

Abstract
Cytochrome c-initiated activation of apoptotic protease activating factor-1 (Apaf-1) is a key step in the mitochondrial-signaling pathway for the activation of death-executing caspases in apoptosis. This signaling pathway has been implicated in the pathophysiology of various neurological disorders, including ischemic brain injury. In this study, we have cloned a novel rat gene product, designated as Apaf-1-interacting protein (AIP), which functions as a dominant-negative inhibitor of the Apaf-1-caspase-9 pathway. AIP is constitutively expressed in the brain, but at substantially lower levels than Apaf-1 and caspase-9. AIP can directly bind to Apaf-1 in vitro through its N-terminal caspase-recruiting domain, and this protein interaction was increased in cells undergoing apoptosis. Cytosolic extracts from cells overexpressing AIP were highly resistant to cytochrome c- dATP-induced activation of caspase-9 and caspase-3. Gene transfection of AIP into cell lines, including the neuronal-differentiated PC12 cells, potently suppressed apoptosis induced by various pro-apoptotic stimuli. To further investigate the functional role of AIP in primary neurons and in the brain, an adeno-associated virus (AAV) vector carrying the AIP cDNA was constructed. AAV-mediated overexpression of AIP in primary cortical- hippocampal neurons markedly reduced cell death and caspase-3 activation triggered by protein kinase C inhibition, DNA damage, or oxygen- glucose deprivation. Moreover, intracerebral infusion of the AAV vector resulted in robust AIP expression in the hippocampus and significantly promoted CA1 neuronal survival after transient global cerebral ischemia. These results suggest that molecular targeting of the Apaf-1-caspase-9 signaling pathway may be a feasible neuroprotective strategy to enhance the endogenous threshold for caspase activation and prevent neuronal loss in stroke and related disorders.
Keywords: cerebral ischemia, mitochondria, AAV, Apaf-1, apoptosome, caspase-9
Introduction
Apoptosis, or programmed cell death, plays an essential role in the homeostasis of multicellular organisms (Steller, 1995) and is implicated in the pathophysiology of various neurodegenerative disorders and stroke (Thompson, 1995; Hara et al., 1997; Lipton, 1999; Robertson et al., 2000; Graham and Chen, 2001; Lo et al., 2003). Apoptosis is a tightly controlled cell-suicide process requiring the activation of caspases (Li and Yuan, 1999; Yuan and Yankner, 2000). The apoptotic signaling cascade generally consists of two main parts: the specific apoptosis initiation pathways and the common execution pathway. Individual stimulus-specific signaling pathways are activated initially by various pro-apoptotic stimuli such as DNA damage, activation of cell membrane death receptors, or mitochondrial oxidative stress, and eventually converge into a common mechanism that activates the death-execution (or effector) caspases. An essential step in the activation of effector caspases by the intrinsic pathway appears to be the formation of a multimeric caspase-activating complex, so-called apoptosome (Li et al., 1997; Hu et al., 1999). In this cytochrome c-initiated process, a multistep reaction results in the oligomerization of apoptotic protease activating factor-1 (Apaf-1). Once the apoptosome is formed, procaspase-9 is recruited to the complex and becomes activated through autocatalysis (Srinivasula et al., 1998), which in turn activates effector caspases such as caspase-3, -6, and -7. This mechanism of apoptosis is evolutionarily conserved and may play an important role in mediating neuronal death after cerebral ischemia or traumatic brain injury (Fujimura et al., 1999; Krajewski et al., 1999; Benchoua et al., 2001; Noshita et al., 2001; Plesnila et al., 2001; Yakovlev et al., 2001; Cao et al., 2002; Sugawara et al., 2002; Tanaka et al., 2004).
Cells contain various endogenous inhibitory proteins that directly or indirectly inhibit the activation of pro-apoptotic signaling pathways. These molecules set up endogenous threshold levels for caspase activation, thus providing an important safeguard mechanism against unwanted induction of apoptosis (Budihardjo et al., 1999). The cellular levels of apoptosis inhibitor proteins may determine the differences in sensitivity to apoptotic stimuli in different cell types, and the intracellular regulation of these molecules may substantially affect the outcome of apoptosis under pathological conditions. The major endogenous apoptosis-inhibitory proteins identified so far include the anti-apoptotic members of the Bcl-2 family (Daugas et al., 2000; Kroemer and Reed, 2000), which negatively regulate apoptosis mainly at the mitochondrial levels by inhibiting the release of apoptogenic factors such as cytochrome c and apoptosis-inducing factor (AIF), and the inhibitor of apoptosis (IAP) family members, which interfere directly with the effector caspases (Deveraux et al., 1997, 1998). Several studies have demonstrated that enhanced expression of either an anti-apoptotic member of the Bcl-2 family or an IAP family member in the brain offers marked neuroprotection against ischemic neuronal death (Martinou et al., 1994; Linnik et al., 1995; Lawrence et al., 1996; Xu et al., 1999; Cao et al., 2002). Thus, molecular approaches that increase the endogenous threshold levels for caspase activation may be a potentially important strategy for minimizing neuronal loss in stroke and other neurodegenerative disorders.
In this study, we have cloned a novel apoptosis inhibitor, a member of the newly discovered group of Apaf-1-caspase-9 analogs with dominant-negative activity (Seol and Billiar, 1999; Srinivasula et al., 1999; Angelastro et al., 2001), that inhibits the Apaf-1-caspase-9 pathway by directly binding to Apaf-1. This gene product is expressed endogenously in the brain and shows potent cell death-suppressing effects in cell lines and primary neurons against various apoptosis-inducing stimuli. Finally, when overexpressed via a viral vector, this Apaf-1-interacting protein (AIP) promotes the survival of hippocampal neurons after transient global ischemia.
Materials and Methods
cDNA cloning of the Apaf-1-interacting protein. For cDNA cloning, a rat brain cDNA library was constructed as described previously (Chen et al., 1998). The Marathon cDNA adaptor was ligated to both ends of the double-strand cDNA using a T4 DNA ligase and then subjected to rapid amplification of cDNA 5′ and 3′ ends (5′- and 3′-RACE) using standard procedures (Chen et al., 2000; Cao et al., 2002).
The RACE primers were designed according to the rat sequence (Cao et al., 2002) of the caspase-recruiting domain (CARD) that is conserved between Apaf-1 and caspase-9, on the basis of the assumption that the endogenous Apaf-1-binding protein interacts with Apaf-1 via CARD and itself should contain the CARD sequence. Therefore, the following primers were used for RACE: for 5′RACE: 5′-CCA GAA CCA ATG TCC ACC GGC CTG-3′ and 5′ nested primer: 5′-CAG CTT CTG GAT CCT GCT TGG CTG CTT-3′; for 3′RACE: 5′-AAG CAG CCA AGC AGG ATC CAG AAG CTG-3′ and 3′ nested primer: 5′-CAG GCC GGT GGA CAT TGG TTC TGG-3′. The adapter-ligated double-strand cDNA served as templates for RACE. After the first round of PCR using adapter primer 1 (AP1) and 5′ or 3′ RACE primer, the resulting PCR product was used as a template for the second round of PCR using adapter primer 2 (AP2) and 5′ or 3′ nested primer. The 5′-RACE- and 3′-RACE-amplified fragments were subcloned into pGEM-T Easy Vector (Promega, Madison, WI), and the cDNAs were sequenced on both strands.
In vitro translation and production of recombinant protein. To confirm that the cloned cDNA, designated as AIP, contains the deduced open reading frame, and to study protein-protein interactions between AIP and Apaf-1, coupled transcription-translation was performed to obtain protein product for each cDNA tested using the TNT quick coupled transcription-translation systems (Promega). To perform the assay, the AIP, Apaf-1, or caspase-9 cDNA was subcloned into pcDNA3.1 or pcDNA-Flag, and then 1 μg of the NotI linearized plasmid DNA was incubated at 30°C for 2 hr in 40 μl of TNT Quick Master Mix and 20 μCi of [35S]methionine. Translated protein was detected by electrophoresis and autoradiography.
AIP recombinant protein was generated using standard methods (Chen et al., 2000; Cao et al., 2001). In brief, the AIP cDNA was fused into the glutathione S-transferase (GST) gene in PGEX-2T vector (Amersham Biosciences, Arlington Heights, IL). The GST-AIP fusion protein was expressed in Escherichia coli BL21 cells and absorbed to a glutathione-Sepharose 4B column. The fusion proteins were then cleaved by thrombin for 16 hr to remove GST and further purified.
RNase protection assay. The RNase protection assay (RPA) was performed to confirm the endogenous expression of AIP mRNA in rat brain. Two cRNA probes were prepared for the assay: a cRNA probe contained the sequence that was unique to AIP (containing part of the unique 3′ untranslated region); the other cRNA probe contained the sequence that was shared between caspase-9 and AIP. For the preparation of the AIP cRNA probe, a DNA fragment of 351 bp was amplified from the AIP cDNA using PCR (sense primer: GGA ATT CGA CGT GGA CTC TTC CGA TCA GTC TAT AG; antisense primer: CAA GCT TCA CAT TCT GCT GCC TTG ACA TCT TAG) and then subcloned into the HindIII and EcoR I cloning site of the pcDNA3.1 plasmid. The plasmid was linearized using EcoR I and subsequently used as a template to prepare the antisense cRNA probe using the following procedures. The linearized pcDNA3.1 plasmid (0.5 μg) containing the AIP fragment was incubated for 60 min at 37°C in the transcription buffer containing 1 μl of 200 mm DTT, 2 μl of nucleotide triphosphate mix (ATP, UTP, and GTP at 4 mm each), 1 μl of placental ribonuclease inhibitor, 10 μl of α- 32P-CTP (10 mCi/ml), and 10 U of T7 RNA polymerase. The DNA template was then digested with 10 U of RNase-free DNase I for 15 min at 37°C, extracted with phenol/chloroform, and then precipitated with ammonium acetate and ethanol. The specific activity of the probe was measured using a liquid scintillation counter. For the preparation of the caspase-9 cRNA probe, a DNA fragment of 534 bp was amplified using the following primers: sense GGA ATT CAT GGA GGA GGC TGA CCG GCA ACT CCT; antisense CAA GCT TGA CAT CAT GAG CTC TGC CAG AAC CAA.
To perform RPA, total RNA (60 μg) prepared from rat brain was precipitated with ethanol and lyophilized and then resuspended in 100 μl of hybridization buffer containing 2 × 106 cpm of the RNA probes described above. The reaction mixture containing tRNA (instead of the brain total RNA) was used as negative control. The reaction was incubated for 5 min at 85°C to denature RNA and rapidly transferred to 45°C for 12 hr. The reaction was then incubated with 400 μl of ribonuclease digestion buffer containing 4 μg/ml ribonuclease T1 for 30 min at 30°C, extracted with phenol/chloroform, and precipitated with ethanol. The pellet was dissolved in 10 μl of RNA loading buffer, denatured at 85°C for 5 min, and analyzed on a 12% denaturing polyacrylamide/8 m urea gel.
In situ hybridization. To determine whether AIP mRNA expression is altered in the brain after ischemia, in situ hybridization was performed using an AIP-specific oligonucleotide probe derived from the 3′ untranslated region of the AIP cDNA (antisense 5′-CAT CTC CTA TAG ACT GAT CGG AAG AGT CCA CGT CTC TC-3′). Rats were anesthetized and decapitated at the indicated time points after ischemia, and coronal sections (15 μm thick) were cut on a cryostat at -20°C. The AIP oligonucleotide was 35S labeled as described previously (Chen et al., 1998). The sections were hybridized with the labeled probe (1 × 107 cpm/ml) in hybridization mixture for 18 hr at 55°C. After the washing procedures, the slides were dehydrated, air dried, and exposed to Kodak film for 3 weeks.
