface Pressure of High Density Lipoprotein, Low Density Lipoprotein, and Related Lipid Droplets. Biophys. J. 2012, 103, 1236–1244. 10.1016/j.bpj.2012.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prévost C.; Sharp M. E.; Kory N.; Lin Q.; Voth G. A.; Farese R. V.; Walther T. C. Mechanism and Determinants of Amphipathic Helix-Containing Protein Targeting to Lipid Droplets. Dev. Cell 2018, 44, 73–86.e4. 10.1016/j.devcel.2017.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacle A.; Gautier R.; Jackson C. L.; Fuchs P. F. J.; Vanni S. Interdigitation between Triglycerides and Lipids Modulates Surface Properties of Lipid Droplets. Biophys. J. 2017, 112, 1417–1430. 10.1016/j.bpj.2017.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulovesi P.; Telenius J.; Koivuniemi A.; Brezesinski G.; Rantamäki A.; Viitala T.; Puukilainen E.; Ritala M.; Wiedmer S. K.; Vattulainen I.; et al. Molecular Organization of the Tear Fluid Lipid Layer. Biophys. J. 2010, 99, 2559–2567. 10.1016/j.bpj.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telenius J.; Koivuniemi A.; Kulovesi P.; Holopainen J. M.; Vattulainen I. Role of Neutral Lipids in Tear Fluid Lipid Layer: Coarse-Grained Simulation Study. Langmuir 2012, 28, 17092–17100. 10.1021/la304366d. [DOI] [PubMed] [Google Scholar]
- Khandelia H.; Duelund L.; Pakkanen K. I.; Ipsen J. H. Triglyceride Blisters in Lipid Bilayers: Implications for Lipid Droplet Biogenesis and the Mobile Lipid Signal in Cancer Cell Membranes. PLoS One 2010, 5, e12811. 10.1371/journal.pone.0012811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaban V. V.; Khandelia H. Lipid Structure in Triolein Lipid Droplets. J. Phys. Chem. B 2014, 118, 10335–10340. 10.1021/jp503223z. [DOI] [PubMed] [Google Scholar]
- Chaban V. V.; Khandelia H. Distribution of Neutral Lipids in the Lipid Droplet Core. J. Phys. Chem. B 2014, 118, 11145–11151. 10.1021/jp506693d. [DOI] [PubMed] [Google Scholar]
- Pan L.; Segrest J. P. Computational Studies of Plasma Lipoprotein Lipids. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 2401–2420. 10.1016/j.bbamem.2016.03.010. [DOI] [PubMed] [Google Scholar]
- Koivuniemi A.; Vattulainen I.. Modeling of Lipid Membranes and Lipoproteins. In A Systems Biology Approach to Study Metabolic Syndrome; Orešič M., Vidal-Puig A., Eds.; Springer International Publishing: Cham, 2014; pp 299–318, 10.1007/978-3-319-01008-3_15. [DOI] [Google Scholar]
- Koivuniemi A.; Vattulainen I. Revealing Structural and Dynamical Properties of High Density Lipoproteins through Molecular Simulations. Soft Matter 2012, 8, 1262–1267. 10.1039/C1SM06742G. [DOI] [Google Scholar]
- Hevonoja T.; Pentikäinen M. O.; Hyvönen M. T.; Kovanen P. T.; Ala-Korpela M. Structure of Low Density Lipoprotein (LDL) Particles: Basis for Understanding Molecular Changes in Modified LDL. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2000, 1488, 189–210. 10.1016/S1388-1981(00)00123-2. [DOI] [PubMed] [Google Scholar]
- Murtola T.; Vuorela T. A.; Hyvönen M. T.; Marrink S.-J.; Karttunen M.; Vattulainen I. Low Density Lipoprotein: Structure, Dynamics, and Interactions of ApoB-100 with Lipids. Soft Matter 2011, 7, 8135–8141. 10.1039/c1sm05367a. [DOI] [Google Scholar]
- Rosenson R. S.; Brewer H. B.; Ansell B. J.; Barter P.; Chapman M. J.; Heinecke J. W.; Kontush A.; Tall A. R.; Webb N. R. Dysfunctional HDL and Atherosclerotic Cardiovascular Disease. Nat. Rev. Cardiol. 2016, 13, 48–60. 10.1038/nrcardio.2015.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih A. Y.; Sligar S. G.; Schulten K. Maturation of High-Density Lipoproteins. J. R. Soc., Interface 2009, 6, 863–871. 10.1098/rsif.2009.0173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gogonea V. Structural Insights into High Density Lipoprotein: Old Models and New Facts. Front. Pharmacol. 2016, 6, 318. 10.3389/fphar.2015.00318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melchior J. T.; Walker R. G.; Cooke A. L.; Morris J.; Castleberry M.; Thompson T. B.; Jones M. K.; Song H. D.; Rye K.-A.; Oda M. N.; et al. A Consensus Model of Human Apolipoprotein A-I in Its Monomeric and Lipid-Free State. Nat. Struct. Mol. Biol. 2017, 24, 1093–1099. 10.1038/nsmb.3501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda M. N. Lipid-Free ApoA-I Structure - Origins of Model Diversity. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2017, 1862, 221–233. 10.1016/j.bbalip.2016.11.010. [DOI] [PubMed] [Google Scholar]
- Silva R. A. G. D.; Hilliard G. M.; Fang J.; Macha S.; Davidson W. S. A Three-Dimensional Molecular Model of Lipid-Free Apolipoprotein A-I Determined by Cross-Linking/Mass Spectrometry and Sequence Threading. Biochemistry 2005, 44, 2759–2769. 10.1021/bi047717+. [DOI] [PubMed] [Google Scholar]
- Pollard R. D.; Fulp B.; Samuel M. P.; Sorci-Thomas M. G.; Thomas M. J. The Conformation of Lipid-Free Human Apolipoprotein A-I in Solution. Biochemistry 2013, 52, 9470–9481. 10.1021/bi401080k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segrest J. P.; Jones M. K.; Shao B.; Heinecke J. W. An Experimentally Robust Model of Monomeric Apolipoprotein A-I Created from a Chimera of Two X-Ray Structures and Molecular Dynamics Simulations. Biochemistry 2014, 53, 7625–7640. 10.1021/bi501111j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Lei D.; Zhang L.; Rames M.; Zhang S. A Model of Lipid-Free Apolipoprotein A-I Revealed by Iterative Molecular Dynamics Simulation. PLoS One 2015, 10, e0120233. 10.1371/journal.pone.0120233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagerstedt J. O.; Budamagunta M. S.; Liu G. S.; DeValle N. C.; Voss J. C.; Oda M. N. The “Beta-Clasp” Model of Apolipoprotein A-I — A Lipid-Free Solution Structure Determined by Electron Paramagnetic Resonance Spectroscopy. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2012, 1821, 448–455. 10.1016/j.bbalip.2011.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker R. G.; Deng X.; Melchior J. T.; Morris J.; Tso P.; Jones M. K.; Segrest J. P.; Thompson T. B.; Davidson W. S. The Structure of Human Apolipoprotein A-IV as Revealed by Stable Isotope-Assisted Cross-Linking, Molecular Dynamics, and Small Angle X-Ray Scattering. J. Biol. Chem. 2014, 289, 5596–5608. 10.1074/jbc.M113.541037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todorova N.; Hung A.; Yarovsky I. Lipid Concentration Effects on the Amyloidogenic ApoC-II60–70 Peptide: A Computational Study. J. Phys. Chem. B 2010, 114, 7974–7982. 10.1021/jp102142x. [DOI] [PubMed] [Google Scholar]
- Gordon S. M.; Pourmousa M.; Sampson M.; Sviridov D.; Islam R.; Perrin B. S.; Kemeh G.; Pastor R. W.; Remaley A. T. Identification of a Novel Lipid Binding Motif in Apolipoprotein B by the Analysis of Hydrophobic Cluster Domains. Biochim. Biophys. Acta, Biomembr. 2017, 1859, 135–145. 10.1016/j.bbamem.2016.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu F.; Jones M. K.; Chen J.; Patterson J. C.; Catte A.; Jerome W. G.; Li L.; Segrest J. P. Structures of Discoidal High Density Lipoproteins: A Combined Computational-Experimental Approach. J. Biol. Chem. 2010, 285, 4652–4665. 10.1074/jbc.M109.069914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L.; Li S.; Jones M. K.; Segrest J. P. Rotational and Hinge Dynamics of Discoidal High Density Lipoproteins Probed by Interchain Disulfide Bond Formation. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2012, 1821, 481–489. 10.1016/j.bbalip.2011.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caulfield T. R. Inter-Ring Rotation of Apolipoprotein A-I Protein Monomers for the Double-Belt Model Using Biased Molecular Dynamics. J. Mol. Graphics Modell. 2011, 29, 1006–1014. 10.1016/j.jmgm.2011.04.005. [DOI] [PubMed] [Google Scholar]
- Gursky O.; Jones M. K.; Mei X.; Segrest J. P.; Atkinson D. Structural Basis for Distinct Functions of the Naturally Occurring Cys Mutants of Human Apolipoprotein A-I. J. Lipid Res. 2013, 54, 3244–3257. 10.1194/jlr.R037911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagerstedt J. O.; Cavigiolio G.; Budamagunta M. S.; Pagani I.; Voss J. C.; Oda M. N. Structure of Apolipoprotein A-I N Terminus on Nascent High Density Lipoproteins. J. Biol. Chem. 2011, 286, 2966–2975. 10.1074/jbc.M110.163097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones M. K.; Gu F.; Catte A.; Li L.; Segrest J. P. Sticky” and “Promiscuous”, the Yin and Yang of Apolipoprotein A-I Termini in Discoidal High-Density Lipoproteins: A Combined Computational–Experimental Approach. Biochemistry 2011, 50, 2249–2263. 10.1021/bi101301g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segrest J. P.; Jones M. K.; Catte A.; Thirumuruganandham S. P. Validation of Previous Computer Models and MD Simulations of Discoidal HDL by a Recent Crystal Structure of ApoA-I. J. Lipid Res. 2012, 53, 1851–1863. 10.1194/jlr.M026229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pourmousa M.; Song H. D.; He Y.; Heinecke J. W.; Segrest J. P.; Pastor R. W. Tertiary Structure of Apolipoprotein A-I in Nascent High-Density Lipoproteins. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 5163–5168. 10.1073/pnas.1721181115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segrest J. P.; Jones M. K.; Catte A.; Manchekar M.; Datta G.; Zhang L.; Zhang R.; Li L.; Patterson J. C.; Palgunachari M. N.; et al. Surface Density-Induced Pleating of a Lipid Monolayer Drives Nascent High-Density Lipoprotein Assembly. Structure 2015, 23, 1214–1226. 10.1016/j.str.2015.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koivuniemi A.; Vattulainen I. Biogenesis of Nascent High Density Lipoprotein Particles. Structure 2015, 23, 1153–1154. 10.1016/j.str.2015.06.006. [DOI] [PubMed] [Google Scholar]
- Islam R. M.; Pourmousa M.; Sviridov D.; Gordon S. M.; Neufeld E. B.; Freeman L. A.; Perrin B. S.; Pastor R. W.; Remaley A. T. Structural Properties of Apolipoprotein A-I Mimetic Peptides That Promote ABCA1-Dependent Cholesterol Efflux. Sci. Rep. 2018, 8, 2956. 10.1038/s41598-018-20965-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pourmousa M.; Pastor R. W. Molecular Dynamics Simulations of Lipid Nanodiscs. Biochim. Biophys. Acta, Biomembr. 2018, 1860, 2094–2107. 10.1016/j.bbamem.2018.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sei Y. J.; Ahn J.; Kim T.; Shin E.; Santiago-Lopez A. J.; Jang S. S.; Jeon N. L.; Jang Y. C.; Kim Y. Detecting the Functional Complexities between High-Density Lipoprotein Mimetics. Biomaterials 2018, 170, 58–69. 10.1016/j.biomaterials.2018.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gogonea V.; Wu Z.; Lee X.; Pipich V.; Li X.-M.; Ioffe A. I.; DiDonato J. A.; Hazen S. L. Congruency between Biophysical Data from Multiple Platforms and Molecular Dynamics Simulation of the Double-Super Helix Model of Nascent High-Density Lipoprotein. Biochemistry 2010, 49, 7323–7343. 10.1021/bi100588a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones M. K.; Zhang L.; Catte A.; Li L.; Oda M. N.; Ren G.; Segrest J. P. Assessment of the Validity of the Double Superhelix Model for Reconstituted High Density Lipoproteins: A Combined Computational-Experimental Approach. J. Biol. Chem. 2010, 285, 41161–41171. 10.1074/jbc.M110.187799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yetukuri L.; Söderlund S.; Koivuniemi A.; Seppänen-Laakso T.; Niemelä P. S.; Hyvönen M.; Taskinen M.-R.; Vattulainen I.; Jauhiainen M.; Orešič M. Composition and Lipid Spatial Distribution of HDL Particles in Subjects with Low and High HDL-Cholesterol. J. Lipid Res. 2010, 51, 2341–2351. 10.1194/jlr.M006494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yetukuri L.; Huopaniemi I.; Koivuniemi A.; Maranghi M.; Hiukka A.; Nygren H.; Kaski S.; Taskinen M.-R.; Vattulainen I.; Jauhiainen M.; et al. High Density Lipoprotein Structural Changes and Drug Response in Lipidomic Profiles Following the Long-Term Fenofibrate Therapy in the FIELD Substudy. PLoS One 2011, 6, e23589. 10.1371/journal.pone.0023589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuorela T.; Catte A.; Niemelä P. S.; Hall A.; Hyvönen M. T.; Marrink S.-J.; Karttunen M.; Vattulainen I. Role of Lipids in Spheroidal High Density Lipoproteins. PLoS Comput. Biol. 2010, 6, e1000964. 10.1371/journal.pcbi.1000964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segrest J. P.; Jones M. K.; Catte A. MD Simulations Suggest Important Surface Differences between Reconstituted and Circulating Spherical HDL. J. Lipid Res. 2013, 54, 2718–2732. 10.1194/jlr.M039206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lähdesmäki K.; Ollila O. H. S.; Koivuniemi A.; Kovanen P. T.; Hyvönen M. T. Membrane Simulations Mimicking Acidic PH Reveal Increased Thickness and Negative Curvature in a Bilayer Consisting of Lysophosphatidylcholines and Free Fatty Acids. Biochim. Biophys. Acta, Biomembr. 2010, 1798, 938–946. 10.1016/j.bbamem.2010.01.020. [DOI] [PubMed] [Google Scholar]
- Koivuniemi A.; Sysi-Aho M.; Orešič M.; Ollila S. Interfacial Properties of High-Density Lipoprotein-like Lipid Droplets with Different Lipid and Apolipoprotein A-I Compositions. Biophys. J. 2013, 104, 2193–2201. 10.1016/j.bpj.2013.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inazu A.; Brown M. L.; Hesler C. B.; Agellon L. B.; Koizumi J.; Takata K.; Maruhama Y.; Mabuchi H.; Tall A. R. Increased High-Density Lipoprotein Levels Caused by a Common Cholesteryl-Ester Transfer Protein Gene Mutation. N. Engl. J. Med. 1990, 323, 1234–1238. 10.1056/NEJM199011013231803. [DOI] [PubMed] [Google Scholar]
