

Abstract
This review will focus on the role of the tumor microenvironment (TME) in the development of drug resistance in melanoma. Resistance to mitogen-activated protein kinase inhibitors (MAPKi) in melanoma is observed months after treatment, a phenomenon that is often attributed to the incredible plasticity of melanoma cells but may also depend on the TME. The TME is unique in its cellular composition – it contains fibroblasts, immune cells, endothelial cells, adipocytes and amongst others. In addition, the TME provides “non-homeostatic” levels of oxygen, nutrients (hypoxia and metabolic stress) and extracellular matrix proteins, creating a pro-tumorigenic niche that drives resistance to MAPKi treatment. In this review, we will focus in how changes in the tumor microenvironment regulate MAPKi resistance.
Keywords: Drug Resistance, Melanoma, Hypoxia, Mechanotransduction, Oxidative stress, Immunology
1. Introduction
Melanoma is a malignancy of melanocytes. The transformation of melanocytes into melanoma requires a burden of mutations that can be initiated by exogenous and endogenous cues. Sporadic melanomas (approximately 90% of all melanoma cases) are frequently driven by low- to moderate-risk alleles that have high prevalence and low penetrance, indicating that environmental cues are key for malignant transformation (Chhabra et al., 2018; Eggermont, Spatz, & Robert, 2014; Hawryluk & Tsao, 2014; Schadendorf, Fisher, et al., 2015; Ward, Lazovich, & Hordinsky, 2012). Currently however, the pathway that has the highest oncogenic and therapeutic relevance in melanoma is the mitogen-activated protein kinase (MAPK) cascade, which is not attributable to direct UV damage (Hodis et al., 2012). The MAPK pathway is associated with cellular proliferation, differentiation, survival and mechanotransduction, and is activated by GTP bound Ras under normal conditions. Upon binding GTP, Ras begins a phosphorylation cascade, in which RAF, MEK, and ERK are consecutively activated, which ultimately results in the phosphorylation of cytoplasmic targets or transcription factors that alter gene expression (De Luca, Maiello, D’Alessio, Pergameno, & Normanno, 2012).
Recently, melanoma tumors were classified into four different types based on the pattern of the most prevalent genes mutated (all components of MAPK pathway): BRAF (detectable in ~52% of all melanomas), RAS (~28%), NF-1 (~14%) and Triple-WT (wild-type) (“Genomic Classification of Cutaneous Melanoma.,” 2015). Compounds targeting this pathway (i.e. BRAF and MEK inhibitors, denoted as BRAFi and MEKi, respectively) have been introduced to treat BRAF-mutated melanoma patients, which effectively lead to a regression of the tumor for few months. Unfortunately, tumor cells overcome MAPK and patients undergo relapse after a median of ~5–7 months, ultimately leading to patient’s death (Chapman et al., 2011; Gadiot, Hooijkaas, Deken, & Blank, 2013; Haferkamp et al., 2013; Hauschild et al., 2012; J. T. Lee et al., 2010; McArthur et al., 2014). Since then, many efforts have been undertaken to understand how melanomas resist therapy.
Resistance to MAPK blockade emerges from a combination of intrinsic and acquired resistance mechanisms. These include genetic alterations that reactivate MAPK signaling such as NRAS mutations (Nazarian et al., 2010), MEK mutations (Wagle et al., 2011) or mutant BRAF amplification (Shi et al., 2012). Resistant melanoma cells have upregulated levels of receptor tyrosine kinases (RTKs), such as epidermal growth factor receptor (EGFR), platelet derived growth factor receptor B (PDGFRB), insulin growth factor 1 receptor (IGF1R), activated TGFβ pathway, hyper phosphorylated ERK, amongst others (Nazarian et al., 2010; Sun et al., 2014; Villanueva et al., 2010). The ERK pathway interacts with other pathways, such as WNT/β-catenin, c-Jun N-terminal kinase (JNK), microphthalmia-associated transcription factor (MITF) and mechanistic target of rapamycin (mTOR), which may collaborate to maintain ERK activity under drug pressure. Such networks of signaling pathways are complex and stochastic in nature, and recent efforts in identifying key players are starting to emerge in the literature. JUN and a protein kinase C (PKC) isoform were recently identified as main drivers of BRAFi resistance (Titz et al., 2016), whereas p-21-activated kinase (PAK) was found to be pivotal in resistance to combinatory MEKi and BRAFi therapy (Zhang et al., 2017). These studies reveal important insights into the biology of melanoma, and cell-intrinsic mechanisms of therapy resistance. However, it is also important to consider the cell-extrinsic, or microenvironmental cues that govern therapy resistance. In this review we will focus on resistance to MAPK blockade driven fibroblast driven changes, both in the extracellular matrix (ECM) and in the oxidative makeup of the TME. We will then examine how changes in the immune microenvironment may also affect targeted therapy. Overall, this review is designed to draw attention to the role that the tumor microenvironment plays in driving therapy resistance.
2. The Stromal Microenvironment in Resistance to MAPK Blockade.
Melanomas are highly heterogenous and comprise a vast number of cancer-associated cells of different origins. Within the TME, melanoma cells interact with surrounding cells through cell-cell contact, adhesion molecules, as well as secreted molecules such as growth factors, cytokines, chemokines, ECM proteins, protease inhibitors and lipids (Piérard, Piérard-Franchimont, & Delvenne, 2012; Ruiter, Bogenrieder, Elder, & Herlyn, 2002). These complex interactions are established between different cell types, including fibroblasts, adipocytes, endothelial and immune cells, which potentially regulate the capacity of tumors to overcome MAPK blockade. In addition, these interactions often spur changes in more global alterations such as changes in oxidative stress, including ROS and hypoxia.
2.1. Fibroblasts as orchestrators of MAPKi Resistance.
Of the multiple cell types encountered by the tumor cell in its microenvironment, fibroblasts are one of the most studied cancer-associated cell types. From the early stages of tumorigenesis, CAFs are observed in the tumor microenvironment, and distinguish themselves from normal skin fibroblasts by their upregulated expression of α-smooth-muscle actin (SMA), fibroblast-activation protein-1 (FAP1), PDGFRs, TGFβ, Vimentin and other proteins. CAFs do not only support tumor growth and metastases (Barcellos-Hoff & Ravani, 2000; Krtolica, Parrinello, Lockett, Desprez, & Campisi, 2001; Ohuchida et al., 2004), they are also implicated in therapy resistance. To date, several groups have shown that fibroblasts protect melanoma cells against MAPK. Upon BRAFi, CAFs secrete factors that contribute to melanoma cell survival and resistance, such as HGF (Straussman et al., 2012) and NRG1 (Capparelli, Rosenbaum, Berger, & Aplin, 2015). Aged fibroblasts, which have CAF-like properties, also protect melanoma cells from BRAFi via secretion of sFRP2 (Kaur et al, 2016). Other secreted proteins include those involved the modeling of the extracellular matrix (Fedorenko et al., 2016; Fedorenko, Wargo, Flaherty, Messina, & Smalley, 2015).
Changes in matrix stiffness, such as loss of pliability, affect the metastatic properties of tumor cells. This occurs not only by providing optimal contractile forces for the migration of tumor cells, but also by affecting signaling (mechanotransduction), which can alter growth and even responses to drugs. Work from the Weaver laboratory and others has shown that in the breast cancer setting, increasing stiffness of the ECM can drive increased metastasis and resistance to chemotherapy through different processes as reviewed elsewhere (Kaushik, Pickup, & Weaver, 2016). Surprisingly, and in contrast to existing studies in other cancers, recent modeling experiments from our own laboratory suggest that this may be different in melanoma. We identified a non-linear relationship between collagen stiffness and invasion, whereby extremely loose and extremely stiff collagen both restrict invasion of melanoma cells in vitro, whereas collagen of an intermediate stiffness provides the optimal conditions for facilitating invasion (Ahmadzadeh et al., 2017). Additional published data supports these observations in various cancers (Goetz et al., 2011; Kwon, Cukierman, & Godwin, 2011; H.-O. Lee et al., 2011). These data suggest that breast cancer cells may start out in a softer matrix, and need a stiffer matrix to invade, where melanoma cells start out in a stiffer matrix and need to soften it to invade. We hypothesize that this may mirror the biological need for plasticity in the breast (lactation, hormonal changes) that likely requires a soft matrix and the need for a firm protective barrier in the skin, that requires a tightly cross-linked matrix.
