

Abstract
Phosphorylation of cytoplasmic polyadenylation element binding protein (CPEB) regulates protein synthesis in hippocampal dendrites. CPEB binds the 3′ untranslated region (UTR) of cytoplasmic mRNAs and, when phosphorylated, initiates mRNA polyadenylation and translation. We report that, of the protein kinases activated in the hippocampus during synaptic plasticity, calcium/calmodulin-dependent protein kinase II (CaMKII) robustly phosphorylated the regulatory site (threonine 171) in CPEB in vitro. In postsynaptic density fractions or hippocampal neurons, CPEB phosphorylation increased when CaMKII was activated. These increases in CPEB phosphorylation were attenuated by a specific peptide inhibitor of CaMKII and by the general CaM-kinase inhibitor KN-93. Inhibitors of protein phosphatase 1 increased basal CPEB phosphorylation in neurons; this was also attenuated by a CaM-kinase inhibitor. To determine whether CaM-kinase activity regulates CPEB-dependent mRNA translation, hippocampal neurons were transfected with luciferase fused to a 3′ UTR containing CPE-binding elements. Depolarization of neurons stimulated synthesis of luciferase; this was abrogated by inhibitors of protein synthesis, mRNA polyadenylation, and CaMKII. These results demonstrate that CPEB phosphorylation and translation are regulated by CaMKII activity and provide a possible mechanism for how dendritic protein synthesis in the hippocampus may be stimulated during synaptic plasticity.
Keywords: CPEB, CaMKII, translation, synaptic plasticity, protein phosphatase, protein synthesis, polyadenylation
Introduction
Neurons are capable of robust changes in synaptic strength, typified by long-term potentiation (LTP) and long-term depression in the hippocampus. Short-term synaptic plasticity, lasting from minutes to hours, is dependent on protein kinases, such as α-calcium/calmodulin-dependent protein kinase II (CaMKII), which modify the functional properties of existing proteins involved in synaptic transmission (Soderling and Derkach, 2000; Lisman et al., 2002). Synaptic plasticity lasting from hours to days requires changes in gene transcription and protein synthesis (Frey et al., 1988; Nguyen et al., 1994; Woo and Nguyen, 2003). However, an unresolved question is how newly synthesized proteins resulting from gene transcription and mRNA translation in the soma are targeted to only the synapses undergoing potentiation.
Previous studies indicate that select proteins are synthesized in dendrites (Steward and Schuman, 2001). mRNAs for a number of proteins, including α-CaMKII, are targeted to dendrites by elements in their 3′ untranslated regions (UTRs) (Mayford et al., 1996; Mori et al., 2000; Rook et al., 2000). Dendrites transected from their cell bodies are also capable of protein synthesis (Job and Eberwine, 2001a). For example, isolated dendrites transfected with green fluorescent protein (GFP) flanked by CaMKII 5′ and 3′ UTRs increase synthesis of GFP when stimulated by BDNF (Aakalu et al., 2001). During LTP, polyribosomes translocate into the base of spines (Ostroff et al., 2002), CaMKII mRNA accumulates in dendrites (Thomas et al., 1994; Roberts et al., 1998), and CaMKII protein is elevated in dendrites 150 μm from the soma within 5 min of LTP induction (Ouyang et al., 1999). Furthermore, knock-out mice with the 3′ UTR of CaMKII disrupted do not transport CaMKII mRNA to dendrites, which results in reduced dendritic CaMKII protein levels and attenuated late-phase LTP (Miller et al., 2002). Together, these findings suggest that during LTP, protein synthesis is stimulated in dendrites at potentiated synapses.
The mechanisms that regulate dendritic protein synthesis during synaptic plasticity are just beginning to be delineated. One possible mechanism involves cytoplasmic polyadenylation element binding protein isoform 1 (CPEB). CPEB binds to the 3′ UTR of mRNAs that contain CPE domains (Wells et al., 2000). This has been extensively studied in Xenopus oocytes, in which progesterone stimulates CPEB phosphorylation by Aurora kinase, resulting in polyadenylation and translation of CPE-containing mRNAs (Mendez et al., 2000a,b). The 3′ UTR of CaMKII mRNA, as well as the mRNAs for several other proteins, contains CPE-binding domains, and activity-dependent polyadenylation and translation of CaMKII mRNA occurs in the visual cortex during synaptic plasticity (Wu et al., 1998; Wells et al., 2001).
The sequence surrounding Xenopus CPEB Serine 174 (Ser174), the regulatory site phosphorylated by Aurora kinase, is highly homologous to threonine 171 (Thr171) in mouse CPEB (Mendez et al., 2000b). This sequence has an arginine three amino acids N terminal of Thr171, making it a possible substrate for several protein kinases, including CaMKII and protein kinase A (PKA). Here, we determined whether protein kinases activated in the hippocampus during LTP such as CaMKII, CaMKIV, PKA, and protein kinase C (PKC) could phosphorylate CPEB and thereby regulate protein synthesis.
Materials and Methods
CPEB phosphorylation assays. Nested PCR was used to clone CPEB from mouse brain cDNA using the following primers: 5′TCCAGTCCCTTAGGGCCACG3′ and 5′AGGCCAGCTCCACTAGCTGG3′, followed by 5′GGTCGGCCATAGCGGCCG CGAAGTCGACATGGCTTTCTCTCTGGAAGAAG3′ and 5′GGATAGCTAGCGG CCGTCAGTTCTTCGGTTTCCTCATTA3′. The PCR product was subcloned into plasmid (p) GEX-4T-3 (SalI/NotI; Amersham Biosciences, Arlington Heights, IL) to create a CPEB fusion with glutathione transferase (GST) on the N terminus. The Thr171Ala mutation was created using Stratagene (La Jolla, CA) Quikchange kit (5′CTCGCCTGGACGCCCGGCCCATC3′). GST-CPEB fusion protein was expressed in E. coli BL21(DE3)pLysS cells that were induced at optical density600 0.45 for 3 hr with 1 mm isopropyl-β-d-thiogalactopyranoside at 20°C. GST-CPEB was purified using glutathione-Sepharose beads according to the instructions of the manufacturer (Amersham Biosciences). GST-CPEB (6 μg/reaction) was phosphorylated with CaMKII (50 nm; purified from baculovirus), CaMKK plus CaMKI (50 nm each; purified from baculovirus), CaMKK plus CaMKIV (50 nm each; purified from baculovirus), PKC (0.025 U; P7956; Sigma, St. Louis, MO), PKA (50 nm; catalytic subunit purified from baculovirus), or MAPK (mitogen-activated protein kinase) (20 nm; Stratagene) for 30 min at 30°C. CaMKK plus CaMKI, and CaMKK plus CaMKIV were preincubated with Ca2+/calmodulin to pre-activate the kinases (Tokumitsu et al., 1994). To activate Aurora kinase with targeting protein for Xenopus kinesin-like protein 2 (TPX2), rat or Xenopus Aurora kinase (500 nm) were incubated with 4 μg of TPX2(1–364) at 4°C for 30 min as described previously (Eyers and Maller, 2004). All protein kinases were tested against myelin basic protein (MBP; 10 ng/reaction) in parallel reactions to ensure activity. Final phosphorylation conditions were: 125 mm HEPES, pH 7.5, 25 mm Mg(Ac)2, 20 μm ATP, 40,000 cpm/pmol [γ-32P]ATP, 2.5 mm CaCl2,5 μm calmodulin, 25 μg/ml aprotinin, 25 μg/ml leupeptin, and 1 μm PMSF, with 320 μg/ml l-α-phosphatidylserine and 30 μg/ml 1,2-dioctanoyl-sn-glycerol added for PKC. For phospho-imaging, the gel was exposed to a phosphor-imager (Molecular Dynamics, Sunnyvale, CA) and scanned using Scanner Control 445 SI software (Molecular Dynamics).
