

Abstract
We demonstrated previously that dynamic stretch injury of cultured axons induces structural changes and Ca2+ influx modulated by tetrodotoxin (TTX)-sensitive voltage-gated sodium channels (NaChs). In the present study, we evaluated potential damage to the NaCh α-subunit, which can cause noninactivation of NaChs. In addition, we explored the effects of pre-injury and post-injury treatment with TTX and protease inhibition on proteolysis of the NaCh α-subunit and intra-axonal calcium levels ([Ca2+]i) over 60 min after trauma. After stretch injury, we found that [Ca2+]i continued to increase in untreated axons for at least 60 min. We also observed that the III-IV intra-axonal loop of the NaCh α-subunit was proteolyzed between 5 and 20 min after trauma. Pre-injury treatment of the axons with TTX completely abolished the posttraumatic increase in [Ca2+]i and proteolysis of the NaCh α-subunit. In addition, both pre-injury and post-injury inhibition of protease activity attenuated long-term increases in [Ca2+]i as well as mitigating degradation of the NaCh α-subunit. These results suggest a unique “feed-forward” deleterious process initiated by mechanical trauma of axons. Na+ influx through NaChs resulting from axonal deformation triggers initial increases in [Ca2+]i and subsequent proteolysis of the NaChα-subunit. In turn, degradation of the α-subunit promotes persistent elevations in [Ca2+]i, fueling additional pathologic changes. These observations may have important implications for developing therapeutic strategies for axonal trauma.
Keywords: axon trauma, diffuse axonal injury, sodium channels, calcium, proteolysis, protease inhibitors, noninactivation, tetrodotoxin, traumatic brain injury
Introduction
Diffuse axonal injury (DAI) is thought to be the most common and important pathology in mild, moderate, and severe traumatic brain injury (Adams et al., 1982, 1989; Graham et al., 1988; Povlishock, 1992; Maxwell and Graham, 1997; Smith and Meaney, 2000). In severe cases of DAI, shearing forces can cause primary disconnection of axons. However, the vast majority of posttraumatic axonal pathologies evolve over time because of a series of deleterious cascades that include activation of proteases, second messengers, and mitochondrial failure (Povlishock et al., 1983; Banik et al., 1987; Povlishock, 1992; Gitler and Spira, 1998; Buki et al., 1999, 2000). We demonstrated previously that dynamic mechanical stretch injury of cultured axons replicates many of the morphological and ultrastructural changes found in DAI in vivo (Smith et al., 1999). With this model, we found the first evidence that rapid stretch of axons induces an immediate increase in intra-axonal calcium levels ([Ca2+]i), and that this response could be completely reversed with the voltage-gated sodium channel (NaCh) blocker tetrodotoxin (TTX) (Wolf et al., 2001). Thus, although sustained elevated [Ca2+]i may be an important mediator of secondary damage to axons after trauma, as proposed previously (George et al., 1995; Saatman et al., 1996, 2003; Buki et al., 1999; Wolf et al., 2001), this increase in [Ca2+]i is dependent on trauma-induced Na+ influx through NaChs. However, the disposition of NaChs after dynamic stretch injury has not been examined previously.
Noninactivation of NaChs has been shown to cause pathological Na+ influx and membrane depolarization, a state that could potentiate Ca2+ influx through voltage-gated Ca2+ channels and reversal of the Na+-Ca2+ exchanger (Stys et al., 1991, 1992, 1993; Fern et al., 1995; Stys and Lopachin, 1998; Imaizumi et al., 1999; Wolf et al., 2001). Persistent noninactivation of NaChs can result from damage to their α-subunit (Armstrong et al., 1973; Vassilev et al., 1988; Stuhmer et al., 1989; Benz et al., 1997). In the present study, we used the model of dynamic stretch injury of axons from primary cortical neurons to investigate potential proteolysis of discrete regions of the NaCh α-subunit after trauma. In addition, we examined the role of potential NaCh proteolysis on prolonged posttraumatic increases in [Ca2+]i.
Materials and Methods
Cell culture. In the present study, we used rat primary neuronal cell cultures in place of the N-Tera 2 cl/D2 (NT2) neurons used previously in the axon stretch-injury model (Smith et al., 1999; Wolf et al., 2001). The primary neurons were purified from neocortices of embryonic day (E) 17 Sprague Dawley rats (Charles River, Wilmington, MA). The cells were maintained in culture with NeuroBasal media (Invitrogen, Gaithersburg, MD) supplemented with B-27 neural supplement (Invitrogen), 5% fetal bovine serum (HyClone, Logan, UT), and 1% penicillin-streptomycin (Invitrogen). These primary neurons were seeded on a poly-l-lysine- and laminin-treated deformable substrate (Specialty Manufacturing, Saginaw, MI) in custom-designed culture wells (Smith et al., 1999). A 2 × 16 mm clear silicone barrier (specially modified from flexiPERM slide; Sigma, St. Louis, MO) was placed on the membrane in the center of the well before plating of the primary neurons to create a 2 mm “gap” through the center of the membrane. Cells were allowed to attach for 24 hr before the barrier was removed. The temporary barrier prevented neurons from seeding in the gap region, creating a cell-free area for growth of isolated axons. After the barrier was removed, axons traversed the gap, ultimately integrating with neurons on the other side (see Fig. 1 A). The cells were plated at 375,000 cells/cm2. The experiments were performed at 12 d in vitro (DIV).
Figure 1.

A, B, Schematic illustration of axonal stretch injury with membrane pre-stretch (A) and post-stretch (B). Note the downward deflection of the membrane resulting in tensile elongation of axons crossing a cell-free gap. C, D, Neurites in the gap are entirely axons demonstrated in representative photomicrographs of immunocytochemical staining of MAP2 (C), a specific marker for the dendrites and neuronal somata, and NaCh protein (D) found on axons as well as the dendrites and neuronal somata. Scale bar, 50 μm.
Support cell culture. To maintain the cultured axons until testing, support cultures were prepared from neocortices of Sprague Dawley E17 rat embryos (Charles River). The cells were plated at 300,000 cells/cm2 on 12 mm Millicell wells (Millipore, Billerica, MA) 1 week before the neuronal cultures for stretch injury (SILASTIC cultures) were plated on custom-designed culture wells. The support cell cultures (10 DIV) were transferred onto the SILASTIC cultures (3 DIV). Once the Millicell wells were placed over the SILASTIC cultures, they received the same feeding and media as the SILASTIC culture.
