

Abstract
Dendritic targeting of mRNA and local protein synthesis are mechanisms that enable neurons to deliver proteins to specific postsynaptic sites. Here, we demonstrate that epileptogenic stimuli induce a dramatic accumulation of BDNF mRNA and protein in the dendrites of hippocampal neurons in vivo. BDNF mRNA and protein accumulate in dendrites in all hippocampal subfields after pilocarpine seizures and in selected subfields after other epileptogenic stimuli (kainate and kindling). BDNF accumulates selectively in discrete dendritic laminas, suggesting targeting to synapses that are active during seizures. Dendritic targeting of BDNF mRNA occurs during the time when the cellular changes that underlie epilepsy are occurring and is not seen after intense stimuli that are non-epileptogenic, including electroconvulsive seizures and high-frequency stimulation. MK801, an NMDA receptor antagonist that can prevent epileptogenesis but not acute seizures, prevents the dendritic accumulation of BDNF mRNA, indicating that dendritic targeting is mediated via NMDA receptor activation. Together, these results suggest that dendritic accumulation of BDNF mRNA and protein play a critical role in the cellular changes leading to epilepsy.
Keywords: neurotrophins, rat hippocampus, epilepsy, plasticity, mRNA trafficking, AMPA receptors, NMDA receptors, muscarinic acetylcholine receptors
Introduction
The cellular events that underlie epileptic hyperexcitability have not been identified definitively but may include reorganization of synaptic connections, neuronal death, microgliosis, neurogenesis, astrogliosis, and axonal sprouting (McNamara, 1999; Mody, 1999). After the epileptogenic stimulus that brings about these cellular alterations, critical mediators are produced, as suggested by the induction of complex changes in gene expression (Brooks-Kayal et al., 1998; Chiang et al., 2001), including a dramatic increase of BDNF (Ernfors et al., 1991; Isackson et al., 1991; Bengzon et al., 1993; Murray et al., 2000; Huang et al., 2002).
BDNF is a secreted trophic factor that potentiates excitatory synapses (Lu and Chow, 1999; Schuman, 1999) through its high-affinity receptor TrkB (Kaplan and Miller, 2000; Thoenen, 2000; Patapoutian and Reichardt, 2001). Several lines of evidence implicate BDNF in epileptogenesis (Binder et al., 2001). Epileptogenesis is reduced in BDNF+/- mutant mice (Kokaia et al., 1995), in rats receiving injections of trkB-receptor bodies (Binder et al., 1999), and in mice overexpressing truncated TrkB, a dominant-negative BDNF receptor (Lahteinen et al., 2002). The pro-epileptic effect of BDNF may be related to its action at synapses, because BDNF increases neuronal excitability (Binder et al., 2001), whereas the signaling pathway activated by the Shc site of TrkB (implicated in survival, differentiation, and neurite outgrowth) is apparently not involved in epileptogenesis (He et al., 2002).
If BDNF plays a pro-epileptic role by potentiating excitatory synapses, the question then becomes how it is delivered to synapses. In hippocampal granule cells, most of the BDNF synthesized in the cell body is transported anterogradely in the axons (the mossy fibers) and stored in their terminal boutons, which synapse with CA3 pyramidal cells (Elmer et al., 1998; Altar and Di Stefano, 1998). However, there is no evidence that such an anterograde mechanism takes place at other synapses.
Another means of delivering BDNF would be via dendritic targeting of BDNF protein or mRNA (the latter followed by local protein synthesis at postsynaptic sites). Such locally synthesized BDNFs could regulate synaptic strength in a site-specific manner (Steward and Schuman, 2001; Lu, 2003). Indeed, BDNF mRNA accumulates in the dendrites of cultured hippocampal neurons in response to electrical activity, in a glutamate receptor-, Ca2+ influx-, and TrkB receptor-dependent manner (Tongiorgi et al., 1997; Righi et al., 2000), and this dendritic mRNA seems to be locally translated into protein (Tongiorgi et al., 1997).
Whether these phenomena occur in vivo is uncertain. BDNF mRNA has been found in proximal dendrites of hippocampal neurons in vivo (Dugich-Djordjevic et al., 1992; Schmidt-Kastner et al., 1996), although these results were not confirmed in other studies (Miranda et al., 1993; Lauterborn et al., 1996; Conner et al., 1997). Furthermore, although limbic seizures cause dramatic increases in BDNF mRNA levels (see above), its dendritic localization has not been studied under these conditions. On this basis, we investigated whether dendritic targeting of BDNF mRNA is part of the mechanisms underlying epileptogenesis. A preliminary account of part of these data was presented at a meeting (Simonato et al., 2002).
Materials and Methods
Preparation of the animals. Male Sprague Dawley rats were used for all experiments. Procedures involving animals and their care were performed in accordance with the European Community and national laws and policies. All efforts were made to minimize animal suffering.
Pilocarpine (300 mg/kg) and kainate (10 mg/kg) were administered intraperitoneally. The rat's behavior was observed for 6 hr thereafter. Within the first hour after injection, all pilocarpine-treated and 80% of kainate-treated animals developed seizures evolving into recurrent generalized convulsions (status epilepticus). These rats were killed at different times after injection of the chemoconvulsant (3, 6, or 24 hr or 1 month for pilocarpine; 3, 6, or 24 hr for kainate), and their brains were used for in situ hybridization or immunohistochemistry, as described below. The remaining 20% of kainate-treated animals either did not seize or died within the first 3 hr and were not included in the study. Controls were vehicle-injected or naive (real control) rats.
For kindling, a twisted bipolar electrode was implanted in the right amygdala (coordinates: 4.8 mm lateral and 0.8 mm posterior to bregma, 8.3 mm deep from dura) under ketamine (100 mg/kg, i.p.) anesthesia. Animals were allowed 7 d to recover. Beginning the following day, rats were stimulated once daily with a single 1 sec train of bipolar pulses [1 msec, 60 Hz; intensity 25% above afterdischarge (AD) threshold]. Behavior [staging according to Racine (1972)] and the duration of AD measured in the right amygdala were recorded after each stimulation. A group of animals was killed 3 hr after the first class 2 seizure. Kindling criteria were three consecutive class 4 or 5 seizures. One week after reaching these criteria, kindled rats were killed either without any further stimulus or after a stimulation evoking a class 4-5 seizure. Sham-stimulated rats were aged-matched littermates that had undergone surgery and daily handling but had not been stimulated.
Electroconvulsive seizures (ECS) were induced by delivering AC current (60 Hz, 40 mA for 0.5 sec) via ear clip electrodes (Wallace et al., 1998). Animals were killed humanely 2 hr later by anesthetic overdose (Nembutal) and were perfused with 4% paraformaldehyde and prepared for in situ hybridization. To block AMPA and NMDA receptors (NMDARs), 30 mg/kg 2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo(f) quinoxaline-7-sulfonamide (NBQX; Tocris, Bristol, UK) and 1 mg/kg MK801 (Dizocilpine; Tocris) were injected intraperitoneally 20 min before pilocarpine.
Neurophysiological techniques. Adult male Sprague Dawley rats were anesthetized by intraperitoneal injection of 20% urethane (500 mg/kg body weight). Supplemental injections of urethane were given approximately every 10 min until the animal was completely unresponsive to tail pinch. The animal was positioned in a stereotaxic apparatus, a craniotomy was performed, and stimulating and recording electrodes were positioned stereotaxically to selectively activate medial perforant path projections while recording in the dentate gyrus, as described by Steward and Worley (2001). The stimulating electrode (an insulated tungsten micro-electrode) was positioned at 4.0 lateral to the midline and 1.0 mm anterior to the transverse sinus. The depth of the stimulating electrode was adjusted to obtain a maximal evoked response in the dentate gyrus at minimal stimulus intensity. Recording electrodes (glass micropipettes) were lowered into the brain at 3.5 posterior to bregma and 1.5-2.0 mm lateral to the midline and positioned in the cell body layer. Stimulus intensity was set to evoke an ∼1-3 mV population spike. Single test pulses were then delivered at a rate of 1/10 sec for 5-10 min to determine baseline response amplitude. Then, three bouts of high-frequency stimulation (10 trains of eight pulses at 400 Hz) were delivered at a rate of 1/10 sec. Between each bout of 10 trains, 10 test responses were collected to determine the extent of synaptic potentiation. After the third bout, high-frequency trains were delivered at a rate of 1/10 sec for 2 hr. At the end of the period of high-frequency stimulation, animals received an anesthetic overdose and were perfused with 4% paraformaldehyde for in situ hybridization.
In situ hybridization. Rats were perfused transcardially with 4% paraformaldehyde under ketamine anesthesia, and their brains were removed and kept in 4% paraformaldehyde/20% sucrose at 4°C for at least 3 d before sectioning. Riboprobes were prepared from a BDNF cDNA insert (nucleotides 74-525) (Maisonpierre et al., 1991) cloned in a pBluescript plasmid. In situ hybridization was performed as described previously (Tongiorgi et al., 1998) on free-floating, 40 μm coronal sections at the level of the dorsal hippocampus. After hybridization, high-stringency washes were performed in 0.1× SSC/0.1% Tween 20 at 60°C. To obtain reproducible and comparable results and to avoid saturation of the reaction, alkaline phosphatase development was always performed for 4 hr at room temperature. All in situ hybridizations have been conducted side by side with control sections.