- Zhong S.; Sharp D. S.; Grove J. S.; Bruce C.; Yano K.; Curb J. D.; Tall A. R. Increased Coronary Heart Disease in Japanese-American Men with Mutation in the Cholesteryl Ester Transfer Protein Gene despite Increased HDL Levels. J. Clin. Invest. 1996, 97, 2917–2923. 10.1172/JCI118751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Äijänen T.; Koivuniemi A.; Javanainen M.; Rissanen S.; Rog T.; Vattulainen I. How Anacetrapib Inhibits the Activity of the Cholesteryl Ester Transfer Protein? Perspective through Atomistic Simulations. PLoS Comput. Biol. 2014, 10, e1003987. 10.1371/journal.pcbi.1003987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cilpa-Karhu G.; Jauhiainen M.; Riekkola M.-L. Atomistic MD Simulation Reveals the Mechanism by Which CETP Penetrates into HDL Enabling Lipid Transfer from HDL to CETP. J. Lipid Res. 2015, 56, 98–108. 10.1194/jlr.M054288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M.; Charles R.; Tong H.; Zhang L.; Patel M.; Wang F.; Rames M. J.; Ren A.; Rye K.-A.; Qiu X.; et al. HDL Surface Lipids Mediate CETP Binding as Revealed by Electron Microscopy and Molecular Dynamics Simulation. Sci. Rep. 2015, 5, 8741. 10.1038/srep08741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karilainen T.; Vuorela T.; Vattulainen I. How Well Does BODIPY-Cholesteryl Ester Mimic Unlabeled Cholesteryl Esters in High Density Lipoprotein Particles?. J. Phys. Chem. B 2015, 119, 15848–15856. 10.1021/acs.jpcb.5b10188. [DOI] [PubMed] [Google Scholar]
- Karilainen T.; Timr Š.; Vattulainen I.; Jungwirth P. Oxidation of Cholesterol Does Not Alter Significantly Its Uptake into High-Density Lipoprotein Particles. J. Phys. Chem. B 2015, 119, 4594–4600. 10.1021/acs.jpcb.5b00240. [DOI] [PubMed] [Google Scholar]
- Neuroscience, 2nd ed.; Purves D., Augustine G. J., Fitzpatrick D., Katz L. C., LaMantia A.-S., McNamara J. O., Williams S. M., Eds.; Sinauer Associates: Sunderland, Mass, 2001. [Google Scholar]
- Wikström M.; Sharma V.; Kaila V. R. I.; Hosler J. P.; Hummer G. New Perspectives on Proton Pumping in Cellular Respiration. Chem. Rev. 2015, 115, 2196–2221. 10.1021/cr500448t. [DOI] [PubMed] [Google Scholar]
- Wikström M.; Sharma V. Proton Pumping by Cytochrome c Oxidase – A 40 Year Anniversary. Biochim. Biophys. Acta, Bioenerg. 2018, 1859, 692–698. 10.1016/j.bbabio.2018.03.009. [DOI] [PubMed] [Google Scholar]
- Musatov A.; Sedlák E. Role of Cardiolipin in Stability of Integral Membrane Proteins. Biochimie 2017, 142, 102–111. 10.1016/j.biochi.2017.08.013. [DOI] [PubMed] [Google Scholar]
- Lemmin T.; Bovigny C.; Lançon D.; Dal Peraro M. Cardiolipin Models for Molecular Simulations of Bacterial and Mitochondrial Membranes. J. Chem. Theory Comput. 2013, 9, 670–678. 10.1021/ct300590v. [DOI] [PubMed] [Google Scholar]
- Dahlberg M.; Marini A.; Mennucci B.; Maliniak A. Quantum Chemical Modeling of the Cardiolipin Headgroup. J. Phys. Chem. A 2010, 114, 4375–4387. 10.1021/jp9110019. [DOI] [PubMed] [Google Scholar]
- Aguayo D.; González-Nilo F. D.; Chipot C. Insight into the Properties of Cardiolipin Containing Bilayers from Molecular Dynamics Simulations, Using a Hybrid All-Atom/United-Atom Force Field. J. Chem. Theory Comput. 2012, 8, 1765–1773. 10.1021/ct200849k. [DOI] [PubMed] [Google Scholar]
- Pöyry S.; Róg T.; Karttunen M.; Vattulainen I. Mitochondrial Membranes with Mono- and Divalent Salt: Changes Induced by Salt Ions on Structure and Dynamics. J. Phys. Chem. B 2009, 113, 15513–15521. 10.1021/jp905915m. [DOI] [PubMed] [Google Scholar]
- Róg T.; Martinez-Seara H.; Munck N.; Orešič M.; Karttunen M.; Vattulainen I. Role of Cardiolipins in the Inner Mitochondrial Membrane: Insight Gained through Atom-Scale Simulations. J. Phys. Chem. B 2009, 113, 3413–3422. 10.1021/jp8077369. [DOI] [PubMed] [Google Scholar]
- Dahlberg M.; Maliniak A. Molecular Dynamics Simulations of Cardiolipin Bilayers. J. Phys. Chem. B 2008, 112, 11655–11663. 10.1021/jp803414g. [DOI] [PubMed] [Google Scholar]
- Dahlberg M.; Maliniak A. Mechanical Properties of Coarse-Grained Bilayers Formed by Cardiolipin and Zwitterionic Lipids. J. Chem. Theory Comput. 2010, 6, 1638–1649. 10.1021/ct900654e. [DOI] [PubMed] [Google Scholar]
- Boyd K. J.; Alder N. N.; May E. R. Buckling Under Pressure: Curvature-Based Lipid Segregation and Stability Modulation in Cardiolipin-Containing Bilayers. Langmuir 2017, 33, 6937–6946. 10.1021/acs.langmuir.7b01185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan J.; Cheng X.; Sharp M.; Ho C.-S.; Khadka N.; Katsaras J. Structural and Mechanical Properties of Cardiolipin Lipid Bilayers Determined Using Neutron Spin Echo, Small Angle Neutron and X-Ray Scattering, and Molecular Dynamics Simulations. Soft Matter 2015, 11, 130–138. 10.1039/C4SM02227K. [DOI] [PubMed] [Google Scholar]
- Boscia A. L.; Treece B. W.; Mohammadyani D.; Klein-Seetharaman J.; Braun A. R.; Wassenaar T. A.; Klösgen B.; Tristram-Nagle S. X-Ray Structure, Thermodynamics, Elastic Properties and MD Simulations of Cardiolipin/Dimyristoylphosphatidylcholine Mixed Membranes. Chem. Phys. Lipids 2014, 178, 1–10. 10.1016/j.chemphyslip.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaurola P.; Sharma V.; Vonk A.; Vattulainen I.; Róg T. Distribution and Dynamics of Quinones in the Lipid Bilayer Mimicking the Inner Membrane of Mitochondria. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 2116–2122. 10.1016/j.bbamem.2016.06.016. [DOI] [PubMed] [Google Scholar]
- Wolf M. G.; Grubmüller H.; Groenhof G. Anomalous Surface Diffusion of Protons on Lipid Membranes. Biophys. J. 2014, 107, 76–87. 10.1016/j.bpj.2014.04.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita T.; Voth G. A. Properties of Hydrated Excess Protons near Phospholipid Bilayers. J. Phys. Chem. B 2010, 114, 592–603. 10.1021/jp908768c. [DOI] [PubMed] [Google Scholar]
- Pöyry S.; Cramariuc O.; Postila P. A.; Kaszuba K.; Sarewicz M.; Osyczka A.; Vattulainen I.; Róg T. Atomistic Simulations Indicate Cardiolipin to Have an Integral Role in the Structure of the Cytochrome bc1 Complex. Biochim. Biophys. Acta, Bioenerg. 2013, 1827, 769–778. 10.1016/j.bbabio.2013.03.005. [DOI] [PubMed] [Google Scholar]
- Zhang M.; Mileykovskaya E.; Dowhan W. Gluing the Respiratory Chain Together: Cardiolipin Is Required For Supercomplex Formation In The Inner Mitochondrial Membrane. J. Biol. Chem. 2002, 277, 43553–43556. 10.1074/jbc.C200551200. [DOI] [PubMed] [Google Scholar]
- Lange C. Specific Roles of Protein-Phospholipid Interactions in the Yeast Cytochrome bc1 Complex Structure. EMBO J. 2001, 20, 6591–6600. 10.1093/emboj/20.23.6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnarez C.; Marrink S. J.; Periole X. Identification of Cardiolipin Binding Sites on Cytochrome c Oxidase at the Entrance of Proton Channels. Sci. Rep. 2013, 3, 1263. 10.1038/srep01263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma V.; Ala-Vannesluoma P.; Vattulainen I.; Wikström M.; Róg T. Role of Subunit III and Its Lipids in the Molecular Mechanism of Cytochrome c Oxidase. Biochim. Biophys. Acta, Bioenerg. 2015, 1847, 690–697. 10.1016/j.bbabio.2015.04.007. [DOI] [PubMed] [Google Scholar]
- Mileykovskaya E.; Dowhan W. Cardiolipin-Dependent Formation of Mitochondrial Respiratory Supercomplexes. Chem. Phys. Lipids 2014, 179, 42–48. 10.1016/j.chemphyslip.2013.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnarez C.; Mazat J.-P.; Elezgaray J.; Marrink S.-J.; Periole X. Evidence for Cardiolipin Binding Sites on the Membrane-Exposed Surface of the Cytochrome bc1. J. Am. Chem. Soc. 2013, 135, 3112–3120. 10.1021/ja310577u. [DOI] [PubMed] [Google Scholar]
- Arnarez C.; Marrink S. J.; Periole X. Molecular Mechanism of Cardiolipin-Mediated Assembly of Respiratory Chain Supercomplexes. Chem. Sci. 2016, 7, 4435–4443. 10.1039/C5SC04664E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammadyani D.; Yanamala N.; Samhan-Arias A. K.; Kapralov A. A.; Stepanov G.; Nuar N.; Planas-Iglesias J.; Sanghera N.; Kagan V. E.; Klein-Seetharaman J. Structural Characterization of Cardiolipin-Driven Activation of Cytochrome c into a Peroxidase and Membrane Perturbation. Biochim. Biophys. Acta, Biomembr. 2018, 1860, 1057–1068. 10.1016/j.bbamem.2018.01.009. [DOI] [PubMed] [Google Scholar]
- Sui X.; Arlt H.; Brock K. P.; Lai Z. W.; DiMaio F.; Marks D. S.; Liao M.; Farese R. V.; Walther T. C. Cryo–Electron Microscopy Structure of the Lipid Droplet–Formation Protein Seipin. J. Cell Biol. 2018, 217, 4080–4091. 10.1083/jcb.201809067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan R.; Qian H.; Lukmantara I.; Gao M.; Du X.; Yan N.; Yang H. Human SEIPIN Binds Anionic Phospholipids. Dev. Cell 2018, 47, 248–256.e4. 10.1016/j.devcel.2018.09.010. [DOI] [PubMed] [Google Scholar]
- Segrest J. P.; Jones M. K.; Loof H. D.; Dashti N. Structure of Apolipoprotein B-100 in Low Density Lipoproteins. J. Lipid Res. 2001, 42, 1346–1367. [PubMed] [Google Scholar]
- Sherman M. B.; Orlova E. V.; Decker G. L.; Chiu W.; Pownall H. J. Structure of Triglyceride-Rich Human Low-Density Lipoproteins According to Cryoelectron Microscopy. Biochemistry 2003, 42, 14988–14993. 10.1021/bi0354738. [DOI] [PubMed] [Google Scholar]
- Kumar V.; Butcher S. J.; Öörni K.; Engelhardt P.; Heikkonen J.; Kaski K.; Ala-Korpela M.; Kovanen P. T. Three-Dimensional CryoEM Reconstruction of Native LDL Particles to 16Å Resolution at Physiological Body Temperature. PLoS One 2011, 6, e18841. 10.1371/journal.pone.0018841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D.; Stansfeld P. J.; Sansom M. S. P.; Keogh A.; Vogeley L.; Howe N.; Lyons J. A.; Aragao D.; Fromme P.; Fromme R.; et al. Ternary Structure Reveals Mechanism of a Membrane Diacylglycerol Kinase. Nat. Commun. 2015, 6, 10140. 10.1038/ncomms10140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafsson C.; Vassiliev S.; Kürten C.; Syrén P.-O.; Brinck T. MD Simulations Reveal Complex Water Paths in Squalene–Hopene Cyclase: Tunnel-Obstructing Mutations Increase the Flow of Water in the Active Site. ACS Omega 2017, 2, 8495–8506. 10.1021/acsomega.7b01084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haapanen O.; Sharma V. A Modeling and Simulation Perspective on the Mechanism and Function of Respiratory Complex I. Biochim. Biophys. Acta, Bioenerg. 2018, 1859, 510–523. 10.1016/j.bbabio.2018.04.001. [DOI] [PubMed] [Google Scholar]
- Liang R.; Swanson J. M. J.; Peng Y.; Wikström M.; Voth G. A. Multiscale Simulations Reveal Key Features of the Proton-Pumping Mechanism in Cytochrome c Oxidase. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 7420–7425. 10.1073/pnas.1601982113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira A. S. F.; Damas J. M.; Baptista A. M.; Soares C. M. Exploring O2 Diffusion in A-Type Cytochrome c Oxidases: Molecular Dynamics Simulations Uncover Two Alternative Channels towards the Binuclear Site. PLoS Comput. Biol. 2014, 10, e1004010. 10.1371/journal.pcbi.1004010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oteri F.; Baaden M.; Lojou E.; Sacquin-Mora S. Multiscale Simulations Give Insight into the Hydrogen In and Out Pathways of [NiFe]-Hydrogenases from Aquifex Aeolicus and Desulfovibrio Fructosovorans. J. Phys. Chem. B 2014, 118, 13800–13811. 10.1021/jp5089965. [DOI] [PubMed] [Google Scholar]
- Padayatti P. S.; Leung J. H.; Mahinthichaichan P.; Tajkhorshid E.; Ishchenko A.; Cherezov V.; Soltis S. M.; Jackson J. B.; Stout C. D.; Gennis R. B.; et al. Critical Role of Water Molecules in Proton Translocation by the Membrane-Bound Transhydrogenase. Structure 2017, 25, 1111–1119. 10.1016/j.str.2017.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma V.; Enkavi G.; Vattulainen I.; Róg T.; Wikström M. Proton-Coupled Electron Transfer and the Role of Water Molecules in Proton Pumping by Cytochrome c Oxidase. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 2040–2045. 10.1073/pnas.1409543112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma V.; Jambrina P. G.; Kaukonen M.; Rosta E.; Rich P. R. Insights into Functions of the H Channel of Cytochrome c Oxidase from Atomistic Molecular Dynamics Simulations. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, E10339–E10348. 10.1073/pnas.1708628114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terasaka E.; Yamada K.; Wang P.-H.; Hosokawa K.; Yamagiwa R.; Matsumoto K.; Ishii S.; Mori T.; Yagi K.; Sawai H.; et al. Dynamics of Nitric Oxide Controlled by Protein Complex in Bacterial System. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 9888. 10.1073/pnas.1621301114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wikström M.; Krab K.; Sharma V. Oxygen Activation and Energy Conservation by Cytochrome c Oxidase. Chem. Rev. 2018, 118, 2469–2490. 10.1021/acs.chemrev.7b00664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L.; Skjevik Å. A.; Han Du W.-G.; Noodleman L.; Walker R. C.; Götz A. W. Water Exit Pathways and Proton Pumping Mechanism in B-Type Cytochrome c Oxidase from Molecular Dynamics Simulations. Biochim. Biophys. Acta, Bioenerg. 2016, 1857, 1594–1606. 10.1016/j.bbabio.2016.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W.; Marinelli F.; Nief C.; Faraldo-Gómez J. D. Atomistic Simulations Indicate the C-Subunit Ring of the F1Fo ATP Synthase Is Not the Mitochondrial Permeability Transition Pore. eLife 2017, 6, e23781. 10.7554/eLife.23781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma V.; Belevich G.; Gamiz-Hernandez A. P.; Róg T.; Vattulainen I.; Verkhovskaya M. L.; Wikström M.; Hummer G.; Kaila V. R. I. Redox-Induced Activation of the Proton Pump in the Respiratory Complex I. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 11571–11576. 10.1073/pnas.1503761112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postila P. A.; Kaszuba K.; Kuleta P.; Vattulainen I.; Sarewicz M.; Osyczka A.; Róg T. Atomistic Determinants of Co-Enzyme Q Reduction at the Qi-Site of the Cytochrome bc1 Complex. Sci. Rep. 2016, 6, 33607. 10.1038/srep33607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haapanen O.; Sharma V. Role of Water and Protein Dynamics in Proton Pumping by Respiratory Complex I. Sci. Rep. 2017, 7, 7747. 10.1038/s41598-017-07930-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postila P. A.; Kaszuba K.; Sarewicz M.; Osyczka A.; Vattulainen I.; Róg T. Key Role of Water in Proton Transfer at the Qo-Site of the Cytochrome bc1 Complex Predicted by Atomistic Molecular Dynamics Simulations. Biochim. Biophys. Acta, Bioenerg. 2013, 1827, 761–768. 10.1016/j.bbabio.2013.02.005. [DOI] [PubMed] [Google Scholar]