Transient cDNA transfection in human 293 cells. The AIP cDNA or cDNA encoding rat caspase-3 (Chen et al., 1998), caspase-9, or Apaf-1 (Cao et al., 2002) was amplified by PCR and then subcloned into the multiple cloning site of the PcDNA3.1 expression vector containing the cytomegalovirus (CMV) promoter (Invitrogen, Gaithersburg, MD). For transient transfection, the green fluorescent protein (GFP)-expressing vector, together with the empty vector or the vector containing the gene of interest, were cotransfected into human 293 cells using LipofectAMINE (Invitrogen). At the indicated time after transfection, the percentages of green (GFP-expressing) apoptotic cells of the total number of green cells were determined under each condition as described previously (Seol and Billiar, 1999). Under selective experimental conditions, cells infected with the genes of interest were subjected to protein extraction and subsequently to Western blot analysis for caspase-3 and caspase-9 activation.
Stable cDNA transfection in neuronal PC12 cells. Stable cell lines overexpressing AIP were produced in PC12 cells using the method described previously (Chen et al., 2000). Briefly, the PcDNA3.1 expression vector containing AIP or the empty vector was transfected into PC12 cells with the assistance of LipofectAMINE reagent. Forty-eight hours after the transfection, the cells were passaged at a ratio of 1:3, and on the next day, Geneticin (G418) (Invitrogen) was added at a concentration of 450 ng/ml. Cells were kept on the G418 for 1 month to ensure selection of stable cell lines.
To determine the cell death-regulatory effect of AIP, gene transfected or nontransfected PC12 cells were subjected to NGF-induced neuronal differentiation for 6 d (Chen et al., 2000), and then the cultures were exposed to 2 hr of transient hypoxia (Cao et al., 2001), the PKC inhibitor staurosporin (STS) (1 μm) (Chen et al., 2000), or the mitochondrial toxin rotenone (1.5 μm) (Pei et al., 2003). At the indicated time after insults, cell death was quantified using the LIVE/DEAD viability quantitation kit (Molecular Probes, Eugene, OR). Apoptotic DNA fragmentation was analyzed using DNA gel electrophoresis (Chen et al., 2000).
In vitro cell-free apoptosis assay. The biochemical process of Apaf-1-dependent activation of caspase-9 and caspase-3 can be reconstituted in vitro by adding cytochrome c- dATP into cytosolic protein extracts (Liu et al., 1996). This assay was performed in the present study to examine the potential inhibitory effects of AIP on the induction of the apoptotic program in cell-free extracts. Cytosolic extracts were prepared from nontransfected PC12 cells and cells stably expressing AIP, respectively, and 100 μg of the protein was incubated with cytochrome c (100 μg/ml) and dATP (1 mm) in 100 μl of the reaction buffer (25 mm HEPES, 10% sucrose, 0.1% CHAPS, 5 mm dithiothreitol, and 5 mm EDTA) containing 0.5 mm Ac-LEHD-pNA (for caspase-9-like activity) or Ac-DEVD-pNA (for caspase-3-like activity) at 30°C. The enzyme-catalyzed release of p-nitroanilide was measured at 405 nm every 15 min for 150 min using a microtiter plate reader (Molecular Devices).
Protein binding assay. Protein-protein binding between Apaf-1 and AIP or caspase-9 was studied using in vitro translation products (Seol and Billiar, 1999). In brief, 1 ml of S35-Apaf-1 was incubated with 0.5-1.0 ml of Flag-AIP or Flag-caspase-9 (in the absence or presence of in vitro-translated AIP) at 30°C for 2 hr in the protein binding buffer containing 150 mm NaCl, 5 mm MgCl2, 1 mm DTT, and 10 μg/ml each of pepstatin, leupeptin, and PMSF. The mixture was then immunoprecipitated with 2 μg of anti-Flag polyclonal antibody (Sigma, St. Louis, MO), and the immunoprecipitates were then subjected to autoradiography for the detection of S35-Apaf-1.
Immunoprecipitation and Western blot analysis. To determine whether the endogenous AIP interacts with Apaf-1, cytosolic protein was prepared from the PC12 cell line stably expressing the Flag-AIP, and immunoprecipitation was performed using standard procedures (Cao et al., 2001). This was done using the rabbit anti-Apaf-1 antibody (Chemicon, Temecula, CA), and the resulting immunoprecipitates were then analyzed by immunoblotting with anti-Flag (PharMingen, San Diego, CA) and anti-Apaf-1 antibodies, respectively. Immunoblotting was performed using the standard method (Cao et al., 2001).
Production of an adeno-associated virus vector. To further study the cell death-regulatory role of AIP in primary neurons and in the in vivo model of cerebral ischemia, an adeno-associated virus (AAV) vector carrying the AIP cDNA was constructed. The AAV expressing vector plasmid, which contains an SV40 splicing site and a polyadenylation signal, was flanked by the AAV inverted terminal repeats. The 534 bp AIP cDNA with an hemagglutinin (HA) tag was isolated by EcoRV and NotI from its parental plasmid and then inserted into the expressing vector plasmid using T4 DNA ligase. The expression of the AIP transgene was under the transcriptional control of the CMV promoter. A control AAV vector was also constructed that contained a 258 bp truncated AIP cDNA lacking the CARD domain sequence.
Large-scale production of the AAV vector was performed using the adenovirus-free triple-plasmid cotransfection method described previously (Xiao et al., 1998). AAV vectors were purified using an FPLC affinity chromatography system in conjunction with the HiTrap Heparin columns (Amersham Biosciences). The titers of the virus particles were determined by the DNA dot-blot method. The FPLC-purified AAV-AIP has a concentration of 1 × 1013 virus particles per milliliter.
Primary neuronal culture and induction of apoptosis. Primary cultures of hippocampal-cortical neurons were prepared from 17-d-old Sprague Dawley rat embryos as described previously (Cao et al., 2001). Experiments were conducted at 14 d in vitro (DIV), when cultures consisted primarily of neurons (∼95%) as determined using cell phenotype-specific immunocytochemistry (Cao et al., 2001). The cultures were infected with the AAV-AIP vector or the control vector at the particle/cell ratio of 1 × 105/1 for 6 hr in serum-free media, and then incubated in vector-free normal media for 3 d. Expression of HA-tagged AIP in neurons was verified by Western blotting using antibodies against the HA tag and the C-terminal sequence of AIP, respectively.
Apoptosis was induced in cultured neurons by incubating the cells at the indicated concentrations with staurosporin or bleomycin. Apoptosis was evaluated 24 hr after the insults using Hoechst 33258 nuclear staining. The percentage of cells showing apoptotic changes (chromatin condensation or fragmentation, or both) was quantified by counting at least 3000 cells under each experimental condition (three randomly selected fields per well, four to six wells per condition per experiment, and three independent experiments using different embryos).
To determine the role of AIP in regulating neuronal death induced by ischemia-like insults, the cultures infected with AAV-AIP or the control vector were subjected to oxygen- glucose deprivation (OGD) for 90 min, which in the presence of a low concentration of the NMDA receptor antagonist MK801 (1 nm) induces a significant portion of caspase-dependent apoptosis (Cao et al., 2001). At 24 hr after OGD, cell death was quantitatively measured using the LIVE/DEAD viability/cytotoxicity kit (Molecular Probes). In addition, the effects of AAV-AIP on OGD-induced caspase activation were studied by immunoblotting detection of the cleavage of caspase-9 and caspase-3 (Cao et al., 2001).
Intracerebral infusion of AAV and transient cerebral ischemia. AAV vectors were infused into the CA1 sector in the dorsal hippocampus two injection spots; coordination: anteroposterior (AP) -3.5 mm and lateral (L) 2.0/3.5 mm from the bregma,) and the caudate-putamen (coordination: AP -1.0 mm and L 4.0 mm from the bregma), respectively, as described previously (Xiao et al., 1997). Briefly, male Sprague Dawley rats weighing 300-325 gm were anesthetized using isofluorane. The AAV vectors (AAV-AIP or control vector) dissolved in physiologic saline (2 × 1010 DNase-resistant particles per microliter) were infused at the rate of 0.5 μl/min over 10 min via a glass micropipette that was connected to an air pressure injection system (Harvard Apparatus, Holliston, MA). The animals were allowed to recover for 3 d to 8 weeks to enable sufficient gene expression. Expression of AIP in the brain was examined by immunohistochemistry and Western blots using the anti-HA antibody.
At 5 d after AAV infusion, global ischemia was induced for 12 min in isofluorane-anesthetized rats using the four-vessel occlusion method (Chen et al., 1998). In the present study, three groups of rats (nontreated group, AAV-AIP infusion group, and control AAV infusion group), each consisting of nine rats, were studied for cell death assessments. Rectal and brain temperature and physiological variables were measured and controlled at normal ranges throughout the experiments (Chen et al., 1998). EEG was measured in all animals to ensure isoelectricity after the induction of ischemia. Rats were killed 4 d after ischemia, and the brains were processed for histological assessments and immunological staining.
For the hippocampal cell death assessments, series coronal sections (20 μm thick) were cut throughout the dorsal hippocampal formation at the coronal levels between -2.5 and -4.5 mm from the bregma. For every 10 adjacent sections, the first 2 sections were stained for cresyl violet. The second pair of sections were immunostained using the anti-HA antibody (for the detection of HA-tagged AIP) and counterstained with cresyl violet. The third pair of sections were immunostained with the anti-HA antibody and counterstained with terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL). The forth pair of sections were immunostained with the anti-HA antibody and counterstained with the antibody against active caspase-3 (Cell Signaling) as described previously (Cao et al., 2002). Cell survival in the CA1 sector was quantified by an investigator who was blinded to the experimental conditions in cresyl violet-stained sections using the criteria described previously (Chen et al., 1998).
Statistical analysis. Results are reported as mean values ± SE. The significance of difference between means was assessed by Student's t test (single comparisons) or by ANOVA and post hoc Fisher's PLSD tests, with p < 0.05 considered statistically significant.
Results
Cloning of a novel rat cDNA
In a search for rat brain Apaf-1-interacting proteins, we performed 5′- and 3′-RACE based on the CARD sequence that is essential for Apaf-1 to interact with caspase-9 and endogenous binding proteins (Hofmann et al., 1997). On the basis of sequences of the RACE products, three cDNAs of 2066 bp (clone 1), 2391 bp (clone 2), and 1199 bp (clone 3) were subsequently obtained by PCR from the rat brain cDNA library. Although clones 1 and 2 share an identical open reading frame (455 amino acids) encoding the rat homolog of full-length caspase-9 (Cao et al., 2002), clone 3 contains an open reading frame of 531 bp that encodes 177 amino acids (see supplemental Fig. 1, available at www.jneurosci.org). Sequence alignment reveals that clone 3 is a novel gene product, and its deduced amino acid sequence contains the CARD domain at its N terminus (91 amino acids), which is followed by an 86 amino acid peptide that shares homology with the larger subunit of rat caspase-9 (Fig. 1). Moreover, clone 3 contains a 3′-UTR (616 base pairs) that shows no significant homology to any other reported sequences.
Figure 1.