Identification of axons in the cell-free zone. We performed immunocytochemistry to detect the identity of the processes traversing the cell-free 2 mm gap in the wells, evaluating immunostaining for microtubule-associated protein 2 (MAP2), a specific marker for the dendrites and neuronal somata, and NaCh protein, which stains the axons in addition to the dendrites and neuronal somata. Cultures were fixed in 4% paraformaldehyde and 0.1 m PBS for 20 min, permeabilized with 0.1% Triton X-100 (PBST) for 20 min at room temperature (RT), and double labeled with monoclonal mouse anti-MAP2 antibody (Ab) (AP20; 1:500; Sigma) (Binder et al., 1986) and polyclonal rabbit anti-pan NaCh Ab (06-811; 1:80; Upstate Biotechnology, Lake Placid, NY) specific for the intracellular III-IV loop of the NaCh α-subunit [amino acid (aa) 1491-1508 of full length, aa 1-2005; P04775), a highly conserved segment of the intracellular III-IV loop (Miller et al., 1983; Dugandzija-Novakovic et al., 1995; Vabnick et al., 1996; Meier et al., 1997; Rasband et al., 1999) at 4°C overnight. After rinsing with PBS, the cultures were incubated with Alexa 594-conjugated anti-mouse IgG (Molecular Probes, Eugene, OR) and Alexa 488-conjugated anti-rabbit IgG (Molecular Probes) for 60 min at RT. Fluorescence microscopy was performed on a Nikon (Tokyo, Japan) Diaphot inverted microscope with a Cooke SensiCam QE CCD camera (Auburn Hills, MI) attached.
Axonal stretch injury. In this study, we used a specially designed axon stretch injury apparatus and technique that we characterized previously (Smith et al., 1999). This method closely mimics mechanical loading conditions of DAI in humans, using dynamic uniaxial stretch or “tensile elongation” of axons to induce injury at strains and strain rates below those that might induce disconnection. To induce stretch injury, the culture wells were placed in a device that consists of an aluminum cover block, a stainless steel plate with a machined 2 × 18 mm slit, and an air pulse-generating system. The culture well was inserted into the cover block and then placed on the slit plate so that the area of the deformable substrate contained the cultured axons. The top plate was attached to the microscope stage, creating a sealed chamber. The top plate had a quartz viewing window in the center, an air inlet for compressed air, and a dynamic pressure transducer (model EPX-V01-25P-/l6F-RF; Entran, Fairfield, NJ) to monitor internal chamber pressure. The introduction of compressed air into the chamber was gated by a solenoid (Parker General Valve, Elyria, OH). The solenoid and the pressure transducer were controlled and monitored by an analog-to-digital board (Metrabyte; Keithley Instruments, Cleveland, OH) integrated with a computer data acquisition system (Capital Equipment Corporation, Bellerica, MA). The device was mounted on the stage of a Nikon inverted microscope (Optical Apparatus, Ardmore, PA), allowing for continuous observation of the axons throughout the experiments. A controlled air pulse was used to induce stretch to only the cultured axons traversing the gap in the well (see Fig. 1 A). A rapid change in chamber pressure deflected downward only the portion of the substrate that contained the cultured axons, inducing tensile elongation (see Fig. 1 B). The rate at which this strain was applied to the axon was between 20 and 35 sec-1, well within the range for traumatic injury experienced by the human brain during rotational acceleration. Measurement of nominal uniaxial strain (ϵ) was calculated by determining the centerline membrane deflection (δ) relative to the slit width (w) and substituting into the geometric relationship:
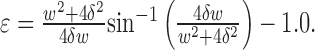 |
For the experiments presented here, peak internal chamber pressure was set at 13 ψ to induce a transient uniaxial strain on the axons calculated at 1.70-1.75 or 70-75% beyond their initial length.
Immunocytochemical detection of NaCh proteolysis and its modulation. We used specific antibodies to the intracellular I-II loop on the NaCh α-subunit (aa 476-485 of full length aa 1-2005; P04775), polyclonal rabbit anti-brain type II NaCh Ab (AB5206; 1:50; Chemicon, Temecula, CA) (Noda et al., 1986; Gordon et al., 1987; Westenbroek et al., 1989), and intracellular III-IV loop of the NaCh α-subunit (aa 1491-1508 of full length aa 1-2005; P04775), and polyclonal rabbit anti-pan NaCh Ab (06-811; 1:80; Upstate Biotechnology) (Miller et al., 1983; Dugandzija-Novakovic et al., 1995; Vabnick et al., 1996; Meier et al., 1997; Rasband et al., 1999) to evaluate immunoreactive changes with and without pharmacologic modulation. Treatment groups included control saline solution (CSS), TTX (Sigma), and a protease inhibitor (PI) mixture tablet Complete (Roche Diagnostics, Indianapolis, IN), which inhibits >90% of each protease activity of serine and cysteine (including calpain I and II) proteases, metalloproteases, Pronase, thermolysin, chymotrypsin, trypsin, and papain. TTX and PI were solubilized separately and added to the culture 10 min before injury or 5 min after injury. Injuries with no treatment were performed using CSS with no modifications (n = 24 wells; no treatment). TTX was used at 1 μm (n = 12, pre-injury treatment; n = 12, post-injury treatment). The PI was used according to the manufacturer instructions (n = 12, pre-injury treatment; n = 12, post-injury treatment). Sham injuries (no stretch injury) with no treatment (n = 6), and pre-injury TTX (n = 3), post-injury TTX (n = 3), pre-injury PI (n = 3), and post-injury PI (n = 3) treatments also were performed.
After injury, the cultures were fixed at 0, 5, 20, and 60 min after injury (n = 3 wells/group/fixation time point) and permeabilized. Sham-injured wells had a single fixation time point of 20 min after sham injury (n = 3 wells/group). After fixation, the cultures were incubated with polyclonal rabbit anti-pan NaCh Ab (06-811; 1:80), specific for a segment of the III-IV intracellular loop of the NaCh α-subunit or polyclonal rabbit anti-brain type II NaCh Ab (AB5206; 1:50) specific for a segment of the intracellular I-II loop on the NaCh α-subunit. Fluorescent labeling of these antibodies was performed as above.
Western blot analysis of NaCh proteolysis and its modulation. To corroborate immunocytochemical evidence of a posttraumatic loss of immunoreactivity to the III-IV loop of the NaCh α-subunit, we performed Western blot analysis. For this analysis, we used protein exclusively extracted from the 2 × 16 mm cell-free zone (i.e., almost entirely derived from axons). Because of the extremely low yield of protein from this region, the number of experimental groups was limited to include only those relevant to the observed changes in immunoreactivity found with immunocytochemical analysis. For each experimental group, extracts from 15 wells were pooled to produce sufficient protein to run three immunoblots for each analysis. Protein extractions of injury with no treatment were evaluated 5 and 20 min after injury. Protein extractions to evaluate the effects of pre-injury TTX treatment and post-injury PI treatment were performed at 20 min after injury. Protein extractions from sham injuries were also collected from no treatment, TTX treatment, and PI treatments at a time point equivalent to the duration of treatment for the injured groups at 20 min after injury.