Understanding the contribution of matrix stiffness to tumor cell progression is important, because ECM alignment associated with assorted cancers effectively predicts patient outcomes and metastasis 16. Melanoma cells in varying collagen concentrations undergo a morphology change reminiscent of an epithelial to mesenchymal transition (EMT), which has recently been linked to chemoresistance (Fischer et al., 2015; Zheng et al., 2015). Indeed, several studies have shown that CAFs are highly involved in EMT: they can deposit pro-metastatic ECM; they can generate the leading edge for EMT melanoma cells; and they release pro-invasive factors that encourage EMT. Growing evidence shows that BRAFi causes a biomechanical adaptation of the tumor niche. In melanoma cells, the blockade of MAPK pathway using BRAFi alone leads to F-actin remodeling via te mechanosensing pathway YAP/TAZ, therefore resulting in more aggressive melanoma cells (Kim et al., 2016). The upregulation of this signaling pathway may be attributed to the increase of ECM proteins secreted by melanoma cells alone. In fact, BRAFi treatment leads to the overexpression of ECM proteins in melanoma cells, such as COL1A1 and FN1 (Titz et al., 2016), which can positively feedback the recruitment of FA and remodeling of F-actin. Furthermore, Hirata and co-workers have found that the biophysical microenvironment created by CAFs is essential for melanoma cell survival in the presence of BRAFi. The authors showed that melanoma survival is in fact promoted by the activation of focal adhesion kinase (FAK) and Src by the CAF-dependent microenvironment, a response that can be reversed by embedding melanoma cells alone in stiff hydrogels composed by a mixture of fibronectin and collagen type I (Hirata et al., 2015). Nevertheless, paracrine factors released by melanoma cells stimulate the differentiation of fibroblasts, leading to increased expression of ECM molecules (Fedorenko et al., 2016, 2015). It appears, therefore, that melanoma cells and fibroblasts enroll in a collaborative effort to form a biomechanical tumor niche that is rich in ECM proteins under drug pressure, which further accelerates acquired resistance. Understanding how these biomechanical changes drive resistance to targeted therapy will be critical in deconstructing the contributions of the TME to therapy resistance, and this is schematically mapped in Figure 1.
Figure 1 – Schematic of the biomechanical adaptation of the TME to BRAFi as consequence of complex melanoma-stroma cross-talk.
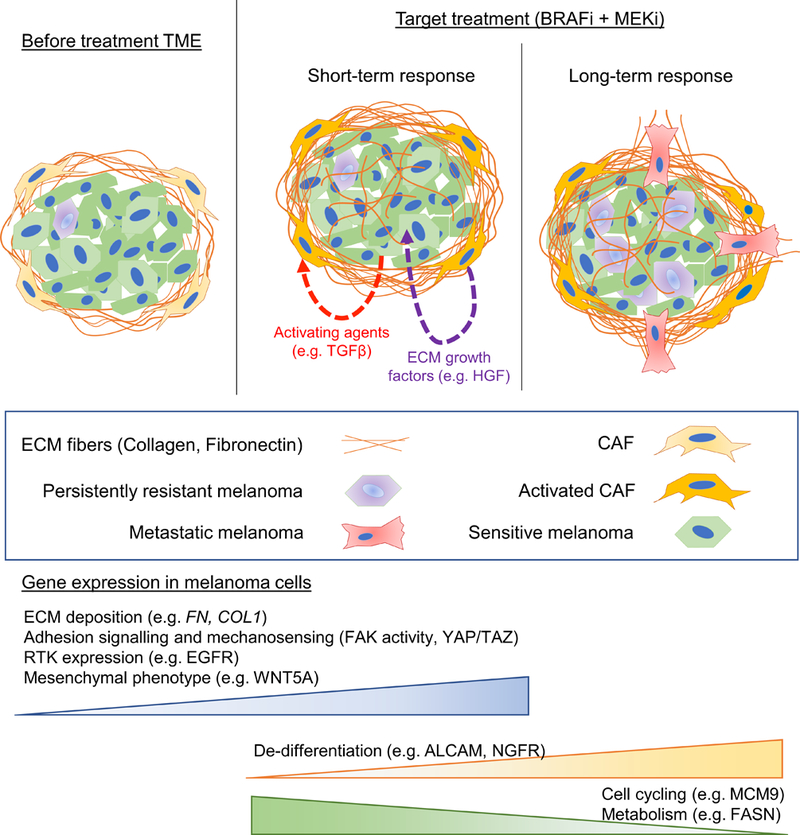
Cancer-associated fibroblasts (CAF) create an ECM-rich TME. Upon BRAFi, melanoma cells release CAF-activating factors and ECM proteins, whereas CAFs release ECM growth factors that confer melanoma cells resistance in the long-term. A phenotype shift in melanoma cells is observed: melanoma cells that resist therapy can become de-differentiated and less proliferative, which ultimately results in metastasis.
2.2. Oxidative stress in drug resistance
In addition to driving biomechanical changes, fibroblasts can also contribute to changes in the oxidative microenvironment of a tumor. We have previously shown that aged fibroblasts lose the expression of anti-oxidants such as SOD3, and peroxiredoxin, resulting in an increase in ROS in the microenvironment of the tumor, creating a genetic instability in the tumor cell that drives tumor metastasis. However, the role of ROS in melanoma progression is quite confusing. In keeping with the data above, it has been shown that reactive oxygen species (ROS) levels that drive oxidative stress are often high in many primary tumors, particularly in melanoma given the burden placed upon the skin by UV radiation, and these increased levels are associated with increased neoplastic transformation and tumor progression (Chandel & Tuveson, 2014; Gao et al., 2007; Gorrini, Harris, & Mak, 2013; Liu-Smith, Dellinger, & Meyskens Jr., 2014; Rinnerthaler, Bischof, Streubel, Trost, & Richter, 2015)(Chandel & Tuveson, 2014; Gao et al., 2007; Gorrini et al., 2013; Liu-Smith et al., 2014; Rinnerthaler et al., 2015). But other studies show that increased oxidative stress placed on disseminated melanoma cells and other tumors has been shown to inhibit distant metastasis and progression (Herraiz et al., 2016; Peiris-Pages, Martinez-Outschoorn, Sotgia, & Lisanti, 2015; Piskounova et al., 2015). Whether this is a reflection of the differing microenvironments in immunocompetent vs incompetent mice is still a subject of debate. Indeed, the inflammatory response initiated by immune cells within primary tumors also significantly increases ROS, suggesting that innate differences involving the immune microenvironment exist.
The balance between ROS, and the anti-oxidants that modulate it may play a key role in understanding the discrepancies in the literature. One hypothesis is that it is important for melanoma cells to adapt to allow survival (Piskounova et al., 2015; Rodrigues et al., 2016). As such, many metastatic populations following dissemination increase production of antioxidants and increased dependence on NADPH-generating enzymes in the folate pathway to help counteract oxidative stress (Piskounova et al., 2015). The expression and activity of antioxidant enzyme catalases such as Mn-SOD2, Zn-SOD1, and the ROS scavenger GSH is much higher when compared with other skin tumors (Wittgen & van Kempen, 2007). Interestingly, the increased resistance of melanoma cells to ROS through these enhanced pathways is not seen in melanocytes, suggesting that the acquisition of an increased antioxidant network is required for primary tumor development (Rinnerthaler et al., 2015; Wittgen & van Kempen, 2007). Metastatic melanoma cells also have increased Ferritin expression, a ferroxidase important in reducing oxidative stress, when compared with primary melanomas, suggesting that further antioxidant pathways are required to be upregulated to allow dissemination and metastasis (Baldi et al., 2005). Intriguingly however, as mentioned above, anti-oxidants in the microenvironment seem to play opposing roles both in metastasis as described above, and in therapy resistance as described below.
