

Abstract
Chromatin-associated proteins are instrumental for controlling spatiotemporal gene expression. Determining where these proteins bind across the genome is critical for understanding gene regulation. A widely used technique at present is ChIP-seq, which leverages chromatin fragmentation, antibody-mediated enrichment, next-generation sequencing and data analysis to uncover the genomic sequences and patterns of protein-DNA interactions. In this review, we will provide an overview of how ChIP-seq was developed, the key elements of the experimentation and data analysis pipeline, as well as the recent variations that push the boundaries of precision and cell number requirement. We will also briefly discuss how future development of ChIP-seq may further advance our understanding of chromatin biology.
Keywords: Chromatin, ChIP-seq, genome, DNA, nucleus, gene regulation
Introduction
Chromatin associated proteins play vital roles in controlling spatial and temporal gene expression to modulate cellular activities. To provide insight into the function of these chromatin associated proteins and to evaluate their direct gene targets, a key method is Chromatin Immunoprecipitation Sequencing (ChIP-seq). ChIP-seq is designed to map genome-wide protein binding sites leveraging next-generation sequencing technologies. This technology has already revolutionized our understanding of gene regulation during transcription through characterizing dynamic protein-DNA interactions across organisms and cell types. In this review, we will discuss the scientific discoveries leading up to the development of ChIP-seq, classical ChIP-seq methods, and variations of ChIP-seq currently being used to overcome limitations of the technique.
History and Development of ChIP-seq
Modern ChIP-seq technology is built upon a series of conceptual and technological breakthroughs in genomic research over the last seventy years (Figure 1) (Sanger, Nicklen, & Coulson, 1977; Schena, Shalon, Davis, & Brown, 1995; Watson & Crick, 1953). The idea of chromatin immunoprecipitation (ChIP) was initially based on the fact that protein-DNA interactions could be stabilized using crosslinking. In a classic experiment in 1969, Brutlag et al. discovered that once chromatin was treated with formaldehyde, histones remained bound to DNA even in the presence of extremely high salt concentration (4M CsCl) or acid (0.2N H2SO4) (Brutlag, Schlehuber, & Bonner, 1969). Building on this, Gilmour and Lis reported the ChIP technique for the first time in 1984 and applied it to identify protein-DNA interaction sites in intact bacteria cells. With ChIP, they were able to show RNA polymerase II binding at highly transcribed genes (Gilmour & Lis, 1984). Subsequently in 1985, they optimized and extended this method to higher eukaryotic systems, with demonstrations of altered RNA polymerase II occupancy after heat shock in Drosophila S2 cells (Gilmour & Lis, 1985). In these early experiments, low-intensity UV crosslinking was used (Gilmour & Lis, 1984; Gilmour & Lis, 1985). To determine the DNA sequences of protein binding sites, these experiments relied on dot blot hybridization or southern blotting using probes designed specifically to sequences of interest (Gilmour & Lis, 1984; Gilmour & Lis, 1985; Orlando & Paro, 1993; Thomas, 1980). The entire hybridization and detection process typically involves several days. The development of polymerase chain reaction (PCR) drastically simplified the sequence-detection component of ChIP, allowing sampling of representative DNA sequences within a few hours (Hecht, Strahl-Bolsinger, & Grunstein, 1996; Kaunitz, 2015; Orlando, 2000).
Figure 1. Milestones of ChIP-seq development.
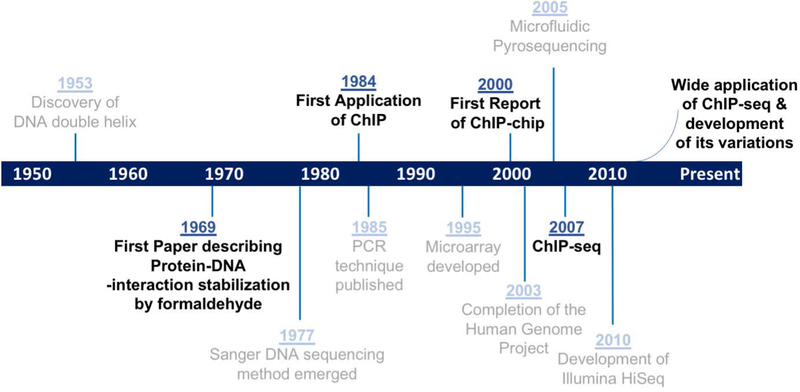
Critical discoveries leading up to ChIP-seq technologies are marked in black. Relevant technological advances are labelled in grey.
Higher throughput analysis was made possible initially with the innovation of microarray technology (Ren et al., 2000). ChIP-on-chip (ChIP-chip) was developed in 2000. With this method, the immunoprecipitated DNA is labeled with one fluorescent color, while DNA from sample prior to immunoprecipitation, referred to as input DNA, is labeled with a different color as control. Input and immunoprecipitated samples are mixed together and hybridized to a microarray containing probes tiling genomic regions. Output can then be measured by comparing the relative fluorescence enrichment of immunoprecipitated DNA versus that of the input control (Buck & Lieb, 2004; Kim & Ren, 2006; Ren et al., 2000). A limitation of ChIP-chip lies in the design of microarray probes and the holding capacity of tiling arrays. Results are limited by the total percent of the genome covered on the array and the resolution or distance between probes. Using microarray, it was also virtually impossible to investigate repetitive regions or regions with strong homology in the genome. These inherent technological limitations made it nearly impossible to identify protein-binding DNA sequences with high precision and in a genome-wide scale for higher eukaryotic organisms (Barski & Zhao, 2009; Ho et al., 2011; Kim & Ren, 2006; Park, 2009).
With the revolutionary next-generation sequencing technologies that first emerged in early 2000 and are still rapidly evolving (Gužvić, 2013; Liu et al., 2011; Margulies et al., 2005; National Institutes of Health, 2003), ChIP-seq was developed and first reported in 2007 (Johnson, Mortazavi, Myers, & Wold, 2007; Robertson et al., 2007). ChIP-seq circumvented the dependency on DNA tiling arrays, and allowed for whole genome data analysis for virtually any organism with a sequenced genome (Johnson et al., 2007). Since the first application of ChIP-seq, many variations have been developed to address limitations and answer specific biological questions (Furey, 2012). We will discuss the experimentation and data analysis of classic ChIP-seq, several examples of ChIP-seq variations, as well as potential areas for further development of understanding gene regulation.
