

Abstract
Dynamic changes in synaptic connectivity and strength, which occur during both embryonic development and learning, have the tendency to destabilize neural circuits. To overcome this, neurons have developed a diversity of homeostatic mechanisms to maintain firing within physiologically defined limits. In this study, we show that activity-dependent control of mRNA for a specific voltage-gated Na+ channel [encoded by paralytic (para)] contributes to the regulation of membrane excitability in Drosophila motoneurons. Quantification of para mRNA, by real-time reverse-transcription PCR, shows that levels are significantly decreased in CNSs in which synaptic excitation is elevated, whereas, conversely, they are significantly increased when synaptic vesicle release is blocked. Quantification of mRNA encoding the translational repressor pumilio (pum) reveals a reciprocal regulation to that seen for para. Pumilio is sufficient to influence para mRNA. Thus, para mRNA is significantly elevated in a loss-of-function allele of pum (pumbemused), whereas expression of a full-length pum transgene is sufficient to reduce para mRNA. In the absence of pum, increased synaptic excitation fails to reduce para mRNA, showing that Pum is also necessary for activity-dependent regulation of para mRNA. Analysis of voltage-gated Na+ current (INa) mediated by para in two identified motoneurons (termed aCC and RP2) reveals that removal of pum is sufficient to increase one of two separable INa components (persistent INa), whereas overexpression of a pum transgene is sufficient to suppress both components (transient and persistent). We show, through use of anemone toxin (ATX II), that alteration in persistent INa is sufficient to regulate membrane excitability in these two motoneurons.
Keywords: aCC, excitability, neural activity, paralytic, Pumilio, RP2
Introduction
Central neurons must face and adapt to changing patterns of synaptic drive. These adaptations are essential to prevent neurons from either falling silent as synaptic excitation falls or, conversely, becoming saturated during periods of intense neuronal activity. Changes in exposure to synaptic excitation are particularly extreme during early embryogenesis when neurons first form synaptic contacts, but also arise as a consequence of the synaptic remodeling that underpins memory and learning (for review, see Turrigiano, 1999; Burrone and Murthy, 2003; Turrigiano and Nelson, 2004]. Although such adaptive mechanisms, often termed homeostatic compensation, have been well documented in both mammals and invertebrates, the underlying mechanisms remain less well understood.
Mammalian cortical neurons maintained in culture compensate for changes in exposure to synaptic excitation by alteration of both intrinsic membrane excitability and responsiveness to excitatory neurotransmitters. Changes in membrane excitability are mediated by alterations in voltage-gated conductances, whereas change in responsiveness to neurotransmitter is mediated by altered AMPA receptor density and/or localization (O'Brien et al., 1998; Turrigiano et al., 1998; Desai et al., 1999). More recently, the same phenomenon has been observed in Drosophila motoneurons in vivo, which offers the significant opportunity to exploit molecular genetics to elucidate the underlying mechanism (Baines et al., 2001; Baines, 2003). Changes in synaptic excitation of two motoneurons (termed aCC and RP2) are compensated for by altered membrane excitability primarily mediated through changes in INa (Baines et al., 2001; Baines, 2003). Activity-dependent regulation of the cAMP-protein kinase A (PKA) pathway is both necessary and sufficient to mediate rapid changes in INa in aCC/RP2 (Baines, 2003), consistent with in vitro studies that show phosphorylation of rat Na+ channels to be an effective determinant of channel conductance (Li et al., 1992; Smith and Goldin, 1997; Catterall, 2000). However, whereas rapid change in INa is predicted to compensate for equally rapid fluctuations in synaptic excitation, longer-term changes in neuronal activity might be better compensated for by changes in gene expression.
In addition to transcriptional control, regulation of translation plays a critical role in gene expression (Gavis, 2001). In Drosophila, the translational repressor Pumilio (Pum) is required for establishment of polarity during early embryogenesis (Tautz, 1988; Wharton and Struhl, 1991; Wharton et al., 1998). Binding of Pum to the nanos response element (NRE) motif located in the 3′ untranslated region (3′-UTR) of hunchback (hb) is the initial step in translation repression, setting up a gradient of transcript across the embryo. The ability of motoneurons to synaptically excite muscle is also reported to be influenced by levels of pum in the Drosophila CNS, which is indicative that this repressor might also contribute to regulation of neuronal excitability (Schweers et al., 2002).
A bioinformatic screen for NRE-like sequences in either 3′- or 5′-UTRs of Drosophila ion channel genes (our unpublished data) identified paralytic (para), the protein products of which carry the voltage-gated Na+ current in embryonic and larval motoneurons (Baines and Bate, 1998). In this study, we show that para mRNA levels are activity dependent, increasing as synaptic excitation falls and vice versa. Levels of pum mRNA are also activity dependent but are reciprocal to that observed for para. Overexpression of a pum transgene is sufficient to reduce para mRNA and to reduce the magnitude of INa in aCC/RP2. The absence of pum [pumbemused (pumbem)] results in a complementary phenotype: increased para mRNA pan-neuronally and increased INa in aCC/RP2. In the absence of pum, genetic manipulations that result in increased synaptic excitation of aCC/RP2 fail to repress para mRNA levels, which is indicative that this translational repressor is both necessary and sufficient for activity-dependent regulation of INa in Drosophila motoneurons.
Materials and Methods
Fly stocks. Flies were fed on apple juice agar supplemented with yeast at 25°C. Wild type (WT) was Canton-S [except in Fig. 6C,D, in which RRC-GAL4 × UAS-GFPn was the WT control (Baines et al., 1999)]. 1407-GAL4 was used to express UAS transgenes in all of the CNS neurons. RN2-O GAL4 (homozygous viable second chromosome) was used to selectively express UAS transgenes in aCC and RP2 (Fujioka et al., 1999; Baines, 2003). Expression of RN2 GAL4 begins in early stage 16 embryos, a stage that precedes the onset of synaptogenesis (Baines and Bate, 1998). The bemused allele of pumilio (rebalanced over a TM3 GFP balancer) and full-length UAS-pumilio transgene used are detailed by Schweers et al. (2002). The isogenic wild type from which the bemused allele was produced served as control and is denoted bem+. Synaptic transmission was blocked by either expression of UAS-tetanus toxin light chain (TeTx-A) or by use of a genetic null of n-synaptobrevin (n-syb), rebalanced over a GFP balancer (Sweeney et al., 1995). Synaptic transmission was increased by expression of UAS-rutabaga (a type I, Ca2+-calmodulin-dependent, adenylate cyclase) (Zars et al., 2000) or by use of well characterized allele of dunce [a cAMP-specific phosphodiesterase (EC:3.1.4)] (Dudai et al., 1976; Davis et al., 1995; Baines, 2003). Expression of para was increased using a characterized genetic duplication: Tp(1;2)r+75c (Stern et al., 1990). PKA activity was promoted using UAS-PKAact1 and inhibited using UAS-PKAinh1 (formerly termed PKABDK22); these transgenes are described by Davis et al. (1998). Para was removed using a small deficiency [Df(1) D34] (Baines and Bate 1998).
Figure 6.