To collect the protein at the appropriate time point, cultures were frozen on dry ice, and only the axons on the 2 × 16 mm cell-free gap were collected under the microscope. The collected samples were lysed in 150 μl of radioimmunoprecipitation assay buffer (1× PBS, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS) with a mixture of protease inhibitor (Roche Diagnostics). After sonicating cells until clear, the protein concentration was determined by DC Assay Kit (Bio-Rad, Hercules, CA) according to the instructions of the manufacturer. The samples were frozen at -80°C until use. The samples were heated to 37°C for 30 min in 2× SDS sample buffer (1 ml of glycerol, 0.5 ml of β-mercaptoethanol, 3 ml of 10% SDS, 1.25 ml of 1.0 m Tris-HCI, pH 6.7, and 2 mg of bromophenol blue) and run on 7% NuPAGE gel (Invitrogen). The polypeptides were electrotransferred to immunobilon membranes (Millipore). Non-specific binding was blocked using 5% nonfat milk in PBST (9.1 mm dibasic sodium phosphate, 1.7 mm monobasic sodium phosphate, 150 mm NaCl, and 0.1% Tween 20) for 30 min at RT. The membranes were incubated with the polyclonal rabbit anti-pan NaCh Ab (06-811; as above), specific for a segment of the III-IV intracellular loop of the NaCh α-subunit at 1:200, the polyclonal rabbit anti-NaCh type II Ab (AB5206; as above), specific for a segment of the I-II intracellular loop of the NaCh α-subunit at 1:200, or the monoclonal mouse anti-NaCh type II Ab (K69/3; Upstate Biotechnology), specific for a segment of the IV loop to carboxyl terminal of the NaCh α-subunit (aa 1882-2005 of full length aa 1-2005; P04775) at 1:200 overnight at 4°C and then by biotinylated secondary Ab (1:250; Vector Laboratories, Burlingame, CA) as well as Vectastain ABC kit (1:1000; Vector Laboratories) at 1: 1000 for 1 hr each at RT. The membranes were rinsed three times with Tween TBS (100 mm Tris, 0.9% NaCl, and 0.1% Tween 20) for 5 min at RT and then visualized by DAB (Vector Laboratories).
The relative immunoreactivity of NaCh blots was examined using NIH Image software. The optical densities of the band regions corresponding to 220 kDa NaCh protein were determined, and the background (optical density of the membrane between lanes) was subtracted from those of each band of NaCh protein. The optical density measurement was performed five times for each of three blots per group and averaged. All values are presented as the means ± SE. Statistical analysis was performed using one-way ANOVA followed by Fisher's test for multiple comparison. A p value <0.05 was considered significant.
Analysis of changes in [Ca2+]i after injury. Intra-axonal Ca2+ levels were determined using a method for NT2N cells that we previously described in detail (Wolf et al., 2001). The cortical cells were loaded with 2 μm fluo-4 AM ester (Molecular Probes) solubilized in DMSO (0.05% final) with pluronic F-127 [0.004% (w/v) final] in a CSS (120 mm NaCl, 5.4 mm KCl, 0.8 mm MgCl2, 1.8 mm CaCl2, 15 mm glucose, and 25 mm HEPES, pH 7.4, adjusted to 330 mOsm with sorbitol) in which all experiments were run (Takahashi et al., 1999). Because of the small diameter and volume of the axons, fluo-4 AM, an analog of the widely used fluo-3 AM, was used to achieve the maximum fluorescence after binding Ca2+. Fluo-4 is superior in this system because of its increased fluorescence excitation at 488 nm compared with fluo-3 AM. The ion dissociation constant Kd (Ca2+) is reported to be similar for the two dyes under identical conditions (fluo-3, 325 nm; fluo-4, 345 nm; manufacturer specifications). In addition, we evaluated axonal fascicles of 5-10 μm to improve total fluorescence. Pluronic F-127 (Molecular Probes), a nonionic detergent, was used to further disperse the dye in the CSS and allow greater access to the cytoplasm by altering membrane fluidity. The dye solution was loaded at 37°C for 30 min; the cells were rinsed, allowed to sit for another 30 min to allow for additional de-esterification of the dye, and then rinsed once more before injury. The ionophore 4-bromo-A23187 at 50 μm (Molecular Probes) served as a positive control for dye response to Ca2+ influx and was used under all treatment conditions to ensure that proper dye loading had occurred and that the treatments had not altered the Ca2+ affinity of the dye. Because of the brightness of the dye at high Ca2+ concentrations and the sensitivity setting of the camera necessary to image axons, the ionophore-treated axons uniformly reached the maximum detectable fluorescence in our system. Fluorescence microscopy was performed on a Nikon Diaphot inverted microscope with a Hamamatsu Orca CCD camera attached (Optical Apparatus, Ardmore, PA). A xenon light source excited the dye at 488 nm, and the emitted fluorescence was collected at 515 nm. Fluorescence images (1024 × 768 pixels) were collected and analyzed using the MetaFluor software package on a personal computer to which the camera was attached (Universal Imaging, West Chester, PA). Images were taken at 1 sec intervals for 20 sec, at which time the injury was induced (see above). Sampling continued at 1 sec intervals for the first minute after injury. Sampling continued once per minute until the end of 60 min experiments. During the experiments, the temperature on the microscope stage was controlled with a specially designed heater at 37°C. Analysis of changes in fluorescence of the fluo-4 dye was performed on six representative axons from each culture. Axons were excluded from analysis if they had undergone primary axotomy (<5% of injured fibers). Three random regions from each axon were analyzed and then averaged. The axons were continuously sampled, except for a 2-10 sec period after injury during which the microscope was refocused. To account for potential variation in dye loading among axons or experiments, we used a standard procedure for nonratioable indicators in which self-ratios were taken (F/F0) between the measured fluorescence (F) and initial fluorescence (F0). Background fluorescence subtraction was accomplished by continuously sampling three areas in the field that had no axons in them for the duration of the experiment. The mean of these values was obtained at every time point and subtracted from the raw value obtained at each analyzed region of the axon before analysis.
Modulation of [Ca2+]i changes after injury. To evaluate modulation of [Ca2+]i relative to the degradation of the NaCh, we used the same treatment groups (CSS, TTX, and PI) and time points of treatment (10 min before, 5 and 20 min after injury). Injuries were performed using CSS (n = 6 wells; no treatment), TTX treatment (n = 6, pre-injury treatment; n = 6, 5 min post-injury treatment; n = 6, 20 min post-injury treatment), and PI treatment (n = 6, pre-injury treatment; n = 6, post-injury treatment). Sham injuries were also performed (n = 3 for each group). To compare experimental groups, mean values for three areas of each axon analyzed in the culture were then averaged to obtain a mean value for each experiment. Mean values of fluorescence over initial fluorescence (F/F0) for the experimental groups were then compared, and statistical significance was calculated using the post hoc Newman-Keuls test.
Results
Morphological response to stretch injury
The neurites clearly visualized by light microscopy in the cell-free gap (region of injury) were comprised only of axons, demonstrated by a complete absence of MAP2 immunoreactivity while staining for NaCh proteins (Fig. 1C,D). In comparison with previous studies using NT2 neurons (Smith et al., 1999; Wolf et al., 2001), axons in the gap region from primary cortical neurons were of smaller caliber (0.5 to 0.9 μm in diameter). In addition, whereas axons of the NT2 neurons that crossed the cell-free zone rarely formed fasciculations, axons from primary cortical neurons formed many fasciculations crossing the gap (5-10 μm in diameter). Because of the larger diameter of fasciculations, changes in calcium fluorescence or immunofluorescence were easier to visualize. In corroboration with our previous observations using NT2 neurons immediately after stretch injury, the axons of primary neurons demonstrated a delayed elastic response, becoming severely undulated but gradually regained most of their pre-stretch orientation by 20-60 min. However, the fasciculated axons appeared less undulated than single axons. Few axons were found disconnected (primary axotomy) immediately after the stretch, limited to approximately <5% per well.
