

Abstract
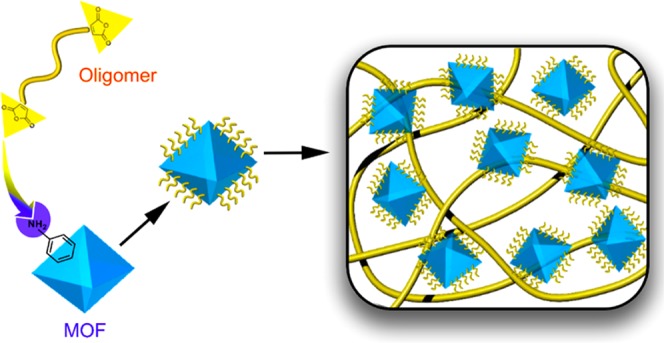
Mixed-matrix membranes (MMMs) formed by dispersing metal–organic framework (MOF) particles in polymers have attracted significant attention because these composite systems can potentially surpass the separation performance of pure polymers alone. However, performance improvements are often unrealized because of poor interfacial compatibility between the MOF and the polymer, which results in interfacial defects. From a practical perspective, strategies are needed to address these defects so that MMMs can be deployed in real-world separation processes. From a fundamental perspective, strategies are needed to reliably form defect-free MMMs so that transport models can be applied to estimate pure MOF property sets, thereby enabling the development of robust structure–property relationships. To address these interfacial challenges, we have developed a method to surface-functionalize a UiO-66-NH2 MOF with a nanoscopic shell of covalently tethered 4,4′-(hexafluoroisopropylidene)diphthalic anhydride–Durene oligomers. When combined with a high-molecular-weight polymer of identical chemical structure to that of the imide-functional MOF surface, defect-free MMMs with uniform particle dispersions can be formed. With this technique, both permeabilities and selectivities of select gases in the MMMs were improved at loadings ranging from 5 to 40 wt %. At a 40 wt % loading, CO2 permeability and CO2/CH4 selectivity were enhanced by 48 and 15%, respectively. Additionally, pure MOF permeabilities for H2, N2, O2, CH4, and CO2 were predicted by the Maxwell model.
Keywords: metal−organic frameworks, postsynthetic modification, gas separations, polyimides, mixed-matrix membranes
1. Introduction
Membrane separations have been identified as promising alternatives to traditional unit operations.1,2 In particular, membranes have several potential advantages such as reduced energy consumption, compact module size, low capital investment, stability at high pressures, and operational simplicity (i.e., no moving parts).3 Several classes of polymers have been commercialized for a suite of gas separation application;1 however, conventional polymeric membranes are limited by a trade-off between permeability and selectivity. Polymers with higher permeability generally have lower selectivity and vice versa.4 This trade-off was first quantified for a large database of polymers by Robeson in 19915 and more recently updated in 2008.6 Tremendous efforts have been undertaken to develop membranes that can surpass the upper bound through the synthesis of new polymers,7,8 post-treatment of polymeric films by thermal annealing near the glass-transition temperature,9 and combining organic or inorganic fillers with polymers to produce mixed-matrix membranes (MMMs).10 Among these techniques, the development of MMMs has attracted significant attention because of the versatility this approach offers to combine high-performance but difficult-to-process fillers with a variety of processable polymers.
In general, MMMs are prepared by dispersing fillers, such as metal–organic framework (MOFs), zeolites, carbon molecular sieves, etc., into a continuous organic polymer phase. By combining these materials, MMMs can potentially have permeability and selectivity property sets that surpass those of the pure polymer. Among the various types of inorganic fillers considered in the literature, MOFs are a relatively new class of materials that have been rapidly growing in interest because of their enormous internal surface areas and micropores that are often sized for gas separations.11,12 The chemical and physical structures of MOFs can be tuned with relative synthetic ease to target the separation of molecules based on their size, shape, and chemical composition. Despite these many potential benefits, an unsolved challenge is identifying methods to improve the interfacial compatibility between the MOF and polymer to prevent the formation of nonselective defects, which leads to a deterioration of gas selectivity.13
To address this interfacial challenge, researchers have focused extensive efforts on developing techniques such as thermal annealing,9 priming casting,14 surface modification of filler particles,15 reducing the size of the fillers,16 in situ synthesis of MOF particles in an already-cast porous polymeric membrane,17 in situ polymerization of the polymeric matrix in the presence of the already-synthesized MOF particles,18 and in situ modification of MOFs in MMMs.19 This report focuses on the formation of MMMs using an amine-functionalized zirconium-based MOF known as UiO-66-NH2, which has been shown to have beneficial separation performance for CO2-based separations.20 The following select examples demonstrate a few successful UiO-66-NH2 modifications or polymer modifications that have resulted in improved interfacial compatibility.
Molavi et al. demonstrated that an improvement in both permeability and selectivity in CO2/CH4 separation can be achieved by grafting either poly(methyl methacrylate) (PMMA) or glycidyl methacrylate (GMA) from UiO-66-NH2,21,22 which forms an interfacial layer on the MOF that is compatible with a pure PMMA polymer matrix due to enhanced interfacial compatibility. Huang et al. observed similar results when they modify UiO-66-NH2 with imidazole-2-carbaldehyde. A concomitant increase in both permeability and selectivity resulted when the modified MOF was added into Matrimid.23 Venna et al. showed that UiO-66-NH2 can be functionalized by molecules with acetyl groups to seal off the interfacial defects, thus improving the selectivity of MMMs.24 In contrast to modifying the MOF particles, the polymer matrices can likewise be modified to improve compatibility. Wang et al. demonstrated that PIM-1 can be functionalized to induce hydrogen bonds with MOF fillers, thereby improving interfacial contact.26 Another approach is in situ polymerization. Kaliaguine and co-workers reacted PIM-1 monomers with UiO-66-NH2 to form covalently tethered hybrid MMM systems.18 Interestingly, the authors performed this in situ polymer–MOF polymerization using stoichiometric equivalents of PIM-1 monomers without regard to the molar contribution of surface amine functionality in UiO-66-NH2. Considering that PIM-1 is formed through a step polymerization reaction, the extra amine functionality from UiO-66-NH2 would necessarily result in a stoichiometric imbalance, and such imbalances often preclude the formation of high-molecular-weight polymers needed to form films. Therefore, although films can be formed via direct in situ polymerization, scaling such reactions to form MMMs with varying degrees of accessible amine functionality is expected to remain a challenge, especially for targeting the formation of MMMs with high MOF loadings.
Given these challenges, this study presents an alternative method to form defect-free MMMs with UiO-66-NH2, as illustrated in Scheme 1. Amine groups on the surface of UiO-66-NH2 nanoparticles are imidized to form a covalently tethered imide layer on the particles. Next, these imide-functional particles are dispersed in a high-molecular-weight polyimide of identical chemistry to that of the functionalized surface, enabling the reproducible formation of defect-free MMMs with loadings up to 40 wt %.
Scheme 1. Steps Outlining Approach of Postsynthetic Modification of UiO-66-NH2 and Formation of MMMs.
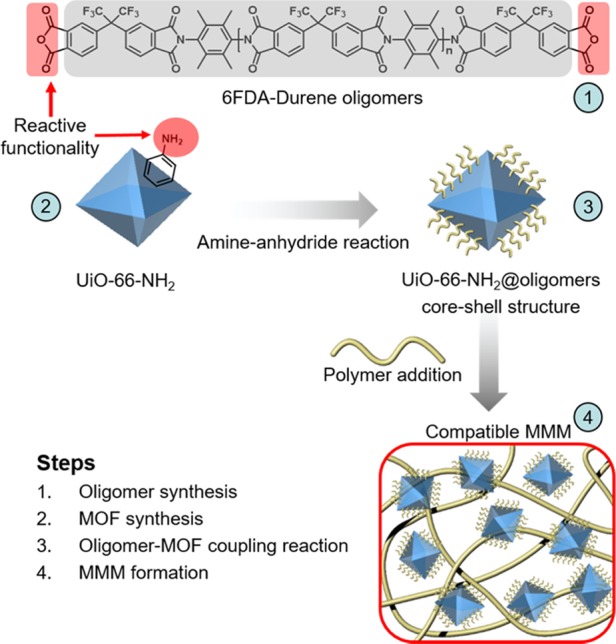
The MOF used in this study was UiO-66-NH2 because of its high thermal and chemical stabilities, as well as its strong affinity toward CO2 over other gases such as CH4 and N2.25 UiO-66-NH2 is also a good candidate for postsynthetic modification (PSM) reactions due to the presence of the −NH2-functional group that can be used as a reactive nucleophile. The polyimide, 6FDA–Durene, which is formed from 2,3,5,6-tetramethyl-1,4-phenylenediamine (Durene diamine) and 4,4′-(hexafluoroisopropylidene)diphthalic anhydride (6FDA), was chosen as the polymer matrix due to its high thermal stability and high glass-transition temperature, as well as its intrinsic CO2/CH4 separation performance.28 Similar to the Durene diamine monomer, UiO-66-NH2 contains accessible amine groups, which should likewise react with dianhydrides to form polyimide linkages. Oligomers of 6FDA–Durene with dianhydride end groups were synthesized by stoichiometric imbalance to form telechelic imide oligomers with molecular weights of approximately 3700 g mol–1. Oligomers of this molecular weight are too large to diffuse into the MOF pores, thereby preserving the internal pore chemistry and structure of UiO-66-NH2. Such an approach enables the formation of a 6FDA–Durene polymer interface on the MOF. It was observed that after the reaction, modified MOF particles could be uniformly dispersed in 6FDA–Durene polymers without interfacial defects. With this technique, both permeability and selectivity of MMMs improved monotonically with increased loadings for select gases and gas pairs up to loadings of 40 wt %.