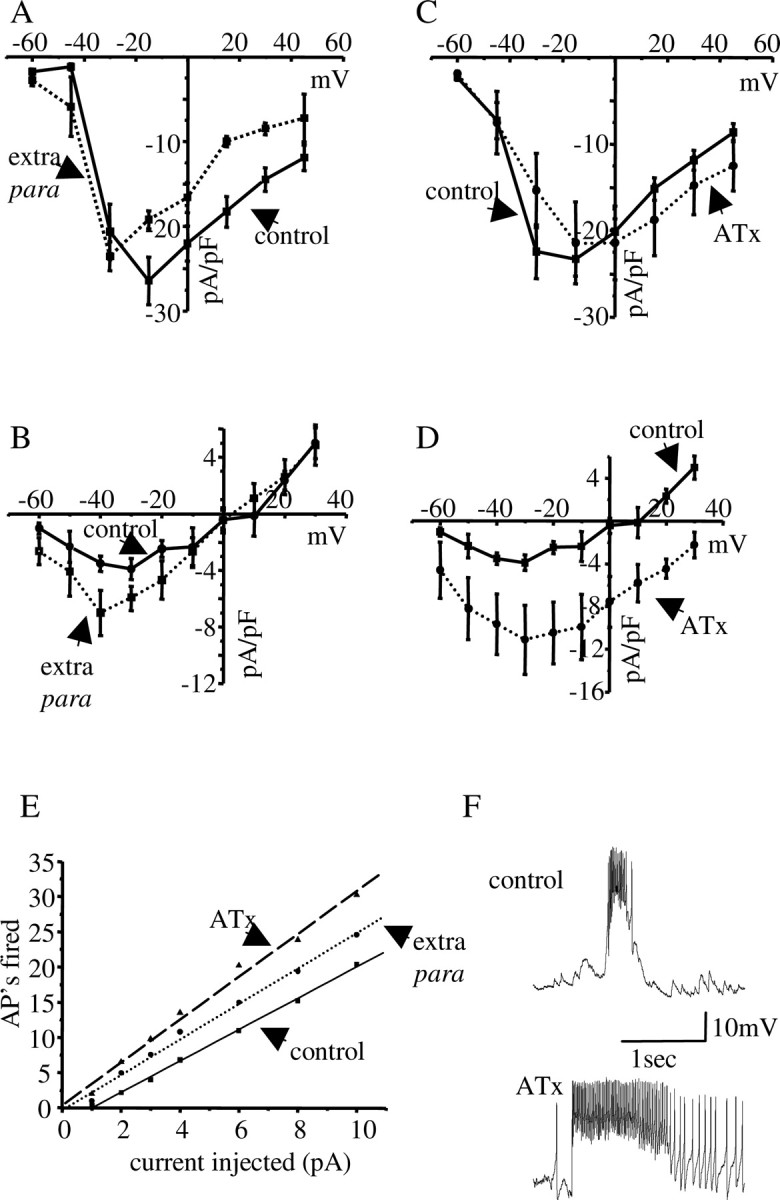
Increased INa(p) is sufficient to increase membrane excitability in aCC/RP2. A, B, Increasing the copy number of para using a genetic duplication [Tp(1;2)r+75c; extra para] results in no significant change in peak amplitude for INa(t) (A) but a significant increase in peak INa(p) (B) compared with a WT control (p ≤ 0.05). C, D, Brief exposure (<5 min) to ATX II (300 nm) similarly fails to affect INa(t) (C), but significantly increases peak INa(p) (p ≤ 0.01) (D). Control is RRC GAL4 × UAS-GFPn. All of the values shown are means ± SE for n ≥ 8. The reversal of INa(p) in the presence of ATX differs from control because the increase in current magnitude produced is sufficient to overcome a nonspecific Icat that is also present in these neurons (Fig. 5C) (E. C. G. Pym and R. A. Baines, unpublished data). Icat normally acts to oppose INa(p) but, because of its slower activation kinetics, does not significantly influence INa(t). E, Membrane excitability in aCC/RP2 is significantly increased (p ≤ 0.05) in both the genetic duplication Tp(1;2)r+75c (extra para) and in the presence of ATX II. Control is WT. All of the values shown are means (n ≥ 8). AP, Action potential. F, Endogenous synaptic inputs to aCC/RP2 (for details, see Baines, 2003) are potentiated in the presence of ATX II (300 nm). Both traces are from the same WT aCC neuron (i.e., before and after ATX II exposure).
Embryo dissection. Larvae were dissected, and central neurons were accessed as described by Baines and Bate (1998). The larva was viewed using a water immersion lens (total magnification, 800×) combined with Nomarski optics (BX51 WI microscope; Olympus Optical, Tokyo, Japan).
Electrophysiology. All of the recordings were performed in young first instar larvae, 1-4 hr after hatching, at room temperature (22-24°C). Whole-cell recordings (current and voltage clamp) were achieved using thick-walled borosilicate glass electrodes (GC100TF-10; Harvard Apparatus, Edenbridge, UK), fire polished to resistances of between 15 and 20 MΩ. Cells were initially identified based on both size and dorsal position in the ventral nerve cord. Unequivocal identification was determined by labeling with sulfur rhodamine (0.3%; Molecular Probes, Eugene, OR), which was included in the patch saline. Recordings were made using an Axopatch-1D amplifier controlled by pClamp 8.1 (Axon Instruments, Foster City, CA). Only cells with an input resistance >1 GΩ were accepted for analysis. Traces were filtered at 2 kHz and sampled at 20 kHz. To better resolve Na+ currents, an on-line leak subtraction protocol was used (P/4). Currents shown are the averages of five trials for each cell. To separate the persistent current (INa(p)), a voltage protocol was used that first stepped to +30 mV (50 msec) to inactivate the transient current (INa(t)). Determination of membrane excitability was performed using injection of depolarizing current (1-10 pA/500 msec) from a resting membrane potential (RMP) of -60 mV. RMPs were maintained at -60 mV by injection of a small amount of hyperpolarizing current (WT RMP without current injection =-44 ± 1.4 mV). Input resistance, which was determined by injection of 1 pA hyperpolarizing current, remained statistically unchanged in all of the genetic backgrounds tested (WT = 7.2 ± 0.9 GΩ).
Solutions. External saline for dissection and current clamp analysis of excitability consisted of the following (in mm): 135 NaCl, 5 KCl, 4 MgCl2·6H2O, 2 CaCl2·2H2O, 5 N-Tris[hydroxymethyl]methyl-2-aminoethanesulfonic acid, 36 sucrose, and 1 mecamylamine (used in the determination of excitability to block synaptic currents). For isolation of Na+ currents, the following solution was used (in mm): 100 NaCl, 5 KCl, 50 TEA, 10 4-AP, 10 HEPES, 10 glucose, and 0.5 CaCl2·2H2O. All of the solutions were pH 7.15. ATX II (Alomone Labs, Jerusalem, Israel) was applied, dissolved in the relevant saline.
Internal patch solution consisted of (in mm): 140 K+ methylsulfonate (KCH3SO3), 2 MgCl2·6H2O, 2 EGTA, 5 KCl, and 20 HEPES, pH 7.4. When recording Na+ currents, CsCl2 was substituted for KCH3SO3.