Cloning of a novel Apaf-1 interacting protein. Molecular structure of rat AIP (GenBank accession number AF293333) and various caspase-9 short variants are shown schematically (Seol and Billiar, 1999; Srinivasula et al., 1999; Angelastro et al., 2001). Compared with the full-length rat caspase-9 (GenBank accession numbers AF262319 and AF286006), AIP lacks both large and small subunits of caspase-9, which is distinctive from the two previously reported caspase-9 short variants with dominant-negative phenotypes, including rat caspase-9-CTD (GenBank accession number AY008275) and human caspase-9 (GenBank accession number AF110376).
Using the clone 3 cDNA as a template, the in vitro transcription-translation assay produced a protein at ∼19 kDa, consistent with the predicted sizes from the deduced amino acid sequence. Subsequently, a recombinant protein was made from this cDNA. Both this recombinant protein and the in vitro translation product were immunoreacted with the antibody against a sequence conserved between the rat caspase-9 larger subunit and the C-terminal of clone 3 (data not shown). These results indicate that we have obtained a novel caspase-9 short variant, which appears to be a splice variant of the caspase-9 gene and is structurally different from the caspase-9 short variants reported previously (Seol and Billiar, 1999; Srinivasula et al., 1999; Angelastro et al., 2001). Because we found that the protein encoded by the clone 3 cDNA functions by directly interacting with Apaf-1 (see results below), we designated this novel gene product as AIP.
Endogenous expression of AIP
RT-PCR was performed to determine the endogenous levels of AIP mRNA expression in neuronal tissues. Because caspase-9 is a homolog gene of AIP, we directly compared their expression levels. The primers were designed as shown in Figure 2a, which allowed the direct comparison of the expression between AIP and caspase-9 mRNA. AIP and caspase-9 mRNAs were readily detectable in various brain regions (Fig. 2a); however, in all regions examined, the levels of AIP mRNA were much lower (<10%) than those of caspase-9 mRNA.
Figure 2.
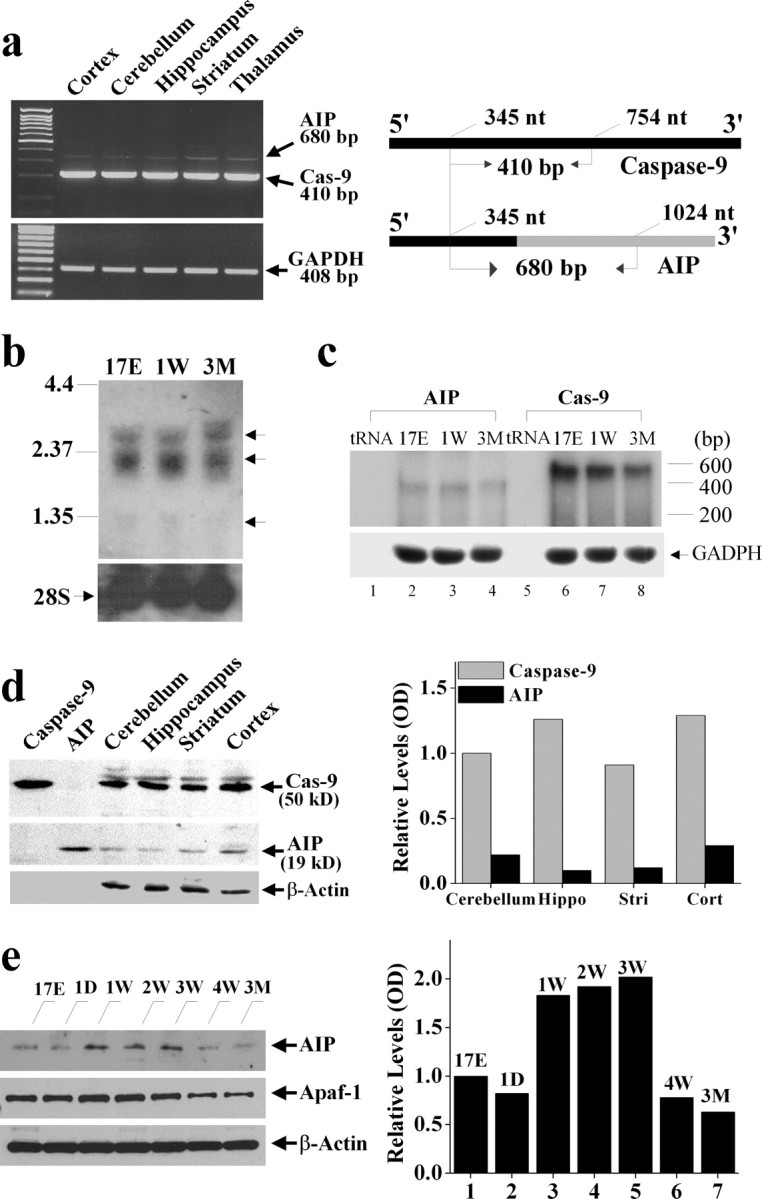
Characterization of endogenous AIP expression in the brain. a, RT-PCR was performed to detect AIP and caspase-9 mRNA in various brain regions in adult rats. The primers were designed to allow the detection of both species in the same reaction. The schematic diagram in the right panel shows that the sense primer (gtt aca ccc cta gac cac ctg gtg c) used in the reaction was the same between AIP and caspase-9, whereas the antisense primers were unique to either species (for AIP: cgc atg cat gcg cac agc aca tac ac; for caspase-9: agc cag cag aag cgg tgc tga agc). The black bars represent the open reading frames, and the gray bar indicates the 3′-untranslated sequence that is unique to AIP. b, Northern blotting of total RNA isolated from rat brain of different ages (17E, embryonic day 17; 1W, postnatal 1 week; 3M, postnatal 3 months) with a cDNA probe recognizing both caspase-9 and AIP. Note that at least three bands are detected (arrows). c, RNase protection assay performed using specific probes confirmed the expression of AIP mRNA in the rat brain of different ages. d, Western blot analysis of AIP protein expression in various brain regions of adult rats. The in vitro translation products of AIP or caspase-9 cDNA served as positive controls. The blot is representative of two independent experiments with similar results. e, Western blot analysis of AIP protein expression in the cerebellum during brain development. An equal amount of cerebellum protein extracts (60 μg) obtained from different ages was immunoreacted with antibodies against AIP, Apaf-1, or β-actin. 17E, Embryonic day 17; 1D, postnatal 1 d; 1-4W, postnatal 1-4 week; 3M, postnatal 3 months. The graphs in b and c illustrate the relative levels of AIP expression, determined from two independent experiments.
To further detect the endogenous expression of AIP mRNA in the brain, Northern blot analysis was performed using the 32P-labeled AIP cDNA probe. As shown (Fig. 2b), two major bands were detected in the size range of 2-3 kb that were consistent with the sizes of mRNA encoding the full-length caspase-9 in the rat brain (Cao et al., 2002). A third band, however, was faintly detectable at ∼1.2-1.3 kb (Fig. 2b). We suspected that this minor species might represent the signals from the AIP mRNA. To address this, we performed the highly sensitive and specific RPA using brain samples from three different ages. The results from RPA confirmed the endogenous expression of AIP mRNA in the rat brain (Fig. 2c). Furthermore, consistent with the RT-PCR data, RPA demonstrated a low ratio of AIP mRNA versus caspase-9 mRNA expression in the brain.
Using Western blot analysis, the levels of AIP protein expression were examined in rat brains. In each experiment, we used in vitro translated AIP protein as the positive control. The 19 kDa AIP protein was detected in various brain regions in adult rats, although the levels were much lower than those of caspase-9 (Fig. 2d) or Apaf-1 (Fig. 2e). Because both caspase-9 and Apaf-1 are required for the induction of apoptosis in neuronal progenitor cells during brain development (Kuida et al., 1998) and both gene products show marked age-dependent regulation of expression in the brain (Yakovlev et al., 2001; Cao et al., 2002), we characterized the temporal profiles of AIP protein expression in the rat cerebellum of various ages (Fig. 2e). AIP was expressed at moderate levels in embryonic cerebellum and at PD1, but the levels were increased at postnatal 1-3 weeks. The expression of AIP subsided to very low levels at postnatal 4 weeks and in the adults, which was similar to the downregulation pattern of Apaf-1 protein at the same ages.
AIP expression is upregulated in resistant neurons after cerebral ischemia
To investigate the potential role of AIP in ischemic injury, we examined the expression of AIP mRNA in normal and ischemic brains, focusing on the hippocampus, where neurons are particularly vulnerable to transient global ischemia. Using in situ hybridization with the AIP-specific oligonucleotide probe, we detected increased AIP mRNA expression in ischemic brains (Fig. 3a) in comparison with the extremely low levels of AIP mRNA in the sham controls. The induced AIP mRNA was localized in the dentate gyrus (DG) granular cell layers, a region that is highly resistant to cell death in this injury model, but not in the highly vulnerable CA1 sector. Optical density measurements of the autoradiographic signals showed that AIP mRNA began to increase in the DG at 8 hr after ischemia (1.7-fold) and reached peaks at 24 (3.6-fold) and 72 (2.8-fold) hours after ischemia (Fig. 3b). In contrast, Apaf-1 mRNA, which is highly constitutively expressed in the entire hippocampal formation in the normal brain, showed no detectable changes after ischemia.
Figure 3.
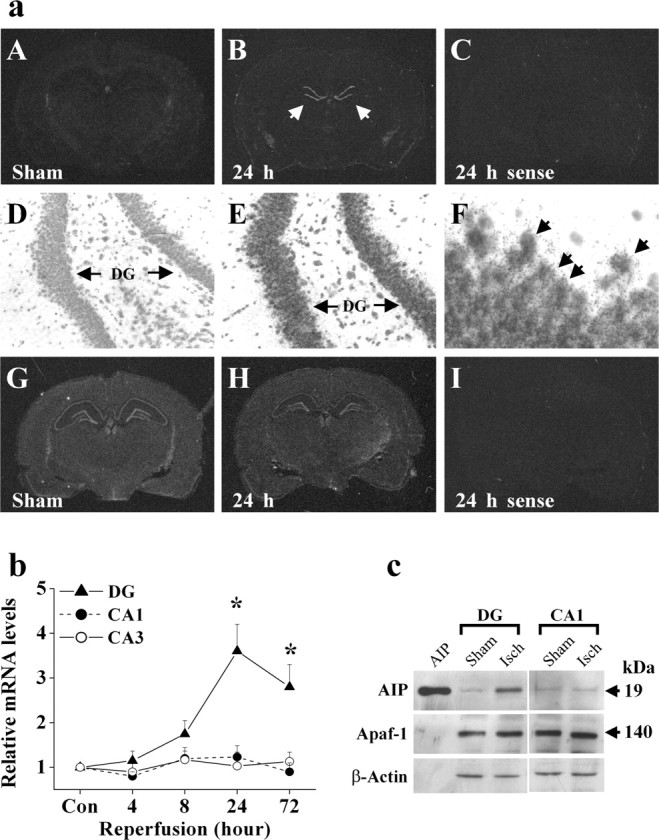
Upregulation of AIF expression in the hippocampus after ischemia. a, In situ hybridization analysis of AIP (A-C) and Apaf-1 (G-I) mRNA expression after ischemia or sham operation. Representative autoradiographs show increased AIP mRNA in the DG sector 24 hr after ischemia (B, arrows), whereas Apaf-1 mRNA expression was not changed; sections from the same brains presented in B and H hybridized with the sense probes for AIP and Apaf-1, respectively, show weak homogenous signals (C, I). Emulsion coating and cresyl violet counterstaining of the section presented in B confirmed the increased AIP mRNA in the DG granular cell layers (E, F, arrows). b, The graph illustrates the relative AIP mRNA changes in the hippocampal CA1, CA3, and DG sectors 4, 8, 24, and 72 hr after ischemia versus sham controls (n = 4 per time point). Data are reported as mean ± SEM and represent fold changes in ischemic brains versus sham controls; *p < 0.05 versus sham controls. c, Representative Western blots show increased AIP protein expression in the DG sector, but not in CA1, at 24 hr after ischemia. Incontrast, the expression of Apaf-1 was not changed in either sector.