2. Experimental/Theory
2.1. Materials
Zirconium(IV) chloride (ZrCl4, >99.5%), 2-aminoterephthalic acid (99%), 1-methyl-2-pyrrolidinone (NMP, 99.5%), acetic anhydride (>98%), 1,4-dichlorobenzene (>99%), and acetic acid (>99.7%) were purchased from Sigma-Aldrich and used as received. Chloroform (>99.8%) and N,N-dimethylformamide (DMF, >99.8%) were purchased from VWR and used as received. The monomer, 2,3,5,6-tetramethyl-1,4-phenylenediamine (Durene diamine, >98%), was purchased from TCI Co. and purified through methanol recrystallization before use. The monomer, 4,4′-(hexafluoroisopropylidene)diphthalic anhydride (6FDA, >99%), was purchased from Sigma-Aldrich and purified through vacuum sublimation at around 235 °C before use. High-molecular-weight 6FDA–Durene polymers (Mn = 230 kDa) were purchased from Akron Polymer Systems and used as received.
2.2. Preparation of 6FDA–Durene Oligomers
6FDA–Durene oligomers were synthesized using a modified procedure based on a 6FDA–Durene polymer synthesis method reported earlier by Chung and co-worker.29 Reactions were carried out in three-necked flasks with overhead mechanical stirrers attached in the presence of a flowing N2 (Airgas UHP grade, purity >99.999%), which was treated with a Drierite drying column first to remove moisture. The monomer, 6FDA (6.277 g, 14.137 mmol, 7 mol % excess of dianhydride), was fully dissolved in 20 mL of NMP while rapidly stirring for approximately 30 min, after which the Durene diamine (2.167 g, 13.213 mmol) was added to the reaction solution. The mixture was then stirred overnight at room temperature for 24 h to form a poly(amic acid). Acetic anhydride (4.526 mL, 52.854 mmol) and pyridine (3.856 mL, 52.854 mmol) were added subsequently, and the reaction solution was stirred overnight to convert the poly(amic acid) into a polyimide. The imide oligomers were precipitated by pouring the reaction mixture slowly into methanol that was stirring in a blender. Oligomers were collected by vacuum filtration, after which they were redissolved in chloroform, reprecipitated in methanol, and filtered by vacuum filtration. Oligomers were then washed in fresh methanol for three consecutive days and filtered by vacuum filtration at the end of each day. The final product was dried in a vacuum oven at 150 °C and stored for future use.
2.3. Preparation of UiO-66-NH2 Nanoparticles
UiO-66-NH2 nanoparticles were synthesized by a modified procedure based on a method reported by Farha and co-workers.30 ZrCl4 (0.032 g, 0.1373 mmol) was dissolved in 20 mL of DMF in a 50 mL Teflon-lined, stainless steel autoclave. Next, 2-aminoterephthalic acid (0.040 g, 0.1373 mmol) was added and fully dissolved. Acetic acid (0.06 mL, 8.238 mmol, 60 equiv to ZrCl4) was added into the solution while stirring until all components were fully mixed. The autoclave was then sealed and placed in an oven at 100 °C for 24 h. The precipitate was isolated by centrifugation (rpm = 12 000, 15 min), washed three times with 15 mL DMF, and then washed three times with 15 mL methanol. The obtained solid was dried in the vacuum oven at 150 °C overnight before characterization.
2.4. Preparation of Oligomer-Coated MOF Particles
UiO-66-NH2 (0.07 g, ∼0.25 mmol total −NH2-functional groups) was dispersed in 5 mL of NMP by sonication. In a separate container, the 6FDA–Durene oligomer (0.625 g, ∼0.25 mmol dianhydride-functional group) was dissolved in 5 mL of NMP before mixing the oligomer with the MOF suspension. Because only a small portion of amine groups in the UiO-66-NH2 MOF are accessible for surface reactions, the dianhydride-functionalized oligomer was effectively added in large excess. The mixture was sonicated indirectly in a water bath for 1 h and then stirred for 3 days at room temperature to form poly(amic acid) linkages. An azeotropic agent, 1,4-dichlorobenzene (30 mL), was then added, and the mixture was heated to 180 °C and stirred overnight to convert the poly(amic acid) to polyimide linkages through a thermal imidization reaction. The yellow suspension was isolated from the solution by centrifugation and washed with 15 mL of fresh NMP three times to remove any unreacted oligomer. The same washing procedure was repeated using chloroform three times to suspend the oligomer-coated UiO-66-NH2 in chloroform for film casting.
2.5. Film Casting
Films were formed via a widely reported solution-casting technique.29 Filler suspensions (oligomer-coated UiO-66-NH2 in chloroform) were mixed with 6FDA–Durene polymer solutions in chloroform at specific ratios to make 2 wt/vol % casting solutions. To ensure uniform dispersion, suspensions were sonicated directly using a probe sonicator for 1 min and then sonicated indirectly using a water bath sonicator for 1 h. Suspensions were then stirred for 1 h. This process was repeated three times. Afterwards, suspensions were stirred overnight at room temperature before casting. Next, suspensions were poured into flat-bottomed glass Petri dishes and covered by glass plates to prevent contamination from dust and to control the evaporation rate. Solutions were left for 24 h at ambient temperature and pressure to form freestanding films. Films were carefully peeled from the glass dish with the assistance of deionized H2O. To remove the solvent, films were dried in a fume hood for 24 h and then dried under a dynamic vacuum in a vacuum oven overnight at 60 °C. Finally, the residual solvent was removed by drying films in a vacuum oven at 150 °C overnight.
2.6. Characterization
The chemical structure of the 6FDA–Durene oligomer was confirmed by a Varian Mercury 300 nuclear magnetic resonance (NMR) spectrometer. The glass-transition temperature (Tg) and thermal stability of the 6FDA–Durene oligomer were evaluated using a TA Instruments differential scanning calorimeter (DSC) 250 and thermogravimetric analyzer (TGA) 550, respectively. The morphology of the UiO-66-NH2 and PSM–MOF was investigated using an FEI Tecnai multipurpose transmission electron microscope (TEM). The chemical structure of the MOF particles before and after PSM was evaluated in the range of 400–4000 cm–1 on a Thermo Fisher FTIR6700 Fourier transform infrared (FTIR) spectrometer in transmission mode. The covalent linkage between UiO-66-NH2 and oligomers was characterized using a 500 MHz Varian Inova-500 1H NMR after MOF digestion in dimethyl sulfoxide/hydrofluoric (aq.) solution. The cross-sectional morphologies of all films were observed on a Zeiss Merlin high-resolution scanning electron microscope (SEM) after fracturing films under liquid nitrogen. The crystallinity of the MOF particles in pure form and in mixed-matrix membrane films was examined by a Rigaku Smartlab multipurpose X-ray diffractometer (XRD) in the 2θ region from 5 to 40°. Nitrogen adsorption isotherms were obtained with a Micromeritics 3Flex system to determine the Brunauer–Emmett–Teller (BET) surface areas of nanoparticles. Viscoelastic behavior of MMMs was characterized with a Q800 dynamic mechanical analyzer (DMA) from TA Instruments using a 3 °C/min ramp rate, an applied frequency of 1 Hz, and a constant amplitude of 15 μm, in accordance with the parameters considered in an earlier study.31
2.7. Pure-Gas Permeation Tests
An automated, constant-volume/variable-pressure system from Maxwell Robotics was used to determine the pure-gas permeabilities of H2, O2, N2, CH4, and CO2. Film areas of approximately 15 mm2 were cut from the as-prepared films and placed over the top of a small hole on a circular brass-supporting disk. The edge of the film was sealed by epoxy glue (Devcon 5 min Epoxy), leaving a small active area of the sample exposed for permeation. The disk was then inserted into a stainless steel permeation cell, sealed, and immersed in a water bath with temperature controlled by an immersion circulator (Thermo Fisher SC150L). All gases tested were ultrahigh-purity gases purchased from Airgas. Before switching to a new permeating gas, the entire system was dosed with approximately 1 bar of helium and then held under a dynamic vacuum for 1 h to ensure no residual gas remained in the tubing. In all cases, the films were tested at 35 °C for upstream pressures varying from 1 to 40 bar except CO2, which was tested from 1 to 50 bar. Each film was tested at least twice to confirm reproducibility. Three films (6FDA–Durene, 10% PSM–MOF MMM, and 10% pure MOF MMM) were chosen to conduct a 5 week aging test. These films were placed on a benchtop after the casting and thermal-annealing step. Pure-gas permeability was measured for these membranes at the start of each week for 5 consecutive weeks after casting.
2.8. Mixed-Gas Permeation Tests
A similar automated, constant-volume/variable-pressure system from Maxwell Robotics was used to determine the mixed-gas permeabilities of CH4 and CO2. An Agilent 7890B GC system was used to analyze the gas composition of both feed and permeant streams. The same sample support disk as that used in pure-gas permeation tests was inserted into a sealed stainless steel permeation cell, and the temperature was controlled by a built-in air-heating system. A gas mixture of 50:50 CO2/CH4 was mixed upstream of the permeation cell for a certain amount of time before the actual permeation step until the upstream composition was stable at 50:50. In all cases, films were tested at 35 °C for total upstream pressures varying from 2 to 30 bar.
2.9. Maxwell Model Prediction
The Maxwell model was used to mathematically assess gas transport in MMMs. This model was first developed to analyze dielectric properties of a dilute conducting suspension of identical particles and later extended to describe gas transport in MMMs based on the close analogy between electrical conduction and gas permeation.32 To describe gas transport in a MMM formed by a continuous polymer matrix and dispersed filler phase, the Maxwell model is given by
| 1 |
where PMMM is the bulk permeability of the MMM, Pp is the permeability of the pure polymer, Pf is the permeability of the filler particles, and ⌀f is the volume fraction of filler particles in the MMM. The Maxwell model was used in this study to estimate the permeability of pure UiO-66-NH2 (i.e., Pf) based on the permeabilities of the neat polyimide films and MMMs with low MOF loadings. Permeabilities of MMMs with high MOF loadings were then predicted by extrapolation and compared with experimental results. Note that the predicted permeabilities for the MOF phase (i.e., Pf) exclude any contribution from the oligomer shell. The volume of the oligomer shell is therefore considered part of the bulk polymer phase (i.e., Pp). More details about the Maxwell model procedure and volume fraction calculations of MOF fillers can be found in the Supporting Information (SI) section C.