RNA isolation. RNA was extracted from 50 late stage 17 embryos (para) or from 50 isolated embryonic late stage 17 CNSs (pumilio) using the Qiagen RNeasy mini kit (Qiagen, Hilden, Germany) with minor modifications. Briefly, 50 μl of RLT solution containing 0.1 m β-mercaptoethanol was added to the embryos or isolated CNSs before homogenizing on ice using a sterile plastic Eppendorf (Hamburg, Germany) homogenizer. The homogenate was centrifuged (3 min at 13,000 rpm) to remove cell debris before addition of 50 μl of 70% ethanol (v/v). Homogenate was loaded onto an RNeasy column and washed, and RNA was eluted as per the manufacturer's protocol. RNA concentration was calculated by spectral analysis at 260 nm and quality assessed by agarose gel (1%) electrophoresis under denaturing conditions. Samples were subsequently stored at -80°C.
cDNA synthesis. cDNA synthesis was performed in a total volume of 20 μl. RNA (up to 5 μg) was mixed with random hexamer primers (0.2 μg; Roche, Mannheim, Germany) and made up to 11 μl with RNase-free water. After incubation (5 min at 65°C), the mix was chilled on ice (2 min). Reaction buffer [4 μl; (in mm): 250 Tris-HCl, 250 KCl, 20 MgCl2, 50 DTT, pH 8.3] and deoxyribonucleoside triphosphates (dNTPs; 2 μl; 10 mm) made up to a final volume of 19 μl (RNase-free water) were added and incubated for 5 min at 25°C. Reverse transcriptase (200 U; RevertAid H Minus M-MuLV; Fermentas, Vilnius, Lithuania) was added before incubation steps of 10 min at 25°C, 60 min at 42°C, and 20 min at 65°C. From the total reaction volume of 20 μl, 1 μl of cDNA was used for each PCR.
PCR conditions and controls. Clone Manager Software (Scied Software, Durham, NC) was used to design PCR primers for para, pumilio, and ribosomal protein 49 (rp49): rp49 forward and reverse primers in 5′ to 3′ orientation, CCAAGGACTTCATCCGCCACC and GCGGGTGCGCTTGTTCGATCC; para forward and reverse primers, GATCTATATGGGCGTGCTCACCGAGAAGTG and TGCAGGCACACGTAATCGTCGTCGGATTG; and pumilio forward and reverse primers, TGAAGAGCCGGCGAGCTGAA and CTGGCCACCAGCTGACCAAT, respectively. Each reaction contained 5 μl of PCR Master Mix (3 mm MgCl2, dNTPs, Taq polymerase; Biogene, Cambridge, UK), 500 nm forward and reverse primers, 1 μl of cDNA, and 1:1000 dilution of SYBR Gold (Biogene) made up to 10 μl with PCR-grade water. PCR was performed using a Roche LightCycler (Roche). After an initial denaturation step at 94°C for 60 sec, temperature cycling was initiated. Each cycle consisted of denaturation at 94°C for 0 sec, hybridization at 54°C for 5 sec for para and 57°C for pumilio, and elongation at 72°C for 10 sec. Determination of rp49 was performed at either temperature. The fluorescent signal was acquired at the end of each hybridization step (F2/F1 channels, fluorimeter gains regulated on 1 for F1, 1 for F2, and 1 for F3). A total of 30 cycles was performed. The authenticity of the PCR products was verified by melting-curve analysis and the presence of a single band of appropriate molecular size by agarose gel electrophoresis.
Real-time PCR quantification. mRNA levels are expressed as relative fold change normalized against rp49 mRNA.
The comparative cycle threshold (Ct) method (User Bulletin 2, 1997; Applied Biosystems, Foster City, CA) was used to analyze the data by generating relative values of the amount of target cDNA. Relative quantification for any given gene, expressed as fold variation over control, was calculated from the determination of the difference between the Ct of the given gene (para, pumilio) and that of the calibrator gene (rp49). Ct values used were the means of triplicate replicates. Experiments were repeated at least five times.
Statistics. Data were compared using a nonpaired t test. Results were deemed significant at p ≤ 0.05. All of the values shown are mean ± SE.
Results
Synaptic excitation regulates membrane excitability in Drosophila motoneurons
The synaptic drive to aCC/RP2 consists of large (50-300 pA) inward synaptic currents that are relatively long-lived (500-1000 msec) and mediated by acetylcholine (Baines et al., 1999) (Fig. 1C). Together with the relatively slow kinetics of these currents, these characteristics are indicative that these currents arise from the synchronous releases of acetylcholine from multiple convergent presynaptic inputs [for a more complete description of these currents, see Baines et al. (2001) and Baines (2003)]. Our previous studies demonstrate that endogenous membrane excitability (i.e., the ability to fire action potentials) of these two motoneurons is regulated by exposure to excitatory synaptic drive (Baines et al., 2001; Baines, 2003). Figure 1 shows additional data that reinforce these previous observations. In a range of genetic backgrounds that affect the magnitude of excitatory synaptic currents recorded in aCC/RP2 (Fig. 1C), the averaged current amplitude [these two neurons show no differences in synaptic inputs indicative of shared presynaptic interneurons (Baines, 2003)] shows a very strong correlation to the number of action potentials elicited by injection of a constant depolarizing current (10 pA/500 msec, a measure of membrane excitability) (Fig. 1A,B). The correlation is inverse in nature; increasing synaptic current amplitude results in reduced membrane excitability and vice versa (for more details, see Baines, 2003).
Figure 1.

Exposure to synaptic excitation regulates membrane excitability in aCC/RP2. A, The number of action potentials (APs) fired, for a given depolarizing input (10 pA/500 msec), is inversely related to endogenous synaptic excitation in aCC/RP2 (line coefficient, 0.92). All of the determinations of AP firing were from a maintained RMP of -60 mV (see Materials and Methods). Input resistances did not differ significantly between genotypes shown. Synaptic current amplitude shown is the average current magnitude recorded from at least eight cells for each genotype (n > 200 currents). Neurons exposed to greater than normal synaptic excitation (WT, 76 ± 3.3 pA) exhibit reduced membrane excitability and vice versa. APs fired are means (n ≥ 8cells). Pan-neuronal expression of TeTx-A results in aCC/RP2 neurons firing 28 ± 2.4 APs while exhibiting no evoked synaptic currents (see also Baines et al., 2001). This “nonphysiological” condition is not shown because it markedly deviates from the line of best fit shown. This is most likely because this frequency of AP firing represents the physiological maximum for these cells at this developmental stage. B, Representative traces for injection of 10 pA/500 msec for the two genotypes stated. C, Endogenous synaptic currents, recorded in voltage clamp (Vh =-60 mV) for the same two genotypes. Three excitatory synaptic currents are shown overlaid in each example. Genotypes are as follows: (1) RN2-O × UAS-PKAinh, (2) B19-GAL4, (3) Canton-S, (4) RN2-O × UAS-dCREBact (1157-19), (5) rut1, (6) dnc2, (7) RN2-O × UAS-PKAact, (8) RN2-O × UAS-dCREBact (1157-51), (9) dnc2, (10) RN2-O × UAS-dCREBinh (9), and (11) 1407-GAL4 × UAS-rut.
