

Abstract
The upregulation of voltage-gated sodium channel Nav1.3 has been linked to hyperexcitability of axotomized dorsal root ganglion (DRG) neurons, which underlies neuropathic pain. However, factors that regulate delivery of Nav1.3 to the cell surface are not known. Contactin/F3, a cell adhesion molecule, has been shown to interact with and enhance surface expression of sodium channels Nav1.2 and Nav1.9. In this study we show that contactin coimmunoprecipitates with Nav1.3 from postnatal day 0 rat brain where this channel is abundant, and from human embryonic kidney (HEK) 293 cells stably transfected with Nav1.3 (HEK-Nav1.3). Purified GST fusion proteins of the N and C termini of Nav1.3 pull down contactin from lysates of transfected HEK 293 cells. Transfection of HEK-Nav1.3 cells with contactin increases the amplitude of the current threefold without changing the biophysical properties of the channel. Enzymatic removal of contactin from the cell surface of cotransfected cells does not reduce the elevated levels of the Nav1.3 current. Finally, we show that, similar to Nav1.3, contactin is upregulated in axotomized DRG neurons and accumulates within the neuroma of transected sciatic nerve. We propose that the upregulation of contactin and its colocalization with Nav1.3 in axotomized DRG neurons may contribute to the hyper-excitablity of the injured neurons.
Keywords: axotomy, sensory neurons, cell adhesion molecule, neuroma, tetrodotoxin sensitive, GPI-anchor
Introduction
Voltage-gated sodium channels mediate a rapid and transient increase in Na+ permeability in response to membrane depolarization, thereby contributing to the generation and conduction of action potentials. Mutations of pore-forming α-subunits underlie a number of human and animal disorders (Goldin, 2001; Keating and Sanguinetti, 2001; Meisler et al., 2001), and dysregulated sodium channel expression may contribute to neuropathic pain (Waxman et al., 2000).
The tetrodotoxin-sensitive (TTX-S) channel Nav1.3 is abundantly expressed in neuronal tissues during embryonic and neonatal stages of development and is rare in adult tissues (Beckh et al., 1989; Felts et al., 1997; Shah et al., 2001; Lindia and Abbadie, 2003). After axonal transection, Nav1.3 is upregulated in dorsal root ganglia (DRG) neurons (Waxman et al., 1994; Dib-Hajj et al., 1996; Black et al., 1999; Kim et al., 2001), and a rapidly repriming TTX-S current emerges in these neurons (Cummins and Waxman, 1997). Nav1.3 produces a similar rapidly-repriming TTX-S current in human embryonic kidney (HEK) 293 cells (Cummins et al., 2001), adding to the evidence that upregulation of Nav1.3 may play a role in rendering axotomized DRG neurons hyperexcitable, thus contributing to neuropathic pain. Within neuromas, Nav1.3 protein accumulates within the transected axon tips (Black et al., 1999) and may thus participate in the generation of ectopic discharges (Matzner and Devor, 1994; Kim et al., 2001). Growth factors suppress the expression of Nav1.3 in axotomized DRG neurons (Boucher et al., 2000; Leffler et al., 2002), leading to the suppression of the rapidly repriming TTX-S sodium currents (Leffler et al., 2002) and ameliorating neuropathic pain symptoms (Boucher et al., 2000). Similarly, Nav1.3 is upregulated in dorsal horn nociceptive neurons after spinal cord injury, and antisense knock-down of Nav1.3 ameliorates the pain syndrome that accompanies the injury (Hains et al., 2003).
Contactin, a glycosyl-phosphatidylinositol (GPI)-anchored protein that is expressed in neurons and glia (Ranscht, 1988; Brummendorf et al., 1989; Gennarini et al., 1989), associates with Nav1.2 in vivo via the β1-subunit and enhances the channel density at the plasma membrane (Kazarinova-Noyes et al., 2001). The interaction of contactin and Nav1.9, which is mediated by the C terminus of the channel (C. Liu et al., 2001), probably requires a cellular linker, for example a β-subunit. The deletion of β1 and β1B subunits abolishes the coimmunoprecipitation of sodium channels and contactin from brains of postnatal day 17 (P17)-P19 mice (Chen et al., 2004). The role of contactin in enhancing sodium channel density at the cell surface is reminiscent of the role of β-subunits (Isom, 2001), and is consistent with the homology of the extracellular domains of β-subunits and Ig domains of cell adhesion molecules, including contactin (Isom et al., 1995; Isom and Catterall, 1996).
In this study, we show that contactin associates with Nav1.3 in P0 brain tissue and interacts with both the N and C termini of Nav1.3. The expression of contactin and Nav1.3 in the HEK-Nav1.3 stable cell line significantly increases the current density. We also show that contactin is upregulated in parallel with Nav1.3 in axotomized DRG neurons and accumulates together with Nav1.3 at the injured axon tips within the neuroma.
Materials and Methods
Antibodies and cell lines. Affinity-purified polyclonal Nav1.3- and Nav1.9-specific antibodies against channel peptides (Nav1.3, aa 511-524; Nav1.9, aa 1748-1765) have been described previously (Fjell et al., 2000; Tyrrell et al., 2001; Hains et al., 2002). The Nav1.3 antibody was used for immunohistochemistry and immunoprecipitation assays but was not sufficiently robust for Western blot analysis under current experimental conditions. Monoclonal anti-pan sodium channel antibody (Sigma, St. Louis, MO) was used for immunocytochemistry of cell cultures. Anti-pan sodium channel antibody (Upstate Laboratories, Waltham, MA) was used for Western blot analysis. Polyclonal goat anti-contactin antibody (R & D Systems, Minneapolis, MN) was used for immunocytochemistry and Western blotting, but was not effective in immunoprecipitation assays. Anti-green fluorescent protein (GFP) polyclonal antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). HEK 293 cells were transfected with a pcDNA3 construct carrying rat Nav1.3 insert (Cummins et al., 2001). HEK 293 cells stably expressing Nav1.3 (termed HEK-Nav1.3) were selected using G418, and a stable line producing robust Nav1.3 current was used in this study.
Plasmid constructs. The bacterial expression vector pGEX-3X (Invitrogen, Carlsbad, CA) was used to produce recombinant proteins in Escherichia coli. The N-terminal (N; aa 1-123), intracellular loop 1 (L1; aa 428-705), intracellular loop 2 (L2; aa 938-1151), intracellular loop 3 (L3; aa 1368-1472), and C-terminal (C; aa 1722-1952) fragments of rat Nav1.3 (GenBank accession number Y00766) were subcloned in frame into the BamHI-EcoRI sites of pGEX-3X (Invitrogen) to produce recombinant proteins GST-Nav1.3N, GST-Nav1.3L1, GST-Nav1.3L2, GST-Nav1.3L3, and GST-Nav1.3C. Rat contactin (Ctn; GenBank accession number D38492) was cloned in frame into the XhoI-KpnI sites of pEGFP-N1 (Clontech, Palo Alto, CA) as described previously (C. Liu et al., 2001) to produce Ctn-GFP fusion protein. The plasmid pEGFP-N1 (Clontech) was used to produce the GFP protein. The open reading frame of rat contactin was subcloned into the mammalian expression vector pTarget (Promega, Madison, WI) to produce recombinant full-length Ctn. The Ctn-GFP construct was used in several assays to facilitate detection of transfected cells and in immunoprecipitation experiments using anti-GFP antibodies to precipitate the contactin channel complex. The pTarget contactin construct was used to test the effect of contactin removal from the cell surface on the elevated levels of the Nav1.3 current. Both contactin and Ctn-GFP were equally efficient in increasing the Nav1.3 current density in HEK 293 cells. The sodium channel auxiliary subunit β1 (Isom et al., 1992) was cloned in frame into the HinDIII-BamHI sites of pDs-Red2-N1 vector (Clontech) to produce the plasmid pβ1-Red. The β1-Red was used together with Ctn-GFP in electrophysiological studies to allow for red-green identification of cotransfected HEK-Nav1.3 cells. In separate experiments to demonstrate coimmunoprecipitation of Nav1.3 and contactin from the HEK-Nav1.3 cell line, we tested the effect of cotransfection of contactin and a human β1 construct (Lossin et al., 2002).
Expression and purification of glutathione S-transferase fusion proteins. For expression of glutathione S-transferase (GST) fusion proteins, Nav1.3 derivatives in pGEX3 (pGEX-Nav1.3N, pGEX-Nav1.3L1, pGEXNav1.3L2, pGEX-Nav1.3L3, and pGEX-Nav1.3C) were transformed into E. coli DH5α (Invitrogen) and grown in culture. Protein expression was induced by the addition of 0.1 mm isopropyl-β-d-thiogalactopyranoside for 2 hr. Cells were lysed in 1% Triton X-100, and fusion proteins were affinity-purified on glutathione-agarose beads. Beads were washed extensively in PBS, and fusion proteins were eluted in 5 mm glutathione. To verify recombinant protein production, 10 μl of the eluted proteins were separated by SDS-PAGE, and the gel was stained with Coomassie blue.
Real-time RT-PCR.Details are provided in the supplemental material on-line, available at www.jneurosci.org/cgi/content/full/24/33/9387/DC1.
In vitro GST binding assay. To examine the binding of contactin to the N and C termini and the intracellular loops of Nav1.3 in vitro, glutathione-Sepharose beads loaded with 0.5 μg of protein of GST, serving as a negative control, GST-Nav1.3N, GST-Nav1.3L1, GST-Nav1.3L2, GST-Nav1.3L3, or GST-Nav1.3C were incubated with extracts (500 μg) from HEK 293 cells transfected with an expression plasmid encoding either GFP alone or full-length Ctn-GFP, as indicated, in an equal volume of buffer AM (2 mm Tris-HCl, pH 7.9; 0.1 mm EDTA; 10% glycerol; 5 mm MgCl2; 0.5 mm DTT; protease inhibitors) supplemented with 100 mm KCl and 0.5 mg/ml bovine serum albumin. The beads were washed extensively in wash buffer (PBS and 0.1% Triton X-100), and bound proteins were separated by 7.5% SDS-PAGE. GFP fusion proteins were detected by immunoblotting with anti-GFP antibodies (Santa Cruz Biotechnology).