ROS levels have been associated with BRAFi resistance (Corazao-Rozas et al., 2013; Kaur et al., 2016; Yu et al., 2014). Studies have shown that melanoma cells exposed to an aged microenvironment are exposed to increased ROS, largely due to the fact that aged fibroblasts lose expression of superoxide dismutase-3 (SOD3), a key anti-oxidant. Melanoma cells in a young microenvironment also express b-catenin, which activates the base excision repair enzyme, apurinic/apyrimidinic endonuclease APE1 (Kaur et al., 2016). APE1 is critical for modulating DNA damage downstream of ROS. Aged fibroblasts secrete the frizzled related protein 2 (sFRP2) which signals to inhibit beta-catenin, and subsequently APE1, rendering the melanoma cells more susceptible to ROS. This increase in ROS is related to an increase in the resistance of melanoma cells to BRAF inhibition. Interestingly, when treated with anti-oxidants, resistant melanoma cells in the aged microenvironment were more sensitive to BRAFi (Kaur et al., 2016). This is also true in studies of BRAF inhibition in colon cancer, where Vitamin C could be used to target and kill BRAF and KRAS mutant colorectal cancer (Yun et al., 2015). The administration of anti-oxidants have, however, failed to prevent tumorigenesis in clinical trials (Fortmann et al., 2013); in fact, in lung and prostate cancer, anti-oxidants increased the incidence of metastasis and deaths (Goodman et al., 2004; Klein et al., 2011; The Alpha-Tocopherol Beta Carotene Cancer Prevention Study Group, 1994). How this reflects the balance between the ROS generated in the microenvironment, vs that within the tumor cell is a subject for further investigation.
2.3. Hypoxia in drug resistance
In melanoma, hypoxia is a key feature associated with the microenvironment and is a well-known driver of heterogeneity (Fluegen et al., 2017; O’Connell et al., 2013; O’Connell & Weeraratna, 2013; Pucciarelli et al., 2016; Widmer et al., 2013). As the tumor continues to develop, the formation of non-functional blood vessels throughout leads to regions of mild (hypoxia) to severe (anoxia) oxygen deprivation, which induces the epigenetic reprogramming of cells to drive adaptation and survival, and is of clinical significance given the strong correlation of hypoxia with poor patient prognosis, increased tumor resistance and metastasis (Eales, Hollinshead, & Tennant, 2016; Wigerup, Pahlman, & Bexell, 2016). To date, the hypoxia inducible factor (HIF) family of transcription factors are central mediators of the adaptive response and are heavily associated with chemo- and radiotherapy resistance, poor patient prognosis and patient relapse and are considered potential molecular targets for the treatment of metastasis (Mills, Joshi, & Niles, 2009; Wigerup et al., 2016).
Many other pathways both upstream and downstream of hypoxia-induced factor (HIF) in melanoma tumors are now being recognized to upregulate the expression of drug resistant and survival genes in response to hypoxia. HIF is a well-known suppressor of MITF (Cheli et al., 2012; Feige et al., 2011), which drives an MITFlow, slow cycling cell state that has been well characterized to have increased therapeutic resistance to a large number of therapeutics (Carreira et al., 2006; Hartman & Czyz, 2015; Hoek & Goding, 2010; Wellbrock & Arozarena, 2015). Cytotoxicity associated with chemo- and radiotherapy is much greater in proliferating populations, thus giving slow-cycling cells a specific survival advantage against more general treatments. MITFlow populations are also characterized to be intrinsically resistant to more specific therapies, particularly those defined by high AXL expression (Fane et al., 2017; Konieczkowski et al., 2014; Muller et al., 2014). Hypoxia also plays a prevalent role in the phenotype switching model of melanoma progression, as it has been shown to act as a central mediator of a HIF-1α dependent switch from an ROR1 positive proliferative cell state to an ROR2 positive invasive cell state (O’Connell et al., 2013). The expression of the ROR2 receptor induces a 10-fold decrease in sensitivity to BRAF inhibition, with this population being induced via the upregulation of WNT5a, which stabilizes HIF-1α levels during hypoxic conditions via increased SIAH2 expression (O’Connell et al., 2013). Recent studies using 3D melanoma spheroid models have also found that hypoxia-driven upregulation of HGF/MET plays an important role in vemurafenib resistance and is prevalent in drug resistant melanoma patients and xenograft models (Qin et al., 2016). Together, the various studies strongly suggest that resistance to targeted therapy in melanoma can be driven by hypoxia. Whether hypoxia impacts fibroblasts such that they produce more factors that drive resistance has not yet been explored.
2. The Immune Microenvironment and Resistance to MAPK Blockade.
In addition to targeted therapy against the MAPK signaling pathway, the other standard of care therapy for melanoma is immunotherapy. Immune checkpoints are pathways that are critical for self-tolerance. During an immune response, the activation of T-cells is a well-defined multistage process designed to prevent the primed T-cell from attacking normal-self tissues. The initial stage of neoantigen processing and presentation by MHC complexes on APCs to the T-cell receptor is a key step in the immune attack response, however, the subsequent binding of the activating co-receptor, CD28, to CD80/CD86 molecules is an equally crucial step for full activation. T-cells also possess inhibitory signals in the form of CTLA-4 (found on regulatory T-cells and activated T-cells) and PD-1 that will bind in place of CD28 preventing T-cell activation, among others. These inhibitory signals are termed checkpoints, and inhibiting these checkpoints is a key angle of immunotherapy. Immunotherapies targeted against these checkpoints (namely PD1 and CTLA4) are now FDA-approved for the treatment of fifteen forms of malignancy (Sharma & Allison, 2015), including tumors that have been previously classed to be insensitive to other immunotherapies (Zou, Wolchok, & Chen, 2016). Immunotherapies like these have similar clinical response rate compared to MAPKi such as vemurafenib with an added long-term success which ensures a more durable response. In addition, the delivery of chimeric antigen receptors (CARs) (Sadelain, 2016) and adoptive cell-transfer (ACT) (Sadelain, 2016) are additional immunotherapies that now offer patients with difficult to treat, advanced metastatic melanoma a significantly improved prognosis. However, for the purpose of this review, we will focus on the interaction between the immune microenvironment and the tumor in the context of targeted therapy efficacy.
3.1. The Immune Tumor Microenvironment.
CD4+ and CD8+ T-cells infiltrating the TME have long been associated with strong anti-tumor effects and a favorable clinical outcome (Clark, 1991; Hadrup, Donia, & thor Straten, 2013). However, the effectiveness of these cells relies on their fundamental ability to recognize neoantigens expressed on tumor cells and their ability to become activated, despite a suppressive microenvironment. This balance is maintained by a balance across a wide range of immune cells including T-cells, regulatory T-cells (Tregs), Myeloid-derived suppressor cells (MDSCs), dendritic cells (DCs) and tumor-associated macrophages (TAMs), as well as secreted cytokines such as TGF-β, IFN-γ, and TNF-α. Tregs are well-described mediators of peripheral self-tolerance. These unique lymphocytes suppress the effects of T-cells recruited to the tumor through mechanisms including cell-cell contact and the secretion of IL-10, IL-35 and TGF-β (Sakaguchi, Yamaguchi, Nomura, & Ono, 2018; Sojka, Huang, & Fowell, 2008). MDSCs, a heterogenous group of immature myeloid cells activated in the presence of inflammatory cytokines are also contributors to immune suppression. Both cell types are reported in high frequencies within numerous malignancies and correlate with a worse patient prognosis and a reduced sensitivity to immunotherapies (Meyer et al., 2014; Mohos et al., 2013).
Macrophages also play an indispensable role in innate and adaptive immunity. They are activated by a number of stimuli which allows them to differentiate into multiple subtypes displaying various phenotypes which are dependent on the microenvironment (Gordon & Taylor, 2005; Solinas, Germano, Mantovani, & Allavena, 2009). Two main states of polarized activation for macrophages have been described: the classically activated M1 macrophage and the alternatively activated M2 macrophage (Mantovani, Sica, & Locati, 2005). Mainly because of their opposing cytokine profiles, the phenotypes of the polarized macrophages are reported to have differing roles within the cancer environment and tumor growth. Classically activated M1 macrophages express a number of pro-inflammatory cytokines, allowing them to act in an anti-tumor manner (Solinas et al., 2009), whereas, the majority of TAMs exhibit M2 phenotypes which express a variety of anti-inflammatory molecules that are over expressed within suppressed TME and drug resistant tumors (Solinas et al., 2009). One such molecule is the CSF1 receptor, expressed on TAMs, that responds to CSF secreted by the tumor. Therapies targeted against CSF1R have failed, and recent data implicate the role of cancer-associated fibroblasts (CAFs) in this failure. This is because blocking CSF1/CSF1R signaling induces the migration of CAFs to the tumor site, where they then secrete a wide variety of chemokines that attract PMN-MDSCs, creating an immune-suppressive microenvironment (Kumar et al., 2017). This study demonstrates how the stromal microenvironment can affect immune components of the tumor microenvironment to drive therapy failure.