3. Results and Discussion
3.1. Synthesis of the 6FDA–Durene Oligomer
The 6FDA–Durene oligomer was synthesized via thermal imidization. Adding a 7% excess stoichiometric imbalance of 6FDA dianhydride to the reaction mixture resulted in both ends of the oligomers being capped with dianhydride functionality. To confirm its structure, the oligomer was characterized by NMR, TGA, and DSC, as shown in Figure 1. The NMR results matched those reported in the literature,28 indicating the successful preparation of the pure 6FDA–Durene oligomer. Additionally, no protons were detected from aryl amines, which are typically in the range of 3–4 ppm for the Durene diamine.33 This finding confirmed that the telechelic oligomer was quantitatively end-capped with dianhydrides within the resolution of NMR. The decomposition temperature (temperature with a 5% weight loss, Td, 0.95) and glass-transition temperature were also similar to those reported in the literature.28 Gel permeation chromatography revealed that the number average molecular weight was 3700 g mol–1 with a polydispersity index (PDI) of 6.97. For ideal, high-molecular-weight step polymerizations, PDI approaches a limiting value of 2.34 Deviations in PDI that are higher than this theoretical prediction are possible for functional groups characterized by unequal reactivities, even for low-molecular-weight oligomers,33 and it is possible that the broad PDI observed here relates to this effect.
Figure 1.

(a) NMR, (b) TGA, and (c) DSC of 6FDA–Durene oligomer. For clarity, the dianhydride end group functionality is not included in the chemical structure in (a). Td,0.95 in (b) is the temperature with a 5% weight loss. The DSC trace is for the second scan.
3.2. Synthesis of UiO-66-NH2 Nanocrystals and PSM Reaction
The as-synthesized UiO-66-NH2 nanoparticles had the characteristic octahedral shape with effective diameters between 40 and 60 nm, as confirmed by the TEM presented in Figure 2a,b. These sizes and dimensionalities are similar to those reported in other studies.26,27,30,38 Following the PSM reaction with the imide oligomer, a thin and uniform layer on the surface of UiO-66-NH2 crystals was formed with a thickness of approximately 3–5 nm, as shown in Figure 2c. As shown in Figure 2d, occasionally, these PSM–MOFs became interconnected through the covalent bridging of the telechelic oligomer. More TEM images of oligomer-coated MOFs at a different magnification can be found in Figure S1.
Figure 2.

TEM images of (a, b) as-synthesized and (c, d) oligomer-functionalized UiO-66-NH2 particles.
Further characterization was required to confirm that the oligomer physically coated the MOF and that covalent bonds between the MOF and oligomer had indeed formed. To verify the physical adhesion of the oligomer on the MOF, TEM–energy-dispersive X-ray (EDX) experiments were conducted, and these results are presented in Figure 3a. A quantifiable deconvolution of elemental composition is challenging with this approach because of the instability of UiO-66-NH2 under the electron beam, which leads to serious thermal-induced drifting issues.35 Nevertheless, it is clear that the PSM–MOF contains fluorine, which is attributed to the fluorine content present in the 6FDA–Durene oligomer but absent from that of UiO-66-NH2. As a control, TEM–EDX results of the as-synthesized MOF are also presented in Figure 3a, revealing no fluorine content before the MOF–oligomer reaction. Addition of the oligomer to the PSM–MOF is further supported by TGA and derivative thermogravimetry (DTG) results in Figures 3b and S6, which shows thermal degradation at a higher temperature for the PSM–MOF compared to that of the pure MOF. The TGA profile for the pure oligomer, which is presented in Figure 3b, demonstrates the higher thermal stability of the oligomer compared to that of the MOF.
Figure 3.

(a) TEM–EDX and (b) TGA of the as-synthesized MOF and PSM–MOF. TGA results also include a heating profile for the pure oligomer. All three curves were normalized based on a 100% starting weight.
To verify covalent attachment between the MOF and oligomer, FTIR was also considered. FTIR results are presented in Figure 4 to highlight three characteristic chemical changes expected for the MOF and oligomer reaction. After the PSM reaction, a decrease in intensity is observed for the ν(−NH2) band centered at 3490 cm–1. This diminishing intensity corresponds with the appearance of new bands for ν(−Ar–C=O) centered at 1700 cm–1 and ν(−C–N) centered at 1290 cm–1. The change in the intensity of the abovementioned peaks suggests that certain −NH2-functional groups on the MOF were reacted to form polyimide linkages. Furthermore, the oligomer coating could not be removed after thoroughly washing the PSM–MOF with chloroform. Chloroform is an excellent solvent for 6FDA–Durene but does not dissolve the MOF, indicating that a covalent bond was formed, as any unreacted oligomer would be removed before FTIR characterization. As a result, these intensity changes were brought about by the covalent linkage between UiO-66-NH2 and the 6FDA–Durene oligomer, rather than simply the physical adhesion of the oligomer on the MOF. 1H NMR after MOF digestion in hydrofluoric acid was conducted to further evaluate the covalent linkage between the MOF and oligomer, as shown in Figure S3. The decrease in peak intensities associated with protons on UiO-66-NH2, as well as the downfield shift of peaks associated with protons on the PSM–MOF, suggests a chemical modification of the 2-amino-1,4-benzenedicarboxylate ligand with the 6FDA–Durene oligomer. The remaining peaks associated with UiO-66-NH2 prove that the reaction is only partial. According to peak integration, approximately 30% conversion of all amine-functional groups was achieved after the PSM step. This analysis allows us to test our hypothesis of oligomer reactions happening exclusively on the MOF surface. Under the assumption that the oligomer is unable to penetrate deep into the UiO-66-NH2 framework, and assuming our particles to be perfectly spherical with uniform diameters of 50 nm, a 30% conversion corresponds with a reaction penetration depth of approximately 3 nm into the MOF core. This limited internal modification of the MOF framework is supported by our surface area characterization and pore size analysis, which will be discussed in greater detail in the following paragraph.
Figure 4.

FTIR comparison between the as-synthesized MOF and the PSM–MOF for different wavenumber ranges: (a) 3600–2400 cm–1, (b) 1800–1650 cm–1, and (c) 1220–1180 cm–1. Bands of interest are highlighted in blue.
Following the PSM reaction, BET surface area decreased from 980 to 750 m2 g–1 (N2 adsorption isotherms can be found in Figure S2). This decrease is attributed to the nonporous contribution of the oligomer on the PSM–MOF particles. Pore size distribution analysis in Figure S2 shows that, after PSM, the pore width of the smaller cavity did not change while the effective pore volume decreased slightly, a result consistent with the added mass from the nonporous oligomer coating. These results further strengthen our claim that PSM does not significantly alter the internal pore structure of UiO-66-NH2. However, the larger cavity shows a slight decrease in the average pore width, which may suggest that the oligomer chains protrude to a limited extent into some of the larger pores. Assuming the PSM–MOF particles to be perfect spheres, 50 nm in diameter for the MOF with uniform 3 nm coatings for the oligomer layer, no internal reactions, and no pore blocking, the polymer shell would account for approximately 28% of the PSM–MOF mass. Since the polymer is nonporous, the resulting surface area of the PSM–MOF would be approximately 700 m2 g–1. This estimation is roughly equivalent to our experimental findings from BET. Given these similarities between our experimental findings and calculated changes in the surface area, these results suggest that our PSM–MOF has an unblocked and unreacted UiO-66-NH2 core, making it an ideal system for studying and predicting transport properties for pure UiO-66-NH2 using models such as the Maxwell model.
3.3. Gas Transport Properties
3.3.1. Characterization of PSM–MOF/6FDA–Durene MMMs
The imide-functionalized MOF particles were dispersed into a 6FDA–Durene polymer matrix to form MMMs for gas transport characterization. The 6FDA–Durene polymer was chosen since it is a well-studied polyimide with good combinations of CO2 permeability and CO2/CH4 selectivity relative to the polymer upper bound.29,36−39 However, this polymer is highly rigid, as characterized by its glass-transition temperature of approximately 424 °C, and forming defect-free MMMs with polymers of limited intrinsic elasticity is notoriously challenging.31 As hypothesized, the oligomer coating helped to form a uniform dispersion of oligomer–MOF hybrid particles in the polymer solution because chloroform is an excellent solvent for 6FDA–Durene,40 and after the PSM reaction, both the polymer and surface of the MOF contain the same functionality. As shown in Figure 5a, suspensions of the polymer and PSM–MOF were visibly stable on the benchtop without any noticeable settling even after 24 h, enabling a facile formation of MMMs with a uniform MOF dispersion. One the other hand, pure MOF particles without any modification settled down in chloroform after only a few hours.
Figure 5.

(a) Pictures of pure MOF and PSM–MOF suspensions in chloroform at 0 and 24 h after leaving suspensions on the benchtop without stirring; (b) pictures of the neat polyimide film and MMMs with (c) 10 wt %, (d) 20 wt %, (e) 30 wt %, and (f) 40 wt % PSM–MOF as fillers.
