

SUMMARY
Phosphatase and tensin homolog deleted on chromosome 10 (PTEN) is a tumor suppressor and bifunctional lipid and protein phosphatase. We report that the metabolic regulator pyruvate dehydrogenase kinase1 (PDHK1) is a synthetic-essential gene in PTEN-deficient cancer and normal cells. The PTEN protein phosphatase dephosphorylates nuclear factor κB (NF-κB)-activating protein (NKAP) and limits NFκB activation to suppress expression of PDHK1, a NF-κB target gene. Loss of the PTEN protein phosphatase upregulates PDHK1 to induce aerobic glycolysis and PDHK1 cellular dependence. PTEN-deficient human tumors harbor increased PDHK1, a biomarker of decreased patient survival. This study uncovers a PTEN-regulated signaling pathway and reveals PDHK1 as a potential target in PTEN-deficient cancers.
Graphical Abstract
In Brief
The tumor suppressor PTEN is widely inactivated in cancers and tumor syndromes. Currently, there is no effective therapeutic strategy in the clinic for PTEN-deficient cancers. Chatterjee et al. found that PTEN-deficient cells and cancers are uniquely sensitive to PDHK1 inhibition and propose PDHK1 as a potential therapeutic target in PTEN-deficient cancers.
INTRODUCTION
Phosphatase and tensin homolog deleted on chromosome 10 (PTEN) is a tumor suppressor with both lipid and protein phosphatase activities (Li and Sun, 1997; Li et al., 1997; Maehama and Dixon, 1998, 1999, 2000; Myers et al., 1997, 1998; Steck et al., 1997). As a lipid phosphatase, PTEN antagonizes PI3K/AKT/mTOR signaling through de-phosphorylation of the lipid second messenger PIP3 (phosphatidylinositol-3,4,5 triphosphate) to PIP2 (phosphatidylinositol-4,5 bisphosphate), regulating cellular metabolism, growth, and survival (Chalhoub and Baker, 2009; Lee et al., 1999; Maehama and Dixon, 1998; Myers et al., 1998; Stambolic et al., 1998).
PTEN can also function through protein phosphatase-dependent mechanisms (Davidson et al., 2010; Gildea et al., 2004; Leslie et al., 2009; Myers et al., 1997). A growing body of literature supports biological roles for the PTEN protein phosphatase (Dey et al., 2008; Gu et al., 2011; Hlobilkova et al., 2000; Leslie et al., 2007; Shi et al., 2014; Shinde and Maddika, 2016; Tibarewal et al., 2012; Wozniak et al., 2017; You et al., 2015). The modest clinical activity of PI3K and AKT inhibitors in PTEN-deficient cancers (Chandarlapaty et al., 2011; Ghosh et al., 2013; Rodon et al., 2013) coupled with the existence of tumor-derived PTEN Y138 mutants, which specifically lack the protein phosphatase activity (Davidson et al., 2010; Tibarewal et al., 2012), suggest that protein phosphatase-dependent cellular processes may contribute to PTEN-mediated tumor suppression. We investigated activity-specific PTEN functions to identify vulnerabilities for therapeutic exploitation to improve the treatment of PTEN-deficient cancers.
RESULTS
PTEN Deficiency Upregulates PDHK1 Expression in Normal and Cancer Cells
To uncover molecular events through which PTEN functions, we investigated gene expression changes in PTEN-deficient human lung adenocarcinoma H1650 cells (Table S1) in comparison with the matched cell line into which wild-type (WT) PTEN was re-introduced. Stable PTEN re-expression suppressed phospho-AKT levels (Figure 1A), as expected (Sos et al., 2009). By unbiased comparative gene expression profiling analysis, we identified 52 significantly differentially regulated genes (fold change > 2, p < 0.05, q < 0.2) between the PTEN re-expressing H1650 and PTEN-deficient H1650-GFP control cells (Figure 1A; Table S2). Re-introduction of PTEN in H1650 cells caused significant down-regulation of 11 genes and upregulation of 41 genes. We found several energy metabolism genes including pyruvate dehydrogenase kinase 1 (PDHK1; gene name PDK1) among the genes that showed significant upregulation (>2-fold increase, p < 0.05, q < 0.2) in the PTEN-deficient H1650-GFP cells (Figure 1A, red boxes).
Figure 1. PTEN Loss or Inactivation Upregulates PDHK1 in Cancer and Normal Cells.

(A) Left: western blots showing PTEN and phospho-AKT expression in PTEN-deficient H1650 cancer cells stably expressing PTEN or empty vector (EV). Right: list of genes significantly upregulated (fold change > 2, multiple t tests, *p < 0.05, q < 0.2, n = 3 replicates) in GFP-expressing H1650 cells compared with H1650 cells stably re-expressing PTEN, by microarray analysis. Highlighted in red boxes are the top upregulated energy metabolism genes, including PDHK1, in H1650-GFP cells.
(B) Western blots showing PTEN, phospho-AKT, and PDHK1 expression in PTEN-deficient cancer cell lines stably expressing PTEN or empty vector (EV). (C and D) Same as (B) in PTEN-proficient cancer (C) or normal (non-cancer) (D) cell lines with stable PTEN knockdown. shPTEN, shRNA to PTEN; SC, scrambled control shRNA.
PDHK1 is a regulator of energy metabolism in cells (Schulze and Downward, 2011) with no known connection to PTEN, unlike the other top hit, 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 4 (PFKFB4) (Figure 1A, red boxes), which regulates AKT, a known downstream effector of PTEN (Chesney et al., 2014; Figueiredo et al., 2017; Houddane et al., 2017; Sun et al., 1999). PDHK1 and other PDHK isoenzymes (PDHK2–4) phosphorylate the E1 alpha subunit (PDHA1) of the pyruvate dehydrogenase complex (PDC) that catalyzes oxidative decarboxylation of pyruvate to acetyl-CoA in mitochondria (Patel and Roche, 1990; Popov et al., 1997; Teague et al., 1979). Phosphorylation of PDHA1 inhibits PDC activity and blocks pyruvate entry into the tricarboxylic acid (TCA) cycle to uncouple glycolysis from the TCA cycle, contributing to the Warburg effect (Korotchkina and Patel, 2001; Linn et al., 1969; Vander Heiden et al., 2009; Warburg, 1956). Analysis of an independently conducted large-scale RNAi screening project (http://www.broadinstitute.org/achilles) showed that three of four PDHK1-targeted short hairpin RNAs (shRNAs) were depleted in H1650 cells, in which we observed PDHK1 upregulation (Figure 1A). These findings suggest that PDHK1 expression may be essential for PTEN-deficient H1650 cell growth. We hypothesized PDHK1 is an unrecognized PTEN effector through which PTEN regulates cellular energy metabolism and survival.
We assessed whether PDHK1 expression was regulated by PTEN across a panel of cancer and normal cell lines that either lack or stably express PTEN (Table S1) (Aguissa-Touré and Li, 2012; Koul, 2008; Lee et al., 2014; Pourmand et al., 2007; Sos et al., 2009). PDHK1 expression was decreased by PTEN re-expression in multiple PTEN-deficient models, such as lung adenocarcinoma (H1650), lung squamous cell carcinoma (H520), glioblastoma (U87MG), melanoma (A2058), prostate adenocarcinoma (PC3), and renal carcinoma cells (786-O) (Figures 1B and S1A; Table S1). PDHK1 expression was increased upon PTEN silencing in cancer cells and normal (non-cancer) cells that are otherwise PTEN proficient, such as lung adenocarcinoma (HCC827, H1975) and non-cancer cells (Beas2B, HEK293T) (Figures 1C, 1D, and S1A; Table S1). These findings indicate an inverse relationship in which PTEN status controls PDHK1 expression. Unlike PDHK1, other PDHK isoforms, including PDHK2, 3, and 4, exhibited lower expression in cancer cells, consistent with earlier studies (Grassian et al., 2011), and were not significantly modulated by PTEN levels (Figure S1B), suggesting specificity in the link between PTEN and PDHK1. Thus, PTEN loss specifically induces PDHK1 expression in cells.
Using an independently generated dataset (Pathak et al., 2013), we corroborated these findings in vivo and in non-malignant tissue. PDHK1 levels were higher in normal murine lung epithelium in which PTEN was genetically inactivated compared with PTENWT-expressing control mice (Figure S1C), suggesting physiological regulation of PDHK1 expression by PTEN in vivo.
We investigated whether PTEN-deficient human cancer specimens harbor increased PDHK1 levels. By immunohistochemistry (IHC) analysis, we detected a significant inverse relationship between PDHK1 and PTEN expression levels in multiple human tumor types with frequent PTEN inactivation, including glioblastoma, prostate adenocarcinoma, and lung squamous cell carcinoma (Figures S2A–S2F). We found a strong association between PTEN inactivation and increased PDHK1 expression in a pan-cancer analysis across 12 different tumor types in The Cancer Genome Atlas (TCGA) dataset (Figure S2G). These data suggest clinical relevance of the PTEN/PDHK1 inverse relationship.
Analysis of the range of PDHK1 expression levels in these PTEN-deficient cancers showed that higher PDHK1 expression was a biomarker of decreased patient survival, suggesting that PDHK1 may contribute to PTEN’s tumor suppressor function (Figure S2H). Our data reveal that PTEN loss promotes PDHK1 expression in malignant and normal cells and tissues and uncover increased PDHK1 levels in PTEN-deficient tumors as a biomarker of worse patient survival.
PTEN and PDHK1 Co-suppression Confers Synthetic Lethality in Normal and Cancer Cells
We investigated whether the PDHK1 upregulation induced by PTEN inactivation in cells is important for growth and survival. By silencing PDHK1 (using shRNAs to knockdown PDHK1) in PTEN-deficient and PTEN-proficient cells (Figures 2A, 2B, S3A, and S3B), we found that PDHK1 was essential for the survival of PTEN-deficient but not PTEN-expressing normal and cancer cells. The synthetic lethality conferred upon by PDHK1 co-suppression with PTEN loss in PTEN-deficient cells was rescued by re-expression of shRNA-resistant PDHK1 (Figure 2C).
Figure 2. PTEN Loss or Inactivation Induces Cellular Dependence on PDHK1 for Survival.

(A and B) Effects of stable PDHK1 knockdown in PTEN-deficient cancer (H1650) (A) or PTEN-proficient cancer (HCC827) and normal (Beas2B) cell lines (B) on cell growth by crystal violet staining assay (left) and apoptosis induction as measured by cleaved PARP levels by immunoblot analysis (right) are shown. shPDHK1#1 and shPDHK1#2, shRNAs to PDHK1; shScramble, scrambled control shRNA.
(C) Effects of stable PDHK1 knockdown and adenovirus-based shRNA resistant PDHK1 re-expression in PTEN-deficient H1650 cancer cells on cell growth by crystal violet staining assay (top) or CellTiter-Glo luminescent assay (middle) and apoptosis induction as measured by cleaved PARP levels by immunoblot analysis (bottom) are shown. shPDHK1#1 and shPDHK1#2, shRNAs to PDHK1; SC, scrambled control shRNA. Data are shown as mean ± SEM (n = 8 replicates). *p < 0.05 and ****p < 0.0001; n.s., not significant compared with “scrambled control shRNA expressing PTEN-deficient cells” by Dunn’s multiple-comparisons one-way ANOVA.
(D and E) Effects of transient PDHK1 knockdown in PTEN-proficient cancer and normal cell lines with or without stable PTEN knockdown on cell viability by crystal violet staining assay (D) and apoptosis induction as measured by cleaved PARP levels by immunoblotting (E) are shown. siPDHK1, PDHK1 specific small interfering RNAs; sc, scrambled control siRNA; shPTEN, shRNA to PTEN; SC, scrambled control shRNA. Data are shown as mean ± SEM (n = 4 replicates). *p < 0.05, **p < 0.01, and ****p < 0.0001; n.s., not significant compared with “scrambled control siRNA and shRNA expressing PTEN-proficient cells” by Tukey’s multiple-comparisons one-way ANOVA.
(F) Effects of stable PDHK1 knockdown in PTEN-deficient cancer cell lines stably expressing PTEN or empty vector (EV) on apoptosis induction as measured by cleaved PARP levels by immunoblotting are shown. shPDHK1, shRNA to PDHK1; SC, scrambled control shRNA. See also Figures S3–S6.
We confirmed that treatment with the PDHK1 inhibitor DCA (dichloroacetate) (Kato et al., 2007; Stacpoole, 1989) suppressed phosphorylation of the PDHK1 target PDHA1, as expected (Whitehouse et al., 1974), in PTEN-deficient cells (Figure S3C). Although DCA may have off-target effects, the primary activity in the PTEN-deficient cells is likely via PDHK1 inhibition in these cells, as other known targets of DCA (e.g., PDHK2–4) were not expressed or not PTEN responsive in these systems (Figure S1B). PTEN status specifically dictated sensitivity to PDHK1 inhibition but not to cytotoxic chemotherapies (Figure S3D).
We next used the genetically controlled system of paired cell lines that stably lack or express PTEN. PDHK1 knockdown significantly decreased survival in normal and cancer cells specifically during PTEN co-suppression (Figure 2D). PDHK1 and PTEN co-suppression was lethal in cancer and normal cells, with evidence of enhanced apoptosis (Figures 2E and 2F). PDHK1 inhibition with DCA decreased phospho-PDHA1 levels, as expected (Whitehouse et al., 1974), and increased apoptosis specifically in PTEN-deficient cells with increased PDHK1 (Figure S3E). Thus, PDHK1 is essential for survival in PTEN-deficient cells.