3.2. MAPKi and the Immune Response
Reports now demonstrate that melanoma cells carrying mutant BRAFV600E generate their own immune-escape mechanisms. These mechanisms involve increased levels of pro-inflammatory cytokines such as IL-6, IL-10 and VEGF that drive the recruitment of Tregs and MDSCs into the TME (Sumimoto, Imabayashi, Iwata, & Kawakami, 2006). Moreover, BRAF mutant cells inhibited the maturation of DCs (Kumar, Patel, Tcyganov, & Gabrilovich, 2016) and their production of TNF-α and IL-12 (Sumimoto et al., 2006). The ability to present antigen through MHC class I molecules on melanoma cells is also abrogated in mutant BRAF cell lines (Sapkota, Hill, & Pollack, 2013) thus resulting in decreased CD8+ T-cell primed attacks. While MAPK blockade is not designed to specifically increase tumor immunogenicity, increasing numbers of reports suggest that this may indeed be an effect of MAPKi. Tumor associated macrophages (TAMs) and CAFs are the major source of IL-1 in the TME (Young et al., 2017), and IL-1is also secreted from BRAFV600E mutant cells (Khalili et al., 2012). IL-1 further stimulated CAFs to up-regulate COX-2, PD-1 ligands, GROα and IL8 (Khalili et al., 2012; Young et al., 2017). Early treatments with BRAFi such as vemurafenib and dabrafenib have also been demonstrated to increase CD4+ and CD8+ T-cells (Hong et al., 2012; Wilmott et al., 2012), reduce the levels of MDSCs (Schilling et al., 2013) and Tregs and restore the maturation of DCs and levels of TNF-α and IL-12 (Sumimoto et al., 2006).
The recruitment of immunosuppressive cells is influenced through the production of chemokines by the tumor including CCL5 (Schlecker et al., 2012), CCL7, CXCL8 and CXCL12 (Highfill et al., 2014; Kumar et al., 2016) and CCL2 (Kudo-Saito, Shirako, Ohike, Tsukamoto, & Kawakami, 2013). The cognate receptors for CCL2, CCR2 and CCR4 are known to be expressed on MDSCs and Tregs. Treatment of vemurafenib in mouse models was able to reduce the production of CCL2 by melanoma cells and inhibit growth through a change in ratio of effector T-cells to immunosuppressive cells in the TME (Knight et al., 2013). Another study showed similar findings using anti-CCR4 treatment which inhibited Treg recruitment to the TME and promoted anti-tumor T-cell responses through ADCC (Sugiyama et al., 2013). Reports have now demonstrated that depleting these suppressive cells from entering the TME results in tumor immunogenicity, and the use of monoclonal antibodies to deplete Tregs have had success within murine studies (Arce Vargas et al., 2017; Viehl et al., 2006). However, as these cells are very important in self-tolerance and normally patrol the blood of healthy individuals, the targeting of intratumoral Tregs remains a difficult obstacle and often results in high level toxicities with increased risk of auto-immunity. Melanoma tumors harboring BRAF mutations are associated with a 2-fold increase in intratumoral Tregs, but is not considered a predictive factor for the success of BRAFi (Leslie et al., 2015).
As described, constitutive MAPK activation works to subdue tumor immunogenicity, whereas, MAPKi can override some of these factors, restoring the balance by reducing TME suppression and also mounting an immune response against the tumor. Early data from clinical trials reported that although immunotherapies have a lower success rate, they do achieve a more durable response, however, MAPKi have a far higher response rate but also a significantly reduced long term success (Hodi et al., 2010; Plimack et al., 2018; Schadendorf, Hodi, et al., 2015). To this end, it was originally thought that combinations consisting of MAPKi with immune checkpoint therapies may work synergistically. However, following a phase 1 clinical trial, the hepatoxicity witnessed was excessive and the study was discontinued (Ribas, Hodi, Callahan, Konto, & Wolchok, 2013). Immunotherapy combinations have achieved far greater success however. More recently, the use of nivolumab and ipilimumab as a combination has been shown to be more effective than monotherapy. In fact, this combination has an equivalent success rate to MAPKi, being reported at 3 years a 58% survival (Wolchok et al., 2017). Although toxicities are high, they provided acceptable safety profiles leading to FDA-approval for stage IV melanoma (Larkin et al., 2015), and unlike MAPK inhibitors, there is durable response in a subset of patients.
3. Concluding remarks and perspective
The TME is a key modulator of the ability of a melanoma cell to overcome MAPK blockade. It has been proposed that MAPK re-activation involves complex interactions of MAPK pathway with non-canonical WNT pathway, TGFβ signaling, PI3K/AKT pathway or the adenyl cyclase/cAMP/PKA pathway. As discussed above, the melanoma-CAF crosstalk may be central in orchestrating drug resistance. CAFs not only secrete factors that help activate these signaling pathways, but they are also responsible for the abnormal deposition of extracellular proteins (e.g. collagen and fibronectin) that can increase therapy resistance. It has been proposed that this enhanced deposition of ECM proteins leads to the increase of melanoma adhesive sites through focal adhesion and remodeling of cytoskeleton, which in turn results in an invasive and resistant phenotype. In addition, melanoma cells secrete ECM proteins, which amplify the activation of focal adhesion. However, the molecular mechanisms that drive cytoskeletal remodeling, and how they result in resistance to MAPK inhibitors, remain poorly understood.
ECM composition and structure change dramatically with age. After 65 years of age, the dermal ECM exhibits decreased fiber area and thickness, which results in impaired mechanical properties (Diridollou et al., 2001; H.-O. Lee et al., 2011; Marcos-Garces et al., 2014; Oh et al., 2011; Panwar et al., 2015). We have recently identified age as a key promoter of melanoma metastasis and resistance to MAPKi (Kaur et al., 2016). Although not yet fully demonstrated, it is possible that with age, dermal fibroblasts have enhanced CAF-like phenotype, which may further contribute to resistance. Other resident cells in skin also intervene in tumorigenesis, and potentially resistance to targeted therapy. Adipocytes and keratinocytes, for instance, were identified to interact with melanoma cells even at a distant, driving their invasiveness and promote resistance (Fattore et al., 2016; Milhas et al., 2016; Romano & Kwong, 2017). Further studies investigating the crosstalk between melanoma and these cancer-associated cells are necessary for the development of efficient drugs for the treatment of advanced melanoma. Nevertheless, future research in cancer should characterize the stochastic nature of the oncogenic-signaling pathways and their cross-talk at molecular level. This requires multi-disciplinary research across disciplines such as Medicine, Biology, Engineering, Mathematics, Chemistry and Physics.
Footnotes
CONFLICT OF INTEREST STATEMENT: The Authors declare no conflict of interest.