Phosphorylation of postsynaptic density fractions. Postsynaptic density (PSD) fractions (8 μg/reaction; generous gift from Dr. Ayse Dosemeci, National Institute of Neurlogical Disorders and Stroke, Bethesda, MD) were preincubated as described previously to decrease basal phosphorylation levels (Vinade and Dosemeci, 2000). Final reaction conditions (30 min at 30°C) were: 20 mm HEPES, pH 7.5, 10 mm Mg(Ac)2, 20 mm DTT, 1 mm ATP, 20 μg/ml aprotinin, 20 μg/ml leupeptin, 100 μm PMSF, and, where indicated, 1.4 mm CaCl2, 5 μm calmodulin, 50 nm calyculin A (CA), and 10 μm CaMKIINtide.
Generation of CPEB monoclonal antibodies. Phospho-Thr171-specific and total CPEB monoclonal antibodies were generated against the synthetic peptide CGSRLDT171RPILDSRSSSPSD corresponding to amino acids 165–184 of mouse CPEB with or without Thr171 phosphorylated. Peptides were conjugated with keyhole limpet hemocyanin via the N terminus Cysteine and were injected into BALB/c mice as described previously (Kimura et al., 1994). The phospho-Thr171 antibody is specific for phosphorylated CPEB, recognizing recombinant CPEB only if Thr171 is phosphorylated (supplemental Fig. 1 A, B, available at www.jneurosci.org). These CPEB antibodies are sensitive enough to detect CPEB at the appropriate molecular weight (∼75 kDa) in PSD fractions (see Fig. 2 A) and cultured hippocampal neurons (see Figs. 3C, 5). The phospho-Thr171 immunoreactivity was abolished when the antibody was preabsorbed with the phospho-peptide, but not the nonphospho-peptide, used to generate the phospho-specific antibody (supplemental Fig. 1C, available at www.jneurosci.org).
Figure 2.
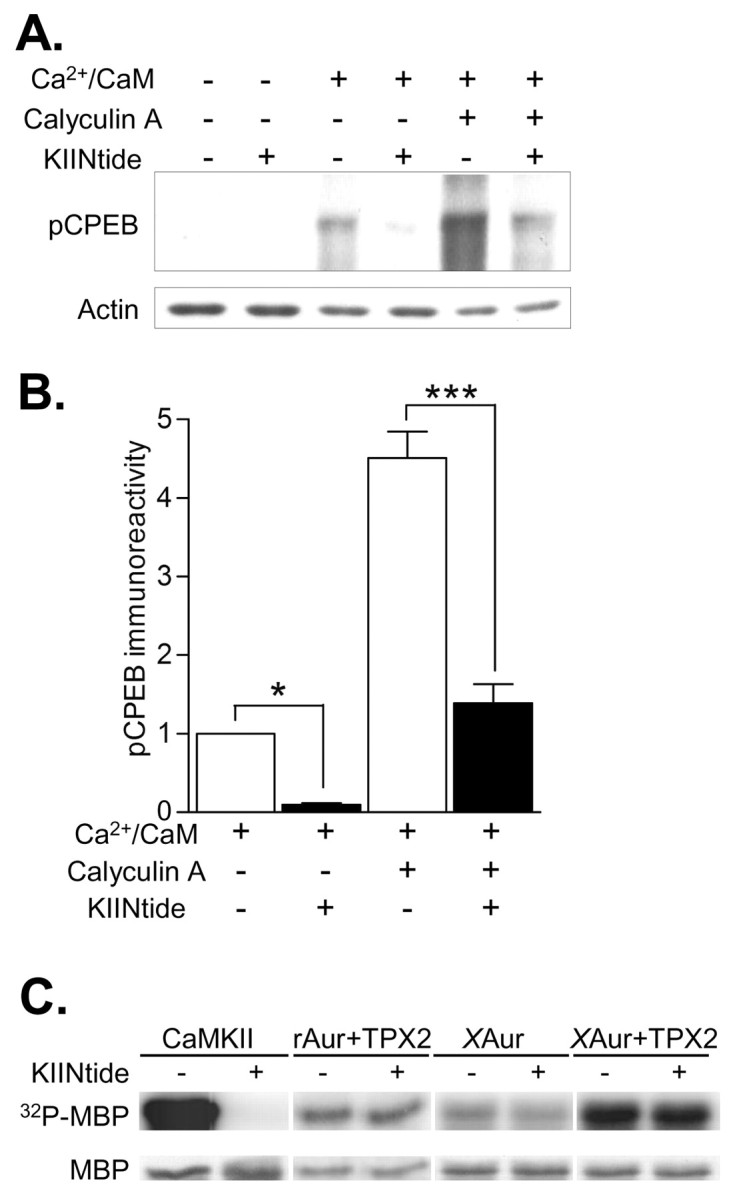
Endogenous CPEB is phosphorylated by CaMKII in the PSD. A, PSD fractions were phosphorylated with or without 1.4 mm Ca2+ and 5 μm CaM, 50 nm CA, or the specific CaMKII inhibitor CaMKIINtide (KIINtide; 10 μm) and then Western blotted for pCPEB. Basal phosphorylation levels were negligible. Ca2+/CaM and CA increased pCPEB levels. Protein levels were equivalent as shown by reprobing with an actin antibody. B, Densitized results, normalized to the Ca2+/CaM stimulation condition (n = 3 for each). CA increased CPEB phosphorylation significantly above the increase seen with Ca2+/CaM (n = 3; p < 0.001). The selective, potent peptide inhibitor of CaMKII, CaMKIINtide, significantly blocked pCPEB in PSD fractions treated with Ca2+/CaM (n = 3; p < 0.05) or CA (n = 3; p < 0.001). C, To test whether another CPEB kinase found in the PSD, Aurora, could be attenuated by CaMKIINtide, phosphorylation of an Aurora kinase substrate, MBP, was assayed. Shown is a representative phosphor-imaging blot for 32P-MBP and the corresponding Coomassie Blue stained gel (MBP). MBP was phosphorylated with CaMKII, rat Aurora kinase (rAur), or Xenopus Aurora kinase (XAur). CaMKIINtide (KIINtide, 10 μm) abolished CaMKII activity. Rat Aurora kinase stimulated by the Aurora kinase chaperone, TPX2, and basal and stimulated Xenopus Aurora A kinase were not affected by CaMKIINtide. Thus, another CPEB kinase found in the PSD, Aurora kinase, was not inhibited by CaMKIINtide.
Figure 3.

Time course of CPEB phosphorylation in neurons stimulated by depolarization. A, Representative Western blots of pCPEB and pCaMKII from neurons depolarized with 90 mm KCl for the indicated time points. For the 10 and 60 min time points, neurons were depolarized for 5 min and then incubated in control solution to assess whether phosphorylation levels were prolonged beyond the initial depolarization. B, Densitized results of the fold increase in pCPEB. pCPEB levels were significantly elevated at 10 sec (0.17 min; n = 18; p < 0.001) and 0.5 min (n = 10; p < 0.001) but not at 5 min (n = 6), 10 min (n = 6), or 60 min (n = 6). There was no significant change in CPEB total levels. C, Densitized fold increase in pCaMKII levels. Similar to pCPEB, increases in pCaMKII were significant at 10 sec (0.17 min; n = 18; p < 0.001) and 0.5 min (n = 10; p < 0.001). D, Densitized results of the fold increase in pCPEB (left y-axis) and pCaMKII (right y-axis) when neurons were stimulated with glutamate (100 μm) for the indicated times. Both pCPEB (n = 3) and pCaMKII (n = 3) increased at 10 sec (0.17 min; p < 0.001) but returned to baseline levels after 5 min.
Figure 5.