To determine whether the expression of AIP protein is increased after ischemia, Western blot analysis was performed using cell extracts prepared from the DG-CA3 and CA1 sectors, respectively, at 24 hr after ischemia or sham operation. The results confirmed that AIP protein was increased in the DG-CA3 but not in the CA1 sector after ischemia (Fig. 3c).
AIP is an anti-apoptotic molecule
Because AIP is expressed endogenously, its potential cell-death regulatory role was investigated. Initially, transient cDNA transfection was performed in human 293 cells. Although transfection of AIP alone was not cytotoxic, even with an extended incubation for 4 d, transfection of either caspase-9 or Apaf-1 markedly induced apoptosis in cultures within 24 hr. When cotransfected, however, AIP inhibited caspase-3 and caspase-9 activation and significantly decreased apoptosis induced by either caspase-9 or Apaf-1 (Fig. 4a,b). The apoptosis-suppressing effect of AIP appeared to be compatible with that of the caspase-3/7 inhibitor z-DEVD.fmk (optimally at 100 μm). Furthermore, transfection of AIP had no effect on apoptosis induced by caspase-3 cDNA transfection (Fig. 4a), which was consistent with the speculation that AIP might function at the level upstream of caspase-3 activation.
Figure 4.
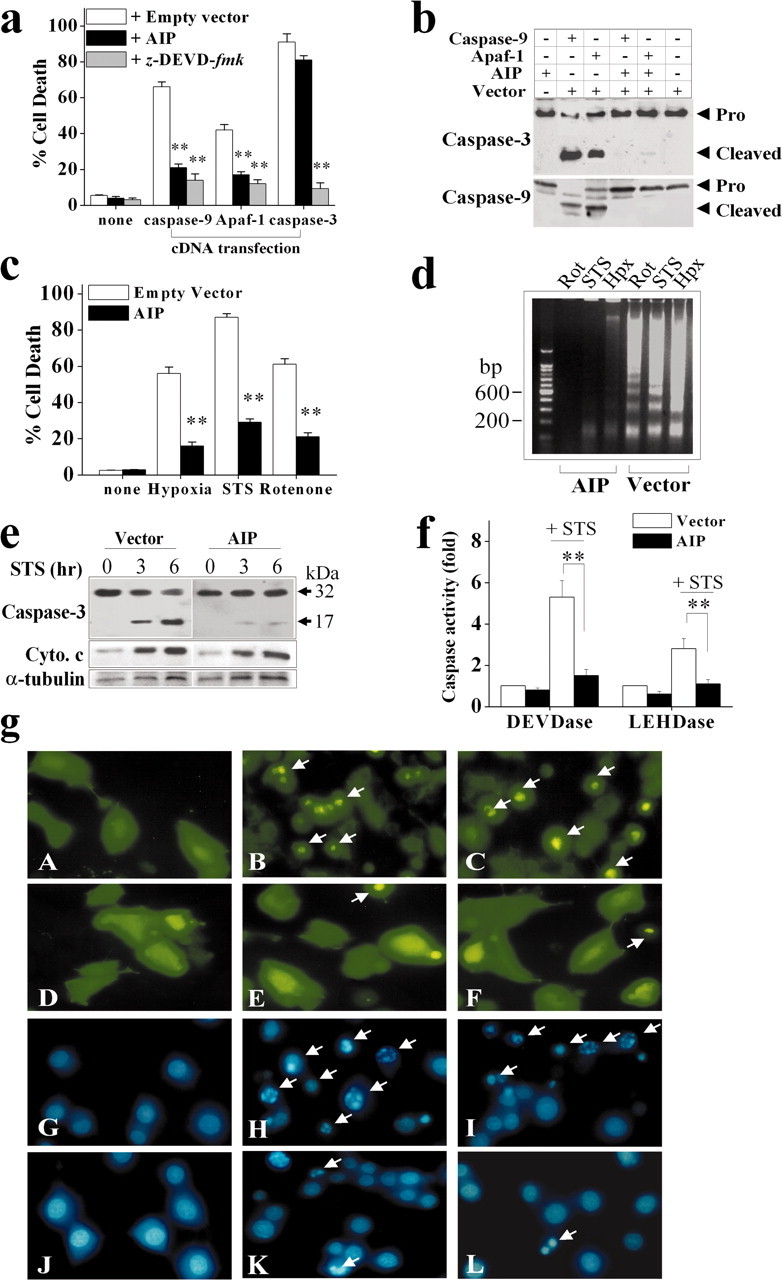
Characterization of the anti-apoptotic effect of AIP in cell lines. a, Transfection of AIP cDNA inhibited apoptosis in human 293 cells induced by transient cDNA transfection of rat caspase-9 or Apaf-1. Cell death was analyzed at 24 hr after transfection. The caspase-3 and caspase-7 inhibitor z-DEVD. fmk, which served as a positive control for caspase inhibition, was applied at 100 μm spontaneously with gene transfection. Data are mean ± SE; n = 12 per condition from two independent experiments. **p < 0.01 versus empty vector controls. b, Western blotting for caspase-3 and caspase-9 was performed 16 hr after gene transfection in 293 cells. Transfection of rat Apaf-1 or caspase-9 cDNA resulted in marked activation of caspase-3 and caspase-9. These effects were blocked by cotransfection with AIP cDNA. c, Anti-apoptotic effect of AIP in PC12 cells. Stable transfection of AIP but not the empty vector significantly decreased cell death in neuronal-differentiated PC12 cells induced by hypoxia (2 hr), STS (1 μm), or rotenone (1.5 μm). Cell death was analyzed at 16 hr after the insults. Data are mean ± SE; n = 12 per condition from two independent experiments. **p < 0.01 versus empty vector controls. d, Transfection of AIP inhibited apoptotic DNA fragmentation in PC12 cells at 16 hr after hypoxia, STS, or rotenone exposure. e, Western blot analysis of caspase-3 and cytochrome c in cytosolic extracts prepared from PC12 cells at 3 or 6 hr after STS treatment (1.5 μm). f, Transfection of AIP inhibited STS-induced (6 hr) caspase-3-like (DEVDase) and caspase-9-like (LEHDase) activities in PC12 cells. g, Morphological changes of 293 cells (A-F) and PC12 cells (G-L) in response to apoptotic challenges in the absence or presence of AIP overexpression. Cells transfected with the GFP vector (A) or GFP-AIP vector (D) showed normal morphology. Cultures transfected with either GFP-caspase-9 (B) or GFP-Apaf-1 (C) for 24 hr showed robust cell death (B, C, arrows), which was attenuated by AIP overexpression (E, F). G and J show nuclei of PC12 cells stably transfected with the empty vector (G) or AIP (J). Nuclear apoptosis (arrows) was induced in empty vector-transfected cells at 16 hr after either STS (H) or rotenone (I). In cells stably transfected with AIP, STS-induced (K) or rotenone-induced (L) nuclear apoptosis was markedly decreased.
To determine the anti-apoptotic effect of AIP in neural cells, stable transfection cell lines overexpressing AIP were established in neuronal-differentiated PC12 cells. Stable transfection of AIP cDNA resulted in 3.5-6-fold increases in the expression of AIP, as determined using Western blotting (data not shown). To determine the impact of AIP gene expression on the susceptibility of PC12 cells to apoptosis, cells were subjected to various death-inducing stimuli. AIP markedly inhibited cell death and apoptotic DNA fragmentation induced by the protein kinase C inhibitor STS, the mitochondrial complex I inhibitor rotenone, or hypoxia (Fig. 4c,d). These results thus establish that AIP is a potent anti-apoptotic molecule. As determined by Western blots (Fig. 4e) and caspase activity assays (Fig. 4f), AIP inhibited STS-induced activation of caspase-3 and caspase-9 but had no inhibitory effect on cytochrome c release, consistent with our prediction that AIP might function at the levels downstream of mitochondrial damage.
Figure 4g illustrates the morphological changes of human 293 cells and PC12 cells in response to apoptotic challenges in the absence or presence of AIP overexpression.
AIP inhibits the Apaf-1-caspase-9 pathway by directly interacting with Apaf-1
Because AIP inhibited caspase-9- or Apaf-1-transfection-induced apoptosis but had no effect on caspase-3-transfection-induced apoptosis (Fig. 4a), we speculated that AIP might inhibit apoptosis by intervening with the Apaf-1-caspase-9 complex-dependent apoptotic cascade, which is upstream of caspase-3 activation. To test this hypothesis, we performed the cell-free apoptosis assays as described by Liu et al. (1996), using cytosolic protein extracts derived from empty vector- and AIP-transfected PC12 cells, respectively. The additions of purified cytochrome c and dATP into the extracts from normal cells (empty vector-transfected) triggered marked increases in caspase-9-like (LEHD-cleavage) and caspase-3/7-like (DEVD-cleavage) activities, whereas this effect was greatly decreased in AIP-transfected cells (Fig. 5a,b). Similarly, the cytochrome c- dATP-triggered caspase-9-like and caspase-3/7-like activities in cytosolic extracts prepared from normal cells were inhibited by the addition of purified AIP recombinant protein in a dose-dependent manner (Fig. 5c,d). In contrast, the DEVD-cleavage activity from the purified recombinant active caspase-3 (PharMingen) or caspase-9 (MBL International, Woburn, MA) was not inhibited by either the cytosolic extracts from AIP-transfected cells or the purified AIP recombinant protein (data not shown). These results suggest that AIP might inhibit Apaf-1-dependent proteolytic processing of caspase-9 and caspase-3 but not the activated caspases per se. This speculation was confirmed by the results from another cell-free assay using in vitro translated 35S-labeled caspase-9 as the reaction substrate, in which the recombinant AIP protein dose-dependently inhibited the cytochrome c-dATP-initiated cleavage of 35S-caspase-9 into its active forms (Fig. 5e).
Figure 5.

AIP directly interacts with Apaf-1 and inhibits Apaf-1-dependent caspase-9 and caspase-3 activation. a, b, Cytochrome c-dATP-triggered (Apaf-1 dependent) caspase-9- or caspase-3-like activity was abolished in PC12 cells stably overexpressing AIP. The in vitro cell-free assay was performed using cytosolic proteins from empty vector- or AIP-transfected cells, and the reaction was initiated by adding purified cytochrome c and dATP. Caspase activities were measured using the colorimetric substrates Ac-LEHD-pNA and Ac-DEVD-pNA, respectively. Data are mean ± SE from three experiments. c, d, Cytochrome c- dATP-triggered caspase-9- or caspase-3-like activity in cytosolic extracts was abolished by exogenous recombinant AIP protein. The cell-free assay was performed using cytosolic proteins from nontransfected PC12 cells, which showed that exogenous AIP protein inhibited caspase activities in a dose-dependent manner. e, In the assays alternative to c and d, caspase-9 activation was determined by autoradiography using the in vitro translated 35S-labeled caspase-9 as the cleavage substrate for Apaf-1 apoptosome. Note that the exogenous AIP protein dose-dependently inhibited caspase-9 cleavage. f, Autoradiograph shows direct protein-protein interaction between AIP and Apaf-1. The protein-binding assay was performed using in vitro translated proteins (the Flag-AIP and 35S-Apaf-1). The reaction products were immunoprecipitated using the anti-Flag antibody, electrophoresed, and then autoradiographed. g, Autoradiograph for 35S-Apaf-1 shows that Flag-caspase-9 can directly bind to Apaf-1, and that AIP, when added in excessive amounts, inhibited Apaf-1-Flag-caspase-9 binding. h, Increased interaction between endogenously expressed AIP and Apaf-1 in apoptotic PC12 cells. Cytosolic proteins were extracted from cells stably overexpressing Flag-AIP or the empty vector, immunoprecipitated with the anti-Apaf-1 antibody, and then immunoblotted with antibodies against Flag and Apaf-1, respectively. Note that exposure of the cells to STS (1 μm) for 2 hr increased the contents of Flag-AIP in the immunoprecipitates, indicating the increased formation of the Apaf-1-Flag-AIP complex.