- Manikandan P.; Nagini S. Cytochrome P450 Structure, Function and Clinical Significance: A Review. Curr. Drug Targets 2018, 19, 38–54. 10.2174/1389450118666170125144557. [DOI] [PubMed] [Google Scholar]
- Hasler J. A.; Estabrook R.; Murray M.; Pikuleva I.; Waterman M.; Capdevila J.; Holla V.; Helvig C.; Falck J. R.; Farrell G.; et al. Human Cytochromes P450. Mol. Aspects Med. 1999, 20, 1–137. 10.1016/S0098-2997(99)00005-9. [DOI] [Google Scholar]
- Burkina V.; Rasmussen M. K.; Pilipenko N.; Zamaratskaia G. Comparison of Xenobiotic-Metabolising Human, Porcine, Rodent, and Piscine Cytochrome P450. Toxicology 2017, 375, 10–27. 10.1016/j.tox.2016.11.014. [DOI] [PubMed] [Google Scholar]
- Shalan H.; Kato M.; Cheruzel L. Keeping the Spotlight on Cytochrome P450. Biochim. Biophys. Acta, Proteins Proteomics 2018, 1866, 80–87. 10.1016/j.bbapap.2017.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanger U. M.; Schwab M. Cytochrome P450 Enzymes in Drug Metabolism: Regulation of Gene Expression, Enzyme Activities, and Impact of Genetic Variation. Pharmacol. Ther. 2013, 138, 103–141. 10.1016/j.pharmthera.2012.12.007. [DOI] [PubMed] [Google Scholar]
- Backman J. T.; Filppula A. M.; Niemi M.; Neuvonen P. J. Role of Cytochrome P450 2C8 in Drug Metabolism and Interactions. Pharmacol. Rev. 2016, 68, 168–241. 10.1124/pr.115.011411. [DOI] [PubMed] [Google Scholar]
- Monk B. C.; Tomasiak T. M.; Keniya M. V.; Huschmann F. U.; Tyndall J. D. A.; O’Connell J. D.; Cannon R. D.; McDonald J. G.; Rodriguez A.; Finer-Moore J. S.; et al. Architecture of a Single Membrane Spanning Cytochrome P450 Suggests Constraints That Orient the Catalytic Domain Relative to a Bilayer. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 3865–3870. 10.1073/pnas.1324245111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousefpour A.; Modarress H.; Goharpey F.; Amjad-Iranagh S. Interaction of Drugs Amlodipine and Paroxetine with the Metabolizing Enzyme CYP2B4: A Molecular Dynamics Simulation Study. J. Mol. Model. 2018, 24, 1–11. 10.1007/s00894-018-3617-8. [DOI] [PubMed] [Google Scholar]
- Lonsdale R.; Rouse S. L.; Sansom M. S. P.; Mulholland A. J. A Multiscale Approach to Modelling Drug Metabolism by Membrane-Bound Cytochrome P450 Enzymes. PLoS Comput. Biol. 2014, 10, e1003714. 10.1371/journal.pcbi.1003714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X.; Nandekar P.; Mustafa G.; Cojocaru V.; Lepesheva G. I.; Wade R. C. Ligand Tunnels in T. Brucei and Human CYP51: Insights for Parasite-Specific Drug Design. Biochim. Biophys. Acta, Gen. Subj. 2016, 1860, 67–78. 10.1016/j.bbagen.2015.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeřábek P.; Florián J.; Martínek V. Lipid Molecules Can Induce an Opening of Membrane-Facing Tunnels in Cytochrome P450 1A2. Phys. Chem. Chem. Phys. 2016, 18, 30344–30356. 10.1039/C6CP03692A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Y.-L.; Xue Q.; Zheng Q.-C.; Zhang J.-L.; Kong C.-P.; Fan J.-R.; Zhang H.-X. Structural Features and Dynamic Investigations of the Membrane-Bound Cytochrome P450 17A1. Biochim. Biophys. Acta, Biomembr. 2015, 1848, 2013–2021. 10.1016/j.bbamem.2015.05.017. [DOI] [PubMed] [Google Scholar]
- Hackett J. C. Membrane-Embedded Substrate Recognition by Cytochrome P450 3A4. J. Biol. Chem. 2018, 293, 4037–4046. 10.1074/jbc.RA117.000961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navrátilová V.; Paloncýová M.; Berka K.; Otyepka M. Effect of Lipid Charge on Membrane Immersion of Cytochrome P450 3A4. J. Phys. Chem. B 2016, 120, 11205–11213. 10.1021/acs.jpcb.6b10108. [DOI] [PubMed] [Google Scholar]
- Navrátilová V.; Paloncýová M.; Kajšová M.; Berka K.; Otyepka M. Effect of Cholesterol on the Structure of Membrane-Attached Cytochrome P450 3A4. J. Chem. Inf. Model. 2015, 55, 628–635. 10.1021/ci500645k. [DOI] [PubMed] [Google Scholar]
- Cui Y.-L.; Wu R.-L. Molecular Dynamics Investigations of Membrane-Bound CYP2C19 Polymorphisms Reveal Distinct Mechanisms for Peripheral Variants by Long-Range Effects on the Enzymatic Activity. Mol. BioSyst. 2017, 13, 1070–1079. 10.1039/C6MB00827E. [DOI] [PubMed] [Google Scholar]
- Navrátilová V.; Paloncýová M.; Berka K.; Mise S.; Haga Y.; Matsumura C.; Sakaki T.; Inui H.; Otyepka M. Molecular Insights into the Role of a Distal F240A Mutation That Alters CYP1A1 Activity towards Persistent Organic Pollutants. Biochim. Biophys. Acta, Gen. Subj. 2017, 1861, 2852–2860. 10.1016/j.bbagen.2017.08.002. [DOI] [PubMed] [Google Scholar]
- Martin L. L.; Holien J. K.; Mizrachi D.; Corbin C. J.; Conley A. J.; Parker M. W.; Rodgers R. J. Evolutionary Comparisons Predict That Dimerization of Human Cytochrome P450 Aromatase Increases Its Enzymatic Activity and Efficiency. J. Steroid Biochem. Mol. Biol. 2015, 154, 294–301. 10.1016/j.jsbmb.2015.09.006. [DOI] [PubMed] [Google Scholar]
- Jeřábek P.; Florián J.; Martínek V. Membrane-Anchored Cytochrome P450 1A2–Cytochrome b5 Complex Features an X-Shaped Contact between Antiparallel Transmembrane Helices. Chem. Res. Toxicol. 2016, 29, 626–636. 10.1021/acs.chemrestox.5b00349. [DOI] [PubMed] [Google Scholar]
- Ramsay R. R. Molecular Aspects of Monoamine Oxidase B. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2016, 69, 81–89. 10.1016/j.pnpbp.2016.02.005. [DOI] [PubMed] [Google Scholar]
- Fišar Z. Drugs Related to Monoamine Oxidase Activity. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2016, 69, 112–124. 10.1016/j.pnpbp.2016.02.012. [DOI] [PubMed] [Google Scholar]
- Apostolov R.; Yonezawa Y.; Standley D. M.; Kikugawa G.; Takano Y.; Nakamura H. Membrane Attachment Facilitates Ligand Access to the Active Site in Monoamine Oxidase A. Biochemistry 2009, 48, 5864–5873. 10.1021/bi900493n. [DOI] [PubMed] [Google Scholar]
- Allen W. J.; Bevan D. R. Steered Molecular Dynamics Simulations Reveal Important Mechanisms in Reversible Monoamine Oxidase B Inhibition. Biochemistry 2011, 50, 6441–6454. 10.1021/bi200446w. [DOI] [PubMed] [Google Scholar]
- Larit F.; Elokely K. M.; Chaurasiya N. D.; Benyahia S.; Nael M. A.; León F.; Abu-Darwish M. S.; Efferth T.; Wang Y. H.; Belouahem-Abed D.; et al. Inhibition of Human Monoamine Oxidase A and B by Flavonoids Isolated from Two Algerian Medicinal Plants. Phytomedicine 2018, 40, 27–36. 10.1016/j.phymed.2017.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riessland M.; Kolisnyk B.; Greengard P. Reactive Dopamine Leads to Triple Trouble in Nigral Neurons. Biochemistry 2017, 56, 6409–6410. 10.1021/acs.biochem.7b01057. [DOI] [PubMed] [Google Scholar]
- Orłowski A.; St-Pierre J.-F.; Magarkar A.; Bunker A.; Pasenkiewicz-Gierula M.; Vattulainen I.; Róg T. Properties of the Membrane Binding Component of Catechol-O-Methyltransferase Revealed by Atomistic Molecular Dynamics Simulations. J. Phys. Chem. B 2011, 115, 13541–13550. 10.1021/jp207177p. [DOI] [PubMed] [Google Scholar]
- Magarkar A.; Parkkila P.; Viitala T.; Lajunen T.; Mobarak E.; Licari G.; Cramariuc O.; Vauthey E.; Róg T.; Bunker A. Membrane Bound COMT Isoform Is an Interfacial Enzyme: General Mechanism and New Drug Design Paradigm. Chem. Commun. 2018, 54, 3440–3443. 10.1039/C8CC00221E. [DOI] [PubMed] [Google Scholar]
- Dzieciuch-Rojek M.; Poojari C.; Bednar J.; Bunker A.; Kozik B.; Nowakowska M.; Vattulainen I.; Wydro P.; Kepczynski M.; Róg T. Effects of Membrane PEGylation on Entry and Location of Antifungal Drug Itraconazole and Their Pharmacological Implications. Mol. Pharmaceutics 2017, 14, 1057–1070. 10.1021/acs.molpharmaceut.6b00969. [DOI] [PubMed] [Google Scholar]
- Wilkosz N.; Rissanen S.; Cyza M.; Szybka R.; Nowakowska M.; Bunker A.; Róg T.; Kepczynski M. Effect of Piroxicam on Lipid Membranes: Drug Encapsulation and Gastric Toxicity Aspects. Eur. J. Pharm. Sci. 2017, 100, 116–125. 10.1016/j.ejps.2017.01.007. [DOI] [PubMed] [Google Scholar]
- Markiewicz M.; Pasenkiewicz-Gierula M. Comparative Model Studies of Gastric Toxicity of Nonsteroidal Anti-Inflammatory Drugs. Langmuir 2011, 27, 6950–6961. 10.1021/la200499p. [DOI] [PubMed] [Google Scholar]
- Lajunen T.; Kontturi L.-S.; Viitala L.; Manna M.; Cramariuc O.; Róg T.; Bunker A.; Laaksonen T.; Viitala T.; Murtomäki L.; et al. Indocyanine Green-Loaded Liposomes for Light-Triggered Drug Release. Mol. Pharmaceutics 2016, 13, 2095–2107. 10.1021/acs.molpharmaceut.6b00207. [DOI] [PubMed] [Google Scholar]
- Cramariuc O.; Rog T.; Javanainen M.; Monticelli L.; Polishchuk A. V.; Vattulainen I. Mechanism for Translocation of Fluoroquinolones across Lipid Membranes. Biochim. Biophys. Acta, Biomembr. 2012, 1818, 2563–2571. 10.1016/j.bbamem.2012.05.027. [DOI] [PubMed] [Google Scholar]
- Dzieciuch M.; Rissanen S.; Szydłowska N.; Bunker A.; Kumorek M.; Jamróz D.; Vattulainen I.; Nowakowska M.; Róg T.; Kepczynski M. Pegylated Liposomes as Carriers of Hydrophobic Porphyrins. J. Phys. Chem. B 2015, 119, 6646–6657. 10.1021/acs.jpcb.5b01351. [DOI] [PubMed] [Google Scholar]
- Stepniewski M.; Kepczynski M.; Jamróz D.; Nowakowska M.; Rissanen S.; Vattulainen I.; Róg T. Interaction of Hematoporphyrin with Lipid Membranes. J. Phys. Chem. B 2012, 116, 4889–4897. 10.1021/jp300899b. [DOI] [PubMed] [Google Scholar]
- Khandelia H.; Witzke S.; Mouritsen O. G. Interaction of Salicylate and a Terpenoid Plant Extract with Model Membranes: Reconciling Experiments and Simulations. Biophys. J. 2010, 99, 3887–3894. 10.1016/j.bpj.2010.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rissanen S.; Kumorek M.; Martinez-Seara H.; Li Y. C.; Jamróz D.; Bunker A.; Nowakowska M.; Vattulainen I.; Kepczynski M.; Róg T. Effect of PEGylation on Drug Entry into Lipid Bilayer. J. Phys. Chem. B 2014, 118, 144–151. 10.1021/jp4105745. [DOI] [PubMed] [Google Scholar]
- Kopeć W.; Telenius J.; Khandelia H. Molecular Dynamics Simulations of the Interactions of Medicinal Plant Extracts and Drugs with Lipid Bilayer Membranes. FEBS J. 2013, 280, 2785–2805. 10.1111/febs.12286. [DOI] [PubMed] [Google Scholar]
- Cramariuc O.; Róg T.; Vattulainen I. Drug-Lipid Membrane Interaction Mechanisms Revealed Through Molecular Simulations. Curr. Phys. Chem. 2012, 2, 379–400. 10.2174/1877946811202040379. [DOI] [Google Scholar]
- Jodko-Piorecka K.; Litwinienko G. First Experimental Evidence of Dopamine Interactions with Negatively Charged Model Biomembranes. ACS Chem. Neurosci. 2013, 4, 1114–1122. 10.1021/cn4000633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matam Y.; Ray B. D.; Petrache H. I. Direct Affinity of Dopamine to Lipid Membranes Investigated by Nuclear Magnetic Resonance Spectroscopy. Neurosci. Lett. 2016, 618, 104–109. 10.1016/j.neulet.2016.02.052. [DOI] [PubMed] [Google Scholar]
- Drolle E.; Kučerka N.; Hoopes M. I.; Choi Y.; Katsaras J.; Karttunen M.; Leonenko Z. Effect of Melatonin and Cholesterol on the Structure of DOPC and DPPC Membranes. Biochim. Biophys. Acta, Biomembr. 2013, 1828, 2247–2254. 10.1016/j.bbamem.2013.05.015. [DOI] [PubMed] [Google Scholar]
- Choi Y.; Attwood S. J.; Hoopes M. I.; Drolle E.; Karttunen M.; Leonenko Z. Melatonin Directly Interacts with Cholesterol and Alleviates Cholesterol Effects in Dipalmitoylphosphatidylcholine Monolayers. Soft Matter 2014, 10, 206–213. 10.1039/C3SM52064A. [DOI] [PubMed] [Google Scholar]
- Huang X.; Gu H. H.; Zhan C. G. Mechanism for Cocaine Blocking the Transport of Dopamine: Insights from Molecular Modeling and Dynamics Simulations. J. Phys. Chem. B 2009, 113, 15057–15066. 10.1021/jp900963n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedlová K.; Nekardová M.; Kačer P.; Syslová K.; Vazdar M.; Jungwirth P.; Kudová E.; Cwiklik L. Distributions of Therapeutically Promising Neurosteroids in Cellular Membranes. Chem. Phys. Lipids 2017, 203, 78–86. 10.1016/j.chemphyslip.2016.12.004. [DOI] [PubMed] [Google Scholar]
