

Abstract
In biological water oxidation, a redox-active tyrosine residue (D1-Tyr161 or YZ) mediates electron transfer between the Mn4CaO5 cluster of the oxygen-evolving complex and the charge-separation site of photosystem II (PSII), driving the cluster through progressively higher oxidation states Si (i = 0–4). In contrast to lower S-states (S0, S1), in higher S-states (S2, S3) of the Mn4CaO5 cluster, YZ cannot be oxidized at cryogenic temperatures due to the accumulation of positive charge in the S1 → S2 transition. However, oxidation of YZ by illumination of S2 at 77–190 K followed by rapid freezing and charge recombination between YZ• and the plastoquinone radical QA•– allows trapping of an S2 variant, the so-called S2trapped state (S2t), that is capable of forming YZ• at cryogenic temperature. To identify the differences between the S2 and S2t states, we used the S2tYZ• intermediate as a probe for the S2t state and followed the S2tYZ•/QA•– recombination kinetics at 10 K using time-resolved electron paramagnetic resonance spectroscopy in H2O and D2O. The results show that while S2tYZ•/QA•– recombination can be described as pure electron transfer occurring in the Marcus inverted region, the S2t → S2 reversion depends on proton rearrangement and exhibits a strong kinetic isotope effect. This suggests that YZ oxidation in the S2t state is facilitated by favorable proton redistribution in the vicinity of YZ, most likely within the hydrogen-bonded YZ–His190–Asn298 triad. Computational models show that tautomerization of Asn298 to its imidic acid form enables proton translocation to an adjacent asparagine-rich cavity of water molecules that functions as a proton reservoir and can further participate in proton egress to the lumen.
1. Introduction
Photosystem II (PSII) catalyzes the light-driven oxidation of water that is coupled to plastoquinone reduction in plants, algae, and cyanobacteria. The site of water oxidation, called the oxygen-evolving complex (OEC), comprises an oxo-bridged cluster of four Mn and one Ca2+ ions. X-ray and more recently femtosecond X-ray free electron laser crystallography of PSII isolated from thermophilic cyanobacteria yielded structures of up to 1.9 Å resolution of the dark-adapted S1 state1−4 and more recently of higher states.5 A structure of spinach photosystem II at 3.2 Å resolution was also reported by cryoelectron microscopy.6 The catalytic cycle of the OEC involves four light-driven one-electron oxidation steps, S0 → S1, S1 → S2, S2 → S3, and S3 → (S4) → S0, accompanied by the progressive removal of four protons from two bound water molecules (Figure 1). Each step is initiated by light absorption that results in charge separation; a special chlorophyll dimer species called P680 is photo-oxidized to P680+, while the electron is transferred to a series of electron acceptors, pheo (pheophytin), QA, and QB (primary and secondary plastoquinone acceptors). QA is a single-electron acceptor, whereas QB is a two-electron/two-proton acceptor. P680+ in turn oxidizes YZ (Tyr161 of polypeptide D1) in a time scale of nanoseconds to microseconds. The resulting YZ• radical regains its electron by oxidizing the Mn4CaO5 cluster within tens of microseconds for the S0 → S1 transition to ca. 1 ms for the S3 → (S4) → S0 transition.7 Oxygen evolves from the transient S4 state during the latter, experimentally unresolved transition (see refs (8−19) and references therein).
Figure 1.

(a) Redox-active components and electron-transfer chain in PSII; (b) cycle of S-states; (c) OEC and endpoints of three channel systems; (d) schematic depiction of the inorganic cluster with labeling of the atoms and first-sphere ligands.
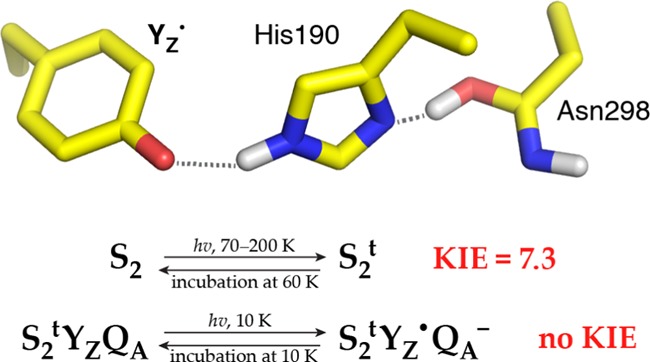
A critical aspect of water oxidation is the requirement for efficient removal of protons along with the four one-electron oxidation steps. The removal of protons and electrons is known to occur in a strictly alternate fashion at each S-state transition.12 However, identification of the proton removal pathways remains elusive. Several water channels have been identified in PSII20−26 forming a complex network24 that connects the OEC with the lumen. Apparent water channels that are associated with the Mn4CaO5 cluster and might be suitable for proton transfer include a channel with an endpoint close to the O4 bridge of the cluster that leads to the PsbU/PsbO protein interface (A in Figure 1c), a channel associated with the proximal chloride ion that leads to PsbO (B in Figure 1c), and a branched channel system with endpoints around the calcium ion and the YZ–His190–Asn298 triad that is suggested to connect the OEC with PsbV (C in Figure 1c).3 Different studies have proposed different functions for each channel and raised the possibility of a given channel serving a different role depending on the particular S-state transition; therefore, the precise role of each channel in proton egress remains inconclusive.17 The Asp61 residue, and hence channel B of Figure 1c, has often been implicated in proton removal,1,20,27−39 but channel A facing the O4 bridge has also been suggested to serve in proton egress at least in the S0 → S1 transition.40 More recent studies have additionally proposed a possible role for channel C,3,39,41−45 particularly for the S2 → S3 transition.
Significant insights into proton rearrangement and proton translocation can be gained by examining the different S-state transitions. For example, examination of the S2 → S3 and S3 → S0 transitions at ambient temperatures indicated that induction of YZ• resulted in proton expulsion from the Mn cluster or its immediate environment with a time constant τ of about 30 μs in the former39 and about 200 μs in the latter transition7 prior to the electron-transfer step from the cluster to the YZ• radical. Reaction rates, activation energies, and kinetic isotope effects (KIEs) have been obtained for the proton and electron-transfer steps of both transitions39,46 for the activation energy of S3 → S0 state transition.
Moreover, a crucial source of insight can be the properties of the observable SiYZ• metalloradical intermediates since they constitute true chemical intermediates of the Si → Si+1 transitions and report on the nature of the changes that occur in each transition. Various SiYZ• intermediates have been extensively studied by electron paramagnetic resonance (EPR) spectroscopy.47−49 At liquid helium temperatures, the common feature of the SiYZ• intermediates is the splitting of the EPR spectrum of YZ• owing to the magnetic interaction of the latter with the Mn4CaO5 cluster. As the temperature increases, the relaxation rate of Mn4CaO5 increases gradually, causing the weakening of the magnetic interaction between the Mn cluster and the YZ•. This effect results in the progressive narrowing of the metalloradical EPR signals,50 whereas at T > 100 K the unperturbed YZ• spectrum is revealed.50 Crucially, the temperature profile of SiYZ• formation gives information about barriers of the respective Si → Si+1 transition. Previous EPR studies showed that YZ is readily oxidized at liquid helium temperatures in the S0 and S1 states of active samples47−49 but not in the more electropositive S2 and S3 states. The S2YZ• intermediate can be created only at T > 77 K,51 whereas S3YZ• can be trapped at temperatures near and above the half inhibition temperature of the S3 → S0 transition (ca. 230 K).52 Elevated temperatures are presumably needed to overcome thermal barriers caused by local pKa changes during the S1 → S2 transition.51,53
The S2 → S3 transition is not only the most complex and most intensively studied among the observable transitions of the catalytic cycle but also the most contested one because of the multitude of intermediates and the still unresolved heterogeneous nature of the S3 state. Regarding the early stages of the transition, the S2YZ• intermediate is trapped by flash illumination in the temperature range 77–190 K, followed by rapid freezing at 10 K. This metalloradical signal decays by charge recombination with QA•– within minutes, but the resulting S2t state is able to form again the S2YZ• intermediate by visible light illumination directly at 10 K. This recombination gives rise to a phenolate structure of the YZ tyrosine. This S2YZ• signal (more appropriately termed S2tYZ• henceforth) can be reinduced several times after its decay at 10 K, implying that a light-adapted configuration of S2, termed S2t (S2trapped), is trapped by this method.51 The EPR signal of S2t shows no alteration compared to the normal multiline signal of S2; thus, the electronic configuration of the Mn4CaO5 cluster does not change upon formation of S2t.
More detailed EPR experiments show that the S2tYZ• signal consists of two components: a wide one with a splitting of ca. 170 G and a narrow one characterized by a splitting of ca. 120 G (Figure 2). Lower temperatures of illumination in the 77–190 K range favor the wide component, which at 10 K decays faster than the narrow one. Reillumination at 10 K after decay of the signal trapped at 77–190 K induces only the narrow component, which can be considered as the characteristic metalloradical signal of S2t. The wide S2tYZ• is assigned to an intermediate that lies between S2 and S2t. Slow and rapid scan EPR experiments in the temperature range 77–190 K revealed high-resolution spectra of the isolated YZ• radical and no evidence of alternative radicals. The two signals of the S2tYZ• intermediate were tentatively attributed to two sequential proton transfers during oxidation of YZ: a primary proton transfer from YZ to D1-His190 resulting in a transient electropositive configuration of S2tYZ• (wide signal) and a secondary one from D1-His190 to D1-Asn298 resulting in a relaxed configuration characterized by the narrow signal.54 The above interpretation of the EPR data implies a role for Asn298 in proton rearrangement and possibly involvement of channel C in proton egress.