Neat 6FDA–Durene films and MMMs with loadings ranging from 5 to 40 wt % were successfully cast, and all of them had qualitative flexibility, as shown in Figure 5b–e. The highest MOF loading achieved in this study was 40 wt %. Attempts to form MMMs with higher MOF loadings were unsuccessful, since films were too brittle to be easily tested or characterized. According to the Lewis–Nielson model, the maximum density for close-packed uniform spheres in a lattice is 64 vol %, although this value reduces to 59 vol % for the random packing of uniform spheres.40 With the enhanced interfacial compatibility brought about by the oligomer-coated surface, the highest MOF loading achieved in this study was approximately 40 vol %. However, this loading only accounts for the MOF core and not the oligomer coating. If the oligomer coating is also included in the volume percent calculation, the PSM–MOF MMM contains approximately 52 vol % of dispersed PSM–MOF particles in the MMM, which is remarkably close to the theoretical upper limit of packing for a random sphere-packed model (see SI section C for more details). Although the particles considered here are not completely uniform in shape and size and are not perfectly spherical, these upper limits to loadings are helpful metrics in assessing how closely our MMMs are to the fundamental geometric limitations of using this approach.
To investigate if MOF crystallinity was preserved after the formation of the MMMs, XRD patterns were collected both before and after the incorporation of the MOF into the MMMs. As shown in Figure 6, the pristine 6FDA–Durene film had a broad XRD peak centered at 13°, which is a characteristic of amorphous polymers and has been previously reported for 6FDA–Durene.37 With increasing MOF loading, peaks corresponding to UiO-66-NH2 grew in intensity, suggesting that MOF crystallinity was retained during MMM formation and subsequent activation. Cross-sectional SEM images were taken to analyze interfacial morphology and compatibility between the MOF filler phase and the polymer phase. Compared to unmodified UiO-66-NH2, which has significant MOF aggregation and interfacial void defects when formed into MMMs (cf., Figure S4), the PSM–MOF had significantly better interfacial properties and uniform particle distributions in their MMMs, as presented in Figure 7. Aggregation and interfacial void spaces were much less prominent, indicating a better affinity between the polymer phase and oligomer-functionalized MOF particles.
Figure 6.
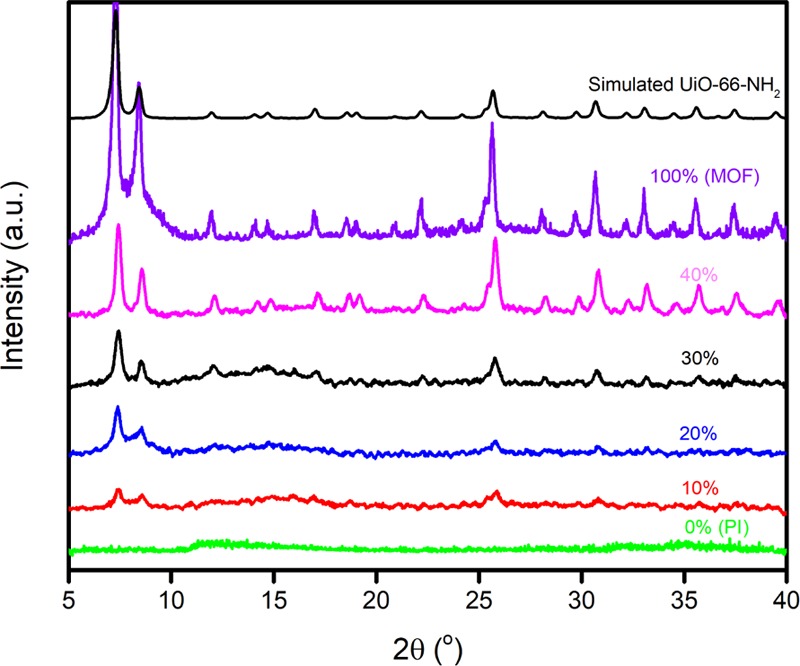
XRD of neat polyimide film, pure MOF particles (synthesized and simulated), and MMMs with different weight percents of PSM–MOF loading.
Figure 7.

Cross-sectional SEM images of MMMs with PSM–MOF loadings of (a) 10 wt %, (b) 20 wt %, (c) 30 wt %, and (d) 40 wt %.
3.3.2. Pure-Gas Permeation Tests
To clearly elucidate the effect PSM–MOF fillers have on polymer–MOF interfacial interactions, the separation performance of the pristine 6FDA–Durene film and MMMs with various weight loadings was tested for pure-gas permeation at 1 bar and 35 °C. Permeabilities of all gases considered (i.e., H2, N2, O2, CH4, and CO2) increased with increasing PSM–MOF loading, and the largest relative increase occurred for CO2. Table 1 summarizes the pure-gas permeabilities of these five gases at different filler loadings. A concomitant increase in permeability and selectivity suggests that all samples contained minimal defects, which typically results in a decreased selectivity at higher filler loadings,2,24,25,41 as shown by the pure MOF MMM data in Figure 8. This finding is attributed to the improved interfacial interaction between the polymer phase and the oligomer-functionalized MOF particles.
Table 1. Tabulated Pure-Gas Permeation Results for the Neat Polyimide and MMMs with Various PSM–MOF Loadingsa.
| permeability (Barrer) |
selectivity |
|||||||
|---|---|---|---|---|---|---|---|---|
| loading (wt %) | CH4 | N2 | O2 | H2 | CO2 | CO2/CH4 | CO2/N2 | H2/CH4 |
| 0% | 83 ± 3 | 86 ± 3 | 218 ± 7 | 820 ± 20 | 1280 ± 40 | 15.4 ± 0.4 | 14.8 ± 0.4 | 9.8 ± 0.3 |
| 2.5% | 84 ± 3 | 86 ± 3 | 222 ± 7 | 840 ± 30 | 1310 ± 40 | 15.7 ± 0.4 | 15.5 ± 0.4 | 10.0 ± 0.3 |
| 5% | 84 ± 3 | 86 ± 3 | 227 ± 7 | 860 ± 30 | 1340 ± 40 | 15.9 ± 0.4 | 15.5 ± 0.4 | 10.2 ± 0.3 |
| 7.5% | 85 ± 3 | 87 ± 3 | 231 ± 7 | 880 ± 30 | 1370 ± 40 | 16.0 ± 0.4 | 15.7 ± 0.4 | 10.3 ± 0.3 |
| 10% | 86 ± 3 | 87 ± 3 | 240 ± 7 | 890 ± 30 | 1400 ± 40 | 16.2 ± 0.4 | 16.0 ± 0.4 | 10.4 ± 0.3 |
| 15% | 90 ± 3 | 92 ± 3 | 249 ± 7 | 940 ± 30 | 1470 ± 40 | 16.4 ± 0.4 | 16.0 ± 0.4 | 10.4 ± 0.3 |
| 20% | 94 ± 3 | 97 ± 3 | 259 ± 8 | 980 ± 30 | 1540 ± 40 | 16.5 ± 0.5 | 15.9 ± 0.4 | 10.5 ± 0.3 |
| 25% | 98 ± 3 | 103 ± 3 | 270 ± 9 | 1040 ± 30 | 1640 ± 50 | 16.7 ± 0.5 | 15.9 ± 0.4 | 10.6 ± 0.3 |
| 30% | 100 ± 3 | 106 ± 3 | 281 ± 9 | 1070 ± 30 | 1710 ± 50 | 17.1 ± 0.5 | 16.1 ± 0.4 | 10.7 ± 0.3 |
| 35% | 104 ± 3 | 108 ± 3 | 300 ± 10 | 1150 ± 40 | 1810 ± 60 | 17.5 ± 0.5 | 16.8 ± 0.5 | 11.1 ± 0.3 |
| 40% | 107 ± 3 | 109 ± 3 | 310 ± 10 | 1180 ± 40 | 1890 ± 60 | 17.7 ± 0.5 | 17.4 ± 0.5 | 11.1 ± 0.3 |
Selectivities are included for CO2/CH4, CO2/N2, and H2/CH4 separations.
Figure 8.

(a) CO2/CH4 and (b) CO2/N2 upper-bound plots (black circle: experimental data for 6FDA–Durene; blue circles: experimental data for PSM–MOF MMMs; pink stars: Maxwell model prediction; red circles: pure MOF MMMs).
In Table 1, all uncertainties were calculated based on error propagation from individual samples.45 Propagation of error analysis yields instrumental uncertainties instead of statistical uncertainties. We anticipate that such instrumental uncertainties (e.g., transducer errors, film thickness measurement, etc.) would be consistent across multiple samples because the same transducers and micrometers were used each time. Additionally, sample-to-sample uncertainties were estimated by preparing and testing at least two films for each scenario, and the results can be found in SI Section B.
The incorporation of UiO-66-NH2 leads to faster permeation of all gas molecules compared to permeation in the neat polyimide due to the introduction of large permanent pores from the MMM filler. Such features provide lower mass transfer resistance to molecular diffusion and are therefore expected to increase the effective diffusion rates of all gases relative to diffusion rates in the pure polymer.10 This diffusion enhancement effect is more pronounced for smaller molecules.54 Thus, a larger diffusion selectivity can be expected for gas pairs involving gas molecules of very different kinetic diameters such as H2 and CH4. Comparing the 40 wt % PSM–MOF MMM to the pristine polyimide film, there is an enhancement of 44% in H2 permeability and 12% in H2/CH4 selectivity.
Moreover, the high internal surface area of UiO-66-NH2 is expected to favor the adsorption of polarizable gases such as CO2. Therefore, for CO2 separations, both diffusion and sorption contributions to transport are expected to bolster selectivity as long as the energetics of sorption do not hinder diffusion.42 Furthermore, the −NH2 linker increases the CO2 selectivity of MMMs through a dipole–quadrupole interaction that exists with CO2 but is not present with the other gases considered.43 With the presence of acetic acid being used as a modulator during the MOF synthesis, it is known that missing linker defects are possible,20 and such defects change the coordination environment in the MOF structure, potentially exposing unsaturated metal sites to the internal pore structure. While not quantified in this study, these sites can further increase CO2 adsorption, resulting in a higher CO2 selectivity over CH4 or N2 compared to defect-free UiO-66-NH2.44 Sorption isotherms of CO2 and CH4 for various types of films were collected, and the data can be found in Figure S9. It was shown that the incorporation of UiO-66-NH2 as fillers enhanced CO2 solubility, as well as CO2/CH4 sorption selectivity, and a larger increase was observed for higher MOF loadings.