We investigated the underlying mechanisms of the observed synthetic lethality and apoptosis induction by PTEN and PDHK1 co-suppression in normal and cancer cells. This lethality induced by PDHK1 suppression in PTEN-deficient cells is likely due to a loss of mitochondrial membrane potential (measured by mitochondrial matrix pH using an established ratiometric pH-sensitive probe SypHer-dmito; Perry et al., 2011; Poburko et al., 2011; Suzuki et al., 2014) upon PDHK1 inhibition, consistent with prior work (Bonnet et al., 2007) (Figure S4A). We found that A2058 PTEN-null melanoma cells exhibited higher mitochondrial matrix pH (higher SypHer-dmito ratio [F470/F430]), indicative of higher mitochondrial membrane potential, compared with A2058 cells with stable PTEN re-expression. These data are consistent with studies showing that PTEN-null mouse embryonic fibroblasts (MEFs) have higher mitochondrial membrane potential compared with MEFs with PTEN reconstitution (Goo et al., 2012). Upon acute inhibition of PDHK1 by DCA in PTEN-deficient cells, the mitochondrial membrane potential decreased (Figure S4A), consistent with data in other cancer cells (Bonnet et al., 2007). This decreased mitochondrial membrane potential upon PDHK1 suppression in PTEN-deficient cells likely underlies cell death (Figures 2 and S3). As a consequence of the mitochondrial membrane potential depolarization upon PDHK1 suppression in PTEN-deficient cells (Figure S4A), we found release of the apoptotic effector cytochrome c into the cytosol from the mitochondria (Figure S4B), activation of caspases 9, 7, and 3 (Figure S4C), and enhanced ROS (reactive oxygen species) generation (Figure S4D) that induce apoptosis (Figures 2 and S3). We also found that the decreased cell viability upon PDHK1 inhibition in PTEN-deficient cells was rescued by the stable overexpression of anti-apoptotic factors Bcl-xL or Bcl2 (Figures S4E and S4F), which can prevent cell death by limiting loss of mitochondrial membrane potential, release of cytochrome c into the cytoplasm, and caspase activation (Chinnaiyan et al., 1996; Shimizu et al., 1996a, 1996b, 1998; Tsujimoto, 1998). These data reveal a role for PTEN in controlling mitochondrial integrity and cell survival via PDHK1 regulation. When PDHK1 is inhibited in PTEN-deficient cells, multiple mechanisms contribute to cell death.
PDHK1 inhibition by DCA treatment also suppressed colony formation in vitro specifically in PTEN-deficient lung adenocarcinoma cells (Figure S5A) and tumor growth in vivo in PTEN-deficient melanoma xenograft models (Figure S5B). These data indicate PDHK1 is conditionally essential for survival specifically in PTEN-deficient cells and tumors.
PDHK1 Upregulation and Activation upon PTEN Loss Promotes Aerobic Glycolysis
We explored the functional consequences of PDHK1 upregulation induced by PTEN inactivation. PTEN and PDHK1 each can regulate energy metabolism but are not known to function together (Gottlob et al., 2001; Jang et al., 2013; Semenza, 2008). PDHK1 activation can promote aerobic glycolysis, through which pyruvate is converted to lactate to produce ATP, by phosphorylating and inactivating PDHA1 and blocking pyruvate entry into the TCA cycle (Patel et al., 2014). We investigated the metabolic effects of PDHK1 upregulation in cells with PTEN deficiency.
Silencing PTEN in cells induced L-lactate secretion, a hallmark of aerobic glycolysis (Figure S6A). PTEN-deficient cells exhibited a marked (4- to 5-fold) increase in L-lactate production in comparison with when these cells were grown in the presence of 2-DG (2-deoxyglucose, a glucose derivative that cannot undergo further glycolysis; Schulze and Harris, 2012; Zhao et al., 2013) that suppresses lactate production independently (Figure S6B). PDHK1 suppression by shRNA or by DCA treatment or by stable PTEN re-expression in PTEN-deficient cell lines decreased L-lactate production (Figure S6B). These data suggest that PDHK1 upregulation and activation that occurs upon loss of PTEN promotes aerobic glycolysis in cells.
Treatment with rotenone (Palmer et al., 1968), an inhibitor of mitochondrial electron transport chain complex I (Figure S6C), or with oligomycin (Lardy et al., 1958), an inhibitor of mitochondrial ATP synthase (Figure S6D), resulted in greater inhibition of ATP production among PTEN-proficient cells but not PTEN-deficient cells. These data suggest that PTEN-deficient cells rely less on oxidative phosphorylation for ATP production compared with PTEN-proficient cells.
Because increased PDHK1 promoted aerobic glycolysis in PTEN-deficient cells (Figure S6B), we tested whether PTEN inactivation enhances cellular glucose dependence, a hallmark of aerobic glycolysis (Lunt and Vander Heiden, 2011). The growth of PTEN-deficient but not PTEN-proficient cells was significantly decreased in glucose-deficient media, an effect rescued by stable PTEN expression (Figures S6E and S6F). These collective data indicate a metabolic shift toward aerobic glycolysis upon PTEN loss.
PTEN Protein Phosphatase Deficiency Activates PDHK1 Independent of PI3K/AKT and Induces Vulnerability to PDHK1 Inhibition
We next investigated the mechanism by which PTEN regulates PDHK1 expression. We determined the requirement of the distinct lipid and protein phosphatase activities of PTEN in the regulation of PDHK1. We leveraged established cancer-derived PTEN mutants that abrogate either PTEN’s lipid phosphatase activity (PTENG129E) or its protein phosphatase activity (PTENY138L) or both (PTENC124S) (Davidson et al., 2010; Myers et al., 1997, 1998; Rodríguez-Escudero et al., 2011; Tibarewal et al., 2012). By stably expressing each of these PTEN mutants or PTENWT in a controlled system of PTEN-deficient cells, we found that the protein phosphatase, but not the lipid phosphatase, activity of PTEN regulates PDHK1 mRNA expression (Figure 3A). We confirmed that both mRNA and protein expression levels of PDHK1 were diminished upon expression of PTENWT or the PTEN mutant that retains the protein phosphatase activity only (PTENG129E, lipid phosphatase mutant) in comparison with PTEN-deficient parental cells (Figures 3A and S7A). PDHK1 protein levels were unaffected by expression of the PTEN mutant lacking both phosphatase activities (PTENC124S), suggesting that PDHK1 regulation by PTEN is phosphatase activity dependent and not via other non-enzymatic properties of PTEN (Freeman et al., 2003; Planchon et al., 2008; Shen et al., 2007)(Figure S7A).
Figure 3. PTEN Protein Phosphatase Represses PDHK1 Independent of PI3K/AKT, and PTEN Protein Phosphatase Deficiency Renders PDHK1 Essential for Cell Survival.

(A) Real-time qRT-PCR analysis of PDHK1 mRNA expression in PTEN-deficient A2058 cancer cells stably expressing PTENWT or PTENG129E or PTENY138L or GFP. Data are shown as mean ± SD (n = 2 replicates). ****p < 0.0001; n.s., not significant compared with “GFP-expressing PTEN-deficient cells” by Tukey’s multiple-comparisons one-way ANOVA.
(B) Western blots showing phospho-AKT and PDHK1 expression in PTEN-deficient cancer cell lines in response to 1 μM BKM-120 (PI3-kinase inhibitor) or vehicle (DMSO) treatment for 24 h.
(C) Western blots showing phospho-AKT, phospho-S6(mTOR effector), and PDHK1 expression in PTEN-proficient cancer cell lines expressing empty vector (EV) or myristoylated-AKT (Myr-AKT) to constitutively activate AKT signaling.
(D) Effects of pharmacologic inhibition of PDHK1 with DCA (dose response: 5, 10, and 20 mM) in PTEN-deficient cancer cell lines stably expressing PTENW7 or PTENG129E or PTENY138L or GFP on cell growth by crystal violet staining assay are shown, with quantification of cell viability in 20 mM DCA treatment relative to vehicle (water) treatment reported as rescue score (STAR Methods).
See also Figure S7.
Because PDHK1 levels were unaffected by the presence or absence of the PTEN lipid phosphatase, we investigated whether PDHK1 activation in PTEN-deficient cells was dependent upon PI3K/AKT signaling. Pharmacologic inhibition of PI3K or AKT in PTEN-deficient cells treated with BKM-120 (Burger et al., 2011) or MK2206 (Hirai et al., 2010), respectively, suppressed AKT activation (phospho-AKT levels) and expression of hexokinase 2 (HK2) (Figures S7B and S7C), which is upregulated in PTEN-deficient cells (Wang et al., 2014). In contrast, the expression of PDHK1 was unaffected under these conditions of PI3K/AKT blockade (Figures 3B and S7B–S7D).
AKT inactivates glycogen synthase kinase 3 α/β (GSK3α/β) through phosphorylation (Cross et al., 1995). Phospho-GSK3α/β levels were suppressed in PTEN-deficient cell lines by BKM-120 treatment, confirming the efficacy of the inhibitor; PDHK1 expression was not significantly affected (Figure S7D). Treatment with BKM-120 (Burger et al., 2011) and another PI3K inhibitor, GDC0941 (Folkes et al., 2008), suppressed phosphorylation of ribosomal protein S6, which is downstream of both AKT and mTORC1, in three different PTEN-deficient cell lines tested and consistent with prior studies (Neshat et al., 2001), without decreasing PDHK1 levels (Figures S7D and S7E).
Ectopic expression of a constitutively active form of AKT (Myr-AKT) (Fulton et al., 1999; Sun et al., 2014), while increasing phospho-S6 levels (Wittenberg et al., 2016), did not increase PDHK1 levels in PTEN-expressing cells (Figure 3C). Thus, PDHK1 is upregulated specifically by PTEN protein phosphatase inactivation, and in a PI3K/AKT-independent manner.
We further found that it was the protein phosphatase, but not the lipid phosphatase, activity of PTEN that was required to rescue PTEN-deficient cells from the lethal effects of PDHK1 inhibition (Figure 3D). These collective data indicate that PDHK1 is conditionally essential for the survival of specifically PTEN protein phosphatase-deficient cells.
PTEN Protein Phosphatase Suppresses Nuclear Factor κB (NF-κB) Activation to Repress PDHK1
We next investigated the mechanism by which PTEN protein phosphatase inactivation increases PDHK1 gene (and thus protein) expression. Although PDHK1 expression can be upregulated by the transcription factor hypoxia-inducible factor 1 (HIF-1) (Kim et al., 2006), silencing the HIF-1α subunit of HIF-1 failed to suppress PDHK1 expression in a panel of PTEN-deficient cell lines, unlike the effects of HIF-1κ silencing in these cells on carbonic anhydrase 9 (CA9), which is known to be regulated by HIF-1 (Figure S8A) (Wykoff et al., 2000). Although we observed HIF-1 independency on PDHK1 induction in PTEN-null cell lines under normoxia (Figure S8A), PTEN deficiency further increased PDHK1 expression in hypoxic conditions concurrently with HIF-1 induction, compared with normoxia (Figure S8B).
To understand the molecular basis of the transcriptional upregulation of PDHK1 induced by PTEN inactivation in cells, we sought to identify the transcription factor(s) involved. We analyzed putative transcription factor binding sequences and/or motifs in or upstream of the PDHK1 promoter using an in silico promoter analysis. We identified a putative nuclear factor kappa light chain enhancer of activated B cells (NF-κB) transcription factor (Gilmore, 2006) consensus DNA binding site (GGGRNNYYCC; R is purine, Y is pyrimidine, and N is any base) (Martone et al., 2003) GGGACGCTCC at nucleotide position 173420479 in chromosome 2, at ~300 bp upstream of the TSS (transcription start site) in the PDHK1 promoter (Figures 4A and S8C; Table S3). We tested the hypothesis that PDHK1 is a transcriptional target of the transcription factor NF-κB.
Figure 4. PTEN Protein Phosphatase Represses PDHK1 by Suppressing NF-κB Activation.

(A) Top: identification of NF-κB consensus binding site in the promoter of PDHK1 gene located between nucleotide positions 173420479 and 173420488 in chromosome 2, ~300 bp upstream of the transcription start site (TSS; red arrow) at nucleotide position 173420779. Bottom: NF-κB (RELA) recruitment at PDHK1 promoter in PTEN-deficient cancer cells by ChIP assay. Primers (forward and reverse arrows) used to amplify a 118 bp region (spanning nucleotide position 173420380) ~40 bp upstream of the NF-κB binding site in the PDHK1 promoter are shown. Fold enrichment (RELA ChIP DNA [pg] to IgG DNA [pg]) data are shown as mean ± SEM (n = 3 replicates). *p < 0.05 and **p < 0.01 compared with “IgG control” by two-tailed unpaired t test with Welch’s correction.
(B) Western blots showing NF-κB (RELA), PDHK1, and phospho-PDHA1 expression in PTEN-deficient cancer cell lines with or without stable RELA knockdown. shRELA, shRNA to RELA.
(C) Effects of PTENWT or PTENG129E or PTENY138L or empty vector (EV) expression in PTEN-deficient cancer cell lines on NF-κB activity by luciferase reporter assays (STAR Methods) are shown. Data are shown as mean ± SD (n = 2 replicates). **p < 0.01 and ***p < 0.001 compared with “empty vector control expressing PTEN-deficient cells” by Tukey’s multiple-comparisons one-way ANOVA.
(D) Effects of PTENWT or PTENG129E or PTENY138L or GFP expression in PTEN-deficient cancer cell lines on NF-κB (RELA) subcellular localization by nuclear-cytoplasmic fractionation and immunoblotting are shown. Western blots were also probed with anti-LaminB1 and anti-actin β antibodies as nuclear and cytoplasmic markers, respectively.
(E) Western blots showing PTEN, phospho-RELA, PDHK1, and phospho-AKT expression in PTEN-deficient cancer cell lines stably expressing PTENWT, PTENG129E, PTEN138L, or GFP. See also Figure S8 and Table S3.