Coimmunoprecipitation of Nav1.3 and contactin. HEK-Nav1.3 cells were grown in 100 mm dishes in DMEM containing 10% fetal bovine serum. After reaching 50% confluence, the cultures were transfected with the mammalian Ctn-GFP or contactin construct. Control HEK 293 cells (not expressing Nav1.3) were also transfected with the Ctn-GFP construct. Cells were harvested 48 hr later in cell lysis buffer (50 mm Tris-HCl, pH 8.0; 10 mm EDTA; 1% Triton X-100; protease inhibitors) and sonicated for 3 sec on ice. Cell lysates were incubated at 4°C for 2 hr, and the insoluble pellet was removed by centrifugation in a microcentrifuge for 5 min at 4°C. After 1 hr of incubation with either Nav1.3-specific antibody (5 μg/ml) or GFP monoclonal antibody (4 μg/ml), the cell extracts were incubated with 60 μl of protein A-agarose (Invitrogen) at 4°C overnight. After washing five times with PBS, the bound proteins were released by incubation in 20 μl of 2× SDS sample buffer for 20 min at 37°C. The released proteins were examined by Western blotting with either anti-GFP (0.2 μg/ml) or anti-pan (0.8 μg/ml) sodium channel antibodies.
Contactin and Nav1.3 were coimmunoprecipitated from rat brain tissue. Rat brains from newborn litters (P0) were quickly dissected and homogenized in ice-cold lysis buffer. The homogenate was treated as described above for cell lysates and precipitated with anti-Nav1.3 antibody and protein A-agarose. Immunoprecipitated proteins were examined by Western blotting with anti-contactin and pan sodium channel antibodies.
Immunocytochemistry: immunostaining of HEK 293 and HEK-Nav1.3.
HEK 293 and HEK-Nav1.3 cells transfected with the contactin construct were cultured on coverslips and processed for immunocytochemistry. Contactin antibody (2 μg/ml) was used to immunolabel live cells for 30 min at 4°C. Coverslips were then incubated sequentially: (1) complete saline solution, two times for 1 min each; (2) 4% paraformaldehyde in 0.14 m Sorensen's phosphate buffer, 10 min; (3) PBS, three times for 3 min each; (4) PBS containing 5% normal horse serum, 2% bovine serum albumin, and 0.1% Triton X-100, 15 min; (5) monoclonal anti-pan sodium channel (1 μg/ml; Sigma) in blocking solution for 2 hr at room temperature; (6) PBS, six times for 5 min each; (7) secondary antibodies (0.5 μg/ml goat anti-mouse IgG-Cy3, Amersham Biosciences, Piscataway, NJ; 2 μg/ml donkey anti-goat IgG-Alexa Fluor 488, Molecular Probes, Eugene, OR), in blocking solution for 2 hr at room temperature; and (8) PBS, six times for 5 min each.
Imaging. Cells were examined with a confocal laser scanning microscope (Nikon PCM 2000) using 20× numerical aperture (NA) 0.75 or 60× NA 1.4 objectives. Cells were optically sectioned in the xy plane with a minimum slice thickness of 0.5 μm with multiple scan averaging. Images were taken through the central planes of all cells and processed with Adobe Photoshop.
Image analysis. Signal intensities for Nav1.3 in HEK-Nav1.3 or HEK-Nav1.3 cells transfected with contactin were quantified using IPLAB image-processing software (Scanalytics, Fairfax, VA). Densitometric measurements were taken through the cytoplasmic section of the cell profile outside of the nucleus; fluorescence intensity in arbitrary units was plotted against the sectional length in pixels (Okuse et al., 2002).
Enzymatic removal of contactin from cell surface of transfected HEK-Nav1.3 cells. HEK-Nav1.3 cells were transfected with the contactin construct. After 48 hr, cells were incubated in DMEM containing 0.4 U/ml phosphatidyl inositol phospholipase C (PI-PLC; Sigma) at 37°C overnight, followed by an additional 0.4 U/ml PI-PLC for 1 hr before processing of the sample for immunocytochemical analysis, or up to 5 hr for patch-clamp experiments. Cells were washed three times in DMEM, and contactin immunolabeling (2 μg/ml) was performed on live cells for 30 min at 4°C. Rabbit anti-goat IgG Cy-3 (6 μg/ml; Zymed, San Francisco, CA) was used as secondary antibody.
Surgery. Adult male Sprague Dawley rats were anesthetized with ketamine-xylazine (80/5 mg/kg, i.p.), right sciatic nerves were exposed at the mid-thigh level, tightly ligated with 4-0 silk sutures, and transected ∼1 mm distal to the ligature. To further promote neuroma formation and prevent regeneration of the proximal stump, ∼5 mm of the distal nerve stump was excised (Black et al., 1999). Contralateral sciatic nerves served as controls. These experiments were performed in accordance with National Institutes of Health guidelines for the care and use of animals in laboratory research.
Tissue processing. Nine to 12 d after sciatic nerve transection, rats were anesthetized and perfused transcardially with PBS followed by 4% paraformaldehyde in 0.14 m Sorensen's phosphate buffer. Contralateral (control) and ipsilateral (transected) sciatic nerves and L4 and L5 DRG were excised and postfixed in 4% paraformaldehyde for 20-30 min, rinsed in PBS, cryoprotected in 30% sucrose in PBS, and processed for immunocytochemistry as previously described (Black et al., 1999). Briefly, tissue was cryosectioned, incubated in blocking solution (PBS containing 5% normal horse serum and 1% BSA) containing 0.1% Triton X-100 and 0.02% sodium azide at room temperature for 30 min, and then incubated with rabbit anti-Nav1.3 (1 μg/ml) (Hains et al., 2002) and goat anti-contactin (2 μg/ml) antibodies overnight at 4°C. Sections were washed in PBS and incubated with anti-rabbit IgG-Cy3 (0.5 μg/ml; Amersham Biosciences) and anti-goat IgG-Alexa Fluor 488 (2 μg/ml; Molecular Probes) in blocking solution for 3 hr, washed in PBS, and mounted with Aqua-PolyMount (Polysciences, Warrington, PA). Control experiments without primary antibody or secondary antibodies showed no staining (data not shown). Images of contralateral and ipsilateral sciatic nerves and DRG were captured with a Nikon Eclipse E800 light microscope or Nikon PCM 2000 confocal microscope, and images were assembled in Adobe Photoshop.
Immunoblot analysis. The ipsilateral and contralateral L4-L5 DRG of six male rats were pooled, homogenized in lysis buffer (50 mm Tris-HCl, pH 8.0; 10 mm EDTA; 1% Triton X-100; protease inhibitors) and spun at 15,000 × g for 20 min at 4°C. The protein concentration of the supernatants was determined by the Bradford protein assay, and 100 μg of total protein was separated by SDS-PAGE. The proteins were examined by Western blotting using goat anti-contactin antibody (2 μg/ml). The membrane was then stripped in a buffer containing 62.5 mm Tris-HCl, pH 6.8, 2% SDS, and 100 mm 2-mercaptoethanol for 30 min at 55°C and reprobed with polyclonal anti-Nav1.9 antibody (0.5 μg/ml). The signal was detected using ECL chemiluminescence reagent (Perkin-Elmer, Boston, MA).
Electrophysiology. Whole-cell voltage clamp recordings were made using an Axopatch 200B amplifier (Axon Instruments, Foster City, CA) using standard techniques. The pipette solution contained (in mm): 140 CsF, 1 EGTA, 10 NaCl, and 10 HEPES, pH 7.3, adjusted to 310 mOsm/l with glucose. The external solution contained (in mm): 140 NaCl, 3 KCl, 1 MgCl2, 1 CaCl2, 10 HEPES, pH 7.3, adjusted to 320 mOsm/l with glucose. The pipette potential was zeroed before seal formation, and the voltages were not corrected for liquid junction potential. Capacity transients were canceled, and series resistance was compensated by 85-90%. Leakage current was digitally subtracted online using hyperpolarizing potentials applied after the test pulse (P/6 procedure). Currents were acquired using Clampex 8.1 software, filtered at 5 kHz and at a sampling rate of 20 kHz via a Digidata 1200 series interface (Axon Instruments). For current density measurements, the currents were divided by the cell capacitance, as read from the amplifier. All experiments were performed at room temperature (21-25°C). Data are expressed as mean ± SEM, and statistical analyses were performed using the Student's t test (significance at least p < 0.05).
Voltage protocols were performed at predetermined times after going whole-cell and were as follows. Standard current-voltage (I-V) families were obtained using 40 msec pulses from a holding potential of -120 mV, to a range of potentials (-65 to +60 mV) every 5 sec. The peak values at each potential were plotted to form I-V curves. Activation curves were fitted with the following Boltzmann distribution equation:
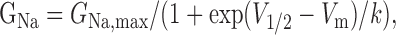 |
where GNa is the voltage-dependent sodium conductance, GNa, max is the maximal sodium conductance, V1/2 is the potential at which activation is half-maximal, Vm is the membrane potential, and k is the slope. The voltage that gave peak current was noted for subsequent protocols. Availability protocols consisted of a series of prepulses (-140 to 0 mV) lasting 500 msec, from the holding potential of -120 mV, followed by a 40 msec depolarization to the voltage that previously produced the peak current, every 10 sec. The normalized curves were fitted using a Boltzmann distribution equation:
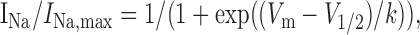 |
where INa, max is the peak sodium current elicited after the most hyper-polarized prepulse, Vm is the preconditioning pulse potential, V1/2 is the half maximal sodium current, and k is the slope factor. For recovery from inactivation experiments, two 40 msec stimuli were given to the voltage that previously produced peak current from the holding potential of -120 mV, with a variable recovery time period in the range of 0.1-400 msec. Curves were fitted with a double rising exponential function.
Results
Contactin-GFP fusion protein is transported to the plasma membrane
The complete ORF of contactin, including the terminal hydro-phobic C terminus, which is required to add the GPI anchor (Berglund and Ranscht, 1994), was cloned in-frame with GFP (C. Liu et al., 2001) to produce the fusion protein Ctn-GFP. To verify the presence of contactin at the cell surface and to test the avidity of the commercial contactin antibody, we tested the immunolabeling of control and HEK 293 cells transfected with contactin and Ctn-GFP constructs. Figure 1 shows the results of immunolabeling of live control HEK 293 cells (Fig. 1A,B), and HEK 293 cells transfected with contactin (Fig. 1C) and Ctn-GFP (Fig. 1D). The field of control HEK 293 cells as defined by the nuclear stain DAPI (Fig. 1B) does not show contactin immunolabeling consistent with the lack of endogenous contactin in these cells, and demonstrating that the contactin antibody does not cross-react with other cellular proteins. In contrast, HEK 293 cells transfected with contactin (Fig. 1C) or Ctn-GFP (Fig. 1D) show the surface immunolabeling using the contactin antibody. Because nonpermeabilized cells were used in this assay, the contactin antibody must have reacted with contactin at the cell surface (Fig. 1C,D), and this explains the lack of the contactin signal in the cytoplasm of HEK 293 cells transfected with contactin (Fig. 1C), whereas the green fluorescence seen in Figure 1D reflects the cytosolic pool of the Ctn-GFP fusion protein. Therefore, the Ctn-GFP construct was used in a series of experiments to facilitate the detection of HEK-Nav1.3 cells expressing contactin and to permit the use of anti-GFP antibody in reciprocal coimmunoprecipitation experiments because the contactin antibody does not work in this assay.