Figure 2.
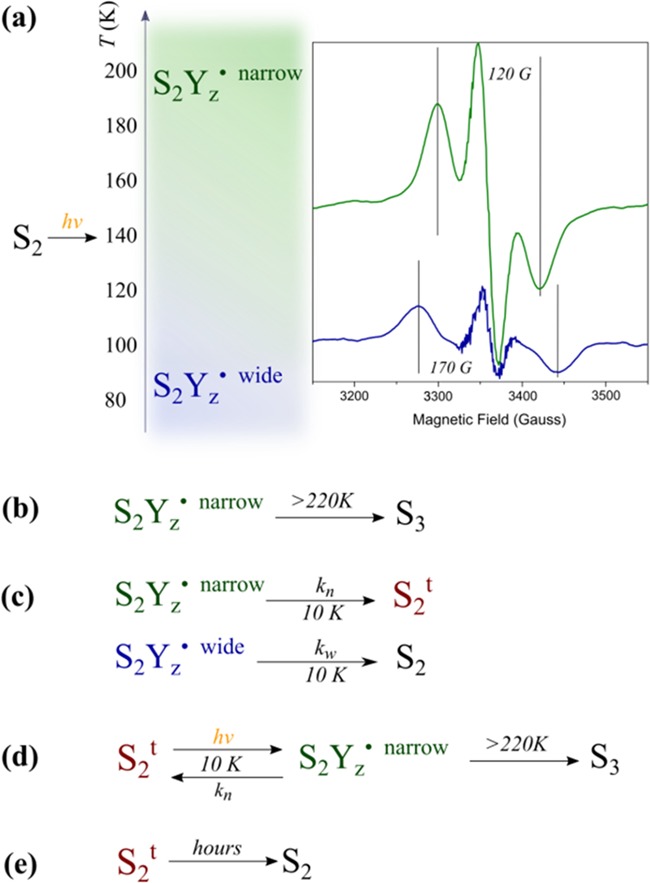
Summary of previous EPR observations related to the S2t intermediate.51,54 (a) At the S2 state, YZ cannot be oxidized at 10 K due to the positive charge in the vicinity of the OEC. However, YZ• is formed by illumination at T > 77 K and can be trapped by rapid freezing to 10 K. Two distinct EPR signals have been observed: the “narrow” in green and the “wide” in blue; the former is favored by higher temperatures of illumination and the latter by lower temperatures. (b) The narrow component of the YZ• proceeds to the S3 state upon annealing at T > 220 K, and thus it is an intermediate of the S-cycle. (c) At 10 K, both signals decay within minutes due to recombination with QA–. The wide signal decays to the normal S2 state or to a spectroscopically similar state. The narrow signal decays to the S2t state (S2trapped), which is distinguished by the normal S2 because it is capable of forming YZ• by illumination directly at 10 K. (d) The narrow signal can be formed several times at 10 K as soon as S2t is present. Interestingly, the S2tYZ• intermediate represented by the narrow signal that has been formed at 10 K can proceed to S3 at T > 220 K. (e) S2t decays slowly to S2.
In this work, we aim to characterize better the features of the S2t state by identifying the chemical nature of its differences with the relaxed S2 state and to examine possible molecular mechanisms for involvement of Asn298 in proton rearrangement. Assuming a two-state model (S2 ↔ S2t) according to which only the S2t, but not the normally prepared S2 state, can give rise to YZ• by illumination at 10 K, the equilibrium would be expected to shift toward S2t upon increasing induction temperature. Furthermore, one would expect the recombination reaction rate of S2tYZ• with QA•– to remain indifferent to changes in the temperature of S2t induction. To investigate these points, we followed the recombination kinetics of S2tYZ• with QA•– by time-resolved EPR spectroscopy at 10 K for S2t induced by illumination in the 77–212 K range upon H2O/D2O exchange. Spectroscopically, the same narrow S2tYZ• signal is obtained at 10 K, irrespective of the S2t induction temperature. However, the recombination reaction rates decrease with increasing induction temperature, whereas the driving force of the reaction increases. The results show that the recombination reaction between S2tYZ• and QA•– involves only electron transfer and shows no dependence on H2O/D2O exchange, whereas the S2t to S2 state reversion depends on proton rearrangement and exhibits a strong primary kinetic isotope effect. This establishes that the S2t is in a constrained configuration that can be gradually relieved by raising the temperature and is formed after proton movement in the YZ environment without proton egress to the bulk. Using computational models, it was found that proton transfer could occur from His190 via Asn298 to a vicinal asparagine-rich water cavity, which acts as a proton reservoir. This proton movement is enabled by tautomerization of Asn298 from the amide to the imidic acid form and can be an intermediate step in further translocation and release of the proton to the bulk.
2. Methods
2.1. Isolation of PSII Samples
PSII-enriched thylakoid membranes were isolated from spinach.55,56 Samples for EPR measurements were suspended in 0.4 M sucrose, 15 mM NaCl, 40 mM 2-N-morpholineethanesulfonic acid (MES), pH 6.5, at about 6–8 mg Chl mL–1 (Chl: chlorophyll) and stored in liquid nitrogen. For experiments performed in D2O, the above buffer was prepared in D2O (99.9% purity, obtained from Deutero GmbH) and the pD was adjusted to 6.5 (pH meter showed 6.1). The samples were subsequently supplemented with 1 mM 2,6-dichloro-p-benzoquinone (DCBQ), dissolved in dimethyl sulfoxide, as an exogenous electron acceptor.
2.2. Illumination Conditions
For illumination purposes, two studio photographic power supplies were used: a 600 W device with pulse duration of 2.1 ms, at temperatures below ca. 220 K, and a 200 W device with pulse duration of 1.2 ms, at temperatures above 220 K. The latter produced single turnovers (i.e., no double hits) at −5 °C. The S2 state was produced typically by single-flash illumination of the S1 state at −5 °C and kept at the same temperature for 30 s to allow electron transfer from QA– to DCBQ. This single flash also resulted in advancement to the S1 state of any small amounts of the S0 state invariably present along with the S1 state in freshly prepared PSII samples. Furthermore, no S3 state could be detected (from double hits on the S1 state). The S2 state was maximized by applying the following illumination cycle three times: one strong flash at 190 K followed by incubation for 1 min at the same temperature and 30 s at −10 °C. This flash caused the advancement of the remaining S1 population to the S2 state. Spectroscopically, the population of the S2 state achieved is very close to 100% since no S3 state is observed. For the induction of the S2t state, which can give rise to the S2tYZ• intermediate by reillumination at 10 K, samples were flash-excited at the appropriate temperatures and, within 0.5–1 s, transferred to liquid nitrogen and from there into the EPR cryostat at 10 K. Following incubation of about 15–20 min at 10 K, to allow for the decay of the trapped radicals,54 the intermediates were reinduced at 10 K by excitation with 3 flashes spaced by 2 s.
2.3. EPR Measurements
EPR measurements were obtained with an extensively upgraded former Bruker ER-200D spectrometer interfaced to a personal computer and equipped with an Oxford ESR 900 cryostat, an Anritsu MF76A frequency counter, a Bruker 035M NMR gaussmeter, and an SR830 digital lock-in amplifier by Stanford Research. The perpendicular 4102ST cavity was used, and the microwave frequency was 9.41 GHz. In the rapid scan experiments, the data were recorded by a multifunction NI 6251 pci card by National Instruments (16 bit/1.25 MS/s) mounted on a personal computer running appropriate software in the LabView programming environment. Synchronization of the data acquisition with the magnetic field ramp produced by the time base unit was achieved by triggering the analog-to-digital converter with a transistor–transistor logic pulse produced by the time base unit at the beginning of each scan. The minimum duration possible for each scan was 20 ms with a delay of 20 ms between the scans.
2.4. Density Functional Theory (DFT) Calculations
Geometry optimizations were carried out with the ORCA program57 using the BP86 density functional.58,59 Scalar relativistic effects were included with the zeroth-order regular approximation (ZORA).60,61 The ZORA-recontracted62 TZVP basis sets63 were used for all atoms, except for C and H, in which the ZORA-SVP basis sets were applied. The resolution of identity approximation was used for the Coulomb integrals with fully decontracted auxiliary def2/J basis sets.64 Atom-pairwise dispersion corrections according to Grimme65 were applied, and implicit solvation was considered with CPCM,66 with a dielectric constant of ε = 4.0. Increased integration grids (Grid4) and tight SCF convergence criteria were used.