We found NF-κB transcription factor RELA (p65) subunit recruitment at the PDHK1 promoter in multiple PTEN-deficient cancer cell types by chromatin immunoprecipitation (ChIP) and real-time qPCR analysis (Figure 4A). NF-κB suppression using either an shRNA to knockdown RELA or the established NF-κB small-molecule inhibitor PBS-1086 (Blakely et al., 2015; Fabre et al., 2012) inhibited NF-κB, decreased PDHK1 expression, and suppressed phosphorylation of the PDHK1 protein substrate PDHA1 in a panel of PTEN-deficient cell lines (Figures 4B and S8D). Although prior studies have shown that PTEN loss and consequent AKT activation can activate NF-κB (Chiao and Ling, 2011; Dan et al., 2008; Gustin et al., 2001; Koul et al., 2001; Mayo et al., 2002), our data reveal that NF-κB hyperactivation upon PTEN loss promotes PDHK1 expression and establish PDHK1 as a NF-κB target gene.
NF-κB inhibitor treatment (Blakely et al., 2015; Fabre et al., 2012) decreased cell survival specifically in PTEN-deficient cells (Figure S8E), phenocopying the effects of PDHK1 inhibition (Figure 2). Thus, PTEN loss renders cells dependent on PDHK1 for survival via NF-κB activation. Stable expression of PDHK1 in PTEN-deficient A2058 (melanoma) cells partially rescued loss of cell viability and decreased apoptosis upon NF-κB inhibition (with Bay 11-7085; Berger et al., 2007) (Figures S8F and S8G).
Both the lipid, as expected (Gustin et al., 2001), and protein phosphatase activities of PTEN decreased NF-κB transcriptional activity (Aoki and Kao, 1997; Blakely et al., 2015) and NF-κB nuclear localization (Figures 4C and 4D). However, although the lipid phosphatase activity of PTEN specifically suppressed AKT activation (phospho-AKT levels), as expected (Gustin et al., 2001), the protein phosphatase activity of PTEN was specifically required to suppress PDHK1 expression via decreased NF-κB activation (phospho-RELA levels) (Figure 4E). Thus, although both the lipid and protein phosphatase activities of PTEN regulate NF-κB, the expression of PDHK1 is regulated predominantly by the protein phosphatase activity of PTEN via NF-κB.
PTEN Protein Phosphatase Regulates NF-κB and PDHK1 via NKAP
We investigated how the PTEN protein phosphatase regulates its downstream effectors NF-κB and PDHK1. De-phosphorylation of a specific protein target of the PTEN protein phosphatase activity could link PTEN to downstream NF-κB and PDHK1 regulation. The full compendium of PTEN protein or lipid phosphatase specific effectors and substrates remains incompletely characterized. To identify the potential PTEN protein phosphatase-specific effector that links PTEN to downstream NF-κB and PDHK1 regulation, we used global mass spectrometry-based phospho-proteomics profiling (Beltrao et al., 2012; Jäger et al., 2011) in a genetically controlled system of PTEN-deficient cell lines with stable GFP or PTENWT or PTENG129E (lipid phosphatase mutant) or PTENY138L (protein phosphatase mutant) re-expression (Figure 5A). We identified phospho-peptides and proteins significantly differentially regulated (log2 fold change < −0.5, q < 0.05) by each PTEN phosphatase activity, in comparison with PTEN-null GFP-expressing control cells (Figure 5A). We prioritized the phospho-proteins that exhibited conserved and specific regulation by the PTEN protein-phosphatase activity in both H1650 (lung adenocarcinoma, EGFR mutant) and A2058 (melanoma, BRAF mutant) cells. The different tissue type and genetic background of these two cell lines provided a filter to identify a potentially conserved target. Forty-two proteins exhibited PTEN protein phosphatase-specific regulation in both cell lines, including NF-κB-activating protein (NKAP) (Figures 5A and S9A).
Figure 5. PTEN Dephosphorylates NF-κB-Activating Protein (NKAP).

(A) Left: workflow of the unbiased, global phospho-proteomic profiling in PTEN-deficient cancer cell lines stably expressing PTENWT or PTENG129E or PTENY138L or GFP to identify the phospho-peptides (and corresponding proteins) affected specifically by the protein or lipid phosphatase (PP or LP) activity of PTEN. Right: Venn diagram showing the number of proteins with phospho-sites specifically affected by the protein phosphatase activity of PTEN in H1650 (n = 169) and A2058 (n = 248) cells, with the phospho-proteins (including NF-κB-activating protein, NKAP highlighted in white) affected in both cell lines shown in the overlap (n = 42).
(B) Schematic representation of NKAP phospho-sites at serines 9 and 149 affected by the protein phosphatase activity of PTEN.
(C and D) Phosphate released (μM) from a phospho-S9-NKAP or phospho-S149-NKAP or phospho-S72-Rab7 (control) peptide after incubation without or with recombinant WT PTEN (C) or protein phosphatase mutant Y138L PTEN (D) enzyme in an in vitro malachite green-based colorimetric assay. Data are shown as mean ± SD (n = 2 replicates). *p < 0.05, **p < 0.01, and ****p < 0.0001 compared with “no PTEN” control by two-tailed unpaired t test with Welch’s correction.
(E) Detection of phospho-NKAP and its de-phosphorylated species (indicated by the red dotted inset) in PTEN-deficient cancer cell lines stably expressing either PTENWT or PTENG129E or PTENY138L or GFP by Phos-tag PAGE and immunoblotting. De-phosphorylation score indicates the extent to which expression of each PTEN mutant in PTEN-deficient cells suppresses NKAP de-phosphorylation relative to PTENWT (set at 1). A lower de-phosphorylation score indicates less de-phosphorylation of NKAP.
(F) Co-immunoprecipitation of NKAP (indicated by the red dotted inset) with PTEN upon overexpression of both NKAP-V5 and FLAG-PTEN and not NKAP-V5 overexpression alone or no overexpression in 293T cells followed by IP-FLAG is shown. Arrowhead denotes NKAP-V5, while the dark band below is background due to secondary antibody cross-reactivity to the immunoglobulin heavy chain (IgH) used in IP.
See also Figure S9.
NKAP, originally identified as a regulator of NF-κB activation (Chen et al., 2003), has various cellular functions (Pajerowski et al., 2009; Thapa et al., 2016; Burgute et al., 2014; Li et al., 2016). NKAP is not known to be linked to PTEN or the regulation of cellular metabolism or cancer. Given NKAP’s established ability to activate NF-κB (Chen et al., 2003), which regulates PDHK1 expression (Figure 4), NKAP was prioritized as a prime candidate PTEN effector arising from our phospho-proteomics screen (Figures 5A and S9A).
We examined whether NKAP is a phospho-protein target of PTEN in biochemical assays. The phospho-proteomics profiling revealed the predominant phosphorylation sites in NKAP in the two cell lines tested were at serines 9 and 149 (Figures 5B and S9A), consistent with other protein phospho-site mapping data (available at phosphosite.org). Using Ser 9 or Ser 149 phosphorylated NKAP peptides in an established malachite green-based in vitro phosphatase assay with recombinant PTENWT and PTENY138L enzymes (Figures 5C and 5D), we found that WT PTEN (PTENWT) enzyme de-phosphorylated NKAP at serines 9 and 149 with similar efficiency (as quantified by the amount of Pi released) as a known phospho-serine substrate of PTEN, phospho-S72 Rab7 (Shinde and Maddika, 2016), used as a positive control (Figure 5C). The protein phosphatase defective PTEN (PTENY138L) enzyme had little or no activity for either phospho-NKAP or phospho-Rab7 and was comparable with the “no enzyme” control (Figure 5D).
By phosphate affinity (Phos-tag) PAGE analysis (Kinoshita et al., 2009), wherein de-phosphorylated NKAP migrates faster than its phosphorylated form, we found enhanced NKAP dephosphorylation in PTEN-deficient cells stably expressing PTENWT or PTENG129E (lipid phosphatase mutant) in comparison with those with stable PTENY138L (protein phosphatase mutant) or GFP expression (Figure 5E). These cellular Phos-tag assays independently confirmed that the protein phosphatase activity of PTEN predominantly regulates NKAP phosphorylation. We noted no significant effect of PTEN re-expression on NKAP protein levels in PTEN-deficient cells (Figure 5E), suggesting that PTEN primarily regulates NKAP phosphorylation and not its expression. No effect of PTEN expression on PDHK1 phosphorylation was observed (Figure S9A), suggesting that although PTEN regulates PDHK1 gene expression via NF-κB, it does not dephosphorylate PDHK1 protein.
NKAP co-immunoprecipitated with PTEN when overexpressed in 293T cells (Figure 5F, red box) under identical conditions in which the known PTEN interactor NHERF2 (Takahashi et al., 2006) was co-immunoprecipitated with PTEN (Figure S9B). An independent affinity purification-mass spectrometry analysis (St-Denis et al., 2016) provided evidence for this PTEN-NKAP interaction in cells. These data reveal NKAP as a protein interactor of PTEN and that PTEN dephosphorylates NKAP at two conserved serine residues, S9 and S149.
To further investigate whether NKAP is a molecular link between PTEN and PDHK1, we examined whether NF-κB activation and PDHK1 expression are suppressed by silencing NKAP in PTEN-deficient cells. NKAP knockdown in different PTEN-deficient cell lines suppressed NF-κB transcriptional activity (Aoki and Kao, 1997; Blakely et al., 2015) (Figure 6A) without affecting phospho-AKT levels (Figure 6C), consistent with prior work (Thapa et al., 2016). In concordance, NKAP knockdown suppressed transcriptional activation of the PDHK1 promoter (Figure 6B) and decreased expression of PDHK1 (Figure 6C), which we identified as a NF-κB target gene (Figure 4). Stable expression of WT NKAP (NKAPWT), but not de-phosphorylation-deficient mutant NKAP (NKAPS9A-S149A), in PTEN-null cells enhanced NF-κB transcriptional activity (Figure S10A) and NF-κB nuclear localization (Figure S10B) and increased PDHK1 expression (Figure 6D). Thus, loss of the PTEN protein phosphatase induces NKAP phosphorylation (on S9 and S149) to activate NF-κB and increase PDHK1 levels, in a PI3K-AKT-independent manner.
Figure 6. Depletion of NKAP Decreases NF-κB Activation and PDHK1 Expression and Induces Synthetic Lethality Specifically upon PTEN Protein Phosphatase Loss.

(A and B) Effects of stable NKAP knockdown in PTEN-deficient cancer cell lines on NF-κB activity (A) or PDHK1 promoter activation (B) by luciferase reporter assays (STAR Methods) are shown. shA and shB, shRNAs to NKAP; SC, scrambled control shRNA. Data are shown as mean ± SEM (n = 3 replicates). **p < 0.01, ***p < 0.001, and ****p < 0.0001 compared with “scrambled control shRNA expressing PTEN-deficient cells” by Tukey’s multiple-comparisons one-way ANOVA.
(C) Western blots showing NKAP, PDHK1, and phospho-AKT expression in PTEN-deficient cancer cell lines with or without stable NKAP knockdown. shA and shB, shRNAs to NKAP; SC, scrambled control shRNA.
(D) Western blots showing HA-NKAP and PDHK1 expression in PTEN-deficient A2058 cancer cells stably expressing empty vector (EV) or wild-type NKAP (HA-NKAPWT) or de-phosphorylation-deficient mutant NKAP (HA-NKAPS9A-S149A).
(E and F) Effects of stable NKAP knockdown in PTEN-deficient cancer cell lines without (E) or with stable PTENWT or PTENG129E or PTENY138L or GFP expression (F) on cell growth by crystal violet staining assay are shown, with quantification of cell viability under each condition relative to cells expressing the scrambled control shRNA. shA and shB, shRNAs to NKAP; SC, scrambled control shRNA.
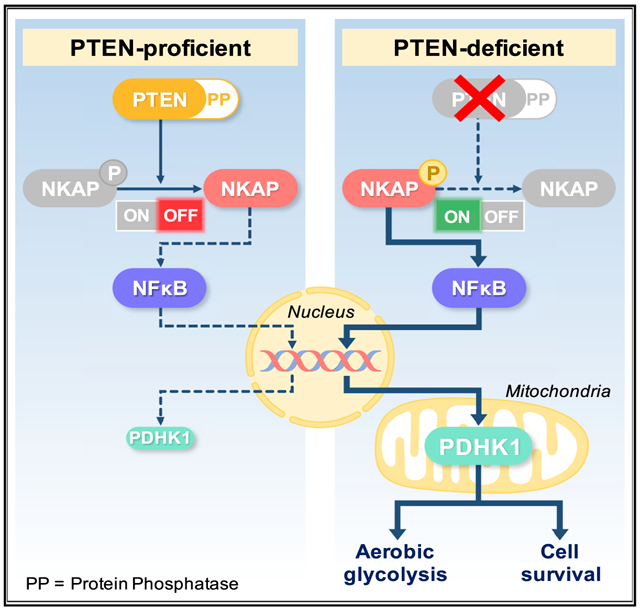
(G) Model of cellular survival and energy metabolism regulation specifically by the protein-phosphatase activity of PTEN via a NKAP-NF-κB-PDHK1-driven signaling axis. Loss of the PTEN protein-phosphatase activity promotes NKAP phosphorylation, NF-κB activation, and PDHK1 upregulation, thereby enhancing aerobic glycolysis and rendering PTEN protein-phosphatase deficient cells dependent on NKAP and PDHK1 for survival.
See also Figure S10.