Sodium channel protein immunoreactivity in untreated stretch-injured axons
Immunocytochemical analysis in sham-injured axons without treatment demonstrated a distinct pattern for both the intracellular III-IV and I-II loops of the NaCh α-subunit. In axons that underwent injury without treatment, the III-IV loop of the NaCh α-subunit was detected at 5 min after injury, but it subsequently almost completely disappeared at 20-60 min after injury (Fig. 2A). However, in axons that underwent injury without treatment, immunoreactivity for the I-II loop of the NaCh α-subunit did not appear to change after stretch at any time point within the first hour (t = 5, 20, 60 min) (Fig. 2C). Nonetheless, the axonal fibers appeared more irregular and tortuous over time after injury, with some displaying swollen regions.
Figure 2.

Proteolytic damage to the NaCh α-subunit on axonal fascicles after stretch injury (without treatment). A, Immunocytochemical staining for the III-IV loop region of the NaCh α-subunit was detected in sham injury and at 5 min after injury but disappeared by 20-60 min after injury. C, In contrast, staining for the I-II loop of NaCh α-subunit was found at all post-injury time points. Scale bars, 20 μm. B, Representative Western blot demonstrates 220 kDa bands corresponding to the NaCh α-subunit III-IV loop in sham injury and at 5 min after injury but not at 20 min after injury (left). Graphic representation of the staining intensities of these bands demonstrates relative changes over time (**p < 0.01 vs sham) (B, right). D, In contrast, representative Western blot analysis of the I-II loop of the NaCh protein demonstrates that the 220 kDa band found at 5 min after injury almost disappeared by 20 min after injury, whereas a new 55 kDa band is found (§§p < 0.01 vs sham). E, Similarly, the 220 kDa MW band found with Western blot analysis of the IV loop of the NaCh protein for sham axons also almost disappeared by 20 min after injury, whereas a new 55 kDa immunoreactive band was found (§§p < 0.01 vs sham). All values are presented as means ± SE.
On Western blot analysis of the III-IV loop of the NaCh protein, we found 220 kDa molecular weight (MW) bands, consistent with the size of α-subunit of NaCh protein. In axons that underwent injury without treatment, immunoblotting appeared to follow the same temporal pattern as was found with immunocytochemical analysis. Staining intensities significantly decreased at 20 min after injury compared with those in sham-injured axons (p < 0.01) (Fig. 2B). In contrast, Western blot analysis of the I-II loop of the NaCh protein revealed 220 kDa MW bands, consistent with the size of α-subunit of NaCh protein in sham axons and at 5 min after injury. At 20 min after injury, this band almost completely disappeared with a significant reduction in staining intensity (p < 0.01). However, at this time point, a new immunoreactive band was found that ranged in MW of 45-55 kDa, not found in sham axons or those 5 min after injury (p < 0.01) (Fig. 2D). Similar to the results with the I-II loop of the NaCh, the 220 kDa MW band found with Western blot analysis of the IV loop of the NaCh protein for sham axons also almost disappeared at 20 min after injury (p < 0.01), whereas a new 55 kDa immunoreactive band was found (p < 0.01) (Fig. 2E). Thus, by 20 min after injury, proteolysis resulted in the production of large fragments of the NaCh.
Sodium channel immunoreactivity in stretch-injured axons treated with TTX
On immunocytochemical analysis for the III-IV loop of the NaCh α-subunit, in axons that were pretreated with TTX (1 μm) and then injured, immunoreactivity remained for at least 60 min after injury (Fig. 3A). However, in axons that were injured and treated with TTX beginning at 5 min after injury, immunoreactivity almost completely disappeared at 20-60 min (Fig. 3B).
Figure 3.
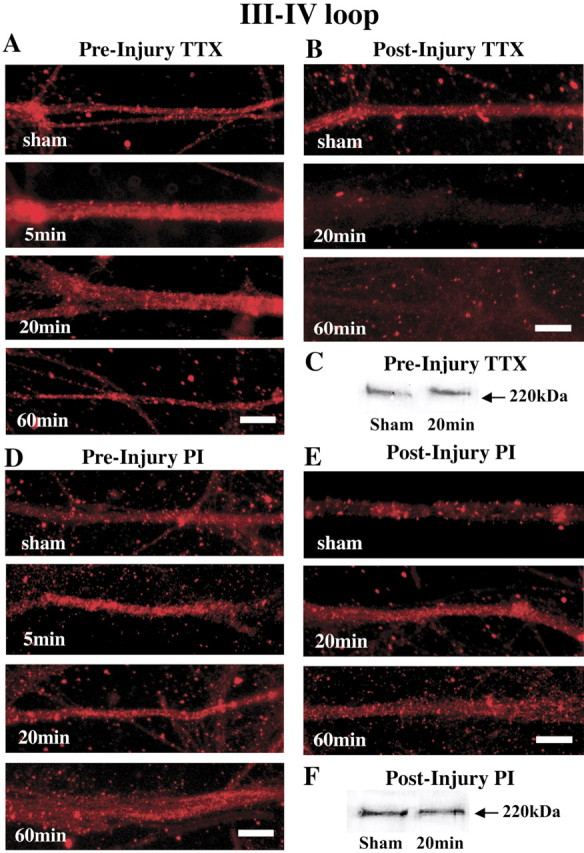
Modulation of NaCh proteolytic damage with TTX and PIs after stretch injury of axon fascicles. A, Representative photomicrographs show that pre-injury treatment with TTX maintains immunoreactivity for the III-IV loop region of the NaCh α-subunit over 60 min after injury. B, However, delaying TTX treatment does not preserve immunoreactivity to the III-IV loop of the NaCh α-subunit. C, Representative Western blot demonstrates that pre-injury TTX treatment prevented the loss of the 220 kDa bands corresponding to the III-IV loop of the NaCh α-subunit after trauma. There was no significant difference of staining intensities between sham and 20 min groups. Scale bars: A, B, 20 μm. D, E, With both pre-injury and post-injury PI treatment, staining for the III-IV loop region of the NaCh α-subunit does not disappear over 60 min after injury. F, Representative Western blot demonstrates no loss of 220 kDa bands corresponding to the III-IV loop of the NaChα-subunit in sham injury and at 20 min after injury. There was no significant difference of staining intensities between sham and 20 min groups. Scale bars: D, E, 20 μm.
On Western blot analysis of the III-IV loop of the NaCh α-subunit, axons that were pretreated with TTX and then injured, distinct immunoreactive bands at 220 kDa were still visible from protein preparations taken 20 min after injury (Fig. 3C). No significant difference in staining intensities was found at 20 min after injury compared with those in sham-treated axons.
Sodium channel immunoreactivity in stretch-injured axons treated with protease inhibitors
On immunocytochemical analysis for the III-IV loop of the NaCh α-subunit, in axons that were either pretreated with PI (Fig. 3D) or post-treated starting 5 min after injury (Fig. 3E), staining was preserved for at least 60 min in both groups.
On Western blot analysis of the III-IV loop of the NaCh α-subunit, axons that were injured and then treated with PI at 5 min after injury, strong immunoreactive bands at 220 kDa were found for at least 20 min after injury. No significant difference in staining intensities was found at 20 min after injury compared with those in sham-treated axons (Fig. 3F).