References
- Ahmadzadeh H, Webster MR, Behera R, Jimenez Valencia AM, Wirtz D, Weeraratna AT, & Shenoy VB (2017). Modeling the two-way feedback between contractility and matrix realignment reveals a nonlinear mode of cancer cell invasion. Proceedings of the National Academy of Sciences, 201617037. 10.1073/pnas.1617037114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arce Vargas F, Furness AJS, Solomon I, Joshi K, Mekkaoui L, Lesko MH, … Quezada SA (2017). Fc-Optimized Anti-CD25 Depletes Tumor-Infiltrating Regulatory T Cells and Synergizes with PD-1 Blockade to Eradicate Established Tumors. Immunity, 46(4), 577–586. 10.1016/j.immuni.2017.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldi A, Lombardi D, Russo P, Palescandolo E, De Luca A, Santini D, … Paggi MG (2005). Ferritin contributes to melanoma progression by modulating cell growth and sensitivity to oxidative stress. Clin Cancer Res, 11(9), 3175–3183. 10.1158/1078-0432.CCR-04-0631 [DOI] [PubMed] [Google Scholar]
- Barcellos-Hoff MH, & Ravani SA (2000). Irradiated mammary gland stroma promotes the expression of tumorigenic potential by unirradiated epithelial cells. Cancer Research, 60(5), 1254–1260. [PubMed] [Google Scholar]
- Capparelli C, Rosenbaum S, Berger AC, & Aplin AE (2015). Fibroblast-derived Neuregulin 1 Promotes Compensatory ErbB3 Receptor Signaling in Mutant BRAF Melanoma. The Journal of Biological Chemistry, 290(40), 24267–24277. 10.1074/jbc.M115.657270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreira S, Goodall J, Denat L, Rodriguez M, Nuciforo P, Hoek KS, … Goding CR (2006). Mitf regulation of Dia1 controls melanoma proliferation and invasiveness. Genes Dev, 20(24), 3426–3439. 10.1101/gad.406406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandel NS, & Tuveson DA (2014). The promise and perils of antioxidants for cancer patients. N Engl J Med, 371(2), 177–178. 10.1056/NEJMcibr1405701 [DOI] [PubMed] [Google Scholar]
- Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, … McArthur GA (2011). Improved survival with vemurafenib in melanoma with BRAF V600E mutation. The New England Journal of Medicine, 364(26), 2507–2516. 10.1056/NEJMoa1103782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheli Y, Giuliano S, Fenouille N, Allegra M, Hofman V, Hofman P, … Ballotti R (2012). Hypoxia and MITF control metastatic behaviour in mouse and human melanoma cells. Oncogene, 31(19), 2461–2470. 10.1038/onc.2011.425 [DOI] [PubMed] [Google Scholar]
- Chhabra Y, Yong HXL, Fane ME, Soogrim A, Lim W, Mahiuddin DN, … Smith AG (2018). Genetic variation in IRF4 expression modulates growth characteristics, tyrosinase expression and interferon-gamma response in melanocytic cells. Pigment Cell & Melanoma Research, 31(1), 51–63. 10.1111/pcmr.12620 [DOI] [PubMed] [Google Scholar]
- Clark WH (1991). Tumour progression and the nature of cancer. British Journal of Cancer, 64(4), 631–644. Retrieved from http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1977704/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corazao-Rozas P, Guerreschi P, Jendoubi M, Andre F, Jonneaux A, Scalbert C, … Marchetti P (2013). Mitochondrial oxidative stress is the achille’s heel of melanoma cells resistant to Braf-mutant inhibitor. Oncotarget, 4(11), 1986–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Luca A, Maiello MR, D’Alessio A, Pergameno M, & Normanno N (2012). The RAS/RAF/MEK/ERK and the PI3K/AKT signalling pathways: role in cancer pathogenesis and implications for therapeutic approaches. Expert Opinion on Therapeutic Targets, 16 Suppl 2, S17–27. 10.1517/14728222.2011.639361 [DOI] [PubMed] [Google Scholar]
- Diridollou S, Vabre V, Berson M, Vaillant L, Black D, Lagarde JM, … Patat F (2001). Skin ageing: changes of physical properties of human skin in vivo. International Journal of Cosmetic Science, 23(6), 353–362. 10.1046/j.0412-5463.2001.00105.x [DOI] [PubMed] [Google Scholar]
- Eales KL, Hollinshead KE, & Tennant DA (2016). Hypoxia and metabolic adaptation of cancer cells. Oncogenesis, 5, e190. 10.1038/oncsis.2015.50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggermont AMM, Spatz A, & Robert C (2014). Cutaneous melanoma. The Lancet, 383(9919), 816–827. 10.1016/S0140-6736(13)60802-8 [DOI] [PubMed] [Google Scholar]
- Fane ME, Chhabra Y, Hollingsworth DE, Simmons JL, Spoerri L, Oh TG, … Smith AG (2017). NFIB Mediates BRN2 Driven Melanoma Cell Migration and Invasion Through Regulation of EZH2 and MITF. EBioMedicine, 16, 63–75. 10.1016/j.ebiom.2017.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fattore L, Mancini R, Acunzo M, Romano G, Laganà A, Pisanu ME, … Ciliberto G (2016). miR-579–3p controls melanoma progression and resistance to target therapy. Proceedings of the National Academy of Sciences of the United States of America, 113(34), E5005–E5013. 10.1073/pnas.1607753113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedorenko IV, Abel EV, Koomen JM, Fang B, Wood ER, Chen YA, … Smalley KSM (2016). Fibronectin induction abrogates the BRAF inhibitor response of BRAF V600E/PTEN-null melanoma cells. Oncogene, 35(10), 1225–1235. 10.1038/onc.2015.188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedorenko IV, Wargo JA, Flaherty KT, Messina JL, & Smalley KSM (2015). BRAF Inhibition Generates a Host–Tumor Niche that Mediates Therapeutic Escape. Journal of Investigative Dermatology, 135(12), 3115–3124. 10.1038/jid.2015.329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feige E, Yokoyama S, Levy C, Khaled M, Igras V, Lin RJ, … Fisher DE (2011). Hypoxia-induced transcriptional repression of the melanoma-associated oncogene MITF. Proc Natl Acad Sci U S A, 108(43), E924–33. 10.1073/pnas.1106351108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer KR, Durrans A, Lee S, Sheng J, Li F, Wong STC, … Gao D (2015). Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature, 527(7579), 472–476. 10.1038/nature15748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fluegen G, Avivar-Valderas A, Wang YR, Padgen MR, Williams JK, Nobre AR, … Aguirre-Ghiso JA (2017). Phenotypic heterogeneity of disseminated tumour cells is preset by primary tumour hypoxic microenvironments. Nature Cell Biology, 19(2), 120–132. 10.1038/ncb3465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortmann SP, Burda BU, Senger CA, Lin JS, Beil TL, O’Connor E, & Whitlock EP (2013, November). Vitamin, Mineral, and Multivitamin Supplements for the Primary Prevention of Cardiovascular Disease and Cancer: A Systematic Evidence Review for the U.S. Preventive Services Task Force U.S. Preventive Services Task Force Evidence Syntheses, formerly Syste. [PubMed] [Google Scholar]
- Gadiot J, Hooijkaas AI, Deken MA, & Blank CU (2013). Synchronous BRAF(V600E) and MEK inhibition leads to superior control of murine melanoma by limiting MEK inhibitor induced skin toxicity. OncoTargets and Therapy, 6, 1649–1658. 10.2147/OTT.S52552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao P, Zhang HF, Dinavahi R, Li F, Xiang Y, Raman V, … Dang CV (2007). HIF-dependent antitumorigenic effect of antioxidants in vivo. Cancer Cell, 12(3), 230–238. 10.1016/j.ccr.2007.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genomic Classification of Cutaneous Melanoma. (2015). Cell, 161(7), 1681–1696. 10.1016/j.cell.2015.05.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetz JG, Minguet S, Navarro-Lerida I, Lazcano JJ, Samaniego R, Calvo E, … Del Pozo MA (2011). Biomechanical remodeling of the microenvironment by stromal caveolin-1 favors tumor invasion and metastasis. Cell, 146(1), 148–163. 10.1016/j.cell.2011.05.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman GE, Thornquist MD, Balmes J, Cullen MR, Meyskens FLJ, Omenn GS, … Williams JHJ (2004). The Beta-Carotene and Retinol Efficacy Trial: incidence of lung cancer and cardiovascular disease mortality during 6-year follow-up after stopping beta-carotene and retinol supplements. Journal of the National Cancer Institute, 96(23), 1743–1750. 10.1093/jnci/djh320 [DOI] [PubMed] [Google Scholar]