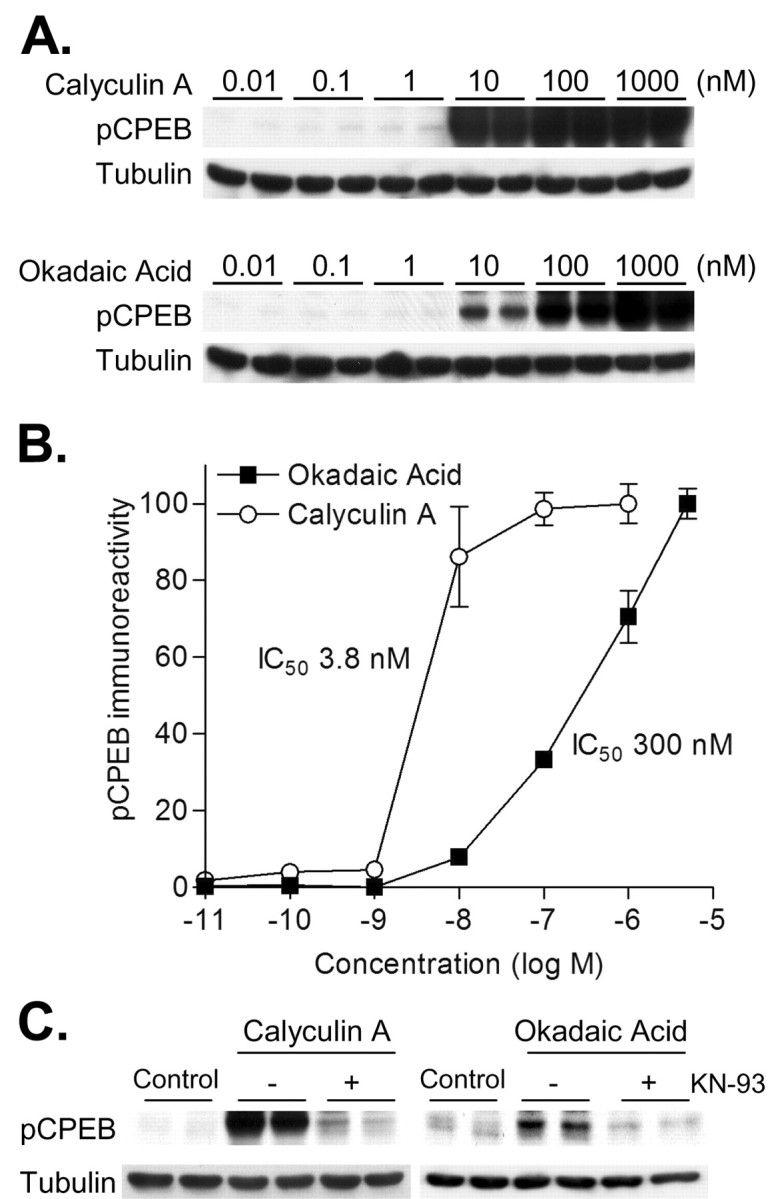
Identification of the protein phosphatase acting on CPEB. A, Representative Western blots of pCPEB and corresponding control blots for total protein (Tubulin). Hippocampal neurons were treated (60 min) with increasing concentrations of the protein phosphatase 1 and 2a inhibitors, calyculin A or okadaic acid, respectively. B, Densitized changes in pCPEB in response to calyculin A and okadaic acid (n = 6 for all concentrations except for 5 μm; n = 4). Lower concentrations of calyculin A (IC50 3.81 nm) were needed to increase pCPEB levels compared with okadaic acid (IC50 301 nm). C, Representative Western blots of pCPEB and total protein (Tubulin). Hippocampal neurons were treated with calyculin A (2.5 nm) or okadaic acid (5 nm) for 60 min. The CaM-kinase inhibitor KN-93 (10 μm) attenuated the increase in pCPEB with calyculin A or okadaic acid. Results are representative of experiments repeated in triplicate.
Western blot analysis. Lysates were electrophoresed (10% SDS-PAGE) and Western blotted using the following antibodies: phospho-Thr171 CPEB (pCPEB) (1:20 of hybridoma supernatant), total CPEB (1:20 of hybridoma supernatant), pCaMKII Thr286 (1:1000; Affinity BioReagents, Golden, CO), CaMKII (1:2000; Affinity BioReagents), β-tubulin (1:5000; Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA), or actin (1:1000; Sigma A2066). Epitopes were visualized with HRP-conjugated secondary antibodies (Amersham Biosciences; 1:1000 to 1:5000) using ECL or ECL plus (Amersham Biosciences) and developed on film (Kodak Biomax MR; Kodak, Rochester, NY). Film was developed to be in a linear range and densitized using Kodak digital science 1D software. Levels of phospho-protein immunoreactivity (e.g., pCPEB) were normalized to total protein immunoreactivity (e.g., CPEB) and then to β-tubulin or actin immunoreactivity to control for total protein levels. Statistical analyses were one-way ANOVAs with Tukey t tests. Data presented are mean ± SEM values.
Hippocampal cultures. Hippocampal neurons were cultured according to the procedure of Brewer (1997) from postnatal day (P) 1 to P2 Sprague Dawley rats. At 7 days in vitro, neurons were preincubated for 30 min at 37°C with 1 μm tetrodotoxin in Tyrodes's solution (25 mm HEPES, pH 7.5, 119 mm NaCl, 2.5 mm KCl, 2 mm MgCl2, 2 mm CaCl2, 30 mm glucose) and with inhibitors where described. Depolarization was delivered in isotonic Tyrode's (90 mm KCl, 1 μm BayK 8644, 10 mm CaCl2). After stimulation, neurons were homogenized in 2× sample buffer supplemented with 1 μm microcystin-lr, 100 mm NaF, 100 mm β-glycerol phosphate, 20 mm EDTA, 20 mm EGTA, 1 μm PMSF, 25 μg/ml aprotinin, and 25 μg/ml leupeptin.
Luciferase translation assays. The reporter plasmid pLuciferase-CPE was constructed as described by Wells et al. (2001) with minor modifications. An EcoRI site was introduced into pEGFP-C1 (Clontech, Cambridge, UK) and pGL3-Promoter (Promega, Madison, WI) by site-directed mutagenesis (pEGFP 5′GGTTACAAATAAAGAATTCGCATCACAAATTTC3′; pGL3 5′CATTATAAGCTGCAATAAAGAATTCAACAACAACAATT GCATTC3′). The polyadenylation sequence of mouse CaMKII 3′ UTR (bp 3075–3217; kindly provided by Dr. Mark Mayford, The Scripps Research Institute, La Jolla, CA) containing the two CPEs, and hexanucleotide motif was PCR amplified (5′GCACTCTAGACTCTCTCTTTCTTTTTTAATCTGTG3′, 5′GCACGAATTCTG TGTAAATTTGTAGCTATTTATTCCACTG3′) and subcloned into the modified pGL3 vector (XbaI/EcoRI) to replace the SV40 early polyadenylation sequence. From this vector, luciferase fused to the CaMKII 3′ UTR was PCR amplified (5′GCACCACCGGTCGCCACCATGGAAGACGCC3′, 5′GCACGAATTCTGTGTAAATTTGTAGCTATTTATTCCACTG3′) and subcloned into the modified pEGFP vector (AgeI/EcoRI) to create pLuciferase-CPE. The CPE-binding domains were mutated as described previously (Wu et al., 1998; Wells et al., 2001). Cultured hippocampal neurons (7–9 days in vitro; 1 × 105 cells/cm2) in 24-well plates were transfected with pLuciferase-CPE (0.4 μg/well) and pRenilla luciferase-TK (0.2 μg/well; Promega) using LipofectAMINE 2000 (1.8–2.5 μl/well; Invitrogen, San Diego, CA) according to the protocol of the manufacturer. For experiments using CaMKIIN, the plasmid CaMKIIN (Chang et al., 1998) was cotransfected (0.8 μg/well), or an equivalent amount of empty vector was used. After transfection for 1 hr, media was replaced with Tyrodes's and 1 μm tetrodotoxin with inhibitors where stated. Cells were incubated for 1 hr and then depolarized (90 mm KCl, 1 μm BayK 8644, 10 mm CaCl2, 0.5 μm actinomycin d) for 5 min in isotonic Tyrode's solution. After stimulation, cells were incubated in Tyrode's with actinomycin d (0.5 μm) for 3 hr with inhibitors where stated and then frozen with liquid nitrogen. Luciferase was measured on a luminometer (Berthold Analytical Instruments, Nashua, NH) using the dual-luciferase reporter assay system (Promega). Firefly luciferase levels were normalized to Renilla luciferase levels to control for transfection efficiency.