Quantification of in situ hybridization data. Images were acquired through a Panasonic F15HS (Panasonic Italia Spa, Milan, Italy) video camera mounted on an Olympus BX50 microscope [Olympus Italia srl, Segrate (Milan), Italy] and analyzed with the program Image Pro Plus 4.0 for Windows (Media Cybernetics, Silver Spring, MD). Images were captured with a 10× objective for measures in CA3 and with a 20× for dentate gyrus and CA1. Illumination was adjusted to obtain optimal staining signal over the dendritic fields (this implied signal saturation over the cell somas) and then was kept constant throughout all experiments. Densitometric analysis was performed on a 400 × 315 μm frame under the 20× objective (which represents the entire frame observed through the camera) and on an 800 × 630 μm frame with the 10× objective, and both were subdivided in 721 × 569 pixels. For the densitometric analysis of the CA3, the stratum pyramidalis of which is curved, a 50 × 630 μm frame was chosen. Therefore, for each animal and hippocampal subfield, we generated curves in which each data point represents the average gray level over a line of pixels (400 μm long for CA1 and dentate gyrus, 50 μm long for CA3) at a given distance from the cell soma. Data obtained from each animal were normalized on the average gray level in the white matter (callosum) of their controls. To compare the different animals, each section was positioned to align the cell layer parallel to one border of the field visualized by the camera.
For statistical analysis, we used the average gray level measured in 20 μm intervals of the different layers in the different hippocampal subfields. The intervals chosen were (as a distance from the cell body layer): for CA1, 100-120 μm (stratum radiatum) and 300-320 μm (stratum lacunosum-moleculare); for CA3, 50-70 μm (stratum lucidum), 180-200 μm (stratum radiatum), and 400-420 μm (stratum lacunosummoleculare); for the dentate gyrus, 40-60 μm (inner molecular layer), 130-150 μm (middle molecular layer), and 220-240 μm (outer molecular layer). The data obtained were analyzed using one-way ANOVA and the post hoc Newman-Keuls test.
Electron microscopy in situ hybridization. Rats were perfused transcardially with 1.25% glutaraldehyde and 0.2% picric acid in PBS. Hippocampi were then sliced manually, postfixed for 24 hr at 4°C in 2% glutaraldehyde, 0.1 m sodium cacodylate, 0.4 m sucrose at pH 7.6, dehydrated through an ascending ethanol series, and, finally, embedded in the resin LR-White at 52°C (Sigma, Milan, Italy). Sections were cut on an ultramicrotome Pabisch Top Ultra 150 and collected on 300-mesh Parlodion (Electron Microscopy Sciences, Fort Washington, PA)-coated gold grids. In situ hybridization was performed with the same digoxigenin-labeled riboprobes described above. In brief, grids were rehydrated 1 min in ultrapure water at 37°C, incubated 10 min at 37°C in reaction buffer (20 mm Tris/HCl, pH 8.0, and 2 mm CaCl2), and then digested 14 min at 37°C in the same buffer with 5 μg/ml proteinase K (Roche Diagnostics, Mannheim, Germany). Grids were then prehybridized at 55°C for 60 min with a mixture containing 50% deionized formamide, 20 mm Tris-HCl, 1 mm EDTA, 300 mm NaCl, 100 mm DTT, 1 μm Denhart's solution, 0.5 mg/ml salmon sperm DNA, and 0.5 mg/ml polyadenylic acid (all reagents were from Sigma-Aldrich, Milan, Italy). In situ hybridization was performed overnight at 55°C in the prehybridization mix, to which 10% dextrane sulfate and the riboprobe (500 ng/ml) were added. After hybridization, grids were washed twice for 30 min with 3× SSC/0.1% Tween 20 at 60°C. After three washes in PBS and one in ultrapure water, grids were incubated overnight at 4°C with goat anti-digoxigenin IgG 1-nm colloidal gold-labeled antibody (SAD1; British Biocell International, Cardiff, UK) diluted 1:100 in PBS, 1% normal goat serum, and 1% BSA (Sigma-Aldrich). Afterward, the grids were washed in PBS, then in ultrapure water, and finally developed with silver-enhancing system HQ (Nanoprobes, Yaphank, NY) for 6 min in a dark room, stained with uranyl acetate for 5 min, and observed with a transmission electron microscope (Philips EM 208; Philips Industrial Electronics, Monza, Italy).
Immunohistochemistry. Rats were killed by decapitation after an anesthetic overdose. Their brains were removed rapidly, immersed in 10% formalin, and then paraffin embedded. Coronal sections (7 μm thick) were cut at the level of the dorsal hippocampus (plate 40) (Pellegrino et al., 1979) and mounted onto polylysine-coated slides.
Sections were dewaxed (two washes in xylene for 10 min, 5 min in 100% ethanol, 5 min in 95% ethanol) and then rehydrated in distilled water for 5 min and in 1× PBS (0.01 m PO42- and 0.15 m NaCl) for 10 min. After an incubation in 0.3% H2O2 for 15 min at room temperature, sections were rinsed rapidly in distilled water and washed twice in PBS. They were then incubated with Ultra V Block (Ultra Vision Detection System; Lab Vision, Fremont, CA) for 5 min at room temperature, to block nonspecific background staining. After washing in 1× PBS for 5 min, sections were incubated overnight at 4°C in a humid atmosphere with the primary antibody for BDNF (5 μg/ml, sc-546; Santa Cruz Biotechnology, Santa Cruz, CA). After rinsing in 1× PBS, they were incubated with biotinylated goat anti-polyvalent serum (Ultra Vision Detection System; Lab Vision) at room temperature for 10 min, washed in 1× PBS for 5 min, and then incubated in streptavidine peroxidase (Ultra Vision Detection System; Lab Vision) at room temperature for 10 min. After another wash in PBS, the reaction product was detected as a brown substrate using a 10 mg tablet of DAB (Sigma) in a solution containing 16.6 ml of 1× PBS and 130 μl of 7% H2O2. Finally, sections were washed three times in 1× PBS (5 min each), counterstained with hematoxylin for 2 min, and washed again in 1× PBS (5 min). Coverslips were mounted using Gel/mount (Biomeda, Foster City, CA). Because of initial concerns about the suitability of this anti-BDNF antibody for immunohistochemical analysis (Fawcett et al., 1997), a series of control experiments was performed. First, in a Western blot analysis, the antibody was found to recognize a BDNF-immunoreactive band of ∼14 kDa (data not shown); in a previous, detailed analysis, the same antibody has been shown to recognize recombinant BDNF, to discriminate it from the other neurotrophins, and to identify it in wild-type (+/+), but not in BDNF null (-/-), mouse brains (Causing et al., 1997). Second, preadsorption with BDNF overnight blocked all immunostaining, and immunolabeling was absent when the primary antibody was omitted (data not shown). Third, in keeping with a previous report (Schwartz et al., 1997), the antibody detected BDNF in BDNF+/+, but not in BDNF-/-, mice (a generous gift from Dr. James O. McNamara, Duke University, Durham, NC; data not shown).
Results
Pilocarpine seizures induce dendritic targeting of BDNF mRNA
A sensitive nonradioactive in situ hybridization procedure (Tongiorgi et al., 1998) was used to study the subcellular distribution of BDNF mRNA in the rat hippocampus. In untreated rats (n = 10) (Fig. 1, control), BDNF mRNA was present in the soma and the proximal dendritic compartment of CA1 and CA3 pyramidal neurons (up to ∼50-70 μm from the cell body) (Fig. 2A, arrowheads), whereas in dentate gyrus granule cells, labeling was present in the soma only. A quantitative densitometric analysis (Fig. 2C-E) confirmed that levels of labeling in distal dendrites were not above background. Sections hybridized with the BDNF sense riboprobe yielded no signal (Fig. 1, sense).
Figure 1.

Pilocarpine seizures induce dendritic targeting of BDNF mRNA. Shown are representative coronal brain sections at the level of the dorsal hippocampus (plate 39) (Pellegrino et al., 1979), exhibiting total hybridization of the digoxigenin-labeled riboprobes. Nonspecific hybridization, estimated using the BDNF sense probe, is shown in the left column (sense). Under control conditions (control), BDNF mRNA is localized in the proximal dendrites of CA1 and CA3 pyramidal cells. Three hours after pilocarpine injection (300 mg/kg, i.p.), BDNF mRNA is found in the stratum radiatum of CA1, in the stratum lucidum, and in the radiatum of CA3 and in the proximal third of dentate gyrus (DG) granule cell dendrites. At 6 and 24 hr after injection, the dendritic staining appears to be less intense and more diffuse over the entire molecular layer in the DG and over the stratum radiatum only in CA1 and CA3. In chronic animals that did not experience seizures during the last 3 hr before being killed, dendritic staining was not different from control levels. These representative sections will not fully correlate with the mean changes in dendritic BDNF mRNA levels shown in Figure 2 because of slight differences among the four to six animals of each group. Scale bar: whole hippocampus, 750 μm; CA1, 75 μm; CA3, 150 μm; DG, 300 μm.