Figure 1.
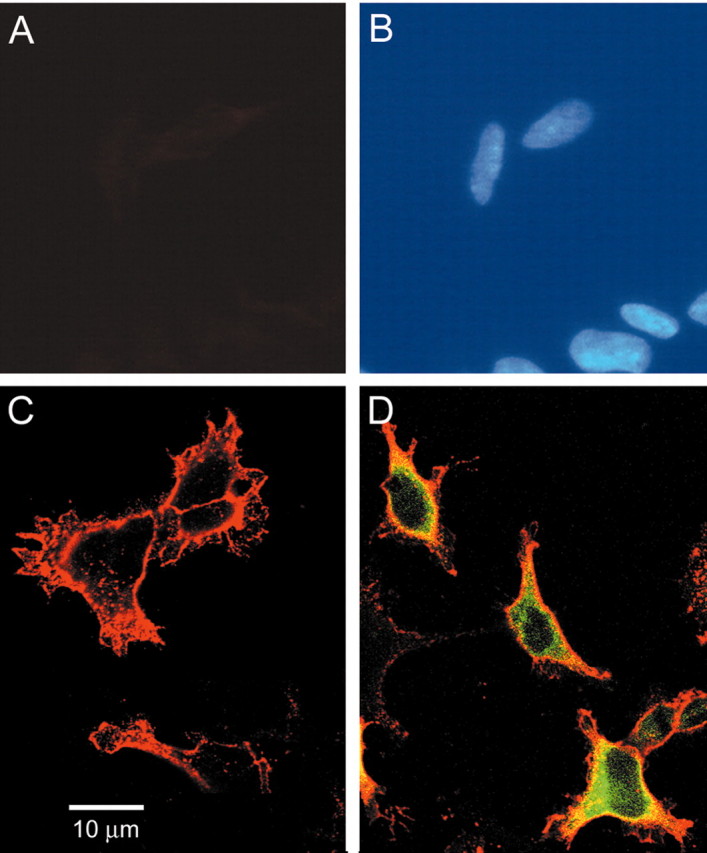
GPI-anchored contactin and contactin-GFP proteins are present at the surface of HEK-Nav1.3 cells. Live nonpermeabilized HEK-Nav1.3 cells were probed with anti-contactin antibody (A) and nuclei labeled with DAPI (B). No contactin-immunofluorescent signal is detected. Contactin (C) or contactin-GFP (D) constructs were transfected into HEK-Nav1.3 cells, and the live cells were reacted with anti-contactin antibody. Surface labeling of contactin (red) is apparent for both contactin and contactin-GFP constructs. HEK-Nav1.3 cells transfected with contactin-GFP also show GFP signal (green).
Contactin binds to both N-terminal and C-terminal polypeptides of Nav1.3 in vitro
Pull-down assays were used to determine if contactin binds to different intracellular polypeptide regions of the Nav1.3 sodium channel (Fig. 2A). Figure 2B shows the affinity-purified recombinant proteins that were used in this assay: GST, GST-Nav1.3N, GST-Nav1.3L1, GST-Nav1.3L2, GST-Nav1.3L3, and GST-Nav1.3C. GST and GST-Nav1.3 derivatives were conjugated to glutathione-Sepharose beads and incubated with lysates of HEK 293 cells that were transfected with the Ctn-GFP construct. Only the GST-Nav1.3N and GST-Nav1.3C fusion proteins captured Ctn-GFP (Fig. 2C). Purified GST does not capture Ctn-GFP in this assay (Fig. 2C, last lane). The N-terminal and C-terminal GST-fusion polypeptides of Nav1.3 pull down Ctn-GFP in this assay, whereas the other cytoplasmic segments of the channel (L1-L3) fail to capture Ctn-GFP. Additionally, neither GST (Fig. 2D, lane 2) nor GST-Nav1.3N/C (Fig. 2D, lanes 3, 4, respectively) bind to GFP alone. Thus, the N and C termini of the Nav1.3 channel interact specifically with contactin. Because total cell lysates of transfected HEK 293 cells were used as the source of Ctn-GFP in this assay, we cannot rule out the possibility that the interaction between contactin and Nav1.3 termini is mediated via cellular protein or proteins.
Figure 2.

Contactin binds to Nav1.3N and Nav1.3C in vitro. Recombinant proteins GST, GST-Nav1.3N, GST-Nav1.3L1, GST-Nav1.3L2, GST-Nav1.3L3, or GST-Nav1.3C (0.5 μg) immobilized on glutathione-Sepharose beads were incubated with extracts prepared from HEK 293 cells transfected with either Ctn-GFP or GFP. Immunoblot analysis of the bound proteins and cell extracts were probed with anti-GFP antibody. A, Schematic diagram of the sodium channel showing the intracellular regions used for the GST pull down assay. B, Coomassie blue-stained gel showing the GST-fusion proteins separated by SDS-PAGE. C, GST pull down assay using cell extracts from HEK 293 cells transfected with Ctn-GFP. Only GST-Nav1.3N and GST-Nav1.3C captured Ctn-GFP. D, GST-Nav1.3N and GST-Nav1.3C were used in a pull down assay using cell extracts from HEK 293 cells transfected with GFP. Neither GST alone nor the GST-Nav1.3 derivatives bind to GFP.
Contactin forms a complex with full-length Nav1.3 in a mammalian cell line
The expression of full-length sodium channel Nav1.3 in HEK 293 cells stably expressing the channel (HEK-Nav1.3) and Ctn-GFP was determined by immunoblot analysis using anti-pan sodium channel and anti-GFP antibodies (Fig. 3A, lanes 1-3). In this assay, lysates of control, nontransfected HEK 293 do not show endogenous sodium channels (Fig. 3A, top panel, lane 1), whereas the expected Nav1.3-immunoreactive band is detected in the HEK-Nav1.3 cell lysates (Fig. 3A, top panel, lanes 2 and 3). Ctn-GFP fusion protein is detected only in lysates of cells transfected with the Ctn-GFP construct: HEK 293 (Fig. 3A, bottom panel, lane 1) and HEK-Nav1.3 cells (Fig. 3A, bottom panel, lane 3). No Ctn—GFP-immunoreactive band was detected in lysates of control HEK 293 cells (Fig. 3A, bottom panel, lane 2), consistent with the lack of contactin surface immunolabeling of control HEK 293 cells (Fig. 1A).
Figure 3.
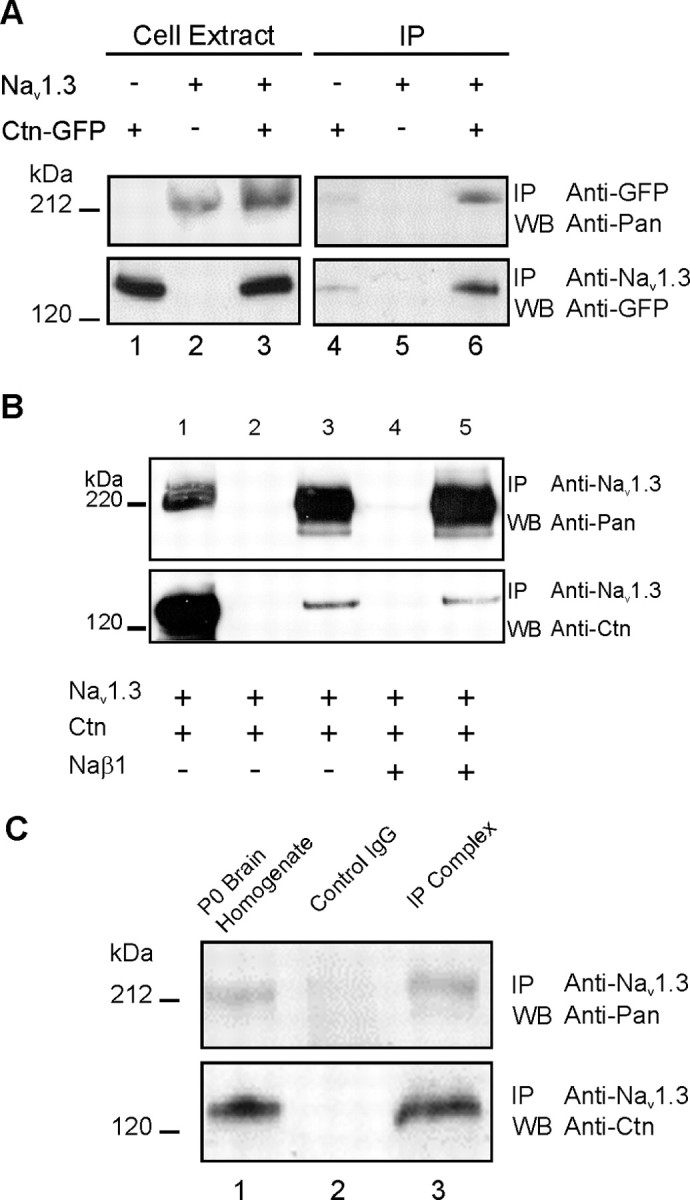
Contactin forms a complex with Nav1.3 in vivo and in HEK 293 cells. A, Interaction of contactin and Nav1.3 in HEK-Nav1.3 cells transfected with the Ctn-GFP construct. Immunoblot analysis (lanes 1-3) of cell extracts prepared from control HEK 293 cells transfected with Ctn-GFP, HEK-Nav1.3 cells, or HEK-Nav1.3 cells transfected with Ctn-GFP, using pan sodium channel (top panel) or GFP (bottom panel) antibodies show that Nav1.3 is detected only in HEK-Nav1.3 cell lysates (top panel, lanes 2, 3), whereas Ctn-GFP is detected only in lysates of cells transfected with Ctn-GFP construct (bottom panel, lanes 1, 3). Endogenous sodium channels (top panel, lane 1) or contactin (bottom panel, lane 2) are not detected in the cell lysates. Reciprocal immunoprecipitation analysis using Nav1.3-specific and GFP-specific antibodies (lanes 4-6). Anti-GFP antibodies coimmunoprecipitated Nav1.3 channels from HEK-Nav1.3-Ctn-GFP cell lysates (top panel, lane 6), whereas Nav1.3-specific antibodies coimmunoprecipitated Ctn-GFP (bottom panel, lane 6). B, Interaction of contactin and Nav1.3 in HEK-Nav1.3 cells is not dependent on the GFP tag or exogenous β1 subunit. HEK-Nav1.3 cell line was transfected with wild-type contactin alone or together with human β1 subunit, as indicated. Western blot analysis of HEK-Nav1.3 cells expressing Nav1.3 and Ctn was performed using pan sodium channel (top panel) and contactin (bottom panel) antibodies. This analysis shows robust expression of Nav1.3 channel (top panel, lane 1) and wild-type contactin (bottom panel, lane 1). Channel complexes were immunoprecipitated using Nav1.3-specific antibodies (lanes 3, 5) or control, nonspecific IgG (lanes 2, 4). Nav1.3 is detected only in the immunoprecipitation analysis using Nav1.3-specific antibody (lanes 3, 5). Control IgG did not immunoprecipitate proteins that reacted with the pan sodium channel or the contactin antibodies (lanes 2, 4). The coexpression of β1 subunit was not necessary for the interaction of Nav1.3 and wild-type contactin (compare lanes 3, 5). C, Contactin interacts with Nav1.3 in P0 rat brain. Western blot analysis using pan sodium channel antibodies and contactin-specific antibodies detect a sodium channel (top panel, lane 1) and contactin-immunoreactive (bottom panel, lane 1) bands of the expected sizes in P0 rat brain homogenate. The immunoprecipitated protein complexes were probed by pan sodium channel antibody (top panel, lane 3) and contactin antibody (bottom panel, lane 3). The volume of homogenate loaded on the gel in lane 1 is 5% of the volume that was used for the immunoprecipitation assay. Anti-Nav1.3 antibodies coimmunoprecipitated contactin. Control IgG antibodies failed to immunoprecipitate the channel or contactin (lane 2). IP, Immunoprecipitate; WB, Western blot.