Materials. KN-93, KN-92, calmidazolium (CZ), and U0126 were obtained from Calbiochem (La Jolla, CA). BayK 8644, CA, okadaic acid, H-89, anisomycin, and actinomycin d were obtained from Alexis Biochemicals (San Diego, CA). Cordycepin was obtained from Sigma. STO-609 and APV were obtained from Tocris Cookson (Bristol, UK).
Results
CPEB is phosphorylated by CaMKII in vitro
To determine whether any of the protein kinases known to be activated during hippocampal synaptic plasticity phosphorylate CPEB, we cloned mouse CPEB isoform 1, the only isoform containing the known regulatory phosphorylation site Thr171 (Mendez et al., 2000b; Theis et al., 2003). CPEB was expressed as wild-type (WT) or Thr171Ala mutant protein fused to GST. All protein kinases tested incorporated [γ-32P]ATP into full-length CPEB (Fig. 1A), but only CaMKII robustly phosphorylated CPEB. The activities of the protein kinases in the phosphorylation reactions were verified using a ubiquitous substrate, MBP. CaMKII phosphorylation of MBP was similar to CaMKI, PKA, and MAPK, whereas CaMKIV and PKC were more potent (Fig. 1A). Thus, the ratio of CaMKII activity toward CPEB, relative to MBP, was much greater than the other kinases. Furthermore, only phosphorylation by CaMKII was significantly attenuated by the Thr171Ala mutation, the site found in Xenopus oocytes to regulate CPEB function (Fig. 1B).
Figure 1.

Phosphorylation of CPEB by CaMKII in vitro. A, Purified CPEB-GST fusion protein or a ubiquitous kinase substrate, MBP, was phosphorylated in vitro with [γ-32P]ATP and the indicated protein kinases. Representative phosphor-imaging blots for 32P-CPEB and 32P-MBP are shown with the corresponding Coomassie Blue stained gels (CPEB and MBP). All protein kinases were active as demonstrated by 32P-incorporation into MBP, but only CaMKII robustly phosphorylated CPEB. B, Densitized results of 32P-incorporation into WT CPEB versus Thr171Ala CPEB, normalizing phosphorylation of WT CPEB to 100% for each protein kinase. CaMKII phosphorylation of CPEB was significantly affected by Thr171Ala mutation (KII, 50 nm; n = 10; p < 0.001), but PKC (0.0125 U; n = 4), PKA (50 nm; n = 10), MAPK (20 nm; n = 7), or CaMKI (KI, 50 nm; n = 4) and CaMKIV (KIV, 50 nm; n = 12), both activated by CaMKK, were unaffected by the mutation. GST phosphorylation was negligible. C, Purified CPEB-GST fusion proteins, WT or Thr171Ala (A), were phosphorylated in vitro and then Western blotted with a pCPEB monoclonal antibody or a total CPEB antibody. D, Densitized results, normalizing the phosphorylation level of WT CPEB by CaMKII to 100%. Phosphorylation by CaMKII, but not activated CaMKI or CaMKIV, was significantly affected by Thr171Ala mutation (n = 3 for each; p < 0.001).
To confirm these results and to specifically detect CPEB phosphorylated at Thr171 (designated as pCPEB), we generated a phospho-specific CPEB monoclonal antibody and a total CPEB monoclonal antibody (supplemental Fig. 1, available at www.jneurosci.org). CPEB fused to GST was phosphorylated in vitro and Western blotted with the pCPEB antibody. CaMKII strongly phosphorylated CPEB (Fig. 1C,D), and this phospho-immunoreactivity was completely abolished in the Thr171Ala mutant. None of the other protein kinases tested, PKC, PKA, MAPK, or CaMKI and CaMKIV (both activated by CaMKK) significantly phosphorylated CPEB Thr171. These results demonstrate that besides Aurora kinase (Mendez et al., 2000b), CaMKII can also phosphorylate CPEB at Thr171.
CPEB is phosphorylated by CaMKII in the postsynaptic density
In hippocampal neurons, CaMKII is highly enriched in the PSD (Kennedy et al., 1983). The PSD, an electron-dense area at the postsynaptic membrane closely apposed to the presynaptic release site, contains several CaMKII substrates (Yoshimura et al., 2002), including CPEB (Huang et al., 2002). Using a PSD preparation from rat cortex, we determined whether CPEB in the PSD could be phosphorylated by activation of endogenous CaMKII (Fig. 2A,B). Basal CPEB phosphorylation in the PSD was negligible, but addition of Ca2+ and CaM to activate CaMKII significantly increased pCPEB levels. This increase in pCPEB was attenuated by inclusion of CaMKIINtide, a specific peptide inhibitor of CaMKII derived from CaMKIIN, an endogenous inhibitor of CaMKII (Chang et al., 1998). Phosphorylation of CPEB in the presence of Ca2+/CaM was also potentiated by addition of calyculin A (CA), a protein phosphatase inhibitor; this increase was also attenuated by CaMKIINtide. CaMKIINtide does not inhibit CaMKK, CaMKI, CaMKIV, PKC, PKA (Chang et al., 1998), or MAPK (S. Nygaard and T. Soderling, personal communication), but it has not been tested with Aurora kinase. To check this, Aurora kinase activity was assayed using an Aurora kinase substrate, MBP, with CaMKIINtide (Fig. 2C). Aurora kinase is activated by the chaperone protein TPX2, which stimulates autophosphorylation and protects the kinase from dephosphorylation (Eyers et al., 2003; Eyers and Maller, 2004). Thus, we tested whether rat Aurora kinase stimulated by TPX2, basal Xenopus Aurora kinase, or Xenopus Aurora kinase stimulated by TPX2 could be inhibited by CaMKIINtide. Although CaMKIINtide abolished CaMKII phosphorylation of MBP, neither rat nor Xenopus Aurora kinase were affected by CaMKIINtide. These results indicate that the inhibition of endogenous CPEB phosphorylation in the PSD by CaMKIINtide was most likely mediated by CaMKII.
CPEB phosphorylation is regulated dynamically in neurons
To determine whether CaM-kinase activity regulates CPEB phosphorylation in neurons, cultured hippocampal neurons were depolarized and then Western blotted for pCPEB and pCaMKII. This protocol clamps the membrane potential at ∼0 mV, activates voltage-gated calcium channels, and robustly activates CaMKII in dendrites (Wu et al., 2001; Menegon et al., 2002). A time course of CPEB phosphorylation during neuronal depolarization revealed that pCPEB increased nearly fourfold within 1 min and then rapidly decayed by 5 min (Fig. 3A,C). A comparison of pCPEB with pCaMKII indicated a similar temporal relationship (Fig. 3B,C). Glutamate stimulation gave a modest but significant increase in CPEB phosphorylation, which paralleled the activation of CaMKII (Fig. 3D). These results suggest that CPEB phosphorylation is tightly controlled by protein kinase and phosphatase activities in hippocampal neurons, similar to our observations in isolated PSDs.