Figure 2.

Densitometric analysis of the effects of pilocarpine seizures. A, High-magnification of the CA1 region of a control animal (shown also in Fig. 1, control) showing proximal dendritic in situ staining for BDNF mRNA in pyramidal neurons and in interneurons within the stratum radiatum. B, CA1 region of a pilocarpine-treated rat (3 hr) showing dramatic enhancement of the BDNF mRNA in situ staining. Several strongly labeled dendrites are visible in the stratum radiatum but not in the stratum oriens. The white lines in A and B indicate the distance 0 from the cell soma used for the densitometric analysis. The left panels in C-E show the densitometric analysis of the dendritic labeling, expressed as pixel intensity (0-255 gray levels; 255 = white; 0 = black) as a function of the distance from the cell soma (in micrometers). Analysis was performed as described in Materials and Methods. Data are the means ± SE of four to six animals per group: control (thin line) and pilocarpine 3 hr (pilo 3h; thick line). Note that, under control conditions, the average gray level in CA1 and CA3 reaches background between 50 and 100 μm from the cell somas, whereas in the dentate gyrus it drops to background levels ∼40 μm from the cell bodies. *p < 0.05; ***p < 0.001 versus control values (□); ANOVA and post hoc Newman-Keuls test. lac-mol, Stratum lacunosum-moleculare; luc, stratum lucidum; ML, molecular layer; rad, stratum radiatum.
The intracellular distribution of BDNF mRNA was strikingly different in animals undergoing seizures induced by pilocarpine (300 mg/kg), a muscarinic acetylcholine receptor agonist that rapidly (latency, 16 ± 2 min) induces a robust status epilepticus lasting several hours (Turski et al., 1989). BDNF mRNA was strongly induced after pilocarpine-induced seizures, confirming previous studies (da Penha Berzaghi et al., 1993; French et al., 1999). The novel finding, however, was that there was also a dramatic accumulation of BDNF mRNA in the dendrites of all hippocampal principal neurons (Fig. 1). In the CA1 subfield, the labeling extended into the stratum radiatum up to 200 μm from the cell soma (Figs. 1, 2C) (n = 10). Maximal accumulation of BDNF mRNA in these dendrites was observed at 3 hr, and then the levels gradually decreased toward background, although they were still significantly elevated at 24 hr (Figs. 1, 2C) (n = 4). In CA3, intense labeling was observed in the strata lucidum and radiatum, but not in the stratum lacunosum-moleculare (Figs. 1, 2D). BDNF mRNA levels in CA3 pyramidal cell dendrites were still high at 6 hr (n = 3) but then returned to essentially basal levels at 24 hr (Figs. 1, 2D). Interestingly, both in CA1 and CA3, pilocarpine seizures did not lead to labeling for BDNF mRNA in the stratum oriens, which contains the basal dendrites of the pyramidal neurons. In the dentate gyrus, BDNF mRNA levels were significantly increased at 3 hr after pilocarpine administration, and only in the inner third of the molecular layer (Figs. 1, 2E). Interestingly, at 24 hr a diffuse and slightly enhanced staining was observable over the entire molecular layer, albeit in the densitometric analysis, this increase did not reach statistical significance (Figs. 1, 2E).
Importantly, the pattern of labeling corresponds closely to the distribution of particular types of synapses. The sharp boundary of labeling in CA1 at the stratum lacunosum-moleculare marks the border between the terminal field of the commissural-associational (C/A) system, which terminates in stratum radiatum and the temporo-ammonic projection from the entorhinal cortex, which terminates in stratum lacunosum-moleculare. In CA3, labeling is high over the stratum lucidum (the terminal field of mossy fibers) and over the stratum radiatum, but low levels of labeling were seen over distal dendritic laminas that receive their input from the entorhinal cortex. In the dentate gyrus, levels of labeling were high over the inner one-third of the molecular layer (the site of termination of C/A projections), and there was a sharp boundary at the transition between this system and the outer two-thirds of the molecular layer, which receives input from the entorhinal cortex via the perforant path. Taken together, these observations suggest that pilocarpine-induced seizures cause BDNF mRNA to accumulate selectively at particular types of synapses.
To explore the possibility that dendritic targeting of BDNF mRNA occurred in chronically epileptic rats (i.e., that it could be implicated in the maintenance of hyperexcitability), a group of animals (here termed “chronic”; n = 6) was killed 3-4 weeks after pilocarpine administration, when spontaneous seizures were observed. These animals were killed after a 3 hr observation period, during which no behavioral seizures occurred (i.e., if an animal displayed behavioral seizures in this observation period, it was not killed and observed again on the following day). In these animals, the staining for dendritic BDNF mRNA was similar to that seen in untreated animals in all hippocampal subfields (Figs. 1, 2).
Dendritic targeting does not occur for all mRNAs
To investigate whether pilocarpine seizure-induced accumulation of BDNF mRNA in dendrites was attributable to a general, unspecific rearrangement of cellular mRNA localization, we analyzed the subcellular distribution of the mRNA coding for the TrkC receptor after pilocarpine administration. This mRNA, in analogy with that coding for BDNF, is localized in the proximal dendritic compartment under basal conditions (Tongiorgi et al., 2000). Three hours after pilocarpine injection, the dendritic distribution of TrkC mRNA in hippocampal neurons was not changed (data not shown). This finding demonstrates that the effect of pilocarpine on BDNF mRNA distribution is not attributable to a general rearrangement of mRNA subcellular distribution.
Ultrastructural localization of BDNF mRNA
To further characterize the effects of pilocarpine seizures on the subcellular localization of BDNF mRNA, in situ hybridization was performed at the electron microscopic (EM) level. In CA1 neurons, no staining with the BDNF sense probe was detectable in the proximal (Fig. 3A)(<15 μm from the cell body) and distal (Fig. 3B) (∼50 μm from the cell body) dendrites. With the anti-sense probe, in untreated rats, gold granules for BDNF mRNA were observed in the proximal dendrites of CA1 neurons (Fig. 3C) in a polyribosome-rich region close to the cell membrane; the gold granules were often associated with polyribosome rosettes. In the distal dendrites of untreated animals (Fig. 3D), a few gold particles for BDNF mRNA were found along the dendritic shaft associated with microtubules. In addition, gold particles were found at postsynaptic specializations (Fig. 3G) (∼70 μm from the cell body). Gold particles were particularly abundant at dendritic bifurcations and were often found in association with polyribosomal structures adjacent to regions containing large dense core vesicles (Fig. 3H) (∼45 μm from the cell body).
Figure 3.
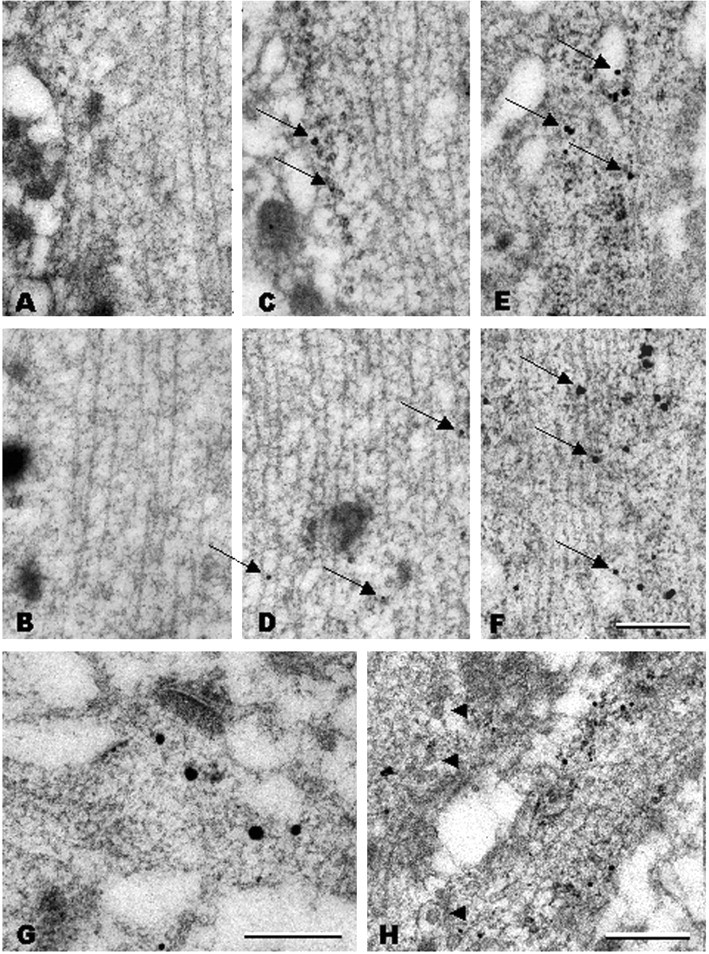
Postembedding in situ hybridization electron microscopy analysis of BDNF mRNA localization in CA1 apical dendrites. A, B, No staining was observed in proximal (A;<15 μm from cell soma) or distal (B;∼50 μm) dendrites with a BDNF sense probe. C, D, In control animals, a few gold grains for BDNF mRNA are associated with polyribosomes in proximal dendrites (C, arrows), whereas they are associated with microtubules in the distal dendrites (D, arrows). E, F, Three hours after pilocarpine administration, gold grains for BDNF mRNA are more abundant and larger both in proximal (E, arrows) and distal (F, arrows) dendrites and are mostly associated with microtubules and less frequently with polyribosomes. Under control conditions, gold grains for BDNF mRNA are also present at distal synapses (G, large black dots; ∼70 μm from the cell soma) close to the postsynaptic density and are accumulated at dendritic branchings (H; ∼45 μm from the cell soma), often in association with polyribosomes and in proximity to large dense core vesicles (arrowheads). Scale bars: (in F) A-F, 300 nm; G, 300 nm; H, 500 nm.