In this study, the best-performing film for CO2/CH4 separation was the 40 wt % PSM–MOF–MMM, which enhanced the CO2 permeability and CO2/CH4 selectivity by 48 and 15%, respectively, at 1 bar and 35 °C. To prove that the change in transport performance is due to the modified MOF fillers rather than the oligomer itself, oligomers were dispersed directly into the polymers without being covalently attached to the MOF. Permeation tests show that the results are identical to those found for the pure polymers. This finding matches theoretical expectations, since 6FDA–Durene, which is formed through step polymerization, already contains significant amounts of short-chain oligomers within its as-synthesized polydispersity.34
3.3.3. Maxwell Model Prediction
The Maxwell model was used to predict permeabilities in the pure MOF, as well as composite membrane separation performance based on experimental data. This model was originally developed for dielectric systems and then applied to MMMs for gas separations.46 As a general guideline, the model is commonly used to predict permeability in the pure filler if defect-free MMMs can be formed with filler loadings that are characteristic of dilute mixtures without filler–filler interactions (i.e., filler loadings below approximately 20 vol %).47 In this study, since defect-free MMMs can be formed with MOFs containing polymer surface coatings that have high loadings up to approximately 40 wt %, it was hypothesized that the application of the Maxwell model could be extended to higher-loading regimes than are typically considered and would still provide accurate predictions of permeability.
To assess this hypothesis, experimental data at low loadings (below 20 wt %) were first used to calculate the pure MOF permeability for each gas. These values were then used together with the pure polymer permeability to predict MMM permeabilities at all loadings. Results are presented in Figure 8 for CO2/CH4 and CO2/N2 separations. Pure MOF permeability data calculated from this study are tabulated in Table 2. For CO2/CH4 separation, the Maxwell model provides an accurate prediction of gas permeabilities within the error bars of all experimental data points presented in Figure 8a. The predication of CO2/N2 performance shown in Figure 8b had a small deviation for loadings above 20 wt %, but the overall trend was similar to that of CO2/CH4. The exact origins behind this deviation are still unknown; however, a potential explanation could relate to nonidealities in the physical oligomer packing morphology created from oligomer–oligomer contact points between functional MOF particles at the point of particle percolation. In this loading regime, oligomer chain entanglement may be inefficient, leading to a lower-localized 6FDA–Durene density within the interfacial layer and, therefore, a corresponding increase in permeabilities and reduced selectivities from hindered chain packing. As noted by the deviation in the Maxwell model fit at higher loadings for CO2/N2 compared to those for CO2/CH4 separations, the subtle difference in the molecular size of CH4 and N2 may indicate that the length scale of this nonideal packing structure is on the same length scale as that of CH4 and N2 probe molecules. However, this hypothesis is highly speculative and requires additional spectroscopic analysis and characterization experiments for validation. We therefore refrain from further interpretation. The Maxwell model predictions of other gas pairs such as H2/CH4 and H2/O2 are shown in Figure S5 for reference.
Table 2. Tabulated Gas Permeabilities in the Pure MOF Calculated from Experimental Results of Pristine 6FDA–Durene and MMMs.
| gas | calculated permeability in pure UiO-66-NH2 (Barrer) |
|---|---|
| CO2 | 3515 |
| H2 | 3450 |
| O2 | 863 |
| N2 | 173 |
| CH4 | 161 |
Finally, a broader comparison with literature data on pure-gas CO2/CH4 separations for other PSM-UiO-66-NH2 MMMs is presented in Figure 9. Tabulated values of the permeability and selectivity for these references can be found in Table S1. In general, the most permeable samples are formed from highly permeable polymer matrices such as PIM-1. Jin and co-workers showed that by using partially −NH2-functionalized PIM-1 as the polymer matrix, CO2 permeability can be as high as 8126 Barrer at 30 wt % UiO-66-NH2 loading, and increased hydrogen bonding between NH2-PIM-1 and UiO-66-NH2 prevents a significant drop in selectivity: CO2/CH4 selectivity decreased from 24 to 18.3.26 Later, Kaliaguine et al. showed that by in situ polymerization of PIM-1 monomers and UiO-66-NH2, a standalone film could be formed from the reaction mixture, and the CO2 permeability reached 15 815 Barrer with a CO2/CH4 selectivity of 19.1.18 Even though the permeability from this approach is among the highest reported in the open literature for UiO-66-based MMMs, it may be challenging to control the stoichiometry needed to consistently synthesize a high-molecular-weight PIM-1 phase needed to form strong and ductile films.
Figure 9.
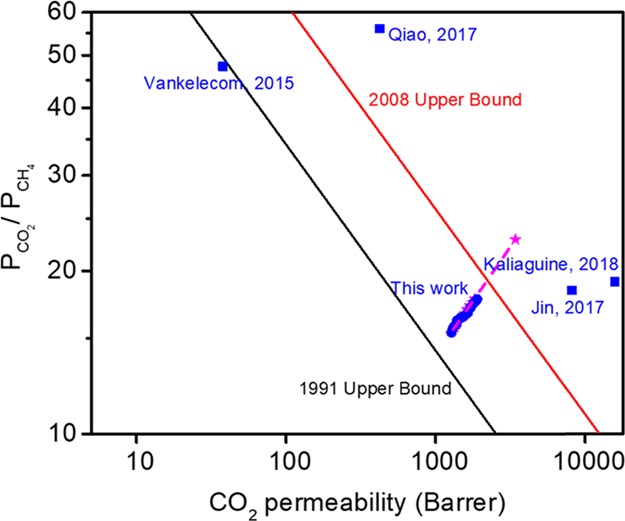
CO2/CH4 separation performance for UiO-66-NH2-based MMMs18,26,48,49 (blue circles: experimental data from this work; pink stars: Maxwell model prediction from this work; blue squares: data from the literature).
In addition to pure UiO-66-NH2 MOFs, transport data from a variety of pore-functionalized UiO-66-NH2 MOFs are also presented in Figure 9. By functionalizing the amine, smaller pore apertures and therefore improved selectivity can be accessed. Molecules reactive to amines, such as 4-aminobenzoic acid (ABA), have been used for this type of approach. Vankelecom and co-workers showed that fillers composed of ABA-functionalized UiO-66-NH2 in Matrimid resulted in a MMM with a CO2/CH4 selectivity of 47.7.48 Similar to the study considered here, Qiao and co-workers functionalized a UiO-66-NH2 surface with poly(ethylene glycol) methacrylate (PEGMA), and after dispersing the functionalized particles in Pebax 2533, CO2/N2 selectivity increased to 56 at a loading of 40 wt %.49
If trend lines were drawn for these four literature approaches, as well as for our data with increasing UiO-66-NH2 loading, they all should converge to a single data point representative of the property set of pure UiO-66-NH2 on the upper-bound plot. However, since only a single MOF loading was reported for each of these literature studies, a cross comparison to validate our pure MOF permeability calculations from the Maxwell model prediction could not be achieved. Indeed, more research at forming defect-free MMMs is required to assess the true theoretical limits in transport for a variety of MOF materials.
3.3.4. Pressure Effects on Permeability and Plasticization Behavior
A variety of challenges must be considered when deploying membranes for industrial gas separations, such as separation performance in the presence of plasticizing gases like CO2 at various pressures.50 Therefore, to better understand the stability of the MMMs considered in this study, pressure effects on pure-gas permeability were investigated, and results for the neat polyimide and 40 wt % PSM–MOF MMM are presented in Figure 10a,b, respectively. For gases other than CO2, permeation tests were conducted with pressures between 1 and 40 bar. To study the effect of CO2-induced plasticization, CO2 permeation tests were conducted with increasing pressures from 1 to 50 bar followed by depressurization from 50 to 1 bar. Finally, the same pressure ramp was tested again with increasing pressures from 1 to 50 bar. Permeabilities for all gases decreased initially with increasing pressure, consistent with a combination of dual-mode and Langmuir adsorption-type effects.51 However, permeability increased at higher pressures for CO2, which is consistent with CO2-induced plasticization.50
Figure 10.

Permeation at different pressures and 35 °C for (a) the neat polyimide and (b) the 40 wt % PSM–MOF MMM (solid circles: pressurization data; empty circles: depressurization data for CO2; solid squares: repressurization data for CO2).
To more carefully investigate the role of the PSM modification on plasticization stability, the CO2 pressure response is considered in greater detail in Figure 11. For the neat polyimide, a plasticization pressure point was observed at approximately 10 bar. This response, whereby permeability increases with increasing pressure, is indicative of sufficient CO2 sorption needed to swell the polymer matrix to increase free volume and therefore increase permeability.9 Despite the higher CO2 capacity of UiO-66-NH2 compared to that of 6FDA–Durene, the plasticization pressure response for the MMMs was significantly mitigated upon the addition of pure UiO-66-NH2 and PSM-UiO-66-NH2 fillers. For the pure MOF sample with 15 wt % filler, the plasticization pressure point did not significantly shift from that of the pure polymer. However, the increase in permeability for each successive data point after 10 bar was less than that observed for the pure polymer, indicating a slower rate of cooperative polymer chain relaxation for the polymer phase in the composite relative to that of the bulk polymer.52 The pure MOF particles have −NH2 surface functionalization, which forms hydrogen-bonding interactions with the −C=O and −NH2 groups in the 6FDA–Durene polymer.28 These secondary forces restrict polymer chain mobility, acting effectively as weak physical cross-links that can decrease the rate of polymer swelling and therefore endow MMMs with plasticization resistance.51 Similar phenomena have been observed by Shariff and co-workers when they found improved plasticization resistance in MMMs based on zeolite T and a 6FDA–Durene polyimide due to improved interfacial interactions.53
Figure 11.