We next examined the regulatory mechanisms of CPEB phosphorylation in depolarized hippocampal neurons (Fig. 4A). Removal of extracellular calcium completely abolished the increases in pCPEB and pCaMKII. Treatment with calmiclazolium (CZ) to inhibit calmodulin, or CdCl2 to block voltage-gated calcium channels, also blocked the increases in pCPEB and pCaMKII. However, an inhibitor of NMDA receptors, APV, did not inhibit CPEB or CaMKII activation with depolarization. Inhibitors of CaMKK (STO-609), PKA (H-89), PKC (chelerythrine), or MAPK (U0126) had no effect on pCPEB levels (Fig. 4B). However, the general CaM-kinase inhibitor, KN-93, significantly decreased pCPEB and pCaMKII levels (Fig. 4C), whereas the inactive analog, KN-92, did not significantly affect pCPEB and pCaMKII. Thus, neuronal depolarization increased CPEB phosphorylation, and this was dependent on extracellular calcium, calmodulin, voltage-gated calcium channels, and CaM-kinase activity. Accordingly, the changes in CPEB phosphorylation correlated well with changes in CaMKII activity as measured by autophosphorylation of Thr286 (Fig. 4D).
Figure 4.

CPEB phosphorylation in depolarized neurons requires extracellular calcium, calmodulin, and voltage-gated calcium channels; all of which lead to activation of CaMKII. A, Densitized results of the fold increase in pCPEB levels (left y-axis) and pCaMKII levels (right y-axis). The increases in pCPEB (KCl; n = 24; p < 0.001) and pCaMKII (KCl; n = 24; p < 0.001) during neuronal depolarization were blocked when calcium was removed from the extracellular solution and 1 mm EGTA was included (0 Ca2+ pCPEB, n = 8; pCaMKII, n = 8) or by 5 μm CZ (pCPEB, n = 9; pCaMKII, n = 10). A voltage-gated calcium channel blocker, 300 μm CdCl2, also blocked the increases in pCPEB (n = 10) and partially attenuated the increases in pCaMKII (n = 10; p < 0.001), but an NMDA receptor antagonist, APV (50 μm), did not affect pCPEB levels (n = 8; p < 0.001) or pCaMKII levels (n = 8; p < 0.001). B, Application of the CaMKK inhibitor STO-609 (STO; 5 μm; n = 11), the PKA inhibitor H-89 (5 μm; n = 6), the PKC inhibitor chelerythrine (Chele; 1 μm; n = 4), or the MAPK inhibitor U0126 (5 μm; n = 6) had no effect on CPEB phosphorylation. C, The CaM-kinase inhibitor, KN-93 (10 μm), attenuated CPEB phosphorylation when neurons were depolarized (n = 16; p < 0.01). This attenuation was comparable with the attenuation in CaMKII activation by KN-93 (n = 16; p < 0.001). The inactive analog KN-92 had no significant effect on either pCPEB or pCaMKII. D, Correlation analysis of the inhibition of pCPEB and pCaMKII with increasing concentrations of KN-93 (0, 2.5, 5, and 10 μm). When hippocampal neurons were depolarized, the increases in pCPEB and pCaMKII were inhibited to a similar extent by KN-93 (R = 0.901).
Given the temporal dynamics of CPEB phosphorylation in neurons, we next examined which phosphatase regulates CPEB in hippocampal neurons using the protein phosphatase inhibitors calyculin A and okadaic acid. Calyculin A inhibits protein phosphatase 1 (PP1; IC50 0.3–0.7 nm) and protein phosphatase 2a (PP2a; IC50 0.2–1nm) at similar concentrations (Fernandez et al., 2002). However, ∼50-fold higher concentrations of okadaic acid are required to inhibit PP1 (IC50 20 nm) compared with PP2a (IC50 0.2–1 nm). Neither phosphatase inhibitor has any significant effect on calcineurin, PP2b (Fernandez et al., 2002). We took advantage of these protein phosphatase inhibitors and their IC50 values to identify the protein phosphatase that regulates CPEB in hippocampal neurons. Cultured hippocampal neurons were incubated in the absence of depolarization with these protein phosphatase inhibitors (Fig. 5A). Calyculin A (IC50 3.8 nm) was 100-fold more potent than okadaic acid (IC50 300 nm) at inhibiting the protein phosphatase that dephosphorylates CPEB in hippocampal neurons (Fig. 5B). Furthermore, the increase in CPEB phosphorylation with calyculin A or okadaic acid was attenuated by the CaM-kinase inhibitor KN-93 (Fig. 5C). However, neither protein phosphatase inhibitor significantly increased CaMKII autophosphorylation (data not shown). These results suggest that basal CaMKII phosphorylates CPEB, and this phosphorylation is strongly suppressed by PP1 by acting on CPEB directly or on CaMKII to modulate basal calcium-dependent activity of CaMKII.
CaMKII stimulates CPE-dependent protein synthesis in neurons
To determine whether CaM-kinase activity regulates CPE-dependent protein synthesis in hippocampal neurons, we used a translation assay in which 180 bp of CaMKII 3′ UTR, which contains two CPEs and the polyadenylation site, were preceded by the reporter gene luciferase. A similar translation reporter assay, with enhanced GFP in lieu of luciferase, has been used previously by Richter and colleagues to assess CPEB-dependent mRNA translation in cultured hippocampal neurons (Wells et al., 2001). Transfected neurons were depolarized for 5 min, as described above, to elicit increases in pCPEB (Figs. 3, 4, 5). Luciferase levels were assayed 3 hr later to determine stable translation rates, because late-phase LTP is protein synthesis dependent at this time point (Frey et al., 1988; Huang and Kandel, 1994). Actinomycin d, a transcription inhibitor, was included to reduce any transcription effects during and after the depolarization. A 1.8-fold increase in luciferase expression was observed in neurons stimulated by depolarization (Fig. 6A). When the CPE-binding domains in the 3′ UTR were mutated to eliminate CPEB binding (Wu et al., 1998; Wells et al., 2001), no increase in luciferase expression was observed. This is a critical control, indicating that the increase in luciferase was CPE dependent. Anisomycin (0.8 μm) and cordycepin (80 μm; a polyadenylation inhibitor), at concentrations that suppressed basal translation by 40–50% (data not shown), blocked the increases in luciferase expression when neurons were depolarized (Fig. 6A). Thus, the increase in luciferase levels observed after neuronal depolarization was attributable to CPE-dependent mRNA polyadenylation and translation.
Figure 6.

Regulation of CPE-mediated protein synthesis by CaMKII in neurons. A, Cultured hippocampal neurons were transfected with plasmids expressing firefly luciferase fused to a portion of CaMKII 3′ UTR that contained two WT CPE domains or mutated CPE domains (MT). Neurons were stimulated with depolarization by 90 mm KCl for 5 min, incubated for 3 hr in control saline, and then assayed for luciferase levels. Luciferase expression significantly increased in depolarized neurons only when the CPE-binding domains were intact (n = 100; p < 0.001). Treatment with 0.8 μm anisomycin (n = 15) or 80 μm cordycepin (n = 17) decreased basal translation levels by 40–50% (data not shown) and blocked the increases in luciferase expression when neurons were depolarized. B, CPE-mediated luciferase translation was blocked by a specific inhibitor of CaMKII (KIIN). Cotransfection of neurons with luciferase fused to the 3′ UTR of CaMKII and empty vector elicited significant increases in luciferase when the CPE domains were not mutated (control, n = 24; p < 0.001). However, cotransfection of CaMKIIN, an endogenous protein inhibitor of CaMKII, blocked the increases in luciferase (n = 22), whereas the PKA inhibitor 5 μm H-89 (n = 11; p < 0.01) or the MAPK inhibitor 5 μm U0126 (n = 16; p < 0.001) did not block the increases in luciferase translation.