Classic ChIP-seq Methodology
From Cells to Sequencing Libraries
Classic ChIP-seq involves multiple steps that typically take several days from the preparation of cells to library construction (Figure 2). The first step in classic ChIP-seq is crosslinking protein to DNA (Park, 2009). Although early experiments used UV to mediate protein-DNA crosslinking, later studies identified formaldehyde crosslinking as a superior method with higher sensitivity, which is especially useful for mammalian cells (Boyd & Farnham, 1997; Boyd, Wells, Gutman, Bartley, & Farnham, 1998). Thus, chemical crosslinking with formaldehyde is most commonly used in current ChIP-seq protocols (Farnham, 2009; Furey, 2012; Park, 2009; Truax & Greer, 2012). After crosslinking, chromatin is usually fragmented to 200–600bp fragments by sonication or enzymatic digestion (Farnham, 2009; Park, 2009). The efficiency of chromatin fragmentation can be assessed by agarose gel electrophoresis. If sonication is used, it is critical to optimize sonication time so that protein is not degraded, but chromatin is sufficiently sheared. The integrity of protein during the sonication process can be determined using Coomassie Blue staining or Western blotting (Pchelintsev, Adams, & Nelson, 2016).
Figure 2. Classical ChIP-seq method.
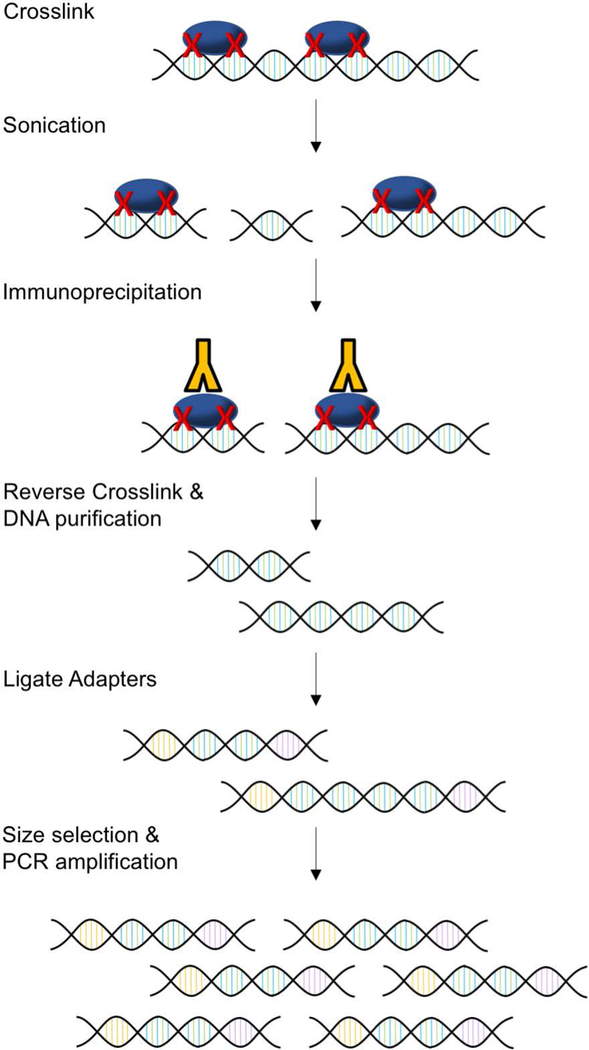
First, protein is crosslinked to DNA with formaldehyde. Sonication is then used to fragment chromatin. Protein-DNA complexes are immunoprecipitated. Crosslinking is reversed, and DNA is purified. DNA is then prepared for sequencing. Adapters are ligated to both 5’ and 3’ ends. Lastly, DNA is size selected and PCR amplified. Libraries are then ready to be sent for sequencing.
Once chromatin has been fragmented, the next step is DNA-protein complex immunoprecipitation. Antibodies for immunoprecipitation must be carefully chosen so that they are specific and sensitive to the proteins of interest. The Encyclopedia of DNA Elements (ENCODE) group recommends both primary and secondary characterization of ChIP-seq antibodies (Landt et al., 2012). Primary characterization includes immunoblot or immunofluorescence to verify correct protein size and cellular localization. Secondary characterization can be completed by knocking down the target gene to further verify the antigen. Alternatively, immunoprecipitation followed by mass spectrometry or probing with an antibody for a different epitope of the protein of interest can be performed to determine specificity (Furey, 2012; Landt et al., 2012). Under circumstances where high quality antibodies cannot be obtained, proteins of interest are usually tagged with an epitope and immunoprecipitated with an antibody for that epitope (Kidder, Hu, & Zhao, 2011). In this scenario, it is critical to verify that the tagged protein is expressed at a level below or near endogenous expression and is also incorporated into the endogenous protein complex. As a negative control for immunoprecipitation, a fraction of input or immunoprecipitated immunoglobulin proteins should be included in the following steps of library construction and data analysis (Furey, 2012; Park, 2009; Truax & Greer, 2012).
Immunoprecipitation can be performed with protein-A-bearing “Staph A” cells or protein A/G conjugated beads. Staph A cells are fixed, heat-inactivated Staphylococcus aureus cells with a Protein A coat on the surface to mediate immunoprecipitation (Palhan, Kreader, & Mueller, 2017). This technique carries the advantage of high sensitivity owing to the high amounts of protein A as well as the small surface of the Staph A cells. This method works well for basic ChIP and ChIP-chip experiments when specific probes are being used; however, using Staph A cells for ChIP-seq is not ideal as DNA from Staph A cells may confound results (Palhan et al., 2017; Wen et al., 2014). For this reason, agarose or magnetic beads are most commonly used for ChIP-seq (Barski & Zhao, 2009; Dahl & Gilfillan, 2018; Park, 2009; Wen et al., 2014). Magnetic beads are often preferable as they tend to be easier to work with, and they generally have higher specificity and lower background as compared to agarose beads (Kim & Ren, 2006; Schmidt et al., 2009).
After immunoprecipitation, the following step is reverse crosslinking and DNA purification. Crosslinking is reversed using high-salt concentration and heat (Kim & Ren, 2006; Orlando, 2000). Proteins are removed using Protease K. DNA purification is subsequently completed using columns, beads, or phenol-chloroform extraction followed by ethanol precipitation (Dahl & Gilfillan, 2018; Truax & Greer, 2012). Phenol-chloroform extraction has been demonstrated to recover higher amounts of purified ChIP DNA, which is usually in the nanogram range, as compared to columns or beads (Dahl, Reiner, & Collas, 2009; Truax & Greer, 2012; Zhong et al., 2017).
The last stage of ChIP-seq experimentation is library construction and sequencing. Commercially available kits are available for library construction from purified ChIP DNA (Dahl & Gilfillan, 2018; Schmidt et al., 2009). In brief, adapters are ligated to both 5’ and 3’ ends of fragments. Fragments are size selected by gel electrophoresis or DNA-purification magnetic beads. Libraries are then amplified by PCR and ready to be sent for sequencing (Linnarsson, 2010; Schmidt et al., 2009). Often multiple libraries are combined in the same high-throughput sequencing lane which is referred to as multiplexing. This can be easily achieved by adding unique barcode adapters to each library. Libraries can then be bioinformatically de-multiplexed after sequencing (Barski & Zhao, 2009; Park, 2009).
