

Abstract
Brain-derived neurotrophic factor (BDNF) is a versatile neurotrophic factor that has been implicated in cell survival, cell differentiation, axonal growth, and activity-dependent synaptic plasticity. Changes in BDNF expression have also been reported during the course of several neurological disorders, including Alzheimer's disease (AD). The role of BDNF in AD, however, has remained elusive. To learn more about this neurotrophic factor, we investigated BDNF expression in brain of amyloid precursor protein overexpressing mice (APP23 transgenic mice). In situ hybridization revealed BDNF mRNA signals associated with amyloid plaques. Laser microdissection in combination with quantitative RT-PCR demonstrated a sixfold increase of BDNF mRNA in the immediate plaque vicinity, a threefold increase in a tissue ring surrounding the plaque, and control levels in interplaque areas comparable with those measured in age-matched nontransgenic mice. Double immunofluorescence localized BDNF to microglial cells and astrocytes surrounding the plaque. Cortical BDNF protein levels were quantified by ELISA demonstrating a >10-fold increase compared with age-matched controls. This upregulation of BDNF protein significantly correlated with the β-amyloid load in the transgenic animals. Taken together, our data demonstrate a plaque-associated upregulation of BDNF in APP23 transgenic mice and implicate this neurotrophin in the regulation of inflammatory and axonal growth processes in the plaque vicinity.
Keywords: Alzheimer's disease, axonal sprouting, neurotrophins, laser microdissection, amyloid precursor protein
Introduction
Brain-derived neurotrophic factor (BDNF) is a versatile member of the neurotrophin family that is expressed widely and highly in brain (Barde et al., 1982; Barde, 1989; Ernfors et al., 1990; Hofer et al., 1990; Ghosh et al., 1994; Murer et al., 2001). Although initially considered a neuronal differentiation and survival-promoting molecule, recent research has implicated BDNF in immune processes (Neumann et al., 1998), axonal growth (Alsina et al., 2001; Tucker et al., 2001), activity-dependent synaptic plasticity (Korte et al., 1995; Vicario-Abejon et al., 2002; Zhang and Poo, 2002), dendritic plasticity (McAllister et al., 1995, 1997; Murphy et al., 1998), and learning and memory (Linnarsson et al., 1997; Ma et al., 1998; Mu et al., 1999). After CNS injury, BDNF expression is increased (Lindvall et al., 1992; Hughes et al., 1999). After a lesion, it plays a role in rescuing neurons from damage (Sendtner et al., 1996) and in promoting neuronal network reorganization (Fusco et al., 1993; Mamounas et al., 1995). This spectrum of biological actions, as well as the fact that BDNF levels are reduced in neurodegenerative diseases (Phillips et al., 1991; Murray et al., 1994; Mogi et al., 1999), has raised considerable interest in BDNF as a potential therapeutic agent for neurodegenerative disorders (Thoenen and Sendtner, 2002).
In brains of patients suffering from Alzheimer's disease (AD), BDNF has been studied in considerable detail. Nevertheless, its role in the pathogenesis of the disease and its potential therapeutic value have remained unclear. Overall decreases in BDNF mRNA and protein have been reported for the hippocampus and several cortical areas (Phillips et al., 1991; Murray et al., 1994; Connor et al., 1997; Holsinger et al., 2000). Additionally, some authors described an association of BDNF with amyloid plaques (Ferrer et al., 1999; Murer et al., 1999), whereas others published negative results (Soontornniyomkij et al., 1999). Because the detailed analysis of cellular and molecular events in human postmortem tissue is intricate and often impossible, data on BDNF in AD have not yet been sufficiently conclusive.
The genetic mouse model is a useful tool for learning more about BDNF in AD. Mice overexpressing the human β-amyloid precursor protein (APP23 transgenic mice) carrying the Swedish mutation (Sturchler-Pierrat et al., 1997; Staufenbiel and Sommer, 1998; Sturchler-Pierrat and Staufenbiel, 2000) impressively replicate the amyloid pathology of the human disease. These animals show increased β-amyloid plaque formation during aging, region-specific neuron loss (Calhoun et al., 1998; Bondolfi et al., 2002), cholinergic changes (Boncristiano et al., 2002), progressive age-related impairment of cognitive behavior (Lalonde et al., 2002; Kelly et al., 2003; Van Dam et al., 2003), amyloid-associated activation of glial cells (Stalder et al., 1999; Sturchler-Pierrat and Staufenbiel, 2000; Bornemann et al., 2001), and aberrant axonal sprouting in the immediate plaque vicinity (Phinney et al., 1999). Because of their prominent amyloid pathology, these mice are particularly suited to clarify the relationship between BDNF and amyloid plaques. Therefore, this study analyzes the expression of BDNF protein as well as of BDNF mRNA in the brain of these mice.
Materials and Methods
Animals and tissue preparation
Generation and genotyping of APP23 transgenic mice has been described previously (Sturchler-Pierrat et al., 1997). These mice express human APP751 cDNA with the Swedish double mutation under control of the neuron-specific mouse Thy-1 promoter fragment. APP23 mice were established on a B6D2 background and continuously back-crossed to C57BL/6. For the most part, male transgenic mice (1 and 21 months old, heterozygous; n = 16) and age-matched nontransgenic C57BL/6 mice of the same breed (controls; n = 13) were used. For the BDNF ELISA in brain homogenates and Aβ determinations, we used 5-, 10.5-, and 20-month-old transgenic mice (heterozygous; n = 21) and the corresponding number of nontransgenic littermate controls. For the BDNF ELISA in mouse serum and cerebrospinal fluid, 4- and 20-month-old control and transgenic mice were used (n = 5 for each group). All animals were housed under standard laboratory conditions.
For histochemical staining and subsequent laser microdissection, mice were killed by an overdose injection of pentobarbital (300 mg/kg body weight, i.p.). The brain was then removed, embedded in tissue-freezing medium (Leica, Bensheim, Germany), and immediately flash-frozen in 2-methyl-butane at -40°C for short-term storage at -70°C or instant sectioning. Cryostat sections (thickness, 12 μm) were cut and mounted on polytarthalene (PET)-foil stretched on a metal frame (Leica). Sections were then fixed in 100% ethanol for 20 sec, dried on a heater at 40°C for 10 min, and subjected to histochemical staining.
For in situ hybridization histochemistry, immunohistochemistry, and double-immunofluorescence studies, mice were anesthetized deeply with pentobarbital (300 mg/kg body weight) and transcardially perfused with 0.9% saline followed by 4% paraformaldehyde in 0.1 m phosphate buffer (PB), pH 7.4. For in situ hybridization studies, all solutions were prepared in RNase-free DEPC water. Brains were removed and postfixed in 4% paraformaldehyde in 0.1 m PB for 5 hr followed by cryoprotection in 20% sucrose in 0.1 m PB at 4°C overnight. Brain tissue was then flash-frozen in 2-methyl-butane at -40°C for short-term storage or instant sectioning. Cryostat sections (thickness, 40 μm) were collected in 2× SSC (1× SSC = 0.15 m NaCl, 0.015 m sodium citrate, pH 7.0) for further processing.
For immunostaining and double-immunofluorescence studies, brains were fixed overnight in 4% paraformaldehyde in 0.1 m PB, rinsed in PB, and embedded in 5% agar. Coronal sections (50 μm) were cut on a Vibratome (VT 1000S; Leica).
Histochemistry
A 0.2% solution of Congo Red (Merck, Darmstadt, Germany) was prepared in 0.9% NaCl in 80% ethanol in RNase-free DEPC water. The solution was then applied directly onto the mounted section, incubated for 12 min at room temperature, and rinsed briefly in DEPC water. After another brief rinse in 80% ethanol in DEPC water, the sections were dried on a heater at 40°C for 10 min and immediately subjected to laser microdissection.
Immunohistochemistry
The monoclonal Aβ antibody 4G8 (Senetek, Maryland Heights, MO) recognizing Aβ17-24 fragments is known to strongly and specifically stain compact and diffuse β-amyloid deposits (Thal et al., 2002). To avoid reactivity with endogenous mouse immunoglobulin, the Dako ARK Animal Research Kit (Dako, Hamburg, Germany) was used. Briefly, free-floating sections were quenched in a 0.3% hydrogen peroxide-methanol solution for 30 min, incubated in formic acid for 10 min, and subsequently blocked in a 5% bovine serum albumin solution. Before application of the primary Aβ antibody to the specimen, the antibody was biotinylated according to the manufacturer's recommendations. The biotinylated antibody (1:2500) was then applied to the section for 18 hr at room temperature. After several rinses in PB, sections were incubated with streptavidin-peroxidase for 30 min. Immunoreactivity was visualized using a 0.8% 3,3-diaminobenzidine solution (Sigma, St. Louis, MO) in conjunction with nickel-cobalt enhancement (1% NH4NiSo4 and 1% CoCl2). Sections were photographed with a digital camera mounted on an Olympus BX61 microscope (Olympus, Hamburg, Germany).