Having established that our translation assay measures CPE-dependent changes in luciferase translation, we next determined whether CaMKII can regulate CPE-dependent protein synthesis. Transfection of neurons with our reporter construct allowed us to cotransfect the CaMKII inhibitor protein CaMKIIN, which is more specific and potent than KN-93 (Chang et al., 1998). We previously observed that transfection of hippocampal neurons with CaMKIIN blocks CaMKII activation (i.e., autophosphorylation on Thr286) after depolarization (Wayman et al., 2004). Importantly, CaMKIINtide, the inhibitory peptide derived from CaMKIIN, does not inhibit Aurora kinase (Fig. 2C). Inhibition of CaMKII with CaMKIIN blocked the increase in luciferase translation compared with neurons cotransfected with empty vector (Fig. 6B). Moreover, inhibitors of PKA or MAPK did not block the increase in luciferase expression. These results demonstrate a potent, selective inhibitor of CaMKII blocked CPE-mediated protein synthesis in hippocampal neurons.
Discussion
CaMKII is a multifunctional modulator of neuronal development and synaptic plasticity (Lisman et al., 2002). We have now described a potential new role for CaMKII: regulation of protein synthesis through CPEB. CaMKII phosphorylated the regulatory site, Thr171, of CPEB in vitro and also endogenous CPEB in the PSD. CPEB phosphorylation in neurons required extracellular calcium, voltage-gated calcium channels, calmodulin, and CaM-kinase activity. Furthermore, CPE-dependent protein synthesis in hippocampal neurons was blocked by a potent specific inhibitor of CaMKII, CaMKIIN, which does not inhibit Aurora kinase. Together, these data demonstrate that CPEB phosphorylation and CPE-dependent protein synthesis are regulated by CaM-kinase activity in hippocampal neurons, providing a possible mechanism for how dendritic protein synthesis can be regulated in various forms of Ca2+-dependent synaptic plasticity.
Previous studies in Xenopus oocytes have shown that CPEB is phosphorylated by Aurora kinase at Ser174 (Mendez et al., 2000b), the site analogous to Thr171 in mouse CPEB. This phosphorylation event recruits polyadenylation factors to the mRNA and initiates polyadenylation and translation (Stebbins-Boaz et al., 1999; Mendez et al., 2000a; Huang et al., 2002). These results and our own demonstrate that CPEB phosphorylation is regulated by Aurora kinase and CaMKII activity in hippocampal neurons. We demonstrated that depolarization of hippocampal neurons, which activated CaMKII, increased CPEB phosphorylation. This was blocked by removal of extracellular calcium, inhibition of calmodulin, and voltage-gated calcium channels; all of which also blocked CaMKII activation. Accordingly, a CaM-kinase inhibitor, KN-93, attenuated endogenous CPEB phosphorylation in hippocampal neurons. CPEB phosphorylation was inhibited by up to 40% by the CaM-kinase inhibitor KN-93. Similarly, CaMKII was also inhibited by only 50%, with a 20-fold activation of CaMKII remaining, which could easily account for the residual CPEB phosphorylation. More complete inhibition by KN-93 could not be tested because the control compound KN-92 also began to modestly affect CaMKII above 10 μm. Although our studies clearly demonstrate that CaMKII phosphorylates CPEB Thr171 and regulates CPE-dependent protein synthesis when neurons are depolarized, other stimulation paradigms may also involve Aurora kinase. Detailed mechanisms for how Aurora kinase may be regulated in neurons have yet to be determined. In our studies, we demonstrate that when hippocampal neurons are depolarized, the increase in CPEB phosphorylation was inhibited by removal of extracellular calcium, calmidazolium, CdCl2, and the CaM-kinase inhibitor KN-93, which all blocked CaMKII activation as well. Additional studies are required to assess the relative roles of CaMKII and Aurora kinase in regulating CPEB-mediated protein synthesis in hippocampal neurons.
At first glance, the rapid and transient phosphorylation of CPEB by CaMKII seems paradoxical to its potential role in regulation of protein synthesis during long-term synaptic plasticity. Intriguingly, protein synthesis occurs within 15 min of LTP induction (Otani et al., 1989; Woo and Nguyen, 2003), suggesting that CPEB could be operative during the first minutes of synaptic plasticity. CPEB phosphorylation results in recruitment of poly(A) polymerase factors to the mRNA, and polyadenylation of the mRNA is thought to disrupt the inhibitory multiprotein complex that suppresses translation initiation (Stebbins-Boaz et al., 1999; Mendez et al., 2000a; Cao and Richter, 2002). We speculate that continued CPEB phosphorylation may not be necessary to complete polyadenylation of the mRNA. That is, CPEB phosphorylation stimulates CPE-dependent mRNA polyadenylation, but CPEB dephosphorylation may not be able to switch off CPE-dependent mRNA translation once polyadenylation is initiated. In oocytes, CPEB-mediated translation is terminated by hyper-phosphorylation and targeted degradation of CPEB (Mendez et al., 2002; Thom et al., 2003). Additional experiments are required to identify the mechanism(s) that switches off CPE-dependent translation in hippocampal neurons.
In agreement with previous in vitro studies (Tay et al., 2003), we found that CPEB was rapidly dephosphorylated in hippocampal neurons. This suggests that to prolong CPEB phosphorylation, a sufficiently high calcium influx may be necessary for simultaneous activation of CaMKII and PKA through calcium-dependent adenylyl cyclases. Activated PKA inhibits PP1 by phosphorylating inhibitor 1 or dopamine- and cAMP-regulated phosphoprotein 32 (DARPP-32) (Winder and Sweatt, 2001). When phosphorylated, these inhibitors suppress PP1 activity. During early-phase LTP, activation of CaMKII is accompanied by PP1 inhibition through this mechanism, and this could result in prolonged phosphorylation of CPEB and stronger stimulation of protein synthesis than occurs with depolarizing stimulation alone (Blitzer et al., 1998; Makhinson et al., 1999; Brown et al., 2000). DARPP-32 is dephosphorylated by calcineurin, a calcium-dependent protein phosphatase, which responds to moderate levels of calcium (Winder and Sweatt, 2001). These signaling pathways may tightly control CPE-dependent protein synthesis such that moderate calcium increases are insufficient to activate translation; rather, high calcium influxes in dendritic spines may be necessary to overcome calcineurin and induce PKA phosphorylation of DARPP-32, consequently inactivating PP1. Thus, CPEB may gate various forms of Ca2+-dependent neuronal plasticity by acting as a coincidence detector for CaMKII activation and PP1 inhibition.
CPEB is found in many species, from Aplysia to humans (Mendez and Richter, 2001). Of the four CPEB isoforms cloned, only isoform 1 has the known regulatory phosphorylation site, Thr171 (Theis et al., 2003). Multiple lines of evidence clearly demonstrate that Thr171 is the regulatory site for CPEB isoform 1-dependent translation. Its phosphorylation enhances coimmunoprecipitation of CPEB with polyadenylation factors (Mendez et al., 2000a) (i.e., cleavage and polyadenylation specificity factor) and breaks the sequestering of translation proteins by the inhibitory protein maskin (Stebbins-Boaz et al., 1999). Finally, phosphorylation of CPEB has been directly shown to increase mRNA polyadenylation and translation in a reconstituted assay, and this is blocked by mutation of the phosphorylation site to Ala (Mendez et al., 2000a). There are at least 28 predicted phosphorylation consensus sequences in CPEB isoform 1, suggesting that other protein kinases may phosphorylate alternative sites and regulate CPEB by other mechanisms. For example, phosphorylation by PKA of Aplysia CPEB, which lacks a homologous Thr171, increased actin mRNA polyadenylation and protein synthesis during long-term facilitation (Liu and Schwartz, 2003). Besides phosphorylation, CPEB is also transcriptionally regulated (Theis et al., 2003). Application of a glutamate agonist increases transcription of CPEB isoforms 3 and 4 in the hippocampus. Recent studies have identified a novel mechanism to regulate CPEB. CPEB can be converted into a prion-like state, stimulating CPE-dependent translation and providing a potential mechanism for how long-term increases in protein synthesis are maintained during synaptic plasticity (Si et al., 2003). Thus, there are multiple mechanisms to regulate CPEB, and it remains to be determined how these isoforms and their mechanisms of regulation interact to control polyadenylation and translation of dendritic mRNAs.