The interaction of contactin and full-length Nav1.3 was tested by coimmunoprecipitation from HEK-Nav1.3 cells transfected with a plasmid encoding Ctn-GFP (Fig. 3A, lanes 4-6). Nav1.3-specific and GFP-specific antibodies were used in reciprocal immunoprecipitation experiments. The Nav1.3 channel complex, which was immunoprecipitated by the anti-Nav1.3 antibody, was probed with the anti-GFP antibody on a Western blot (Fig. 3A, bottom panel, lanes 4-6). A robust signal for Ctn-GFP was detected in the samples from HEK-Nav1.3 cells that were transfected with Ctn-GFP construct (Fig. 3A, bottom panel, lane 6). In the reciprocal experiment, the Ctn-GFP complex, which was immunoprecipitated by anti-GFP antibodies, was probed with pan sodium channel antibody (Fig. 3A, top panel, lanes 4-6). A robust signal for Nav1.3 was detected in the samples from HEK-Nav1.3 cells that were transfected with Ctn-GFP construct (Fig. 3A, top panel, lane 6). Thus, full-length Nav1.3 channel and contactin associate in a mammalian cell line.
We also tested the interaction of the full-length Nav1.3 channel and wild-type contactin in the presence or absence of recombinant human β1 subunit in the HEK-Nav1.3 cell line (Fig. 3B). Sodium channel β1 subunit has been shown to be necessary for the interaction of Nav1.2 and contactin (Kazarinova-Noyes et al., 2001; McEwen et al., 2004). The Nav1.3-specific antibody was used in the immunoprecipitation experiments, and the immunoprecipitated complex was probed with pan sodium channel (Fig. 3B, top panel) and contactin (Fig. 3B, bottom panel) antibodies. A sample of the transfected cells (1/20th volume) was loaded on the gel and probed with pan sodium channel (Fig. 3B, top panel, lane 1) and contactin (Fig. 3B, bottom panel, lane 1). Robust signals for the Nav1.3 and contactin were detected in the samples from extracts of HEK-Nav1.3 cells that were transfected with contactin (Fig. 3B, lane 1). The Nav1.3 channel complex shows a robust signal when probed with the pan sodium channel antibody (Fig. 3B, top panel, lane 3) and when probed with the contactin antibody (Fig. 3B, bottom panel, lane 3). Nonspecific IgG did not immunoprecipitate a complex that reacts with either the pan sodium channel antibody or the contactin antibody (Fig. 3B, lane 2). The intense signal of Nav1.3-immunoreactive band in lane 3, compared with the lysate sample in lane 1, is caused by the concentration effect of loading the whole immunoprecipitate complex derived from the 20-fold volume sample used in the immunoprecipitation assay.
The coexpression of human β1 subunit does not appear to be necessary for the interaction of Nav1.3 and contactin in the HEK-Nav1.3 cell line. A human β1 construct (Lossin et al., 2002) was cotransfected with contactin into the HEK-Nav1.3 cell line. The immunoreactive bands in lane 5 show comparable intensities of both Nav1.3 and contactin to those in lane 3. Control IgG does not immunoprecipitate a complex that reacts with the pan sodium channel or contactin antibodies (Fig. 3B, lane 4).
The coimmunoprecipitation of Nav1.3 and contactin (Fig. 3B), or contactin-mediated increase in the Nav1.3 current density (see below), does not depend on recombinant β1 subunit. It is possible that the interaction between the channel and contactin is mediated by saturatable adaptor protein or proteins that may include endogenous β subunits. Using quantitative real-time RTPCR to measure endogenous β1 and β2 transcripts in the HEK-Nav1.3 cell line, we found transcript levels that are ∼0.6 and 0.16%, respectively, of those in the cerebellum (see supplementary material on-line, available at www.jneurosci.org/cgi/content/full/24/33/9387/DC1). However, the primer-probe set that was used to detect the β1 transcript does not detect the β1B splice variant of human β1 subunit (Qin et al., 2003). In an independent set of experiments we measured the levels of endogenous β1B levels in the HEK-Nav1.3 cell line using quantitative real-time RT-PCR. A primer-probe set that is specific to β1B was designed based on the sequence of this alternative isoform (Qin et al., 2003) and used for this analysis (see supplementary material on-line, available at www.jneurosci.org/cgi/content/full/24/33/9387/DC1). The analysis shows that levels of endogenous β1B are ∼5% of the transcript levels in the brain. Thus, it is possible that the endogenous β1B subunits mediate the interaction of Nav1.3 and contactin.
Nav1.3 forms a complex with contactin in neonatal rat brain
The interaction of Nav1.3 and contactin in vivo was tested by immunoprecipitation of the channel complex from P0 rat brain (Fig. 3C). P0 rat brains were homogenized, and the Nav1.3-specific antibody was used to immunoprecipitate the channel complex. Western blot analysis of a 5% sample of total brain homogenate (lane 1) using pan sodium channel antibody (top panel) and anti-contactin antibody (bottom panel) show sodium channel and contactin-immunoreactive bands, respectively, of the expected sizes. The remainder of the homogenate were split into two samples and incubated with control IgG and Nav1.3-specific antibody. The complex that was immunoprecipitated using anti-Nav1.3 antibody revealed that contactin was specifically pulled down by Nav1.3 antibody (bottom panel, compare lanes 1 and 3). As expected, a sodium channel immunoreactive band was also detected using anti-pan sodium channel antibody (top panel, lane 3). Control rabbit IgG did not immunoprecipitate contactin or Nav1.3 (lane 2).
Contactin expression induces redistribution of Nav1.3 in HEK 293 Cells
We tested the hypothesis that the interaction between contactin and Nav1.3 influences the subcellular distribution of the channel. Monoclonal anti-pan sodium channel antibody produces robust sodium channel immunolabeling in HEK-Nav1.3 and in HEK-Nav1.3 transfected with contactin (Fig. 4), whereas the Nav1.3-specific antibody produced a similar pattern but with an attenuated intensity (data not shown). Neither antibody, however, produced a fluorescence signal in nontransfected HEK 293 cells. Thus, the monoclonal anti-pan sodium channel antibody was used for the densitometric analysis of the Nav1.3 signal in the experiments shown in Figure 4. Control experiments included incubation without primary antibody and preadsorption of the antibody with 100-500 molar excess of immunizing peptide. Only background levels of fluorescence were detected in the control experiments (data not shown).
Figure 4.

Contactin enhances Nav1.3 at the cell surface in transfected HEK 293 cells. HEK-Nav1.3 cells or HEK-Nav1.3 cells transfected with contactin were stained with anti-pan sodium channel and anti-contactin antibodies. HEK-Nav1.3 cells show prominent cytoplasmic Nav1.3 staining. HEK-Nav1.3 transfected with contactin show enhanced peripheral Nav1.3 labeling. B-D show HEK-Nav1.3 cells stained with anti-pan sodium channel antibody (B), contactin (C), and overlay (D). HEK-Nav1.3 cells transfected with contactin show Nav1.3 staining (F), contactin staining (G), and overlay (H). Confocal microscopic images were quantified using Scion Image software. A, E, Typical intensity measurements of Nav1.3 immunoreactivity in HEK-Nav1.3 cells (A) or HEK-Nav1.3 cells transfected with contactin (E) are shown. Densitometric measurements of Nav1.3 signal were taken through the cell axis, as indicated by the white line (B, F). The fluorescence density in arbitrary units was plotted against the sectional length in pixels. Scale bar, 10 μm.
HEK-Nav1.3 cells (Fig. 4A-D), HEK-Nav1.3 cells transfected with plasmid encoding the full-length contactin construct (Fig. 4E-H) were analyzed by immunocytochemistry using anti-pan sodium channel and anti-contactin antibodies to determine the effect of contactin on the subcellular distribution of Nav1.3. HEK293-Nav1.3 cells probed with the anti-pan antibody exhibited diffuse channel immunoreactivity throughout the cytoplasm (Fig. 4B) and, as expected, no contactin staining (Fig. 4C). The lack of a signal in Figure 4C demonstrates that there is no bleed-through fluorescence between Cy2 and Cy3 labels. In contrast, coexpression of Nav1.3 and contactin resulted in the enhancement of peripheral Nav1.3 distribution in these cells (Fig. 4F). Live cell staining by contactin antibody clearly shows the surface expression of contactin (Fig. 4G). Densitometric measurements (along axes marked in Fig. 4B) of Nav1.3 immunofluorescence in untransfected HEK293-Nav1.3 cells shows a generally uniform distribution of the signal within the cytoplasm (outside the nucleus) of several cells (Fig. 4A). In contrast, similar measurements (along axes marked in Fig. 4F) of Nav1.3 staining in HEK293-Nav1.3 cells transfected with contactin shows the redistribution of the signal to the cell periphery (Fig. 4E). This data are consistent with enhanced delivery of Nav1.3 to the cell surface when contactin is coexpressed with this channel in HEK 293 cells, similar to contactin-induced delivery of Nav1.2 to the plasma membrane of Chinese hamster lung (CHL) cells (Kazarinova-Noyes et al., 2001).