Synaptic excitation regulates abundance of para mRNA
The ability of a neuron to alter intrinsic membrane excitability in response to changing synaptic excitation is advantageous for the maintenance of global activity levels within neural circuits (Turrigiano and Nelson, 2000). Our previous work implicates a PKA-mediated regulation of INa as a mechanism able to compensate for rapid fluctuations in synaptic excitation (Baines, 2003). Undoubtedly, however, neurons must also adapt to changes in synaptic excitation that persist over the longer term, and we speculate that additional mechanisms might exist to compensate for such chronic changes. One conceivable mechanism is that of activity-dependent regulation of Na+ channel gene expression. To test this hypothesis, we used real-time PCR [quantitative reverse-transcription PCR (QRT-PCR)] to quantify the abundance of para mRNA in the CNS of late stage 17 (19-21 hr after egg laying) Drosophila embryos, in which levels of synaptic excitation have been genetically manipulated. The voltage-gated Na+ current in Drosophila central neurons is exclusively encoded by para during this early developmental stage (Hong and Ganetzky, 1994; Thackeray and Ganetzky, 1994; Baines and Bate, 1998).
To block synaptic neurotransmitter release, thereby removing synaptic excitation, we used two complementary approaches. First, we used pan-neuronal expression of TeTx-A, which is sufficient to block the evoked release of neurotransmitter (Sweeney et al., 1995; Baines et al., 1999). Second, we used a null allele of n-synaptobrevin that is essential for evoked synaptic vesicle release (Broadie et al., 1995). We observe that, in both genetic backgrounds, the abundance of para mRNA is significantly increased (2.5 ± 0.5- and 3.9 ± 0.4-fold, respectively; p ≤ 0.01) relative to controls (inactive tetanus toxin and n-syb/+, respectively) (Fig. 2). These observations correlate to our previous finding that INa is significantly increased in aCC/RP2 when TeTx-A is expressed in all of the central neurons (Baines et al., 2001). To potentiate synaptic excitation, we again used two independent but complementary approaches. Elevation of cAMP is seemingly sufficient to increase excitatory synaptic release in the Drosophila CNS (Baines, 2003). Thus, cAMP was elevated using an allele of dunce (dnc2) that lacks a cAMP-specific phosphodiesterase (Dudai et al., 1976; Davis et al., 1995) and by pan-neuronal expression of rutabaga (rut; a type I, Ca2+-calmodulin-dependent, adenylate cyclase) (Zars et al., 2000). Both genetic backgrounds exhibit increased synaptic excitation of aCC/RP2 (Fig. 1A), whereas para mRNA was significantly reduced in both (-3.9 ± 0.2- and -3.4 ± 0.5-fold; p ≤ 0.01) relative to controls (WT and 1407 GAL4, respectively) (Fig. 2B). Measurement of INa in aCC/RP2 in the dnc2 background shows a significant reduction compared with WT (Baines, 2003).
Figure 2.

Abundance of para mRNA is activity dependent. A, Typical QRT-PCR output for determination of para mRNA isolated from whole embryos that expressed either active (TeTx-A) or inactive (TeTx-I) tetanus toxin in all of the neurons. For quantification, all of the values are normalized to RP49 expression (for details of analysis, see Materials and Methods). B, Averaged changes in para mRNA, relative to controls (set to 0), show that abundance increases in backgrounds in which synaptic transmission is absent (TeTx-A and n-syb), and decreases when synaptic excitation is greater than normal (dnc2 and 1407 GAL4 × UAS-rut). Values given are means ± SE (n ≥ 6). All of the changes are significant from respective controls (inactive toxin expression, n-syb/+, WT, and 1407 GAL4; p = 0.008, 0.01, 0.002, and 0.01, respectively).
Manipulation of pumilio affects para mRNA
A characterization of a newly identified loss-of-function allele of pumilio (bemused) indicates, albeit indirectly, that this translational repressor is capable of altering excitability at the Drosophila neuromuscular junction (Schweers et al., 2002). Levels of Pum are greatly reduced in pumbem, and, moreover, two pum transcripts are absent. In such mutants, stimulation of the axon innervating muscle 6 results in a hyperexcitability phenotype, whereas overexpression of full-length UAS-pum in the CNS results in an opposite phenotype, hypoexcitability (Schweers et al., 2002). To test whether activity-dependent changes in para mRNA abundance are downstream of pum in the CNS, we first quantified the levels of para mRNA in these same pum backgrounds. Figure 3A shows that, in the loss-of-function pumbem background, para mRNA is significantly increased (2 ± 0.1-fold; p ≤ 0.01) relative to control (bem+). By comparison, overexpression of full-length UAS-pum in all of the neurons of the CNS resulted in a significant reduction of para mRNA (-3.5 ± 0.06-fold; p ≤ 0.01) relative to control (1407 GAL4). Thus, in genetic backgrounds in which pum is significantly reduced (pumbem), para expression is increased, whereas overexpression of pum is sufficient to suppress para mRNA. Given that Pum is a known translational repressor, these results are consistent with the hypothesis that Pum might act to suppress translation of, and target for degradation, para mRNA.
Figure 3.

Expression of pum mRNA is activity dependent and regulates para mRNA abundance. A, The abundance of para mRNA in isolated CNS is significantly greater in the loss-of-function pumbem allele. By comparison, overexpression of UAS-pum in all of the neurons of the CNS (1407 GAL4) is sufficient to significantly reduce para mRNA. Values given are means (n ≥ 6). All of the changes are significant from respective controls (pum+ and 1407-GAL4; p = 0.001 and 0.0006, respectively). B, In genetic backgrounds in which evoked release of synaptic transmitter is blocked (TeTx-A and n-syb), pum mRNA is significantly reduced compared with controls (inactive toxin and n-syb/+; p = 0.007 and 0.01, respectively). In contrast, CNS isolated from dnc2 embryos, which is known to enhance synaptic excitation of aCC/RP2 (Baines, 2003), results in an opposite and significant increase in pum mRNA compared with control (WT; p = 0.007). Controls are set to zero. Values given are means ± SE (n ≥ 6).
Expression of pum is activity dependent
At the simplest level, if pum is a regulator of para in central neurons of Drosophila, then pum itself would be predicted to be regulated by synaptic activity. We tested this prediction by quantifying pum mRNA in the same synaptic activity backgrounds used to examine para mRNA abundance. Because pum is widely expressed in Drosophila embryos and not restricted to the CNS as in the case of para, this analysis used isolated CNSs. CNSs taken from late stage 17 embryos, which expressed either active TeTx-A in all of the neurons or a loss-of-function n-syb genotype, show a significant reduction in pum mRNA abundance (-2.5 ± 0.3- and -2.9 ± 0.1-fold, respectively; p ≤ 0.01) (Fig. 3B) compared with controls (TeTx-inactive and n-syb/+). In contrast, CNS isolated from dnc2 embryos, in which synaptic excitation of aCC/RP2 is increased (Baines, 2003), exhibited a significant increase in pum mRNA (3.5 ± 0.4-fold; p ≤ 0.01) compared with WT control. Thus, these data strongly implicate that expression of pum is itself regulated in an activity-dependent manner. Moreover, the polarity of this regulation (activity increasing pum and vice versa) is reciprocal to that observed for activity-dependent changes in para mRNA and, as such, is consistent with a model that links increased pum to repression of para mRNA.