The initial geometry was based on the XFEL coordinates of Suga et al.4 (PDB 4UB6), considering the residues bonded to the OEC (Asp170, Glu189, His332, Glu333, Asp342, Ala344, and CP43-Glu354), second sphere ligands (Asp61, Tyr161, His190, His337, CP43-Arg357), and the backbones of Ser169, Asn191, Asn298, Gly299, Phe300, Asn301, Leu343, Val407, Gly408, Gly409, Val410, Ala411, and Thr412, as well as 17 water molecules. Of these, four are ligands to the OEC (one of them deprotonated as the W2 = OH ligand to Mn4), whereas the others are involved in H-bond networks around the OEC, the Tyr161, and the asparagine-rich cavity that lies next to Asn298.3 The model contains a total of 343 atoms. The positions of α-carbon atoms were kept fixed, as in previous studies.35,67−69 Additional constraints on backbone atoms defining the Asn-rich cavity were applied during exploration of proton storage states in the water pocket adjacent to Asn298 as described in the text.
3. Results and Discussion
3.1. Recombination Kinetics of Photoinduced S2tYZ• with QA•–
Given that between the S2 and S2t configurations, only the latter is able to form the S2tYZ• intermediate by visible illumination at 10 K, the S2t state was first induced by flash illumination on the S2 state at 148 K. Subsequently, the S2tYZ• was formed by flash illumination at 10 K, whereas its detailed study of the decay kinetics was followed at the same temperature by rapid scanning EPR. In Figure 3, we present the experimental data, along with the fitting curve. In the inset of Figure 3, the EPR spectrum of the S2tYZ• is shown; the two arrows indicate the magnetic field positions at which their intensity difference is plotted against time. A single exponential decay curve with k = 5.31 × 10–3 s–1 was fitted to the data. This means that the recombination of the S2tYZ• with QA•– can be adequately described by a single kinetic event.
Figure 3.

Decay kinetics of the S2tYZ• EPR signal followed at 10 K. The S2t state was flash-induced at 148 K. Data points represent the intensity difference at 3285 G minus 3410 G. A single exponential curve (solid line) is fitted to the data with a reaction rate constant k = 0.00531 s–1. Inset: transient S2tYZ• EPR spectrum recorded at 10 K; it represents the average of the first 10 spectra recorded after the flash minus the average of 10 spectra recorded at time = 700 s after the flash. Arrows indicate the positions on the spectrum used for the decay kinetics recording. Rapid-mode EPR conditions: modulation amplitude, 25 Gpp; microwave power, 31.7 mW; microwave frequency, 9.41 GHz; modulation frequency, 100 kHz; sweep field width, 200 G; sweep time, 2 s; time constant, 10 ms.
The charge recombination kinetics of the S2tYZ• with QA•– were followed at 10 K for the S2t state induced by illumination in the temperature range 77–212 K. The temperature dependence of the reaction rate constants of recombination thus obtained is shown in Figure 4. If one assumed a two-state model for the S2 state, that is, S2 ↔ S2t, according to which the S2 state, as normally prepared, could not give rise to YZ• by illumination at 10 K, but the S2t state could, then one would expect the equilibrium between the two to shift toward the S2t state as the induction temperature increased. Our results show that the latter holds true. As mentioned in the Introduction, the S2tYZ• radical obtained after illumination of the S2t state at 10 K represents the narrow signal (∼120 Gpp) and is spectroscopically homogeneous. Hence, at first, one would anticipate that by increasing the temperature of S2t induction, its population should increase; this is indeed observed experimentally (not shown), and a maximum is reached at roughly 150 K. Above this temperature, the rate of deactivation of the S2t state presumably becomes higher than the rate of trapping, and its population diminishes.
Figure 4.
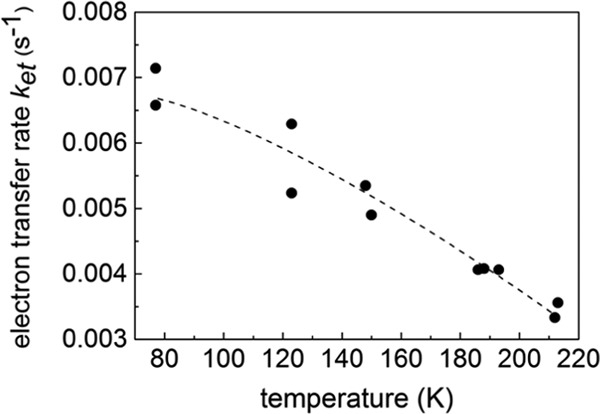
Variation of the recombination reaction rate of the S2tYZ•/QA•– pair with temperature of induction of the S2t state. The recombination reaction kinetics were followed at 10 K. The dashed line is drawn only as an aid to the eye.
It is also expected that the rate of recombination of the reinduced S2tYZ• with QA•– at 10 K should have been constant and independent of the temperature of S2t induction. However, as shown in Figure 4, the reaction rate constant decreases with increasing temperature of S2t induction. The recombination reaction rate constants vary from 7.14 × 10–3 s–1 (at 77 K) to 3.33 × 10–3 s–1 (at 212 K) (see Table S1 in the Supporting Information). Nonetheless, after S2t induction at a certain temperature, each rate constant of S2tYZ•/QA•– recombination was found to be temperature independent from 4.2 K to about 40 K. For example, for S2t that was induced at 145 K, a mean recombination k = 5 × 10–3 s–1 was measured in the range 4.2–35 K. In a separate experiment, the S0YZ•/QA•– rate constant was calculated at 5.26 × 10–3 s–1, the same order as with the S2 state. Also, from our previous study,70 we can observe that the rate constant obtained at 70 K (slow phase accounted for 82% of centers) was 9.72 × 10–3 s–1. It appears that the rate constants do not vary significantly with temperature from 10 K to about 70 K. Also, these rate constants are similar to those previously reported for the recombination of S0YZ•, S1YZ•, and S2tYZ• (S2t was induced at 200 K) with QA•– in the study of PSII from Thermosynechococcus elongatus.71 The observation that S2tYZ•/QA•– charge recombination kinetics (of S2t induced at a certain temperature) appear virtually temperature independent (in agreement with ref (71)) indicates that the reaction can be described as electron tunneling between YZ• and QA•– (at a distance of ∼32 Å).
In a previous investigation,71 it was observed that the reaction rate decreased upon increasing of the driving force of the reaction. The investigators used the Hopfield equation72 (a semiclassical version of the Marcus equation73,74) for electron transfer
where HAB is the electronic coupling between the electron donor and acceptor, ΔG0 is the standard free reaction energy (i.e., −ΔG0 is the driving force), λ is the reorganization energy, T is the temperature, kB is the Boltzmann constant, and ω is a single high-frequency vibrational mode, which is coupled to the electron-transfer event to fit their data. By assuming that ℏω = 70 meV (a value taken from the P+/QA•– charge recombination in bacterial reaction centers),75 they deduced values of 547 meV for λ and 7.6 × 10–6 meV for HAB. Allowing for variation in driving force and ℏω, it was concluded that λ remained small and did not change much.
We used the same expression for electron transfer and the values ℏω = 70 meV, HAB = 7.6 × 10–6 meV, and λ = 550 meV as in ref (71) and calculated the driving force −ΔG0 for all our measured respective reaction rates; the dependence of the thus-calculated driving force (see Table S1 in the Supporting Information) on the rate constants is depicted in Figure 5.
Figure 5.

Driving force dependence of reaction rate constants. The continuous line was drawn using the Hopfield expression with ℏω = 70 meV, HAB = 7.6 × 10–6 meV, λ = 550 meV, and T = 10 K.
Our experimentally determined reaction rate constants, k, decrease with increasing driving force, −ΔG0, and at the same time the latter is noticeably greater than λ (−ΔG0 ≈ 2λ); hence, the reaction takes place in the inverted region of the Marcus curve. The effect of having a large −ΔG0 into the inverted region is to effectively cause the retardation of the QA•– to YZ• back-reaction, ensuring efficient forward electron transfer from the Mn cluster to YZ•.
3.2. Kinetic Isotope Effect of the S2t to S2 Reversion
To probe whether there is an effect of possible proton rearrangement or even removal that occurred upon S2t induction, we performed the recombination reactions in buffer solutions prepared in D2O. Following induction of the S2t state at various temperatures, examination of the reaction rate constants of the S2tYZ•/QA•– charge recombination showed that they were indifferent to H/D exchange, in accord with Boussac et al.71 This result shows that in the rate-determining step of the recombination reaction, only electron transfer is involved and there is no proton transfer. However, a large kinetic isotope effect (KIE) was found in the reversion of the S2t state back to the normal S2 state. This reversion was prompted by incubating the S2t state at higher temperatures for various time intervals. After the incubation period, the sample was cooled down to 10 K and illuminated to form the S2tYZ• state. Its EPR signal amplitude was compared to the maximal one obtained prior to incubation. In each experiment performed, the maximal signal was obtained by induction of the S2t state at 150 K. It was found that the S2t in H2O remained stable for at least an hour at around 40 K, whereas in D2O it was stable for an hour at around 50 K. Figure 6 depicts the time course of the S2t to S2 reversion by incubation at 62 K. The rate constants deduced by fitting a single exponential function to the data were 0.415 min–1 in H2O and 0.057 min–1 in D2O. In the first case, the reversion reaction was completed to about 80%, whereas in the latter case, the reaction was completed to about 60%. The reversion reactions do not appear to reach completion. It is tentatively assumed that the S2t state as prepared comprises more than one conformers, in which the proton is delocalized on different locations, and these conformers need to overcome somewhat different thermal barriers. The temperature dependence of the S2t to S2 reversion is currently under investigation. The sizeable primary KIE of 7.3 indicates that proton translocation(s) are involved in the rate-determining step(s) of the S2t to S2 reversion.