Contactin increases the amplitude of Nav1.3 current in HEK 293 cells
To examine the functional consequences of coexpression of contactin together with Nav1.3, we analyzed the Na current density in the stable cell line HEK-Nav1.3 transfected with GFP or with Ctn-GFP. HEK-Nav1.3 cells transfected with either GFP or Ctn-GFP were analyzed by whole-cell voltage clamp electrophysiology (Fig. 5). Only cells with a robust GFP fluorescence signal were used for electrophysiological recording. HEK 293 cells not expressing recombinant Nav1.3 were transfected with Ctn-GFP construct to serve as a negative control. These cells showed a very low sodium current density (Fig. 5A, top) (14.9 ± 3.7 pA/pF; n = 8). HEK-Nav1.3 cells transfected with GFP construct produced a robust sodium current (Fig. 5A, middle) (287.9 ± 36.8 pA/pF; n = 17), similar to the current previously obtained by the transient expression of Nav1.3 in HEK 293 cells (Cummins et al., 2001). Coexpression of Ctn-GFP in HEK-Nav1.3 cells produced a threefold higher current density (Fig. 5A, bottom) (873 ± 9 pA/pF; n = 20); the increase in current density was statistically significant (p < 0.01). The expression of Ctn-GFP in HEK-Nav1.3 did not significantly change the capacitance of these cells (23.7 ± 3.6 pF; n = 20) compared with HEK-Nav1.3 transfected with GFP (26.0 ± 2.6 pF; n = 17). Thus, in comparison to HEK-Nav1.3, the coexpression of Ctn-GFP with Nav1.3 causes a substantial increase in current density (Fig. 5B).
Figure 5.

Contactin increases functional cell surface expression of Nav1.3 in transfected HEK-Nav1.3 cells. A, Representative families of traces from control HEK 293 cells transfected with Ctn-GFP that do not express recombinant Nav1.3 (top); HEK-Nav1.3 cells transfected with GFP (middle); and HEK-Nav1.3 cells transfected with Ctn-GFP (bottom). B, Current densities (in picoamperes per picofarad) recorded from control HEK 293 cells transfected with Ctn-GFP showed only very low levels of current. HEK-Nav1.3 cells transfected with GFP displayed a sodium current of 287.9 ± 36.8 pA/pF. HEK-Nav1.3 cells stably expressing Nav1.3 transfected with Ctn-GFP showed a significant (*p < 0.01) and nearly threefold increase in current density (873 ± 87 pA/pF). C, Representative families of traces from HEK-Nav1.3 cells transfected with Ctn-GFP (top), β1-red (middle), or Ctn-GFP and β1-red (bottom). D, Current densities recorded from HEK-Nav1.3 cells transfected with Ctn-GFP, β1-red, or Ctn-GFP plus β1-red. HEK 293 cells stably expressing Nav1.3 cotransfected with Ctn-GFP and β1-red showed no significant difference in current density in comparison to HEK-Nav1.3 cells transfected with Ctn-GFP alone. β1-red transfection alone showed no effect on current density.
Although we found that exogenous β1 subunit is not necessary for the coimmunoprecipitation of Nav1.3 and contactin from the HEK 293 cell line, a role of β1-subunit has been demonstrated in the contactin-induced increase of Nav1.2 current amplitude (Kazarinova-Noyes et al., 2001). We reasoned that measurements of the Nav1.3 current density may be more sensitive than the biochemical assay to detect an effect of recombinant β1 coexpression with contactin on the current amplitude, thus a separate set of transfections were performed with the HEK-Nav1.3 stable cell line. HEK-Nav1.3 cells were transfected with either Ctn-GFP alone, β1-Red alone, or Ctn-GFP together with β1-Red. HEK-Nav1.3 cells that showed a robust green and/or red fluorescence signal, indicating the presence of the appropriate construct or constructs, were used for electrophysiological recordings. Figure 5C shows representative currents of HEK-Nav1.3 transfected with Ctn-GFP (Fig. 5C, top), β1-Red (Fig. 5C, middle), or Ctn-GFP and β1-Red (Fig. 5C, bottom). Comparison of the average current density (Fig. 5D) shows that the cotransfection of β1-Red had no significant additional effect on the contactin-induced increase in the Nav1.3 current: 608 ± 65 pA/pF (n = 21) and 547 ± 81 pA/pF (n = 15), respectively. In addition, transfection of β1-Red alone had no effect on the Nav1.3 current density (270 ± 44 pA/pF; n = 20).
We also investigated the effects of contactin on the biophysical properties of the Nav1.3 current. HEK-Nav1.3 cells exhibited sodium currents characterized by fast activation and inactivation and fast recovery from inactivation (repriming) similar to the current that is recorded from axotomized small DRG neurons and transient Nav1.3 currents in HEK 293 cells (Cummins et al., 2001). Figure 6 shows the normalized current-voltage relationship (Fig. 6A), steady-state inactivation (Fig. 6B), recovery from inactivation (Fig. 6C), and current kinetics (Fig. 6C, inset) for Nav1.3 coexpressed with either GFP or Ctn-GFP in the stable line HEK-Nav1.3. Table 1 summarizes the electrophysiological properties of the HEK-Nav1.3 current when coexpressed with GFP or Ctn-GFP. These data demonstrate that there is not a significant change in Nav1.3 steady-state activation, inactivation, recovery from inactivation, or current kinetics when HEK-Nav1.3 cells are transfected with Ctn-GFP versus GFP.
Figure 6.

Contactin does not change steady-state characteristics of Nav1.3 in transfected HEK-Nav1.3 cells. Normalized current-voltage relationships are shown (A). Steady-state, voltage-dependent inactivation (500 msec prepulses) (B), and recovery from inactivation (C) were determined as described in Materials and Methods. The time course for recovery from inactivation of peak current at -120 mV is shown. Squares, HEK-Nav1.3 cells transfected with GFP. Circles, HEK-Nav1.3 cells transfected with Ctn-GFP. No differences in these properties were seen in HEK-Nav1.3 cells transfected with either Ctn-GFP or GFP. C, Inset, Averaged normalized currents were elicited by a depolarizing pulse from -120 to -10 mV and are overlaid. The currents are indistinguishable from one another.
Table 1.
Comparison of voltage-dependent properties of Nav 1.3 expressed in HEK 293 cells coexpressing GFP or contactin-GFP
|
|
|
|
Recovery from inactivation |
|
|
|---|---|---|---|---|---|
| Condition |
Activation V1/2 (mV) |
Inactivation V1/2 (mV) |
τ1 at - 120 mV (msec) |
τ2 at - 120 mV (msec) |
τ1 fraction |
| GFP | −30 ± 1.6 (n = 17) | −72 ± 2.0 (n = 12) | 4.7 ± 0.6 | 74.7 ± 9.7 | 0.81 ± 0.02 (n = 11) |
| Contactin-GFP |
−33 ± 1.6 (n = 20) |
−70 ± 1.4 (n = 18) |
4.4 ± 0.6 |
60.5 ± 9.8 |
0.81 ± 0.03 (n = 13) |
Summary of fitted electrophysiological parameters for activation, inactivation (Boltzmann fits), and recovery from inactivation (double exponential fit). No significant differences were seen in any of the measured properties.
Removal of contactin from the cell surface by PI-PLC treatment does not reduce the elevated Nav1.3 current density
To determine whether the association of contactin and Nav1.3 at the cell surface is required to maintain the contactin-induced increase in the Nav1.3 current density, HEK-Nav1.3 cells transfected with the full-length contactin construct were treated with PI-PLC to cleave the GPI anchor and remove contactin from the cell surface. The immunolabeling of live cells (not permeabilized) with the anti-contactin antibody demonstrates the presence of contactin at the cell surface of transfected HEK-Nav1.3 cells (Fig. 7A). Overnight treatment of these live cells with PI-PLC causes the disappearance of the contactin immunolabeling (Fig. 7B-D). To ascertain the time course of contactin re-emergence at the cell surface, PI-PLC-treated HEK-Nav1.3-Ctn cells were assayed at different time points after overnight treatment: 16 hr (Fig. 7B), 19 hr (Fig. 7C), and 21 hr (Fig. 7D). Live immunolabeling of treated cells shows the disappearance of a contactin signal at the cell surface at the 16 hr mark and only attenuated signal as late as 21 hr.
Figure 7.

Removal of surface contactin does not reduce the elevated Nav1.3 density at the cell surface in HEK-Nav1.3-Ctn cells. HEK-Nav1.3 cells were transfected with contactin construct (HEK-Nav1.3-Ctn) and treated with PI-PLC or were untreated (see Materials and Methods). Live cells (nonpermeabilized) were immunostained for contactin using a polyclonal anti-contactin antibody in untreated (A), and 16 hr (B), 19 hr (C), and 21 hr (D) after initial treatment with PI-PLC to ascertain level of cell surface expression of contactin over time. Scale bar, 10 μm. E, Peak current density for HEK-Nav1.3 treated with PI-PLC and HEK-Nav1.3-Ctn treated with PI-PLC or untreated was plotted in picoamperes per picofarad. HEK-Nav1.3-Ctn treated with PI-PLC do not show significant difference in the Nav1.3 current density compared with untreated cells.
To test the effect of contactin removal from the surface of PI-PLC-treated HEK-Nav1.3-Ctn on the elevated Nav1.3 current density, electrophysiological recordings were conducted after overnight treatment with PI-PLC. Cells were treated with PI-PLC overnight and, to ensure continued absence of surface contactin during the 5 hr long recording session, a fresh aliquot of the enzyme was added on the morning of the recording. Just before recording, the culture medium was replaced by the bath solution, which was devoid of the enzyme. Thus, the electrophysiological recording was done at a time point in which detectable contactin labeling is not observed at the cell surface. PI-PLC treatment of HEK-Nav1.3 coexpressing GFP did not affect the current density (263 ± 44 pA/pF; n = 10) (Fig. 7E), compared with untreated HEK-Nav1.3 cells (287 ± 36.8 pA/pF; n = 17) (Fig. 4). Thus, endogenous GPI-anchored proteins do not appear to play a role in maintaining functional Nav1.3 at the surface of HEK 293 in this time frame. As expected, HEK-Nav1.3 cells cotransfected with contactin plus GFP constructs (to identify transfected cells) yielded a higher current density (720 ± 108 pA/pF; n = 19) (Fig. 7E), which was not significantly (p > 0.05) changed after treatment with PI-PLC (632 ± 186 pA/pF; n = 19) (Fig. 7E). Both untreated HEK-Nav1.3-Ctn and PI-PLC-treated HEK-Nav1.3-Ctn groups showed a statistically significant increase in Nav1.3 current density compared with HEK-Nav1.3 cells not expressing contactin (p < 0.05). Additionally, the similar levels of increased Nav1.3 current density when HEK-Nav1.3 cells coexpress contactin plus GFP (Fig. 7E) (720 ± 108 pA/pF; n = 19) or Ctn-GFP fusion protein (Fig. 5) (873 ± 188.6 pA/pF; n = 20), are consistent with the view that the presence of the GFP tag at the C terminus of contactin in the Ctn-GFP fusion protein does not interfere with the functional association of contactin with Nav1.3, and further validate the use of the Ctn-GFP construct in the biochemical and immunoprecipitation experiments discussed earlier (Figs. 3, 5).