Calcium response in untreated stretch-injured axons
In noninjured axons, baseline Fluo-4 fluorescence remained stable for at least 60 min. In nondisconnected axons, we observed a large increase in the measured fluorescence immediately after injury (F/F0 = 1.96; p < 0.01). After immediate increase in the mean fluorescence, there was a downward trend 2 min after stretch (t = 2 min; F/F0 = 1.85). At 20 and 60 min after stretch, however, the fluorescence significantly increased (t = 20 min, F/F0 = 2.41; t = 60 min, F/F0 = 2.94; p < 0.01) (Fig. 4A,B).
Figure 4.
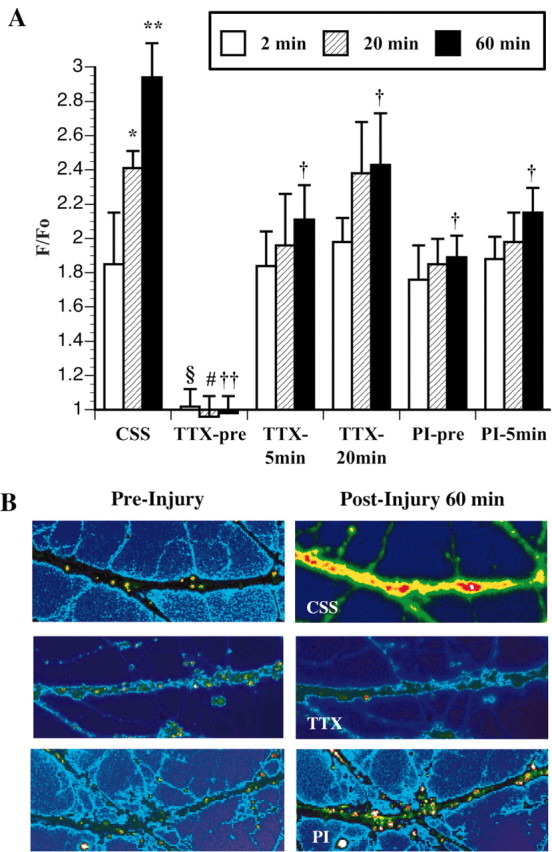
Graphic representation of changes in intra-axonal Ca2+ fluorescence at 2, 20, and 60 min on axonal fascicles after stretch injury in CSS compared with pre-injury treatment of TTX (TTX-pre), 5 min post-injury treatment of TTX (TTX-5 min), 20 min post-injury treatment of TTX (TTX-20 min), pre-injury treatment of PI (PI-pre), and 5 min post-injury treatment of PI (PI-5 min). A, F/F0 = change in Ca2+ fluorescence over initial fluorescence. Comparison with 2 min after injury in CSS: *p < 0.05; **p < 0.01. Comparison with 20 min after injury in CSS: #p < 0.05. Comparison with 60 min after injury in CSS: †p < 0.05; ††p < 0.01. B, Representative photomicrographs demonstrate changes in Ca2+ fluorescence before (left) and 60 min after (right) axonal injury in CSS, with pre-injury treatment of TTX and with pre-injury treatment of PI.
Calcium response in stretch-injured axons with pharmacological manipulation
In axons that were pretreated with TTX (1 μm) and then injured, we found a complete attenuation of the Ca2+ influx (F/F0 = 1.02; NS) response immediately after injury compared with untreated axons that underwent stretch injury (F/F0 = 1.96; p < 0.01). In axons that were injured and then treated with TTX at 5 min after injury, we found significant attenuation of the Ca2+ influx response at 60 min (F/F0 = 2.11) (Fig. 4B) after injury compared with untreated axons that underwent stretch injury (F/F0 = 2.94; p < 0.05), whereas there was no significant attenuation of the Ca2+ influx response at 20 min (F/F0 = 1.96) (Fig. 4A). Surprisingly, in axons that were injured and then treated with TTX at 20 min after injury, we also found significant attenuation of the Ca2+ influx response at 60 min (F/F0 = 2.11) (Fig. 4A) after injury compared with untreated axons that underwent stretch injury (F/F0 = 2.94; p < 0.05). Importantly, there was no significant increase of the Ca2+ influx between 20 and 60 min after injury (p > 0.05) in 20 min post-injury TTX-treated axons.
In axons that were pretreated with PI and then injured, we found no attenuation of the Ca2+ influx response 2 min after injury compared with untreated axons that underwent stretch injury. However, pretreatment with the PI did result in a significant attenuation of the Ca2+ influx response at 60 min after injury (F/F0 = 1.86) (Fig. 4B) compared with untreated axons that underwent stretch injury (F/F0 = 2.94; p < 0.05) (Fig. 4A). In axons that were injured and then treated with PI at 5 min after injury, we found a significant attenuation of the Ca2+ influx response at 60 min (F/F0 = 1.98) after injury compared with untreated axons that underwent stretch injury (F/F0 = 2.94; p < 0.05), whereas there was no significant attenuation of the Ca2+ influx response at 20 min (F/F0 = 1.98) (Fig. 4A).
Discussion
In this study, we report the first evidence that dynamic stretch injury of axons induces selective proteolysis of NaChs linked to progressively increasing levels of intra-axonal Ca2+. A key region of the NaCh that was degraded after axonal trauma included intracellular domains III and IV of the α-subunit. Blockade of the NaCh with TTX before injury completely inhibited posttraumatic increases in [Ca2+]i and prevented proteolysis of the NaCh α-subunit. In addition, protease inhibition attenuated long-term increases in [Ca2+]i and degradation of the III-IV linker of the NaCh α-subunit. These data suggest a sequence of deleterious events after axonal trauma that is coupled to NaCh damage. Mechanical trauma of axons results in Na+ influx through the NaCh, triggering rapid increases in [Ca2+]i. This initial increase in [Ca2+]i then induces proteolysis of the NaCh α-subunit, which in turn pushes already high [Ca2+]i to continue to increase. These observations may have important implications for the development of therapeutic strategies for axonal damage in traumatic brain injury.
It has been shown previously that proteolysis of the α-subunit at the intracellular loop of the III-IV linker domain of the NaCh will prevent the normal inactivation of NaChs leading to persistent leakage of Na+ (Vassilev et al., 1988; Stuhmer et al., 1989; Benz et al., 1997). Without inactivation, pathologic Na+ influx can induce sustained depolarization, opening of voltage-gated Ca2+ channels (Fern et al., 1995; Imaizumi et al., 1999; Wolf et al., 2001), and reversal of the Na+-Ca2+ exchanger (Stys et al., 1992, 1993; Stys and Lopachin, 1998; Wolf et al., 2001; Stys and Waxman, 2004). The resulting increased intracellular Ca2+ concentrations may induce several deleterious cascades including activation of proteases and degradation of cytoskeletal elements (George et al., 1995; Saatman et al., 1996, 2003; Buki et al., 1999). Based on the current results, a similar process initiated by loss of normal inactivation of NaChs may occur in axonal trauma with one important distinction. Initial NaCh dysfunction after stretch injury is caused by direct or indirect mechanical deformation rather than through enzymatic cleavage, because the initial TTX-dependent Ca2+ rise could not be blunted by broad spectrum protease inhibition.