The presence of the CARD domain in the AIP protein strongly suggests that AIP may directly interact with Apaf-1 via this domain. As the first effort to address this issue, we performed the protein-binding assay using in vitro translated proteins. As shown in Figure 5f, the Flag-tagged AIP was able to bind to 35S-labeled Apaf-1 in the cell-free reaction system, similar to Flag-caspase-9 (Fig. 5g). Truncated AIP with the deletion of the CARD domain completely lost its ability to form dimerization with Apaf-1 (data not shown), consistent with the notion that the CARD domain is essential for such a reaction. Furthermore, when AIP was added in excessive amounts (ratio of AIP/Flag-caspase-9 = 2-4:1) in the reaction mixture, it nearly completely blocked the binding between Apaf-1 and Flag-caspase-9 (Fig. 5g). These results strongly suggest that AIP functions by competing with caspase-9 for binding to Apaf-1, thus preventing the formation of a functional apoptosome essential for the proteolytic activation of caspase-9.
To determine whether endogenous Apaf-1 and AIP form a dimerization complex under a normal or apoptotic condition, the PC12 stable transfection cell line overexpressing Flag-AIP was treated with 1 μm STS for 2 hr. The cytosolic protein extracts were subjected to immunoprecipitation using the antibody against Apaf-1, followed by immunoblotting with the anti-Flag antibody. Coimmunoprecipitation of Apaf-1 and AIP was detectable only in STS-treated cells (Fig. 5h). These results suggest that the interaction between Apaf-1 and AIP is accelerated by the active status of Apaf-1 under apoptotic conditions. This mechanism is consistent with the observations by Srinivasula et al. (1998) that in Apaf-1-dependent apoptosis, cytochrome c-dATP induces the conformational changes of Apaf-1 that allow its CARD domain to be exposed for protein-protein interactions (Srinivasula et al., 1998). Therefore, it is likely that AIP may not be able to bind to Apaf-1 (such as under nonstressed conditions) unless Apaf-1 is activated and its CARD becomes accessible.
The cell death-suppressing effect of AIP in primary neurons
The ultimate goal of this study was to explore the role of AIP as a potential neuroprotectant against the neuronal apoptosis that occurs in cerebral ischemia. To achieve highly efficient gene transfection of AIP in primary cultures of neurons and in the rat brain in vivo, we constructed an AAV vector (Fig. 6a). The successful construction of an AIP expression vector was first verified in human 293 cells, in which infection with either AIP expression plasmid (before AAV packaging) or AAV-AIP (after packaging) resulted in overexpression of AIP that was recognized by antibodies against AIP and HA, respectively (Fig. 6b). The infection efficiency of AAV-AIP was then examined in primary cultures of hippocampal-cortical neurons. Incubation of AAV-AIP in neuronal cultures resulted in robust expression of AIP in a time-dependent manner, reaching a transfection efficiency of 81.7 ± 4.6% (mean ± SE of total cells; n = 9)by 3 d of incubation; the transfection efficiency of AAV-AIP was compatible with the AAV vectors expressing the Bcl-2 gene or the reporter gene β-gal (data not shown). Double-label immunofluorescent staining for the HA tag and mitogen-associated protein 2 (MAP-2) revealed that the expression of AIP exhibited mainly a cytosolic localization (Fig. 6c).
Figure 6.
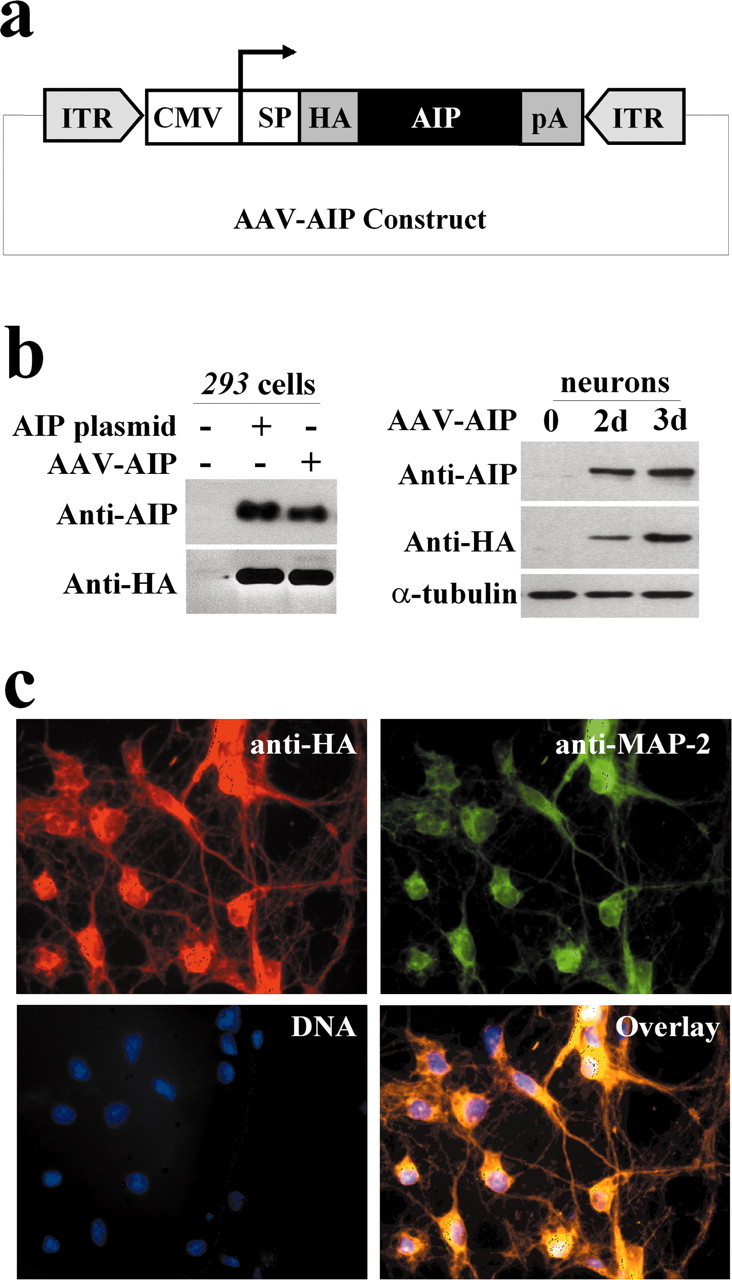
Construction of an AAV vector carrying the AIP expression cassette. a, The construct contains the CMV promoter and an HA tag, which allows distinction of AIP from endogenous AIP or caspase-9. SP and pA are the SV40 mRNA splicing site and polyadenylation signal, respectively; ITR is the inverted terminal repeats of AAV. b, Verification of the AIP expression plasmid (before AAV packaging) and AAV-AIP (after packaging) in human 293 cells and rat primary neurons. Left panel, Human 293 cells were transfected for 24 hr with the AIP expression plasmid or AAV-AIP, and cell extracts were subjected to Western blotting with antibodies against HA and AIP (caspase-9), respectively. Right panel, Western blots demonstrate that AAV-AIP infection for 3 d induced robust AIP overexpression in primary hippocampal-cortical neurons. c, Double-label immunofluorescent staining (red, HA; green, MAP-2) shows AIP overexpression in hippocampal-cortical neurons 3 d after AAV-AIP infection. AIP expression is mainly cytosolic.
Primary neurons infected with AAV-AIP showed highly increased resistance to apoptotic induction. STS-induced or the DNA-damaging agent bleomycin-induced neuronal apoptosis was significantly and markedly attenuated by AAV-AIP infection (Fig. 7a-c). These anti-apoptotic effects were not observed in cultures transfected with the truncated AIP lacking the CARD domain, emphasizing the importance of CARD as the primary functional domain of AIP. Therefore, in subsequent studies the latter vector was used as a control AAV vector (AAV-CON).
Figure 7.
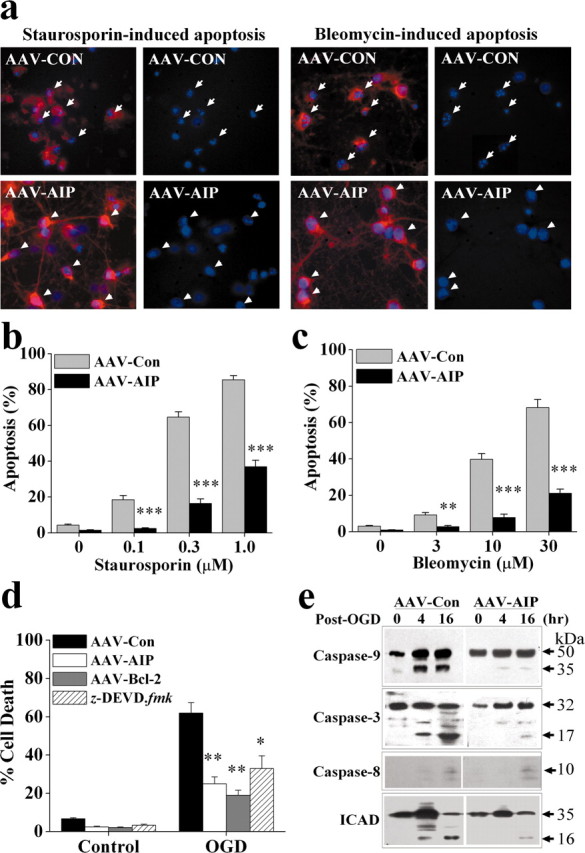
Neuroprotective effect of AAV-AIP against apoptosis and oxygen- glucose deprivation injury in primary hippocampal-cortical neurons. a, Primary neurons were infected for 3 d with AAV-AIP or AAV-CON carrying the truncated AIP cDNA that lacks the CARD domain, and then subjected to apoptosis induction by STS (1 μm) or bleomycin (30 μm). Twenty-four hours later, neurons were subjected to double-label immunofluorescent staining for HA (red), to detect the HA-tagged AIP protein, and Hoechst33258 (blue). AAV-CON infection had no effect on STS- or bleomycin-induced apoptosis (arrows in the top panel point to apoptotic nuclei), whereas AAV-AIP-mediated AIP overexpression inhibited apoptosis (arrowheads in the bottom panel point to nuclei with normal morphology). b, c, Quantitative data show the protective effect of AAV-AIP against neuronal apoptosis induced by STS (b) or bleomycin (c) at the indicated concentrations, measured at 24 hr after induction. n = 12-18 per experimental condition. **p < 0.01; ***p < 0.001 versus AAV-CON-infected neurons. d, AIP overexpression significantly attenuated OGD-induced neuronal cell death. Neurons were infected for 3 d with AAV-AIP, AAV-Bcl-2, or AAV-CON or treated with z-DEVD-fmk (100 μm) for 1 hr and then subjected to OGD for 90 min. Cell death was measured at 24 hr after OGD. Data are mean ± SE; n = 12 per experimental condition from three independent experiments. *p < 0.05; **p < 0.01 versus AAV-CON-infected neurons. e, Western blots show that AIP overexpression attenuated the proteolytic activation of caspase-9 and caspase-3 at 4 and 16 hr after OGD but had no effect on caspase-8 cleavage. AIP overexpression also decreased OGD-induced cleavage of ICAD into its inactive form (∼16 kDa).