- Gordon S, & Taylor PR (2005). Monocyte and macrophage heterogeneity. Nature Reviews. Immunology, 5(12), 953–964. 10.1038/nri1733 [DOI] [PubMed] [Google Scholar]
- Gorrini C, Harris IS, & Mak TW (2013). Modulation of oxidative stress as an anticancer strategy. Nature Reviews Drug Discovery, 12(12), 931–947. 10.1038/nrd4002 [DOI] [PubMed] [Google Scholar]
- Hadrup S, Donia M, & thor Straten P (2013). Effector CD4 and CD8 T Cells and Their Role in the Tumor Microenvironment. Cancer Microenvironment, 6(2), 123–133. 10.1007/s12307-012-0127-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haferkamp S, Borst A, Adam C, Becker TM, Motschenbacher S, Windhovel S, … Meierjohann S (2013). Vemurafenib induces senescence features in melanoma cells. The Journal of Investigative Dermatology, 133(6), 1601–1609. 10.1038/jid.2013.6 [DOI] [PubMed] [Google Scholar]
- Hartman ML, & Czyz M (2015). MITF in melanoma: mechanisms behind its expression and activity. Cell Mol Life Sci, 72(7), 1249–1260. 10.1007/s00018-014-1791-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauschild A, Grob J-J, Demidov LV, Jouary T, Gutzmer R, Millward M, … Chapman PB (2012). Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet (London, England), 380(9839), 358–365. 10.1016/S0140-6736(12)60868-X [DOI] [PubMed] [Google Scholar]
- Hawryluk EB, & Tsao H (2014). Melanoma: clinical features and genomic insights. Cold Spring Harbor Perspectives in Medicine, 4(9), a015388. 10.1101/cshperspect.a015388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herraiz C, Calvo F, Pandya P, Cantelli G, Rodriguez-hernandez I, Orgaz JL, … Sanz-moreno V (2016). Reactivation of p53 by a Cytoskeletal Sensor to Control the Balance Between DNA Damage and Tumor Dissemination, 108, 1–14. 10.1093/jnci/djv289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Highfill SL, Cui Y, Giles AJ, Smith JP, Zhang H, Morse E, … Mackall CL (2014). Disruption of CXCR2-mediated MDSC tumor trafficking enhances anti-PD1 efficacy. Science Translational Medicine, 6(237), 237ra67. 10.1126/scitranslmed.3007974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirata E, Girotti MR, Viros A, Hooper S, Spencer-Dene B, Matsuda M, … Sahai E (2015). Intravital imaging reveals how BRAF inhibition generates drug-tolerant microenvironments with high integrin β1/FAK Signaling. Cancer Cell, 27(4), 574–588. 10.1016/j.ccell.2015.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, … Urba WJ (2010). Improved survival with ipilimumab in patients with metastatic melanoma. The New England Journal of Medicine, 363(8), 711–723. 10.1056/NEJMoa1003466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat J-P, … Chin L (2012). A landscape of driver mutations in melanoma. Cell, 150(2), 251–263. 10.1016/j.cell.2012.06.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoek KS, & Goding CR (2010). Cancer stem cells versus phenotype-switching in melanoma. Pigment Cell Melanoma Res, 23(6), 746–759. 10.1111/j.1755-148X.2010.00757.x [DOI] [PubMed] [Google Scholar]
- Hong DS, Vence L, Falchook G, Radvanyi LG, Liu C, Goodman V, … Hwu P (2012). BRAF(V600) Inhibitor GSK2118436 Targeted Inhibition of Mutant BRAF in Cancer Patients Does Not Impair Overall Immune Competency. Clinical Cancer Research, 18(8), 2326 LP-2335. Retrieved from http://clincancerres.aacrjournals.org/content/18/8/2326.abstract [DOI] [PubMed] [Google Scholar]
- Kaur A, Webster MR, Marchbank K, Behera R, Ndoye A, Kugel CH 3rd, … Weeraratna AT (2016). sFRP2 in the aged microenvironment drives melanoma metastasis and therapy resistance. Nature, 532(7598), 250–254. 10.1038/nature17392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushik S, Pickup MW, & Weaver VM (2016). From transformation to metastasis: deconstructing the extracellular matrix in breast cancer. Cancer and Metastasis Reviews, 35(4), 655–667. 10.1007/s10555-016-9650-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalili JS, Liu S, Rodríguez-Cruz TG, Whittington M, Wardell S, Liu C, … Lizée G (2012). Oncogenic BRAF(V600E) promotes stromal cell-mediated immunosuppression via induction of interleukin-1 in melanoma. Clinical Cancer Research : An Official Journal of the American Association for Cancer Research, 18(19), 5329–5340. 10.1158/1078-0432.ccr-12-1632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MH, Kim J, Hong H, Lee S-H, Lee J-K, Jung E, & Kim J (2016). Actin remodeling confers BRAF inhibitor resistance to melanoma cells through YAP/TAZ activation. The EMBO Journal, 35(5), 462–478. 10.15252/embj.201592081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein EA, Thompson IMJ, Tangen CM, Crowley JJ, Lucia MS, Goodman PJ, … Baker LH (2011). Vitamin E and the risk of prostate cancer: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA, 306(14), 1549–1556. 10.1001/jama.2011.1437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight DA, Ngiow SF, Li M, Parmenter T, Mok S, Cass A, … Smyth MJ (2013). Host immunity contributes to the anti-melanoma activity of BRAF inhibitors. The Journal of Clinical Investigation, 123(3), 1371–1381. 10.1172/jci66236 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Konieczkowski DJ, Johannessen CM, Abudayyeh O, Kim JW, Cooper ZA, Piris A, … Garraway LA (2014). A melanoma cell state distinction influences sensitivity to MAPK pathway inhibitors. Cancer Discovery, 4(7), 816–827. 10.1158/2159-8290.CD-13-0424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krtolica A, Parrinello S, Lockett S, Desprez PY, & Campisi J (2001). Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proceedings of the National Academy of Sciences of the United States of America, 98(21), 12072–12077. 10.1073/pnas.211053698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudo-Saito C, Shirako H, Ohike M, Tsukamoto N, & Kawakami Y (2013). CCL2 is critical for immunosuppression to promote cancer metastasis. Clinical & Experimental Metastasis, 30(4), 393–405. 10.1007/s10585-012-9545-6 [DOI] [PubMed] [Google Scholar]
- Kumar V, Donthireddy L, Marvel D, Condamine T, Wang F, Lavilla-Alonso S, … Gabrilovich DI (2017). Cancer-Associated Fibroblasts Neutralize the Anti-tumor Effect of CSF1 Receptor Blockade by Inducing PMN-MDSC Infiltration of Tumors. Cancer Cell, 32(5), 654–668.e5. 10.1016/j.ccell.2017.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V, Patel S, Tcyganov E, & Gabrilovich DI (2016). The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends in Immunology, 37(3), 208–220. 10.1016/j.it.2016.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon Y, Cukierman E, & Godwin AK (2011). Differential expressions of adhesive molecules and proteases define mechanisms of ovarian tumor cell matrix penetration/invasion. PloS One, 6(4), e18872. 10.1371/journal.pone.0018872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, … Wolchok JD (2015). Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. The New England Journal of Medicine, 373(1), 23–34. 10.1056/NEJMoa1504030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H-O, Mullins SR, Franco-Barraza J, Valianou M, Cukierman E, & Cheng JD (2011). FAP-overexpressing fibroblasts produce an extracellular matrix that enhances invasive velocity and directionality of pancreatic cancer cells. BMC Cancer, 11, 245 10.1186/1471-2407-11-245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JT, Li L, Brafford PA, van den Eijnden M, Halloran MB, Sproesser K, … Herlyn M (2010). PLX4032, a potent inhibitor of the B-Raf V600E oncogene, selectively inhibits V600E-positive melanomas. Pigment Cell & Melanoma Research, 23(6), 820–827. 10.1111/j.1755-148X.2010.00763.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie C, Bowyer SE, White A, Grieu-Iacopetta F, Trevenen M, Iacopetta B, … Millward M (2015). FOXP3+ T regulatory lymphocytes in primary melanoma are associated with BRAF mutation but not with response to BRAF inhibitor. Pathology, 47(6), 557–563. 10.1097/PAT.0000000000000314 [DOI] [PubMed] [Google Scholar]
