

Abstract
Voltage-gated tetrodotoxin-sensitive sodium channels of Purkinje neurons produce “resurgent” current with repolarization, which results from relief of an open-channel block that terminates current flow at positive potentials. The associated recovery of sodium channels from inactivation is thought to facilitate the rapid firing patterns characteristic of Purkinje neurons. Resurgent current appears to depend primarily on NaV1.6 α subunits, because it is greatly reduced in “med” mutant mice that lack NaV1.6. To identify factors that regulate the susceptibility of α subunits to open-channel block, we voltage clamped wild-type and med Purkinje neurons before and after slowing conventional inactivation with β-pompilidotoxin (β-PMTX). β-PMTX increased resurgent current in wild-type neurons and induced resurgent current in med neurons. In med cells, the resurgent component of β-PMTX-modified sodium currents could be selectively abolished by application of intracellular alkaline phosphatase, suggesting that, like in NaV1.6-expressing cells, the open-channel block of NaV1.1 and NaV1.2 subunits is regulated by constitutive phosphorylation. These results indicate that the endogenous blocker exists independently of NaV1.6 expression, and conventional inactivation regulates resurgent current by controlling the extent of open-channel block. In Purkinje cells, therefore, the relatively slow conventional inactivation kinetics of NaV1.6 appear well adapted to carry resurgent current. Nevertheless, NaV1.6 is not unique in its susceptibility to open-channel block, because under appropriate conditions, the non-NaV1.6 subunits can produce robust resurgent currents.
Keywords: sodium current, Scn8a, med, open-channel block, cerebellum, CA3
Introduction
The tetrodotoxin- (TTX-) sensitive sodium channels of cerebellar Purkinje neurons open with depolarization to produce transient current and reopen with repolarization to produce “resurgent” current (Raman and Bean, 1997). These kinetics rely on two distinct modes of inactivation. “Conventional” inactivation, mediated by the cytoplasmic linker between domains III and IV (Vassilev et al., 1988, 1989; Stühmer et al., 1989), dominates at moderately negative potentials, whereas a rapid, voltage-dependent inactivation, mediated by an endogenous but unidentified open-channel blocker, dominates at more positive potentials (Raman and Bean, 2001). Depolarizations above 0 mV, therefore, promote channel opening followed by channel block, whereas repolarizations allow resurgent current to flow through channels that reopen as the block is relieved (Raman and Bean, 2001).
The molecular basis for resurgent current is only partly known. The sodium channel α subunit NaV1.6 appears important, because resurgent current is reduced by 90% in Purkinje neurons of mutant mice that lack expression of NaV1.6 (Scn8amed/med or “med” mice) (Burgess et al., 1995; Raman et al., 1997). NaV1.6 alone is not sufficient to generate resurgent kinetics, however, because several cell types that express NaV1.6 have no resurgent component to their sodium currents (Raman and Bean, 1997; Smith et al., 1998; Pan and Beam, 1999). Our working hypothesis is that, although these cells express NaV1.6, they lack other factors that are required to produce sodium channels with resurgent kinetics, including the presence of an endogenous open-channel blocker and/or constitutive phosphorylation of an unknown substrate (Grieco et al., 2002).
The requirement of NaV1.6 for resurgent current is not absolute, however, because the resurgent component of sodium currents of med Purkinje cells is greatly reduced but not abolished (Raman et al., 1997). The small resurgent current that remains in med cells suggests that the other sodium channels expressed by Purkinje cells, NaV1.1 and NaV1.2 (Felts et al., 1997; Vega-Saenz de Miera et al., 1997; Shah et al., 2001), may bind the putative open-channel blocker, although relatively weakly. The question arises, therefore, of what factors regulate the sensitivity of the different α subunits to open-channel block, thereby controlling their ability to carry resurgent current.
A potential clue comes from the observation that wild-type and med Purkinje neurons differ in their transient as well as resurgent currents: currents evoked by depolarization inactivate more rapidly in med than in wild-type neurons (Raman et al., 1997). Incorporating this difference in inactivation into a kinetic model of Purkinje sodium current demonstrated that the faster the rate of conventional inactivation, the less successfully the open-channel blocker competed with the inactivation gate, thereby reducing the amount of resurgent current (Khaliq et al., 2003).
In these experiments, by slowing conventional inactivation with the wasp venom β-pompilidotoxin (β-PMTX) (Kinoshita et al., 2001), we tested whether the rate of conventional inactivation significantly influences the susceptibility of non-NaV1.6 subunits to open-channel block. We also explored whether Purkinje neurons retain a functional blocker even in the absence of NaV1.6. The results support the idea that open-channel block and conventional inactivation compete with one another and suggest that, under the appropriate conditions, non-NaV1.6 as well as NaV1.6 α subunits may carry resurgent current.
Materials and Methods
Cell preparation. Experiments were performed on C57BL/6 mice (Charles River Laboratories, Wilmington, MA), unaffected Scn8a+/+ or Scn8a+/med mice, or affected Scn8amed/med mice (The Jackson Laboratory, Bar Harbor, ME). The med mice, which lack expression of the sodium channel α subunit NaV1.6 (Burgess et al., 1995), could be identified by their severe ataxia, which was evident after postnatal day 12. Genotypes were verified using standard Northern blot analyses (Khaliq et al., 2003). Recordings from Purkinje cells from C57BL/6 and from Scn8a+/+ or Scn8a+/med mice were indistinguishable, and the data were pooled and classified as representing wild-type Purkinje cells.
Cerebellar Purkinje neurons and hippocampal CA3 pyramidal neurons (“CA3 cells”) were acutely isolated from mice according to published methods (Regan, 1991; Raman et al., 1997). In accordance with institutional guidelines, mice were anesthetized with halothane before decapitation. For Purkinje neurons, the superficial layers of the cerebellum of 13- to 20-d-old mice were removed and minced in ice-cold, oxygenated dissociation solution containing the following (in mm): 82 Na2SO4, 30 K2SO4, 5 MgCl2, 10 HEPES, 10 glucose, and 0.001% phenol red, buffered to pH 7.4 with NaOH. For CA3 neurons, the hippocampus of 8- to 13-d-old mice were cut into 350 μm slices with a tissue chopper (The Mickle Laboratory Engineering Co. Ltd., Gomshall, Surrey, UK). In both dissections, the tissue was then incubated for 7 min in 10 ml of dissociation solution containing 3 mg/ml protease XXIII at 31°C (pH readjusted), with 100% oxygen blown over the surface of the fluid. The tissue was then washed in warmed, oxygenated dissociation solution containing 1 mg/ml bovine serum albumin and 1 mg/ml trypsin inhibitor (pH readjusted), in which the tissue was microdissected to excise the regions of interest. The pieces were transferred to Tyrode's solution containing the following (in mm): 150 NaCl, 4 KCl, 2 CaCl2, 2 MgCl2, 10 HEPES, and 10 glucose, buffered to pH 7.4 with NaOH, at room temperature and then triturated with a series of fire-polished Pasteur pipettes to liberate individual neurons. Cells were allowed to settle in extracellular solution in the recording chamber after trituration. Recordings were made between 1 and 6 hr after trituration.