An examination of dendritically localized mRNAs, such as Arc, α-CaMKII, β-actin, fragile X mental retardation protein, MAP2, and PSD95 (postsynaptic density-95), reveals that some, but not all, contain CPE domains (Job and Eberwine, 2001b). All of the necessary factors for CPE-dependent polyadenylation and translation are present in hippocampal dendrites (Huang et al., 2002). Interestingly, many of these other translation factors (e.g., eukartyotic translation initiation factor 4E, eIF4E-binding proteins can also be phosphorylated, layering another level of control to dendritic protein synthesis (Gingras et al., 1999; Kelleher et al., 2004). For example, phosphorylation of eukaryotic elongation factor 2, an initiation factor that binds the 5′ UTR, increases CaMKII mRNA translation in synaptosomes in response to glutamate stimulation (Scheetz et al., 2000). Future studies will continue to reveal the full complexity of the regulation and coordination of dendritic protein synthesis during synaptic plasticity.
The present work evokes the hypothesis that induction of hippocampal LTP, which requires postsynaptic elevations of calcium and produces a prolonged (>1 hr) activation of CaMKII and inactivation of PP1, may promote protein synthesis from CPE-containing dendritic mRNAs through phosphorylation of CPEB.
Footnotes
This work was supported by National Institutes of Health (NIH) Grant NS 27037 (T.R.S.), an N. L. Tartar Trust Fellowship (C.M.A.), and NIH Training Grants NS07381 and DK07680. We thank Dr. Ayse Dosemeci for providing the postsynaptic density preparation, Dr. Mark Mayford for providing the CaMKII 3′ UTR DNA, Dr. James Maller for providing the Xenopus Aurora kinase and targeting protein for Xenopus kinesin-like protein 2, Sean Nygaard for technical assistance, Dr. Victor Derkach and Dr. Anthony Oliva for critical reading of this manuscript, and the Soderling lab for helpful discussions.
Correspondence should be addressed to Dr. Thomas R. Soderling, Vollum Institute, Oregon Health and Science University, 3181 Southwest Sam Jackson Park Road, Portland, OR 97239. E-mail: soderlit@ohsu.edu.
DOI:10.1523/JNEUROSCI.0854-04.2004
Copyright © 2004 Society for Neuroscience 0270-6474/04/245193-09$15.00/0
References
- Aakalu G, Smith WB, Nguyen N, Jiang C, Schuman EM (2001) Dynamic visualization of local protein synthesis in hippocampal neurons. Neuron 30: 489–502. [DOI] [PubMed] [Google Scholar]
- Blitzer RD, Connor JH, Brown GP, Wong T, Shenolikar S, Iyengar R, Landau EM (1998) Gating of CaMKII by cAMP-regulated protein phosphatase activity during LTP. Science 280: 1940–1942. [DOI] [PubMed] [Google Scholar]
- Brewer GJ (1997) Isolation and culture of adult rat hippocampal neurons. J Neurosci Methods 71: 143–155. [DOI] [PubMed] [Google Scholar]
- Brown GP, Blitzer RD, Connor JH, Wong T, Shenolikar S, Iyengar R, Landau EM (2000) Long-term potentiation induced by theta frequency stimulation is regulated by a protein phosphatase-1-operated gate. J Neurosci 20: 7880–7887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Q, Richter JD (2002) Dissolution of the maskin-eIF4E complex by cytoplasmic polyadenylation and poly(A)-binding protein controls cyclin B1 mRNA translation and oocyte maturation. EMBO J 21: 3852–3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang BH, Mukherji S, Soderling TR (1998) Characterization of a calmodulin kinase II inhibitor protein in brain. Proc Natl Acad Sci USA 95: 10890–10895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyers PA, Maller JL (2004) Regulation of Xenopus Aurora A activation by TPX2. J Biol Chem 279: 9008–9015. [DOI] [PubMed] [Google Scholar]
- Eyers PA, Erikson E, Chen LG, Maller JL (2003) A novel mechanism for activation of the protein kinase Aurora A. Curr Biol 13: 691–697. [DOI] [PubMed] [Google Scholar]
- Fernandez JJ, Candenas ML, Souto ML, Trujillo MM, Norte M (2002) Okadaic acid, useful tool for studying cellular processes. Curr Med Chem 9: 229–262. [DOI] [PubMed] [Google Scholar]
- Frey U, Krug M, Reymann KG, Matthies H (1988) Anisomycin, an inhibitor of protein synthesis, blocks late phases of LTP phenomena in the hippocampal CA1 region in vitro Brain Res 452: 57–65. [DOI] [PubMed] [Google Scholar]
- Gingras AC, Raught B, Sonenberg N (1999) eIF4 initiation factors: effectors of mRNA recruitment to ribosomes and regulators of translation. Annu Rev Biochem 68: 913–963. [DOI] [PubMed] [Google Scholar]
- Huang YS, Jung MY, Sarkissian M, Richter JD (2002) N-methyl-D-aspartate receptor signaling results in Aurora kinase-catalyzed CPEB phosphorylation and alpha CaMKII mRNA polyadenylation at synapses. EMBO J 21: 2139–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YY, Kandel ER (1994) Recruitment of long-lasting and protein kinase A-dependent long-term potentiation in the CA1 region of hippocampus requires repeated tetanization. Learn Mem 1: 74–82. [PubMed] [Google Scholar]
- Job C, Eberwine J (2001a) Identification of sites for exponential translation in living dendrites. Proc Natl Acad Sci USA 98: 13037–13042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Job C, Eberwine J (2001b) Localization and translation of mRNA in dendrites and axons. Nat Rev Neurosci 2: 889–898. [DOI] [PubMed] [Google Scholar]
- Kelleher III RJ, Govindarajan A, Jung H-Y, Kang H, Tonegawa S (2004) Translational control by MAPK signaling in long-term synaptic plasticity and memory. Cell 116: 467–479. [DOI] [PubMed] [Google Scholar]
- Kennedy MB, Bennett MK, Erondu NE (1983) Biochemical and immunochemical evidence that the “major postsynaptic density protein” is a subunit of a calmodulin-dependent protein kinase. Proc Natl Acad Sci USA 80: 7357–7361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura K, Nozaki N, Saijo M, Kikuchi A, Ui M, Enomoto T (1994) Identification of the nature of modification that causes the shift of DNA topoisomerase II beta to apparent higher molecular weight forms in the M phase. J Biol Chem 269: 24523–24526. [PubMed] [Google Scholar]
- Lisman J, Schulman H, Cline H (2002) The molecular basis of CaMKII function in synaptic and behavioural memory. Nat Rev Neurosci 3: 175–190. [DOI] [PubMed] [Google Scholar]
- Littlepage LE, Wu H, Andresson T, Deanehan JK, Amundadottir LT, Ruderman JV (2002) Identification of phosphorylated residues that affect the activity of the mitotic kinase Aurora-A. Proc Natl Acad Sci USA 99: 15440–15445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makhinson M, Chotiner JK, Watson JB, O'Dell TJ (1999) Adenylyl cyclase activation modulates activity-dependent changes in synaptic strength and Ca2+/calmodulin-dependent kinase II autophosphorylation. J Neurosci 19: 2500–2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayford M, Baranes D, Podsypanina K, Kandel ER (1996) The 3′-untranslated region of CaMKII alpha is a cis-acting signal for the localization and translation of mRNA in dendrites. Proc Natl Acad Sci USA 93: 13250–13255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez R, Richter JD (2001) Translational control by CPEB: a means to the end. Nat Rev Mol Cell Biol 2: 521–529. [DOI] [PubMed] [Google Scholar]