- Juhola H.; Postila P. A.; Rissanen S.; Lolicato F.; Vattulainen I.; Rog T. Negatively Charged Gangliosides Promote Membrane Association of Amphipathic Neurotransmitters. Neuroscience 2018, 384, 214–223. 10.1016/j.neuroscience.2018.05.035. [DOI] [PubMed] [Google Scholar]
- Mouchlis V. D.; Bucher D.; McCammon J. A.; Dennis E. A. Membranes Serve as Allosteric Activators of Phospholipase A 2, Enabling It to Extract, Bind, and Hydrolyze Phospholipid Substrates. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, E516–E525. 10.1073/pnas.1424651112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouchlis V. D.; Chen Y.; McCammon J. A.; Dennis E. A. Membrane Allostery and Unique Hydrophobic Sites Promote Enzyme Substrate Specificity. J. Am. Chem. Soc. 2018, 140, 3285–3291. 10.1021/jacs.7b12045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riccardi L.; Arencibia J. M.; Bono L.; Armirotti A.; Girotto S.; De Vivo M. Lid Domain Plasticity and Lipid Flexibility Modulate Enzyme Specificity in Human Monoacylglycerol Lipase. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2017, 1862, 441–451. 10.1016/j.bbalip.2017.01.002. [DOI] [PubMed] [Google Scholar]
- Willems N.; Lelimousin M.; Koldsø H.; Sansom M. S. P. Interfacial Activation of M37 Lipase: A Multi-Scale Simulation Study. Biochim. Biophys. Acta, Biomembr. 2017, 1859, 340–349. 10.1016/j.bbamem.2016.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cozza G.; Rossetto M.; Bosello-Travain V.; Maiorino M.; Roveri A.; Toppo S.; Zaccarin M.; Zennaro L.; Ursini F. Glutathione Peroxidase 4-Catalyzed Reduction of Lipid Hydroperoxides in Membranes: The Polar Head of Membrane Phospholipids Binds the Enzyme and Addresses the Fatty Acid Hydroperoxide Group toward the Redox Center. Free Radical Biol. Med. 2017, 112, 1–11. 10.1016/j.freeradbiomed.2017.07.010. [DOI] [PubMed] [Google Scholar]
- Chatron N.; Chalmond B.; Trouvé A.; Benoît E.; Caruel H.; Lattard V.; Tchertanov L. Identification of the Functional States of Human Vitamin K Epoxide Reductase from Molecular Dynamics Simulations. RSC Adv. 2017, 7, 52071–52090. 10.1039/C7RA07463H. [DOI] [Google Scholar]
- Gumbart J. C.; Chipot C. Decrypting Protein Insertion through the Translocon with Free-Energy Calculations. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 1663–1671. 10.1016/j.bbamem.2016.02.017. [DOI] [PubMed] [Google Scholar]
- Bonardi F.; Halza E.; Walko M.; Du Plessis F.; Nouwen N.; Feringa B. L.; Driessen A. J. M. Probing the SecYEG Translocation Pore Size with Preproteins Conjugated with Sizable Rigid Spherical Molecules. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 7775–7780. 10.1073/pnas.1101705108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumbart J.; Chipot C.; Schulten K. Free-Energy Cost for Translocon-Assisted Insertion of Membrane Proteins. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 3596–3601. 10.1073/pnas.1012758108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori T.; Ishitani R.; Tsukazaki T.; Nureki O.; Sugita Y. Molecular Mechanisms Underlying the Early Stage of Protein Translocation through the Sec Translocon. Biochemistry 2010, 49, 945–950. 10.1021/bi901594w. [DOI] [PubMed] [Google Scholar]
- Wickles S.; Singharoy A.; Andreani J.; Seemayer S.; Bischoff L.; Berninghausen O.; Soeding J.; Schulten K.; van der Sluis E. O.; Beckmann R. A Structural Model of the Active Ribosome-Bound Membrane Protein Insertase YidC. eLife 2014, 3, e03035. 10.7554/eLife.03035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondar A. N.; del Val C.; Freites J. A.; Tobias D. J.; White S. H. Dynamics of SecY Translocons with Translocation-Defective Mutations. Structure 2010, 18, 847–857. 10.1016/j.str.2010.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson O. B.; Hedman R.; Marino J.; Wickles S.; Bischoff L.; Johansson M.; Müller-Lucks A.; Trovato F.; Puglisi J. D.; O’Brien E. P.; et al. Cotranslational Protein Folding inside the Ribosome Exit Tunnel. Cell Rep. 2015, 12, 1533–1540. 10.1016/j.celrep.2015.07.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulmschneider M. B.; Koehler Leman J.; Fennell H.; Beckstein O. Peptide Folding in Translocon-Like Pores. J. Membr. Biol. 2015, 248, 407–417. 10.1007/s00232-015-9808-7. [DOI] [PubMed] [Google Scholar]
- Gumbart J. C.; Teo I.; Roux B.; Schulten K. Reconciling the Roles of Kinetic and Thermodynamic Factors in Membrane-Protein Insertion. J. Am. Chem. Soc. 2013, 135, 2291–2297. 10.1021/ja310777k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rychkova A.; Warshel A. Exploring the Nature of the Translocon-Assisted Protein Insertion. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 495–500. 10.1073/pnas.1220361110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niesen M. J. M.; Wang C. Y.; Van Lehn R. C.; Miller T. F. Structurally Detailed Coarse-Grained Model for Sec-Facilitated Co-Translational Protein Translocation and Membrane Integration. PLoS Comput. Biol. 2017, 13, e1005427. 10.1371/journal.pcbi.1005427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crespo-Otero R.; Barbatti M. Recent Advances and Perspectives on Nonadiabatic Mixed Quantum–Classical Dynamics. Chem. Rev. 2018, 118, 7026–7068. 10.1021/acs.chemrev.7b00577. [DOI] [PubMed] [Google Scholar]
- Yu L.-J.; Suga M.; Wang-Otomo Z.-Y.; Shen J.-R. Structure of Photosynthetic LH1–RC Supercomplex at 1.9 Å Resolution. Nature 2018, 556, 209–213. 10.1038/s41586-018-0002-9. [DOI] [PubMed] [Google Scholar]
- Qian P.; Siebert C. A.; Wang P.; Canniffe D. P.; Hunter C. N. Cryo-EM Structure of the Blastochloris Viridis LH1–RC Complex at 2.9 Å. Nature 2018, 556, 203–208. 10.1038/s41586-018-0014-5. [DOI] [PubMed] [Google Scholar]
- Niwa S.; Yu L.-J.; Takeda K.; Hirano Y.; Kawakami T.; Wang-Otomo Z.-Y.; Miki K. Structure of the LH1–RC Complex from Thermochromatium Tepidum at 3.0 Å. Nature 2014, 508, 228–232. 10.1038/nature13197. [DOI] [PubMed] [Google Scholar]
- Gisriel C.; Sarrou I.; Ferlez B.; Golbeck J. H.; Redding K. E.; Fromme R. Structure of a Symmetric Photosynthetic Reaction Center–Photosystem. Science 2017, 357, 1021–1025. 10.1126/science.aan5611. [DOI] [PubMed] [Google Scholar]
- Nogi T.; Fathir I.; Kobayashi M.; Nozawa T.; Miki K. Crystal Structures of Photosynthetic Reaction Center and High-Potential Iron-Sulfur Protein from Thermochromatium Tepidum: Thermostability and Electron Transfer. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 13561–13566. 10.1073/pnas.240224997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saer R. G.; Pan J.; Hardjasa A.; Lin S.; Rosell F.; Mauk A. G.; Woodbury N. W.; Murphy M. E. P.; Beatty J. T. Structural and Kinetic Properties of Rhodobacter Sphaeroides Photosynthetic Reaction Centers Containing Exclusively Zn-Coordinated Bacteriochlorophyll as Bacteriochlorin Cofactors. Biochim. Biophys. Acta, Bioenerg. 2014, 1837, 366–374. 10.1016/j.bbabio.2013.11.015. [DOI] [PubMed] [Google Scholar]
- Koepke J.; Krammer E.-M.; Klingen A. R.; Sebban P.; Ullmann G. M.; Fritzsch G. pH Modulates the Quinone Position in the Photosynthetic Reaction Center from Rhodobacter Sphaeroides in the Neutral and Charge Separated States. J. Mol. Biol. 2007, 371, 396–409. 10.1016/j.jmb.2007.04.082. [DOI] [PubMed] [Google Scholar]
- Katona G.; Andréasson U.; Landau E. M.; Andréasson L.-E.; Neutze R. Lipidic Cubic Phase Crystal Structure of the Photosynthetic Reaction Centre from Rhodobacter Sphaeroides at 2.35 Å Resolution. J. Mol. Biol. 2003, 331, 681–692. 10.1016/S0022-2836(03)00751-4. [DOI] [PubMed] [Google Scholar]
- Stowell M. H. Light-Induced Structural Changes in Photosynthetic Reaction Center: Implications for Mechanism of Electron-Proton Transfer. Science 1997, 276, 812–816. 10.1126/science.276.5313.812. [DOI] [PubMed] [Google Scholar]
- Chirino A. J.; Lous E. J.; Huber M.; Allen J. P.; Schenck C. C.; Paddock M. L.; Feher G.; Rees D. C. Crystallographic Analyses of Site-Directed Mutants of the Photosynthetic Reaction Center from Rhodobacter Sphaeroides. Biochemistry 1994, 33, 4584–4593. 10.1021/bi00181a020. [DOI] [PubMed] [Google Scholar]
- Chang C. H.; El-Kabbani O.; Tiede D.; Norris J.; Schiffer M. Structure of the Membrane-Bound Protein Photosynthetic Reaction Center from Rhodobacter Sphaeroides. Biochemistry 1991, 30, 5352–5360. 10.1021/bi00236a005. [DOI] [PubMed] [Google Scholar]
- Johansson L. C.; Arnlund D.; Katona G.; White T. A.; Barty A.; DePonte D. P.; Shoeman R. L.; Wickstrand C.; Sharma A.; Williams G. J.; et al. Structure of a Photosynthetic Reaction Centre Determined by Serial Femtosecond Crystallography. Nat. Commun. 2013, 4, 2911. 10.1038/ncomms3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wöhri A. B.; Wahlgren W. Y.; Malmerberg E.; Johansson L. C.; Neutze R.; Katona G. Lipidic Sponge Phase Crystal Structure of a Photosynthetic Reaction Center Reveals Lipids on the Protein Surface. Biochemistry 2009, 48, 9831–9838. 10.1021/bi900545e. [DOI] [PubMed] [Google Scholar]
- Deisenhofer J.; Epp O.; Sinning I.; Michel H. Crystallographic Refinement at 2.3 Å Resolution and Refined Model of the Photosynthetic Reaction Centre FromRhodopseudomonas Viridis. J. Mol. Biol. 1995, 246, 429–457. 10.1006/jmbi.1994.0097. [DOI] [PubMed] [Google Scholar]
- Standfuss J.; Terwisscha van Scheltinga A. C.; Lamborghini M.; Kühlbrandt W. Mechanisms of Photoprotection and Nonphotochemical Quenching in Pea Light-Harvesting Complex at 2.5 Å Resolution. EMBO J. 2005, 24, 919–928. 10.1038/sj.emboj.7600585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X.; Li M.; Wan T.; Wang L.; Jia C.; Hou Z.; Zhao X.; Zhang J.; Chang W. Structural Insights into Energy Regulation of Light-Harvesting Complex CP29 from Spinach. Nat. Struct. Mol. Biol. 2011, 18, 309–315. 10.1038/nsmb.2008. [DOI] [PubMed] [Google Scholar]
- Liu Z.; Yan H.; Wang K.; Kuang T.; Zhang J.; Gui L.; An X.; Chang W. Crystal Structure of Spinach Major Light-Harvesting Complex at 2.72 Å Resolution. Nature 2004, 428, 287–292. 10.1038/nature02373. [DOI] [PubMed] [Google Scholar]
- Koepke J.; Hu X.; Muenke C.; Schulten K.; Michel H. The Crystal Structure of the Light-Harvesting Complex II (B800–850) from Rhodospirillum Molischianum. Structure 1996, 4, 581–597. 10.1016/S0969-2126(96)00063-9. [DOI] [PubMed] [Google Scholar]
- Papiz M. Z.; Prince S. M.; Howard T.; Cogdell R. J.; Isaacs N. W. The Structure and Thermal Motion of the B800–850 LH2 Complex from Rps. Acidophila at 2.0 Å Resolution and 100 K: New Structural Features and Functionally Relevant Motions. J. Mol. Biol. 2003, 326, 1523–1538. 10.1016/S0022-2836(03)00024-X. [DOI] [PubMed] [Google Scholar]
- Prince S. M.; Papiz M. Z.; Freer A. A.; McDermott G.; Hawthornthwaite-Lawless A. M.; Cogdell R. J.; Isaacs N. W. Apoprotein Structure in the LH2 Complex from Rhodopseudomonas Acidophila Strain 10050: Modular Assembly and Protein Pigment Interactions 1 1Edited by R. Huber. J. Mol. Biol. 1997, 268, 412–423. 10.1006/jmbi.1997.0966. [DOI] [PubMed] [Google Scholar]
- Ago H.; Adachi H.; Umena Y.; Tashiro T.; Kawakami K.; Kamiya N.; Tian L.; Han G.; Kuang T.; Liu Z.; et al. Novel Features of Eukaryotic Photosystem II Revealed by Its Crystal Structure Analysis from a Red Alga. J. Biol. Chem. 2016, 291, 5676–5687. 10.1074/jbc.M115.711689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suga M.; Akita F.; Sugahara M.; Kubo M.; Nakajima Y.; Nakane T.; Yamashita K.; Umena Y.; Nakabayashi M.; Yamane T.; et al. Light-Induced Structural Changes and the Site of O = O Bond Formation in PSII Caught by XFEL. Nature 2017, 543, 131–135. 10.1038/nature21400. [DOI] [PubMed] [Google Scholar]
- Suga M.; Akita F.; Hirata K.; Ueno G.; Murakami H.; Nakajima Y.; Shimizu T.; Yamashita K.; Yamamoto M.; Ago H.; et al. Native Structure of Photosystem II at 1.95 Å Resolution Viewed by Femtosecond X-Ray Pulses. Nature 2015, 517, 99–103. 10.1038/nature13991. [DOI] [PubMed] [Google Scholar]
- Koua F. H. M.; Umena Y.; Kawakami K.; Shen J.-R. Structure of Sr-Substituted Photosystem II at 2.1 Å Resolution and Its Implications in the Mechanism of Water Oxidation. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 3889–3894. 10.1073/pnas.1219922110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umena Y.; Kawakami K.; Shen J.-R.; Kamiya N. Crystal Structure of Oxygen-Evolving Photosystem II at a Resolution of 1.9 Å. Nature 2011, 473, 55–60. 10.1038/nature09913. [DOI] [PubMed] [Google Scholar]