Contactin expression is upregulated in axotomized DRG neurons
Finally, we investigated whether the expression of contactin in DRG neurons is altered after sciatic nerve transection, an in vivo paradigm that is known to trigger the upregulation of Nav1.3 in DRG neurons and their transected axon tips within the neuroma (Waxman et al., 1994; Dib-Hajj et al., 1996; Black et al., 1999; Boucher et al., 2000; Leffler et al., 2002). Sections of contralateral (sciatic nerve not transected) and ipsilateral (sciatic nerve transected at mid-thigh level) L4-5 DRG were probed with specific antibodies against contactin and Nav1.3. In contralateral DRG, contactin labeling was present in most neurons regardless of cell size (Fig. 8A); in most neurons, the contactin labeling was peripherally distributed. Twelve days after transection of the sciatic nerve, there was a substantial increase in contactin immunofluorescence within ipsilateral L4-5 DRG (Fig. 8B). The increase in contactin labeling was most pronounced in small-diameter (<30 μm) DRG neurons, but some larger diameter neurons also exhibited increased contactin levels. In general, the increased contactin immunolabeling was not peripherally localized but occurred diffusely within the cytoplasm.
Figure 8.

Contactin expression is increased in axotomized DRG neurons. A, In control DRG, contactin is present in most neurons and is peripherally localized. B, In axotomized DRG neurons, there is a substantial increase in contactin immunostaining. C, Western blot analysis shows that transection of the sciatic nerve causes an increase in the contactin protein levels in the DRG. The Western blot was probed with anti-contactin antibody (lanes 1, 2). The increase in contactin protein signal can be seen in ipsilateral (I) DRGs (lane 2) in comparison to contralateral (C) DRGs (lane 1). The blot was then stripped and reprobed with anti-Nav1.9 antibodies (lanes 3, 4). The expected decrease in Nav1.9 protein signal can be seen in ipsilateral DRGs (lane 4) in comparison to contralateral DRGs (lane 3). D, Nav1.3 is not detectable in control DRG neurons. E, There is a substantial increase in Nav1.3 immunofluorescence in DRG neurons (same section as shown in B) after transection of the sciatic nerve. F, Merged images of B and E. Scale bar, 50 μm.
The upregulation of contactin after transection of the sciatic nerve was also determined using immunoblot analysis. The ipsilateral and contralateral L4-L5 DRG of six male rats were pooled and analyzed by immunoblot analysis for the change in the steady-state level of contactin protein (Fig. 8C). In agreement with the immunohistochemical results (Fig. 8A,B), the intensity of the contactin-immunoreactive band is higher in the ipsilateral DRG sample (Fig. 8C, lane 1) than in the contralateral DRG sample (Fig. 8C, lane 2). To ensure that the elevated contactin signal in the ipsilateral sample is not an artifact of sample preparation or gel loading, the blot was stripped and reprobed with anti-Nav1.9 antibody. We have shown previously that Nav1.9 expression at the levels of RNA and protein are significantly down-regulated after transection of the sciatic nerve (Dib-Hajj et al., 1998; Sleeper et al., 2000; Tyrrell et al., 2001). As expected, Nav1.9 protein level in the ipsilateral DRG sample (Fig. 8C, lane 3) is substantially lower than in the contralateral DRG sample (Fig. 8C, lane 4). The opposite changes in Nav1.9 and contactin protein levels within the same sample validate the conclusion that the elevated level of contactin in DRG after axotomy is injury-induced and is not caused by artifacts of sample preparation and gel loading. Furthermore, the apparent increase in contactin levels in ipsilateral versus contralateral DRG in Figure 8C is an underestimation because ∼40% of L4-L5 DRG neurons send axons that branch before the site of ligation in the sciatic nerve (Yip et al., 1984; Devor et al., 1985), and are thus unaffected by the injury.
Consistent with our published data (Black et al., 1999), there was no detectable Nav1.3 immunolabeling signal in control DRG (Fig. 8D). Axotomy caused a substantial increase in the Nav1.3 immunostaining in axotomized DRG neurons (Fig. 8E), and Nav1.3 and contactin were colocalized in many cells (Fig. 8F).
Contactin colocalizes with Nav1.3 at transected axon tips within the neuroma
To determine whether the increase in contactin and Nav1.3 immunoreactivity within axotomized DRG neurons was paralleled by increased immunoreactivity for contactin and Nav1.3 within the neuroma formed at the proximal stump of the transected sciatic nerve, we examined neuromas after they were reacted with antibodies to sodium channel Nav1.3 and contactin. Figure 9 shows a representative montage of low-magnification images of the contralateral sciatic nerve and transected nerve, extending from the neuroma to several millimeters proximal. As shown in Figure 9, a stronger contactin signal (green) is present in the ipsilateral compared with the contralateral nerve, and a strong contactin signal was present within axon-like profiles at the neuroma just proximal to the ligature, and less frequently, in axon-like profiles more proximally within the nerve. Focal accumulations of contactin suggesting its accumulation at paranodes or nodes of Ranvier were not observed in the neuroma. Similar to previous findings of Nav1.3 accumulation within neuromas (Black et al., 1999), enhanced Nav1.3 immunofluorescence was present at transected axon tips within the neuroma, with lower levels of Nav1.3 in proximal regions of the nerve (Fig. 9, red). Overlay of Nav1.3 and contactin images (Fig. 9, right), demonstrates considerable colocalization of Nav1.3 and contactin within the neuroma (short arrows).
Figure 9.

Nav1.3 and contactin colocalize in sciatic nerve neuroma. Images of uninjured control (contralateral) and transected (ipsilateral) sciatic nerve show that contactin (left) and Nav1.3 (middle) accumulate within axon-like profiles within the neuroma (right, short arrows), with less immunostaining in proximal regions of the transected nerve. Contactin and Nav1.3 exhibit extensive colocalization (right, yellow) within the neuroma. Black arrow “ligature” indicates site of ligation. Scale bar, 500 μm.
At increased magnification (Fig. 10), individual axon tips within the neuroma can be discerned, and many of these axon tips exhibit colocalization of contactin (Fig. 10A) and Nav1.3 (Fig. 10B).
Figure 10.
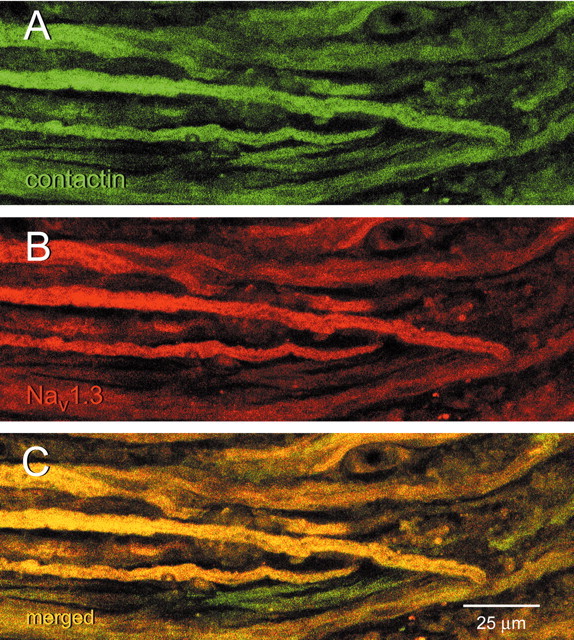
Nav1.3 and contactin colocalize in axons within neuroma. This section shows axons, at the level indicated by white short arrows in Figure 9, at higher magnification. Contactin (A) and Nav1.3 (B) exhibit substantial colocalization (C, yellow) in most axons within sciatic nerve neuroma. Scale bar, 25 μm.
Discussion
We have used multiple independent approaches in this study to show that contactin associates with the voltage-gated sodium channel Nav1.3 under normal and pathologic conditions. Contactin interacts with the N and C termini of Nav1.3 in pull down assays and coimmunoprecipitates with this channel from P0 rat brain and in a stable HEK-Nav1.3 cell line. The expression of contactin in HEK-Nav1.3 cells causes a threefold increase in the amplitude of the sodium current without affecting the biophysical properties of the channel. However, the continued presence of contactin at the cell surface is not necessary to maintain the higher Nav1.3 current density. Finally, Nav1.3 and contactin colocalize in sensory neurons of the DRG and at neuromas within the sciatic nerve after nerve transection.
Nav1.3 and contactin may form a complex in a cytoplasmic compartment that is then translocated to the cell surface. Contactin-dependent translocation of Caspr to the plasma membrane follows a similar mechanism in which the two proteins form a cytosolic complex via their extracellular domains, which is subsequently recruited to lipid rafts and translocated to the plasma membrane (Faivre-Sarrailh et al., 2000; Rios et al., 2000). We have previously shown that contactin and Nav1.9 C-terminal polypeptide interact directly based on a far-western assay and concluded that the interaction between contactin and the full-length channel may be transient (C. Liu et al., 2001). Although a transient interaction of the unprocessed contactin and sodium channel N and C termini cannot be formally ruled out at this time, it is more likely that the interaction between contactin and sodium channels in vivo requires an adapter protein. The pull down of contactin by the N and C termini of Nav1.3 suggests the presence of such an adaptor in HEK 293 cells. This is consistent with recent findings that HEK 293 cells express a large number of neuronal markers (Shaw et al., 2002).
The sodium channel β1 subunit has been shown to mediate the interaction of contactin and Nav1.2 (Kazarinova-Noyes et al., 2001); thus β subunits may act as a bridge between sodium channels and contactin, with the cytosolic C terminus of β subunits binding to the cytosolic termini of the channel, whereas the extracellular domain binds to contactin. Indeed, the C terminus of the β1 subunit has been shown to be essential for the efficient association of the β1 and Nav1.2 in heterologous expression systems (Meadows et al., 2001). The inability of anti-contactin antibodies to coimmunoprecipitate sodium channels from brain tissue of Scn1b null mice (Chen et al., 2004) provides strong evidence for an essential role of β1 and/or β1B in the association of sodium channels and contactin. Whether the association of Nav1.3 and contactin could be mediated by other cellular linkers, in addition to β1 or β1B, remains to be determined because the mice that were used for the coimmunoprecipitation experiments were P17-P19, a developmental time when only very low levels of Nav1.3 expression are detected in brain tissues (Beckh et al., 1989; Felts et al., 1997).