Pum is necessary for activity-dependent para regulation
To test directly whether Pum is required for activity-dependent changes in para mRNA abundance, we overexpressed UAS-rut in all of the neurons of the CNS (1407-GAL4) in the pumbem background. Our prediction was that the reduction in para mRNA that normally results from overexpression of UAS-rut in a WT background (Fig. 2) would not occur if Pum is required. Figure 4 shows that this is indeed the case. In the absence of normal levels of pum, overexpression of rut in all of the neurons of the CNS is unable to reduce para mRNA levels. Thus, in addition to being sufficient to repress para mRNA when overexpressed, pum is also necessary for suppression of para mRNA that results from the overexpression of rut.
Figure 4.

Pumilio is necessary for rut-dependent reduction of para mRNA. Overexpression of UAS-rut in all of the neurons of the CNS in the loss-of-function pumbem allele fails to reduce the level of para mRNA. Changes in para mRNA observed in 1407-GAL4 × UAS-rut (wild-type background) and in pumbem are shown for comparative purposes (gray boxes). Values given are means ± SE (n ≥ 6). The change in para mRNA observed in 1407 × rut (pumbem) is significant from that seen in 1407 × rut (WT) at p = 0.01.
Pum regulates INa in aCC/RP2
Our previous electrophysiological studies show that altered synaptic excitation, cAMP, and/or PKA are sufficient to regulate INa in aCC/RP2 (Baines et al., 2001; Baines, 2003). To address whether manipulation of pum is equally able to regulate INa in these motoneurons, as our model predicts, we used whole-cell voltage clamp to measure voltage-gated INa while manipulating the level of pum expression. Voltage-gated INa in aCC/RP2 is composed of two principal components. In addition to a typical, rapidly inactivating transient current (INa(t)), there is an additional slower inactivating persistent current (INa(p)) (Fig. 5A,B). To show that both of these components are encoded by the para gene, we repeated these recordings in a para null (Df(1) D34). In this background, both the transient and persistent components of INa are absent (Fig. 5C), confirming previous reports that para encodes INa at early stages of development (O'Dowd et al., 1989). Recordings in this para null uncovers an outward current that exhibits similarities to the cation current (Icat), which has been described in rat neocortical neurons (Alzheimer, 1994). This current has relatively slow activation kinetics and, as such, is unable to significantly affect INa(t) but is able to counteract INa(p) (see also Fig. 6D). Isolation of Icat by addition of TTX (1 μm) to the Na+ isolation saline used to record INa shows that it is unaffected by manipulation of pum (E. C. G. Pym and R. A. Baines, unpublished data).
Figure 5.

Voltage-gated INa is regulated by Pum in aCC/RP2. A, B, Voltage-gated INa in aCC/RP2 has at least two components. The first is a rapidly inactivating transient current (shown in A), whereas the second is a persistent current that inactivates slowly (shown in B). The outward current component in both traces is caused by a contamination by an Icat-like cation current (Alzheimer, 1994) that we are unable to block at present. This current activates at approximately +20 mV and is not changed after manipulation of pum (determined by adding TTX to the Na+-isolation saline that isolates just Icat). C, Both components of INa are absent in recordings in a para-null [Df(1) D34] late stage 17 embryo. These recordings isolate Icat. D, E, In the loss-of-function allele Pumbem, the magnitude of INa(t) does not change compared with control (Pum+) (D), whereas the magnitude of peak INa(p) is significantly increased (E) (p ≤ 0.05). F, G, Overexpression of UAS-pum in aCC/RP2 (RN2-O GAL4) is sufficient to reduce both components of INa (control is RN2-O GAL4). H, Membrane excitability of aCC/RP2, determined by injection of constant current (1-10pA/500 msec), shows a significant increase in Pumbem and a significant decrease in overexpression of UAS-pum, respectively (p ≤ 0.05). Control is WT. All of the values shown are means (± SE in D-G) for n ≥ 8. AP, Action potential. I, Representative traces showing the firing of action potentials by successively greater depolarizing current injections (3, 6, and 10 pA/500 msec) in a WT aCC neuron.
We used established voltage protocols to separate out each INa current type (see Materials and Methods). In the loss-of-function pumbem allele, the magnitude of INa(p) but not INa(t) is significantly increased relative to controls (bem+) (Fig. 5D,E). Thus, in backgrounds in which pum is reduced, which our QRT-PCR analysis shows to result in increased para expression, at least one component of INa in aCC/RP2 is significantly increased. In comparison, overexpression of UAS-pum in just aCC/RP2 (using RN2-O GAL4) is sufficient to reduce both components of INa (INa(t) and INa(p)) in these two motoneurons (Fig. 5F,G). Again, this effect is predictable based on our QRT-PCR analysis. Measurement of endogenous membrane excitability in aCC/RP2 [which is identical for both neurons (Baines, 2003)] shows that a reduction in pum expression (pumbem) is sufficient to significantly increase membrane excitability (i.e., increased number of action potentials fired by injection of depolarizing current). In contrast, increased expression of pum (UAS-pum) is sufficient to reduce membrane excitability (Fig. 5H,I). Both effects are entirely consistent with the changes observed in INa (see below).
Increased para expression increases only the persistent Na+ current
It is remarkable that, in the absence of pum (pumbem), only the persistent component of INa is increased. That this should occur is indicative of an additional level of control of this current. To test this idea, we used the genetic duplication Tp(1;2)r+75c (Stern et al., 1990) to increase para expression in the CNS (an approximately twofold increase in para mRNA was observed by QRT-PCR in this genetic background; data not shown). Analysis of INa in aCC/RP2, in Tp(1;2)r+75c, shows that only INa(p), not INa(t), is significantly increased in amplitude (Fig. 6A,B), thereby phenocopying the effect of reduced pum expression. Injection of depolarizing current shows that this genetic duplication is, moreover, sufficient to increase membrane excitability in aCC/RP2 (Fig. 6E). To independently validate these results, we used the anemone toxin, ATX II, which is reported to increase only INa(p), not INa(t), in a wide variety of species (Mantegazza et al., 1998). In agreement with these previous studies, the presence of ATX II (300 nm) is sufficient to significantly increase INa(p), but not INa(t), in aCC/RP2 (Fig. 6C,D). Injection of depolarizing current shows that an ATX II-dependent increase in INa(p) [we observed no change to other currents present in aCC/RP2 (E. C. G. Pym and R. A. Baines, unpublished data)] is sufficient to increase membrane excitability (Fig. 6E). Consistent with this conclusion is our additional observation that application of ATX II to our electrophysiological preparation potentiates endogenous synaptic depolarizations that are sufficient to fire action potentials (Baines, 2003) in aCC/RP2 (Fig. 6F). Thus, we conclude that increased INa(p) in aCC/RP2, which we show results from either increases in para expression or a reduction in pum expression, mediates an increase in membrane excitability in these neurons. By comparison, reductions in para or increases in pum are sufficient to suppress membrane excitability through reductions in both INa(t) and INa(p).
Discussion
Embryonic neurons face rapidly changing synaptic excitation as neuronal circuits are first formed and then subjected to modulation by sensory feedback. To maintain stable circuit output while individual synapses are being either strengthened or weakened requires that the constituent neurons of these embryonic circuits exhibit adaptation to changing synaptic excitation. Known homeostatic mechanisms include activity-dependent gene expression (West et al., 2002), fixed ratio co-translation of ion channel mRNAs (MacLean et al., 2003), and posttranslational modification of ion channel conductance (Li et al., 1992; Smith and Goldin, 1997; Catterall, 2000). Our results are of importance in that they increase this repertoire to include activity-dependent regulation of an ion channel mRNA by Pumilio, a known translational repressor.