It has long been suggested that elevated intra-axonal Ca2+ levels play a pivotal role in the secondary damage to axons after mechanical deformation (Povlishock et al., 1983; Banik et al., 1987; Povlishock, 1992; Gitler et al., 1998; Buki et al., 1999, 2000). However, we only recently found direct evidence that Ca2+ enters axons shortly after dynamic stretch injury in culture using the neuronal cell line NT2N (Wolf et al., 2001). It was observed that the posttraumatic rise in intra-axonal Ca2+ was completely dependent on Na+ entering through TTX-sensitive NaChs, as corroborated in the present study using primary cortical cells in the same model. It was also found that after trauma, intra-axonal Ca2+ increased by entry through voltage-gated Ca2+ channels and reversal of the Na+-Ca2+ exchanger. These observations are consistent with mechanisms of Ca2+ influx in models of noninactivation of NaChs after proteolytic damage to the α-subunit.
The present data suggest that although mechanical deformation is the watershed event triggering increases in [Ca2+]i, proteolysis of the NaCh α-subunit plays an important role in the pathologic sequelae of axonal trauma. These sequelae appear to include a unique feed-forward process after axonal trauma, whereby mechanical trauma leads to NaCh proteolysis, in turn, perpetuating pathologic Ca2+ influx (Fig. 5). Specifically, increases in [Ca2+]i were found shortly after injury, yet loss of immunoreactivity to domains III and IV of the α-subunit was not observed until between 5 and 20 min after trauma. Both the posttraumatic increase in [Ca2+]i and proteolysis of the NaCh α-subunit could be abolished by pre-injury treatment with TTX. This demonstrates that Na+ influx through NaChs after axonal trauma induced initial increases in [Ca2+]i, triggering degradation of the NaCh α-subunit. Although TTX treatment at 5 or 20 min after injury had no effect on proteolysis of the NaCh α-subunit, both completely abolished any additional increases in [Ca2+]i until at least 60 min after trauma. Thus, although Ca2+-dependent proteolysis of the NaCh α-subunit is a rapid event after trauma, attenuation of additional Ca2+ influx into the axon can still be achieved with post-injury blockade of the NaCh. The observation that TTX eliminated Ca2+ influx even after proteolytic damage to the NaCh had occurred suggests that the overall NaCh structure remained sufficiently intact to allow pathologic Na+ influx.
Figure 5.
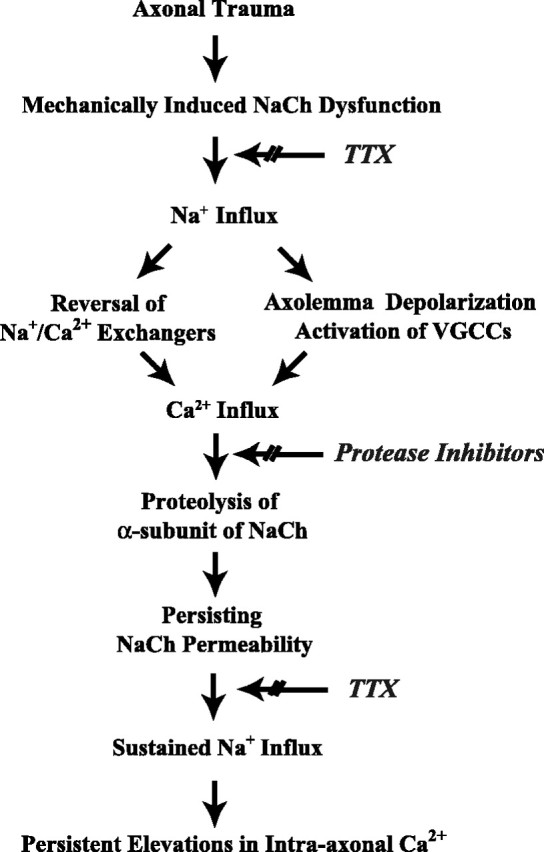
Proposed feed-forward pathway of Ca2+ entry and NaCh proteolysis resulting from traumatic mechanical deformation of axons. VGCCs, Voltage-gated Ca2+ channels.
It was also found that pre-injury and post-injury inhibition of protease activity attenuated delayed increases in [Ca2+]i as well as mitigating degradation of the NaCh α-subunit. These data demonstrate that protease activity plays an important role in maintaining increases in [Ca2+]i after axonal trauma. In particular, proteolytic degradation of the III and IV linker of the NaCh α-subunit may ensure long-term influx of Na+ into axons known to be linked with persistent elevations in [Ca2+]i (Stys et al., 1991, 1992, 1993; Fern et al., 1995; Stys and Lopachin, 1998; Imaizumi et al., 1999; Wolf et al., 2001). This feed-forward cycle may proceed with the high [Ca2+]i fueling progressive proteolysis, in turn, inducing even additional increases in [Ca2+]i. It is important to note that we found [Ca2+]i continued to increase over at least 1 hr after axonal trauma. Although we did not identify specific calcium-mediated proteases responsible for NaCh degradation, one likely candidate is calpain, which has been shown to play an important role in generalized proteolysis in axonal trauma (George et al., 1995; Saatman et al., 1996, 2003; Buki et al., 1999, 2000).
It is not presently clear why the NaCh α-subunit is so rapidly proteolyzed after axonal trauma. One possibility is that traumatic deformation may induce a conformational change in domains III and IV, rendering this region a target for rapid proteolysis. Previously and in the current study, we found direct evidence that the mechanical loading conditions of axon stretch injury can induce immediate changes to other axonal structures (Smith et al., 1999; Wolf et al., 2001). In particular, the loss of axon elasticity after trauma demonstrates an immediate transformation of cytoskeletal elements. Likewise, conformational changes of the NaCh may be responsible for both the pathologic influx of Na+ immediately after injury and predisposition to proteolytic degradation of the α-subunit. However, direct physical disruption of the NaCh at the time of trauma remains to be determined.
To date, all phase III clinical trials evaluating treatments for human brain trauma have failed to identify an efficacious agent (Bullock et al., 1999; Morris et al., 1999; Narayan et al., 2002). Reasons for this are certainly multifactorial, but it must be taken into account that few of these therapies specifically targeted one of the most important pathologic features of human brain injury, DAI. The development of therapeutic strategies for DAI rests primarily on the understanding of the initiating events of axonal trauma. Our current results support previous proposals that sustained intra-axonal Ca2+ increases may initiate a series of deleterious cascades such as activation of proteases (George et al., 1995; Saatman et al., 1996, 2003; Buki et al., 1999, 2000). However, it remains to be established whether NaCh dysregulation and proteolysis play a global role in the pathogenesis of axonal trauma in vivo. For example, differences in NaCh distribution between axons of varying caliber and extent of myelination may result in a differential response to trauma.