After pilocarpine seizures, BDNF mRNA gold granules were present in abundance in proximal (Fig. 3E) and distal (Fig. 3F) (∼50 μm from the cell soma) dendritic compartments. The dendrites also contained large intracellular vacuoles (Fig. 3E). In CA1 neurons of pilocarpine-treated rats, the BDNF mRNA gold particles were found mostly in the core of the dendrite and were often associated with microtubules (Fig. 3E, F). No labeling was found over presynaptic terminals or in the distal dendritic domains in the stratum lacunosum-moleculare of CA1 (Fig. 3G, presynaptic terminal). EM analysis of in situ hybridizations performed on dentate gyrus sections confirmed that pilocarpine-induced seizures also trigger the accumulation of BDNF mRNA in the dendrites of granule cells (Fig. 4A). In the proximal dendrites, BDNF mRNA gold particles were mostly associated with polyribosomes (Fig. 4A-D). Thus, the ultrastructural analysis (1) demonstrates that a baseline amount of BDNF mRNA is normally present in the dendrites of untreated rats, at synapses and in association with ribosomes, and (2) suggests that pilocarpine induces BDNF mRNA targeting to distal dendrites through transport along the microtubules.
Figure 4.

Postembedding in situ hybridization electron microscopy in the dentate gyrus after pilocarpine seizures. BDNF mRNA is targeted to dendrites of dentate gyrus granule cells. A, Three hours after pilocarpine administration, gold grains for BDNF mRNA can be visualized in dentate gyrus dendrites associated with a large number of polyribosomes at dendritic hillock (A, B), proximal dendrite (A, C), and dendritic branchings at more distal dendritic domains (A, D). A few grains are also associated with microtubules (A, C, D). Scale bars: A, 2 μm; (in D) B-D, 1 μm.
Pilocarpine increases BDNF protein levels in dendrites
To determine whether the dramatic accumulation of BDNF mRNA in dendrites in pilocarpine-treated animals was accompanied by a corresponding increase in protein levels, an immunohistochemical study was performed (Fig. 5). Control animals did not display a significant labeling along the dendritic length of the principal neurons in CA1 (supplemental Fig. A, available at www.jneurosci.org/cgi/content/full/24/30/6842/DC1), CA3, and the dentate gyrus. In pilocarpine-treated animals, the dendritic immunolabeling for BDNF increased dramatically in a time-dependent manner after seizure induction, reaching the maximal intensity 6 hr after injection. This peak was followed by a marked decrease at 24 hr to staining levels that were still elevated in comparison with control animals, especially in CA1. Remarkably, in the pilocarpine-treated animals, the staining in the dentate gyrus was restricted to the inner molecular layer, matching exactly the mRNA localization. Increased BDNF-like immunoreactivity was also observed in the dendrites in the chronic animals (3-4 weeks after pilocarpine-induced status epilepticus): again, dendritic staining was observed especially in CA1 but also in CA3 and in the dentate gyrus.
Figure 5.
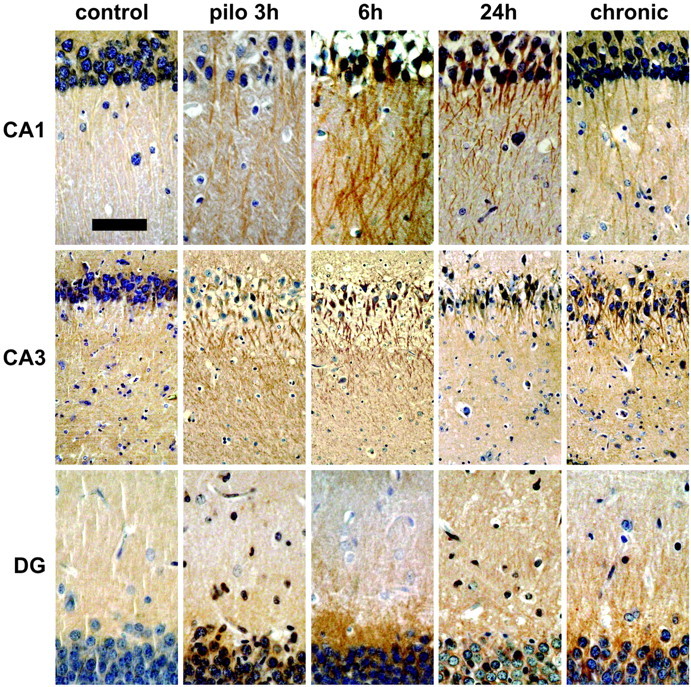
Pilocarpine seizure-induced dendritic BDNF mRNA is translated into protein. Shown are representative hematoxylin counterstained coronal brain sections at the level of the dorsal hippocampus (plate 39) (Pellegrino et al., 1979), exhibiting DAB-labeled BDNF-like immunoreactivity (LI). Omitting the primary antibody to estimate nonspecific signal yielded completely negative labeling (data not shown). Under control conditions (control), BDNF-LI is localized in the proximal dendrites. Peaking 6 hr after pilocarpine injection (pilo 6h; 300 mg/kg, i.p.), BDNF-LI is found in the stratum radiatum of CA1 pyramidal neurons, in the stratum lucidum, and in the radiatum of CA3 pyramidal neurons and in the proximal third of dentate gyrus granule cell dendrites. Increased dendritic BDNF-LI is also observed in spontaneously seizing animals killed 3-4 weeks after pilocapine administration. Scale bar: CA1 and DG, 75 μm; CA3, 120 μm.
BDNF mRNA targeting in other seizure models
To determine whether the pattern of mRNA localization depended on the type of epileptogenic stimulus, we assessed BDNF mRNA localization after kainate seizures and in the kindling model of epilepsy, in which particular pathways are activated by indwelling stimulating electrodes. Animals treated with kainate (10 mg/kg, i.p.) displayed the characteristic progression of behavioral seizure activity, reaching class 5 and entering status epilepticus 90 ± 9 min after injection. There was no detectable increase in BDNF mRNA dendritic levels 3 hr after kainate administration (Fig. 6, KA 3h) (n = 10), whereas by 6 hr after injection, there was a striking accumulation of BDNF mRNA in the dendrites of CA1 pyramidal neurons (Fig. 6, KA 6h) (n = 3), Interestingly, however, levels of labeling were not increased in other hippocampal subfields (Fig. 6B). Levels of labeling in CA1 returned to baseline by 24 hr (Fig. 6, KA 24h) (n = 3).
Figure 6.