Figure 6.
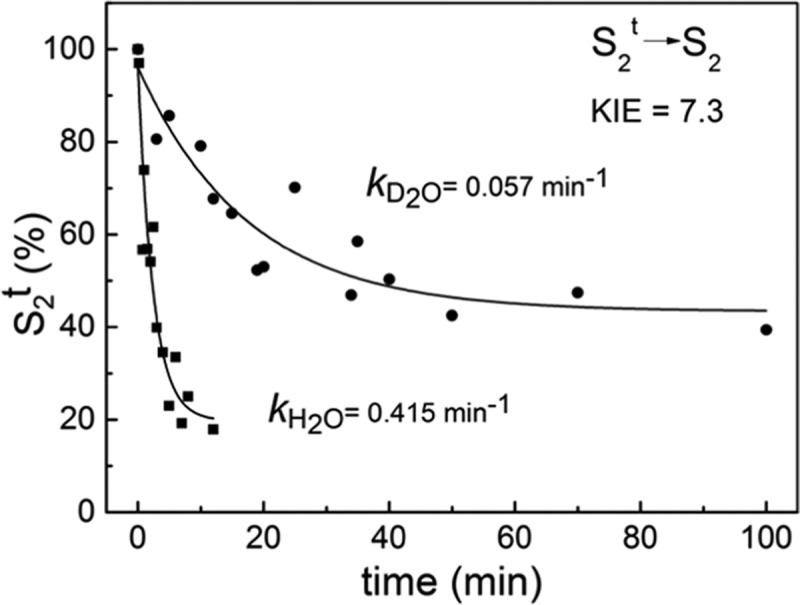
Time-dependent reversion of the S2t to the S2 state upon incubation at 62 K in the presence of H2O (rectangles) or D2O (circles). The maximal S2t occupancy was obtained by illumination of the S2 state at 150 K. Single exponential functions plus offsets were fitted to the experimental points with rate constants of 0.415 min–1 in H2O and 0.057 min–1 in D2O.
A similarly strong KIE of 5.6 was found in the “S2+” to “S2n” reaction, which represents the first elementary step in the S2 → S3 state transition, where S2+ represents the normal S2 state (the extra charge denotes that there was no proton release in the previous S1 → S2 state transition)76−79 and S2n represents the ensuing neutral S2 state. This elementary step was attributed to deprotonation of the water environment of the Mn cluster, triggered by the oxidation of YZ by P680+.39 The deprotonation event was monitored by time-resolved photothermal beam deflection experiments and was characterized by high activation energy (Ea = 0.46 eV) and fast kinetics (τ = 30 ms) at 20 oC. It appears that the S2t state is similar to the S2n state39 since both represent the S2 state, which has lost a proton, prior to electron transfer from the Mn4CaO5 cluster to YZ•. However, the fact that the S2t state is able to revert back to the normal S2 state simply by incubation at temperatures above ca. 60 K indicates that it does not represent a deprotonated S2 state. Instead, the positive charge is rearranged but maintained in close proximity. This is in accord with our previous study70 where we examined the spectral characteristics of YZ• at temperatures around 200 K, where the radical is magnetically uncoupled from the Mn4CaO5 cluster. In that study, we concluded that the YZ• radical was involved in a strong H bond and/or it was found in an electropositive environment because its EPR spectrum was characterized by a small gx (estimated to 2.00689) and a considerably high value for ρc1 (∼0.41) (where ρc1 is the spin density of carbon 1 of the YZ ring). Hence, we suggest that the S2t state lies temporally prior to S2n, in which proton translocation to the bulk has presumably occurred.
According to the EPR results, the proton movement does not appear to originate from the Mn4CaO5 cluster itself since we could not detect any changes in the Mn multiline EPR signal in the S2t configuration, that is, in the state that is capable of producing the narrow signal at 10 K. That is to say, the Mn multiline signal is the same in the S2 and S2t states. However, changes are detected on the multiline signal only in the presence of the S2t YZ• signal; they manifest as spectral line shifts and are attributed to the magnetic interaction of the manganese cluster with the tyrosine radical.51 Since the origin of the spectral shifts is not found on the Mn4CaO5 cluster, it follows that they most likely originate from changes in the environment of YZ. We note that an alternative hypothesis has postulated the involvement of a radical centered on His190 as the origin of one of the different signals.80 However, this can be excluded since the hyperfine structure of both EPR signals at elevated temperatures, where the magnetic interaction with the Mn cluster is relieved, clearly identifies them with the tyrosyl radical.54
It is evident that by changing the temperature of S2t induction, that is, flash illumination of the S2 state in the range of 77–212 K, we managed to trap different conformations of S2t. These, upon illumination at 10 K, give rise to S2tYZ• intermediates that are indistinguishable spectroscopically yet exhibit varied recombination kinetics. To explain the experimental finding that the S2tYZ• intermediate comprised two EPR signals, a wide one with width 170 Gpp and a narrow one with width ∼120 Gpp, we had previously proposed a model involving proton movement from YZ• to His190 to form His190+ (attributed to the wide signal) and from His190+ to Asn298 to form Asn298+ (attributed to the narrow signal).54 Upon illumination of the S2t state at 10 K, only the narrow signal is observed. Hence, according to the above hypothesis, at each temperature of S2t induction, although proton translocation to Asn298 has been completed, the YZ•–His198–Asn298+ state may have not reached a relaxed configuration. These variably strained states may account for the temperature-dependent recombination reaction rate of the S2tYZ•/QA•– pair, being faster and in a more strained conformation at lower temperatures, whereas at higher temperatures, the strain is relieved and the recombination rate is decreased (vide infra). A similarly strained tyrosine radical–histidine pair (YD•–D2-His189) was described previously, where the strain was relieved by raising the temperature to 77 K.81 In the following, the possible involvement of Asn298 in accepting and transferring a proton from His190+ will be investigated on the basis of structural analysis and quantum chemical calculations.
3.3. Asn298 and Its Adjacent Asn-Rich Water Cavity
Asn298 is a conserved residue in cyanobacteria and plants, and mutations at this site have been shown to significantly impair oxygen-evolving activity.82 If Asn298 plays a direct role in proton transfer, it is unclear how this can be achieved at the atomic level. Whether our previous suggestion holds true or not, namely, that Asn298 may be able to accept a proton from His190+, any further proton movement requires a molecular mechanism for bond rearrangement and for proton stabilization in a position that lies beyond the YZ–His190–Asn298 triad. With respect to the latter requirement, water clusters can serve the purpose of stabilizing protons as hydrogen-bonded hydronium ions; therefore, we investigated the region around Asn298 for extensively hydrogen-bonded water clusters. The most likely candidate is a pocket of water molecules within a cavity directly proximal to Asn298 (Figure 7). This water cluster has been already suggested as a possible proton reservoir.80 A highly unusual feature of this cavity is that it is lined almost exclusively by asparagine residues: in addition to Asn298, the cavity is defined by the side chains of Asn191, Asn301, Asn303, Asn322, Asn325, and Gln187. Following the nomenclature of the 4UB6 structure, the main water molecules in this pocket are W631, W558, W604, and W628; additional hydrogen-bonded water molecules include W563, W592, and W632.
Figure 7.

Asn298 water pocket that can serve as a proton reservoir (W631, W558, W604, and W628) and selected residues and water molecules forming a possible hydrogen-bond network that extends beyond the cavity toward the lumen. The labeling of water molecules follows the 4UB6 model.
With respect to the first requirement stated above, that is, a molecular mechanism of bond rearrangement to enable proton translocation, it is suggested that this can occur via tautomerization of Asn298 from the amide to the imidic acid form. This type of tautomerization has already been shown to mediate proton transfer through a H-bond network in cellulase PcCel45A from the basidiomycete Phanerochaete chrysosporium. Neutron and X-ray diffraction studies confirmed that an asparagine residue (Asn92), via tautomerization to the imidic acid form, accepts a proton from a water molecule during hydrolysis of cellulose, while at the same time an aspartate (Asp114) on the other side of the reaction center loses a proton; the two residues are recovered by proton hopping through an H-bond network.83 The definition of the water cavity and the hypothesis of Asn tautomerization provide the basis for examining the feasibility of an analogous tautomerization-assisted proton transfer mechanism in the case of Asn298 of PSII.
3.4. Tautomerization-Assisted Proton Translocation
Large quantum mechanics (QM) models were constructed, explicitly including the OEC together with the complete YZ–His190–Asn298 triad and the adjacent cavity. The structures of the Mn cluster and the Mn oxidation states were assigned according to the “open cubane” structure of the S2 state that is associated with the multiline g = 2 EPR signal of spin S = 1/2. Upon oxidation of this model, a YZ• tyrosyl radical is formed, and the proton is transferred to the Nε atom of His190, resulting in a positively charged imidazolium. This electronic structure is the starting point for all models discussed below. First of all, the possibility of proton transfer from the Nδ of His190+ to the CO group of Asn298 was examined with DFT calculations. Assuming that no major backbone rearrangements occur, our efforts to locate a minimum with a neutral His190 and a positively charged Asn298 reverted to a doubly protonated His190. This suggests that proton movement toward Asn298 most likely cannot lead to a stable minimum without additional structural modifications, which are probably not captured automatically by our models. On the other hand, if Asn298 does accept a proton from His190+, it can be stabilized by tautomerization into its imidic acid form, donating a proton from its NH2 group further to a water acceptor in the vicinal Asn-rich cavity. In this way, Asn298 would avoid the unfavorable buildup of positive charge, and the proton would be stabilized within the highly connected H-bonding network of the water cavity. DFT calculations that incorporate the imidic acid form of Asn298 confirm that this is indeed the case (Scheme S1 in the Supporting Information shows a possible pathway for the above chemical events).