Electrophysiological recording. Purkinje neurons were identified by their large size and characteristic tear shape, and CA3 pyramidal cell bodies were identified by their pyramidal morphology. Voltage-clamp recordings were made with an Axopatch 200B amplifier (Axon Instruments, Foster City, CA). Data were recorded with an InstruTech (Great Neck, NY) ITC-18 interface and PULSE software (HEKA Elektronik, Lambrecht, Germany). Borosilicate pipettes (A-M Systems, Carlsborg, WA) were wrapped with parafilm or coated with Sylgard to minimize capacitance. For whole-cell recordings from Purkinje cells and some CA3 cells, the pipettes were filled with a “CsCH3SO3” internal solution containing the following (in mm): 120 CsCH3SO3, 10 NaCl, 2 MgCl2, 10 HEPES, 2 EGTA, 53 sucrose, 14 Tris-creatinePO4, 4 MgATP, and 0.3 Tris-GTP, buffered to pH 7.4 with CsOH. Additional recordings from CA3 neurons were made with internal solution containing the following (in mm): 90 KH2PO4, 20 tetraethylammonium (TEA)-Cl, 1 NaCl, 2 MgCl2, 10 HEPES, and 2 EGTA, buffered to pH 7.2 with TEA-OH. The different internal solutions did not significantly alter the CA3 sodium currents, and the data were pooled.
For whole-cell recordings, cells were positioned in front of a series of three gravity-driven flow pipes that contained a “control” solution (in mm): 50 NaCl, 110 TEA-Cl, 2 CoCl2, and 10 HEPES, buffered to pH 7.4 with TrisOH; control solution plus 10 μm β-PMTX; or control solution plus 300 nm TTX. Recordings were made in each solution, and subtractions gave the TTX-sensitive sodium current with or without β-PMTX. The series resistance and capacitance were compensated, and the voltages reported in Table 1 include a correction for the liquid junction potentials (3 mV).
Table 1.
Effect of β-PMTX on activation parameters of sodium currents
|
|
|
Control |
β-PMTX |
n |
p value |
|---|---|---|---|---|---|
| Wild-type Purkinje | V½ (mV) | −39 ± 1 | −35 ± 1 | 10 | 0.02 |
| k (mV) | 4.5 ± 0.7 | 5.9 ± 0.7 | 0.02 | ||
| Gmax (nS) | 61 ± 5 | 54 ± 3 | 0.01 | ||
| Wild-type CA3 | V½ (mV) | −35 ± 2 | −38 ± 2 | 5 | 0.04 |
| k (mV) | 4.7 ± 0.4 | 5.2 ± 0.4 | 0.4 | ||
| Gmax (nS) | 34 ± 9 | 30 ± 8 | 0.1 | ||
| med Purkinje | V½ (mV) | −39 ± 2 | −42 ± 2 | 7 | 0.03 |
| k (mV) | 6.5 ± 0.3 | 7.8 ± 0.6 | 0.03 | ||
|
|
Gmax (nS) |
25 ± 4.3 |
23 ± 4.3 |
|
0.15 |
For inside-out patch recordings, the pipettes were filled with an extracellular solution containing Tyrode's solution, 10 mm TEA-Cl and 3 μm CdCl2, with or without 10 μm β-PMTX. Inside-out patches were positioned in front of a series of three gravity-driven flow pipes that contained the control CsCH3SO3 intracellular solution, CsCH3SO3 plus 3 mg/ml alkaline phosphatase, or CsCH3SO3 plus 400 μm QX-314. Recordings were made in each solution, and subtractions gave the QX-314-sensitive sodium current with or without alkaline phosphatase.
Drugs. Drugs were obtained from Sigma-Aldrich (St. Louis, MO), except TTX (Alomone Labs, Jerusalem, Israel), QX-314 (Calbiochem, San Diego, CA), and β-PMTX (Tocris Cookson, Ellisville, MO).
Analysis. Data were analyzed with IgorPro 4.02 software (WaveMetrics, Lake Oswego, OR). Current decays were fit with a single exponential of the form I = A * exp (-t/τdecay) + Iss, where I is current, A is the amplitude at the beginning of the voltage step, t is time, τdecay is the time constant, and Iss is the steady-state component of the current. Activation curves were measured from peak currents evoked by depolarization and normalized by the driving force. The resulting conductances were plotted against voltage and fit with a Boltzmann equation of the form G = Gmax/(1 + exp(-(V - V1/2)/k)), where G is conductance, Gmax is the maximal conductance, V is voltage, V1/2 is the half-maximal voltage of activation, and k is the slope factor.
Capacitative artifacts were digitally reduced or blanked in most figures. Data are reported as mean ± SE. Statistical significance between parameters of currents measured in control and drug-containing solutions was assessed with Student's two-tailed paired t tests, and p values are reported.
Results
Recordings were made in three classes of whole-cell voltage-clamped neurons that differ in the composition of their sodium channel complexes, as well as in their ability to produce resurgent sodium current. These included the following: (1) Purkinje cells from wild-type mice, which express NaV1.1, NaV1.2, and NaV1.6, as well as an endogenous voltage-dependent open-channel blocker, and produce large resurgent currents; (2) CA3 pyramidal cells from wild-type mice, which also express NaV1.1, NaV1.2, and NaV1.6 (Shah et al., 2001) but have no evidence of block and have no resurgent component to their sodium currents (Raman and Bean, 1997); and (3) Purkinje neurons from med mice, which express NaV1.1 and NaV1.2 but lack NaV1.6 and whose resurgent currents are greatly reduced relative to wild type. The decreased resurgent current amplitude in med Purkinje cells may indicate either a reduced availability of α subunits that can pass resurgent current or a reduced efficacy of an open-channel blocker.
TTX-sensitive sodium currents were recorded before and after inactivation was modulated by β-PMTX. In all three cell types, β-PMTX had small but consistent effects on the parameters of activation, summarized in Table 1. Consistent with studies in expression systems (Kinoshita et al., 2001), application of 10 μm β-PMTX to dissociated neurons slowed the rate of inactivation of transient sodium currents and enhanced steady-state currents, although the extent of these effects differed across the three classes of cells. In wild-type Purkinje cells, β-PMTX modestly but significantly increased the decay time constant (τdecay) of currents evoked by a step from -90 to 0 mV from 0.52 ± 0.01 to 0.73 ± 0.05 msec (n = 10; p < 0.01) (Fig. 1A, top). In contrast, in CA3 cells, inactivation of transient sodium currents in control solutions was relatively slow (τdecay = 1.1 ± 0.07 msec; n = 5), and β-PMTX nearly tripled the τdecay (to 3.0 ± 0.54 msec; p = 0.03) (Fig. 1A, middle). Because wild-type Purkinje and CA3 cells express the same sodium channel α subunits, these results raise the possibility that the effect of β-PMTX is influenced by the additional rapid mode of inactivation, namely open-channel block, that is present in Purkinje but not in CA3 cells. Specifically, in addition to accelerating inactivation in control solutions, open-channel block may compensate for the slowing of conventional inactivation by β-PMTX.