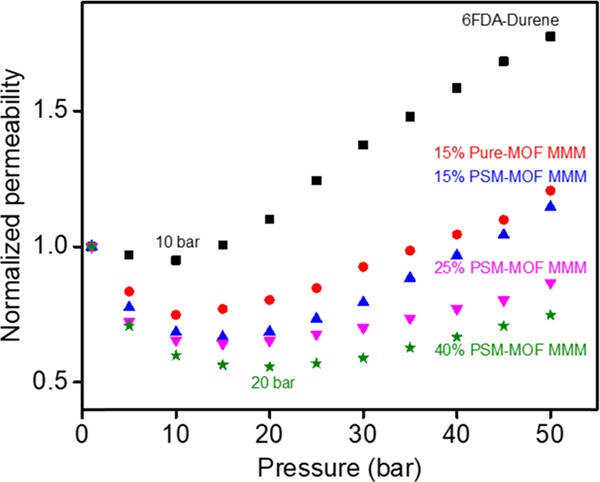
CO2 plasticization isotherms at 35 °C for the neat polyimide and MMMs with various types and loadings of fillers. Permeability is normalized to the data collected at approximately 1 bar. Plasticization pressure points of 10 and 20 bar are indicated for the neat polyimide and 40 wt % PSM–MOF MMM.
Interestingly, the surface functionalization had an effect on the plasticization response for the MMMs. With the PSM–MOF as the filler, higher plasticization pressures and significant reductions in the after-plasticization change in permeability were observed for the imide-functionalized MOF compared to that of the unfunctionalized MOF. For example, at 15 wt %, the PSM–MOF MMM had a plasticization pressure point of approximately 15 bar compared to approximately 10 bar for the pure MOF MMM. Additionally, the permeability change after the plasticization pressure was consistently lower for the PSM–MOF MMM compared to that of the pure MOF MMM. Considering that surface functionalization eliminates interfacial imide–amine hydrogen-bonding effects, the origins behind the increased stability of the polymer phase to CO2 for the PSM–MOF required further investigation. Therefore, DMA experiments were considered for MMMs formed with 15 wt % of pure UiO-66-NH2 and 15 wt % of PSM-UiO-66-NH2. The tan delta comparison is presented in Figure 12. The PSM–MOF MMM has a greater intensity for its tan delta response than the pure MOF MMM, which indicates that the PSM reaction induces stronger viscous contributions relative to elastic contributions for the mechanical response near the glass transition.54 As a result, polymer chains in PSM–MOF MMMs are expected to be less mobile, leading to improved resistance toward swelling caused by CO2 sorption. Such a result indicates that the combined contributions of weak intermolecular dispersion forces between the imide-functional surface of the MOF and the high-molecular-weight polymer matrix are greater than that of single hydrogen-bonding contact points characteristic for −C=O and −NH2. Therefore, a reduced plasticization response can be expected for MMMs formed from PSM–MOFs compared to those formed from pure MOFs. Shifts to higher plasticization pressure points and a reduced pressure dependence on permeability after the plasticization pressure point was monotonic with increasing PSM–MOF loading, as shown by the data points for the 25 and 40 wt %-loaded samples in Figure 11.
Figure 12.
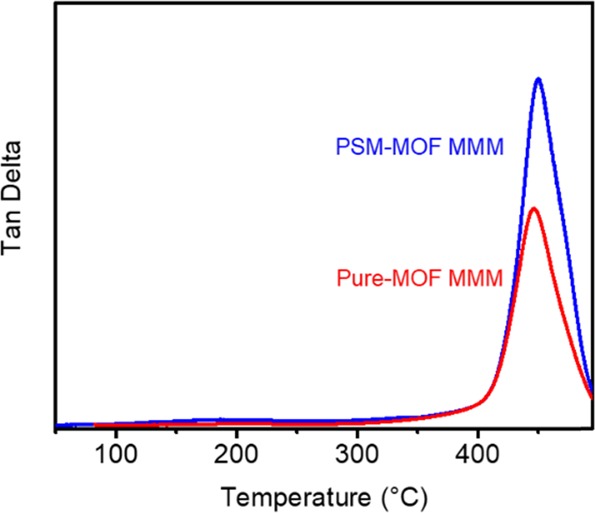
DMA analysis showing tan delta (viscous response/elastic response) as a function of temperature for the PSM–MOF MMM and Pure-MOF MMM. Both films have a 15 wt % MOF loading.
3.3.5. Mixed-Gas Permeation Tests and Physical Aging
To validate this technique’s potential in real industrial application such as natural gas upgrading, a 50:50 CO2/CH4 mixed-gas permeation test was conducted on the best-performing 40% PSM–MOF MMM, as well as the pure 6FDA–Durene film. Table 3 and Figure 13 summarize the result of this mixed-gas permeation test. In 50:50 CO2/CH4 mixed-gas permeation test, the selectivity of both films increased as compared to that of pure-gas data. When CO2 and CH4 are permeating together through the film, there is competitive sorption effect between CO2 and CH4. More sorbing sites inside the film were occupied by CO2 due to greater solubility, resulting in lower CH4 permeability. CO2 permeability decreased due to fewer available sorbing sites but the percentage decrease was smaller than that of CH4. A more direct comparison was made between mixed-gas results of 6FDA–Durene and 40% PSM–MOF MMM. Due to enhanced diffusion through MOF porosity and sorption interaction between CO2- and −NH2-functional groups, mixed-gas permeability and selectivity for the 40% PSM–MOF MMM increased by 36.8 and 13.5% at 1 bar partial pressure, respectively, as compared to pure 6FDA–Durene film. At a total pressure of 20 bar where the CO2 partial pressure was at 10 bar, the plasticization pressure of the 6FDA–Durene, a significant reduction in selectivity was observed for 6FDA–Durene film, whereas the 40% PSM–MOF MMM retained its high selectivity, proving that the addition of PSM–MOF as fillers increased the plasticization resistance of the MMM. At a partial pressure of 10 bar, sorbed CO2 molecules swell the polymer chains in pure 6FDA–Durene film, leading to larger free volume and allowing more CH4 to pass. Due to its larger size, CH4 was more significantly influenced due to this increase in free volume, and, as a result, perm-selectivity decreased to 12.9. When the partial pressure increased to 15 bar, more plasticization occurred with the 6FDA–Durene film and a large increase in CH4 permeability was observed, leading to a drastic decrease of CO2/CH4 selectivity to 8.1. In contrast, the 40% PSM–MOF MMM still retained its selectivity. The plasticization phenomenon is more directly reflected by the sudden change in trend between 10 and 15 bar for 6FDA–Durene in Figure 13.
Table 3. Tabulated Mixed-Gas Permeation Results for 0 and 40% PSM–MOF MMM at 35 °C.
| total pressure | 2 bar |
10 bar |
20 bar |
30 bar |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| permeability
(Barrer) |
selectivity | permeability (Barrer) |
selectivity | permeability (Barrer) |
selectivity | permeability (Barrer) |
selectivity | |||||
| loading (wt %) | CH4 | CO2 | CO2/CH4 | CH4 | CO2 | CO2/CH4 | CH4 | CO2 | CO2/CH4 | CO2 | CH4 | CO2/CH4 |
| 0% | 70 | 1180 | 17.0 | 63 | 990 | 15.7 | 63 | 810 | 12.9 | 870 | 107 | 8.1 |
| 40% | 84 | 1610 | 19.3 | 72 | 1380 | 19.1 | 61 | 1090 | 17.8 | 930 | 56 | 16.5 |
Figure 13.
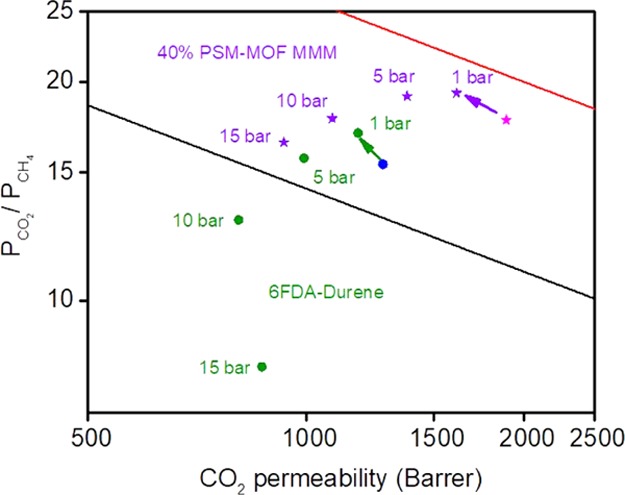
Mixed-gas test results on CO2/CH4 upper-bound plot (blue circle: pure-gas data for 6FDA–Durene; pink star: pure-gas data for 40% PSM–MOF MMM; purple stars: mixed-gas data for 40% PSM–MOF MMM; green circles: mixed-gas data for 6FDA–Durene).
Physical aging is another important consideration industrially. Polymer chains in a nonideal entangled structure after casting slowly rearrange into their ideal packing state over time.44 It was hypothesized that the insertion of PSM–MOF fillers would increase resistance to physical aging. A 1 month physical aging experiment was performed on 6FDA–Durene, 10% Pure-MOF MMM, and 10% PSM–MOF MMM by measuring pure-gas permeability at the start of each week for 5 consecutive weeks. As shown in Figure S10, the addition of MOF fillers suppressed physical aging as reflected by the normalized helium permeability over time. MOF insertion disrupts the packing structure of polymer chains and hinders the process of polymer chain rearrangement.18 For the PSM–MOF MMM, an extra interaction between MOF particles and the polymer matrix due to the oligomer coating causes the polymer chain to rigidify further at the point of contact between the two phases. This rigidification does not only resist plasticization, as shown above in Figure 12, but also resists physical aging, leading to a slower deterioration of separation performance of the film over time.