- Liu-Smith F, Dellinger R, & Meyskens FL Jr. (2014). Updates of reactive oxygen species in melanoma etiology and progression. Arch Biochem Biophys, 563, 51–55. 10.1016/j.abb.2014.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani A, Sica A, & Locati M (2005). Macrophage polarization comes of age. Immunity, 23(4), 344–346. 10.1016/j.immuni.2005.10.001 [DOI] [PubMed] [Google Scholar]
- Marcos-Garces V, Molina Aguilar P, Bea Serrano C, Garcia Bustos V, Benavent Segui J, Ferrandez Izquierdo A, & Ruiz-Sauri A (2014). Age-related dermal collagen changes during development, maturation and ageing - a morphometric and comparative study. Journal of Anatomy, 225(1), 98–108. 10.1111/joa.12186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McArthur GA, Chapman PB, Robert C, Larkin J, Haanen JB, Dummer R, … Hauschild A (2014). Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation-positive melanoma (BRIM-3): extended follow-up of a phase 3, randomised, open-label study. The Lancet. Oncology, 15(3), 323–332. 10.1016/S1470-2045(14)70012-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer C, Cagnon L, Costa-Nunes CM, Baumgaertner P, Montandon N, Leyvraz L, … Speiser DE (2014). Frequencies of circulating MDSC correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunology, Immunotherapy : CII, 63(3), 247–257. 10.1007/s00262-013-1508-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milhas D, Lazar I, Clement E, Soldan V, Ducoux-petit M, & Legonidec S (2016). Adipocyte Exosomes Promote Melanoma Aggressiveness through Fatty Acid Oxidation : A Novel Mechanism Linking Obesity and Cancer, 4051–4058. 10.1158/0008-5472.CAN-16-0651 [DOI] [PubMed] [Google Scholar]
- Mills CN, Joshi SS, & Niles RM (2009). Expression and function of hypoxia inducible factor-1 alpha in human melanoma under non-hypoxic conditions. Mol Cancer, 8, 104 10.1186/1476-4598-8-104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohos A, Sebestyén T, Liszkay G, Plótár V, Horváth S, Gaudi I, & Ladányi A (2013). Immune cell profile of sentinel lymph nodes in patients with malignant melanoma – FOXP3(+) cell density in cases with positive sentinel node status is associated with unfavorable clinical outcome. Journal of Translational Medicine, 11, 43 10.1186/1479-5876-11-43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller J, Krijgsman O, Tsoi J, Robert L, Hugo W, Song C, … Peeper DS (2014). Low MITF/AXL ratio predicts early resistance to multiple targeted drugs in melanoma. Nat Commun, 5, 5712 10.1038/ncomms6712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, … Lo RS (2010). Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature, 468(7326), 973–977. 10.1038/nature09626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell MP, Marchbank K, Webster MR, Valiga AA, Kaur A, Vultur A, … Weeraratna AT (2013). Hypoxia induces phenotypic plasticity and therapy resistance in melanoma via the tyrosine kinase receptors ROR1 and ROR2. Cancer Discov, 3(12), 1378–1393. 10.1158/2159-8290.CD-13-0005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell MP, & Weeraratna AT (2013). Change is in the air: the hypoxic induction of phenotype switching in melanoma. J Invest Dermatol, 133(10), 2316–2317. 10.1038/jid.2013.208 [DOI] [PubMed] [Google Scholar]
- Oh J-H, Kim YK, Jung J-Y, Shin J, Kim KH, Cho KH, … Chung JH (2011). Intrinsic aging- and photoaging-dependent level changes of glycosaminoglycans and their correlation with water content in human skin. Journal of Dermatological Science, 62(3), 192–201. 10.1016/j.jdermsci.2011.02.007 [DOI] [PubMed] [Google Scholar]
- Ohuchida K, Mizumoto K, Murakami M, Qian L-W, Sato N, Nagai E, … Tanaka M (2004). Radiation to stromal fibroblasts increases invasiveness of pancreatic cancer cells through tumor-stromal interactions. Cancer Research, 64(9), 3215–3222. [DOI] [PubMed] [Google Scholar]
- Panwar P, Lamour G, Mackenzie NCW, Yang H, Ko F, Li H, & Bromme D (2015). Changes in Structural-Mechanical Properties and Degradability of Collagen during Aging-associated Modifications. The Journal of Biological Chemistry, 290(38), 23291–23306. 10.1074/jbc.M115.644310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peiris-Pages M, Martinez-Outschoorn UE, Sotgia F, & Lisanti MP (2015). Metastasis and Oxidative Stress: Are Antioxidants a Metabolic Driver of Progression? Cell Metab, 22(6), 956–958. 10.1016/j.cmet.2015.11.008 [DOI] [PubMed] [Google Scholar]
- Piérard GE, Piérard-Franchimont C, & Delvenne P (2012). Malignant melanoma and its stromal nonimmune microecosystem. Journal of Oncology, 2012. 10.1155/2012/584219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piskounova E, Agathocleous M, Murphy MM, Hu Z, Huddlestun SE, Zhao Z, … Morrison SJ (2015). Oxidative stress inhibits distant metastasis by human melanoma cells. Nature, 527(7577), 186–191. 10.1038/nature15726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plimack ER, Bellmunt J, Gupta S, Berger R, Chow LQM, Juco J, … O’Donnell PH (2018). Safety and activity of pembrolizumab in patients with locally advanced or metastatic urothelial cancer (KEYNOTE-012): a non-randomised, open-label, phase 1b study. The Lancet Oncology, 18(2), 212–220. 10.1016/S1470-2045(17)30007-4 [DOI] [PubMed] [Google Scholar]
- Pucciarelli D, Lengger N, Takacova M, Csaderova L, Bartosova M, Breiteneder H, … Hafner C (2016). Hypoxia increases the heterogeneity of melanoma cell populations and affects the response to vemurafenib. Mol Med Rep, 13(4), 3281–3288. 10.3892/mmr.2016.4888 [DOI] [PubMed] [Google Scholar]
- Qin Y, Roszik J, Chattopadhyay C, Hashimoto Y, Liu C, Cooper ZA, … Grimm EA (2016). Hypoxia-Driven Mechanism of Vemurafenib Resistance in Melanoma. Mol Cancer Ther, 15(10), 2442–2454. 10.1158/1535-7163.MCT-15-0963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas A, Hodi FS, Callahan M, Konto C, & Wolchok J (2013). Hepatotoxicity with Combination of Vemurafenib and Ipilimumab. New England Journal of Medicine, 368(14), 1365–1366. 10.1056/NEJMc1302338 [DOI] [PubMed] [Google Scholar]
- Rinnerthaler M, Bischof J, Streubel MK, Trost A, & Richter K (2015). Oxidative stress in aging human skin. Biomolecules, 5(2), 545–589. 10.3390/biom5020545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues MF, Obre E, de Melo FH, Santos GC Jr., Galina A, Jasiulionis MG, … Amoedo ND (2016). Enhanced OXPHOS, glutaminolysis and beta-oxidation constitute the metastatic phenotype of melanoma cells. Biochem J, 473(6), 703–715. 10.1042/BJ20150645 [DOI] [PubMed] [Google Scholar]
- Romano G, & Kwong LN (2017). miRNAs, Melanoma and Microenvironment: An Intricate Network. International Journal of Molecular Sciences, 18(11), 2354 10.3390/ijms18112354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiter D, Bogenrieder T, Elder D, & Herlyn M (2002, January 1). Melanoma-stroma interactions: Structural and functional aspects. Lancet Oncology. Elsevier. 10.1016/S1470-2045(01)00620-9 [DOI] [PubMed] [Google Scholar]
- Sadelain M (2016). Chimeric antigen receptors: driving immunology towards synthetic biology. Current Opinion in Immunology, 41, 68–76. 10.1016/J.COI.2016.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi S, Yamaguchi T, Nomura T, & Ono M (2018). Regulatory T Cells and Immune Tolerance. Cell, 133(5), 775–787. 10.1016/j.cell.2008.05.009 [DOI] [PubMed] [Google Scholar]
- Sapkota B, Hill CE, & Pollack BP (2013). Vemurafenib enhances MHC induction in BRAF(V600E) homozygous melanoma cells. Oncoimmunology, 2(1), e22890. 10.4161/onci.22890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schadendorf D, Fisher DE, Garbe C, Gershenwald JE, Grob J-J, Halpern A, … Hauschild A (2015, April). Melanoma. Nature Reviews. Disease Primers. England. 10.1038/nrdp.2015.3 [DOI] [PubMed] [Google Scholar]