Figure 1.
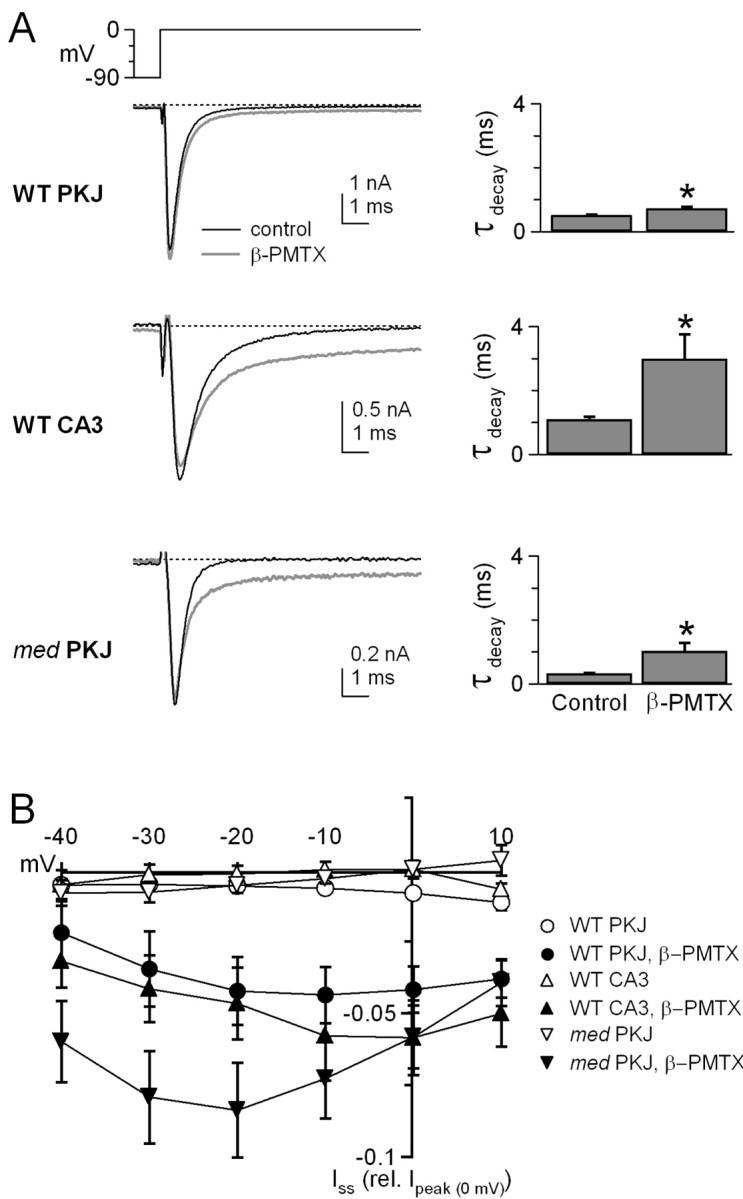
β-PMTX slows conventional sodium channel inactivation and increases steady-state sodium current in three cell types. A, Cells were held at -90 mV. Left panels, Transient currents evoked by steps from -90 to 0 mV in control (black traces) and β-PMTX (10 μm; gray traces). Dotted lines on all traces indicate 0 pA. Right panels, Time constants of inactivation (τdecay) in wild-type Purkinje (WT PKJ, top), wild-type CA3 (WT CA3, middle), and med Purkinje (med PKJ, bottom) neurons. Asterisks in all figures indicate p < 0.05. B, Steady-state sodium current (Iss) in control conditions (open symbols) and in β-PMTX (filled symbols) for WT PKJ (circles), WT CA3 (triangles), and med PKJ (inverted triangles) cells. Mean Iss at each potential was normalized to peak sodium current at 0 mV (Ipeak (0 mV)) in each cell.
As described previously (Raman et al., 1997), sodium currents of med Purkinje cells inactivated extremely rapidly in control solutions (τdecay = 0.33 ± 0.01 msec; n = 7) (Fig. 1A, bottom), indicative of kinetic differences between non-NaV1.6 and NaV1.6 subunits in Purkinje cells. In β-PMTX, the τdecay was prolonged to 1.0 ± 0.2 msec (p = 0.02) (Fig. 1A, bottom). This threefold increase in decay time is suggestive of an effective modulation of sodium channels by β-PMTX; nevertheless, the absolute decay time of β-PMTX-modulated currents in med cells remained quite brief, resembling wild-type Purkinje cells more than CA3 cells.
In addition to slowing the rate of sodium current inactivation at 0 mV, β-PMTX increased the amplitude of steady-state sodium currents measured in the last 20 msec of a 200 msec step in all three cell types. To allow comparison of cells with different current densities, the steady-state current at each potential was normalized by the total transient current evoked at 0 mV in each cell (Fig. 1B). In control solutions, the steady-state currents of all three cell types were small, and, occasionally, inactivation was so profound that the steady-state current was often lost in the noise (Fig. 1B, open symbols). Exposure to β-PMTX greatly increased the steady-state current amplitudes in all classes of cells, although the extent of this enhancement appeared to vary across potentials (Fig. 1B, filled symbols).
To quantify the β-PMTX-mediated increase in steady-state current, we calculated the steady-state current as a percentage of the peak current (100 * Iss/Ipeak) at each potential between -20 and +10 mV, with and without β-PMTX (Fig. 2A). In the absence of β-PMTX, Iss/Ipeak in all neuronal types was very small (<1% at -20 mV) and varied only slightly with voltage, such that a linear fit to the data gave a slope of 0.03 ± 0.01% per millivolt for wild-type Purkinje and CA3 cells and -0.03 ± 0.03% per millivolt for med Purkinje cells (Fig. 2A, open symbols, B). In the presence of β-PMTX, Iss/Ipeak measured at -20 mV increased significantly in wild-type Purkinje and CA3 neurons (to 3.0 ± 0.7 and 3.8 ± 1%, respectively; p < 0.01) and even more dramatically in med Purkinje neurons (to 6.8 ± 1.4%; p < 0.01). The larger increase in med Purkinje neurons raises the possibility that different sodium channel α subunits differ in their sensitivity to the toxin.
Figure 2.