Immunofluorescence studies and confocal laser scanning microscopy
Because the polyclonal anti-BDNF antibody used (Santa Cruz Biotechnology, Santa Cruz, CA) was raised in the same species (rabbit) as the anti-glial fibrillary acidic protein (GFAP) antibody (Dako) for labeling astrocytes and the ionized calcium-binding adaptor molecule 1 (Iba1) antibody (generously provided by Dr. Yoshinori Imai, National Institute for Neuroscience, Tokyo, Japan) (Imai et al., 1996; Ito et al., 1998) as a marker for microglial cells, the tyramide signal amplification (TSA) method was used (TSA-Kit, MoBiTec, Göttingen, Germany). Tyramide deposition catalyzed by horseradish peroxidase (HRP) is generally used to strongly amplify signals in various applications. A modification of the TSA protocol has been developed for the simultaneous immunofluorescence determination of two antigens with antisera raised in the same species (Shindler and Roth, 1996; Botchkarev et al., 1999). Briefly, after quenching of endogenous peroxidases, unspecific binding sites were blocked by incubating sections in 10% goat serum for 30 min. Next, antisera against GFAP (1:128,000) or Iba1 (1:4000) were applied overnight. At these dilutions, antigen-antibody complexes were undetectable with a fluorochrome-labeled secondary goat anti-rabbit antibody. Antigen-antibody complexes became visible when using HRP-labeled goat anti-rabbit antiserum and the TSA Kit. Sections were incubated with the HRP-labeled secondary antibody overnight followed by incubation with an Alexa488-labeled tyramide derivative. Next, sections were washed, again blocked with 10% goat serum for 30 min, and then incubated with the BDNF antibody (1:100) overnight. After incubation with an Alexa568-labeled goat anti-rabbit antibody (MoBiTec), sections were scanned with a confocal laser scanning system (LSM 510, Zeiss, Jena, Germany). To ensure the specificity of the double-immunostaining procedure, control experiments were performed in which the two primary antibodies were omitted. No staining occurred under these conditions. In addition, double immunostaining was considered specific only if the two chromogens could be visualized independently in plaque-free areas of the brain (supplemental Fig. 1, available at www.jneurosci.org). Immunofluorescence studies for BDNF only were performed on free-floating sections without using the tyramide amplification method. Briefly, sections were blocked in 10% goat serum for 30 min and incubated with the BDNF antibody (Santa Cruz Biotechnology; 1:100) overnight. Antigen-antibody complexes were visualized by incubation with an Alexa568-labeled goat anti-rabbit antibody (1:1000) for 2 hr and embedded in fluorescent mounting medium (Dako).
In situ hybridization studies
Preparation of cRNA probes. Digoxigenin-labeled cRNA probes were generated by in vitro transcription from appropriate plasmids. A 0.52 kb rat BDNF cDNA fragment (Hofer et al., 1990) inserted into the ApaI site of pBluescript SK- (Stratagene, La Jolla, CA) was linearized with either HindIII to serve as template for T7 RNA polymerase (sense orientation) or with KpnI for T3 RNA polymerase (antisense orientation). The restricted DNA was purified by phenol extraction and ethanol precipitation.
In vitro transcription was performed with 1 μg of linearized plasmid template (50 μl reaction) in the presence of ATP, GTP, CTP, and digoxigenin (DIG)-11-UTP (Roche, Mannheim, Germany), RNasin, transcription buffer, and T3 or T7 RNA polymerase (Roche) for 2 hr at 37°C according to the manufacturer's recommendations. DIG-labeled cRNAs were purified by ethanol precipitation with LiCl and resuspended in 40 μl of DEPC-treated H2O. The yield of both BDNF cRNA probe preparations was determined by dot blot analysis and was usually ∼20 μg.
In situ hybridization histochemistry. Cryostat sections of aged APP23 transgenic (n = 5) and age-matched control mouse brains (n = 5) were pretreated in hybridization buffer (50% formamide, 4× SSC, 50 mm NaH2PO4, 250 μg/ml heat-denatured salmon sperm DNA, 100 μg/ml tRNA, 5% dextransulfate, and 1% Denhardt's solution) diluted with 2× SSC (1:1) for 15 min and prehybridized in hybridization buffer for 60 min at 55°C. Hybridization was performed in the same buffer with the addition of 250 ng/ml of DIG-labeled BDNF antisense or sense cRNA probes at 55°C overnight. After hybridization, the brain sections were washed in 2× SSC (two times for 15 min each) at room temperature, 2× SSC and 50% formamide, 0.1× SSC and 50% formamide for 15 min each, and in 0.1× SSC (two times for 15 min each) at 65°C. For the immunological detection of DIG-labeled hybrids, the sections were rinsed in buffer 1 (100 mm Tris/HCl, pH 7.5) two times for 10 min each at room temperature, blocked in buffer 2 (1% blocking reagent in buffer 1) for 60 min, and incubated overnight with the anti-DIG antibody from sheep conjugated with alkaline phosphatase (Roche) diluted 1:1500 in buffer 2. For the color reaction, tissue sections were equilibrated in buffer 1 (two times for 10 min each) and in buffer 3 (100 mm Tris/HCl, pH 9.5, 100 mm NaCl, 50 mm MgCl2) for 10 min before the addition of nitroblue tetrazolium salt (0.34 mg/ml) and 5-bromo-4-chloro-3-indolylphosphate (0.17 mg/ml) diluted in buffer 3. Development of the color reaction was performed in the dark and stopped by transfer into buffer 4 (10 mm Tris/HCl, pH 8.0; 1 mm EDTA) when the desired intensity of the precipitate was reached. Tissue sections were mounted on glass slides, air-dried, and embedded in Kaiser's glycerol gelatin (Merck).
Laser microdissection, RNA isolation, and reverse transcription
Laser microdissection (LMD) was performed as described (Burbach et al., 2003), although study-specific adaptations of the published protocol were required. Briefly, PET-foil metal frames were mounted on a Leica AS LMD system with the section facing downward. After intensity, aperture, and cutting velocity were adjusted, the pulsed UV laser beam was carefully directed along the borders of Congo Red-stained cerebral cortex amyloid-β plaques in 21-month-old transgenic mice (n = 3). In addition, 30-μm-wide tissue rings around each individual plaque as well as plaque-free tissue commensurate to the plaque area were defined for separate collection. A total of ∼200 plaques were picked in serial sections. Proportional tissue amounts were isolated in the aforementioned regions as well as in the plaque-free cortex of transgenic (1 month old; n = 3) and nontransgenic mice (1 and 21 months old; n = 3, respectively). The cut area was then transferred by gravity alone into a microcentrifuge tube cap placed directly underneath the section. The tube cap was filled with a guanidine isothiocyanate-containing buffer (Buffer RLT, RNeasy Mini Kit, Qiagen, Hilden, Germany) to ensure isolation of intact RNA. Tissue collection was verified by inspecting the tube cap. Microcentrifuge tubes were immediately transferred on ice after tissue collection followed by three freeze-thaw cycles in a dry ice-ethanol bath. Total RNA was isolated using the RNeasy Mini Kit (Qiagen) according to the manufacturer's recommendations, including on-column DNase digestion and a 2 × 30 μl elution step. Integrity of the isolated RNA was determined using the Agilent RNA 6000 Pico LabChip (Agilent Technologies, Waldbronn, Germany). Only RNA with clearly distinguishable ribosomal peaks and without a shift in RNA size distribution to small size fragments was used. RNA was then reverse transcribed using TaqMan Reverse Transcription Reagents (Applied Biosystems, Darmstadt, Germany).
Quantitative PCR
cDNAs were subjected to PCR using the Abi Prism 7000 Sequence Detection System (Applied Biosystems) and TaqMan Universal PCR Master Mix (Applied Biosystems). The following BDNF primers within the 3′ exon of the mouse BDNF gene encoding the mature protein were selected using Primer Express software (Applied Biosystems): BDNF sense primer 5′-GGGTCACAGCGGCAGATAAA-3′; BDNF antisense primer 5′-GCCTTTGGATACCGGGACTT-3′; BDNF probe 5′-TCTGGCGGGACGGTCACAGTCCTA-3′. For normalization of cycle threshold values to an endogenous control, the following eukaryotic 18S ribosomal RNA primers were used: 18S rRNA sense primer 5′-CGGCTACCACATCCAAGGAA-3′; 18S rRNA antisense primer 5′-GCTGGAATTACCGCGGCT-3′; 18S rRNA probe 5′-TGCTGGCACCAGACTTGCCCTC-3′. Probes were labeled with 6-carboxyfluorescein at the 3′ end and with 6-carboxy-tetramethyl-rhodamine at the 5′ end. For the amplification of both mouse BDNF and 18S cDNA, a standard amplification program was used (1 cycle of 50° for 2 min, 1 cycle of 95°C for 10 min, 45 cycles of 95°C for 15 sec and 60°C for 1 min). Direct detection of the PCR product was enabled by the 5′→ 3′ exonuclease activity of Taq polymerase, thereby terminating the close proximity of the reporter dye to the quencher dye by reporter fluorochrome cleavage and leading to an increase of reporter dye fluorescence equivalent to the increase of amplified PCR product. Using standard curves of serial cDNA dilutions, a relative quantitation of target cDNA expressed in x-fold differences was performed. All quantitations were normalized to an endogenous control (18S rRNA) to account for variability in the initial concentration and quality of total RNA and in the conversion efficiency of the reverse transcription reaction (Burbach et al., 2003). Statistical analysis was performed using SPSS for Windows (SPSS, München, Germany).