Homeoststatic mechanisms maintain neuronal excitability
As nervous systems mature, neurons may change shape, lose or gain synaptic inputs, be exposed to changing patterns of synaptic drive, and experience constant turnover of ion channel proteins. Despite these changes, neurons are able to maintain relatively constant firing properties, indicative of intrinsic mechanisms that strive to maintain stability (Turrigiano and Nelson, 2004). A pertinent example is provided by neurons of the stomatogastric nervous system (STG) of Crustacea. STG neurons exhibit bursting behavior that is a consequence of phasic inhibitory inputs combined with intrinsic conductances. When isolated from these inputs in culture, these same neurons initially exhibit tonic firing. If isolation is continued for several days, however, these neurons regain an ability to fire in bursts (Turrigiano et al., 1994, 1995). Clearly, input and the lack of it have the effect of triggering a homeostatic mechanism that restores bursting behavior to these neurons. Although the particular aspect of synaptic input that coordinates this homeostatic response remains obscure, a likely possibility is that of altered Ca2+ influx across the neuronal membrane as a direct consequence of depolarization. Although direct evidence for the involvement of Ca2+ is lacking, manipulation of membrane potential is sufficient to evoke homeostatic responses in both neurons and muscle (Leslie et al., 2001; Paradis et al., 2001; Burrone et al., 2002). In Drosophila, for example, genetically induced hyperpolarization of somatic muscle is sufficient to evoke increased presynaptic neurotransmitter release from innervating motoneurons to maintain depolarization within physiological limits (Paradis et al., 2001).
Mechanisms identified to mediate homeostatic control in mature neurons are likely to have features in common with the regulation of electrical properties in differentiating embryonic neurons. Our previous observations show that Drosophila embryonic motoneurons regulate intrinsic membrane excitability when faced with changing synaptic excitation (Baines et al., 2001; Baines, 2003). At least part of this mechanism involves activity-dependent regulation of the cAMP-PKA pathway (Baines, 2003). PKA activity in these motoneurons is sufficient to reduce membrane excitability through a reduction in INa (Baines, 2003). Our present results indicate that activity is also sufficient to regulate mRNA abundance for the same voltage-gated Na+ channel. The genetic manipulations (dnc2 and overexpression of UAS-rut) that we use to increase synaptic excitation in the CNS probably do so through elevation of cAMP. Thus, a potential caveat to our use of these genetic manipulations is that the changes seen in para mRNA might not be a direct consequence of increased synaptic excitation but instead a consequence of increased cAMP. To counter this, we examined para mRNA in a third genetic background not predicted to influence cAMP directly, in which synaptic excitation of aCC/RP2 is increased. The mutation slamdanceiso7.8 (sda) encodes an aminopeptidase N, the loss of which is linked to induction of a seizure phenotype (Zhang et al., 2002). Synaptic excitation of aCC/RP2 is markedly increased in this genetic background, and analysis of para mRNA shows a significant reduction compared with WT (C. J. Mee and R. A. Baines, unpublished data). Together, our data are supportive of a model whereby exposure to synaptic excitation regulates INa in aCC/RP2 through activity-dependent changes in both activation of PKA (Baines, 2003) and abundance of para mRNA.
Posttranscriptional repression is a control mechanism of gene expression
The importance of RNA-binding proteins as regulators of cellular gene expression is underscored by the diverse roles that such proteins are implicated to control. In addition to the establishment of embryonic polarity (Tautz, 1988; Wharton and Struhl, 1991), posttranscriptional gene regulation is central to cell cycle control (Antic and Keene, 1997; Wang et al., 2000), neuronal differentiation (Blichenberg et al., 1999), cytokine expression (Atasoy et al., 1998), and DNA recombination (Hicks et al., 2000). In Drosophila, Pum is also implicated in the control of both long-term memory (Dubnau et al., 2003) and neuronal dendrite morphogenesis (Ye et al., 2004). Where studied, regulation is enabled by cis-regulatory sequences located in either the 3′- or 5′-UTRs of the transcript. The identities of these sequences are known to include the canonical NRE sequence recognized by Pum (Zamore et al., 1997) and a U-rich motif (U4-6A1-2U) designated the cytoplasmic polyadenylation element (Walker et al., 1996).
Our analysis of para mRNA shows an NRE-like sequence located in the 5′-UTR (our unpublished data) indicative that this mRNA might be subject to Pum-dependent translational repression. However, the presence of a cis-regulatory region homologous to the hb NRE motif is, by itself, insufficient evidence to implicate translational repression. To show this beyond doubt, a number of criteria must first be satisfied. These include a demonstration of specific binding of Pum to mRNA containing the cis-regulatory motif. Although gel-shift assays have been used to show an interaction between Pum and the 3′-UTR of hb (Zamore et al., 1997; Sonoda and Wharton, 1999), we have been unable, thus far, to show a strong interaction between Pum and the para NRE-like sequence. This lack of binding requires that we consider alternate mechanisms for the observed effect of Pum. One such mechanism could be that Pum is a transcriptional regulator of para, although there is no evidence from previous work to indicate that Pum has any other activities in addition to that of a translational repressor. Our observation of altered para mRNA after manipulation of pum is consistent with both transcriptional and translational mechanisms. This is because Pum-dependent translational repression of hb may also increase the rate of degradation of hb mRNA by removal of the poly(A) tail (Wharton and Struhl, 1991; Wreden et al., 1997; Chagnovich and Lehmann, 2001). Of course, Pum could regulate para mRNA through interaction with an intermediate factor. Clearly, additional studies are required to establish the true nature of this regulatory mechanism.
Persistent Na+ current influences neuronal excitability
Voltage-gated Na+ channels are major determinants of neuronal excitability and as such have been identified as a convergent locus for intracellular regulation through PKA- and PKC-dependent mechanisms (Li et al., 1992; Smith and Goldin, 1997; Catterall, 2000). In a majority of neurons examined, phosphorylation mediates a reduction in maximal conductance in INa that, in Drosophila, has been shown to be sufficient to downregulate membrane excitability in vivo (Catterall, 2000; Cantrell and Catterall, 2001; Baines, 2003). Analysis of INa in aCC/RP2 shows two distinct components, a fast transient current that inactivates rapidly and a smaller persistent current that slowly inactivates. Although the transient current is suited to the initiation of single spikes, its function is compromised if membrane potentials remain depolarized. Motoneurons, including those in Drosophila, produce plateau potentials that amplify and sustain their motor output (Li and Bennett, 2003; Rohrbough et al., 2003; Li et al., 2004). Persistent sodium and calcium currents are principal contributing conductances that underlie these plateau potentials (Li and Bennett, 2003; Li et al., 2004). In the light of this, it is satisfying that we observe regulation of INa(p) in a Pum-dependent manner. However, it is intriguing that, in the absence of pum (Pumbem), or when para is upregulated [Tp(1;2)r+75c], only INa(p) is increased, whereas INa(t) seemingly remains unchanged. This is even more puzzling when one considers that overexpression of pum is sufficient to downregulate both current components. That these two current components show differential regulation is suggestive that they may arise from different splice variants of para. Para is a highly complex gene with the capacity to produce multiple splice variants (Thackeray and Ganetzky, 1994, 1995). It is quite probable that these isoforms will, through differing kinetics and regulation, contribute to neuronal signaling in unique ways. Indeed, alternative splicing of exons a and i within the first intracellular loop is sufficient to alter INa(t) (INa(p) was not analyzed) (O'Dowd et al., 1995), and it is therefore not inconceivable that other isoforms will preferentially affect INa(p).