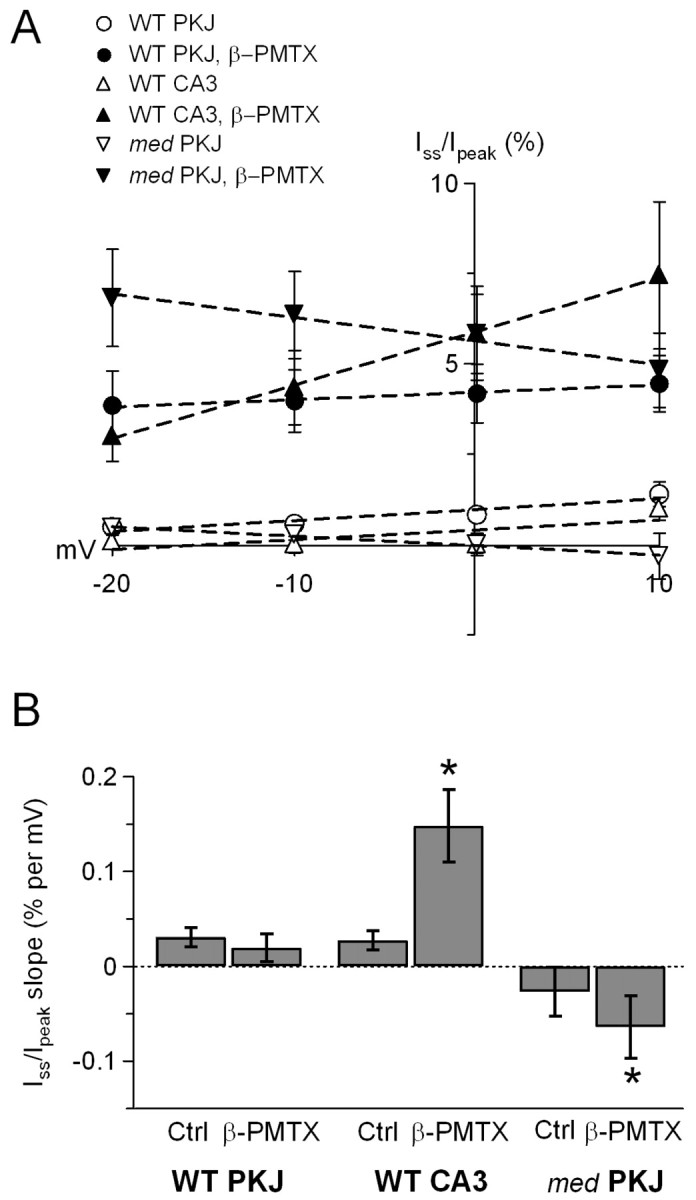
Effects of β-PMTX are voltage dependent. A, The percentage of steady-state relative to peak sodium current (100 * Iss/Ipeak) in wild-type Purkinje (WT PKJ), wild-type CA3 (WT CA3), and med Purkinje (med PKJ) cells in control and β-PMTX-containing solutions, at potentials between -20 and 10 mV. Symbols are as in Figure 1B. Dashed lines indicate linear fits to the data. B, Plot of the slope of Iss/Ipeak fitted in A for all classes of cells. Dotted line indicates zero slope. Ctrl, Control.
Previous studies have shown that the binding site of β-PMTX is located on the S3-S4 linker of domain IV of sodium channel α subunits (Kinoshita et al., 2001). Because the accessibility of this site is likely to increase at depolarized potentials (Jiang et al., 2003), the Iss/Ipeak in β-PMTX is expected to increase with voltage, predicting a positive slope to the plot of Iss/Ipeak versus voltage. The β-PMTX-modified sodium currents of CA3 cells, like expressed NaV1.2 channels (Kinoshita et al., 2001), conformed to this pattern of voltage dependence. Iss/Ipeak became larger at more positive potentials, increasing the slope of the fit to the data fivefold (to 0.15 ± 0.03% per millivolt; p = 0.04) (Fig. 2A, filled triangles, B).
In β-PMTX-treated wild-type Purkinje neurons, however, Iss/Ipeak remained nearly constant across all potentials, keeping the slope of the fit to the data near zero (0.02 ± 0.01% per millivolt; p = 0.5) (Fig. 2A, filled circles, B). This result reflects the observation that the current decays substantially at positive potentials in Purkinje cells, even in the presence of the toxin. Because wild-type Purkinje and CA3 cells express the same α subunits, it is possible that, in Purkinje cells, the putative blocking factor partially compensates at positive potentials for the β-PMTX-mediated destabilization of conventional inactivation.
In med Purkinje neurons, despite the 17-fold increase in Iss/Ipeak at -20 mV during application of β-PMTX, the Iss/Ipeak actually decreased with potential, reaching a level at +10 mV similar to that measured in wild-type Purkinje cells. As a result, the plot of Iss/Ipeak versus voltage for med cells was more steeply negative in β-PMTX relative to control solutions (-0.06 ± 0.03% per millivolt; p = 0.04) (Fig. 2A, filled inverted triangles, B), and the relief of inactivation by β-PMTX at positive potentials was less than predicted. This observation is again consistent with the idea that, even in the absence of NaV1.6 expression, the β-PMTX-induced destabilization of conventional inactivation may be overcome at positive potentials, although incompletely, by an increased efficacy of an alternative mechanism of inactivation present in Purkinje neurons, possibly open-channel block.
If, in the presence of a functional open-channel blocker, β-PMTX permits a greater extent of block at positive potentials, then the amplitude of resurgent current, which results from the relief of open-channel block with repolarization, should be increased. To test this possibility, we recorded currents evoked by repolarization, in the absence and presence of β-PMTX. Cells were held at -90 mV, depolarized to +30 mV for 5 msec, and then repolarized to -30 mV. In wild-type Purkinje neurons, the decay of transient current at +30 mV was rapid and profound, and repolarization from +30 to -30 mV elicited resurgent sodium current in control solutions (Fig. 3A, top, black trace). The slow rise of this current (2-5 msec) (Raman and Bean, 1997) is thought to reflect unbinding of the channel blocker, whereas the slow decay corresponds to the onset of conventional inactivation. Tail currents are seen only rarely under these recording conditions, partly because of the nearly complete inactivation and partly because of the extremely rapid deactivation kinetics of the currents, which often remain unresolved or are masked by imperfectly subtracted capacitative transients. When wild-type Purkinje neurons were exposed to β-PMTX, resurgent current was greatly enhanced, and, in some records, rapid tail currents that decayed within 300 μsec were also visible (Fig. 3A, top, red trace). Although the amplitude of the resurgent current was clearly different in β-PMTX, the kinetics and voltage dependence of the current were similar to those in control solutions, showing a slow rise and a peak current between -30 and -40 mV (n = 10) (Fig. 3C, top). These data are therefore consistent with the hypothesis that the PMTX-induced destabilization of inactivation results in an increased occupancy not only of the open state, producing a larger Iss/Ipeak, but also of the blocked state, producing a larger resurgent current with repolarization from positive potentials.
Figure 3.

β-PMTX increases the amplitude of resurgent sodium current in wild-type Purkinje neurons. A, Currents elicited by step depolarizations from -90 to +30 mV, followed by step repolarizations to -30 mV in control (black traces) and β-PMTX (red traces) for wild-type Purkinje (WT PKJ, top) and wild-type CA3 (WT CA3, bottom) neurons. Transient currents are off scale (tildes). Calibration bars refer to both sets of traces. Vertical dashed lines indicate region expanded in B. B, Currents elicited by 100 msec repolarizing steps to potentials between -70 and -10 mV in 20 mV increments, as indicated by arrows. Calibration bars refer to both sets of traces. For the illustrated cells, the transient current at -30 mV in β-PMTX was 67% (wild-type Purkinje) and 108% (CA3) of control. C, Mean current-voltage relationship for currents recorded in control (black symbols) and β-PMTX (red symbols) for wild-type Purkinje (top) and wild-type CA3 neurons (bottom). Peak current was measured as the maximal current occurring >300 μsec after the repolarization, which excluded the extremely fast tail that was evident in a few wild-type Purkinje cells, but included the wild-type CA3 tail current.