ELISA studies
Frontal cortex as well as cerebellum as control tissue were dissected from one brain hemisphere of APP23 mice (5, 10.5, and 20 months old). The tissue pieces were weighed, immediately frozen on dry ice, and stored at -80°C until use. The samples were homogenized by ultrasonication in 10-20 vol of lysing buffer containing 0.1 m Tris-HCl, pH 7.0, 0.4 m NaCl, 0.1% NaN3, and various protease inhibitors (Hellweg et al., 1989). In addition, mouse blood and cerebrospinal fluid were collected. The blood was left at ambient temperature for 30 min and centrifuged (500 × g for 15 min) to obtain serum. Samples were stored at -80°C until analysis. Endogenous concentrations of BDNF were measured in the rethawed homogenates or rethawed serum and cerebrospinal fluid samples (diluted with assay buffer 2:7 and 1:200, respectively) using commercial ELISA kits, in principle according to the manufacturer's instructions (Promega, Mannheim, Germany) but adapted as described previously in detail (Hellweg et al., 2003). Recombinant human BDNF (Sigma) was used as a standard. The detection limit of the assay was 1 pg/ml. Determination of recovery and specific and unspecific neurotrophin binding (the latter against mouse IgG1 obtained from MOPC 21) involved quadruplicate fluorescence determinations for each tissue sample. The BDNF concentration was expressed as picograms per milligram (wet weight; brain tissue) or picograms per milliliter (cerebrospinal fluid and serum). Student's t test was used to determine significance (p ≤ 0.05).
Determination of Aβ concentrations
Aβ1-40 and Aβ1-42 concentrations were measured by quantitative Western blotting in the remainder of the homogenates that had been prepared for ELISA analysis (10.5- and 20-month-old mice only). Homogenate proteins were separated on 10% Tris-Bicine gels containing 8 m urea (Staufenbiel and Paganetti, 1999). For standard curve generation, known amounts of synthetic Aβ1-40 and Aβ1-42 peptides in control mouse brain homogenate were loaded on each gel. After blotting, Aβ peptides were detected using 6E10 antibody (Signet Laboratories, Dedham, MA), goat anti-mouse IgG coupled to horseradish peroxidase (Chemicon, Temecula, CA), and the chemiluminescent substrate ECL (Amersham Biosciences, Little Chalfont, UK). Signals on film were quantified using the MCID software (Imaging Research, St. Catherines, Ontario, Canada). The correlation of Aβ and BDNF concentrations was tested after logarithmic transformation of the data using the Pearson method and Systat software (Systat Software, London, UK).
Digital illustrations
Figures were prepared digitally using a commercially available graphics software (Photoshop 6.0, Adobe). Fluorescent images were acquired using a Zeiss LSM 510 laser scanning microscope (Zeiss). Single fluorescent images of the same section were digitally superimposed. Images were adjusted for contrast, brightness, and sharpness.
Results
Amyloid plaque pathology in APP23 transgenic mouse cortex
The distribution of β-amyloid plaques in aged APP23 transgenic mice was similar to the one described previously (Sturchler-Pierrat et al., 1997). β-amyloid immunostaining revealed several intensely stained dense-core plaques as well as numerous blotchy stains in the brain parenchyma representing diffuse β-amyloid (Fig. 1a). Furthermore, numerous blood vessels were also β-amyloid positive (Calhoun et al., 1999; Burgermeister et al., 2000). Nontransgenic control mice from the same breed did not show β-amyloid staining (Fig. 1b).
Figure 1.
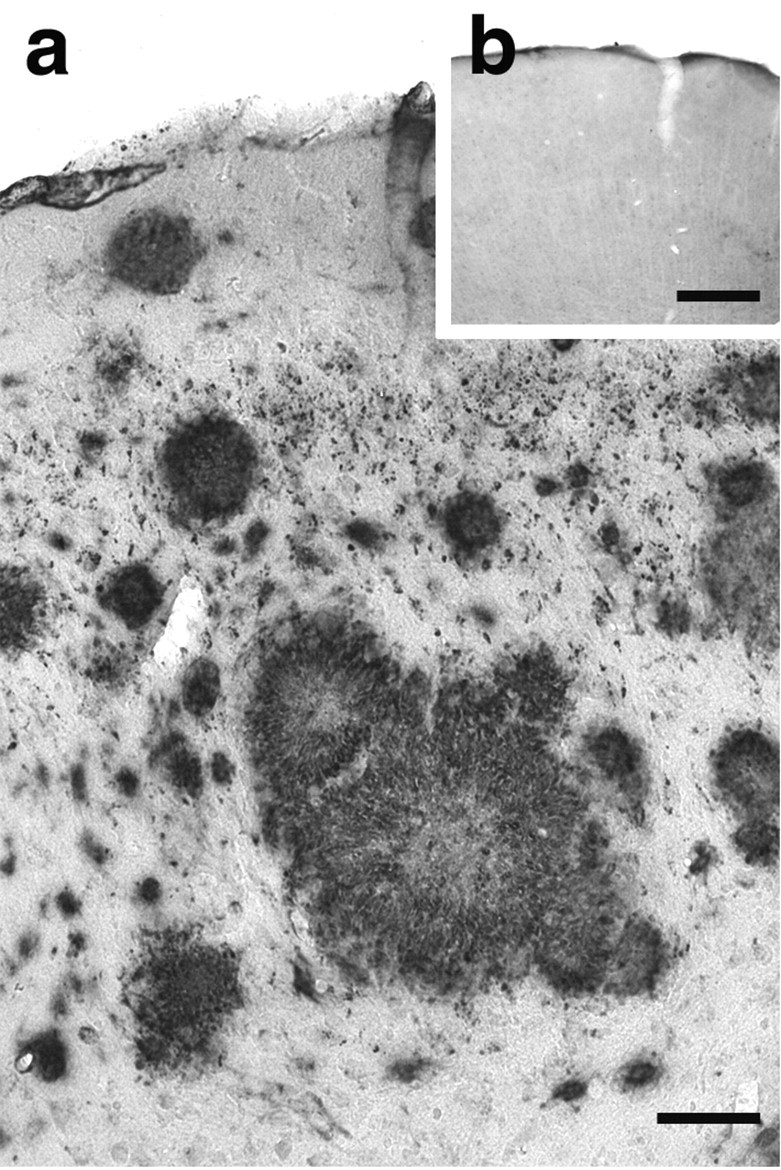
Amyloid plaque pathology in cortex of APP23 transgenic mice. Cerebral cortex from 21-month-old APP23 transgenic (a) and age-matched C57BL/6 (b) mice immunostained for β-amyloid (4G8 antibody). Although C57BL/6 mouse tissue showed no specific staining (b), cortex from APP23 transgenic mice revealed numerous amyloid plaques (a). Scale bars: a, 50 μm; b, 100 μm.
Plaque-associated BDNF mRNA expression: evidence for a BDNF mRNA gradient
In situ hybridization for BDNF mRNA revealed a normal BDNF mRNA expression pattern in brain of 21-month-old control mice (Fig. 2a,c). BDNF mRNA was widely and strongly expressed throughout the entire brain, as described previously (Hofer et al., 1990). BDNF mRNA signals were found primarily in neuronal cell layers, which is compatible with the predominantly neuronal localization of this neurotrophin (Hofer et al., 1990). Staining was absent in the sense control.
Figure 2.
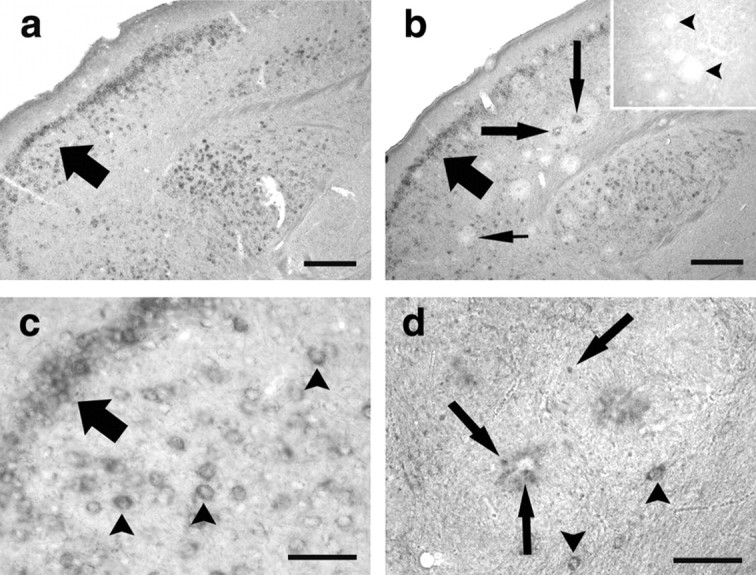
BDNF mRNA is associated with amyloid plaques. In situ hybridization histochemistry for BDNF was performed on cortical sections from 21-month-old C57BL/6 (a, c) and age-matched APP23 transgenic (b, d) mice. BDNF in situ hybridization in C57BL/6 mice demonstrated distinct BDNF mRNA signals in cortical neurons (a, c, arrow, arrowheads). In APP23 transgenic mice, a comparable BDNF mRNA expression was observed (b, arrow; d, arrowheads). In addition, however, BDNF mRNA staining was also found within amyloid plaques, which differed in their staining intensity. Although some plaques showed a strong BNDF mRNA signal (b, long arrows), others were BDNF mRNA negative (b, pointed arrow). Inset shows sense control with unlabeled plaques (arrowheads). Higher magnification of individual plaques (d) showed small mRNA-positive cells (arrows) within the plaque as well as in the plaque periphery. Note that numerous BDNF mRNA-positive neurons were also observed (arrowheads), demonstrating the specificity of the in situ hybridization. Scale bars: a, b, 100 μm; c, d, 25 μm.