Figure 8 depicts models in which the proton is stabilized at different positions within the cavity (see the Supporting Information for complete atomic coordinates). Maintaining the crystallographic assignment for the orientation of all Asn side chains, an optimized structure was obtained with a hydronium ion stabilized at the W558 position, with hydrogen bonds to W628, W604, and the backbone carbonyl of Gly299 (Figure 8a). Rotations of the side chains of the asparagine residues that line the cavity play a direct role in stabilizing the cation at different positions. Upon rotation of Asn191, the hydronium ion is stabilized at W628, with hydrogen bonds to Asn191, Asn301, W558, and the backbone carbonyl of Phe300 (Figure 8b). If the side chain of Asn301 is rotated, then the hydronium ion can be transiently stabilized at W604, and subsequently a more stable minimum is reached at which the proton is mostly shifted toward the amide carbonyl of Asn301 (Figure 8c). Thus, with the exception of W631, all other waters in the cavity can accommodate the proton.
Figure 8.

Computational models with tautomerized Asn298 where the proton is translocated in different positions within the vicinal water cavity ((a) W558; (b) W628; (c) W604/Asn301). Red numbers indicate labeling of water molecules according to 4UB6. For clarity, only a small part of the models is depicted; full structures are provided in the Supporting Information.
The above results demonstrate that the water cavity next to Asn298 can function as a proton reservoir, and they support the fact that proton translocation from Tyr161 to the water cluster is enabled by Asn298 tautomerization. It is likely that multiple water clusters around the OEC could have different functions at different steps of the S-cycle. From this perspective, the present models rationalize the unusual high concentration of asparagines in the cavity: simple rotations of the amide side chains can create multiple hydrogen-bonding arrangements and thus assist in “delocalizing” the positive charge. It is noted that our models are not large or flexible enough to deliver reliable energetics for the possible proton transfer steps and cannot be considered to have sampled the conformational space sufficiently—both tasks would be best tackled with large-scale ab initio molecular dynamics. For reference, the relative energies of models (a)–(c) shown in Figure 8 are 0.0, 8.0, and 3.7 kcal mol–1, respectively, suggesting that charge shift within and across the cavity is facile. Despite intrinsic uncertainties in energetics, the models do provide clear suggestions toward interpreting the experimental observations discussed above: the multiple, temperature-dependent, and variously strained configurations attributed to the S2t state may result from the various proton transfer steps involved in Asn298 tautomerism and translocation to the water cavity.
A more precise role for the multiple asparagine residues might be to direct the transfer of proton toward specific pathways. The fact that in one of the models the Asn301 easily accepts a proton from a hydronium at W604 implies that this can be another candidate for tautomerization; hence, by rotation of the side chain or by donation of its NH2 proton to another proton acceptor, it could transfer the proton outside the cavity or push it further along the hydrogen-bond network. Alternatively, a proton accommodated at W628 could be transferred to one of W592 or W632 (see Figure 7). Although our QM models are already too large to extend beyond the initial section of this cavity, it is possible to identify possible pathways for proton release to the lumen by correlating these models with crystallographic data. Asn322 was suggested by Umena et al. to be part of a hydrogen-bond network that extends from the YZ–His190–Asn298 site via His304, Asp319, Asn322, and Arg323 to the PsbV residues Tyr137, Asp128, and Lys129, terminating at the lumen-exposed Arg55. The proton transfer possibilities examined here converge to the above suggestion at Asn322 but explicitly involve tautomerization of Asn298 and proton hopping through W631, W558, W604, and W628. This can be followed by involvement of other water molecules or by another tautomerization or amide rotation and proton shifting through the tightly bound Asn301–Asn303–Asn322 triad. Closer inspection of the crystal structure around the Asn322 residue suggests an additional pathway alternative to that proposed by Umena et al. If Asn322 is assumed to gate proton transfer to W614, then there is only a very short distance with two intervening water molecules (W613 and (D2)W628) until the hydrogen-bonded pair of D1-Asn315 and D2-Gln322 is reached (see Figure 7), which appears to be already in contact with the lumen. Proton release in that case would be at the interface of PsbA (D1) and PsbD (D2), which is significant because it does not implicate an extrinsic protein (PsbV) that is specific to cyanobacteria. Regardless of the exit points, the Asn residues may be functioning as gates: continuity in the hydrogen-bonding network that would facilitate proton transfer can be enabled either by rotation of the amide side chain or by tautomerization from the amide to the imidic acid form.
In addition to the structure-based suggestion of Umena et al. regarding a possible role of the Asn298-related channel C in proton translocation, Dau and co-workers based on PBD experiments39 in combination with mutations of the Asp61 residue33,84 suggested that the elementary proton transfer step during S2 → S3, that is, formation of S2n from S2+, is realized via the water circuit between the Mn cluster and YZ toward the lumen, that is, via the Asn298 proton pathway. Eventually, electron transfer to YZ• coupled with proton transfer to the deprotonated water close to YZ• takes place. Interestingly, the authors suggested that this pathway serves in proton removal during the S2 → S3 transition, whereas in S3 → S0 the Asp61 pathway plays this role.39 This suggestion is in agreement with our observations for the S2YZ• and S3YZ• intermediates: gradual increase of S2YZ• signal intensity upon increasing induction temperature and the phenomenology described previously is attributed to proton movement from YZ• toward Asn298 and the adjacent water cavity, which is favored by increasing temperature. By contrast, no such effect has been observed during the S3 → S0 transition; S3YZ• can be trapped only at ca. 230 K,52 which implies that the first deprotonation step follows a different mechanism in the two transitions.
The Noguchi group based on the Fourier-transform infrared (FTIR) observation of a polarizable proton between YZ• and His190+ in Mn-depleted samples, together with QM/molecular mechanics simulations, proposed that during the S2 → S3 transition the proton trapped between YZ• and His190 can hop to a water molecule that is hydrogen-bonded to the calcium-bound water W4.41 This water molecule that acts as an immediate proton acceptor is connected to the channel near Asn298 that leads to the lumen, but is outside the Asn-rich cavity described in the present work. Time-resolved FTIR experiments in the S2 → S3 transition of active PSII were also consistent with proton removal from this channel coupled with YZ reduction by the Mn cluster.43 FTIR studies of the Asn298Ala PSII mutant suggested that this proton abstraction pathway acts during S2 → S3 and also the first deprotonation step in the S3 → S0 transition.42 In these studies, Asn298 itself is considered part of the hydrogen-bond network but has no direct involvement in proton removal. The present work therefore is consistent with the above suggestions regarding proton translocation originating from the YZ site but supports that Asn298 itself plays a direct gating role through its tautomerization, which enables proton translocation from His190 to the vicinal water cluster. Molecular dynamics simulations of PSII by Ishikita and co-workers independently suggested that the water molecules assigned here to the asparagine-rich crypt are not easily exchangeable with bulk water, consistent with a role in proton transfer.44
An important consequence of the idea that Asn298 and the associated H-bond pathway may serve in proton transfer is that YZ• may play a dual role in both electron and proton abstraction from the Mn cluster, at least for specific steps of the cycle. This would be consistent with the original (now largely abandoned) concept of hydrogen-atom abstraction by YZ• proposed by Babcock85 and with ideas explored in more recent works on the S2 → S3 transition.39,41 The likelihood of different proton storage sites and translocation pathways in the OEC, and their mechanistic implications should be further investigated in the light of the new findings.
4. Conclusions
In the present paper, we investigate the differences between the S2 and S2t configurations and the role of the Asn298 in the proton transfer process during the S2 to S2t step of the OEC. The metalloradical EPR signal of S2tYZ• was used as a probe of the S2t state since between the S2 and S2t configurations only the latter is able to form the S2tYZ• that is proportional to the S2t population. By using time-resolved EPR spectroscopy, we have undertaken a detailed kinetic study of both S2tYZ•/QA•– → S2tYZ/QA and S2t → S2 upon H2O/D2O exchange. The results show that the recombination reaction S2tYZ•/QA•– → S2tYZ/QA presents no dependence on the H2O/D2O exchange and can be described as pure electron transfer occurring in the Marcus inverted region. By contrast, the S2t to S2 state reversion depends on the proton rearrangement and exhibits a strong kinetic isotope effect. This establishes that the S2t is in a constrained configuration that can be gradually relieved to the S2 state by raising the temperature. Our observations strongly indicate that the modification of the proton arrangement of the Mn4O5Ca during the S2 to S2t step results in proton redistribution in the vicinity of YZ, presumably along the YZ–His190–Asn298 pathway. This effect facilitates the oxidation of YZ in the S2t constrained configuration, reflecting the difference between the latter configuration and the S2. To examine the possibility of proton transfer with the involvement of Asn298 upon oxidation of YZ, we have performed DFT calculations, which suggest that proton transfer can occur from His190 via Asn298 to a vicinal asparagine-rich water cavity, which acts as a proton reservoir. This proton movement is enabled by tautomerization of Asn298 from the amide to the imidic acid form and can be an intermediate step in further translocation and release of the proton to the bulk.