The voltage dependence of β-PMTX binding may complicate this analysis, however; as β-PMTX binding becomes less stable with repolarization, many β-PMTX-modulated channels that are open at positive potentials are predicted to relax into conventional inactivated states. These transitions from open to inactivated states at moderately negative potentials might produce a current decay reminiscent of resurgent sodium current, regardless of the extent of block. Therefore, to test the effect of β-PMTX on currents evoked by repolarizing steps in the absence of a blocking element, we repeated the experiments in CA3 cells. In control solutions, transient currents of CA3 cells inactivated almost completely. With repolarization, no additional current was elicited after the decay of a small tail current, consistent with the channels being stably inactivated (Fig. 3A, bottom, black trace). In the presence of β-PMTX, however, a steady-state current was evident at +30 mV, and repolarization elicited an instantaneous current that decayed rapidly (Fig. 3A, bottom, red trace). When CA3 cells were repolarized to several different potentials, the resulting currents showed no evidence of either a rising phase or the non-monotonic current-voltage relationships that typify resurgent sodium currents (Fig. 3B, bottom). Instead, analysis of the repolarization-evoked currents suggested that they were classical tail currents; they had an instantaneous onset, their amplitude increased with larger driving forces, and their extrapolated reversal potential was near ENa (+40 mV; n = 5) (Fig. 3B,C, bottom). The fast component of decay of these instantaneous currents is likely to reflect deactivation, because channels are only approximately half-activated at this potential (Table 1), whereas the slower component is likely to reflect inactivation as a result of the relief of the β-PMTX-modulation at negative voltages. Thus, comparison of the data from wild-type Purkinje and CA3 cells suggests that the presence or absence of a rising phase on repolarization-evoked currents in β-PMTX serves as an indicator of whether or not a cell possesses a functional open-channel blocker.
The observation that β-PMTX increased resurgent current in wild-type Purkinje cells suggests a direct relationship between the rate of conventional inactivation and the extent of open-channel block. Specifically, the binding of the III-IV linker appears to hinder binding of the open-channel blocker and vice versa. If so, then the resurgent current occasionally recorded in med Purkinje cells in control solutions may be small simply because the rapid, stable inactivation of non-NaV1.6 channels limits the binding of an otherwise functional blocking element. To test this possibility, we recorded sodium currents elicited in med neurons by step repolarizations with and without β-PMTX. In control solutions, repolarizing steps from +30 to -30 mV evoked either a very small, but detectable, resurgent sodium current (n = 2 of 7 cells) or no measurable current at all (n = 5 of 7 cells) (Fig. 4A, black trace). In the presence of β-PMTX, however, a robust resurgent current was evoked in all seven cells (Fig. 4A, red trace, B), with kinetics and amplitudes that were similar to those of wild-type Purkinje neurons exposed to β-PMTX (Fig. 4C). These data provide evidence, first, that med Purkinje neurons contain a functional blocker that is independent of NaV1.6 expression, and second, that NaV1.1 and/or NaV1.2 α subunits may be susceptible to open-channel block if their inactivation kinetics are sufficiently slow.
Figure 4.
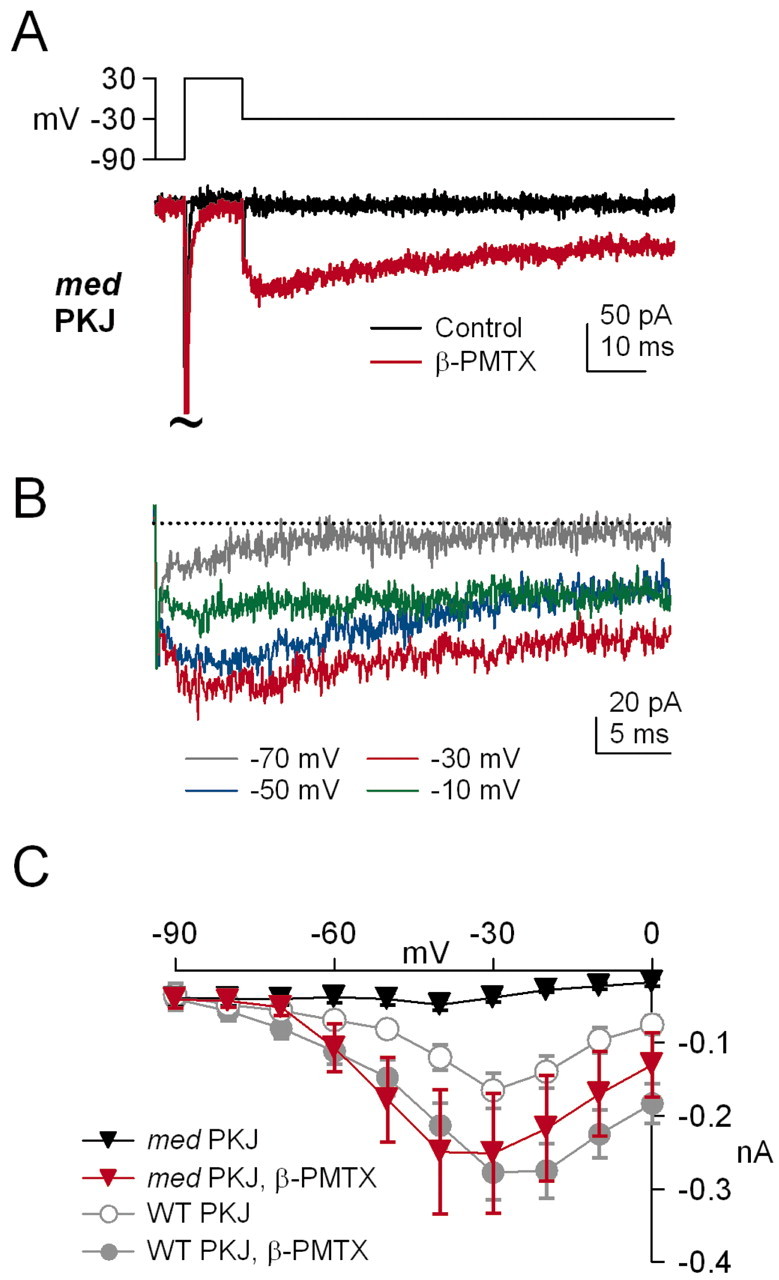
β-PMTX induces resurgent sodium current in med Purkinje neurons. A, Currents elicited by step depolarizations from -90 to +30 mV, followed by step repolarizations to -30 mV in control (black traces) and β-PMTX (red traces) for med Purkinje neurons (med PKJ). B, Currents elicited by 100 msec repolarizing steps to potentials between -70 and -10 mV in 20 mV increments, as labeled. Transient currents are off scale (tildes). For the illustrated cell, the med Purkinje transient current at -30 mV in β-PMTX was 112% of control. C, The I-V relationship of repolarization-evoked currents in med Purkinje neurons recorded in control (black squares) and β-PMTX (red squares). Wild-type Purkinje (WT PKJ) data from Figure 3C is illustrated in gray for comparison.