- Wiwczar J. M.; LaFountain A. M.; Wang J.; Frank H. A.; Brudvig G. W. Chlorophyll a with a Farnesyl Tail in Thermophilic Cyanobacteria. Photosynth. Res. 2017, 134, 175–182. 10.1007/s11120-017-0425-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupitz C.; Basu S.; Grotjohann I.; Fromme R.; Zatsepin N. A.; Rendek K. N.; Hunter M. S.; Shoeman R. L.; White T. A.; Wang D.; et al. Serial Time-Resolved Crystallography of Photosystem II Using a Femtosecond X-Ray Laser. Nature 2014, 513, 261–265. 10.1038/nature13453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami K.; Umena Y.; Kamiya N.; Shen J.-R. Location of Chloride and Its Possible Functions in Oxygen-Evolving Photosystem II Revealed by X-Ray Crystallography. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 8567–8572. 10.1073/pnas.0812797106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiya N.; Shen J.-R. Crystal Structure of Oxygen-Evolving Photosystem II from Thermosynechococcus Vulcanus at 3.7-Å Resolution. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 98–103. 10.1073/pnas.0135651100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young I. D.; Ibrahim M.; Chatterjee R.; Gul S.; Fuller F. D.; Koroidov S.; Brewster A. S.; Tran R.; Alonso-Mori R.; Kroll T.; et al. Structure of Photosystem II and Substrate Binding at Room Temperature. Nature 2016, 540, 453–457. 10.1038/nature20161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayyer K.; Yefanov O. M.; Oberthür D.; Roy-Chowdhury S.; Galli L.; Mariani V.; Basu S.; Coe J.; Conrad C. E.; Fromme R.; et al. Macromolecular Diffractive Imaging Using Imperfect Crystals. Nature 2016, 530, 202–206. 10.1038/nature16949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guskov A.; Kern J.; Gabdulkhakov A.; Broser M.; Zouni A.; Saenger W. Cyanobacterial Photosystem II at 2.9-Å Resolution and the Role of Quinones, Lipids, Channels and Chloride. Nat. Struct. Mol. Biol. 2009, 16, 334–342. 10.1038/nsmb.1559. [DOI] [PubMed] [Google Scholar]
- Loll B.; Kern J.; Saenger W.; Zouni A.; Biesiadka J. Towards Complete Cofactor Arrangement in the 3.0 Å Resolution Structure of Photosystem II. Nature 2005, 438, 1040–1044. 10.1038/nature04224. [DOI] [PubMed] [Google Scholar]
- Ferreira K. N. Architecture of the Photosynthetic Oxygen-Evolving Center. Science 2004, 303, 1831–1838. 10.1126/science.1093087. [DOI] [PubMed] [Google Scholar]
- Qin X.; Suga M.; Kuang T.; Shen J.-R. Structural Basis for Energy Transfer Pathways in the Plant PSI-LHCI Supercomplex. Science 2015, 348, 989–995. 10.1126/science.aab0214. [DOI] [PubMed] [Google Scholar]
- Amunts A.; Toporik H.; Borovikova A.; Nelson N. Structure Determination and Improved Model of Plant Photosystem I. J. Biol. Chem. 2010, 285, 3478–3486. 10.1074/jbc.M109.072645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan P.; Fromme P.; Witt H. T.; Klukas O.; Saenger W.; Krauß N. Three-Dimensional Structure of Cyanobacterial Photosystem I at 2.5 Å Resolution. Nature 2001, 411, 909–917. 10.1038/35082000. [DOI] [PubMed] [Google Scholar]
- Brunger A. T.; Adams P. D.; Fromme P.; Fromme R.; Levitt M.; Schröder G. F. Improving the Accuracy of Macromolecular Structure Refinement at 7 Å Resolution. Structure 2012, 20, 957–966. 10.1016/j.str.2012.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman H. N.; Fromme P.; Barty A.; White T. A.; Kirian R. A.; Aquila A.; Hunter M. S.; Schulz J.; DePonte D. P.; Weierstall U.; et al. Femtosecond X-Ray Protein Nanocrystallography. Nature 2011, 470, 73–77. 10.1038/nature09750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X.; Ma J.; Su X.; Cao P.; Chang W.; Liu Z.; Zhang X.; Li M. Structure of the Maize Photosystem I Supercomplex with Light-Harvesting Complexes I and II. Science 2018, 360, 1109–1113. 10.1126/science.aat1156. [DOI] [PubMed] [Google Scholar]
- Pi X.; Tian L.; Dai H.-E.; Qin X.; Cheng L.; Kuang T.; Sui S.-F.; Shen J.-R. Unique Organization of Photosystem I–Light-Harvesting Supercomplex Revealed by Cryo-EM from a Red Alga. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 4423–4428. 10.1073/pnas.1722482115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons J. A.; Aragão D.; Slattery O.; Pisliakov A. V.; Soulimane T.; Caffrey M. Structural Insights into Electron Transfer in caa3-Type Cytochrome Oxidase. Nature 2012, 487, 514–518. 10.1038/nature11182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrenga A.; Michel H. The Cytochrome c Oxidase from Paracoccus Denitrificans Does Not Change the Metal Center Ligation upon Reduction. J. Biol. Chem. 1999, 274, 33296–33299. 10.1074/jbc.274.47.33296. [DOI] [PubMed] [Google Scholar]
- Ishigami I.; Zatsepin N. A.; Hikita M.; Conrad C. E.; Nelson G.; Coe J. D.; Basu S.; Grant T. D.; Seaberg M. H.; Sierra R. G.; et al. Crystal Structure of CO-Bound Cytochrome c Oxidase Determined by Serial Femtosecond X-Ray Crystallography at Room Temperature. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 8011–8016. 10.1073/pnas.1705628114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo F.; Shinzawa-Itoh K.; Hagimoto K.; Shimada A.; Shimada S.; Yamashita E.; Yoshikawa S.; Tsukihara T. Structure of Bovine Cytochrome c Oxidase in the Ligand-Free Reduced State at Neutral pH. Acta Crystallogr., Sect. F: Struct. Biol. Commun. 2018, 74, 92–98. 10.1107/S2053230X17018532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada A.; Hatano K.; Tadehara H.; Yano N.; Shinzawa-Itoh K.; Yamashita E.; Muramoto K.; Tsukihara T.; Yoshikawa S. X-Ray Structural Analyses of Azide-Bound Cytochrome c Oxidases Reveal That the H-Pathway Is Critically Important for the Proton-Pumping Activity. J. Biol. Chem. 2018, 293, 14868–14879. 10.1074/jbc.RA118.003123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukihara T.; Shimokata K.; Katayama Y.; Shimada H.; Muramoto K.; Aoyama H.; Mochizuki M.; Shinzawa-Itoh K.; Yamashita E.; Yao M.; et al. The Low-Spin Heme of Cytochrome c Oxidase as the Driving Element of the Proton-Pumping Process. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 15304–15309. 10.1073/pnas.2635097100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan S. S.; Yamashita E.; Baniulis D.; Cramer W. A. Quinone-Dependent Proton Transfer Pathways in the Photosynthetic Cytochrome b6f Complex. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 4297–4302. 10.1073/pnas.1222248110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroebel D.; Choquet Y.; Popot J.-L.; Picot D. An Atypical Haem in the Cytochrome B6f Complex. Nature 2003, 426, 413–418. 10.1038/nature02155. [DOI] [PubMed] [Google Scholar]
- Yamashita E.; Zhang H.; Cramer W. A. Structure of the Cytochrome b6f Complex: Quinone Analogue Inhibitors as Ligands of Heme cn. J. Mol. Biol. 2007, 370, 39–52. 10.1016/j.jmb.2007.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J.; Kurisu G.; Cramer W. A. Intraprotein Transfer of the Quinone Analogue Inhibitor 2,5-Dibromo-3-Methyl-6-Isopropyl-p-Benzoquinone in the Cytochrome b6f Complex. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 69–74. 10.1073/pnas.0504909102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurisu G. Structure of the Cytochrome b6f Complex of Oxygenic Photosynthesis: Tuning the Cavity. Science 2003, 302, 1009–1014. 10.1126/science.1090165. [DOI] [PubMed] [Google Scholar]
- Esser L.; Gong X.; Yang S.; Yu L.; Yu C.-A.; Xia D. Surface-Modulated Motion Switch: Capture and Release of Iron-Sulfur Protein in the Cytochrome bc1 Complex. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 13045–13050. 10.1073/pnas.0601149103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solmaz S. R. N.; Hunte C. Structure of Complex III with Bound Cytochrome c in Reduced State and Definition of a Minimal Core Interface for Electron Transfer. J. Biol. Chem. 2008, 283, 17542–17549. 10.1074/jbc.M710126200. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Huang L.; Shulmeister V. M.; Chi Y.-I.; Kim K. K.; Hung L.-W.; Crofts A. R.; Berry E. A.; Kim S.-H. Electron Transfer by Domain Movement in Cytochrome bc1. Nature 1998, 392, 677–684. 10.1038/33612. [DOI] [PubMed] [Google Scholar]
- Crowley P. J.; Berry E. A.; Cromartie T.; Daldal F.; Godfrey C. R. A.; Lee D.-W.; Phillips J. E.; Taylor A.; Viner R. The Role of Molecular Modeling in the Design of Analogues of the Fungicidal Natural Products Crocacins A and D. Bioorg. Med. Chem. 2008, 16, 10345–10355. 10.1016/j.bmc.2008.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L.; Cobessi D.; Tung E. Y.; Berry E. A. Binding of the Respiratory Chain Inhibitor Antimycin to the Mitochondrial Bc1 Complex: A New Crystal Structure Reveals an Altered Intramolecular Hydrogen-Bonding Pattern. J. Mol. Biol. 2005, 351, 573–597. 10.1016/j.jmb.2005.05.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L.; Sun G.; Cobessi D.; Wang A. C.; Shen J. T.; Tung E. Y.; Anderson V. E.; Berry E. A. 3-Nitropropionic Acid Is a Suicide Inhibitor of Mitochondrial Respiration That, upon Oxidation by Complex II, Forms a Covalent Adduct with a Catalytic Base Arginine in the Active Site of the Enzyme. J. Biol. Chem. 2006, 281, 5965–5972. 10.1074/jbc.M511270200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L.-S.; Shen J. T.; Wang A. C.; Berry E. A. Crystallographic Studies of the Binding of Ligands to the Dicarboxylate Site of Complex II, and the Identity of the Ligand in the “Oxaloacetate-Inhibited” State. Biochim. Biophys. Acta, Bioenerg. 2006, 1757, 1073–1083. 10.1016/j.bbabio.2006.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun F.; Huo X.; Zhai Y.; Wang A.; Xu J.; Su D.; Bartlam M.; Rao Z. Crystal Structure of Mitochondrial Respiratory Membrane Protein Complex II. Cell 2005, 121, 1043–1057. 10.1016/j.cell.2005.05.025. [DOI] [PubMed] [Google Scholar]
- Horsefield R.; Yankovskaya V.; Sexton G.; Whittingham W.; Shiomi K.; O̅mura S.; Byrne B.; Cecchini G.; Iwata S. Structural and Computational Analysis of the Quinone-Binding Site of Complex II (Succinate-Ubiquinone Oxidoreductase): A MECHANISM OF ELECTRON TRANSFER AND PROTON CONDUCTION DURING UBIQUINONE REDUCTION. J. Biol. Chem. 2006, 281, 7309–7316. 10.1074/jbc.M508173200. [DOI] [PubMed] [Google Scholar]
- Yankovskaya V. Architecture of Succinate Dehydrogenase and Reactive Oxygen Species Generation. Science 2003, 299, 700–704. 10.1126/science.1079605. [DOI] [PubMed] [Google Scholar]
- Jormakka M. Molecular Basis of Proton Motive Force Generation: Structure of Formate Dehydrogenase-N. Science 2002, 295, 1863–1868. 10.1126/science.1068186. [DOI] [PubMed] [Google Scholar]
- Agip A.-N. A.; Blaza J. N.; Bridges H. R.; Viscomi C.; Rawson S.; Muench S. P.; Hirst J. Cryo-EM Structures of Complex I from Mouse Heart Mitochondria in Two Biochemically Defined States. Nat. Struct. Mol. Biol. 2018, 25, 548–556. 10.1038/s41594-018-0073-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parey K.; Brandt U.; Xie H.; Mills D. J.; Siegmund K.; Vonck J.; Kühlbrandt W.; Zickermann V. Cryo-EM Structure of Respiratory Complex I at Work. eLife 2018, 7, e39213. 10.7554/eLife.39213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothery R. A.; Bertero M. G.; Cammack R.; Palak M.; Blasco F.; Strynadka N. C. J.; Weiner J. H. The Catalytic Subunit of Escherichia Coli Nitrate Reductase A Contains a Novel [4Fe-4S] Cluster with a High-Spin Ground State. Biochemistry 2004, 43, 5324–5333. 10.1021/bi049938l. [DOI] [PubMed] [Google Scholar]
- Bertero M. G.; Rothery R. A.; Palak M.; Hou C.; Lim D.; Blasco F.; Weiner J. H.; Strynadka N. C. J. Insights into the Respiratory Electron Transfer Pathway from the Structure of Nitrate Reductase A. Nat. Struct. Mol. Biol. 2003, 10, 681–687. 10.1038/nsb969. [DOI] [PubMed] [Google Scholar]
- Abe K.; Irie K.; Nakanishi H.; Suzuki H.; Fujiyoshi Y. Crystal Structures of the Gastric Proton Pump. Nature 2018, 556, 214–218. 10.1038/s41586-018-0003-8. [DOI] [PubMed] [Google Scholar]
- Andersson M.; Mattle D.; Sitsel O.; Klymchuk T.; Nielsen A. M.; Møller L. B.; White S. H.; Nissen P.; Gourdon P. Copper-Transporting P-Type ATPases Use a Unique Ion-Release Pathway. Nat. Struct. Mol. Biol. 2014, 21, 43–48. 10.1038/nsmb.2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa H.; Cornelius F.; Hirata A.; Toyoshima C. Sequential Substitution of K+ Bound to Na+,K+-ATPase Visualized by X-Ray Crystallography. Nat. Commun. 2015, 6, 8004. 10.1038/ncomms9004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa H.; Shinoda T.; Cornelius F.; Toyoshima C. Crystal Structure of the Sodium-Potassium Pump (Na+,K+-ATPase) with Bound Potassium and Ouabain. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 13742–13747. 10.1073/pnas.0907054106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinoda T.; Ogawa H.; Cornelius F.; Toyoshima C. Crystal Structure of the Sodium–Potassium Pump at 2.4 Å Resolution. Nature 2009, 459, 446–450. 10.1038/nature07939. [DOI] [PubMed] [Google Scholar]
- Laursen M.; Gregersen J. L.; Yatime L.; Nissen P.; Fedosova N. U. Structures and Characterization of Digoxin- and Bufalin-Bound Na+,K+-ATPase Compared with the Ouabain-Bound Complex. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 1755–1760. 10.1073/pnas.1422997112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanai R.; Ogawa H.; Vilsen B.; Cornelius F.; Toyoshima C. Crystal Structure of a Na+-Bound Na+,K+-ATPase Preceding the E1P State. Nature 2013, 502, 201–206. 10.1038/nature12578. [DOI] [PubMed] [Google Scholar]
- Nyblom M.; Poulsen H.; Gourdon P.; Reinhard L.; Andersson M.; Lindahl E.; Fedosova N.; Nissen P. Crystal Structure of Na+, K +-ATPase in the Na+-Bound State. Science 2013, 342, 123–127. 10.1126/science.1243352. [DOI] [PubMed] [Google Scholar]
- Laursen M.; Yatime L.; Nissen P.; Fedosova N. U. Crystal Structure of the High-Affinity Na+,K+-ATPase-Ouabain Complex with Mg2+ Bound in the Cation Binding Site. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 10958–10963. 10.1073/pnas.1222308110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morth J. P.; Pedersen B. P.; Toustrup-Jensen M. S.; Sørensen T. L.-M.; Petersen J.; Andersen J. P.; Vilsen B.; Nissen P. Crystal Structure of the Sodium–Potassium Pump. Nature 2007, 450, 1043–1049. 10.1038/nature06419. [DOI] [PubMed] [Google Scholar]