We report in this study the detection of only low levels of β1 and β2 transcripts in the HEK-Nav1.3 isolate (<1% of the levels in the brain), and the lack of further enhancement of the effect of contactin on the current density by recombinant β1. We detected, however, higher levels of β1B transcripts (∼5% of the levels in the brain) in the HEK-Nav1.3 cell line. Although it is likely that endogenous β1B (Moran et al., 2000; Qin et al., 2003) mediates the contactin-induced increase in the current density in the HEK-Nav1.3 cell line, we cannot rule out the possibility that the increase is attributable in part to the presence of other factor or factors. The presence of channel partners other than β subunits in mammalian expression systems, including the HEK 293 cell line, is consistent with two lines of evidence. First, antisense knock-down of β1B in HEK 293 cells did not change the current properties of recombinant Nav1.4 channels (Moran et al., 2003). Second, recombinant human Nav1.3 channels activate and inactivate normally, without a slow inactivating component, in Chinese hamster ovary (CHO) cells despite the absence of β1-β1A, β2 and β3 transcripts (Meadows et al., 2002).
The increase in Nav1.3 current amplitude when the channel is coexpressed with contactin is likely to be caused by the increased insertion of functional channels at the cell surface. The increase in the Nav1.3 current amplitude could be attributed, in theory, to changes to the biophysical properties of the channel or to the release from the effects of an endogenous blocker. Our results show, however, that the presence of contactin has no detectable effect on the voltage-dependent activation, steady-state inactivation, recovery from inactivation, or current kinetics of the Nav1.3 channel. Instead, we show that the coexpression of contactin and Nav1.3 results in a redistribution of the immunostaining of the channel to the cell periphery. The coexpression of contactin and Nav1.2 in CHL cells caused an increase in the channel density at the cell membrane leading to higher current amplitude without significantly altering the current properties (Kazarinova-Noyes et al., 2001). Thus, contactin appears to play a role in the trafficking of sodium channels but not in modulating sodium currents.
Previous studies have reported that, in CHO cells cotransfected with Caspr and contactin, PI-PLC treatment, which removes the GPI-anchored contactin from the cell surface, produced no effect on Caspr distribution at the cell surface (Faivre-Sarrailh et al., 2000). Instead, the retention of the contactin-Caspr complex at the cell membrane in neurons is dependent on the interaction of the cytosolic tail of Caspr with the cytoskeleton-associated protein 4.1B (Gollan et al., 2002). We show in this study that the treatment of HEK-Nav1.3-Ctn cells with PI-PLC for up to 20 hr did not diminish the elevated levels of Nav1.3 current density. Thus, similar to Caspr, Nav1.3 delivery, but not its retention at the cell surface, is enhanced by the presence of contactin. The lack of an effect of the relatively long PI-PLC treatment on the Nav1.3 current density may indicate that the rate of channel internalization is not affected by the absence of contactin from the channel complex.
The interaction of Nav1.3 and contactin may play an important role in shaping the activity-dependent remodeling of neural circuits during early development when this channel is abundant. Contactin plays multiple essential roles in the CNS (Durbec et al., 1992; Boyle et al., 2001; Perrin et al., 2001) probably via interaction with multiple proteins both in cis- and in trans. Deletion of contactin leads to juvenile lethality characterized by cerebellar defects (Berglund et al., 1999) and a decrease in sodium channel clusters at nodes of Ranvier (Bergstrom et al., 2002). The role of contactin in establishing normal nodes is linked to its role in delivering Caspr and sodium channels to the cell surface (Faivre-Sarrailh et al., 2000; Bhat et al., 2001; Kazarinova-Noyes et al., 2001). The potential role of contactin in delivering Nav1.2 to maturing nodes may be impaired in contactin-null mice, possibly contributing to the juvenile lethality phenotype of the strain. A role of contactin in enhancing the expression of Nav1.3 at the neuronal cell surface might be more critical at earlier ontogenetic stages, when neuronal activity is required for the normal development of synaptic connections (Katz and Shatz, 1996).
The putative interaction of contactin and Nav1.3 in transected sciatic nerve may accentuate the role of this channel in rendering injured neurons hyperexcitable. Nav1.3 protein accumulates within the blindly ending axons that form neuromas in ligated nerves (Black et al., 1999), where it is likely to contribute to ectopic firing (Matzner and Devor, 1994). Injured DRG neurons manifest ectopic discharges that are significantly attenuated by application of ∼20 nm TTX (Liu et al., 1999, 2001). Delivery of a similar dose of TTX to L5 after spinal nerve ligation (Lyu et al., 2000) significantly reduces neuropathic pain behavior in injured rats and administration of glial-derived neurotrophic factor (GDNF), which reduces the levels of Nav1.3 transcripts and the rapidly repriming TTX-S sodium current in small DRG neurons (Boucher et al., 2000; Leffler et al., 2002) has a similar effect. We show in this study that contactin is upregulated in axotomized DRG neurons and within the tips of their transected axons at the neuroma, and is colocalized at this site with Nav1.3. It has been shown that axonal-surface recruitment of contactin in hypothalamic neurons is activity-dependent (Pierre et al., 2001). Therefore, it might be predicted that the hyperactivity of axotomized DRG neurons can increase trafficking of contactin to the injured neuronal surface, promoting an additional increase in the number of Nav1.3 channels at the cell surface of these injured neurons. This positive feedback loop would constitute a maladaptive recapitulation of the possible relationship of contactin and Nav1.3 early in development, and could exacerbate neuropathic pain that ensues after injury to the neurons.
Footnotes
This work was supported in part by grants from the Rehabilitation Research Service and Medical Research Service, Department of Veterans Affairs, and from the National Multiple Sclerosis Society. The Center for Neuroscience and Regeneration Research is a Collaboration of the Paralyzed Veterans of America and The United Spinal Association with Yale University. We thank Dr. Ted Cummins and Bart Toftness for valuable discussions and technical assistance.
Correspondence should be addressed to Dr. Stephen G. Waxman, Department of Neurology, Yale University School of Medicine, LCI 707, New Haven, CT 06510. E-mail: stephen.waxman@yale.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/249387-13$15.00/0
B.S.S. and A.M.R. contributed equally to this work.
References
- Beckh S, Noda M, Lübbert H, Numa S (1989) Differential regulation of three sodium channel messenger RNAs in the rat central nervous system during development. EMBO J 8: 3611-3616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berglund EO, Ranscht B (1994) Molecular cloning and in situ localization of the human contactin gene (CNTN1) on chromosome 12q11-q12. Genomics 21: 571-582. [DOI] [PubMed] [Google Scholar]
- Berglund EO, Murai KK, Fredette B, Sekerkova G, Marturano B, Weber L, Mugnaini E, Ranscht B (1999) Ataxia and abnormal cerebellar microorganization in mice with ablated contactin gene expression. Neuron 24: 739-750. [DOI] [PubMed] [Google Scholar]
- Bergstrom U, Berglund EO, Kazarinova-Noyes K, Peles E, Brophy PJ, Shrager PBR (2002) Contactin supports the organization of central paranode junctions. Soc Neurosci Abstr 28: 819.10. [Google Scholar]
- Bhat MA, Rios JC, Lu Y, Garcia-Fresco GP, Ching W, Martin MS, Li J, Einheber S, Chesler M, Rosenbluth J, Salzer JL, Bellen HJ (2001) Axon-glia interactions and the domain organization of myelinated axons requires neurexin iv/caspr/paranodin. Neuron 30: 369-383. [DOI] [PubMed] [Google Scholar]
- Black JA, Cummins TR, Plumpton C, Chen YH, Hormuzdiar W, Clare JJ, Waxman SG (1999) Upregulation of a silent sodium channel after peripheral, but not central, nerve injury in DRG neurons. J Neurophysiol 82: 2776-2785. [DOI] [PubMed] [Google Scholar]
- Boucher TJ, Okuse K, Bennett DL, Munson JB, Wood JN, McMahon SB (2000) Potent analgesic effects of GDNF in neuropathic pain states. Science 290: 124-127. [DOI] [PubMed] [Google Scholar]
- Boyle ME, Berglund EO, Murai KK, Weber L, Peles E, Ranscht B (2001) Contactin orchestrates assembly of the septate-like junctions at the paranode in myelinated peripheral nerve. Neuron 30: 385-397. [DOI] [PubMed] [Google Scholar]
- Brummendorf T, Wolff JM, Frank R, Rathjen FG (1989) Neural cell recognition molecule F11: homology with fibronectin type III and immunoglobulin type C domains. Neuron 2: 1351-1361. [DOI] [PubMed] [Google Scholar]
- Chen C, Westenbroek RE, Xu X, Edwards CA, Sorenson DR, Chen Y, McEwen DP, O'Malley HA, Bharucha V, Meadows LS, Knudsen GA, Vilay-thong A, Noebels JL, Saunders TL, Scheuer T, Shrager P, Catterall WA, Isom LL (2004) Mice lacking sodium channel beta1 subunits display defects in neuronal excitability, sodium channel expression, and nodal architecture. J Neurosci 24: 4030-4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins TR, Waxman SG (1997) Downregulation of tetrodotoxin-resistant sodium currents and upregulation of a rapidly repriming tetrodotoxin-sensitive sodium current in small spinal sensory neurons after nerve injury. J Neurosci 17: 3503-3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins TR, Aglieco F, Renganathan M, Herzog RI, Dib-Hajj SD, Waxman SG (2001) Nav1.3 sodium channels: rapid repriming and slow closed-state inactivation display quantitative differences after expression in a mammalian cell line and in spinal sensory neurons. J Neurosci 21: 5952-5961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devor M, Govrin-Lippmann R, Frank I, Raber P (1985) Proliferation of primary sensory neurons in adult rat dorsal root ganglion and the kinetics of retrograde cell loss after sciatic nerve section. Somatosens Res 3: 139-167. [DOI] [PubMed] [Google Scholar]