We have further investigated the anti-death effect of AAV-AIP in primary neurons subjected to transient OGD, which simulates the major pathologic components of ischemic injury (Cao et al., 2001). As shown in Figure 7d, AIP overexpression significantly decreased OGD-induced cell death in primary neuronal cultures. The level of protection by AIP was compatible with that by AAV-mediated Bcl-2 overexpression or the caspase-3/7 inhibitor z-DEVD.fmk, indicating that the cytochrome c-Apaf-1-dependent caspase-3 activation may be an important mechanism for cell death after transient OGD. Furthermore, in support of the role of AIP in inhibiting the caspase-9-caspase-3 pathway, OGD-induced proteolytic activation of caspase-9 and caspase-3, but not caspase-8, was attenuated in cultures infected with AAV-AIP (Fig. 7e). Finally, OGD-induced cleavage of inhibitor of caspase-activated deoxyribonuclease (ICAD), a hallmark of caspase-3- and caspase-7-dependent nuclear DNA degradation (Cao et al., 2001), was also attenuated by AAV-AIP. Taken together, these data are in agreement with the recent report suggesting a predominant role for the intrinsic pathway (caspase-9 dependent) but not the extrinsic pathway (caspase-8 dependent) in the execution of cell death in the OGD model (Zhang et al., 2003).
AIP gene infection promotes cell survival in the rat model of transient global ischemia
Microinfusion of AAV-AIP into the rat dorsal hippocampus at two spots or striatum time-dependently resulted on AIP overexpression. HA immunohistochemical staining on brain sections 5 d after AAV infusion detected robust expression of AIP in almost the entire CA1 sector and striatal cells (Fig. 8a). Western blot analysis (Fig. 8b) confirms the time-dependent AIP expression in the hippocampal CA1 sector after AAV-AIP infusion. Overexpression of AIP was readily detectable in CA1 3 d after infusion; the levels were increased thereafter, and the high levels of expression lasted for at least 8 weeks.
Figure 8.
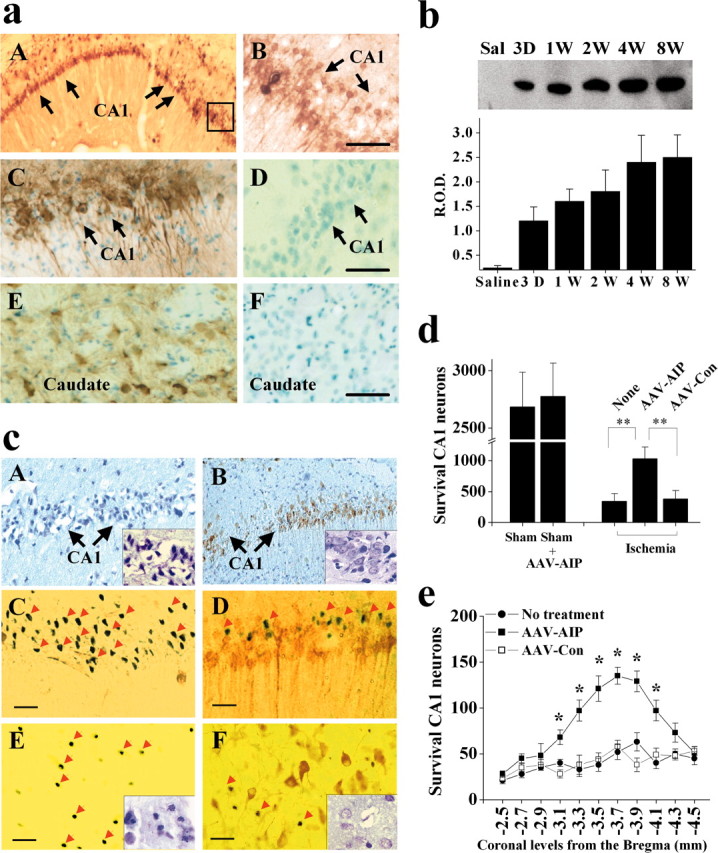
AIP overexpression promotes CA1 cell survival after transient global ischemia. a, In vivo expression of AIP after AAV vector infection in the rat brain. AAV was infused at two coordinates in to the hippocampus to achieve widespread expression in the CA1 sector. Images show AIP immunoreactivity, detected using the monoclonal anti-HA antibody, in the hippocampal CA1 (A, C) and caudate-putamen (E) at 5 d after AAV-AIP infection; no AIP expression is seen in these regions after empty vector infection (D, F). B is a high-power view (scale bars, 50 μm) of CA1 from the marked area in A. C-F show AIP immunoreactivity counter-stained with methyl Green. b, Western blot (anti-HA) confirms the expression of AIP protein in rat hippocampus at various time points (3 d to 8 weeks) after AAV-AIP infection. Saline (Sal) refers to hippocampal sample obtained 4 weeks after saline injection. The graph summarizes results from three to four hippocampi for each time point. c, Representative micrographs from CA1 (A-D) and caudate-putamen (E, F) at 4 d after transient global ischemia. AAV-AIP (B, D, F) or the control vector (A, C, E) was infused into the brain 5 d before ischemia. Brain sections were immunostained for HA and counterstained with cresyl violet (A, B, low power, 100×) or TUNEL (C-F, high power, 400×). Scale bars, 30 μm. Many AIP-infected neurons (D, F, brown staining) survived the ischemic insult. C-F, Arrows point to TUNEL-positive cells that are in contrast to AIP-infected neurons. A, B, E, F, Insets are cresyl violet staining in CA1 and caudate-putamen, respectively, that show improved histological morphology in the ischemic brain after AAV-AIP infection. d, e, CA1 cell survival in the dorsal hippocampus at 4 d after ischemia after AAV-AIP infection, calculated as the sum of survival neurons in 11 coronal sections taken every 0.2 mm at the levels of -2.5 to -4.5 mm from the bregma (n = 9 per group). e, Graph shows the number of CA1 cell survivals at each coronal level. Data are mean ± SE; *p < 0.05, **p < 0.01 versus nontreated or control vector-treated brains.
The cell death-suppressing effect of AIP was studied in the model of transient global cerebral ischemia produced by the four-vessel occlusion method. Previous studies with this model have suggested that ischemic neurodegeneration of CA1 is mediated via the sequential activation of caspase-9 and caspase-3 (Chen et al., 1998; Krajewski et al., 1999; Cao et al., 2001, 2002; Sugawara et al., 2002). To determine whether AIP overexpression could inhibit ischemic neuronal death, the AAV-AIP vector or the AAV control vector was infused into the CA1 sector in the dorsal hippocampus and caudate-putamen, respectively; 5 d later, the animals were subjected to 12 min of global ischemia. The brains were processed at 4 d after ischemia for histological assessments and TUNEL staining. AIP overexpression significantly enhanced the survival of CA1 neurons (Fig. 8c-e) and striatal neurons (Fig. 8c) after ischemia. As determined in 11 coronal levels in the dorsal hippocampus (-2.5 to -4.5 from the bregma, every 0.2 mm), only 13.6 ± 4.7% of CA1 neurons in nontreated brains survived the ischemic insult; however, the number of survival CA1 neurons was increased to 38.4 ± 7.6% in brains infected with AAV-AIP (p < 0.01 vs noninfected or control AAV-infected brains), representing a 2.8-fold increase in neuronal survival.
To determine the relationship between AIP overexpression and caspase-3 activation in the ischemic CA1 neurons, double-label staining was performed to colocalize HA-AIP immunoreactivity with active caspase-3. Nearly all HA-AIP immunoreactive neurons showed an absence of active caspase-3 (Fig. 9a), suggesting that caspase-3 was prevented by AIP overexpression. Furthermore, as demonstrated by both Western blot analysis and caspase activity assays, AIP overexpression significantly decreased ischemia-induced activation of both caspase-9 and caspase-3 at 48 hr after ischemia (Fig. 9b,c), a time point that precedes CA1 cell death in this model. Finally, consistent with the deduced role for AIP in blocking the formation of apoptosome, coimmunoprecipitation results demonstrated that AIP overexpression in hippocampal CA1 neurons attenuated the dimerization between Apaf-1 and caspase-9, a hallmark of apoptosome formation (Cao et al., 2002), after ischemia.
Figure 9.

AIP overexpression attenuates caspase activation after transient global ischemia. a, AIP overexpression inhibited caspase-3 activation in CA1 (A, B) and caudate-putamen (C, D) after ischemia. Double-label immunofluorescent staining for HA (green) and active caspase-3 (red) was performed 4 d after ischemia in brains infected with the control vector (A, C) or AAV-AIP (B, D). B, D, Arrows point to AIP-infected neurons that are in contrast to caspase-3-positive cells. Scale bars, 30 μm. b, Representative Western blots showing decreased activation of caspase-3 and caspase-9 in the CA1 sector after ischemia in AIP-infected brains. The CA1 sector was dissected at 2 d after ischemia, and the protein extracts were immunoblotted with antibodies against active caspase-9 and caspase-3, respectively. c, AIP overexpression decreased caspase-9-like (LEHDase) and caspase-3-like (DEVDase) activities after ischemia. Caspase activities were measured in cell extracts prepared from the CA1 sector with or without AAV-AIP infection at 2 d after ischemia (n = 6 per group). Data are mean ± SE; *p < 0.05 versus noninfected brains. d, AIP overexpression attenuated the Apaf-1-caspase-9 dimerization after ischemia. Isotonic, detergent-free hippocampal homogenates were prepared from brains 24 hr after ischemia. The cytosolic fraction was subjected to immunoprecipitation using anti-Apaf-1 antibody followed by immunoblot analysis of caspase-9 (top) and Apaf-1 (bottom). The in vitro translation product of the caspase-9 cDNA was used as the positive control (PC; last lane). The blot was representative of two independent experiments with similar results.
Discussion
The role of Apaf-1 and caspase-9 as the essential components of the intracellular execution machinery responsible for neuronal apoptosis via the intrinsic pathway is well established (Cecconi et al., 1998; Kuida et al., 1998; Yoshida et al., 1998; Deshmukh et al., 2000; Kermer et al., 2000; D'Sa-Eipper et al., 2001; Gerhardt et al., 2001). We report here the cloning and characterization of a small AIP in rat brain. AIP is a novel short variant of caspase-9, which contains the caspase-recruiting domain but lacks the catalytic domains that caspase-9 possesses. Functionally, AIP is an anti-apoptotic molecule, showing potent cell death-suppressing activity in neuronal and non-neuronal cells against various apoptosis-inducing stimuli. Mechanistically, AIP inhibits the Apaf-1-caspase-9 pathway for caspase activation by directly binding to Apaf-1 via the CARD domain. Moreover, when overexpressed in the brain via an AAV vector, AIP significantly promoted the survival of hippocampal CA1 neurons after transient global ischemia. Taken together, these observations suggest that AIP is a potent death-suppressing molecule and thus can be a good candidate for neuroprotective interventions in cerebral ischemia and, possibly, other forms of brain injury.