4. Conclusions
MMMs were formed by combining CO2-selective, imide-functionalized UiO-66-NH2 nanoparticles in a 6FDA–Durene polymer. UiO-66-NH2 nanoparticles with uniform size distributions and diameters of approximately 50 nm were first synthesized using an acetic acid modulator approach and then postsynthetically modified to be encapsulated by a thin layer of covalently bound 6FDA–Durene oligomers. After forming MMMs, both CO2 permeability and CO2/CH4 selectivity increased with increasing MOF loading, and the separation performance moved closer to the Robeson upper bound for CO2/CH4. The best separation performance was observed for a 40 wt % MOF loading with a CO2 permeability of 1890 Barrer and a CO2/CH4 selectivity of 18. Maxwell model predictions indicate that the pure UiO-66-NH2 MOF has a combination of permeability and selectivity beyond that of the upper bounds for CO2/CH4, H2/CH4, and H2/O2 separations. Additionally, the PSM–MOF MMMs reported in this study show improved resistance to CO2-induced swelling. This phenomenon relates to a localized reduction in polymer chain flexibility at the interface between the imide-functionalized MOF and the polyimide. The predicted permeabilities and selectivities for pure UiO-66-NH2 reported in this study permit the application of models, such as the Maxwell model, to predict the limit of performance for other MMMs formed from unique polymeric species, assuming that such MMMs can be formed with defect-free interfaces.
Acknowledgments
The authors gratefully acknowledge financial support from the MIT Tata Center for Technology and Design. The authors are also grateful for discussions and experimental guidance related to DMA experiments from Drs. David Hopkinson and Victor Kusuma at the National Energy Technology Laboratory (NETL) in Pittsburgh, PA. This material is based, in part, upon the work supported by the National Science Foundation Graduate Research Fellowship Program under grant no. 1122374 for A.W. and P.A. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the National Science Foundation.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsami.9b07500.
TEM images of PSM–MOF, pore distribution and N2 adsorption isotherms for pure MOF and PSM–MOF, 1H NMR on digested PSM-MOF particles, cross-sectional SEM images of pure MOF and PSM–MOF MMMs, H2/CH4 and H2/O2 upper-bound plots, tabulated CO2/CH4 separation data from the literature and upper-bound plot, DTG curves for pure MOF and PSM–MOF, tabulated data for reproducibility test, permeation results for pure MOF MMMs, CO2 and CH4 sorption isotherms for MMMs, physical aging plots of MMMs, and Maxwell model predication calculation can be found in the Supporting Information (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Galizia M.; Chi W. S.; Smith Z. P.; Merkel T. C.; Baker R. W.; Freeman B. D. 50th Anniversary Perspective: Polymers and Mixed Matrix Membranes for Gas and Vapor Separation: A Review and Prospective Opportunities. Macromolecules 2017, 50, 7809–7843. 10.1021/acs.macromol.7b01718. [DOI] [Google Scholar]
- Zornoza B.; Tellez C.; Coronas J.; Gascon J.; Kapteijn F. Metal Organic Framework Based Mixed Matrix Membranes: An Increasingly Important Field of Research with a Large Application Potential. Microporous Mesoporous Mater. 2013, 166, 67–78. 10.1016/j.micromeso.2012.03.012. [DOI] [Google Scholar]
- Koros W. J.; Zhang C. Materials for Next-Generation Molecularly Selective Synthetic Membranes. Nat. Mater. 2017, 16, 289–297. 10.1038/nmat4805. [DOI] [PubMed] [Google Scholar]
- Robeson L. M. Correlation of Separation Factor versus Permeability for Polymeric Membranes. J. Membr. Sci. 1991, 62, 165–185. 10.1016/0376-7388(91)80060-J. [DOI] [Google Scholar]
- Freeman B. D. Basis of Permeability/Selectivity Tradeoff Relations in Polymeric Gas Separation Membranes. Macromolecules 1999, 32, 375–380. 10.1021/ma9814548. [DOI] [Google Scholar]
- Robeson L. M. The Upper Bound Revisited. J. Membr. Sci. 2008, 320, 390–400. 10.1016/j.memsci.2008.04.030. [DOI] [Google Scholar]
- Stern S. A. Polymers for Gas Separations: the Next Decade. J. Membr. Sci. 1994, 94, 1–65. 10.1016/0376-7388(94)00141-3. [DOI] [Google Scholar]
- Thomas S.; Pinnau I.; Du N.; Guiver M. D. Pure- and Mixed-Gas Permeation Properties of a Microporous Spirobisindane-Based Ladder Polymer (PIM-1). J. Membr. Sci. 2009, 333, 125–131. 10.1016/j.memsci.2009.02.003. [DOI] [Google Scholar]
- Swaidan R. J.; Ma X.; Litwiller E.; Pinnau I. Enhanced Propylene/Propane Separation by Thermal Annealing of an Intrinsically Microporous Hydroxyl-Functionalized Polyimide Membrane. J. Membr. Sci. 2015, 495, 235–241. 10.1016/j.memsci.2015.08.015. [DOI] [Google Scholar]
- Dechnik J.; Gascon J.; Doonan C. J.; Janiak C.; Sumby C. J. Mixed-Matrix Membranes. Angew. Chem., Int. Ed. 2017, 56, 9292–9310. 10.1002/anie.201701109. [DOI] [PubMed] [Google Scholar]
- Seoane B.; Coronas J.; Gascon I.; Benavides M. E.; karvan O.; Caro J.; Kapteijn F.; Gascon J. Metal-Organic Framework Based Mixed Matrix Membranes: A Solution for Highly Efficient CO2 Capture. Chem. Soc. Rev. 2015, 44, 2421–2454. 10.1039/C4CS00437J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y.; Zhou H. C. Recent Progress in the Synthesis of Metal-Organic Frameworks. Sci. Technol. Adv. Mater. 2015, 16, 54202 10.1088/1468-6996/16/5/054202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y.; Ciston J.; Zheng B.; Miao X.; Czarnik C.; Pan Y.; Sougrat R.; Lai Z.; Hsiung C. E.; Yao K.; et al. Unravelling Surface and Interfacial Structures of a Metal-Organic Framework by Transmission Electron Microscopy. Nat. Mater. 2017, 16, 532–536. 10.1038/nmat4852. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Feng X.; Yuan S.; Zhou J.; Wang B. Challenges and Recent Advances in MOF-Polymer Composite Membranes for Gas Separation. Inorg. Chem. Front. 2016, 3, 896–909. 10.1039/C6QI00042H. [DOI] [Google Scholar]
- Cohen S. M. The Postsynthetic Renaissance in Porous Solids. J. Am. Chem. Soc. 2017, 139, 2855–2863. 10.1021/jacs.6b11259. [DOI] [PubMed] [Google Scholar]
- Sabetghadam A.; Seoane B.; Keskin D.; Duim N.; Rodenas T.; Shahid S.; Sorribas S.; Le Guillouzer C.; Clet G.; Tellez C.; et al. Metal Organic Framework Crystals in Mixed-Matrix Membranes: Impact of the Filler Morphology on the Gas Separation Performance. Adv. Funct. Mater. 2016, 26, 3154–3163. 10.1002/adfm.201505352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaraju D.; Bhagat D. G.; Banerjee R.; Kharul U. K. In Situ Growth of Metal-Organic Frameworks on a Porous Ultrafiltration Membrane for Gas Separation. J. Mater. Chem. A 2013, 1, 8828–8835. 10.1039/c3ta10438a. [DOI] [Google Scholar]
- Tien-Binh N.; Rodrigue D.; Kaliaguine S. In-Situ Cross Interface Linking of PIM-1 Polymer and UiO-66-NH2 for Outstanding Gas Separation and Physical Aging Control. J. Membr. Sci. 2018, 548, 429–438. 10.1016/j.memsci.2017.11.054. [DOI] [Google Scholar]
- Denny M. S.; Cohen S. M. In Situ Modification of Metal-Organic Frameworks in Mixed-Matrix Membranes. Angew. Chem., Int. Ed. 2015, 54, 9029–9032. 10.1002/anie.201504077. [DOI] [PubMed] [Google Scholar]
- Katz M. J.; Brown Z. J.; Colón Y. J.; Siu P. W.; Scheidt K. A.; Snurr R. Q.; Hupp J. T.; Farha O. K. A Facile Synthesis of UiO-66, UiO-67 and Their Derivatives. Chem. Commun. 2013, 49, 9449–9451. 10.1039/c3cc46105j. [DOI] [PubMed] [Google Scholar]
- Molavi H.; Shojaei A.; Mousavi A. Improving Mixed-Matrix Membrane Performance Via PMMA Grafting From Functionalized NH2-UiO-66. J. Mater. Chem. A. 2018, 6, 2775–2791. 10.1039/C7TA10480D. [DOI] [Google Scholar]
- Molavi H.; Eskandari A.; Shojaei A.; Mousavi S. A. Enhancing CO2/N2 Adsorption Selectivity via Post-Synthetic Modification of NH2-UiO-66(Zr). Microporous Mesoporous Mater. 2018, 257, 193–201. 10.1016/j.micromeso.2017.08.043. [DOI] [Google Scholar]