- Dib-Hajj S, Black JA, Felts P, Waxman SG (1996) Down-regulation of transcripts for Na channel alpha-SNS in spinal sensory neurons following axotomy. Proc Natl Acad Sci USA 93: 14950-14954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dib-Hajj SD, Tyrrell L, Black JA, Waxman SG (1998) NaN, a novel voltage-gated Na channel, is expressed preferentially in peripheral sensory neurons and down-regulated after axotomy. Proc Natl Acad Sci USA 95: 8963-8968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbec P, Gennarini G, Goridis C, Rougon G (1992) A soluble form of the F3 neuronal cell adhesion molecule promotes neurite outgrowth. J Cell Biol 117: 877-887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faivre-Sarrailh C, Gauthier F, Denisenko-Nehrbass N, Le Bivic A, Rougon G, Girault JA (2000) The glycosylphosphatidyl inositol-anchored adhesion molecule F3/contactin is required for surface transport of paranodin/contactin-associated protein (caspr). J Cell Biol 149: 491-502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felts PA, Yokoyama S, Dib-Hajj S, Black JA, Waxman SG (1997) Sodium channel alpha-subunit mRNAs I, II, III, NaG, Na6 and HNE (PN1): different expression patterns in developing rat nervous system. Mol Brain Res 45: 71-82. [DOI] [PubMed] [Google Scholar]
- Fjell J, Hjelmstrom P, Hormuzdiar W, Milenkovic M, Aglieco F, Tyrrell L, Dib-Hajj S, Waxman SG, Black JA (2000) Localization of the tetrodotoxin-resistant sodium channel NaN in nociceptors. NeuroReport 11: 199-202. [DOI] [PubMed] [Google Scholar]
- Gennarini G, Cibelli G, Rougon G, Mattei MG, Goridis C (1989) The mouse neuronal cell surface protein F3: a phosphatidylinositol-anchored member of the immunoglobulin superfamily related to chicken contactin. J Cell Biol 109: 775-788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldin A (2001) Resurgence of sodium channel research. Annu Rev Physiol 63: 871-894. [DOI] [PubMed] [Google Scholar]
- Gollan L, Sabanay H, Poliak S, Berglund EO, Ranscht B, Peles E (2002) Retention of a cell adhesion complex at the paranodal junction requires the cytoplasmic region of Caspr. J Cell Biol 157: 1247-1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hains BC, Black JA, Waxman SG (2002) Primary motor neurons fail to upregulate voltage-gated sodium channel Nav1.3/brain type III following axotomy resulting from spinal cord injury. J Neurosci Res 70: 546-552. [DOI] [PubMed] [Google Scholar]
- Hains BC, Klein JP, Saab CY, Craner MJ, Black JA, Waxman SG (2003) Upregulation of sodium channel Nav1.3 and functional involvement in neuronal hyperexcitability associated with central neuropathic pain after spinal cord injury. J Neurosci 23: 8881-8892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isom LL (2001) Sodium channel beta subunits: anything but auxiliary. Neuroscientist 7: 42-54. [DOI] [PubMed] [Google Scholar]
- Isom LL, Catterall WA (1996) Na+ channel subunits and Ig domains. Nature 383: 307-308. [DOI] [PubMed] [Google Scholar]
- Isom LL, De Jongh KS, Patton DE, Reber BF, Offord J, Charbonneau H, Walsh K, Goldin AL, Catterall WA (1992) Primary structure and functional expression of the beta 1 subunit of the rat brain sodium channel. Science 256: 839-842. [DOI] [PubMed] [Google Scholar]
- Isom LL, Ragsdale DS, De Jongh KS, Westenbroek RE, Reber BF, Scheuer T, Catterall WA (1995) Structure and function of the beta 2 subunit of brain sodium channels, a transmembrane glycoprotein with a CAM motif. Cell 83: 433-442. [DOI] [PubMed] [Google Scholar]
- Katz LC, Shatz CJ (1996) Synaptic activity and the construction of cortical circuits. Science 274: 1133-1138. [DOI] [PubMed] [Google Scholar]
- Kazarinova-Noyes K, Malhotra JD, McEwen DP, Mattei LN, Berglund EO, Ranscht B, Levinson SR, Schachner M, Shrager P, Isom LL, Xiao ZC (2001) Contactin associates with Na+ channels and increases their functional expression. J Neurosci 21: 7517-7525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keating MT, Sanguinetti MC (2001) Molecular and cellular mechanisms of cardiac arrhythmias. Cell 104: 569-580. [DOI] [PubMed] [Google Scholar]
- Kim CH, Oh Y, Chung JM, Chung K (2001) The changes in expression of three subtypes of TTX sensitive sodium channels in sensory neurons after spinal nerve ligation. Mol Brain Res 95: 153-161. [DOI] [PubMed] [Google Scholar]
- Leffler A, Cummins TR, Dib-Hajj SD, Hormuzdiar WN, Black JA, Waxman SG (2002) GDNF and NGF reverse changes in repriming of TTX-sensitive Na(+) currents following axotomy of dorsal root ganglion neurons. J Neurophysiol 88: 650-658. [DOI] [PubMed] [Google Scholar]
- Lindia JA, Abbadie C (2003) Distribution of the voltage gated sodium channel Na(v)1.3-like immunoreactivity in the adult rat central nervous system. Brain Res 960: 132-141. [DOI] [PubMed] [Google Scholar]
- Liu C, Dib-Hajj SD, Black JA, Greenwood J, Lian Z, Waxman SG (2001) Direct interaction with contactin targets voltage-gated sodium channel Nav1.9/NaN to the cell membrane. J Biol Chem 276: 46553-46561. [DOI] [PubMed] [Google Scholar]
- Liu X, Chung K, Chung JM (1999) Ectopic discharges and adrenergic sensitivity of sensory neurons after spinal nerve injury. Brain Res 849: 244-247. [DOI] [PubMed] [Google Scholar]
- Liu X, Zhou JL, Chung K, Chung JM (2001) Ion channels associated with the ectopic discharges generated after segmental spinal nerve injury in the rat. Brain Res 900: 119-127. [DOI] [PubMed] [Google Scholar]
- Lossin C, Wang DW, Rhodes TH, Vanoye CG, George Jr AL (2002) Molecular basis of an inherited epilepsy. Neuron 34: 877-884. [DOI] [PubMed] [Google Scholar]
- Lyu YS, Park SK, Chung K, Chung JM (2000) Low dose of tetrodotoxin reduces neuropathic pain behaviors in an animal model. Brain Res 871: 98-103. [DOI] [PubMed] [Google Scholar]
- Matzner O, Devor M (1994) Hyperexcitability at sites of nerve injury depends on voltage-sensitive Na+ channels. J Neurophysiol 72: 349-359. [DOI] [PubMed] [Google Scholar]
- McEwen DP, Meadows LS, Chen C, Thyagarajan V, Isom LL (2004) Sodium channel beta 1 subunit-mediated modulation of Nav1.2 currents and cell surface density is dependent on interactions with contactin and ankyrin. J Biol Chem 279: 16044-16049. [DOI] [PubMed] [Google Scholar]
- Meadows L, Malhotra JD, Stetzer A, Isom LL, Ragsdale DS (2001) The intracellular segment of the sodium channel beta 1 subunit is required for its efficient association with the channel alpha subunit. J Neurochem 76: 1871-1878. [DOI] [PubMed] [Google Scholar]
- Meadows LS, Chen YH, Powell AJ, Clare JJ, Ragsdale DS (2002) Functional modulation of human brain Nav1.3 sodium channels, expressed in mammalian cells, by auxiliary beta 1, beta 2 and beta 3 subunits. Neuroscience 114: 745-753. [DOI] [PubMed] [Google Scholar]
- Meisler MH, Kearney J, Ottman R, Escayg A (2001) Identification of epilepsy genes in human and mouse. Annu Rev Genet 35: 567-588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran O, Nizzari M, Conti F (2000) Endogenous expression of the beta1A sodium channel subunit in HEK-293 cells. FEBS Lett 473: 132-134. [DOI] [PubMed] [Google Scholar]
- Moran O, Conti F, Tammaro P (2003) Sodium channel heterologous expression in mammalian cells and the role of the endogenous beta1-subunits. Neurosci Lett 336: 175-179. [DOI] [PubMed] [Google Scholar]
- Okuse K, Malik-Hall M, Baker MD, Poon WY, Kong H, Chao MV, Wood JN (2002) Annexin II light chain regulates sensory neuron-specific sodium channel expression. Nature 417: 653-656. [DOI] [PubMed] [Google Scholar]
- Perrin FE, Rathjen FG, Stoeckli ET (2001) Distinct subpopulations of sensory afferents require F11 or axonin-1 for growth to their target layers within the spinal cord of the chick. Neuron 30: 707-723. [DOI] [PubMed] [Google Scholar]
- Pierre K, Dupouy B, Allard M, Poulain DA, Theodosis DT (2001) Mobilization of the cell adhesion glycoprotein F3/contactin to axonal surfaces is activity dependent. Eur J Neurosci 14: 645-656. [DOI] [PubMed] [Google Scholar]
- Qin N, D'Andrea MR, Lubin ML, Shafaee N, Codd EE, Correa AM (2003) Molecular cloning and functional expression of the human sodium channel beta1B subunit, a novel splicing variant of the beta1 subunit. Eur J Biochem 270: 4762-4770. [DOI] [PubMed] [Google Scholar]
- Ranscht B (1988) Sequence of contactin, a 130-kD glycoprotein concentrated in areas of interneuronal contact, defines a new member of the immunoglobulin supergene family in the nervous system. J Cell Biol 107: 1561-1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rios JC, Melendez-Vasquez CV, Einheber S, Lustig M, Grumet M, Hemperly J, Peles E, Salzer JL (2000) Contactin-associated protein (Caspr) and contactin form a complex that is targeted to the paranodal junctions during myelination. J Neurosci 20: 8354-8364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah BS, Stevens EB, Pinnock RD, Dixon AK, Lee K (2001) Developmental expression of the novel voltage-gated sodium channel auxiliary subunit beta3, in rat CNS. J Physiol (Lond) 534: 763-776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw G, Morse S, Ararat M, Graham FL (2002) Preferential transformation of human neuronal cells by human adenoviruses and the origin of HEK 293 cells. FASEB J 16: 869-871. [DOI] [PubMed] [Google Scholar]
- Sleeper AA, Cummins TR, Dib-Hajj SD, Hormuzdiar W, Tyrrell L, Waxman SG, Black JA (2000) Changes in expression of two tetrodotoxin-resistant sodium channels and their currents in dorsal root ganglion neurons after sciatic nerve injury but not rhizotomy. J Neurosci 20: 7279-7289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyrrell L, Renganathan M, Dib-Hajj SD, Waxman SG (2001) Glycosylation alters steady-state inactivation of sodium channel Nav1.9/NaN in dorsal root ganglion neurons and is developmentally regulated. J Neurosci 21: 9629-9637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman SG, Kocsis JD, Black JA (1994) Type III sodium channel mRNA is expressed in embryonic but not adult spinal sensory neurons, and is reexpressed following axotomy. J Neurophysiol 72: 466-470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman SG, Dib-Hajj S, Cummins TR, Black JA (2000) Sodium channels and their genes: dynamic expression in the normal nervous system, dys-regulation in disease states(1). Brain Res 886: 5-14. [DOI] [PubMed] [Google Scholar]
- Yip HK, Rich KM, Lampe PA, Johnson Jr EM (1984) The effects of nerve growth factor and its antiserum on the postnatal development and survival after injury of sensory neurons in rat dorsal root ganglia. J Neurosci 4: 2986-2992. [DOI] [PMC free article] [PubMed] [Google Scholar]