We propose that specific loss of the PTEN protein phosphatase activates a downstream NKAP-NF-κB signaling axis to drive PDHK1 expression and cellular dependence on PDHK1 for survival (Figure 6G). NKAP knockdown significantly decreased cell viability in PTEN-deficient cells compared with PTEN-expressing cells (Figure 6E), phenocopying the effects of both PDHK1 and NF-κB inhibition in PTEN-deficient cells (Figures 2, S4, and S8C). Similar to PDHK1 inhibition (Figure 3D), NKAP silencing significantly decreased the viability of cells specifically lacking the PTEN protein phosphatase activity (Figure 6F). NKAP silencing affected viability of cells expressing PTENWT or PTENG129E to some extent which may be due to other effects of NKAP knockdown. PTEN-deficient cells re-constituted with PTENY138L (protein phosphatase mutant) were generally as sensitive as PTEN-deficient cells to NKAP knockdown (Figure 6F). In contrast, cells re-constituted with PTEN that harbors the protein phosphatase activity (either PTENWT or PTENG129E, the lipid phosphatase mutant) were generally less affected by NKAP suppression (Figure 6F). These data show a role for the PTEN protein phosphatase in controlling cell survival via regulation of a PTEN/NKAP/NF-κB/PDHK1 signaling axis.
DISCUSSION
Our findings unveil molecular events specifically regulated by the protein phosphatase activity of PTEN in mammalian cells and cancer and uncover a vulnerability in PTEN-deficient cancers (i.e., PDHK1 dependence; Figure 6G). Our findings highlight PDHK1 as a promising therapeutic target for evaluation for improving the treatment of PTEN-deficient cancers, an unmet clinical need. Our study illustrates the importance of defining activity-specific PTEN functions that contribute to physiologic regulation in normal cells and that when dysregulated contribute to genetically driven human diseases such as cancer.
Our data suggest that PTEN’s different enzymatic activities can be used by cells to impart specificity in the regulation of cell signaling and transcriptional output (here, via NKAP/NF-κB/PDHK1). The differential recruitment of NF-κB to genomic loci such as the PDHK1 promoter may be guided not only by the underlying DNA sequence in canonical gene regulatory elements, but also by co-factor proteins that influence preferential recruitment of NF-κB to one target gene promoter or another. Different co-factor proteins can regulate NF-κB binding at specific target genes under certain conditions (Wan and Lenardo, 2009). NKAP may act as a co-factor specifically regulated by the PTEN protein phosphatase activity to direct differential NF-κB target promoter binding and gene expression only upon loss of the PTEN protein phosphatase. This model may help explain how cells finely tune NF-κB’s broad transcriptional repertoire via activity-specific upstream inputs, such as the bi-functional phosphatase PTEN.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Trever Bivona (trever.bivona@ucsf.edu). This study did not generate new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
Female, 4-5 weeks old NOD/SCID mice were obtained from Taconic Biosciences. All animal studies followed the NIH Guidelines and AAALAC International standards set for the ethical use and care of laboratory animals, NRC 2011 and were conducted under a UCSF IACUC approved animal protocol (AN107889).
Cell cultures
All cell lines were grown in RPMI-1640 or DMEM (GE Healthcare) supplemented with 10% fetal bovine serum (Sigma-Aldrich) and 100 U/ml penicillin G and 100 μg/ml streptomycin (GE Healthcare) at 37°C under 5% CO2. For hypoxia experiments, 24 hours after cell seeding cells were incubated at 37°C for indicated times in a sealed hypoxia chamber filled with 1% O2, 4% CO2 and 95% N2.
METHOD DETAILS
Transfection and viral infection
293T cells were transfected with various plasmids using TransIT-LT1 transfection reagent (Mirus Bio) according to manufacturer’s instructions. Briefly, plasmid DNA diluted in serum-free OptiMEM medium was mixed with TransIT-LT1 transfection reagent at 1:3 ratio. After incubating the DNA-TransIT-LT1 mixture at room temperature for 15 min, the complexes were added to cells to allow the transfection of plasmid DNA.
293T cells were transfected with various lentiviral constructs and packaging plasmids using calcium phosphate or TransIT-LT1 (Mirus Bio) transfection reagent according to manufacturer’s instructions. Three days post-transfection lentiviral supernatant supplemented with 6 μg/ml of polybrene was used to infect the indicated cell lines. 48 hours post-infection cells were replated with antibiotic containing media and selection was continued for an additional 2-7 days. Experiments were performed with the cells that survived antibiotic selection (stable cell lines).
293-GPG viral packaging cells (Ory et al., 1996) were transfected with various p-BABE-Puro retroviral constructs using Lipofectamine-2000 (Invitrogen) according to manufacturer’s instructions. Three days post-transfection retroviral supernatant supplemented with 6 μg/ml polybrene was used to transduce various cancer and normal cell lines. 48 hours post-infection cells were replated with Puromycin (1-2 μg/ml) containing media and selection was continued for an additional 4 days. Cells that survived puromycin selection (stable cell lines) were used in subsequent experiments.
High titer pre-made adenovirus expressing PTEN (Applied Biological Materials) was used to transduce the indicated PTEN-deficient cell lines to transiently overexpress PTEN. High titer pre-made adenovirus expressing PDHK1-HA (Applied Biological Materials) was used to infect PTEN-deficient H1650 cells with stable PDHK1 knockdown for transient overexpression of shRNA-resistant PDHK1-HA.
Cell viability and growth assays
Cells were plated at a density of 5,000 cells per well in 96-well plates 24 hours before drug treatment. The number of viable cells was determined 72 hours after the initiation of drug treatment using CellTiter-Glo luminescent cell viability reagent according to the manufacturer’s instructions (Promega). Each assay consisted of five to eight replicate wells. Data are expressed as percentage of the cell viability of control cells. Data were graphically displayed using GraphPad Prism version 6.0 for Mac (GraphPad Software).
When cell viability was measured in an ATP independent assay, cells (200,000) were plated per well in 6-well plates 24 hours prior to drug treatment. 72 hours post-drug treatment cells were fixed with 4% paraformaldehyde. 0.05% Crystal violet was used to stain the viable cells and imaged using an ImageQuant LAS 4000 instrument (GE Healthcare). The crystal violet staining based viability assay measures the anchorage-dependent growth of adherent cells and hence whether the targeted cells are dying or not could be determined from this assay. To quantify the number of viable cells, stained cells were solubilized in 1% SDS and absorbance was measured at 590 nm in a plate reader (SpectraMax M5, Molecular devices).
For the determination of rescue score, cells were treated with a dose-response of DCA and stained with crystal violet 72 hours after treatment. For each cell line, the degree of cell viability at the highest concentration of DCA (20 mM) was normalized to the vehicle control for each genetic background (cells expressing GFP or PTENWT or PTENMutant). This normalized cell viability value was then used to calculate the rescue score as: normalized viability in the PTEN-reconstitution condition/normalized viability in the PTEN-deficient, GFP, condition. A rescue score > 1 indicates the degree of rescue from lethality upon PDHK1 inhibition.
Intracellular ATP and ROS measurement
Intracellular levels of ATP were measured by using CellTiter-Glo luminescent assay (Promega), according to manufacturer’s instructions. For each cell line, 5,000 cells were plated per well of 96 well plates. After 24 hours, cells were treated with 100 nM of rotenone for 2 hours or with 100 μM of oligomycin for 1 hour at 37°C to quickly achieve the maximal inhibition of complex I or V, respectively, while not to induce cell death during the experimental period. Relative ATP levels (% of untreated) were calculated.
For measurement of intracellular ROS levels, 20,000 cells of A2058 or PC3 cells or 15,000 cells of H1650 cells were plated per well of 96 well plates. After 24 hours, cells were washed once with serum free RPMI media without phenol red and incubated with 2.5 μM of 2′,7’-dichlorodihydrofluorescein diacetate (H2DCFDA) solution in serum free RPMI media without phenol red for 1 hour at 37°C. The H2DCFDA solution was removed and cells were treated with 40 or 50 mM DCA with or without 1 mM NAC or with only 1 mM NAC in serum free RPMI media without phenol red. After 24 hours of indicated treatment, the 2′,7′-dichlorofluorescein (DCF) fluorescence was measured with a plate reader (SpectraMax M5, Molecular devices) at Ex/Em: 485 nm/538 nm. Relative ROS levels (% of untreated) were calculated.
L-lactate measurement
Extracellular L-lactate was measured using a lactate assay kit (Cayman) following manufacturer’s instructions. Briefly, cells were plated at equal numbers in at least 8 micro wells per condition and cultured with the same volume of media. The assay employs the feature of NAD+ reduction to NADH which occurs concomitantly with the oxidation of lactate to pyruvate. NADH reacts with the fluorescent substrate to produce high fluorescence at Ex/Em: 540 nm/590 nm. The greater the signal the higher is the lactate concentration in the culture media. For glucose conditioned media, RPMI or DMEM without glucose was used (GIBCO) and 2DG (2-deoxyglucose) was supplemented at 10 mM final concentration.
Protein blot analysis
Cells (200,000) were seeded per well in 6-well plates 24 h before drug treatment, and whole-cell lysates were prepared using RIPA buffer (10 mM Tris-HCl (pH 8.0), 1 mM EDTA, 0.1% sodium deoxycholate, 0.1% SDS, 140 mM NaCl) supplemented with protease inhibitor and phosphatase inhibitor (Pierce) and clarified by sonication and centrifugation. Nuclear and cytosolic fractionation was performed as described before (Suzuki et al., 2010). Mitochondrial and cytosolic fractionation was performed using Cytochrome c release assay reagents (Abcam). Equal amounts of protein were separated by 4%–15% gradient SDS-PAGE and were transferred onto nitrocellulose membranes (Bio-Rad) for protein blot analysis. Membranes were incubated with primary antibody overnight, washed and incubated with secondary antibody. Membranes were developed using either a fluorescence system (Li-Cor) or a chemiluminescent reagent; images were captured, and bands were quantified using an ImageQuant LAS 4000 instrument (GE Healthcare).
Gene expression profiling
Three biological replicate retroviral infections of H1650 cells were performed with pBABE-PTEN or pBABE-GFP as described above, followed by selection in 2 μg/ml puromycin for 2 days. Stable PTEN expression was confirmed by immunoblot analysis 5 days post-infection. Total RNA was isolated for each replicate using the RNeasy kit (QIAGEN) according to manufacturer’s instructions. After quality control validation, 2 μg of total RNA was processed for hybridization and image quantification of U133A 2.0 gene expression arrays (Affymetrix).
Real time qRT-PCR
PDHK1 gene expression changes observed by microarray analysis were validated by real-time qRT-PCR in paired cell lines engineered to stably lack or express PTEN. PDHK1 gene expression regulation by different phosphatase activities of PTEN was also measured by real-time qRT-PCR in PTEN-deficient cell lines stably expressing PTENWT, PTENG129E, PTENY138L or GFP. Briefly, for real time qRT-PCR analysis total RNA was extracted from cultured cells using the RNeasy kit (QIAGEN). cDNA was synthesized with SuperScript III reverse transcriptase using random hexamer primers (Invitrogen) and Real time qRT-PCR was performed on the QuantStudio 12K Flex real-time qPCR system using Taqman probes (human PDK1: Assay ID, Hs01561850_m1, Applied Biosystems). GAPDH expression was used as an internal control to normalize input cDNA (human GAPDH: Assay ID, Hs99999905_m1, Applied Biosystems). Ratios of the expression level of each gene to that of the reference gene were then calculated.
Chromatin immunoprecipitation (ChIP) assays
ChIP assays were performed using the chromatin IP kit (Cell Signaling Technology) following manufacturer’s instructions. Briefly, 4×107 cells were treated with formaldehyde to crosslink proteins to DNA and nuclear/cytosolic fractionation was performed. Nuclear lysates were sonicated using the Covaris S2 sonicator and an aliquot of DNA was purified to check for sonication efficiency (fragments of ~500bp). Anti-p65, anti-IgG and anti-H3 (positive control) antibodies were used to pull down the indicated protein-bound chromatin. Washing, elution and reverse crosslink followed by DNA purification concluded the sample preparation. Quantification of NF-κB (or RELA) enrichment was done using real-time qPCR with primers designed to amplify a 118 bp region spanning nucleotide position 173420380 in the PDHK1 promoter (EpiTect ChIP qPCR Primer Assay for human PDK1, NM002610.3 (−)01Kb, QIAGEN). Assay position: Chromosome 2, 173420380, TSS position: Chromosome 2, 173420779, Assay tile: (−) 01Kb). A standard curve was generated to examine the efficiency of amplification by plotting Ct (threshold cycle) versus DNA quantity (Log10 scale). The fold enrichment was calculated using the following two formulas: Formula 1: Y = M(Log10X) + B (X = DNA quantity, Y = Ct, M = slope of the curve, B = Ct value where X = 1). Formula 2: Fold enrichment = (ChIP DNA Quantity)/(IgG DNA Quantity).
Luciferase reporter assays
pGreenFire1-NF-κB (EF1α-Puro) lentiviral plasmid containing four NF-κB transcription factor response elements fused in tandem upstream of a luciferase gene under a minimal CMV promoter was transfected into PTEN-deficient cancer cells stably expressing either PTENWT or PTENG129E or PTENY138L or GFP to measure the effects of PTEN’s protein and / or lipid phosphatase activities on the transcriptional activity of NF-κB. pGreenFire1-NF-κB (EF1α-Puro) reporter plasmid was also transfected into PTEN-deficient cells stably expressing NKAPWT or NKAPs9AS149A or with stable NKAP knockdown to study the effects of NKAP phosphorylation on NF-κB activity. PDK1 promoter reporter plasmid containing the PDHK1 promoter fused to luciferase was transfected into PTEN-deficient cancer cell lines with stable NKAP knockdown to measure the effects of NKAP downregulation on PDHK1 promoter activation. Luciferase activity was measured 24 hours after transfection. Cells were washed with phenol-red minus DMEM media once and luciferase detection reagent was added (Bright-Glo, Promega, Cat# E2610) at 100 μL per well and luminescence was measured after 5 min of incubation at RT in a plate reader (SpectraMax M5, Molecular devices).