Data Analysis
Once the ChIP-seq libraries are sequenced, the data processing can be completed using a fairly well-established work flow (Figure 3A). A variety of software has been developed for fast and simple analysis of ChIP-seq data. First, software such as deML and FASTX-Toolkit can be used to demultiplex libraries (“FASTX-Toolkit,” ; Renaud, Stenzel, Maricic, Wiebe, & Kelso, 2015). Raw reads are in FASTQ format (Figure 3B) which can be directly input into software such as SOAP2, BWA, Bowtie, or Bowtie2 (Langmead & Salzberg, 2012; Langmead, Trapnell, Pop, & Salzberg, 2009; Li & Durbin, 2009; R. Li et al., 2009) to be aligned to a reference genome. Among these pieces of software, Bowtie2 has been demonstrated to run faster and have more aligned reads with fewer incorrect alignments (Langmead & Salzberg, 2012).
Figure 3. ChIP-seq data processing and analysis.
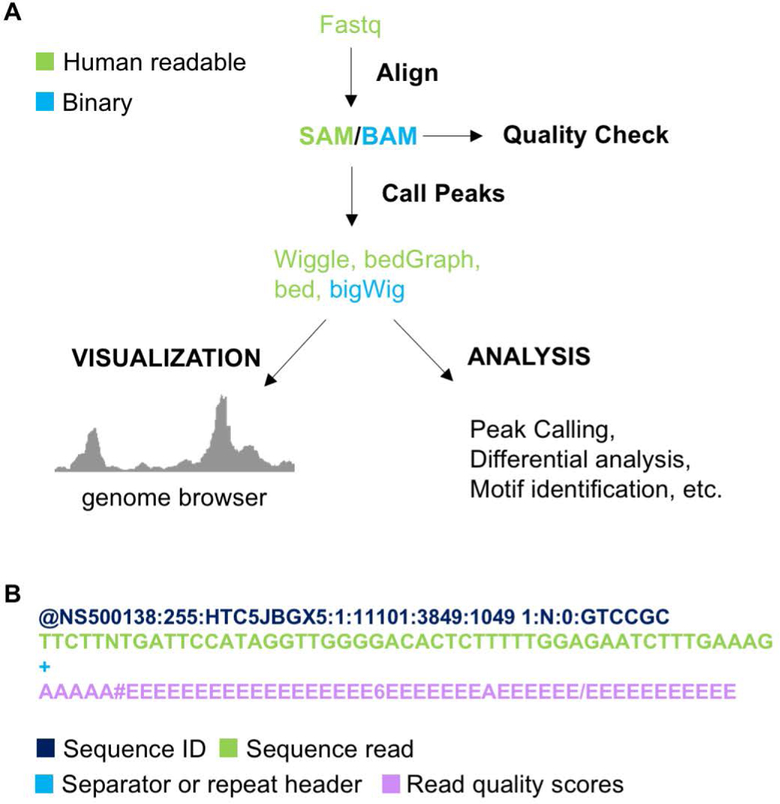
A) Data processing and analysis pipeline for ChIP-seq data. B) Fastq file format.
After alignment, measures should be taken to ensure libraries are of high quality. Alignment files should be checked for the total percentage of reads aligned to the reference genome. High quality libraries should have high percent alignment (Bailey et al., 2013; Nakato & Shirahige, 2017). Greater than 70% alignment is usually expected for high quality ChIP-seq libraries (Bailey et al., 2013). Additionally, duplicates can be marked using bioinformatic tools such as PICARD or SAMBLASTER (Faust & Hall, 2014; “Picard toolkit,”). The primary purpose of marking duplicates is to assess the complexity of libraries (Furey, 2012).
Sequencing reads can be filtered based on the quality of alignment. Alignment output files are in SAM file format and contain a MAPQ value. A higher MAPQ score is indicative of a higher quality read. Using software such as SAMtools (H. Li et al., 2009), reads can be filtered by MAPQ score to ensure only high quality reads are kept for further analysis (Jia et al., 2012). The chosen MAPQ cut off score varies by study but generally falls in the range of five to thirty (Anand et al., 2016; Brind’Amour et al., 2015; Guo et al., 2012; Rubin et al., 2017; Xu, DiCarlo, Satya, Peng, & Wang, 2014).
Once any desired filtering steps are complete, read depth of libraries should be checked. Libraries must be of sufficient read depth for robust data analysis. Recommended sequencing depth for transcription factors with human samples is 20 million aligned reads (Landt et al., 2012). For fly and worm libraries, around 5–10 million reads is generally the minimum (Furey, 2012; Landt et al., 2012) due to the smaller genome sizes.
A number of bioinformatic tools and packages are available for downstream processing and analysis of ChIP-seq data. ChIP-seq data can be visualized using genome browsers. Sam files from alignment can be converted to wiggle, bigWig, or bedGraph format. These files are readable by Integrative Genomics Viewer (IGV), UCSC genome browser and Ensemble genome browser (J. T. Robinson et al., 2011; Schmidt et al., 2009). In successful ChIP-seq experiments, sites where the protein of interest is stably binding should have sequence enrichment. Enriched regions can be visualized by scanning through browser tracks but can be more systematically identified by calling peaks using genomic tools. Peak calling identifies genomic locations where protein is enriched based on total sequences accumulated in the sample library relative to control (Farnham, 2009; Park, 2009). MACS, MACS2, PeakSeq, and SICER are software available for peak calling (Rozowsky et al., 2009; Zang et al., 2009; Zhang et al., 2008). Peak calling parameters should be carefully considered for each library as settings will influence accuracy of results. Peaks can be further filtered by P-value, FDR, and fold enrichment (Landt et al., 2012; Rozowsky et al., 2009; Zhang et al., 2008). The HOMER software suite can then be used for annotating peaks and for both de novo and known transcription-factor-binding motif identification (Heinz et al., 2010). To identify altered binding of chromatin-associated proteins in different conditions, differential peak calling software such as DESeq and edgeR are also available (Anders & Huber, 2010; M. D. Robinson, McCarthy, & Smyth, 2010).