The similar profiles of β-PMTX-enhanced resurgent current in wild-type and med Purkinje cells raise the possibility that the nature of channel block is the same in both cell types. In wild-type Purkinje cells, constitutive phosphorylation is necessary to maintain a functional open-channel blocker, because outside-out patches exposed to intracellular alkaline phosphatase, a broad-spectrum phosphatase, lack resurgent current (Grieco et al., 2002). To verify that the resurgent component of sodium current could be selectively abolished by dephosphorylation, we first recorded sodium currents from inside-out patches from wild-type Purkinje cells. In control solutions, the kinetics of transient and resurgent currents in inside-out patches were similar to those measured in whole-cell recordings and outside-out patches (Grieco et al., 2002). Application of alkaline phosphatase (3 mg/ml) to the intracellular face of the patch consistently slowed inactivation of transient currents at 0 mV (from τ = 0.42 ± 02 to 0.68 ± 12 msec; p = 0.04) and abolished resurgent currents (n = 5 of 5 patches) (Fig. 5A, top traces).
Figure 5.
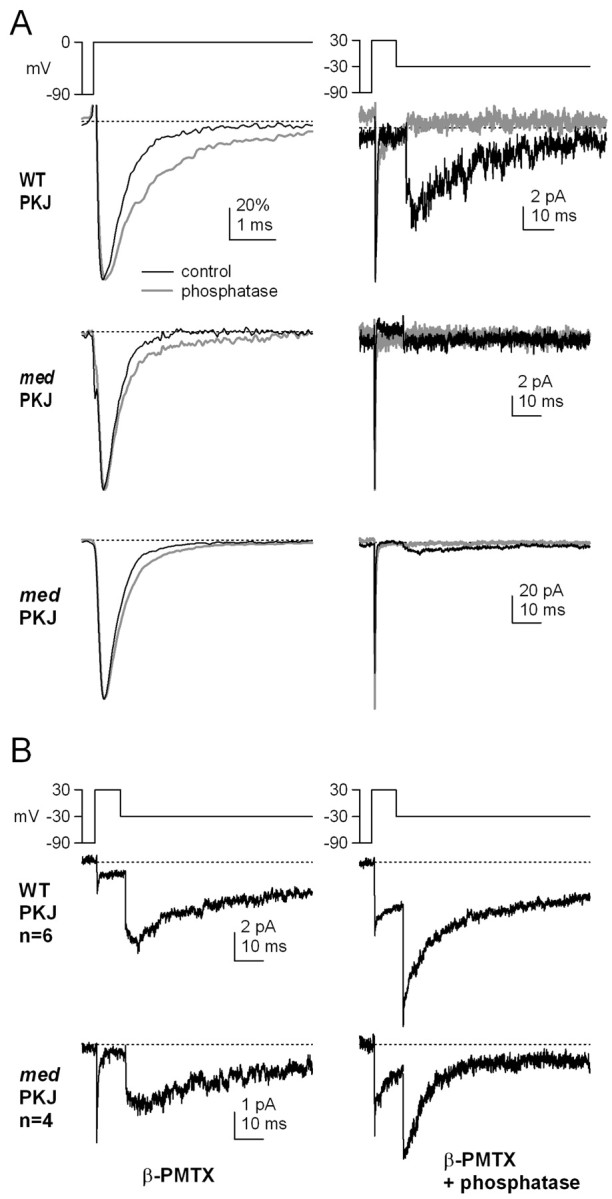
Sensitivity of resurgent current to dephosphorylation in both wild-type and med Purkinje inside-out patches. A, Currents elicited in inside-out patches by depolarizations from -90 to 0 mV (left) and by repolarizations from +30 to -30 mV (right). Data from one wild-type Purkinje (WT PKJ, top traces) and two med Purkinje (med PKJ, middle and bottom traces) patches before (black lines) and after (gray lines) exposure to alkaline phosphatase (3 mg/ml). Transient currents (left) were normalized to reveal differences in inactivation kinetics. Calibration bar in top left panel applies to all left panels. B, Inside-out patches from wild-type Purkinje (WT PKJ, top) and med Purkinje (med PKJ, bottom) neurons with 10 μm β-PMTX included in the extracellular (pipette) solution before (left) and after (right) exposure to phosphatase. Traces are averaged data from six wild-type or four med patches. Calibration bars apply to both left and right panels.
In inside-out patches from med Purkinje cells, transient currents were also evident with depolarization from -90 to 0 mV; inactivation of these currents slowed only slightly after application of alkaline phosphatase (from τ = 0.32 ± 0.006 to 0.52 ± 0.05 msec; n = 3; p = 0.07) (Fig. 5A, left middle traces). With repolarization from +30 to -30 mV, resurgent current was detectable only occasionally, consistent with data from whole cells. Two patches showed no resurgent current, and the currents were not measurably modified by application of alkaline phosphatase (Fig. 5A, right middle traces). In the third patch, resurgent current was clearly visible, although the relative amplitude of the resurgent to transient current was much smaller than in wild-type Purkinje cells. As in wild-type patches, application of alkaline phosphatase to the med patch abolished the resurgent current (Fig. 5A, bottom traces).
Next, we recorded the effects of the phosphatase on wild-type sodium currents modulated by β-PMTX. Inside-out patches were excised from wild-type and med Purkinje neurons with 10 μm β-PMTX in the extracellular (pipette) solution. With control intracellular solutions, extracellular β-PMTX slowed the decay of transient currents in wild-type patches and induced large steady-state as well as resurgent currents (Fig. 5B, top left trace; average data from six patches), effects that were similar to those measured in whole cells. To estimate the time course of the fast component of the decay of transient current evoked by a step to 0 mV, the first 10 msec of the inactivating phase was fit with a single exponential, giving a τdecay of 0.94 ± 0.21 msec (n = 6; data not shown). When the same patches were exposed to intracellular alkaline phosphatase, the peak amplitude of transient current increased (-7.8 ± 1.6 to -13.4 ± 1.8 pA; p < 0.01), and the time course of inactivation was significantly slowed (τdecay = 2.8 ± 0.55 msec; p = 0.02). Because of the relatively large steady-state currents in patches exposed to both alkaline phosphatase and β-PMTX, repolarization evoked a large tail current (Fig. 5B, top right trace), reminiscent of the β-PMTX-induced tail currents in CA3 cells. The instantaneous component of the current evoked with repolarization increased 2.5-fold in alkaline phosphatase (n = 6). The slowly rising phase of current that is typical of resurgent current was absent from the phosphatase-treated patches, however, consistent with a loss of open-channel block and unblock. Resurgent currents in β-PMTX as well as tail currents in alkaline phosphatase and β-PMTX decayed with a similar time course to a similar steady-state level; on the basis of the data from CA3 cells, we interpret this decay as reflecting entry into conventional inactivated states.