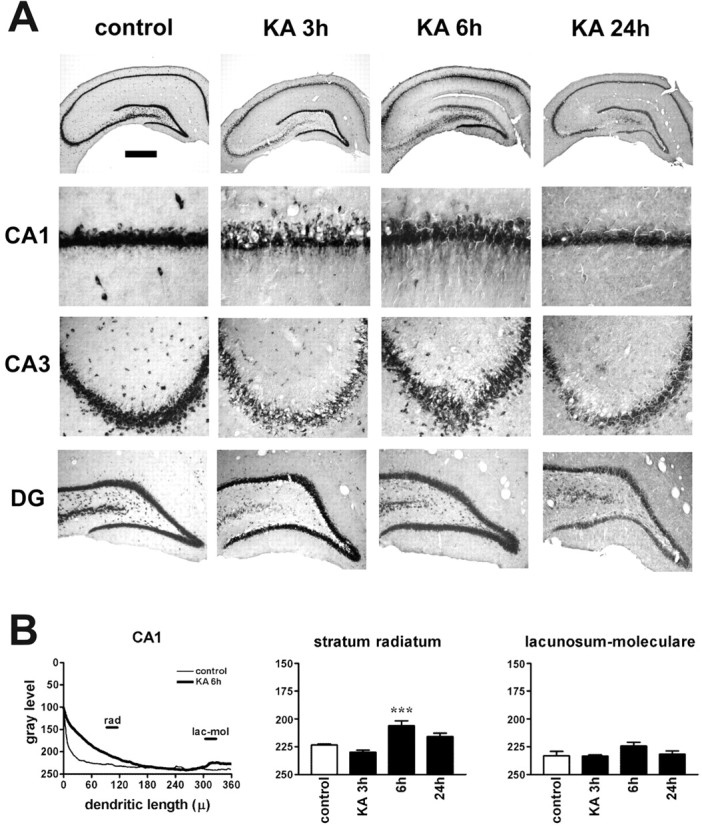
Kainate seizures induce dendritic targeting of BDNF mRNA in CA1 pyramidal neurons. A, Representative coronal brain sections at the level of the dorsal hippocampus, exhibiting total hybridization of the digoxigenin-labeled riboprobes. Six hours after kainate injection (10 mg/kg, i.p.), BDNF mRNA is found in the stratum radiatum of CA1. The serepresentative sections will not fully correlate with the mean changes in the dendritic BDNF mRNA levels shown in B because of slight differences in the three to seven animals of each group. DG, Dentate gyrus. Scale bar: whole hippocampus, 750 μm; CA1, 75 μm; CA3, 150 μm; DG, 300 μm. B, Densitometric analysis of the effects of kainate. The left panel shows the densitometric analysis of the dendritic labeling, expressed as pixel intensity (0-255 gray levels; 255 = white, 0 = black) as a function of the distance from the cell soma (in micrometers). Data are the mean of control (thin line; n = 5) and kainate 6 hr (KA 6h; thick line; n = 3). The other panels report the changes in BDNF mRNA levels in the different layers of CA1. No significant changes were observed in other hippocampal subfields. Analysis was performed as described in Materials and Methods. Data are the mean ± SE of three to seven animals per group. ***p < 0.001 versus control values (□); ANOVA and post hoc Newman-Keuls test. lac-mol, Stratum lacunosum-moleculare; rad, stratum radiatum.
A still different pattern of dendritic targeting was seen in rats that had received kindling stimulation. In this experiment, rats (n = 6) received daily kindling stimulation via an electrode implanted in the amygdala. Animals were killed 3 hr after the first class 2 kindled seizure was induced. This seizure class was reached after 4 ± 1 stimulations (cumulative AD duration, 158 ± 16 sec; last AD duration, 59 ± 6 sec). After a class 2 kindled seizure, there was a striking accumulation of BDNF mRNA in the dendrites of CA3 pyramidal neurons. This effect was more pronounced ipsi-lateral to the stimulation and was observed in the strata lucidum and radiatum, but not in the stratum lacunosum-moleculare (Fig. 7A,B). When animals that had experienced class 2 seizures were analyzed at later time points (6 hr, n = 4; 24 hr, n = 4), a progressive normalization of the dendritic staining in CA3 was observed (data not shown). In animals that were implanted with electrodes but left unstimulated, the distribution of BDNF mRNA was identical to control animals (Fig. 7A). These results demonstrate that accumulation of BDNF mRNA in the dendrites occurs in different epilepsy models, although different neuronal populations exhibit induction and dendritic targeting in the different models.
Figure 7.
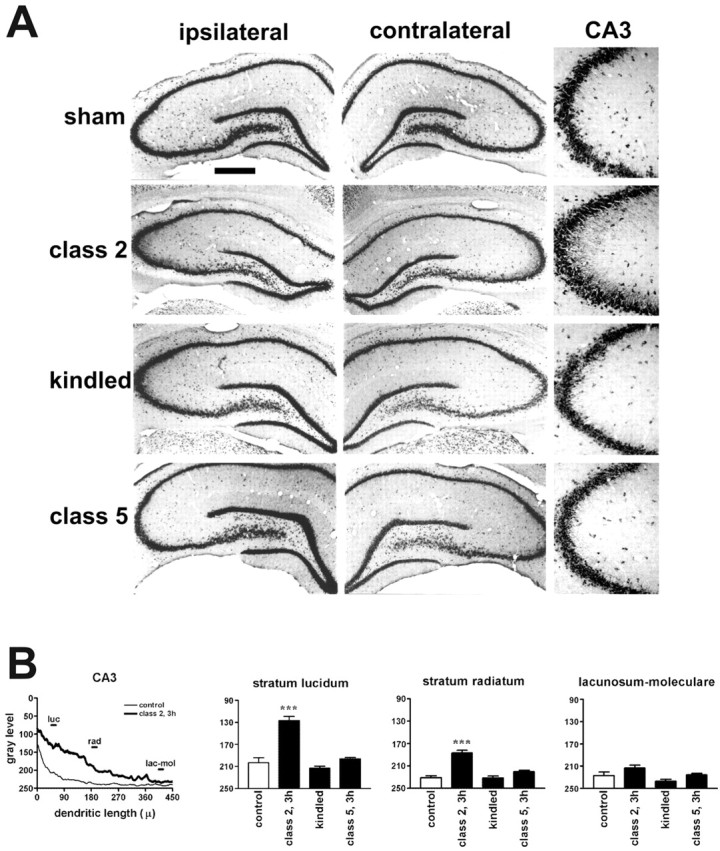
Class 2 kindled seizures induce dendritic targeting of BDNF mRNA in CA3 pyramidal neurons. A, Representative coronal brain sections at the level of the dorsal hippocampus, exhibiting total hybridization of the digoxigenin-labeled riboprobes. Three hours after class 2 kindled seizures, BDNF mRNA is found in the strata lucidum and radiatum of CA3. These representative sections will not fully correlate with the mean changes in the dendritic BDNF mRNA levels shown in B because of slight differences among the three to six animals of each group. B, Densitometric analysis of the effects of kindling. The left panel shows the densitometric analysis of the dendritic labeling, expressed as pixel intensity (0-255 gray levels; 255 = white; 0 = black) as a function of the distance from the cell soma (in micrometers). Data are the mean of sham-stimulated (control; thin line; n = 4) and class 2, 3 hr (class 2, 3h; thick line; n = 5). The other panels report the changes in BDNF mRNA levels in the different layers of CA3. No significant changes were observed in other hippocampal subfields nor at other stages of kindling development. Analysis was performed as described in Materials and Methods. Data are the mean ± SE of three to six animals per group. ***p < 0.001 versus control values (□); ANOVA and posthoc Newman-Keuls test.lac-mol, Stratum lacunosum-moleculare; luc, stratum lucidum; rad, stratum radiatum. Scale bar: whole hippocampus, 750 μm; CA3, 150 μm.
To determine whether dendritic targeting occurred after seizures in chronically epileptic animals, another group of rats was stimulated until fully kindled (three consecutive class 4 or 5 seizures) and then killed 1 week later, either without any additional stimulation (Fig. 7, kindled) (n = 6) or 3 hr after a new stimulus-evoked seizure (Fig. 7, class 5) (n = 5). Kindling criteria were reached after 13 ± 1 stimulations (cumulative AD duration, 705 ± 54 sec). The expression pattern of BDNF mRNA in kindled rats (left unstimulated for 1 week) was identical to that observed in control and sham-stimulated rats, as also confirmed by the densitometric analysis (Fig. 7B). Importantly, there was no evidence of dendritic targeting of BDNF mRNA after inducing a class 5 seizure (AD duration, 79 ± 8 sec) in a fully kindled animal (Fig. 7A, class 5, B). Thus, intense seizures per se are not sufficient stimuli for inducing the dendritic targeting. Instead, dendritic targeting occurs only after seizures that induce the cellular and molecular cascades that underlie epileptogenesis.
Non-epileptogenic stimuli do not induce dendritic accumulation of BDNF mRNA
To test whether other, non-epileptogenic types of stimuli (including non-epileptogenic seizures) would also induce a dendritic accumulation of BDNF mRNA, a group of animals (n = 3) was killed 2 hr after a single ECS, and a second group (n = 3) was killed after high-frequency stimulation (400 Hz trains, eight pulses per train, delivered at a rate of 1/10 sec for 2 hr) of the medial entorhinal cortex, a procedure that reliably induces long-term potentiation at the perforant path synapses of the dentate gyrus middle molecular layer (Steward, 1976; Steward et al., 1998). Neither one of these stimulation protocols is epileptogenic (i.e., they do not lead to spontaneous seizures). The former stimulation procedure, however, activates a diffuse dendritic targeting of Arc (Steward et al., 1998), and the latter a selective targeting of Arc mRNA (Steward et al., 1998), and an increase in the levels of α-CaMKII protein (Steward and Halpain, 1999) in the middle portion of the dendrite contacted by the activated synapses. Neither stimulation protocol produced dendritic accumulation of BDNF mRNA in any hippocampal subfield (supplemental Fig. B, available at www.jneurosci.org/cgi/content/full/24/30/6842/DC1). Thus, intense but non-epileptogenic stimuli do not induce measurable accumulation of BDNF mRNA in the dendrites.