While ChIP-seq has become a widely used technique, there are still limitations to its use. One limitation comes from formaldehyde as the choice of crosslinker. Formaldehyde has advantages including cell permeability, rapid reactivity, and reversibility as a crosslinker for protein-protein and protein-DNA interactions (Farnham, 2009; Hoffman, Frey, Smith, & Auble, 2015); however, its short spacer length makes it difficult to capture the chromatin association of many proteins that indirectly bind to DNA. Another limitation is resolution. Classic ChIP-seq is limited by the size of fragments generated by sonication making it difficult to determine exact, narrow protein binding sites (Fan, Lamarre-Vincent, Wang, & Struhl, 2008; Kadosh, 1998; Skene, Hernandez, Groudine, & Henikoff, 2014). ChIP-seq is also limited by total amount of input for successful library construction. For a successful ChIP-seq experiment, classic methods require use of millions of cells which makes it challenging to study small cell populations (Brind’Amour et al., 2015; Mikkelsen et al., 2007). Lastly, ChIP-seq is only able to indicate a two-dimensional interaction between protein and chromatin (Furey, 2012). To combat these limitations, there has been a push for development of ChIP-seq variations and an expansion of available ChIP-seq-quality antibodies. Several representative ChIP-seq variations that have been developed to address these challenges are discussed below (Table 1).
Table 1.
Summary of ChIP-seq variations and extensions.
| ChIP-seq Variations | Abbreviations | Technical Advances | References |
|---|---|---|---|
| Double crosslinking ChIP-seq | NA | Facilitate profiling of genomic binding sites for transient/indirect chromatin binding proteins | Zeng, Vakoc, Chen, Blobel, & Berger, 2006; Nowak, Tian, & Brasier, 2005 |
| Native ChIP-seq | N-ChIP-seq | Minimize indirect interactions, provide single-nucleosome-level resolution | Barski et al., 2007 |
| ChIP and lambda exonuclease digestion | ChIP-exo | Provide up to single-base-pair resolution for protein binding locations | Rhee & Pugh, 2011; Rossi, Lai, & Pugh, 2018 |
| High resolution crosslinking ChIP-seq | X-ChIP-seq | Profile protein binding sites at high resolution without crosslinking | Skene & Henikoff, 2015; Skene et al., 2014 |
| Recovery via protection ChIP-seq | RP-ChIP-seq | Include yeast chromatin as carrier to protect from DNA loss from using low input (~500 cells) | Zheng et al., 2015 |
| Favored amplification recovery via protection ChIP-seq | FARP-ChIP-seq | Include synthetic Biotin-DNA, which is amplifiable during library preparation, as carrier to protect from DNA loss using low input | Zheng et al., 2015 |
| Tn5 transposase tagmentation ChIP-seq | ChIPmentation | Incorporate on-bead tagmentation for fast and low-input (104-105 cells) ChIP-seq | Schmidl, Rendeiro, Sheffield, & Bock, 2015 |
| Microfluidic based ChIP-seq | MOWChIP-seq | Compatible with as few as 100 cells, allows library construction without preamplification | Cao, Chen, He, Tan, & Lu, 2015; Rotem et al., 2015 |
| Chromosome conformation capture | 3C | Characterize chromatin interactions using quantitative PCR, between single pairs of loci | Dekker et al., 2002 |
| Chromosome conformation capture-on-chip | 4C | Characterize chromatin interactions between one locus and other regions | Simonis et al., 2006 |
| Whole genome chromosome conformation capture | Hi-C | Characterize genome-wide chromatin interactions | Lieberman-Aiden et al., 2009 |
| ChIP mass spectrometry | ChIP-MS | Identify protein-protein interactions on chromatin, involving double crosslinking | Engelen et al., 2015 |
| Rapid immunoprecipitation mass spectrometry | RIME | Identify protein-protein interactions on chromatin, involving robust washing and on-bead digestion | Mohammed et al., 2016 |
| Selective isolation of chromatin associated proteins and ChIP | ChIP-SICAP | Simultaneously identify both interacting proteins and binding sequences for chromatin-associated proteins | Rafiee, Girardot, Sigismondo, & Krijgsveld, 2016 |
Variations of ChIP-seq
Modified crosslinking conditions
Many chromatin-associated proteins, such as histone modifiers, bind dynamically to chromatin without stable contact with DNA. To efficiently capture the genomic locations of these co-factors, a second crosslinker, usually with a longer spacer arm, is typically used in conjunction with formaldehyde (2 Å spacer arm) for “double crosslinking”. Several types of crosslinking reagents, such as the NHS-ester EGS (16.1 Å spacer arm) (Zeng, Vakoc, Chen, Blobel, & Berger, 2006) and DSG (7.7 Å spacer arm) (Bao et al., 2017; Nowak, Tian, & Brasier, 2005) have already been successfully incorporated in ChIP experiments.
On the other hand, Native ChIP-seq (N-ChIP-seq) (Figure 4A) is designed to minimize the indirect interactions identified by crosslinking. N-ChIP-seq requires no crosslinking of DNA (Park, 2009). Instead, native chromatin is sheared by Micrococcal nuclease (MNase) digestion. This allows for increased antibody specificity and eliminates any indirect interactions that may be identified using formaldehyde; however, this technique is generally only applicable to proteins with very strong affinity for DNA, such as histones (Barski et al., 2007). Most chromatin-associated proteins do not remain bound to DNA during digestion (O’Neill, 2003). Further advances with N-ChIP-seq have recently been made to allow for low input with as few as 103 cells, but this too is largely limited to identifying histone marks and nucleosome positions (Brind’Amour et al., 2015; Dahl & Gilfillan, 2018; Gilfillan et al., 2012).
Figure 4. Principles of Representative ChIP-seq Variations.
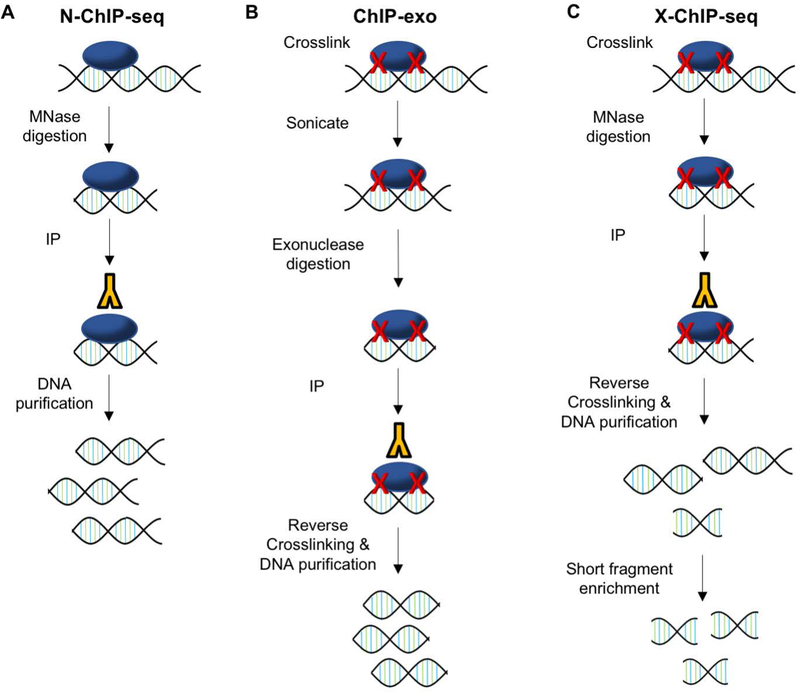
A) N-ChIP-seq uses MNase digestion of chromatin with no crosslinking or sonication step. B) ChIP-exo requires exonuclease digestion of crosslinked and sonicated chromatin. C) X-ChIP-seq starts with crosslinking but is followed by MNase digestion with no sonication. Libraries are also enriched for short fragments.