In vitro phosphatase assays
5-14 amino acid (SGSRpSPDREA) p-S9-NKAP or 145-154 amino acid (VWGLpSPKNPE) p-S149-NKAP or 68-77 amino acid (ERFQpSLGVAF) p-S72-Rab7 peptide (used as a +ve control) was incubated without or with bacterially expressed and purified (as described before (Taylor and Dixon, 2003)) PTENWT or mutant Y138L PTEN enzyme in reaction buffer (25mM HEPES, pH 7.5, 10 mM MgCl2, 10 mM DTT) at 37°C for 90 min. Following incubation, the released phosphate from each phospho-peptide ± PTENWT or mutant Y138L PTEN enzyme was detected using Malachite Green Assay Kit (Echelon) by measuring the absorbance at 620 nm in a plate reader (SpectraMax M5, Molecular devices).
Phos-tag assays
Phos-tag PAGE analysis was carried out to detect phosphorylated-NKAP and its de-phosphorylated species according to manufacturer’s instructions (Wako Chemicals, Japan). Briefly, PTEN-deficient cancer cells stably expressing either PTENWT or PTENG129E or PTENY138L or GFP were lysed and subjected to Zinc-based polyacrylamide gel electrophoresis. After electrophoresis, Phos-tag acrylamide gels were washed with gentle shaking in transfer buffer containing 10 mM EDTA for 30 min and then incubated in transfer buffer without EDTA for 15 min. Proteins were then transferred to nitrocellulose membrane for further analysis by standard immunoblotting protocol using specific antibodies.
Immunoprecipitation
For immunoprecipitation assays, 48 hours post-transfection of different plasmids in 293T cells, the cells were resuspended in lysis buffer (200 mM NaCl, 0.5 mM EDTA, 0.5% NP40, 50 mM Tris.Cl, pH 7.5) containing 250 U Benzonase (SIGMA)/5 mL and protease inhibitors and lysed by a rapid freeze-thaw cycle (10 min on dry ice followed by 1 min at 37°C). The whole-cell extracts (WCE) obtained by centrifugation were incubated overnight with M2 agarose-FLAG beads (SIGMA) at 4°C. The immunocomplexes were washed once with wash buffer I (50 mM Tris (pH 7.4, 150 mM NaCl, 0.05% NP40), three more times with wash buffer II (50 mM Tris (pH 7.4, 150 mM NaCl), eluted from the beads with elution buffer (150 ng/ μl FLAG peptide in wash buffer II) and applied to SDS-PAGE.
Time-lapse imaging
A2058 empty vector or stable PTEN expressing cells were plated at 50,000 cells per well in a 24 well plate on Day zero. Cells were transfected with SypHer-dmito construct (Suzuki et al., 2014) using Xfect (Clontech) on Day 1 and treated without or with 50 mM DCA on Day 2 for 2 hr at 37°C in normal culture medium. Immediately before image acquisition by time-lapse microscopy, media was replaced with physiological salt solution (PSS) containing 150 mM NaCl, 4 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 5.6 mM glucose and 25 mM HEPES (pH 7.4) ± 50 mM DCA at 37°C. Images were acquired with CMOS image sensor, ORCA-Flash 4.0 (Hamamatsu, Japan), equipped with 418-442 nm and 450-490 nm excitation filters, and 510-540 nm emission filter. SypHer-dmito fluorescence (F470/F430) ratio was calculated for each cell after subtracting background signal. All images were analyzed with ImageJ software (Schneider et al., 2012). For each cell type, cells with F470/F430 ratio values greater than two s.d. from the mean value were excluded.
Immunohistochemistry
For immunohistochemistry, 5-15 μm thick formalin-fixed paraffin embedded (FFPE) human tissue sections were purchased from US Biomax (Glioblastoma multiforme, Prostate adenocarcinoma and Lung Carcinoma) and stained with PTEN or PDHK1 antibodies following manufacturer’s instructions. Stained slides were digitized using the Aperio ScanScope slide scanner (Aperio Technologies) with a 40× objective. Scanned images from the immunohistochemistry staining were used to score expression of PTEN or PDHK1 in paired samples (scoring expression on a scale of 1 to 3, with 1 indicating ‘low’, 2 ‘intermediate’, and 3 ‘high’ expression of each protein). Bars indicate 50 μm and arrows indicate representative tumor cells scored for PTEN and PDHK1 expression.
Phospho-proteomics study
GFP or PTENWT or PTENG129E or PTENY138L was stably introduced in two different PTEN-deficient cancer cell lines H1650 and A2058 with different genetic and histologic backgrounds. Cells were lysed in a buffer containing 8M urea, 100 mM Tris.Cl pH 8.0, 150 mM NaCl, and protease inhibitors (EDTA free complete cocktail tablet, Roche). Lysates were reduced with 4 mM TCEP for 30 minutes at room temperature and alkylated with 10 mM iodoacetamide for 30 minutes at room temperature in the dark. Samples were diluted 1:4 in 100 mM Tris pH 8.0 to reduce urea concentration to 2M and digested with trypsin (1:100 enzyme: substrate ratio) overnight at 37°C. Peptides were desalted with Sep-Pak reversed phase C18 solid phase extraction (Waters) according the manufacturer’s instructions and phosphopeptides were purified using an immobilized metal affinity chromatography approach. Purified phosphopeptides were analyzed in an LTQ Orbitrap Elite mass spectrometry system (Thermo Scientific) equipped with a Proxeon Easy nLC 1000 ultra high-pressure liquid chromatography and auto sampler system. Samples were injected onto a C18 column (25 cm × 75 um I.D. packed with ReproSil Pur C18 AQ 1.9 um particles) in 0.1% formic acid and then subjected to a 4-hour gradient from 0.1% formic acid to 30% ACN/0.1% formic acid. The mass spectrometer collected data in a data-dependent fashion, collecting one full scan in the Orbitrap at 120,000x resolution followed by 20 collision-induced dissociation MS/MS scans in the dual linear ion trap for the 20 most intense peaks from the full scan. Dynamic exclusion was enabled for 30 s with a repeat count of 1. Charge state screening was employed to reject analysis of singly charged species or species for which a charge could not be assigned.
Colony formation assays
50,000 cells were seeded in 0.35% Noble-agar overlaying 0.6% Noble agar base in 60 mm dishes (Falcon). Cells were grown for 21-28 days and media was changed after every 4 days. Colonies were visualized after staining with 0.005% crystal violet, imaged and quantified.
Animal studies
A2058-GFP and A2058-PTEN tumor xenografts were generated by injection of 2×106 cells/tumor mixed with matrigel in NOD/SCID mice. Stable PTEN re-expression in PTEN-deficient A2058 cells was confirmed by immunoblotting before in vivo transplantation. Tumors were allowed to grow until they reached a minimum volume of 200 mm3 at Day 17 when both group of animals (with GFP and PTEN expressing tumors) received treatment with DCA (750 mg/L) in drinking water. Tumor growth was assessed at the indicated time-points by caliper measurements over 31 days post in vivo transplantation and data shown represent the endpoint of the study. A minimum of 6 tumors per treatment group was assessed for the duration of the study.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses
Statistical differences between two experimental groups were calculated using the unpaired two-tailed Student’s t test. Statistical differences between multiple experimental groups were calculated using the multiple comparisons one-way ANOVA test. Statistical differences of tumor volumes were calculated using the two-way ANOVA test. A significance level of p < 0.05 or less was used throughout the study. Replicate information is reported in the figure legends.
TCGA data analysis
Data preparation: TCGA pan-cancer gene expression by RNaseq using the Illumina HiSeq technology was downloaded (https://toil.xenahubs.net/download/tcga_RSEM_Hugo_norm_count.gz). Data from all TCGA cohorts (N = 10535) were combined to produce this dataset. Values are Log2 (x+1) transformed RSEM values.
TCGA pan-cancer gene-level copy number variation (CNV) estimated using the GISTIC2 threshold method was downloaded. Data from all TCGA cohorts (N = 10845) were combined to produce this dataset. GISTIC2 further thresholded the estimated values to −2, −1, 0, 1, 2, representing homozygous deletion, single copy deletion, diploid normal copy, low-level copy number amplification, or high-level copy number amplification. Genes were mapped onto the human genome coordinates using UCSC cgData HUGO probeMap (https://tcga.xenahubs.net/download/TCGA.PANCAN.sampleMap/Gistic2_CopyNumber_Gistic2_all_thresholded.by_genes.gz).
Samples were subtyped based on PTEN copy number status, samples with CNV = 0 were assigned to PTEN normal (N = 6640), samples with CNV = −1 or −2 were assigned to PTEN copy number loss (N = 3422). PDHK1 gene expression was extracted from TCGA pan-cancer normalized RNaseq RFKM matrix and matched with samples with PTEN normal group (N = 5995) or PTEN copy number loss group (N = 2845).
Expression analysis of patient samples: Expression levels of PDHK1mRNA after normalization were compared between patient samples with single or double copy number loss of PTEN to patients with copy number neutral PTEN by boxplot analysis. The minimum, first quartile, median, third quartile and maximum were shown with horizontal lines from lower to upper with 1.5 interquartile range. Unfilled circles are outlier expression of PDHK1 in these two subgroups. Reported p-values are for two-tailed unpaired t test with Welch’s correction comparing the expression samples with PTEN copy number loss to the normal samples.
Survival analysis: Patient survival outcomes were downloaded from the Firehose web portal. We excluded from survival analysis patients over 75 years of age (N = 416) or patients who died less than 30 days from diagnosis (N = 184). Reported p-values are for a cox proportional hazards likelihood ratio test comparing a baseline model to a three-category model indicating a split of the patient cohort into three bins according to lower quartile (25 percentile and below), inter-quartile range (25-75 percentiles) and upper quartile (75 percentile and above) PDHK1 normalized expression values.
Gene expression array data analysis
Raw data was processed using R/Bioconductor package with Robust Multichip Average (RMA) normalization to identify differentially expressed genes between stable PTEN or GFP expressing H1650 cells. The normalized expression intensities were Log2 transformed and multiple t test for null hypothesis was applied and adjusted with Benjamini-Hochberg’s false discovery rate (FDR). The significantly differentially expressed genes were selected by applying fold change cut-off > 2, *p < 0.05 and FDR adjusted p-value or q-value < 0.2.
Phospho-proteomics Data Analysis
Raw mass spectrometry data were analyzed using the MaxQuant software package (version 1.3.0.5). Data were matched to SwissProt reviewed entries for Homo sapiens in the UniProt protein database. MaxQuant was configured to generate and search against a reverse sequence database for false discovery rate calculations. Variable modifications were allowed for methionine oxidation, protein N terminus acetylation, and serine, threonine, and tyrosine phosphorylation. A fixed modification was indicated for cysteine carbamidomethylation. Full trypsin specificity was required. The first search was performed with a mass accuracy of ± 20 parts per million and the main search was performed with a mass accuracy of ± 6 parts per million. A maximum of 5 modifications were allowed per peptide. A maximum of 2 missed cleavages were allowed. The maximum charge allowed was 7+. Individual peptide mass tolerances were allowed. For MS/MS matching, a mass tolerance of 0.5 Da was allowed and the top 6 peaks per 100 Da were analyzed. MS/MS matching was allowed for higher charge states, water and ammonia loss events. The data were filtered to obtain a peptide, protein, and site-level false discovery rate of 0.01. The minimum peptide length was 7 amino acids. Results were matched between runs with a time window of 2 minutes for technical duplicates. The label-free PTEN phosphoproteomics data was analyzed using an in-house computational pipeline built for the analysis of post-translationally modified peptides with mixed effect models, implemented in the MSstats (v2.3.4) Bioconductor package(Choi et al., 2014). First, protein identifiers were converted into modification site identifiers, contaminant and false positive MaxQuant search results were removed and all samples were normalized per cell line by median-centering the log2-transformed MS1-intensity distributions. Next, the MSstats group Comparison function was run with the following options: no interaction terms for missing values, no interference, unequal intensity feature variance, restricted technical and biological scope of replication. Statistically significant changing sites between PTEN variants and the GFP control were selected by applying a Log2 fold-change < − 0.5 and q-value < 0.05 threshold.