- Schadendorf D, Hodi FS, Robert C, Weber JS, Margolin K, Hamid O, … Wolchok JD (2015). Pooled Analysis of Long-Term Survival Data From Phase II and Phase III Trials of Ipilimumab in Unresectable or Metastatic Melanoma. Journal of Clinical Oncology, 33(17), 1889–1894. 10.1200/JCO.2014.56.2736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilling B, Sucker A, Griewank K, Zhao F, Weide B, Gorgens A, … Paschen A (2013). Vemurafenib reverses immunosuppression by myeloid derived suppressor cells. International Journal of Cancer, 133(7), 1653–1663. 10.1002/ijc.28168 [DOI] [PubMed] [Google Scholar]
- Schlecker E, Stojanovic A, Eisen C, Quack C, Falk CS, Umansky V, & Cerwenka A (2012). Tumor-Infiltrating Monocytic Myeloid-Derived Suppressor Cells Mediate CCR5-Dependent Recruitment of Regulatory T Cells Favoring Tumor Growth. The Journal of Immunology, 189(12), 5602 LP-5611. Retrieved from http://www.jimmunol.org/content/189/12/5602.abstract [DOI] [PubMed] [Google Scholar]
- Sharma P, & Allison JP (2015). The future of immune checkpoint therapy. Science, 348(6230), 56 LP–61. Retrieved from http://science.sciencemag.org/content/348/6230/56.abstract [DOI] [PubMed] [Google Scholar]
- Shi H, Moriceau G, Kong X, Lee M-K, Lee H, Koya RC, … Lo RS (2012). Melanoma whole-exome sequencing identifies (V600E)B-RAF amplification-mediated acquired B-RAF inhibitor resistance. Nature Communications, 3, 724 10.1038/ncomms1727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sojka DK, Huang Y-H, & Fowell DJ (2008). Mechanisms of regulatory T-cell suppression – a diverse arsenal for a moving target. Immunology, 124(1), 13–22. 10.1111/j.1365-2567.2008.02813.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solinas G, Germano G, Mantovani A, & Allavena P (2009). Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. Journal of Leukocyte Biology, 86(5), 1065–1073. 10.1189/jlb.0609385 [DOI] [PubMed] [Google Scholar]
- Straussman R, Morikawa T, Shee K, Barzily-Rokni M, Qian ZR, Du J, … Golub TR (2012). Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature, 487(7408), 500–504. 10.1038/nature11183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama D, Nishikawa H, Maeda Y, Nishioka M, Tanemura A, Katayama I, … Sakaguchi S (2013). Anti-CCR4 mAb selectively depletes effector-type FoxP3+CD4+ regulatory T cells, evoking antitumor immune responses in humans. Proceedings of the National Academy of Sciences of the United States of America, 110(44), 17945–17950. 10.1073/pnas.1316796110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumimoto H, Imabayashi F, Iwata T, & Kawakami Y (2006). The BRAF-MAPK signaling pathway is essential for cancer-immune evasion in human melanoma cells. The Journal of Experimental Medicine, 203(7), 1651–1656. 10.1084/jem.20051848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C, Wang L, Huang S, Heynen GJJE, Prahallad A, Robert C, … Bernards R (2014). Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature, 508(7494), 118–122. 10.1038/nature13121 [DOI] [PubMed] [Google Scholar]
- The Alpha-Tocopherol Beta Carotene Cancer Prevention Study Group. (1994). The effect of vitamin E and beta carotene on the incidence of lung cancer and other cancers in male smokers. The New England Journal of Medicine, 330(15), 1029–1035. 10.1056/NEJM199404143301501 [DOI] [PubMed] [Google Scholar]
- Titz B, Lomova A, Le A, Hugo W, Kong X, Hoeve J, … Ate M (2016). JUN dependency in distinct early and late BRAF inhibition adaptation states of melanoma. 10.1038/celldisc.2016.28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viehl CT, Moore TT, Liyanage UK, Frey DM, Ehlers JP, Eberlein TJ, … Linehan DC (2006). Depletion of CD4+CD25+ regulatory T cells promotes a tumor-specific immune response in pancreas cancer-bearing mice. Annals of Surgical Oncology, 13(9), 1252–1258. 10.1245/s10434-006-9015-y [DOI] [PubMed] [Google Scholar]
- Villanueva J, Vultur A, Lee JT, Somasundaram R, Fukunaga-Kalabis M, Cipolla AK, … Herlyn M (2010). Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell, 18(6), 683–695. 10.1016/j.ccr.2010.11.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagle N, Emery C, Berger MF, Davis MJ, Sawyer A, Pochanard P, … Garraway LA (2011). Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. Journal of Clinical Oncology : Official Journal of the American Society of Clinical Oncology, 29(22), 3085–3096. 10.1200/JCO.2010.33.2312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward KA, Lazovich D, & Hordinsky MK (2012). Germline melanoma susceptibility and prognostic genes: a review of the literature. Journal of the American Academy of Dermatology, 67(5), 1055–1067. 10.1016/j.jaad.2012.02.042 [DOI] [PubMed] [Google Scholar]
- Wellbrock C, & Arozarena I (2015). Microphthalmia-associated transcription factor in melanoma development and MAP-kinase pathway targeted therapy. Pigment Cell Melanoma Res, 28(4), 390–406. 10.1111/pcmr.12370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widmer DS, Hoek KS, Cheng PF, Eichhoff OM, Biedermann T, Raaijmakers MIG, … Levesque MP (2013). Hypoxia contributes to melanoma heterogeneity by triggering HIF1alpha-dependent phenotype switching. The Journal of Investigative Dermatology, 133(10), 2436–2443. 10.1038/jid.2013.115 [DOI] [PubMed] [Google Scholar]
- Wigerup C, Pahlman S, & Bexell D (2016). Therapeutic targeting of hypoxia and hypoxia-inducible factors in cancer. Pharmacol Ther, 164, 152–169. 10.1016/j.pharmthera.2016.04.009 [DOI] [PubMed] [Google Scholar]
- Wilmott JS, Long GV, Howle JR, Haydu LE, Sharma RN, Thompson JF, … Scolyer RA (2012). Selective BRAF inhibitors induce marked T-cell infiltration into human metastatic melanoma. Clinical Cancer Research : An Official Journal of the American Association for Cancer Research, 18(5), 1386–1394. 10.1158/1078-0432.CCR-11-2479 [DOI] [PubMed] [Google Scholar]
- Wittgen HG, & van Kempen LC (2007). Reactive oxygen species in melanoma and its therapeutic implications. Melanoma Research, 17(6), 400–409. 10.1097/CMR.0b013e3282f1d312 [DOI] [PubMed] [Google Scholar]
- Wolchok JD, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob J-J, Cowey CL, … Larkin J (2017). Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. The New England Journal of Medicine, 377(14), 1345–1356. 10.1056/NEJMoa1709684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young HL, Rowling EJ, Bugatti M, Giurisato E, Luheshi N, Arozarena I, … Hurlstone A (2017). An adaptive signaling network in melanoma inflammatory niches confers tolerance to MAPK signaling inhibition. Journal of Experimental Medicine. 10.1084/jem.20160855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, Gao LX, Ma XQ, Hu FX, Li CM, & Lu Z (2014). Involvement of superoxide and nitric oxide in BRAF(V600E) inhibitor PLX4032-induced growth inhibition of melanoma cells. Integr Biol (Camb), 6(12), 1211–1217. 10.1039/c4ib00170b [DOI] [PubMed] [Google Scholar]
- Yun J, Mullarky E, Lu C, Bosch KN, Kavalier A, Rivera K, … Cantley LC (2015). Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science (New York, N.Y.), 350(6266), 1391–1396. 10.1126/science.aaa5004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Liu X, Sadek N, Zhang W, Chen G, Cheng C, … Herlyn M (2017). MAPK inhibitors in BRAF -mutant melanomas. 10.1038/nature24040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Carstens JL, Kim J, Scheible M, Kaye J, Sugimoto H, … Kalluri R (2015). Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature, 527(7579), 525–530. 10.1038/nature16064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou W, Wolchok JD, & Chen L (2016). PD-L1 (B7-H1) and PD-1 Pathway Blockade for Cancer Therapy: Mechanisms, Response Biomarkers and Combinations. Science Translational Medicine, 8(328), 328rv4–328rv4. 10.1126/scitranslmed.aad7118 [DOI] [PMC free article] [PubMed] [Google Scholar]