For Improved Resolution
Most transcription-factor binding sites are only 6–20 base pairs while DNA fragments in classical ChIP-seq are hundreds of base pairs long (Furey, 2012). Thus, a number of techniques have been developed to improve ChIP-seq resolution. One of the techniques made to address this issue is ChIP-exo (Figure 4B). ChIP-exo follows classic methods by starting with formaldehyde cross-linking and sonication, but after the sonication step, a 5’ to 3’ exonuclease is used to cut DNA sequences proximal to where protein was crosslinked to DNA. This method achieves close to single-nucleotide resolution and was shown to have low noise (Rhee & Pugh, 2011, 2012). The original ChIP-exo technique was published in 2011, and has since been updated to simplify methods and allow for lower input (Rossi, Lai, & Pugh, 2018). Similarly, X-ChIP-seq uses enzymatic digestion to improve resolution, but instead of sonicating first, this method applies MNase digestion to cells immediately after crosslinking, skipping the sonication step altogether. In addition, after DNA purification, X-ChIP enriches for short fragments prior to library preparation. This results in ~35bp resolution (Figure 4C) (Skene & Henikoff, 2015; Skene et al., 2014).
For Low Input
There has been a recent drive to reduce the required input for ChIP-seq, as many cell types are currently available only in low quantities. Several new methods have been recently developed to address this technical barrier. Recovery via protection (RP-ChIP-seq) uses carrier DNA from a different organism to prevent loss of DNA. A further modified version of this, favored amplification RP-ChIP-seq (FARP-ChIP-seq), uses synthetic, biotinylated DNA in place of carrier DNA from another organism. These methods were shown to be successful for identifying histone mark locations using as few as 500 cells (Zheng et al., 2015). Another recently developed method for low input ChIP is ChIPmentation. This method uses a hyperactive Tn5 transposase to perform on-bead chromatin fragmentation and sequencing adaptor integration, which simplifies the library construction workflow and improves ChIP-seq efficiency. ChIPmentation is a promising new technique for low input ChIP as it has worked not only for histone marks but also for GATA1 and CTCF transcription factors using only 100,000 cells as input (Schmidl, Rendeiro, Sheffield, & Bock, 2015).
Microfluidic based methods are also now being developed for low input ChIP-seq (Dahl & Gilfillan, 2018). Using only 100 cells, a microfluidic based ChIP-seq technique was successful for identifying the genomic locations of histone modifications. This technique is also known as microfluidic-oscillatory-washing-based ChIP-Seq (MOWChIP-seq), which leverages packed beads and oscillatory washing to achieve high ChIP efficiency, bypassing the pre-amplification step in library construction (Cao, Chen, He, Tan, & Lu, 2015). Finally, at the pinnacle of low input methods, ChIP-seq is being modified for analysis of single cells. Using a microfluidic system, single cell ChIP-seq analysis has been completed for histone marks (Rotem et al., 2015). While this is not an exhaustive list, it provides a summary of evolving technology for low input ChIP-seq.
Extensions of ChIP-seq
Higher-order chromatin structure plays an essential role in gene expression regulation (Dekker, Rippe, Dekker, & Kleckner, 2002). Built on the principle of ChIP, which was originally designed to identify two-dimensional protein-DNA interactions, chromosome conformation capture (3C) was developed in 2002 to characterize chromatin folding in the nucleus using quantitative PCR (Dekker et al., 2002). In 2006, chromosome conformation capture-on-chip (4C) was developed. Leveraging microarray technology, 4C provided a bigger picture of how one region of chromatin interacts with other chromatin regions (Simonis et al., 2006). Parallel to the development of ChIP-seq, chromosome capture evolved away from microarray and toward a genome wide analysis approach with the development of Hi-C which allowed for an unbiased view of all chromatin interactions (Lieberman-Aiden et al., 2009).
In combination with mass spectrometry, the principle of ChIP has also been adapted to identify protein-protein interactions on chromatin. Both ChIP mass spectrometry (ChIP-MS) and rapid immunoprecipitation mass spectrometry (RIME) have been developed to identify proteins associated with chromatin (Engelen et al., 2015; Mohammed et al., 2016). ChIP-MS involves crosslinking chromatin-associated proteins by both DSG and formaldehyde, as well as separating proteins on SDS-PAGE before protein identification by mass spectrometry. RIME uses only formaldehyde to crosslink interacting proteins, but incorporates vigorous washing and on-bead digestion before mass spectrometry. Furthermore, a method for simultaneously identifying interacting proteins and chromatin binding sites, Selective Isolation of Chromatin Associated Proteins combined with ChIP (ChIP-SICAP), has been developed. ChIP-SICAP involves biotin labeling of DNA fragments, and purification of protein complexes by both immunoprecipitation and subsequent DNA purification using streptavidin beads. Both DNA and peptides can be recovered from the same assay to be used for both ChIP-seq and mass spectrometry (Rafiee, Girardot, Sigismondo, & Krijgsveld, 2016). These methods expand on ChIP-seq by elucidating protein interaction networks at protein-DNA interaction sites.
The Future of ChIP-seq and Characterization of Chromatin Associated Proteins
While many variations have been developed to address the limitations of ChIP-seq, limitations of the method still exist. The application for low cell numbers still remains challenging, as both Native ChIP and low-input ChIP techniques were primarily optimized for abundant histone marks (Dahl & Gilfillan, 2018; Gilfillan et al., 2012). These methods will require further optimization before they can be applied to transcription factors or DNA binding cofactors. Advances in library construction and further development of sequencing technologies may begin to combat such limitations. Additionally, there remains the limitation of antibody quality and availability. Antibodies need be carefully chosen so that they are both efficient and specific (Furey, 2012; Kidder et al., 2011; Landt et al., 2012). At present, many chromatin-associated proteins still do not have ChIP-grade antibodies available commercially. In this case, tagging proteins of interest may be considered. It is important to point out that the location of the tag on the protein, N-terminal or C-terminal, may impact results by interfering with protein-protein or protein-DNA interactions. Tagged proteins must also be carefully regulated so that expression matches or is below endogenous levels (Kidder et al., 2011). Finally, determining precise spatial localization of protein-DNA interactions still presents a major challenge. While tools have been developed to identify three-dimensional interactions, we are still limited in our ability to determine where dynamic interactions are taking place within the three-dimensional nucleus. Continued efforts to enhance existing technologies will be imperative to advance our current understanding of chromatin organization and regulation of gene expression.