In mammals, the kinetics of INa are also reported to be influenced by subunit composition. For example, the presence of the Nav1.6 Na+ channel subunit, in rat Purkinje neurons, confers a greater degree of persistent Na+ current than in its absence (Raman et al., 1997). Thus, the increase in INa(p) that we observe in aCC/RP2 may be accounted for by additional regulatory mechanisms that alter the predominant splice variants (in lieu of changing subunit composition) of the functionally expressed channels. Regardless of the precise mechanism, application of ATX II shows clearly that an increase in INa(p) is sufficient to increase membrane excitability, whereas a reduction in total INa is sufficient to reduce excitability (Baines, 2003). In addition to Na+ conductance, membrane excitability is also dependent on K+ conductances and a complete understanding of how Pum regulates excitability in aCC/RP2 will require an analysis of how this protein regulates such conductances.
In summary, we present data to show that exposure to synaptic activity is able to regulate neuronal excitability in two identified Drosophila motoneurons through a Pum-dependent mechanism. That this mechanism is able to affect the abundance of para mRNA, together with the known function of this protein, implicates activity-dependent translational repression as a mechanism through which neurons maintain stability in firing when faced with changing synaptic excitation within the CNS.
Footnotes
This study was funded by the Wellcome Trust (R.A.B.). We are grateful to J. Jaynes and M. Fujioka for providing RN2 GAL4 flies. We thank B. Ganetzky, K. O'Dell, M. Stern, T. Zars, and the Bloomington Stock Center for providing flies.
Correspondence should be addressed to Dr. Richard A. Baines, Department of Biological Sciences, University of Warwick, Coventry CV4 7AL, UK. E-mail: RBaines@bio.warwick.ac.uk.
Copyright © 2004 Society for Neuroscience 0270-6474/04/248695-09$15.00/0
References
- Alzheimer C (1994) A novel voltage-dependent cation current in rat neocortical neurones. J Physiol (Lond) 479: 199-204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antic D, Keene JD (1997) Embryonic lethal abnormal visual RNA-binding proteins involved in growth, differentiation, and post-transcriptional gene expression. Am J Hum Genet 61: 273-278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atasoy U, Watson J, Patel D, Keene JD (1998) ELAV protein HuA (HuR) can redistribute between nucleus and cytoplasm and is up-regulated during serum stimulation and T cell activation. J Cell Sci 111: 3145-3156. [DOI] [PubMed] [Google Scholar]
- Baines RA (2003) Postsynaptic protein kinase A reduces neuronal excitability in response to increased synaptic excitation in the Drosophila CNS. J Neurosci 23: 8664-8672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baines RA, Bate M (1998) Electrophysiological development of central neurons in the Drosophila embryo. J Neurosci 18: 4673-4683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baines RA, Robinson SG, Fujioka M, Jaynes JB, Bate M (1999) Postsynaptic vesicle release is essential for synaptogenesis in Drosophila Curr Biol 9: 1267-1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baines RA, Uhler JP, Thompson A, Sweeney ST, Bate M (2001) Altered electrical properties in Drosophila neurons developing without synaptic transmission. J Neurosci 21: 1523-1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blichenberg A, Schwanke B, Rehbein M, Garner CC, Richter D, Kindler S (1999) Identification of a cis-acting dendritic targeting element in MAP2 mRNAs. J Neurosci 19: 8818-8829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broadie K, Prokop A, Bellen HJ, O'Kane CJ, Schulze KL, Sweeney ST (1995) Syntaxin and synaptobrevin function downstream of vesicle docking in Drosophila Neuron 15: 663-673. [DOI] [PubMed] [Google Scholar]
- Burrone J, Murthy VK (2003) Synaptic gain control and homeostasis. Curr Opin Neurobiol 13: 560-567. [DOI] [PubMed] [Google Scholar]
- Burrone J, O'Byrne M, Murthy VN (2002) Multiple forms of synaptic plasticity triggered by selective suppression of activity in individual neurons. Nature 420: 414-418. [DOI] [PubMed] [Google Scholar]
- Cantrell AR, Catterall WA (2001) Neuromodulation of Na+ channels: an unexpected form of cellular plasticity. Nat Rev Neurosci 2: 397-407. [DOI] [PubMed] [Google Scholar]
- Catterall WA (2000) From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron 26: 13-25. [DOI] [PubMed] [Google Scholar]
- Chagnovich D, Lehmann R (2001) Poly(A)-independent regulation of maternal hunchback translation in the Drosophila embryo. Proc Natl Acad Sci USA 98: 11359-11364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis GW, DiAntonio A, Petersen SA, Goodman CS (1998) Postsynaptic PKA controls quantal size and reveals a retrograde signal that regulates presynaptic transmitter release in Drosophila Neuron 20: 305-315. [DOI] [PubMed] [Google Scholar]
- Davis RL, Cherry J, Dauwalder B, Han PL, Skoulakis E (1995) The cyclic AMP system and Drosophila learning. Mol Cell Biochem 149/150: 271-278. [DOI] [PubMed] [Google Scholar]
- Desai NS, Rutherford L, Turrigiano GG (1999) Plasticity in the intrinsic excitability of cortical pyramidal neurons. Nat Neurosci 2: 515-520. [DOI] [PubMed] [Google Scholar]
- Dubnau J, Chiang AS, Grady L, Barditch J, Gossweiler S, McNeil J, Smith P, Buldoc F, Scott R, Certa U, Broger C, Tully T (2003) The staufen/pumilio pathway is involved in Drosophila long-term memory. Curr Biol 13: 286-296. [DOI] [PubMed] [Google Scholar]
- Dudai Y, Jan YN, Byers D, Quinn WG, Benzer S (1976) dunce, a mutant of Drosophila deficient in learning. Proc Natl Acad Sci USA 73: 1684-1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujioka M, Emi-Sarker Y, Yusibova GL, Goto T, Jaynes JB (1999) Analysis of an even-skipped rescue transgene reveals both composite and discrete neuronal and early blastoderm enhancers, and multi-stripe positioning by gap gene repressor gradients. Development 126: 2527-2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavis ER (2001) Over the rainbow to translational control. Nat Struct Biol 8: 387-389. [DOI] [PubMed] [Google Scholar]
- Hicks GG, Singh N, Nashabi A, Mai S, Bozek G, Klewes L, Arapovic D, White EK, Kroury MJ, Oltz EM, Van Kaer L, Ruley HE (2000) Fus deficiency in mice results in defective B-lymphocyte development and activation, high levels of chromosomal instability and perinatal death. Nat Genet 24: 175-179. [DOI] [PubMed] [Google Scholar]
- Hong CS, Ganetzky B (1994) Spatial and temporal expression patterns of two sodium channel genes in Drosophila J Neurosci 14: 5160-5169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie KR, Nelson SB, Turrigiano GG (2001) Postsynaptic depolarization scales quantal amplitude in cortical pyramidal neurons. J Neurosci 21: RC170(1-6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, West JW, Lai Y, Scheuer T, Catterall WA (1992) Functional modulation of brain sodium channels by cAMP-dependent phosphorylation. Neuron 8: 1151-1159. [DOI] [PubMed] [Google Scholar]