- Norimatsu Y.; Hasegawa K.; Shimizu N.; Toyoshima C. Protein–Phospholipid Interplay Revealed with Crystals of a Calcium Pump. Nature 2017, 545, 193–198. 10.1038/nature22357. [DOI] [PubMed] [Google Scholar]
- Clausen J. D.; Bublitz M.; Arnou B.; Montigny C.; Jaxel C.; Møller J. V.; Nissen P.; Andersen J. P.; le Maire M. SERCA Mutant E309Q Binds Two Ca 2+ Ions but Adopts a Catalytically Incompetent Conformation: Structure and Function of SERCA Mutant E309Q. EMBO J. 2013, 32, 3231–3243. 10.1038/emboj.2013.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoshima C.; Iwasawa S.; Ogawa H.; Hirata A.; Tsueda J.; Inesi G. Crystal Structures of the Calcium Pump and Sarcolipin in the Mg2+-Bound E1 State. Nature 2013, 495, 260–264. 10.1038/nature11899. [DOI] [PubMed] [Google Scholar]
- Obara K.; Miyashita N.; Xu C.; Toyoshima I.; Sugita Y.; Inesi G.; Toyoshima C. Structural Role of Countertransport Revealed in Ca2+ Pump Crystal Structure in the Absence of Ca2+. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 14489–14496. 10.1073/pnas.0506222102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saroussi S.; Schushan M.; Ben-Tal N.; Junge W.; Nelson N. Structure and Flexibility of the C-Ring in the Electromotor of Rotary FoF1-ATPase of Pea Chloroplasts. PLoS One 2012, 7, e43045. 10.1371/journal.pone.0043045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata T. Structure of the Rotor of the V-Type Na+-ATPase from Enterococcus Hirae. Science 2005, 308, 654–659. 10.1126/science.1110064. [DOI] [PubMed] [Google Scholar]
- Huang C.-S.; Pedersen B. P.; Stokes D. L. Crystal Structure of the Potassium-Importing KdpFABC Membrane Complex. Nature 2017, 546, 681–685. 10.1038/nature22970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson Z. L.; Chen J. ATP Binding Enables Substrate Release from Multidrug Resistance Protein 1. Cell 2018, 172, 81–89.e10. 10.1016/j.cell.2017.12.005. [DOI] [PubMed] [Google Scholar]
- Shintre C. A.; Pike A. C. W.; Li Q.; Kim J.-I.; Barr A. J.; Goubin S.; Shrestha L.; Yang J.; Berridge G.; Ross J.; et al. Structures of ABCB10, a Human ATP-Binding Cassette Transporter in Apo- and Nucleotide-Bound States. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 9710–9715. 10.1073/pnas.1217042110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldham M. L.; Chen S.; Chen J. Structural Basis for Substrate Specificity in the Escherichia Coli Maltose Transport System. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 18132–18137. 10.1073/pnas.1311407110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S.; Oldham M. L.; Davidson A. L.; Chen J. Carbon Catabolite Repression of the Maltose Transporter Revealed by X-Ray Crystallography. Nature 2013, 499, 364–368. 10.1038/nature12232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldham M. L.; Chen J. Snapshots of the Maltose Transporter during ATP Hydrolysis. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 15152–15156. 10.1073/pnas.1108858108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho H.; Miu A.; Alexander M. K.; Garcia N. K.; Oh A.; Zilberleyb I.; Reichelt M.; Austin C. D.; Tam C.; Shriver S.; et al. Structural Basis for Dual-Mode Inhibition of the ABC Transporter MsbA. Nature 2018, 557, 196–201. 10.1038/s41586-018-0083-5. [DOI] [PubMed] [Google Scholar]
- Hu N.-J.; Iwata S.; Cameron A. D.; Drew D. Crystal Structure of a Bacterial Homologue of the Bile Acid Sodium Symporter ASBT. Nature 2011, 478, 408–411. 10.1038/nature10450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruprecht J. J.; Hellawell A. M.; Harding M.; Crichton P. G.; McCoy A. J.; Kunji E. R. S. Structures of Yeast Mitochondrial ADP/ATP Carriers Support a Domain-Based Alternating-Access Transport Mechanism. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, E426–E434. 10.1073/pnas.1320692111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nury H.; Dahout-Gonzalez C.; Trézéguet V.; Lauquin G.; Brandolin G.; Pebay-Peyroula E. Structural Basis for Lipid-Mediated Interactions between Mitochondrial ADP/ATP Carrier Monomers. FEBS Lett. 2005, 579, 6031–6036. 10.1016/j.febslet.2005.09.061. [DOI] [PubMed] [Google Scholar]
- Pebay-Peyroula E.; Dahout-Gonzalez C.; Kahn R.; Trézéguet V.; Lauquin G. J.-M.; Brandolin G. Structure of Mitochondrial ADP/ATP Carrier in Complex with Carboxyatractyloside. Nature 2003, 426, 39–44. 10.1038/nature02056. [DOI] [PubMed] [Google Scholar]
- Wöhlert D.; Kühlbrandt W.; Yildiz Ö. Structure and Substrate Ion Binding in the Sodium/Proton Antiporter PaNhaP. eLife 2014, 3, e03579. 10.7554/eLife.03579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baradaran R.; Wang C.; Siliciano A. F.; Long S. B. Cryo-EM Structures of Fungal and Metazoan Mitochondrial Calcium Uniporters. Nature 2018, 559, 580–584. 10.1038/s41586-018-0331-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshy C.; Schweikhard E. S.; Gärtner R. M.; Perez C.; Yildiz Ö.; Ziegler C. Structural Evidence for Functional Lipid Interactions in the Betaine Transporter BetP: Structural Evidence for Functional Lipid Interactions in the BetP. EMBO J. 2013, 32, 3096–3105. 10.1038/emboj.2013.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez C.; Koshy C.; Yildiz Ö.; Ziegler C. Alternating-Access Mechanism in Conformationally Asymmetric Trimers of the Betaine Transporter BetP. Nature 2012, 490, 126–130. 10.1038/nature11403. [DOI] [PubMed] [Google Scholar]
- Li F.; Liu J.; Zheng Y.; Garavito R. M.; Ferguson-Miller S. Crystal Structures of Translocator Protein (TSPO) and Mutant Mimic of a Human Polymorphism. Science 2015, 347, 555–558. 10.1126/science.1260590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wöhlert D.; Grötzinger M. J.; Kühlbrandt W.; Yildiz Ö. Mechanism of Na+-Dependent Citrate Transport from the Structure of an Asymmetrical CitS Dimer. eLife 2015, 4, e09375. 10.7554/eLife.09375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman J. A.; Gouaux E. Structural Basis for Recognition of Diverse Antidepressants by the Human Serotonin Transporter. Nat. Struct. Mol. Biol. 2018, 25, 170–175. 10.1038/s41594-018-0026-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungnickel K. E. J.; Parker J. L.; Newstead S. Structural Basis for Amino Acid Transport by the CAT Family of SLC7 Transporters. Nat. Commun. 2018, 9, 550. 10.1038/s41467-018-03066-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penmatsa A.; Wang K. H.; Gouaux E. X-Ray Structures of Drosophila Dopamine Transporter in Complex with Nisoxetine and Reboxetine. Nat. Struct. Mol. Biol. 2015, 22, 506–508. 10.1038/nsmb.3029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heng J.; Zhao Y.; Liu M.; Liu Y.; Fan J.; Wang X.; Zhao Y.; Zhang X. C. Substrate-Bound Structure of the E. Coli Multidrug Resistance Transporter MdfA. Cell Res. 2015, 25, 1060–1073. 10.1038/cr.2015.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oswald C.; Tam H.-K.; Pos K. M. Transport of Lipophilic Carboxylates Is Mediated by Transmembrane Helix 2 in Multidrug Transporter AcrB. Nat. Commun. 2016, 7, 13819. 10.1038/ncomms13819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai X.; Yan C.; Yang G.; Lu P.; Ma D.; Sun L.; Zhou R.; Scheres S. H. W.; Shi Y. An Atomic Structure of Human γ-Secretase. Nature 2015, 525, 212–217. 10.1038/nature14892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quigley A.; Dong Y. Y.; Pike A. C. W.; Dong L.; Shrestha L.; Berridge G.; Stansfeld P. J.; Sansom M. S. P.; Edwards A. M.; Bountra C.; et al. The Structural Basis of ZMPSTE24-Dependent Laminopathies. Science 2013, 339, 1604–1607. 10.1126/science.1231513. [DOI] [PubMed] [Google Scholar]
- Vinothkumar K. R. Structure of Rhomboid Protease in a Lipid Environment. J. Mol. Biol. 2011, 407, 232–247. 10.1016/j.jmb.2011.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Shem A.; Fass D.; Bibi E. Structural Basis for Intramembrane Proteolysis by Rhomboid Serine Proteases. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 462–466. 10.1073/pnas.0609773104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo P.; Yu X.; Wang W.; Fan S.; Li X.; Wang J. Crystal Structure of a Phosphorylation-Coupled Vitamin C Transporter. Nat. Struct. Mol. Biol. 2015, 22, 238–241. 10.1038/nsmb.2975. [DOI] [PubMed] [Google Scholar]
- Yang J.; Kulkarni K.; Manolaridis I.; Zhang Z.; Dodd R. B.; Mas-Droux C.; Barford D. Mechanism of Isoprenylcysteine Carboxyl Methylation from the Crystal Structure of the Integral Membrane Methyltransferase ICMT. Mol. Cell 2011, 44, 997–1004. 10.1016/j.molcel.2011.10.020. [DOI] [PubMed] [Google Scholar]
- Mao G.; Zhao Y.; Kang X.; Li Z.; Zhang Y.; Wang X.; Sun F.; Sankaran K.; Zhang X. C. Crystal Structure of E. Coli Lipoprotein Diacylglyceryl Transferase. Nat. Commun. 2016, 7, 10198. 10.1038/ncomms10198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diver M. M.; Pedi L.; Koide A.; Koide S.; Long S. B. Atomic Structure of the Eukaryotic Intramembrane RAS Methyltransferase ICMT. Nature 2018, 553, 526–529. 10.1038/nature25439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrou V. I.; Herrera C. M.; Schultz K. M.; Clarke O. B.; Vendome J.; Tomasek D.; Banerjee S.; Rajashankar K. R.; Belcher Dufrisne M.; Kloss B.; et al. Structures of Aminoarabinose Transferase ArnT Suggest a Molecular Basis for Lipid A Glycosylation. Science 2016, 351, 608–612. 10.1126/science.aad1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai L.; Wang T.; Zhao G.; Kovach A.; Li H. The Atomic Structure of a Eukaryotic Oligosaccharyltransferase Complex. Nature 2018, 555, 328–333. 10.1038/nature25755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wild R.; Kowal J.; Eyring J.; Ngwa E. M.; Aebi M.; Locher K. P. Structure of the Yeast Oligosaccharyltransferase Complex Gives Insight into Eukaryotic N-Glycosylation. Science 2018, 359, 545–550. 10.1126/science.aar5140. [DOI] [PubMed] [Google Scholar]
- Basak S.; Gicheru Y.; Samanta A.; Molugu S. K.; Huang W.; Fuente M. la de; Hughes T.; Taylor D. J.; Nieman M. T.; Moiseenkova-Bell V.; et al. Cryo-EM Structure of 5-HT3A Receptor in Its Resting Conformation. Nat. Commun. 2018, 9, 514. 10.1038/s41467-018-02997-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S.; Noviello C. M.; Teng J.; Walsh R. M.; Kim J. J.; Hibbs R. E. Structure of a Human Synaptic GABAA Receptor. Nature 2018, 559, 67–72. 10.1038/s41586-018-0255-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X.; Jensen M. Ø.; Jogini V.; Stein R. A.; Lee C.-H.; Mchaourab H. S.; Shaw D. E.; Gouaux E. Mechanism of NMDA Receptor Channel Block by MK-801 and Memantine. Nature 2018, 556, 515–519. 10.1038/s41586-018-0039-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh R. M.; Roh S.-H.; Gharpure A.; Morales-Perez C. L.; Teng J.; Hibbs R. E. Structural Principles of Distinct Assemblies of the Human Α4β2 Nicotinic Receptor. Nature 2018, 557, 261–265. 10.1038/s41586-018-0081-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan J.; Chen Q.; Willenbring D.; Mowrey D.; Kong X.-P.; Cohen A.; Divito C. B.; Xu Y.; Tang P. Structure of the Pentameric Ligand-Gated Ion Channel GLIC Bound with Anesthetic Ketamine. Structure 2012, 20, 1463–1469. 10.1016/j.str.2012.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauguet L.; Poitevin F.; Murail S.; Van Renterghem C.; Moraga-Cid G.; Malherbe L.; Thompson A. W.; Koehl P.; Corringer P.-J.; Baaden M.; et al. Structural Basis for Ion Permeation Mechanism in Pentameric Ligand-Gated Ion Channels. EMBO J. 2013, 32, 728–741. 10.1038/emboj.2013.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan C.; Choi W.; Sun W.; Du J.; Lu W. Structure of the Human Lipid-Gated Cation Channel TRPC3. eLife 2018, 7, e36852. 10.7554/eLife.36852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strugatsky D.; McNulty R.; Munson K.; Chen C.-K.; Soltis S. M.; Sachs G.; Luecke H. Structure of the Proton-Gated Urea Channel from the Gastric Pathogen Helicobacter Pylori. Nature 2012, 493, 255–258. 10.1038/nature11684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin E. J.; Cao Y.; Enkavi G.; Quick M.; Pan Y.; Tajkhorshid E.; Zhou M. Structure and Permeation Mechanism of a Mammalian Urea Transporter. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 11194–11199. 10.1073/pnas.1207362109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J.; Daniels B. V.; Fu D. Crystal Structure of AqpZ Tetramer Reveals Two Distinct Arg-189 Conformations Associated with Water Permeation through the Narrowest Constriction of the Water-Conducting Channel. J. Biol. Chem. 2006, 281, 454–460. 10.1074/jbc.M508926200. [DOI] [PubMed] [Google Scholar]
- Tani K.; Mitsuma T.; Hiroaki Y.; Kamegawa A.; Nishikawa K.; Tanimura Y.; Fujiyoshi Y. Mechanism of Aquaporin-4’s Fast and Highly Selective Water Conduction and Proton Exclusion. J. Mol. Biol. 2009, 389, 694–706. 10.1016/j.jmb.2009.04.049. [DOI] [PubMed] [Google Scholar]