Significance Statement.
Chromatin structure and function is controlled by a myriad of proteins that dynamically associate with the genomic DNA. These proteins often occupy selective regions across the genome, modulating spatiotemporal gene expression. To determine the regulatory functions of these chromatin-associated proteins, a widely used technique at present is chromatin-immunoprecipitation sequencing (ChIP-seq). ChIP-seq involves chromatin fragmentation, antibody-mediated enrichment, and next-generation sequencing to pinpoint the genomic locations of protein-DNA interaction. In this manuscript, we will review the history, the methodology, and recent variations of ChIP-seq. We will also discuss how future development of ChIP-seq may further advance our understanding of chromatin biology.
Acknowledgments
This work is supported by a K99/R00 Award (R00AR065480) and Searle Leadership Fund to X. B., as well as a NIH CMBD T32 training grant (T32 GM008061) that supports S.M.L. as a trainee.
References:
- Anand S, Mangano E, Barizzone N, Bordoni R, Sorosina M, Clarelli F, … De Bellis G (2016). Next Generation Sequencing of Pooled Samples: Guideline for Variants’ Filtering. Sci Rep, 6, 33735. doi: 10.1038/srep33735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, & Huber W (2010). Differential expression analysis for sequence count data. Genome Biology, 11(R106). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey T, Krajewski P, Ladunga I, Lefebvre C, Li Q, Liu T, … Zhang J (2013). Practical guidelines for the comprehensive analysis of ChIP-seq data. PLoS Comput Biol, 9(11), e1003326. doi: 10.1371/journal.pcbi.1003326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao X, Siprashvili Z, Zarnegar BJ, Shenoy RM, Rios EJ, Nady N, … Khavari PA (2017). CSNK1a1 Regulates PRMT1 to Maintain the Progenitor State in Self-Renewing Somatic Tissue. Dev Cell, 43(2), 227–239 e225. doi: 10.1016/j.devcel.2017.08.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, … Zhao K (2007). High-resolution profiling of histone methylations in the human genome. Cell, 129(4), 823–837. doi: 10.1016/j.cell.2007.05.009 [DOI] [PubMed] [Google Scholar]
- Barski A, & Zhao K (2009). Genomic location analysis by ChIP-Seq. J Cell Biochem, 107(1), 11–18. doi: 10.1002/jcb.22077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd KE, & Farnham PJ (1997). Myc versus USF: Discrimination at the cad gene is determined by core promoter elements. Molecular and Cellular Biology, 17(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd KE, Wells J, Gutman J, Bartley SM, & Farnham PJ (1998). c-Myc target gene specificity is determined by a post-DNA-binding mechanism. Proc. Natl. Acad. Sci, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brind’Amour J, Liu S, Hudson M, Chen C, Karimi MM, & Lorincz MC (2015). An ultra-low-input native ChIP-seq protocol for genome-wide profiling of rare cell populations. Nat Commun, 6, 6033. doi: 10.1038/ncomms7033 [DOI] [PubMed] [Google Scholar]
- Brutlag D, Schlehuber C, & Bonner J (1969). Properties of Formaldehyde-Treated Nucleohistone. Biochemistry, 8(8), 3214–3218. [DOI] [PubMed] [Google Scholar]
- Buck MJ, & Lieb JD (2004). ChIP-chip: considerations for the design, analysis, and application of genome-wide chromatin immunoprecipitation experiments. Genomics, 83(3), 349–360. doi: 10.1016/j.ygeno.2003.11.004 [DOI] [PubMed] [Google Scholar]
- Cao Z, Chen C, He B, Tan K, & Lu C (2015). A microfluidic device for epigenomic profiling using 100 cells. Nat Methods, 12(10), 959–962. doi: 10.1038/nmeth.3488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl JA, & Gilfillan GD (2018). How low can you go? Pushing the limits of low-input ChIP-seq. Brief Funct Genomics, 17(2), 89–95. doi: 10.1093/bfgp/elx037 [DOI] [PubMed] [Google Scholar]
- Dahl JA, Reiner AH, & Collas P (2009). Fast genomic muChIP-chip from 1,000 cells. Genome Biol, 10(2), R13. doi: 10.1186/gb-2009-10-2-r13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker J, Rippe K, Dekker M, & Kleckner N (2002). Capturing Chromosome Conformation. Science, 295. [DOI] [PubMed] [Google Scholar]
- Engelen E, Brandsma JH, Moen MJ, Signorile L, Dekkers DH, Demmers J, … Poot RA (2015). Proteins that bind regulatory regions identified by histone modification chromatin immunoprecipitations and mass spectrometry. Nat Commun, 6, 7155. doi: 10.1038/ncomms8155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan X, Lamarre-Vincent N, Wang Q, & Struhl K (2008). Extensive chromatin fragmentation improves enrichment of protein binding sites in chromatin immunoprecipitation experiments. Nucleic Acids Res, 36(19), e125. doi: 10.1093/nar/gkn535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farnham PJ (2009). Insights from genomic profiling of transcription factors. Nat Rev Genet, 10(9), 605–616. doi: 10.1038/nrg2636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- FASTX-Toolkit. Retrieved from http://hannonlab.cshl.edu/fastx_toolkit/
- Faust GG, & Hall IM (2014). SAMBLASTER: fast duplicate marking and structural variant read extraction. Bioinformatics, 30(17), 2503–2505. doi: 10.1093/bioinformatics/btu314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furey TS (2012). ChIP-seq and beyond: new and improved methodologies to detect and characterize protein-DNA interactions. Nat Rev Genet, 13(12), 840–852. doi: 10.1038/nrg3306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilfillan GD, Hughes T, Sheng Y, Hjorthaug HS, Straub T, Gervin K, … Lyle R (2012). Limitations and possibilities of low cell number ChIP-Seq. BMC Genomics, 13(645). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmour DS, & Lis JT (1984). Detecting protein-DNA interactions in vivo: Distribution of RNA Polymerase on specific bacterial genes. Proc. Natl. Acad. Sci, 81, 4275–4279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmour DS, & Lis JT (1985). In vivo interactions of RNA Polymerase II with Genes of Drosophila melanogaster. Molecular and Cellular Biology, 5(8), 2009–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Long J, Li C, Cai Q, Shu X, Zheng W, & Li C (2012). Exome sequencing generates high quality data in non-target regions. BMC Genomics, 13(194). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gužvić M (2013). The History of DNA Sequencing / ISTORIJAT SEKVENCIRANJA DNK. Journal of Medical Biochemistry, 32(4), 301–312. doi: 10.2478/jomb-2014-0004 [DOI] [Google Scholar]