In situ hybridization for BDNF mRNA in aged APP23 transgenic animals revealed an expression pattern similar to the one observed in controls (Fig. 2b). In addition, strong plaque-associated BDNF mRNA signals were observed. Small cell-shaped hybridization signals could be identified in the plaque and in the plaque periphery (Fig. 2d). Because BDNF mRNA-expressing cells were also observed within and around white matter plaques, where BDNF mRNA-expressing neurons are absent, these cells probably represented glial cells. Interestingly, a subpopulation of plaques was observed that was consistently BDNF mRNA negative. These plaques were morphologically identical to the BDNF mRNA-positive subgroup, and it remains unclear why these plaques do not show a hybridization signal.
Although plaque-associated hybridization signals had a somewhat fuzzy appearance, in situ hybridization quality was excellent, because the cellular resolution of BDNF mRNA was high in plaque-free brain areas and in cell layers close to the plaques (Fig. 2d). In addition, we can exclude staining caused by unspecific attachment of riboprobes to amyloid because the hybridization signal was not observed around all plaques, and plaque-associated staining was absent in the sense control (Fig. 2b, inset). Nevertheless, we decided to verify the presence of BDNF mRNA in the plaque vicinity with a different technique and used quantitative PCR (qPCR) for BDNF mRNA detection and quantification. Therefore, congophilic plaques in aged APP23 transgenic mice were collected by noncontact laser microdissection (Fig. 3a,b). Furthermore, a 30-μm-wide tissue ring surrounding the individual plaques (Fig. 3c) as well as plaque-free tissue (Fig. 3d) was microdissected. Additionally, corresponding regions in nontransgenic mice of the same breed that did not bear any β-amyloid plaques were collected and subjected to qPCR analysis. Using this methodology, we could confirm a plaque-associated BDNF mRNA expression that was sixfold higher in congophilic β-amyloid plaques, threefold higher in the tissue ring surrounding the plaque, and close to control levels in interplaque areas in aged APP23 transgenic mice compared with corresponding areas in nontransgenic mice (Fig. 3e). These findings demonstrate an increasing BDNF mRNA gradient from plaque-free tissue to amyloid plaques. This gradient may be even more pronounced, however, because BDNF mRNA-positive as well as BDNF mRNA-negative plaques could not be distinguished by Congo Red staining and therefore were collected indiscriminately.
Figure 3.
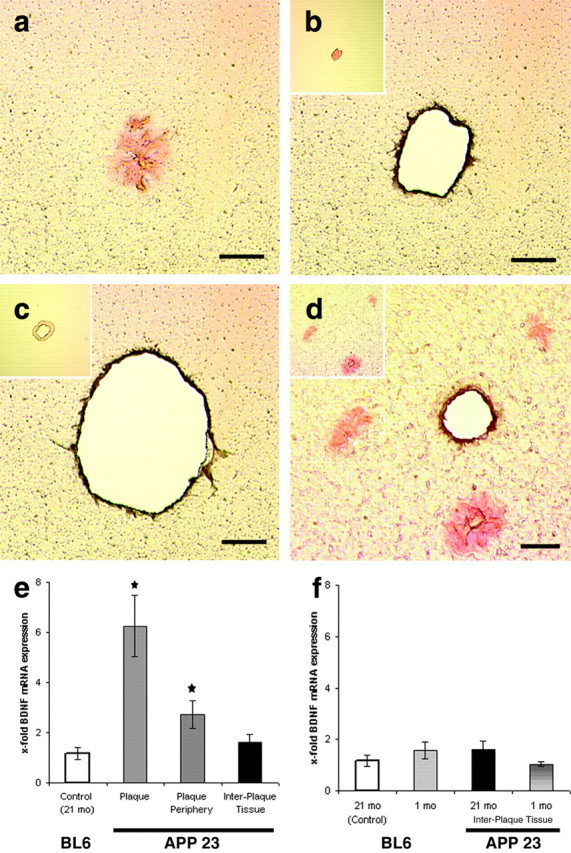
Increasing BDNF mRNA expression toward amyloid plaques. Cortex from 21-month-old APP23 transgenic and age-matched C57BL/6 mice subjected to Congo Red staining. Congo Red-stained plaques were identified in APP23 transgenic mice (a). These plaques (b), a 30-μm-wide tissue ring around these plaques (c), and interplaque tissues (d) were collected using laser microdissection. Corresponding plaque-free regions were isolated in controls. Furthermore, cortical tissue from 1-month-old APP23 transgenic mice, which had not yet developed plaques, and age-matched C57BL/6 mice was microdissected. Total RNA was isolated, reverse transcribed, and subjected to quantitative PCR analysis. Insets in b and c demonstrate the tissue collected after completion of the laser microdissection procedure. The inset in d shows the section before microdissection. Histograms in e and f illustrate quantitative changes in BDNF mRNA expression. Statistical analysis was performed using SPPS for Windows (p ≤ 0.05). Scale bars: a-d, 50 μm.
To investigate whether BDNF mRNA expression levels are affected by the expression of the transgene in the APP23 transgenic animals, BDNF mRNA expression levels were also quantified in 1-month-old APP23 transgenic animals as well as in age-matched C57BL/6 mice. No significant differences were observed in BDNF mRNA expression levels in these mice (Fig. 3f). In addition, BDNF mRNA expression levels in the cortex of 1-month-old APP23 transgenic animals and in the interplaque tissue of 21-month-old APP23 transgenic animals were comparable. Thus, it can be excluded that an unknown effect of the transgene or aging accounts for the increases in BDNF mRNA levels observed in the present study.
Correlation between BDNF protein concentration and amyloid deposition in the frontal cortex of APP23 mice
The small amount of BDNF protein precludes its quantification in and around isolated amyloid plaques as done for the mRNA. We therefore determined BDNF concentrations during aging of APP23 mice just before amyloid deposition (5 months) and at an intermediate (10.5 months) and a high (20 months) amyloid load. The frontal cortex and cerebellum, which do not develop amyloid deposits, were prepared as control tissue from transgenic and littermate mice, and the BDNF content was determined by ELISA. BDNF concentrations in frontal cortex remained relatively unchanged in control animals at all ages (Fig. 4). Comparable values were found for APP23 mice aged 5 and 10.5 months, although BDNF appeared slightly elevated in the young group. In contrast, at 20 months all APP23 mice contained higher BDNF concentrations than the nontransgenic animals, resulting in a 17-fold increase over control. No changes during aging or differences between transgenic and control mice were found for BDNF in the cerebellum (data not shown).
Figure 4.

Increased BDNF protein concentrations in the frontal cortex of aged APP23 mice. BDNF was measured in APP23 and nontransgenic littermate (control) mice at the different ages indicated using a sensitive ELISA. The top panel shows the values for individual animals (n = 7 per group) as well as the median (horizontal bar). Significant changes (p ≤ 0.005) are indicated by brackets and asterisks. Note the increase in the old group as well as the individual variation. The bottom panel shows the correlation of BDNF and Aβ1-40 and Aβ1-42 concentrations, respectively. Aβ was determined in amyloid plaque-containing mice (10.5 and 20 months) as a measure of the amyloid content. A log scale was chosen to account for the large differences in the values.
The large variation in BDNF concentrations between individual animals may be related to the known variation in amyloid deposition in APP transgenic mice. To test this hypothesis, Aβ1-40 and Aβ1-42 concentrations were determined in forebrain homogenates of the plaque-containing groups (10.5- and 20-month-old mice) using Western blotting. Comparison with the BDNF content showed a highly significant correlation with both Aβ1-40 (Pearson correlation after log-transformation: 0.869; p = 0.00005) and Aβ1-42 (correlation: 0.882; p = 0.00003) (Fig. 4). These data further indicate that amyloid plaque deposition leads to an increased level of BDNF in neocortex.
In addition, BDNF concentrations were measured in the serum and cerebrospinal fluid of young and aged control mice as well as APP23 transgenic mice. Although the very sensitive ELISA assay made it possible to measure very low serum levels of BDNF (30-40 pg/ml), BDNF concentrations in the cerebrospinal fluid were below the detection limit. No differences in the BDNF serum levels were observed between the groups [control mice: 32 ± 11 (4 months), 35 ± 4 (20 months); APP23 tg mice: 39 ± 12 (4 months), 29 ± 11 pg/ml (20 months)]. Thus, elevated brain BDNF levels in APP23 transgenic mice do not lead to a detectable increase in serum or cerebrospinal fluid BDNF levels, which supports the concept that BDNF changes are local, i.e., plaque associated, rather than global.