DATA AND CODE AVAILABILITY
Microarray data from this study have been deposited in GEO database (GEO: GSE121217). Phospho-proteomics data from this study have been deposited in PRIDE database (PRIDE: PXD014707).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-PTEN | Cell Signaling Technology | Cat# 9188; RRID: AB_2253290 |
| Mouse anti-AKT (pan) | Cell Signaling Technology | Cat# 2920; RRID: AB_1147620 |
| Rabbit anti-p-AKT (S473) | Cell Signaling Technology | Cat# 4060; RRID: AB_2315049 |
| Mouse anti-β Actin | Sigma-Aldrich | Cat# A2228; RRID: AB_476697 |
| Rabbit anti-PDHK1 | Cell Signaling Technology | Cat# 3820; RRID: AB_1904078 |
| Mouse polyclonal anti-PDK2 | Abcam | Cat# ab172065 |
| Rabbit polyclonal anti-PDK3 | Abcam | Cat# ab154549 |
| Goat polyclonal anti-PDK4 | Santa Cruz Biotechnology | Cat# sc-14495 (discontinued) |
| Rabbit polyclonal anti-p-PDHE1A (S293) | Millipore | Cat# ABS204; RRID: AB_11205754 |
| Mouse anti-cleaved PARP | Cell Signaling Technology | Cat# 9546; RRID: AB_2160593 |
| Mouse anti-GAPDH | Santa Cruz Biotechnology | Cat# sc-365062; RRID: AB_10847862 |
| Rabbit polyclonal anti-p-S6 (S240/S244) | Cell Signaling Technology | Cat# 2215; RRID: AB_331682 |
| Rabbit anti-NFκB p65 | Cell Signaling Technology | Cat# 8242; RRID: AB_10859369 |
| Rabbit anti-p-NFκB p65 (S536) | Cell Signaling Technology | Cat# 3033; RRID: AB_331284 |
| Rabbit anti-Lamin B1 | Cell Signaling Technology | Cat# 12586; RRID: AB_2650517 |
| Rabbit polyclonal anti-HSP90 | Cell Signaling Technology | Cat# 4874; RRID: AB_2121214 |
| Rabbit polyclonal anti-HSP60 | Cell Signaling Technology | Cat# 4870; RRID: AB_2295614 |
| Rabbit polyclonal anti-H3 | Cell Signaling Technology | Cat# 9715; RRID: AB_331563 |
| Rabbit polyclonal anti-NKAP | Abcam | Cat# ab121121; RRID: AB_11139756 |
| Mouse anti-NKAP (K85-80-5) | Burgute et al., 2014 | N/A |
| Rabbit polyclonal anti-V5 | Sigma-Aldrich | Cat# V8137; RRID: AB_261889 |
| Mouse anti-FLAG M2 | Sigma-Aldrich | Cat# F1804; RRID: AB_262044 |
| Mouse anti-GST | Cell Signaling Technology | Cat# 2624; RRID: AB_2189875 |
| Rabbit anti-HA | Cell Signaling Technology | Cat# 3724; RRID: AB_1549585 |
| Mouse anti-Cytochrome c | Abcam | Cat# ab13575; RRID: AB_300470 |
| Rabbit anti-VDAC | Cell Signaling Technology | Cat# 4661; RRID: AB_10557420 |
| Rabbit polyclonal anti-Caspase 9 | Cell Signaling Technology | Cat# 9502; RRID: AB_2068621 |
| Rabbit polyclonal anti-Caspase 7 | Cell Signaling Technology | Cat# 9492; RRID: AB_2228313 |
| Mouse monoclonal anti-Caspase 3 | Cell Signaling Technology | Cat# 9668; RRID: AB_206987 |
| Rabbit polyclonal anti-cleaved Caspase 9 | Cell Signaling Technology | Cat# 9505; RRID: AB_2290727 |
| Rabbit polyclonal anti-cleaved Caspase 7 | Cell Signaling Technology | Cat# 9491; RRID: AB_2068144 |
| Rabbit polyclonal anti-cleaved Caspase 3 | Cell Signaling Technology | Cat# 9661; RRID: AB_2341188 |
| Rabbit anti-BCL-xL | Cell Signaling Technology | Cat# 2764; RRID: AB_2228008 |
| Rabbit anti-BCL2 | Cell Signaling Technology | Cat# 4223; RRID: AB_1903909 |
| Rabbit polyclonal anti-p-GSK-3α/β (S21/9) | Cell Signaling Technology | Cat# 9331; RRID: AB_329830 |
| Rabbit anti-HK2 | Cell Signaling Technology | Cat# 2867; RRID: AB_2232946 |
| Rabbit polyclonal anti-HIF-1α | Cell Signaling Technology | Cat# 3716 |
| Rabbit anti-CA9 | Cell Signaling Technology | Cat# 5649; RRID: AB_10706355 |
| Bacterial and Virus Strains | ||
| PTEN adenovirus (human) | Applied Biological Materials | Cat# 000028A |
| PDK1-HA adenovirus (human) | Applied Biological Materials | Cat# 119140A |
| Biological Samples | ||
| Brain tumor tissue microarray | US Biomax | Cat# GL1921 |
| Prostate adenocarcinoma tissue microarray | US Biomax | Cat# PR803a |
| Lung Carcinoma tissue microarray | US Biomax | Cat# LC801 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| BKM120 (PI3K inhibitor) | Selleckchem | Cat# S2247 |
| GDC-0941 (PI3K inhibitor) | Selleckchem | Cat# S1065 |
| MK-2206 (AKT inhibitor) | Selleckchem | Cat# S1078 |
| DCA (PDHK inhibitor) | Sigma-Aldrich | Cat# 347795 |
| PBS-1086 (NF-κB inhibitor) | Blakely et al., 2015 | N/A |
| Bay 11-7085 (NF-κB inhibitor) | Selleckchem | Cat# S7352 |
| Cisplatin | Selleckchem | Cat# S1166 |
| Docetaxel | Selleckchem | Cat# S1148 |
| NAC | Sigma-Aldrich | Cat# A7250 |
| H2DCFDA | Thermo Fisher Scientific | Cat# D399 |
| Rotenone (ETC complex I inhibitor) | Abcam | Cat# ab143145 |
| Oligomycin (ATP synthase inhibitor) | Abcam | Cat# ab141829 |
| 2-DG | Sigma-Aldrich | Cat# D8375 |
| Human TNF-α | Peprotech | Cat# 300-01A |
| Human NKAP-p-S9 peptide (SGSR-p-SPDREA) | Elim Biopharmaceuticals | This paper |
| Human NKAP-pS149 peptide (VWGL-p-SPKNPE) | Elim Biopharmaceuticals | This paper |
| Human Rab7-p-S72 peptide (ERFQ-p-SLGVAF) | Elim Biopharmaceuticals | This paper |
| Critical Commercial Assays | ||
| DC Protein Assay | Bio-Rad | Cat# 500-0116 |
| Trans-Blot Turbo RTA Midi Nitrocellulose Transfer kit | Bio-Rad | Cat# 170-4271 |
| RNeasy kit | QIAGEN | Cat# 74106 |
| CellTiter-Glo Cell Viability Assay | Promega | Cat# G7573 |
| L-lactate Assay | Cayman | Cat# 700510 |
| SimpleChIP Enzymatic Chromatin IP Kit | Cell Signalling Technology | Cat# 9003 |
| EpiTect ChIP qPCR Primer Assay for human PDK1, NM_002610.3 (−)01Kb | Qiagen | Cat# GPH1007880 (−)01A |
| TaqMan Gene Expression Assay (FAM) for human PDK1, Hs01561850_m1 | Thermo Fisher Scientific | Cat# 4331182 |
| TaqMan Gene Expression Assay (FAM) for human GAPDH, Hs99999905_m1 | Thermo Fisher Scientific | Cat# 4331182 |
| Bright-Glo Luciferase Assay | Promega | Cat# E2610 |
| Malachite Green Assay | Echelon | Cat# K1500 |
| Cytochrome c Release Assay | Abcam | Cat# ab65311 |
| Deposited Data | ||
| Raw and analyzed microarray data | This paper | GEO: GSE121217 |
| Raw and analyzed phospho-proteomics data | This paper | PRIDE: PXD014707 |
| Experimental Models: Cell Lines | ||
| Human lung adenocarcinoma H1650 | Laboratory of William Pao | N/A |
| Human lung Adenocarcinoma H1975 | Laboratory of William Pao | N/A |
| Human lung Adenocarcinoma HCC827 | Laboratory of William Pao1 | N/A |
| Human prostate Adenocarcinoma PC3 | ATCC | Cat# CRL-1435 |
| Human renal clear cell carcinoma 786-O | Laboratory of M. Celeste Simon | N/A |
| Human lung squamous carcinoma H520 | ATCC | Cat# HTB-182 |
| Human skin cutaneous melanoma A2058 | ATCC | Cat# CRL-11147 |
| Human glioblastoma multiforme U87MG | ATCC | Cat# HTB-14 |
| Human embryonic kidney cells 293T | ATCC | Cat# CRL-3216 |
| Human bronchial epithelial cells BEAS2B | ATCC | Cat# CRL-9609 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C.B-Igh-1b/IcrTac-Prkdcscid | Taconic Biosciences | Cat# CB17SC-F |
| Oligonucleotides | ||
| PTEN Mission shRNA (3’-UTR): CCGGCCACAAATGAAGGGATATAAACTCGAGTTTATATCCCTTCATTTGTGGTTTTTG |
Sigma-Aldrich | Cat# TRCN0000230370 |
| PDHK1 Mission shRNA (CDS) #1: CCGGGCTCTGTCAACAGACTCAATACTCGAGTATTGAGTCTGTTGACAGAGCTTTTT |
Sigma-Aldrich | Cat# TRCN0000006261 |
| PDHK1 Mission shRNA (CDS) #2: CCGGCCAGGGTGTGATTGAATACAACTCGAGTTGTATTCAATCACACCCTGGTTTTT |
Sigma-Aldrich | Cat# TRCN0000006263 |
| PDHK1 Mission shRNA (3’-UTR): CCGGGAAGTAGAAGTCTACCATATTCTCGAGAATATGGTAGACTTCTACTT CTTTTTTG |
Sigma-Aldrich | Cat# TRCN0000196728 |
| RELA Mission shRNA (3’-UTR): CCGGGCCTTAATAGTAGGGTAAGTTCTCGAGAACTTACCCTACTATTAAGGCTTTTT |
Sigma-Aldrich | Cat# TRCN0000014683 |
| NKAP Mission shRNA (3’-UTR): CCGGCTGATTGTCCAGAAGACATTTCTCGAGAAATGTCTTCTGGACAATCAGTTTTTTG |
Sigma-Aldrich | Cat# TRCN0000145605 |
| NKAP Mission shRNA (CDS): CCGGGCTGAAGAACCATCAGATTTACTCGAGTAAATCTGATGGTTCTTCAGCTTTTTTG |
Sigma-Aldrich | Cat# TRCN0000145475 |
| ON-TARGETplus Non-targeting Pool siRNA | Dharmacon | Cat# D-001810-10 |
| SMARTpool: ON-TARGETplus PDK1 siRNA | Dharmacon | Cat# L-005019-00-0005 |
| SMARTpool: ON-TARGETplus HIF1A siRNA | Dharmacon | Cat# L-004018-00-0005 |
| Recombinant DNA | ||
| pLKO.1-puro Non-Target shRNA | Sigma-Aldrich | Cat# SHC016 |
| pBABE-Puro-EV | Addgene | Cat# 1764 |
| pBABE-GFP | Addgene | Cat# 10668 |
| pBABE-PTEN | Addgene | Cat# 10785 |
| pBABE-PTENC124S | Addgene | Cat# 10931 |
| pBABE-PTENG129E | Addgene | Cat# 10771 |
| pBABE-PTENY138L | This paper | N/A |
| pCDH-Puro-EV | This paper | N/A |
| pCDH-puro-Bcl2 | Addgene | Cat# 46971 |
| pCDH-puro-Bcl-xL | Addgene | Cat# 46972 |
| pCW22-TRE-Blast-EV | This paper | N/A |
| pCW22-TRE-blast-NKAP-HA-FLAG | This paper | N/A |
| pCW22-TRE-blast-NKAPS9AS149A-HA-FLAG | This paper | N/A |
| pLV-EF1a-IRES-Hygro | Addgene | Cat# 85134 |
| pLV-EF1a-IRES-Hygro-PDHK1 | This paper | N/A |
| pCMV-2xFLAG-PTEN | Addgene | Cat# 28298 |
| pLX304-NKAP-V5 | Dharmacon | Cat# OHS6085-213576668 |
| pEBG-GST-NHERF2 | Addgene | Cat# 28292 |
| pET-30 b-PTEN-6xHis | Addgene | Cat# 20741 |
| pET-30 b-PTENY138L-6xHis | This paper | N/A |
| pGreenFire1-NF-kB (EF1α-Puro) | System Biosciences | Cat# TR012PA-P |
| Plasmid: LightSwitch PDK1 Promoter Reporter | SwitchGear Genomics | Cat# S721832 |
| Software and Algorithms | ||
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| GraphPad Prism 6 | GraphPad | RRID:SCR_002798 |
Highlights.
PDHK1 is a synthetic-essential gene in PTEN-deficient normal and cancer cells
PTEN-PP dephosphorylates NKAP to suppress NF-κB activation and PDHK1 expression
PTEN-PP loss upregulates PDHK1 and promotes glycolysis and PDHK1 cellular dependence
High PDHK1 levels in PTEN-deficient tumors correlate with inferior patient survival
ACKNOWLEDGMENTS
We would like to thank all members of the Bivona laboratory for critical review of the manuscript and acknowledge funding support from NIH/NCI grants U54CA224081, R01CA204302, R01CA211052, and R01CA169338 and the Pew Foundation and Stewart Foundation (to T.G.B.); U54 CA209891 (to N.J.K.); R01GM123159 (to J.S.F.); R01GM118939 (to Y.K.); and NIH/NCI T32CA108462-14 (to N.C.). We thank Timothy Fouts and Jeffrey Meshulam at Profectus Biosciences, Inc., for generously providing PBS-1086. We thank Bhagyashri Burgute and Angelika Noegel at the University of Cologne, Germany, for kindly providing the NKAP mouse mAb (K85-80-5). We thank Albertas Navickas and Hani Goodarzi at UCSF for the hypoxia chamber.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.07.063.
DECLARATION OF INTERESTS
T.G.B. is an advisor to Array Biopharma, Revolution Medicines, Novartis, AstraZeneca, Takeda, Springworks, and Jazz Pharmaceuticals and receives research funding from Novartis and Revolution Medicines.
SUPPORTING CITATIONS
The following references appear in the Supplemental Information: Millán-Uclés et al. (2016); Soria et al. (2002); Sun (2010); Tristan et al. (2011); Tsunoda and Takagi (1999).