In inside-out patches from med Purkinje neurons, currents recorded in β-PMTX were qualitatively similar to those of wild-type patches, showing a relatively slowly inactivating transient current (τdecay = 0.71 ± 0.1 msec; n = 4; data not shown) and large resurgent currents (Fig. 5B, bottom left trace; average of four patches). Application of alkaline phosphatase increased the peak amplitude in three of four patches (-6.9 ± 1.9 to -9.0 ± 3.0 pA; p = 0.37) and further slowed the transient current at 0 mV to 3.6 ± 0.7 msec (p = 0.04). In med patches, as in wild-type patches, alkaline phosphatase also converted the β-PMTX-induced slowly rising resurgent current into an instantaneous tail current (Fig. 5C, bottom right trace) that was 2.7-fold larger in phosphatase-treated patches (n = 4). This result supports the idea that the resurgent current induced in non-NaV1.6 channels by β-PMTX relies on phosphorylation-dependent block, similar to that seen in wild-type neurons.
Discussion
These results suggest that the two inactivation mechanisms in sodium channels of Purkinje neurons, namely the domain III-IV linker and an endogenous open-channel blocker, compete with one another for the same, or overlapping, binding sites. The resulting dependence of the extent of channel block on the rate of conventional inactivation not only accounts for the small residual resurgent currents observed in NaV1.6-lacking Purkinje neurons but also offers a resolution to the question of the necessity of NaV1.6 in the production of resurgent sodium current.
NaV1.6 α subunits have been associated with several physiological specializations: not only are they responsible for the large resurgent currents of wild-type Purkinje neurons (Raman et al., 1997), but they are also the primary sodium channel α subunits in peripheral nodes (Caldwell et al., 2000) and initial segments of retinal ganglion cells (Boiko et al., 2003), and they provide the majority of steady-state sodium current in prefrontal cortex neurons (Maurice et al., 2001). The questions remain, however, what molecular characteristics make NaV1.6 particularly well adapted for these functions, and to what extent NaV1.6 is uniquely able to assume these roles. In the present experiments, we explored what characteristics of NaV1.6 subunits may optimize them for the generation of resurgent current in Purkinje cells. Our results suggest that, at a physiological level, wild-type Purkinje cells express NaV1.6 subunits with relatively slow inactivation kinetics, which make them highly susceptible to the open-channel block that is necessary for production of resurgent current. At a biophysical level, however, NaV1.6 is not unique in its ability to bind the open-channel blocker, because PMTX-induced slowing of inactivation of non-NaV1.6 subunits in Purkinje cells can induce a resurgent current that is indistinguishable from that in NaV1.6-expressing cells.
Previous studies have suggested that sodium channels that carry resurgent current must fulfill two criteria. First, the sodium channel must have a closely associated open-channel blocker, and second, the α subunit, the blocker, and/or an associated protein must be constitutively phosphorylated; both of these criteria are fulfilled by NaV1.6 channels of Purkinje cells (Raman and Bean, 2001; Grieco et al., 2002). From those experiments, however, we could resolve neither whether the blocking factor was part of NaV1.6 itself nor whether the site of phosphorylation was unique to NaV1.6. The present observation however, that NaV1.1 and/or NaV1.2 can efficiently carry resurgent current in med Purkinje cells when inactivation is slowed by β-PMTX indicates that the open-channel blocker can exist independently of NaV1.6 and that it is in fact present and functional in NaV1.6-lacking Purkinje cells. The observation that the blocker is retained in excised membrane patches suggests that it can associate tightly with (or is a part of) any or all of the α subunits present in Purkinje cells. Furthermore, the ability of alkaline phosphatase to abolish endogenous as well as β-PMTX-induced resurgent current in both wild-type and med Purkinje neurons suggests that the important site(s) of phosphorylation is either present on a variety of subunits or altogether distinct from the α subunit.
The ability of non-NaV1.6 subunits to carry resurgent current under certain conditions reveals a third requirement for the production of resurgent current, namely, that the channel must inactivate sufficiently slowly to permit open-channel block. Again, NaV1.6 channels of Purkinje cells fulfill this criterion. The inactivation rates of sodium channel α subunits, however, vary widely across cells, depending on the intracellular milieu and other expression conditions. For example, when expressed in Xenopus oocytes, NaV1.1, NaV1.2, and NaV1.6 α subunits inactivate slowly (τdecay > 10 msec at 0 mV), but these inactivation rates can be significantly accelerated by the coexpression of β subunits and/or by expression in mammalian cell lines (Isom et al., 1992, 1995; Smith and Goldin, 1998; Smith et al., 1998; Qu et al., 2001). Because most mammalian neurons express β subunits (Shah et al., 2001), most natively expressed sodium channels inactivate in a few milliseconds at potentials near 0 mV. Within this time scale, however, the inactivation rates of specific sodium channels can vary across or even within cells. For instance, the ultra-fast inactivation of NaV1.1 and NaV1.2 observed in med Purkinje cells may not be widespread, because the transient sodium currents of prefrontal cortex neurons from med mice, which are carried by NaV1.1 and NaV1.2, inactivate at rates similar to those of NaV1.6-expressing wild-type cells (Maurice et al., 2001). Additionally, the rate of inactivation of NaV1.2 channels expressed in mammalian cell lines can vary with phosphorylation state (Ratcliffe et al., 2000). These observations leave open the possibility that, in non-Purkinje cell types that contain a functional open-channel blocker, non-NaV1.6 subunits might inactivate sufficiently slowly to produce resurgent current.
Resurgent sodium current has been most extensively described in cerebellar Purkinje neurons, in which it appears to facilitate the high rates of firing that are typical of Purkinje neurons by allowing a rapid recovery of sodium channel availability (Raman and Bean, 1997; Khaliq et al., 2003). Resurgent current is not unique to Purkinje cells, however. It is also present in neurons of the subthalamic nuclei (Do and Bean, 2003) and has been observed or hypothesized to exist in other cerebellar neurons, including unipolar brush cells (Mossadeghi and Slater, 1998), granule cells (D'Angelo et al., 2001), and some neurons of the cerebellar nuclei (Raman et al., 2000). Although the profile of resurgent current is qualitatively similar across cell types, different neurons may or may not achieve resurgent kinetics by identical mechanisms. For instance, the resurgent current in neurons other than Purkinje cells may or may not strictly require NaV1.6, depending on the rates of conventional inactivation of the α subunits expressed in each neuron. Additionally, the open-channel blocker itself, as well as the extent of phosphorylation, may be different in different cells. By generalizing the properties necessary for the production of resurgent kinetics, our results may provide a context for exploring the extent to which NaV1.6 is the dominant channel that carries resurgent current under physiological conditions in other regions of the brain.
Footnotes
This work was supported by National Institutes of Health Grants F31 NS045483 (T.M.G.) and NS39395 (I.M.R.) and the Klingenstein Foundation (I.M.R.). We thank Andrea Frassetto for genotyping the mice and Zayd Khaliq, Jason Pugh, Fatemeh Afshari, Nathan Gouwens, and Petra Telgkamp for helpful discussions.