Plaque-associated astroglial and microglial cells express BDNF protein
Immunostaining of BDNF, GFAP, and Iba1 was performed to localize BDNF protein in the plaque vicinity (Fig. 5). Confocal laser scanning microscopy revealed an abundance of BDNF-positive structures surrounding amyloid plaques in APP23 transgenic mouse cortex. Often, these BDNF-immunopositive structures resembled glial cells and their processes (Fig. 5a,b). Double immunofluorescence with the microglial marker Iba1 revealed that some of these structures were BDNF-positive microglial cells (Fig. 5a,c,e). Almost all microglial cells close to the plaque were BDNF positive (Fig. 5), whereas microglial cells found in plaque-free areas of the brain (supplemental Fig. 1a-f, available at www.jneurosci.org) did not show immunolabeling for BDNF. Specificity of the double-immunostaining procedure was verified, as described in Materials and Methods. Because Iba1 did not label all plaque-associated BDNF-positive structures, double immunofluorescence was also performed for the astrocytic marker molecule GFAP. This approach revealed numerous BDNF-positive astrocytes in addition to BDNF-positive microglial cells. Astroglial cells found in plaque-free areas of the brain were BDNF negative (supplemental Fig. 1g-l, available at www.jneurosci.org). In addition, thin BDNF-positive processes were observed extending from both microglial and astroglial cells toward the plaque core. Although our immunocytochemical data and immunocytochemical control experiments show specific immunolabeling of glial cells in the plaque region, cross-reactivity with an unknown antigen cannot be excluded completely. On the whole, however, our data indicate that plaque-associated glial cells express BDNF.
Figure 5.
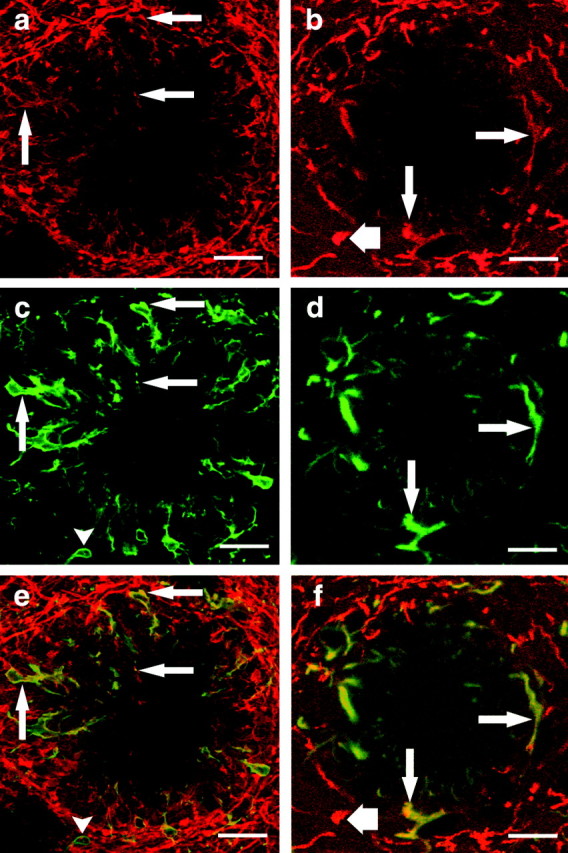
Plaque-associated glial cells express BDNF protein. Double-immunofluorescence studies for BDNF-Iba1 (a, c, e) and BDNF-GFAP (b, d, f) were performed on APP23 transgenic mouse cerebral cortex (21 months old). High-magnification confocal laser scanning microscopy revealed amyloid plaques surrounded by BDNF-positive fibers (a, b), Iba1-positive microglial cells (c), and GFAP-positive astroglial cells (d). Digital merging of these fluorescent images (e, f) demonstrated BDNF-Iba1 double-labeled microglial cells (e, arrows), as well as BDNF-GFAP double-labeled astrocytes (f, arrows). Note a single BDNF-negative microglial cell in the plaque periphery (c, e, arrowhead) and BDNF-negative peripheral astroglial processes (d, f), which demonstrate the specificity of the double-immunolabeling procedure. In addition, BDNF-positive dystrophic boutons were observed at the border of several plaques (b, f, short arrow). Scale bars: a-f, 25 μm.
BDNF-positive putative boutons surrounding amyloid plaques
BDNF immunostaining around amyloid plaques also revealed BDNF-positive balloon-like structures in the plaque periphery (Fig. 6a-c). These structures were unusually large (5-15 μm), spherical or ovoid, and did not extend any processes. Their location in the periphery of the plaque and their morphology closely resembled that of dystrophic axonal boutons that we had observed previously in APP23 transgenic animals using anterograde tracing (Phinney et al., 1999). The number and size of immunopositive boutons varied considerably between individual plaques (Figs. 5b,f, 6a,c,d). High magnification confocal imaging was used to reconstruct a large BDNF-positive bouton in three dimensions (Fig. 6b). In this case, BDNF immunostaining was unevenly distributed, and the center of the bouton contained several unstained patches. In contrast, BDNF-positive dendrites were not affected by plaques in their vicinity. This could be readily seen in the CA1 region of the hippocampus, where BDNF-positive pyramidal cells extend apical dendrites into stratum radiatum and stratum lacunosum moleculare. These dendrites are oriented in parallel and can be followed from the neuronal soma to the plaque vicinity. Although BDNF-immunopositive dendrites of CA1 pyramidal cells were clearly displaced by the plaque, they did not form any pathological BDNF-positive boutons. These observations are in line with previous reports that demonstrated only limited dendritic pathology in APP23 mice using MAP2 immunostaining (Phinney et al., 1999). Taken together, these data indicate that BDNF-positive dystrophic axonal boutons surround amyloid plaques in APP23 mice.
Figure 6.
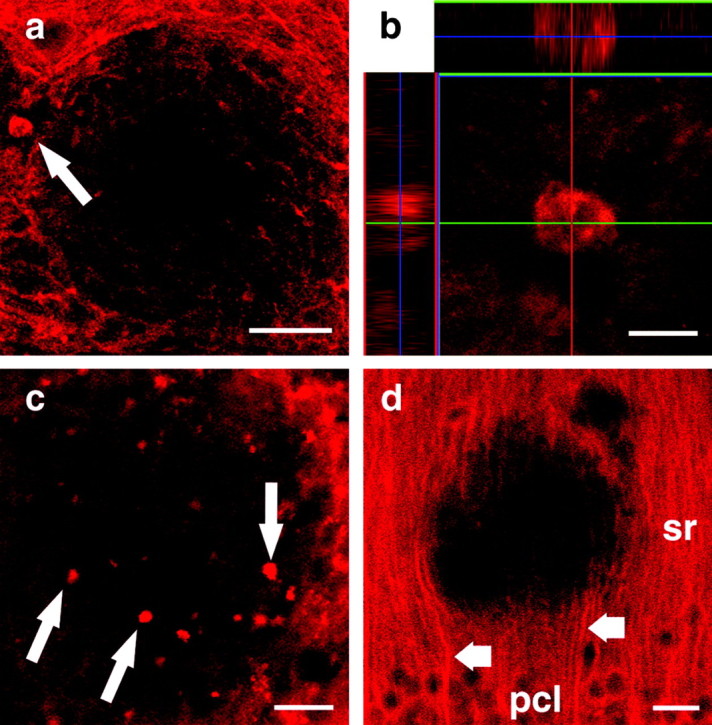
BDNF-positive dystrophic boutons surrounding amyloid plaques. Confocal laser scanning microscopy revealed dystrophic axonal boutons (a-c) surrounding amyloid plaques in cortical sections of 21-month-old APP23 transgenic mice. A large BDNF-positive dystrophic bouton was observed in the plaque periphery (a, arrow). The bouton was reconstructed three-dimensionally using a confocal stack (b). Note the patchy quality of BDNF immunostaining. Other plaques were surrounded by similar but smaller BDNF-positive boutons (c). No dendritic pathology was observed in the stratum radiatum (sr) of the hippocampal CA1 subfield (d). In this region, BDNF-positive apical dendrites of pyramidal neurons (d, short arrows) located in the pyramidal cell layer (pcl) could be readily followed from the cell soma to the plaque vicinity. Scale bars: a, 50 μm; b, 10 μm; c, d, 25 μm.
Discussion
In this study using a genetic model of AD we report plaque-associated BDNF mRNA and protein expression. Our data indicate increasing BDNF mRNA expression from plaque-free tissue to amyloid plaques. A cortical increase in BDNF protein was detected that correlated with the β-amyloid load. BDNF immunostaining indicates that microglial and astroglial cells, as well as putative dystrophic boutons surrounding the plaques, contain BDNF.
Plaque-associated BDNF mRNA expression: evidence for a BDNF mRNA gradient
The most striking changes were associated with amyloid plaques, in which a strong BDNF mRNA in situ hybridization signal was found (Fig. 2). Laser microdissection and qPCR confirmed these qualitative findings and revealed a more than sixfold BDNF mRNA upregulation in isolated amyloid plaques, a threefold increase in a tissue ring surrounding the plaques, and control levels in the interplaque area comparable with BDNF mRNA levels obtained from control tissue (Fig. 3e). In young APP23 mice as well as young control mice, BDNF mRNA levels were similar to the levels in interplaque tissue of aged APP23 transgenic and plaque-free cortex of aged control animals (Fig. 3f). Taken together, these data demonstrate that an increasing BDNF mRNA gradient exists from plaque-free tissue toward an amyloid plaque. Because all age groups and all genotypes showed comparable levels of BDNF mRNA expression in plaque-free tissue, the steady increase of BDNF mRNA toward an amyloid plaque cannot be attributed to an unknown effect of the transgene or to aging. Thus, the increases in BDNF mRNA observed in APP23 transgenic animals have to be considered plaque dependent.