- Jiang Y.; Liu C.; Caro J.; Huang A. A New UiO-66-NH2 Based Mixed-matrix Membranes With High CO2/CH4 Separation Performance. Microporous Mesoporous Mater. 2019, 274, 203–211. 10.1016/j.micromeso.2018.08.003. [DOI] [Google Scholar]
- Venna S. R.; Lartey M.; Li T.; Spore A.; Kumar S.; Nulwala H. B.; Luebke D. R.; Rosi L.; Albenze E. Fabrication of MMMs with Improved Gas Separation Properties Using Externally-Functionalized MOF Particles. J. Mater. Chem. A 2015, 3, 5014–5022. 10.1039/C4TA05225K. [DOI] [Google Scholar]
- Ahmad M.; Navarro M.; Lhotka M.; Zornoza B.; Tellez C.; Vos W. M.; Benes N. E.; Konnertz N. M.; Visser T.; Semino R.; Maurin G.; Fila V.; Coronas J. Enhanced gas separation performance of 6FDA-DAM based mixed matrix membranes by incorporating MOF UiO-66 and its derivatives. J. Membr. Sci. 2018, 558, 64–77. 10.1016/j.memsci.2018.04.040. [DOI] [Google Scholar]
- Wang Z.; Ren H.; Zhang S.; Zhang F.; Jin J. Polymers of Intrinsic Microporosity/Metal–organic Framework Hybrid Membranes with Improved Interfacial Interaction for High-Performance CO2 Separation. J. Mater. Chem. A. 2017, 5, 10968–10977. 10.1039/C7TA01773A. [DOI] [Google Scholar]
- Khdhayyer M. R.; Esposito E.; Fuoco A.; Monteleone M.; Giorno L.; Jansen J. C.; Attfield M. P.; Budd P. M. Mixed Matrix Membranes Based on UiO-66 MOFs in the Polymer of Intrinsic Microporosity PIM-1. Sep. Purif. Technol. 2017, 173, 304–313. 10.1016/j.seppur.2016.09.036. [DOI] [Google Scholar]
- Xu Y. M.; Japip S.; Chung T. S. Mixed Matrix Membranes with Nano-Sized Functional UiO-66-Type MOFs Embedded in 6FDA-HAB/DABA Polyimide for Dehydration of C1-C3 Alcohols via Pervaporation. J. Membr. Sci. 2018, 549, 217–226. 10.1016/j.memsci.2017.12.001. [DOI] [Google Scholar]
- Lin W.; Vora R. H.; Chung T. Gas Transport Properties of 6FDA-Durene/1,4- Phenylenediamine (PPDA) Copolyimides. J. Appl. Polym. Sci. 2000, 38, 2703–2713. . [DOI] [Google Scholar]
- Katz M. J.; Brown Z. J.; Colón Y. J.; Siu P. W.; Scheidt K. A.; Snurr R. Q.; Hupp J. T.; Farha O. K. A Facile Synthesis of UiO-66, UiO-67 and Their Derivatives. Chem. Commun. 2013, 49, 9449–9451. 10.1039/c3cc46105j. [DOI] [PubMed] [Google Scholar]
- Smith Z. P.; Bachman J. E.; Li T.; Gludovatz B.; Kusuma V. A.; Xu T.; Hopkinson D. P.; Ritchie R. O.; Long J. R. Increasing M2(dobdc) Loading in Selective Mixed-Matrix Membranes: A Rubber Toughening Approach. Chem. Mater. 2018, 30, 1484–1495. 10.1021/acs.chemmater.7b02908. [DOI] [Google Scholar]
- Bouma R. H. B.; Checchetti A.; Chidichimo G.; Drioli E. Permeation through a Heterogeneous Membrane: The Effect of the Dispersed Phase. J. Membr. Sci. 1997, 128, 141–149. 10.1016/S0376-7388(96)00303-1. [DOI] [Google Scholar]
- Flory P. J.Principles of Polymer Chemistry; Cornell University Press: 1953. [Google Scholar]
- Odian G.Principles of Polymerization; John Wiley & Sons, 2004. [Google Scholar]
- Nellist P. D.; Chisholm M. F.; Dellby N.; Krivanek O. L.; Murfitt M. F.; Szilagyi Z. S.; Lupini A. R.; Borisevich A.; Sides W. H.; Pennycook S. J. Direct Sub-Angstrom Imaging of a Crystal Lattice. Science 2004, 305, 1741. 10.1126/science.1100965. [DOI] [PubMed] [Google Scholar]
- Nafisi V.; Hägg M. B. Gas Separation Properties of ZIF-8/6FDA-Durene Diamine Mixed Matrix Membrane. Sep. Purif. Technol. 2014, 128, 31–38. 10.1016/j.seppur.2014.03.006. [DOI] [Google Scholar]
- Jusoh N.; Yeong Y. F.; Lau K. K.; Shariff A. M. Transport Properties of Mixed Matrix Membranes Encompassing Zeolitic Imidazolate Framework 8 (ZIF-8) Nanofiller and 6FDA-Durene Polymer: Optimization of Process Variables for the Separation of CO2 from CH4. J. Cleaner Prod. 2017, 149, 80–95. 10.1016/j.jclepro.2017.02.069. [DOI] [Google Scholar]
- Friebe S.; Mundstock A.; Volgmann K.; Caro J. On the Better Understanding of the Surprisingly High Performance of Metal-Organic Framework-Based Mixed-Matrix Membranes Using the Example of UiO-66 and Matrimid. ACS Appl. Mater. Interfaces 2017, 9, 41553–41558. 10.1021/acsami.7b13037. [DOI] [PubMed] [Google Scholar]
- Kammakakam I.; Nam S. Y.; Kim T. H. Ionic Group-Mediated Crosslinked Polyimide Membranes for Enhanced CO2 Separation. RSC Adv. 2015, 5, 69907–69914. 10.1039/C5RA13081F. [DOI] [Google Scholar]
- Dong G.; Li H.; Chen V. Challenges and Opportunities for Mixed-Matrix Membranes for Gas Separation. J. Mater. Chem. A 2013, 1, 4610–4630. 10.1039/c3ta00927k. [DOI] [Google Scholar]
- Su N. C.; Sun D. T.; Beavers C. M.; Britt D. K.; Queen W. L.; Urban J. J. Enhanced Permeation Arising from Dual Transport Pathways in Hybrid Polymer-MOF Membranes. Energy Environ. Sci. 2016, 9, 922–931. 10.1039/C5EE02660A. [DOI] [Google Scholar]
- Jing X.; Zou D.; Cui P.; Ren H.; Zhu G. Facile Synthesis of Cost-Effective Porous Aromatic Materials with Enhanced Carbon Dioxide Uptake. J. Mater. Chem. A 2013, 1, 13926–13931. 10.1039/c3ta13115g. [DOI] [Google Scholar]
- Poloni R.; Lee K.; Berger R. F.; Smit B.; Neaton J. B. Understanding Trends in CO2 adsorption in Metal-Organic Frameworks with Open-Metal Sites. J. Phys. Chem. Lett. 2014, 5, 861–865. 10.1021/jz500202x. [DOI] [PubMed] [Google Scholar]
- Lin W. H.; Chung T. S. Gas Permeability, Diffusivity, Solubility, and Aging Characteristics of 6FDA-Durene Polyimide Membranes. J. Membr. Sci. 2001, 186, 183–193. 10.1016/S0376-7388(01)00333-7. [DOI] [Google Scholar]
- Bevington P. R.; Robinson D. K.. Data Reduction and Error Analysis: for the Physical Sciences; McGraw-Hill: New York, 1992 10.1063/1.4823194 [DOI] [Google Scholar]
- Vinh-Thang H.; Kaliaguine S. Predictive Models for Mixed-Matrix Membrane Performance: A Review. Chem. Rev. 2013, 113, 4980–5028. 10.1021/cr3003888. [DOI] [PubMed] [Google Scholar]
- Kammakakam I.; Nam S. Y.; Kim T. H. Ionic Group-Mediated Crosslinked Polyimide Membranes for Enhanced CO2 Separation. RSC Adv. 2015, 5, 69907–69914. 10.1039/C5RA13081F. [DOI] [Google Scholar]
- Anjum M. W.; Vermoortele F.; Khan A. L.; Bueken B.; De Vos D. E.; Vankelecom I. F. J. Modulated UiO-66-Based Mixed-Matrix Membranes for CO2 Separation. ACS Appl. Mater. Interfaces 2015, 7, 25193–25201. 10.1021/acsami.5b08964. [DOI] [PubMed] [Google Scholar]
- Xie K.; Fu Q.; Kim J.; Lu H.; He Y.; Zhao Q.; Scofield J.; Webley P. A.; Qiao G. G. Increasing Both Selectivity and Permeability of Mixed-Matrix Membranes: Sealing the External Surface of Porous MOF Nanoparticles. J. Membr. Sci. 2017, 535, 350–356. 10.1016/j.memsci.2017.04.022. [DOI] [Google Scholar]
- Wessling M.; Schoeman S.; van der Boomgaard T.; Smolders C. A. Plasticization of Gas Separation Membranes. Gas Sep. Purif. 1991, 5, 222–228. 10.1016/0950-4214(91)80028-4. [DOI] [Google Scholar]
- Kanehashi S.; Nagai K. Analysis of Dual-Mode Model Parameters for Gas Sorption in Glassy Polymers. J. Membr. Sci. 2005, 253, 117–138. 10.1016/j.memsci.2005.01.003. [DOI] [Google Scholar]
- Shahid S.; Nijmeijer K. Performance and Plasticization Behavior of Polymer-MOF Membranes for Gas Separation at Elevated Pressures. J. Membr. Sci. 2014, 470, 166–177. 10.1016/j.memsci.2014.07.034. [DOI] [Google Scholar]
- Jusoh N.; Yeong Y. F.; Lau K. K.; Shariff A. M. Transport Properties of Mixed Matrix Membranes Encompassing Zeolitic Imidazolate Framework 8 (ZIF-8) Nanofiller and 6FDA-Durene Polymer: Optimization of Process Variables for the Separation of CO2 from CH4. J. Cleaner Prod. 2017, 149, 80–95. 10.1016/j.jclepro.2017.02.069. [DOI] [Google Scholar]
- Budd P. M.; Elabas E. S.; Ghanem B. S.; Makhseed S.; McKeown N. B.; Msayib K. J.; Tattershall C. E.; Wang D. Solution-Processed, Organophilic Membrane Derived from a Polymer of Intrinsic Microporosity. Adv. Mater. 2004, 16, 456–459. 10.1002/adma.200306053. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.