Acknowledgments
We thank Dr Y. Sanakis for helpful discussions and critical reading of the manuscript and M. Tzifas for technical assistance. We also acknowledge support of this work by the project MIS 5002567, implemented under the “Action for the Strategic Development on the Research and Technological Sector”, funded by the Operational Programme “Competitiveness, Entrepreneurship and Innovation” (NSRF 2014-2020) and cofinanced by Greece and the European Union (European Regional Development Fund). MC, JCdMS, and DAP gratefully acknowledge financial support by the Max Planck Society and by the project MANGAN (03EK3545) funded by the Bundesministeriums für Bildung und Forschung (BMBF).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jpcb.9b02317.
Charge recombination kinetics of the S2YZ• with QA•− at 10 K, schematic illustration of the suggested deprotonation events that lead to the proton’s exit, figure depicting the full size of the 343-atom models, Cartesian coordinates of all structures (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Ferreira K. N.; Iverson T. M.; Maghlaoui K.; Barber J.; Iwata S. Architecture of the Photosynthetic Oxygen-Evolving Center. Science 2004, 303, 1831–1838. 10.1126/science.1093087. [DOI] [PubMed] [Google Scholar]
- Guskov A.; Kern J.; Gabdulkhakov A.; Broser M.; Zouni A.; Saenger W. Cyanobacterial Photosystem II at 2.9 Å Resolution and the Role of Quinones, Lipids, Channels and Chloride. Nat. Struct. Mol. Biol. 2009, 16, 334–342. 10.1038/nsmb.1559. [DOI] [PubMed] [Google Scholar]
- Umena Y.; Kawakami K.; Shen J.-R.; Kamiya N. Crystal Structure of the Oxygen-Evolving Photosystem II at a Resolution of 1.9 Å. Nature 2011, 473, 55–60. 10.1038/nature09913. [DOI] [PubMed] [Google Scholar]
- Suga M.; Akita F.; Hirata K.; Ueno G.; Murakami H.; Nakajima Y.; Shimizu T.; Yamashita K.; Yamamoto M.; Ago H.; et al. Native Structure of Photosystem II at 1.95 Å Resolution Viewed by Femtosecond X-ray Pulses. Nature 2015, 517, 99–103. 10.1038/nature13991. [DOI] [PubMed] [Google Scholar]
- Suga M.; Akita F.; Sugahara M.; Kubo M.; Nakajima Y.; Nakane T.; Yamashita K.; Umena Y.; Nakabayashi M.; Yamane T.; et al. Light-Induced Structural Changes and the Site of O=O bond Formation in PSII Caught by XFEL. Nature 2017, 543, 131–135. 10.1038/nature21400. [DOI] [PubMed] [Google Scholar]
- Wei X.; Su X.; Cao P.; Liu X.; Chang W.; Li M.; Zhang X.; Liu Z. Structure of spinach photosystem II–LHCII supercomplex at 3.2 Å resolution. Nature 2016, 534, 69–74. 10.1038/nature18020. [DOI] [PubMed] [Google Scholar]
- Haumann M.; Liebisch P.; Müller C.; Barra M.; Grabolle M.; Dau H. Photosynthetic O2 Formation Tracked by Time-Resolved X-ray Experiments. Science 2005, 310, 1019–1021. 10.1126/science.1117551. [DOI] [PubMed] [Google Scholar]
- Britt R. D.; Campbell K. A.; Peloquin J. M.; Gilchrist M. L.; Aznar C. P.; Dicus M. M.; Robblee J.; Messinger J. Recent Pulsed EPR Studies of the Photosystem II Oxygen-Evolving Complex: Implications as to Water Oxidation Mechanisms. Biochim. Biophys. Acta, Bioenerg. 2004, 1655, 158–171. 10.1016/j.bbabio.2003.11.009. [DOI] [PubMed] [Google Scholar]
- McEvoy J. P.; Brudvig G. W. Water-Splitting Chemistry of Photosystem II. Chem. Rev. 2006, 106, 4455–4483. 10.1021/cr0204294. [DOI] [PubMed] [Google Scholar]
- Hillier W.; Messinger J.. Mechanism of Photosynthetic Oxygen Production. In Photosystem II. The Light-Driven Water:Plastoquinone Oxidoreductase; Wydrzynski T., Satoh K., Eds.; Springer: Dordrecht, 2005; Vol. 22, pp 567–608. [Google Scholar]
- Yachandra V. K.The Catalytic Manganese Cluster: Organization of the Metal Ions. In Photosystem II: The Light-Driven Water:Plastoquinone Oxidoreductase; Wydrzynski T. J., Satoh K., Freeman J. A., Eds.; Springer: Dordrecht, The Netherlands, 2005; pp 235–260. [Google Scholar]
- Dau H.; Haumann M. Eight Steps Preceding O-O Bond Formation in Oxygenic Photosynthesis–A Basic Reaction Cycle of the Photosystem II Manganese Complex. Biochim. Biophys. Acta, Bioenerg. 2007, 1767, 472–483. 10.1016/j.bbabio.2007.02.022. [DOI] [PubMed] [Google Scholar]
- Meyer T. J.; Huynh M. H. V.; Thorp H. H. The Possible Role of Proton-Coupled Electron Transfer (PCET) in Water Oxidation by Photosystem II. Angew. Chem., Int. Ed. 2007, 46, 5284–5304. 10.1002/anie.200600917. [DOI] [PubMed] [Google Scholar]
- Renger G. Light Induced Oxidative Water Splitting in Photosynthesis: Energetics, Kinetics and Mechanism. J. Photochem. Photobiol., B 2011, 104, 35–43. 10.1016/j.jphotobiol.2011.01.023. [DOI] [PubMed] [Google Scholar]
- Krewald V.; Retegan M.; Pantazis D. A. Principles of Natural Photosynthesis. Top. Curr. Chem. 2016, 371, 23–48. 10.1007/128_2015_645. [DOI] [PubMed] [Google Scholar]
- Pérez-Navarro M.; Neese F.; Lubitz W.; Pantazis D. A.; Cox N. Recent developments in biological water oxidation. Curr. Opin. Chem. Biol. 2016, 31, 113–119. 10.1016/j.cbpa.2016.02.007. [DOI] [PubMed] [Google Scholar]
- Pantazis D. A. Missing Pieces in the Puzzle of Biological Water Oxidation. ACS Catal. 2018, 8, 9477–9507. 10.1021/acscatal.8b01928. [DOI] [Google Scholar]
- Dau H.; Zaharieva I.; Haumann M. Recent developments in research on water oxidation by photosystem II. Curr. Opin. Chem. Biol. 2012, 16, 3–10. 10.1016/j.cbpa.2012.02.011. [DOI] [PubMed] [Google Scholar]
- Shen J.-R. The Structure of Photosystem II and the Mechanism of Water Oxidation in Photosynthesis. Annu. Rev. Plant Biol. 2015, 66, 23–48. 10.1146/annurev-arplant-050312-120129. [DOI] [PubMed] [Google Scholar]
- Murray J. W.; Barber J. Structural Characteristics of Channels and Pathways in Photosystem II Including the Identification of an Oxygen Channel. J. Struct. Biol. 2007, 159, 228–237. 10.1016/j.jsb.2007.01.016. [DOI] [PubMed] [Google Scholar]
- Ho F. M.; Styring S. Access Channels and Methanol Binding Site to the CaMn4 Cluster in Photosystem II Based on Solvent Accessibility Simulations, with Implications for Substrate Water Access. Biochim. Biophys. Acta, Bioenerg. 2008, 1777, 140–153. 10.1016/j.bbabio.2007.08.009. [DOI] [PubMed] [Google Scholar]
- Vassiliev S.; Zaraiskaya T.; Bruce D. Molecular Dynamics Simulations Reveal Highly Permeable Oxygen Exit Channels Shared with Water Uptake Channels in Photosystem II. Biochim. Biophys. Acta, Bioenerg. 2013, 1827, 1148–1155. 10.1016/j.bbabio.2013.06.008. [DOI] [PubMed] [Google Scholar]
- Vassiliev S.; Zaraiskaya T.; Bruce D. Exploring the Energetics of Water Permeation in Photosystem II by Multiple Steered Molecular Dynamics Simulations. Biochim. Biophys. Acta, Bioenerg. 2012, 1817, 1671–1678. 10.1016/j.bbabio.2012.05.016. [DOI] [PubMed] [Google Scholar]
- Bondar A.-N.; Dau H. Extended Protein/Water H-bond Networks in Photosynthetic Water Oxidation. Biochim. Biophys. Acta, Bioenerg. 2012, 1817, 1177–1190. 10.1016/j.bbabio.2012.03.031. [DOI] [PubMed] [Google Scholar]
- Retegan M.; Pantazis D. A. Interaction of methanol with the oxygen-evolving complex: atomistic models, channel identification, species dependence, and mechanistic implications. Chem. Sci. 2016, 7, 6463–6476. 10.1039/C6SC02340A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Retegan M.; Pantazis D. A. Differences in the Active Site of Water Oxidation among Photosynthetic Organisms. J. Am. Chem. Soc. 2017, 139, 14340–14343. 10.1021/jacs.7b06351. [DOI] [PubMed] [Google Scholar]