The existence of a BDNF mRNA gradient implies a source of BDNF mRNA in the tissue areas that were microdissected. Evidence for the cellular source was obtained by in situ hybridization, which revealed small cells in the plaque area as well as in the tissue rim surrounding the plaque (Fig. 2d). The shape and size of these cells, which were smaller than BDNF mRNA-expressing neurons and the fact that similar cells were observed around white matter plaques, point to glial cells as the main source of plaque-associated BDNF mRNA. Because glial activation is strongest in the immediate plaque vicinity (Sturchler-Pierrat et al., 1997), the BDNF mRNA gradient seen in our laser microdissection experiment could be caused by different numbers as well as expression levels of BDNF mRNA containing glial cells in the different tissue areas that were analyzed.
Plaque-associated BDNF protein expression
Using BDNF immunostaining, the most evident alterations were observed in the vicinity of amyloid plaques, where BDNF-immunoreactive glial cells as well as BDNF-positive putative dystrophic boutons could be identified (Figs. 5, 6). Our data demonstrate that microglial as well as astroglial cells express BDNF in the immediate plaque vicinity. Although almost all cells close to the plaque were double-immunopositive for glial markers and BDNF, the more peripheral glial cells were usually BDNF negative. In the interplaque area, BDNF immunostaining of glial cells was also essentially absent (supplemental Fig. 1, available at www.jneurosci.org). These observations correspond nicely to the in situ hybridization data as well as the qPCR results.
In addition, large balloon-shaped BDNF-positive structures were found around some but not all plaques. These structures did not arise from BDNF-positive dendrites, because hippocampal pyramidal cells did not show a plaque-associated dendritic pathology (Fig. 6d). Because the BDNF-positive globular structures correspond in size, shape, and location to dystrophic axonal boutons surrounding plaques in APP23 mice (Phinney et al., 1999), our data indicate that some of the dystrophic boutons surrounding plaques contain BDNF.
ELISA measurements revealed a >10-fold increase in BDNF protein in the frontal cortex of old APP23 mice (Fig. 4). This corresponds to the mRNA data and shows that plaque-associated BDNF mRNA is translated into biologically relevant amounts of protein. Because mRNA levels are normal in the interplaque tissue and the increase in BDNF protein correlated with the amyloid plaque load, the increase of BDNF protein is dependent on amyloid deposition.
Changes of BDNF expression in AD
Several investigators have studied BDNF in brains of AD patients. With only one notable exception (Durany et al., 2000), decreased BDNF levels were reported (Phillips et al., 1991; Murray et al., 1994; Connor et al., 1997; Ferrer et al., 1999; Hock et al., 2000; Holsinger et al., 2000; Michalski and Fahnestock, 2003). In all studies reporting decreased BDNF levels, however, late stages of AD were studied in which considerable neuron loss has already occurred (Braak and Braak, 1991; Hof et al., 1999). Because this cell loss affects BDNF mRNA-expressing cells and non-BDNF mRNA-expressing cells indiscriminately (Murer et al., 1999), decreased BDNF levels are a logical consequence. Most importantly, a decrease in neuronal BDNF expression may completely mask local, i.e., plaque-associated, increases in glial BDNF. Whether BDNF is decreased in early AD patients, in which neuronal cell loss is not yet pronounced, needs to be established. In contrast, APP23 transgenic mice were analyzed in this study. These mice show only mild neurodegeneration (Calhoun et al., 1998) and have normal neuronal BDNF expression levels in interplaque areas (Fig. 3e). Thus, plaque-associated increases in glial BDNF expression can be detected because they are not masked by a loss of neuronal BDNF.
As far as plaque-associated upregulation of BDNF is concerned, data from human AD patients is controversial. Although some have observed BDNF immunoreactivity closely associated with plaques (Murer et al., 1999) or with plaques and dystrophic boutons (Ferrer et al., 1999), others have reported negative results (Soontornniyomkij et al., 1999). In the present study, BDNF mRNA as well as BDNF protein were unequivocally associated with amyloid plaques. Immunocytochemistry indicates that BDNF protein is present in glial cells and dystrophic boutons in the immediate plaque vicinity. Our observations most closely resemble those of Ferrer et al. (1999), who reported plaque-associated BDNF immunoreactivity in dystrophic boutons, and Murer et al. (1999, 2001), who reported BDNF-immunoreactive cells, most likely glial cells, around plaques in human AD. Although these authors could not unequivocally localize the BDNF signals to specific glial cells in the postmortem tissue, their data indicate that a plaque-associated glial BDNF upregulation is present in human brain.
BDNF in AD: a role in inflammation and axonal sprouting?
So far, the relevance of BDNF changes in AD patients and APP23 transgenic mice is only poorly understood. Because BDNF has been discussed as a potential therapeutic for AD patients (Ferrer et al., 1999; Murer et al., 2001; Thoenen and Sendtner, 2002; Russo-Neustadt, 2003), however, it is important to unravel the biological role of this neurotrophin in animal models. As far as changes in BDNF in the immediate plaque vicinity are concerned, the expression of BDNF by microglial and astroglial cells links BDNF to inflammatory processes (Rogers et al., 2002). Activated microglial and astroglial cells are found regularly in the plaque vicinity (Stalder et al., 1999; Bamberger et al., 2003) and are believed to participate in the processing of amyloid (Rogers et al., 2002). Because BDNF inhibits microglial major histocompatibility complex class II molecule expression (Neumann et al., 1998), an immunomodulatory function of BDNF in AD is conceivable. A similar upregulation of BDNF in glial cells has been observed in other neurological diseases, such as multiple sclerosis (Stadelmann et al., 2002; Gielen et al., 2003) and human immunodeficiency virus-encephalitis (Soontornniyomkij et al., 1998), and after brain injury (Batchelor et al., 1999; Dougherty et al., 2000; Batchelor et al., 2002). Thus, changes in BDNF expression are part of the inflammatory response of the brain, although the precise role of BDNF in the inflammatory cascade has yet to be unraveled.
Because of its role in neuronal plasticity and axonal growth, BDNF may also be linked to aberrant regenerative processes occurring around senile plaques. In AD brains (Geddes et al., 1985, 1986; Masliah et al., 1996) and brains of APP23 mice (Phinney et al., 1999), extensive axonal sprouting has been reported, although the molecular basis of this process has remained unclear. Because BDNF induces axonal sprouting and synapse formation in vivo (Mamounas et al., 2000; Alsina et al., 2001; Tucker et al., 2001), it is also a prime candidate for the regulation of aberrant sprouting in AD.
Taken together, our data regarding BDNF expression in APP23 transgenic mice support the following sequence of events. After deposition of amyloid in the brain, activated glial cells are recruited to the plaque periphery. These cells synthesize and release BDNF to neighboring axons. BDNF, possibly in concert with other factors, could act as a trophic stimulus that induces axonal growth along a BDNF gradient toward the amyloid plaque. In an inflamed and potentially toxic environment, this growth becomes aberrant. Furthermore, because BDNF has been implicated in synaptic differentiation, local BDNF release could explain why we observed an abortive synaptic differentiation of axon terminals in the plaque vicinity (Phinney et al., 1999). Through this hypothesized mechanism, BDNF could contribute significantly to the pathogenesis of AD and thereby to the cognitive decline seen in this neurodegenerative disorder. Whether such a local inflammatory mechanism involving BDNF could limit the usefulness of BDNF as a potential therapeutic in AD will require further study.
Footnotes
This work was supported by grants from the Deutsche Forschungsgemeinschaft (SFB 269; T.D.), the Volkswagen-Stiftung (T.D.), the Alzheimer Forschung Initiative (T.D., M.J.), and by a Förderung Nachwuchs Kommission grant of Freie Universität (R.H., U.D.). Parts of this work were incorporated into a doctoral thesis presented to the Freie Universität Berlin by U.D. We thank Charlotte Nolte-Uhl and Susanne Huber for excellent technical assistance. Anke Bicyszko, Ute Fertig, Barbara Nagel, Ina Schettler, Uwe Otten, and Hilary Jackson also contributed to this study by assisting in laser microdissection, immunostaining procedures, brain dissection, and Aβ determination. We also thank Udo Rüb and Estifanos Ghebremedhin for helpful comments on this manuscript and Heidi Danker-Hopfe for help with statistical analysis and graphic art.
Correspondence should be addressed to Dr. Thomas Deller, Institute of Clinical Neuroanatomy, J. W. Goethe-University, Theodor-Stern-Kai 7, D-60590 Frankfurt, Germany. E-mail: t.deller@em.uni-frankfurt.de.