- Li Y, Bennett DJ (2003) Persistent sodium and calcium currents cause plateau potentials in motoneurons of chronic spinal rats. J Neurophysiol 90: 857-869. [DOI] [PubMed] [Google Scholar]
- Li Y, Gorassini MA, Bennett DJ (2004) Role of persistent sodium and calcium currents in motoneuron firing and spasticity in chronic spinal rats. J Neurophysiol 91: 767-783. [DOI] [PubMed] [Google Scholar]
- MacLean JN, Zhang Y, Johnson BR, Harris-Warrick RM (2003) Activity-independent homeostasis in rhythmically active neurons. Neuron 37: 109-120. [DOI] [PubMed] [Google Scholar]
- Mantegazza M, Franceschetti S, Avanzini G (1998) Anemone toxin (ATX II)-induced increase in persistent sodium current: effects on the firing properties of rat neocortical pyramidal neurons. J Physiol (Lond) 507: 105-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien RJ, Kamboj S, Ehlers MD, Rosen KR, Fischbach GD, Huganir RL (1998) Activity-dependent modulation of synaptic AMPA receptor accumulation. Neuron 21: 1067-1078. [DOI] [PubMed] [Google Scholar]
- O'Dowd DK, Germeraad SE, Aldrich RW (1989) Alterations in the expression and gating of Drosophila sodium channels by mutations in the para gene. Neuron 2: 1301-1311. [DOI] [PubMed] [Google Scholar]
- O'Dowd DK, Gee JR, Smith MA (1995) Sodium current density correlates with expression of specific alternatively spliced sodium channel mRNAs in single neurons. J Neurosci 15: 4005-4012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis S, Sweeney ST, Davis GW (2001) Homeostatic control of presynaptic release is triggered by postsynaptic membrane depolarization. Neuron 30: 737-749. [DOI] [PubMed] [Google Scholar]
- Raman IM, Sprunger LK, Meisler MH, Bean BP (1997) Altered subthreshold sodium currents and disrupted firing patterns in Purkinje neurons of Scn8a mutant mice. Neuron 19: 881-891. [DOI] [PubMed] [Google Scholar]
- Rohrbough J, O'Dowd DK, Baines RA, Broadie K (2003) Cellular bases of behavioral plasticity: establishing and modifying synaptic circuits in the Drosophila genetic system. J Neurobiol 54: 254-271. [DOI] [PubMed] [Google Scholar]
- Schweers BA, Walters KJ, Stern M (2002) The Drosophila melanogaster translational repressor pumilio regulates neuronal excitability. Genetics 161: 1177-1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RD, Goldin AL (1997) Phosphorylation at a single site in the rat brain sodium channel is necessary and sufficient for current reduction by protein kinase A. J Neurosci 17: 6086-6093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonoda J, Wharton RP (1999) Recruitment of Nanos to hunchback mRNA by Pumilio. Genes Dev 13: 2704-2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern M, Kreber R, Ganetzky B (1990) Dosage effects of a Drosophila sodium channel gene on behavior and axonal excitability. Genetics 124: 133-143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney ST, Broadie K, Keane J, Niemann H, O'Kane J (1995) Targeted expression of tetanus toxin light chain in Drosophila specifically eliminates synaptic transmission and causes behavioral defects. Neuron 14: 341-351. [DOI] [PubMed] [Google Scholar]
- Tautz D (1988) Regulation of Drosophila segmentation gene hunchback by two maternal morphogenetic centres. Nature 332: 281-284. [DOI] [PubMed] [Google Scholar]
- Thackeray JR, Ganetzky B (1994) Developmentally regulated alternative splicing generates a complex array of Drosophila para sodium channel isoforms. J Neurosci 14: 2569-2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thackeray JR, Ganetzky B (1995) Conserved alternative splicing patterns and splicing signals in the Drosophila sodium channel gene para Genetics 141: 203-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG (1999) Homeostatic plasticity in neural networks: the more things change, the more they stay the same. Trends Neurosci 22: 221-227. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Nelson SB (2000) Hebb and homeostasis in neuronal plasticity. Curr Opin Neurobiol 10: 358-364. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Nelson SB (2004) Homeostatic plasticity in the developing nervous system. Nat Rev Neurosci 5: 97-107. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Abbot LF, Marder E (1994) Activity-dependent changes in the intrinsic properties of cultured neurons. Science 264: 974-977. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, LeMasson G, Marder E (1995) Selective regulation of current densities underlies spontaneous changes in activity in cultured neurons. J Neurosci 15: 3640-3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG, Leslie KR, Desai NS, Rutherford LC, Nelson SB (1998) Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature 391: 892-896. [DOI] [PubMed] [Google Scholar]
- Walker J, Dale M, Standart N (1996) Unmasking mRNA in clam oocytes: role of phosphorylation of a 3′ UTR masking element-binding protein at fertilization. Dev Biol 11: 2510-2521. [DOI] [PubMed] [Google Scholar]
- Wang W, Caldwell MC, Lin S, Furneaux H, Gorospe M (2000) HuR regulates cyclin A and cyclin B1 mRNA stability during cell proliferation. EMBO J 19: 2340-2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AE, Griffith EC, Greenberg ME (2002) Regulation of transcription factors by neuronal activity. Nat Rev Neurosci 3: 921-931. [DOI] [PubMed] [Google Scholar]
- Wharton RP, Struhl G (1991) RNA regulatory elements mediate control of Drosophila body pattern by the posterior morphogen nanos. Cell 67: 955-967. [DOI] [PubMed] [Google Scholar]
- Wharton RP, Sonoda J, Lee T, Patterson M, Murata Y (1998) The Pumilio RNA-binding domain is also a translational repressor. Mol Cell 1: 863-872. [DOI] [PubMed] [Google Scholar]
- Wreden C, Verotti AC, Schisa JA, Lieberfarb ME, Strickland S (1997) Nanos and pumilio establish embryonic polarity in Drosophila by promoting posterior deadenylation of hunchback mRNA. Development 124: 3015-3023. [DOI] [PubMed] [Google Scholar]
- Ye B, Petritsch C, Clark IE, Gavis ER, Jan LY, Jan YN (2004) Nanos and Pumilio are essential for dendrite morphogenesis in Drosophila peripheral neurons. Curr Biol 14: 314-321. [DOI] [PubMed] [Google Scholar]
- Zamore PD, Williamson JR, Lehmann R (1997) The Pumilio protein binds RNA through a conserved domain that defines a new class of RNA-binding proteins. RNA 3: 1421-1433. [PMC free article] [PubMed] [Google Scholar]
- Zars T, Wolf R, Davis R, Heisenberg M (2000) Tissue-specific expression of a type I adenylyl cyclase rescues the rutabaga mutant memory defect: in search of the engram. Learn Mem 7: 18-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Tan J, Reynolds E, Kuebler D, Faulhaber S, Tanouye M (2002) The Drosophila slamdance gene: a mutation in an aminopeptidase can cause seizure, paralysis and neuronal failure. Genetics 162: 1283-1299. [DOI] [PMC free article] [PubMed] [Google Scholar]