- Ishikita H.; Saenger W.; Loll B.; Biesiadka J.; Knapp E.-W. Energetics of a Possible Proton Exit Pathway for Water Oxidation in Photosystem II. Biochemistry 2006, 45, 2063–2071. 10.1021/bi051615h. [DOI] [PubMed] [Google Scholar]
- Debus R. J. Evidence from FTIR Difference Spectroscopy That D1-Asp61 Influences the Water Reactions of the Oxygen-Evolving Mn4CaO5 Cluster of Photosystem II. Biochemistry 2014, 53, 2941–2955. 10.1021/bi500309f. [DOI] [PubMed] [Google Scholar]
- Service R. J.; Hillier W.; Debus R. J. Network of Hydrogen Bonds near the Oxygen-Evolving Mn4CaO5 Cluster of Photosystem II Probed with FTIR Difference Spectroscopy. Biochemistry 2014, 53, 1001–1017. 10.1021/bi401450y. [DOI] [PubMed] [Google Scholar]
- Service R. J.; Hillier W.; Debus R. J. Evidence from FTIR Difference Spectroscopy of an Extensive Network of Hydrogen Bonds near the Oxygen-Evolving Mn4Ca Cluster of Photosystem II Involving D1-Glu65, D2-Glu312, and D1-Glu329. Biochemistry 2010, 49, 6655–6669. 10.1021/bi100730d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dau H.; Haumann M. The Manganese Complex of Photosystem II in its Reaction Cycle–Basic Framework and Possible Realization at the Atomic Level. Coord. Chem. Rev. 2008, 252, 273–295. 10.1016/j.ccr.2007.09.001. [DOI] [Google Scholar]
- Rivalta I.; Amin M.; Luber S.; Vassiliev S.; Pokhrel R.; Umena Y.; Kawakami K.; Shen J. R.; Kamiya N.; Bruce D.; et al. Structural-Functional Role of Chloride in Photosystem II. Biochemistry 2011, 50, 6312–6315. 10.1021/bi200685w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dilbeck P. L.; Hwang H. J.; Zaharieva I.; Gerencser L.; Dau H.; Burnap R. L. The D1-D61N Mutation in Synechocystis sp. PCC 6803 Allows the Observation of pH-Sensitive Intermediates in the Formation and Release of O2 from Photosystem II. Biochemistry 2012, 51, 1079–1091. 10.1021/bi201659f. [DOI] [PubMed] [Google Scholar]
- Retegan M.; Cox N.; Lubitz W.; Neese F.; Pantazis D. A. The First Tyrosyl Radical Intermediate Formed in the S2–S3 Transition of Photosystem II. Phys. Chem. Chem. Phys. 2014, 16, 11901–11910. 10.1039/c4cp00696h. [DOI] [PubMed] [Google Scholar]
- Retegan M.; Krewald V.; Mamedov F.; Neese F.; Lubitz W.; Cox N.; Pantazis D. A. A Five-Coordinate Mn(IV) Intermediate in Biological Water Oxidation: Spectroscopic Signature and a Pivot Mechanism for Water Binding. Chem. Sci. 2016, 7, 72–84. 10.1039/C5SC03124A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawashima K.; Takaoka T.; Kimura H.; Saito K.; Ishikita H. O2 Evolution and Recovery of the Water-Oxidizing Enzyme. Nat. Commun. 2018, 9, 1247 10.1038/s41467-018-03545-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narzi D.; Bovi D.; Guidoni L. Pathway for Mn-Cluster Oxidation by Tyrosine-Z in the S2 State of Photosystem II. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, 8723–8728. 10.1073/pnas.1401719111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegbahn P. E. M. Mechanisms for Proton Release During Water Oxidation in the S2 to S3 and S3 to S4 Transitions in Photosystem II. Phys. Chem. Chem. Phys. 2012, 14, 4849–4856. 10.1039/c2cp00034b. [DOI] [PubMed] [Google Scholar]
- Klauss A.; Haumann M.; Dau H. Alternating Electron and Proton Transfer Steps in Photosynthetic Water Oxidation. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 16035–16040. 10.1073/pnas.1206266109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito K.; Rutherford A. W.; Ishikita H. Energetics of proton release on the first oxidation step in the water-oxidizing enzyme. Nat. Commun. 2015, 6, 8488 10.1038/ncomms9488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura S.; Nagao R.; Takahashi R.; Noguchi T. Fourier Transform Infrared Detection of a Polarizable Proton Trapped between Photooxidized Tyrosine YZ and a Coupled Histidine in Photosystem II: Relevance to the Proton Transfer Mechanism of Water Oxidation. Biochemistry 2014, 53, 3131–3144. 10.1021/bi500237y. [DOI] [PubMed] [Google Scholar]
- Nagao R.; Ueoka-Nakanishi H.; Noguchi T. D1-Asn-298 in Photosystem II is Involved in a Hydrogen-Bond Network Near the Redox-Active Tyrosine YZ for Proton Exit During Water Oxidation. J. Biol. Chem. 2017, 292, 20046–20057. 10.1074/jbc.M117.815183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto H.; Shimizu T.; Nagao R.; Noguchi T. Monitoring the Reaction Process During the S2→S3 Transition in Photosynthetic Water Oxidation Using Time-Resolved Infrared Spectroscopy. J. Am. Chem. Soc. 2017, 139, 2022–2029. 10.1021/jacs.6b11989. [DOI] [PubMed] [Google Scholar]
- Sakashita N.; Watanabe H. C.; Ikeda T.; Saito K.; Ishikita H. Origins of Water Molecules in the Photosystem II Crystal Structure. Biochemistry 2017, 56, 3049–3057. 10.1021/acs.biochem.7b00220. [DOI] [PubMed] [Google Scholar]
- Noguchi T. Fourier Transform Infrared Difference and Time-Resolved Infrared Detection of the Electron and Proton Transfer Dynamics in Photosynthetic Water Oxidation. Biochim. Biophys. Acta, Bioenerg. 2015, 1847, 35–45. 10.1016/j.bbabio.2014.06.009. [DOI] [PubMed] [Google Scholar]
- Rappaport F.; Ishida N.; Sugiura M.; Boussac A. Ca2+ Determines the Entropy Changes Associated with the Formation of Transition States During Water Oxidation by Photosystem II. Energy Environ. Sci. 2011, 4, 2520–2524. 10.1039/c1ee01408k. [DOI] [Google Scholar]
- Nugent J. H. A.; Muhiuddin I. P.; Evans M. C. W. Electron Transfer from the Water Oxidizing Complex at Cryogenic Temperatures: The S1 to S2 Step. Biochemistry 2002, 41, 4117–4126. 10.1021/bi011320d. [DOI] [PubMed] [Google Scholar]
- Nugent J. H. A.; Muhiuddin I. P.; Evans M. C. W. Effect of Hydroxylamine on Photosystem II: Reinvestigation of Electron Paramagnetic Resonance Characteristics Reveals Possible S State Intermediates. Biochemistry 2003, 42, 5500–5507. 10.1021/bi034116u. [DOI] [PubMed] [Google Scholar]
- Zhang C.; Styring S. Formation of Split Electron Paramagnetic Resonance Signals in Photosystem II Suggests That Tyrosine-Z Can Be Photooxidized at 5 K in the S0 and S1 States of the Oxygen-Evolving Complex. Biochemistry 2003, 42, 8066–8076. 10.1021/bi0269299. [DOI] [PubMed] [Google Scholar]
- Zahariou G.; Ioannidis N.; Sioros G.; Petrouleas V. The Collapse of the Tyrosine Z•–Mn Spin–Spin Interaction above ∼100 K Reveals the Spectrum of Tyrosine Z•. An Application of Rapid-Scan EPR to the Study of Intermediates of the Water Splitting Mechanism of Photosystem II. Biochemistry 2007, 46, 14335–14341. 10.1021/bi7018767. [DOI] [PubMed] [Google Scholar]
- Ioannidis N.; Zahariou G.; Petrouleas V. Trapping of the S2 to S3 State Intermediate of the Oxygen-Evolving Complex of Photosystem II. Biochemistry 2006, 45, 6252–6259. 10.1021/bi060520s. [DOI] [PubMed] [Google Scholar]
- Zahariou G.; Chrysina M.; Petrouleas V.; Ioannidis N. Can we Trap the S3YZ• Metalloradical Intermediate During the S-State Transitions of Photosystem II? An EPR Investigation. FEBS Lett. 2014, 588, 1827–1831. 10.1016/j.febslet.2014.03.045. [DOI] [PubMed] [Google Scholar]
- Petrouleas V.; Koulougliotis D.; Ioannidis N. Trapping of Metalloradical Intermediates of the S-States at Liquid Helium Temperatures. Overview of the Phenomenology and Mechanistic Implications. Biochemistry 2005, 44, 6723–6728. 10.1021/bi0503201. [DOI] [PubMed] [Google Scholar]