Copyright © 2004 Society for Neuroscience 0270-6474/04/242421-10$15.00/0
References
- Alsina B, Vu T, Cohen-Cory S (2001) Visualizing synapse formation in arborizing optic axons in vivo: dynamics and modulation by BDNF. Nat Neurosci 4: 1093-1101. [DOI] [PubMed] [Google Scholar]
- Bamberger ME, Harris ME, McDonald DR, Husemann J, Landreth GE (2003) A cell surface receptor complex for fibrillar beta-amyloid mediates microglial activation. J Neurosci 23: 2665-2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barde YA (1989) Trophic factors and neuronal survival. Neuron 2: 1525-1534. [DOI] [PubMed] [Google Scholar]
- Barde YA, Edgar D, Thoenen H (1982) Purification of a new neurotrophic factor from mammalian brain. EMBO J 1: 549-553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batchelor PE, Liberatore GT, Wong JY, Porritt MJ, Frerichs F, Donnan GA, Howells DW (1999) Activated macrophages and microglia induce dopaminergic sprouting in the injured striatum and express brain-derived neurotrophic factor and glial cell line-derived neurotrophic factor. J Neurosci 19: 1708-1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batchelor PE, Porritt MJ, Martinello P, Parish CL, Liberatore GT, Donnan GA, Howells DW (2002) Macrophages and microglia produce local trophic gradients that stimulate axonal sprouting toward but not beyond the wound edge. Mol Cell Neurosci 21: 436-453. [DOI] [PubMed] [Google Scholar]
- Boncristiano S, Calhoun ME, Kelly PH, Pfeifer M, Bondolfi L, Stalder M, Phinney AL, Abramowski D, Sturchler-Pierrat C, Enz A, Sommer B, Staufenbiel M, Jucker M (2002) Cholinergic changes in the APP23 transgenic mouse model of cerebral amyloidosis. J Neurosci 22: 3234-3243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondolfi L, Calhoun M, Ermini F, Kuhn HG, Wiederhold KH, Walker L, Staufenbiel M, Jucker M (2002) Amyloid-associated neuron loss and gliogenesis in the neocortex of amyloid precursor protein transgenic mice. J Neurosci 22: 515-522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornemann KD, Wiederhold KH, Pauli C, Ermini F, Stalder M, Schnell L, Sommer B, Jucker M, Staufenbiel M (2001) Abeta-induced inflammatory processes in microglia cells of APP23 transgenic mice. Am J Pathol 158: 63-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botchkarev VA, Botchkareva NV, Welker P, Metz M, Lewin GR, Subramaniam A, Bulfone-Paus S, Hagen E, Braun A, Lommatzsch M, Renz H, Paus AR (1999) A new role for neurotrophins: involvement of brain-derived neurotrophic factor and neurotrophin-4 in hair cycle control. FASEB J 13: 395-410. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E (1991) Neuropathological staging of Alzheimer-related changes. Acta Neuropathol (Berl) 82: 239-259. [DOI] [PubMed] [Google Scholar]
- Burbach G, Dehn D, Del Turco D, Deller T (2003) Quantification of layer-specific gene expression in the hippocampus: effective use of laser microdissection in combination with quantitative RT-PCR. J Neurosci Methods 131: 83-91. [DOI] [PubMed] [Google Scholar]
- Burgermeister P, Calhoun ME, Winkler DT, Jucker M (2000) Mechanisms of cerebrovascular amyloid deposition. Lessons from mouse models. Ann NY Acad Sci 903: 307-316. [DOI] [PubMed] [Google Scholar]
- Calhoun ME, Wiederhold KH, Abramowski D, Phinney AL, Probst A, Sturchler-Pierrat C, Staufenbiel M, Sommer B, Jucker M (1998) Neuron loss in APP transgenic mice. Nature 395: 755-756. [DOI] [PubMed] [Google Scholar]
- Calhoun ME, Burgermeister P, Phinney AL, Stalder M, Tolnay M, Wiederhold KH, Abramowski D, Sturchler-Pierrat C, Sommer B, Staufenbiel M, Jucker M (1999) Neuronal overexpression of mutant amyloid precursor protein results in prominent deposition of cerebrovascular amyloid. Proc Natl Acad Sci USA 96: 14088-14093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor B, Young D, Yan Q, Faull RL, Synek B, Dragunow M (1997) Brain-derived neurotrophic factor is reduced in Alzheimer's disease. Brain Res Mol Brain Res 49: 71-81. [DOI] [PubMed] [Google Scholar]
- Dougherty KD, Dreyfus CF, Black IB (2000) Brain-derived neurotrophic factor in astrocytes, oligodendrocytes, and microglia/macrophages after spinal cord injury. Neurobiol Dis 7: 574-585. [DOI] [PubMed] [Google Scholar]
- Durany N, Michel T, Kurt J, Cruz-Sanchez FF, Cervas-Navarro J, Riederer P (2000) Brain-derived neurotrophic factor and neurotrophin-3 levels in Alzheimer's disease brains. Int J Dev Neurosci 18: 807-813. [PubMed] [Google Scholar]
- Ernfors P, Ibanez CF, Ebendal T, Olson L, Persson H (1990) Molecular cloning and neurotrophic activities of a protein with structural similarities to nerve growth factor: developmental and topographical expression in the brain. Proc Natl Acad Sci USA 87: 5454-5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer I, Marin C, Rey MJ, Ribalta T, Goutan E, Blanco R, Tolosa E, Marti E (1999) BDNF and full-length and truncated TrkB expression in Alzheimer disease. Implications in therapeutic strategies. J Neuropathol Exp Neurol 58: 729-739. [DOI] [PubMed] [Google Scholar]
- Fusco M, Vantini G, Schiavo N, Zanotti A, Zanoni R, Facci L, Skaper SD (1993) Gangliosides and neurotrophic factors in neurodegenerative diseases: from experimental findings to clinical perspectives. Ann NY Acad Sci 695: 314-317. [DOI] [PubMed] [Google Scholar]
- Geddes JW, Monaghan DT, Cotman CW, Lott IT, Kim RC, Chui HC (1985) Plasticity of hippocampal circuitry in Alzheimer's disease. Science 230: 1179-1181. [DOI] [PubMed] [Google Scholar]
- Geddes JW, Anderson KJ, Cotman CW (1986) Senile plaques as aberrant sprout-stimulating structures. Exp Neurol 94: 767-776. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Carnahan J, Greenberg ME (1994) Requirement for BDNF in activity-dependent survival of cortical neurons. Science 263: 1618-1623. [DOI] [PubMed] [Google Scholar]
- Gielen A, Khademi M, Muhallab S, Olsson T, Piehl F (2003) Increased brain-derived neurotrophic factor expression in white blood cells of relapsing-remitting multiple sclerosis patients. Scand J Immunol 57: 493-497. [DOI] [PubMed] [Google Scholar]
- Hellweg R, Hock C, Hartung HD (1989) An improved and rapid highly sensitive enzyme immunoassay for nerve growth factor. Technique. J Methods Cell Mol Biol 1: 43-48. [Google Scholar]
- Hellweg R, von Arnim CA, Buchner M, Huber R, Riepe MW (2003) Neuroprotection and neuronal dysfunction upon repetitive inhibition of oxidative phosphorylation. Exp Neurol 183: 346-354. [DOI] [PubMed] [Google Scholar]
- Hock C, Heese K, Hulette C, Rosenberg C, Otten U (2000) Region-specific neurotrophin imbalances in Alzheimer disease: decreased levels of brain-derived neurotrophic factor and increased levels of nerve growth factor in hippocampus and cortical areas. Arch Neurol 57: 846-851. [DOI] [PubMed] [Google Scholar]
- Hof PR, Bouras C, Morrison JH (1999) Cortical neuropathology in aging and dementing disorders. In: Cerebral cortex (Peters A, Morrison JH, eds), pp 175-311. New York: Kluwer/Plenum.