- Hecht A, Strahl-Bolsinger S, & Grunstein M (1996). Spreading of transcriptional repressor SIR3 from telomeric heterochromatin. Nature, 383(5). [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, … Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell, 38(4), 576–589. doi: 10.1016/j.molcel.2010.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho JWK, E., B., V., K. P., N., N., White KP, & Park PJ (2011). ChIP-chip versus ChIP-seq: Lessons for experimental design and data analysis. BMC Genomics, 12(134). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman EA, Frey BL, Smith LM, & Auble DT (2015). Formaldehyde crosslinking: a tool for the study of chromatin complexes. J Biol Chem, 290(44), 26404–26411. doi: 10.1074/jbc.R115.651679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia P, Li F, Xia J, Chen H, Ji H, Pao W, & Zhao Z (2012). Consensus rules in variant detection from next-generation sequencing data. PLoS One, 7(6), e38470. doi: 10.1371/journal.pone.0038470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DS, Mortazavi A, Myers RM, & Wold BJ (2007). Genome-Wide mapping of in vivo protein-DNA interactions. Science, 316, 1497–1502. [DOI] [PubMed] [Google Scholar]
- Kadosh D a. K. S. (1998). Targeted Recruitment of the Sin3-Rpd3 Histone Deacetylase Complex Generates a Highly Localized Domain of Represssed Chromatin In Vivo. Molecular and Cellular Biology, 18(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaunitz JD (2015). The Discovery of PCR: ProCuRement of Divine Power. Dig Dis Sci, 60(8), 2230–2231. doi: 10.1007/s10620-015-3747-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidder BL, Hu G, & Zhao K (2011). ChIP-Seq: technical considerations for obtaining high-quality data. Nat Immunol, 12(10), 918–922. doi: 10.1038/ni.2117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TH, & Ren B (2006). Genome-wide analysis of protein-DNA interactions. Annu Rev Genomics Hum Genet, 7, 81–102. doi: 10.1146/annurev.genom.7.080505.115634 [DOI] [PubMed] [Google Scholar]
- Landt SG, Marinov GK, Kundaje A, Kheradpour P, Pauli F, Batzoglou S, … Snyder M (2012). ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res, 22(9), 1813–1831. doi: 10.1101/gr.136184.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, & Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods, 9(4), 357–359. doi: 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, & Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol, 10(3), R25. doi: 10.1186/gb-2009-10-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, & Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics, 25(14), 1754–1760. doi: 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, … Subgroup, G. P. D. P. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics, 25(16), 2078–2079. doi: 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Yu C, Li Y, Lam TW, Yiu SM, Kristiansen K, & Wang J (2009). SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics, 25(15), 1966–1967. doi: 10.1093/bioinformatics/btp336 [DOI] [PubMed] [Google Scholar]
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, … Dekker J (2009). Comprehensive Mapping of Long-Range Interactions Reveals Folding Principles of the Human Genome. Science, 326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linnarsson S (2010). Recent advances in DNA sequencing methods - general principles of sample preparation. Exp Cell Res, 316(8), 1339–1343. doi: 10.1016/j.yexcr.2010.02.036 [DOI] [PubMed] [Google Scholar]
- Liu L, Hu N, Wang B, Chen M, Wang J, Tian Z, … Lin D (2011). A brief utilization report on the Illumina HiSeq 2000 sequencer. Mycology, 2(3). doi: 10.1080/21501203.2011.615871 [DOI] [Google Scholar]
- Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, … Rothberg JM (2005). Genome sequencing in microfabricated high-density picolitre reactors. Nature, 437(7057), 376–380. doi: 10.1038/nature03959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, … Bernstein BE (2007). Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature, 448(7153), 553–560. doi: 10.1038/nature06008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammed H, Taylor C, Brown GD, Papachristou EK, Carroll JS, & D’Santos CS (2016). Rapid immunoprecipitation mass spectrometry of endogenous proteins (RIME) for analysis of chromatin complexes. Nat Protoc, 11(2), 316–326. doi: 10.1038/nprot.2016.020 [DOI] [PubMed] [Google Scholar]
- Nakato R, & Shirahige K (2017). Recent advances in ChIP-seq analysis: from quality management to whole-genome annotation. Brief Bioinform, 18(2), 279–290. doi: 10.1093/bib/bbw023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Institutes of Health. (2003). International Consortium Completes Human Genome Project. All Goals Achieved; New Vision for Genome Research Unveiled Retrieved from https://www.genome.gov/11006929/2003-release-international-consortium-completes-hgp/
- Nowak DE, Tian B, & Brasier AR (2005). Two-step cross-linking method for identification of NF-kappaB gene network by chromatin immunoprecipitation. Biotechniques, 39(5), 715–725. doi: 10.2144/000112014 [DOI] [PubMed] [Google Scholar]
- O’Neill L (2003). Immunoprecipitation of native chromatin: NChIP. Methods, 31(1), 76–82. doi: 10.1016/s1046-2023(03)00090-2 [DOI] [PubMed] [Google Scholar]
- Orlando V (2000). Mapping chromosomal proteins in vivo by formaldehyde-crosslinked-chromatin immunoprecipitation. Trends in Biochemical Sciences, 25, 99–104. [DOI] [PubMed] [Google Scholar]
- Orlando V, & Paro R (1993). Mapping Polyomb-repressed domains in the bithorax complex using in vivo formaldehyde cross-linked chromatin. Cell, 75, 1187–1198. [DOI] [PubMed] [Google Scholar]
- Palhan VB, Kreader C, & Mueller E (2017). Cells for chromatin immunoprecipitation and methods for making [Google Scholar]
- Park PJ (2009). ChIP-seq: advantages and challenges of a maturing technology. Nat Rev Genet, 10(10), 669–680. doi: 10.1038/nrg2641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pchelintsev NA, Adams PD, & Nelson DM (2016). Critical Parameters for Efficient Sonication and Improved Chromatin Immunoprecipitation of High Molecular Weight Proteins. PLoS One, 11(1), e0148023. doi: 10.1371/journal.pone.0148023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard toolkit. Retrieved from http://broadinstitute.github.io/picard/