As determined by sequence alignment, the AIP cDNA described here is a novel short isoform of caspase-9, which is structurally distinct from previously reported caspase-9 short variants that possess dominant-negative properties. The first of such short variants, the 30 kDa caspase-9, identified in human cell lines (Seol and Billiar, 1999; Srinivasula et al., 1999), is missing most of the large subunit of caspase-9, including the catalytic site, but has the intact small subunit. Later, a rat C-terminal divergent caspase-9, designated as casp-9-CTD, was discovered (Angelastro et al., 2001). Casp-9-CTD encodes a protein of 45 kDa that contains the entire large subunit of caspase-9 but has an alternative C-terminal sequence (Angelastro et al., 2001). In contrast, the protein encoded by the AIP cDNA is missing most of the large subunit and the entire small subunit of caspase-9, yet shows potent anti-apoptotic properties. As suggested previously (Seol and Billiar, 1999; Angelastro et al., 2001) and further supported in this study, the N-terminal CARD domain, which is identical among all three variants, may be the principal functional domain of this new class of apoptosis-inhibitor proteins.
The data presented here demonstrate that AIP is expressed endogenously in neuronal tissues. RT-PCR, RPA, and Western blots showed that both AIP mRNA and protein could be detected in various regions in the brain. Like several other endogenous apoptosis inhibitory proteins such as Bcl-2 and XIAP (X-linked inhibitor of apoptosis), the basal levels of AIP expression were substantially lower than those of its pro-apoptotic counterparts (caspase-9 and Apaf-1), consistent with the notion that in non-stressed cells it is the combined effect of these low-expressing inhibitory proteins that sets up a threshold for preventing the unwanted activation of apoptosis (Budihardjo et al., 1999). AIP, however, appears to be an inducible gene after injury in the brain, because its mRNA and protein levels were upregulated several fold in the hippocampal DG and DG-CA3, respectively, after transient global ischemia. The DG-CA3 represents a distinctive region in the hippocampus that is highly resistant to cell death after global ischemia; these results indicate that the inducible expression of AIP in DG neurons could be a contributing factor to the death-resistant nature of this neuronal population. It is indeed interesting that despite the fact that the open reading frame sequence of AIP is identical to the N terminus of caspase-9, two distinctive expression patterns are presented in the ischemic hippocampus for AIP (increased in the DG but not in the CA1) and caspase-9 (increased in the CA1 but not in the DG) (Cao et al., 2002). The mechanism responsible for this difference is unknown; however, it has been suggested that the differences in 3′UTR of two closely related mRNA species may markedly influence their stability, translation efficiency, or location in a tissue-specific manner (Edwalds-Gilbert et al., 1997).
The functional role of AIP as a potent anti-apoptotic molecule is established in the present study using gene transfection strategies. Transient gene transfection of AIP in human 293 cells potently inhibited Apaf-1- or caspase-9-induced apoptosis. Furthermore, stable gene transfection in neuronal-differentiated PC12 cells or AAV-mediated gene infection of AIP in primary neurons offered marked protection against apoptosis induced by various pro-death stimuli, including PKC inhibition, DNA damage, mitochondrial complex I inhibition, hypoxia, and OGD. Such broad-spectrum cell death-suppressing effects exhibited by AIP in neuronal and non-neuronal cells crossing different species (human and rat) indicate that this death suppressor inhibits apoptosis via interference with a common death-execution pathway. In support of this notion, on several occasions the death-stimulus-induced activation of terminal caspases such as caspase-3 was markedly attenuated by AIF overexpression, whereas mitochondrial cytochrome c release that occurred during the early stages of apoptosis was not prevented. These results place the anti-apoptotic actions of AIP within the component of the death-execution cascade that is upstream of terminal caspase activity but downstream of mitochondrial injury.
The mechanism by which AIP interferes with the Apaf-1-caspase-9 signaling pathway was investigated using several assays. As predicted from its molecular structure, in vitro translation-generated AIP clearly showed the ability of direct binding to Apaf-1 through its N-terminal CARD domain. The endogenous protein-protein interaction between Apaf-1 and AIP was also confirmed in cultured cells undergoing STS-induced apoptosis, in which coimmunoprecipitation of Apaf-1 and AIP was markedly increased. Because CARD is present in both Apaf-1 and procaspase-9 and is the essential domain for procaspase-9 to be recruited to the apoptosome at a ratio to Apaf-1 of ∼1:1 (Srinivasula et al., 1998; Yoshida et al., 1998), it is conceivable that the dimerization between AIP and Apaf-1 through the CARD would decrease the recruitment of procaspase-9 by Apaf-1. The results from the in vitro binding assays strongly support this speculation, in which the addition of an excessive amount of AIP to the reaction system nearly completely prevented the binding of procaspase-9 to Apaf-1. Therefore, we further speculate that AIP may inhibit the Apaf-1-caspase-9 pathway by directly binding to Apaf-1 and thus preventing the activation of caspase-9. It is equally possible, however, that AIP may also bind directly to procaspase-9 through CARD. In either scenario, AIP likely acts to interrupt the interaction between Apaf-1 and caspase-9 and prevent the formation of a functional apoptosome. Accordingly, the intracellular ratio between AIP and caspase-9 or Apaf-1 may be an important intrinsic factor determining the susceptibility of the cell to the activation of apoptosis-execution machinery via the mitochondrial signaling pathway. Consistent with this notion, we found that cytochrome c-ATP-initiated activation of caspase-9 and caspase-3 was greatly attenuated in cells overexpressing AIP.
Because AIP showed potent cell death-suppressing activity in neuronal cultures, we further studied its potential death-regulatory role in the rat model of transient global ischemia. Several sequential components of the mitochondrial apoptosis-signaling pathway, including cytochrome c release, second mitochondria-derived activator of caspase release, caspase-9 and Apaf-1 activation, and caspase-3 activation, have been well documented previously in this model (Chen et al., 1998; Himi et al., 1998; Gillardon et al., 1999; Sugawara et al., 1999; Cao et al., 2001, 2002; Tanaka et al., 2004). To achieve sufficient AIP gene delivery in the hippocampal CA1 neurons in vivo, we constructed an AAV vector. This vector was chosen on the basis of its nonpathogenic, noncytotoxic, and replication-defective features and its powerful capacity to deliver gene products into neurons in vitro and in vivo (Kaplitt et al., 1994; McCown et al., 1996; Mandel et al., 1997; Xiao et al., 1997, 1998; Davidson et al., 2000). In this study, we demonstrate that infusion of AAV-AIP into the hippocampus resulted in robust AIP expression in CA1 neurons in a time-dependent manner. Furthermore, the data show that AAV-mediated AIP overexpression significantly promoted CA1 neuronal survival after transient global ischemia. These results thus confirm the deduced role of the Apaf-1-caspase-9 signaling pathway in mediating CA1 neurodegeneration after transient global ischemia (Cao et al., 2002; Sugawara et al., 2002), and provide the first experimental evidence that direct molecular targeting of the Apaf-1-caspase-9 pathway using a small Apaf-1-interacting protein offers significant neuroprotection against ischemic neuronal death.
The significance of the findings documented in this report is twofold. First, the cloning and characterization of AIP provide a novel and specific molecular tool that can help elucidate the role of the Apaf-1-caspase-9 apoptosis signaling pathway in neuronal death associated with various neurological disorders. Second, the demonstrated neuroprotection by AIP in primary neuronal cultures and in the in vivo model of cerebral ischemia enforce the argument that molecular approaches that enhance the endogenous threshold levels for caspase activation may be a feasible and potentially effective therapeutic intervention in ischemic neuronal injury and related diseases.
Footnotes
This project was supported by National Institutes of Health-National Institute of Neurological Disorders and Stroke Grants NS43802, NS45048, and NS36736 (J.C.) and NS35965 (J.C., S.H.G., R.P.S.), American Heart Association Established Investigator Award 240135N (J.C.), and Beginning Grant-in-Aid Award 0265414U (G.C.). F.S. was supported by grants from the Chinese Natural Science Foundation (39825109 and 30028006). J.C. and S.H.G. were also supported in part by the Geriatric Research, Education, and Clinical Center, Veterans Affairs Pittsburgh Health Care System, (Pittsburgh, PA). We thank Liping Sheng and Cristine O'Horo for technical assistance, Carol Culver for editorial assistance, and Pat Strickler for secretarial support.
Correspondence should be addressed to Dr. Jun Chen, Department of Neurology, S-507, Biomedical Science Tower, University of Pittsburgh School of Medicine, Pittsburgh, PA 15213. E-mail: chenj2@upmc.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/246189-13$15.00/0
G.C. and M.X. contributed equally to this work.