Seizure-induced dendritic targeting of BDNF mRNA is NMDAR dependent
The data shown thus far suggest that dendritic targeting of BDNF mRNA is triggered by epileptogenic, but not other, stimuli. To begin to determine whether this phenomenon was, in fact, part of the mechanism of epileptogenesis, and to begin to define the key signal transduction pathways that trigger the accumulation of BDNF mRNA in dendrites after seizures, we attempted to prevent the dendritic targeting of BDNF mRNA without interfering with the acute seizures or with BDNF expression. Toward this aim, NMDAR antagonists (like MK801) represent an interesting tool because it has been shown that NMDAR antagonists do not prevent pharmacologically induced status epilepticus or kindled seizures (McNamara et al., 1988; Ormandy et al., 1989) but do prevent pilocarpine-induced epileptogenesis (Rice and DeLorenzo, 1998) and kindling development (McNamara et al., 1988). Moreover, NMDAR activation does not increase BDNF mRNA transcription (Zafra et al., 1990; Wetmore et al., 1994). We compared the effect of MK801 to that of the AMPA receptor antagonist NBQX, an effective anticonvulsant agent (Lees, 2000).
NBQX (30 mg/kg, i.p.) prevented pilocarpine-induced status epilepticus in two of four animals, whereas MK801 (1 mg/kg, i.p.; n = 4) had no effect on seizure activity. Nevertheless, treatment with MK801 drastically reduced the effect of pilocarpine on the dendritic targeting of BDNF mRNA in all animals (Fig. 8, pilo 3h + NBQX). NBQX reduced dendritic targeting in the animals in which it prevented status epilepticus (Fig. 8, pilo 3h + NBQX). These data demonstrate that the accumulation of BDNF mRNA in the dendrites requires activation of NMDARs and suggest that this phenomenon is causally related to epileptogenesis.
Figure 8.

Pilocarpine seizure-induced dendritic targeting of BDNF mRNA is glutamate receptor dependent. Animal pretreatment with the AMPA receptor antagonist NBQX [30 mg/kg, i.p.; administered 20 min before pilocarpine (pilo 3h + NBQX)] and with the NMDAR antagonist MK801 [1 mg/kg i.p.; administered 20 min before pilocarpine (pilo 3h + MK801)] prevented BDNF mRNA dendritic accumulation induced by pilocarpine (300 mg/kg, i.p.). Higher magnification of the CA1 subfield is shown in the bottom panels. Scale bar: whole hippocampus, 750 μm; CA1, 75 μm.
Discussion
BDNF mRNA localization under basal conditions
This study provides the first unequivocal demonstration that BDNF mRNA has a somatodendritic distribution in vivo. A number of previous studies using radioactive in situ hybridization did not specifically address the question of BDNF mRNA subcellular localization, and none was performed at the EM level. High magnification pictures were published previously, suggesting, but not conclusively demonstrating, the localization in the proximal dendritic domain of hippocampal neurons from young rats treated with kainate (Dugich-Djordjevich et al., 1992) or adult rats treated with pilocarpine (Schmidt-Kastner et al., 1996).
The EM analysis performed in this study provides new insights in the localization of BDNF mRNA in dendrites. Under control conditions, we found BDNF mRNA at distances of 70 μm from the soma, a localization that should be considered as genuinely dendritic (Peters et al., 1991). This finding is in agreement with our previous data on hippocampal neurons in vitro showing that, in unstimulated cultures, BDNF mRNA is found in apical dendrites of pyramidal neurons up to 80 μm from the cell soma (Tongiorgi et al., 1997). Remarkably, we also found an occasional presence of BDNF mRNA in the proximity of the postsynaptic compartment of excitatory synapses, which suggests its potential availability for local protein synthesis. In addition, we found the dendritic BDNF mRNA often associated with polyribosomes, suggesting active translation (Job and Eberwine, 2001).
Epileptogenic stimuli, not seizures per se, trigger dendritic localization of BDNF
Seizures induced by pilocarpine and kainate and the initial seizures induced by kindling are epileptogenic in that they trigger a process that leads to hyperexcitability and an epileptic state. These seizures cause a dramatic accumulation of BDNF in the dendrites. The time course of induction differed depending on the epileptogenic stimuli: dendritic targeting occurred somewhat later after kainate-induced, than after pilocarpine-induced, status epilepticus. Most likely, this depends on the long latency to status after kainate injection.
Importantly, class 5 seizures in the fully kindled animal did not induce dendritic targeting of BDNF mRNA. It should be emphasized that fully kindled seizures were much more intense (class 5) than those (class 2) occurring early in the course of kindling, yet class 5 seizures did not induce dendritic targeting of BDNF mRNA, whereas class 2 did. Thus, targeting is induced by stimuli that trigger the epileptogenic process, but not by seizures per se. These data suggest that the targeting of BDNF mRNA to distal dendrites is associated with epileptogenesis and not with the maintenance of the epileptic condition.
This conclusion is further supported by the fact that dendritic accumulation of BDNF mRNA does not occur after nonepileptogenic stimuli (single maximal electroshock seizures and high-frequency perforant path stimulation). These stimuli involve intense neuronal activity and trigger dendritic localization and synapse-specific targeting of Arc mRNA (Steward and Worley, 2001), but are not followed by spontaneous seizures, and are thus not epileptogenic. Although we cannot exclude that these stimulations may have effects on BDNF mRNA motility, subcellular localization, or local translation in dendrites, they do not induce the dramatic dendritic targeting seen with epileptogenic stimuli, again supporting the hypothesis that distal dendritic localization is induced exclusively by stimuli that launch the epileptogenic process.
Epileptogenic seizures cause BDNF mRNA to localize at particular sets of synapses
In the first 6 hr after pilocarpine-induced seizures, BDNF mRNA was localized in particular laminas that correspond closely to the terminal fields of the mossy fibers or of the C/A projection system. C/A projections to the dentate gyrus terminate in the inner one-third of the molecular layer, whereas C/A projections to CA1 terminate in the stratum radiatum (Raisman et al., 1965). In both areas, more distal dendritic laminas are occupied by projections from the entorhinal cortex (Steward, 1976). The neurons that give rise to the C/A projections (CA3 pyramidal and dentate gyrus hilar neurons) are highly activated during seizures (indeed, they eventually degenerate after pilocarpine-induced status) (Mello et al., 1993). The selective targeting of BDNF mRNA to the sites of termination of the C/A pathway thus suggests selective targeting to the most active synapses. Our quantitative analysis also suggests dendritic (i.e., postsynaptic) accumulation of BDNF mRNA at specific synaptic contacts after kainate-induced seizures (stratum radiatum) and at the early stages of kindling development (in the mossy and in the C/A fiber-CA3 synaptic area of the stratum lucidum and stratum radiatum, respectively).
It remains unclear why both in vivo (this study) and in vitro (Tongiorgi et al., 1997) BDNF mRNA staining extended up to two-thirds of the length of pyramidal neurons apical dendrites or one-third of the dentate gyrus molecular layer. It is conceivable that the strong activation of specific sets of synapses may create a docking signal that keeps the mRNA from traveling on to distal dendrites, as is the case with Arc mRNA (Steward and Worley, 2001).
Dendritic targeting is not related to excitotoxic injury
Both pilocarpine and kainate induce excitotoxic death of neurons in the hippocampus. Thus, it is reasonable to consider the possibility that the dendritic targeting of BDNF mRNA is related to injury. This possibility can be excluded because dendritic targeting of BDNF clearly occurs in situations that do not involve excitotoxic injury. First, neither pilocarpine nor kainate induce the degeneration of dentate granule cells, yet BDNF is targeted to dendrites in these neurons. Second, kindling stimulation, which does not induce detectable neuronal degeneration, induces dendritic targeting of BDNF mRNA.
Localization of BDNF mRNA may allow local synthesis of BDNF protein
After pilocarpine-induced seizures, BDNF protein accumulates in dendrites with a distribution that closely matches the distribution of BDNF mRNA, suggesting that dendritic BDNF mRNA may be locally translated into protein. In contrast, in chronic, spontaneously seizing pilocarpine-treated animals, increased dendritic protein localization did not correlate with increased dendritic mRNA levels. This discrepancy may be explained considering that chronic animals were killed at least 3 hr after the last seizure and, therefore, dendritic BDNF mRNA (but not protein) levels may have already returned to baseline. Alternative explanations include increased translation of dendritic mRNA, or increased transport of protein synthesized in the cell soma, or both.
The observation that BDNF protein levels are increased in the dentate gyrus inner molecular layer of chronic pilocarpine-treated animals is consistent with the observations of Scharfman et al. (1999). However, whereas these authors suggested an axonal localization of BDNF in sprouted mossy fibers, our data suggest that at least part of the staining within the inner molecular layer is in the dendrites of granule cells. This idea is supported by the observation that BDNF protein accumulation in the inner molecular layer can be observed as early as 3 hr after pilocarpine injection, a time at which no axonal sprouting is observed. It should be noted that the mossy fibers are poorly labeled by the anti-BDNF antibody used in this study, whereas they are strongly labeled by another anti-BDNF antibody (Conner et al., 1997). Because both these antibodies have been validated in various studies, differences in staining patterns may depend on a different capacity of recognizing different isoforms or post-translational modifications of BDNF.