- Chrysina M.; Zahariou G.; Sanakis Y.; Ioannidis N.; Petrouleas V. Conformational changes of the S2YZ• intermediate of the S2 to S3 transition in photosystem II. J. Photochem. Photobiol., B 2011, 104, 72–79. 10.1016/j.jphotobiol.2011.02.012. [DOI] [PubMed] [Google Scholar]
- Berthold D. A.; Babcock G. T.; Yocum C. F. A Highly Resolved, Oxygen-Evolving Photosystem II Preparation from Spinach Thylakoid Membranes. FEBS Lett. 1981, 134, 231–234. 10.1016/0014-5793(81)80608-4. [DOI] [Google Scholar]
- Ford R. C.; Evans M. C. W. Isolation of a Photosystem 2 Preparation from Higher Plants with Highly Enriched Oxygen Evolution Activity. FEBS Lett. 1983, 160, 159–164. 10.1016/0014-5793(83)80957-0. [DOI] [Google Scholar]
- Neese F. The ORCA Program System. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2012, 2, 73–78. 10.1002/wcms.81. [DOI] [Google Scholar]
- Perdew J. P. Density-Functional Approximation for the Correlation-Energy of the Inhomogeneous Electron-Gas. Phys. Rev. B 1986, 33, 8822–8824. 10.1103/PhysRevB.33.8822. [DOI] [PubMed] [Google Scholar]
- Becke A. D. Density-Functional Exchange-Energy Approximation with Correct Asymptotic-Behavior. Phys. Rev. A 1988, 38, 3098–3100. 10.1103/PhysRevA.38.3098. [DOI] [PubMed] [Google Scholar]
- van Lenthe E.; Baerends E. J.; Snijders J. G. Relativistic Regular Two-component Hamiltonians. J. Chem. Phys. 1993, 99, 4597–4610. 10.1063/1.466059. [DOI] [Google Scholar]
- van Lenthe E.; Baerends E. J.; Snijders J. G. Relativistic Total-Energy Using Regular Approximations. J. Chem. Phys. 1994, 101, 9783–9792. 10.1063/1.467943. [DOI] [Google Scholar]
- Pantazis D. A.; Chen X. Y.; Landis C. R.; Neese F. All-electron scalar relativistic basis sets for third-row transition metal atoms. J. Chem. Theory Comput. 2008, 4, 908–919. 10.1021/ct800047t. [DOI] [PubMed] [Google Scholar]
- Schäfer A.; Huber C.; Ahlrichs R. Fully Optimized Contracted Gaussian-Basis Sets of Triple Zeta Valence Quality for Atoms Li to Kr. J. Chem. Phys. 1994, 100, 5829–5835. 10.1063/1.467146. [DOI] [Google Scholar]
- Weigend F. Accurate Coulomb-Fitting Basis Sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. 10.1039/b515623h. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Antony J.; Ehrlich S.; Krieg H. A Consistent and Accurate ab initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H–Pu. J. Chem. Phys. 2010, 132, 154104 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- Cossi M.; Rega N.; Scalmani G.; Barone V. Energies, Structures, and Electronic Properties of Molecules in Solution with the C-PCM Solvation Model. J. Comput. Chem. 2003, 24, 669–681. 10.1002/jcc.10189. [DOI] [PubMed] [Google Scholar]
- Siegbahn P. E. M. The Effect of Backbone Constraints: The Case of Water Oxidation by the Oxygen-Evolving Complex in PSII. ChemPhysChem 2011, 12, 3274–3280. 10.1002/cphc.201100475. [DOI] [PubMed] [Google Scholar]
- Pantazis D. A.; Ames W.; Cox N.; Lubitz W.; Neese F. Two Interconvertible Structures that Explain the Spectroscopic Properties of the Oxygen-Evolving Complex of Photosystem II in the S2 State. Angew. Chem., Int. Ed. 2012, 51, 9935–9940. 10.1002/anie.201204705. [DOI] [PubMed] [Google Scholar]
- Krewald V.; Retegan M.; Cox N.; Messinger J.; Lubitz W.; DeBeer S.; Neese F.; Pantazis D. A. Metal Oxidation States in Biological Water Splitting. Chem. Sci. 2015, 6, 1676–1695. 10.1039/C4SC03720K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioannidis N.; Zahariou G.; Petrouleas V. The EPR Spectrum of Tyrosine Z• and Its Decay Kinetics in O2-Evolving Photosystem II Preparations. Biochemistry 2008, 47, 6292–6300. 10.1021/bi800390r. [DOI] [PubMed] [Google Scholar]
- Boussac A.; Rappaport F.; Brettel K.; Sugiura M. Charge Recombination in SnTyrZ•QA–• Radical Pairs in D1 Protein Variants of Photosystem II: Long Range Electron Transfer in the Marcus Inverted Region. J. Phys. Chem. B 2013, 117, 3308–3314. 10.1021/jp400337j. [DOI] [PubMed] [Google Scholar]
- Hopfield J. J. Electron Transfer Between Biological Molecules by Thermally Activated Tunneling. Proc. Natl. Acad. Sci. U.S.A. 1974, 71, 3640–3644. 10.1073/pnas.71.9.3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus R. A. Chemical and Electrochemical Electron-Transfer Theory. Annu. Rev. Phys. Chem. 1964, 15, 155–196. 10.1146/annurev.pc.15.100164.001103. [DOI] [Google Scholar]
- Marcus R. A.; Sutin N. Electron Transfers in Chemistry and Biology. Biochim. Biophys. Acta, Bioenerg. 1985, 811, 265–322. 10.1016/0304-4173(85)90014-X. [DOI] [Google Scholar]
- Moser C. C.; Keske J. M.; Warncke K.; Farid R. S.; Dutton P. L. Nature of Biological Electron Transfer. Nature 1992, 355, 796–802. 10.1038/355796a0. [DOI] [PubMed] [Google Scholar]
- Lavergne J.; Junge W. Proton Release During the Redox Cycle of the Water Oxidase. Photosynth. Res. 1993, 38, 279–296. 10.1007/BF00046752. [DOI] [PubMed] [Google Scholar]
- Barber J.; Anderson J. M.; Junge W.; Haumann M.; Ahlbrink R.; Mulkidjanian A.; Clausen J. Electrostatics and Proton Transfer in Photosynthetic Water Oxidation. Philos. Trans. R. Soc., B 2002, 357, 1407–1418. 10.1098/rstb.2002.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlodder E.; Witt H. T. Stoichiometry of Proton Release from the Catalytic Center in Photosynthetic Water Oxidation - Reexamination by a Glass Electrode Study at pH 5.5-7.2. J. Biol. Chem. 1999, 274, 30387–30392. 10.1074/jbc.274.43.30387. [DOI] [PubMed] [Google Scholar]
- Suzuki H.; Sugiura M.; Noguchi T. Monitoring Proton Release during Photosynthetic Water Oxidation in Photosystem II by Means of Isotope-Edited Infrared Spectroscopy. J. Am. Chem. Soc. 2009, 131, 7849–7857. 10.1021/ja901696m. [DOI] [PubMed] [Google Scholar]
- Kawashima K.; Saito K.; Ishikita H. Mechanism of Radical Formation in the H-Bond Network of D1-Asn298 in Photosystem II. Biochemistry 2018, 57, 4997–5004. 10.1021/acs.biochem.8b00574. [DOI] [PubMed] [Google Scholar]
- Faller P.; Goussias C.; Rutherford A. W.; Un S. Resolving Intermediates in Biological Proton-Coupled Electron Transfer: A Tyrosyl Radical Prior to Proton Movement. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 8732–8735. 10.1073/pnas.1530926100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroda H.; Kodama N.; Sun X.-Y.; Ozawa S.-i.; Takahashi Y. Requirement for Asn298 on D1 Protein for Oxygen Evolution: Analyses by Exhaustive Amino Acid Substitution in the Green Alga Chlamydomonas reinhardtii. Plant Cell Physiol. 2014, 55, 1266–1275. 10.1093/pcp/pcu073. [DOI] [PubMed] [Google Scholar]
- Nakamura A.; Ishida T.; Kusaka K.; Yamada T.; Fushinobu S.; Tanaka I.; Kaneko S.; Ohta K.; Tanaka H.; Inaka K.; et al. “Newton’s Cradle” Proton Relay with Amide–Imidic Acid Tautomerization in Inverting Cellulase Visualized by Neutron Crystallography. Sci. Adv. 2015, 1, e1500263 10.1126/sciadv.1500263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hundelt M.; Hays A.-M. A.; Debus R. J.; Junge W. Oxygenic Photosystem II: The Mutation D1–D61N in Synechocystis sp. PCC 6803 Retards S-State Transitions without Affecting Electron Transfer from YZ to P680+. Biochemistry 1998, 37, 14450–14456. 10.1021/bi981164j. [DOI] [PubMed] [Google Scholar]
- Hoganson C. W.; Babcock G. T. A Metalloradical Mechanism for the Generation of Oxygen from Water in Photosynthesis. Science 1997, 277, 1953–1956. 10.1126/science.277.5334.1953. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.