Although TTX and generalized protease inhibition used in the present study may not be good candidates for treatment in humans, their collective ability that suppresses Ca2+-induced intraaxonal proteolysis with post-injury application demonstrates a therapeutic opportunity. It has been reported that protease inhibition suppressed the degradation of neurofilament proteins at the site of mechanical insult and secondary axonal degeneration and improved motor function in acute spinal cord injury in rats (Iwasaki et al., 1985, 1987; Iizuka et al., 1986; Schumacher et al., 2000). Likewise, calpain inhibition suppressed axonal pathology in traumatic brain injury (Buki et al., 1999), anoxic optic nerve (Jiang and Stys, 2000), and traumatic optic nerve stretch injury (Witgen et al., 2001). It has also been shown that NaCh blockers improve functional outcome and reduce axonal pathology in models of spinal cord crush injury (Teng and Wrathall, 1997; Rosenberg et al., 1999; Schwartz and Fehlings, 1999) as well as functional outcome in traumatic brain injury (McIntosh et al., 1996; Zhang et al., 1998). Taken in context with the present results, these therapeutic approaches may show benefit by interrupting the feed-forward process initiated by NaCh dysfunction after traumatic axonal injury.
In summary, these experiments are the first to demonstrate the exquisite sensitivity of NaChs to axonal trauma. As a consequence of mechanical deformation of axons, sustained Na+ influx through NaChs triggers initial increases in [Ca2+]i linked to subsequent proteolysis within domains III and IV of the NaCh α-subunit. In turn, this degradation of the α-subunit maintains long-term increases in [Ca2+]i, potentially fueling additional pathology. The observation that these deleterious responses can be mitigated after injury suggests a therapeutic window of opportunity and potential strategies to treat traumatic axonal injury in vivo.
Footnotes
This work was supported by National Institutes of Health Grants AG21527 and NS38104 (D.H.S.), by a Heart and Stroke Foundation of Ontario Career Investigator Award (P.K.S.) and Grants NS35712 and NS41699, and by Center for Disease Control Grant CCR312712 (D.F.M.). We thank Anastasia Melissaratos, David W. Matthews, and Tony Chen for their excellent technical assistance, Dr. Kathryn Saatman for her thoughtful advice on this manuscript, and Dr. Susan S. Margulies for providing her fluorescence microscope system.
Correspondence should be addressed to Douglas H. Smith, University of Pennsylvania, 3320 Smith Walk, 105 Hayden Hall, Philadelphia, PA 19104-6316. E-mail: smithdou@mail.med.upenn.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/244605-09$15.00/0
References
- Adams JH, Graham DI, Murray LS, Scott G (1982) Diffuse axonal injury due to nonmissile head injury in humans: an analysis of 45 cases. Ann Neurol 12: 557-563. [DOI] [PubMed] [Google Scholar]
- Adams JH, Doyle D, Ford I, Gennarelli TA, Graham DI, McClellan DR (1989) Diffuse axonal injury in head injury: definition, diagnosis, and grading. Histopathology 15: 49-59. [DOI] [PubMed] [Google Scholar]
- Armstrong CM, Bezanilla F, Rojas E (1973) Destruction of sodium conductance inactivation in squid axons perfused with pronase. J Gen Physiol 62: 375-391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banik NL, Hogan EL, Hsu CY (1987) The multimolecular cascade of spinal cord injury. Studies on prostanoids, calcium, and proteinases. Neurochem Pathol 7: 57-77. [DOI] [PubMed] [Google Scholar]
- Benz I, Beck W, Kraas W, Stoll D, Jung G, Kohlhardt M (1997) Two types of modified cardiac Na+ channels after cytosolic interventions at the alpha-subunit capable of removing Na+ inactivation. Eur Biophys J 25: 189-200. [DOI] [PubMed] [Google Scholar]
- Binder LI, Frankfurter A, Rebhun LI (1986) Differential localization of MAP-2 and tau in mammalian neurons in situ Ann NY Acad Sci 466: 145-166. [DOI] [PubMed] [Google Scholar]
- Buki A, Siman R, Trojanowski JQ, Povlishock JT (1999) The role of calpain-mediated spectrin proteolysis in traumatically induced axonal injury. J Neuropathol Exp Neurol 58: 365-375. [DOI] [PubMed] [Google Scholar]
- Buki A, Okonkwo DO, Wang KK, Povlishock JT (2000) Cytochrome c release and caspase activation in traumatic axonal injury. J Neurosci 20: 2825-2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullock MR, Lyeth BG, Muizelaar JP (1999) Current status of neuroprotection trials for traumatic brain injury: lessons from animal models and clinical studies. Neurosurgery 45: 207-217. [DOI] [PubMed] [Google Scholar]
- Dugandzija-Novakovic S, Koszowski AG, Levinson SR, Shrager P (1995) Clustering of Na+ channels and node of Ranvier formation in remyelinating axons. J Neurosci 15: 492-503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fern R, Ransom BR, Waxman SG (1995) Voltage-gated calcium channels in CNS white matter: role in anoxic injury. J Neurophysiol 74: 369-377. [DOI] [PubMed] [Google Scholar]
- George EB, Glass JD, Griffin JW (1995) Axotomy-induced axonal degeneration is mediated by calcium influx through ion-specific channels. J Neurosci 15: 6445-6452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitler D, Spira ME (1998) Real time imaging of calcium-induced localized proteolytic activity after axotomy and its relation to growth cone formation. Neuron 20: 1123-1135. [DOI] [PubMed] [Google Scholar]
- Gordon D, Merrick D, Auld V, Dunn R, Goldin AL, Davidson N, Catterall WA (1987) Tissue-specific expression of the RI and RII sodium channel subtypes. Proc Natl Acad Sci USA 84: 8682-8686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham DI, Adams JH, Gennarelli TA (1988) Mechanisms of nonpenetrating head injury. Prog Clin Biol Res 264: 159-168. [PubMed] [Google Scholar]
- Iizuka H, Iwasaki Y, Yamamoto T, Kadoya S (1986) Morphometric assessment of drug effects in experimental spinal cord injury. J Neurosurg 65: 92-98. [DOI] [PubMed] [Google Scholar]
- Imaizumi T, Kocsis JD, Waxman SG (1999) The role of voltage-gated Ca2+ channels in anoxic injury of spinal cord white matter. Brain Res 817: 84-92. [DOI] [PubMed] [Google Scholar]
- Iwasaki Y, Iizuka H, Yamamoto T, Konno H, Kadoya S (1985) Alleviation of axonal damage in acute spinal cord injury by a protease inhibitor: automated morphometric analysis of drug-effects. Brain Res 347: 124-126. [DOI] [PubMed] [Google Scholar]
- Iwasaki Y, Yamamoto H, Iizuka H, Yamamoto T, Konno H (1987) Suppression of neurofilament degradation by protease inhibitors in experimental spinal cord injury. Brain Res 406: 99-104. [DOI] [PubMed] [Google Scholar]
- Jiang Q, Stys PK (2000) Calpain inhibitors confer biochemical, but not electrophysiological, protection against anoxia in rat optic nerves. J Neurochem 74: 2101-2107. [DOI] [PubMed] [Google Scholar]