- Hite R. K.; Li Z.; Walz T. Principles of Membrane Protein Interactions with Annular Lipids Deduced from Aquaporin-0 2D Crystals. EMBO J. 2010, 29, 1652–1658. 10.1038/emboj.2010.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonen T.; Cheng Y.; Sliz P.; Hiroaki Y.; Fujiyoshi Y.; Harrison S. C.; Walz T. Lipid–Protein Interactions in Double-Layered Two-Dimensional AQP0 Crystals. Nature 2005, 438, 633–638. 10.1038/nature04321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin P.; Bulkley D.; Guo Y.; Zhang W.; Guo Z.; Huynh W.; Wu S.; Meltzer S.; Cheng T.; Jan L. Y.; et al. Electron Cryo-Microscopy Structure of the Mechanotransduction Channel NOMPC. Nature 2017, 547, 118–122. 10.1038/nature22981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkes M.; Madej M. G.; Kreuter L.; Rhinow D.; Heinz V.; De Sanctis S.; Ruppel S.; Richter R. M.; Joos F.; Grieben M.; et al. Molecular Insights into Lipid-Assisted Ca2+ Regulation of the TRP Channel Polycystin-2. Nat. Struct. Mol. Biol. 2017, 24, 123–130. 10.1038/nsmb.3357. [DOI] [PubMed] [Google Scholar]
- Hirschi M.; Herzik M. A. Jr; Wie J.; Suo Y.; Borschel W. F.; Ren D.; Lander G. C.; Lee S.-Y. Cryo-Electron Microscopy Structure of the Lysosomal Calcium-Permeable Channel TRPML3. Nature 2017, 550, 411–414. 10.1038/nature24055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z.; Tóth B.; Szollosi A.; Chen J.; Csanády L. Structure of a TRPM2 Channel in Complex with Ca2+ Explains Unique Gating Regulation. eLife 2018, 7, e36409. 10.7554/eLife.36409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan J.; Li J.; Zeng B.; Chen G.-L.; Peng X.; Zhang Y.; Wang J.; Clapham D. E.; Li Z.; Zhang J. Structure of the Mouse TRPC4 Ion Channel. Nat. Commun. 2018, 9, 3102. 10.1038/s41467-018-05247-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinayagam D.; Mager T.; Apelbaum A.; Bothe A.; Merino F.; Hofnagel O.; Gatsogiannis C.; Raunser S. Electron Cryo-Microscopy Structure of the Canonical TRPC4 Ion Channel. eLife 2018, 7, e36615. 10.7554/eLife.36615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang L.; Gamal El-Din T. M.; Swanson T. M.; Pryde D. C.; Scheuer T.; Zheng N.; Catterall W. A. Structural Basis for Inhibition of a Voltage-Gated Ca2+ Channel by Ca2+ Antagonist Drugs. Nature 2016, 537, 117–121. 10.1038/nature19102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang L.; Gamal El-Din T. M.; Payandeh J.; Martinez G. Q.; Heard T. M.; Scheuer T.; Zheng N.; Catterall W. A. Structural Basis for Ca2+ Selectivity of a Voltage-Gated Calcium Channel. Nature 2014, 505, 56–61. 10.1038/nature12775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H.; Hu M.; Guo J.; Ou X.; Cai T.; Liu Z. Pore Architecture of TRIC Channels and Insights into Their Gating Mechanism. Nature 2016, 538, 537–541. 10.1038/nature19767. [DOI] [PubMed] [Google Scholar]
- Ahuja S.; Mukund S.; Deng L.; Khakh K.; Chang E.; Ho H.; Shriver S.; Young C.; Lin S.; Johnson J. P.; et al. Structural Basis of Nav1.7 Inhibition by an Isoform-Selective Small-Molecule Antagonist. Science 2015, 350, aac5464–aac5464. 10.1126/science.aac5464. [DOI] [PubMed] [Google Scholar]
- Pan X.; Li Z.; Zhou Q.; Shen H.; Wu K.; Huang X.; Chen J.; Zhang J.; Zhu X.; Lei J.; et al. Structure of the Human Voltage-Gated Sodium Channel Nav1.4 in Complex with Β1. Science 2018, 362, eaau2486. 10.1126/science.aau2486. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Ren W.; DeCaen P.; Yan C.; Tao X.; Tang L.; Wang J.; Hasegawa K.; Kumasaka T.; He J.; et al. Crystal Structure of an Orthologue of the NaChBac Voltage-Gated Sodium Channel. Nature 2012, 486, 130–134. 10.1038/nature11054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenaeus M. J.; Gamal El-Din T. M.; Ing C.; Ramanadane K.; Pomès R.; Zheng N.; Catterall W. A. Structures of Closed and Open States of a Voltage-Gated Sodium Channel. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, E3051–E3060. 10.1073/pnas.1700761114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payandeh J.; Gamal El-Din T. M.; Scheuer T.; Zheng N.; Catterall W. A. Crystal Structure of a Voltage-Gated Sodium Channel in Two Potentially Inactivated States. Nature 2012, 486, 135–139. 10.1038/nature11077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payandeh J.; Scheuer T.; Zheng N.; Catterall W. A. The Crystal Structure of a Voltage-Gated Sodium Channel. Nature 2011, 475, 353–358. 10.1038/nature10238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang D.; Gamal El-Din T. M.; Ing C.; Lu P.; Pomès R.; Zheng N.; Catterall W. A. Structural Basis for Gating Pore Current in Periodic Paralysis. Nature 2018, 557, 590–594. 10.1038/s41586-018-0120-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan J.; Li Z.; Li J.; Hulse R. E.; Santa-Cruz A.; Valinsky W. C.; Abiria S. A.; Krapivinsky G.; Zhang J.; Clapham D. E. Structure of the Mammalian TRPM7, a Magnesium Channel Required during Embryonic Development. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, E8201–E8210. 10.1073/pnas.1810719115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whorton M. R.; MacKinnon R. X-Ray Structure of the Mammalian GIRK2−Βγ G-Protein Complex. Nature 2013, 498, 190–197. 10.1038/nature12241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen S. B.; Tao X.; MacKinnon R. Structural Basis of PIP2 Activation of the Classical Inward Rectifier K+ Channel Kir2.2. Nature 2011, 477, 495–498. 10.1038/nature10370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hite R. K.; Tao X.; MacKinnon R. Structural Basis for Gating the High-Conductance Ca2+-Activated K+ Channel. Nature 2017, 541, 52–57. 10.1038/nature20775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao X.; Hite R. K.; MacKinnon R. Cryo-EM Structure of the Open High-Conductance Ca2+-Activated K+ Channel. Nature 2017, 541, 46–51. 10.1038/nature20608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whicher J. R.; MacKinnon R. Structure of the Voltage-Gated K+ Channel Eag1 Reveals an Alternative Voltage Sensing Mechanism. Science 2016, 353, 664–669. 10.1126/science.aaf8070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pau V.; Zhou Y.; Ramu Y.; Xu Y.; Lu Z. Crystal Structure of an Inactivated Mutant Mammalian Voltage-Gated K+ Channel. Nat. Struct. Mol. Biol. 2017, 24, 857–865. 10.1038/nsmb.3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao X.; Lee A.; Limapichat W.; Dougherty D. A.; MacKinnon R. A Gating Charge Transfer Center in Voltage Sensors. Science 2010, 328, 67–73. 10.1126/science.1185954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long S. B.; Tao X.; Campbell E. B.; MacKinnon R. Atomic Structure of a Voltage-Dependent K+ Channel in a Lipid Membrane-like Environment. Nature 2007, 450, 376–382. 10.1038/nature06265. [DOI] [PubMed] [Google Scholar]
- Rheinberger J.; Gao X.; Schmidpeter P. A.; Nimigean C. M. Ligand Discrimination and Gating in Cyclic Nucleotide-Gated Ion Channels from Apo and Partial Agonist-Bound Cryo-EM Structures. eLife 2018, 7, e39775. 10.7554/eLife.39775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- She J.; Guo J.; Chen Q.; Zeng W.; Jiang Y.; Bai X. Structural Insights into the Voltage and Phospholipid Activation of the Mammalian TPC1 Channel. Nature 2018, 556, 130–134. 10.1038/nature26139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y. Y.; Pike A. C. W.; Mackenzie A.; McClenaghan C.; Aryal P.; Dong L.; Quigley A.; Grieben M.; Goubin S.; Mukhopadhyay S.; et al. K2P Channel Gating Mechanisms Revealed by Structures of TREK-2 and a Complex with Prozac. Science 2015, 347, 1256–1259. 10.1126/science.1261512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kintzer A. F.; Green E. M.; Dominik P. K.; Bridges M.; Armache J.-P.; Deneka D.; Kim S. S.; Hubbell W.; Kossiakoff A. A.; Cheng Y.; et al. Structural Basis for Activation of Voltage Sensor Domains in an Ion Channel TPC1. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, E9095–E9104. 10.1073/pnas.1805651115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuello L. G.; Cortes D. M.; Perozo E. The Gating Cycle of a K+ Channel at Atomic Resolution. eLife 2017, 6, e28032. 10.7554/eLife.28032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matulef K.; Annen A. W.; Nix J. C.; Valiyaveetil F. I. Individual Ion Binding Sites in the K+ Channel Play Distinct Roles in C-Type Inactivation and in Recovery from Inactivation. Structure 2016, 24, 750–761. 10.1016/j.str.2016.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenaeus M. J.; Burdette D.; Wagner T.; Focia P. J.; Gross A. Structures of KcsA in Complex with Symmetrical Quaternary Ammonium Compounds Reveal a Hydrophobic Binding Site. Biochemistry 2014, 53, 5365–5373. 10.1021/bi500525s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matulef K.; Komarov A. G.; Costantino C. A.; Valiyaveetil F. I. Using Protein Backbone Mutagenesis to Dissect the Link between Ion Occupancy and C-Type Inactivation in K+ Channels. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 17886–17891. 10.1073/pnas.1314356110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson A. N.; Kim I.; Panosian T. D.; Iverson T. M.; Allen T. W.; Nimigean C. M. Mechanism of Potassium-Channel Selectivity Revealed by Na+ and Li+ Binding Sites within the KcsA Pore. Nat. Struct. Mol. Biol. 2009, 16, 1317–1324. 10.1038/nsmb.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y.; Morais-Cabral J. H.; Kaufman A.; MacKinnon R. Chemistry of Ion Coordination and Hydration Revealed by a K+ Channel–Fab Complex at 2.0 Å Resolution. Nature 2001, 414, 43–48. 10.1038/35102009. [DOI] [PubMed] [Google Scholar]
- Labro A. J.; Cortes D. M.; Tilegenova C.; Cuello L. G. Inverted Allosteric Coupling between Activation and Inactivation Gates in K+ Channels. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 5426–5431. 10.1073/pnas.1800559115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong X.; Qian H.; Cao P.; Zhao X.; Zhou Q.; Lei J.; Yan N. Structural Basis for the Recognition of Sonic Hedgehog by Human Patched1. Science 2018, 361, eaas8935. 10.1126/science.aas8935. [DOI] [PubMed] [Google Scholar]
- Wu H.; Wang C.; Gregory K. J.; Han G. W.; Cho H. P.; Xia Y.; Niswender C. M.; Katritch V.; Meiler J.; Cherezov V.; et al. Structure of a Class C GPCR Metabotropic Glutamate Receptor 1 Bound to an Allosteric Modulator. Science 2014, 344, 58–64. 10.1126/science.1249489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenstein K.; Kean J.; Bortolato A.; Cheng R. K. Y.; Doré A. S.; Jazayeri A.; Cooke R. M.; Weir M.; Marshall F. H. Structure of Class B GPCR Corticotropin-Releasing Factor Receptor 1. Nature 2013, 499, 438–443. 10.1038/nature12357. [DOI] [PubMed] [Google Scholar]
- Zhang D.; Gao Z.-G.; Zhang K.; Kiselev E.; Crane S.; Wang J.; Paoletta S.; Yi C.; Ma L.; Zhang W.; et al. Two Disparate Ligand-Binding Sites in the Human P2Y1 Receptor. Nature 2015, 520, 317–321. 10.1038/nature14287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.; Zhang K.; Gao Z.-G.; Paoletta S.; Zhang D.; Han G. W.; Li T.; Ma L.; Zhang W.; Müller C. E.; et al. Agonist-Bound Structure of the Human P2Y12 Receptor. Nature 2014, 509, 119–122. 10.1038/nature13288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K.; Zhang J.; Gao Z.-G.; Zhang D.; Zhu L.; Han G. W.; Moss S. M.; Paoletta S.; Kiselev E.; Lu W.; et al. Structure of the Human P2Y12 Receptor in Complex with an Antithrombotic Drug. Nature 2014, 509, 115–118. 10.1038/nature13083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacker D.; Wang S.; McCorvy J. D.; Betz R. M.; Venkatakrishnan A. J.; Levit A.; Lansu K.; Schools Z. L.; Che T.; Nichols D. E.; et al. Crystal Structure of an LSD-Bound Human Serotonin Receptor. Cell 2017, 168, 377–389.e12. 10.1016/j.cell.2016.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W.; Wacker D.; Gati C.; Han G. W.; James D.; Wang D.; Nelson G.; Weierstall U.; Katritch V.; Barty A.; et al. Serial Femtosecond Crystallography of G Protein-Coupled Receptors. Science 2013, 342, 1521–1524. 10.1126/science.1244142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacker D.; Wang C.; Katritch V.; Han G. W.; Huang X.-P.; Vardy E.; McCorvy J. D.; Jiang Y.; Chu M.; Siu F. Y.; et al. Structural Features for Functional Selectivity at Serotonin Receptors. Science 2013, 340, 615–619. 10.1126/science.1232808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua T.; Vemuri K.; Nikas S. P.; Laprairie R. B.; Wu Y.; Qu L.; Pu M.; Korde A.; Jiang S.; Ho J.-H.; et al. Crystal Structures of Agonist-Bound Human Cannabinoid Receptor CB1. Nature 2017, 547, 468–471. 10.1038/nature23272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W.; Manglik A.; Venkatakrishnan A. J.; Laeremans T.; Feinberg E. N.; Sanborn A. L.; Kato H. E.; Livingston K. E.; Thorsen T. S.; Kling R. C.; et al. Structural Insights into Μ-Opioid Receptor Activation. Nature 2015, 524, 315–321. 10.1038/nature14886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manglik A.; Kruse A. C.; Kobilka T. S.; Thian F. S.; Mathiesen J. M.; Sunahara R. K.; Pardo L.; Weis W. I.; Kobilka B. K.; Granier S. Crystal Structure of the μ-Opioid Receptor Bound to a Morphinan Antagonist. Nature 2012, 485, 321–326. 10.1038/nature10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Che T.; Majumdar S.; Zaidi S. A.; Ondachi P.; McCorvy J. D.; Wang S.; Mosier P. D.; Uprety R.; Vardy E.; Krumm B. E.; et al. Structure of the Nanobody-Stabilized Active State of the Kappa Opioid Receptor. Cell 2018, 172, 55–67.e15. 10.1016/j.cell.2017.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thal D. M.; Sun B.; Feng D.; Nawaratne V.; Leach K.; Felder C. C.; Bures M. G.; Evans D. A.; Weis W. I.; Bachhawat P.; et al. Crystal Structures of the M1 and M4Muscarinic Acetylcholine Receptors. Nature 2016, 531, 335–340. 10.1038/nature17188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shihoya W.; Nishizawa T.; Yamashita K.; Inoue A.; Hirata K.; Kadji F. M. N.; Okuta A.; Tani K.; Aoki J.; Fujiyoshi Y.; et al. X-Ray Structures of Endothelin ETB Receptor Bound to Clinical Antagonist Bosentan and Its Analog. Nat. Struct. Mol. Biol. 2017, 24, 758–764. 10.1038/nsmb.3450. [DOI] [PubMed] [Google Scholar]