Correspondence should be addressed to Indira M. Raman, Department of Neurobiology and Physiology, 2205 Tech Drive, Northwestern University, Evanston, IL 60208. E-mail: i-raman@northwestern.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/230035-08$15.00/0
References
- Boiko T, Van Wart A, Caldwell JH, Levinson SR, Trimmer JS, Matthews G (2003) Functional specialization of the axon initial segment by isoform-specific sodium channel targeting. J Neurosci 23: 2306-2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess DL, Kohrman DC, Galt J, Plummer NW, Jones JM, Spear B, Meisler MH (1995) Mutation of a new sodium channel gene, Scn8a, in the mouse mutant “motor endplate disease.” Nat Genet 10: 461-465. [DOI] [PubMed] [Google Scholar]
- Caldwell JH, Schaller KL, Lasher RS, Peles E, Levinson SR (2000) Sodium channel NaV1.6 is localized at nodes of Ranvier, dendrites, and synapses. Proc Natl Acad Sci USA 97: 5616-5620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Angelo E, Nieus T, Maffei A, Armano S, Rossi P, Taglietti V, Fontana A, Naldi G (2001) Theta-frequency bursting and resonance in cerebellar granule cells: experimental evidence and modeling of a slow K+-dependent mechanism. J Neurosci 21: 759-770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do MT, Bean BP (2003) Subthreshold sodium currents and pacemaking of subthalamic neurons: modulation by slow inactivation. Neuron 39: 109-120. [DOI] [PubMed] [Google Scholar]
- Felts PA, Yokoyama S, Dib-Hajj S, Black JA, Waxman SG (1997) Sodium channel alpha-subunit mRNAs I, II, III, NaG, Na6 and hNE (PN1): different expression patterns in developing rat nervous system. Brain Res Mol Brain Res 45: 71-82. [DOI] [PubMed] [Google Scholar]
- Grieco TM, Afshari F, Raman IM (2002) A role for phosphorylation in the maintenance of resurgent sodium current in cerebellar Purkinje neurons. J Neurosci 22: 3100-3107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isom LL, De Jongh KS, Patton DE, Reber BF, Offord J, Charbonneau H, Walsh K, Goldin AL, Catterall WA (1992) Primary structure and functional expression of the beta 1 subunit of the rat brain sodium channel. Science 256: 839-842. [DOI] [PubMed] [Google Scholar]
- Isom LL, Scheuer T, Brownstein AB, Ragsdale DS, Murphy BJ, Catterall WA (1995) Functional co-expression of the beta 1 and type IIA alpha subunits of sodium channels in a mammalian cell line. J Biol Chem 270: 3306-3312. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Ruta V, Chen J, Lee A, MacKinnon R (2003) The principle of gating charge movement in a voltage-dependent K+ channel. Nature 423: 42-48. [DOI] [PubMed] [Google Scholar]
- Khaliq ZM, Gouwens NW, Raman IM (2003) The contribution of resurgent sodium current to high-frequency firing in Purkinje neurons: an experimental and modeling study. J Neurosci 23: 4899-4912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita E, Maejima H, Yamaoka K, Konno K, Kawai N, Shimizu E, Yokote S, Nakayama H, Seyama I (2001) Novel wasp toxin discriminates between neuronal and cardiac sodium channels. Mol Pharmacol 59: 1457-1463. [PubMed] [Google Scholar]
- Maurice N, Tkatch T, Meisler M, Sprunger LK, Surmeier DJ (2001) D1/D5 dopamine receptor activation differentially modulates rapidly inactivating and persistent sodium currents in prefrontal cortex pyramidal neurons. J Neurosci 21: 2268-2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mossadeghi B, Slater NT (1998) Persistent and resurgent sodium currents in cerebellar unipolar brush cells. Soc Neurosci Abstr 24: 1078. [Google Scholar]
- Pan F, Beam KG (1999) The absence of resurgent current in mouse spinal neurons. Brain Res 849: 162-168. [DOI] [PubMed] [Google Scholar]
- Qu Y, Curtis R, Lawson D, Gilbride K, Ge P, DiStefano PS, Silos-Santiago I, Catterall WA, Scheuer T (2001) Differential modulation of sodium channel gating and persistent sodium currents by the beta1, beta2, and beta3 subunits. Mol Cell Neurosci 18: 570-580. [DOI] [PubMed] [Google Scholar]
- Raman IM, Bean BP (1997) Resurgent sodium current and action potential formation in dissociated cerebellar Purkinje neurons. J Neurosci 17: 4517-4526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman IM, Bean BP (2001) Inactivation and recovery of sodium currents in cerebellar Purkinje neurons: evidence for two mechanisms. Biophys J 80: 729-737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman IM, Sprunger LK, Meisler MH, Bean BP (1997) Altered subthreshold sodium current and disrupted firing patterns in Purkinje neurons of Scn8a mutant mice. Neuron 19: 881-891. [DOI] [PubMed] [Google Scholar]
- Raman IM, Gustafson AE, Padgett D (2000) Ionic currents and spontaneous firing in neurons isolated from the cerebellar nuclei. J Neurosci 20: 9004-9016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratcliffe CM, Qu Y, McCormick KA, Tibbs VC, Dixon JE, Scheuer T, Catterall WA (2000) A sodium channel signaling complex: modulation by associated receptor protein tyrosine phosphatase beta. Nat Neurosci 3: 437-444. [DOI] [PubMed] [Google Scholar]
- Regan LJ (1991) Voltage-dependent calcium currents in Purkinje cells from rat cerebellar vermis. J Neurosci 11: 2259-2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah BS, Stevens EB, Pinnock RD, Dixon AK, Lee K (2001) Developmental expression of the novel voltage-gated sodium channel auxiliary subunit beta 3, in rat CNS. J Physiol (Lond) 534: 763-776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MR, Smith RD, Plummer NM, Meisler MH, Goldin AL (1998) Functional analysis of the mouse Scn8a sodium channel. J Neurosci 18: 6093-6102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RD, Goldin AL (1998) Functional analysis of the rat I sodium channel in Xenopus oocytes. J Neurosci 18: 811-820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stühmer W, Conti F, Suzuki H, Wang XD, Noda M, Yahagi N, Kubo H, Numa S (1989) Structural parts involved in activation and inactivation of the sodium channel. Nature 339: 597-603. [DOI] [PubMed] [Google Scholar]
- Vassilev PM, Scheuer T, Catterall WA (1988) Identification of an intracellular peptide segment involved in sodium channel inactivation. Science 241: 1658-1661. [DOI] [PubMed] [Google Scholar]
- Vassilev P, Scheuer T, Catterall WA (1989) Inhibition of inactivation of single sodium channels by a site-directed antibody. Proc Natl Acad Sci USA 86: 8147-8151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega-Saenz de Miera E, Rudy B, Sugimori M, Llinás R (1997) Molecular characterization of the sodium channel subunits expressed in mammalian cerebellar Purkinje neurons. Proc Natl Acad Sci USA 94: 7059-7064. [DOI] [PMC free article] [PubMed] [Google Scholar]