- Maxwell WL, Graham DI (1997) Loss of axonal microtubules and neurofilaments after stretch-injury to guinea pig optic nerve fibers. J Neurotrauma 14: 603-614. [DOI] [PubMed] [Google Scholar]
- McIntosh TK, Smith DH, Voddi M, Perri BR, Stutzmann JM (1996) Riluzole, a novel neuroprotective agent, attenuates both neurologic motor and cognitive dysfunction following experimental brain injury in the rat. J Neurotrauma 13: 767-780. [DOI] [PubMed] [Google Scholar]
- Meier T, Hauser DM, Chiquet M, Landmann L, Ruegg MA, Brenner HR (1997) Neural agrin induces ectopic postsynaptic specializations in innervated muscle fibers. J Neurosci 17: 6534-6544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JA, Agnew WS, Levinson SR (1983) Principal glycopeptide of the tetrodotoxin/saxitoxin binding protein from electrophorus electricus: isolation and partial chemical and physical characterization. Biochemistry 22: 462-470. [DOI] [PubMed] [Google Scholar]
- Morris GF, Bullock R, Marshall SB, Marmarou A, Maas A, Marshall LF (1999) Failure of the competitive N-methyl-D-aspartate antagonist Selfotel (CGS 19755) in the treatment of severe head injury: results of two phase III clinical trials. The selfotel investigators. J Neurosurg 91: 737-743. [DOI] [PubMed] [Google Scholar]
- Narayan RK, Michel ME, Ansell B, Baethmann A, Biegon A, Bracken MB, Bullock MR, Choi SC, Clifton GL, Contant CF, Coplin WM, Dietrich WD, Ghajar J, Grady SM, Grossman RG, Hall ED, Heetderks W, Hovda DA, Jallo J, Katz RL, et al. (2002) Clinical trials in head injury. J Neurotrauma 19: 503-557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda M, Ikeda T, Suzuki H, Takeshima H, Takahashi T, Kuno M, Numa S (1986) Expression of functional sodium channels from cloned cDNA. Nature 322: 826-828. [DOI] [PubMed] [Google Scholar]
- Povlishock JT (1992) Traumatically induced axonal injury: pathogenesis and pathobiological implications. Brain Pathol 2: 1-12. [PubMed] [Google Scholar]
- Povlishock JT, Becker DP, Cheng CL, Vaughan GW (1983) Axonal change in minor head injury. J Neuropathol Exp Neurol 42: 225-242. [DOI] [PubMed] [Google Scholar]
- Rasband MN, Peles E, Trimmer JS, Levinson SR, Lux SE, Shrager P (1999) Dependence of nodal sodium channel clustering on paranodal axoglial contact in the developing CNS. J Neurosci 19: 7516-7528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg LJ, Teng YD, Wrathall JR (1999) Effects of the sodium channel blocker tetrodotoxin on acute white matter pathology after experimental contusive spinal cord injury. J Neurosci 19: 6122-6133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saatman KE, Bozyczko-Coyne D, Marcy V, Siman R, McIntosh TK (1996) Prolonged calpain-mediated spectrin breakdown occurs regionally following experimental brain injury in the rat. J Neuropathol Exp Neurol 55: 850-860. [DOI] [PubMed] [Google Scholar]
- Saatman KE, Abai B, Grosvenor A, Vorwerk CK, Smith DH, Meaney DF (2003) Traumatic axonal injury results in biphasic calpain activation and retrograde transport impairment in mice. J Cereb Blood Flow Metab 23: 34-42. [DOI] [PubMed] [Google Scholar]
- Schumacher PA, Siman RG, Fehlings MG (2000) Pretreatment with calpain inhibitor CEP-4143 inhibits calpain I activation and cytoskeletal degradation, improves neurological function, and enhances axonal survival after traumatic spinal cord injury. J Neurochem 74: 1646-1655. [DOI] [PubMed] [Google Scholar]
- Smith DH, Meaney DF (2000) Axonal damage in traumatic brain injury. The Neuroscientist 6: 483-495. [Google Scholar]
- Smith DH, Wolf JA, Lusardi TA, Lee VM, Meaney DF (1999) High tolerance and delayed elastic response of cultured axons to dynamic stretch injury. J Neurosci 19: 4263-4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuhmer W, Conti F, Suzuki H, Wang XD, Noda M, Yahagi N, Kubo H, Numa S (1989) Structural parts involved in activation and inactivation of the sodium channel. Nature 339: 597-603. [DOI] [PubMed] [Google Scholar]
- Stys PK, Lopachin RM (1998) Mechanisms of calcium and sodium fluxes in anoxic myelinated central nervous system axons. Neuroscience 82: 21-32. [DOI] [PubMed] [Google Scholar]
- Stys PK, Waxman SG (2004) Ischemic white matter damage. In: Myelin and its diseases (Griffin J, Lassman H, Nave K-A, Trapp B, Lazzarini R, Miller R, eds), pp 985-1007. San Diego: Academic.
- Stys PK, Waxman SG, Ransom BR (1991) Na(+)-Ca2+ exchanger mediates Ca2+ influx during anoxia in mammalian central nervous system white matter. Ann Neurol 30: 375-380. [DOI] [PubMed] [Google Scholar]
- Stys PK, Waxman SG, Ransom BR (1992) Ionic mechanisms of anoxic in jury in mammalian CNS white matter: role of Na+ channels and Na+/Ca2+ exchanger. J Neurosci 12: 430-439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stys PK, Sontheimer H, Ransom BR, Waxman SG (1993) Noninactivating, tetrodotoxin-sensitive Na+ conductance in rat optic nerve axons. Proc Natl Acad Sci USA 90: 6976-6980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi A, Camacho P, Lechleiter JD, Herman B (1999) Measurement of intracellular calcium. Physiol Rev 79: 1089-1125. [DOI] [PubMed] [Google Scholar]
- Teng YD, Wrathall JR (1997) Local blockade of sodium channels by tetrodotoxin ameliorates tissue loss and long-term functional deficits resulting from experimental spinal cord injury. J Neurosci 17: 4359-4366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vabnick I, Novakovic SD, Levinson SR, Schachner M, Shrager P (1996) The clustering of axonal sodium channels during development of the peripheral nervous system. J Neurosci 16: 4914-4922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilev PM, Scheuer T, Catterall WA (1988) Identification of an intracellular peptide segment involved in sodium channel inactivation. Science 241: 1658-1661. [DOI] [PubMed] [Google Scholar]
- Westenbroek RE, Merrick DK, Catterall WA (1989) Differential subcellular localization of the RI and RII Na+ channel subtypes in central neurons. Neuron 3: 695-704. [DOI] [PubMed] [Google Scholar]
- Witgen BM, Abai B, Grosvenor AE, Vorwerk CK, Saatman KE (2001) Calpain inhibition restores retrograde axonal transport after axonal injury. J Neurotrauma 18: 1171. [Google Scholar]
- Wolf JA, Stys PK, Lusardi T, Meaney D, Smith DH (2001) Traumatic axonal injury induces calcium influx modulated by tetrodotoxin-sensitive sodium channels. J Neurosci 21: 1923-1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Raghupathi R, Saatman KE, Smith DH, Stutzmann JM, Wahl F, McIntosh TK (1998) Riluzole attenuates cortical lesion size, but not hippocampal neuronal loss, following traumatic brain injury in the rat. J Neurosci Res 52: 342-349..0 [DOI] [PubMed] [Google Scholar]