- Hofer M, Pagliusi SR, Hohn A, Leibrock J, Barde YA (1990) Regional distribution of brain-derived neurotrophic factor mRNA in the adult mouse brain. EMBO J 9: 2459-2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holsinger RM, Schnarr J, Henry P, Castelo VT, Fahnestock M (2000) Quantitation of BDNF mRNA in human parietal cortex by competitive reverse transcription-polymerase chain reaction: decreased levels in Alzheimer's disease. Brain Res Mol Brain Res 76: 347-354. [DOI] [PubMed] [Google Scholar]
- Hughes PE, Alexi T, Walton M, Williams CE, Dragunow M, Clark RG, Gluckman PD (1999) Activity and injury-dependent expression of inducible transcription factors, growth factors and apoptosis-related genes within the central nervous system. Prog Neurobiol 57: 421-450. [DOI] [PubMed] [Google Scholar]
- Imai Y, Ibata I, Ito D, Ohsawa K, Kohsaka S (1996) A novel gene iba1 in the major histocompatibility complex class III region encoding an EF hand protein expressed in a monocytic lineage. Biochem Biophys Res Commun 224: 855-862. [DOI] [PubMed] [Google Scholar]
- Ito D, Imai Y, Ohsawa K, Nakajima K, Fukuuchi Y, Kohsaka S (1998) Microglia-specific localisation of a novel calcium binding protein, Iba1. Brain Res Mol Brain Res 57: 1-9. [DOI] [PubMed] [Google Scholar]
- Kelly PH, Bondolfi L, Hunziker D, Schlecht HP, Carver K, Maguire E, Abramowski D, Wiederhold KH, Sturchler-Pierrat C, Jucker M, Bergmann R, Staufenbiel M, Sommer B (2003) Progressive age-related impairment of cognitive behavior in APP23 transgenic mice. Neurobiol Aging 24: 365-378. [DOI] [PubMed] [Google Scholar]
- Korte M, Carroll P, Wolf E, Brem G, Thoenen H, Bonhoeffer T (1995) Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc Natl Acad Sci USA 92: 8856-8860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalonde R, Dumont M, Staufenbiel M, Sturchler-Pierrat C, Strazielle C (2002) Spatial learning, exploration, anxiety, and motor coordination in female APP23 transgenic mice with the Swedish mutation. Brain Res 956: 36-44. [DOI] [PubMed] [Google Scholar]
- Lindvall O, Ernfors P, Bengzon J, Kokaia Z, Smith ML, Siesjo BK, Persson H (1992) Differential regulation of mRNAs for nerve growth factor, brain-derived neurotrophic factor, and neurotrophin 3 in the adult rat brain following cerebral ischemia and hypoglycemic coma. Proc Natl Acad Sci USA 89: 648-652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linnarsson S, Bjorklund A, Ernfors P (1997) Learning deficit in BDNF mutant mice. Eur J Neurosci 9: 2581-2587. [DOI] [PubMed] [Google Scholar]
- Ma YL, Wang HL, Wu HC, Wei CL, Lee EH (1998) Brain-derived neurotrophic factor antisense oligonucleotide impairs memory retention and inhibits long-term potentiation in rats. Neuroscience 82: 957-967. [DOI] [PubMed] [Google Scholar]
- Mamounas LA, Blue ME, Siuciak JA, Altar CA (1995) Brain-derived neurotrophic factor promotes the survival and sprouting of serotonergic axons in rat brain. J Neurosci 15: 7929-7939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamounas LA, Altar CA, Blue ME, Kaplan DR, Tessarollo L, Lyons WE (2000) BDNF promotes the regenerative sprouting, but not survival, of injured serotonergic axons in the adult rat brain. J Neurosci 20: 771-782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Sisk A, Mallory M, Mucke L, Schenk D, Games D (1996) Comparison of neurodegenerative pathology in transgenic mice overexpressing V717F beta-amyloid precursor protein and Alzheimer's disease. J Neurosci 16: 5795-5811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister AK, Lo DC, Katz LC (1995) Neurotrophins regulate dendritic growth in developing visual cortex. Neuron 15: 791-803. [DOI] [PubMed] [Google Scholar]
- McAllister AK, Katz LC, Lo DC (1997) Opposing roles for endogenous BDNF and NT-3 in regulating cortical dendritic growth. Neuron 18: 767-778. [DOI] [PubMed] [Google Scholar]
- Michalski B, Fahnestock M (2003) Pro-brain-derived neurotrophic factor is decreased in parietal cortex in Alzheimer's disease. Brain Res Mol Brain Res 111: 148-154. [DOI] [PubMed] [Google Scholar]
- Mogi M, Togari A, Kondo T, Mizuno Y, Komure O, Kuno S, Ichinose H, Nagatsu T (1999) Brain-derived growth factor and nerve growth factor concentrations are decreased in the substantia nigra in Parkinson's disease. Neurosci Lett 270: 45-48. [DOI] [PubMed] [Google Scholar]
- Mu JS, Li WP, Yao ZB, Zhou XF (1999) Deprivation of endogenous brain-derived neurotrophic factor results in impairment of spatial learning and memory in adult rats. Brain Res 835: 259-265. [DOI] [PubMed] [Google Scholar]
- Murer MG, Boissiere F, Yan Q, Hunot S, Villares J, Faucheux B, Agid Y, Hirsch E, Raisman-Vozari R (1999) An immunohistochemical study of the distribution of brain-derived neurotrophic factor in the adult human brain, with particular reference to Alzheimer's disease. Neuroscience 88: 1015-1032. [DOI] [PubMed] [Google Scholar]
- Murer MG, Yan Q, Raisman-Vozari R (2001) Brain-derived neurotrophic factor in the control human brain, and in Alzheimer's disease and Parkinson's disease. Prog Neurobiol 63: 71-124. [DOI] [PubMed] [Google Scholar]
- Murphy DD, Cole NB, Segal M (1998) Brain-derived neurotrophic factor mediates estradiol-induced dendritic spine formation in hippocampal neurons. Proc Natl Acad Sci USA 95: 11412-11417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray KD, Gall CM, Jones EG, Isackson PJ (1994) Differential regulation of brain-derived neurotrophic factor and type II calcium/calmodulin-dependent protein kinase messenger RNA expression in Alzheimer's disease. Neuroscience 60: 37-48. [DOI] [PubMed] [Google Scholar]
- Neumann H, Misgeld T, Matsumuro K, Wekerle H (1998) Neurotrophins inhibit major histocompatibility class II inducibility of microglia: involvement of the p75 neurotrophin receptor. Proc Natl Acad Sci USA 95: 5779-5784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips HS, Hains JM, Armanini M, Laramee GR, Johnson SA, Winslow JW (1991) BDNF mRNA is decreased in the hippocampus of individuals with Alzheimer's disease. Neuron 7: 695-702. [DOI] [PubMed] [Google Scholar]
- Phinney AL, Deller T, Stalder M, Calhoun ME, Frotscher M, Sommer B, Staufenbiel M, Jucker M (1999) Cerebral amyloid induces aberrant axonal sprouting and ectopic terminal formation in amyloid precursor protein transgenic mice. J Neurosci 19: 8552-8559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers J, Strohmeyer R, Kovelowski CJ, Li R (2002) Microglia and inflammatory mechanisms in the clearance of amyloid beta peptide. Glia 40: 260-269. [DOI] [PubMed] [Google Scholar]
- Russo-Neustadt A (2003) Brain-derived neurotrophic factor, behavior, and new directions for the treatment of mental disorders. Semin Clin Neuropsychiatry 8: 109-118. [DOI] [PubMed] [Google Scholar]
- Sendtner M, Holtmann B, Hughes RA (1996) The response of motoneurons to neurotrophins. Neurochem Res 21: 831-841. [DOI] [PubMed] [Google Scholar]
- Shindler KS, Roth KA (1996) Double immunofluorescent staining using two unconjugated primary antisera raised in the same species. J Histochem Cytochem 44: 1331-1335. [DOI] [PubMed] [Google Scholar]
- Soontornniyomkij V, Wang G, Pittman CA, Wiley CA, Achim CL (1998) Expression of brain-derived neurotrophic factor protein in activated microglia of human immunodeficiency virus type 1 encephalitis. Neuropathol Appl Neurobiol 24: 453-460. [DOI] [PubMed] [Google Scholar]
- Soontornniyomkij V, Wang G, Pittman CA, Hamilton RL, Wiley CA, Achim CL (1999) Absence of brain-derived neurotrophic factor and trkB receptor immunoreactivity in glia of Alzheimer's disease. Acta Neuropathol (Berl) 98: 345-348. [DOI] [PubMed] [Google Scholar]
- Stadelmann C, Kerschensteiner M, Misgeld T, Bruck W, Hohlfeld R, Lassmann H (2002) BDNF and gp145trkB in multiple sclerosis brain lesions: neuroprotective interactions between immune and neuronal cells? Brain 125: 75-85. [DOI] [PubMed] [Google Scholar]
- Stalder M, Phinney A, Probst A, Sommer B, Staufenbiel M, Jucker M (1999) Association of microglia with amyloid plaques in brains of APP23 transgenic mice. Am J Pathol 154: 1673-1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staufenbiel M, Sommer B (1998) Transgenic animal models in the development of therapeutic strategies for Alzheimer's disease. In: The molecular biology of Alzheimer's disease: genes and mechanisms involved in amyloid generation (Haass C, ed), pp 309-326. Amsterdam: Harwood.
- Staufenbiel M, Paganetti PA (1999) Electrophoretic separation and immunoblotting of Abeta1-40 and Abeta1-42. In: Alzheimer's disease: methods and protocols (Hooper NM, ed), pp 91-99. Totowa, NJ: Humana.
- Sturchler-Pierrat C, Staufenbiel M (2000) Pathogenic mechanisms of Alzheimer's disease analyzed in the APP23 transgenic mouse model. Ann NY Acad Sci 920: 134-139. [DOI] [PubMed] [Google Scholar]
- Sturchler-Pierrat C, Abramowski D, Duke M, Wiederhold KH, Mistl C, Rothacher S, Ledermann B, Burki K, Frey P, Paganetti PA, Waridel C, Calhoun ME, Jucker M, Probst A, Staufenbiel M, Sommer B (1997) Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci USA 94: 13287-13292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thal DR, Rub U, Orantes M, Braak H (2002) Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 58: 1791-1800. [DOI] [PubMed] [Google Scholar]
- Thoenen H, Sendtner M (2002) Neurotrophins: from enthusiastic expectations through sobering experiences to rational therapeutic approaches. Nat Neurosci [Suppl] 5: 1046-1050. [DOI] [PubMed] [Google Scholar]
- Tucker KL, Meyer M, Barde YA (2001) Neurotrophins are required for nerve growth during development. Nat Neurosci 4: 29-37. [DOI] [PubMed] [Google Scholar]
- Van Dam D, D'Hooge R, Staufenbiel M, Van Ginneken C, Van Meir F, De Deyn PP (2003) Age-dependent cognitive decline in the APP23 model precedes amyloid deposition. Eur J Neurosci 17: 388-396. [DOI] [PubMed] [Google Scholar]
- Vicario-Abejon C, Owens D, McKay R, Segal M (2002) Role of neurotrophins in central synapse formation and stabilization. Nat Rev Neurosci 3: 965-974. [DOI] [PubMed] [Google Scholar]
- Zhang X, Poo MM (2002) Localized synaptic potentiation by BDNF requires local protein synthesis in the developing axon. Neuron 36: 675-688. [DOI] [PubMed] [Google Scholar]