References
- Angelastro JM, Moon NY, Liu DX, Yang AS, Greene LA, Franke TF (2001) Characterization of a novel isoform of caspase-9 that inhibits apoptosis. J Biol Chem 276: 12190-12200. [DOI] [PubMed] [Google Scholar]
- Benchoua A, Guegan C, Couriaud C, Hosseini H, Sampaio N, Morin D, Onteniente B (2001) Specific caspase pathways are activated in the two stages of cerebral infarction. J Neurosci 21: 7127-7134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budihardjo I, Oliver H, Lutter M, Luo X, Wang X (1999) Biochemical pathways of caspase activation during apoptosis. Annu Rev Cell Dev Biol 15: 269-290. [DOI] [PubMed] [Google Scholar]
- Cao G, Pei W, Lan J, Stetler RA, Luo Y, Nagayama T, Graham SH, Yin XM, Simon RP, Chen J (2001) Caspase-activated DNase/DNA fragmentation factor 40 mediates apoptotic DNA fragmentation in transient cerebral ischemia and in neuronal cultures. J Neurosci 21: 4678-4690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao G, Luo Y, Nagayama T, Pei W, Stetler RA, Graham SH, Chen J (2002) Cloning and characterization of rat caspase-9: implications for a role in mediating caspase-3 activation and hippocampal cell death after transient cerebral ischemia. J Cereb Blood Flow Metab 22: 534-546. [DOI] [PubMed] [Google Scholar]
- Cecconi F, Alvarez-Bolado G, Meyer BI, Roth KA, Gruss P (1998) Apaf1 (CED-4 homolog) regulates programmed cell death in mammalian development. Cell 94: 727-737. [DOI] [PubMed] [Google Scholar]
- Chen D, Stetler RA, Cao G, Pei W, O'Horo C, Yin XM, Chen J (2000) Characterization of the rat DNA fragmentation factor 35/inhibitor of caspase-activated DNase (short form). The endogenous inhibitor of caspase-dependent DNA fragmentation in neuronal apoptosis. J Biol Chem 275: 38508-38517. [DOI] [PubMed] [Google Scholar]
- Chen J, Nagayama T, Jin K, Stetler RA, Zhu RL, Graham SH, Simon RP (1998) Induction of caspase-3-like protease may mediate delayed neuronal death in the hippocampus after transient cerebral ischemia. J Neurosci 18: 4914-4928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daugas E, Susin SA, Zamzami N, Ferri KF, Irinopoulou T, Larochette N, Prevost MC, Leber B, Andrews D, Penninger J, Kroemer G (2000) Mitochondrio-nuclear translocation of AIF in apoptosis and necrosis. FASEB J 14: 729-739. [PubMed] [Google Scholar]
- Davidson BL, Stein CS, Heth JA, Martins I, Kotin RM, Derksen TA, Zabner J, Ghodsi A, Chiorini JA (2000) Recombinant adeno-associated virus type 2, 4, and 5 vectors: transduction of variant cell types and regions in the mammalian central nervous system. Proc Natl Acad Sci USA 97: 3428-3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshmukh M, Kuida K, Johnson EM Jr (2000) Caspase inhibition extends the commitment to neuronal death beyond cytochrome c release to the point of mitochondrial depolarization. J Cell Biol 150: 131-143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deveraux QL, Takahashi R, Salvesen GS, Reed JC (1997) X-linked IAP is a direct inhibitor of cell-death proteases. Nature 388: 300-304. [DOI] [PubMed] [Google Scholar]
- Deveraux QL, Roy N, Stennicke HR, Van Arsdale T, Zhou Q, Srinivasula SM, Alnemri ES, Salvesen GS, Reed JC (1998) IAPs block apoptotic events induced by caspase-8 and cytochrome c by direct inhibition of distinct caspases. EMBO J 17: 2215-2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Sa-Eipper C, Leonard JR, Putcha G, Zheng TS, Flavell RA, Rakic P, Kuida K, Roth KA (2001) DNA damage-induced neural precursor cell apoptosis requires p53 and caspase 9 but neither Bax nor caspase 3. Development 128: 137-146. [DOI] [PubMed] [Google Scholar]
- Edwalds-Gilbert G, Veraldi KL, Milcarek C (1997) Alternative poly(A) site selection in complex transcription units: means to an end? Nucleic Acids Res 25: 2547-2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimura M, Morita-Fujimura Y, Kawase M, Copin JC, Calagui B, Epstein CJ, Chan PH (1999) Manganese superoxide dismutase mediates the early release of mitochondrial cytochrome c and subsequent DNA fragmentation after permanent focal cerebral ischemia in mice. J Neurosci 19: 3414-3422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhardt E, Kugler S, Leist M, Beier C, Berliocchi L, Volbracht C, Weller M, Bahr M, Nicotera P, Schulz JB (2001) Cascade of caspase activation in potassium-deprived cerebellar granule neurons: targets for treatment with peptide and protein inhibitors of apoptosis. Mol Cell Neurosci 17: 717-731. [DOI] [PubMed] [Google Scholar]
- Gillardon F, Kiprianova I, Sandkuhler J, Hossmann KA, Spranger M (1999) Inhibition of caspases prevents cell death of hippocampal CA1 neurons, but not impairment of hippocampal long-term potentiation following global ischemia. Neuroscience 93: 1219-1222. [DOI] [PubMed] [Google Scholar]
- Graham SH, Chen J (2001) Programmed cell death in cerebral ischemia. J Cereb Blood Flow Metab 21: 99-109. [DOI] [PubMed] [Google Scholar]
- Hara H, Friedlander RM, Gagliardini V, Ayata C, Fink K, Huang Z, Shimizu-Sasamata M, Yuan J, Moskowitz MA (1997) Inhibition of interleukin 1beta converting enzyme family proteases reduces ischemic and excitotoxic neuronal damage. Proc Natl Acad Sci USA 94: 2007-2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himi T, Ishizaki Y, Murota S (1998) A caspase inhibitor blocks ischaemia-induced delayed neuronal death in the gerbil. Eur J Neurosci 10: 777-781. [DOI] [PubMed] [Google Scholar]
- Hofmann K, Bucher P, Tschopp J (1997) The CARD domain: a new apoptotic signaling motif. Trends Biochem Sci 22: 155-156. [DOI] [PubMed] [Google Scholar]
- Hu Y, Benedict MA, Ding L, Nunez G (1999) Role of cytochrome c and dATP/ATP hydrolysis in Apaf-1-mediated caspase-9 activation and apoptosis. EMBO J 18: 3586-3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplitt MG, Leone P, Samulski RJ, Xiao X, Pfaff DW, O'Malley KL, During MJ (1994) Long-term gene expression and phenotypic correction using adeno-associated virus vectors in the mammalian brain. Nat Genet 8: 148-154. [DOI] [PubMed] [Google Scholar]
- Kermer P, Ankerhold R, Klocker N, Krajewski S, Reed JC, Bahr M (2000) Caspase-9: involvement in secondary death of axotomized rat retinal ganglion cells in vivo. Brain Res Mol Brain Res 85: 144-150. [DOI] [PubMed] [Google Scholar]
- Krajewski S, Krajewska M, Ellerby LM, Welsh K, Xie Z, Deveraux QL, Salvesen GS, Bredesen DE, Rosenthal RE, Fiskum G, Reed JC (1999) Release of caspase-9 from mitochondria during neuronal apoptosis and cerebral ischemia. Proc Natl Acad Sci USA 96: 5752-5757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer G, Reed JC (2000) Mitochondrial control of cell death. Nat Med 6: 513-519. [DOI] [PubMed] [Google Scholar]
- Kuida K, Haydar TF, Kuan CY, Gu Y, Taya C, Karasuyama H, Su MS, Rakic P, Flavell RA (1998) Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell 94: 325-337. [DOI] [PubMed] [Google Scholar]
- Lawrence MS, Ho DY, Sun GH, Steinberg GK, Sapolsky RM (1996) Over-expression of Bcl-2 with herpes simplex virus vectors protects CNS neurons against neurological insults in vitro and in vivo J Neurosci 16: 486-496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Yuan J (1999) Deciphering the pathways of life and death. Curr Opin Cell Biol 11: 261-266. [DOI] [PubMed] [Google Scholar]
- Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X (1997) Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 91: 479-489. [DOI] [PubMed] [Google Scholar]
- Linnik MD, Zahos P, Geschwind MD, Federoff HJ (1995) Expression of bcl-2 from a defective herpes simplex virus-1 vector limits neuronal death in focal cerebral ischemia. Stroke 26: 1670-1674. [DOI] [PubMed] [Google Scholar]
- Lipton P (1999) Ischemic cell death in brain neurons. Physiol Rev 79: 1431-1568. [DOI] [PubMed] [Google Scholar]
- Liu X, Kim CN, Yang J, Jemmerson R, Wang X (1996) Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell 86: 147-157. [DOI] [PubMed] [Google Scholar]
- Lo EH, Dalkara T, Moskowitz MA (2003) Mechanisms, challenges and opportunities in stroke. Nat Rev Neurosci 4: 399-415. [DOI] [PubMed] [Google Scholar]
- Mandel RJ, Spratt SK, Snyder RO, Leff SE (1997) Midbrain injection of recombinant adeno-associated virus encoding rat glial cell line-derived neurotrophic factor protects nigral neurons in a progressive 6-hydroxydopamine-induced degeneration model of Parkinson's disease in rats. Proc Natl Acad Sci USA 94: 14083-14088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinou JC, Dubois-Dauphin M, Staple JK, Rodriguez I, Frankowski H, Missotten M, Albertini P, Talabot D, Catsicas S, Pietra C, Huarte J (1994) Overexpression of BCL-2 in transgenic mice protects neurons from naturally occurring cell death and experimental ischemia. Neuron 13: 1017-1030. [DOI] [PubMed] [Google Scholar]
- McCown TJ, Xiao X, Li J, Breese GR, Samulski RJ (1996) Differential and persistent expression patterns of CNS gene transfer by an adeno-associated virus (AAV) vector. Brain Res 713: 99-107. [DOI] [PubMed] [Google Scholar]
- Noshita N, Sugawara T, Fujimura M, Morita-Fujimura Y, Chan PH (2001) Manganese superoxide dismutase affects cytochrome c release and caspase-9 activation after transient focal cerebral ischemia in mice. J Cereb Blood Flow Metab 21: 557-567. [DOI] [PubMed] [Google Scholar]
- Pei W, Liou AK, Chen J (2003) Two caspase-mediated apoptotic pathways induced by rotenone toxicity in cortical neuronal cells. FASEB J 17: 520-522. [DOI] [PubMed] [Google Scholar]
- Plesnila N, Zinkel S, Le DA, Amin-Hanjani S, Wu Y, Chiarugi A, Thomas SS, Kohane DS, Moskowitz MA (2001) BID mediates neuronal cell death after oxygen/glucose deprivation and focal cerebral ischemia. Proc Natl Acad Sci USA 98: 15318-15323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson GS, Crocker SJ, Nicholson DW, Schulz JB (2000) Neuroprotection by the inhibition of apoptosis. Brain Pathol 10: 283-292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seol DW, Billiar TR (1999) A caspase-9 variant missing the catalytic site is an endogenous inhibitor of apoptosis. J Biol Chem 274: 2072-2076. [DOI] [PubMed] [Google Scholar]
- Srinivasula SM, Ahmad M, Fernandes-Alnemri T, Alnemri ES (1998) Auto-activation of procaspase-9 by Apaf-1-mediated oligomerization. Mol Cell 1: 949-957. [DOI] [PubMed] [Google Scholar]
- Srinivasula SM, Ahmad M, Guo Y, Zhan Y, Lazebnik Y, Fernandes-Alnemri T, Alnemri ES (1999) Identification of an endogenous dominant-negative short isoform of caspase-9 that can regulate apoptosis. Cancer Res 59: 999-1002. [PubMed] [Google Scholar]
- Steller H (1995) Mechanisms and genes of cellular suicide. Science 267: 1445-1449. [DOI] [PubMed] [Google Scholar]
- Sugawara T, Fujimura M, Morita-Fujimura Y, Kawase M, Chan PH (1999) Mitochondrial release of cytochrome c corresponds to the selective vulnerability of hippocampal CA1 neurons in rats after transient global cerebral ischemia. J Neurosci 19: RC39(1-6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara T, Noshita N, Lewen A, Gasche Y, Ferrand-Drake M, Fujimura M, Morita-Fujimura Y, Chan PH (2002) Overexpression of copper/zinc su-peroxide dismutase in transgenic rats protects vulnerable neurons against ischemic damage by blocking the mitochondrial pathway of caspase activation. J Neurosci 22: 209-217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka H, Yokota H, Jover T, Cappuccio I, Calderone A, Simionescu M, Bennett MVL, Zukin RS (2004) Ischemic preconditioning: neuronal survival in the face of caspase-3 activation. J Neurosci 24: 2750-2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson CB (1995) Apoptosis in the pathogenesis and treatment of disease. Science 267: 1456-1462. [DOI] [PubMed] [Google Scholar]
- Xiao X, Li J, Samulski RJ (1998) Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. J Virol 72: 2224-2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao X, Li J, McCown TJ, Samulski RJ (1997) Gene transfer by adeno-associated virus vectors into the central nervous system. Exp Neurol 144: 113-124. [DOI] [PubMed] [Google Scholar]
- Xu D, Bureau Y, McIntyre DC, Nicholson DW, Liston P, Zhu Y, Fong WG, Crocker SJ, Korneluk RG, Robertson GS (1999) Attenuation of ischemia-induced cellular and behavioral deficits by X chromosome-linked inhibitor of apoptosis protein overexpression in the rat hippocampus. J Neurosci 19: 5026-5033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakovlev AG, Ota K, Wang G, Movsesyan V, Bao WL, Yoshihara K, Faden AI (2001) Differential expression of apoptotic protease-activating factor-1 and caspase-3 genes and susceptibility to apoptosis during brain development and after traumatic brain injury. J Neurosci 21: 7439-7446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Kong YY, Yoshida R, Elia AJ, Hakem A, Hakem R, Penninger JM, Mak TW (1998) Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell 94: 739-750. [DOI] [PubMed] [Google Scholar]
- Yuan J, Yankner BA (2000) Apoptosis in the nervous system. Nature 407: 802-809. [DOI] [PubMed] [Google Scholar]
- Zhang WH, Wang X, Narayanan M, Zhang y, Huo C, Reed JC, Friedlander RM (2003) Fundamental role of the Rip2/caspase-1 pathway in hypoxia and ischemia-induced neuronal cell death. Proc Natl Acad Sci USA 100: 16012-16017. [DOI] [PMC free article] [PubMed] [Google Scholar]