REFERENCES
- Aguissa-Touré AH, and Li G (2012). Genetic alterations of PTEN in human melanoma. Cell. Mol. Life Sci. 69, 1475–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki Y, and Kao PN (1997). Cyclosporin A-sensitive calcium signaling represses NFkappaB activation in human bronchial epithelial cells and enhances NFkappaB activation in Jurkat T-cells. Biochem. Biophys. Res. Commun. 234, 424–431. [DOI] [PubMed] [Google Scholar]
- Beltrao P, Albanèse V, Kenner LR, Swaney DL, Burlingame A, Villén J, Lim WA, Fraser JS, Frydman J, and Krogan NJ (2012). Systematic functional prioritization of protein posttranslational modifications. Cell 150, 413–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger N, Ben Bassat H, Klein BY, and Laskov R (2007). Cytotoxicity of NF-kappaB inhibitors Bay 11-7085 and caffeic acid phenethyl ester to Ramos and other human B-lymphoma cell lines. Exp. Hematol. 35, 1495–1509. [DOI] [PubMed] [Google Scholar]
- Blakely CM, Pazarentzos E, Olivas V, Asthana S, Yan JJ, Tan I, Hrustanovic G, Chan E, Lin L, Neel DS, et al. (2015). NF-κB-activating complex engaged in response to EGFR oncogene inhibition drives tumor cell survival and residual disease in lung cancer. Cell Rep. 11, 98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Bonnet S, et al. (2007). A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 11, 37–51. [DOI] [PubMed] [Google Scholar]
- Burger MT, Pecchi S, Wagman A, Ni ZJ, Knapp M, Hendrickson T, Atallah G, Pfister K, Zhang Y, Bartulis S, et al. (2011). Identification of NVP-BKM120 as a potent, selective, orally bioavailable class I PI3 kinase inhibitor for treating cancer. ACS Med. Chem. Lett. 2, 774–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgute BD, Peche VS, Steckelberg AL, Glöckner G, Gaßen B, Gehring NH, and Noegel AA (2014). NKAP is a novel RS-related protein that interacts with RNA and RNA binding proteins. Nucleic Acids Res. 42, 3177–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalhoub N, and Baker SJ (2009). PTEN and the PI3-kinase pathway in cancer. Annu. Rev. Pathol. 4, 127–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandarlapaty S, Sawai A, Scaltriti M, Rodrik-Outmezguine V, Grbovic-Huezo O, Serra V, Majumder PK, Baselga J, and Rosen N (2011). AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell 19, 58–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Li Z, Yang Q, Zhang J, Zhai Z, and Shu HB (2003). Identification of a nuclear protein that promotes NF-kappaB activation. Biochem. Biophys. Res. Commun. 310, 720–724. [DOI] [PubMed] [Google Scholar]
- Chesney J, Clark J, Klarer AC, Imbert-Fernandez Y, Lane AN, and Telang S (2014). Fructose-2,6-bisphosphate synthesis by 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 4 (PFKFB4) is required for the glycolytic response to hypoxia and tumor growth. Oncotarget 5, 6670–6686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiao PJ, and Ling J (2011). Kras, Pten, NF-κB, and inflammation: dangerous liaisons. Cancer Discov. 1, 103–105. [DOI] [PubMed] [Google Scholar]
- Chinnaiyan AM, Orth K, O’Rourke K, Duan H, Poirier GG, and Dixit VM (1996). Molecular ordering of the cell death pathway. Bcl-2 and Bcl-xL function upstream of the CED-3-like apoptotic proteases. J. Biol. Chem. 271, 4573–4576. [DOI] [PubMed] [Google Scholar]
- Choi M, Chang CY, Clough T, Broudy D, Killeen T, MacLean B, and Vitek O (2014). MSstats: an R package for statistical analysis of quantitative mass spectrometry-based proteomic experiments. Bioinformatics 30, 2524–2526. [DOI] [PubMed] [Google Scholar]
- Cross DA, Alessi DR, Cohen P, Andjelkovich M, and Hemmings BA (1995). Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 378, 785–789. [DOI] [PubMed] [Google Scholar]
- Dan HC, Cooper MJ, Cogswell PC, Duncan JA, Ting JP, and Baldwin AS (2008). Akt-dependent regulation of NF-kappaB is controlled by mTOR and Raptor in association with IKK. Genes Dev. 22, 1490–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson L, Maccario H, Perera NM, Yang X, Spinelli L, Tibarewal P, Glancy B, Gray A, Weijer CJ, Downes CP, and Leslie NR (2010). Suppression of cellular proliferation and invasion by the concerted lipid and protein phosphatase activities of PTEN. Oncogene 29, 687–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey N, Crosswell HE, De P, Parsons R, Peng Q, Su JD, and Durden DL (2008). The protein phosphatase activity of PTEN regulates SRC family kinases and controls glioma migration. Cancer Res. 68, 1862–1871. [DOI] [PubMed] [Google Scholar]
- Fabre C, Mimura N, Bobb K, Kong SY, Gorgun G, Cirstea D, Hu Y, Minami J, Ohguchi H, Zhang J, et al. (2012). Dual inhibition of canonical and noncanonical NF-kappaB pathways demonstrates significant antitumor activities in multiple myeloma. Clin. Cancer Res. 18, 4669–4681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueiredo AL, Maczkowiak F, Borday C, Pla P, Sittewelle M, Pegoraro C, and Monsoro-Burq AH (2017). PFKFB4 control of AKT signaling is essential for premigratory and migratory neural crest formation. Development 144, 4183–4194. [DOI] [PubMed] [Google Scholar]
- Folkes AJ, Ahmadi K, Alderton WK, Alix S, Baker SJ, Box G, Chuckowree IS, Clarke PA, Depledge P, Eccles SA, et al. (2008). The identification of 2-(1H-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-thieno[3,2-d]pyrimidine (GDC-0941) as a potent, selective, orally bioavailable inhibitor of class I PI3 kinase for the treatment of cancer. J. Med. Chem. 51, 5522–5532. [DOI] [PubMed] [Google Scholar]
- Freeman DJ, Li AG, Wei G, Li HH, Kertesz N, Lesche R, Whale AD, Martinez-Diaz H, Rozengurt N, Cardiff RD, et al. (2003). PTEN tumor suppressor regulates p53 protein levels and activity through phosphatase-dependent and -independent mechanisms. Cancer Cell 3, 117–130. [DOI] [PubMed] [Google Scholar]
- Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K, Franke TF, Papapetropoulos A, and Sessa WC (1999). Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature 399, 597–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Varela L, Sood A, Park BH, and Lotan TL (2013). mTOR signaling feedback modulates mammary epithelial differentiation and restrains invasion downstream of PTEN loss. Cancer Res. 73, 5218–5231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gildea JJ, Herlevsen M, Harding MA, Gulding KM, Moskaluk CA, Frierson HF, and Theodorescu D (2004). PTEN can inhibit in vitro organotypic and in vivo orthotopic invasion of human bladder cancer cells even in the absence of its lipid phosphatase activity. Oncogene 23, 6788–6797. [DOI] [PubMed] [Google Scholar]
- Gilmore TD (2006). Introduction to NF-kappaB: players, pathways, perspectives. Oncogene 25, 6680–6684. [DOI] [PubMed] [Google Scholar]
- Goo CK, Lim HY, Ho QS, Too HP, Clement MV, and Wong KP (2012). PTEN/Akt signaling controls mitochondrial respiratory capacity through 4E-BP1. PLoS ONE 7, e45806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlob K, Majewski N, Kennedy S, Kandel E, Robey RB, and Hay N (2001). Inhibition of early apoptotic events by Akt/PKB is dependent on the first committed step of glycolysis and mitochondrial hexokinase. Genes Dev. 15, 1406–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassian AR, Metallo CM, Coloff JL, Stephanopoulos G, and Brugge JS (2011). Erk regulation of pyruvate dehydrogenase flux through PDK4 modulates cell proliferation. Genes Dev. 25, 1716–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu T, Zhang Z, Wang J, Guo J, Shen WH, and Yin Y (2011). CREB is a novel nuclear target of PTEN phosphatase. Cancer Res. 71, 2821–2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustin JA, Maehama T, Dixon JE, and Donner DB (2001). The PTEN tumor suppressor protein inhibits tumor necrosis factor-induced nuclear factor kappa B activity. J. Biol. Chem. 276, 27740–27744. [DOI] [PubMed] [Google Scholar]
- Hirai H, Sootome H, Nakatsuru Y, Miyama K, Taguchi S, Tsujioka K, Ueno Y, Hatch H, Majumder PK, Pan BS, and Kotani H (2010). MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol. Cancer Ther. 9, 1956–1967. [DOI] [PubMed] [Google Scholar]
- Hlobilkova A, Guldberg P, Thullberg M, Zeuthen J, Lukas J, and Bartek J (2000). Cell cycle arrest by the PTEN tumor suppressor is target cell specific and may require protein phosphatase activity. Exp. Cell Res. 256, 571–577. [DOI] [PubMed] [Google Scholar]
- Houddane A, Bultot L, Novellasdemunt L, Johanns M, Gueuning MA, Vertommen D, Coulie PG, Bartrons R, Hue L, and Rider MH (2017). Role of Akt/PKB and PFKFB isoenzymes in the control of glycolysis, cell proliferation and protein synthesis in mitogen-stimulated thymocytes. Cell. Signal. 34, 23–37. [DOI] [PubMed] [Google Scholar]
- Jäger S, Cimermancic P, Gulbahce N, Johnson JR, McGovern KE, Clarke SC, Shales M, Mercenne G, Pache L, Li K, et al. (2011). Global landscape of HIV-human protein complexes. Nature 481, 365–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang M, Kim SS, and Lee J (2013). Cancer cell metabolism: implications for therapeutic targets. Exp. Mol. Med. 45, e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M, Li J, Chuang JL, and Chuang DT (2007). Distinct structural mechanisms for inhibition of pyruvate dehydrogenase kinase isoforms by AZD7545, dichloroacetate, and radicicol. Structure 15, 992–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JW, Tchernyshyov I, Semenza GL, and Dang CV (2006). HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 3, 177–185. [DOI] [PubMed] [Google Scholar]
- Kinoshita E, Kinoshita-Kikuta E, and Koike T (2009). Separation and detection of large phosphoproteins using Phos-tag SDS-PAGE. Nat. Protoc. 4, 1513–1521. [DOI] [PubMed] [Google Scholar]
- Korotchkina LG, and Patel MS (2001). Probing the mechanism of inactivation of human pyruvate dehydrogenase by phosphorylation of three sites. J. Biol. Chem. 276, 5731–5738. [DOI] [PubMed] [Google Scholar]
- Koul D (2008). PTEN signaling pathways in glioblastoma. Cancer Biol. Ther. 7, 1321–1325. [DOI] [PubMed] [Google Scholar]
- Koul D, Yao Y, Abbruzzese JL, Yung WK, and Reddy SA (2001). Tumor suppressor MMAC/PTEN inhibits cytokine-induced NFkappaB activation without interfering with the IkappaB degradation pathway. J. Biol. Chem. 276, 11402–11408. [DOI] [PubMed] [Google Scholar]
- Lardy HA, Johnson D, and McMURRAY WC (1958). Antibiotics as tools for metabolic studies. I. A survey of toxic antibiotics in respiratory, phosphorylative and glycolytic systems. Arch. Biochem. Biophys. 78, 587–597. [DOI] [PubMed] [Google Scholar]
- Lee JO, Yang H, Georgescu MM, Di Cristofano A, Maehama T, Shi Y, Dixon JE, Pandolfi P, and Pavletich NP (1999). Crystal structure of the PTEN tumor suppressor: implications for its phosphoinositide phosphatase activity and membrane association. Cell 99, 323–334. [DOI] [PubMed] [Google Scholar]
- Lee HJ, Lee HY, Lee JH, Lee H, Kang G, Song JS, Kang J, and Kim JH (2014). Prognostic significance of biallelic loss of PTEN in clear cell renal cell carcinoma. J. Urol. 192, 940–946. [DOI] [PubMed] [Google Scholar]
- Leslie NR, Yang X, Downes CP, and Weijer CJ (2007). PtdIns(3,4,5)P(3)-dependent and -independent roles for PTEN in the control of cell migration. Curr. Biol. 17, 115–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie NR, Maccario H, Spinelli L, and Davidson L (2009). The significance of PTEN’s protein phosphatase activity. Adv. Enzyme Regul. 49, 190–196. [DOI] [PubMed] [Google Scholar]
- Li DM, and Sun H (1997). TEP1, encoded by a candidate tumor suppressor locus, is a novel protein tyrosine phosphatase regulated by transforming growth factor beta. Cancer Res. 57, 2124–2129. [PubMed] [Google Scholar]
- Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R, et al. (1997). PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 275, 1943–1947. [DOI] [PubMed] [Google Scholar]
- Li T, Chen L, Cheng J, Dai J, Huang Y, Zhang J, Liu Z, Li A, Li N, Wang H, et al. (2016). SUMOylated NKAP is essential for chromosome alignment by anchoring CENP-E to kinetochores. Nat. Commun. 7, 12969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linn TC, Pettit FH, and Reed LJ (1969). Alpha-keto acid dehydrogenase complexes. X. Regulation of the activity of the pyruvate dehydrogenase complex from beef kidney mitochondria by phosphorylation and dephosphorylation. Proc. Natl. Acad. Sci. U S A 62, 234–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunt SY, and Vander Heiden MG (2011). Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 27, 441–464. [DOI] [PubMed] [Google Scholar]
- Maehama T, and Dixon JE (1998). The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 273, 13375–13378. [DOI] [PubMed] [Google Scholar]
- Maehama T, and Dixon JE (1999). PTEN: a tumour suppressor that functions as a phospholipid phosphatase. Trends Cell Biol. 9, 125–128. [DOI] [PubMed] [Google Scholar]
- Maehama T, and Dixon JE (2000). [PTEN tumor suppressor: functions as a lipid phosphatase]. Tanpakushitsu Kakusan Koso 45, 2405–2414. [PubMed] [Google Scholar]