- Burg J. S.; Ingram J. R.; Venkatakrishnan A. J.; Jude K. M.; Dukkipati A.; Feinberg E. N.; Angelini A.; Waghray D.; Dror R. O.; Ploegh H. L.; et al. Structural Basis for Chemokine Recognition and Activation of a Viral G Protein-Coupled Receptor. Science 2015, 347, 1113–1117. 10.1126/science.aaa5026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oswald C.; Rappas M.; Kean J.; Doré A. S.; Errey J. C.; Bennett K.; Deflorian F.; Christopher J. A.; Jazayeri A.; Mason J. S.; et al. Intracellular Allosteric Antagonism of the CCR9 Receptor. Nature 2016, 540, 462–465. 10.1038/nature20606. [DOI] [PubMed] [Google Scholar]
- Cheng R. K. Y.; Segala E.; Robertson N.; Deflorian F.; Doré A. S.; Errey J. C.; Fiez-Vandal C.; Marshall F. H.; Cooke R. M. Structures of Human A1 and A2A Adenosine Receptors with Xanthines Reveal Determinants of Selectivity. Structure 2017, 25, 1275–1285.e4. 10.1016/j.str.2017.06.012. [DOI] [PubMed] [Google Scholar]
- Liu X.; Ahn S.; Kahsai A. W.; Meng K.-C.; Latorraca N. R.; Pani B.; Venkatakrishnan A. J.; Masoudi A.; Weis W. I.; Dror R. O.; et al. Mechanism of Intracellular Allosteric β2AR Antagonist Revealed by X-Ray Crystal Structure. Nature 2017, 548, 480–484. 10.1038/nature23652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum D. M.; Zhang C.; Lyons J. A.; Holl R.; Aragao D.; Arlow D. H.; Rasmussen S. G. F.; Choi H.-J.; DeVree B. T.; Sunahara R. K.; et al. Structure and Function of an Irreversible Agonist-β2 Adrenoceptor Complex. Nature 2011, 469, 236–240. 10.1038/nature09665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacker D.; Fenalti G.; Brown M. A.; Katritch V.; Abagyan R.; Cherezov V.; Stevens R. C. Conserved Binding Mode of Human β2 Adrenergic Receptor Inverse Agonists and Antagonist Revealed by X-Ray Crystallography. J. Am. Chem. Soc. 2010, 132, 11443–11445. 10.1021/ja105108q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warne T.; Moukhametzianov R.; Baker J. G.; Nehmé R.; Edwards P. C.; Leslie A. G. W.; Schertler G. F. X.; Tate C. G. The Structural Basis for Agonist and Partial Agonist Action on a β1-Adrenergic Receptor. Nature 2011, 469, 241–244. 10.1038/nature09746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami M.; Kouyama T. Crystallographic Analysis of the Primary Photochemical Reaction of Squid Rhodopsin. J. Mol. Biol. 2011, 413, 615–627. 10.1016/j.jmb.2011.08.044. [DOI] [PubMed] [Google Scholar]
- Murakami M.; Kouyama T. Crystal Structure of Squid Rhodopsin. Nature 2008, 453, 363–367. 10.1038/nature06925. [DOI] [PubMed] [Google Scholar]
- Mattle D.; Kuhn B.; Aebi J.; Bedoucha M.; Kekilli D.; Grozinger N.; Alker A.; Rudolph M. G.; Schmid G.; Schertler G. F. X.; et al. Ligand Channel in Pharmacologically Stabilized Rhodopsin. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 3640–3645. 10.1073/pnas.1718084115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Standfuss J.; Edwards P. C.; D’Antona A.; Fransen M.; Xie G.; Oprian D. D.; Schertler G. F. X. The Structural Basis of Agonist-Induced Activation in Constitutively Active Rhodopsin. Nature 2011, 471, 656–660. 10.1038/nature09795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenkamp R. E. Alternative Models for Two Crystal Structures of Bovine Rhodopsin. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2008, 64, 902–904. 10.1107/S0907444908017162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman B.; Kelly B.; McMillan B. J.; Seegar T. C. M.; Dror R. O.; Kruse A. C.; Blacklow S. C. Crystal Structure of a Full-Length Human Tetraspanin Reveals a Cholesterol-Binding Pocket. Cell 2016, 167, 1041–1051.e11. 10.1016/j.cell.2016.09.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.; Clarke O. B.; Kim J.; Stowe S.; Kim Y.-K.; Assur Z.; Cavalier M.; Godoy-Ruiz R.; von Alpen D. C.; Manzini C.; et al. Structure of the STRA6 Receptor for Retinol Uptake. Science 2016, 353, aad8266–aad8266. 10.1126/science.aad8266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan S. K.; Kitajima-Ihara T.; Fujii R.; Gotoh T.; Murakami M.; Ihara K.; Kouyama T. Crystal Structure of Cruxrhodopsin-3 from Haloarcula Vallismortis. PLoS One 2014, 9, e108362. 10.1371/journal.pone.0108362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran T.; Ozorowski G.; Gao Y.; Sineshchekov O. A.; Wang W.; Spudich J. L.; Luecke H. Cross-Protomer Interaction with the Photoactive Site in Oligomeric Proteorhodopsin Complexes. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2013, 69, 1965–1980. 10.1107/S0907444913017575. [DOI] [PubMed] [Google Scholar]
- Wada T.; Shimono K.; Kikukawa T.; Hato M.; Shinya N.; Kim S. Y.; Kimura-Someya T.; Shirouzu M.; Tamogami J.; Miyauchi S.; et al. Crystal Structure of the Eukaryotic Light-Driven Proton-Pumping Rhodopsin, Acetabularia Rhodopsin II, from Marine Alga. J. Mol. Biol. 2011, 411, 986–998. 10.1016/j.jmb.2011.06.028. [DOI] [PubMed] [Google Scholar]
- Luecke H.; Schobert B.; Stagno J.; Imasheva E. S.; Wang J. M.; Balashov S. P.; Lanyi J. K. Crystallographic Structure of Xanthorhodopsin, the Light-Driven Proton Pump with a Dual Chromophore. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 16561–16565. 10.1073/pnas.0807162105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouyama T.; Fujii R.; Kanada S.; Nakanishi T.; Chan S. K.; Murakami M. Structure of Archaerhodopsin-2 at 1.8 Å Resolution. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2014, 70, 2692–2701. 10.1107/S1399004714017313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gushchin I.; Reshetnyak A.; Borshchevskiy V.; Ishchenko A.; Round E.; Grudinin S.; Engelhard M.; Bldt G.; Gordeliy V. Active State of Sensory Rhodopsin II: Structural Determinants for Signal Transfer and Proton Pumping. J. Mol. Biol. 2011, 412, 591–600. 10.1016/j.jmb.2011.07.022. [DOI] [PubMed] [Google Scholar]
- Vogeley L. Anabaena Sensory Rhodopsin: A Photochromic Color Sensor at 2.0 Å. Science 2004, 306, 1390–1393. 10.1126/science.1103943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouyama T.; Kanada S.; Takeguchi Y.; Narusawa A.; Murakami M.; Ihara K. Crystal Structure of the Light-Driven Chloride Pump Halorhodopsin from Natronomonas Pharaonis. J. Mol. Biol. 2010, 396, 564–579. 10.1016/j.jmb.2009.11.061. [DOI] [PubMed] [Google Scholar]
- Nogly P.; Weinert T.; James D.; Carbajo S.; Ozerov D.; Furrer A.; Gashi D.; Borin V.; Skopintsev P.; Jaeger K.; et al. Retinal Isomerization in Bacteriorhodopsin Captured by a Femtosecond X-Ray Laser. Science 2018, eaat0094. 10.1126/science.aat0094. [DOI] [PubMed] [Google Scholar]
- Nango E.; Royant A.; Kubo M.; Nakane T.; Wickstrand C.; Kimura T.; Tanaka T.; Tono K.; Song C.; Tanaka R.; et al. A Three-Dimensional Movie of Structural Changes in Bacteriorhodopsin. Science 2016, 354, 1552–1557. 10.1126/science.aah3497. [DOI] [PubMed] [Google Scholar]
- Nakane T.; Hanashima S.; Suzuki M.; Saiki H.; Hayashi T.; Kakinouchi K.; Sugiyama S.; Kawatake S.; Matsuoka S.; Matsumori N.; et al. Membrane Protein Structure Determination by SAD, SIR, or SIRAS Phasing in Serial Femtosecond Crystallography Using an Iododetergent. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 13039–13044. 10.1073/pnas.1602531113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogly P.; James D.; Wang D.; White T. A.; Zatsepin N.; Shilova A.; Nelson G.; Liu H.; Johansson L.; Heymann M.; et al. Lipidic Cubic Phase Serial Millisecond Crystallography Using Synchrotron Radiation. IUCrJ 2015, 2, 168–176. 10.1107/S2052252514026487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T.; Sessions A. O.; Lunde C. S.; Rouhani S.; Glaeser R. M.; Duan Y.; Facciotti M. T. Deprotonation of D96 in Bacteriorhodopsin Opens the Proton Uptake Pathway. Structure 2013, 21, 290–297. 10.1016/j.str.2012.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spudich E. N.; Ozorowski G.; Schow E. V.; Tobias D. J.; Spudich J. L.; Luecke H. A Transporter Converted into a Sensor, a Phototaxis Signaling Mutant of Bacteriorhodopsin at 3.0 Å. J. Mol. Biol. 2012, 415, 455–463. 10.1016/j.jmb.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borshchevskiy V. I.; Round E. S.; Popov A. N.; Büldt G.; Gordeliy V. I. X-Ray-Radiation-Induced Changes in Bacteriorhodopsin Structure. J. Mol. Biol. 2011, 409, 813–825. 10.1016/j.jmb.2011.04.038. [DOI] [PubMed] [Google Scholar]
- Luecke H.; Schobert B.; Richter H.-T.; Cartailler J.-P.; Lanyi J. K. Structural Changes in Bacteriorhodopsin During Ion Transport at 2 Angstrom Resolution. Science 1999, 286, 255–260. 10.1126/science.286.5438.255. [DOI] [PubMed] [Google Scholar]
- Luecke H.; Schobert B.; Richter H.-T.; Cartailler J.-P.; Lanyi J. K. Structure of Bacteriorhodopsin at 1.55 Å Resolution. J. Mol. Biol. 1999, 291, 899–911. 10.1006/jmbi.1999.3027. [DOI] [PubMed] [Google Scholar]
- Belrhali H.; Nollert P.; Royant A.; Menzel C.; Rosenbusch J. P.; Landau E. M.; Pebay-Peyroula E. Protein, Lipid and Water Organization in Bacteriorhodopsin Crystals: A Molecular View of the Purple Membrane at 1.9 Å Resolution. Structure 1999, 7, 909–917. 10.1016/S0969-2126(99)80118-X. [DOI] [PubMed] [Google Scholar]
- Facciotti M. T.; Rouhani S.; Burkard F. T.; Betancourt F. M.; Downing K. H.; Rose R. B.; McDermott G.; Glaeser R. M. Structure of an Early Intermediate in the M-State Phase of the Bacteriorhodopsin Photocycle. Biophys. J. 2001, 81, 3442–3455. 10.1016/S0006-3495(01)75976-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schobert B.; Cupp-Vickery J.; Hornak V.; Smith S. O.; Lanyi J. K. Crystallographic Structure of the K Intermediate of Bacteriorhodopsin: Conservation of Free Energy after Photoisomerization of the Retinal. J. Mol. Biol. 2002, 321, 715–726. 10.1016/S0022-2836(02)00681-2. [DOI] [PubMed] [Google Scholar]
- Tanaka K.; Caaveiro J. M. M.; Morante K.; González-Mañas J. M.; Tsumoto K. Structural Basis for Self-Assembly of a Cytolytic Pore Lined by Protein and Lipid. Nat. Commun. 2015, 6, 6337. 10.1038/ncomms7337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary O. P.; Paz A.; Adelman J. L.; Colletier J.-P.; Abramson J.; Grabe M. Structure-Guided Simulations Illuminate the Mechanism of ATP Transport through VDAC1. Nat. Struct. Mol. Biol. 2014, 21, 626–632. 10.1038/nsmb.2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ujwal R.; Cascio D.; Colletier J.-P.; Faham S.; Zhang J.; Toro L.; Ping P.; Abramson J. The Crystal Structure of Mouse VDAC1 at 2.3 Å Resolution Reveals Mechanistic Insights into Metabolite Gating. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 17742–17747. 10.1073/pnas.0809634105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruss F.; Zähringer F.; Jakob R. P.; Burmann B. M.; Hiller S.; Maier T. The Structural Basis of Autotransporter Translocation by TamA. Nat. Struct. Mol. Biol. 2013, 20, 1318–1320. 10.1038/nsmb.2689. [DOI] [PubMed] [Google Scholar]
- Su C.-C.; Radhakrishnan A.; Kumar N.; Long F.; Bolla J. R.; Lei H.-T.; Delmar J. A.; Do S. V.; Chou T.-H.; Rajashankar K. R.; et al. Crystal Structure of the Campylobacter Jejuni CmeC Outer Membrane Channel: Crystal Structure of the C. Jejuni CmeC Outer Membrane Channel. Protein Sci. 2014, 23, 954–961. 10.1002/pro.2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eren E.; Vijayaraghavan J.; Liu J.; Cheneke B. R.; Touw D. S.; Lepore B. W.; Indic M.; Movileanu L.; van den Berg B. Substrate Specificity within a Family of Outer Membrane Carboxylate Channels. PLoS Biol. 2012, 10, e1001242. 10.1371/journal.pbio.1001242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monk B. C.; Tomasiak T. M.; Keniya M. V.; Huschmann F. U.; Tyndall J. D. A.; O’Connell J. D.; Cannon R. D.; McDonald J. G.; Rodriguez A.; Finer-Moore J. S.; et al. Architecture of a Single Membrane Spanning Cytochrome P450 Suggests Constraints That Orient the Catalytic Domain Relative to a Bilayer. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 3865–3870. 10.1073/pnas.1324245111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magotti P.; Bauer I.; Igarashi M.; Babagoli M.; Marotta R.; Piomelli D.; Garau G. Structure of Human N-Acylphosphatidylethanolamine-Hydrolyzing Phospholipase D: Regulation of Fatty Acid Ethanolamide Biosynthesis by Bile Acids. Structure 2015, 23, 598–604. 10.1016/j.str.2014.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray L. C.; Das D.; Entova S.; Lukose V.; Lynch A. J.; Imperiali B.; Allen K. N. Membrane Association of Monotopic Phosphoglycosyl Transferase Underpins Function. Nat. Chem. Biol. 2018, 14, 538–541. 10.1038/s41589-018-0054-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan J.; Xia S.; Liu X.; Lieberman J.; Wu H. Cryo-EM Structure of the Gasdermin A3Membrane Pore. Nature 2018, 557, 62–67. 10.1038/s41586-018-0058-6. [DOI] [PMC free article] [PubMed] [Google Scholar]