- Mendez R, Barnard D, Richter JD (2002) Differential mRNA translation and meiotic progression require Cdc2-mediated CPEB destruction. EMBO J 21: 1833–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez R, Murthy KG, Ryan K, Manley JL, Richter JD (2000a) Phosphorylation of CPEB by Eg2 mediates the recruitment of CPSF into an active cytoplasmic polyadenylation complex. Mol Cell 6: 1253–1259. [DOI] [PubMed] [Google Scholar]
- Mendez R, Hake LE, Andresson T, Littlepage LE, Ruderman JV, Richter JD (2000b) Phosphorylation of CPE binding factor by Eg2 regulates translation of c-mos mRNA. Nature 404: 302–307. [DOI] [PubMed] [Google Scholar]
- Menegon A, Verderio C, Leoni C, Benfenati F, Czernik AJ, Greengard P, Matteoli M, Valtorta F (2002) Spatial and temporal regulation of Ca2+/calmodulin-dependent protein kinase II activity in developing neurons. J Neurosci 22: 7016–7026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller S, Yasuda M, Coats JK, Jones Y, Martone ME, Mayford M (2002) Disruption of dendritic translation of CaMKIIalpha impairs stabilization of synaptic plasticity and memory consolidation. Neuron 36: 507–519. [DOI] [PubMed] [Google Scholar]
- Mori Y, Imaizumi K, Katayama T, Yoneda T, Tohyama M (2000) Two cis-acting elements in the 3′ untranslated region of alpha-CaMKII regulate its dendritic targeting. Nat Neurosci 3: 1079–1084. [DOI] [PubMed] [Google Scholar]
- Nguyen PV, Abel T, Kandel ER (1994) Requirement of a critical period of transcription for induction of a late phase of LTP. Science 265: 1104–1107. [DOI] [PubMed] [Google Scholar]
- Ostroff LE, Fiala JC, Allwardt B, Harris KM (2002) Polyribosomes redistribute from dendritic shafts into spines with enlarged synapses during LTP in developing rat hippocampal slices. Neuron 35: 535–545. [DOI] [PubMed] [Google Scholar]
- Otani S, Marshall CJ, Tate WP, Goddard GV, Abraham WC (1989) Maintenance of long-term potentiation in rat dentate gyrus requires protein synthesis but not messenger RNA synthesis immediately post-tetanization. Neuroscience 28: 519–526. [DOI] [PubMed] [Google Scholar]
- Ouyang Y, Rosenstein A, Kreiman G, Schuman EM, Kennedy MB (1999) Tetanic stimulation leads to increased accumulation of Ca(2+)/calmodulin-dependent protein kinase II via dendritic protein synthesis in hippocampal neurons. J Neurosci 19: 7823–7833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts LA, Large CH, Higgins MJ, Stone TW, O'Shaughnessy CT, Morris BJ (1998) Increased expression of dendritic mRNA following the induction of long-term potentiation. Brain Res Mol Brain Res 56: 38–44. [DOI] [PubMed] [Google Scholar]
- Rook MS, Lu M, Kosik KS (2000) CaMKIIalpha 3′ untranslated region-directed mRNA translocation in living neurons: visualization by GFP linkage. J Neurosci 20: 6385–6393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheetz AJ, Nairn AC, Constantine-Paton M (2000) NMDA receptor-mediated control of protein synthesis at developing synapses. Nat Neurosci 3: 211–216. [DOI] [PubMed] [Google Scholar]
- Si K, Lindquist S, Kandel ER (2003) A neuronal isoform of the aplysia CPEB has prion-like properties. Cell 115: 879–891. [DOI] [PubMed] [Google Scholar]
- Soderling TR, Derkach VA (2000) Postsynaptic protein phosphorylation and LTP. Trends Neurosci 23: 75–80. [DOI] [PubMed] [Google Scholar]
- Stebbins-Boaz B, Cao Q, de Moor CH, Mendez R, Richter JD (1999) Maskin is a CPEB-associated factor that transiently interacts with elF-4E. Mol Cell 4: 1017–1027. [DOI] [PubMed] [Google Scholar]
- Steward O, Schuman EM (2001) Protein synthesis at synaptic sites on dendrites. Annu Rev Neurosci 24: 299–325. [DOI] [PubMed] [Google Scholar]
- Tay J, Hodgman R, Sarkissian M, Richter JD (2003) Regulated CPEB phosphorylation during meiotic progression suggests a mechanism for temporal control of maternal mRNA translation. Genes Dev 17: 1457–1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theis M, Si K, Kandel ER (2003) Two previously undescribed members of the mouse CPEB family of genes and their inducible expression in the principal cell layers of the hippocampus. Proc Natl Acad Sci USA 100: 9602–9607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thom G, Minshall N, Git A, Argasinska J, Standart N (2003) Role of cdc2 kinase phosphorylation and conserved N-terminal proteolysis motifs in cytoplasmic polyadenylation-element-binding protein (CPEB) complex dissociation and degradation. Biochem J 370: 91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas KL, Laroche S, Errington ML, Bliss TV, Hunt SP (1994) Spatial and temporal changes in signal transduction pathways during LTP. Neuron 13: 737–745. [DOI] [PubMed] [Google Scholar]
- Tokumitsu H, Brickey DA, Glod J, Hidaka H, Sikela J, Soderling TR (1994) Activation mechanisms for Ca2+/calmodulin-dependent protein kinase IV. Identification of a brain CaM-kinase IV kinase. J Biol Chem 269: 28640–28647. [PubMed] [Google Scholar]
- Vinade L, Dosemeci A (2000) Regulation of the phosphorylation state of the AMPA receptor GluR1 subunit in the postsynaptic density. Cell Mol Neurobiol 20: 451–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayman GA, Kaech S, Grant WF, Davare M, Impey S, Tokumitsu H, Nozaki N, Banker G, Soderling TR (2004) Regulation of axonal extension and growth cone motility by calmodulin-dependent protein kinase I. J Neurosci 24: 3786–3794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells DG, Richter JD, Fallon JR (2000) Molecular mechanisms for activity-regulated protein synthesis in the synapto-dendritic compartment. Curr Opin Neurobiol 10: 132–137. [DOI] [PubMed] [Google Scholar]
- Wells DG, Dong X, Quinlan EM, Huang YS, Bear MF, Richter JD, Fallon JR (2001) A role for the cytoplasmic polyadenylation element in NMDA receptor-regulated mRNA translation in neurons. J Neurosci 21: 9541–9548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winder DG, Sweatt JD (2001) Roles of serine/threonine phosphatases in hippocampal synaptic plasticity. Nat Rev Neurosci 2: 461–474. [DOI] [PubMed] [Google Scholar]
- Woo NH, Nguyen PV (2003) Protein synthesis is required for synaptic immunity to depotentiation. J Neurosci 23: 1125–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu GY, Deisseroth K, Tsien RW (2001) Spaced stimuli stabilize MAPK pathway activation and its effects on dendritic morphology. Nat Neurosci 4: 151–158. [DOI] [PubMed] [Google Scholar]
- Wu L, Wells D, Tay J, Mendis D, Abbott MA, Barnitt A, Quinlan E, Heynen A, Fallon JR, Richter JD (1998) CPEB-mediated cytoplasmic polyadenylation and the regulation of experience-dependent translation of alpha-CaMKII mRNA at synapses. Neuron 21: 1129–1139. [DOI] [PubMed] [Google Scholar]
- Yoshimura Y, Shinkawa T, Taoka M, Kobayashi K, Isobe T, Yamauchi T (2002) Identification of protein substrates of Ca(2+)/calmodulin-dependent protein kinase II in the postsynaptic density by protein sequencing and mass spectrometry. Biochem Biophys Res Commun 290: 948–954. [DOI] [PubMed] [Google Scholar]