- Martone R, Euskirchen G, Bertone P, Hartman S, Royce TE, Luscombe NM, Rinn JL, Nelson FK, Miller P, Gerstein M, et al. (2003). Distribution of NF-kappaB-binding sites across human chromosome 22. Proc. Natl. Acad. Sci. U S A 100, 12247–12252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayo MW, Madrid LV, Westerheide SD, Jones DR, Yuan XJ, Baldwin AS Jr., and Whang YE (2002). PTEN blocks tumor necrosis factor-induced NF-kappa B-dependent transcription by inhibiting the transactivation potential of the p65 subunit. J. Biol. Chem. 277, 11116–11125. [DOI] [PubMed] [Google Scholar]
- Millán-Uclés Á, Zuluaga S, Marqués M, Vallejo-Díaz J, Sanz L, Cariaga-Martínez AE, Real FX, and Carrera AC (2016). E-cadherin downregulation sensitizes PTEN-mutant tumors to PI3Kβ silencing. Oncotarget 7, 84054–84071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers MP, Stolarov JP, Eng C, Li J, Wang SI, Wigler MH, Parsons R, and Tonks NK (1997). P-TEN, the tumor suppressor from human chromosome 10q23, is a dual-specificity phosphatase. Proc. Natl. Acad. Sci. U S A 94, 9052–9057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers MP, Pass I, Batty IH, Van der Kaay J, Stolarov JP, Hemmings BA, Wigler MH, Downes CP, and Tonks NK (1998). The lipid phosphatase activity of PTEN is critical for its tumor supressor function. Proc. Natl. Acad. Sci. U S A 95, 13513–13518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neshat MS, Mellinghoff IK, Tran C, Stiles B, Thomas G, Petersen R, Frost P, Gibbons JJ, Wu H, and Sawyers CL (2001). Enhanced sensitivity of PTEN-deficient tumors to inhibition of FRAP/mTOR. Proc. Natl. Acad. Sci. U S A 98, 10314–10319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ory DS, Neugeboren BA, and Mulligan RC (1996). A stable human-derived packaging cell line for production of high titer retrovirus/vesicular stomatitis virus G pseudotypes. Proc. Natl. Acad. Sci. U S A 93, 11400–11406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajerowski AG, Nguyen C, Aghajanian H, Shapiro MJ, and Shapiro VS (2009). NKAP is a transcriptional repressor of notch signaling and is required for T cell development. Immunity 30, 696–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer G, Horgan DJ, Tisdale H, Singer TP, and Beinert H (1968). Studies on the respiratory chain-linked reduced nicotinamide adenine dinucleotide dehydrogenase. XIV. Location of the sites of inhibition of rotenone, barbiturates, and piericidin by means of electron paramagnetic resonance spectroscopy. J. Biol. Chem. 243, 844–847. [PubMed] [Google Scholar]
- Patel MS, and Roche TE (1990). Molecular biology and biochemistry of pyruvate dehydrogenase complexes. FASEB J. 4, 3224–3233. [DOI] [PubMed] [Google Scholar]
- Patel MS, Nemeria NS, Furey W, and Jordan F (2014). The pyruvate dehydrogenase complexes: structure-based function and regulation. J. Biol. Chem. 289, 16615–16623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathak RR, Grover A, Malaney P, Quarni W, Pandit A, Allen-Gipson D, and Davé V (2013). Loss of phosphatase and tensin homolog (PTEN) induces leptin-mediated leptin gene expression: feed-forward loop operating in the lung. J. Biol. Chem. 288, 29821–29835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry SW, Norman JP, Barbieri J, Brown EB, and Gelbard HA (2011). Mitochondrial membrane potential probes and the proton gradient: a practical usage guide. Biotechniques 50, 98–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planchon SM, Waite KA, and Eng C (2008). The nuclear affairs of PTEN. J. Cell Sci. 121, 249–253. [DOI] [PubMed] [Google Scholar]
- Poburko D, Santo-Domingo J, and Demaurex N (2011). Dynamic regulation of the mitochondrial proton gradient during cytosolic calcium elevations. J. Biol. Chem. 286, 11672–11684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popov KM, Hawes JW, and Harris RA (1997). Mitochondrial alpha-ketoacid dehydrogenase kinases: a new family of protein kinases. Adv. Second Messenger Phosphoprotein Res. 31, 105–111. [PubMed] [Google Scholar]
- Pourmand G, Ziaee AA, Abedi AR, Mehrsai A, Alavi HA, Ahmadi A, and Saadati HR (2007). Role of PTEN gene in progression of prostate cancer. Urol. J. 4, 95–100. [PubMed] [Google Scholar]
- Rodon J, Dienstmann R, Serra V, and Tabernero J (2013). Development of PI3K inhibitors: lessons learned from early clinical trials. Nat. Rev. Clin. Oncol. 10, 143–153. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Escudero I, Oliver MD, Andrés-Pons A, Molina M, Cid VJ, and Pulido R (2011).A comprehensive functional analysis of PTEN mutations: implications in tumor- and autism-related syndromes. Hum. Mol. Genet. 20, 4132–4142. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze A, and Downward J (2011). Flicking the Warburg switch-tyrosine phosphorylation of pyruvate dehydrogenase kinase regulates mitochondrial activity in cancer cells. Mol. Cell 44, 846–848. [DOI] [PubMed] [Google Scholar]
- Schulze A, and Harris AL (2012). How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature 491, 364–373. [DOI] [PubMed] [Google Scholar]
- Semenza GL (2008). Tumor metabolism: cancer cells give and take lactate. J. Clin. Invest. 118, 3835–3837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen WH, Balajee AS, Wang J, Wu H, Eng C, Pandolfi PP, and Yin Y (2007). Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell 128, 157–170. [DOI] [PubMed] [Google Scholar]
- Shi Y, Wang J, Chandarlapaty S, Cross J, Thompson C, Rosen N, and Jiang X (2014). PTEN is a protein tyrosine phosphatase for IRS1. Nat. Struct. Mol. Biol. 21, 522–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu S, Eguchi Y, Kamiike W, Matsuda H, and Tsujimoto Y (1996a). Bcl-2 expression prevents activation of the ICE protease cascade. Oncogene 12, 2251–2257. [PubMed] [Google Scholar]
- Shimizu S, Eguchi Y, Kamiike W, Waguri S, Uchiyama Y, Matsuda H, and Tsujimoto Y (1996b). Bcl-2 blocks loss of mitochondrial membrane potential while ICE inhibitors act at a different step during inhibition of death induced by respiratory chain inhibitors. Oncogene 13, 21–29. [PubMed] [Google Scholar]
- Shimizu S, Eguchi Y, Kamiike W, Funahashi Y, Mignon A, Lacronique V, Matsuda H, and Tsujimoto Y (1998). Bcl-2 prevents apoptotic mitochondrial dysfunction by regulating proton flux. Proc. Natl. Acad. Sci. U S A 95, 1455–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinde SR, and Maddika S (2016). PTEN modulates EGFR late endocytic trafficking and degradation by dephosphorylating Rab7. Nat. Commun. 7, 10689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soria JC, Lee HY, Lee JI, Wang L, Issa JP, Kemp BL, Liu DD, Kurie JM, Mao L, and Khuri FR (2002). Lack of PTEN expression in non-small cell lung cancer could be related to promoter methylation. Clin. Cancer Res. 8, 1178–1184. [PubMed] [Google Scholar]
- Sos ML, Koker M, Weir BA, Heynck S, Rabinovsky R, Zander T, Seeger JM, Weiss J, Fischer F, Frommolt P, et al. (2009). PTEN loss contributes to erlotinib resistance in EGFR-mutant lung cancer by activation of Akt and EGFR. Cancer Res. 69, 3256–3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St-Denis N, Gupta GD, Lin ZY, Gonzalez-Badillo B, Veri AO, Knight JDR, Rajendran D, Couzens AL, Currie KW, Tkach JM, et al. (2016). Phenotypic and interaction profiling of the human phosphatases identifies diverse mitotic regulators. Cell Rep. 17, 2488–2501. [DOI] [PubMed] [Google Scholar]
- Stacpoole PW (1989). The pharmacology of dichloroacetate. Metabolism 38, 1124–1144. [DOI] [PubMed] [Google Scholar]
- Stambolic V, Suzuki A, de la Pompa JL, Brothers GM, Mirtsos C, Sasaki T, Ruland J, Penninger JM, Siderovski DP, and Mak TW (1998). Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell 95, 29–39. [DOI] [PubMed] [Google Scholar]
- Steck PA, Pershouse MA, Jasser SA, Yung WK, Lin H, Ligon AH, Langford LA, Baumgard ML, Hattier T, Davis T, et al. (1997). Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat. Genet. 15, 356–362. [DOI] [PubMed] [Google Scholar]
- Sun SY (2010). N-acetylcysteine, reactive oxygen species and beyond. Cancer Biol. Ther. 9, 109–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H, Lesche R, Li DM, Liliental J, Zhang H, Gao J, Gavrilova N, Mueller B, Liu X, and Wu H (1999). PTEN modulates cell cycle progression and cell survival by regulating phosphatidylinositol 3,4,5,-trisphosphate and Akt/protein kinase B signaling pathway. Proc. Natl. Acad. Sci. U S A 96, 6199–6204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C, Wang L, Huang S, Heynen GJ, Prahallad A, Robert C, Haanen J, Blank C, Wesseling J, Willems SM, et al. (2014). Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature 508, 118–122. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Bose P, Leong-Quong RY, Fujita DJ, and Riabowol K (2010). REAP: a two minute cell fractionation method. BMC Res. Notes 3, 294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki J, Kanemaru K, Ishii K, Ohkura M, Okubo Y, and Iino M (2014). Imaging intraorganellar Ca2+ at subcellular resolution using CEPIA. Nat. Commun. 5, 4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y, Morales FC, Kreimann EL, and Georgescu MM (2006). PTEN tumor suppressor associates with NHERF proteins to attenuate PDGF receptor signaling. EMBO J. 25, 910–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor GS, and Dixon JE (2003). PTEN and myotubularins: families of phosphoinositide phosphatases. Methods Enzymol. 366, 43–56. [DOI] [PubMed] [Google Scholar]
- Teague WM, Pettit FH, Yeaman SJ, and Reed LJ (1979). Function of phosphorylation sites on pyruvate dehydrogenase. Biochem. Biophys. Res. Commun. 87, 244–252. [DOI] [PubMed] [Google Scholar]
- Thapa P, Chen MW, McWilliams DC, Belmonte P, Constans M, Sant’Angelo DB, and Shapiro VS (2016). NKAP regulates invariant NKT cell proliferation and differentiation into ROR-γt-expressing NKT17 cells. J. Immunol. 196, 4987–4998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibarewal P, Zilidis G, Spinelli L, Schurch N, Maccario H, Gray A, Perera NM, Davidson L, Barton GJ, and Leslie NR (2012). PTEN protein phosphatase activity correlates with control of gene expression and invasion, a tumor-suppressing phenotype, but not with AKT activity. Sci. Signal. 5, ra18. [DOI] [PubMed] [Google Scholar]
- Tristan C, Shahani N, Sedlak TW, and Sawa A (2011). The diverse functions of GAPDH: views from different subcellular compartments. Cell. Signal. 23, 317–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujimoto Y (1998). Role of Bcl-2 family proteins in apoptosis: apoptosomes or mitochondria? Genes Cells 3, 697–707. [DOI] [PubMed] [Google Scholar]
- Tsunoda T, and Takagi T (1999). Estimating transcription factor bindability on DNA. Bioinformatics 15, 622–630. [DOI] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, and Thompson CB (2009). Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan F, and Lenardo MJ (2009). Specification of DNA binding activity of NF-kappaB proteins. Cold Spring Harb. Perspect. Biol. 1, a000067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Xiong H, Wu F, Zhang Y, Wang J, Zhao L, Guo X, Chang LJ, Zhang Y, You MJ, et al. (2014). Hexokinase 2-mediated Warburg effect is required for PTEN- and p53-deficiency-driven prostate cancer growth. Cell Rep. 8, 1461–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O (1956). On the origin of cancer cells. Science 123, 309–314. [DOI] [PubMed] [Google Scholar]
- Whitehouse S, Cooper RH, and Randle PJ (1974). Mechanism of activation of pyruvate dehydrogenase by dichloroacetate and other halogenated carboxylic acids. Biochem. J. 141, 761–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittenberg AD, Azar S, Klochendler A, Stolovich-Rain M, Avraham S, Birnbaum L, Binder Gallimidi A, Katz M, Dor Y, and Meyuhas O (2016). Phosphorylated ribosomal protein S6 is required for Akt-driven hyperplasia and malignant transformation, but not for hypertrophy, aneuploidy and hyperfunction of pancreatic β-cells. PLoS ONE 11, e0149995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniak DJ, Kajdacsy-Balla A, Macias V, Ball-Kell S, Zenner ML, Bie W, and Tyner AL (2017). PTEN is a protein phosphatase that targets active PTK6 and inhibits PTK6 oncogenic signaling in prostate cancer. Nat. Commun. 8, 1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wykoff CC, Beasley NJ, Watson PH, Turner KJ, Pastorek J, Sibtain A, Wilson GD, Turley H, Talks KL, Maxwell PH, et al. (2000). Hypoxia-inducible expression of tumor-associated carbonic anhydrases. Cancer Res. 60, 7075–7083. [PubMed] [Google Scholar]
- You D, Xin J, Volk A, Wei W, Schmidt R, Scurti G, Nand S, Breuer EK, Kuo PC, Breslin P, et al. (2015). FAK mediates a compensatory survival signal parallel to PI3K-AKT in PTEN-null T-ALL cells. Cell Rep. 10, 2055–2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Butler EB, and Tan M (2013). Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis. 4, e532. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Microarray data from this study have been deposited in GEO database (GEO: GSE121217). Phospho-proteomics data from this study have been deposited in PRIDE database (PRIDE: PXD014707).