Significance of dendritic BDNF for epileptogenesis
The present results demonstrate that BDNF mRNA and protein are accumulated in dendrites by events that trigger cellular processes leading to epilepsy. Might this targeting actually be a key step of epileptogenesis? One clue in this regard is that NMDAR antagonists (MK801) block dendritic targeting and prevent pilocarpine-induced epileptogenesis (Rice and DeLorenzo, 1998) and kindling development (McNamara et al., 1988) but do not prevent pharmacologically induced status epilepticus or kindled seizures (McNamara et al., 1988; Ormandy et al., 1989). In contrast, an AMPA antagonist (NBQX) prevented accumulation of BDNF mRNA in the dendrites only in animals in which it prevented seizures. Because NMDA agonists do not increase BDNF gene expression (Zafra et al., 1990; Wetmore et al., 1994), NMDAR activation may represent the trigger for a program that controls BDNF mRNA targeting without requiring new mRNA transcription. Interestingly, NMDARs have been also implicated in the activity-dependent dendritic targeting of Arc mRNA (Steward and Worley, 2001).
The mechanism(s) by which dendritic BDNF may play a causal role in epileptogenesis remains to be investigated. Local accumulation of BDNF is capable of potentiating excitatory synaptic transmission on a long-term basis (Kang and Schuman, 1995, 1996), which could account for increased network excitability in epilepsy. In addition, one particularly potentiated synapse in kindled animals is the one between mossy fibers and the CA3 pyramidal cell (King et al., 1985) (i.e., the synaptic contact at which we detected dendritic accumulation of BDNF mRNA during kindling development). If dendritic BDNF-mediated synaptic potentiation was a paramount mechanism of epileptogenesis, our data would predict that pilocarpine status epilepticus will potentiate many synapses, whereas one (Schaffer to CA1) should be potentiated the most after kainate. However, BDNF may be only one of the factors involved in the synaptic potentiation of epileptogenesis.
In conclusion, we propose that dendritic targeting of BDNF mRNA may be causally involved in synaptic potentiation associated with epileptogenesis and thus may represent a new therapeutic target for some forms of partial epilepsy.
Footnotes
This work was supported by grants from Telethon-Italy (E0954 to E.T. and M.S.), the Italian National Council for Research (Strategico “Genomica Funzionale” to M.S. and E.T.), SISSA, the Ministero della Ricerca Scientifica e Tecnologica (COFIN 1997 to A.C.), and the National Institutes of Health (NS12333 to O.S.). We are grateful to Dr. Helen Scharfman (Helen Hayes Hospital, New York, NY) for useful discussions and to Prof. Andrea Nistri (SISSA, Trieste, Italy) for advice on pharmacological treatments. We also thank Prof. James McNamara (Duke University, Durham, NC) for providing sections from BDNF knock-out animals for control experiments to verify antibody specificity. In addition, we thank Cristina Chiaruttini and Claudio Gamboz (University of Trieste, Trieste, Italy) for technical support.
Correspondence should be addressed to Dr. Enrico Tongiorgi, BRAIN Centre for Neuroscience, Department of Biology, University of Trieste, Via Giorgieri, 10, 34127 Trieste, Italy. E-mail: tongi@units.it.
Copyright © 2004 Society for Neuroscience 0270-6474/04/246842-11$15.00/0
References
- Altar CA, Di Stefano PS (1998) Neurotrophin trafficking by anterograde transport. Trends Neurosci 21: 433-437. [DOI] [PubMed] [Google Scholar]
- Bengzon J, Kokaia Z, Ernfors P, Kokaia M, Leanza G, Nilsson OG, Persson H, Lindvall O (1993) Regulation of neurotrophin and trkA, trkB and trkC tyrosine kinase receptor messenger RNA expression in kindling. Neuroscience 53: 433-446. [DOI] [PubMed] [Google Scholar]
- Binder DK, Routbort MJ, Ryan TE, Yancopoulos DG, McNamara JO (1999) Selective inhibition of kindling development by intraventricular administration of TrkB receptor body. J Neurosci 19: 1424-1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder DK, Croll SD, Gall CM, Scharfman HE (2001) BDNF and epilepsy: too much of a good thing? Trends Neurosci 24: 47-53. [DOI] [PubMed] [Google Scholar]
- Brooks-Kayal AR, Shumate MD, Jin H, Rikhter TY, Coulter DA (1998) Selective changes in single cell GABA(A) receptor subunit expression and function in temporal lobe epilepsy. Nat Med 4: 1166-1172. [DOI] [PubMed] [Google Scholar]
- Causing CG, Gloster A, Aloyz R, Bamji SX, Chang E, Fawcett J, Kuchel G, Miller FD (1997) Synaptic innervation density is regulated by neuron-derived BDNF. Neuron 18: 257-267. [DOI] [PubMed] [Google Scholar]
- Chiang LW, Grenier JM, Ettwiller L, Jenkins LP, Ficenec D, Martin J, Jin F, DiStefano PS, Wood A (2001) An orchestrated gene expression component of neuronal programmed cell death revealed by cDNA array analysis. Proc Natl Acad Sci USA 98: 2814-2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conner JM, Lauterborn JC, Yan Q, Gall CM, Varon S (1997) Distribution of brain-derived neurotrophic factor (BDNF) protein and mRNA in the normal adult rat CNS: evidence for anterograde axonal transport. J Neurosci 17: 2295-2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Penha Berzaghi M, Cooper J, Castren E, Zafra F, Sofroniew M, Thoenen H, Lindholm D (1993) Cholinergic regulation of brain-derived neurotrophic factor (BDNF) and nerve growth factor (NGF) but not neurotrophin-3 (NT-3) mRNA levels in the developing rat hippocampus. J Neurosci 13: 3818-3826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugich-Djordjevic MM, Tocco G, Willoughby DA, Najm I, Pasinetti G, Thompson RF, Baudry M, Lapchak PA, Hefti F (1992) BDNF mRNA expression in the developing rat brain following kainic acid-induced seizure activity. Neuron 8: 1127-1138. [DOI] [PubMed] [Google Scholar]
- Elmer E, Kokaia Z, Kokaia M, Carnahan J, Nawa H, Lindvall O (1998) Dynamic changes of brain-derived neurotrophic factor protein levels in the rat forebrain after single and recurring kindling-induced seizures. Neuroscience 83: 351-362. [DOI] [PubMed] [Google Scholar]
- Ernfors P, Bengzon J, Kokaia Z, Persson H, Lindvall O (1991) Increased levels of messenger RNAs for neurotrophic factors in the brain during kindling epileptogenesis. Neuron 7: 165-176. [DOI] [PubMed] [Google Scholar]
- Fawcett JP, Aloyz R, McLean JH, Pareek S, Miller FD, McPherson PS, Murphy RA (1997) Detection of brain-derived neurotrophic factor in a vesicular fraction of brain synaptosomes. J Biol Chem 272: 8837-8840. [DOI] [PubMed] [Google Scholar]
- French SJ, Humby T, Horner CH, Sofroniew MV, Rattray M (1999) Hippocampal neurotrophin and trk receptor mRNA levels are altered by local administration of nicotine, carbachol and pilocarpine. Brain Res Mol Brain Res 67: 124-136. [DOI] [PubMed] [Google Scholar]
- He XP, Minichiello L, Klein R, McNamara JO (2002) Immunohistochemical evidence of seizure-induced activation of trkB receptors in the mossy fiber pathway of adult mouse hippocampus. J Neurosci 22: 7502-7508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Doherty JJ, Dingledine R (2002) Altered histone acetylation at glutamate receptor 2 and brain-derived neurotrophic factor genes is an early event triggered by status epilepticus. J Neurosci 22: 8422-8428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isackson PJ, Huntsman MM, Murray KD, Gall CM (1991) BDNF mRNA expression is increased in adult rat forebrain after limbic seizures: temporal patterns of induction distinct from NGF. Neuron 6: 937-948. [DOI] [PubMed] [Google Scholar]
- Job C, Eberwine J (2001) Identification of sites for exponential translation in living dendrites. Proc Natl Acad Sci USA 98: 13037-13042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H, Schuman EM (1995) Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science 267: 1658-1662. [DOI] [PubMed] [Google Scholar]
- Kang H, Schuman EM (1996) A requirement for local protein synthesis in neurotrophin-induced hippocampal synaptic plasticity. Science 273: 1402-1406. [DOI] [PubMed] [Google Scholar]
- Kaplan DR, Miller FD (2000) Neurotrophin signal transduction in the nervous system. Curr Opin Neurobiol 10: 381-391. [DOI] [PubMed] [Google Scholar]