- Rafiee MR, Girardot C, Sigismondo G, & Krijgsveld J (2016). Expanding the Circuitry of Pluripotency by Selective Isolation of Chromatin-Associated Proteins. Mol Cell, 64(3), 624–635. doi: 10.1016/j.molcel.2016.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren B, Robert F, Wyrick JJ, Aparicio O, Jennings EG, Simon I, … Young RA (2000). Genome-Wide location and function of DNA binding proteins. Science, 290, 2306–2309. [DOI] [PubMed] [Google Scholar]
- Renaud G, Stenzel U, Maricic T, Wiebe V, & Kelso J (2015). deML: robust demultiplexing of Illumina sequences using a likelihood-based approach. Bioinformatics, 31(5), 770–772. doi: 10.1093/bioinformatics/btu719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee HS, & Pugh BF (2011). Comprehensive genome-wide protein-DNA interactions detected at single-nucleotide resolution. Cell, 147(6), 1408–1419. doi: 10.1016/j.cell.2011.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee HS, & Pugh BF (2012). ChIP-exo method for identifying genomic location of DNA-binding proteins with near-single-nucleotide accuracy. Curr Protoc Mol Biol, Chapter 21, Unit 21 24. doi: 10.1002/0471142727.mb2124s100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson G, Hirst M, Bainbridge M, Bilenky M, Zhao Y, Zeng T, … Jones S (2007). Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat Methods, 4(8), 651–657. doi: 10.1038/nmeth1068 [DOI] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, & Mesirov JP (2011). Integrative genomics viewer. Nature Biotechnology, 29(1). doi: 10.1038/nbt0111-24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, & Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics, 26(1), 139–140. doi: 10.1093/bioinformatics/btp616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi MJ, Lai WKM, & Pugh BF (2018). Simplified ChIP-exo assays. Nat Commun, 9(1), 2842. doi: 10.1038/s41467-018-05265-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotem A, Ram O, Shoresh N, Sperling RA, Goren A, Weitz DA, & Bernstein BE (2015). Single-cell ChIP-seq reveals cell subpopulations defined by chromatin state. Nat Biotechnol, 33(11), 1165–1172. doi: 10.1038/nbt.3383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozowsky J, Euskirchen G, Auerbach RK, Zhang ZD, Gibson T, Bjornson R, … Gerstein MB (2009). PeakSeq enables systematic scoring of ChIP-seq experiments relative to controls. Nat Biotechnol, 27(1), 66–75. doi: 10.1038/nbt.1518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin AJ, Barajas BC, Furlan-Magaril M, Lopez-Pajares V, Mumbach MR, Howard I, … Khavari PA (2017). Lineage-specific dynamic and pre-established enhancer-promoter contacts cooperate in terminal differentiation. Nat Genet, 49(10), 1522–1528. doi: 10.1038/ng.3935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanger F, Nicklen S, & Coulson AR (1977). DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci, 74(12), 5463–5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schena M, Shalon D, Davis RW, & Brown PO (1995). Quantitative Monitoring of Gene Expression Patterns with Complementary DNA Microarray. Science, 270(5235), 467–470. [DOI] [PubMed] [Google Scholar]
- Schmidl C, Rendeiro AF, Sheffield NC, & Bock C (2015). ChIPmentation: fast, robust, low-input ChIP-seq for histones and transcription factors. Nat Methods, 12(10), 963–965. doi: 10.1038/nmeth.3542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt D, Wilson MD, Spyrou C, Brown GD, Hadfield J, & Odom DT (2009). ChIP-seq: using high-throughput sequencing to discover protein-DNA interactions. Methods, 48(3), 240–248. doi: 10.1016/j.ymeth.2009.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonis M, Klous P, Splinter E, Moshkin Y, Willemsen R, de Wit E, … de Laat W (2006). Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C). Nat Genet, 38(11), 1348–1354. doi: 10.1038/ng1896 [DOI] [PubMed] [Google Scholar]
- Skene PJ, & Henikoff S (2015). A simple method for generating high-resolution maps of genome-wide protein binding. Elife, 4, e09225. doi: 10.7554/eLife.09225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skene PJ, Hernandez AE, Groudine M, & Henikoff S (2014). The nucleosomal barrier to promoter escape by RNA polymerase II is overcome by the chromatin remodeler Chd1. Elife, 3, e02042. doi: 10.7554/eLife.02042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas PS (1980). Hybridization of denatured RNA and small DNA fragments transferred to nitrocellulose. Proc. Natl. Acad. Sci, 77(9), 5201–5205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truax AD, & Greer SF (2012). ChIP and Re-ChIP assays: investigating interactions between regulatory proteins, histone modifications, and the DNA sequences to which they bind. Methods Mol Biol, 809, 175–188. doi: 10.1007/978-1-61779-376-9_12 [DOI] [PubMed] [Google Scholar]
- Watson JD, & Crick FHC (1953). Molecular structure of nucleic acids. Nature, 171. [DOI] [PubMed] [Google Scholar]
- Wen H, Li Y, Xi Y, Jiang S, Stratton S, Peng D, … Shi X (2014). ZMYND11 links histone H3.3K36me3 to transcription elongation and tumour suppression. Nature, 508(7495), 263–268. doi: 10.1038/nature13045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, DiCarlo J, Satya R, Peng Q, & Wang Y (2014). Comparison of somatic mutation calling methods in amplicon and whole exome sequence data. BMC Genomics, 15(244). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zang C, Schones DE, Zeng C, Cui K, Zhao K, & Peng W (2009). A clustering approach for identification of enriched domains from histone modification ChIP-Seq data. Bioinformatics, 25(15), 1952–1958. doi: 10.1093/bioinformatics/btp340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng PY, Vakoc CR, Chen ZC, Blobel GA, & Berger SL (2006). In vivo dual cross-linking for identification of indirect DNA-associated proteins by chromatin immunoprecipitation. Biotechniques, 41(6), 694, 696, 698. doi: 10.2144/000112297 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, … Liu XS (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol, 9(9), R137. doi: 10.1186/gb-2008-9-9-r137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Yue S, Chen H, Weber B, Jia J, & Zheng Y (2015). Low-Cell-Number Epigenome Profiling Aids the Study of Lens Aging and Hematopoiesis. Cell Rep, 13(7), 1505–1518. doi: 10.1016/j.celrep.2015.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong J, Ye Z, Lenz SW, Clark CR, Bharucha A, Farrugia G, … Lee JH (2017). Purification of nanogram-range immunoprecipitated DNA in ChIP-seq application. BMC Genomics, 18(1), 985. doi: 10.1186/s12864-017-4371-5 [DOI] [PMC free article] [PubMed] [Google Scholar]