- King GL, Dingledine R, Giacchino JL, McNamara JO (1985) Abnormal neuronal excitability in hippocampal slices from kindled rats. J Neurophysiol 54: 1295-1304. [DOI] [PubMed] [Google Scholar]
- Kokaia M, Ernfors P, Kokaia Z, Elmer E, Jaenisch R, Lindvall O (1995) Suppressed epileptogenesis in BDNF mutant mice. Exp Neurol 133: 215-224. [DOI] [PubMed] [Google Scholar]
- Lahteinen S, Pitkanen A, Saarelainen T, Nissinen J, Koponen E, Castren E (2002) Decreased BDNF signalling in transgenic mice reduces epileptogenesis. Eur J Neurosci 15: 721-734. [DOI] [PubMed] [Google Scholar]
- Lauterborn JC, Rivera S, Stinis CT, Hayes VY, Isackson PJ, Gall CM (1996) Differential effects of protein synthesis inhibition on the activity-dependent expression of BDNF transcripts: evidence for immediate-early gene responses from specific promoters. J Neurosci 16: 7428-7436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees GJ (2000) Pharmacology of AMPA/kainate receptor ligands and their therapeutic potential in neurological and psychiatric disorders. Drugs 59: 33-78. [DOI] [PubMed] [Google Scholar]
- Lu B (2003) BDNF and activity-dependent synaptic modulation. Learn Mem 10: 86-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B, Chow A (1999) Neurotrophins and hippocampal synaptic transmission and plasticity. J Neurosci Res 58: 76-87. [PubMed] [Google Scholar]
- Maisonpierre PC, Le Beau MM, Espinosa RI, Ip NY, Belluscio L, La Monte S, Squinto S, Furth ME, Yancopulos GD (1991) Human and rat BDNF and NT3: gene structures, distributions and chromosomal localization. Genomics 10: 558-568. [DOI] [PubMed] [Google Scholar]
- McNamara JO (1999) Emerging insights into the genesis of epilepsy. Nature [Suppl] 399: A15-A22. [DOI] [PubMed] [Google Scholar]
- McNamara JO, Russell RD, Rigsbee L, Bonhaus DW (1988) Anticonvulsant and antiepileptogenic actions of MK-801 in the kindling and electroshock models. Neuropharmacology 27: 563-568. [DOI] [PubMed] [Google Scholar]
- Mello LE, Cavalheiro EA, Tan AM, Kupfer WR, Pretorius JK, Babb TL, Finch DM (1993) Circuit mechanisms of seizures in the pilocarpine model of chronic epilepsy: cell loss and mossy fiber sprouting. Epilepsia 34: 985-995. [DOI] [PubMed] [Google Scholar]
- Miranda RC, Sohrabji F, Toran-Allerand CD (1993) Neuronal colocalization of mRNAs for neurotrophins and their receptors in the developing central nervous system suggests a potential for autocrine interactions. Proc Natl Acad Sci USA 90: 6439-6443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mody I (1999) Synaptic plasticity in kindling. Adv Neurol 79: 631-643. [PubMed] [Google Scholar]
- Murray KD, Isackson PJ, Eskin TA, King MA, Montesinos SP, Abraham LA, Roper SN (2000) Altered mRNA expression for brain-derived neurotrophic factor and type II calcium/calmodulin-dependent protein kinase in the hippocampus of patients with intractable temporal lobe epilepsy. J Comp Neurol 418: 411-422. [DOI] [PubMed] [Google Scholar]
- Ormandy GC, Jope RS, Snead 3rd OC (1989) Anticonvulsant actions of MK-801 on the lithium-pilocarpine model of status epilepticus in rats. Exp Neurol 106: 172-180. [DOI] [PubMed] [Google Scholar]
- Patapoutian A, Reichardt LF (2001) Trk receptors: mediators of neurotrophin action. Curr Opin Neurobiol 11: 272-280. [DOI] [PubMed] [Google Scholar]
- Pellegrino LJ, Pellegrino AS, Cushman AJ (1979) A stereotaxic atlas of the rat brain. New York: Plenum.
- Peters A, Palay S, Webster H (1991) The fine structure of the nervous system. New York: Oxford UP.
- Racine RJ (1972) Modification of seizure activity by electrical stimulation II. Motor seizure. Electroencephalogr Clin Neurophysiol 32: 281-294. [DOI] [PubMed] [Google Scholar]
- Raisman G, Cowan WM, Powell TP (1965) The extrinsic afferent, commissural, and associational fibers of the hippocampus. Brain 88: 963-996. [Google Scholar]
- Righi M, Tongiorgi E, Cattaneo A (2000) Brain-derived neurotrophic factor (BDNF) induces dendritic targeting of BDNF and tyrosine kinase B mRNAs in hippocampal neurons through a phosphatidylinositol-3 kinase-dependent pathway. J Neurosci 20: 3165-3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice AC, DeLorenzo RJ (1998) NMDA receptor activation during status epilepticus is required for the development of epilepsy. Brain Res 782: 240-247. [DOI] [PubMed] [Google Scholar]
- Scharfman HE, Goodman JH, Sollas AL (1999) Actions of brain-derived neurotrophic factor in slices from rats with spontaneous seizures and mossy fiber sprouting in the dentate gyrus. J Neurosci 19: 5619-5631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt-Kastner R, Humpel C, Wetmore C, Olson L (1996) Cellular hybridization for BDNF, trkB, and NGF mRNAs and BDNF-immunoreactivity in rat forebrain after pilocarpine-induced status epilepticus. Exp Brain Res 107: 331-347. [DOI] [PubMed] [Google Scholar]
- Schuman E (1999) Neurotrophin regulation of synaptic transmission. Curr Opin Neurobiol 9: 105-109. [DOI] [PubMed] [Google Scholar]
- Schwartz PM, Borghesani PR, Levy RL, Pomeroy SL, Segal RA (1997) Abnormal cerebellar development and foliation in BDNF-/- mice reveals a role for neurotrophins in CNS patterning. Neuron 19: 269-281. [DOI] [PubMed] [Google Scholar]
- Simonato M, Bregola G, Armellin M, Del Piccolo P, Rodi D, Zucchini S, Tongiorgi E (2002) Dendritic targeting of mRNAs for plasticità genes in experimental models of temporal lobe epilepsy. Epilepsia [Suppl] 43: 153-158. [DOI] [PubMed] [Google Scholar]
- Steward O (1976) Topographic organization of the projections from the entorhinal area to the hippocampal formation of the rat. J Comp Neurol 167: 285-314. [DOI] [PubMed] [Google Scholar]
- Steward O, Halpain S (1999) Lamina-specific synaptic activation causes domain-specific alterations in dendritic immunostaining for MAP2 and CAM kinase II. J Neurosci 19: 7834-7845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steward O, Schuman EM (2001) Protein synthesis at synaptic sites on dendrites. Annu Rev Neurosci 24: 299-325. [DOI] [PubMed] [Google Scholar]
- Steward O, Worley PF (2001) Selective targeting of newly synthesized Arc mRNA to active synapses requires NMDA receptor activation. Neuron 30: 227-240. [DOI] [PubMed] [Google Scholar]
- Steward O, Wallace CS, Lyford GL, Worley PF (1998) Synaptic activation causes the mRNA for the IEG Arc to localize selectively near activated postsynaptic sites on dendrites. Neuron 21: 741-751. [DOI] [PubMed] [Google Scholar]
- Thoenen H (2000) Neurotrophins and activity-dependent plasticity. Prog Brain Res 128: 183-191. [DOI] [PubMed] [Google Scholar]
- Tongiorgi E, Righi M, Cattaneo A (1997) Activity-dependent dendritic targeting of BDNF and TrkB mRNAs in hippocampal neurons. J Neurosci 17: 9492-9505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tongiorgi E, Righi M, Cattaneo A (1998) A non-radioactive in situ hybridization method that does not require RNAse-free conditions. J Neurosci Methods 85: 129-139. [DOI] [PubMed] [Google Scholar]
- Tongiorgi E, Armellin M, Cattaneo A (2000) Differential somato-dendritic localization of TrkA, TrkB, TrkC and p75 mRNAs in vivo. NeuroReport 11: 3265-3268. [DOI] [PubMed] [Google Scholar]
- Turski L, Ikonomidou C, Turski W, Bortolotto Z, Cavalheiro E (1989) Rev: cholinergic mechanisms and epileptogenesis. The seizures induced by pilocarpine: a novel experimental model of intractable epilepsy. Synapse 3: 154-171. [DOI] [PubMed] [Google Scholar]
- Wallace CS, Lyford GL, Worley PF, Steward O (1998) Differential intracellular sorting of immediate early gene mRNAs depends on signals in the mRNA sequence. J Neurosci 18: 26-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetmore C, Olson L, Bean AJ (1994) Regulation of brain-derived neurotrophic factor (BDNF) expression and release from hippocampal neurons is mediated by non-NMDA type glutamate receptors. J Neurosci 14: 1688-1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zafra F, Hengerer B, Leibrock J, Thoenen H, Lindholm D (1990) Activity dependent regulation of BDNF and NGF mRNAs in the rat hippocampus is mediated by non-NMDA glutamate receptors. EMBO J 9: 3545-3550. [DOI] [PMC free article] [PubMed] [Google Scholar]